TECHNISCHE UNIVERSITÄT MÜNCHEN II. Medizinische Klinik und Poliklinik des Klinikums rechts der Isar Die Bedeutung von Socs3 im in-vivo-Modell der pankreatischen Karzinogenese Thuy Trang Phan
Transcript
TECHNISCHE UNIVERSITÄT MÜNCHEN
II. Medizinische Klinik und Poliklinik
des Klinikums rechts der Isar
Die Bedeutung von Socs3 im in-vivo-Modell der pankreatischen Karzinogenese
Thuy Trang Phan
TECHNISCHE UNIVERSITÄT MÜNCHEN
II. Medizinische Klinik und Poliklinik
des Klinikums rechts der Isar
(Direktor: Univ.-Prof. Dr. Roland M. Schmid)
Die Bedeutung von Socs3 im in-vivo-Modell der pankreatischen Karzinogenese
Thuy Trang Phan
Vollständiger Abdruck der von der Fakultät für Medizin
der Technischen Universität München
zur Erlangung des akademischen Grades eines
Doktors der Medizin
genehmigten Dissertation.
Vorsitzender: Univ.-Prof. Dr. Ernst J. Rummeny
Prüfer der Dissertation: 1. Priv.-Doz. Dr. Hana Algül
2. Univ.-Prof. Dr. Helmut Friess
Die Dissertation wurde am 10.07.2014 bei der Technischen Universität München
eingereicht und durch die Fakultät für Medizin am 15.04.2015 angenommen.
Meinen Eltern gewidmet
INHALTSVERZEICHNIS
INHALTSVERZEICHNIS
I. ABKÜRZUNGEN ......................................................................................................... 3
II. EINLEITUNG ............................................................................................................... 5
1. Das Pankreaskarzinom ................................................................................................ 5
IV. ERGEBNISSE ............................................................................................................. 41
II. EINLEITUNG
2
1. Pankreasspezifische Inaktivierung von Socs3 in der Maus .................................... 41 1.1. Generierung einer pankreasspezifisch Socs3-defizienten Mauslinie Socs3
Δpanc unter Anwendung des Cre/loxP-Rekombinationssystems ..................................................... 41
1.2. Morphologische Charakterisierung des exokrinen und endokrinen Pankreaskompartiments von Soc3
4.1. Beschleunigung der PanIN-Progression in KrasG12D
;Socs3Δpanc-Mäusen .................... 56
4.1.1. Morphologische und quantitative Charakterisierung der PanIN-Läsionen ............... 56
V. DISKUSSION ............................................................................................................. 66
1. Rolle von Socs3 für die pankreatische Karzinogenese im KrasG12D-Mausmodell . 66
2. Einfluss von Socs3 auf die Apoptose und Proliferation im KrasG12D-Mausmodell
...................................................................................................................................... 68 3. Entzündliche Prozesse fördern die Initiierung und Progression der
pankreatischen Vorläuferläsionen zu duktalem Pankreaskarzinom .................... 71
4. Rolle von Socs3 bei der Pankreasfibrosierung/Desmoplasie in KrasG12D-
Insulinom, Gastrinom und Glukagonom treten selten auf (Mulkeen et al., 2006).
In diesem Abschnitt der Arbeit soll besonders auf das PDA und dessen Vorläuferläsionen
eingegangen werden. Begriffe wie Pankreaskarzinom, Pankreastumor und duktales
Adenokarzinom werden in diesem Zusammenhang synonym verwendet.
In den letzten Jahren haben klinische, histopathologische und molekulargenetische
Untersuchungen drei wichtige Typen von Vorläuferläsionen identifiziert, die sich über einen
schrittweisen Transformationsprozess zum duktalen Adenokarzinom entwickeln können. Zu
diesen Vorstufen gehören zum einen die muzinös-zystischen Neoplasien (MCN), die
intraduktalen papillär-muzinösen Neoplasien (IPMN) und zum anderen die pankreatisch
intraepithelialen Neoplasien (PanINs) (Brugge et al., 2004; Maitra et al., 2005).
Das auf molekularer und histopathologischer Ebene am besten untersuchte und charakterisierte
Tumorprogressionsmodell für das Pankreaskarzinom beruht auf der Entwicklung und
Progression der PanINs zum pankreatischen duktalen Adenokarzinom. Demnach werden drei
PanIN-Stadien klassifiziert, die je nach Schwere der zellulären und architektonischen Atypien
im Bereich der duktalen Strukturen aufsteigend eingestuft werden (PanIN-1 bis PanIN-3, siehe
Abbildung 1).
Abbildung 1: Morphologische Tumorprogression über PanIN-Vorläuferläsionen. Die H.E.-Färbung zeigt die unterschiedlichen PanIN-Stufen mit aufsteigenden zellulären und architektonischen Atypien. Das PanIN-1A-Stadium weist ein verlängertes Zylinderepithel auf, das im Stadium-1B in ein papilläres Wachstum übergeht. Moderate Kernatypien mit Polaritätsverlust, Hyperchromatismus, verminderte Mitoserate sind charakteristisch für Stadium PanIN-2. PanIN-3 Läsionen zeigen neben zunehmender Schwere der Kernatypien Abknospung von Epithelzellen und intraluminale Nekrosen (400-fache Vergrößerung). Der Stern kennzeichnet Azini.
PanIN-1A und PanIN-1B sind durch eine Zellkörperverlängerung und eine vermehrte
Schleimproduktion gekennzeichnet. Die im PanIN-1A-Stadium noch vorhandene flache
epitheliale Architektur des Pankreasganges geht im Stadium-1B in ein papilläres
Wachstumsmuster über. Erstaunlicherweise lassen sich diese frühesten Vorläuferformen in
40% aller adulten Pancreata nachweisen, ohne dass ein malignes Geschehen vorliegt. Das
PanIN-2-Stadium weist moderate Kernatypien in Form von Polaritätsverlust,
Hyperchromatismus, nukleärer Vergrößerung und verminderter Mitoserate auf, die im
Stadium-3 an Schwere zunehmen. Zusätzlich beobachtet man bei diesem terminalen
Läsionsgrad weit in das Ganglumen ragende epitheliale Zellknospen sowie luminale Nekrosen
als Zeichen von Zelluntergang und –abstoßung. PanIN-Läsionen der Stufe 3 findet man in der
Regel in weniger als 5% der gesunden Organe, während 30-50% aller invasiven Karzinome
diese typischen schweren Veränderungen aufzeigen.
Pankreasadenokarzinome weisen darüber hinaus ein an das Tumorgewebe angrenzendes
reaktives desmoplastisches Stroma auf. Dieses „Tumor-Microenvironment“ setzt sich aus einer
Vielzahl unterschiedlicher Zellen, extrazellulären Matrixproteinen und neugebildeten Gefäßen
zusammen, die das Wachstum und die Progression des PDAC fördern. Infiltrierende
inflammatorische Zellen und Makrophagen setzen Chemokine und Zytokine frei, die wiederum
eine reaktive Aktivierung von Fibroblasten und pankreatische Sternzellen (pancreatic stellate
cells, PSC) bewirken. Fibroblasten und aktivierte PSCs produzieren daraufhin Fibronektin und
Kollagen, was die Fibrosierung des Pankreasparenchyms verstärkt und das Tumorwachstum
begünstigt (Algül et al., 2007b).
Molekulare Untersuchungen haben gezeigt, dass die morphologischen Auffälligkeiten
einzelner PanIN-Progressionsstufen bis zum invasiven Karzinom bestimmte genetische
Veränderungen aufweisen, die entsprechend ihrer Anzahl und Schwere mit dem Grad der
Läsion korrelieren (siehe Abbildung 2). Wie bei anderen Tumorentitäten kommt in diesem
Zusammenhang Onkogenen und Tumorsuppressorgenen eine besondere Bedeutung zu
(Sakorafas et al., 2001).
II. EINLEITUNG
9
Abbildung 2: Morphologisches und genetisches Progressionsmodell des duktalen Pankreasadenokarzinoms (Hruban et al., 2000).
Die Kras-Punktmutation am Kodon 12 ist die wohl bedeutendste mit der Tumorgenese
assoziierte Mutation eines Onkogens. Initiale PanIN-Läsionen und Pankreasadenokarzinome
weisen eine Mutationsrate von bis zu 44% bzw. 100% auf (Shibata et al., 1990; Magee et al.,
2001). Das K-Ras-Protein stellt das Genprodukt von Kras dar und gehört zu den kleinen GTP-
bindenden Proteinen der Ras-Familie. Diese auch als G-Proteine genannten Moleküle sind an
der Innenseite der Plasmamembran lokalisiert und werden nach Bindung von Zytokinen,
Hormonen, Wachstumsfaktoren an die extrazelluläre Domäne der G-Protein-gekoppelten
Transmembranrezeptoren aktiviert. Hierbei kommt es zur Umwandlung von inaktivem, GDP-
gebundenen K-Ras in die aktive, GTP-assoziierte Form. Über nachgeschaltete intrazelluläre
Signaltransduktionswege wie zum Beispiel die Raf-mitogen-activated protein kinase (Raf-
MAP-Kinase)- oder die Phosphoinositol-3-Kinase-Signalkaskade wird der extrazelluläre
Stimulus in den Nucleus übertragen, wo eine Aktivierung weiterer Transkriptionsfaktoren
initiiert wird. Somit übernehmen die R-Proteine eine bedeutende Funktion in der
Zellproliferation, Differenzierung und Apoptose (Friday et al., 2008).
Bei Kras-Mutationen handelt es sich meist um eine Punktmutation eines einzelnen Nukleotids,
wodurch ein Aminosäureaustausch (Glycin wird durch Valin, Arginin oder Aspartat ersetzt)
stattfindet und infolge dessen ein Genprodukt mit Verlust der intrinsischen katalytischen
Eigenschaft entsteht. Aufgrund der Unfähigkeit zur Hydrolyse des gebundenen GTP bleibt das
„falsche“ G-Protein konstitutiv aktiv, sodass durch permanente Stimulierung der Downstream-
Kaskade kontinuierlich Signale an den Zellkern vermittelt werden, die zu dereguliertem
unkontrollierbaren Zellwachstum, Zellteilung und Differenzierung führen (Downward 2003).
II. EINLEITUNG
10
Molekulare Analysen haben eine signifikant verstärkte Amplifikation von Her2/neu mit
permanenter Aktivierung der entsprechenden Signaltransduktion und erhöhte EGF-
Konzentrationen beim PDA nachgewiesen. Bereits in frühen PanIN-Stadien ist die
Überexpression vorzufinden, sodass sie offenbar neben der Kras-Mutation zu den frühzeitig im
Ablauf der Tumorprogression auftretenden genetischen Veränderungen zählt (Lei et al., 1995;
Tsiambas et al., 2006; Hudis 2007).
Neben den Onkogenen werden die Tumorsuppressorgene als zweithäufigste mutierte
Genklasse im Pankreasadenokarzinom betrachtet. Mutationen an den jeweiligen Genloki
bewirken entweder eine Funktionsreduktion oder sogar einen vollständigen Funktionsverlust
der entsprechenden Genprodukte.
Zu den wichtigsten Vertretern der Tumorsuppressorgene gehören INK4A (inhibitor of cyclin
dependent kinase 4A) und ARF (Alternative Reading Frame), die sich beide auf der gleichen
Region 9p21 des Chromosoms 9 befinden. Das auch als CDKN2A (cyclin dependent kinase
inhibitor 2A) bezeichnete INK4A kodiert für das tumorsuppressive Protein p16, dessen
Inaktivierung in etwa 98% aller sporadischen Pankreaskarzinome auftritt (Schutte et al., 1997).
Über eine inhibitorische Bindung von p16 an die Cyklin-abhängigen Kinasen Cdk4 und Cdk6,
wodurch die funktionelle Phosphorylierung des Retinoblastom-Proteins verhindert wird,
kommt es zur Hemmung der Progression des Zellzyklus vor dem Eintritt in die S-Phase. Aus
der INK4A-Mutation resultiert konsequenterweise eine unkontrollierte Zellproliferation durch
Störungen in der Zellzyklusregulation. Der zweite vom gleichen Genlokus kodierte
Tumorsuppressor ist das Transkript der ARF-Gensequenz und wird als p19 bezeichnet. Die
Aufgabe dieses Proteins besteht in der Hemmung der MDM2-abhängigen Ubiquitinierung und
somit der Verhinderung des proteasomalen Abbaus von p53. Konsequenterweise führt die
Loss-of-function-Mutation des betroffenen Genabschnitts zur Reduktion des p19-Spiegels und
zum Abfall des p53-Levels (Wilentz et al., 1998; Lowe et al., 2003).
Das auf dem Chromosom 17 ansässige Tumorsuppressorgen TP53 kodiert einen weiteren für
die Kontrolle der Zellzyklus-Progression wichtigen „Wächter des Genoms“ p53. TP53 ist das
am häufigsten mutierte Gen in malignen Tumoren des Menschen. Beim Pankreaskarzinom
beträgt die Mutationsrate 50 bis 75%, wobei diese genetische Veränderung zumeist in
fortgeschrittenen PanIN-3-Läsionen vorzufinden ist (Talar-Wojnarowska et al., 2006). Das
Protein p53 stimuliert als Transkriptionsfaktor bei Replikationsstress mit Akkumulation von
gravierenden DNA-Schäden eine Reihe von Genen, die einen Zellzyklusarrest einleiten. Bei
Funktionsverlust des Proteins durch homozygote Mutation kommt es zum Ausfall der
tumorsuppressiven Eigenschaft des Genprodukts und das Wachstum von Zellen mit
II. EINLEITUNG
11
prokarzinogenen chromosomalen Aberrationen wird gefördert (Sherr 2004; Efeyan et al.,
2007).
Ein weiterer an der späteren Pankreastumorgenese beteiligter Tumorsuppressor ist SMAD4
bzw. DPC4 (deleted in pancreatic cancer locus 4), dessen Genprodukt SMAD4 bzw. DPC4
eine besondere Bedeutung in der Transforming Growth Faktor-ß (TGF-ß)-Signalkaskade
besitzt. Über dieses Messenger-Protein löst TGF-ß die Transkription spezieller Zielgene mit
antiproliferativer Wirkung aus. Infolgedessen kommt es bereits in der G1-Phase des Zellzyklus
zu einem Arrest, wodurch das Wachstum und die Differenzierung epithelialer Zellen zum
Stillstand kommen. Läsionen in diesem Genlokus führen zu unkontrollierter Proliferation
duktaler Zellen im Pankreas (Bardeesy et al. 2002; Miyaki et al., 2003). Ebenso sind
genetische Veränderungen des BRCA2-Gens in fortgeschrittenen Stadien der
Tumorprogression beschrieben worden. Eine Inaktivierung bedeutet Dysregulation der DNA-
Reparaturprozesse und daraus resultierende zelluläre Entartung (Hahn et al., 2003; Couch et
al., 2007).
2. Das murine Pankreaskarzinommodell
Trotz des zunehmenden Verständnisses der Signale und Mechanismen der Kanzerogenese
gehört das duktale Pankreasadenokarzinom zu den sehr schwer therapierbaren
Tumorerkrankungen. Deshalb hat sich die Pankreaskarzinom-Forschung zum Ziel gesetzt, die
humane Erkrankung und ihre Charakteristika in möglichst identischen präklinischen
Modellorganismen widerzuspiegeln und somit eine effizientere Therapie zu entwickeln.
Erstaunlicherweise lassen sich die molekularbiologischen und –genetischen Veränderungen
des humanen Pankreaskarzinoms und dessen Vorstufen in der Maus exakt nachbilden. Auf der
Grundlage dieser Erkenntnis wurden in den letzten Jahren einige Mausmodelle entwickelt, bei
denen mittels moderner molekularer Technik die Aktivierung von relevanten Onkogenen bzw.
das Ausschalten von Tumorsuppressorgenen ermöglicht wird (Hruban et al., 2006; Olive et al.,
2006). Zur Generierung genetisch gezielt modifizierter Mäuse bietet sich idealerweise das
Cre/loxP-Rekombinationssystem an, welches zur Etablierung des in dieser Arbeit verwendeten
KrasG12D-Mausmodells essentiell war. Demzufolge werden im Folgenden sowohl die Cre/loxP-
Technologie als auch das KrasG12D-Mausmodell ausführlich beschrieben.
II. EINLEITUNG
12
2.1. Das Cre/loxP-Rekombinationssystem
Die Rekombination ist definiert als ein durch spezifische Enzyme (Rekombinasen)
katalysierter Prozess der Spaltung und Neuverknüpfung der DNA. Auf diesem Prinzip
basierend ermöglicht die Cre/loxP-Technik eine gezielte Entfernung von DNA-Sequenzen in
relevanten Gewebe- oder Zellarten, ohne dass andere davon betroffen sind. Ein in der
molekulargenetischen Forschung häufig verwendetes Rekombinationsverfahren beruht auf der
Aktivität der Cre (cyclization recombination)-Rekombinase des Bakteriophagen P1. Dieses 38
kD schwere, in allen Organismen vorkommende Protein katalysiert die ortsspezifische
Rekombination zwischen zwei angrenzenden locus of x-over of P1 (loxP)-
Erkennungssequenzen. Das loxP-Motiv besteht aus einer 8 bp Spacer-Region, die wiederum
von zwei, jeweils 13 bp langen, invertierten Wiederholungen (inverted repeats) flankiert wird.
Die invers repetitiven Komponenten dienen der Erkennung und DNA-Bindung von Cre. Die
zwei eingebauten loxP-Sequenzen ermöglichen eine effiziente Exzision des loxP-flankierten
(„gefloxten“) DNA-Abschnitts. Das herausgeschnittene DNA-Segment wird als zirkuläres
Rekombinationsprodukt intrazellulär abgebaut. Lediglich verbleibt eine einzelne loxP-Sequenz
im modifizierten Genmaterial. Mittlerweile ist das Cre/loxP-Rekombinationssystem als eine
gängige molekulargenetische Methode zur Herstellung gewebespezifischer Knockout-Mäuse
etabliert. Zur Generierung dieser transgenen Tiere werden zwei genetisch veränderte
Mauslinien benötigt (siehe Abbildung 3).
Abbildung 3: Gewebespezifische Deletion von Mausgenen mittels Cre/loxP-Technologie. Der grüne Pfeil kennzeichnet den Vorgang der Transkription. In Zelltypen mit aktiver Cre-Rekombinase in der Cre/loxP-Maus ist die Genfunktion durch Exzision zerstört.
II. EINLEITUNG
13
Die Mauslinie 1 der F0-Generation trägt den gefloxten Genabschnitt. Hierzu werden die
gleichgerichteten loxP-Sequenzen vor und nach dem betroffenen Allel in die flankierenden
Intronbereiche integriert, somit bleibt das Zielexon funktionsfähig. Die Mauslinie 2 der
Parentalgeneration exprimiert die Cre-Rekombinase selektiv in bestimmten Geweben bzw.
Zelltypen. Zur Generierung der Cre-Maus wird ein Cre-Transgen in das Genom eingebracht.
Die Auswahl des vorgeschalteten Promotors, unter dessen Kontrolle das Cre-Transgen gestellt
wird, bestimmt über Zeitpunkt und Gewebespezifität der Cre-Expression. Daher wird die Cre-
Rekombinase nur von denjenigen Zellen gebildet, die auch über den entsprechenden Promotor
verfügen (Claudine 2004).
Nach Verpaarung der parentalen Cre-exprimierenden und gefloxten Mauslinien geht eine
Maus (Mauslinie 3) in der F1-Generation hervor, die beide genetische Veränderungen in ihrem
Erbgut trägt. Cre-Rekombinase exprimierende Zelltypen weisen eine Exzision des loxP-
markierten Zielexons und daraus resultierend eine Deletion des definierten Gens auf. Im
Gegensatz dazu bleibt in allen anderen Geweben ohne Cre-Transkription die entsprechende
Genfunktion unbeeinflusst. Somit lässt sich mithilfe der Cre/loxP-basierenden Methode ein
gewebespezifischer Gen-Knockout in der Mausgenetik etablieren.
Eine weitere Bedeutung gewinnt das Cre/loxP-System in der Induktion gewebespezifischer
Mutationen über die Verwendung loxP-flankierter STOP-Kassetten. Diese Art der
Genommodifikation wurde im KrasG12D-Mausmodell zur Herstellung onkogener Kras-
Mutationen angewendet und wird im folgenden Abschnitt detailliert beschrieben.
2.2. Das Kras
G12D-Mausmodell
Mit der Entwicklung des LSL-KrasG12D Mausmodells für das PDA gelang dem Forscherteam
unter David Tuveson und Tyler Jacks die Etablierung eines der bedeutendsten murinen
pankreatischen Karzinommodelle, welches auch in dieser Arbeit eine wesentliche Stellung
einnimmt. Grundidee dieses Tiermodells ist die vom endogenen Kras-Locus ausgehende
heterozygote pankreasspezifische Aktivierung des mutierten Kras(G12D)-Onkogens. Über die
Transfektion eines Plasmids in einen Mausstamm wurde das Kras-Allel so verändert, sodass
dieses ein modifiziertes Exon 1 mit einer Guanin-zu-Adenin-Transition auf dem Kodon 12
(G12D) und folglich einen Glycin-Aspartat-Aminosäureaustausch im Genprodukt aufweist
(Jackson et al., 2001). Unter Anwendung des bereits beschriebenen Cre/loxP-
Rekombinationssystems wird die gezielte Expression des veränderten Kras-Allels spezifisch in
Pankreaszellen erreicht. Hierzu wird dem das Kodon 12 enthaltende Exon 1 eine Lox-STOP-
Lox (LSL)-Sequenz vorgeschaltet (siehe Abbildung 4). Dieser LSL-Konstrukt verhindert die
II. EINLEITUNG
14
Transkription des veränderten Strukturgens und somit die Überexpression des onkogenen K-
Ras-Proteins in der LSL-KrasG12D-Mauslinie. Um eine Kras
G12D-Aktivierung selektiv im
Pankreas vorzunehmen, wird die LSL-KrasG12D-Maus mit einer Mauslinie gekreuzt, in der die
Cre-Rekombinase unter pankreassspezifischem Promotor Pdx-1 oder als Knock-in heterozygot
durch Entfernung des Exons 1 in einen Ptf1a-Lokus eingebaut exprimiert wird. Die daraus
resultierende Mauslinie bezeichnet man als Ptf1a-Creex1 bzw. Pdx1-Cre (Hingorani et al.,
2003a; Nakkai et al., 2007).
Pdx1 kodiert für den Transkriptionsfaktor Pankreatischer und duodenaler Homebox Faktor 1
(Pdx1), der auch als Insulin-Promotor-Faktor 1 bezeichnet und ab dem Entwicklungstag 8,5
produziert wird. Eine homozygote Deletion von Pdx1 endet letal in der Embryonalperiode. Das
von Ptf1a-Genbereich kodierte Ptf1a-Protein, welches auch als p48 genannt wird, stellt die
Untereinheit des heterotrimerischen Pankreastranskriptionsfaktorkomplex 1 dar (Rose et al.,
2001). P48 wird ab Tag 9,5 der Embryonalentwicklung exprimiert und ist gemeinsam mit Pdx1
für die pankreasspezifische Differenzierung der Zellen verantwortlich. Das Vorhandensein
beider Faktoren ist essenziell für die morphologische und funktionelle Entwicklung der
Vorläuferzellen zu Pankreasgewebszellen, wobei erwachsene Mäuse Pdx1 zur Bildung der
Inselzellen und p48 zur Initiierung der Azinuszelldifferenzierung benötigen (Rose et al., 2001;
Beres et al., 2006). Demzufolge führt die vollständige Deletion der p48-Gene zu einer
Agenesie des exokrinen Pankreas und konsequenterweise zum Tod der Mäuse früh postnatal
(Krapp et al., 1998; Sellik et al., 2004).
Nach Kreuzung der transgenen Mauslinien LSL-KrasG12D und Ptf1-Cre
ex1 bzw. Pdx1-Cre geht
nun eine Mausmutante LSL-Kras+/G12D;Ptf1a-Cre
ex1 bzw. LSL-Kras+/G12D ;Pdx1-Cre hervor,
die eine Heterozygotie für das KrasG12D-Allel aufweist. Aufgrund der Ptf1a- bzw. Pdx1-Cre-
Rekombinase assoziierten Exzision der loxP-flankierten STOP-Sequenz in diesem auch als
KrasG12D bezeichneten Tumormausmodell entfällt die transkriptionsinhierende Funktion der
STOP-Kassette, was wiederum zur deregulierten hochfrequenten Transkription des mutierten
Exon 1 führt (siehe Abbildung 4).
II. EINLEITUNG
15
Abbildung 4: Systematik der Generierung des Kras
G12D-Tumormausmodells. Grüne Pfeile beschreiben die Transkription der Strukturgene. Roter Balken kennzeichnet die transkriptionsinhibierende Wirkung der STOP-Kassette (modifiziert nach Hingorani et al 2003).
Mithilfe der LSL-KrasG12D Mauslinie ist die selektive endogene Expression des onkogenen K-
Ras-Proteins in Ptf1a bzw. Pdx1 positiven und Cre-Rekombinase positiven
Pankreasgewebszellen möglich. Somit kann die Tumorentwicklung pankreasspezifisch initiiert
und die Entstehung des humanen Pankreaskarzinoms exakt in der Maus rekapituliert werden.
Die genetisch veränderten KrasG12D-Mäuse zeigen daraufhin alle Stadien der PanIN-Läsionen,
die analog zum humanen PDA in ihrer Anzahl und Schwere mit dem Alter der Mäuse
zunehmen. In 10% der Fälle und nach einer Latenz von etwa neun Monaten entwickeln sich
auch invasive und metastasierende Karzinome. Der Nachweis der Kras-Mutation bei allen
PanIN-Stufen in den Progenitorzellen bestärkt dessen Bedeutung in der Induktion und
Progression der Pankreaskarzinogenese. Bei LSL-KrasG12D;Pdx1-Cre-Mäusen treten zusätzlich
andere Tumoren mit variierender Penetranz auf, wie die intestinale Hyperplasie des
Magenepithels, monokutane Papillome und hyperplastische Polypen des Duodenums. Die
simultane Existenz dieser Malignome beweist die embryonale Expression von Pdx1 in
endothelialen Vorläuferzellen (Rose et al., 2001). Ausgehend von diesem etablierten KrasG12D-
Tumormausmodell sind verschiedene komplexe Tiermodellsysteme entwickelt worden, die
additiv zu dem mutierten Kras-Allel andere genetische Veränderungen aufzeigen. Damit lässt
sich der Einfluss einzelner Genmutationen auf die Karzinomentwicklung und –progression
genauer analysieren und mögliche Ansatzpunkte für ein gezieltes medizinisches Eingreifen
finden.
II. EINLEITUNG
16
3. Suppressors of cytokine signaling (SOCS)
Die Mitglieder der SOCS-Familie stellen wichtige Feedback-Inhibitoren des Zytokin-
Signaltransduktionswegs dar. Mittlerweile sind acht SOCS-Proteine (CIS, Socs1-Socs7)
beschrieben worden, die auf ein Zytokinsignal gewebeabhängig exprimiert werden. Von diesen
auch als STAT-induzierte STAT-Inhibitoren (SSI) bezeichneten negativen Regulatoren der
Zytokin-Signalkaskade sind die Proteine Socs1, Socs2 und Socs3 besonders gut charakterisiert
und nehmen eine wichtige Stellung in der Interleukin-6 (IL-6) Signalkaskade ein. Im
Gegensatz dazu sind Socs4 bis Socs7 bislang noch wenig erforscht (Krebs et al., 2000).
Aufgrund der tragenden Rolle von Socs3 in dieser Arbeit, soll im Folgenden besonders auf
diesen inhibitorischen Regulationsfaktor eingegangen werden.
3.1. Die Struktur von Socs3
Socs3 stellt ein aus 225 Aminosäuren (AS) bestehendes Molekül dar, das aus unterschiedlichen
Domänen zusammengesetzt ist (vergleiche Abbildung 5). Die einzelnen Abschnitte erfüllen im
Rahmen der Signaltransduktion bestimmte Funktionen, die in ihrer Gesamtheit für den
negativen Feedback-Mechanismus des Socs3-Proteins von großer Bedeutung sind. Die N-
terminale Kinase-Inhibitor-Region (KIR) bindet als Pseudosubstrat an das aktive Zentrum von
Janus-Kinasen (Jak), wodurch das Andocken nativer Substrate blockiert wird. Eine zentrale
Stellung nimmt die SH2 (Scr-homology-2)-Domäne im Socs3-Molekül ein. Diese bildet die
eigentliche Kinase-Bindungsstelle, die eine hohe Affinität zu phosphorylierten Tyrosinresten
im Aktivierungsloop der Jaks aufweist. Unterstützt wird die SH2-Sequenz von den beiden
flankierenden N-ESS (extended SH2 subdomain) und C-ESS-Domänen, die strukturell die
Assoziation und die Affinität von SH2 an die Tyrosinmotive erhöht. Am C-terminalen Ende
der SH2-Domäne liegt eine PEST-Insertion zwischen zwei konservierten Sekundär-
Strukturelementen. Untersuchungen haben gezeigt, dass dieses PEST-Motiv als wichtiger
Regulator der Proteinstabilität fungiert und seine Entfernung weder zu einer
Strukturveränderung führt noch die Funktion von Socs3 beeinträchtigt. Jedoch zeigen PEST-
Knockout Mäuse eine Zunahme der Socs3-Halbwertszeit durch Beeinflussung des Proteasom-
vermittelten Abbaumechanismus (Babon et al., 2006). Die für die hemmende Wirkung von
Socs3 ebenfalls wichtige SOCS-Box-Region ist C-terminal lokalisiert (siehe Abbildung 5).
II. EINLEITUNG
17
Abbildung 5: Schematische Darstellung der Socs3-Architektur. Die Socs3-Struktur besteht aus einem kleinen N-Terminus, einer Kinase-Inhibitor-Region (KIR), einer SH2-Domäne und einer C-terminalen SOCS-Box. Die zentrale SH2-Domäne setzt sich aus 141 Aminosäuren (AS) zusammen und wird durch ein aus 35 AS bestehendes PEST-Motiv unterbrochen. N-terminal bzw. C-terminal wird die SH2-Domäne durch eine N-ESS- bzw. C-ESS-Sequenz flankiert (modifiziert nach Babon et al. 2006).
Durch Rekrutierung von Elongin B, Elongin C, Cullin 2 und das Ringfinger-Protein Rbx1 an
der SOCS-Box bildet sich ein E3-Ubiquitin-Ligase-Komplex (siehe Abbildung 6), in dem
Socs3 als Adaptormolekül die Ubiquitination und Degradation von bestimmten an der
Signaltransduktion beteiligten Proteinen fördert (Kamura et al., 1998; Kamura et al. 2004).
Abbildung 6: Socs3 als Trägermolekül des E3-Ubiquitin-Ligase-Komplexes (modifiziert nach Krebs et al. 2000).
3.2. Socs3 als Zytokin-Signaltransduktionsinhibitor
Die intrazelluläre Socs3-Proteinkonzentration ist unter normalen Umständen relativ gering.
Für die Aktivierung des auf Chromosom 11 liegenden Socs3-Gens (entspricht dem humanen
Socs3-Gen auf Chromosom 17) ist die Interaktion von Zytokinen besonders der Interleukin
(IL)-6-Familie, Wachstumsfaktoren wie den Insulin-like growth factor oder Hormonen mit
den spezifischen Zelloberflächenrezeptoren essentiell. Innerhalb der Untergruppen zeigen die
Zytokine ähnliche Molekül- und Rezeptorstrukturen. Stellvertretend soll an dieser Stelle der
IL-6-Rezeptor und dessen anschließende Signaltransduktionswege näher erläutert werden.
Der IL-6-Rezeptor verfügt über zwei Untereinheiten. Durch Bindung an das extrazelluläre
Zytokin-bindende Modul, das auch als Glykoprotein (gp)180 bekannt ist, übt IL-6 seine
biologische Aktivität aus. Intrazellulär bilden die ubiquitär exprimierten gp130-
Rezeptoruntereinheiten die signalvermittelte Struktur durch Aktivierung der Signalkaskade
II. EINLEITUNG
18
und stellen gleichzeitig Ziel verschiedener Rückkopplungsmechanismen dar. Weitere
Charakteristika des IL-6-Rezeptors sind die fehlende intrinsische katalytische
Tyrosinkinaseaktivität und die Verankerung in der Plasmamembran über eine singuläre
Transmembrandomäne. Bei Interaktion des Liganden IL-6 mit der extrazellulären
Rezeptorkomponente kommt es zunächst zur Aktivierung rezeptorassoziierter, intrazellulärer
Janus-Kinasen (Jak) durch Autophosphorylierung. Daraufhin phosphorylieren die aktivierten
Jaks die zytosolischen Zytokinrezeptoranteile am Tyrosinrest 759, der als Bindungsstelle
sowohl für diverse Transkriptionsfaktoren wie Stat3 als auch Inhibitoren wie Socs3-Moleküle
fungiert. Die Rezeptor-Ligand-Interaktion setzt somit den Jak/Stat-Signalweg ebenso wie die
PI3K- und die Ras/Raf/MAPK-Signalkaskade in Gang.
Die als Monomere im Zytoplasma lokalisierten Stat3-Moleküle binden an die gp130-
Untereinheit und werden anschließend Jak-vermittelt am Tyrosin 705 phosphoryliert. Die
aktivierten phospho(p)-Stat3-Elemente lagern sich dann zu Homo-oder Heterodimeren
zusammen, die in den Zellkern translozieren und dort an Enhancer-Sequenzen in Promotoren
von IL-6-Zielgenen binden und folglich deren Transkriptionsfrequenz positiv modulieren
(siehe Abbildung 7). In vielen malignen Tumoren kommt es durch eine Hyperaktivität der
Rezeptor-Tyrosinkinasen zu einer konstitutiven Phosphorylierung und Aktivierung von Stat3
und folglich zu einer Überexpression der an verschiedenen physiologischen Prozessen wie
Proliferation, Apoptose, Angiogense, Inflammation beteiligten Zielgene. Dies führt zur
Dysregulation des Zellzyklus und der Zellfunktion, was wiederum das Wachstum und
Überleben transformierter Zellen begünstigt und die Karzinomentwicklung fördert.
Abbildung 7: Die Mechanismen der Aktivierung der IL-6/Jak/Stat3-Signalkaskade und dessen Feedback-Inhibition durch Socs3.
II. EINLEITUNG
19
Die Aktivierung der Ras/Raf/MAPK-Kaskade setzt die Bildung eines Molekülkomplexes
EDTA pH 8,0 HEPES pH 7,9 NaCl Glycerin NP-40 und kurz vor der Lyse supplementieren mit: DTT PMSF Protease Inhibitor Cocktail Phosphatase Inhibitor Cocktail I Phosphatase Inhibitor Cocktail II
1 mM 50 mM 150 mM 10 % 0.5 % 1 mM 0,2 mM 1 % 1 % 1 %
1.2. Morphologische Charakterisierung des exokrinen und endokrinen Pankreaskompartiments von Soc3
Δpanc-Mäusen
Nach dem molekulargenetischen Nachweis der homozygoten Socs3-Deletion in Socs3Δpanc-
Mäusen mittels PCR interessierte uns die Auswirkung dieser Cre-Rekombinase vermittelten
DNA-Mutation auf die Morphologie und die physiologische Funktionalität des exokrinen und
endokrinen Pankreaskompartiments. Hierzu fertigten wir Paraffin-Schnitte aus isolierten
Pankreata von vier Wochen alten Socs3F/F-und Socs3
Δpanc-Mäusen an.
Die H.E.-Färbung der Schnitte diente zur anatomischen Abgrenzung und Differenzierung der
unterschiedlichen Gewebekomponente. Um relevante Proteine zu detektieren, setzten wir die
Technik der Immunhistochemie ein (siehe Abbildung 12).
Abbildung 12: Charakterisierung des exokrinen und endokrinen Pankreaskompartiments. Morphologische und immunhistochemische Analysen des Pankreas vier Wochen alter Socs3
Die Ergebnisse zeigen für die Mauslinie KrasG12D eine deutlich erhöhte mRNA-Expression
des endogenen Inhibitors Socs3 gegenüber der der Kontrolltiere. Konsequenterweise führt die
gesteigerte mRNA-Bildung zu einer verstärkten Socs3-Proteinbiosynthese an den Ribosomen.
Im Folgenden überprüften wir die Expression von Socs3 in der KrasG12D-Mauslinie auf
histochemischer Ebene. Wie Abbildung 16 demonstriert, konnten wir immunhistochemisch
mithilfe eines gegen Socs3 gerichteten Primärantikörpers dessen vermehrte Bildung in Azini-
Zellen und PanIN-Läsionen neun Wochen alter KrasG12D-Mäuse nachweisen. Somit legt die
starke Induktion des endogenen Inhibitors eine regulierende Funktion des Socs3 in der
pankreatischen Onkogenese nahe.
Abbildung 16: Immunhistochemische Detektion von Socs3 in Zellen der Azini (schwarze Pfeile) und in den PanIN-Läsionen (weiße Pfeile) neun Wochen alter Kras
G12D-Mäuse.
IV. ERGEBNISSE
49
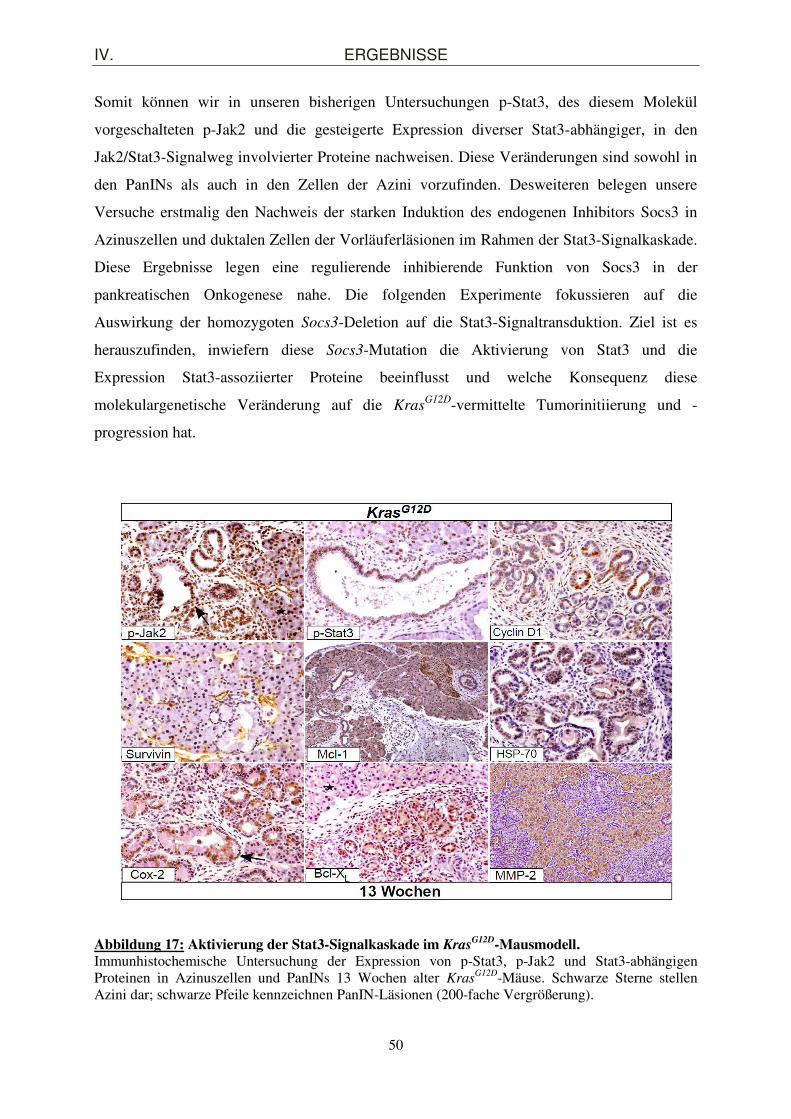
2.3. Aktivierung der an der Jak2/Stat3-Signaltransduktion beteiligten Proteine in Kras
G12D-Mäusen
Neben der Induktion des endogenen Inhibitors Socs3 regulieren phosphorylierte Stat3-Dimere
intranukleär die Transkription bestimmter Gene, die an der Steuerung von Proliferation,
Wachstum, Apoptose und Inflammation beteiligt sind. Der Nachweis der exprimierten
Genprodukte lässt auf die Aktivierung des Jak2/Stat3-Signaltransduktionsweges
rückschließen. Hierzu setzen wir erneut die Methode der Immunhistochemie ein und
detektieren die relevanten Proteine. Das zu färbende Pankreasgewebe stammte von 13
Wochen alten KrasG12D-Mäusen und offenbart histologisch PanIN-Vorläuferläsionen (siehe
Abbildung 17). Charakteristisch für die pankreatischen intraepithelialen Neoplasien in diesem
Wochenalter ist die Zunahme der Anzahl der Ausführungsgänge, eine Verlängerung des
epithelialen Zellkörpers der Ducti und eine erhöhte zytoplasmatische Muzinproduktion. Hinzu
geht das normalerweise flach dominierende Ausführungsgang-Epithel in ein papilläres
Wachstumsmuster über. Diese auf Abbildung 17 aufgeführten Eigenschaften weisen auf
PanIN-Stufen 1A bis 1B hin, die zusätzlich ein umgebendes desmoplasmatisches Stroma
aufweisen. Sowohl in den Azinuszellen als auch in den Pankreasgangzellen innerhalb der
PanIN-Läsionen lässt sich das phosphorylierte Jak2-Protein (p-Jak2) nachweisen. Das aktive
p-Jak2 phosphoryliert das Stat3-Monomer am Tyrosinrest Y705 zu p-Stat3 (Babon et al.,
2006). Dementsprechend konnten wir, wie Abbildung 17 veranschaulicht, sowohl in den
Azini als auch in den duktalen Zellen eine erhöhte p-Stat3-Konzentration detektieren.
Darüber hinaus ist es uns gelungen, die Expression Stat3-abhängiger Proteine in den
präneoplastischen Läsionen und in den Azinuszellen 13 Wochen alter KrasG12D-Mäuse zu
demonstrieren. Hierzu zählen Cyclin D1, Survivin, die antiapoptisch wirkenden und zur
Proteinfamilie der Bcl-2 (B-cell lymphoma-2) gehörigen Mitglieder Mcl-1 (Myloid cell
lymphoma-1) und Bcl-XL. Zusätzlich ließen sich andere Moleküle bei unseren
immunhistochemischen Untersuchungen detektieren, wie die Matrix Metalloproteinase-2
(MMP-2), welche im Abbau der extrazellulären Matrix involviert ist und somit für den
Prozess der Metastasierung eine entscheidende Rolle spielt und das proinflammatorische
Protein Cox-2 (Cyclooxygenase-2). Auch die Expression des von uns nachgewiesenen
Hitzeschockproteins-70 (heat shock protein 70, HSP-70) ist Stat3-abhängig und wird durch
dessen Aktivierung gesteigert. HSP-70 stabilisiert in Situationen des zellulären Stresses
andere Eiweißstrukturen, um diese vor Denaturierung und Abbau zu schützen. Andererseits
unterstützt HSP-70 die proteasomale Degradation funktionsunfähiger Proteinmoleküle.
IV. ERGEBNISSE
50
Somit können wir in unseren bisherigen Untersuchungen p-Stat3, des diesem Molekül
vorgeschalteten p-Jak2 und die gesteigerte Expression diverser Stat3-abhängiger, in den
Jak2/Stat3-Signalweg involvierter Proteine nachweisen. Diese Veränderungen sind sowohl in
den PanINs als auch in den Zellen der Azini vorzufinden. Desweiteren belegen unsere
Versuche erstmalig den Nachweis der starken Induktion des endogenen Inhibitors Socs3 in
Azinuszellen und duktalen Zellen der Vorläuferläsionen im Rahmen der Stat3-Signalkaskade.
Diese Ergebnisse legen eine regulierende inhibierende Funktion von Socs3 in der
pankreatischen Onkogenese nahe. Die folgenden Experimente fokussieren auf die
Auswirkung der homozygoten Socs3-Deletion auf die Stat3-Signaltransduktion. Ziel ist es
herauszufinden, inwiefern diese Socs3-Mutation die Aktivierung von Stat3 und die
Expression Stat3-assoziierter Proteine beeinflusst und welche Konsequenz diese
molekulargenetische Veränderung auf die KrasG12D-vermittelte Tumorinitiierung und -
progression hat.
Abbildung 17: Aktivierung der Stat3-Signalkaskade im Kras
G12D-Mausmodell. Immunhistochemische Untersuchung der Expression von p-Stat3, p-Jak2 und Stat3-abhängigen Proteinen in Azinuszellen und PanINs 13 Wochen alter Kras
3. Homozygote Socs3-Deletion führt zu einer konstitutiven Aktivierung des Stat3-Signalweges im Kras
G12D-Mausmodell
3.1. Generierung der pankreasspezifisch Socs3-defizienten Tumormodellmaus Kras
G12D;Socs3Δpanc
Um der Bedeutung von Socs3 auf die KrasG12D-vermittelte Tumorinitiierung und -progression
genauer nachzugehen, generierten wir zunächst eine Socs3-defiziente KrasG12D-Mauslinie.
Hierzu benötigten wir erneut zwei Mauslinien: die bereits unter I.2.2. beschriebene LSL-
KrasG12D;Ptf1a-Cre
ex1 (KrasG12D)-Tumormaus und die unter III.1.1. charakterisierte
Socs3F/F
;Ptf1a-Creex1 (Socs3
Δpanc)-Mauslinie. Nachdem wir beide Mauslinien miteinander
gekreuzt haben, gingen Nachkommen hervor, die einen Genotyp LSL-KrasG12D
;Socs3F/F
;
Ptf1a-Creex1 aufwiesen. Diese neu entstandene auch als Kras
G12D;Socs3
Δpanc bezeichnete
Tumormauslinie besaß sowohl das aktive pankreasspezifische Onkogen KrasG12D, welches die
Entwicklung und Progression der PanINs förderte, als auch die Ptf1a-Cre-vermittelte
homozygote Deletion des Exons 2 des Socs3-Gens, die zu einer fehlenden Expression von
Socs3 führte. Somit haben wir mit der Etablierung des neuen KrasG12D
;Socs3Δpanc-
Tumormausmodells die Basis für die daran anschließenden Versuche geschaffen. Hierin galt
es die Charakteristika der von uns generierten KrasG12D
;Socs3Δpanc-Maus mit den der nicht
Socs3-defizienten KrasG12D-Kontrollmaus zu vergleichen und so die Bedeutung bzw. die
Relevanz des Inhibitorproteins Socs3 auf die KrasG12D-vermittelte Entwicklung
pankreatischer präneoplastischer Läsionen und die Stat3-Phosphorylierung zu untersuchen.
3.2. Proteinbiochemische und morphologische Charakterisierung von Kras
G12D;Socs3
Δpanc-Mäusen
3.2.1. Proteinbiochemischer Nachweis verstärkter Stat3-Phosphorylierung und Expression Stat3-abhängiger Proteine
Der Tyrosinrest 759 an der zytosolischen gp130-Untereinheit stellt im phosphorylierten
Zustand die Andockstelle für die Stat3-Monomere und Socs3-Moleküle dar. Gebunden an
diesem aktiven Phosphotyrosin werden die Stat3-Proteine anschließend Jak2-vermittelt am
Tyrosin 705 zu p-Stat3 umgewandelt. Das phosphorylierte Tyrosin 759 ist zugleich
Bindungsort von dem endogenen Inhibitor Socs3, an dem dieser den inhibierenden Feedback-
Mechanismus auf den Stat3-Signaltransduktionsweg ausübt (Lang et al., 2000).
Ausgehend von diesen Vorkenntnissen untersuchten wir im folgenden Schritt die Auswirkung
der pankreasspezifischen Socs3-Defizienz auf die Induktion von p-Stat3 und den Stat3-
abhängigen Proteinen in KrasG12D
;Socs3Δpanc-Mäusen. Hierzu verglichen wir die Western
IV. ERGEBNISSE
52
Blot-Analysen der Socs3-defizienten KrasG12D-Maus mit den der Kras
G12D-Kontrollmaus. Die
von uns im Rahmen der Experimente verwendeten Antikörper sind in Tabelle 7 aufgelistet.
Zunächst überprüften wir proteinbiochemisch die Expression von Stat3, p-Stat3 und p-Jak2 in
beiden Mauslinien. Wie Abbildung 18 demonstriert, exprimieren sowohl vier Wochen alte
KrasG12D
;Socs3Δpanc-Mäuse als auch gleich alte Kras
G12D-Kontrolltiere Stat3 und p-Stat3.
Jedoch weist das Pankreasgewebe der KrasG12D
;Socs3Δpanc-Mäuse eine frühzeitige und
prolongierte Stat3-Phosphorylierung auf, was an dem hohen Proteinlevel von p-Stat3 zu
erkennen ist. Auch die Phosphorylierung der dem Stat3-Molekül vorgeschalteten Jak2 ist
ebenso frühzeitig nachweisbar.
Desweiteren interessierte uns die Auswirkung der Socs3-Defizienz auf die Expression Stat3-
abhängiger Proteine. Wie die Western Blot-Analysen in der Abbildung 18 veranschaulichen,
führt die frühzeitige und prolongierte Aktivierung von p-Stat3 ebenfalls zu einer gesteigerten
Expression diverser Stat3-Zielproteine in den exemplarisch dargestellten vier Wochen alten
KrasG12D
;Socs3Δpanc-Mäusen im Vergleich zu den Kras
G12D-Kontrolltumormäusen. Für Bcl-
XL, Mcl-1, Cox-2, HSP-70 und Cyclin D1 weisen die Socs3-deletierten KrasG12D-Mäuse ein
höheres Proteinlevel auf. ß-actin wurde hier als Ladekontrolle eingesetzt.
A B
Abbildung 18: Proteinbiochemischer Nachweis der Socs3-Defizienz in Kras
G12D;Socs3
Δpanc-Mäusen. A: Nachweis der frühzeitigen und prolongierten Stat3-Phosphorylierung im Pankreasgewebe vier Wochen alter Socs3-defizienter Kras
G12D-Mäuse. B: Verstärkte Expression Stat3-abhängiger Proteine in Western Blot-Analysen vier Wochen alter Kras
G12D;Socs3
Δpanc-Mäuse.
IV. ERGEBNISSE
53
3.2.2. Konstitutive Aktivierung des onkogenen K-Ras-Proteins in Kras
G12D;Socs3
Δpanc-Mäusen
Das G-Protein K-Ras bindet im aktiven Zustand Guanosintriphosphat (GTP). Durch die
intrinsische Hydrolyseaktivität wird das GTP-gebundene K-Ras bei Terminierung des
Ligandensignals in die inaktive GDP-assoziierte Form umgewandelt. Bei der KrasG12D-
Mutation liegt eine ungenügende Überführung von GTP-korreliertem K-Ras in die inaktive
GDP-gebundene Konfiguration, wodurch eine konstitutive, wachstumsfördernde K-Ras-
Proteinaktivität entsteht (Shibata et al., 1990). Da in den Socs3-defizienten KrasG12D-Mäusen
sowohl eine KrasG12D-Mutation als auch eine Socs3-Deletion vorliegt, überprüften wir
mithilfe des Ras-Aktivierungsassays mit dem Ras Activation Assay Kit, ob die Inaktivierung
von Socs3 einen Einfluss auf die Ausprägung der G12D-Mutation ausübt. Das Prinzip dieser
eingesetzten Methode beruht auf der hochspezifischen Bindung der Ras-bindenden Domäne
(RBD) des im Kit enthaltenen Proteins Raf-1 (Ras effector kinase) an aktives GTP-
korreliertes Ras-Protein. Nach Herstellung der Proteinlysate aus Pankreasgewebe beider
Mauslinien wurden diese jeweils mit dem Raf-1 RBD Agarose enthaltenden Reaktionsreagenz
versetzt. Anschließend ließ sich der entstandene Raf-RBD/GTP-Ras Komplex im Western
Blot mit einem konjugierten Ras-spezifischen Sekundärantikörper detektieren. Die Western
Blot-Ergebnisse des durchgeführten Ras Pull-Down Assays in der Abbildung 19 zeigen für
beide vier Wochen alte KrasG12D- und Kras
G12D;Socs3
Δpanc-Mauslinien eine annähernd gleich
starke Expression von RBD- und damit von GTP-assoziiertem Ras-Protein im Pankreas. Aus
unseren gewonnenen Daten kann demnach geschlossen werden, dass die pankreasspezifische
Inaktivierung des Socs3-Gens keinen Einfluss auf die Ras-Aktivität bzw. auf die Expression
des konstitutiv aktiven K-Ras-Proteins ausübt. Beide genetische Veränderungen existieren
nebeneinander und scheinen synergistisch die Progression von pankreatischen
intraepithelialen Neoplasien zum duktalen Adenokarzinom zu verstärken.
Abbildung 19: Nachweis der Expression konstitutiv aktiver K-Ras-Proteine. In beiden Mauslinien liegt eine unveränderte Aktivierung des onkogenen K-Ras-Proteins vor.
IV. ERGEBNISSE
54
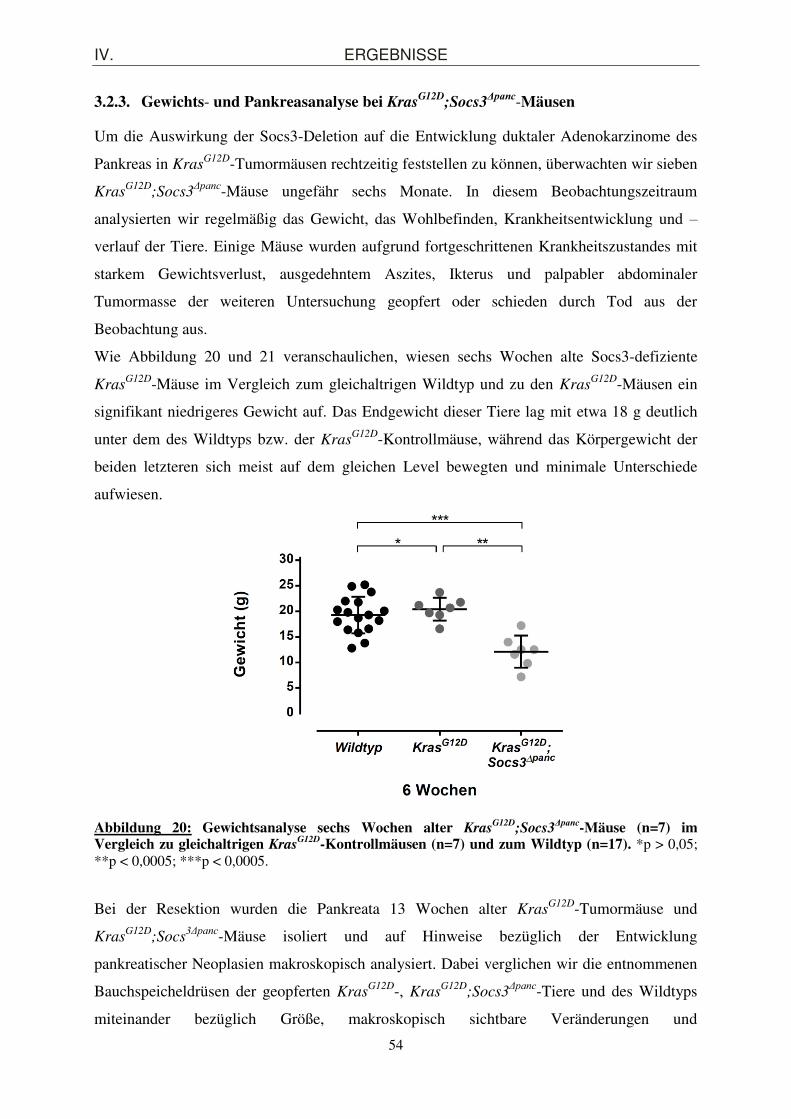
3.2.3. Gewichts- und Pankreasanalyse bei KrasG12D
;Socs3Δpanc-Mäusen
Um die Auswirkung der Socs3-Deletion auf die Entwicklung duktaler Adenokarzinome des
Pankreas in KrasG12D-Tumormäusen rechtzeitig feststellen zu können, überwachten wir sieben
KrasG12D
;Socs3Δpanc-Mäuse ungefähr sechs Monate. In diesem Beobachtungszeitraum
analysierten wir regelmäßig das Gewicht, das Wohlbefinden, Krankheitsentwicklung und –
verlauf der Tiere. Einige Mäuse wurden aufgrund fortgeschrittenen Krankheitszustandes mit
starkem Gewichtsverlust, ausgedehntem Aszites, Ikterus und palpabler abdominaler
Tumormasse der weiteren Untersuchung geopfert oder schieden durch Tod aus der
Beobachtung aus.
Wie Abbildung 20 und 21 veranschaulichen, wiesen sechs Wochen alte Socs3-defiziente
KrasG12D-Mäuse im Vergleich zum gleichaltrigen Wildtyp und zu den Kras
G12D-Mäusen ein
signifikant niedrigeres Gewicht auf. Das Endgewicht dieser Tiere lag mit etwa 18 g deutlich
unter dem des Wildtyps bzw. der KrasG12D-Kontrollmäuse, während das Körpergewicht der
beiden letzteren sich meist auf dem gleichen Level bewegten und minimale Unterschiede
aufwiesen.
Abbildung 20: Gewichtsanalyse sechs Wochen alter KrasG12D
;Socs3Δpanc-Mäuse (n=7) im
Vergleich zu gleichaltrigen KrasG12D-Kontrollmäusen (n=7) und zum Wildtyp (n=17). *p > 0,05;
**p < 0,0005; ***p < 0,0005.
Bei der Resektion wurden die Pankreata 13 Wochen alter KrasG12D-Tumormäuse und
KrasG12D
;Socs3Δpanc-Mäuse isoliert und auf Hinweise bezüglich der Entwicklung
pankreatischer Neoplasien makroskopisch analysiert. Dabei verglichen wir die entnommenen
Bauchspeicheldrüsen der geopferten KrasG12D-, Kras
G12D;Socs3
Δpanc-Tiere und des Wildtyps
miteinander bezüglich Größe, makroskopisch sichtbare Veränderungen und
IV. ERGEBNISSE
55
Gewebebeschaffenheit. Die Abbildung 21 demonstriert ein im Vergleich zum Wildtyp stark
vergrößertes Pankreas der KrasG12D-Tumormaus. Zusätzlich lassen sich Lobulierungen und
Indurationen im Organgewebe nachweisen.
Die zusätzliche Socs3-Defizienz in KrasG12D-Tumormäusen bewirkt jedoch eine starke
Abnahme des Organgewichts (siehe Abbildung 21).
Abbildung 21: Analyse des makroskopischen Pankreasaspekts. Größe, Form und Konsistenz der Pankreata 13 Wochen alter Kras
G12D; Socs3
Δpanc-Mäuse und KrasG12D-Kontrollmäuse im Vergleich
zum Wildtyp. Zum Größenvergleich ist aus jeder Mauslinie ein Tier exemplarisch dargestellt.
Abbildung 21 demonstriert ein im Vergleich zum Wildtyp bis auf ein Fünftel der Organmasse
geschrumpfte Bauchspeicheldrüse mit diffus nodulärer und lobulierter Parenchymstruktur.
Die ausgedehnte Fibrosierung des sonst weichen Gewebes lässt das Organ derb, knotig und
formlos erscheinen. Diese makroskopisch sichtbaren Veränderungen an den Pankreata der
von uns generierten KrasG12D
;Socs3Δpanc-Mauslinie deuten auf vorhandene präneoplastische
und/oder neoplastische Prozesse hin, die sich scheinbar in weiter fortgeschrittenen Stadien
befinden als die der gleichaltrigen Mäuse mit singulärer KrasG12D-Mutation.
Im Beobachtungszeitraum von sechs Monaten entwickelten 28,57% (2/7) der
KrasG12D
;Socs3Δpanc-Mäuse ein Pankreasadenokarzinom, während in der Kras
G12D-Kohorte
keine Karzinomentwicklung zu beobachten war. Die Tumorlatenz von etwa 63 Tagen bei den
KrasG12D
;Socs3Δpanc-Versuchstieren war somit geringer im Vergleich zu den Kras
G12D-
Kontrolltieren.
Aus unseren bisher gewonnen Daten können wir damit annehmen, dass die
pankreasspezifische Deletion des Socs3-Gens und die dadurch aufgehobene negative
Rückkopplung auf den Stat3-Signalweg einen maßgeblichen Effekt auf die Tumorprogression
in der KrasG12D-induzierten pankreatischen Onkogenese haben könnte. Die im Folgenden
4. Einfluss der Inaktivierung des endogenen Inhibitorproteins Socs3 auf die PanIN-Progression im Kras
G12D-Mausmodell
4.1. Beschleunigung der PanIN-Progression in KrasG12D
;Socs3Δpanc-Mäusen
4.1.1. Morphologische und quantitative Charakterisierung der PanIN-Läsionen
In der vergleichenden Analyse der PanIN-Entwicklung in Pankreata von KrasG12D- und
KrasG12D
;Socs3Δpanc-Mäusen im folgenden Abschnitt möchten wir den Einfluss der aus der
Inaktivierung des Inbibitorproteins Socs3 fehlenden Hemmung des Stat3-Signalweges auf die
alterskorrelierte PanIN-Progression in Socs3-defizienten KrasG12D-Mäusen näher beleuchten.
Zunächst untersuchten wir anhand H.E.-gefärbter Gewebefeinschnitte vier, neun, 13 und 18
Wochen alter KrasG12D-Kontrollmäuse und Kras
G12D;Socs3
Δpanc-Mäuse die
Pankreasmorphologie im Vergleich. Die Ergebnisse dieser Übersichtsfärbung sind in zehn-
und 100-facher Vergrößerung in der Abbildung 22 dargestellt.
IV. ERGEBNISSE
57
Abbildung 22: Morphologische Charakterisierung der PanINs und des PDA. H.E.-Übersichtsfärbungen der Pankreasgewebefeinschnitte vier, neun, 13 und 18 Wochen alter Tiere bei zehn- und 100-facher Vergrößerung. Schwarze Pfeile kennzeichnen PanIN-Läsionen; der weiße Pfeil hebt einen reaktiven Ausführungsgang hervor; der weiße Stern markiert regelrechte Azini; der schwarze Stern kennzeichnet das PDA.
KrasG12D-Tumormodellmäuse weisen zum Zeitpunkt von vier Wochen ein nahezu
unauffälliges Pankreas auf. Zwischen den normal konfigurierten Azini sind die
Ausführungsgänge schmal und selten sichtbar. Vereinzelt können wir reaktive
Ausführungsgänge und PanIN-1-Läsionen darstellen. Die Lobuli werden von unauffälligen
Bindegewebssepten separiert, die keine Entzündungszellen enthalten. Das Pankreas der neun
Wochen alten Tiere offenbart bereits bei zehnfacher Vergrößerung suspekte Areale, die diffus
im ganzen Organ lokalisiert vorliegen. Diese weißen Salzkorn-artige Läsionen lassen die
betroffene Bauchspeicheldrüse durchlöchert erscheinen. Bei höherer Vergrößerung eines
betroffenen Bereiches erkennen wir eine zunehmende Anzahl reaktiver Gangstrukturen und
früher Stadien von PanIN-Läsionen. Die jetzt zwischen den basophilen Azinuzellen deutlich
sichtbaren Ausführungsgänge mit stark eosinophilen duktalen Zellen sind erweitert und
treiben die Lobuli auseinander. Die zunehmende Anzahl histologisch erkennbarer Ducti mit
IV. ERGEBNISSE
58
Formanomalie lässt auf einen neoplastischen duktalen Prozess hindeuten. Zudem weist das
vermehrte desmoplastische Stroma auf eine verstärkte Entzündungsreaktion mit fibrotischer
Begleitreaktion hin. Mit zunehmendem Alter nimmt sowohl die Dichte als auch die Schwere
der PanIN-Läsionen zu. Die zehnfache Vergrößerung des Pankreas der 13 Wochen alten
KrasG12D-Kontrollmaus veranschaulicht, dass in diesem Stadium ein dominierender Organteil
von den PanINs eingenommen ist und die zunehmende Ausdehnung dieser eine Verdrängung
der exokrinen Azini bedeutet. Bei 100-facher Vergrößerung erkennen wir im Vergleich zur
neun Wochen alten Maus Areale mit übermäßig dilatierten Gangstrukturen, die statt dem
normal flachen Epithel ein papilläres Wachstumsmuster offenbaren. Zusätzlich bewirkt die
raumfordernde Expansion des umgebenden desmoplastischen Stromas ein Auseinanderdriften
der Azini. Um die 18. Lebenswoche zeigt die 10-fach vergrößerte Pankreasaufnahme der
Mäuse mit KrasG12D-Mutation eine weitere Progression der PanIN-Dichte und des
Schweregrades der Neoplasien. Wie Abbildung 22 verdeutlicht, erstrecken sich
präneoplastische Läsionen und das Stroma über fast das ganze Organ mit daraus
resultierendem deutlichem Rückgang der Azinuszellen. In der höheren Vergrößerung
erkennen wir zusätzlich intraluminale Pankreasgangnekrosen als Charakteristikum der PanINs
im Stadium 3 und atrophierte Azinuszellen. Die exemplarisch dargestellten Pankreata der
KrasG12D-Mäuse unterschiedlichen Alters zeigen somit die für diese Tumormauslinie typische
zeit- bzw. altersabhängige PanIN-Progression.
Die Socs3-Defizienz in KrasG12D
-Mäusen lässt in den H.E.-gefärbten Gewebefeinschnitten
eine Beschleunigung der PanIN-Progression erkennen. Im Vergleich zu gleichaltrigen
KrasG12D-Mäusen ist das Pankreas dieser Mäuse interessanterweise schon nach vier Wochen
morphologisch sehr stark umgewandelt. Bereits bei 10-facher Vergrößerung demaskieren sich
duktale Vorläuferläsionen aufgrund starker Gangdilatation, die annähernd die Hälfte des
Organs eingenommen haben. Während Pankreata vier Wochen alter KrasG12D-Mäuse nahezu
unauffällig erscheinen, fallen bei näherer histologischer Betrachtung der Gewebestruktur
Areale auf. Neben normalen Azinis und reaktiven Gängen sind ausgeprägte hochgradig
veränderte Ausführungsgänge in Form von PanIN-1- bis PanIN-3-Läsionen zu erkennen. Die
schon in der vierten Lebenswoche eingetretenen Veränderungen der Gangstruktur deuten auf
ein im Vergleich zu der KrasG12D-Tumormauslinie sehr früh einsetzendes neoplastisches
duktales Geschehen mit ausgedehnter fibrotischer Begleitreaktion hin.
Um das Ausmaß der PanIN-Progression bei pankreasspezifischer Inaktivierung von Socs3
noch genauer beurteilen zu können, führten wir anschließend einen quantitativ analytischen
IV. ERGEBNISSE
59
Vergleich der Gangstrukturen in Pankreata vier Wochen alter KrasG12D
;Socs3Δpanc-Mäuse und
KrasG12D-Kontrollmäuse durch. Hierfür bestimmten wir zunächst mittels 200-fach
vergrößerter pankreatischer Gewebeschnitte die Gesamtzahl der Ausführungsgänge bei
beiden Mauslinien.
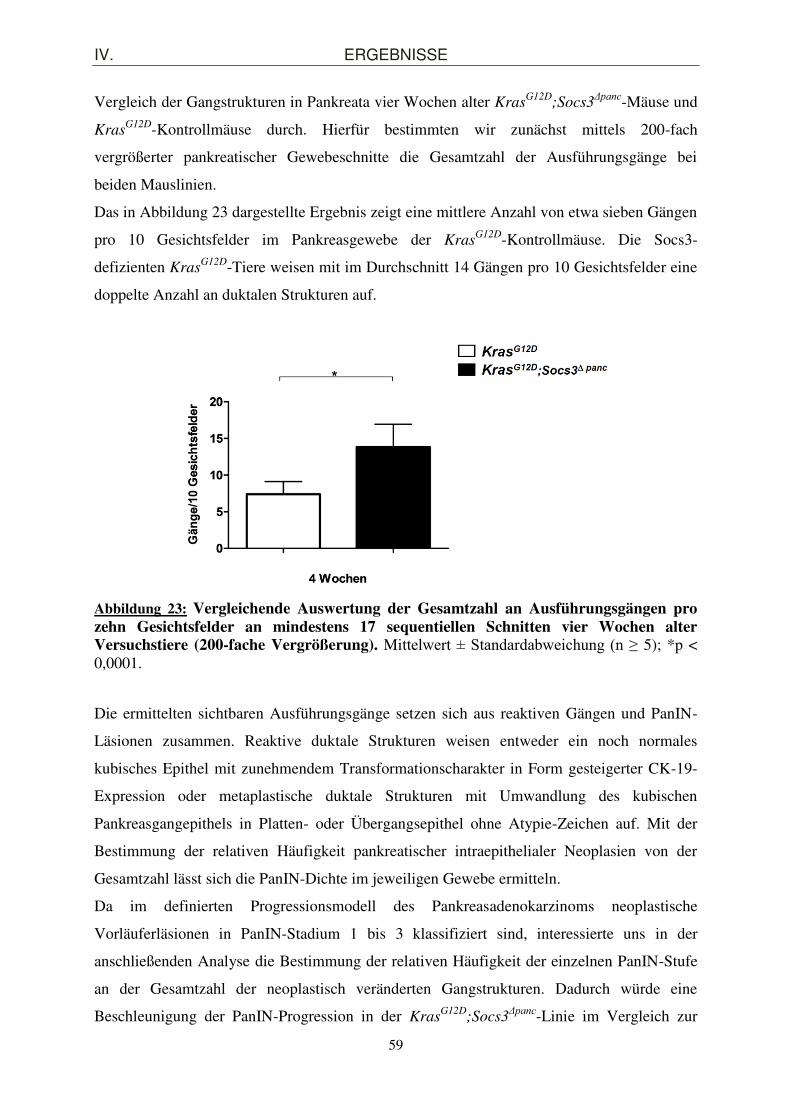
Das in Abbildung 23 dargestellte Ergebnis zeigt eine mittlere Anzahl von etwa sieben Gängen
pro 10 Gesichtsfelder im Pankreasgewebe der KrasG12D-Kontrollmäuse. Die Socs3-
defizienten KrasG12D-Tiere weisen mit im Durchschnitt 14 Gängen pro 10 Gesichtsfelder eine
doppelte Anzahl an duktalen Strukturen auf.
Abbildung 23: Vergleichende Auswertung der Gesamtzahl an Ausführungsgängen pro zehn Gesichtsfelder an mindestens 17 sequentiellen Schnitten vier Wochen alter Versuchstiere (200-fache Vergrößerung). Mittelwert ± Standardabweichung (n ≥ 5); *p < 0,0001.
Die ermittelten sichtbaren Ausführungsgänge setzen sich aus reaktiven Gängen und PanIN-
Läsionen zusammen. Reaktive duktale Strukturen weisen entweder ein noch normales
kubisches Epithel mit zunehmendem Transformationscharakter in Form gesteigerter CK-19-
Expression oder metaplastische duktale Strukturen mit Umwandlung des kubischen
Pankreasgangepithels in Platten- oder Übergangsepithel ohne Atypie-Zeichen auf. Mit der
Bestimmung der relativen Häufigkeit pankreatischer intraepithelialer Neoplasien von der
Gesamtzahl lässt sich die PanIN-Dichte im jeweiligen Gewebe ermitteln.
Da im definierten Progressionsmodell des Pankreasadenokarzinoms neoplastische
Vorläuferläsionen in PanIN-Stadium 1 bis 3 klassifiziert sind, interessierte uns in der
anschließenden Analyse die Bestimmung der relativen Häufigkeit der einzelnen PanIN-Stufe
an der Gesamtzahl der neoplastisch veränderten Gangstrukturen. Dadurch würde eine
Beschleunigung der PanIN-Progression in der KrasG12D
;Socs3Δpanc-Linie im Vergleich zur
IV. ERGEBNISSE
60
KrasG12D-Linie bestätigen lassen. Hierzu wurden erneut 200-fach vergrößerte pankreatische
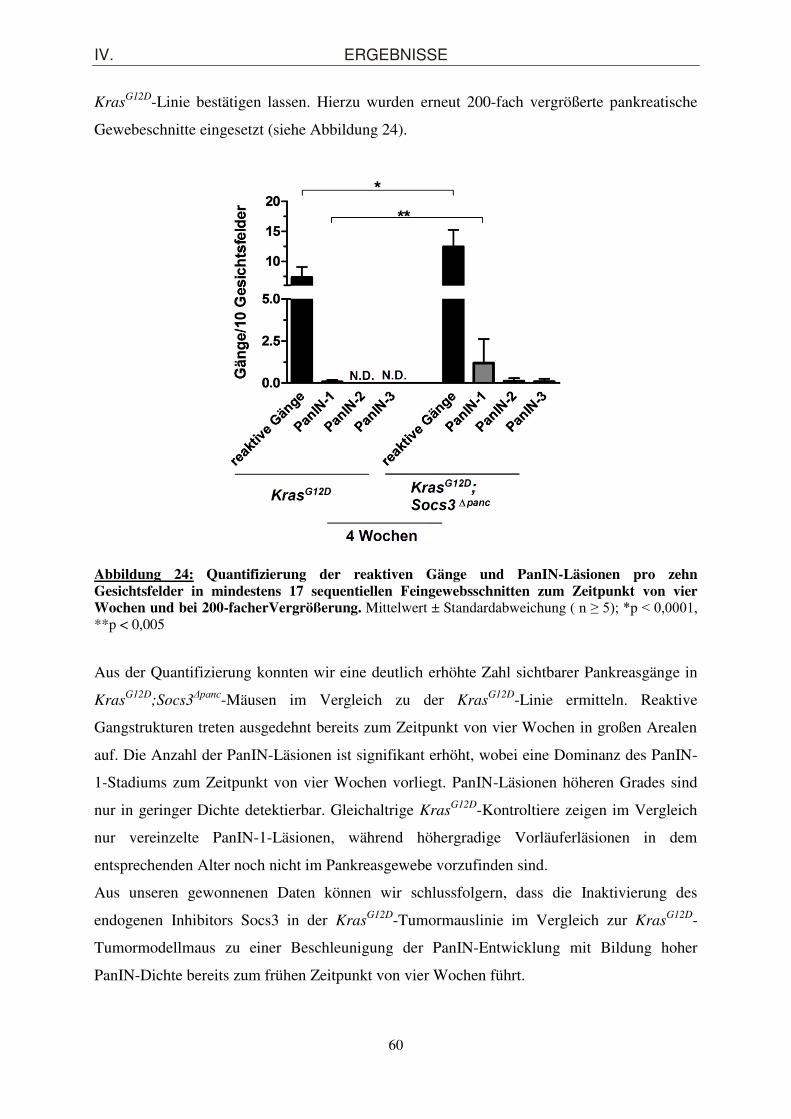
Gewebeschnitte eingesetzt (siehe Abbildung 24).
Abbildung 24: Quantifizierung der reaktiven Gänge und PanIN-Läsionen pro zehn Gesichtsfelder in mindestens 17 sequentiellen Feingewebsschnitten zum Zeitpunkt von vier Wochen und bei 200-facherVergrößerung. Mittelwert ± Standardabweichung ( n ≥ 5); *p < 0,0001, **p < 0,005
Aus der Quantifizierung konnten wir eine deutlich erhöhte Zahl sichtbarer Pankreasgänge in
KrasG12D
;Socs3Δpanc-Mäusen im Vergleich zu der Kras
G12D-Linie ermitteln. Reaktive
Gangstrukturen treten ausgedehnt bereits zum Zeitpunkt von vier Wochen in großen Arealen
auf. Die Anzahl der PanIN-Läsionen ist signifikant erhöht, wobei eine Dominanz des PanIN-
1-Stadiums zum Zeitpunkt von vier Wochen vorliegt. PanIN-Läsionen höheren Grades sind
nur in geringer Dichte detektierbar. Gleichaltrige KrasG12D-Kontroltiere zeigen im Vergleich
nur vereinzelte PanIN-1-Läsionen, während höhergradige Vorläuferläsionen in dem
entsprechenden Alter noch nicht im Pankreasgewebe vorzufinden sind.
Aus unseren gewonnenen Daten können wir schlussfolgern, dass die Inaktivierung des
endogenen Inhibitors Socs3 in der KrasG12D-Tumormauslinie im Vergleich zur Kras
G12D-
Tumormodellmaus zu einer Beschleunigung der PanIN-Entwicklung mit Bildung hoher
PanIN-Dichte bereits zum frühen Zeitpunkt von vier Wochen führt.
IV. ERGEBNISSE
61
Immunhistochemische Zusatzfärbungen mit gegen CK-19, p-Stat3 und Cyclin D1 gerichteten
Primärantikörpern bestätigen die bereits unter III.3.2.1. beschriebene verstärkte Aktivierung
von p-Stat3 und des Stat3-abhängigen Proteins Cyclin D1 in den Azinuszellen, reaktiven
Gängen und PanIN-Läsionen bei der Socs3-defizienten KrasG12D
;Socs3Δpanc-Mauslinie (siehe
Abbildung 25).
Abbildung 25: Immunhistochemischer Nachweis der übermäßigen Expression von CK-19, p-Stat3 und Cyclin D1 in reaktiven Gängen und PanIN-Läsionen vier Wochen alter Kras
G12D;Socs
Δpanc-Mäuse. 100-fache Vergrößerung
Die intensive Expression des Intermediärfilaments CK-19 als Epithelzellmarker ist
charakteristisch für einen malignen duktalen Prozess im Rahmen der PanIN-Progression. In
der weiteren Entwicklung nimmt die Anzahl besonders der höhergradigen Läsionen zu. Hinzu
treten fokale Karzinome auf, die von ausgeprägter fibrotischer Umwandlung des sonst
weichen Pankreasgewebes begleitet werden.
Die 10-fach vergrößerte Übersichtsfärbung der exemplarisch gewählten neun Wochen alten
KrasG12D
;Socs3Δpanc-Maus zeigt ein an Größe abnehmendes Pankreas, welches stark
aufgelockert erscheint. Mehr als die Hälfte des betroffenen Organs ist von neoplastischen
Vorläuferläsionen höheren Grades mit luminaler Nekrose befallen. Morphologisch normale
Azinuszellen sind zugunsten der Karzinomexpansion verdrängt und nur noch vereinzelt
gruppiert detektierbar. In der 13. Lebenswoche weisen die Pankreata der erkrankten Tiere nur
noch ein Drittel der Organgröße gleichaltriger KrasG12D-Kontrollmäuse auf. Das Gewebe wird
IV. ERGEBNISSE
62
weiterhin neoplastisch verändert bis in der 18. Lebenswoche der größte Teil der
Bauchspeicheldrüse vom invasiv duktalen Adenokarzinom (PDA) eingenommen wird.
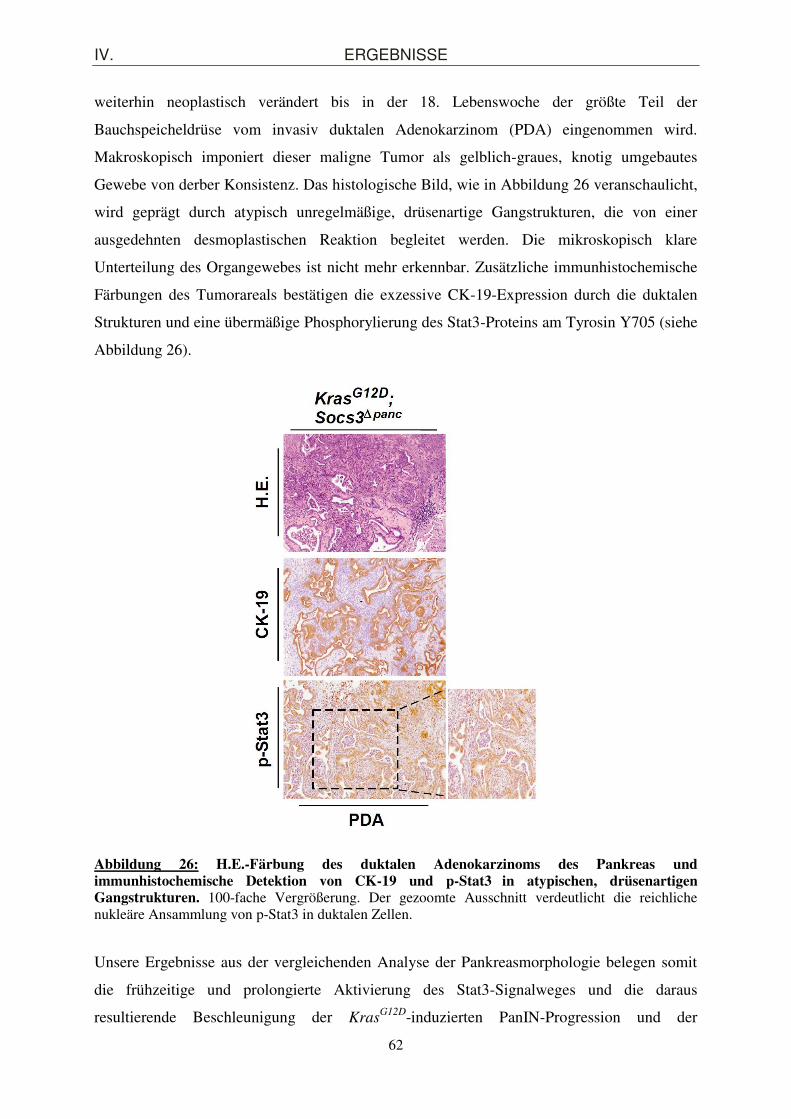
Makroskopisch imponiert dieser maligne Tumor als gelblich-graues, knotig umgebautes
Gewebe von derber Konsistenz. Das histologische Bild, wie in Abbildung 26 veranschaulicht,
wird geprägt durch atypisch unregelmäßige, drüsenartige Gangstrukturen, die von einer
ausgedehnten desmoplastischen Reaktion begleitet werden. Die mikroskopisch klare
Unterteilung des Organgewebes ist nicht mehr erkennbar. Zusätzliche immunhistochemische
Färbungen des Tumorareals bestätigen die exzessive CK-19-Expression durch die duktalen
Strukturen und eine übermäßige Phosphorylierung des Stat3-Proteins am Tyrosin Y705 (siehe
Abbildung 26).
Abbildung 26: H.E.-Färbung des duktalen Adenokarzinoms des Pankreas und immunhistochemische Detektion von CK-19 und p-Stat3 in atypischen, drüsenartigen Gangstrukturen. 100-fache Vergrößerung. Der gezoomte Ausschnitt verdeutlicht die reichliche nukleäre Ansammlung von p-Stat3 in duktalen Zellen.
Unsere Ergebnisse aus der vergleichenden Analyse der Pankreasmorphologie belegen somit
die frühzeitige und prolongierte Aktivierung des Stat3-Signalweges und die daraus
resultierende Beschleunigung der KrasG12D-induzierten PanIN-Progression und der
IV. ERGEBNISSE
63
Entstehung fokaler Karzinome in KrasG12D-Tumormodellmäusen mit Inaktivierung des
Inhibitorproteins Socs3. Diese Tiere zeigen pankreasmorphologisch bereits in der vierten
Lebenswoche neben harmlosen reaktiven Gängen alle Stadien der PanIN-Läsionen, die sich
rasch zu fokalen Karzinomen mit ausgedehnter fibrotischer Umwandlung entwickeln,
während Pankreata KrasG12D-Kontrollmäuse gleichen Alters nahezu regelrecht normal
erscheinen. Aus unseren bisherigen Daten kann somit geschlossen werden, dass Socs3 eine
wichtige Funktion als Tumorsuppressor im Rahmen der pankreatischen Onkogenese ausübt
und über die Regulation der Stat3-Phosphorylierung die PanIN-Progression in
Um die Veränderungen der duktalen Strukturen und dessen Entwicklung genauer
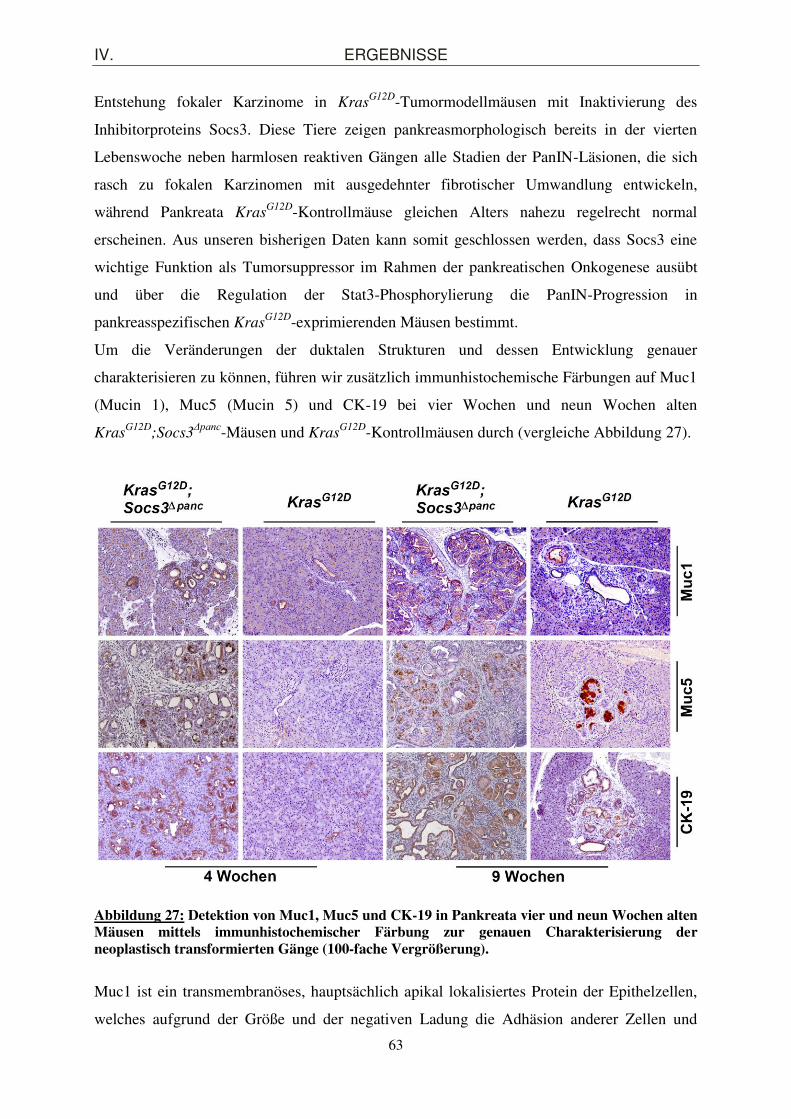
charakterisieren zu können, führen wir zusätzlich immunhistochemische Färbungen auf Muc1
(Mucin 1), Muc5 (Mucin 5) und CK-19 bei vier Wochen und neun Wochen alten
KrasG12D
;Socs3Δpanc-Mäusen und Kras
G12D-Kontrollmäusen durch (vergleiche Abbildung 27).
Abbildung 27: Detektion von Muc1, Muc5 und CK-19 in Pankreata vier und neun Wochen alten Mäusen mittels immunhistochemischer Färbung zur genauen Charakterisierung der neoplastisch transformierten Gänge (100-fache Vergrößerung).
Muc1 ist ein transmembranöses, hauptsächlich apikal lokalisiertes Protein der Epithelzellen,
welches aufgrund der Größe und der negativen Ladung die Adhäsion anderer Zellen und
IV. ERGEBNISSE
64
Mikroorganismen verhindert. Bei zahlreichen Adenokarzinomen oder anderen epithelialen
Tumoren kommt es zu einer Überexpression dieses Moleküls. Durch Polaritätsverlust der
Zelle bei fortgeschrittenen Tumoren wird Muc1 vermehrt auch basolateral gebildet, was eine
verminderte Zelladhäsion hervorruft. Die Abnahme des Zellzusammenhalts stellt somit die
Voraussetzung der Metastasenbildung bei invasiven Karzinomen dar. Muc5 gehört ebenfalls
zu den Glykoproteinen, die jedoch von Schleimhäuten als sekretorische Mucine abgesondert
werden. Eine verstärkte Expression dieses Makromoleküls liegt in PanIN-Läsionen und
Adenokarzinomen vor. Wie die in Abbildung 27 dargestellten Immunhistochemie-Bilder
veranschaulichen, lassen sich zahlreiche sowohl Muc1- als auch Muc5-exprimierende
Ausführungsgänge und PanINs in vier Wochen alten KrasG12D
;Socs3Δpanc-Mäusen
nachweisen, die in der neunten Lebenswoche an Dichte und Schweregrad zunehmen. Im
Vergleich dazu sind im Pankreasgewebe der exemplarisch gewählten KrasG12D-Kontrollmaus
die Mucin-positiven Gänge und intraepithelialen Neoplasien erst zum Zeitpunkt von neun
Wochen detektierbar, während vier Wochen alte Tiere nur vereinzelt Muc1 in den reaktiven
Gängen produzieren. Auch die Anzahl der CK-19-exprimierenden Gangstrukturen und
PanINs in Pankreata der KrasG12D-Linie ist deutlich geringer als im Gewebe gleichaltriger
Socs3-defizienten KrasG12D-Mäuse.
Somit unterstützen die gewonnenen Daten einer frühzeitigen und intensiven Expression
PanIN-spezifischer Markerproteine Muc1, Muc5 und CK-19 in der Bauchspeicheldrüse von
KrasG12D
;Socs3Δpanc-Mäusen die Aussage: Die Deletion von Socs3 in der Kras
G12D;Socs3
Δpanc-
Mauslinie führt zu einer deutlichen Beschleunigung der Progression KrasG12D-induzierter
PanINs mit frühem Auftreten von invasiv duktalen Pankreasadenokarzinomen.
5. Zusammenfassung der Ergebnisse
In Bezug auf die unter I.4. formulierte Zielsetzung können die Ergebnisse der im Rahmen
dieser Arbeit durchgeführten Untersuchungen wie folgt zusammengefasst werden:
1) Im LSL-KrasG12D
;Ptf1a-Creex1(Kras
G12D)-Tumormausmodell konnten wir in den
PanIN-Läsionen eine Aktivierung des Stat3-Signaltransduktionsweges nachweisen.
Das Homodimer aus zwei aktivierten p-Stat3 fungiert als intranukleärer
Transkriptionsfaktor und reguliert die Expression diverser Stat3-Zielproteine. Socs3
als wichtiger Tumorsuppressor lässt sich in KrasG12D-Mäusen in hoher Konzentration
detektieren und reguliert den IL6/Stat3-Signalweg über negative Rückkopplung.
IV. ERGEBNISSE
65
2) Socs3-defiziente KrasG12D-Mäuse weisen im Verlauf der Beobachtung ein im
Vergleich zu KrasG12D-Kontrolltieren deutlich reduziertes Körper- und
Pankreasgewicht auf.
3) Mithilfe des Cre/loxP-Rekombinationssystems gelang es uns die Generierung der
LSL-KrasG12D
;Socs3F/F
;Ptf1a-Creex1(Kras
G12D;Socs3
Δpanc)-Mauslinie. Diese genetisch
veränderten Tiere weisen eine pankreasspezifische Deletion des Exons 2 des Socs3-
Gens auf. Die auf diesem Wege erreichte Inaktivierung des endogenen
Inhibitorproteins Socs3 im Pankreas führt zu einer frühzeitigen und prolongierten
Jak2-vermittelten Stat3-Phosphorylierung am Tyrosinrest Y705 (p-Stat3), die bereits
zum Zeitpunkt von vier Wochen detektierbar ist. Sowohl Morphologie und
Physiologie des endokrinen und exokrinen Pankreas als auch die PanIN-induzierte
konstitutive Aktivierung des onkogenen KrasG12D bleiben von der Socs3-Deletion
unbeeinflusst. Histomorphologische und proteinbiochemische Untersuchungen des
Pankreas der KrasG12D
;Socs3Δpanc-Mäuse im Vergleich zu Kras
G12D-Kontrollmäusen
belegen jedoch die beschleunigte Entwicklung und Progression der altersabhängigen
KrasG12D-induzierten PanIN-Läsionen schon nach der vierten Lebenswoche.
4) Im Beobachtungszeitraum von sechs Monaten entwickelten 28,57% (2/7) der
KrasG12D
;Socs3Δpanc-Mäuse ein Pankreasadenokarzinom, während in der Kras
G12D-
Kohorte keine Karzinomentwicklung zu beobachten war. Die Tumorlatenz von etwa
63 Tagen bei den KrasG12D
;Socs3Δpanc-Versuchstieren war somit kürzer im Vergleich
zu den KrasG12D-Kontrolltieren. Das Pankreasgewebe ist schon zum Zeitpunkt von
vier Wochen morphologisch stark umgewandelt. Es finden sich heterogene Areale mit
normalen Azinis, PanIN-Läsionen unterschiedlichen Grades mit ausgedehnter
desmoplasmatischer Stromareaktion vor.
5) Aus den gewonnenen Ergebnissen lässt sich schlussfolgern, dass die Inaktivierung des
endogenen Tumorsuppressors Socs3 die Progression präneoplastischer Läsionen im
murinen KrasG12D-Tumormodell beschleunigt. Socs3 übt somit durch negative
Rückkopplung eine tragende Funktion im Akzelerationsprozess der PanINs aus und
bestimmt über die Stat3-Phosphorylierung die Regulation der PanIN-Entwicklung und
–Progression zu invasiv duktalem Adenokarzinom.
V. DISKUSSION
66
V. DISKUSSION
1. Rolle von Socs3 für die pankreatische Karzinogenese im Kras
G12D-Mausmodell
Obwohl Socs3 bereits in einigen humanen Tumorzelllinien und –geweben als ein wichtiger
Tumorsuppressor beschrieben wurde, blieb dessen genaue Rolle und Funktion in der
pankreatischen Karzinogenese bisher unbeleuchtet.
In einigen Studien wurde über eine endogene DNA-Hypermethylierung als epigenetischer
Inaktivierungsmechanismus bestimmter Socs-Gene im Rahmen der Karzinogenese spekuliert.
He und Kollegen konnten in humanen NSCLC (Nichtkleinzelliges Karzinom)-Zellinien,
Mesotheliom-Zelllinien und operativ gewonnenem Lungenkarzinomgewebe eine im
Vergleich zu den normalen Kontrollzelllinien supprimierte Expression des Socs3-Transkripts
feststellen. Die Socs3-Defizienz in den Tumorzellen resultierte in einer Hyperaktivität des
Jak/Stat-Signaltransduktionsweges mit proliferativer und onkogener Eigenschaft. Eine
genetische Wiederherstellung der Socs3-Funktion induzierte hingegen durch Inhibition der
Stat3-Aktivität Apoptose und Suppression des Zellwachstums.
Interessanterweise wurden hypermethylierte CpG-Inseln des Socs3-Gens in allen
Tumorzelllinien gefunden, während normale Gewebszellen mit regelrechter Socs3-Expression
keine Veränderung der entsprechenden Genareale zeigten (He et al., 2003).
CpG-Inseln haben ihre Bedeutung in der Regulation der Genexpression und stellen somit
einen Mechanismus der epigenetischen Genregulation dar (Antequera 2003). Die
Methylierung der CpG-Inseln eines Gens hat die Konsequenz, dass dieses Gen nicht mehr
transkribiert wird, was unter anderem in der Tumorentstehung durch das Abschalten
relevanter Tumorsuppressorgene eine große Rolle spielt. Untersuchungen mittels
molekulargenetischer Analysen an ösophagealem Plattenepithelkarzinom und Barrett-
Adenokarzinom (Tischoff et al., 2007), humanem Kopf- und Hals-Plattenepithelkarzinom und
deren dysplastischen Vorläuferläsionen (Weber et al., 2005), hepatozellulären Karzinomen
(HCC) und deren präneoplastischen Läsionen (Niwa et al., 2005) offenbarten ebenfalls eine
Hypermethylierung der CpG-Inseln enthaltenden Socs3-Promotorregion. Auch hier kam es
als Folge der Socs3-Defizienz zu einer konstitutiven Aktivierung des Stat3-Signalweges. Im
Kolon bietet die endogene Induktion der Socs3-Expression demzufolge einen protektiven
Mechanismus gegenüber Verletzung-induzierter Kryptenhyperplasie und Entzündungs-
assoziierter Karzinogenese durch Hemmung der IL-6/Stat3-Signalkaskade (Rigby et al.,
2007).
V. DISKUSSION
67
In den meisten Karzinomzellen der Ovarien und der Mamma kommt es zu einer Inaktivierung
des Socs1- und Socs2-Gens durch CpG-Hypermethylierung in der Promotorregion, wodurch
diese Gene nicht mehr transkribiert werden. Im Gegensatz zu den Ergebnissen von He und
Kollegen war die Promotorregion des Socs3-Gens in den beiden genannten Tumorentitäten
nicht hypermethyliert und das Genprodukt dementsprechend intrazellulär in hoher
Konzentration detektierbar. Darüber hinaus wurde eine erhöhte Stat3-Aktivität in den
betroffenen Tumorzellen registriert, die auf die fehlende Expression des Socs1-Moleküls mit
wachstums- und proliferationsinhibierender Wirkung zurück geführt wurde (Yoshikawa et
al., 2001; Lee et al., 2006). Die konstitutive Stat3-Aktivierung ist mit einer Überexpression
von Socs3 assoziiert und führte zur Zunahme der Proliferationsrate der mamillären
Epithelzellen. Die gewonnenen Ergebnisse ließen Sutherland und Kollegen annehmen, dass
das endogene Inhibitorprotein Socs3 in dieser Zellart keine Tumorsuppressor-Aktivität erfüllt,
was wiederum dessen vielseitige teilweise unerforschte Funktion in verschiedenen
Karzinomarten verdeutlicht (Sutherland et al., 2004).
Drei Jahre nach der Veröffentlichung der Arbeit von Sutherland und Kollegen, wurde die
Studie von der Arbeitsgruppe um Evans publik, in der sie entgegen Sutherlands Aussagen
eine Zunahme der Expression von Socs2 bis Socs7 postulierten. Im Vergleich zu den
normalen mamillären Epithelzellen reagierten die Tumorzelllinien trotz hypermethylierter
CpG-Inseln adäquat auf Zytokin- bzw. Wachstumsfaktor-Signale mit erhöhter Expression von
Socs1 und Socs3, wodurch der Mechanismus der Socs-vermittelten Feedback-Inhibition nicht
gestört wurde (Evans et al., 2007). In Übereinstimmung mit den Ergebnissen von der
Arbeitsgruppe um Sutherland, konnten sie belegen, dass trotz Zunahme des Socs3-Levels in
Mammakarzinomzellen keine effiziente Feedback-Inhibition auf konstitutive Stat3-
Aktivierung erfolgte. Socs3 fungierte stattdessen als Tumorpromotor, welcher die IFN-
α/Stat1-vermittelte Wachstumsinhibition hemmte.
Die intrazelluläre Stat3-Aktivierung im Rahmen der pankreatischen Onkogenese scheint
ebenfalls unter der Socs3-Kontrolle zu stehen. In unseren Versuchen konnten wir erstmalig
zeigen, dass in KrasG12D-Tumormäusen eine erhöhte Socs-3 Expression vorliegt. Um den
Einfluss von Socs3 auf die Pankreaskarzinogenese im KrasG12D-transgenen Mausmodell zu
untersuchen, generierten wir eine weitere Mauslinie mit pankreasspezifischer Socs3-Deletion.
Eine Inaktivierung des Socs3-Gens führte demnach zu einer prolongierten und verstärkten
Stat3-Phosphorylierung, was wiederum die Beschleunigung der PanIN-Progression und die
Entstehung von Pankreaskarzinomen förderte.
V. DISKUSSION
68
Im Einklang mit den aufgeführten Daten konnten wir in unserer Arbeit zeigen, dass innerhalb
des Beobachtungszeitraums von sechs Monaten 28,57% (2/7) der KrasG12D
;Socs3Δpanc-Mäuse
ein Pankreasadenokarzinom entwickelten, während in der KrasG12D-Kohorte keine
Karzinomentwicklung zu beobachten war. Die Tumorlatenz von etwa 63 Tagen bei den
KrasG12D
;Socs3Δpanc-Versuchstieren war somit kürzer im Vergleich zu den Kras
G12D-
Kontrolltieren.
2. Einfluss von Socs3 auf die Apoptose und Proliferation im Kras
G12D-Mausmodell
Proliferation und Apoptose sind zellautonome Effekte, die das Gleichgewicht zwischen
Zellwachstum und –eliminierung gewährleisten. Ein Ungleichgewicht dieser teilweise
komplexen Mechanismen kann eine unkontrollierte Proliferation begünstigen und die
Tumorentstehung fördern. Während in frühen und späten PanIN-Stadien die Apoptose als ein
seltenes Ereignis anzusehen ist, stellt die Proliferation einen dominierenden Mechanismus in
der Tumorprogression dar (Luttges et al., 2003).
Stat3 ist bereits in vielen Untersuchungen als einen wichtigen Regulator des programmierten
Zelltods und des Zellzyklus beschrieben worden (Konnikova et al., 2003; Lin et al., 2005;
Xihong et al., 2008). Aktivierte p-Stat3 Homo- oder Heterodimere aktivieren als
Transkriptionsfaktoren zahlreiche Zielgene. U.a. werden Proteine wie Bcl-XL, Mcl-1,
Survivin und Cyclin D1 vermehrt gebildet. Bcl-XL und Mcl-1 gelten als anti-apoptotische
Signalmoleküle, die durch verstärkte Expression zum Überleben transformierter Zellen in
einigen humanen Tumorerkrankungen beiträgt (Krajewska et al., 1996; Okaro et al., 2001; Ma
et al., 2004; Bhattacharya et al., 2005; Yu et al., 2009; Placzek et al., 2010).
Untersuchungen an humanen Pankreastumorzelllinien haben gezeigt, dass eine Inaktivierung
von Stat3 in einer Abnahme der Transkriptionsrate von Bcl-XL und Mcl-1 resultierte und
folglich die Apoptose induzierte (Huang et al., 2010). In unseren weiterführenden
Experimenten konnten wir zum ersten Mal nachweisen, dass die homozygote Deletion des
negativen Regulators Socs3 in vier Wochen alten KrasG12D-Tumormäusen eine frühe und
prolongierte Stat3-Aktivierung hervorruft und somit eine im Vergleich zu gleichaltrigen
KrasG12D-Tumormäusen stärkere Expression der anti-apoptotischen Proteine Bcl-XL und Mcl-
1 verursacht.
Zusätzlich zu den o.g. Ergebnissen konnten wir proteinbiochemisch eine stärkere Cyclin-D1
Expression in Socs3-defizienten KrasG12D-Tumormäusen im Vergleich zu den Kras
G12D-
Kontrollmäusen nachweisen. Cyclin D1 ist ein proliferationsförderndes, intranukleär
V. DISKUSSION
69
lokalisiertes Protein, welches die G1/S-Progression innerhalb des Zellzyklus fördert und somit
die proliferative Aktivität einer Zelle steuert (Baldin et al., 1993). Die Überexpression von
Cyclin D1 in KrasG12D
;Socs3Δpanc-Mäusen kennzeichnete somit eine Verkürzung des G1/S-
Phasenübergangs, die wiederum die Progression transformierter Zellen innerhalb des
Zellzyklus beschleunigt und die Entwicklung von Karzinomen wesentlich beeinflusst. In
anderen Studien konnte in Übereinstimmung mit unseren Resultaten ebenfalls eine
Überexpression von Cyclin D1 in humanen Pankreaskarzinomzellen nachgewiesen werden,
die mit einer schlechten Prognose assoziiert ist (Gansauge et al., 1997). Die Bedeutung von
Cyclin D1 in der Tumorgenese nicht nur im Pankreasadenokarzinom, sondern auch in
zahlreichen anderen Tumorarten wird auch in anderen Arbeiten betont (Musgrove et al.,
2011).
In unseren Versuchen konnten wir erstmalig zeigen, dass die übermäßige Stat3 Expression in
vier Wochen alten KrasG12D-Mäusen eine starke Aktivierung der genannten Proteine zur
Folge hat. Dieses Ergebnis verdeutlicht die Proliferation als dominierender Prozess in der
PanIN-Progression. Die zusätzliche, homozygote Deletion des Socs3-Gens in diesen
Tumormäusen führt zu einer im Vergleich zu gleichaltrigen KrasG12D-Kontrollmäusen
verstärkten und prolongierten Aktivierung von Stat3 und der Stat3-assoziierten Proteine durch
Wegfall des negativen Inhibitors. Die dadurch hervorgerufene, persistierende Stat3 Aktivität
und die vermehrte Bildung anti-apoptotischer und proliferationsfördernder Moleküle
verursachten eine Beschleunigung der PanIN-Progression zu pankreatischem duktalem
Adenokarzinom.
Im murinen Modell der Hepatitis-assoziierten Hepatokarzinogenese stieg nach Injektion des
Karzinogens DEN bzw. con A die IL-6 Serumkonzentration sowohl im Wildtyp als auch in
den Soc3-Knockouts ohne signifikanten Unterschied dramatisch an. Jedoch war in den Socs3-
defizienten Tieren nach 24 Stunden eine anhaltende und stärkere Stat3-Phosphorylierung im
Vergleich zu den Wildtypen detektierbar. In diesem Zusammenhang nahm die
Transkriptionsfrequenz der Stat3-Zielgene Bcl-XL, Mcl-1, Cyclin D1 zu. Die homozygote
Deletion des Socs3-Gens in den Leberparenchymzellen resultierte in einer verstärkten,
prolongierten Stat3-Aktivierung und beschleunigte infolgedessen die Entwicklung des HCC
(Ogata et al., 2006).
Ein weiteres Ergebnis unserer experimentellen Untersuchungen stellte die
immunhistochemiche Detektion des Enzyms Cyclooxygenase 2 (Cox-2) in präneoplastischen
pankreatischen Läsionen dar. Cox-2 gehört zu den Zielgenen von Stat3 und wird bei dessen
Aktivierung verstärkt transkribiert (Yu et al., 2008). Cox-2-Synthese wird erst bei
V. DISKUSSION
70
Entzündungen bzw. Verletzung des Gewebes induziert und fördert anschließend die
Produktion von pro-inflammatorischen Prostaglandinen, die chronische Entzündungen
unterhalten. Inflammatorische Prozesse sind bedeutende epigenetische Faktoren, die
maßgeblich zur Entstehung und Progression zahlreicher Tumoren führen (Shacter et al.,
2002). Die PanIN-Progression und die Tumorentwicklung in der Pankreaskarzinogenese
scheinen auch entzündungsassoziiert zu sein (Raimondi et al., 2010). Da Cox2 ein Zielgen
von Stat3 darstellt und dessen Genprodukt pro-inflammatorische Wirkung besitzt, kann davon
ausgegangen werden, dass Stat3 maßgeblich als Regulator tumor-assoziierter Entzündung
fungiert. Mittels immunhistochemischer Färbungen konnten wir in PanINs der KrasG12D-
Tumormäuse Cox-2 nachweisen. Unsere Western-Blot Analysen demonstrierten eine bereits
in der vierten Lebenswoche verstärkte und prolongierte Expression von Cox-2 in Socs3-
defizienten KrasG12D-Mäusen im Vergleich zu den gleichaltrigen Kras
G12D-Kontrolltieren.
Diese Ergebnisse deuten darauf hin, dass die homozygote Deletion von Socs3 die Stat3-
vermittelte Cox-2-Synthese verstärkt, die wiederum inflammatorische Mechanismen bereits in
der vierten Lebenswoche aktivieren und so eine tumor-assoziierte Entzündung unterhalten,
die die PanIN-Progression und Karzinomentwicklung beschleunigen. Weiterhin begünstigt
das durch Cox-2 gebildete Prostaglandin E2 in vielen Tumorarten die Bildung des
Angiogenese-Faktors VEGF, das wiederum das Tumorwachstum fördert. Demzufolge wird
vermutet, dass Cox-2 eine wesentliche Rolle in der Tumorgenese spielen könnte (Simmons et
al., 2004).
In KrasG12D-induzierten PanINs war uns ebenfalls der proteinbiochemische Nachweis von
HSP-70 möglich. HSP-70 stellt ein wichtiger Bestandteil des Chaperonsystems dar, das
ubiquitär in eukaryontischen Zellen vorkommt. Die wichtigsten Funktionen von HSP-70
liegen in der richtigen Faltung und Aktivierung vieler Proteine. Zusätzlich ermöglicht dieses
Molekül über anti-apoptotische Mechanismen das Überleben und Wachstum von Tumorzellen
(Hatfield et al., 2012). HSP-70 wird reichlich in malignen Tumoren unterschiedlichen
Ursprungs gebildet und seine Expression korreliert mit erhöhter Zellproliferation, schlechter
Differenzierung, Lymphknotenmetastasen und schlechtem Therapie-Outcome (Ciocca et al.,
1993; Vargas-Roig et al., 1997; Jäättelä, 1999). Das Gen HSP-70 gehört ebenfalls zu den
Zielgenen von Stat3. Analog zur verstärkten und prolongierten Aktivierung von Stat3 in vier
Wochen alten, Socs3-defizienten KrasG12D-Tumormäusen konnten wir mittels Western Blot-
Methode ebenfalls eine im Vergleich zu den Kontrolltieren verstärkte HSP-70 Bildung
nachweisen. Unsere Ergebnisse deuten darauf hin, dass HSP-70 als zytoprotektives Molekül
Über die Inaktivierung von Socs3 in humanen Karzinomen und Tumorzelllinien ist in einigen
Arbeiten berichtet worden (He et al., 2003; Sutherland et al., 2004; Weber et al., 2005; Niwa
et al., 2005). Die Bedeutung von Socs3 im murinen in-vivo-Modell des Pankreaskarzinoms
blieb jedoch bisher unbeleuchtet. In unseren Untersuchungen konnten wir zeigen, dass die
KrasG12D-Mauslinie zeit- und altersabhängig alle PanIN-Stufen entwickelten, die den
humanen pankreatischen Vorläuferläsionen entsprechen. Zudem ließ sich eine starke Stat3-
Phosphorylierung am Tyrosinrest Y705 in den KrasG12D-Tumormäusen nachweisen. Socs3
wird Stat3-abhängig exprimiert und fungiert in den PanIN-Läsionen als essentieller Regulator
der Aktivierung und Inhibition von Stat3 und bestimmt somit über die zeitabhängige
Progression der PanINs im KrasG12D-Tumormausmodell.
Aus unseren Ergebnissen können wir damit annehmen, dass auch in humanen
Pankreaskarzinomen die Ausschaltung der Socs3-Aktivität eine Beschleunigung der malignen
Transformation und Progression bedeutet.
In einigen durchgeführten experimentellen Untersuchungen der letzten Jahre versuchten
Forscher die Socs3-Effizienz auf molekulargenetischer Ebene wiederherzustellen. In humanen
Tumorzelllinien wurde die häufig vorzufindende Methylierung und folglich Inaktivierung des
Socs3-Gens durch die Behandlung mit der demethylierenden Verbindung 5-aza-2-
deoxycytidin (5-AZA-DC; Sutherland et al., 2004; Weber et al., 2005; Niwa et al., 2005) oder
Socs3-Gen tragender viraler (CMV, Adenoviren) Vektoren (Lin et al., 2010; Rossa et al.,
2012) aufgehoben. Die dadurch ermöglichte Reaktivierung der Socs3-Expression reduzierte
die Proliferationsrate und induzierte die Apoptose von humanen Tumorzellen. Ebenfalls kam
es zu einer Down-Regulierung der Stat3-Phosphorylierung sowie der Stat3-abhängigen
Zielproteine, die diverse Mechanismen des Zellwachstums und der Zelldifferenzierung, der
Apoptose und Inflammation steuerten (He et al., 2003; Weber et al., 2005; Iwahori et al.,
2011). Diese Ergebnisse demonstrierten, dass das endogene Inhibitorprotein Socs3 einen
potenziell tumorsuppressiven Effekt besitzt und dessen Inaktivierung eine Beschleunigung der
Tumorprogression bedeutet. Bisher ist der Einfluss von Socs3 auf die Initiierung und
Progression von präneoplastischen Läsionen des Pankreas zu duktalen Adenokarzinomen
kaum beleuchtet worden. Mit den gewonnenen Daten gelang unserer Arbeitsgruppe erstmalig
VI. ZUSAMMENFASSUNG
82
der Nachweis, dass Socs3 die Rolle eines essentiellen Regulators der zeitabhängigen
Progression der PanINs in der KrasG12D-initiirenden Onkogenese des Pankreas einnimmt und
die Inaktivierung dieses endogenen Inhibitors letztlich in einer malignen Entartung duktaler
Zellen endet. Diese Erkenntnisse über die Funktion und Mechanismen des Stat3/Socs3-
Signalweges sollen den Ausgangspunkt für weitere sowohl experimentelle als auch klinische
Untersuchungen zur Validierung eines spezifischen und vielversprechenden Targets in der
Therapie des hochaggressiven Pankreasadenokarzinoms darstellen.
VI. ZUSAMMENFASSUNG
Das Pankreaskarzinom steht an vierter Stelle der tumorbedingten Todesfälle. Mit einer
medianen Überlebenszeit von etwa drei bis fünf Monaten und einer 5-Jahresüberlebensrate
von weniger als 5% hat das Pankreaskarzinom eine düstere Prognose (Hezel et al., 2006).
Wesentliche Ursachen für das schlechte Outcome dieser Tumorerkrankung sind die späte
Diagnosestellung, frühe Metastasierung und mangelnde Therapieoptionen (Lionetto et al.,
1995; Yeo et al., 2002b; Kleef et al., 2007a).
Für die Entstehung des duktalen Pankreasadenokarzinoms wurde auf histopathologischer und
molekulargenetischer Ebene ein spezifisches Tumorprogressionsmodell entwickelt.
Demzufolge entsteht der invasive Tumor aus definierten, präneoplastischen
Vorläuferläsionen, den sogenannten pankreatischen intraepithelialen Neoplasien (PanINs), die
wiederum in drei Stadien eingeteilt sind. Mit steigendem Schweregrad dieser Läsionen
nehmen die architektonischen und zellulären Veränderungen der duktalen Strukturen
ebenfalls zu. Molekulargenetische Untersuchungen haben gezeigt, dass die einzelnen PanIN-
Progressionsstufen bestimmte genetische Veränderungen in Form von Mutationen der
Onkogene und Tumorsuppressorgene aufweisen (Hruban et al., 2000).
Mithilfe des Cre/loxP-Rekombinationssystems gelang David Tuveson und Tyler Jacks die
Etablierung eines Mausmodells, in dem die pankreasspezifische KrasG12D-Mutation am Exon1
des entsprechenden Gens vorliegt. In diesem Tumormausmodell konnte das komplette PanIN-
Spektrum rekapituliert werden, da die genetisch veränderten Mäuse alters- und zeitabhängig
alle Stadien dieser Vorläuferläsionen aufzeigen (Jackson et al., 2001). Ausgehend von diesem
etablierten KrasG12D-Tumormausmodell sind verschiedene komplexe Tiermodellsysteme
entwickelt worden, die additiv zu dem mutierten Kras-Allel andere genetische Veränderungen
aufzeigen. Damit lässt sich der Einfluss einzelner Genmutationen auf die
VI. ZUSAMMENFASSUNG
83
Karzinomentwicklung und –progression genauer analysieren und mögliche Ansatzpunkte für
ein gezieltes medizinisches Eingreifen finden.
Das Socs3-Molekül hat intrazellulär eine auf den IL6/Jak2/Stat3-Signalweg inhibierende
Funktion. Stat3-vermittelt exprimiert, moduliert dieses Inhibitorprotein in einer Feedback-
Schleife die Stat3-Phosphorylierung und somit die Stat3-abhängige Expression von
bestimmten Zielgenen, die Prozesse der Proliferation, Apoptose, Angiogenese und
Inflammation steuerten. In zahlreichen humanen Tumorgeweben bzw. -zelllinien konnte
bereits eine Inaktivierung von Socs3 wie z.B. durch eine Hypermethylierung der
Promotorregion und folglich eine Hemmung der Socs3-Transkription als epigenetische
Veränderung nachgewiesen werden (Sutherland et al., 2004; Weber et al., 2005; Niwa et al.,
2005). Die Auswirkung einer Socs3-Defizienz auf die Progression der Vorläuferläsionen zu
duktalen Adenokarzinomen in der pankreatischen Onkogenese blieb bisher jedoch
unbeleuchtet.
In dieser Arbeit haben wir zunächst die Expression von Stat3 und Stat3-abhängigen Proteinen
v.a. Socs3 in KrasG12D-Tumormäusen auf proteinbio- und immunhistochemischer Ebene
bestätigt. Daraus konnte geschlussfolgert werden, dass Socs3 eine balancierende Funktion auf
die Stat3-Phosphorylierung ausübt.
Um die Auswirkung einer Socs3-Defizienz auf die PanIN-Progression genau beurteilen zu
können, generierten wir in einem weiteren Schritt mithilfe der Cre/loxP-Technologie eine
KrasG12D
;Socs3Δpanc-Mauslinie, die eine Deletion des für die Bindungsdomäne am gp130-
Rezeptor kodierenden Exons 2 im Socs3-Gen aufweisen. Durch diese genetische Mutation ist
die Bindung an den intrazellulären Rezeptorbestandteil und folglich die Feedback-Hemmung
der Stat3-Phosphorylierung nicht mehr gewährleistet.
Socs3-defiziente KrasG12D-Mäuse weisen keine Beeinträchtigung in der morphologischen und
funktionellen Entwicklung des exokrinen und endokrinen Pankreas auf. Ebenso wird die
Ausprägung der KrasG12D-Mutation durch die Socs3-Deletion nicht beeinflusst. Diese
Mauslinie ermöglicht somit die Untersuchung des Inhibitorproteins Socs3 auf die
pankreatische Karzinogenese und die Stat3-Phosphorylierung.
Unsere morphologische und biochemische Forschungsergebnisse demonstrieren für
KrasG12D
;Socs3Δpanc-Mäuse im Vergleich zu den Kras
G12D-Kontrolltieren eine frühzeitige und
prolongierte Stat3-Phosphorylierung, die bereits nach vier Wochen postpartum detektierbar
ist, sowie eine Beschleunigung der KrasG12D-initiierenden PanIN-Progression.
Interessanterweise ist das Pankreas der Socs3-defizienten KrasG12D-Mäuse schon zum
Zeitpunkt von vier Wochen morphologisch sehr stark umgewandelt. In dem fibrotisch
84
umgebauten Organ sind heterogne Areale detektierbar, die neben normalen Azinis ausgeprägt
hochgradig veränderte Gänge in Form von PanINs aufweisen.
Das endogene Inhibitorprotein Socs3 übt somit die Rolle eines Tumorsuppressors in der
Onkogenese des Pankreas aus und bestimmt über die Regulation der Stat3-Phosphorylierung
die PanIN-Progression. Unsere Ergebnisse dienen dazu, einen neuen vielversprechenden
Ansatz in der Therapie und Prophylaxe des Pankreaskarzinoms zu definieren und zu
etablieren, weshalb weitere experimentelle und klinische Studien angeschlossen werden
sollten.
VII. LITERATURVERZEICHNIS
85
VII. LITERATURVERZEICHNIS
Aarnio M., Mecklin J.P., Aaltonen L.A., Nystrom-Lahti M., Jarvinen, H.J. (1995). Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int. J. Cancer 64, 430-433.
Abell K., Bilancio A., Clarkson R.W.E., Tiffen P.G., Altaparmakov A.I., Burdon T.G., Asano
P., Oettle H., Schlag P.M., Schmid R., Schmiegel W., Schlottmann K., Werner J., Wiedenmann
B., Kopp I. (2007). S3- Guidelines ˝Exocrine pancreatic cancer˝ 2007. Z Gastroenterol 45(6), 487-523.
Ahmad N.A., Lewis J.D., Ginsberg G.G., Rosato E.F., Morris J.B., Kochman M.L. (2000).
Endoscopic ultrasound in preoperative staging of pancreatic cancer. Gastrointest Endosc 52,
463-8.
Alexander W.S., Starr R., Metcalf D., Nicholson S.E., Farley A., Elefanty A.G., Brysha M., Kile B.T., Richardson R., Baca M., Zhang J.-G., Willson T.A., Viney E.M., Sprigg N.S., Rakar S., Corbin J., Mifsud S., DiRago L., Cary D., Nicola N.A., Hilton D.J. (1999). Suppressors of cytokine signaling (SOCS): negative regulators of signal transduction. Journal of Leukocyte Biology 66(4), 588-592.
Algül H., Treiber M:, Lesina M., Schmid R.M. (2007b). Mechanisms of Disease: chronic inflammation and cancer in the pancreas-a potential role for pancreatic stellate cells? Nat Clin Pract Gastroenterol Hepatol 4, 454-462.
American Cancer Society 2011.
Antequera F. (2003). Structure, function and evolution of CpG island promoters. Cell Mol Life Sci 60(8), 1647-58.
Assinder S.J., Dong Q., Kovacevic Z., Richardson D.R. (2009). The TGF-β, PI3K/Akt and PTEN pathways: established and proposed biochemical integration in prostate cancer. Biochem J 417, 411–421.
Babon J.J., McManus E.J., Yao S., DeSouza D.P., Mielke L.A., Sprigg N.S., Willson T.A., Hilton D.J., Nicola N.A., Baca M., Nicholson S.E., Norton R.S. (2006). The Structure of SOCS3 Reveals the Basis of the Extended SH2 Domain Function and Identifies an Unstructured Insertion That Regulates Stability. Molecular Cell 22, 205–216.
Baldin V., Lukas J., Marcote M.J., Pagano M., Draetta G. (1993). Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 7(5), 812-21.
Bardeesy N., DePinho R.A. (2002). Pancreatic cancer biology and genetics. Nat Rev Cancer 2, 897-909.
Barton G.M. (2008). A calculated response: control of inflammation by the innate immune system. J Clin Invest 118(2), 413-420.
Beres T.M., Masui T., Swift G.H., Shi L., Henke R.M., MacDonald R.J. (2006). PTF1 is an Organ-Specific and Notch-Independent Basic Helix-Loop-Helix complex containing the mammalian suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol Cell Biol 26(1), 117-130.
Bhatia M., Brady M., Kang Y.K., Costello E., Newton D.J., Christmas S.E., Neoptolemos J.P., Slavin J. (2002). MCP-1 but not CINC synthesis is increased in rat pancreatic acini in response to cerulein hyperstimulation. Am J Physiol Gastrointest Liver Physiol 282(1), G77-85.
Bhattacharya S., Ray R.M., Johnson L.R. (2005). STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J 392, 335–344.
Bissell D.M., Wang S.S., Jarnagin W.R., Roll F.J. (1995). Cell-specific expression of transforming growth factor-ß in rat liver: evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 96, 447-455.
Blinman T.A., Gukovsky I., Mouria M., Zaninovic V., Livingston E., Pandol S.J., Gukovskaya A.S. (2000). Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol Cell Physiol 279, C1993–2003.
Bollrath J., Phesse T.J., von Burstin V.A., Putoczki T., Bennecke M., Bateman T., Nebelsiek T., Lundgren-May T., Canli O., Schwitalla S., Matthews V., Schmid R.M., Kirchner T., Arkan M.C., Ernst M., Greten F.R. (2009). gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15(2), 91-102.
Brembeck F.H., Schreiber F.S., Deramaudt T.B., Craig L., Rhoades B., Swain G.; Grippo P., Stoffers D.A., Silberg D.G., Rustgi A.K. (2003). The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res 63, 2005-2009.
Bromberg, J. (2002). Stat proteins and oncogenesis. J Clin Invest 109, 1139-1142.
Brugge W.R., Lauwers G.Y., Sahani D., Fernandez-del Castillo C., Warshaw A.L. (2004). Cystic neoplasms of the pancreas. N Engl J Med 351, 1218-1226.
Burris H.A., Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P., Nelson R., Dorr F.A., Stephens C.D., Von Hoff D.D. (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15, 2403-13.
Cappell M.S. (2005). The pathophysiology, clinicalpresentation, and diagnosis of colon cancer and adenomatous polyps. Med Clin N Am 89, 1-42.
Ciocca D.R., Clark G.M., Tandon A.K., Fuqua S.A.W., Welch W.J., McGuire W.L. (1993). Heat shock protein hsp70 in patients with axillary lymph node-negative breast cancer: Prognostic Implications. JNCI J Natl Cancer Inst 85(7), 570-574.
Claudine H. (2004). Methods in Nutrition Science: Cre/loxP System for Generating Tissue-specific Knockout Mouse Models. Nutrition Reviews 62(6), 243-246.
VII. LITERATURVERZEICHNIS
87
Corcoran R.B., Contino G., Deshpande V., Tzatsos A., Conrad C., Benes C.H. Levy D.E., Settleman J., Engelman J.A., Bardeesy N. (2011). Stat3 plays a critical role in Kras-induced pancreatic tumorigenesis. Cancer Res 71, 5020-5029.
Couch F.J., Johnson M.R., Rabe K.G., Brune K., de Andrade M., Goggins M., Rothenmund H., Gallinger S., Klein A., Petersen G.M., Hruban R.H.(2007). Cancer Epidemiol Biomarkers Prev 16, 342-346.
Croker B.A., Krebs D.L., Zhang J.-G., Wormald S., Willson T.A., Stanley E.G., Robb L., Greenhalgh D.J., Förster I., Clausen B.E.,Nicola N.A., Metcalf D., Hilton D.J. , Roberts A.W., Alexander W.S. (2003). SOCS3 negatively regulates IL-6 signaling in vivo. Nature Immunology 4, 540-545.
Dang C.V., O'Donnell K.A., Zeller K.I., Nguyen T., Osthus R.C., Li F. (2006). The c-Myc target gene network. Semin Cancer Biol 16(4), 253-64.
De La O J.-P., Emerson L.L., Goodman J.L., Froebe S.C., Illum B.E., Curtis A.B., Murtaugh L.C. (2008). Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. PNAS 105(48), 18907-18912.
Desmouliere A., Geinoz A., Gabbiani F., Gabbiani G. (1993). Transforming growthfactor-beta 1 induces alpha-smooth muscle acin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. JBC 122(1), 103-111.
Domagk D., Wessling J., Reimer P., Hertel L., Poremba C., Senninger N., Heinecke A., Domschke W., Menzel J. (2004). Endoscopic retrograde cholangiopancreatography, intraductal ultrasonography, and magnetic resonance cholangiopancreatography in bile duct strictures: a prospective comparison of imaging diagnostics with histopathological correlation. Am J Gastroenterol 99, 1684-9.
Downward J. (2003). Targeting RAS signalling pathways in cancer therapy. Nature Reviews Cancer 3, 11-22.
Dubrovska A., Kim S., Salamone R.J., Walker J.R., Maira S.-M., García-Echeverría C., Schultz P.G., Reddy V.A. (2008). The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. PNAS 106(1), 268-273.
Ebrahimi B., Tucker S.L., Li D., Abbruzzese J.L., Kurzrock R. (2004). Cytokines in pancreatic carcinoma: correlation with phenotypic characteristics and prognosis. Cancer 101(12), 2727-36.
Efeyan A., Serrano M. (2007). p53: Guardian of the Genome and Policeman of the Oncogenes. Cell Cycle 6(9), 1006-1010.
Emanuelli B., Peraldi P., Filloux C., Chavey C., Freidinger K., Hilton D.J., Hotamisligil G.S., Van Obberghen E. (2001). SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-α in the adipose tissue of obese mice. J Biol Chem 276, 47944-47949.
Emanuelli B., Peraldi P., Filloux C., Sawka-Verhelle D., Hilton D., Van Obberghen E. (2000). SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem 275, 15985-15991.
Erkan M., Hausmann S., Michalski C.W., Fingerie A.A., Dobritz M., Kleeff J., Friess H. (2012). The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol and Hepatol 9, 454-467.
Erkan M., Michalski C.W., Rieder S., Reiser-Erkan C., Abiatari I., Kolb A., Giese N.A., Esposito I., Friess H., Kleef J. ( 2008). The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocacinoma. Clin Gastroenterol Hepatol 6, 1155-1161.
Ernst M., Najdovska M., Grai D., Lundgren-May T., Buchert M., Tye H., Matthews V.B., Armes J., Bhathal P.S., Hughes N.R., Marcusson E.G., Karras J.G., Na S., Sedgwick J.D., Hertzog P.J., Jenkins B.J. (2008). STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest 118(5), 1727-1738.
Evans M.K., Yu C.-R., Lohani A., Mahdi R.M., Liu X., Trzeciak A.R. Egwuagu C.E. (2007). Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene 26, 1941-1948.
Fendrich V., Esni F., Garay M.V., Feldmann G., Habbe N., Jensen J.N., Dor Y., Stoffers D., Jensen J., Leach S.D., Maitra A. (2008). Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology 135(2), 621-31.
Ferrara N. (2002). VEGF and the quest for tumour angiogenesis factors. Nature Reviews Cancer 2, 795-803.
Fléjou J.F. (2005). Barrett’s oesophagus: from metaplasia to dysplasia and cancer. Gut 54, i6-i12.
Friday B.B., Adjei A.A. (2008). Advances in Targeting the Ras/Raf/MEK/Erk Mitogen-Activated Protein Kinase Cascade with MEK Inhibitors for Cancer Therapy. Clin Cancer Res 14, 342.
Friess H., Kleeff J., Fischer L., Muller M., Buchler M.W. (2003). Chirurgische Standardtherapie beim Pankreaskarzinom. Chirurg 74, 183-190.
Fukuda A., Wang S.C., Morris J.P., Folias A.E., Liou A., Kim G.E., Akira S., Boucher K.M., Firpo M.A., Mulvihill S.J., Hebrok M. (2011). Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 19(4), 441-55.
Gansauge S., Gansauge F., Ramadani M., Stobbe H., Rau B., Harada N, Beger H.G. (1997). Overexpression of Cyclin Dl in Human Pancreatic Carcinoma is associated with poor prognosis. Cancer Research 57, 1634-1637.
Glienke W., Maute L., Wicht J., Bergmann L. (2010). Curcumin inhibits constitutive STAT3 phosphorylation in human pancreatic cancer cell lines and downregulation of survivin/BIRC5 gene expression. Cancer Invest 28(2), 166-71.
Grady T., Liang P., Ernst S.A., Logsdon C.D. (1997). Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 113, 1966-1975.
Grivennikov S., Karin E., Terzic J., Mucida D., Yu G.Y., Vallabhapurapu S., Scheller J, Rose-John S., Cheroutre H., Eckmann L., Karin M. (2009). IL-6 and Stat3 are required for
survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15(2), 103-13.
Guerra C., Schuhmacher A.J., Canamero M., Grippo P.J., Verdaguer L., Perez-Gallego L., Dubus P., Sandgren E.P., Barbacid M. (2007). Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11(3), 291-302.
Gugliotta P., Sapino A., Macri L., Skalli O., Gabbiani G., Bussolati G. (1988). Specific demonstration of myoepithelial cells by anti-alpha smooth muscle actin antibody. J Histochem Cytochem 36(6), 659-663.
Hahn S.A., Greenhalf B., Ellis I., Sina-Frey M., Rieder H., Korte B., Gerdes B., Kress R., Ziegler A., Raeburn J.A., Campra D., Grützmann R., Rehder H., Rothmund M., Schmiegel W., Neoptolemos J.P.,Bartsch D.K. (2003). BRCA2 Germline Mutations in Familial Pancreatic Carcinoma. JNCI J Natl Cancer Inst 95(3), 214-221.
Hamilton K., Lund, Kay P., Galanko, J.; Sandler R. Keku T. (2010). Suppressor of cytokine signaling 3 (SOCS3) is not an independent biomarker of colorectal adenoma risk. BMC Research Notes 1, 144.
Hatfield M.P.D., Lovas S. (2012). Role of Hsp70 in cancer growth and survival. Protein & Peptide Letters 19, 616-624.
He B., You L., Uematsu K., Zang K., Xu Z., Lee A.Y., Costello J.F., McCormick F., Jablons D.M. (2003). SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A 100(24), 14133-8.
Heinrich P.C., Behrmann I., Müller-Newen G., Schaper F., Graeve L. (1998). IL-6-type cytokine signalling through the gp130/JAK/STAT pathway. Biochem J 334, 297.
Hezel A.F., KimmelmannA.C., Stanger B.Z., Bardeesy N., Depinho R.A. (2006). Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 20, 1218-49.
Hingorani S.R., Petricoin III E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A., Kawaguchi Y., Johann D., Liotta L.A., Crawford H.C., Putt M.E., Jacks T., Wright C.V.E., Hruban R., Lowy A.M., Tuveson D.A. (2003a). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437-450.
Hirai I., Kimura W., Ozawa K., Kudo S., Suto K., Kuzu H., Fuse A.(2002). Perineural invasion in pancreatic cancer. Pancreas 24, 15-25.
Hodge D.R., Hurt E.M., Farrar W.L. (2005). The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41(16), 2502-12.
Hong S., Park J., Hruban R., Goggins M. (2011). Molecular Signatures of Pancreatic Cancer. Archives of Pathology & Laboratory Medicine 135(6), 716-727.
Horton K.M. (2002). Multidetector CT and three-dimensional imaging of the pancreas: state of the art. J Gastrointest Surg 6, 126-128.
House M.G., Choti M.A. (2005). Palliative therapy for pancreatic/biliary cancer. The Surgical Clinics of North America 85(2), 359-371.
Hruban R.H., Adsay N.V., Albores-Saavedra J., Anver M.R., Biankin A.V., Boivin G.P., Furth E.E., Furukawa T., Klein A., Klimstra D.S., Kloppel G., Lauwers G.Y., Longnecker D.S., Luttges J., Maitra A., Offerhaus G.J., Pérez-Gallego L., Redston M., Tuveson D.A. (2006). Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res 66, 95-106.
Hruban R.H., Goggins M., Parsons J., Kern S.E. (2000). Progression Model for Pancreatic Cancer. Clin Cancer Res 6, 2969.
Hruban R.H., Takaori K., Klimstra D.S., Adsay N.V., Albores-Saavedra J., Biankin A.V., Biankin S.A., Compton C., Fukushima N., Furukawa T., Goggins M., Kato Y., Kloppel G., Longnecker D.S., Luttges J., Maitra A., Offerhaus G.J., Shimizu M., Yonezawa S. (2004). An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J of Surg Pathol 28(8), 977-987.
Huang S., Sinicrope F.A. (2010). Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol Cancer Ther 9(3), 742-750.
Hudis C.A. (2007). Trastuzumab- Mechanism of Action and Use in Clinical Practice. N Engl J Med 357, 39-51.
Hutzen B., Friedman L., Sobo M., Lin L., Cen L., De Angelis S., Yamakoshi H., Shibata H., Iwabuchi Y., Lin J. (2009). Curcumin analogue GO-Y030 inhibits STAT3 activity and cell growth in breast and pancreatic carcinomas. Int J Oncol 35, 867-872.
Iwahori K., Serada S., Fujimoto M., Nomura S., Osaki T., Lee C.M., Mizuguchi H., Takahashi T., Ripley B., Okumura M., Kawase I., Kishimoto T., Naka T. (2011). Overexpression of SOCS3 exhibits preclinical antitumor activity against malignant pleural mesothelioma. Int J Cancer 129(4), 1005-17.
Jackson E.L., Willis N., Mercer K., Bronson R.T., Crowley D., Montoya R., Jacks T., Tuveson D.A. (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243-3248.
Jäättelä M. (1999). Heat shock proteins as cellular lifeguards. Ann Med 31(4), 261-71.
Jo D., Liu D., Yao S., Collins R.D., Hawiger J. (2005). Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med 11(8), 892-8.
Kamura T., Maenaka K., Kotoshiba S., Matsumoto M., Kohda D., Conaway R.C., Conaway J.W., Nakayama K.I. (2004). VHL‐box and SOCS‐box domains determine binding specificity for Cul2‐Rbx1 and Cul5‐ Rbx2 modules of ubiquitin ligases. Genes Dev 18, 3055-3065.
Kamura T., Sato S., Haque D., Liu L., Kaelin W.G. Jr., Conaway R.C., Conaway J.W. (1998). The Elongin BC complex interacts with the conserved SOCS‐box motif present in members of the SOCS, ras. WD‐40 repeat, and ankyrin repeat families. Genes Dev 12, 3872‐3881.
Kim J.E., Lee K.T., Lee J.K., Paik S.W., Rhee J.C., Choi K.W. (2004). Clinical usefulness of carbohydrate antigen 19-9 as a screening test for pancreatic cancer in an asymptomatic population. J Gastroenterol Hepatol 19, 182-186.
Kim S., Keku T.O., Martin C., GalankoJ., Woosley J.T., Schroeder J.C., Satia J.A., Halabi S.,
Sandler R.S. (2008). Circulating Levels of Inflammatory Cytokines and Risk of Colorectal
Kile B.T., Schulman B.A., Alexander W.S., Nicola N.A., Martin H.M., Hilton D.J. (2002). The SOCS box: a tale of destruction and degradation. Trends Biochem Sci 27(5), 235-41.
Kleeff J., Beckhove P., Esposito I., Herzig S., Huber P.E., Lohr J.M., Friess H. (2007a). Pancreatic cancer microenvironment. Int J Cancer 121, 699-705.
Klingler A., Klocker J., Springer P., Kober F., Glaser K. (2000). Combined laparoscopy and laparoscopic ultrasonography in the oncologic diagnostics of pancreas and liver. European Surgery 32(5), 228-232.
Kong F., Guo X., Noel J.G., Wells D.A., Lovell G.J., Ogle C.K. (2002). Thermal injury-induced increases of hepatocyte SOCS3 lead to decreases in STAT3. Shock 18(4), 374-9.
Konnikova L., Kotecki M., Kruger M.M., Cochran B.H. (2003). Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer 3, 23.
Kopp J.L., von Figura G., Mayes E., Liu F.-F., Dubois C.L., Morris J.P., Pan F.C., Akiyama H., Wright C.V.E., Jensen K., Hebrok M., Sander M. (2012). Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737-750.
Kos C.H. (2004). Cre/loxP System for Generating Tissue-specific Knockout Mouse Models. Nutrition Reviews 62(6), 243-246.
Krajewska M., Krajewski S., Epstein J.I., Shabaik A., Sauvageot J., Song K., Kitada S, Reed J.C. (1996). Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol 148(5), 1567-76.
Krapp A., Knofler M., Ledermann B., Burki K., Berney C., Zoerkler N., Hagebuchle O., Wellauer P.K. (1998). The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev 12, 3752-3763.
Lang R., Pauleau A.-L., Parganas E., Takahashi Y., Mages J., Ihle J.N., Rutschman R., Murray P.J. (2003). SOCS3 regulates the plasticity of gp130 signaling. Nature Immunology 4(6), 546-550.
Lin L., Hutzen B., Zuo M., Ball S., Deangelis S., Foust E., Pandit B., Ihnat M.A., Shenoy S.S., Kulp S., Li P.K., Li C., Fuchs J., Lin J. (2010b). Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res 70(6), 2445-54.
Lee T.L., Yeh J., Van Waes C., Chen Z. (2006). Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther 5(1), 8-19.
Lei S., Appert H.E., Nakata B., Domenico D.R.,Kim K., Howard J.M. (1995). Overexpression of HER2/neu Oncogene in Pancreatic Cancer Correlates with Shortened Survival. Int J Pancreatol 17(1), 15-21.
Lesina M., Kurkowski M.U., Ludes K., Rose-John S., Treiber M., Klöppel G., Yoshimura A., Reindl W., Sipos B., Akira S., Schmid R.M., Algül H. (2011). Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19(4), 456-69.
Li Y., Du H., Qin Y., Roberts J., Cummings O.W., Yan C. (2007). Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res 67, 8494-8503.
Lim K.-H., Counte C.M. (2005). Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell 8, 381-392.
Lin Y.-C., Lin C.-K., Tsai Y.-H., Weng H.-H.; Li Y.-C., Liang Y., Chen J.-K., Jablons D.M., Yang C.-T. (2010). Adenovirus-mediated SOCS3 gene transfer inhibits the growth and enhances the radiosensitivity of human non-small cell lung cancer cells. Oncology reports 24(6), 1605-1612
Lionetto R., Pugliese V., Bruzzi P., Rosso R. (1995). No Standard treatment is available for advanced pancreatic Cancer. Eur J Cancer 31A, 882-887.
Lin Q., Lai R., Chirieac L.R., Li C., Thomazy V.A., Grammatikakis I., Rassidakis G.Z., Zhang W., Fujio Y., Kunisada K., Hamilton S.R.,Amin H.M. (2005). Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol 167(4), 969-80.
Lowe S.W., Sherr C.J. (2003). Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev 13(1), 77-83.
Lowenfels A. B., Maisonneuve P. (2006). Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 20, 197-209.
Lowenfels A.B., Maisonneuve P., Cavallini G., Ammann R.W., Lankisch P.G., Andersen J.R., Dimagno E.P., Andren-Sandberg A., Domellof L. and the International Pancreatitis Study Group (1993). Pancreatitis and the Risk of Pancreatic Cancer. N Engl J Med 328, 1433-1437.
Lu Y., Fukuyama S., Yoshida R., Kobayashi T., Saeki K., Shiraishi H., Yoshimura A., Takaesu G. (2006). Loss of SOCS3 Gene Expression Converts STAT3 Function from Anti-apoptotic to Pro-apoptotic. The Journal of Biological Chemistry 281(48), 36683-36690.
Lüttges J., Neumann S., Jesenofsky R., Borries V., Löhr M., Klöppel G. (2003). Lack of apoptosis in PanIN-1 and PanIN-2 lesions associated with pancreatic ductal adenocarcinoma is not dependent on K-ras status. Pancreas 27(3), e57-62.
Ma X.T.,Wang S., Ye Y.J., Du R.Y., Cui Z.R., Somsouk M. (2004). Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World J Gastroenterol 10(11), 1569-73.
Magee C.J., Greenhalf W., Howes N. Ghaneh P., Neoptolemos J.P. (2001). Molecular pathogenesis of pancreatic ductal adenocarcinoma and clinical implications. Surg Oncol 10, 1–23.
Maitra A., Fukushima N., Takaori K., Hruban R.H. (2005). Precursors to invasive pancreatic cancer. Adv Anat Pathol 12, 81-91.
Malka D., Hammel P., Maire F., Rufat P., Madeira I., Pessione F., Lévy P., Ruszniewski P. (2002). Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 51, 849-852.
Means A.L., Meszoely I.M., Suzuki K., Miyamoto Y., Rustgi A.K., Coffey R.J. Jr, Wright C.V., Stoffers D.A., Leach S.D. (2005). Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132(16), 3767-76.
Medzhitov R. (2008). Origin and physiological roles of inflammation. Nature 454, 428-435.
Menssen A., Hermeking H. (2002). Characterization of the c-MYC-regulated transcriptome by SAGE: Identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 99(9), 6274-6279.
Miyaki M., Kuroki T. (2003). Role of Smad4 (DPC4) inactivation in human cancer. Biochemical and Biophysical Research Communications 306(4), 799-804.
Miyatsuka T., Kaneto H., Shiraiwa T., Matsuoka T.A., Yamamoto K., Kato K., Nakamura Y., Akira S., Takeda K., Kajimoto Y., Yamasaki Y.,Sandgren E.P., Kawaguchi Y., Wright C.V., Fujitani Y. (2006). Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev 20(11), 1435-40.
Mori H., Hanada R., Hanada T., Aki D., Mashima R., Nishinakamura H., Torisu T., Chien K. R., Yasukawa H., Yoshimura A. (2004). Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nature Medicine 10, 739-743.
Morris J.P., Cano D.A., Sekine S., Wang S.C., Hebrok M. (2010). β-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 120(2), 508-20.
Mulkeen A.L., Yoo P.S., Cha C. (2006). Less common neoplasms of the pancreas. World J Gastroenterol 12(20), 3180-3185.
Musgrove E.A., Caldon C.E., Barraclough J., Stone A., Sutherland R.L. (2011). Cyclin D as a therapeutic target in cancer. Nature Reviews Cancer 11, 558-572.
Nakhai H., Sel S., Favor J., Mendoza-Torres L., Paulsen F., Duncker G.I., Schmid R.M. (2007). Ptf1a is essentialnfor the differentiation of GABAergic and glycinergic amacrine cells and horizontal cells in the mouse retina. Development 134, 1151-1160.
Nazli O., Bozdag A.D., Tansug T., Kir R., Kaymak E. (2000). The diagnostic importance of CEA and CA 19-9 for the early diagnosis of pancreatic carcinoma. Hepatogastroenterology 47, 1750-2.
Niwa Y., Kanda H., Shikauchi Y., Saiura A., Matsubara K., Kitagawa T., Yamamoto J., Kubo T., Yoshikawa H. (2005). Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 24(42), 6406-17.
Oettle H., Post S., Neuhaus P., Gellert K., Langrehr J., Ridwelski K., Harald Schramm H., Fahlke J., Zuelke C., Burkart C., Gutberlet K., Kettner E., Schmalenberg H., Weigang-Koehler K., Bechstein W.-O., Niedergethmann M., Schmidt-Wolf I., Roll L.; Doerken B., Riess H. (2007). Adjuvant Chemotherapy with Gemcitabine vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic CancerA Randomized Controlled Trial. JAMA 297(3), 267-277.
Ogata H., Chinen T., Yoshida T., Kinjyo I., Takaesu G., Shiraishi H., Iida M., Kobayashi T. (2006). Loss of Socs3 in the liver promotes fibrosis by enhancing Stat3-mediated TGF-ß1 production. Oncogene 25, 2520-2530.
Ogata H., Kobayashi T., Chinen T., Takaki H., Sanada T., Minoda Y., Koga K., Takaesu H., Maehara Y., Iida M., Yoshimura A. (2006). Deletion of the SOCS3 Gene in Liver Parenchymal Cells Promotes Hepatitis-Induced Hepatocarcinogenesis. Gastroenterol 131, 179-193.
Okada S., Nakamura M., Katoh H., Miyao T., Shimazaki T., Ishii K., Yamane J., Yoshimura
A., Iwamoto Y., Toyama Y., Hideyuki Okano H. (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nature Medicine 12, 829-834.
Okaro A.C., Deery A.R., Hutchins R.R., Davidson B.R. (2001). The expression of antiapoptotic proteins Bcl-2, Bcl-XL, and Mcl-1 in benign, dysplastic, and malignant biliary epithelium. J Clin Pathol 54, 927-932.
Okusaka T., Okada S., Ueno H., Ikeda M., Shimada K., Yamamoto J., Kosuge T., Yamasaki S., Fukushima N., Sakamoto M. (2001). Abdominal pain in patients with resectable pancreatic cancer with reference to clinicopathologic findings. Pancreas 22, 279-84.
Olive K.P., Tuveson D.A. (2006). The Use of Targeted Mouse Models for Preclinical Testing of Novel Cancer Therapeutics. Clin Cancer Res 12, 5277.
Pandol S., Edderkaoui M., Gukovsky I., Lugea A., Gukovskaya A. (2009). Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol 7(11), 44-47.
Pasca di Magliano M., Sekine S., Ermilov A., Ferris J., Dlugosz A.A., Hebrok M. (2006). Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev 20(22), 3161-73.
Pensa S., Regis G., Boselli D., Novelli F., Poli V. (2009). STAT1 and STAT3 in Tumorigenesis: Two Sides of the Same Coin? Madame Curie Bioscience Database (Internet).
Placzek W.J., Wei J., Kitada S., Zhai D., Reed J.C., Pellecchia M. (2010). A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death and Disease 1, e40.
Pour P.M., Egami H., Takiyama Y. (1991). Patterns of growth and metastases of induced pancreatic cancer in relation to the prognosis and its clinical implications. Gastroenterology 100, 529-36.
Raimondi S., Lowenfels A.B., Morselli-Labate A.M., Maisonneuve P., Pezzilli R. (2010). Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol 24(3), 349-58.
Riehle K.J., Campbell J.S., McMahan R.S., Johnson M.M., Beyer R.P., Bammler T.K., Fausto N. (2007). Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. JEM 205(1), 91-103.
Rigby R.J., Simmons J.G., Greenhalgh C.J., Alexander W.S., Lund P.K. (2007). Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyperproliferation and inflammation-associated tumorigenesis in the colon. Oncogene 26, 4833-4841.
Roberts A.W., Robb L., Rakar S., Hartley L., Cluse L., Nicola N.A., Metcalf D., Hilton D.J., Alexander W.S. (2001). Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. PNAS 98(16), 9324-9329.
Rose S.D., Swift G.H., Peyton M. J., Hammer R.E., McDonald R.J. (2001). The role of PTF1-P48 in pancreatic acinar gene expression .J Biol Chem 276, 44018-44026.
Rosewicz S., Wiedenmann B. (1997). Pancreatic carcinoma. Lancet 349(9050), 485-9.
Rösch T., Dittler H.J., Strobel K., Meining A., Schusdziarra V., Lorenz R., Allescher H.-D., Kassem A.M., Gerhardt P., Siewert J.-R., Höfler H., Classen M.( 2000). Endoscopic ultrasound criteria for vascular invasion in the staging of cancer of the head of the pancreas: a blind reevaluation of videotapes. Gastrointest Endosc 52, 469-77.
Rossa C., Sommer G., Spolidorio L.C., Rosenzweig S.A., Watson D.K., Kirkwood K.L. (2012). Loss of Expression and Function of SOCS3 Is an Early Event in HNSCC: Altered Subcellular Localization as a Possible Mechanism Involved in Proliferation, Migration and Invasion. PLoS ONE 7(9), e45197.
Rui L., Yuan M., Frantz D., Shoelson S., White, M.F. (2002). SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Bio Chem 277, 42394-42398.
Saad E.D., Machado M.C.,Wajsbrot D., Abramoff R., Hoff P.M., Tabacof J., Katz A., Simon S.D., Gansl S.C. (2002). Pretreatment Ca 19-9 level as a prognostic factor in patients with advanced pancreatic cancer treated with gemcitabine. Int J Gastrointest Cancer 32(1), 35-41.
Saxena N.K., Ikeda K., Rockey K.I., Friedmann S.L., Anania F.A. (2002). Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology 35(4), 762-771.
Schenk M., Schwartz A.G., O’Neal E., Kinnard M., Greenson J.K., Fryzek J.P., Ying G.S., Garabrant D.H. (2001). Familial risk of pancreatic cancer. J Natl Cancer Inst 93, 640-644.
Schindeler A. ,Kolind M., Little D.G. (2013). Cellular transitions and tissue engineering. Cell Reprogram 15(2), 101-6.
Scholz A., Heinze S., Detjen K.M., Peters M., Welzel M., Hauff P., Schirner M., Wiedenmann B., Rosewicz S. (2003). Activated signal transducer and activator of
transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 125(3), 891-905.
Schutte M., Hruban R.H., Geradts J., Maynard R., Hilgers W., Rabindran S.K., Moskaluk C.A., Hahn S.A., Schwarte-Waldhoff I., Schmiegel W., Baylin S.B., Kern S.E., Herman J.G. (1997). Abrogation of the Rb/p16 Tumor-suppressive Pathway in Virtually All Pancreatic Carcinomas. Cancer Research 57, 3126-313.
Sellik G.S., Barker K.T., Stolte-Dijkstra I., Fleischmann C., Coleman R.J., Garrett C., Gloyn A.L., Edghill E.L., Hattersley A.T., Wellauer P.K., Goodwin G., Houlston R.S. (2004). Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 36, 1301-1305.
Senn J.J., Klover P.J., Nowak I.A., Zimmers T.A., Koniaris L.G., Furlanetto R.W. Mooney R.A. (2003). Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 278, 13740-13746.
Shacter E., Weitzman S.A. (2002). Chronic inflammation and cancer. Oncology (Williston Park) 16(2), 217-26.
Shaib Y., Davila J., Naumann C., El-Serag H. (2007). The impact of curative intent surgery on the survival of pancreatic cancer patients: a U.S. Population based study. Am J Gastroenterol 102, 1377-82.
Sherr C.J. (2004). Principles of Tumor Suppression. Cell 116(2), 235-246.
Shibata D., Capella G., Perucho M. (1990). Mutational activation of the c-K-ras gene in human pancreatic carcinoma. Baillière's Clinic Gastroenterol 4(1), 151-169.
Shimizu K. (2008). Mechanism of pancreatic fibrosis and applications to the treatment of chronic pancreatitis. J Gastroenterol 43, 823-832.
Simmons D.L., Botting R.M., Hla T. (2004). Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol Rev 56, 387-437.
Song M.M., Shuai K. (1998). The Suppressor of Cytokine Signaling (SOCS) 1 and SOCS3 but Not SOCS2 Proteins Inhibit Interferon-mediated Antiviral and Antiproliferative Activities. Journal of Biological Chemistry 273, 35056-35062.
Starr R., Willson T.A., Viney E.M., Murray L.J.L., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A., Hilton D.J. (1997). A family of cytokine inducible inhibitors of signalling. Nature 387, 917-921.
Stocken D.D., Büchler M.W., Dervenis C., Bassi C., Jeekel H., Klinkenbijl J.H.G., Bakkevold K.E., Takada T., Amano H., Neoptolemos J.P. (2005). Meta-analysis of randomised adjuvant therapy trials for pancreatic cancer. British Journal of Cancer 92, 1372-1381.
Su L.K., Steinbach G., Sawyer J.C., Hindi M., Ward P.A., Lynch P.M. (2000). Genomic rearrangements of the APC tumor-suppressor gene in familial adenomatous polyposis. Hum Genet 106, 101-107.
Sutherland K.D., Lindeman G.J., Choong D.Y.H., Wittlin S., Brentzell L., Phillips W., Campbell I.G., Visvader J.E. (2004). Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 23, 7726-7733.
Suzuki A., Hanada T., Mitsuyama K., Yoshida T., Kamizono S., Hoshino T., Kubo M., Yamashita A., Okabe M., Takeda K., Akira S., Matsumoto S., Toyonaga A., Sata M., Yoshimura A. (2001). CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med 193(4), 471-81.
Takahashi Y., Carpino1 N., Cross J.C., Torres M., Parganas E., Ihle J.N. (2003). SOCS3: an essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. The EMBO Journal 22(3), 372-384.
Talar-Wojnarowska R., Gasiorowska A., Smolarz B., Romanowicz-Makowska H., Kulig A., Malecka-Panas E. (2009). Clinical significance of interleukin-6 (IL-6) gene polymorphism and IL-6 serum level in pancreatic adenocarcinoma and chronic pancreatitis. Dig Dis Sci. 54(3), 683-9.
Talar-Wojnarowska R., Malecka-Panas E. (2006). Molecular pathogenesis of pancreatic adenocarcinoma: potential clinical implications. Med Sci Monit 12, 186-193.
Testini M., Gurrado A., Lissidini G., Venezia P., Greco L., Piccinni G. (2010). Management of mucinous cystic neoplasms of the pancreas. World J Gastroenterol 16(45), 5682-5692.
Tischoff I., Hengge U.R., Vieth M., Ell C., Stolte M., Weber A., Schmidt W.E., Tannapfel A. (2007). Methylation of SOCS‐3 and SOCS‐1 in the carcinogenesis of Barrett's adenocarcinoma. Gut. 56(8), 1047-1053.
Torisu T., Sato N., Yoshiga D., Kobayashi T., Yoshioka T., Mori H., Iida M., Yoshimura A. (2007). The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes to Cells 12, 143-154.
Toth K.G., McKay B.R., De Lisio M., Little J.P., Tarnopolsky M.A., Parise G. (2011). IL-6 Induced STAT3 Signalling Is Associated with the Proliferation of Human Muscle Satellite Cells Following Acute Muscle Damage. PLoS ONE 6(3), 1-12.
Tsiambas E., Karameris A., Dervenis C., Lazaris A.C., Giannakou N., Gerontopoulos K., Patsouris E. (2006). HER2/neu Expression and Gene Alterations in Pancreatic Ductal Adenocarcinoma: A Comparative Immunohistochemistry and Chromogenic in Situ Hybridization Study Based on Tissue Microarrays and Computerized Image Analysis. JOP J Pancreas 7(3), 283-294.
Ueki. K., Kondo T., Kahn, C.R. (2004a). Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 24, 5434-5446.
Ueki, K., Kondo T., Tseng Y.H., Kahn, C.R. (2004b). Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Pro Natl Acad Sci USA 101, 10422-10427.
Vargas-Roig L.M., Fanelli M.A., López L.A., Gago F.E., Tello O., Aznar J.C., Ciocca D.R. (1997). Heat shock proteins and cell proliferation in human breast cancer biopsy samples. Cancer Detect Prev 21(5), 441-51.
Velasco J.M., Rossi H., Hieken T.J., Fernandez M. (2000). Laparoscopic ultrasound enhances diagnostic laparoscopy in the staging of intra-abdominal neoplasms. Am Surg 66(4), 407-411.
Vona-Davis L.C., Frankenberry K.A., Waheed U., Peterson E., McFadden D.W. (2005). Expression of STAT3 and SOCS3 in pancreatic acinar cells. J Surg Res 127(1), 14-20.
Weber A., Hengge U.R., Bardenheuer W., Tischoff I., Sommerer F., Markwarth A., Dietz A., Wittekind C., Tannapfel A. (2005). SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene 24, 6699-6708.
White G.E., Cotterill A., Addley M.R., Soilleux E.J., Greaves D.R. (2011). Suppressor of cytokine signalling protein SOCS3 expression is increased at sites of acute and chronic inflammation. J Mol Histol 42(2), 137-51.
Wilentz R.E., Geradts J., Maynard R., Offerhaus G.J.A., Kang M., Goggins M., Yeo C.J., Kern S.E., Hruban R.H. (1998). Inactivation of the p16 (INK4A) Tumor-suppressor Gene in Pancreatic Duct Lesions: Loss of Intranuclear Expression. Cancer Research 5, 4740-4744.
Xiong H., Zhang Z.G., Tian X.Q., Sun D.F., Liang Q.C., Zhang Y.J., Lu R., Chen Y.X., Fang J.Y. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. 2008. Neoplasia, 10(3), 287-97.
Xu Q., Briggs J., Park S., Niu G., Kortylewski M., Zhang S., Gritsko T., Turkson J., KayH., Semenza G.L., Cheng J.Q., Jove R., Yu H. (2005). Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552-5560.
Yeo C.J., Cameron J.L., Lillemoe K.D., Sitzmann J.V., Hruban R.H., Goodman S.N., Dooley W.C., Coleman J., Pitt H.A. (1995). Pancreaticoduodenectomy for cancer of the head of the pancreas. 201 patients. Ann Surg 221, 721-31.
Yeo T.P., Hruban R.H., Leach S.D., Wilentz R.E., Sohn T.A., Kern S.E., Iacobuzio-Donahue C.A., Maitra A., Goggins M., Canto M.I., Abrams R.A., Laheru D., Jaffee E.M., Hidalgo M., Yeo C.J. (2002b). Pancreatic cancer. Curr Probl Cancer 26, 176-275.
Yoshikawa H., Matsubara K., Qian G.S., Jackson P., Groopman J.D., Manning J.E., Harris C.C., Herman J.G.(2001). SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet 28(1), 29-35.
Yu H., Kortylewski M., Pardoll D. (2007). Crosstalk between cancer and immune cells: role of Stat3 in the tumor microenvironment. Nat Rev Immunol 7(1), 41-51.
Yu H., Pardoll D., Jove R. (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9, 798-809.
Yu L.-J., Wu M.-L., Li H., Chen X.-Y., Wang Q., Sun Y., Kong Q.-K., Liu J. (2008). Inhibition of STAT3 Expression and Signaling in Resveratrol-Differentiated Medulloblastoma Cells. Neoplasia 10(7), 736-744.
Abbildung 1: Morphologische Tumorprogression über PanIN-Vorläuferläsionen ................... 8
Abbildung 2: Morphologisches und genetisches Progressionsmodell des duktalen Pankreaskarzinoms. .................................................................................................................. 10
Abbildung 3: Gewebespezifische Deletion von Mausgenen mittels Cre/loxP-Technologie. .. 13
Abbildung 4: Systematik der Generierung des KrasG12D-Tumormausmodells. ....................... 16
Abbildung 5: Schematische Darstellung der SOCS3-Architektur. .......................................... 18
Abbildung 6: SOCS3 als Trägermolekül des E3-Ubiquitin-Ligase-Komplexes ..................... 18
Abbildung 7: Die Mechanismen der Aktivierung der IL-6/Jak/Stat3-Signalkaskade und dessen Feedback-Inhibition durch SOCS3-Moleküle .......................................................................... 19
Abbildung 8: Die Aktivierung der Ras/Raf/MAPK- und PI3K/AKT-Signalkaskade durch IL-6 und dessen negative Regulation durch Socs3........................................................................... 20
Abbildung 9: Schematische Darstellung der Generierung der Socs3Δpanc-Mauslinie. ............. 42
Abbildung 10: Nachweis der Deletion von Exon 2 im Socs3-Gen mittels mRNA Konzentrationsbestimmung ...................................................................................................... 43
Abbildung 11: PCR-Analysen der möglichen Genotypen aus der Kreuzung von Socs3F/F-und
Abbildung 16: Immunhistochemische Detektion von Socs3 in Zellen der Azini (schwarze Pfeile) und in den PanIN-Läsionen (weiße Pfeile) neun Wochen alter Kras
G12D-Mäuse. ....... 49
Abbildung 17: Aktivierung der Stat3-Signalkaskade im KrasG12D-Mausmodell. .................... 51
Abbildung 18: Proteinbiochemischer Nachweis der Socs3-Defizienz in KrasG12D
Abbildung 19: Nachweis der Expression konstitutiv aktiver K-Ras-Proteine. ........................ 54
Abbildung 20: Gewichtsanalyse sechs Wochen alter KrasG12D
;Socs3Δpanc-Mäuse (n=7) im
Vergleich zu gleichaltrigen KrasG12D-Kontrollmäusen (n=7) und zum Wildtyp (n=17). ........ 55
Abbildung 21: Analyse des makroskopischen Pankreasaspekts. ............................................. 56
100
Abbildung 22: Morphologische Charakterisierung der PanINs und des PDA ......................... 58
Abbildung 23: Vergleichende Auswertung der Gesamtzahl an Ausführungsgängen pro zehn Gesichtsfelder an mindestens 17 sequentiellen Schnitten vier Wochen alter Versuchstiere (200-fache Vergrößerung) ........................................................................................................ 60
Abbildung 24: Quantifizierung der reaktiven Gänge und PanIN-Läsionen pro zehn Gesichtsfelder in mindestens 17 sequentiellen Feingewebsschnitten zum Zeitpunkt von vier Wochen und bei 200-facher Vergrößerung .............................................................................. 61
Abbildung 25: Immunhistochemischer Nachweis der übermäßigen Expression von CK-19, p-Stat3 und Cyclin D1 in reaktiven Gängen und PanIN-Läsionen vier Wochen alter Kras
Abbildung 26: H.E.-Färbung des duktalen Adenokarzinoms des Pankreas und immunhistochemische Detektion von CK-19 und p-Stat3 in atypischen, drüsenartigen Gangstrukturen. .............................. 63
Abbildung 27: Detektion von Muc1, Muc5 und CK-19 in Pankreata vier und neun Wochen alten Mäusen mittels immunhistochemischer Färbung zur genauen Charakterisierung der neoplastisch transformierten Gänge. 100-fache Vergrößerung. .......................................................................... 64
IX. TABELLENVERZEICHNIS
101
IX. TABELLENVERZEICHNIS Tabelle 1: Chemikalien ............................................................................................................ 24
Tabelle 2: Geräte und Hilfsmittel ............................................................................................. 26
Tabelle 3: Puffer und Lösungen ............................................................................................... 28