Der Mensch besitzt etwa 4-6 Liter Blut. Die Menge an Blut, bezogen auf die fettfreie Körpermasse, entspricht etwa 6-8% bei Erwachsenen und 8-9% bei Kindern. Blut setzt sich aus Blutplasma und aus zellulären Bestandteilen zusammen. Blutplasma besteht hauptsächlich aus Wasser in dem Elektrolyte, Nährstoffe, Vitamine, Proteine und Gase gelöst sind. Blutzellen (Leukozyten) und zelluläre Bestandteile (Erythrozyten, Thrombozyten) machen die festen Bestandteile des Blutes aus. Ein normales Blutvolumen ist wichtig, da es den Druck in den zentralen Venen und dadurch das Füll- und Auswurfvolumen des Herzens bestimmt (s. Praktika: „Herz“ und „Kreislauf“). Das Blutvolumen kann mithilfe des Prinzips der Indikator-Verdünnung gemessen werden. Hierbei gilt: Verteilungsvolumen = injizierte Menge/ Konzentration im Blut, wobei man entweder mittels radioaktivem Chrom die Erythrozyten, oder mittels radioaktivem Jod das Albumin markiert. Die vielfältigen Funktionen des Bluts können in drei Gebiete eingeteilt werden:
• Transport- und Kommunikationssystem: Austausch von Stoffwechsel-Edukten und -Produkten, Sauerstoff, Nährstoffen, Hormonen, Wärme etc. Die Zusammensetzung des Bluts bleibt trotz des Austauschs relativ konstant, sodass man bei Abweichungen auf pathologische Veränderungen schließen kann (s. Kreatinin-Werte, Praktikum „Niere“).
• Abwehr: von Viren, Bakterien, Pilzen, veränderten Körperzellen. ( III. Immunsystem) • Blutgerinnung: Schützt den Menschen vor zu großem Blutverlust ( II. Gerinnung) 1. Das Blutplasma
Das Blutplasma besteht zu 90% aus Wasser und 10% festen Bestandteilen. Blutserum erhält man, wenn man Blut gerinnen lässt und anschließend zentrifugiert. Der flüssige Überstand, das Serum, ist durch das Fehlen gerinnungsaktiver Substanzen gekennzeichnet ( II. Gerinnung). Am vielfältigsten sind die Plasmaproteine, von denen bisher über hundert verschiedene Spezies nachgewiesen wurden. Mittels Trägerelektrophorese werden fünf große Gruppen unterschieden, die sich durch ihre Zusammensetzung und quantitativen Verteilung in einer typischen Kurve darstellen (s. Biochemie-Lehrbücher): Albumin dient als Vehikel für wasserunlösliche Bestandteile (z.B. Fettsäuren) und der Aufrechterhaltung des kolloidosmotischen (= onkotischen) Drucks (~ 80% des Drucks). Hieraus resultiert ein intravasaler KOD, der dem KOD des Interstitiums und dem
hydrostatischen Druck der Gefäße entgegenwirkt und so die Flüssigkeit des Kreislaufsystems zurückhält. Die restlichen vier Elektrophorese-Banden der Plasmaproteine sind heterogene Gruppen die durch verschiedene Proteine gebildet werden. Hierunter fasst man u.a. spezifische Transport-proteine zusammen (z.B. Apo-Transferrin, Trans-cobalamin, Transcortin), die sowohl ein Transport-, als auch ein Vorratssystem darstellen. Physiologisch und medizinisch relevant ist auch die Gruppe der Lipoproteine, die für den Transport von Lipiden nötigt sind. Hierbei unterscheidet man HDL, IDL, LDL, VLDL und Chylomikronen (s. Lehrbücher Biochemie). Zu den Plasmaproteinen gehören auch Proteine der Blutgerinnung und Immunabwehr ( II. Gerinnung, III. Immunsystem).
BLUT 4
Abb. 2: Übersicht der Plasmaproteine
Neben den Proteinen sind als weitere feste Bestandteile des Plasmas die Elektrolyte zu nennen, die zusammen eine osmolale Konzentration von 290 mosm/kg Wasser haben und im Blut als extrazellulärem Raum in charakteristischer Weise verteilt sind (siehe Tabelle )
Da die Gefäßwand für die Kationen und Anionen permeabel ist, kommt es zwischen Interstitium und dem intra-vasalem Raum zu einem Ausgleich der Elektrolyt-Konzentrationen, sodass an dieser Stelle auch kein Elektrolyt-bedingter osmotischer Druck wirksam wird. Über der Kapillarmembran herrscht also nur die mittlere kolloidosmotische Druckdifferenz von 20 mmHg (25 mmHg – 5 mmHg), die durch Plasmaproteine gegeben ist. Zwischen dem extra- und intrazellulären Milieu entsteht allerdings durch die Elektrolyte ein osmotischer Druck, da die Zellmembranen für die Kationen und Anionen weitgehend impermeambel sind. Daher müssen Infusionen isoton sein, d.h. der osmotische Druck der Infusionslösung muss dem des Plasmas entsprechen, da es sonst zu Flüssigkeitsverschiebungen über der Zellmembran kommt: Ist die Infusionslösung hyperton (osmotischer Druck größer als im Plasma), tritt
Wasser aus den Zellen aus (Zellschrumpfung). Ist die Infusionslösung dagegen hypoton (osmotischer Druck kleiner als im Plasma), strömt Wasser in die Zellen ein. CAVE: eine Lösung mit gleicher Osmolalität wie das Blutplasma ist nur dann isoton, wenn die Zellmembran für das betrachtete Teilchen impermeabel ist (trifft z.B. für Na+ zu, nicht aber für eine isoosmolale Harnstofflösung). Eine 0,9%ige NaCl-Lösung ist isoton und wird als physiologische Kochsalzlösung bezeichnet. Nach großen Blutverlusten durch z.B. Unfälle, werden isotone Plasma-Ersatzflüssigkeiten verwendet um den Druck in den Gefäßen konstant zu halten. Solche Flüssigkeiten können zusätzlich mit Kolloiden versetzt werden (sog. Plasmaexpander).
Praktikumsversuch : „Osmotische Resistenz“: Entsprechend handelt es sich bei dem Versuch zum einen um eine isotone und zum anderen um eine hypertone Flüssigkeit!
BLUT 5 2. Die zellulären Bestandteile
Man unterscheidet bei den zellulären Blutbestandteilen Erythrozyten, Leukozyten und Thrombozyten. Alle zellulären Bestandteile gehen aus den Stammzellen des hämatopoetischen Gewebes hervor (Abb. 3). Diese Stammzellen haben für die Blutbildung zwei besondere Eigenschaften:
• Pluripotenz: aus einer Stammzelle können verschiedene Zellen entstehen. • Selbsterneuerung: sie können sich unbegrenzt mitotisch teilen.
Aus den Stammzellen entwickeln sich unter dem Einfluss von Wachstumsfaktoren über verschiedene Zwischenstufen die Zellen des Bluts. Solche Wachstumsfaktoren sind z.B.:
Interleukin 3 Erythropoietin: fördert die Erythrozytenbildung (s. Praktikum „Niere“) Thrombopoietin: fördert die Thrombozytenbildung Colony stimulating factors: fördern die B- und T-Lymphozytenbildung
Die Entwicklung bis zur reifen Blutzelle wird „terminale Differenzierung“ genannt, da sie nicht umkehrbar ist und nur in Richtung Reifung ablaufen kann.
Erythrozyten sind kernlose, bikonkave Zellen mit einem Durchmesser von ~7,5 μm und einer Dicke von ~1,5 μm (Abb. 5: Price-Jones-Kurve, klinische Bezüge). Erythrozyten entstehen aus Retikulozyten und besitzen keine Organellen, sodass sie zur Energie-bereitstellung nur anaerobe Glykolyse betreiben können. Die Reifung der Erythrozyten dauert ~7 Tage, die Lebensdauer beträgt ~120 Tage. Danach werden sie vom mononukleären Phagozytensystem (MPS) der Milz und Leber aufgenommen und abgebaut. Entsprechend müssen ständig neue Erythrozyten gebildet werden, d.h. eine hohe Syntheserate an DNA und Hämoglobin ist notwendig. Liegt ein Mangel an einem der für die Synthese benötigten Stoffe (Eisen, Cobalamin, Folsäure) vor, kommt es zu charakteristischen Veränderungen der Erythrozyten, die unter dem Begriff „Anämie“ zusammengefasst sind (s. klinische Bezüge). Für die Form und Verformbarkeit der Erythrozyten ist das Membran- und Zytoskelett verantwortlich. Die hierfür essenziellen Proteine sind Spectrindimer, Bande-4.1-Protein, Aktin und das Ankyrin (Abb. 4). Weitere Bestandteile der Erythrozytenmembran sind Glykophorin, GLUT1-Transporter, Aquaporine und das Bande-3-Protein (Cl-/HCO3
--Aus-
Abb. 3: Blutbildung Abb. 4: Erythrozytenaufbau
BLUT 6 tauscher). Zudem befinden auf der Membranoberfläche Glykolipide, die antigen wirken falls sie in einen nicht-kompatiblen Organismus gelangen ( IV. Blutgruppen). Für den Transport von Sauerstoff in den Erythrozyten ist das Hämoglobin (Hb) verantwortlich, das als sog. Chromoproteid aus einem Proteinanteil und einem Nicht-Proteinanteil (Häm) besteht. Das Häm trägt zentral ein Eisen-Atom an dem der Sauerstoff gebunden ist. Da jedes Hämoglobin-Molekül vier Häm-Gruppen besitzt, können pro Mol Hämoglobin vier Mol Sauerstoff transportiert werden. Erythrozyten können auch CO2 transportieren, doch wird dies nicht am zentralen Eisen-Atom gebunden, sondern an einer der Aminogruppen als sog. Carbamino-Hämoglobin. Retikulozyten sind die Vorläuferzellen der Erythrozyten im Blut, die zwar kernlos sind, aber noch Reste von Organellen besitzen, d.h. noch in geringem Maß Hämoglobin bilden können. CAVE: nicht mit Retikulumzellen verwechseln !! Leukozyten sind die zellulären Bestandteile der spezifischen und unspezifischen Abwehr. Sie stellen eine sehr differenzierte Gruppe von Blutzellen dar ( III. Immunsystem). Thrombozyten auch Blutplättchen genannt, sind kernlose Zellbruchstücke, die durch Abschnürung vom Megakaryozyten entstehen. Funktion ( II. Blutstillung, Gerinnung). Anämie („Blutarmut“) bedeutet, dass bei normalem Blutvolumen die Hämoglobin-Konzentration, die Erythrozytenzahl und/oder der Hämatokrit unterhalb des Referenzbereichs liegen. Mittels der Erythrozytenindices MCH und MCV können Anämien klassifiziert
werden. Bei erhöhtem bzw. erniedrigtem MCH liegt eine hyperchrome oder hypochrome Anämie vor. Bei verändertem MCV, liegt eine makrozytäre oder eine mikrozytäre Anämie vor. Klinische Bezüge: Meist liegen Anämieformen in Kombinationen vor (s. Farben). Man spricht dann
Die perniziöse Anämie (Morbus Biermer) ist eine makrozytäre, hyperchrome Anämie die durch einen Cobalamin-Mangel bedingt ist. Charakteristisch sind die erniedrigte Erythrozytenzahl und die Makrozytose (MCV 100 fl), durch die der Hämoglobingehalt des einzelnen Erythrozyten (MCH) auf bis zu 50 pg ansteigen kann. Eine solche Situation betrifft alle sich teilenden Zellen, doch macht sie sich besonders in den schnell teilenden Knochenmarkzellen bemerkbar. Ursache kann eine jahrelange Mangelernährung an Cobalamin bzw. eine Resorptionsstörung aufgrund eines Mangels an Intrinsic Factor sein, der für die Resorption des Extrinsic factor (= Cobalamin = Vitamin B12) essenziell ist. Bei einer mikrozytären, hypochromen Anämie wie der Eiselmangelanämie ist das mittlere Hämoglobin pro Erythrozyt vermindert (MCH 15-27 pg) und es liegen Mikrozyten vor (MCV erniedrigt). Der Eisenmangel kann z.B. durch starke Menstruationsblutungen, Blutungen im Bereich des Gastrointestinaltraktes bedingt sein. Wegen der begrenzten Eisenresorption im Darm führt bereits ein Verlust von wenigen Millilitern Blut pro Tag zu einer Eisenmangelanämie.
BLUT 7
in µm Abb. 5: Price-Jones Kurve
Bei der sphärozytären Anämie kommt es durch den Defekt von Ankyrin (s.o.) zu einer angeborenen kugelförmigen Auftreibung der Erythrozyten. Diese Sphärozyten werden aufgrund ihrer reduzierten Membranflexibilität vorzeitig in der Milz aussortiert und haben somit eine verminderte Lebensdauer ( 10 Tage). Wenn der vermehrte Abbau nicht kompensiert werden kann kommt es zur Ausprägung einer Anämie. Therapeutisch kann durch eine Splenektomie (Milzentfernung) die Lebensdauer der Sphärozyten auf ~80 Tage verlängert werden. Anämien können auch über die Retikulozytenzahl erkannt werden. Entsprechend kommt es bei Proliferationsstörungen (gestörte Hämoglobin- oder DNA-Synthese) zu einer verminderten Retikulozytenzahl. Dagegen ist die Anzahl erhöht, wenn z.B. nach größeren Blutverlusten, Höhenaufenthalten oder hämolytischen Anämien die Erythropoese gesteigert wird.
Die Price-Jones-Kurve beschreibt die Verteilung des Durchmessers aller Erythrozyten einer Blutprobe. Im gesunden Patienten liegt eine normalverteilte Gauß´sche Kurve vor, mit einer Basisbreite von 6-9 µm und einem Mittelwert und Maximum von ~7,5 μm (Abb.5). Eine Rechtsverschiebung des Mittelwertes bezeichnet man als Makrozytose, eine Links-verschiebung als Mikrozytose. Anormal große Erythrozyten werden als Makrozyten, anormal kleine als Mikrozyten bezeichnet. Liegen diese in zu hoher Konzentration vor, so ist die Price-Jones-Kurve breit und abgeflacht; man spricht von einer Anisozytose.
II. Die BLUTSTILLUNG (Hämostase) II. Die BLUTSTILLUNG (Hämostase) Verletzungen des Blutgefäßsystems müssen schnell abgedichtet werden, um Blutverluste gering zu halten. Hieran sind Thrombozyten, Gerinnungsfaktoren und Gefäßendothel beteiligt. Prinzipiell unterscheidet man bei der Blutgerinnung die primäre Hämostase und die sekundäre Hämostase: Da die Blutgerinnung sehr verwirrend sein kann, rate ich den folgenden Teil, insbesondere die sekundäre Hämostase, Schritt für Schritt und immer anhand der Bilder durchzuarbeiten! 1. Primäre Hämostase: Thrombozytenfunktion und Bildung des „weißen Thrombus“
Die primäre Hämostase ist eine direkte Funktion der Thrombozyten (zelluläre Komponente der Hämostase). Mit einer Konzentration von 170.000 – 400.000/μl gewährleisten sie in weniger als einer Minute den ersten Wundverschluss. Bei einer Verminderung der Thrombozytenzahl ( 50.000/μl) werden Störungen in der Blutstillung deutlich. In normalen, unverletzten Gefäßen zirkulieren die Thrombozyten, ohne dass sie am Gefäßendothel haften bleiben oder aggregieren. Bei einer Gefäßverletzung treten Thrombozyten mit den freigelegten subendothelialen Kollagenfasern in Kontakt. Diese Adhäsion wird durch eine molekulare Brücke, bestehend aus und dem GP Ib/IX Rezeptorkomplex der Thrombozyten und dem von-Willebrand-Faktor, ermöglicht. Thrombozyten können auch über den GP Ia/IIa Rezeptorkomplex direkt an Kollagen binden. Die Thrombozytenadhäsion wird u.a. durch
Praktikumsversuch : „Bestimmung der Zellzahl und Erythrozyten-Parameter“ Hier werden Erythrozyten gezählt und Hämoglobin und Hämatokrit gemessen. Aus diesenParametern werden Erythrozyten-Indizes errechnet, die Aussagen über das eventuelleVorliegen einer Anämie erlauben (s. auch Tabelle „Blutwerte – Normalblut“) .
BLUT 8Fibronektin und Laminin verstärkt. Dadurch kommt es zur Aktivierung der Thrombozyten, wobei es im Wesentlichen zu drei Reaktionen kommt: Sekretion: Die Thrombozyten schütten Granula aus, die Gerinnungsfaktoren, „Klebstoffe“ und Wachstumsfaktoren enthalten, um dadurch weitere Blutplättchen zu aktivieren, eine Vasokonstriktion auszulösen und die Wundheilung einzuleiten. Formänderung: Durch Thrombin (s. sekundäre Hämostase) wird in den Thrombozyten globu-läres in fibrilläres Aktin umgewandelt, so dass die aktivierten Thrombozyten lange Fortsätze (Pseudopodien) ausbilden, über die sie sich mit weiteren aktivierten Thrombozyten verzahnen können. Aggregation: Durch die Formänderung aggregieren und kontrahieren viele Thrombozyten. Damit die Thrombozyten gut aneinander haften bilden sie auf ihrer Membran einen Rezeptorkomplex (GPIIb/IIIa) aus, an dem der „Klebstoff“ Fibrinogen binden kann. Thrombospondin, sowie andere Klebstoffe verstärken den Thrombus (Thrombozytenpfropf = weißer Thrombus).
Damit sich ein genügend großer Thrombus bildet, der die Verletzung effizient verschließt, müssen ausreichend viele Thrombozyten aktiviert werden. Daher sezernieren bereits aktivierte Thrombozyten Stoffe, die weitere Blutplättchen anlocken und aktivieren. Außerdem bilden aktivierte Thrombozyten weitere, synergistisch wirkende Substanzen: Thomboxan A2: wirkt zusammen mit Serotonin stark vasokonstriktorisch, um den
Blutverlust zu minimieren. Platelet Activating Factor (PAF): verstärkt zusammen mit Thromboxan A2 und Thrombin
(s. sekundäre Hämostase) die Thrombozytenaktivierung. PAF ist chemotaktisch wirksam, d.h. es lockt auch Phagozyten (Makrophagen und Granulozyten) an und kann anders herum auch von Phagozyten freigesetzt werden (Kommunikation verschiedene Zelltypen).
2. Sekundäre Hämostase: Bildung des „roten Thrombus“ Der weiße Thrombus ist instabil und kann von der Stelle der Verletzung weggeschwemmt werden. Durch die Bildung eines Maschenwerks aus Fibrinfäden (= roter Thrombus) wird der Thrombus gefestigt und stabilisiert. Zur Bildung der Fibrinfäden sind Ca2+-Ionen, Phospholipide und Plasmaproteine (=Gerinnungsfaktoren, Abb. 2, 6) nötig. Sie bilden zusammen die Gerinnungskaskade. Der Gerinnungskaskade folgend unterscheidet man die Aktivierungs-, Koagulations- und Retraktionsphase. Mittelpunkt der Kaskade ist Faktor X, der in seiner aktiven Form (Xa) zusammen mit weiteren Komponenten den Enzymkomplex Prothrombinase bildet. Dadurch wird inaktives Prothrombin (II) in Thrombin (IIa) überführt. Thrombin ist für die Bildung der vernetzenden Fibrinfäden verantwortlich. Aktivierungsphase: Faktor X kann durch ein exogenes und ein endogenes System aktiviert werden. Für den exogenen Weg (extrinsisches System) ist das Gewebethromboplastin (Tissue Factor : TF)
Praktikumsversuch : „Blutungszeit nach Marx“ Mit der Aggregation und Kontraktion der Thrombozyten und der Gefäßkontraktion ist die primäre Hämostase abgeschlossen (normale Dauer: etwa 2 – 4 Minuten).
Abb. 6: Gerinnungskaskade
BLUT 9
Abb. 7: Faktoren der sekundären Hämostase
entscheidend. Es kommt extravasal in der Adventitia der Blutgefäße vor. Durch eine Verletzung treten TF und der zuvor im Blut aktivierte VIIa miteinander in Kontakt und bilden zusammen mit Ca2+-Ionen und Phospholipiden den VII-TF-Komplex (Abb. 6). Am Anfang des endogenen Wegs (intrinsisches System) steht XII, der durch den Kontakt mit negativ geladenen Oberflächen (Kollagenfasern) aktiviert wird. Am Ende dieses Weges steht der Faktor X-Aktivatorkomplex (des endogenen Weges) bestehend aus IXa, VIIIa, Ca2+-Ionen und Phospholipiden. Der endogene Weg ist weniger in vivo als in der in vitro Diagnostik von Bedeutung. Letzteres dient der Erkennung von Gerinnungsstörungen aufgrund spezifischen Faktorenmangels (s. partielle Thromboplastinzeit, PTT). Neben seiner Rolle in der in vitro Diagnostik (s. Quick-Test), ist der exogene Weg entscheidend für die Gerinnung in vivo. Die Aktivierung von IX und X durch den VII-TF-Komplex wird nach kurzer Zeit durch den Tissue Factor Pathway Inhibitor (TFPI) blockiert, sodass die Aktivierung von Faktor X hauptsächlich über den endogenen Weg weiterläuft (Aktivierung von VII durch VII-TF- Ca2+-Ionen und Phospholipiden = Faktor X-Aktivatorkomplex des exogenen Weges). Die bis dahin aktivierte Menge an Xa (Prothrombinase) bildet zusammen mit Va, Ca2+-Ionen und Phospholipiden den sog. Prothrombin-Aktivatorkomplex, der wiederum Prothrombin (II) zu Thrombin (IIa) umwandelt. Neben der Aktivierung von Fibrinogen (I) zu Fibrinmonomeren (Ia) ist Thrombin ein entscheidender Rückkopplungsmechanismus zur weiteren Aktivierung der Faktoren V, VIII, XI und XIII. Die Faktoren VIIIa und IXa (von XIa aktiviert) bilden den Faktor X-Aktivatorkomplex des endogenen Weges (s.o.), sodass Xa immer wieder neu gebildet werden kann. Dieses Modell erklärt auch, warum die Gerinnung entscheidend von den Faktoren VIII und IX abhängt (klinischer Bezug: „Hämophilie“). CAVE: Letztlich ist der exogene Weg nicht wegen der Menge an gebildetem Thrombin wichtig, seine Bedeutung liegt in Form einer „Startreaktion“ für die Gerinnung! Anmerkung: Eben wegen der verschiedenen Rückkopplungsmechanismen sind der endogene und der exogene Weg nicht wirklich als getrennte Wege zu betrachten. Thrombin ist eine Serinprotease und hat mehrere Aufgaben: Aktivator verschiedener Faktoren in der Gerinnungskaskade (s. sekundäre Hämostase) Verstärkung der Thrombozytenaktivierung (s. primäre Hämostase) Mitogen für die Endothelzellen und glatten Gefäßmuskelzellen (s. Wundheilung) Aktivator der Lymphozyten (Vermittlerrolle)
Koagulationsphase:
In der Koagulationsphase spaltet das Thrombin aus Fibrinogen kleine Fibrinopeptide ab, die sich über instabile Verbindungen wie Wasserstoff-brücken zusammenlagern bzw. koagulieren. Für die Ausbildung von stabilen/kovalenten Verbind-ungen sorgt Faktor XIIIa. Retraktionsphase:
Die Fibrinfäden legen sich über den weißen Thrombus und verbinden sich mit den aktivierten Thrombozyten über den GPIIb/IIIa-Rezeptor. Die Verbindung wird durch Fibronectin stabilisiert. Mittels a) Aktivierung des Aktin/Myosin-Systems
durch Thrombin, b) die feste Verbindung der Thrombozyten durch Fibrin und c) unter Einfluss von Thrombosthenin (Protein der Thrombozyten), zieht sich der Thrombus auf einen Bruchteil seines ursprünglichen Volumens zusammen (Retraktion). So kommt es zur Thrombus-Festigung und zum mechanischen Wundverschluss.
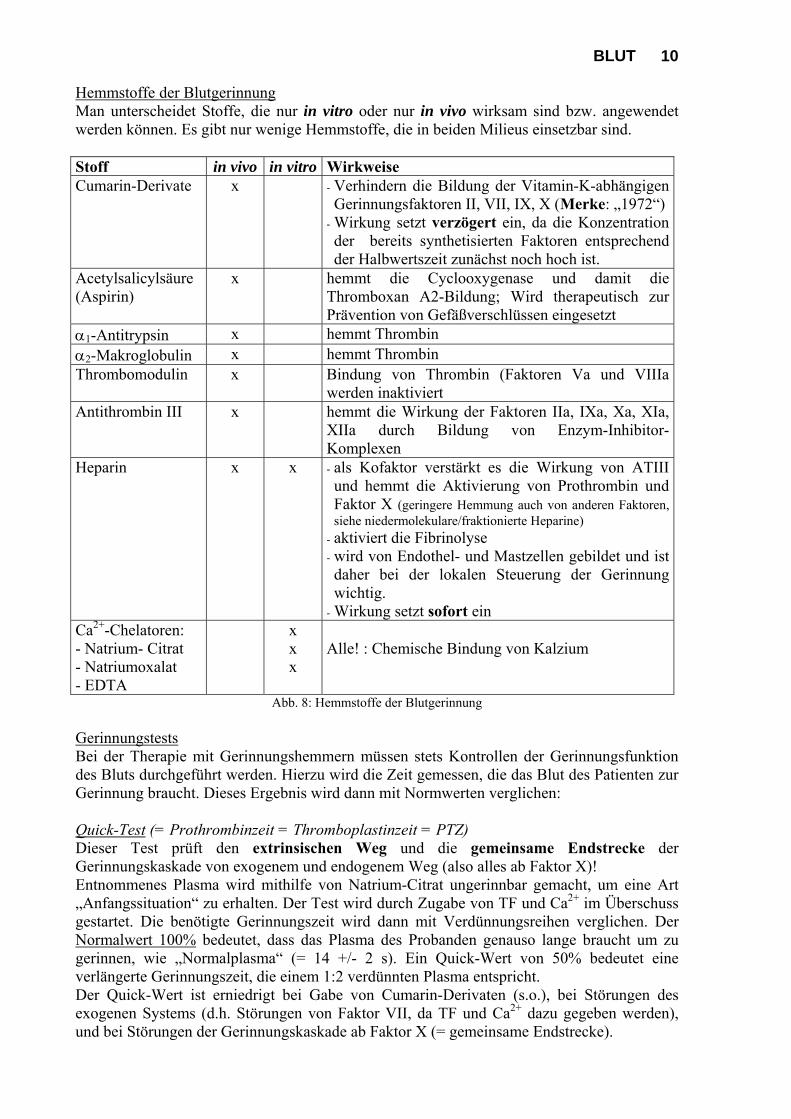
BLUT 10 Hemmstoffe der Blutgerinnung Man unterscheidet Stoffe, die nur in vitro oder nur in vivo wirksam sind bzw. angewendet werden können. Es gibt nur wenige Hemmstoffe, die in beiden Milieus einsetzbar sind. Stoff in vivo in vitro Wirkweise Cumarin-Derivate x - Verhindern die Bildung der Vitamin-K-abhängigen
Gerinnungsfaktoren II, VII, IX, X (Merke: „1972“) - Wirkung setzt verzögert ein, da die Konzentration der bereits synthetisierten Faktoren entsprechend der Halbwertszeit zunächst noch hoch ist.
Acetylsalicylsäure (Aspirin)
x hemmt die Cyclooxygenase und damit die Thromboxan A2-Bildung; Wird therapeutisch zur Prävention von Gefäßverschlüssen eingesetzt
1-Antitrypsin x hemmt Thrombin 2-Makroglobulin x hemmt Thrombin Thrombomodulin x Bindung von Thrombin (Faktoren Va und VIIIa
werden inaktiviert Antithrombin III x hemmt die Wirkung der Faktoren IIa, IXa, Xa, XIa,
XIIa durch Bildung von Enzym-Inhibitor-Komplexen
Heparin x x - als Kofaktor verstärkt es die Wirkung von ATIII und hemmt die Aktivierung von Prothrombin und Faktor X (geringere Hemmung auch von anderen Faktoren, siehe niedermolekulare/fraktionierte Heparine)
- aktiviert die Fibrinolyse - wird von Endothel- und Mastzellen gebildet und ist daher bei der lokalen Steuerung der Gerinnung wichtig.
Abb. 8: Hemmstoffe der Blutgerinnung Gerinnungstests Bei der Therapie mit Gerinnungshemmern müssen stets Kontrollen der Gerinnungsfunktion des Bluts durchgeführt werden. Hierzu wird die Zeit gemessen, die das Blut des Patienten zur Gerinnung braucht. Dieses Ergebnis wird dann mit Normwerten verglichen: Quick-Test (= Prothrombinzeit = Thromboplastinzeit = PTZ) Dieser Test prüft den extrinsischen Weg und die gemeinsame Endstrecke der Gerinnungskaskade von exogenem und endogenem Weg (also alles ab Faktor X)! Entnommenes Plasma wird mithilfe von Natrium-Citrat ungerinnbar gemacht, um eine Art „Anfangssituation“ zu erhalten. Der Test wird durch Zugabe von TF und Ca2+ im Überschuss gestartet. Die benötigte Gerinnungszeit wird dann mit Verdünnungsreihen verglichen. Der Normalwert 100% bedeutet, dass das Plasma des Probanden genauso lange braucht um zu gerinnen, wie „Normalplasma“ (= 14 +/- 2 s). Ein Quick-Wert von 50% bedeutet eine verlängerte Gerinnungszeit, die einem 1:2 verdünnten Plasma entspricht. Der Quick-Wert ist erniedrigt bei Gabe von Cumarin-Derivaten (s.o.), bei Störungen des exogenen Systems (d.h. Störungen von Faktor VII, da TF und Ca2+ dazu gegeben werden), und bei Störungen der Gerinnungskaskade ab Faktor X (= gemeinsame Endstrecke).
BLUT 11
INR (= International Normalized Ratio) Die INR stellt eine Standardisierung des Quick-Tests zur besseren Vergleichbarkeit dar! Der INR-Normwert beträgt 1,0 und entspricht einem Quick-Wert von 100%. Bei einem INR=2,0 ist die Gerinnungszeit entsprechend verlängert. In der Antikoagulanz-Therapie (z.B. durch Cumarin-Derivate) werden INR-Werte zwischen 2,0 und 3,5 angestrebt, um einerseits die Thrombosegefahr gering zu halten, andererseits das Blutungsrisiko nicht zu sehr zu erhöhen.
Thrombinzeit Dieser Test prüft nur die gemeinsame Endstrecke der Gerinnungskaskade von exogenem und endogenem Weg! Die Thrombinzeit ist die benötigte Gerinnungszeit nach Zugabe von Thrombin zu Citratplasma. Sie dient der Überprüfung eines Fibrinogenmangels oder der Überwachung einer Heparintherapie. Der Normalwert beträgt zwischen 17 – 24 s Thrombozytenzählung Eine Gerinnungsstörung kann außerdem vorliegen, wenn die Thrombozytenzahl verändert ist. Wenn die Zahl zu hoch ist, spricht man von einer Thrombozytose. Dagegen wird eine verminderte Anzahl als Thrombozytopenie bezeichnet. Aus den Ergebnissen der verschiedenen Gerinnungstests kann auf eine wahrscheinliche Ursache für eine Gerinnungsstörung geschlossen werden:
Quick PTT TZ-Zahl Blutungszeit mögl. Ursachen einer Störung der Hämostase
↓ ↑ ↔ ↔ Cumaringabe, Vitamin K-Mangel, Faktorenmangel I, II, V, X
↔ ↑** ↔ ↑ ** PTT nur bei v Willebrand-Jürgens-Syndrom Typ 3 erhöht wegen Faktor VIII-Mangel
↓ ↑ ↓ ↑ Leberschaden, Verbrauchskoagulopathie
Abb. 9: Interpretation von Gerinnungstests
Klinische Bezüge Hämophilie (= Bluterkrankheit): Bei Fehlen der Faktoren VIII und IX der Gerinnungskaskade kommt es zu Gerinnungs-störungen. Abhängig davon welcher Faktor fehlt, unterscheidet man die Hämophilie A (F.VIII fehlt) und die Hämophilie B (F.IX fehlt). Statistisch tritt die Hämophilie A 5-mal häufiger auf als die Hämophilie B. Prinzipiell sind die Symptome bei beiden Formen gleich: Es kommt zu großen Blutergüssen (Hämatomen), zu lang andauernden Blutungen nach Verletzungen und zu Blutungen in die Gelenke (Hämarthrosen), die zu Gelenkversteifungen führen können. Da die Aktivität der beiden Faktoren selten komplett ausfällt, ist die klinische Manifestation der Symptome abhängig von der Restaktivität des defekten Gerinnungsfaktors. Die Therapie basiert auf einer Substitution des jeweils fehlenden Faktors.
Praktikumsversuch : „Partielle Thromboplastinzeit (PTT)“ Die PTT testet den intrinsischen Weg und die gemeinsame Endstrecke der Gerinnungskaskade von exogenem und endogenem Weg! Der Normwert beträgt zwischen 30 – 50 s
BLUT 12
Abb. 10: Fibrinolyse
Fibrinolyse Die Fibrinolyse dient der Auflösung von Fibrinfäden. Das Gegenstück zum Thrombin IST das Plasmin, das die Faktoren V, VIII, I und Ia spaltet. Für die Aktivierung von Plasmin aus Plasminogen gibt es ebenfalls ein exogenes (Gewebeaktivatoren) und ein endogenes System (Blutaktivatoren): Blutaktivatoren: - Kallikrein: durch F.XIIa aktiviert CAVE: Faktor XIIa ist also nicht nur über das intrinsische System an der Gerinnung beteiligt, sondern auch an der Fibrinolyse für die er eine große Bedeutung hat! Gewebeaktivatoren: - tissue plasminogen activator (t-Pa): durch Gefäßdehnung und
Katecholamine aktiviert - Urokinase: von Epithelien der ableitenden Harnwege freigesetzt
Plasminogen und ihre Aktivatoren haben eine hohe Affinität zu Fibrinfäden, sodass sich Plasmin in direkter Gegenwart von Fibrin bildet und dieses dann spaltet. Die Spaltprodukte hemmen die Thrombinaktivität, sodass die Neubildung von Fibrin gebremst wird. Die
Fibrinolyse wird durch 2-Anti-plasmin gehemmt. Die Fibrinolyse kann beeinflusst werden, indem man Patienten bakterielle Streptokinase oder auch rekombinantes t-Pa (rt-Pa) verabreicht. Beide fördern die Fibrinolyse (Therapie bei der Auflösung von Thromben).
Wundheilung Ziel der Wundheilung ist die Wiederherstellung der Integrität und der Funktionstüchtigkeit des Gewebes nach einer Verletzung („Restitutio ad integrum“). Ist dies nicht möglich, ver-bleibt eine Narbe. Mit den „Aufräumarbeiten“ (Phagozytose), kommt es makroskopisch zu einer akuten Entzündung die mit den vier typischen Kardinalsymptomen einhergeht: • Calor (Erwärmung) • Rubor (Rötung) • Tumor (Schwellung) • Dolor (Schmerz) Nach den Aufräumarbeiten kann die Reparatur des Gewebes und damit die eigentliche Wundheilung beginnen. Sie ist gekennzeichnet durch:
Angiogenese (Neubildung von Blutgefäßen) Proliferation und verstärkte Kollagenbildung durch Fibroblasten Vermehrung epidermaler Keratinozyten zur Abdeckung der Wunde
Damit die Wundheilung regelgerecht abläuft sind verschiedene Wachstumsfaktoren nötig, die in einem komplexen Wechselspiel miteinander in Beziehung treten. Damit dieses Wechselspiel korrekt abläuft, dient die extrazelluläre Matrix als „Architekturvorlage“. III. DAS IMMUNSYSTEM Grundsätzlich kann dieses Abwehrsystem in unspezifisches (bzw. angeborenes oder natürliches) und spezifisches (bzw. adaptives oder erworbenes) Immunsystem unterteilt werden. Beide Komponenten setzen sich aus humoralen und zellulären Faktoren zusammen. Die unspezifische Immunabwehr antwortet sehr schnell und unterscheidet lediglich zwischen „körpereigen“ und „körperfremd“. Die spezifische Immunabwehr erkennt hingegen spezifisch Erreger. Dieses Erkennen führt zur Differenzierung besonderer immunologischer Abwehr-
BLUT 13
zellen und zur Ausbildung eines immunologischen Gedächtnisses. Deshalb ist die beim Erstkontakt noch langsam anlaufende spezifische Immunantwort bei einem erneuten Kontakt deutlich schneller. Dieses Phänomen wird als Immunität bezeichnet. Beide Abwehrsysteme dürfen allerdings nicht getrennt betrachtet werden. Zur effektiven Immunantwort sind beide Teile, die miteinander kommunizieren, unbedingt nötig. A. Die Leukozyten
Die weißen Blutzellen sind die zellulären Bestandteile sowohl der spezifischen als auch der unspezifischen Abwehr. Sie können sich amöboid fortbewegen und die Blutbahn durch einen Mechanismus, der als Leukodiapedese bezeichnet wird, verlassen, um ihre Aufgabe im Gewebe zu erfüllen. Sie entstehen alle aus einer pluripotenten hämatopoetischen Stammzelle des Knochenmarks. Die Bildung von Granulozyten und Monozyten wird durch Kolonie-Stimulierende Faktoren (CSF) angeregt. Die Lymphozyten werden unreif aus dem Knochenmark entlassen und dann in den primären lymphatischen Organen (Thymus oder Knochenmark) geprägt.
Normwert der Leukozyten im Blut: 4000 – 10000/μl Differenzial-Blutbild siehe Praktikumsanleitung „Blutwerte Normalblut“ Leukopenie: Abfall unter die Norm (meist Abfall neutrophiler Granulozyten (s.u.) durch Bildungsstörung im Knochenmark, vorzeitigen Leukozytenuntergang oder Verteilungs-störung) Leukozytose: Anstieg über die Norm (meist bei bakteriell infektiösen Prozessen) 1. Granulozyten
Die Granulozyten bilden die vorderste Frontlinie beim Eindringen körperfremder Substanzen oder pathogener Keime. Sie werden bei entzündlichen Vorgängen über chemotaktische Mediatoren (Chemokine) aus dem Blut ins Gewebe gelockt. Diese Zellgruppe wird durch die unterschiedliche Farbaufnahme ihrer Granula unterteilt.
Neutrophile Granulozyten (Durchmesser ~10 μm): Rolle: Phagozytose und Entzündungsreaktion durch Ausschüttung von Mediatoren. Sie haben phagozytäre Eigenschaften und können aufgenommenes Material durch verschiedene Enzyme (Phagozytose: s.u.) abbauen. Außerdem senden die Neutrophilen verschiedene Mediatoren aus und steuern so die Entzündungsvorgänge. Sie sind aber keine Antigen präsentierenden Zellen.
Eosinophile Granulozyten (Durchmesser ~12 μm): Rolle: Parasitenbekämpfung Aktivierung: Erkennen der Fremdkörper durch Antikörper (IgE), die sie durch Fc-Rezeptoren an ihrer Oberfläche tragen. Antwort: Degranulation Ausschüttung von Peroxidasen, Katalasen und Proteasen (…) Angelockt werden die eosinophilen Granulozyten vor allem durch Histamin, welches aus Mastzellen freigesetzt wird. Neben Parasitenbefall, auch bei allergischen Reaktionen gehäuftes Auftreten.
Basophile Granulozyten (Durchmesser ~9 μm): Rolle: ist schlecht verstanden; wahrscheinlich Beteiligung an Parasiten-Abwehr. Aktivierung: siehe eosinophile Granulozyten. Antwort: Degranulation Ausschüttung von Heparin, Histamin, Serotonin und Proteasen. Auch sie stehen im Zusammenhang mit allergischen Reaktionen.
BLUT 14
Abb. 11: Klonale Selektion
2. Monozyten / Makrophagen (Durchmesser ~16 μm)
Lokalisation: Monozyten – Blut; Makrophagen – Gewebe Mononukleäres Phagozytensystem (Retikuloendotheliales System) = Monozyten plus Makrophagen. Bildung: Makrophagen wandern entweder während ihrer Entstehung in das Gewebe ein oder differenzieren sich aus Monozyten, die bei einer Entzündungsreaktion in das Gewebe eintreten (Makrophagen: z.B. Kupffer-Sternzellen der Leber, Mikroglia-Zellen des ZNS). Antwort: Phagozytose, T-Zellenaktivierung, Mediatoren-Freisetzung (z.B. Leukotriene, Interferon γ, Interleukin 1) zur Steuerung der Immunantwort. Typische Antigen präsentierenden Zellen. 3. Lymphozyten
Lymphozyten (B- und T-Lymphozyten) sind die Zellen der spezifischen Immunantwort, da sie jeweils nur ein bestimmtes Antigen mit deren B-Zell-Rezeptor (BCR) bzw. T-Zell-Rezeptor (TCR) erkennen. Herkunft: Knochenmark. Nach Entstehung von B- bzw. T- Vorläuferzellen müssen diese diverse Prägungsschritte durchlaufen. Prägung (Zweck): Vernichtung autoaggressiver Zellen.
(Ort): in den jeweiligen primären lymphatischen Organen T-Lymphozyten - Thymus; B-Lymphozyten - Knochenmark (B engl. bone marrow = Bursa Fabricii Äquivalent der Vögel).
(Vorgang): positive Selektion (nur T-Zellen) und negative Selektion (B- und T- Zellen), die zur Beseitigung autoaggressiver Zellen entweder durch Apoptose oder dauerhafte Inaktivierung (= anerge Zellen) führt. Nach erfolgreicher Prägung bewegen sich die Lymphozyten über die Blutbahn zu den sekundären lymphatischen Organen. Trifft z.B. in der Milz oder den Lymphknoten ein Antigen auf einen passenden B- bzw. T-Lymphozyten, wird dieser aktiviert und zur massenhaften Vermehrung angeregt (s.u.) klonale Selektion (jede Tochterzelle besitzt dieselben Antigenrezeptoren, Abb. 11). Die Zellen der Klone differenzieren sich entweder zu Effektorzellen oder zu Gedächtniszellen. Letztere zirkulieren noch Jahre nach dem ersten Antigenkontakt im Blut und können bei erneutem Kontakt sofort eine spezifische Immunantwort initiieren (Grundlage für erworbene Immunität). a. B-Lymphozyten Sie besitzen B-Zellen-Rezeptoren (BCR) auf ihrer Oberfläche und sind nichts anderes als membranständige Immunglobuline (Ig). Sie werden durch den Antigenkontakt zur Reifung in Plasmazellen angeregt, die dann massenweise Antikörper produzieren (s.u.). b. T-Lymphozyten Sie besitzen den T-Zell Rezeptor (TCR) der stets membranständig ist. Es ist ein Protein, dass ähnlich dem BCR zur Erkennung kleiner Antigenbruchstücke dient, aber nur im Kontext der MHC-Moleküle (engl. major histocompatibility complex) von antigenpräsentierenden Zellen angeboten werden. Man unterscheidet hauptsächlich drei T-Zell-Subklassen: - CD4+-T-Zellen (T-Helfer-Zellen): Träger des CD4-Moleküls. Übernehmen wichtige
regulatorische Aufgaben und kommunizieren eng mit Makrophagen und B-Lymphozyten.
BLUT 15 - CD8+-T-Zellen (Killer- oder Zytotoxische T-Zellen): Träger des CD8-Moleküls. Spielen
vor allem bei der Zerstörung von körpereigenen Zellen, die z.B. mit Viren infiziert wurden, eine entscheidende Rolle.
- T-Suppressorzellen: sollen eine überschießende Immunreaktion verhindern. Beide CD-Moleküle sind somit Co-Rezeptoren, die in erster Linie die Aufgabe haben, die Interaktion der TCRs mit den antigenbeladenen MHCs zu stabilisieren.
B. Das unspezifische Immunsystem
Die angeborene Immunität versucht durch verschiedene Mechanismen das Eindringen von Erregern zu verhindern oder, wenn dies nicht gelingt, die stattfindende Infektion einzu-dämmen. Das geschieht ca. in den ersten vier Tagen einer Infektion.
1. Barrieren, die das Eintreten verhindern sollen
a. Physikalische Barrieren unversehrte, verhornte Haut. Guter Schutz vor vielen verschiedenen Erregern. Durch
Wunden (z.B. Biss oder Nadelstich) besteht ein enorm gesteigertes Risiko für den Eintritt infektiöser Einheiten in den Organismus.
Schleimhäute. Bietet nur gewissen Schutz gegen Krankheitserreger, da sie membranständige Rezeptoren besitzen, die die Mikroorganismen (MO) zur Anheftung an Zellen missbrauchen können. Zur besseren Abschirmung werden die Zellen von einer Schleimschicht bestehend aus verschiedenster Glykoproteine umgeben. Im Respirationstrakt führt die Zilien-Bewegung zum Abtransport der eindringenden Partikel. Urogenitaltrakt = Spüleffekt durch Harn; Gastrointestinalbereich = Peristaltik.
b. Chemische Barrieren Oberflächenepithelien bilden eine Reihe von Substanzen mit mikrozider Wirkung: - Säuren aus Talg- und Schweißdrüsen bereiten für MO ungünstiges Milieu (z.B. Magen =
sehr niedriger pH) - Antibakterielle Enzyme wie Lysozym. Schädigt die Zellwand grampositiver Zellen und
mit Hilfe von Komplementfaktoren auch die gramnegativer Bakterien. Folge = Platzen angegriffener Zellen. Ist im Gewebe, sowie in fast jeder Körperflüssigkeit zu finden. Besonders hohe Konzentrationen werden in Tränenflüssigkeit und Speichel erreicht.
- Protease Pepsin (Magen) fügt MO durch Proteinspaltung großen Schaden zu. - Verschiedene kationische Oligopeptide, die vor allem in Darmschleimhaut und Lunge die
Bakterienzellmembranen zerstören.
2. Humorale unspezifische Abwehr
a. Surfactant-Proteine (Protein C) und Teile des Komplementsystems markieren Erreger und erleichtern somit die Phagozytose durch Immunzellen Opsonisierung.
b. Komplementsystem Dazu gehören ~20 verschiedene im Blut zirkulierende Proteine, wobei die Faktoren C1 - C9 die wichtigsten sind. Bedeutend für Abwehr bakterieller Infektionen. Es folgt dem Prinzip der limitierten Proteolyse: die aktivierte Komponente spaltet die Vorstufe einer anderen und aktiviert diese (Vgl. Komplement- und Gerinnungskaskade).
Die Aktivierung kann auf drei Wegen erfolgen:
Klassischer Weg: Da er auf das Vorhandensein von Antigen-Antikörper-Komplexen angewiesen ist, handelt es sich um eine Brücke zum spezifischen Immunsystem.
BLUT 16Die Komponente C1 besitzt 6 Bindungsstellen für die Fc-Region von Antigen-gebundenem Immunglobulin (s.u.). Zur Aktivierung müssen beide Fc-Teile besetzt werden. Über die Zwischenschritte C4 und C2 wird der zentrale Faktor C3 gespalten (Abb. 12).
Alternativer Weg: Polysaccharide der Bakterien-oberflächen stabilisieren ein Spaltprodukt von C3 (C3b) welches dann in einem Komplex (C3bBb) wiederum C3 spaltet (Abb. 12). Da Antikörper unabhängig ist dies ein ehr direkter Aktivierungsweg.
MBL-Weg (Mannan-bindendes Lektin): Mannose (auf Bakterienoberflächen) wird durch im Serum befindliches Lektin gebunden. Lektin ähnelt strukturell C1 und kann dadurch C4 direkt aktivieren. Dieser Weg entspricht somit dem klassischen Weg, kommt allerdings ohne das Vorhandensein von Antigen-Antikörper-Komplexen aus.
Nach der Spaltung von C3 bindet das Spaltprodukt C3b an die Erregeroberfläche, während C3a abdiffundiert. Durch die Bindung wird zum einen der Erreger opsonisiert und zum anderen die Bildung des Membran-Angriff-Komplexes in Gang gesetzt. Dabei aktiviert C3b C5, der wiederum in C5a (diffundiert ab) und C5b gespalten wird. Letzteres bildet zusammen mit C6 bis C9 den Membran-Angriffs-(lytischen) Komplex (Abb. 12), durch den Ca2+, Na+ und H2O einströmen. Dadurch kommt es zur Zelllyse. C3a, C4a und C5a spielen eine wichtige Rolle bei der Entzündungsreaktion.
c. Akute-Phase-Proteine Plasmaproteine, deren Konzentration während Entzündungsreaktionen im Blut steigt. Synthese findet in der Leber statt und wird vor allem durch Interleukin-6 gefördert. Am bekanntesten ist das C-reaktive-Protein (CRP). Es reagiert mit dem C-Polysaccharid aus der Kapsel (engl. capsule) von Pneumokokken. Bindet an Oberflächenstrukturen vieler verschiedener Bakterien (Opsonisierung). Kann dann das Komplementsystem aktivieren. Weitere Akute-Phase-Proteine sind Proteinase-Inhibitoren wie α1-Antitrypsin und α2-Makroglobulin.
3. Zelluläre unspezifische Abwehr
a. Phagozytose Aktive Aufnahme fester Partikel in Zellen. Wird von Phagozyten betrieben. Die wichtigsten Phagozyten sind (v.a. neutrophile) Granulozyten und Monozyten. Die Phagozyten erkennen durch verschiedene Rezeptoren die Fremdsubstanzen: - Mannoserezeptoren und TOLL-like Rezeptoren können ohne weitere Proteine die
Phagozytose auslösen. - Komplementrezeptoren sind für die Phagozytose der opsonisierten Erreger wichtig. - Fc-Rezeptoren leiten die Aufnahme solcher Partikel ein, die bereits durch Antikörper
komplexiert wurden (hier muss das spezifische Immunsystem schon aktiv sein). Zunächst umschließt die Phagozytenzellmembran den Infektionserreger, den er dann aufnimmt. Das resultierende Vesikel wird als Phagosom bezeichnet (Abb. 14). Durch dessen Ansäuerung wird ein bakteriostatisches Milieu geschaffen. Nach der Anlagerung von Lysosomen kommt es zur Verschmelzung und Bildung eines Phagolysosoms. Mit Hilfe verschiedener lysosomaler Enzyme (Defensine, kationische Peptide, Lysozym, Laktoferrin, usw.), sowie toxischen Sauerstoffradikalen und Stickstoffoxide, die durch lysosomale Enzyme beim Prozess des respiratorischen Burst hergestellt werden, werden die
Abb. 12: Komplementsystem
BLUT 17
Abb. 13: Zytokine
aufgenommenen Mikroorganismen abgetötet. Granulozyten sterben nach erfolgter Phagozytose durch Apoptose. Makrophagen hingegen präsentieren den T-Lymphozyten Teile des aufgenommenen Partikels mittels MHC-II.
b. Interferone und natürliche Killerzellen Hier handelt es sich um Effektoren des unspezifischen Immunsystems, die sich vorrangig gegen intrazelluläre Erreger, vor allem Viren, aber auch Bakterien und Parasiten, richten. Interferone sind Zytokine, die man in IFN-α, -β, und -γ unterteilen kann. Sie werden von Virus-befallenen Zellen produziert. IFN-α und -β verhindern die Virusreplikation in infizierten Zellen und schützen unbetroffene Zellen vor Befall. Außerdem steigern sie die Expression von MHC-I-Molekülen. Daneben regen sie die Aktivität der natürlichen Killerzellen an. IFN-γ stimulieren diverse Zellen des Immunsystems. Vor allem in Makrophagen führen diese zur Hochregulierung der MHC-I und -II-Moleküle, wodurch sich die Präsentation antigener Peptide verbessert. Natürliche Killerzellen (Nullzellen) sind eng mit Lymphozyten verwandt. Sie besitzen Rezeptoren, die die normgerechte Dichte und Struktur der MHC-Moleküle auf Zellen prüfen. Bei Abweichungen schütten sie zytotoxische Granula aus und töten diese Zelle.
c. Zytokine Hormonähnliche Botenstoffe zur Steuerung der Immunreaktion. Sie werden entweder von mononukleären Phagozyten (treten also schon früh während der Immunantwort auf) oder von den Lymphozyten (Zellen der spezifischen Antwort) gebildet. Sie haben autokrine und parakrine Funktion. Interleukine sind Zytokine, die hauptsächlich Zellinteraktionen beein-flussen. Neben den in Abb. 13 gelisteten Zytokinen ist TNF-α (Tumor-Nekrosefaktor) von entscheidender Bedeutung. Er wird von Makrophagen gebildet, infolge einer Bindung von bakteriellen Lipopolysacchariden. Die TNF-α-Wirkung ist vielfältig, z.B. Steigerung der Gefäßpermeabilität, Induktion gesteigerter Adhäsionsmolekülbildung, Blutplättchen-stimulation (so wird in kleinen Gefäßen die Ausbreitung bakterieller Erreger begrenzt) und Mobilisierung dendritischer Zellen.
d. Die Entzündungsreaktion Im Laufe einer Entzündungsreaktion auftretende Zeichen wie Schwellung (Ödembildung), Erwärmung und Rötung sind Folgen einer lokalen Veränderung im Blutgefäßsystem. Aktivierte Makrophagen sezernieren Stoffe wie Prostaglandine, Leukotriene und TNF-α, die zusammen mit den Komplementfaktoren C3a und C5a und anderen Immunmodulatoren einen Anstieg der Gefäßpermeabilität, eine Senkung der Blutflussgeschwindigkeit sowie eine Endothelaktivierung bewirken. Außerdem wird eine chemotaktische Wirkung auf Entzündungszellen ausgeübt. Dadurch kommt es zu einem Flüssigkeitseinstrom, zur Leukozytendiapedese und zum Anstieg der lokalen Konzentration von immunologisch wirksamen Plasmaproteinen (Komplement und Antikörper). Da Neutrophile nach erfolgter Phagozytose sterben, ergießt sich ihr Zellinhalt ins Gewebe (Eiterbildung).
BLUT 18Exkurs Endothelaktivierung und Chemotaxis: Interleukin-1 und TNF-α veranlassen das Endothel zur Bildung von Selektinen. Diese Adhäsionsmoleküle bewirken ein Leukozytenrollen auf dem Endothel. Somit wird eine Aktivierung der Phagozyten induziert, welche die Bildung von Integrinen auf ihrer Zellmembran hervorruft. Auch das sind Adhäsionsmoleküle; sie garantieren eine festere Anheftung der Leukozyten an die Gefäßwand und ermöglichen deren transendotheliales Einwandern (Diapedese) in das Gewebe. Der gesamte Vorgang wird als Extravasion bezeichnet. Chemokine wie das IL-8 sowie die oben genannten Komplementfaktoren oder auch Bakterienbestandteile leiten die Entzündungszellen dann zum Infektionsherd.
C. Das spezifische Immunsystem
Das spezifische Immunsystem basiert auf der Antigen-Antikörper-Reaktion. Sie erfolgt mittels Wasserstoffbrückenbindungen und hydrophoben Wechselwirkungen. Da die kompletten Antigene meist zu groß sind für Antigenbindungsstellen, dient meist nur eine kleine Sequenz des Antigens (Determinante oder Epitop) als Erkennungsstelle für das Gesamtmolekül. Trennt man nun diese vom Molekül, erhält man ein Hapten, das von Antikörpern gebunden werden kann, aber alleine keine Immunreaktion auslöst. (Anmerkung: Als Paratop wird der zum Epitop passende Immunglobulinrezeptor bezeichnet) 1. Antigenpräsentation
a. Antigenpräsentierende Zellen (APZ) Prinzipiell präsentieren alle kernhaltigen Zellen Antigene. Betrachtet man aber die Präsentation von extrazellulären Fremdsubstanzen, kommen hier nur verschiedene Zellen des Immunsystems in Frage: Monozyten und die sich daraus differenzierenden Gewebs-makrophagen, B-Lymphozyten und dendritische Zellen. b. Histokompatibilitätskomplex Die Präsentation von Antigenen erfolgt prinzipiell mittels MHC-Molekülen; der TCR kann nämlich nur Antigene erkennen, die im Kontext der MHCs auf der Zelloberfläche dargereicht werden. Dieser Umstand wird als MHC-Restriktion bezeichnet. Synonyme für den major histocompatibility complex sind HLA (human leukocyte antigen) und Transplantations-antigene. Grundsätzlich lassen sich MHC-I (in der Membran aller kernhaltigen Zellen befindlich) und MHC-II (Expression auf Zellen des Immunsystems beschränkt) unterscheiden. Die Beladung der MHCs erfolgt auf zwei unterschiedliche Arten: MHC-I-Moleküle sind für die Präsentation von intrazellulären Erregern (wie z.B. Viren) bedeutsam. Mit ihrer Hilfe werden nämlich Peptidfragmente der gerade von der Zelle produzierten Proteine dargeboten. Solche Komplexe können wie oben beschrieben von einem passenden TCR eines CD8+-T-Lymphozyten erkannt werden. MHC-II-Moleküle sind für die Präsentation von extrazellulären Fremdsubstanzen (wie z.B. Bakterien) verantwortlich. Dem entsprechend befinden sie sich auf den antigen-präsentierenden Zellen des Immunsystems. Ein Komplex aus MHC-II-Molekül und Fremdpeptid kann von CD4+-T-Lymphozyten erkannt werden (Abb. 14). 2. Spezifische zelluläre Abwehr
a. T-Helferzellen (CD4+) Eine antigenpräsentierende Zelle nimmt ein Antigen unspezifisch auf und präsentiert es mittels MHC-II. Diesen Komplex wird durch eine T-Helferzelle, die einen für das präsentierte Epitop des Antigens spezifisch TCR besitzt, erkannt. Nach der Komplex-Rezeptor-Interaktion werden sowohl von der antigenpräsentierenden Zelle als auch von der T-Helferzelle verschiedene Zytokine (s.o.), wobei das wichtigste das IL-2 ist, freigesetzt, wodurch es zu
BLUT 19
Abb. 15: Proliferation B-Zellen
einer klonalen Proliferation der entsprechenden T-Helferzelle kommt, die sich dann in eine Effektorzelle oder eine Gedächnisszelle differenziert (Abb. 14). Die CD4+ Effektorzellen üben ihre Funktion über die Sekretion von Zytokinen aus. Je nachdem, welche Zytokine sie ausschütten üben sie unterschiedliche, ja sogar gegensätzliche, Funktionen aus. Daher werden CD4+ in zwei Untergruppen eingeteilt: TH1-Zellen: Aktivieren durch die Ausschüttung von IFN-γ vor allem Makrophagen und tragen besonders zur Beseitigung von Bakterien bei. Es resultiert eine zellbasierte inflammatorische Immunantwort. IFN-γ aktiviert nicht nur Makrophagen sondern hat eine inhibitorische Wirkung auf TH2-Zellen und reguliert somit die antikörpervermittelte Immunantwort (s.u.). TH2-Zellen: Sie sind im Wesentlichen eine Differenzierungshilfe für B-Lymphozyten. Dadurch resultiert eine humorale, antikörpervermittelte Immunantwort. Beispiele für die durch TH2-Zellen sezernierten Zytokine sind IL-4, IL-10 und TGF-β. IL-4 differenziert naive CD4+ T-Zellen (TH0-Zellen) zu TH2-Zellen, denen generell eine anti-inflammatorische Rolle zugeschrieben wird. IL-4 stimuliert aktivierte B-Zellen und führt zu einem Immunglobulin-Klassenwechsel (IgM zu IgG, IgE). IL-10 wirkt direkt supprimierend auf Makrophagen. TGF-β unterdrückt TH1-Zellen und somit eine Makrophagenstimulierung. b. Zytotoxische T-Zellen (CD8+-T-Effektorzellen) Nach Aktivierung (die ähnlich zur CD4+-T-Zellen Aktivierung verläuft) und klonaler Proliferation lösen sie die Apoptose in der Zielzelle aus. Die Signalvermittlung erfolgt durch Ausschütten von Granula, die aus zwei Hauptkomponenten bestehen. Perforine bilden Poren in der Membran der Zielzelle. Granzymes sind Serinproteasen, die durch Perforinporen in die Zelle eintreten und dort eine Apoptosekaskade auslösen. Außerdem können zytotoxische T-Lymphozyten eine rezeptorvermittelte Apoptose durch „Todesrezeptoren“ (z.B. Fas-Molekül, das von T-Zellen aktiviert werden kann) induzieren. 3. Spezifische humorale Immunabwehr a. B-Lymphozyten
Träger der spezifischen, humoralen Immunantwort. Binden durch den BCR ihr spezifisches Antigen, phagozytieren und präsentieren es nach Zerlegung auf der Zelloberfläche im MHC-II-Komplex. Erkennt eine TH2-Zelle diesen Komplex, aktiviert sie die entsprechende B-Zelle via Zytokinen (v.a. Il-4) wodurch diese sich vermehrt (klonale Expansion) und dann zur Plasmazelle differenziert (Abb. 15). Plasmazellen sind gewebsständig und produzieren Antikörper (Immunglobuline, Ig), die spezifisch sind für das Antigen, welches anfangs der B-Lymphozyt gebunden hatte. Während der Immunreaktion kommt es noch zur Erhöhung der Antikörperspezifität (Somatische Hypermutation) und Isotypen-Switch (s.u.). Aktivierte B-Zellen bilden neben den Plasmazellen auch B-Gedächtniszellen aus. Diese produzieren Ig-G-Moleküle bei ihrer Aktivierung. Da IgM vor allem
Abb. 14: Differenzierung der T-Zellen
BLUT 20
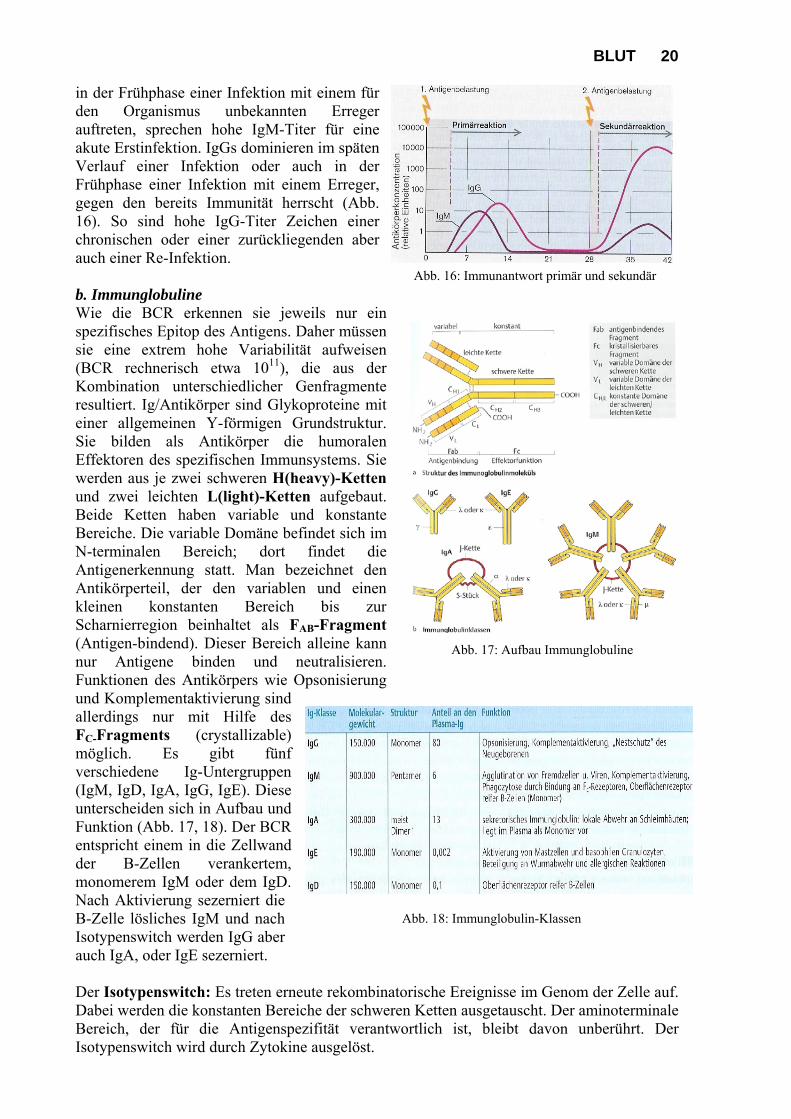
Abb. 16: Immunantwort primär und sekundär
Abb. 17: Aufbau Immunglobuline
Abb. 18: Immunglobulin-Klassen
in der Frühphase einer Infektion mit einem für den Organismus unbekannten Erreger auftreten, sprechen hohe IgM-Titer für eine akute Erstinfektion. IgGs dominieren im späten Verlauf einer Infektion oder auch in der Frühphase einer Infektion mit einem Erreger, gegen den bereits Immunität herrscht (Abb. 16). So sind hohe IgG-Titer Zeichen einer chronischen oder einer zurückliegenden aber auch einer Re-Infektion. b. Immunglobuline Wie die BCR erkennen sie jeweils nur ein spezifisches Epitop des Antigens. Daher müssen sie eine extrem hohe Variabilität aufweisen (BCR rechnerisch etwa 1011), die aus der Kombination unterschiedlicher Genfragmente resultiert. Ig/Antikörper sind Glykoproteine mit einer allgemeinen Y-förmigen Grundstruktur. Sie bilden als Antikörper die humoralen Effektoren des spezifischen Immunsystems. Sie werden aus je zwei schweren H(heavy)-Ketten und zwei leichten L(light)-Ketten aufgebaut. Beide Ketten haben variable und konstante Bereiche. Die variable Domäne befindet sich im N-terminalen Bereich; dort findet die Antigenerkennung statt. Man bezeichnet den Antikörperteil, der den variablen und einen kleinen konstanten Bereich bis zur Scharnierregion beinhaltet als FAB-Fragment (Antigen-bindend). Dieser Bereich alleine kann nur Antigene binden und neutralisieren. Funktionen des Antikörpers wie Opsonisierung und Komplementaktivierung sind allerdings nur mit Hilfe des FC-Fragments (crystallizable) möglich. Es gibt fünf verschiedene Ig-Untergruppen (IgM, IgD, IgA, IgG, IgE). Diese unterscheiden sich in Aufbau und Funktion (Abb. 17, 18). Der BCR entspricht einem in die Zellwand der B-Zellen verankertem, monomerem IgM oder dem IgD. Nach Aktivierung sezerniert die B-Zelle lösliches IgM und nach Isotypenswitch werden IgG aber auch IgA, oder IgE sezerniert. Der Isotypenswitch: Es treten erneute rekombinatorische Ereignisse im Genom der Zelle auf. Dabei werden die konstanten Bereiche der schweren Ketten ausgetauscht. Der aminoterminale Bereich, der für die Antigenspezifität verantwortlich ist, bleibt davon unberührt. Der Isotypenswitch wird durch Zytokine ausgelöst.
BLUT 21 4. Hypersensibilitätsreaktionen
Kommt es bei einem Antigenkontakt zu einer überschießenden Immunreaktion, so spricht man von einer Hypersensitivitätsreaktion. Sie findet erst nach Sensibilisierung durch erstmaligen Antigenkontakt statt. Es gibt vier unterschiedliche Typen (von denen aber nur die Sofortreaktion hier detailliert besprochen wird):
Typ I (Soforttypeaktion; anaphylaktische Reaktion) Sie wird durch IgE-Antikörper vermittelt, die während des Erstkontakts mit dem Allergen von einer Plasmazelle gebildet werden. Binden nun diese Antikörper mittels ihres Fc-Fragments an Mastzellen oder basophilen Granulozyten, ist eine Sensibilisierung erfolgt. Bei einem erneuten Antigenkontakt wird durch eine Überbrückung von mindestens zwei IgE-Molekülen die Degranulation der entsprechenden Zelle ausgelöst. In diesen Granula sind vasoaktive Substanzen wie Prostaglandine, Leukotriene und Histamin. Hiervon ist Histamin zur Auslösung der typischen Allergiesymptomatik einer Sofortreaktion besonders hervorzuheben: In Sekunden bis Minuten kommt es zu einer Vasodilatation (Erythem), Steigerung der Gefäßpermeabilität (Ödem), Steigerung der Kontraktion der glatten Muskulatur (Bronchospasmus und Koliken), Hypersekretion der Schleimhäute (Rhinitis) und Juckreiz. Typische Beispiele sind die allergische Rhinitis und Heuschnupfen. Der allergische Schock stellt die Maximalvariante da. IV. DIE BLUTGRUPPEN
Blutgruppen sind die erblich verschiedene Ausprägung von Oberflächenantigenen der Erythrozytenmembran (meist Glykolipide). Neben den bekannten AB0- und Rhesus-Systemen gibt es über 15 weitere Systeme. Vor allem zur Verhinderung von Transfusionszwischenfällen, die durch inkompatibles Blut hervorgerufen werden, ist eine Feststellung der Blutgruppen unabdingbar.
A. Das AB0-System Die Antigene A und B sind Kohlenhydratreste auf Glykolipiden. Sie werden kodominant vererbt. Wenn keines der Antigene ausgeprägt wird, spricht man von der Blutgruppe 0. Hier ist nur die Grundsubstanz H, die auch bei A und B vorhanden ist, übrig. Dieses Merkmal wird folglich rezessiv gegenüber den anderen vererbt. Im Plasma zirkulieren präformierte Antikörper gegen jeweils die Erythrozytenantigene, die nicht ausgebildet werden (Blutgruppe A: Anti-B-Antikörper; Blutgruppe B: Anti-A-Antikörper; Blutgruppe 0: Anti-A und -B-Antikörper). Diese Immunglobuline sind vom IgM-Typ. Da sie fremde Erythrozyten zur Agglutination bringen, werden sie als Isoagglutinine bezeichnet. Sie werden schon in den ersten Lebensmonaten und ohne Kontakt mit Fremdblut gebildet. Hierfür werden physiologische Darmbakterien, deren Oberflächenstruktur den Blutgruppenantigenen ähnelt, verantwortlich gemacht. Die Bestimmung der AB0-Blutgruppe erfolgt mittels zweier Testseren, die Anti-A- bzw. Anti-B-Antikörper enthalten (Abb 19).
BLUT 22 B. Das Rhesus-System Im Rhesus-System werden sechs Merkmale (C, D, E, c, d, e) unterschieden. Entscheidend ist hier die Ausprägung des Merkmals D; ist dieses Merkmal vorhanden, spricht man von Rhesus-positivem Blut (Rh+ [D]). d bedeutet, dass das Merkmal D nicht ausgeprägt ist (rh- [dd]). Im Gegensatz zum AB0-System gibt es beim Rhesus-System keine präformierten Antikörper. Antikörper treten nur auf, wenn eine Rhesus-negative Person in Kontakt mit Rhesus-positivem Blut kommt. Die gegen die Rhesus-Antigene gebildeten Antikörper sind vom IgG-Typ (plazentagängig!). Praktikumsversuch „Blutgruppen“ C. Bluttransfusionen Bei Bluttransfusionen werden sowohl das Spender- als auch das Empfänger-Blut hinsichtlich seiner Gruppe untersucht. Das geschieht durch Major- und Minor-Test. Beim Major-Test werden Spender-Erythrozyten mit dem Empfänger-Serum vermischt; beim Minor-Test Spender-Serum mit Empfänger-Erythrozyten in Mischung gebracht. Beim Auftreten einer Verklumpung werden die Blutgruppen als inkompatibel bezeichnet und Transfusion darf nicht stattfinden. Kommt es doch zur Transfusion inkompatiblen Blutes, können schwere Transfusionszwischenfälle auftreten: Dabei führen die Isoagglutinine zu einer Verklumpung der fremden Erythrozyten und zur Aktivierung des Komplementsystems. Die klinischen Symptome reichen von Fieber und Schüttelfrost bis Nierenversagen. D. Exkurs Morbus hämolyticus neonatorum In dem Fall, dass eine rh- Mutter mit einem Rh+ Kind schwanger ist und es zu einem Übertritt von kindlichem Blut in den maternalen Kreislauf bei der Geburt kommt, kommt es zur Bildung von Antikörpern gegen die Rhesus-Antigene. Bei erneuter Schwangerschaft mit einem Rh+ Kind können nun die über die Plazenta in den fetalen Kreislauf gelangten Antikörper zur Zerstörung der kindlichen Erythrozyten führen. Man sollte deshalb bei der Situation Rh- Mutter mit Rh+ Kind in Erstschwangerschaft der Mutter nach der Entbindung prophylaktisch ein Antiserum, das sich gegen den Rhesus-Faktor richtet, injizieren. Dadurch kommt es zu einer Zerstörung von kindlichen Erythrozyten, die bei der Geburt in das mütterliche Blut gelangt sind, ohne Aktivierung und ohne Bildung eines Gedächtnisses des Immunsystems.
Praktikumsversuch : „ Blutgruppenbestimmung“ Es werden der Minor- und der Majortest für die AB0-Blutgruppeneigenschaften und der Rhesusfaktor bestimmt.
BLUT 23
HAUSAUFGABEN
1) Zeichnen Sie den schematischen Bau von IgG, IgA, IgM 2) Zeichnen Sie Agglutinationen ein Serum --> | Anti-B | Anti-A | Anti-B | Blutgruppe | + Anti-A| ---------------------------------------------------------- | | | | B ---------------------------------------------------------- | | | | A ---------------------------------------------------------- | | | | AB ---------------------------------------------------------- | | | | 0 ----------------------------------------------------------
BLUT 24
Blutwerte - Normalblut (w = Frauen; m = Männer)
Blutvolumen
:
4-6 Liter
Plasmavolumen : 3-4 Liter
Blut-pH : pH ~ 7,4
Osmolarität : 300 mosmol/l
Osmotischer Druck : 5800 mm Hg (770 kPa)
KOD Blutplasma : 25 mm Hg (3,3 kPa)
KOD Interstitium : 5 mm Hg (0,7 kPa)
Plasmaprotein : 70 g/l
Erythrozyten (Ery) : 4,2–5,4 (w) 4,6–5,9 (m) (106/µl oder 1012/l)
VERSUCH: HÄMOLYSE EINER BLUTPROBE OSMOTISCHE RESISTENZ
In diesem Versuch wird untersucht, wie hoch die Widerstandsfähigkeit der Erythrozytenmembran gegenüber der hämolysierenden Wirkung hypotoner NaCl-Lösungen ist. Die osmotische Resistenz der Erythrozyten hängt von morphologischen Gegebenheiten, sowie ihrem Energiehaushalt ab. Die osmotische Resistenz ist herabgesetzt, wenn das Verhältnis von Erythrozyten-Oberfläche zu Erythrozyten-Volumen zugunsten des Volumens verschoben ist, wie bei der Kugelzellanämie. Wenn ein Mangel in der Enzymausstattung des Erythrozyten besteht, wie bei den verschiedenen angeborenen Formen der hämolytischen Anämien, kann die osmotische Resistenz herabgesetzt sein. Eine erhöhte osmotische Resistenz findet sich bei Retikulozyten und bei krankhaft veränderten Erythrozyten. Zum Beispiel weisen Thalassämien eine erweiterte Resistenzbreite auf: normale bis leicht verminderte Minimalresistenz bei deutlich erhöhter Maximalresistenz. Normalwerte: 0,45% NaCl (beginnende Hämolyse = Minimalresistenz) und 0,35% NaCl (komplette Hämolyse = Maximalresistenz). Die osmotische Resistenz ist erhöht u.a. bei Thalassämie, Sichelzellanämie, schwerer Eisen-Mangel-Anämie, chronischen Lebererkrankungen. Die osmotische Resistenz ist erniedrigt u.a. bei Kugelzellanämie, hämolytischer Anämie, Benzolvergiftung. Durchführung: Befüllen sie je eins von 13 Zentrifugenröhrchen mit 3 ml NaCl-Lösungen folgender Konzentration: 0,1%, 0,2%, 0,25%, 0,3%, 0,35%, 0,4%, 0,45%, 0,5%, 0,6%, 0,7%, 0,8%, 0,9% NaCl. In jedes Röhrchen pipettieren sie drei Tropfen Blut mit einer Plasik-Pasteurpipette. Verschließen sie die Röhrchen mit Parafilm und mischen sie die Lösungen sorgfältig über Kopf (Schaumbildung vermeiden, nicht schütteln!). Die Röhrchen werden erst zum Ende des Praktikums abgelesen. Bei der Ablesung wird die NaCl-Konzentration des Röhrchens ermittelt, in dem die Lösung den ersten gelblichen Farbton aufweist (Minimalresistenz) und die Konzentration des Röhrchens, in dem kein Erythrozytenbodensatz mehr nachweisbar ist (vollständige Hämolyse, Maximalresistenz). Normalerweise beginnt die Hämolyse bei 0,46 - 0,42 % NaCl und ist komplett bei 0,34 - 0,30 % NaCl. Den Bereich zwischen diesen beiden Punkten bezeichnet man als Resistenzbreite. VERSUCH: MIKROSKOPIE VON ERYTHROZYTEN IN ISOTONER
UND HYPERTONER KOCHSALZLÖSUNG Je ein Tropfen Blut werden mit einer Pipette in 5 ml NaCl-Lösung (4 % und 0,9 %) gegeben. Die Proben werden gemischt und je ein Tropfen auf einen Objektträger gegeben und mit einem Deckglas abgedeckt. Beobachten Sie die Erythrozyten durch das Mikroskop (40x Objektiv). Erkennen Sie, die für Erythrozyten typische bikonkave Scheibenform? Wie verhält sich die Erythrozytenform in den verschiedenen NaCl-Konzentrationen?
BLUT 26 VERSUCH: BESTIMMUNG DER ZELLZAHLEN IM BLUT UND
BESTIMMUNG DER ERYTHOZYTEN-PARAMETER In den roten Blutkörperchen, den Erythrozyten, befindet sich Hämoglobin (Hb). Seine funktionelle Bedeutung liegt in der Bindung, dem Transport und der Abgabe von Sauerstoff (O2) und Kohlendioxid (CO2). Ein Zustand, bei dem der Hb-Gehalt im Blut erniedrigt ist, wird als Anämie bezeichnet. Ursachen können eine verminderte Erythrozytenzahl und eine reduzierte Hb-Synthese sein. Da die Anämie keine Krankheit, sondern ein Symptom ist, muss jede über den Hb-Wert diagnostizierte Anämie Anlass weiterer Untersuchungen sein, um die zu Grunde liegende Krankheit zu diagnostizieren und sinnvoll zu therapieren. Zu diesen Untersuchungen zählt die Bestimmung des Hämatokrit (Hkt). Der Hkt gibt den prozentualen Volumenanteil aller Blutzellen, primär den der Erythrozyten, am Gesamtblutvolumen an. Wenn die Zahl der Erythrozyten im Blut, das Hb und der Hkt bekannt sind, können aus diesen Werten Erythrozyten-Eigenschaften, die sog. Erythrozytenindizes, berechnet werden. Nach der Erythrozytengröße unterscheidet man zwischen mikrozytären, normozytären und makro-zytären Anämien; nach dem Hb-Gehalt unterscheidet man zwischen hypochromen, normo-chromen und hyperchromen Anämien. Durchführung: Die Bestimmung der Zellzahl und der Erythrozyten-Parameter wird durch ein Zellzählgerät (ABX Micros 60 HORIBA) automatisch durchgeführt. Verwenden sie die rote Monovette für diese Bestimmung. Die rote Monovette wird direkt vor der Messung mindestens 25-mal über Kopf gemischt. Stellen sie die Monovette in den Probenhalter und schließen sie zum Start der Messung die Probentür. Der Probenhalter wird automatisch nach oben gefahren, der Deckel des Probenröhrchens mit einer Kanüle durchstochen und eine Probenmenge von 10 µl entnommen. Lesen sie die Blutwerte vom Display ab und tragen sie diese in die ausliegende Tabelle ein. Errechnen sie die Erythrozytenindices manuell. Arbeitsprinzip des Gerätes: 1. Bestimmung von Hämoglobin und Leukozytenzahl aus der Primärverdünnung. Die aspirierten 10 μl Blut werden im Gerät mit isotoner Lösung 1:260 verdünnt (Primärverdünnung). Ein Teil der Primärverdünnung wird mit zyanidhaltigem Lyse-Reagenz versetzt und zur Bestimmung der Leukozytenzahl (WBC = white blood cells) verwendet. Durch die Lyse der Zellmembranen werden das Hb der Erythrozyten und die Zellkerne der Leukozyten freigesetzt. Die Hb-Menge wird photometrisch ermittelt (siehe Biochemie). Die Zählung der Leukozytenkerne (WBC) erfolgt auf denselben Grundlagen wie die der Erythrozyten (RBC) und Thrombozyten (PLT); siehe unten. 2. Bestimmung von Erythrozyten- und Thrombozytenzahlen aus der Sekundärverdünnung. Für die Zählung der Erythrozyten und Thrombozyten wird die Primärverdünnung durch das Gerät mit isotoner Lösung weiter verdünnt. Die Sekundärverdünnung beträgt 1:15,000 (Überlegen sie: Warum ist eine Sekundärverdünnung nötig?). RBC (red blood cells, Erythrozyten) und PLT (platelets = Thrombozyten) werden auf Grundlage einer Impedanz-Messung ermittelt. Das heisst: Um die Öffnung der Mikrokapillare, die von den Blutzellen passiert wird, besteht ein elektrisches Feld. Die Zellpassage im elektrischen Feld erzeugt einen Widerstand. Da die Stromstärke konstant gehalten wird, ist der Widerstand der passierenden Zelle proportional zu ihrer Größe (kleine Zelle = kleiner Widerstand; große Zelle großer Widerstand). Gleiches gilt für die gemessene Spannung (kleine Zelle = kleine Spannung; große Zelle = große Spannung). Die Impulsgröße der elektrischen Spannung variiert beim Passieren der Öffnung durch die Zellen. Die Impulse werden entsprechend ihrer Größe kategorisiert, mit bekannten Schwellenwerten verglichen und mathematisch aufbereitet, sodass für die Zahl an Erythrozyten (RBC) und Thrombozyten (PLT) bestimmt werden kann.
BLUT 27
A) Messparameter der Primärverdünnung: WBC (109/l) = Anzahl weißer Blutkörperchen HB/HGB (g/dl) = Hämoglobinkonzentration B) Messparameter der Sekundärverdünnung: RBC (1012/l) = Erythrozytenzahl MCV (fl) = mittleres corpusculäres Volumen eines Erythrozyten Hämatokrit (%) PLT (10/l)
5. Berechnung der Erythrozytenparameter MCH und MCHC: Aus den oben ermittelten Werten für die Blutprobe sollen die Erythrozytenparameter ausgerechnet werden und ebenfalls in die ausliegende Tabelle eingetragen werden. MCH (pg) = mittleres corpuskuläres Hämoglobin eines Erythrozyten: MCH = Hämoglobin ( g / l ) Erythrozytenzahl / l MCHC (g/l) = mittleres corpuskuläres Hämoglobin aller Erythrozyten:Hb(g/l)/Hkt MCHC = Hämoglobin ( g / l ) Hämatokrit VERSUCH: BLUTUNGSZEIT UND PTT Diese Tests sollen bei vorliegender Blutungsneigung Hinweise auf die zugrunde liegende Störung geben. Man unterscheidet zwischen Blutungszeit (Pfropfbildung, primäre Hämostase) und Gerinnungszeit (Fibrinbildung, sekundäre Hämostase). Störungen der Gerinnungszeit werden üblicherweise mit dem so genannten Quick-Test oder der partiellen Thrombo-plastinzeit (PTT) erfasst. Hierbei werden verschiedene Faktoren der exogenen und/oder endogenen Gerinnung getestet. Dem gegenüber steht die Blutungszeit, deren pathologische Veränderungen hauptsächlich auf Thrombozyten-Ebene stattfinden. 1. PARTIELLE TROMBOPLASTIN ZEIT (PTT)
Die Gerinnungszeit wird in diesem Test mit der so genannten "Häkelmethode" gemessen (wird gezeigt). Pipettieren Sie 0,1 ml Citratplasma und 0,1 ml Pathromtin (Siliciumdioxid-Partikel und pflanzliche Phospholipide) in ein Plastikröhrchen (!) und durchmischen Sie den Ansatz sorgfältig durch schwenken. Die Probe wird nun für 3 min bei 37°C im Wasserbad inkubiert. Zum Reaktionsstart (Stoppuhr und Häkelnadel bereithalten!) werden 0,1 ml einer 25 mM CaCl2-Lösung zum Probenansatz pipettiert. Starten Sie die Stoppuhr parallel dazu und "häkeln" Sie mit der Häkelnadel von unten nach oben durch ihre Probe. Stoppen Sie die Gerinnungszeit sobald der erste Fibrinfaden sichtbar ist. Die Gerinnungszeit beträgt in der Norm 30 bis 50 Sekunden. Fragen: Welche Faktoren werden durch die PTT erfasst? Was ist der Unterschied zum Quick-Test?
2. MESSUNG DER BLUTUNGSZEIT NACH MARX
Nach entsprechender Desinfektion mit 70%-igem Alkohol wird mit einem sterilen Hämostilett ein ca. drei Millimeter tiefer Einstich in die seitliche Fingerbeere gesetzt. Die Fingerbeere wird in 37°C warmes, destilliertes Wasser eingetaucht. Beim Erscheinen des ersten Blutfadens
BLUT 28wird die Stoppuhr in Gang gesetzt. Gemessen wir die Zeit bis zum Abreißen des Blutfadens. Die Blutungszeit beträgt bei gesunden Individuen etwa 2-4 Minuten. Frage: Wie kann eine verlängerte Blutungszeit zustande kommen?
VERSUCH: BLUTGRUPPEN-BESTIMMUNG Antigene des ABO-Systems (erythrozytäre Eigenschaften): Die den Antigenen zugehörigen Gene A (A1 oder A2), B und 0 werden unabhängig nach dem Mendelschen Gesetz vererbt. Die drei Gene kombinieren zu den sechs Genotypen AA, BB, 00, AB, A0, B0. 0 ist jedoch ein „stummes“ Gen, das an der Erythrozytenmembran kein Genprodukt (Antigen) bewirkt. Somit lassen sich im Phänotyp nur die vier Gruppen A, B, AB, und 0 nachweisen. Die Antigene des Rhesus-Systems werden mit C,c, D,d, E,e bezeichnet, wobei D die stärkste antigene Wirksamkeit hat; „d“ bedeutet, dass D fehlt. Jeweils drei dieser Merkmale werden als Rhesus-Blutgruppensystem vererbt. Blut, das Erythrozyten mit Antigen D enthält, wird als rhesus-positiv (Rh) bezeichnet; Blut dessen Erythrozyten die D-Eigenschaft fehlt („d“), wird als rhesus-negativ (rh) bezeichnet. Isoagglutinine (Serumeigenschaften): Die Antigene A und B kommen nicht nur auf Erythrozyten, sondern auch auf Darmbakterien vor (heterophile Antigene). So führt die Darmflora im ersten Lebensjahr zur Bildung von Antikörpern (Isoagglutinine Anti-A und Anti-B), wenn die eigenen Erythrozyten diese Antigene nicht tragen. Im Rhesus-System sind normalerweise keine Isoagglutinine nachweisbar. Diese entstehen nur dann, wenn der Organismus mit dem körperfremden Antigen sensibilisiert wurde (z.B. durch Übertragung von D-Erythrozyten in einen d-Organismus durch fehlerhafte Blutübertragung oder während der Geburt über die Wundfläche Plazenta/Uterus). Durchführung: 1.) Blutgruppenbestimmung: Dazu wird das Blut zuerst zentrifugiert. Man erhält zwei Phasen: überstehendes Plasma und die Blutzellen (u.a. Erythrozyten), die sich unten befinden. Überführen Sie das Plasma vorsichtig, ohne die Blutzellen zu berühren, mit einer Plastik- Pasteurpipette in ein sauberes Reagenzglas und beschriften Sie es mit dem Patientennamen. Die Agglutinationsreaktion wird auf vorbereiteten Glasplatten mit Hohlschliff durchgeführt. Die Serumeigenschaften werden bestimmt, indem Blut-Plasma zu Testerythrozyten (Firma Biotest; 5%-ige Aufschwemmung in 0,9 % NaCl-Lösung) mit bekannter Antigenstruktur (A1, A2, B, 0) gegeben wird. Nach leichtem durchmischen der Proben mit einem Rührstäbchen auf der Platte können wir die Agglutination nach etwa 2 min ablesen. Die erythrozytären Eigenschaften werden bestimmt, indem je ein Tropfen Erythrozyten zu den staatlich geprüften Testseren Anti-A, Anti-B, Anti-AB und Anti-D (das letzte enthält Supplement) auf der Glasplatte gegeben wird. Nach leichtem Umrühren der Proben auf der Platte können wir die Agglutination nach etwa 2 min ablesen. Während die Agglutination im ABO-System bereits nach wenigen Sekunden eine deutliche Verklumpung zeigt, kann die Agglutination im Rh-System wesentlich länger dauern und bisweilen nur von Erfahrenen erkannt werden.
Blutgruppenbestimmung
+ 1 Tropfen Erythrozyten
+ 1 Tropfen Plasma
Anti-A Anti-B Anti-AB
A1 A2 B 0
Rh
Test-Seren
Test-Erythrozyten
BLUT 292.) Kreuzprobe: sie soll alle Unverträglichkeits-Reaktionen ausschließen, auch solche außerhalb des ABO und des Rh-Systems. Sie muss, um die kompletten und die inkompletten Antikörper zu erfassen, mit Supplement-Zusatz (20%-ige Rinderalbumin-Lösung) bei 37°C durchgeführt werden. Spender-Erythrozyten und Empfänger-Serum („Major-Test“) sowie Spender-Serum und Empfänger-Erythrozyten („Minor-Test“) werden zusammengebracht. Zeigt sich eine Agglutination, die durch einige Tropfen physiologischer Kochsalzlösung nicht wieder aufgelöst wird (Geldrollen-Bildung), so bezeichnen wir die Kreuzprobe als positiv. Wichtig: Eine Transfusion darf nur bei negativer Kreuzprobe erfolgen!!!
VERSUCH: Minor- und Majortest Führen Sie eine Minor- und einen Majortest mit den von ihnen gewonnenen Blutproben durch. Dazu nehmen sie ihre eigene Blutprobe und wählen eine zweite Blutprobe aus dem Praktikum aus. Die Durchführung ist ähnlich wie zuvor unter „Blutgruppenbestimmung“ beschrieben: Sie vermengen jeweils einen Tropfen Serum mit 1 Tropfen Erythrozyten und dokumentieren das Auftreten von Agglutinationen. Würden sie mit dem erzielten Resultat eine Transfusion anordnen?





































![Erythrozyten mit Plasmodien - Home | MedUni Wien5 Milliarden Menschen (42 %) leben in Risiko-Gebieten [1945: 80 %!] > 500 Millionen sind infiziert 350 Millionen erkranken jährlich](https://static.unterlagen.site/doc/80x56/5cc15d5f88c99315158beecd/erythrozyten-mit-plasmodien-home-meduni-wien-5-milliarden-menschen-42-leben.jpg)






