Pharmakokinetische und pharmakodynamische Untersuchungen zu selektiven und nicht selektiven Cyclooxygenasehemmern Den naturwissenschaftlichen Fakultäten der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades vorgelegt von Olga Cheremina aus Kysyl / Russland
Transcript
Pharmakokinetische und pharmakodynamische
Untersuchungen zu selektiven und nicht selektiven
Cyclooxygenasehemmern
Den naturwissenschaftlichen Fakultäten
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
vorgelegt von
Olga Cheremina
aus Kysyl / Russland
Als Dissertation genehmigt von den Naturwissenschaftlichen Fakultäten der Universität
Die nichtsteroidalen Antirheumatika (NSAR) wurden schon viele Jahre therapeutisch eingesetzt,
bevor ihr Wirkmechanismus erkannt wurde. Ihre Wirkung beruht im Wesentlichen auf einer
unselektiven Hemmung der beiden bekannten Cyclooxygenaseisoformen (COX-1 und COX-2).
Dadurch wird die Synthese von Prostaglandinen und Thromboxanen gehemmt. Diese
Substanzen stellen wichtige Mediatoren für Schmerz- und Entzündungsreaktionen dar. Sie
erfüllen darüber hinaus Schutzfunktionen in zahlreichen Organen des menschlichen Körpers,
z.B in der Magen- Darmschleimhaut, den Nieren und den Blutgefäßen. In den 90-er Jahren
wurden selektiven COX-2-Hemmer (Coxibe), als Analgetika mit niedrigerer gastrointestinalen
Toxizität im Vergleich zu den traditionellen NSARs, entwickelt.
Der Schwerpunkt dieser Dissertation wurde auf die Hemmung der peripheren Cyclooxygenasen
in menschlichen Blutzellen durch die Vertreter von selektiven und nichtselektiven COX-
Inhibitoren gelegt. Die in dieser Dissertation verwendete Methode erlaubt aus der gleichen
Blutprobe (ex-vivo) die Wirkstoffkonzentration (Pharmakokinetik) sowie die durch diese
Konzentration ausgelöste Hemmung beider Cyclooxygenasen (Pharmakodynamik) zu
bestimmen. Die Hemmung der COX-Aktivitäten wurde mittels enzymgekoppelten
Immunadsorptionstest (ELISA) bestimmt, wobei das koagulationsinduzierte Thromboxan B2
(TXB2) und das Lipopolysaccharid-induzierte Prostaglandin E2 (PGE2) ex-vivo und in-vitro im
menschlichen Vollblut als Marker der COX-1 und COX-2 Aktivität herangezogen wurden.
Aufgrund der Analyse von Ausmaß und Dauer der COX-2-Hemmung konnte die kleinste
effektive analgetische Dosis und das entsprechende Dosisintervall bestimmt werden. Durch
diese Erkenntnisse könnte man Überdosierungen und damit verbundene Nebenwirkungen
vermeiden. Hinsichtlich der Analyse der COX-1-Hemmung diente eine relevante Hemmung der
Thrombozytenaggregation (über 95%) als erhöhtes Risiko für Blutungen sowie als Prognose zur
Kardioprotektion eines Medikamentes.
Die gleichzeitige Analyse der Pharmakokinetik und Pharmakodynamik den untersuchten
Wirkstoffen nach der oralen Einnahme von Medikamenten ermöglichte verschiedene
Fragestellungen zu untersuchen.
Zunächst wurden die Untersuchungen zum Wirkungsmechanismus von Metamizol und
Paracetamol als Vertreter der nicht sauren Analgetika durchgeführt. Diese Medikamente üben
analgetische und antipyretische Effekte aus, weisen jedoch keine nennenswerte
entzündungshemmende Wirkkomponente sowie nur geringe gastrointestinale Nebenwirkungen
auf. Dagegen besitzen saure Analgetika potente analgetische, antipyretische und
antiphlogistische Eigenschaften und führen z.T. zu schweren gastrointestinalen und renalen
Nebenwirkungen.
Zusammenfassung 8
Metamizol ist ein Prodrug und wird im Organismus zu 4-Methylaminoantipyrin (4-MAA)
hydrolisiert, das weiter zu 4-Aminoantipyrin (4-AA) demethyliert wird. Die vorliegende Studie mit
Metamizol ist die erste, die ex-vivo beim Menschen die Selektivität von Metamizol zu den
peripheren COX-Enzymen sowie die Bedeutung der entsprechenden Metamizolmetaboliten (4-
MAA und 4-AA) untersuchte. Die Pharmakokinetiken der Metaboliten und die ex-vivo COX-
Hemmungen wurden bei fünf Probanden bestimmt, die einzelne orale Dosen von 500 oder
1000 mg Metamizol erhielten. Es zeigte sich, dass Metamizol in therapeutischer Dosierung eine
anhaltende Hemmung der beiden COX-Isoformen durch 4-MAA verursacht. Die COX-2-
Hemmung über 80% wurde bereits mit einer Dosierung von 500 mg erreicht. Nach Dosierung
von 1000 mg wurde eine über 95%-ige TXB2-Hemmung bei einigen Probanden gemessen.
Höhere Einzeldosen erscheinen aufgrund dieses Befundes nicht effektiver sein, erhöhen aber
das Risiko von unerwünschten Effekten.
Die Tatsache, dass Paracetamol funktionell einem selektiven COX-2 Hemmer ähnelt, führte zur
Überprüfung der Hypothese, dass Paracetamol bevorzugt über eine Hemmung von COX-2
analgetisch wirkt. Dafür wurden die Pharmakokinetik von Paracetamol und die ex-vivo COX-
Hemmung bei fünf Probanden bestimmt, die 1000 mg als einmalige orale Dosis erhielten.
Zusätzlich wurden in-vitro-Versuche durchgeführt, um festzustellen, ob die Expression oder
Aktivität von COX-2 in Blutmonozyten nach oraler Paracetamolgabe beeinflusst wird. In-vitro
sowie ex-vivo zeigte Paracetamol eine 4-fach höhere Selektivität für COX-2-Hemmung. Nach
der oralen Gabe des Medikamentes (1000 mg) wurde COX-2 (83%) im Vergleich zu COX-1
(56%) deutlich effektiver gehemmt. Mittels weiterer in-vitro Versuche konnte nachgewiesen
werden, dass Paracetamol die COX-2-Aktivität beeinflußt jedoch nicht die Expression von COX-
2 in Blutmonozyten.
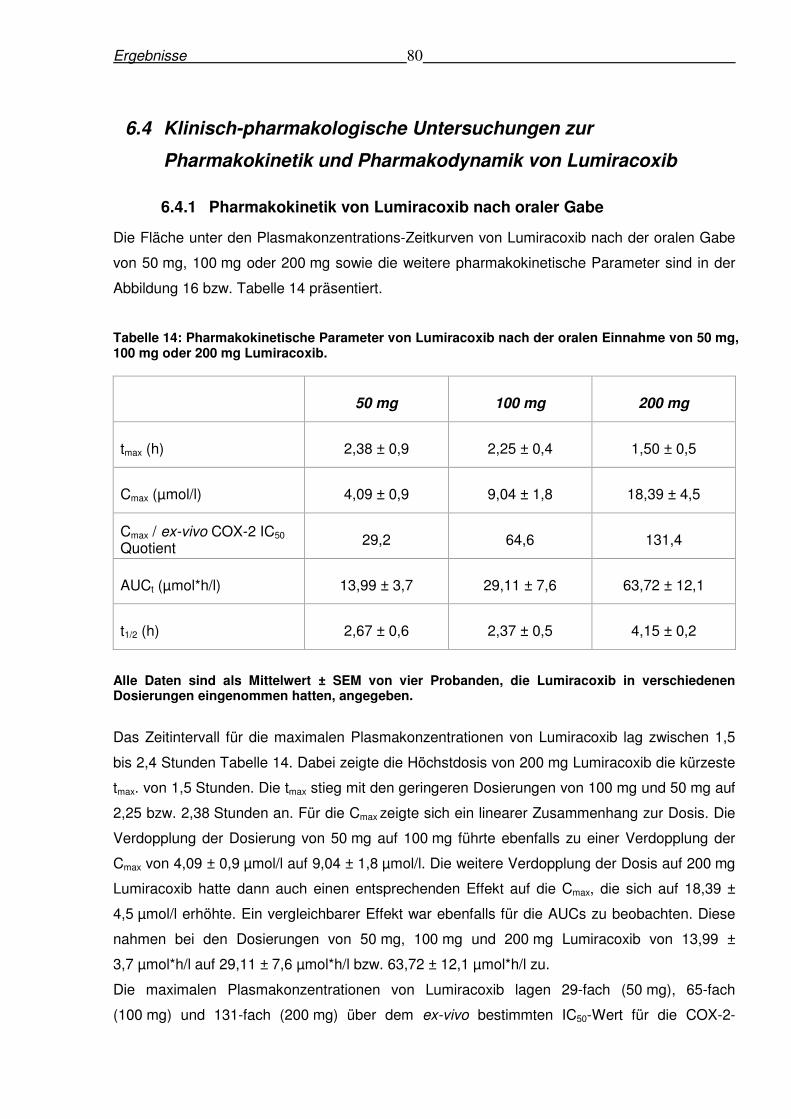
Weiterhin wurde die COX-2-Hemmung durch den selektiven COX-2-Inhibitor Lumiracoxib
untersucht. Unter den im Jahr 2007 verfügbaren COX-2-Inhibitoren war Lumiracoxib der einzige
Arzneistoff für den in einer großen Patientenstudie Vorteile hinsichtlich gastrointestinaler
Nebenwirkungen bei Patienten mit Osteoarthritis demonstriert werden konnten. Dennoch wurde
im August 2007 von Fällen berichtet, in denen sich schwerwiegende lebertoxische
Nebenwirkung nach Lumiracoxibeinnahme in höheren Dosierungen (200 mg, 400 mg pro Tag)
zeigte. In der vorliegenden Studie wurde untersucht, ob eine ausreichende schmerzlindernde
COX-2-Hemmung (80%) auch nach oraler Einnahme von 50 mg erreicht werden kann. Um
dieses Ziel zu erreichen wurde eine Methode zur Bestimmung von Lumiracoxib im
menschlichen Plasma entwickelt und validiert. Anschließend wurde eine randomisierte, dreifach
cross-over Studie mit vier Probanden, die einmalig 50 mg, 100 mg oder 200 mg Lumiracoxib
eingenommen hatten, durchgeführt. Es konnte gezeigt werden, dass Lumiracoxib innerhalb
eines Zeitraums von 1 bis 1,5 Stunden nach Applikation zur kompletten Hemmung der COX-2-
Aktivität bei allen untersuchten Dosierungen führte. Die schmerzlindernde Hemmung der COX-
Zusammenfassung 9
2 (80%) wurde bis zu 6 Stunden bereits bei der kleinsten untersuchten Dosierung festgestellt,
bis zu 8 Stunden bei 100 mg und bis zu 10 Stunden bei 200 mg. Daraus folgt, dass Lumiracoxib
in der Dosierung von 50 mg eine vollständige und anhaltende COX-2-Hemmung hervorruft.
Sinnvolle therapeutische Effekte müssten aufgrund dieser Befunde bereits in der Dosierung von
100 mg erreichbar sein.
Anschließend wurde eine Untersuchung mit dem nicht selektiven COX-Hemmer Naproxen
durchgeführt um die immer wieder postulierte kardioprotektive Wirkung von Naproxen zu
untersuchen. Es ist bekannt, dass Naproxen beide Enzyme effektiv hemmt, er hat dazu eine
lange Halbwertzeit, deswegen könnte eine langandauernde Hemmung von TXB2 Synthese über
95% ein möglicher Mechanismus für die kardioprotektive Wirkung sein. Dafür wurde eine Studie
mit vier Probanden durchgeführt, die 220 mg Naproxen Natrium zweimal täglich für 7 Tage
einnahmen. Die stärkste Hemmung der COX-1 ergab sich mit 94% nach der ersten
Tabletteneinnahme und mit 93% im Steady-state. Stärkere Hemmung als 95% wurde bei zwei
von vier Probanden nach der Einzeldosis und bei einem von vier Probanden im Steady - state
beobachtet. Daraus folgt, dass Naproxen in OTC (rezeptfreie) Dosierung mögliche aber keine
zuverlässige kardioprotektive Wirkung hat.
Summary 10
2 Summary
The nonsteroidal antiinflammatory drugs (NSAIDs) had been in use for years before their mode
of action became known. Their activity is based largely on the non-selective inhibition of the two
known cyclooxygenases (COX-1 and COX-2), which results in a reduction of prostaglandins
and thromboxanes, both of which are important mediators of pain and inflammation. In addition,
they play a protective role in many organs of the human body - for example in gastrointestinal
mucosa, in the kidney and blood vessels. In the 1990s, selective inhibitors of the COX-2
enzyme (coxibs) were developed as analgesics with a lower gastrointestinal toxicity than
traditional NSAIDs.
The major focus of the present dissertation was the investigation of the inhibition of the
peripheral cyclooxygenases in human blood cells by members of selective and nonselective
COX-inhibitors. The method employed in this study makes it possible to determine, in one and
the same blood sample, both the concentration of the active agent (pharmacokinetics) and the
inhibition of the two cyclooxygenases at that concentration (pharmacodynamics). The inhibition
of the COX activities was determined by the enzyme-linked immunosorbent assay (ELISA),
using the coagulation-induced thromboxane B2 (TXB2) and the lipopolysaccharide-induced
prostaglandin E2 (PGE2), measured ex vivo and in vitro in human whole blood, as indices of
COX-1 and COX-2 activity.
An analysis of the extent and duration of COX-2 inhibition made it possible to determine the
minimal analgesic dose and the dosing interval. This knowledge might help to avoid overdosing
related side effects. In the case of COX-1 inhibition a clinically relevant inhibition of platelet
aggregation was used as an indication of whether a drug may be the cause of an elevated risk
of bleeding or provide cardioprotection.
The simultaneous analysis of the pharmacokinetics and pharmacodynamics of the substances
investigated following oral administration of the drugs made it possible to study a number of
questions.
First, the mode of action of metamizol (dipyrone) and paracetamol (acetaminophen) as
representatives of the non-acidic analgesics was investigated. These drugs have analgesic and
antipyretic activities, but no appreciable anti-inflammatory effects and only mild gastrointestinal
side effects. In contrast, acidic analgesics have potent analgesic, antipyretic and anti-
inflammatory properties and in some cases may cause severe gastrointestinal and renal side
effects.
Metamizol is a prodrug and is hydrolysed in the organism to 4-methylamnioantipyrine (4-MAA),
which is then demethylated to 4-aminoantipyrine (4-AA). The present study is the first to
Summary 11
investigate, ex-vivo in humans, the selectivity of metamizol for the peripheral COX enzymes and
the significance of the corresponding metabolites (4-MAA und 4-AA). The pharmacokinetics of
the metabolites and ex vivo COX inhibition were investigated in volunteers receiving metamizol
at single oral doses of 500 or 1000 mg. It was found that metamizol in therapeutic doses brings
about sustained inhibition of both COX isoforms via 4-MAA. Even the lower dose (500 mg)
results in a COX-2 inhibition of more than 80%. At a dose of 1000 mg a greater than 95%
inhibition of TXB2 was seen in some volunteers. Higher single doses would thus not appear to
be more effective, but would increase the risk of undesirable side effects.
The fact that paracetamol (acetaminophen) has an action similar to that of a selective COX-
inhibitor, prompted us to investigate the hypothesis that its analgesic activity might be mediated
by the inhibition of COX-2. To this end ex vivo COX inhibition and the pharmacokinetics of
paracetamol were assessed in five volunteers receiving single oral 1000 mg dose. In addition, in
vitro experiments were done to establish whether the expression or activity of COX-2 in blood
monocytes is influenced by oral paracetamol. Paracetamol showed a 4-fold greater selectivity
towards COX-2 inhibition, both in vitro and ex vivo. After administration of the drug the activity of
COX-2 was inhibited considerably more strongly (83%) than that of COX-1 (56%). Further in
vitro experiments showed that while paracetamol influenced COX-2 activity it had no influence
on the expression of COX-2 in blood monocytes.
COX-2 inhibition by the selective COX-2 inhibitor lumiracoxib was also investigated. In a study
involving a large number of patients, lumiracoxib showed to have advantages in terms of
gastrointestinal side effects in patients with osteoarthritis. Nevertheless, in August 2007 a
number of patients were reported to have developed severe liver problems following high daily
doses of lumiracoxib (200 mg, 400 mg). The present study investigated the question whether an
oral dose of 50 mg could also achieve adequate COX-2 inhibition (80%). For this purpose a
method for the determination of lumiracoxib in human plasma was developed and validated. A
randomised cross-over study was then done on four volunteers who had taken a single dose of
50 mg, 100 mg or 200 mg lumiracoxib. It was shown that all three doses resulted in complete
inhibition of COX-2 activity within a period of 1 to 1.5 hours. The analgesic effect of COX-2
inhibition (over 80%) persisted for 6 hours at the smallest analysed dose investigated, for 8
hours at a dose of 100 mg, and for 10 hours at 200 mg. This implies that Lumiracoxib at a dose
of 50 mg produces complete and sustained COX-2 inhibition. On the basis of these data, a dose
of 100 mg should suffice to achieve an acceptable therapeutic effect.
A further investigation was carried out with a non-selective COX-inhibitor – naproxen, to study
its potential cardioprotective action. It is known that naproxen effectively inhibits both COX-
enzyme isoforms and has a long half life. Hence, a possible mechanism of its cardioprotective
action might be prolonged inhibition of TXB2 synthesis above 95%. To investigate this, four
Summary 12
subjects were given a twice daily dose of 220 mg naproxen sodium for 7 days. The maximum
COX-1 inhibition after the first dose was 94% and at steady state 93%. A greater than 95%
inhibition was observed in two of four volunteers at time of maximal plasma concentration after
the first dose and in one of four volunteers throughout the 12-hour dose interval at steady-state.
Thus, a cardioprotective effect of over-the-counter doses of naproxen is possible but not reliably
certain.
Summary 13
The results may be summarised as follows:
• Via its metabolite 4-MAA, metamizol (dipyrone) dose-dependently inhibits the two COX
enzymes to an appreciable extent in vitro and ex vivo in the blood of volunteers. The
rapid and profound inhibition of COX-2 in human blood monocytes supports the
assumption that the analgesic action of metamizol is mediated by peripheral
mechanisms. In this connection no COX-1/COX-2 selectivity was observed. With regard
to the observed COX-1 inhibition it is assumed that physicochemical factors (lack of
acidity) are responsible for the superior gastrointestinal tolerability of metamizol as
compared with acidic COX inhibitors. The good tissue penetration properties of
metamizol suggest that, in the CNS, too, it inhibits COX-2 sufficiently to produce an
analgesic effect. Since a single dose of 1 g metamizol has a prolonged effect, larger
doses would appear unnecessary.
• Paracetamol (acetaminophen) inhibited COX-2 by more than 80%, i.e. to a degree
comparable with that seen with non-steroidal anti-inflammatory drugs (NSAIDs) and
selective COX-2 inhibitors. On the other hand, a relevant COX-1 inhibition (at least
95%), as is necessary for the suppression of platelet function, was not observed. In vitro
and ex vivo paracetamol revealed an approximately 4-fold higher selectivity for COX-2.
In view of its substantial COX-2 inhibition, recently defined cardiovascular warnings
against the use of COX-2 inhibitors should also be considered for this substance.
• Lumiracoxib at a dose of 50 mg elicits complete and prolonged COX-2 inhibition. It
would appear meaningful to consider using lumiracoxib for pain therapy at therapeutic
doses of not more than 100 mg.
• The administration of naproxen sodium at OTC doses was associated with a profound
inhibition of both COX enzymes. Although in some volunteer an anticoagulant, i.e.
cardioprotective effect of COX-1 inhibition was observed already at OTC dose, reliable
cardioprotection probably requires a dose of ca. 1 g a day.
In the light of the results presented here, a critical review of the “usual” doses of
cyclooxygenase inhibitors would appear justified.
Einleitung 14
3 Einleitung
3.1 Cyclooxygenase
Die Cyclooxygenase (COX), auch Prostaglandin-H-Synthase (PGHS) genannt, ist ein Enzym,
das die Umsetzung der Arachidonsäure (AA) zu wichtigen Botenstoffen katalysiert. Der Name
der Arachidonsäure leitet sich von der Erdnuss (Arachis hypogaea) her, aus der die AA zuerst
isoliert wurde. Arachidonsäure (all-cis-5,8,11,14-Eicosatetraensäure) wird aus den
Membranphospholipiden durch die Phospholipase (PLA2) freigesetzt und kann über die
Cyclooxygenase und Lipoxygenase weiter verstoffwechselt werden. Durch die Katalyse der
Lipoxygenase entstehen Leukotriene, durch die Cyclooxygenase werden Prostaglandine und
Thromboxane synthetisiert. Neben Prostaglandinen werden aus Arachidonsäure auch andere
Fettsäureprodukte, wie z.B. Epoxyeicosatriensäuren (EET), Isoprostane (IP),
Endocannabinoide, Lipoxine (LX) und Hepoxiline (HX) gebildet. Sie alle werden unter dem
Begriff Eicosanoide zusammengefasst, da sie alle von der Fettsäure Prostansäure, die aus 20
(griech: eikosi) Kohlenstoffatomen aufgebaut ist, abgeleitet sind.
Abbildung 1: Schematische Darstellung der Arachidonsäurekaskade. Die Cyclooxygenasen COX-1 und COX-2 verstoffwechseln die Arachidonsäure zu Prostaglandinen, Prostazyklin und Thromboxan in zwei aufeinander folgenden Reaktionen. Dabei wird zuerst die AA durch die Endoperoxidsynthase-Reaktion zu Prostaglandin G2 (PGG2) umgewandelt. Dann vermittelt die Peroxidase-Reaktion die Umwandlung zu Prostaglandin H2. Inhibitoren der Cyclooxygenase hemmen dabei die Endoperoxidsynthase-Reaktion.
Die Cyclooxygenase ist ein bifunktionelles Enzym und besitzt daher zwei aktive Zentren
(Hawkey, 1999). Als Endoperoxid-Synthase (Cyclooxygenase-Reaktion) oxidiert und zyklisiert
PGH2
Arachidonsäure
Endoperoxidsynthase-Reaktion
PGG2
Peroxidase-Reaktion
PGF2α
PGD2
PGE2
PGI2
TXA2
COX
Einleitung 15
sie die AA zum Prostaglandin G2 (PGG2), wobei der charakteristische Cyclopentanring der
Prostaglandine entsteht. Ein zweites katalytisches Zentrum der COX, die Peroxidase, reduziert
die Peroxidgruppe am C-15 zum Alkohol. Das dadurch entstandene Prostaglandin H2 (PGH2)
wird durch gewebespezifische Enzyme weiter zu den verschiedenen Substanzen, wie
Prostaglandinen (PGE, PGF und PGD), Prostazyklin (PGI), und Thromboxan (TXA)
transformiert (Abbildung 1).
3.2 Isoformen der COX
Zurzeit sind zwei COX-Isoformen, die COX-1 und COX-2 bekannt. Beide Isoenzyme
unterscheiden sich in ihrer Gewebeverteilung und der Regulation ihrer Expression. Die COX-1-
Isoform wird in fast allen Geweben einschließlich Gastrointestinaltrakt, Thrombozyten,
Endothelzellen und Nieren konstitutiv exprimiert (Crofford, 1997) und ist vor allem an
physiologischen Funktionen, wie z.B. die Protektion der Magenschleimhaut, die Kontrolle des
renalen Blutflusses und die Plättchenaggregation beteiligt. Im Gegensatz zu der überall
vorkommenden COX-1 findet man COX-2 unter physiologischen Bedingungen nur in wenigen
Geweben wie ZNS (Beiche et al., 1996), Niere (Harris et al., 1994; Catella-Lawson et al., 1999;
McAdam et al., 1999), weibliche Reproduktionsgewebe (Kniss, 1999) und Magen (Zimmermann
et al., 1998). Alle anderen Gewebe produzieren das Enzym lediglich bei Bedarf, so nach
unterschiedliche Stimuli wie z.B. proinflammatorische Zytokine (Mitchell et al., 1994),
Wachstumsfaktoren (DeWitt et al., 1993), Endotoxine und Tumorpromotoren (Lee and Ip, 1992),
sowie als Antwort auf Stress durch chemische Reizung, Verletzung, UV-Bestrahlung, bakterielle
Infektion usw. Da es bei solchen Stressreaktionen meist zu Entzündungen kommt, gilt COX-2
als „proinflammatorisches Notfallenzym“. Zum anderen spielt die COX-2 eine wichtige Rolle bei
der Wund- bzw. Ulkusheilung und bei der Regulierung des Augeninnendruckes (Hinz and Brune,
2002).
Beide Isoformen zeigen eine ca. 60 %-ige Homologie in ihrer Aminosäuresequenz (Vane et al.,
1998). Sie bestehen aus ca. 600 Aminosäuren (Smith et al., 1996), wobei die COX-1 576
Aminosäuren, und die COX-2 587 Aminosäuren enthält. Beide Isoformen besitzen ähnliche
aktive Zentren, einen langen und engen, tunnelartigen, hydrophoben Kanal an dessen Ende die
Enzymreaktionen katalysiert werden (Garavito, 1996). Der wichtigste Unterschied liegt in der
Tertiärstruktur der katalytischen Zentren beider Enzyme. Durch den Austausch von Isoleucin
(Ile) gegen das kleinere Valin (Val) bei der COX-2 an der Position 523 wird die Bindungstasche
der COX-2 gegenüber der COX-1 um 25 % größer (Smith et al., 2000; Garavito et al., 2002;
Gupta et al., 2004). Die größere Bindungstasche der COX-2 wurde bei der Entwicklung von
selektiven COX-2-Inhibitoren (siehe Abschnitt 3.5.2) ausgenutzt (Hinz and Brune, 2002).
Einleitung 16
Zusätzlich zu diesen gut charakterisierten Isoformen wurden mehrere Splicevarianten von
COX-1 und COX-2 in verschiedenen Geweben nachgewiesen (Simmons et al., 2004). Für eine
katalytisch aktive COX-1-Variante, die in Hunden entdeckt wurde und durch Paracetamol
gehemmt wurde (Chandrasekharan et al., 2002), konnten in weiteren genetischen
Untersuchungen jedoch gezeigt werden, dass diese Variante beim Menschen nur ein Protein
mit geringer COX-Homologie und ohne COX-Aktivität darstellt (Kis et al., 2005). Daher ist
dieses Protein beim Menschen vermutlich nicht an der Prostaglandin-Biosynthese beteiligt.
3.3 Produkte des Cyclooxygenaseweges: Prostaglandine und
Thromboxane
Die Produkte, die unter der katalytischen Wirkung der COX-Enzyme gebildet werden, sind unter
anderen die Prostaglandine und Thromboxane. Sie werden auch unter dem Begriff Prostanoide
zusammengefasst. Die Prostanoide sind wegen ihrer raschen metabolischen Inaktivierung nur
für wenige Sekunden bis Minuten am Ort ihrer Bildung wirksam und müssen also ständig neu
synthetisiert werden.
3.3.1 Prostaglandin E2
Das Prostaglandin E2 (PGE2) ist ein primäres Prostaglandin, das aus PGH2 durch die PGE-
Synthase entsteht. Zurzeit sind drei PGE-Synthasen, die Membran-gebundene (mikrosomale)
PGE-Synthase-1 (mPGES-1) (Jakobsson et al., 1999), die Membran-gebundene (mikrosomale)
PGE-Synthase-2 (mPGES-2) (Murakami et al., 2003) und die cytosolische PGE-Synthase
(cPGES) bekannt (Tanioka et al., 2000). mPGES-1 ist funktionell an COX-2 gekoppelt und kann
genauso wie COX-2 durch proinflammatorische Stimuli (IL-1β) hochreguliert werden
(Stichtenoth et al., 2001; Korotkova et al., 2005). Die hauptsächlich im Gehirn vorkommende
zytostolische PGE-Synthase (cPGES) ist dagegen funktionell an COX-1 gebunden und wird wie
diese konstitutiv exprimiert (Tanioka et al., 2000). Die mPGES-2 kann zusammen sowohl mit
COX-1 als auch mit COX-2 vorkommen (Murakami et al., 2003). Es wird vermutet, dass die
physiologische PGE2-Funktionen vorwiegend durch cPGES / mPGES-2 / COX-1 und die
pathophysiologische PGE2-Funktionen durch das mPGES-1 / COX-2-System vermittelt werden
(Helliwell et al., 2004).
PGE2 kann viele und teilweise gegensätzlichen Wirkungen, wie z.B. eine Kontraktion oder
Relaxation von glatten Muskeln verursachen, eine Stimulation oder Suprimierung der Sekretion
von Hormonen hervorrufen. Diese sind auf die PGE-Rezeptor-Subtypen: EP1, EP2, EP3 und
EP4 zurückzuführen. (Bos et al., 2004). Eine Aktivierung des EP1-Rezeptors führt zur
Kontraktion glatter Muskelzellen in den Bronchien und dem Gastrointestinaltrakt (Narumiya et
al., 1999). Eine Aktivierung des EP2-Rezeptors ruft dagegen eine Relaxation von glatten
Einleitung 17
Muskelzellen in den Bronchien, im Gastrointestinaltrakt und in Blutgefäßen hervor. Eine
Aktivierung des EP3-Rezeptors führt zur Hemmung der Magensäureproduktion, einer
verstärkten Uteruskontraktion in der Schwangerschaft (Mutschler E., 2001). Darüber hinaus ist
der EP3-Rezeptor im Hypothalamus an der Induktion des Fiebers beteiligt (Nakamura et al.,
1999). Der EP4-Rezeptor vermittelt eine vermehrte Schleimsekretion des Magens und hält den
Ductus arteriosus Botalli offen (Mutschler E., 2001). Im Bereich des Intestinaltraktes ist u.a. das
PGE2 von zytoproliferativer Bedeutung. Es wurde berichtet, dass exogenes Prostaglandin E2 die
Proliferation von Stammzellen intestinaler Krypten stimuliert (Cohn et al., 1997).
3.3.2 Thromboxan B2
Das Thromboxan B2 (TXB2) ist ein stabiler Metabolit des Thromboxan A2 (TXA2), das in
physiologischer Umgebung eine äußerst instabile Verbindung ist. Das Thromboxan A2 wird von
der Thromboxansynthase, das zu den Cytochrom P450 Isoenzymen gehört (Wang and
Kulmacz, 2002), in den Thrombozyten gebildet. TXA2 vermittelt die Plättchenaggregation und
die Kontraktion glatter Muskelzellen in den Blutgefäßen und den Bronchien über den TXA2-
Rezeptor. Mit einer Halbwertzeit von 30 Sekunden zerfällt es nicht enzymatisch zu dem stabilen,
aber inaktiven TXB2 (Hamberg et al., 1975; Needleman et al., 1976), das man nach
Blutkoagulation messen kann. Im Urin wurden zahlreiche Metabolite des TXB2 nachgewiesen,
vor allem jedoch 2,3-Dinor-TXB2 and 11-Dehydro-TXB2.
3.4 Hemmung der Prostaglandinsynthese als
Wirkungsmechanismus antipyretischer Analgetika
Antipyretische Analgetika sind chemisch heterogene Medikamente, die sich aus
pharmakologischer und physikochemischer Sicht in zwei Gruppen einteilen lassen: saure und
nicht saure Antipyretika.
3.4.1 Saure Antipyretika
Zu den sauren Analgetika gehören die Pharmaka, die neben analgetischen und antipyretischen
Effekten in höherer Dosierung auch durch eine ausgeprägte antiphlogistische Wirkung
charakterisiert sind. Nach der Entdeckung der antiinflammatorischen Wirkung der
Glucocorticoide im Jahre 1949 werden die antiphlogistisch wirkenden sauren antipyretischen
Analgetika auch kollektiv als nicht steroidale Antiphlogistika bezeichnet ("nonsteroidal
antiinflammatory drugs", NSAIDs, NSAR). Die entsprechenden Substanzen sind schwache
Säuren (pKA-Wert 3-5,5) wie z.B. Acetylsalicylsäure, 2-Arylpropionsäuren (Profen),
Arylessigsäuren und heterozyklische Ketoenolsäuren. Sie weisen eine ausgeprägte Polarität
sowie eine hochgradige Bindung an Plasmaproteine (>90%) auf (Hinz and Brune, 2007).
Einleitung 18
1971 klärte John R. Vane den Wirkmechanismus von NSAR auf und teilte sich dafür 1982 den
Nobelpreis für Physiologie und Medizin mit den Drs. Sune Bergstrom, Bengt Samuelson. Vane
zeigte, dass die sauren antiphlogistischen Analgetika durch Hemmung der Cyclooxygenase zur
Reduktion proinflammatorischer Prostaglandine führen (Vane, 1971). Die Prostaglandine
erhöhen im traumatisierten Gewebe die Empfindlichkeit von feiner Nervenendigungen
(Nozizeptoren) auf andere Stimuli, indem sie normalerweise nicht erregbare polymodale
Rezeptoren („silent nociceptors“) in einen Zustand leichter Erregbarkeit überführen. Die NSAR
normalisieren die erhöhte Empfindlichkeit Nozizeptoren im geschädigten Gewebe. Die
entsprechenden analgetischen Substanzen üben insofern im strengen Sinne keinen
analgetischen Effekt aus, sondern wirken durch Modulierung der Sensitivität polymodaler
Nozizeptoren „antihyperalgetisch“ (Schaible and Grubb, 1993).
Die Arbeitsgruppe um Brune zeigte anhand radiographischer Studien, dass sich die sauren
Analgetika besonders im Blut, in Leber, Milz und Knochenmark, aber auch in Kompartimenten
mit saurem extrazellulärem pH-Wert, insbesondere in den entzündeten Geweben, in der Wand
des oberen Gastrointestinaltrakts und in den Sammelrohren der Nieren, anreichern (Brune,
1974; Brune et al., 1976; Rainsford et al., 1981). Die Akkumulation saurer Analgetika im
entzündeten Gewebe wird als entscheidender Faktor ihrer antiinflammatorischen Effekte
angesehen. Im entzündeten Gewebe verhindern die sauren NSAR die pathologische
Überproduktion von Prostaglandinen und damit die vor allem von PGE2 ausgelösten
entzündlichen Reaktionen (Brune and Zeilhofer, 1999).
3.4.2 Nicht saure Antipyretika
Nicht saure Analgetika (Paracetamol, Pyrazolinone) üben analgetische und antipyretische
Effekte aus, besitzen jedoch keine nennenswerte antiphlogistische Wirkkomponente. Ihre
Vertreter sind gering polar, neutral (Paracetamol) oder schwach alkalisch (Pyrazolinone) und
nur geringgradig an Plasmaproteine gebunden. Sie verteilen sich schnell und homogen im
Organismus und penetrieren auch die Blut-Hirn-Schranke (Brune et al., 1980). Ebenso weisen
sie nicht die für saure Analgetika typischen Nebenwirkungen im Magen-Darm-Trakt, in der
Niere und bei der Blutgerinnung auf.
Die genaueren Beschreibungen zum Wirkmechanismus von nicht sauren Analgetika erfolgen in
den Abschnitten 3.4.2.1 und 3.4.2.2. Jedoch ist der genaue analgetische
Wirkungsmechanismus dieser Substanzklasse bisher noch nicht vollständig geklärt.
Abbildung 2: Strukturformel von Metamizol Natrium.
Metamizol ist ein häufig verschriebenes, potentes spasmolytisches Analgetikum und
Antipyretikum (Carlsson et al., 1986; Vlahov et al., 1990). Ein antiphlogistischer Effekt tritt
jedoch erst bei höheren Dosierungen auf (Gelgor et al., 1992). Indikationen für dieses
Medikament sind starke akute und chronische Schmerzen, sowie Tumorschmerzen (Aventis,
2003). Auf Grund der krampflösenden Wirkung wird es zudem häufig bei kolikartigen
Schmerzen im Gastrointestinaltrakt und Urogenitaltrakt eingesetzt. Die Anwendung ist
beschränkt auf Schmerzzustände, bei denen eine Therapie mit NSAR nicht ausreichend ist.
Diese Anwendungsbeschränkung und die Einführung der Verschreibungspflicht im Jahre 1987
sind Folgen des in den 70er Jahren entdeckten Zusammenhangs zwischen der Einnahme von
Metamizol und dem Auftreten einer Agranulozytose. Als Folge der daraus entstandenen
kontroversen Diskussion über das Nutzen-Risiko-Potential von Metamizol ist der Arzneistoff in
vielen Ländern gänzlich vom Markt verschwunden. Währenddessen kommt dieser Arzneistoff
besonders in Entwicklungsländern zur Anwendung und ist dort ohne Verschreibung erhältlich.
Die Inzidenz dieser unerwünschten Effekte ist dennoch seltener als bei den NSAR (Maj and
Centkowski, 2004; Ibanez et al., 2005). Somit ist Metamizol ein weitgehend sicheres
Medikament, dessen analgetische Potenz mit schwachen Opiaten vergleichbar ist.
Metamizol ist in oralen und rektalen Darreichungsformen verfügbar (Aventis, 2003). Durch
Einführung einer Sulfonsäurestruktur und Bildung des Natriumsalzes konnte eine waserlösliche
Formulierung erzielt werden. Dies erlaubt auch intravenöse Applikationen dieses Medikamentes.
Da nach schneller intravenöser Injektion von Metamizol Schockreaktionen mit tödlichem
Ausgang beschrieben worden sind, sollte die Substanz langsam injeziert und unter strenger
Indikationsstellung i.v. eingesetzt werden.
Der Metabolismus von Metamizol im Körper führt über zahlreiche Biotransformationsprodukte.
Nach der oralen Verabreichung von Metamizol-Natriumsalz (Prodrug von Metamizol)
(Abbildung 2) erfolgt eine schnelle nicht enzymatische Hydrolyse im Gastrointestinaltrakt zu 4-
NN
NCH3
CH3 O
CH2SO3Na
CH3
Einleitung 20
Methylaminoantipyrin (4-MAA) (Abbildung 3, links), das nahezu vollständig resorbiert wird.
(Brune, 1988). Anschließend wird 4-Methylaminoantipyrin zu 4-Formylaminoantipyrin (4-FAAP)
enzymatisch oxidiert (Noda et al., 1976), sowie zu 4-Aminoantipyrin (4-AA) (Abbildung 3,
rechts) demethyliert, das durch eine polymorphe N-Acetyl-Transferase in der Leber zum
4-Acetylaminoantipyrin (4-AAA) acetyliert wird. Das Ausmaß dieses Metabolisierungsschrittes
ist vom Acetylierungsphänotyp des Patienten abhängig (Zylber-Katz et al., 1992). Diese vier
Hauptmetaboliten (4-MAA, 4-AA, 4-FAAP, 4-AAA) von Metamizol stellen ungefähr 65% der
verabreichten Dosis dar (Volz and Kellner, 1980). In sehr geringem Maße wird 4-Aminoantipyrin
auch zum atypischen Metaboliten 4-Hydroxyantipyrin umgesetzt (Reinhardt et al., 2006). Nach
hohen Dosen kann im Urin der rot gefärbte, jedoch harmlose Metabolit Rubazonsäure
nachgewiesen werden (Reinhardt et al., 2006).
Abbildung 3: Strukturformeln der untersuchten Metamizolmetaboliten: 4-Methylaminoantipyrin (links) und 4-Aminoantipyrin (rechts).
4-MAA und sein demethyliertes Derivat 4-AA werden als pharmakologisch wichtigste Metabolite
diskutiert (Weithmann and Alpermann, 1985). Sie sind, chemisch gesehen, schwach basische
Amine. Aus diesem Grund findet eine Anreicherung im leicht sauren, entzündeten Gewebe wohl
kaum statt. (Frolich et al., 1986). Daher kann man wahrscheinlich nur bei Dosierungen, die
oberhalb des therapeutischen Bereichs liegen, eine antiphlogistische Wirkung beobachten.
Weiterhin sind 4-MAA und 4-AA hydrophil und verteilen sich gleichmäßig in allen wässrigen
Kompartimenten. Sie werden bis zu 58 % bzw. 48 % an Plasmaproteine gebunden. Ihre
Plasmahalbwertzeiten variieren interindividuell sehr stark. Sie ist im Falle des 4-AA vom
Acetylierungsphänotyp abhängig und liegt deshalb im Bereich von 3,7 bis 8,1 Stunden. Die von
enzymatischen Reaktionen unabhängige Plasmahalbwertzeit des 4-MAA ist mit ca 2,5 Stunden
dagegen relativ kurz (Zylber-Katz et al., 1992).
Bis jetzt wurde angenommen, dass die antinozizeptive Wirkung von Metamizol wenigsten zum
Teil über einen Wirkmechanismus im zentralen Nervensytem vermittelt wird. (Carlsson et al.,
1986; Gelgor et al., 1992; Neugebauer et al., 1994; Shimada et al., 1994; Tortorici and Vanegas,
1995; Beirith et al., 1998). Da 4-MAA als auch 4-AA die Blut-Hirn-Schranke sehr gut
überwinden können, werden nach oraler Einnahme von Metamizol (1000 mg) maximale
Konzentrationen von 5-30 µg/ml (4-MAA) bzw. 1-7 µg/ml (4-AA) in der cerebrospinalen
NN
NCH3
CH3 O
CH3
H
NN
NCH3
CH3 O
H
H
Einleitung 21
Flüssigkeit erreicht. (Cohen et al., 1998). In Übereinstimmung damit wurde eine zeitabhängige
Senkung der Thromboxan B2-Konzentrationen in der cerebrospinalen Flüssigkeit bei Patienten,
die Metamizol erhalten hatten, nachgewiesen (Levy et al., 1998). Das unterstützt ebenfalls
einen zentral vermittelten Wirkungsmechanismus von Metamizol, der erstmalig von Tomek
bereits 1955 vermutet wurde. (Tomek, 1955). Die Hemmung der Prostaglandinsynthese wurde
zusätzlich dazu auch an anderen Orten des zentralen Nervensystems festgestellt. (Dembinska-
Kiec, 1976; Yaksh et al., 2001). Eine Bindung von 4-MAA, 4-AA bzw. Metamizol an Opioid-
Rezeptoren ist dagegen eher unwahrscheinlich. (Masuko et al., 1979; Goldenberg and De Boni,
1983).
Es wurde jedoch gezeigt, dass Metamizol auch die periphere Synthese von Eicosanoiden
einschließlich TXB2 in menschlichen Blutplättchen inhibiert. (Eldor et al., 1984; Weithmann and
Alpermann, 1985; Frolich et al., 1986; Geisslinger et al., 1996).
Eine periphere Wirkungsweise wurde 1985 von Lorenzetti postuliert, da er eine durch Carageen,
PGE2, Isoprenalin und Calciumchlorid ausgelöste Hyperalgesie durch Metamizol unterdrückten
konnte. Dabei wurde hauptsächlich PGE2 die Hauptrolle für die Hyperalgesie zugeschrieben.
(Lorenzetti and Ferreira, 1985). Weitere Studien konnten eine Hemmung der PGE2-Synthese in
menschlicher Magenschleimhaut (Peskar et al., 1986) und eine Absenkung der PGE2-
Konzentration im Magensaft nachweisen. (Frolich et al., 1986). Untersuchungen an Astrozyten
und Makrophagen zeigten, dass 4-MAA ein vergleichbares Hemmpotential der
Prostaglandinsynthese in beiden Zelltypen aufwies. (Lanz et al., 1986). In Übereinstimmung mit
diesen früheren Erkentnissen verhindert Metamizol, wie vor kurzem gezeigt wurde, die
enzymatischen Aktivitäten der beiden Cyclooxygenasen-Isoformen sowohl in gereinigten
Enzymen, als auch in verschiedenen peripheren Zellen (Campos et al., 1999).
Einleitung 22
3.4.2.2 Wirkungmechanismus von Paracetamol
Chemische Bezeichnung: 4´-Hydroxyacetanilid
Synonym: Acetaminophen
Summenformel: C8H9NO2
Molekulargewicht: 151,2 g/mol
Abbildung 4: Strukturformel von Paracetamol
Paracetamol, ist eines der weltweit am meisten verwendeten freiverkäuflichen, antipyretischen
und analgetischen Arzneimittel. Es wird als Therapeutikum erster Wahl gegen den
Osteoarthrose-assoziierten Schmerz (Schnitzer, 2002) empfohlen. Paracetamol gehört zu den
Aniliden und ist ein p-Aminophenol-Derivat. Es kann oral, rektal und intravenös appliziert
werden. Nach oraler Gabe erfolgt eine schnelle und vollständige Resorption sowie
gleichmäßige Verteilung in alle Gewebe. Die Inaktivierung erfolgt in der Leber durch die schnell
sättigbare Konjugation an Glucuron- oder Schwefelsäure. Ein geringerer Teil wird über das
Cytochrom P-450 Enzyme zum giftigen N-Acetyl-p-benzochinonimin metabolisiert, das über
Glutathion entgiftet und an Cystein gebunden ausgeschieden wird. Bei Überdosierung steht
nach gewisser Zeit nicht genügend Cystein für die Entgiftung zur Verfügung. Die Elimination
erfolgt mit einer HWZ von ca. zwei Stunden hauptsächlich renal. Paracetamol besitzt bei
therapeutischer Dosierung (4 g pro Tag bei Erwachsenen, 15 mg pro kg Körpergewicht am Tag
bei Kindern) keine nennenswerten Nebenwirkungen. Bei akuter Überdosierung (bei Dosen über
10 g pro Tag) tritt Hepatotoxizität auf, die zu schweren, meist tödlich verlaufenden
Leberzellnekrosen führt (Prescott et al., 1971). Trotz der Tatsache, dass Paracetamol schon vor
100 Jahren entdeckt wurde sowie auch seit ungefähr 50 Jahren vielfältig verwendet wird, ist der
komplette Wirkungsmechanismus immer noch unklar. Es wird allgemein angenommen, dass
Paracetamol zentral wirkt und bestenfalls ein schwacher Inhibitor der durch COX-1 und COX-2
vermittelten Prostaglandinsynthese ist (Botting, 2000). Dieses Konzept beruht auf der frühen
Arbeit von Flower und Vane, die zeigten, dass Paracetamol die Prostaglandin-Synthese im
Gehirn zehnfach stärker hemmt als in der Milz (Flower et al., 1972). Jedoch wurde diese
Entdeckung durch spätere Studien (Lanz et al., 1986; Warner et al., 2004) nicht bestätigt.
Außerdem stellte es sich heraus, dass Paracetamol keine messbare Inhibition der Bildung von
Prostaglandinen in aufgeschlossenen Zellpräparationen jedoch aber eine deutliche Hemmung
in intakten Zellen hervorruft (Graham and Scott, 2005).
Lucas et al. konnten zeigen, dass die Reduktion der nervalen Prostaglandin-Synthese durch
Paracetamol nicht auf transkriptioneller Ebene sondern auf Proteinebene reguliert wird (Lucas
et al., 2005). Als weitere Einflussgröße konnte die zelluläre Konzentration an Peroxiden
identifiziert werden. Bei niedriger Peroxidkonzentration ist das COX-Inhibitionspotential des
Einleitung 23
Paracetamols deutlich größer, da hohe Peroxidkonzentration die reduzierenden Eigenschaften
von Paracetamol antagonisieren und gleichzeitig die COX in einen höheren Oxidationszustand
überführen (Lucas et al., 2005). Auf Grundlage der pH-Abhängigkeit wurde ebenfalls
vorgeschlagen, dass Paracetamol keine Affinität zum aktiven Zentrum der COX habe, sondern
die COX durch Reduktion der aktiven oxidierten Form in die inaktive Form überführe (Lucas et
al., 2005). Da das Nervensystem über einen niedrigen Oxidationsstatus verfügt, könnte das
eine plausible Erklärung für die Wirkung im zentralen Nervensystem sein.
Auch wurde die Annahme, dass die Wirkung von Paracetamol auf die Hemmung von COX-3
zurückzuführen ist (Chandrasekharan et al., 2002), aus verschiedenen Gründen (Kis et al.,
2005) verworfen. Dies erfolgte vor allem aufgrund der Tatsache, dass die Existenz einer
funktionellen menschlichen COX-3 in Frage gestellt wurde. Im Falle der COX-3, ein Produkt des
COX-1 Genes, führt der Verbleib des Intron 1 in der COX-3 mRNA zu einem kürzeren, nicht
funktionellen Protein (Dinchuk et al., 2003; Schwab et al., 2003a).
Qin et al. berichteten über eine niedrige Expression von drei Splice-Varianten der COX-1 in
menschlichen Geweben (Qin et al., 2005). Es konnte aber kein relevanter Unterschied
zwischen der Paracetamol-vermittelten Hemmung von COX-1 und der Intron 1-enthaltenden
Splicevariante nachgewiesen werden. Obwohl die Aktivität der caninen COX-3 vollständig durch
Paracetamol inhibierbar ist, trifft diese Erklärung für die pharmakologischen Wirkungen des
Paracetamols beim Menschen nicht zu (Schwab et al., 2003b; Lucas et al., 2005). Deshalb wird
die Forschung auf dem Gebiet der Identifizierung der Wirkmechanismen von Paracetamol
weiter fortgesetzt.
Einleitung 24
3.5 Cyclooxygenasehemmer
Die fibersenkend wirkenden Analgetika werden als Cyclooxygenaseinhibitoren (COX-Inhibitoren,
COX-Hemmer) bezeichnet, weil sie ihre Wirkung über eine periphere und / oder zentrale
Hemmung der Cyclooxygenase entfalten. Je nach den bevorzugt gehemmten Isoenzymen
werden nicht selektive und selektive Inhibitoren unterschieden.
Die analgetischen, antipyretischen und antiphlogistischen Wirkungen nicht steroidaler
Antirheumatika (NSAR) beruhen im Wesentlichen auf einer unselektiven Hemmung der beiden
bekannten Cyclooxygenaseisoformen COX-1 und COX-2. Deswegen werden diese
Medikamente auch als nicht selektive Cyclooxygenase-Inhibitoren bezeichnet. Seit einiger Zeit
steht neben den traditionellen nicht steroidalen antiinflammatorischen Substanzen die Klasse
der selektiven COX-2-Hemmer (Coxibe) zur Verfügung. Eine Substanz wird als ein selektiver
COX-2-Inhibitor betrachtet, wenn sie die COX-2 hemmt, aber keine klinisch relevante COX-1-
Hemmung in therapeutischen Dosen verursacht.
Die 3D-Strukturen der COX-1 und COX-2, die mit Hilfe von Röntgenstrukturanalysen ermittelt
wurden, ermöglichten Einblicke in den molekularen Wirkmechanismus von NSAR. Diese
Erkenntnisse führten auch zu einem verbesserten Verständnis darüber wie die Selektivität von
Hemmstoffen erreicht wird. Die Daten zeigten, dass NSAR den hydrophoben Enzymkanal der
COX-1 in der Mitte blockieren. Dies wird über Wasserstoffbrückenbindungen zwischen dem
Arzneistoff und dem polarem Arginin an der Position 120 vermittelt. Arginin ist an dieser
Position sowohl in COX-1 als auch in COX-2 vorzufinden. Kritisch für die Selektivität von COX-
Hemmer ist die Aminosäureposition 523. In COX-1 befindet sich an dieser Stelle ein Isoleucin
wogegen in COX-2 ein Valin vorzufinden ist. Der Unterschied an dieser Position führt dazu,
dass die COX-2 selektiv durch Coxibe gehemmt werden kann (Hawkey, 1999).
Für die Bestimmung der COX-1 / COX-2-Selektivität hat sich der von Patrono und Mitarbeitern
beschriebene „Vollblutassay“ als vorteilhaft erwiesen. (Patrignani et al., 1994). Mit diesem
Assay (siehe Abschnitt 5.2.2) wird die COX-1- und COX-2-Aktivität eines Hemmstoffes an
klinisch relevanten humanen Blutzellen bestimmt (COX-1: Thrombozyten bei der Blutgerinnung,
COX-2: Monozyten nach Lipopolysaccharidstimulation). Dieser Test ermöglicht die
Untersuchung der COX-Selektivität unter physiologischen Bedingungen bei denen die
Plasmaproteinbindung berücksichtigt wird. (Patrignani et al., 1997).
Einleitung 25
3.5.1 Nicht selektive Cyclooxygenasehemmer
Untersuchungen mit dem in Abschnitt 5.2.2 beschriebenen Versuchsystem zeigten, dass keines
der derzeit therapeutisch eingesetzten NSAR die COX-2 selektiv hemmt (Patrignani et al.,
1997). Einige Substanzen (Diclofenac, Aceclofenac, Meloxicam) weisen eine bevorzugte
Hemmung des COX-2-Enzyms auf und können deshalb als präferentielle Hemmer der COX-2
betrachtet werden (Tabelle 1) (Hinz and Brune, 1999).
Tabelle 1: Relative Potenz einiger analgetischer, antipyretischer, antiphlogistischer Wirkstoffe hinsichtlich ihrer COX-1 und COX-2- Hemmungen.
Substanz COX-1 / COX-2 IC50-Ratio
Ibuprofen 0,50
Naproxen 0,56
S-Ketoprofen 0,61
Flurbiprofen 1,00
Indomethacin 1,90
Piroxicam 3,10
Meloxicam 11,16
Nimesulid 17,70
Diclofenac 18,90
Die IC50-Werte wurden ex-vivo mittels des humanen Vollblutassays (siehe Abschnitt 3.5) bestimmt. Die Daten stammen aus der Arbeit von Patrignani (Patrignani et al., 1997). Für die aufgeführten Substanzen ist das Verhältnis der COX-1- zur COX-2-Hemmung angegeben (Quotient IC50 COX-1 bzw. IC50 COX-2). Ist der Quotient größer als 1, dann ist diese Substanz durch eine vergleichsweise stärkeren COX-2-Hemmung gekennzeichnet.
Als unerwünschte Begleiterscheinungen der nicht selektiven COX-Inhibitoren, die sich
besonders im Bereich des oberen Gastrointestinaltraktes manifestieren, treten
Schleimhautschädigungen, petechiale Blutungen, Erosionen sowie Ulzerationen auf (Graham
and Smith, 1988; Langman, 1989; McCarthy, 1989; Cheatum et al., 1999).
Einleitung 26
3.5.1.1 Thrombozytenhemmende und potenziell kardioprotektive Wirkung
Das Arylpropionsäurederivat Naproxen ist ein gut verträgliches NSAR mittlerer Wirkstärke. Es
wird nach oraler Gabe des Natriumsalzes rasch und gut resorbiert, maximale
Plasmakonzentration wird bereits nach einer Stunde erreicht. Infolge der langen
Serumhalbwertzeit von 13-18 Stunden braucht Naproxen auch bei chronischen entzündlichen
Schmerzen nur ein- bis zweimal täglich eingenommen zu werden.
Im Laufe der letzten Jahre entwickelten sich Bedenken hinsichtlich der kardiovaskulären
Sicherheit von selektiven Cyclooxygenase-2-Hemmern und nicht steroidalen Antirheumatika bei
höheren Dosierungen (FitzGerald and Patrono, 2001; Tegeder and Geisslinger, 2006; Hinz and
Brune, 2007). Tatsächlich konnte in gut geplanten, Plazebo-kontrollierten Studien gezeigt
werden, dass die langfristige Einnahme von COX-2-Hemmern bei Patienten, die anamnestisch
ein kolorektales Adenom aufwiesen, mit erhöhten kardiovaskulären Nebenwirkungen in
Verbindung gebracht wurden (Bresalier et al., 2005; Solomon et al., 2005). Außerdem konnte
bei Patienten mit einem erhöhten kardiovaskulären Risiko die Kurzzeitbehandlung mit
Valdecoxib oder Parecoxib mit einer höheren Anzahl von starken thromboembolischen
Ereignissen assoziiert werden (Nussmeier et al., 2005).
Obwohl keine plazebokontrollierten, randomisierten Studien durchgeführt wurden, um das
kardiovaskuläre Risiko von traditionellen NSAR zu untersuchen, zeigten bevölkerungsbasierte
Fall-Kontroll-Studien deutlich, dass der gegenwärtige Gebrauch von COX-2 Hemmstoffen und
traditionellen NSAR mit einer erhöhten Anzahl von Myokardinfarkten assoziiert war (Hippisley-
Cox and Coupland, 2005). Außerdem demonstrierte die kürzlich veröffentlichte MEDAL-Studie,
dass NSAR (Diclofenac) und COX-2-Hemmer (Etoricoxib) ein vergleichbares kardiovaskuläres
Risikopotential aufwiesen (Cannon et al., 2006). Deshalb wurde vorgeschlagen, dass keine
Unterscheidung zwischen diesen Gruppen von Medikamenten hinsichtlich der kardiovaskulären
Toxizität notwendig ist. Die ursprüngliche Annahme, dass NSAR durch die Hemmung der
prothrombotischen COX-1 zur Senkung des kardiovaskulären Risikos führen sollte, konnte nicht
bestätig werden, weil die COX-1-Aktivität in Thrombozyten über 95% unterdrückt werden muss,
um die Hemmung der Thrombozytenaggregation hervorzurufen (Reilly and FitzGerald, 1987).
Einleitung 27
Jedoch wurde in einer Meta-Analyse gezeigt, dass das Auftreten von ernsten vaskulären
Ereignissen von Naproxen mit Placebo vergleichbar ist (Kearney et al., 2006).
3.5.2 Selektive Cyclooxygenasehemmer
Viele unerwünschte Effekte der NSAR wie z.B. Magen- und Darm-Ulzerationen, Blutungen und
Thrombozytenfunktionsstörungen wurden mit einer Hemmung der COX-1-abhängigen
protektiven Prostaglandine assoziiert, wohingegen die erwünschten therapeutischen,
antientzündlichen, schmerzlindernden und fiebersenkenden Effekte dieser Verbindungen mit
einer Hemmung der COX-2-abhängigen proinflammatorischen Prostaglandine in Verbindung
gebracht wurden. Als Folge entstand die Hypothese, dass die selektive Hemmung der COX-2
die traditionellen therapeutischen Wirkungen der NSAR ohne deren unerwünschten
Nebenwirkungen hervorrufen könnte. Diese Idee führte letztlich zur Entwicklung von selektiven
COX-2-Hemmstoffen.
Die selektiven COX-2-Inhibitoren, die sich zurzeit in klinischer Anwendung befinden, sind
Celecoxib (Sulfonamid), Etoricoxib (Methylsulfon) und Parecoxib, ein intravenös applizierbares
Prodrug von Valdecoxib (Sulfonamid).
Tabelle 2: Relative Potenz der selektiven COX-Inhibitoren hinsichtlich ihrer COX-1- und COX-2- Hemmungen.
Substanz COX-1 / COX-2 IC50-Ratio
Celecoxib 30
Etoricoxib 344
Lumiracoxib 515
Die Daten wurden ex-vivo mit dem humanen Vollblutassay generiert (Brune and Hinz, 2004). Lumiracoxib weist dabei die höchste COX-2-Selektivität auf, gefolgt von Etoricoxib und Celecoxib.
Die nicht saure COX-2-Hemmer Celecoxib und Etoricoxib verteilen sich homogen im Körper.
Aufgrund seiner hohen Lipophilie wird Celecoxib relativ langsam und unvollständig resorbiert.
Etoricoxib wird dagegen schnell resorbiert, jedoch langsam aus dem Körper eliminiert (t1/2: ca.
20–26 h). (Hinz et al., 2006). Die unterschiedlichen Pharmakokinetiken der Coxibe sind
Grundlage für die verschiedenen Indikationen der entsprechenden COX-2-Hemmstoffe. So ist
z.B. Celecoxib für die Behandlung von akuten Schmerzen aufgrund seiner langsamen
Resorption und des variablen First-pass-Metabolismus ungeeignet. Hingegen erscheint
Etoricoxib aufgrund seiner langen Plasmahalbwertszeit besonders für Indikationen, bei denen
eine verlängerte Wirkdauer (akute Gichtarthritis, rheumatische Arthritis) erforderlich ist, sehr
Einleitung 28
vielversprechend. Parecoxib, als ein intravenös applizierbares Coxib ist für die Behandlung des
postoperativen Schmerzens gut geeignet. Ein anderer hochselektiver Cyclooxygenase-2-
Hemmer, der für die symptomatische Behandlung von Osteoarthritis und akutem Schmerz
verwendet wurde ist das Phenylacetylsäurederivat Lumiracoxib (Bannwarth and Berenbaum,
2007). Lumiracoxib wurde in Januar 2007 in Deutschland zugelassen, jedoch bereits im
Dezember 2007 wieder vom Markt zurückgenommen.
Einleitung 29
F Cl
NH
COOH
H3C
3.5.2.1 Lumiracoxib ein hochselektiver COX-2-Hemmer
Die Probanden erhielten im Abstand von zwei Wochen einmalig Prüfdosierungen (50 mg,
100 mg bzw. 200 mg) von Lumiracoxib. Die Prüfmedikation wurde nüchtern mit 150 ml Wasser
zwischen 8:00 und 9:00 Uhr verabreicht. Venöse Blutproben wurden mit einer Verweilkanüle
unmittelbar vor der Tabletteeinnahme (Zeitpunkt 0) und 0,25; 0,75; 1,5; 3; 5; 8, 10 und 24
Experimenteller Teil 55
Stunden nach der Medikation abgenommen. Für die Bestimmung von Lumiracoxib wurden die
heparinisierten Blutproben zentrifugiert und die Aliquote vom Plasma wurden bei -20°C bis zu
einer Woche gelagert.
5.3.3.6 Hemmung der COX-2-Aktivität durch Lumiracoxib ex-vivo
Sofort nach den Blutentnahmen wurden heparinisierte Vollblutproben mit 10 µg/ml LPS für 24
Stunden bei 37° inkubiert. Die Blutplättchen COX-1-Aktivität wurde mit Zusatz von Aspirin (10
µg/ml) am Anfang von Inkubation unterdrückt. Das Plasma wurde durch Zentrifugation
abgetrennt und PGE2- Konzentrationen wurden bestimmt.
5.3.3.7 Bestimmung von Lumiracoxib in menschlichem Plasma.
Lumiracoxib wurde nach einer neu entwickelten und validierten Methode mittels HPLC–UV
(Cheremina et al., 2006) analysiert (Abschnitt 6.3).
Für die Probenaufarbeitung und Kalibrierungsstandard wurden 0,5 ml 0,1 M Salzsäure und 0,1
ml interner Standard (100 µg/ml Nifluminsäure in Methanol) zu 1 ml Plasma hinzugefügt.
Anschließend wurde 4 ml Mischung n-Hexan:Diethylether 70:30 (v/v) gegeben. Die
Reagenzgläser mit der Mischung wurden in einem Shaker für 10 min über Kopf geschüttelt und
anschließend in einer Kühlzentrifuge (Eppendorf 5810R) bei 4000 r.p.m. für eine Dauer von 10
Minuten bei 4°C zentrifugiert. Die organische Phase wurde in einen Reagenzglas überführt und
wurde bei Raumtemperatur unter einem Strom des Stickstoffes evaporiert. Der Rückstand
wurde in 150 µl Methanol:Wasser 50:50 (v:v) aufgelöst, vortext und 100 µl wurde in HPLC-
Anlage injeziert. Die Substanzen wurden mittels RP-Säule CC 125/4 Nucleosil 120-3 C8
(Machery-Nagel, Düren, Germany) und eine Vorsäule CC 8/4 Nucleosil C8 3µ getrennt. Die
mobile Phase bestand aus einer Mischung vom Acetonitril und 0,05 % Trichloressigsäure 35:65
(v/v). Der Fluss war 1,0 ml/min. UV-Detektion wurde auf 270 nm gesetzt. Lumiracoxib wurde bei
16,9 min und Nifluminsäure bei 10,4 min eluiert. Linearität der Standardkurven wurde über
einen Konzentrationsbereich von 10 bis 10 000 ng/ml, mit Regressionskoeffizienten größer als
0,997 bewiesen.
5.3.3.8 Statistik
Potenziale Unterschiede in pharmakodynamischen Parametern in verschiedenen Zeitintervalen
(0,25 bis 5, 8, 12 oder 24 Stunden nach Medikamentverabreichung für durchschnittliche
Hemmungen; 0 bis 5, 8, 12 oder 24 Stunden nach der Medikamentverabreichung für AWEC-
Werte) der drei Gruppen (50 mg gegen 100 mg; 50 mg gegen 200 mg; 100 mg gegen 200 mg)
Experimenteller Teil 56
wurden vom Student´s paired t-Test bewertet. A P-Wert weniger als 0,05 wurde als signifikant
betrachtet. Statistische Analysen wurden mit PRISM® Version 3.0 (GraphPad, San Diego, CA,
USA) durchgeführt.
Experimenteller Teil 57
5.3.4 Klinisch-pharmakologische Untersuchungen zur Pharmakokinetik
und Pharmakodynamik von Naproxen
5.3.4.1 Studienziel
Die vorliegende Studie untersuchte, ob Dosierungen von freiverkäuflichem (OTC) Naproxen
Natrium eine für Thrombozytenaggregation ausreichende TXB2-Hemmung nach einmaliger
Gabe und im Steady-state (200 mg Naproxen zweimal am Tag für 7 Tage) und dadurch
mögliche kardioprotektive Wirkung erzeugen können.
5.3.4.2 Studiendesign
Es handelte sich um eine mehrfachdosierte nichtverblindete Probandenstudie, die am Lehrstuhl
für Pharmakologie und Toxikologie der Universität Erlangen-Nürnberg im Dezember 2006 - Mai
2007 durchgeführt wurde.
5.3.4.3 Probandenpopulation
In die Studie wurden 4 Probanden (drei Männer und eine Frau) im Alter von 26 bis 66 Jahren
(durchschnittliches Alter: 44,8 Jahre) mit einer Körpermasse 75,8 ± 7,1 kg (mean ± SEM)
eingeschlossen. Alle Probanden waren die approbierten Ärzte am Institut für Experimentelle
und Klinische Pharmakologie und Toxikologie. Voraussetzung für die Teilnahme war das
Einverständnis des Probanden und keine Einnahme von NSAR zwei Wochen vor der ersten
Blutentnahme. Die Ausschlusskriterien entsprachen den in der Fachinformation für Aleve®
genannten Gegenanzeigen.
5.3.4.4 Studienmedikation
Als Studienmedikation wurde Naproxen Natrium 220 mg (entspricht den 200 mg Naproxen,
Aleve®, Bayer, Germany), das von einer öffentlichen Apotheke bezogen wurde. Die Probanden
nahmen 1 Tablette zweimal täglich für 7 Tage ein.
5.3.4.5 Studienplan
Am 1. Tag wurde die Prüfmedikation nüchtern zwischen 8:00 und 8:30 Uhr verabreicht. Venöse
Blutproben wurden mit einer Verweilkanüle unmittelbar vor der Tabletteeinnahme (Zeitpunkt 0)
und 1, 3, 6 und 12 Stunden nach der Medikamentgabe abgenommen. In den Tagen 2 bis 5
wurde Blut sofort vor der Einnahme der Morgendosis, d.h. 12 Stunden nach der Abenddosis
Experimenteller Teil 58
abgenommen. Um Recovery von COX-Enzymen nach der Beendigung der
Naproxeneinnahmen zu untersuchen, wurden zusätzliche Blutproben am Tag 8 (12 und 24
Stunden nach der letzten Dosis) und Tag 9 (36 Stunden nach der letzten Dosis) abgenommen.
Tabelle 3: Protokoll über Einnahme von Naproxen Natrium 220 mg 2 Mal/Tag und Blutentnahme
Tag 1 Mo
2 Di
3 Mi
4 Do
5 Fr
6 Sa
7 So
8 Mo
9 Di
Zeit der Tabletteeinahme
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
8:00
20:00
Zeit der Blutentnahme
8:00
9:00
11:00
14:00
20:00
8:00
8:00
8:00
8:00
8:00
20:00
8:00
Für die Naproxenbestimmung wurden die heparinisierten Blutproben zentrifugiert und die
Plasmaaliquoten wurden bei -20°C bis zu zwei Wochen gelagert.
5.3.4.6 Hemmung der COX-Aktivitäten durch Naproxen 200 mg zweimal am
Tag für 7 Tage bei menschlichen Probanden.
COX-1: Sofort nach Blutentnahme wurden Vollblutproben ohne Antikoagulans für 1 Stunde bei
37°C inkubiert, anschließend zentrifugiert, und die Serum-TXB2-Konzentrationen wurden
bestimmt.
COX-2: Sofort nach den Blutentnahmen wurden heparinisierte Vollblutproben mit 10 µg/ml LPS
für 24 Stunden bei 37° inkubiert. Die Blutplättchen COX-1-Aktivität wurde mit Zusatz von Aspirin
(10 µg/ml) am Anfang von Inkubation unterdrückt. Das Plasma wurde durch Zentrifugation
abgetrennt und PGE2-Konzentrationen wurden bestimmt.
5.3.4.7 Bestimmung von Naproxen in menschlichem Plasma
Für die Kalibrierungsstandard und Probenaufarbeitung für Naproxenbestimmung wurden 0,1 ml
1 M Salzsäure und 0,025 ml interner Standard (200 µg/ml Flurbiprofen in Methanol) zu 0,25 ml
Plasma hinzugefügt. Anschließend wurde 3 ml Mischung n-Hexan:Diethylether (80:20 v/v)
Experimenteller Teil 59
gegeben, die Reagenzgläser mit der Mischung in einem Shaker für 5 min über Kopf geschüttelt
und anschließend in einer Kühlzentrifuge (Eppendorf 5810R) bei 4000 r.p.m. für eine Dauer von
5 Minuten bei 4°C zentrifugiert. Die organische Phase wurde in einen Reagenzglas überführt
und wurde bei Raumtemperatur unter einem Strom des Stickstoffes evaporiert. Der Rückstand
wurde in 200 µl Fließmittel aufgelöst und 50 µl wurde für HPLC-Analyse benutzt. Die
Substanzen wurden mittels RP-Säule CC 125/4 Nucleosil 120-3 C8 (Machery-Nagel, Düren,
Germany) und eine Vorsäule CC 8/4 Nucleosil C8 3µ getrennt. Die mobile Phase bestand aus
einer Mischung vom Methanol und Wasser 60:40 (v/v) (pH = 3,1). Der Fluss war 0,7 ml/min.
UV-Detektion wurde auf 230 nm gesetzt. Naproxen wurde bei 4,5 min und Ibuprofen bei 6,5 min
eluiert. Linearität der Standardkurven wurde über einen Konzentrationsbereich von 1 bis 50
µg/ml mit Regressionskoeffizienten größer als 0,999 bewiesen.
Ergebnisse 60
6 Ergebnisse
6.1 Klinisch-pharmakologische Untersuchuingen zur
Pharmakokinetik und Pharmakodynamik von Metamizol
6.1.1 In-vitro Wirkungen von 4-MAA und 4-AA auf die COX-1 und COX-2-
Aktivität in menschlichem Vollblut
Durch Zugabe von Metamizol in steigender Konzentration zum Vollblut und anschliessender
Bestimmung von Thromboxan und Prostaglandin E2, lasst sich das Ausmaß der peripheren
COX-1- und COX-2-Inhibition abschätzen. Der Versuch zur Bestimmung der hemmenden
Wirkung der Metamizolmetaboliten (4-MAA und 4-AA) auf die COX-1- bzw. COX-2-Aktivität
zeigte, dass die beiden Metabolite eine konzentrationsabhängige Hemmung der beiden COX-
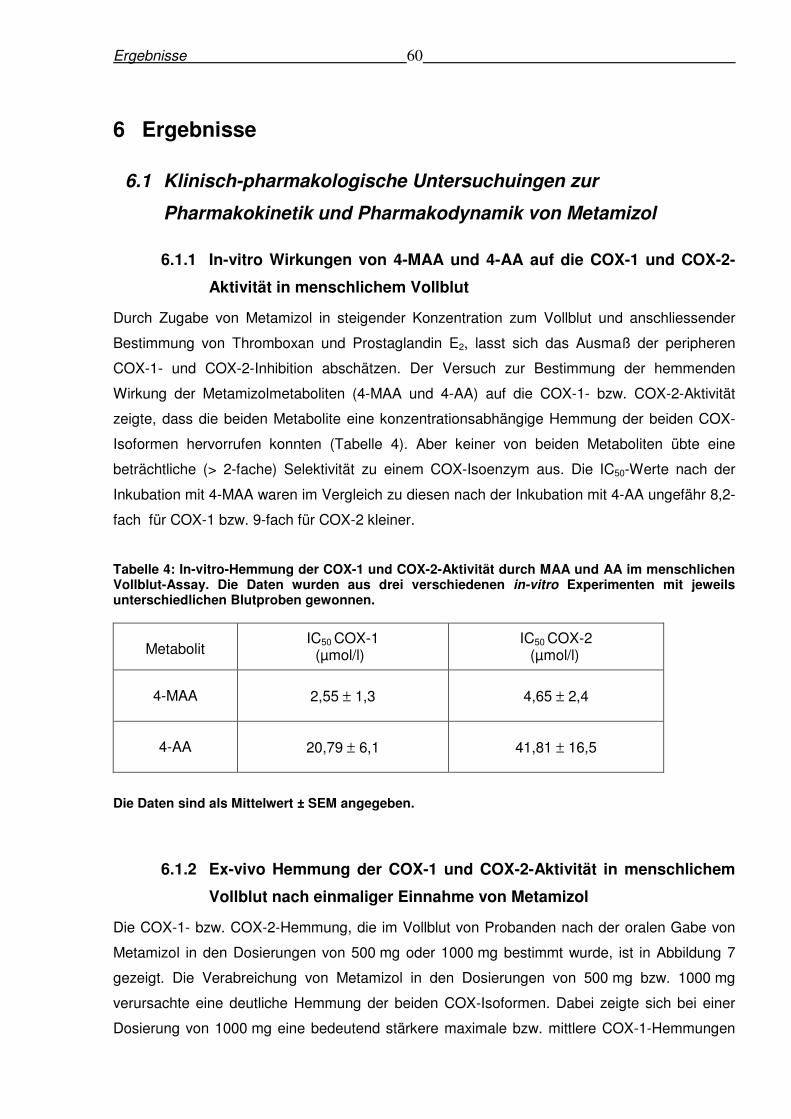
Isoformen hervorrufen konnten (Tabelle 4). Aber keiner von beiden Metaboliten übte eine
beträchtliche (> 2-fache) Selektivität zu einem COX-Isoenzym aus. Die IC50-Werte nach der
Inkubation mit 4-MAA waren im Vergleich zu diesen nach der Inkubation mit 4-AA ungefähr 8,2-
fach für COX-1 bzw. 9-fach für COX-2 kleiner.
Tabelle 4: In-vitro-Hemmung der COX-1 und COX-2-Aktivität durch MAA und AA im menschlichen Vollblut-Assay. Die Daten wurden aus drei verschiedenen in-vitro Experimenten mit jeweils unterschiedlichen Blutproben gewonnen.
Metabolit
IC50 COX-1 (µmol/l)
IC50 COX-2 (µmol/l)
4-MAA
2,55 ± 1,3 4,65 ± 2,4
4-AA
20,79 ± 6,1 41,81 ± 16,5
Die Daten sind als Mittelwert ± SEM angegeben.
6.1.2 Ex-vivo Hemmung der COX-1 und COX-2-Aktivität in menschlichem
Vollblut nach einmaliger Einnahme von Metamizol
Die COX-1- bzw. COX-2-Hemmung, die im Vollblut von Probanden nach der oralen Gabe von
Metamizol in den Dosierungen von 500 mg oder 1000 mg bestimmt wurde, ist in Abbildung 7
gezeigt. Die Verabreichung von Metamizol in den Dosierungen von 500 mg bzw. 1000 mg
verursachte eine deutliche Hemmung der beiden COX-Isoformen. Dabei zeigte sich bei einer
Dosierung von 1000 mg eine bedeutend stärkere maximale bzw. mittlere COX-1-Hemmungen
Ergebnisse 61
0 2 4 6 8 10 12
0
25
50
75
100
500 mg Metamizol1000 mg Metamizol
Zeit (h)
CO
X-1
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
95% Hemmung
im Vergleich zur Gabe von 500 mg Metamizol (Abbildung 7). Überdies war die AWEC in der
500 mg Gruppe im Vergleich zum jeweiligen Wert in der 1000 mg Gruppe (Tabelle 5)
bedeutend niedriger.
Ebenfalls konnte moderate Zunahme der AWEC für die COX-2 in der 1000 mg Gruppe im
Vergleich zur 500 mg Gruppe beobachtet werden (Tabelle 5). Jedoch war dieser Unterschied
statistisch nicht signifikant. Ebenso gab es keinen statistisch signifikanten Unterschied für die
COX-1- und COX-2-Hemmung innerhalb einer Dosierungsgruppe.
A
B
Abbildung 7: Die Hemmung der COX-1- bzw. COX-2-Aktivität nach oraler Einnahme von Metamizol in den Dosierungen von 500 mg und 1000 mg. Die eingefügte horizontale Linie stellt in Abbildung A die COX-1-Hemmung von 95% bzw. in Abbildung B die COX-2-Hemmung von 80% dar.
0 2 4 6 8 10 12
0
20
40
60
80
100
500 mg Metamizol1000 mg Metamizol
80% Hemmung
Zeit (h)
CO
X-2
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
Ergebnisse 62
Tabelle 5: Pharmakodynamische Parameter der Metamizol-vermittelten COX-1- und COX-2-Hemmung bei fünf Probanden nach der oralen Einnahme von entweder 500 mg oder 1000 mg Metamizol.
COX-1-Hemmung
Zeit bis maximale Hemmung
(h)
Maximale Hemmung
(%)
Durchschnittliche Hemmung 0-12 h
(%)
AWEC0-12 h (%⋅h)
Metamizol 500 mg
1,1 ± 0,2
94,4 ± 0,8
72,9 ± 5,0
861,9 ± 72,0
Metamizol 1000 mg
0,9 ± 0,2
97,4 ± 0,4**
85,5 ± 3,4*
1020,3 ± 55,0*
COX-2-Hemmung
Zeit bis maximale Hemmung
(h)
Maximale Hemmung
(%)
Durchschnittliche Hemmung 0-12 h
(%)
AWEC0-12 h (%⋅h)
Metamizol 500 mg
3,0 ± 1,3
86,6 ± 3,6
62,0 ± 7,8
759,4 ± 69,7
Metamizol 1000 mg
1,7 ± 0,4
94,0 ± 1,8
73,8 ± 2,9
860,9 ± 56,9
Die Werte sind als Mittelwerte ± SEM von fünf Probanden angegeben. * P < 0,05; ** P < 0,01 vs. 500 mg Metamizol-Gruppe (Student´s paired t-test).
6.1.3 Pharmakokinetik von Metamizolmetaboliten nach oraler Gabe von
Metamizol in den Dosierungen von 500 mg und 1000 mg
Nach der oralen Applikation von entweder 500 mg oder 1000 mg Metamizol an jeweils fünf
Probanden wurden die AUCs und Plasmakonzentrations-Zeitkurven von 4-MAA und 4-AA
bestimmt. Die Plasmakonzentrations-Zeitkurven sind in Abbildung 8 gezeigt.
Die maximalen Plasmakonzentrationen von 4-MAA wurden bei der Dosierung 500 mg oder
1000 mg Metamizol nach 1,7 und 1,2 Stunden gemessen, wohingegen 4-AA die maximalen
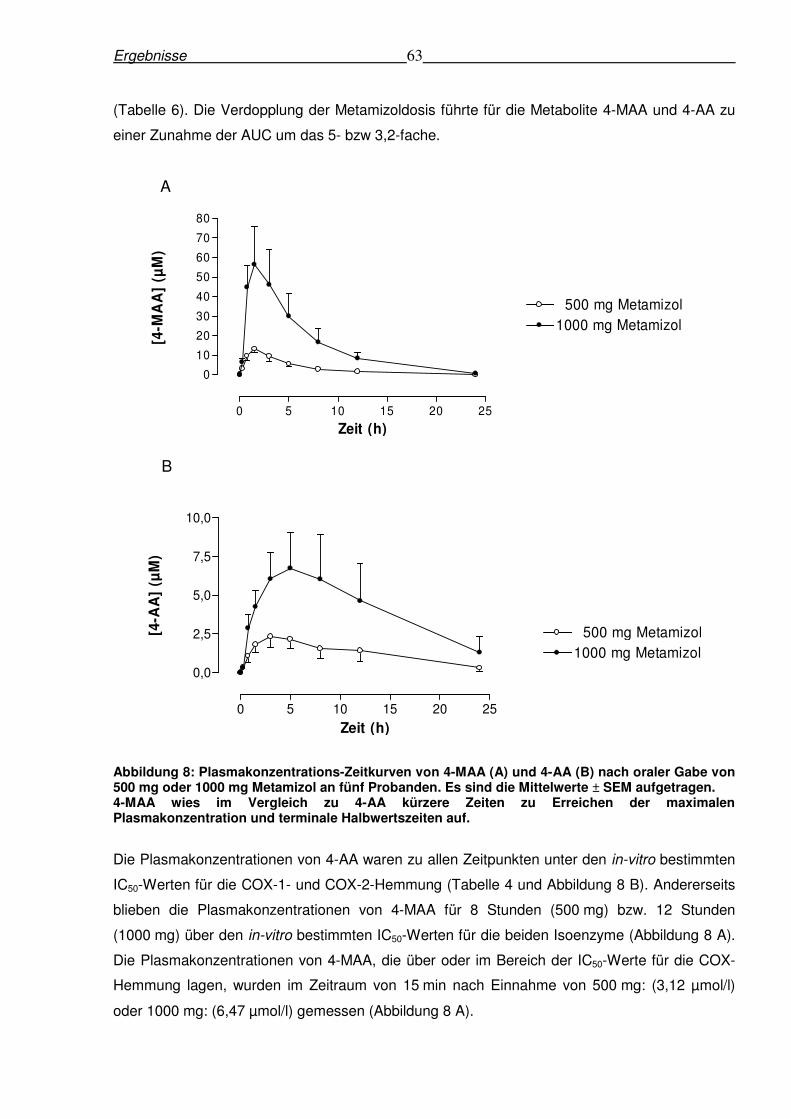
Plasmakonzentrationen erst nach 3,1 bzw. 5,2 Stunden erreichte (Tabelle 6). Überdies fielen im
Vergleich zu 4-MAA die entsprechenden Plasmakonzentrationen von 4-AA aufgrund der
terminalen Halbwertzeiten von 6,3 h. (500 mg) und 5,7 h (1000 mg) deutlich langsamer ab
Ergebnisse 63
(Tabelle 6). Die Verdopplung der Metamizoldosis führte für die Metabolite 4-MAA und 4-AA zu
einer Zunahme der AUC um das 5- bzw 3,2-fache.
A
B
Abbildung 8: Plasmakonzentrations-Zeitkurven von 4-MAA (A) und 4-AA (B) nach oraler Gabe von 500 mg oder 1000 mg Metamizol an fünf Probanden. Es sind die Mittelwerte ± SEM aufgetragen. 4-MAA wies im Vergleich zu 4-AA kürzere Zeiten zu Erreichen der maximalen Plasmakonzentration und terminale Halbwertszeiten auf.
Die Plasmakonzentrationen von 4-AA waren zu allen Zeitpunkten unter den in-vitro bestimmten
IC50-Werten für die COX-1- und COX-2-Hemmung (Tabelle 4 und Abbildung 8 B). Andererseits
blieben die Plasmakonzentrationen von 4-MAA für 8 Stunden (500 mg) bzw. 12 Stunden
(1000 mg) über den in-vitro bestimmten IC50-Werten für die beiden Isoenzyme (Abbildung 8 A).
Die Plasmakonzentrationen von 4-MAA, die über oder im Bereich der IC50-Werte für die COX-
Hemmung lagen, wurden im Zeitraum von 15 min nach Einnahme von 500 mg: (3,12 µmol/l)
oder 1000 mg: (6,47 µmol/l) gemessen (Abbildung 8 A).
0 5 10 15 20 25
0
10
20
30
40
50
60
70
80
500 mg Metamizol1000 mg Metamizol
Zeit (h)
[4-M
AA
] (µ
M)
0 5 10 15 20 25
0,0
2,5
5,0
7,5
10,0
500 mg Metamizol1000 mg Metamizol
Zeit (h)
[4-A
A]
(µM
)
Ergebnisse 64
Tabelle 6: Pharmakokinetische Parameter von 4-MAA und 4-AA von fünf Probanden nach der oralen Einnahme von 500 mg oder 1000 mg Metamizol
4-MAA
Cmax (µmol/l) tmax (h)
AUCt (µmol⋅h/l)
t1/2 (h)
Metamizol 500 mg
13,6 ± 2,0 1,7 ± 0,4 75,7 ± 16,3 3,2 ± 0,6
Metamizol 1000 mg
59,1 ± 19,5 1,2 ± 0,2 381,6 ± 125,9 3,5 ± 0,5
4-AA
Cmax (µmol/l) tmax (h)
AUCt (µmol⋅h/l)
t1/2 (h)
Metamizol 500 mg
2,5 ± 0,7 3,1 ± 0,87 31,0 ± 11,9 6,3 ± 1,0
Metamizol 1000 mg
7,4 ± 2,7 5,2 ± 0,8 99,1 ± 43,6 5,7 ± 1,2
6.1.4 Korrelation zwischen Plasmakonzentration von 4-MAA und ex-vivo-
Hemmung der COX-Isoformen
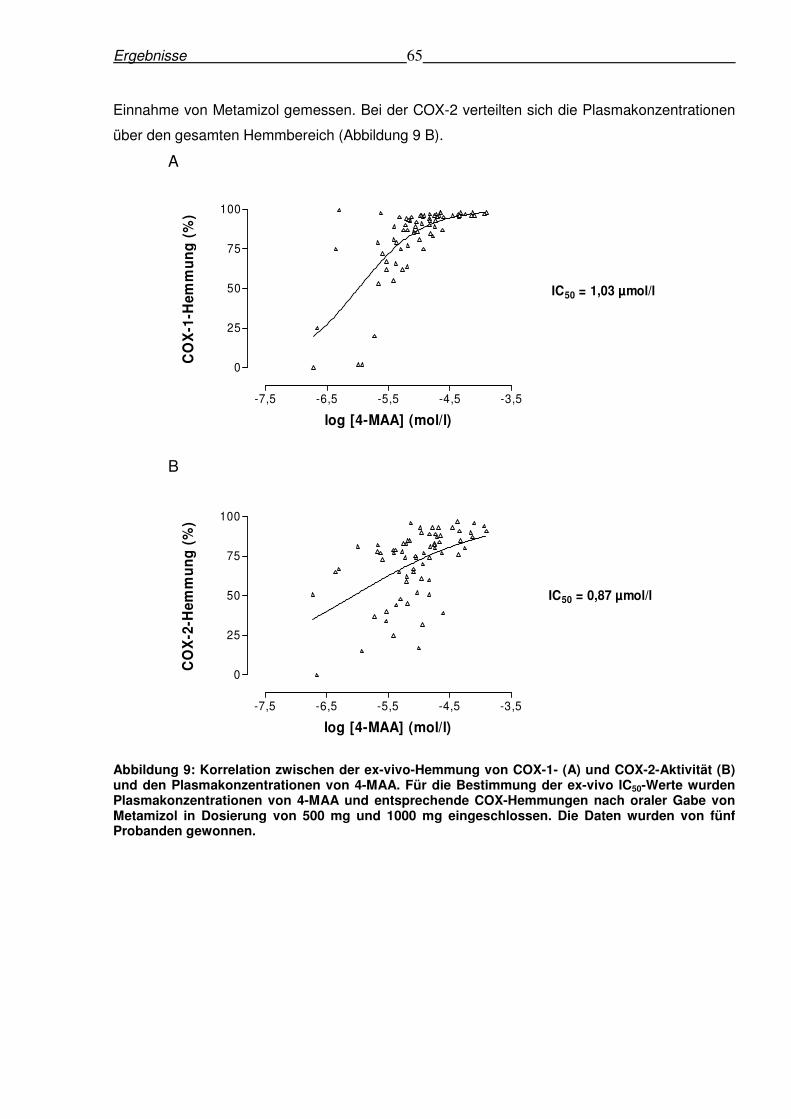
Der Zusammenhang zwischen der 4-MAA Plasmakonzentrationen und der ex-vivo-Hemmung
von COX-1 und COX-2 wurde mittels grafischer Analyse untersucht (Abbildung 9). Dadurch war
die ex-vivo-Bestimmung der notwendigen Plasmakonzentrationen (IC50) für eine 50%-ige
Hemmung der jeweiligen COX-Isoform möglich. Für die COX-1 konnte ein IC50-Wert von 1,03
µmol/l 4-MAA bestimmt werden. Der IC50-Wert für die COX-2-Hemmung war mit 0,87 µmol/l nur
geringfügig niedriger als der IC50-Wert für die COX-1. Die sigmoidale Hemmkurve von 4-MAA
für COX-1 zeigte, dass bei den meisten Plasmakonzentrationen von 4-MAA bereits eine 50 %-
ige Hemmung der COX-1 erreicht wurde. Nur im Falle von fünf Plasmakonzentrationen wurde
eine COX-1-Hemmung unter 50 % beobachtet (Abbildung 9 A). Diese Plasmakonzentrationen
wurden bei zwei Probanden 12 Stunden bzw. bei drei Probanden 24 Stunden nach der
Ergebnisse 65
Einnahme von Metamizol gemessen. Bei der COX-2 verteilten sich die Plasmakonzentrationen
über den gesamten Hemmbereich (Abbildung 9 B).
A
B
Abbildung 9: Korrelation zwischen der ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) und den Plasmakonzentrationen von 4-MAA. Für die Bestimmung der ex-vivo IC50-Werte wurden Plasmakonzentrationen von 4-MAA und entsprechende COX-Hemmungen nach oraler Gabe von Metamizol in Dosierung von 500 mg und 1000 mg eingeschlossen. Die Daten wurden von fünf Probanden gewonnen.
-7,5 -6,5 -5,5 -4,5 -3,5
0
25
50
75
100
IC50 = 1,03 µmol/l
log [4-MAA] (mol/l)
CO
X-1
-Hem
mu
ng
(%
)
-7,5 -6,5 -5,5 -4,5 -3,5
0
25
50
75
100
IC50 = 0,87 µmol/l
log [4-MAA] (mol/l)
CO
X-2
-Hem
mu
ng
(%
)
Ergebnisse 66
6.2 Klinisch-pharmakologische Untersuchuingen zur
Pharmakokinetik und Pharmakodynamik von Paracetamol
6.2.1 In-vitro Wirkung von Paracetamol auf die COX-1- und COX-2-Aktivität
in menschlichem Vollblut
Analog zur Aktivitätsbestimmung der peripheren COX im Vollblut nach Metamizolzugabe wurde
die Suppression der COX-1- und COX-2-Aktivität durch die Zugabe von Paracetamol in
verschiedenen Konzentrationen zu Vollblutproben untersucht (siehe Abschnitt 5.3.2.6.1). Als
Aktivitätsmaß für die COX-1 wurde Thromboxan B2 bzw. für die COX-2 Prostaglandin E2
bestimmt. Der in-vitro-Versuch zeigte, dass Paracetamol eine konzentrationsabhängige
Hemmung der beiden COX-Isoformen hervorruft (Tabelle 7). Paracetamol beeinflusste die
COX-2 jedoch deutlich effektiver als die COX-1. Auf Grundlage des Quotienten aus den IC50-
Werten, dem sogenannten Selektivitätsquotient, zeigte Paracetamol eine 4,4-fach höhere
Selektivität zu COX-2.
Tabelle 7: In-vitro-Hemmung der COX-1 und COX-2-Aktivität durch Paracetamol im menschlichen Vollblut-Assay. Die Daten wurden aus vier verschiedenen in-vitro Experimenten mit jeweils unterschiedlichen Blutproben gewonnen.
COX-1 IC50 (µmol/l) 113,7 ± 17,2
COX-2 IC50 (µmol/l) 25,8 ± 1,8
Selektivitätsquotient (COX-1/COX-2) 4,4
Die Daten sind als Mittelwerte ± SEM angegeben.
6.2.2 Pharmakokinetik von Paracetamol nach einmaliger oraler Gabe von
1000 mg Paracetamol
Nach der oralen Applikation von 1000 mg Paracetamol an jeweils fünf Probanden wurden die
Plasmakonzentrations-Zeitkurven von Paracetamol bestimmt (Abbildung 10).
Die durchschnittlichen Plasmakonzentrationen von Paracetamol blieben unter den in-vitro
bestimmten IC50-Wert für die COX-1-Hemmung innerhalb des gesamten Zeitraums der Studie.
Jedoch lagen die Plasmakonzentrationen von Paracetamol für mindestens 5 Stunden nach der
Medikamenteneinnahme über dem in-vitro bestimmten IC50-Wert für die COX-2-Hemmung
(Abbildung 10).
Ergebnisse 67
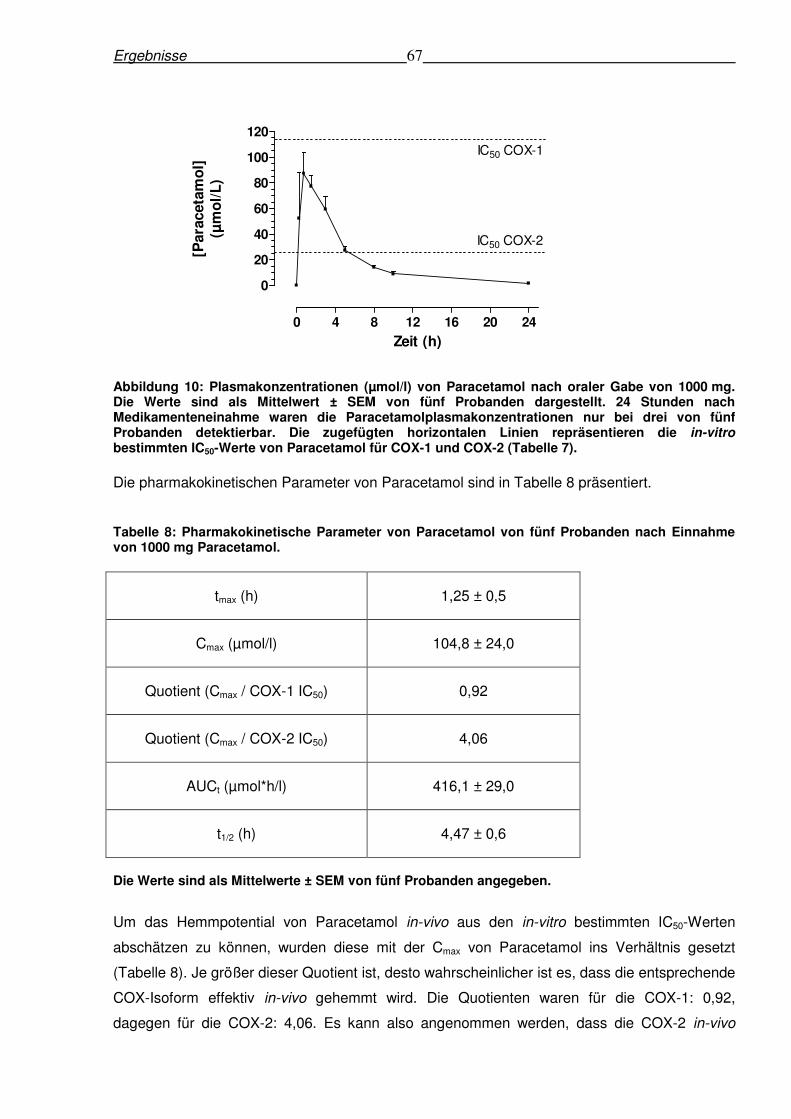
Abbildung 10: Plasmakonzentrationen (µmol/l) von Paracetamol nach oraler Gabe von 1000 mg. Die Werte sind als Mittelwert ± SEM von fünf Probanden dargestellt. 24 Stunden nach Medikamenteneinahme waren die Paracetamolplasmakonzentrationen nur bei drei von fünf Probanden detektierbar. Die zugefügten horizontalen Linien repräsentieren die in-vitro bestimmten IC50-Werte von Paracetamol für COX-1 und COX-2 (Tabelle 7).
Die pharmakokinetischen Parameter von Paracetamol sind in Tabelle 8 präsentiert.
Tabelle 8: Pharmakokinetische Parameter von Paracetamol von fünf Probanden nach Einnahme von 1000 mg Paracetamol.
tmax (h) 1,25 ± 0,5
Cmax (µmol/l) 104,8 ± 24,0
Quotient (Cmax / COX-1 IC50) 0,92
Quotient (Cmax / COX-2 IC50) 4,06
AUCt (µmol*h/l) 416,1 ± 29,0
t1/2 (h) 4,47 ± 0,6
Die Werte sind als Mittelwerte ± SEM von fünf Probanden angegeben.
Um das Hemmpotential von Paracetamol in-vivo aus den in-vitro bestimmten IC50-Werten
abschätzen zu können, wurden diese mit der Cmax von Paracetamol ins Verhältnis gesetzt
(Tabelle 8). Je größer dieser Quotient ist, desto wahrscheinlicher ist es, dass die entsprechende
COX-Isoform effektiv in-vivo gehemmt wird. Die Quotienten waren für die COX-1: 0,92,
dagegen für die COX-2: 4,06. Es kann also angenommen werden, dass die COX-2 in-vivo
0 4 8 12 16 20 24
0
20
40
60
80
100
120IC50 COX-1
IC50 COX-2
Zeit (h)
[Par
acet
amo
l](µ
mo
l/L
)
Ergebnisse 68
durch Paracetamol in therapeutischen Dosierungen gehemmt wird. Dagegen ist keine effektive
Hemmung von COX-1 durch Paracetamol in-vivo zu erwarten.
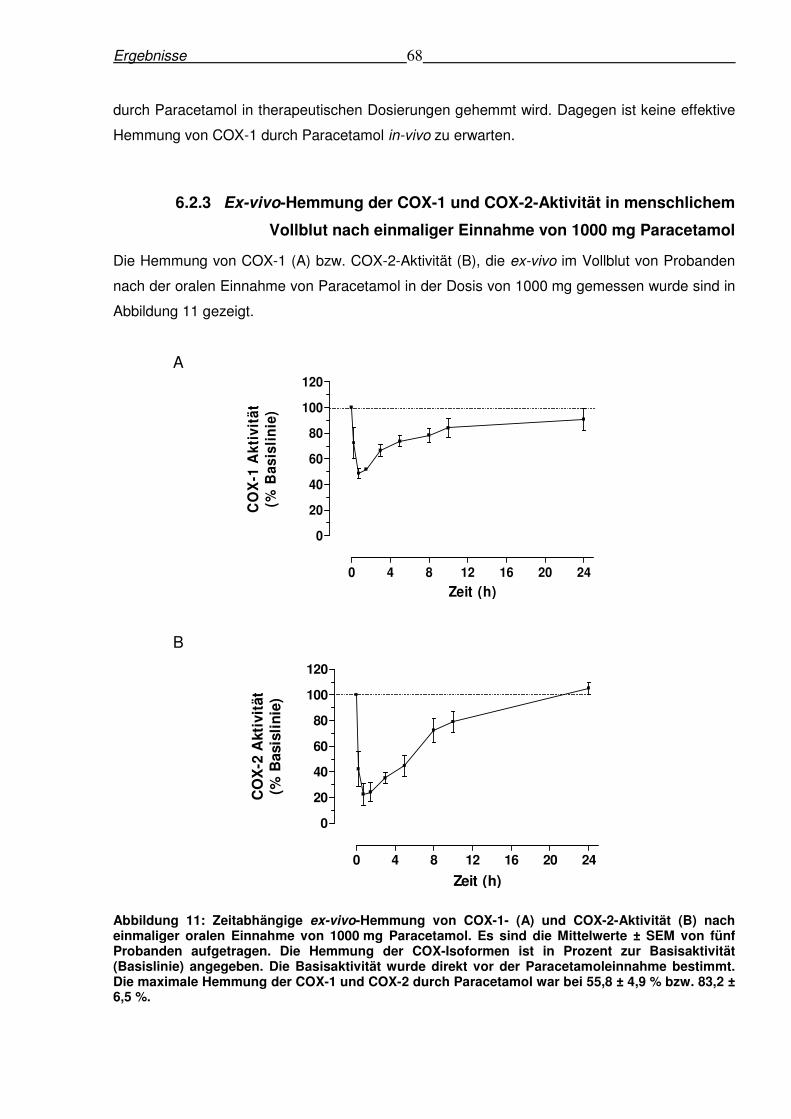
6.2.3 Ex-vivo-Hemmung der COX-1 und COX-2-Aktivität in menschlichem
Vollblut nach einmaliger Einnahme von 1000 mg Paracetamol
Die Hemmung von COX-1 (A) bzw. COX-2-Aktivität (B), die ex-vivo im Vollblut von Probanden
nach der oralen Einnahme von Paracetamol in der Dosis von 1000 mg gemessen wurde sind in
Abbildung 11 gezeigt.
A
B
Abbildung 11: Zeitabhängige ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) nach einmaliger oralen Einnahme von 1000 mg Paracetamol. Es sind die Mittelwerte ± SEM von fünf Probanden aufgetragen. Die Hemmung der COX-Isoformen ist in Prozent zur Basisaktivität (Basislinie) angegeben. Die Basisaktivität wurde direkt vor der Paracetamoleinnahme bestimmt. Die maximale Hemmung der COX-1 und COX-2 durch Paracetamol war bei 55,8 ± 4,9 % bzw. 83,2 ± 6,5 %.
0 4 8 12 16 20 24
0
20
40
60
80
100
120
Zeit (h)
CO
X-1
Akt
ivit
ät(%
Bas
isli
nie
)
0 4 8 12 16 20 24
0
20
40
60
80
100
120
Zeit (h)
CO
X-2
Akt
ivit
ät(%
Bas
isli
nie
)
Ergebnisse 69
Die pharmakodynamischen Parameter sind in der Tabelle 9 präsentiert. Diese Daten zeigten
eine deutliche Hemmung der COX-2 mit einer maximalen Hemmung von 83% sowie einer
durchschnittlichen Hemmung von 66% innerhalb der ersten fünf Stunden nach
Paracetamoleinnahme (Tabelle 9 und Abbildung 11). Die COX-1 wurde dagegen nur bis
maximal 56 % der Basisaktivität gehemmt (Tabelle 9). Die Zeiten zur maximalen Hemmung der
COX-1 (0,95 h) und COX-2 (1,65 h) entsprachen der tmax (1,25 h) von Paracetamol (Tabelle 8
und Tabelle 9).
Tabelle 9: Pharmakodynamische Parameter von fünf Probanden nach der oralen Einnahme von 1000 mg Paracetamol.
COX-1-Hemmung COX-2-Hemmung
Zeit bis zur maximalen Hemmung (h) 0,95 ± 0,24 1,65 ± 0,86
Maximale Hemmung (%) 55,8 ± 4,9 83,2 ± 6,5
Durchschnittliche Hemmung
0,25-5 hours (%) 37,4 ± 4,4 66,2 ± 4,7
Durchschnittliche Hemmung
0,25-24 hours (%) 29,8 ± 4,7 47,5 ± 2,1
AWEC0-5 hours (% · h) 181,9 ± 16,8 323,9 ± 16,5
AWEC0-24 hours (% · h) 500,3 ± 123,7 640,8 ± 81,9
IC50 (µmol/l) 105,2 26,3 Die Werte sind als Mittelwerte ± SEM von fünf Probanden, die 1000 mg Paracetamol eingenommen haben, aufgetragen
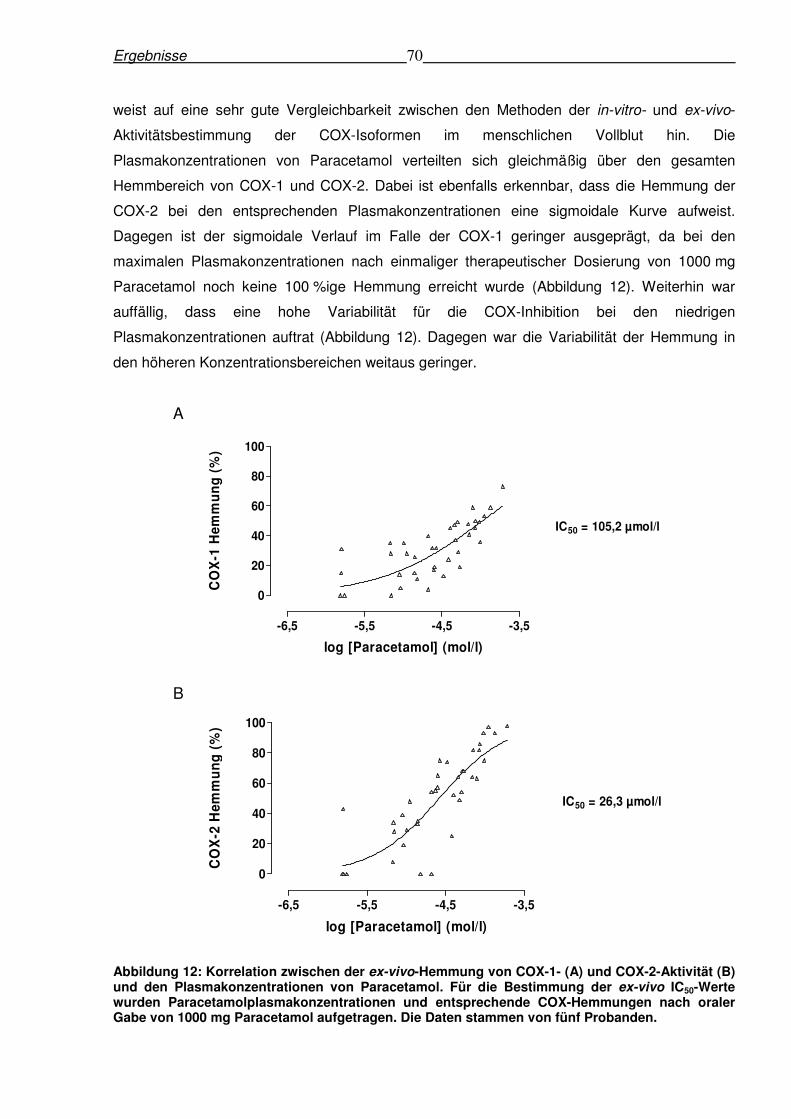
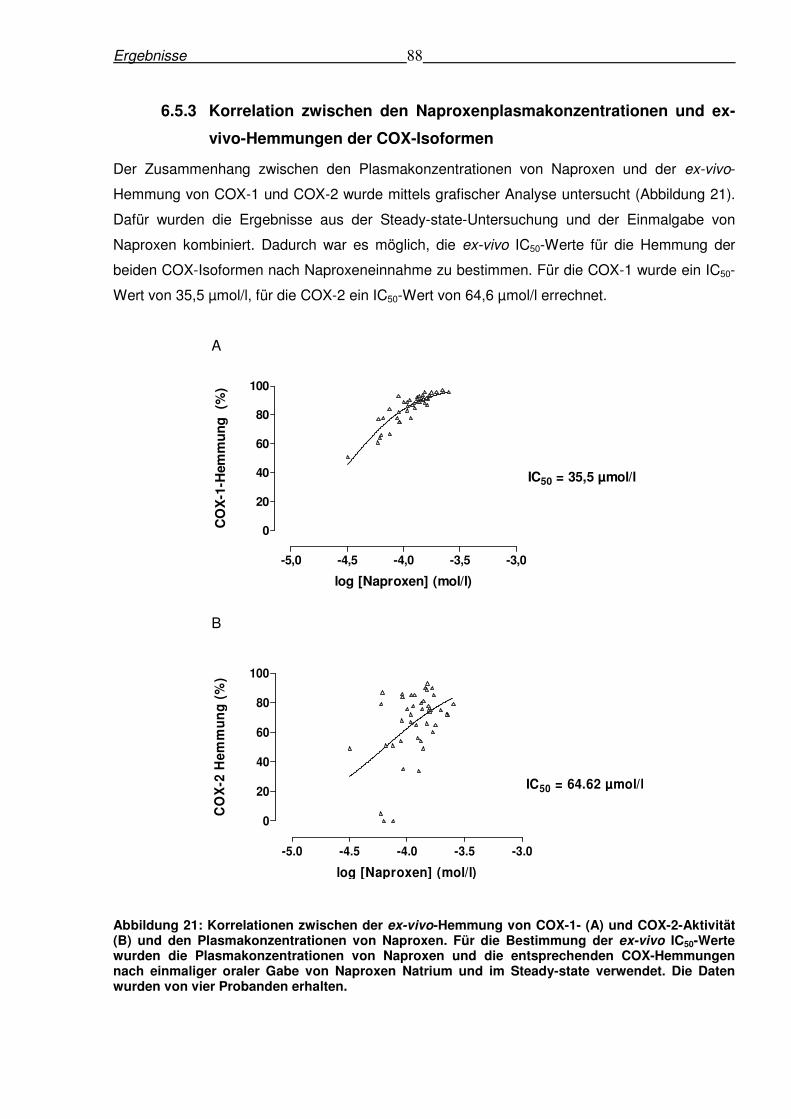
6.2.4 Korrelation zwischen den Plasmakonzentrationen von Paracetamol
und den ex-vivo-Hemmungen von COX-1 und COX-2
Der Zusammenhang zwischen den Plasmakonzentrationen von Paracetamol und der ex-vivo-
Hemmung von COX-1 und COX-2 wurde mittels grafischer Analyse untersucht. Dadurch war es
möglich, die ex-vivo IC50-Werte für die Hemmung der beiden COX-Isoformen durch
Paracetamoleinnahme zu bestimmen. Für die COX-1 wurde ein IC50-Wert von 105,2 µmol/l, für
die COX-2 ein IC50-Wert von 26,3 µmol/l errechnet. Diese IC50-Werte stellen ein Maß für die
Hemmung der COX-Isoformen in-vivo dar, da die Enzymaktivität im Vollblut der Probanden
nach der Medikamenteneinnahme bestimmt wurde. Die dadurch errechneten ex-vivo IC50-Werte
(Abbildung 10) waren vergleichbar mit den in-vitro IC50-Werten von Paracetamol für die
Hemmung der COX-1 (113,7 µmol/l) bzw. COX-2 (25,8 µmol/l) (Tabelle 7). Diese Tatsache
Ergebnisse 70
weist auf eine sehr gute Vergleichbarkeit zwischen den Methoden der in-vitro- und ex-vivo-
Aktivitätsbestimmung der COX-Isoformen im menschlichen Vollblut hin. Die
Plasmakonzentrationen von Paracetamol verteilten sich gleichmäßig über den gesamten
Hemmbereich von COX-1 und COX-2. Dabei ist ebenfalls erkennbar, dass die Hemmung der
COX-2 bei den entsprechenden Plasmakonzentrationen eine sigmoidale Kurve aufweist.
Dagegen ist der sigmoidale Verlauf im Falle der COX-1 geringer ausgeprägt, da bei den
maximalen Plasmakonzentrationen nach einmaliger therapeutischer Dosierung von 1000 mg
Paracetamol noch keine 100 %ige Hemmung erreicht wurde (Abbildung 12). Weiterhin war
auffällig, dass eine hohe Variabilität für die COX-Inhibition bei den niedrigen
Plasmakonzentrationen auftrat (Abbildung 12). Dagegen war die Variabilität der Hemmung in
den höheren Konzentrationsbereichen weitaus geringer.
A
B
Abbildung 12: Korrelation zwischen der ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) und den Plasmakonzentrationen von Paracetamol. Für die Bestimmung der ex-vivo IC50-Werte wurden Paracetamolplasmakonzentrationen und entsprechende COX-Hemmungen nach oraler Gabe von 1000 mg Paracetamol aufgetragen. Die Daten stammen von fünf Probanden.
-6,5 -5,5 -4,5 -3,5
0
20
40
60
80
100
IC50 = 105,2 µmol/l
log [Paracetamol] (mol/l)
CO
X-1
Hem
mu
ng
(%
)
-6,5 -5,5 -4,5 -3,5
0
20
40
60
80
100
IC50 = 26,3 µmol/l
log [Paracetamol] (mol/l)
CO
X-2
Hem
mu
ng
(%
)
Ergebnisse 71
6.2.5 In-vitro-Wirkung von Paracetamol auf die COX-2-Aktivität von
humanem rekombinanten Enzym und Blutmonozyten
Durch weitere Experimente wurde untersucht, ob die beobachtete COX-2-Hemmung (sieh
Abschnitt 6.2.3) die Folge einer kompetetiven Hemmung und / oder einer verringerten
Expression ist. Zunächst wurden deshalb die COX-2-Aktivitäten von humaner rekombinanter
COX-2 (siehe Methoden 5.3.2.6.2) sowie von LPS-stimulierten humanen Monozyten nach einer
Kurzzeitinkubation mit Paracetamol (siehe Methoden 5.3.2.6.3) bestimmt. Wie in Abbildung 13
zu erkennen ist, führte Paracetamol in therapeutischen Konzentrationen (100 µmol/l) zu einer
wesentlichen Hemmung der COX-2 bei sowohl rekombinanten Enzymen als auch in humanen
Monozyten. Die in Abbildung 13 dargestellte ex-vivo-Hemmung der COX-2 in humanem Vollblut
(Tabelle 9) wurde zur besseren visuellen Vergleichbarkeit ebenfalls dargestellt.
Abbildung 13: Wirkung von Paracetamol (100 µmol/l) auf die Aktivität humaner rekombinanter COX-2 sowie die COX-2-Aktivität in LPS-stimulierten humanen Monozyten. Die gezeigte Hemmung der COX-2-Aktivität im Vollblut stellt die im Abschnitt 6.2.3 beschriebene ex-vivo-Hemmung der COX-2 dar. Die Werte sind als Mittelwerte ± SEM (n= 3 bis 6 Experimente) in Prozent der Kontrolle (Inkubation ohne Paracetamol) aufgetragen.
6.2.6 In-vitro-Wirkung von Paracetamol auf die COX-2-Expression
Im Weiteren wurde die Frage untersucht ob die COX-2-Hemmung durch Paracetamol zusätzlich
durch eine verringerte Expression der COX-2 vermittelt wird. Dafür wurde die COX-2-
Expression in humanen mononukleären Zellen mit und ohne LPS-Stimulation nach einer 24-
stündigen Inkubation mit Paracetamol, analysiert (siehe Abschnitt 5.3.2.6.4). Die Expression der
Ergebnisse 72
COX-2 wurde dann mittels Immunoblotanalyse visualisiert und ausgewertet. Es konnte gezeigt
werden, dass sowohl in LPS-stimulierten Zellen als auch in nicht LPS-stimulierten Zellen sich
die COX-2-Expression nicht durch Paracetamol änderte (Abbildung 14). Somit kann
geschlussfolgert werden, dass Paracetamol nur einen Effekt auf die COX-2-Aktivität hat.
Abbildung 14: Repräsentativer Immunoblot zum Einfluss von Paracetamol auf die Expression von COX-2 in humanen mononukleären Zellen. Die Immunoblotanalyse zeigt, dass die COX-2-Expression durch Paracetamol nicht beeinflusst wird. Als interner Standard wurde β-Aktin nach dem „Stripping“ nachgewiesen. Über den Blots sind die densitometrischen bestimmten Expressionsdaten der COX-2 in Prozent der Kontrolle gezeigt (n=4 Immunoblots). Innerhalb der nicht LPS-stimulierten Zellen bzw. der LPS-stimulierten Zellen zeigte sich keine Änderung der COX-2-Expression durch Paracetamol. Die LPS-Stimulation der Zellen führte zu einer Zunahme der COX-2-Expression um durchschnittlich 160 %.
Ergebnisse 73
6.3 Entwicklung und Validierung einer HPLC-UV Methode zur
Bestimmung der Konzentrationen von Lumiracoxib in
menschlichem Plasma
6.3.1 Methodenentwicklung
Zur Bestimmung der Plasmakonzentrationen von Lumiracoxib war es notwendig, eine Methode
zu entwickeln. Die zu entwickelnde Methode müsste für laborübliche Analysengeräte
zugeschnitten sein. Deswegen wurde die Analyse mittels HPLC und UV-Detektion durchgeführt.
Nach der Spektroskopie hat es sich zwei Maximume ergeben. Deswegen wurde Messung von
Lumiracoxib bei Wellenlänge: 210, 212, 270, 280 nm durchgetestet. Dabei wurden bei niedrigen
Wellenlängen zweimal größere Peakflächen von Lumiracoxib, als bei höheren Wellenlängen
gemessen. Jedoch wurden mehrere Störpeaks mitgemessen. Deshalb wurde es für
Wellenlänge 270 nm entschieden. Die Lumiracoxib-Plasmakonzentrationen wurden mithilfe
einer Internen-Standard-Methode bestimmt. Als interner Standard wurde Nifluminsäure und
Diclofenac untersucht. Es wurde für Nifluminsäure entschieden, weil Diclofenac unmittelbar vor
dem Lumiracoxibpeak rausgegangen ist, wodurch schlechtere Auflösung erzielt wurde.
Als mobile Phase für die Trennung von Analyten wurde Gemisch aus 0,05% Trichloressigsäure
und Acetonitril im verschiedenen Verhältnisse überprüft. Bei Mischungsverhältnis von 0,05%
Trichloressigsäure und Acetonitril 35:65 (v/v) war die Trennung von Lumiracoxib und Internen
Standard optimal, ohne dass Interferenzen mit den übrigen Plasmabestandteilen auftraten.
Es hat sich angeboten, ein zweiphasiges Lösungsmittelgemisch bestehend aus einem polaren,
und einem weniger polaren, leicht verdampfbaren Lösungsmittel zu verwenden. Nach diesen
Vorüberlegungen wurden zweiphasige Lösungsmittelsysteme, so wie Pentan:Chloroform und
Hexan:Diethylether auf ihre Eignung als Extraktionsmittel untersucht. Ein Gemisch aus Hexan
und Diethylether stellte sich als das effektivste Extraktionsmittel heraus. Dabei wurden größere
Peakflächen bei der Verhältnis 70:30 (v/v) von Hexan:Diethylether gemessen.
Die Methode ermöglichte die Bestimmung von Lumiracoxib im Konzentrationsbereich von 10 –
10 000 ng/ml.
6.3.1.1 Standardlösungen
4,5 mg Lumiracoxib wurden in 4,5 ml Methanol gelöst so dass eine Standardlösung von 1
mg/ml Lumiracoxib erhalten wurde. Diese Lösung wurde weiter 1:10 mit 50%-igem (v/v)
wässrigen MeOH verdünnt, so dass die eine Standardlösung von 100 µg/ml erhalten wurde.
Ergebnisse 74
Die Standardlösung des internen Standards Nifluminsäure mit einer Konzentration von 10 µg/ml
wurde durch Lösen von 1 mg Nifluminsäure in 100 ml MeOH erhalten. Alle Standardlösungen
wurden bei -20°C aufbewahrt.
6.3.1.2 Probenaufarbeitung und Messung
Zu 1000 µl Plasma wurden 100 µl interner Standard (Nifluminsäure 10 µg/ml Methanol), 500 µl
1M HCl sowie 4 ml Extraktionsmittel (Hexan:Diethylether 70:30) gegeben. Die Proben wurden
10 Minuten über Kopf geschüttelt und anschließend in einer Kühlzentrifuge (Eppendorf 5810R)
bei 4000 r.p.m. für eine Dauer von 10 Minuten bei 4°C zentrifugiert. Organische Phase wurde
abgehoben und unter Stickstoff evaporiert.
Als mobile Phase für die HPLC-Analyse diente ein Gemisch aus 0,05% Trichloressigsäure und
Acetonitril im Verhältnis 35:65 (v/v). Die Fließgeschwindigkeit lag bei 1,0 ml/min. Temperatur
des Säulenofens wurde auf 28°C gestellt. Das Fließmittel wurde vor der Verwendung in
Ultraschalbad gestellt, um gelöste Gase zu beseitigen, die die HPLC-Analytik stören könnten.
Vor Beginn der Messungen wurde die HPLC-Apparatur mindestens eine halbe Stunde lang mit
der mobilen Phase äquilibriert. Die Leitungen des automatischen Probengebers und des
Injektionssystems wurden vor und nach jeder Messung mit entgastem 50%-igem wässrigen
Acetonitril gespült. Die Detektion erfolgte mithilfe eines Ultraviolettdetektors bei einer
Wellenlänge von 270 nm. Die Retentionszeit für Nifluminsäure (IS) lag bei 10,4 Minuten und für
Lumiracoxib bei 16,9 Minuten. Bei jeder Messung während der Lumiracoxibanalytik wurde
Qualitätskontrollproben (10, 50, 100, 500, 1000, 5000 und 10 000 ng/ml) mitgemessen. Die
Schwankungen des Peakhöhenverhältnisses von Lumiracoxib zu Nifluminsäure in den
Kontrollproben, berechnet als Variationskoeffizient, lagen im Lauf der Bestimmungen bei
weniger als 15%, bei Lumiracoxibkonzentration von 10 ng/ml bei weniger als 20%. Das HPLC-
System lief unter Verwendung der Software Borwin (Jasco-Borwin Version 1.50).
Ergebnisse 75
6.3.2 Validierung
In dieser Arbeit wurde die Validierung auf der Basis der von Shah (2000) zusammengefassten
Richtlinien der Konferenz „Analytical methods validation: Bioavailability, bioequivalence and
pharmacokinetic studies“ (Shah et al., 2000) durchgeführt.
6.3.2.1 Spezifität, Linearität und Bestimmungsgrenze
Spezifität (auch Selektivität) wurde nachgewiesen, indem man Leerplasmaproben von sechs
verschiedenen Probanden untersucht hat und keine Störpeaks an der Stelle der Analytes
festgestellt hat.
Linearität der Kalibriergerade für Lumiracoxib ist in Messbereich von 10 ng/ml bis 10000 ng/ml
vorhanden. Diesen Bereich wurde in 3 kleinere Messbereiche aufgeteilt: kleinen 10-100 ng/ml,
mittleren 100-1000 ng/ml und großen 1000-10 000 ng/ml und für jedes Bereich separat validiert.
Die Linearität der Messung wurde durch die Analyse von jeden Messbereich bestehend aus
jeweils fünf Konzentrationen ermittelt. Die Korrelationskoeffiziente waren größer als 0,997. Die
Bestimmungsgrenze lag bei 10 ng/ml.
Ergebnisse 76
6.3.2.2 Repräsentative Chromatogramme
Mit der entwickelten analytischen Methode wurde optimale Trennung von Analyten erreicht.
In Abbildung 15 sind typische Chromatogramme für die Analyse von Leerplasma, einer
Vergleichsprobe mit zugesetztem Lumiracoxib und Nifluminsäure und einer Patientenprobe
dargestellt.
Abbildung 15: Chromatogramme einer Leerplasmaprobe (A), einer Vergleichsprobe mit einer Lumiracoxibkonzentration von 1000 ng/ml (B) and einer Plasmaprobe vom Proband, der einmalig Lumiracoxib 200 mg eingenommen hat (C). Die Blutprobe wurde 15 min nach Medikamenteinnahme entnommen. Die Plasmakonzentration an Lumiracoxib betrug 608,8 ng/ml.
A
B
C
5
5
5
10
10
10 15
15
15 20
20
20
Lumiracoxib
Lumiracoxib
Nifluminsäure
Nifluminsäure
[min]
[min]
[min]
Ergebnisse 77
6.3.2.3 Intra-day und - Inter-day-Präzision
Akzeptable und zuverlässige Präzision und Richtigkeit der Methode wurden in intra-day und
inter-day Validierung mit untere Bestimmungsgrenze von 10 ng/ml gezeigt (nachgewiesen). Zur
Bestimmung der Intra-day-Präzision und -Richtigkeit des Messverfahrens wurden jeweils fünf
Proben bei sieben verschiedenen Lumiracoxibkonzentrationen über den gesamten Messbereich
analysiert. Die Ergebnisse von intra-day-Präzision und Richtigkeit sind in Tabelle 10 präsentiert.
Inter-day Präzision und Richtigkeit wurde an drei verschiedenen Tage bewertet und in Tabelle
11 zusammengefasst.
Tabelle 10: Intra-day-Präzision and Richtigkeit für kleine, mittlere und höhere Konzentrationen von Lumiracoxib.
Intra-day Soll / Konzentration
(ng/ml)
Gemessene Konzentration (ng/ml)
CV (%)
Richtigkeit
(%)
10
9,54 ± 0,71
7,5
4,6
50
52,68 ± 1,04
2,0
5,4
100
110,10 ± 2,26
2,1
10,4
100
100,44 ± 1,77
1,8
0,5
500
513,35 ± 7,41
1,4
2,7
1000
1002,27 ± 22,70
2,3
0,2
1000
919,63 ± 23,73
2,6
-8,0
5000
5069,70 ± 64,48
1,3
1,4
10000
10220,88 ± 246,50
2,4
2,2
CV = Variationskoeffizient. Die Werte sind als Mittelwerte ±±±± SD (n = 5) eingegeben.
Ergebnisse 78
Tabelle 11: Inter-day-Präzision and Richtigkeit für kleine, mittlere und höhere Konzentrationen von Lumiracoxib.
Inter-day Soll / Konzentration
(ng/ml)
Gemessene Konzentration
(ng/ml)
CV (%)
Richtigkeit
(%)
10
9,80 ± 0,59
6,1
-2,0
50
49,04 ± 4,07
8,3
-1,9
100
103,28 ± 7,65
7,4
3,3
100
105,00 ± 8,27
7,9
5,0
500
498,19 ± 31,34
6,3
-0,4
1000
969,51 ± 43,48
4,5
-3,0
1000
925,12 ± 66,16
7,2
-7,5
5000
4942,37 ± 255,92
5,2
-1,2
10000
10140,34 ± 497,99
4,9
1,4
CV = Variationskoeffizient. Die Werte sind als Mittelwerte ±±±± SD (3 Tage, n = 5 je.) eingegeben.
6.3.2.4 Wiederfindung
Wiederfindung (Recovery) von Lumiracoxib wurde bei vier Konzentrationen bestimmt und ist in
Tabelle 12 aufgeführt. Durchschnittliche Widerfindungsrate lag zwischen 82-93%.
Tabelle 12: Wiederfindung von Lumiracoxib in Plasma
Konzentration
(ng/ml)
Wiederfindung
(%)
10 93,2 ± 8,8
100 87,0 ± 4,5
1000 82,4 ± 4,5
10000 85,8 ± 2,5
Die Werte sind als Mittelwerte ±±±± SD (n = 5 für jede Gruppe) eingegeben.
Ergebnisse 79
6.3.2.5 Stabilität
Die Untersuchung der Stabilität wurde über einen fünfwöchigen Zeitraum bei -70°C
durchgeführt. Die Ergebnisse sind in Tabelle 13 präsentiert. Der Gehalt der Lumiracoxib nimmt
weniger als 10%, außer LOQ von 10 ng/ml (15%) ab.
Tabelle 13: Stabilität von Lumiracoxib nach Gefrierzeit von fünf Wochen bei – 70°C.
Soll / Konzentration
(ng/ml)
Ist / Konzentration
(ng/ml)
SD (%)
10
11,5 ± 0,63
14,6
50
47,85 ± 0,84
4,3
100
91,14 ± 0,65
8,9
100
94,94 ± 0,65
5,1
500
465,70 ± 11,53
6,9
1000
1025,33 ± 9,89
2,5
1000
953,39 ± 10,12
4,7
5000
5254,76 ± 94,86
5,1
10000
10164,50 ± 341,56
2,7
Die Werte sind als Mittelwerte ±±±± SD (n = 5 für jede Gruppe) eingegeben.
Ergebnisse 80
6.4 Klinisch-pharmakologische Untersuchungen zur
Pharmakokinetik und Pharmakodynamik von Lumiracoxib
6.4.1 Pharmakokinetik von Lumiracoxib nach oraler Gabe
Die Fläche unter den Plasmakonzentrations-Zeitkurven von Lumiracoxib nach der oralen Gabe
von 50 mg, 100 mg oder 200 mg sowie die weitere pharmakokinetische Parameter sind in der
Abbildung 16 bzw. Tabelle 14 präsentiert.
Tabelle 14: Pharmakokinetische Parameter von Lumiracoxib nach der oralen Einnahme von 50 mg, 100 mg oder 200 mg Lumiracoxib.
Alle Daten sind als Mittelwert ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen eingenommen hatten, angegeben.
Das Zeitintervall für die maximalen Plasmakonzentrationen von Lumiracoxib lag zwischen 1,5
bis 2,4 Stunden Tabelle 14. Dabei zeigte die Höchstdosis von 200 mg Lumiracoxib die kürzeste
tmax. von 1,5 Stunden. Die tmax stieg mit den geringeren Dosierungen von 100 mg und 50 mg auf
2,25 bzw. 2,38 Stunden an. Für die Cmax zeigte sich ein linearer Zusammenhang zur Dosis. Die
Verdopplung der Dosierung von 50 mg auf 100 mg führte ebenfalls zu einer Verdopplung der
Cmax von 4,09 ± 0,9 µmol/l auf 9,04 ± 1,8 µmol/l. Die weitere Verdopplung der Dosis auf 200 mg
Lumiracoxib hatte dann auch einen entsprechenden Effekt auf die Cmax, die sich auf 18,39 ±
4,5 µmol/l erhöhte. Ein vergleichbarer Effekt war ebenfalls für die AUCs zu beobachten. Diese
nahmen bei den Dosierungen von 50 mg, 100 mg und 200 mg Lumiracoxib von 13,99 ±
3,7 µmol*h/l auf 29,11 ± 7,6 µmol*h/l bzw. 63,72 ± 12,1 µmol*h/l zu.
Die maximalen Plasmakonzentrationen von Lumiracoxib lagen 29-fach (50 mg), 65-fach
(100 mg) und 131-fach (200 mg) über dem ex-vivo bestimmten IC50-Wert für die COX-2-
Ergebnisse 81
0 4 8 12 16 20 24
0
5
10
15
20
25
100 mg200 mg
50 mg
Zeit (h)
[Lu
mir
aco
xib
](µ
mo
l/l)
Hemmung (Abbildung 18). Weiterhin waren die durchschnittlichen Plasmakonzentrationen von
Lumiracoxib 8 Stunden (50 mg) oder 12 Stunden (100 mg und 200 mg) nach der
Dosiseinnahme höher als der ex-vivo bestimmte IC50-Wert für die COX-2-Hemmung.
Abbildung 16: Plasmakonzentrationen von Lumiracoxib nach der oralen Verabreichung von 50 mg, 100 mg oder 200 mg. Die Werte sind als Mittelwerte ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen erhielten, angegeben. 24 Stunden nach der Dosisverabreichung von 200 mg Lumiracoxib bei allen Probanden noch quantifizierbar. Jedoch war 24 Stunden nach der Gabe von 100 mg oder 50 mg Lumiracoxib die Substanz nur bei zwei bzw. einem Probanden im Plasma quantifizierbar.
Ergebnisse 82
0 4 8 12 16 20 24
0
20
40
60
80
100
120
140
100 mg200 mg
50 mg
Zeit (h)
CO
X-2
-Akt
ivit
ät(%
vo
n B
asis
lin
ie)
80% Hemmung
6.4.2 Ex-vivo-Hemmung der COX-2-Aktivität nach oraler Gabe von
Lumiracoxib
Die ex-vivo Hemmung der COX-2-Aktivität nach oraler Verabreichung von 50 mg, 100 mg oder
200 mg Lumiracoxib ist in Abbildung 17 gezeigt. Die durchschnittlichen pharmakodynamischen
Parameter sind in Tabelle 15 präsentiert. Lumiracoxib führte im Zeitraum von 1 bis 1,5 Stunden
zur kompletten Hemmung der COX-2-Aktivität bei allen untersuchten Dosisierungen (Tabelle
15).
Abbildung 17: Ex-vivo-Hemmung der COX-2-Aktivität nach einmaliger oraler Verabreichung von 50 mg, 100 mg oder 200 mg Lumiracoxib bis 24 Stunden nach der Einnahme. Alle Daten sind als Mittelwerte ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen erhielten, angegeben. Die eingefügte horizontale Linie stellt die 80%-ige COX-2-Hemmung dar. Die Zeiträume die zur maximalen COX-2-Hemmung notwendig waren lagen für alle
untersuchten Dosierungen im gleichen Bereich für die entsprechenden tmax von Lumiracoxib
(Tabelle 14, Tabelle 15). Die maximale COX-2-Hemmung war in dieser Zeitspanne bei allen
Dosierungen identisch ( ∼99,9 %). Jedoch konnten dosisabhängig bereits nach 5 Stunden
Unterschiede in der Hemmung der COX-2 nachgewiesen werden, die bei der Dosis von 50 mg
bei 90%, bei der Dosis von 100 mg bei 94% und für die 200 mg Dosis bei 97% lag (Tabelle 15).
Diese Tendenz verstärkte sich mit zunehmender Zeit.
Die durchschnittlichen Werte der Fläche innerhalb der Effekt-Zeitkurve (AWEC) unterschieden
sich zwischen den drei Gruppen innerhalb aller analysierten Zeiträume (5, 8, 12 oder 24
Stunden nach Medikation) nicht signifikant (Tabelle 15).
Ergebnisse 83
Tabelle 15: Ex-vivo bestimmte pharmakodynamische Parameter von Lumiracoxib nach der oralen Einnahme von 50 mg, 100 mg oder 200 mg Lumiracoxib.
50 mg 100 mg 200 mg
Zeit zur maximalen COX-2-Hemmung (h) 1,50 ± 0,53 1,06 ± 0,66 1,06 ± 0,66
AWEC: Effekt-Zeitkurve. Alle Daten sind als Mittelwerte ± SEM von vier Probanden, die Lumiracoxib in verschiedenen Dosierungen erhielten, angegeben.
In jeder Dosisgruppe kehrte die durchschnittliche COX-2-Aktivität 24 Stunden nach
Dosisverabreichung auf den Basiswert zurück, wobei eine relativ hohe Variabilität in der COX-2
zu diesem Zeitpunkt (Abbildung 17) beobachtet wurde.
Ergebnisse 84
-6,5 -6,0 -5,5 -5,0 -4,5 -4,0
0
20
40
60
80
100
IC50 = 0,14 µmol/l
log [Lumiracoxib] (mol/l)
CO
X-2
Hem
mu
ng
(%
)
6.4.3 Korrelation zwischen den Lumiracoxibplasmakonzentrationen und
der ex-vivo COX-2-Hemmung
Der berechnete ex-vivo IC50-Wert von 0,14 µmol/l (Abbildung 18) ist vergleichbar mit dem in-
vitro IC50-Wert von Lumiracoxib für die COX-2 (0,13 µmol/l), der bereits veröffentlicht wurde
(Capone et al., 2007).
Abbildung 18: Korrelation zwischen der ex-vivo COX-2-Hemmung und den Lumiracoxibplasmakonzentrationen. Für die Berechnung der ex-vivo IC50-Werte wurden die Lumiracoxibplasmakonzentrationen und die entsprechenden COX-2-Hemmungen nach der einmaliger oraler Verabreichung von 50 mg, 100 mg und 200 mg Lumiracoxib aufgetragen. Die Werte wurden von jeweils vier Probanden pro Dosis erhalten.
Ergebnisse 85
6.5 Klinisch-pharmakologische Untersuchungen zur
Pharmakokinetik und Pharmakodynamik von Naproxen
Die medizinischen Daten aus Untersuchungen mit Naproxen wurden in der Doktorarbeit von Dr.
med. Dominika Besz dargestellt (Besz, 2008).
6.5.1 Pharmakokinetik von Naproxen und ex-vivo-Hemmung der COX-1-
und COX-2-Aktivität nach einmaliger oraler Gabe von 220 mg
Naproxen Natrium bei gesunden Probanden
Die Plasmakonzentrations-Zeitkurven von Naproxen nach der einmaligen oralen Einnahme von
220 mg Naproxen Natrium (A) sowie die entsprechenden COX-1- bzw. COX-2-Hemmungen (B
und C) sind in Abbildung 19 dargestellt.
A
B
0 1 2 3 4 5 6 7 8 9 10 11 12
0
20
40
60
80
100
120
140
95% Hemmung
Zeit nach der Dosiseinnahme (h)
CO
X-1
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
0 1 2 3 4 5 6 7 8 9 10 11 12
0
50
100
150
200
250
300
Zeit nach der Dosiseinnahme (h)
[Na
pro
xe
n]
(µM
)
Ergebnisse 86
C
Abbildung 19: Plasmakonzentrations-Zeitkurven von Naproxen (A) und die entsprechenden COX-1- bzw. COX-2-Hemmungen (B bzw. C) nach einmaliger oraler Gabe von 220 mg Naproxen Natrium. Die Proben für die Bestimmung der Plasmakonzentrationen von Naproxen und für die Bestimmung der COX-Hemmung wurden vor der ersten Tabletteneinnahme sowie 1, 3 und 12 Stunden danach gewonnen. Die untere eingefügte horizontale Linie in Abbildung B stellt die COX-1-Hemmung von 95%dar. Es sind die Mittelwerte ± SEM von vier Probanden aufgetragen. Nach einmaliger Gabe von 220 mg Naproxen Natrium war die maximale Hemmung der COX-1
und COX-2 94,1% ± 1% bzw. 78,8% ± 5,5%. Die Zeiten bis zur maximalen Hemmung der COX-
1 und COX-2 entsprachen der tmax und lagen bei 1 Stunde nach Naproxengabe.
6.5.2 Pharmakokinetik von Naproxen und ex-vivo-Hemmung der COX-1
und COX-2-Aktivität nach oraler Gabe von 220 mg Naproxen Natrium
im Steady-state
Die folgenden Ergebnisse zur Pharmakokinetik von Naproxen (A) und zur Hemmung von COX-
1 bzw. COX-2 (B und C) wurden im Steady-state gewonnen und sind in Abbildung 20
präsentiert. Dabei nahmen die Probanden zweimal täglich 220 mg Naproxen Natrium für sieben
Tage ein. Das detailierte Protokoll ist in Abschnitt 5.3.4.5 beschrieben.
Nach der Beendigung der Medikamenteinnahme änderten sich die Aktivitäten der COX-1 und
COX-2 innerhalb von 36 Stunden auf 28 % bzw. 39 % der Ausgangswerte vor
Tabletteneinnahme (Zeitpunkt 0, Tag 1, Abbildung 20 B, C). Zu diesem Zeitpunkt waren die
Naproxenplasmakonzentrationen noch quantifizierbar und betrugen durchschnittlich 73,2 µM
(Abbildung 20 A).
Im Steady-state, das am dritten Tag der Tabletteneinnahme erreicht wurde, waren die
maximalen Hemmungen für die COX-1 bzw. COX-2 93,1 ± 1,3 % und 84,5 ± 3,1 %. Die
maximalen Hemmungen der COX-1 und COX-2 in dieser Studie wurden 4,5 ± 0,5 Tage bzw.
6,8 ± 1,3 Tage nach der ersten Dosisverabreichung beobachtet.
0 1 2 3 4 5 6 7 8 9 10 11 12
0
20
40
60
80
100
120
140
Zeit nach der Dosiseinnahme (h)
CO
X-2
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
Ergebnisse 87
1 2 3 4 5
0
50
100
150
200
250
300
8 9
Naproxeneinnahme Recovery(Tag 8 - 9)(Tag 1 - 7)
[Nap
roxe
n]
(µM
)
A
B
C
Abbildung 20: Plasmakonzentrations-Zeitkurven von Naproxen (A) und die entsprechenden COX-1- bzw. COX-2-Hemmungen (B und C) im Steady-State nach oraler Gabe von Naproxen Natrium zweimal pro Tag für sieben Tage. Die Blutproben für die Bestimmung der Plasmakonzentrationen von Naproxen und COX-Hemmungen wurden vor der ersten Tabletteeinnahme und zu bestimmten Zeitpunkten am 1. Tag (Abbildung 19) sowie an den Tagen 2, 3, 4, 5 und 8 zwölf Stunden nach der vorherigen Abenddosis (sieh Protokoll Tabelle 3) gewonnen. Die Untersuchungen in der Recovery-Phase erfolgten bis 36 Stunden nach der letzten Tabletteeinnahme. Es sind die Mittelwerte ± SEM von vier Probanden aufgetragen. Die eingefügte horizontale Linie (B) stellt die 95%-ige COX-1-Hemmung dar.
1 2 3 4 5
0
20
40
60
80
100
120
140
8 9
Naproxeneinnahme Recovery(Tag 8 - 9)(Tag 1 - 7)
95% Hemmung
CO
X-1
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
1 2 3 4 5
0
20
40
60
80
100
120
140
8 9
Naproxeneinnahme Recovery(Tag 8 - 9)(Tag 1 - 7)
CO
X-2
Akt
ivit
ät(%
vo
n B
asis
lin
ie)
Ergebnisse 88
6.5.3 Korrelation zwischen den Naproxenplasmakonzentrationen und ex-
vivo-Hemmungen der COX-Isoformen
Der Zusammenhang zwischen den Plasmakonzentrationen von Naproxen und der ex-vivo-
Hemmung von COX-1 und COX-2 wurde mittels grafischer Analyse untersucht (Abbildung 21).
Dafür wurden die Ergebnisse aus der Steady-state-Untersuchung und der Einmalgabe von
Naproxen kombiniert. Dadurch war es möglich, die ex-vivo IC50-Werte für die Hemmung der
beiden COX-Isoformen nach Naproxeneinnahme zu bestimmen. Für die COX-1 wurde ein IC50-
Wert von 35,5 µmol/l, für die COX-2 ein IC50-Wert von 64,6 µmol/l errechnet.
A
B
Abbildung 21: Korrelationen zwischen der ex-vivo-Hemmung von COX-1- (A) und COX-2-Aktivität (B) und den Plasmakonzentrationen von Naproxen. Für die Bestimmung der ex-vivo IC50-Werte wurden die Plasmakonzentrationen von Naproxen und die entsprechenden COX-Hemmungen nach einmaliger oraler Gabe von Naproxen Natrium und im Steady-state verwendet. Die Daten wurden von vier Probanden erhalten.
-5,0 -4,5 -4,0 -3,5 -3,0
0
20
40
60
80
100
IC50 = 35,5 µmol/l
log [Naproxen] (mol/l)
CO
X-1
-Hem
mu
ng
(%
)
-5.0 -4.5 -4.0 -3.5 -3.0
0
20
40
60
80
100
IC50 = 64.62 µmol/l
log [Naproxen] (mol/l)
CO
X-2
Hem
mu
ng
(%
)
Ergebnisse 89
Der sigmoidale Verlauf ist im Falle der COX-1 deutlicher ausgeprägt, da bei den maximalen
Plasmakonzentrationen nach einmaliger therapeutischer Dosierung von 220 mg Naproxen
Natrium eine 100 %ige Hemmung erreicht wurde (Abbildung 21). Weiterhin war auffällig, dass
für die COX-2 der sigmoidale Kurvenverlauf weniger deutlich ausgeprägt war als für die COX-1.
Dies ist auf die hohe Variabilität für die COX-Inhibition über den gesamten Bereich der