DE DE EUROPÄISCHE KOMMISSION Brüssel, den 31.5.2010 KOM(2010)270 endgültig 2010/0146 (NLE) C7- 0057/12 Vorschlag für einen BESCHLUSS DES RATES über den Abschluss eines Abkommens zwischen der Europäischen Union und Australien zur Änderung des Abkommens zwischen der Europäischen Gemeinschaft und Australien über die gegenseitige Anerkennung der Konformitätsbewertung, der Bescheinigungen und der Kennzeichnungen
Transcript
DE DE
EUROPÄISCHE KOMMISSION
Brüssel, den 31.5.2010 KOM(2010)270 endgültig
2010/0146 (NLE)
C7- 0057/12
Vorschlag für einen
BESCHLUSS DES RATES
über den Abschluss eines Abkommens zwischen der Europäischen Union und Australien zur Änderung des Abkommens zwischen der Europäischen Gemeinschaft und
Australien über die gegenseitige Anerkennung der Konformitätsbewertung, der Bescheinigungen und der Kennzeichnungen
DE 2 DE
BEGRÜNDUNG
I. DIE ÄNDERUNG
1. Hintergrund
Das Abkommen zwischen der Europäischen Gem einschaft und Australien („die Vertragsparteien“) über die gegenseitige Anerkennung der Konformitätsbewertung, der Bescheinigungen und der Kennzeichnungen 1 (im Folgenden „Abkomm en über die gegenseitige Anerkennung“ genannt) trat am 1. Januar 19992 in Kraft. Um die Funktionsweise des Abkomm ens über die gegenseitige Anerkennung weiter zu verbessern und zu vereinfachen, haben die Vertragsparteien beschlossen, einige Bestimmungen des Abkommens zu ändern.
Auf der Grundlage der Verhandlungsrichtlinien in dem spezifischen Beschluss des Rates vom 21. September 1992 zur Erm ächtigung der Komm ission, Vereinbarungen zwischen der Europäischen W irtschaftsgemeinschaft und be stimmten Dr ittländern über die gegenseitige Anerkennung der Konform itätsbewertungen auszuha ndeln, geändert durch die vom Rat a m 26. Mai 1997 und am 8. Juli 2002 angenomm enen s pezifischen Beschlüsse, hat die Kommission eine Änderung des Abkomm ens über die ge genseitige Anerkennung (i m Folgenden „Änderung“ genannt) ausgehandelt und paraphiert.
Der Wortlaut der Änderung ist diesem Vorschlag beigefügt. Die Kommission schlägt dem Rat vor, die Unterzeichnung der Änderung im Namen der Union zu genehmigen.
Das Abkommen zwischen der Europäischen Gem einschaft und Neuseeland über die gegenseitige Anerkennung der Konfor mitätsbewertung3 ist m it dem Abkomm en über die gegenseitige Anerkennung mit Australien praktisch identisch. Ein Vorschlag für ein paralleles Abkommen zur Änderung des Abkommens mit Neuseeland wird folgen.
2. Erläuterung der Änderung
Ziel der Änderungen ist es, die Struktur de r Sektoralen Anhänge des Abkomm ens über die gegenseitige Anerkennung flexibler zu m achen, unnötige H andelsbeschränkungen zwischen den Vertragsparteien zu be seitigen, den Verwaltungsaufwand im Zusa mmenhang m it de m Abkommen zu reduzieren und die Durchf ührung des Abkomm ens zu erleichtern und übersichtlicher zu gestalten.
Außerdem sind der S ektorale Anhang über Arzn eimittel, GMP-Kontrolle und Zertifizierun g der Chargen sowie der Sektorale Anhang übe r Medizinprodukte aufgrund von Änderungen in der technischen und adm inistrativen Praxis sowie von Änderungen bei den in diesen Anhängen auf gelisteten Organisationen inzw ischen über holt; s ie wurden bei dieser Gelegenheit überarbeitet.
Der Vorschlag hat keine finanzielle n Auswirkungen. Die Änderung wird im Amtsblatt der Europäischen Union veröffentlicht.
1 ABl. L 229 vom 17.8.1998, S. 3. 2 ABl. L 5 vom 9.1.1999, S. 74. 3 Ebda., S. 62.
DE 3 DE
Es folgt eine ausführliche Erläuterung der Änderung:
1. Zur Beseitigung unnötiger Handelsbeschränkungen wird die in Artikel 4 festgelegte Beschränkung der Anwendung des Abkomm ens auf gewerbliche Ursprungswaren der Vertragsparteien gemäß den nichtpräferentiellen Ursprungsregeln aufgehoben. In seiner geänderten Fassung gilt das Abkommen über die gegenseitige Anerkennung für alle unter das Abkommen fallenden Waren, unabhängig von ihrem Ursprung.
2. Um der Tatsache Rechnung zu tragen, dass der Vorsitz im Ge mischten Ausschuss von den Vertragsparteien gemeinsam geführt wird, werden die Bezugnahmen auf den Vorsitz des Gemischten Ausschusses in den Artikeln 8 und 12 gestrichen.
3. Um die Durchführung des Abkomm ens über die gegenseitig e Anerk ennung zu vereinfachen, wird in Artikel 12 ein einfacheres Verfahren zur Benennung, Rücknahme der Benennung und Aussetzung der Benennung von Konfor mitäts-bewertungsstellen aufgenommen. Infolg edessen m uss de m Be schluss einer benennenden Behörde über die Benennung oder Rücknahm e der Benennung einer Konformitätsbewertungsstelle nicht länger durch eine Änderung eines Sektoralen Anhangs Wirksamkeit verliehen werden; der Gemischte Ausschuss muss nur noch in Fällen einer Anfechtung durch die andere Vertragspartei nach Artikel 8 tätig werden.
4. Damit die Sektoralen Anhänge zeitnah an den technischen Fortschritt angepasst und andere Faktoren wie die Erweiterung der Europäischen Union berücksichtigt werden können, wird Artikel 12 auch dahingehe nd geändert, dass der Gem ischte Ausschuss ausdrücklich dazu erm ächtigt wird, die Sek toralen Anhän ge auch in Fällen zu ändern, in denen es nicht darum geht, dem Beschluss einer benennenden Behörde über die B enennung oder die Rücknahm e der Benennung einer bestimm ten Konformitätsbewertungsstelle W irksamkeit zu verleihen, sowie neue Sektorale Anhänge anzunehmen.
5. Artikel 3 wird geändert, um den Ände rungen des Artikels 12 Rechnung zu tragen und die Struktur der S ektoralen Anhänge des Abkomm ens über die gegenseitige Anerkennung flexibler zu gestalten.
6. Der Wortlaut der Artikel 6, 7, 8, 9 und 15 sowie der Nummern 9 und 10 des Anhangs wurde geändert, um den Änderungen des Artikels 12 Rechnung zu tragen.
7. Der Sektorale Anhang über Arzneim ittel, GMP-Kontrolle und Zertifizierung d er Chargen wurde überarbeitet, um die Entwicklungen in der technischen und administrativen Praxis, die sich aus der Änderung des verfügenden Teils des Abkommens über die gegenseitige An erkennung ergebenden Änderungen, die Aktualisierungen hinsichtlich der au fgelisteten Organisationen sowie die Änderungen der diesen Sektor betreffenden Rechtsvorschrif ten der Vertragsparteien zu berücksichtigen. Die Funktionsweise dieses Sektoralen Anhangs bleibt im Grundsatz unverändert.
8. Der Sektorale Anhang über Medizinpr odukte wurde überarbeitet, um die Entwicklungen in der technischen und adm inistrativen Praxis, die sich aus der Änderung des verfügenden Teils des Abkommens über die gegenseitige Anerkennung ergebenden Änderungen, die Aktualisierungen hinsichtlich der aufgelisteten Organisationen sowie die Ä nderungen der diesen Sektor betreffenden
DE 4 DE
Rechtsvorschriften der Vertragsparteien zu berücksich tigen. Die Funktionsweis e dieses Sektoralen Anhangs bleibt im Grundsatz unverändert.
3. Verhältnis zu EFTA/EWR-Mitgliedsländern
Im Einklang m it den im Abkomm en über den Europäischen W irtschaftsraum und in seine m Protokoll 12 festgelegten Inform ations- und K onsultationsverfahren hat die Komm ission die EFTA/EWR-Mitgliedsländer über die Fortschr itte bei den Verhandlungen und über deren Endergebnis informiert.
II. DER VORSCHLAG FÜR EINEN BESCHLUSS DES RATES
Das Abkommen zwis chen der Europäischen Union und Australien zur Änderung des Abkommens zwischen der Europäischen Gem einschaft und Australien üb er die gegenseitige Anerkennung der Konformitätsbewertung wurde von der Kommission am […] unterzeichnet.
Die Kommission schlägt daher dem Rat vor, de n beigefügten Beschlus s über den Abschluss der Änderung des Abkommens nach Zustimmung des Parlaments anzunehmen.
DE 5 DE
2010/0146 (NLE)
Vorschlag für einen
BESCHLUSS DES RATES
über den Abschluss eines Abkommens zwischen der Europäischen Union und Australien zur Änderung des Abkommens zwischen der Europäischen Gemeinschaft und
Australien über die gegenseitige Anerkennung der Konformitätsbewertung, der Bescheinigungen und der Kennzeichnungen
DER RAT DER EUROPÄISCHEN UNION –
gestützt auf den Vertrag über die Arbeitsweis e der Europäischen Union, insbesondere auf Artikel 207 Absatz 4 Unterabsatz 1 in Verbindung mit Artikel 218 Absatz 6 Buchstabe a,
auf Vorschlag der Kommission,
nach Zustimmung des Europäischen Parlaments4,
in Erwägung nachstehender Gründe:
(1) Das Abkomm en z wischen der Europäischen Gem einschaft und Australien über die gegenseitige Anerkennung der Konform itätsbewertung, der Bescheinigungen und der Kennzeichnungen5 (im Folgenden „Abkomm en übe r die gegenseitige Anerkennung“ genannt) trat am 1. Januar 19996 in Kraft.
(2) Gemäß dem Beschluss 2010/XXX des Rates vo m [… ]7 wurde das Abkomme n zwischen der Europäischen Union und Australien zur Änderung des Abkomm ens zwischen d er Europäis chen Gem einschaft und Australien über die gegenseitige Anerkennung der Konform itätsbewertung (im Folgenden „Abkomm en“ genannt) von der Komm ission vorbehaltlich seines Abschl usses zu einem späteren Zeitpunkt am […] unterzeichnet.
(3) Das Abkommen sollte abgeschlossen werden –
HAT FOLGENDEN BESCHLUSS ERLASSEN:
Artikel 1
Das Abkommen zwis chen der Europäischen Union und Australien zur Änderung des Abkommens zwischen der Europäischen Gem einschaft und Australien üb er die gegenseitige
4 ABl. C […] vom […], S. […]. 5 ABl. L 229 vom 17.8.1998, S. 3. 6 ABl. L 5 vom 9.1.1999, S. 74. 7 ABl. L […] vom […], S. […].
DE 6 DE
Anerkennung der Konform itätsbewertung, der Bescheinigungen und der Kennzeichnungen (im Folgenden „Abkommen“ genannt) wird geschlossen.
Der Wortlaut des abzuschließenden Abkommens ist diesem Beschluss beigefügt.
Artikel 2
Der Ratspräsident bestellt die Person, die bef ugt ist, die in Artikel 14 des Abkommens über die gegens eitige Anerk ennung gen annte dip lomatische Note im Nam en der Europäisch en Union zu übermitteln, um die Zustimmung der Europäischen Union zur Rechtsverbindlichkeit des Abkommens zum Ausdruck zu bringen.
Artikel 3
Dieser Bes chluss tr itt am Tag seiner Annahm e in Kraf t. Er wird im Amtsblatt der Europäischen Union veröffentlicht.
Der Tag des Inkrafttretens des Abkomm ens wird im Amtsblatt der Europäischen Union veröffentlicht.
Geschehen zu Brüssel am […]
Im Namen des Rates Der Präsident […]
DE 7 DE
ANNEX I
ABKOMMEN
zur Änderung des Abkommens zwischen der Europäischen Gemeinschaft und Australien über die gegenseitige Anerkennung der Konformitätsbewertung, der
Bescheinigungen und der Kennzeichnungen
DIE EUROPÄISCHE UNION UND AUS TRALIEN, i m Folgenden „die Vertragsparteien“ genannt –
NACH ABSCHLUSS eines Abkomm ens übe r die gegenseitige Anerkennung der Konformitätsbewertung, der Bescheinigunge n und der K ennzeichnungen (im Folgenden „Abkommen“ genannt), das am 24. Juni 1998 in Canberra unterzeichnet wurde,
IN ANBETRACHT der Notwendigkeit, die Durchführung des Abkommens zu vereinfachen,
IN ANBETRACHT der Notwendigkeit, den Status der Sektoralen Anhänge des Abkomm ens zu klären,
IN DER ERWÄGUNG, dass in Ar tikel 3 des Abkommens die Form der Sektoralen Anhänge detailliert festgelegt wird,
IN DER ERWÄGUNG, dass in Artikel 4 des Abkommens die Anwendung des Abkomm ens auf gewerbliche Ursprungswaren der Vertrags parteien gem äß den nichtpräferentiellen Ursprungsregeln beschränkt wird,
IN DER ER WÄGUNG, dass in Artikel 12 des Abkommens ein Gem ischter Ausschuss eingesetzt wird, der unter anderem de n Beschlüssen über die Aufnahm e von Konformitätsbewertungsstellen in die Sektor alen Anhänge und über ihre Streichung aus diesen Anhängen Wirksamkeit verleiht, und ein diesbezügliches Verfahren festgelegt wird,
IN DER ERWÄGUNG, dass in den Artikeln 8 und 12 des Abko mmens auf den Vorsitz des Gemischten Ausschusses Bezug genommen wird,
IN DER E RWÄGUNG, dass in Artikel 12 des Abkommens der Gem ischte Ausschuss nicht ausdrücklich dazu erm ächtigt wird, die Sektor alen Anhänge zu ändern, es sei denn, um dem Beschluss einer benennenden Behörde übe r die Benennung oder die Rücknahm e der Benennung einer bestimmten Konformitätsbewertungsstelle Wirksamkeit zu verleihen,
EINGEDENK der Tatsache, dass Artikel 3 geä ndert werden sollte, um einerseits den Änderungsvorschlägen zu Artikel 12 Rechnung zu tragen, wonach die Anforderung an den Gemischten Ausschuss, bei der B enennung oder der Rücknahm e der Benennung von Konformitätsbewertungsstellen tätig zu werden, au f diejenigen Fälle besc hränkt werden soll, in denen eine Anfechtung durch die andere Ve rtragspartei nach Ar tikel 8 vorliegt, und u m andererseits die Struktur der Sektoralen Anhänge des Abkommens flexibler zu gestalten,
DE 8 DE
IN ANBETRACHT DESSEN, dass die Urspr ungsbeschränkung in Artikel 4 aufgehoben werden sollte, dam it der Handel zwischen den Vertragsparteien nicht unnötig eingeschränkt wird,
IN DER ERWÄGUNG, dass die Bezugnahm en auf den Vorsitz des Gem ischten Ausschusses in den Artikeln 8 und 12 des Abkomm ens gest richen w erden sollten, um der Tatsache Rechnung zu tragen, dass der Vorsitz im Ge mischten Ausschuss von den Vertragsparteien gemeinsam geführt wird,
EINGEDENK der Tatsache, dass ein inten siverer Informations austausch zwisch en den Vertragsparteien über die Durchführung des Abkommens dessen Durchführung erleichtern wird,
UNTER BERÜCKSICHTIGUNG der Tatsache, dass der Gemischte Ausschuss in Artikel 12 ausdrücklich dazu ermächtigt werden sollte, di e Sektoralen Anhänge auch in anderen Fällen, als um dem Beschluss einer benennenden Be hörde über die Benennung oder die Rücknahme der Benennung einer bestimmten Konformitätsbewertungsstelle Wirksamkeit zu verleihen, zu ändern und auch neue Sektorale Anhänge anzunehmen, damit die Sektoralen Anhänge zeitnah an den technischen F ortschritt angepasst und ande re Faktoren wie die Erweiterung der Europäischen Union berücksichtigt werden können,
IN ANE RKENNUNG der Tatsache , dass die Vertragsparteien m öglicherweise bestimm te nationale Verfahren durchführen müssen, bevor die Änderungen der Sektoralen Anhänge oder die Annahme neuer Sektoraler Anhänge wirksam werden,
IN DER ERWÄGUNG, dass ein Tätigwerden des Gem ischten Ausschusses bei der Benennung oder der Rücknahm e der Benennung von Konfor mitätsbewertungsstellen auf diejenigen Fälle beschränkt werden sollte, bei denen eine Anfechtung durch die andere Vertragspartei nach A rtikel 8 vorliegt , um die Durchführung des Abkomm ens zu vereinfachen,
IN ANBETRACHT der Tatsache, dass im Hinblick auf eine vereinfach te Durchführung des Abkommens in Artikel 12 ein einfacheres Verfahren zur Benennung, Rücknahm e der Benennung und Aussetzung der Benennung von Konfor mitätsbewertungsstellen festgelegt und der Standpunkt zu Konformitätsbewertungen, die von Stellen durchgeführt wurden, deren Benennung im Nachhinein ausgesetzt oder zu rückgenommen wurde, klargestellt werden sollte –
SIND ÜBEREINGEKOMMEN, DAS ABKOMMEN WIE FOLGT ZU ÄNDERN:
Artikel 1
Änderungen des Abkommens
Das Abkommen wird wie folgt geändert:
1. Artikel 3 Absatz 2 erhält folgende Fassung:
DE 9 DE
„2. Jeder Sektorale Anhang enthält im Allgemeinen folgende Informationen:
a) Angaben zu seinem Anwendungs- und Geltungsbereich,
b) die Rechts- und Verw altungsvorschriften für die Konform itätsbewer-tungsverfahren,
c) die benennenden Behörden,
d) die Verfahren für die Benennung von Konformitätsbewertungsstellen und
e) gegebenenfalls zusätzliche Bestimmungen.“
2. Artikel 4 erhält folgende Fassung:
„Artikel 4
Anwendungs- und Geltungsbereich
Die Bestimmungen dieses Abkomm ens gelten für die Konfor mitätsbewertung der in den Angaben zum Anwendungs- und Geltungsbere ich in den einzelnen Sektoralen Anhängen genannten Produkte.“
3. Artikel 6 Absatz 1 erhält folgende Fassung:
„1. Die Vertragsparteien stellen si cher, dass die für die Benennung der Konformitätsbewertungsstellen zustä ndigen benennenden Behörden über die erforderlichen Befugnisse und die erfo rderliche fachliche Kom petenz zur Benennung, zur Aussetzung der Benennung, zum W iderruf der Aussetzung und zur Rücknahme der Benennung dieser Stellen verfügen.“
4. Artikel 6 Absatz 2 erhält folgende Fassung:
„2. Sofern in den Sektoralen Anhängen nichts anderes bestimmt ist, halten sich die benennenden Behörden bei der Bene nnung, der Aussetzung der Benennung, dem Widerruf der Aussetzung und der Rü cknahme der Benennung an die in Artikel 12 und im Anhang vorgesehenen Benennungsverfahren.“
5. Artikel 6 Absatz 3 wird gestrichen.
6. Artikel 7 Absatz 1 erhält folgende Fassung:
„1. Die Vertragsparteien tauschen Informationen über die Verfahren aus, durch die sichergestellt werden so ll, dass die in ihre Zuständigkeit fallenden benannten Konformitätsbewertungsstellen die in de n Sektoralen Anhängen festgelegten Rechts- und Verwaltungsvorschriften beachten und den Anforderungen an ihre fachliche Kompetenz gemäß dem Anhang genügen.“
7. Artikel 8 Absatz 3 erhält folgende Fassung:
DE 10 DE
„3. Diese Anfechtung ist in einem an die andere Vertra gspartei und den Gemischten Ausschuss gerichtete n Schreiben m it objektiven und sachdienlichen Argumenten zu begründen.“
8. Artikel 8 Absatz 6 erhält folgende Fassung:
„6. Sofern der Ge mischte Ausschuss ni chts anderes beschließt, wird die Benennung der betreffenden Konfor mitätsbewertungsstelle von der zuständigen benennenden Behörde ab dem Zeitpunkt, zu dem die Erfüllung der Anforderungen durch diese Stelle oder ih re fachliche Kompetenz angefochten wurde, so lange ausgesetzt, b is im Gemischten Ausschuss eine Einigung über den Status der Stelle erzielt wurde o der bis d ie anfechtende Vertragspartei der anderen Vertragspartei und dem Gemischten Ausschuss notifiziert, dass sie die fachliche K ompetenz der betreffenden Konformitätsbewertungsstelle und die Erfüllung der Anforderungen durch diese Stelle als zufriedenstellend erachtet.“
9. Artikel 9 erhält folgende Fassung:
„Artikel 9
Informationsaustausch
1. Die Vertragsparteien tauschen Informationen über die Durchführung der in den Sektoralen Anhängen aufgeführten Rech ts- u nd Verwaltun gsvorschriften aus und führen eine aktuelle Liste de r im Einklang m it diesem Abkommen benannten Konformitätsbewertungsstellen.
2. Im Einklang m it ihren Verpflichtun gen aufgrund des W TO-Übereinkommens über technische Handelshemmnisse unterrich tet jede Vertragspartei die andere Vertragspartei über die von ihr beabsi chtigten Änderungen der Rechts- und Verwaltungsvorschriften in den Bereichen, die Gegenstand dieses Abkommens sind, und notifiziert, außer in dem in Ar tikel 9 Absatz 3 genannten Fall, der anderen Vertragspartei die neuen Bestimmungen mindestens 60 Tage vor deren Inkrafttreten.
3. Ergreift eine Vertragspartei dri ngende Maßnahm en, die sie aus Gründen der Sicherheit, der Gesundheit oder des Umweltschutzes für gerechtfertigt hält, um eine unm ittelbar drohende Gefahr ab zuwenden, die von einem unter einen Sektoralen Anhang fallenden Produkt au sgeht, so setzt sie die andere Vertragspartei über die Maßnahm en und die Gründe für deren Einleitung unverzüglich oder gem äß anderslautender Bestimmungen in einem Sektoralen Anhang in Kenntnis.“
10. Artikel 12 Absatz 3 erhält folgende Fassung:
„3. Der Gem ischte Ausschuss tritt m indestens einm al jährlich zusammen, s ofern der Gemischte Ausschuss oder die Vertragsparteien nichts anderes beschließen. Wenn dies für die ordnungsgem äße Durchführung dieses Abkommens erforderlich ist, oder auf Antrag einer Vertragspartei, können eine oder mehrere zusätzliche Sitzungen anberaumt werden.“
11. Artikel 12 Absatz 4 erhält folgende Fassung:
DE 11 DE
„4. Der Gem ischte Ausschu ss behande lt alle F ragen im Zusamm enhang mit de r Durchführung dieses Abkommens. Insbesondere ist er für Folgendes zuständig:
a) Änderung der Sektoralen Anhänge nach Maßgabe dieses Abkommens;
b) Austausch von Informationen über die Verfahren, die von den Vertragsparteien angewendet werde n, um sicherzustellen, dass die Konformitätsbewertungsstellen das erforderliche Kom petenzniveau beibehalten;
c) Einsetzung einer oder m ehrerer gemischter Expertengruppen zwecks Überprüfung der fachlichen Kom petenz einer Konfor mitätsbewertungs-stelle und der Erfüllung anderer einschlägiger Anforderungen durch diese Stelle gemäß Artikel 8;
d) Inform ationsaustausch und Notifikation der Änderungen der in den Sektoralen Anhängen aufgeführten R echts- und Verwaltungsvorschriften einschließlich derjenige n, die eine Änderung der Sektoralen Anhänge erfordern, an die Vertragsparteien;
e) Regelung aller Fragen im Zusa mmenhang mit der Durchführung dieses Abkommens und seiner Sektoralen Anhänge;
f) Annahme neuer Sektoraler Anhänge nach Maßgabe dieses Abkommens.“
12. Artikel 12 Absatz 5 erhält folgende Fassung:
„5. Der Gemischte Ausschuss notifiziert jeder Vertragspartei umgehend schriftlich alle im Einklang m it diesem A bkommen vorgenommenen Änderungen der Sektoralen Anhänge sowie alle im Einklang m it diesem Abkomm en angenommenen neuen Sektoralen Anhänge; diese werden für die Vertragsparteien zu dem Zeitpunkt wirksa m, zu de m jede Vertragspartei de m Gemischten Ausschuss den Abschluss ihr er jeweilig en Verf ahren f ür das Wirksamwerden der Änderungen der Sektoralen Anhänge oder neuer Sektoraler Anhänge notifiziert hat, so fern d ie Vertrags parteien nic ht im gegenseitigen Einvernehmen schriftlich etwas anderes festlegen.“
13. Artikel 12 Absatz 6 erhält folgende Fassung:
„6. Für die B enennung einer Konform itätsbewertungsstelle gilt folgendes Verfahren:
a) Eine Vertragspartei, die eine Konformitätsbewertungsstelle benennen möchte, überm ittelt der andere n Vertrags partei ihre n Vorschla g schriftlich mit den vom Gemischten Ausschuss festgelegten Unterlagen.
b) Nachdem die andere Vertragspart ei dem Vorschlag zugestimmt hat oder nach Ablauf von 60 Tagen, sofern innerhalb dieser Frist keine Einwände gemäß den Verfahren des Ge mischten Ausschusses erhoben werden, gilt die Konformitätsbewertungsstelle als benannte Konfor mitätsbewertungs-stelle nach Maßgabe des Artikels 5.
DE 12 DE
c) Bestreitet d ie ande re Vertragsp artei gem äß Artikel 8 die fachliche Kompetenz einer Konform itätsbewertungsstelle oder die E rfüllung der Anforderungen durch diese Stelle i nnerhalb der vorgenannten Frist von 60 Tagen, so kann der Ge mischte Ausschuss ge mäß Ar tikel 8 eine Überprüfung der betreffenden Stelle beschließen.
d) Im Fall der Benennung einer neuen Konformitätsbewertungsstelle sind die von dieser Stelle vorgenommene n Konformitätsbewertungen ab dem Zeitpunkt gültig, zu dem die Konformitätsbewertungsstelle eine benannte Konformitätsbewertungsstelle nach Maßgabe dieses Abkommens wird.
e) Jede Vertragspartei kann die Benennung einer Konfor mitätsbewertungs-stelle in ihrem Zuständigkeitsber eich aussetzen, die Aussetzung der Benennung widerrufen oder die Benennung zurücknehm en. Die betreffende Vertragspartei notifiziert der anderen Vertragspartei und dem Gemischten Ausschuss umgehend schrif tlich ihren Beschluss, zusammen mit dem Zeitpunkt, zu dem dieser Beschluss erging. Die Aussetzung, der Widerruf der Aussetzung oder die Rücknahme der Benennung werden zu dem Zeitpunkt wirksam, zu dem der Beschluss der Vertragspartei erging.
f) Gemäß Artikel 8 hat jede Vertragspartei das Recht, unter außergewöhnlichen Umständen die f achliche Kom petenz einer in d ie Zuständigkeit der anderen Vertra gspartei fallenden benannten Konformitätsbewertungsstelle anzuf echten. In diesem Fall kann der Gemischte Ausschuss ge mäß Artikel 8 eine Überprüfung der betreffenden Stelle beschließen.“
14. Artikel 12 Absatz 7 erhält folgende Fassung:
„7. Wird die Benennung einer Konfor mitätsbewertungsstelle ausgesetzt oder zurückgenommen, so bleiben die Konfor mitätsbewertungen, die von dieser Konformitätsbewertungsstelle vor de m Zeitpunkt vorgenommen wurden, zu dem die Aussetzung oder die Rücknahme der Benennung wirksam wird, gültig, sofern die zuständige Vertragspartei ihre Gültig keit n icht eingeschränk t oder aufgehoben hat oder der Ge mischte Ausschuss nichts anderes beschließt. Die Vertragspartei, in deren Zuständigke itsbereich die Konform itätsbewertungs-stelle tätig war, deren Benennung ausg esetzt oder zurückgenommen wurde, notifiziert der anderen Vertrags partei sch riftlich alle Änderungen im Zusammenhang mit einer Einschränkung oder Aufhebung der Gültigkeit.“
15. Nach Artikel 12 Absatz 8 wird folgender Absatz 9 in das Abkommen eingefügt:
„9. Der Gemischte Ausschuss aktualisiert laufend die Sektoralen Anhänge und legt diese den Vertragsparteien vor, sobald Änderungen wirksam werden.“
16. Artikel 15 Absatz 1 erhält folgende Fassung:
„1. Der Anhang ist Bestandteil dieses Abkommens. Die Sektoralen Anhänge bilden die Verwaltungsvereinbarungen für die Durchführung dieses Abkommens, sie haben keinen Vertragsstatus.“
DE 13 DE
17. Artikel 15 Absatz 3 erhält folgende Fassung:
„3. Der Gem ischte Ausschuss kann Se ktorale Anhänge a nnehmen, auf die Artikel 2 Anwendung findet und die die Durchführungsbestimm ungen für dieses Abkomm en enthalten. Zusätzli che Sektorale Anhänge werden gem äß Artikel 12 Absatz 5 wirksam.“
18. Artikel 15 Absatz 4 erhält folgende Fassung:
„4. Über Änderungen der Sektoralen A nhänge und die Annahme neuer Sektoraler Anhänge entscheidet der Gem ischte Ausschuss, sie werden gem äß Artikel 12 Absatz 5 wirksam.“
19. Nummer 9 des Anhangs erhält folgende Fassung:
„9. Die benennenden Behörden unterrichten die Vertrete r ih rer Vertragsp artei in dem gemäß Artikel 12 dieses Abkommens eingesetzten Gemischten Ausschuss darüber, welche Konformitätsbewertungsstellen benannt werden sollen und für welche Konfor mitätsbewertungsstellen die Benennung ausgesetzt oder zurückgenommen wer den soll. Die Benennung, die Aussetzung oder die Rücknahme der Benennung von Konfor mitätsbewertungsstellen erfolgt im Einklang m it den Bestimmungen dieses Abkommens und der Geschäftsordnung des Gemischten Ausschusses.“
20. Nummer 10 des Anhangs erhält folgende Fassung:
„10. Die benennende Behörde er teilt dem Vertreter ihrer Ve rtragspartei in dem mit diesem Abkomm en e ingesetzten Ge mischten Ausschuss zu jeder zu benennenden Konformitätsbewertungsstelle folgende Angaben:
a) Nam e,
b) Postanschrift,
c) Faxnummer und E-Mail-Adresse,
d) Palette der Produkte, Verfahren, Normen oder Dienstleistungen, für deren Bewertung sie zugelassen ist,
e) Konfor mitätsbewertungsverfahren, für deren Durchführung sie zugelassen ist, und
f) Verfahren zur Feststellung ihrer fachlichen Kompetenz.“
21. Der Sektorale Anhang über Arzneim ittel, GMP-Kontrolle und Zertifizierung der Chargen wird gestrichen und durch folgenden Wortlaut ersetzt:
„SEKTORALER ANHANG ÜBER ARZNEIMITTEL, GMP-KONTROLLE UND ZERTIFIZIERUNG DER CHARGEN
ANWENDUNGS- UND GELTUNGSBEREICH
DE 14 DE
1. Die Vertragsparteien legen einvernehm lich fest, dass dieser Sektorale Anhang für alle Arzneim ittel gilt, die in Australien und in der Europäischen Unio n industriell hergestellt werden und den Anfor derungen an die gute Herstellungspraxis (Good Manufacturing Practice, GMP) genügen müssen.
Für die un ter d iesen Sektora len Anha ng f allenden Arz neimittel er kennt jede Vertragspartei die Ergebnisse der von de n zuständigen Kontrolldiensten der anderen Vertragspartei durchgeführten Kontrollen der Hersteller und die von den zuständigen Behörden der anderen Vertragspartei erteilten Herstellungsgenehmigungen an.
Ferner wird die vom Hersteller vorgenommene Zertifizierung der Konf ormität jeder Charge m it ihren Spezifikationen von der anderen Vertragspart ei ohne erneute Kontrolle bei der Einfuhr anerkannt.
„Arzneimittel“ sind alle Produkte, die unt er die in Abschnitt I aufgeführten Arzneimittelvorschriften der Europäischen Union und Australiens fallen. Die Definition der Arzneimittel umfasst alle Human- und Tierarzneimittel wie chemische und biologische Arzneim ittel, immunologi sche Arzneim ittel, Radiopharm aka, haltbare Ar zneimittel a us m enschlichem Blut oder aus m enschlichem Plasm a, Vormischungen für die Herstellung von Tier arzneifuttermitteln und gegebenenfalls Vitamine, Mineralien, Heilkräuter und homöopathische Arzneimittel.
„GMP“ ist jener Teil der Qualitätss icherung, durch den sich ergestellt wird, dass die Produkte durchweg nach den Qualitätsnor men für ihre beabsichtigte Verwendung und im Einklang mit der von der einführenden Vertragspartei erteilten Genehmigung für das Inverkehrbringen hergestellt und im Laufe der Herste llung kontrolliert werden. Für die Zwecke dieses Sektoralen Anhangs um fasst sie auch das System, nach dem der Hersteller vom Inhaber der Genehm igung für das Inverkehrbringen oder vom Antragsteller die Spezifikati on des Produkts und/ode r des Verfahrens erhält und s icherstellt, dass das Arzneim ittel gemäß dieser Spezifikatio n herges tellt wird (entspricht der Zertifizierung durch eine sachkundige Person in der Europäischen Union).
2. Bei Arzneim itteln, die unter die Rechtsvor schriften d er einen Vertragsp artei („regulierende Vertrag spartei“), nicht ab er unter diejenig en d er anderen Vertragspartei fallen, kann der Herstell er [bei der Behörde, die von der in Abschnitt III unter N ummer 12 auf geführten zuständig en Kontaktste lle d er regulierenden Vertragspartei benannt wurde] für die Zwecke dieses Abkommens eine Kontrolle durch den örtlich zuständi gen Kontrolldienst beantragen. Diese Bestimmung gilt unter anderem fü r die Herstellung von pharmazeutischen Wirkstoffen, Zwischenprodukten und Produkt en, die für klinische Versuche bestimmt s ind, sowie für einvernehm lich festgelegte Kontrollen vor dem Inverkehrbringen. Die Durchführungsbestimmungen sind in Abschnitt III Nummer 3 Buchstabe b enthalten.
Zertifizierung der Hersteller
3. Auf Antrag eines Ausführers, eines Ei nführers oder der zuständigen Behörde der anderen Vertragspartei beschein igen die für die Erteilung der Herstellungsgenehmigungen und di e Überwachung de r Herstellung von Arzneimitteln zuständigen Behörden, dass der Hersteller:
DE 15 DE
– eine ordnungsgem äße Genehm igung zur Herstellung des betreffenden Arzneimittels oder zur Durchführung des betreffenden Herstellungsvo rgangs besitzt,
– regelmäßig von den Behörden kontrolliert wird und
– den nationalen GMP-Anforderungen nach Abschnitt I genügt, die von beiden Vertragsparteien als gleichwertig anerkannt werden. Kann auf unterschiedliche GMP-Anforderungen Bezug genomm en werden (gem äß Abschnitt III Nummer 3 Buchstabe b), so wird dies auf der Bescheinigung vermerkt.
Die Bescheinigungen weisen ferner de n oder die Herstellungsstandorte (und gegebenenfalls die vertraglich verpflichteten Prüflaboratorien) aus. Über das Muster der Bescheinigung befindet die Gemischte Sektorgruppe.
Die Bescheinigungen werden rasch ausgeste llt, spätestens jedoch innerhalb von 30 Kalendertagen. In Ausnahm efällen, wenn z. B. eine neue Kontrolle durchgeführt werden muss, kann diese Frist auf 60 Tage verlängert werden.
Zertifizierung der Chargen
4. Jeder ausgeführten Charge ist eine Besche inigung beigefügt, die der Hersteller nach einer vollständigen qualitativen Analyse, einer quantitativen Analyse aller Wirkstoffe und nach Durchführung aller anderen Test s oder Kontrollen ausstellt, die zur Gewährleistung der Qualität des P rodukts entsprechend den Anforderungen der Genehmigung für das I nverkehrbringen erforderlich sind (Selbstzertifizierung). Mit dieser Bescheinigung wird die Üb ereinstimmung der Charge m it ihren Spezifikationen bestätigt; sie wird vom Einführer der Charge aufbewahrt. Auf Antrag der zuständigen Behörde wird sie vorgelegt.
Der Hersteller stellt die Bescheini gung nach den Bestimmungen des derzeit geltenden W HO-Zertifizierungssystems f ür die Qualitä t der Arz neimittel im internationalen Handelsverkehr aus. Auf de r Bescheinigung werden die detaillierten Spezifikationen des Produkts, die Re ferenz der Analysem ethode und die Analyseergebnisse vermerkt. Ferner wird dam it erklärt, dass die Aufzeichnungen über die Verarbeitung und Verpackung der Charge überprüf t wurden und der GM P entsprechen. Die Bescheinigung wird von der für die Freigabe der Charge zum Verkauf oder zur Auslieferung verantwortlichen Person unterzeichnet, bei der es sich in der Europäischen Union um die in ei nschlägigen EU-Rechtsvorschriften genannte „sachkundige Person“ handelt. Für die Qu alitätskontrolle bei der H erstellung in Australien sind die in den einschlägigen australischen Rechtsvorschriften genannten Personen zuständig.
DE 16 DE
ABSCHNITT I
RECHTS- UND VERWALTUNGSVORSCHRIFTEN
Vorbehaltlich der Durchführungsbestimm ungen in Abschnitt III werden die allgem einen GMP-Kontrollen anhand der GMP-Anforder ungen der ausführenden Vertragspartei durchgeführt. Die für diesen Sektoralen Anhang geltenden Rechts- und Verwaltungsvorschrif-ten sind in der Anlage aufgeführt.
Es gelten jedoch die Q ualitätsanforderungen an die auszuführenden Produkte einschließlich ihrer Herstellungsm ethode und Spe zifikationen, die in der von der einführenden Partei erteilten Genehmigung für das Inverkehrbringen des betreffenden Produkts festgelegt sind.
ABSCHNITT II
AMTLICHE KONTROLLDIENSTE
Die Listen der am tlichen Kontrolldienste fü r diesen Sektoralen Anhang wurden von den Vertragsparteien einvernehmlich erstellt und we rden von ihnen laufend aktualisiert. Ersucht eine Vertragspartei di e andere Vertragspartei um ein Ex emplar der aktuellen L isten ihrer amtlichen Kontrolld ienste, so übe rmittelt d ie ersuch te Vertragsp artei der ersu chenden Vertragspartei innerhalb von 30 Tagen nach Eingang des Ersuchens ein Exem plar dieser Listen.
ABSCHNITT III
DURCHFÜHRUNGSBESTIMMUNGEN
1. Übermittlung der Kontrollberichte
Wenn bestimmte Analysearbeiten vergeben werden, überm itteln die zuständigen Kontrolldienste auf begründeten Antrag eine Kopie des letzten Kontrollberichts über die Herstellungsanlage oder über die kontrollierte Anlage. Es kann ein „vollständiger Kontrollbericht“ od er ein „ausführlicher Be richt“ (siehe Numm er 2) angefordert werden. Die Vertragspartei behandelt diese Kontrollberichte mit der Vertraulichkeit, die von der sie übermittelnden Vertragspartei gefordert wird.
Liegt die Kontrolle des Herstellu ngsverfahrens f ür das betref fende Arzneim ittel längere Zeit zurück, d. h. mehr als zw ei Jahre, oder wird ein besonderer Kontrollbedarf festgestellt, so kann eine spezifische und detaillierte Kontrolle beantragt w erden. Die Vertragsp arteien s tellen sicher, dass die Kontrollberichte innerhalb von 30 Kalendertagen versandt werden, wobei diese Frist auf 60 Tage verlängert wird, wenn eine neue Kontrolle durchgeführt wird.
2. Kontrollberichte
Ein „vollständiger“ Kontrollbericht um fasst die (vom Hersteller oder vom Kontrolldienst zusammengestellten) Stammdaten der Anlage (Site Master File) und einen Bericht des Kontrolldienstes. Ein „au sführlicher Bericht“ beantwortet die von der anderen Vertragspartei gestellten spezifischen Fragen zu einem Unternehmen.
3. Bezugs-GMP
DE 17 DE
a) Die Hersteller werden anhand d er geltenden GMP der ausführenden Vertragspartei (siehe Abschnitt I) kontrolliert.
b) Bei den Arzneimitteln, die unter die Arzneimittelvorschriften der einführenden Vertragspartei, aber nicht unter die de r ausführenden Vertragspartei fallen, kontrolliert der örtl ich zuständige Kontrolldiens t, der zur Durchführung der Kontrolle des betreffenden Herstellungsve rfahrens bereit ist, die betreffenden Herstellungsvorgänge anhand seiner ei genen GMP oder - in Erm angelung spezifischer GMP-Anforderungen - anhand der geltenden G MP der einführenden Vertragspartei. Letzteres gilt auch in dem Fall, in dem die lokale GMP in Bezug auf die Qualitätssicherung des Endprodukts nicht als gleichwertig m it der GMP der einführe nden Vertragspartei angesehen wird. Die Gleichwertigkeit der GMP-Anfo rderungen für spezifische Produkte oder Produktklassen (z. B. Arzneim ittel für Untersuchungszwecke, Ausg angs-materialien) wird nach einem von der Gemischten Sektorgruppe festgelegten Verfahren bestimmt.
4. Art der Kontrollen
a) Die Kontrollen dienen der laufen den Bewertung der Einhaltung der GM P durch die Hersteller. Sie werden als allgemeine GMP-Kontrollen (auch als regelmäßige, periodische oder laufende Kontrollen) bezeichnet.
b) „Produkt- oder verfahrensorientierte“ Kontrollen (in bestimmten Fällen handelt es sich hierbei auch um Kontrollen vor dem Inverkeh rbringen) befassen sich gezielt m it der Herstellung eines oder einer Reihe von Produkten oder m it einem oder einer Reihe von Verf ahren und um fassen eine Bewertung der Validierung von und der Konform ität mit bestimm ten Verfahrens- oder Kontrollaspekten, die in der Genehm igung für das Inverkehrbringen dargelegt sind. Bei Bedarf wird die betreffende Produktinformation (die die Q ualität betreffenden Unterlagen eines Antrags/einer Genehm igung) de m Kontrolldienst auf Vertrauensbasis zur Verfügung gestellt.
5. Kontroll-/Bearbeitungsgebühren
Die Regelung für die Kontroll-/Bearbe itungsgebühren ist vom Standort des Herstellers abhängig. Für unter diesen Se ktoralen Anhang fallende Produkte werden den im Gebiet der andere n Vertragsp artei ansäs sigen Hers tellern ke ine Kontroll-/Bearbeitungsgebühren in Rechnung gestellt.
6. Schutzklausel für die Kontrollen
Die Vertragsparteien erkennen einvernehm lich an, dass sich jede Vertragspartei das Recht vorbehält, aus Gründen, die der ande ren Vertragspartei dargelegt werden, eigene Kontrollen durchzuführen. Diese Kont rollen sind der anderen Vertragspartei im Voraus zu notifizieren, und sie erhält di e Möglichkeit, daran teilzunehmen. Diese Schutzklausel sollte nu r in Ausnah mefällen in Anspruch genommen werden. Für solche Kontrollen kann eine Kostenerstattung gefordert werden.
DE 18 DE
7. Informationsaustausch zwischen den Behörden und Angleichung der Qualitätsanforderungen
Im Einklang m it den allgem einen Bes timmungen des Abkomm ens tauschen die Vertragsparteien alle für die laufende gegenseitige Aner kennung der Kontrollen erforderlichen sachdienlichen Informationen aus. Zum Zwecke des Nachweises der Befähigung im Fall von erheblichen Ä nderungen des Regelungssystem s einer Vertragspartei kann die andere Vertragspart ei zusätzlich gezielte Inform ationen zu einem a mtlichen Kontrolldienst anforder n. Gezielte Inform ationen können unter anderem Be reiche wie Ausbildung, Kontro llverfahren, allgem einer Austausch von Informationen und Unterlag en sowie Transparenz der Audits am tlicher Kontrolldienste durch Agenturen betr effen, die für die Durchführung dieses Sektoralen Anhangs relevant sind. Die ents prechenden Ersuchen sollten im Rahm en der Gem ischten Sektorgruppe als Teil eines Programms zur fortlaufenden Angleichung gestellt und bearbeitet werden.
Ferner unterrichten die betreffenden Behörden in Australien und in der Europäischen Union einander über alle neuen techni schen Leitlinien oder Änderungen der Kontrollverfahren. Vor der Annahme neuer Leitlinien oder Änderungen konsultieren die Vertragsparteien einander.
8. Amtliche Freigabe der Chargen
Das Verfahren zur am tlichen Freigabe der Char gen dient de r zusätzlichen Kontrolle der Sicherheit und der W irksamkeit von immunologischen Arzneim itteln (Impfstoffen) und Blutderivaten und wird von den zuständige n Behörden vor dem Vertrieb jeder Charge des Produkts durchge führt. Die gegenseitige Anerkennung der amtlichen Freigab e der Chargen is t nich t Geg enstand d ieses Abkomm ens. W ird jedoch ein am tliches Verf ahren zur Freigabe d er Chargen angewandt, so legt de r Hersteller auf Antrag der einführenden Ve rtragspartei die Bescheinigung über die amtliche Freigabe d er Charge v or, wenn die betreffende Charge von den Kontrollbehörden der ausführenden Vertragspartei geprüft wurde.
Für die Eu ropäische Union werden die am tlichen Charg enfreigabeverfahren für Humanarzneimittel von der Europ äischen Dir ektion f ür Arzneim ittelqualität und Gesundheitsfürsorge (E uropean Directorat e for the Quality of Medicines and Healthcare) veröffentlicht. Für Australien is t das amtliche Chargenfreigabeverfahren in dem Dokument „WHO Technical Report Series, No 822, 1992“ festgelegt.
9. Ausbildung der Kontrolleure
Gemäß den allgemeinen Bestimmungen des Abkommens sind die von den Behörden veranstalteten Ausbildungslehrgänge für die Kontrolleure auch für die Kontrolleure der ande ren Vertrag spartei zugänglich. Die Vertragspart eien unterrich ten einand er über die Durchführung dieser Lehrgänge.
10. Gemeinsame Kontrollen
Gemäß den allgem einen Bestimmungen des Abkomm ens und im gegenseitige n Einvernehmen der Vertragsparteien können gem einsame Kontrollen genehm igt werden. Diese Kontrollen dienen de r Entwicklung eines gem einsamen
DE 19 DE
Verständnisses und einer gem einsamen Auslegung der Verfahrensweisen und der Anforderungen. Die Organisation und die Form dieser Kontrollen werden nach den von der Gemischten Sektorgruppe genehmigten Verfahren vereinbart.
11. Warnsystem
Die Vertragsparteien vereinbaren die Ei nrichtung von Kontakts tellen, dam it die zuständigen Behörden und die Hersteller die Behörden der jeweils anderen Vertragspartei bei Qual itätsmängeln, beim Rückruf von Chargen, bei N achahmung und anderen Problem en im Zusa mmenhang m it der Qualität, die zusätzliche Kontrollen oder die Aussetzung des Vertriebs der be treffenden Charge erforderlich machen können, so schnell wie m öglich unterrichten können. Es wird einvernehmlich ein detailliertes Warnverfahren vereinbart.
Jede Vertragspartei stellt sicher, dass jede (gänzliche oder teilweise) Aussetzung oder Rücknahme einer Herstellungsg enehmigung wegen Nichtb eachtung der GMP, die den Schutz der öffentlichen Gesundheit beeinträchtigen könnt en, der anderen Vertragspartei mit der gebotenen Dringlichkeit mitgeteilt wird.
12. Kontaktstellen
Für die Zwecke dies es Sektoralen Anha ngs sind folgende Kontaktstellen für technische Fragen wie den Austausc h von Kontrollberichten, die Ausbildungs-lehrgänge für Kontrolleure, technische Anforderungen usw. vorgesehen:
Australien: Humanarzneimittel: The Head of Office Therapeutic Goods Administration Department of Health and Ageing PO Box 100 Woden ACT 2606 Australia Tel: 61-6-232-8622 Fax: 61-6-232-8426
Tierarzneimittel: The Manager, GMP Section Australian Pesticides and Veterinary Medicines Authority PO Box 6182 Kingston ACT 2604 Australia Tel: 61-6210-4803 Fax: 61-6210-4741
Europäische Union: The Director of the European Agency for the Evaluation of Medicinal Products 7 Westferry Circus Canary Wharf London E14 4HB United Kingdom
DE 20 DE
Tel: 44-171-418 8400 Fax: 44-171-418 8416
13. Gemischte Sektorgruppe
Im Rahm en dieses Sektoralen Anhangs wird eine Gem ischte Sektorgruppe aus Vertretern der Vertrag sparteien e ingesetzt. Diese ist für die ordnungsgem äße Durchführung dieses Sektoralen Anhangs verantwortlich. Sie erstattet dem Gemischten Ausschuss nach dessen Vorgaben Bericht.
Die Gemischte Sektorgruppe gibt sich eine Geschäftsordnung. Ih re Beschlüsse und Empfehlungen werden einvern ehmlich angenommen. Sie kann beschließen, ihre Aufgaben an Untergruppen zu delegieren.
14. Meinungsverschiedenheiten
Die Vertragsparteien bemühen sich nach besten Kräften, Meinungsverschiedenhei-ten, unter anderem über die Erfüllung der Anforderungen durch die H ersteller und über die Schlussfolgerungen der Kontro llberichte, auszuräum en. Ungelöst e Meinungsverschiedenheiten werden der Gemischten Sektorgruppe unterbreitet.
DE 21 DE
ABSCHNITT IV
ÄNDERUNGEN DER LISTE DER AMTLICHEN KONTROLLDIENSTE
Die Vertragsparteien erkennen einvernehm lich an, dass dieser Sektorale Anhang für Änderungen offen sein muss, insbesondere für die Aufnahme neuer amtlicher Kontrolldienste oder für Änderungen hinsichtlich bestehender zuständiger Behörden und ihrer Aufgaben. Sind bei amtlichen Kontrolldiensten wes entliche Änderungen eingetreten, so prüft die Gem ischte Sektorgruppe, ob zusätzliche - und gegebenenfa lls welche - Infor mationen erforderlich sind, um Programme zu überprüfen und um die gegenseitige Anerkennung von Kontrollen gem äß Abschnitt III Nummer 7 einzuführen oder aufrechtzuerhalten.
Nach Maßgabe der Bestimmungen des Abkommens werden die australischen Hersteller von Tierarzneimitteln im Nam en der Australian Pes ticides and Veterinary Medicines Authority von der T herapeutic Goods Adm inistration (TGA) auf der Grundl age des aktuellen australischen GMP-Kodex und des GMP-Leitfadens der EG für Tierarzneim ittel kontrolliert. Die Europäische Union erkennt die Schlu ssfolgerungen der von der TGA durchgeführten Kontrollen und die Zertifizier ungen der Konform ität der C hargen durch die australischen Hersteller an. Sollte die Australian Pesticides and Veterinary Medicines Authority (APVMA) beginnen, selbst Kontrollen durchzuführen, so werden die Kontrollberichte von Amts wegen auch an die einführende Vertragspartei übermittelt, bis ein e zuf riedenstellende Überprüfung des GMP-Kontrollsystems der APVMA stattgefunden hat.
______________
DE 22 DE
Anlage 1
LISTE DER GELTENDEN RECHTS- UND VERWALTUNGSVORSCHRIFTEN
Europäische Union:
Richtlinie 65/65/EW G des Rates vom 26. Januar 1965 zur Angleichung der Rechts- und Verwaltungsvorschriften über Arzneispezialitä ten in der verlängert en, erweiterten und geänderten Fassung
Zweite Richtlinie 75/319/EWG des Rates vom 20. Mai 1975 zur Angleichung der Rechts- und Verwaltungsvorschriften über Arzneispezialitä ten in der verlängert en, erweiterten und geänderten Fassung
Richtlinie 81/851/EWG des Rates vom 28. September 1981 zur Angleichung der Rechtsvorschriften de r Mitg liedstaaten übe r Tiera rzneimittel in der erwe iterten und geänderten Fassung
Richtlinie 91/356/EWG der Komm ission vom 13. Juni 1991 zur Festle gung der Grundsätze und Leitlinien der guten Herstellungspraxis für zur Anwendung beim Menschen bestimm te Arzneimittel
Richtlinie 91/412/EW G der Komm ission vom 23. Juli 1991 zur Festlegung der Grundsätze und Leitlinien der guten Herstellungspraxis für Tierarzneimittel
Verordnung (EWG) Nr. 2309/93 des Rate s vom 22. Juli 1993 zur Festlegung von Gemeinschaftsverfahren für die Gene hmigung und Überwachung von Hum an- und Tierarzneimitteln und zur Schaffung einer Eu ropäischen Agentur für die Beurteilung von Arzneimitteln
Richtlinie 92/25/EW G des Rates vom 31. März 1992 über de n Großhandelsvertrieb von Humanarzneimitteln & Leitfaden für die gute Vertriebspraxis
Aktuelle Fassung des L eitfadens für die gute Hers tellungspraxis, Regeln für Arzneim ittel in der Europäischen Gemeinschaft, Anhang IV
Australien:
Humanarzneimittel:
Therapeutic Goods Act 1989 und Durchführungsbe stimmungen (Regulations, Orders and Determinations), insbesondere zur Festle gung der Nor men für die Kennzeichnung, der Herstellungsgrundsätze und des Australischen Kodex für die gute Herstellungspraxis
DE 23 DE
Tierarzneimittel:
Rechtsvorschriften - Commonwealth:
· Agricultural and Veterinary Chemicals (Administration) Act, 1992 · Agricultural and Veterinary Chemicals Act, 1994 · Agricultural and Veterinary Chemicals Code Act, 1994 · Agricultural and Veterinary Chem icals (Adm inistration) Regulation s, 1995 · Agricultural and Veterinary Chem icals Instrum ent No 1 (Manufacturing Principles), 2007 · Agricultural and Veterinary Chemicals Code Regulations, 1995
Rechtsvorschriften - Neusüdwales:
· Stock Foods Act, 1940 · Stock Medicines Act, 1989 · Public Health Act, 1991 · Poisons and Therapeutic Goods Act, 1966 · Pesticides Act, 1979 · Agricultural and Veterinary Chemicals (NSW) Act, 1994
einschließlich alle r im Rahm en dieser Rechts vorschriften erlas senen Durchführungsbestimmungen (regulations, orders or instruments).
Rechtsvorschriften - Victoria:
· Animal Preparations Act, 1987 · Health Act, 1958 · Drugs, Poisons and Controlled Substances Act, 1981 · Agricultural and Veterinary Chemicals (Victoria) Act, 1994
einschließlich alle r im Rahm en dieser Rechts vorschriften erlas senen Durchführungsbestimmungen (regulations, orders or instruments).
einschließlich alle r im Rahm en dieser Rechts vorschriften erlas senen Durchführungsbestimmungen (regulations, orders or instruments).“
DE 25 DE
22. Der Sektorale Anhang über Medizinprodukte wird gestrichen und durch folgenden Wortlaut ersetzt:
„SEKTORALER ANHANG ÜBER MEDIZINPRODUKTE ZUM ABKOMMEN ZWISCHEN DER EUROPÄISCHEN GEMEINSCHAFT UND
AUSTRALIENÜBER DIE GEGENSEITIGE ANERKENNUNG DER KONFORMITÄTSBEWERTUNG, DER BESCHEINIGUNGEN UND DER
KENNZEICHNUNGEN
ANWENDUNGS- UND GELTUNGSBEREICH
Die Vertragsparteien legen ei nvernehmlich fest, dass dieser Sektorale Anhang für folgende Produkte gilt:
Produkte für die Ausfuhr in die Europäische Union
1) Alle Medizinprodukte, die
a) in Australien hergestellt werden und
b) den Konformitätsbewertungsverfahren Dritter unterliegen, sowohl für das Produkt als auch für die Qualitätssicherung, und
c) in der in der Richtlinie 90/385/E WG des Rates vom 20. Juni 1990 zur Angleichung der Rechtsvorschrifte n der Mitgliedstaaten über aktive implantierbare Medizinprodukte in der zuletzt geänderten F assung aufgeführt sind und
d) in der Richtlinie 93/42/EWG des Rates vom 14. Juni 1993 über Medizinprodukte in der zuletzt geänderten Fassung aufgeführt sind.
2) Für die Zwecke von Absatz 1 gilt:
a) In Anlage 1 aufgeführte Medizinprodukte sind ausgenommen, und
b) soweit nicht anders bestimm t oder von den Vertragsparteien einvernehm lich vereinbart, umfasst die „Herstellung“ eines Medizinprodukts Folgendes nicht:
i) Wiederherstellungs- oder Erne uerungsverfahren wie Reparatur, Instandsetzung, Überholung oder Neugestaltung oder
ii) Verfahren wie Pressung, Kenn zeichnung, Etikettierung, Verpackung und Vorbereitung für den Verkauf, die ein zeln oder in Kom bination miteinander durchgeführt werden, oder
iii) nur Prüfungen zur Qualitätskontrolle oder
iv) nur Sterilisation.
Produkte für die Ausfuhr nach Australien
(1) Alle Medizinprodukte, die
DE 26 DE
a) in der Europäischen Union hergestellt werden und
b) Konfor mitätsbewertungsverfahren nach dem Australian T herapeutic Goods Act 1989 und den Therapeutic Goods Regul ations in der zu letzt geänderten Fassung unterliegen, sowohl fü r das Produkt als auch für die Qualitätssicherung.
(2) Für die Zwecke von Absatz 1 gilt:
a) In Anlage 1 aufgeführte Medizinprodukte sind ausgenommen, und b)
b) soweit nicht anders bestimm t oder von den Vertragsparteien einvernehm lich vereinbart, umfasst die „Herstellung“ eines Medizinprodukts Folgendes nicht:
i) Wiederherstellungs- oder Erne uerungsverfahren wie Reparatur, Instandsetzung, Überholung oder Neugestaltung oder
ii) Verfahren wie Pressung, Kenn zeichnung, Etikettierung, Verpackung und Vorbereitung für den Verkauf, die ein zeln oder in Kom bination miteinander durchgeführt werden, oder
iii) nur Prüfungen zur Qualitätskontrolle oder
iv) nur Sterilisation.
ABSCHNITT I
RECHTS- UND VERWALTUNGSVORSCHRIFTEN
Rechts- und Verwaltungsvorschriften der Europäischen Union, aufgrund deren die von Australien benannten Konformitätsbewertungs-
Rechts- und Verwaltungsvorschriften Australiens, aufgrund deren die von de r Europäischen Union benannten Konfor mitätsbewertungsstellen die
DE 27 DE
stellen die Übereinstimmung bewerten Übereinstimmung bewerten
– Richtlinie 90/385/EWG des Rates vom 20. Juni 1990 zur Angleichung der Rechtsvorschriften der Mitg liedstaaten über aktive im plantierbare Medizinprodukte, in der geänderten und ergänzten Fassung
– Richtlinie 93/42/EWG des Rates vom 14. Juni 1993 über Medizinprodukte, in der geänderten und ergänzten Fassung
– und alle auf der Grundlag e dieser Richtlinien erlassenen Rechtsvorschriften
– Therapeutic Goods Act 1989, in der geänderten Fassung
– Therapeutic Goods Regulations 1990, in der geänderten Fassung
– Therapeutic Goods (Medical Devices) Regulations 2002, in der geänderten Fassung
– und alle nachgeordneten Rechtsvorschriften, auf die in den vorgenannten Rechtsvorschriften verwiesen wird, in der geänderten Fassung8
8 Allgemeiner Verweis auf nac hgeordnete Rechtsvorschriften Australiens, auf die im Therapeutic Goods
Act und in den Therapeutic Goods Regulations verwiesen wird, und auf mögliche künftige Änderungen der Rechtsvorschriften.
DE 28 DE
ABSCHNITT II
BENANNTE KONFORMITÄTSBEWERTUNGSSTELLEN
Die Listen der benannten Konform itätsbewer-tungsstellen wurden von den Vertragsparteien einvernehmlich erstellt und werden von ihnen auf dem neuesten Stand gehalten.
Die Listen der benannten Konform itätsbewer-tungsstellen wurden von den Vertragsparteien einvernehmlich erstellt und werden von ihnen auf dem neuesten Stand gehalten.
Von Aus tralien benannte Konfor mitäts-bewertungsstellen für die Bewertung der Produkte aufgrund der Rechts- und Verwaltungs-vorschriften der Europäischen Union
Von der Europäischen Union benannte Konformitätsbewertungsstellen für die Bewertung der Produkte aufgrund der Rechts- und Verwaltungsvorschriften Australiens
DE 29 DE
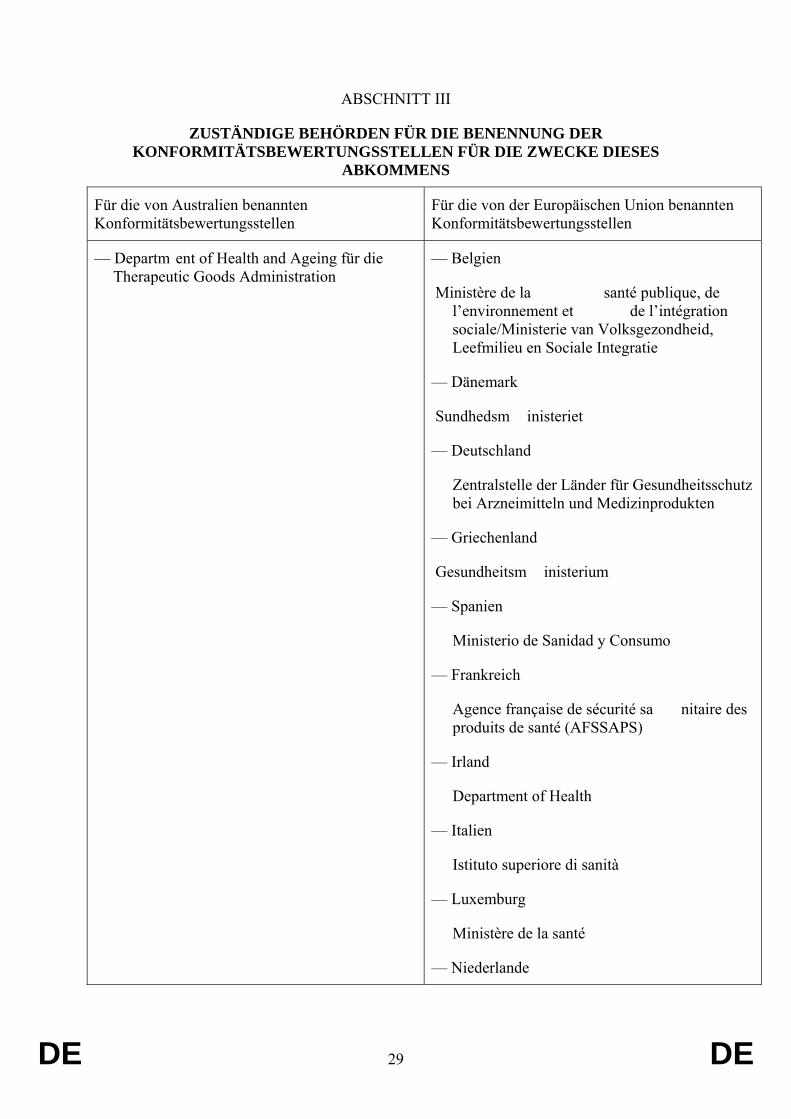
ABSCHNITT III
ZUSTÄNDIGE BEHÖRDEN FÜR DIE BENENNUNG DER KONFORMITÄTSBEWERTUNGSSTELLEN FÜR DIE ZWECKE DIESES
ABKOMMENS
Für die von Australien benannten Konformitätsbewertungsstellen
Für die von der Europäischen Union benannten Konformitätsbewertungsstellen
— Departm ent of Health and Ageing für die Therapeutic Goods Administration
— Belgien
Ministère de la santé publique, de l’environnement et de l’intégration sociale/Ministerie van Volksgezondheid, Leefmilieu en Sociale Integratie
— Dänemark
Sundhedsm inisteriet
— Deutschland
Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten
— Griechenland
Gesundheitsm inisterium
— Spanien
Ministerio de Sanidad y Consumo
— Frankreich
Agence française de sécurité sa nitaire des produits de santé (AFSSAPS)
— Irland
Department of Health
— Italien
Istituto superiore di sanità
— Luxemburg
Ministère de la santé
— Niederlande
DE 30 DE
Staat der Nederlanden
— Österreich
Bundesministerium für Arbeit, Gesundheit und Soziales
— Portugal
Ministério da saúde
— Finnland
Sosiaali- ja te rveysministeriö/Social- oc h hälsovårdsministeriet
— Schweden
Unter der Zuständigkeit der schwedischen Regierung: Styrelsen för ackreditering och teknisk kontroll (SWEDAC)
— Vereinigtes Königreich
Medicines and Healthcare Products Regulatory Agency (MHRA)
— Tschechische Republik
Tschechisches Amt für Normung, Messtechnik und Prüfwesen (Office for Standards, Metrology and Testing)
DE 31 DE
ABSCHNITT IV
VERFAHREN FÜR DIE BENENNUNG DER KONFORMITÄTSBEWERTUNGSSTELLEN
Von Australien einzuhaltende Verfahren bei der Benennung der Konfor mitätsbewertungsstellen für die Bewertung der Produkte aufgrund der Anforderungen der Europäischen Union
Von der Europäischen Union einzuhaltende Verfahren bei der Bene nnung der Konfor mitäts-bewertungsstellen für die Bewertung der Produkte aufgrund der Anforderungen Australiens
Die Therapeutic Go ods Adm inistration des Department of Health and Ageing entspricht den Anforderungen der in Abschnitt I aufgeführten Richtlinien unter Berücksichtigung des Beschlusses 93/465/EWG des Rates vom 22. Juli 1993 über die in den technischen Harmonisierungsrichtlinien zu verwendenden Module für die verschiedenen Phasen der Konformitätsbewertungsverfahren und die Regeln für die Anbringung und Verwendung der C E-Konformitätskennzeichnung und wird für bestimmte Kategorien oder Klassen von Medizinprodukten und Konfor mitätsbewertungs-verfahren benannt. Für unter Abschnitt V fallende Produkte erfolgt die Benennung auf der Grundlage eines P rogramms zur Vertrauens-bildung im Sinne von Abschnitt V Nummer 1.29.
Die Konfor mitätsbewertungsstellen entsprechen den Anforderungen der in Abschnitt I aufgeführten Richtlinien unter Berücksichtigung des Beschlusses 93/465/EWG des Rates vom 22. Juli 1993 über die in den technischen Harmonisierungsrichtlinien zu verwendenden Module für die verschiedenen Phasen der Konformitätsbewertungsverfahren und die Regeln für die Anbringung und Verwendung der C E-Konformitätskennzeichnung und werden für bestimmte Kategorien oder Klassen von Medizinprodukten und Konfor mitätsbewertungs-verfahren benannt. Für unter Abschnitt V fallende Produkte erfolgt die Benennung auf der Grundlage eines P rogramms zur Vertrauens-bildung im Sinne von Abschnitt V Nummer 1.210.
ABSCHNITT V
ZUSÄTZLICHE BESTIMMUNGEN
1. Vertrauensbildung bei mit hohem Risiko behafteten Medizinprodukten
1.1. Zwecks Stärkung des Ve rtrauens in die Be nennungssysteme der Vertragsparteien is t für folgende Medizinprodukte ein Vertrauensbildungsprozess vorgesehen:
– aktive im plantierbare Geräte na ch der Def inition in d en in Abs chnitt I aufgeführten Rechtsvorschriften,
– Medizinprodukte, die nach den in Abschnitt I aufgeführten Rechtsvors chriften der Klasse III zugeordnet werden,
9 Nach er folgreichem Absc hluss de r vertrauensbildenden M aßnahmen i m Falle vo n M edizinprodukten
des Abschnitts V wird davon ausgegangen, dass die Kompetenz gewährleistet ist. 10 Nach er folgreichem Absc hluss de r vertrauensbildenden M aßnahmen i m Falle vo n M edizinprodukten
des Abschnitts V wird davon ausgegangen, dass die Kompetenz gewährleistet ist.
DE 32 DE
– ein Medizinprodukt, bei de m es sich um eine implantierbare Intraokularlinse handelt,
– ein Medizinprodukt, bei dem es sich um eine intraokulare viskoelastische Flüssigkeit handelt,
– ein Medizinprodukt, das zur m echanischen Empfängnisverhütung (Barriere ) oder zur Verhinderung der sexuellen Übertragung von Krankheiten bestimm t ist.
1.2. Die Vertragsparteien erstellen unt er Beteiligung der Therapeutic Goods Administration und der zuständigen Be hörden der Europäischen Union ein ausführliches Programm zu diesem Zweck.
1.3 Der Zeitraum für die vertrauensbildende n Maßnahm en wird zwei Jahre nach dem Datum, zu dem der Sektorale Anhang in der geänderten Fassung wirksam wird, überprüft.
1.4. Zusätzliche besondere Anforderungen für Fortschritte im Regelungsbereich:
1.4.1. Nach Artikel 2, Artikel 7 Absatz 1, Artik el 8 Absatz 1 und Artikel 9 Absatz 1 des Abkommens können die Vertragsparteien zu sätzliche besondere Anforderungen für die Konformitätsbewertungsstellen zum Nachweis ihrer Fachkenntnis im Bereich der sich weiterentwickelnden Regelungssysteme festlegen.
1.4.2. Diese besonderen Anforderungen können Ausbildungsm aßnahmen, „Observed Audits“ der Konfor mitätsbewertungsstellen, Besuche und den Austausch von Informationen und Unterlagen, einschließlich Auditberichte, umfassen.
1.4.3. Diese Anforderungen können gleicherm aßen für die Benennung einer Konfor mitäts-bewertungsstelle im Einklang mit diesem Abkommen gelten.
2. Registrierungs-, Auflistungs- und Aufnahmeverfahren für das Australian Register of Therapeutic Goods (ARTG)
2.1. Die Vertragsparteien erkennen an, dass die im Rahm en des Therapeutic Goods Act 1989 vorgesehenen australischen Verf ahren zur Registrierung, Auflistung oder Aufnahme der Produkte zum Zweck e der Marktüberwachung und die entsprechenden Verfahren der E uropäischen Union von diesem Abkomm en unberührt bleiben.
2.2. Im Rahmen dieses Abkommens trägt die australische Regelungsbehörde ein Produkt der Europäischen Union unverzüglich in das ARTG ein, ohne dass eine weitere Bewertung des Produkts vorgenomm en wird. Vo raussetzung hierfür sind der Erhalt eines Antrags für das betreffende Produkt sowie der vorgeschr iebenen Gebühr und die Vorlage der Bescheinigung der Konfor mitätsbewertungsstelle über die Erfüllung der Anforderungen Australiens.
2.3. Die Gebühren, die die Vertragsparteien fü r die Registrierung erheben, dürfen nur die Kosten berücksichtigen, die ihnen für die Registrierung der Medizinprodukte, die Durchsetzung und die Überwachung nach de m Inverkehrbringen in diesem Sektor entstehen.
DE 33 DE
3. Informationsaustausch
Die Vertragsparteien kommen überein, einander über Folgendes zu unterrichten:
– Rücknahme, Aussetzung, Eins chränkung oder Aufhebung von Bescheinigungen,
– unerwünschte Vorkommnisse im Rahm en des Überwachungsverfahrens der GHTF für Medizinprodukte,
– Fragen im Zusammenhang mit der Produktsicherheit und
– alle Rechtsvorschriften oder Änderunge n bestehender Rechtsvorschriften, die auf der Grundlage der in Abschnitt I aufgeführten Rechtstexte erlassen wurden.
Die Vertragsparteien richten für jeden dieser Zwecke Kontaktstellen ein.
Die Vertragsparteien berücksichtigen die A uswirkungen der Einrichtung von Eudamed.
Darüber hinaus inform iert die Therap eutic Goods Adm inistration über alle ausgestellten Bescheinigungen.
4. Neue Rechtsvorschriften
Die Vertragsparteien nehm en ge meinsam z ur Kenntnis, dass Australien neue Rechtsvorschriften über In-Vit ro-Diagnostika (IVD) zu erla ssen plant und dass jede neue Regelung m it den Grundsätzen de s A bkommens über die gegenseitige Anerkennung (MRA) in Einklang stehen muss.
Die Vertragsparteien erklären einve rnehmlich, dass sie beabsichtigen, den Anwendungsbereich des MRA auf In-Vitro -Diagnostika auszudehnen, sobald die australischen Rechtsvorschriften über IVD erlassen sind.
5. Maßnahmen zum Schutz der öffentlichen Gesundheit und Sicherheit
Die Umsetzung dieses Sektoralen Anhangs hi ndert die Vertragsparteien nicht daran, die zum Schutz de r öf fentlichen Gesund heit und Sicherhe it erf orderlichen Maßnahmen entspre chend den in Abschnitt I auf geführten Rechtsvors chriften zu ergreifen. Jede Vertragspartei unterrichte t die andere Vertragspartei ordnungsgem äß über derartige Maßnahmen.
6. Gemischte Sektorgruppe
Im Rahm en dieses Sektoralen Anhangs wird eine Gem ischte Sektorgruppe aus Vertretern der Vertrag sparteien e ingesetzt. Diese ist für die ordnungsgem äße Durchführung dieses Sektoralen Anhangs verantwortlich. Sie erstattet dem Gemischten Ausschuss nach dessen Vorgaben Bericht.
Die Gemischte Sektorgruppe gibt sich eine Geschäftsordnung. Ih re Beschlüsse und Empfehlungen werden einvern ehmlich angenommen. Sie kann beschließen, ihre Aufgaben an Untergruppen zu delegieren.
DE 34 DE
7. Meinungsverschiedenheiten
Beide Vertragsparteien bem ühen si ch n ach besten Kräften, Meinungsverschieden-heiten auszuräumen. Ungelöste Meinungsvers chiedenheiten werden der Ge mischten Sektorgruppe unterbreitet.
DE 35 DE
ANLAGE 1
Nach Nummer 2 Buchstabe a dieses Sektoralen Anhangs gelten seine Bestimmungen nicht für folgende Medizinprodukte:
• Medizinprodukte, die abgetöte te Zellen, Gewebe oder Ge webederivate tierischen Ursprungs enthalten oder aus solchen herges tellt werden und bei denen durch anerkannte Verfahren zur Ausm erzung oder Inaktiv ierung von Viren im Verlauf des Herstellungsprozesses für den Schutz vor Vire n oder anderen übertragbaren Erregern zu sorgen ist;
• Medizinprodukte, die Gewebe , Zellen oder Stoffe m ikrobiellen, bakteriellen oder rekombinanten Ursprungs enthalten und di e zur Verwendung i m oder am m enschlichen Körper bestimmt sind;
• Medizinprodukte, die Gewebe oder Gewebederivate menschlichen Ursprungs enthalten;
• Medizinprodukte, die stabile Derivate aus m enschlichem Blut oder Blutplasm a enthalten, die in Ergänzung zu dem Produkt eine W irkung auf den m enschlichen Körper entfalten können;
• Medizinprodukte, die al s Bestandteil einen Stoff entha lten oder enthalten sollen, der - gesondert verwendet - als ein Arzneimittel betrachtet werden könnte, das in Ergänzu ng zu dem Produkt eine Wirkung auf den Patienten entfalten soll;
• ein Medizinprodukt, das vom Hersteller sp eziell für den Einsatz zur chem ischen Desinfektion eines anderes Medizinprodukts bestimmt ist, ausgenommen Sterilisatoren, die mit trockener Hitze, feuchter Hitze oder Ethylenoxid arbeiten.
Beide Vertragsparteien können im gegenseitigen Einvernehmen beschließen, die Anwendung dieses Sektoralen Anhangs auf die vorgenannten Medizinprodukte auszudehnen.“
DE 36 DE
Artikel 2
Inkrafttreten
Dieses Abkommen tritt am ersten Tag des zweiten Monats in Kraft, der auf den Tag folgt, an dem die Vertragsparteien diplom atische No ten zur Bestätigung des Abschlusses ihrer jeweiligen f ür das Inkrafttreten des Abkomme ns erforderlichen Verfahren ausgetausch t haben.
Dieses Abkommen ist in zwei Urschriften in b ulgarischer, dänischer, deutscher, englischer, estnischer, finnischer, französischer, griechischer, irischer, italienischer, lettischer, litauischer, maltesischer, niederlän discher, po lnischer, portugiesischer, rum änischer, schwedischer, slowakischer, slowenischer, spanischer, ts chechischer und ungarische r Sprache abgefasst, wobei jeder Wortlaut gleichermaßen verbindlich ist.