

Das Proteom
Vorlesung: Gene und Genome
Proteom -1-
g
Wintersemester 2006 / 2007
Kristian Müller
Institut für Biologie III, Albert-Ludwigs-Universität Freiburg
Ziele der Vorlesung
• Interesse am Proteom wecken
• Einführung in experimentellen Methoden zur Erforschung des Proteoms
• Einführung in bioinformatische Methoden zur Analyse von Proteom Daten
• Vorstellung von bioinformatischen Analysewerkzeugen
• Wege durch den Informationsdschungel aufzeigen
structural biology
geneticssequencinginformation
Proteom -2-
molecularbiology
medicine
physiology toxicologygene
expression

Proteom, was ist das?
Proteom -3-
Vom Gen zum Proteinkomplex
Proteom -4-
Curr Opin Chem Biol 2003, 7, S. 21
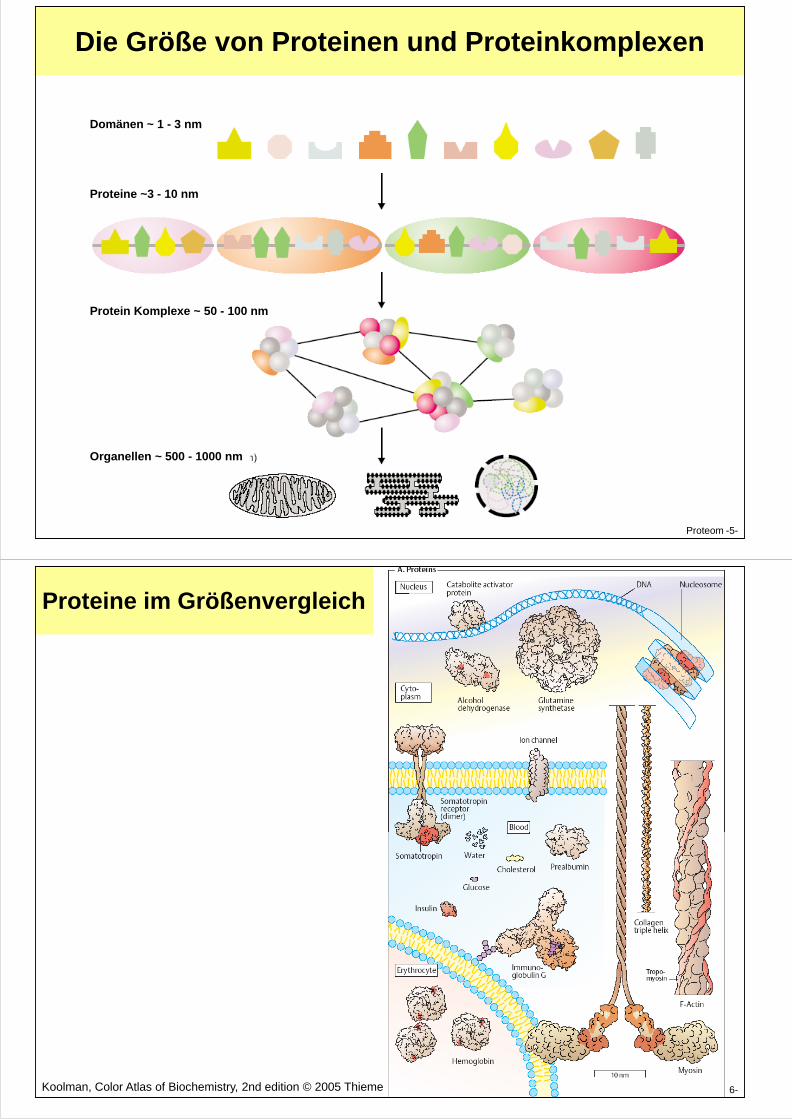
Die Größe von Proteinen und Proteinkomplexen
Domänen ~ 1 - 3 nm
Proteine ~3 - 10 nm
Proteom -5-
Protein Komplexe ~ 50 - 100 nm
Organellen ~ 500 - 1000 nm
Proteine im Größenvergleich
Proteom -6-Koolman, Color Atlas of Biochemistry, 2nd edition © 2005 Thieme

Vom Gen zum Proteinkomplex
Proteom -7-Curr Opin Cell Biol 2003 15, S. 199
Umfassende Ansichten: “…omics”
Genomics: Das Genom und die Gene eines Organismus
Proteomics: Die Proteine eines Organismus
Proteom -8-
Metabolomics: Die Stoffwechselwege eines Organismus
Functomics: Funktion und Interaktion der Proteine eines Organismus
Structural Genomics: Die räumliche Struktur der Moleküle eines Organismus
Systems Biology: Ganzheitliche Biologie der Zelle
Genom, Proteom, Phosphoproteom, Interaktom, Metabolom, ...

Experimentelle Methoden
Wie entstehen Daten zum Proteom?
• S i j kt O i
Proteom -9-
• Sequenzierprojekte von ganzen Organismen
• Viele einzelne Experimente zu den jeweiligen Proteinen
• Proteomics Experimente
Methoden des Proteomics
• 2D Gelelektrophorese (das ursprüngliche Proteomics)Auftrennung von Gesamtzelllysaten in der ersten Dimension nach demisoelektrischen Punkt (isoelektrische Fokussierung) und in der zweitenDimension nach dem Molekulargewicht. Die Proteinflecken können anhandIhres Laufverhaltens mit Datenbanken abgeglichen werden oder aus demGel zur weiteren Charakterisierung (z.B. MS) isoliert werden.
• Protein Sequenzierung (Edman Abbau)Die N-terminale Sequenz eines Proteins kann bis zu einigen Aminosäurenbestimmt werden.
Proteom -10-
• MassenspektrometrieBestimmung der Masse eines Proteins. Proteine werden häufigbiochemisch (Proteolyse) oder physikalisch (MS-MS) zerkleinert. Sokönnen auch große Proteine bestimmt werden. Die Ergebnisse können mitberechneten Massen aus Genomdatenbanken verglichen werden. Bereitsgrob vorgetrennte Zellysate können bestimmt werden.
• Bibliotheken Technologien / Repertoire TechnologienMit molekularbiologischen in vivo Testmethoden können genomorientierteProteinfunktionsanalysen vorgenommen werden. Z.B. wurden alle 6000open reading frames der Hefe mit dem Yeast-two-Hybrid System aufinteraktion getestet.

Proteom Fraktionierung und Analyse
Zellulär
Bestandteile
Biochemisch
2- 3-D Chromatographie2-D ElektrophoreseAffinitätsreinigung
Chemische
Aktivität
Proteom -11-
BestandteileOrganellen
AktivitätPost Translationale ModifikationenAminosäure Reaktivität
Physikalisch
Massenspretrometrie
Proteomics - der Ursprung:2D Gelelektrophorese
2D Gel: Hela Nucleus Extrakt
Proteom -12-
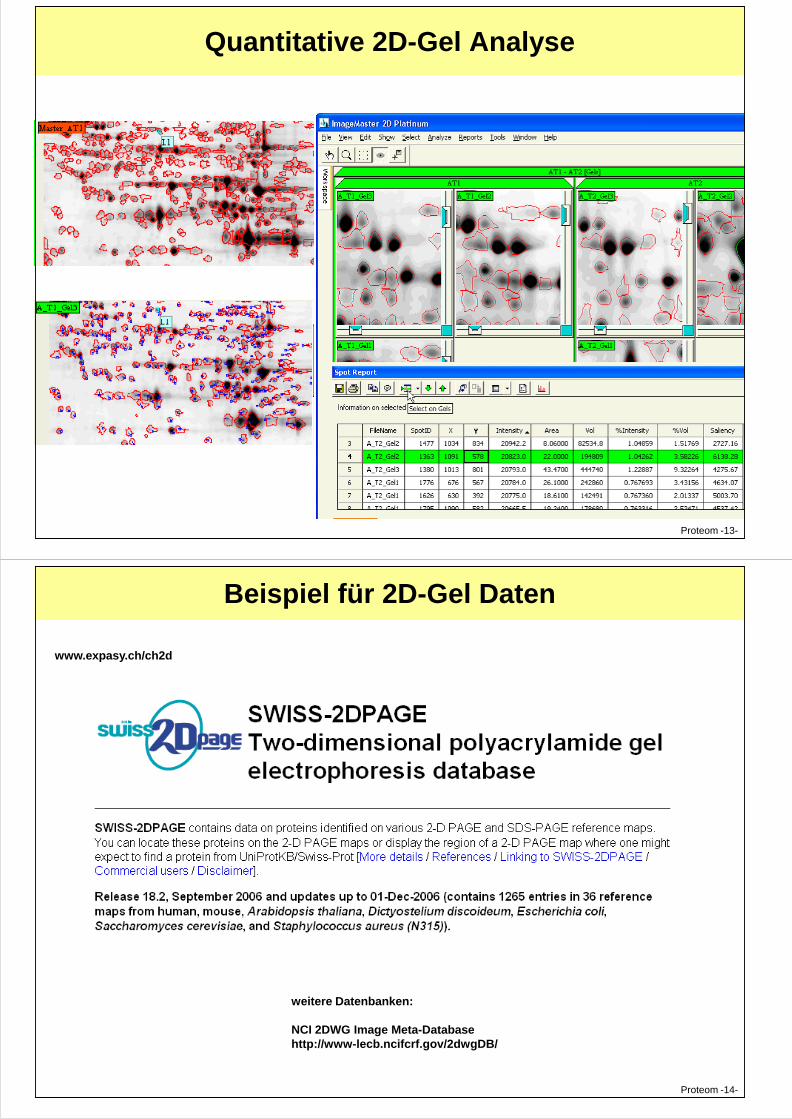
Quantitative 2D-Gel Analyse
Proteom -13-
Beispiel für 2D-Gel Daten
www.expasy.ch/ch2d
Proteom -14-
weitere Datenbanken:
NCI 2DWG Image Meta-Databasehttp://www-lecb.ncifcrf.gov/2dwgDB/
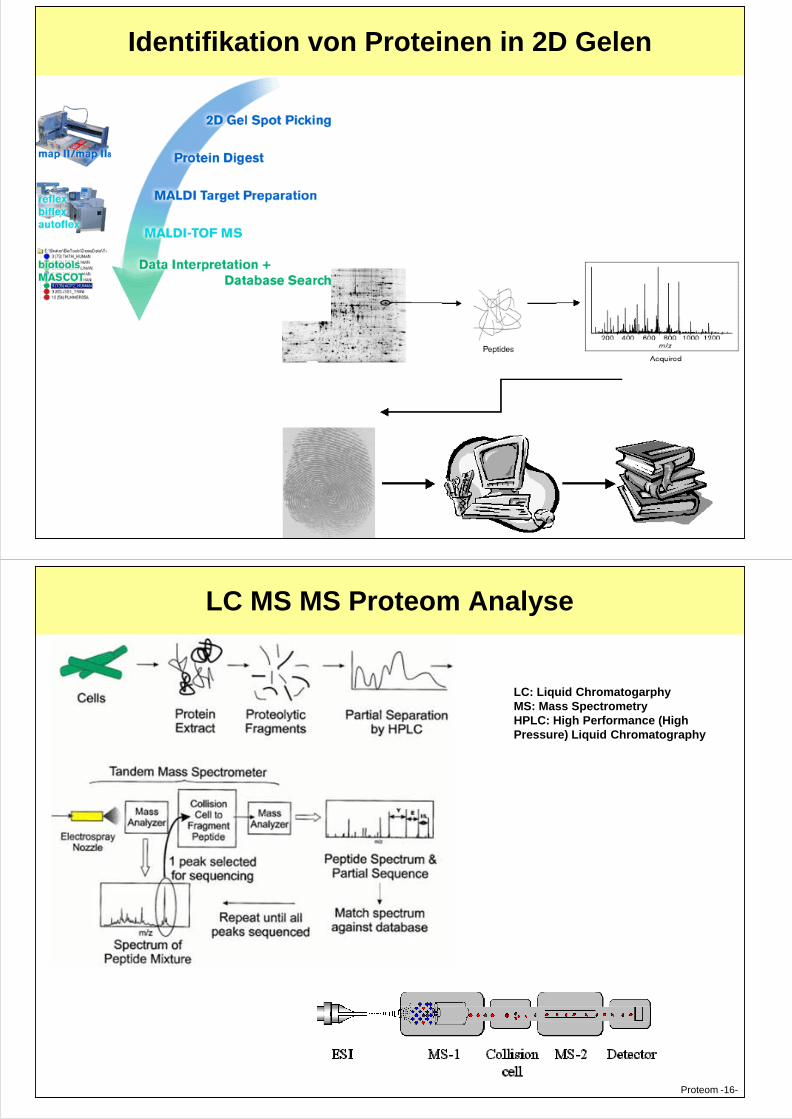
Identifikation von Proteinen in 2D Gelen
Proteom -15-
LC MS MS Proteom Analyse
LC: Liquid ChromatogarphyMS: Mass SpectrometryHPLC: High Performance (High Pressure) Liquid Chromatography
Proteom -16-

Post Translationale Modifikationen (PTM)
Proteom -17-
Nature Reviews Molecular
Cell Biology, 2006, 7, 391-403
• Nitrosylation
• Glycation
• ADP-ribosylation
• O-GlcNAc
Isotope-Coded Affinity Tag (ICAT) für MS
Proteom -18-
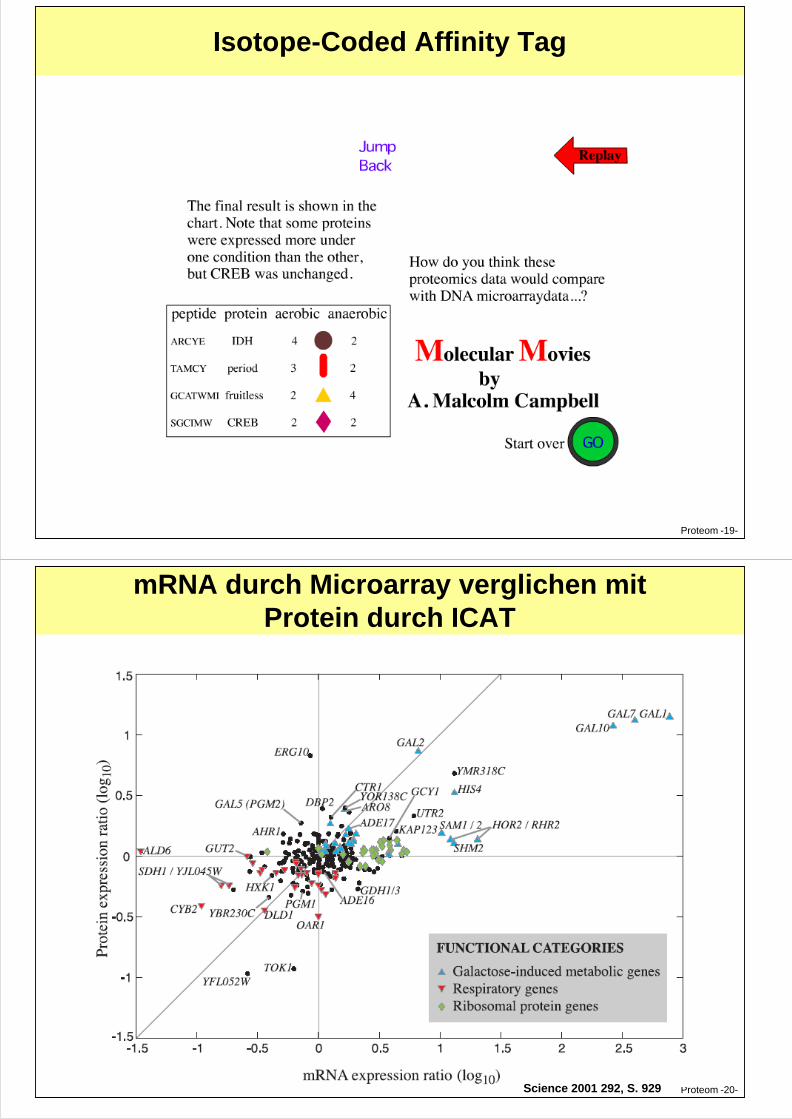
Isotope-Coded Affinity Tag
Proteom -19-
mRNA durch Microarray verglichen mitProtein durch ICAT
Proteom -20-Science 2001 292, S. 929

Proteom Daten durch Genomsequenzierung
Proteom -21-
Wie erhalte und verarbeite ich Daten eines Proteoms ?
Datenbanken auf dem Internet
Primärdatenbanken
• DNA-Sequenzdatenbanken z.B. EMBL, GenBank (weitestgehend automatisierte Datenbanken, Eingabe durch Wissenschaftler und überwiegend Sequenzierprojekte)
• Proteindatenbanken z.B. TrEMBL, Swissprot (automatische und manuelle Eingabe durch C t )
Proteom -22-
Curatoren)
• Strukturdatenbanken z.B. PDB (Eingabe durch Wissenschaftler)
Abgeleitete Datenbanken
• Sequenzorientierte Datenbanken
– z.B. Interpro (Metadatenbank für Proteome)
– z.B. CluSTr, Pfam, Prosite, ProDom ...
• Strukturorientiere Datenbanken
– z.B. SCOP, CATH
Literaturdatenbanken z.B. PubMed
European Bioinformatics Institute (EBI)www.ebi.ac.uk
Proteom -23-
NCBI
http://www.ncbi.nlm.nih.gov/
Proteom -24-
There are approximately 65,369,091,950 bases in 61,132,599 sequence records in the traditional GenBank divisions and 80,369,977,826 bases in 17,960,667 sequence records in the WGS division as of August 2006.

Glycolyse Vergleich mit BioCyc
Escherichia coli
Proteom -25-
Picrophilus Torridus(lebt bei pH 0, 65°C)
Natronomonas pharaonis(liebt 3.5 M NaCl , pH 8.5, kann auch bei pH 11 leben)
Beispiel für Proteom Datenbank: Integr8
Integr8
• is a browser for information relating to completed genomes and proteomes,
• based on data contained in Genome Reviews and the UniProt proteome sets
• provides access to species descriptions, literature, statistical analysis and summary information about each complete proteome
• integrates data from a variety of sources, including InterPro, CluSTr and GO.
Proteom -26-
• integrates data from a variety of sources, including InterPro, CluSTr and GO.
Integr8 Database 2007Integr8 Database 2005
http://www.ebi.ac.uk/integr8/
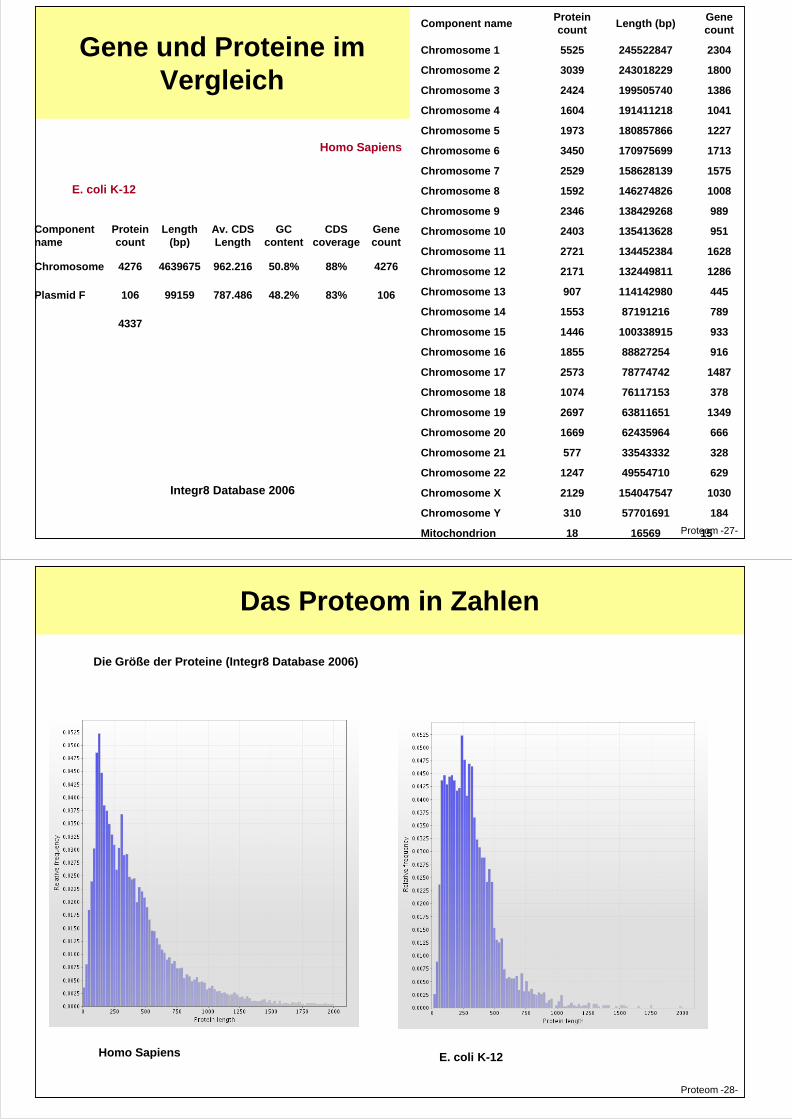
Gene und Proteine im Vergleich
Component name Protein count
Length (bp) Gene count
Chromosome 1 5525 245522847 2304
Chromosome 2 3039 243018229 1800
Chromosome 3 2424 199505740 1386
Chromosome 4 1604 191411218 1041
Chromosome 5 1973 180857866 1227
Chromosome 6 3450 170975699 1713
Chromosome 7 2529 158628139 1575
Chromosome 8 1592 146274826 1008
Chromosome 9 2346 138429268 989
Chromosome 10 2403 135413628 951
Chromosome 11 2721 134452384 1628
Chromosome 12 2171 132449811 1286
Homo Sapiens
Component name
Protein count
Length (bp)
Av. CDS Length
GC content
CDS coverage
Gene count
Chromosome 4276 4639675 962.216 50.8% 88% 4276
E. coli K-12
Proteom -27-
Chromosome 12 2171 132449811 1286
Chromosome 13 907 114142980 445
Chromosome 14 1553 87191216 789
Chromosome 15 1446 100338915 933
Chromosome 16 1855 88827254 916
Chromosome 17 2573 78774742 1487
Chromosome 18 1074 76117153 378
Chromosome 19 2697 63811651 1349
Chromosome 20 1669 62435964 666
Chromosome 21 577 33543332 328
Chromosome 22 1247 49554710 629
Chromosome X 2129 154047547 1030
Chromosome Y 310 57701691 184
Mitochondrion 18 16569 15
Plasmid F 106 99159 787.486 48.2% 83% 106
4337
Integr8 Database 2006
Das Proteom in Zahlen
Die Größe der Proteine (Integr8 Database 2006)
Proteom -28-
Homo Sapiens E. coli K-12

Die häufigsten Proteine
InterPro top 30 entries for H. sapiens
InterProProteins matched(Proteome coverage)
RankName
IPR007110 1191 (3.1%) 1 Immunoglobulin-like
IPR007087 1057 (2.8%) 2 Zinc finger, C2H2-type
IPR003599 987 (2.6%) 3 Immunoglobulin subtype
IPR011009 902 (2.4%) 4 Protein kinase-like
IPR000276 896 (2.4%) 5 Rhodopsin-like GPCR superfamily
Proteom -29-
IPR000719 808 (2.1%) 6 Protein kinase
IPR013106 634 (1.7%) 7 Immunoglobulin V-set
IPR011993 596 (1.6%) 8 Pleckstrin homology-type
IPR000725 510 (1.3%) 9 Olfactory receptor
IPR007086 491 (1.3%) 10 Zinc finger, C2H2-subtype
IPR001909 467 (1.2%) 11 KRAB box
IPR011992 440 (1.2%) 12 EF-Hand type
IPR008271 430 (1.1%) 13 Serine/threonine protein kinase, active site
IPR012677 420 (1.1%) 14 Nucleotide-binding, alpha-beta plait
IPR001680 417 (1.1%) 15 WD-40 repeat
IPR13032 336 (0.9%) 29 EGF-like region
Interaktomdas Yeast-Two-Hybrid System
zur Interaktionsanalyse
Köder (Bait)
Binding Domain (BD)
Proteom -30-
Nature 1989 340, S. 245PNAS 1991 88, S. 9578
Beute (Prey) Activation Domain (AD)
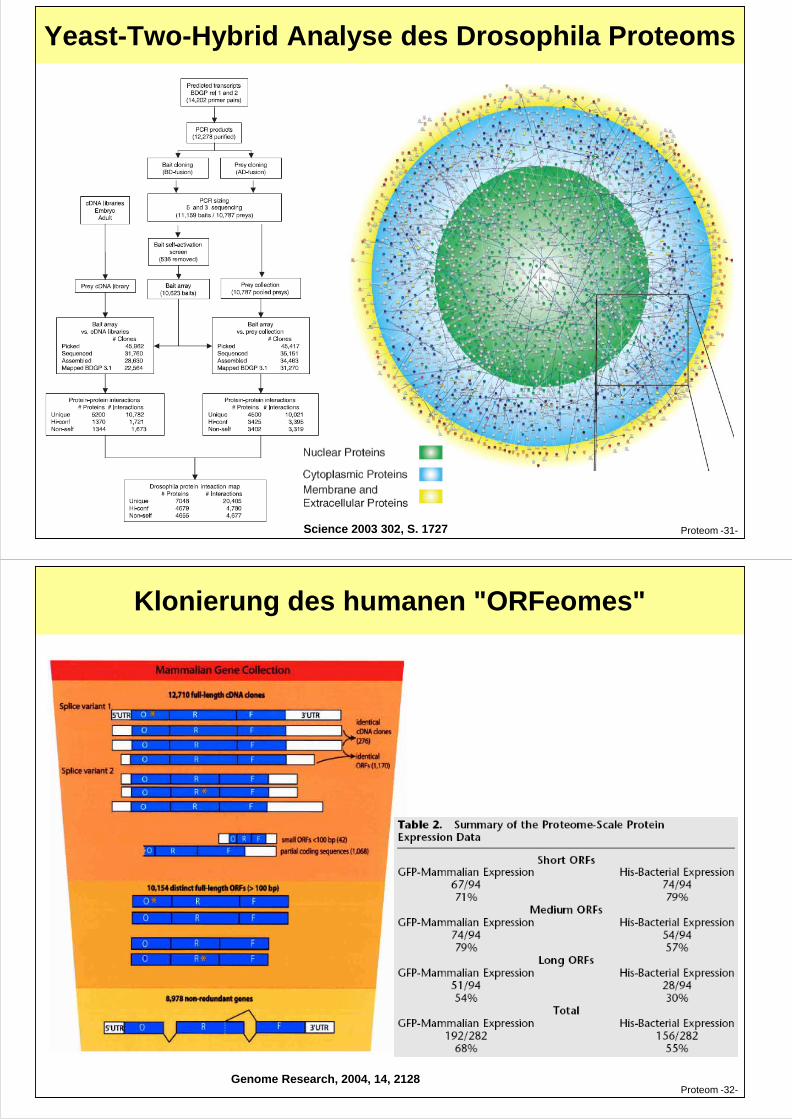
Yeast-Two-Hybrid Analyse des Drosophila Proteoms
Proteom -31-Science 2003 302, S. 1727
Klonierung des humanen "ORFeomes"
Proteom -32-Genome Research, 2004, 14, 2128

Yeast two hybridAnalyse des humanen ORFeomes
Selektionstrategie Ausgewählte Interaktionen
Proteom -33-Nature 2005, 437, 1173
Verknüpfung biochemischer Daten
Proteom -34-Curr Opin Chem Biol 2003 7, S. 44
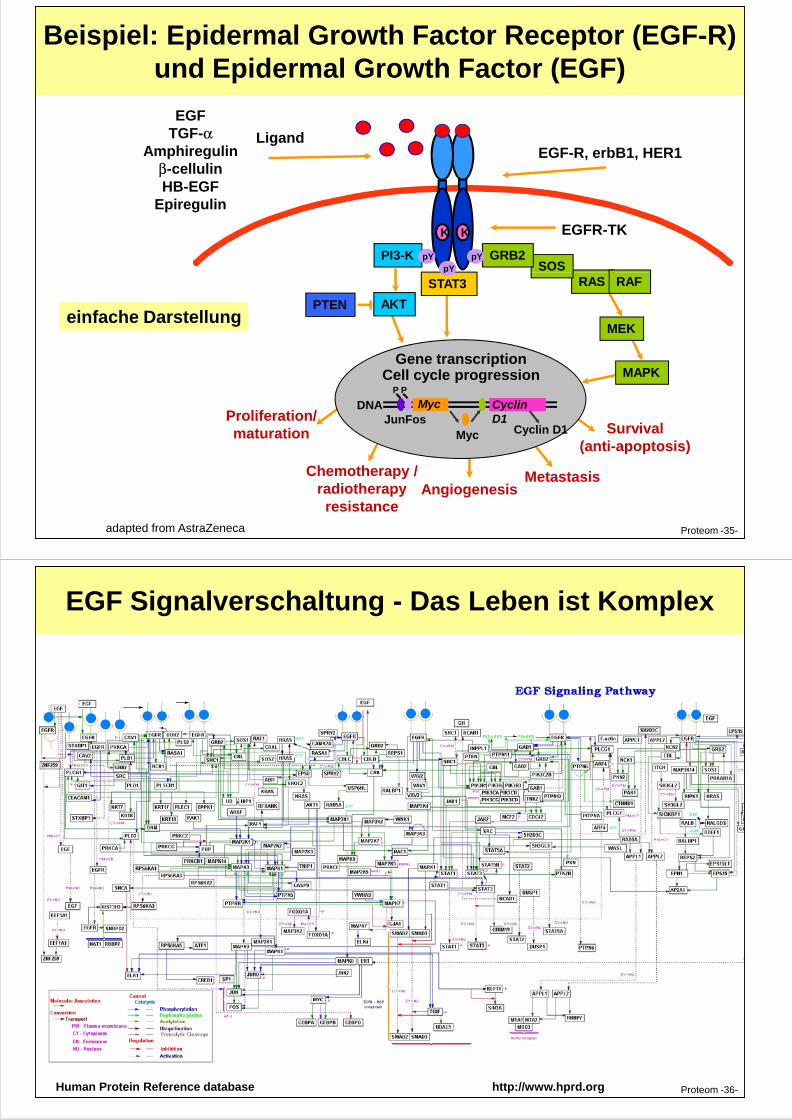
Beispiel: Epidermal Growth Factor Receptor (EGF-R) und Epidermal Growth Factor (EGF)
PI3-K
EGF-R, erbB1, HER1Ligand
EGFR-TKKK
pYpY SOS
GRB2pY
EGFTGF-α
Amphiregulinβ-cellulinHB-EGF
Epiregulin
Proteom -35-
Survival(anti-apoptosis)
PTEN AKT
STAT3
MEK
Gene transcriptionCell cycle progression
DNA Myc
Myc Cyclin D1JunFos
P P
MAPK
Proliferation/maturation
Chemotherapy /radiotherapyresistance
AngiogenesisMetastasis
Cyclin D1
p
RAS RAF
adapted from AstraZeneca
einfache Darstellung
EGF Signalverschaltung - Das Leben ist Komplex
Proteom -36-http://www.hprd.orgHuman Protein Reference database

Suche nach einem bekannten Protein
Proteom -37-
Die Suche nach einem bestimmten Gen / Protein:Sequence Retrieval System (SRS) Oberfläche
http://srs.ebi.ac.uk/
Proteom -38-

Auswahl an Datenbanken im SRS
Proteom -39-
Vernetzung der Protein-Datenbanken
Proteom -40-Verschachtelung der Datenbanken: z.B. PDB - SCOP – Superfamily – Interpro; UniProt – InterPro – UniProt
InterPro 2006

Proteine finden: UniProt
Proteom -41-
Ergebnis der Suche nach “EGF”
Treffer Liste
UniProt Eintrag EGF_Human
Proteom -42-
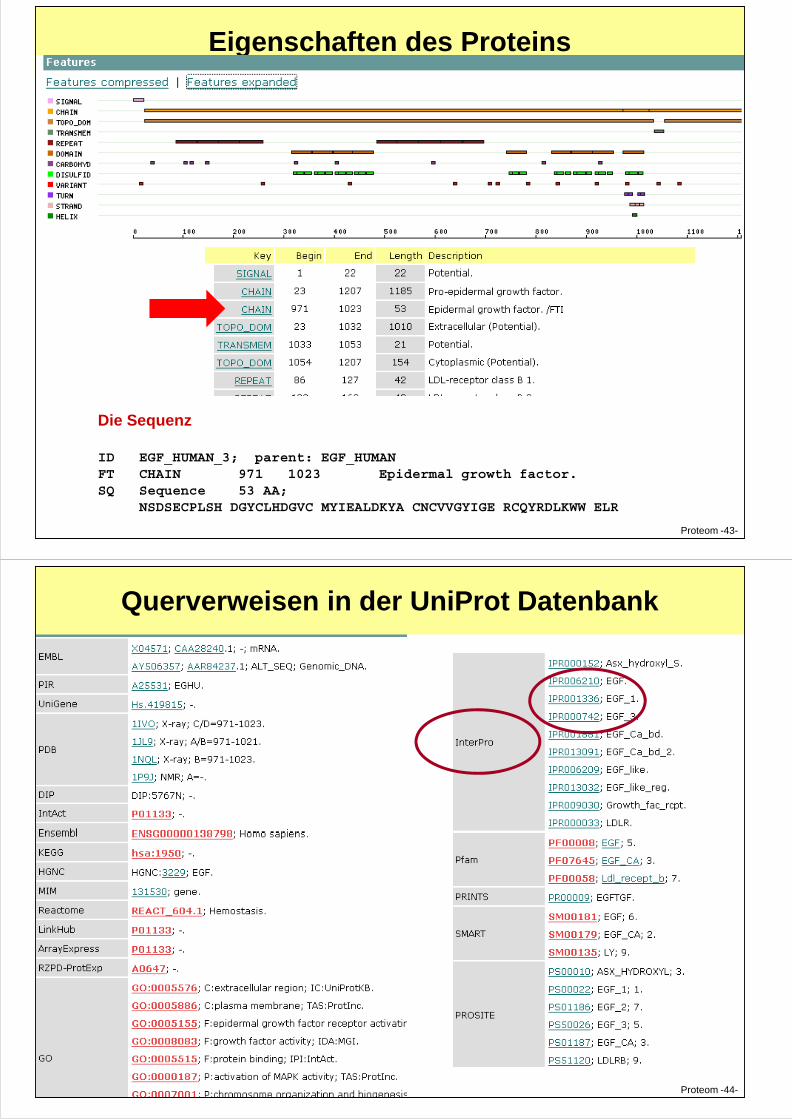
Eigenschaften des Proteins
Proteom -43-
Die Sequenz
ID EGF_HUMAN_3; parent: EGF_HUMANFT CHAIN 971 1023 Epidermal growth factor.SQ Sequence 53 AA;
NSDSECPLSH DGYCLHDGVC MYIEALDKYA CNCVVGYIGE RCQYRDLKWW ELR
Querverweisen in der UniProt Datenbank
Proteom -44-

InterPro Eintrag für EGF Typ 1
Proteom -45-
InterPro Datenbank: EGF Vergleich in verschiedenen Organismen
Proteom -46-

Wo ist der Unterschied?type I EGF, EGF subtype II, EGF like
• Der Abstand der Cysteine variiert.• Die Aminosäure Komposition variiert.• Es gibt verschiedene Ausgangs-Sequenzabgleiche.
• EGF
Proteom -47-
•+-------------------+ +------+| | | |
C-x(7)-C-x(4-5)-C-x(10-13)-C-x-C-x(8)-Cx| | +---------------+
• Ca-binding EGF+------------------+ +---------+| | | |
nxnnC-x(3-14)-C-x(3-7)-CxxbxxxxaxC-x(1-6)-C-x(8-13)-Cx| | +------------------+
Pfam
Proteom -48-http://www.sanger.ac.uk/Software/Pfam/

LTB1_RAT/1462-1497 CQDPN...SCID...GQCVNTEG........SYNCFCTHP.MVLDASEKRCLTB1_RAT/185-212 CVP.....PCQNG..GMCLRPQ...........FCVCKPG.....TKGKACLTBS_HUMAN/77-104 CHL.....PCMNG..GQCSSRD...........KCQCPPN.....FTGKLCO01768/1993-2029 CHQL....ECKNG..GYCLKPDPS...ANRTSPTCLCSPG.....FKGLLCCRB_DROME/1919-1950 CEIT....PCDNG..GLCLTTG........AVPMCKCSLG.....YTGRLCCRB_DROME/1763-1794 CFQS....DCKND..GFCQSPSD........EYACTCQPG.....FEGDDCCRB_DROME/984-1020 CDQN....PCLNG..GACLPYLINE...VTHLYTCTCENG.....FQGDKCCRB_DROME/310-342 CAKN....PCENG..GSCLENSE.......GNYQCFCDPN.....HSGQHCNOTC_XENLA/260-291 CPSN....NCRNG..GTCVDGVN........TYNCQCPPD.....WTGQYCURT2_DESRO/87-120 CSEL....RCFNG..GTCWQAAS......FSDFVCQCPKG.....YTGKQCTENA_CHICK/187-218 CSEPACPRNCLNR..GLCVRG............KCICEEG.....FTGEDCFA12_HUMAN/98-130 CSKHS...PCQKG..GTCVNMP........SGPHCLCPQH.....LTGNHCO01768/1799 1833 CVGH ECLNG GFCSYANS NRSLPHCICPSG FTGDHC
Pfam: EGF Motiv Sequenzabgleich
seed 73representative 1025full 5323
Proteom -49-
O01768/1799-1833 CVGH....ECLNG..GFCSYANS.....NRSLPHCICPSG.....FTGDHCNOTC_XENLA/22-56 CTQTA..EMCLNG..GRCEMTPG.......GTGVCLCGNL.....YFGERCO01768/1878-1912 CQKQP..NWCHNG..GRCLDTPGY.......PGKCKCLPR.....FAGPRCCRB_DROME/2034-2069 CNATD...LCLNG..GRCVESCG.....AKPDYYCECPEG.....FAGKNCCRB_DROME/549-580 CNATN..GKCLNG..GTCSMNG..........THCYCAVG.....YSGDRCNOTC_DROME/100-135 CNSM....RCQNG..GTCQVTFRNG....RPGISCKCPLG.....FDESLCPGH2_HUMAN/22-54 CSH.....PCQNR..GVCMSVG.......FDQYKCDCTRT....GFYGENCPGH1_HUMAN/36-68 CYY.....PCQHQ..GICVRFG.......LDRYQCDCTRT....GYSGPNCDL_DROME/378-415 CSDK....PCHQ...GICRNVRPGLGS.KGQGYQCECPIG.....YSGPNCSLIT_DROME/1052-1085 CSPEF..NPCANG..AKCMDHFT........HYSCDCQAG.....FHGTNCSLIT_DROME/935-967 CFEQ....PCQNQ..AQCVALPQ.......REYQCLCQPG.....YHGKHCMFGM_MOUSE/68-107 CSPN....PCYND..AKCLVTLDTQRGDIFTEYICQCPVG.....YSGIHCO01768/1955-1985 CSE......CSNE..AKCIKKPS.......GTVICQCPQG.....LGGEYCGRFA_VARV/45-80 CGPEG.DRYCFH...GICIHARD......IDGMYCRCSHG.....YTGIRCAGRI_CHICK/1489-1521 CDPT....PCHIS..ATCLVLPE.......GGAMCACPMG.....REGEFCMEPA_MOUSE/675-710 CDPN....PCQNE..GTCVNVKG........MASCRCVSG.HAFFYAGERCAGRI_CHICK/1450-1482 CHPN....PCHHG..ASCHVKEA.......EMFHCECLHS.....YTGPTCFA12_HUMAN/178-209 CRTN....PCLHG..GRCLEVE........GHRLCHCPVG.....YTGPFCAGRI_CHICK/1233-1264 CDSH....PCLHG..GTCEDDG........REFTCRCPAG.....KGGAVCDL_DROME/260-288 CVLEP...NCIH...GTCNKP...........WTCICNEG.....WGGLYCPRTS_BOVIN/121-154 CNPL....PCNEDGFMTCKDGQ........ATFTCICKSG.....WQGEKCAMPR_HUMAN/146-181 CNAEF.QNFCIH...GECKYIEH......LEAVTCKCQQE.....YFGERCEGF_HUMAN/976-1012 CPLSH.DGYCLHD..GVCMYIEALD......KYACNCVVG.....YIGERC
Suche nach dem Gen zum Protein
Proteom -50-
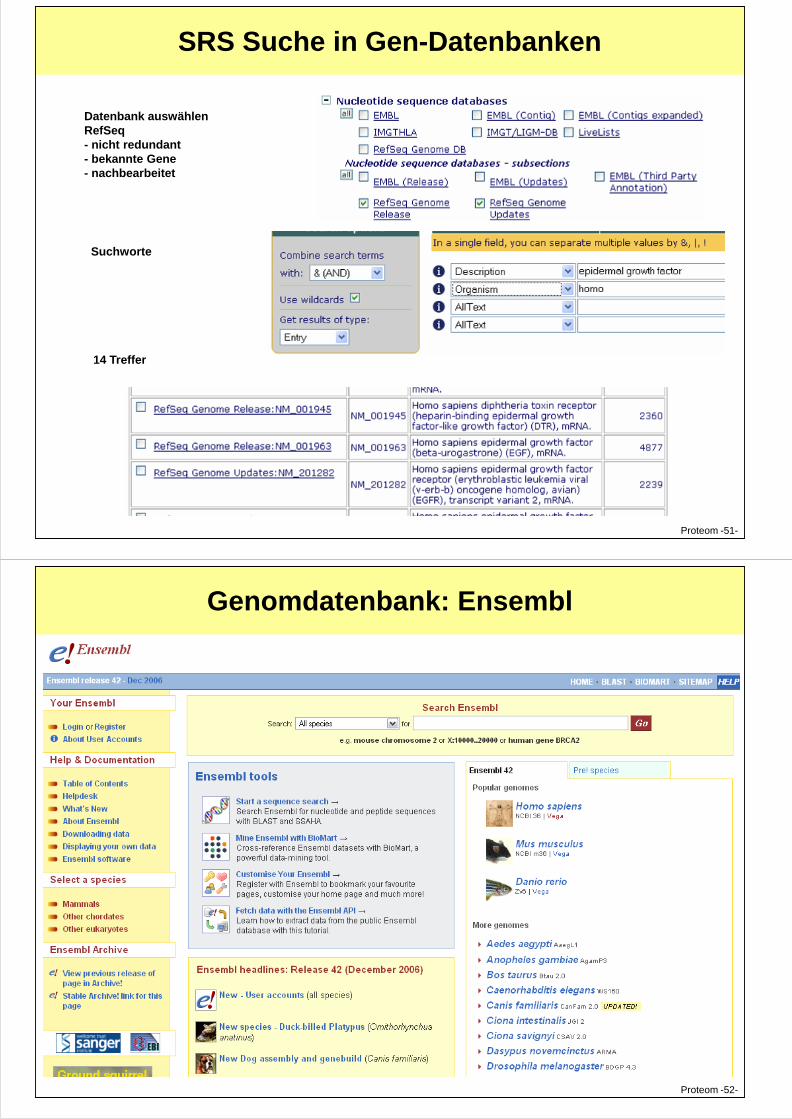
SRS Suche in Gen-Datenbanken
Datenbank auswählenRefSeq - nicht redundant- bekannte Gene- nachbearbeitet
Suchworte
Proteom -51-
14 Treffer
Genomdatenbank: Ensembl
Proteom -52-
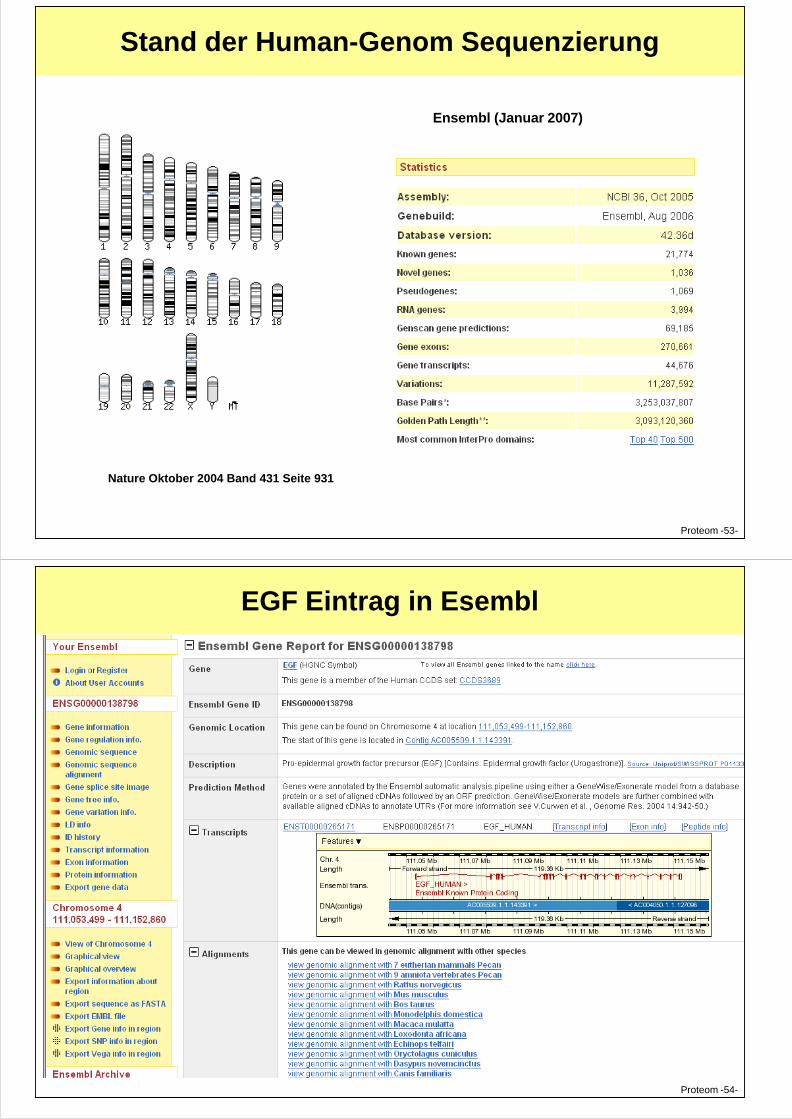
Stand der Human-Genom Sequenzierung
Ensembl (Januar 2007)
Proteom -53-
Nature Oktober 2004 Band 431 Seite 931
EGF Eintrag in Esembl
Proteom -54-

Proteom -55-
Ensembl vom Chromosom zum
Protein
NCBI Map viewer - EGF
Proteom -56-
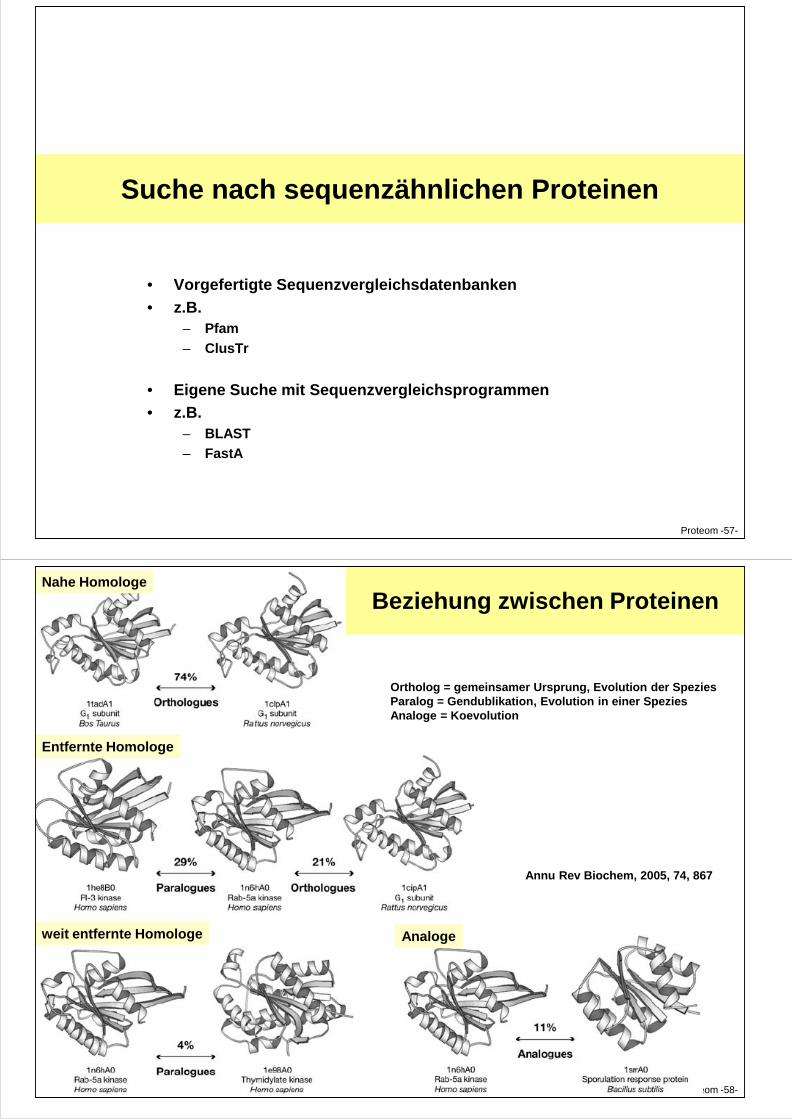
Suche nach sequenzähnlichen Proteinen
Proteom -57-
• Vorgefertigte Sequenzvergleichsdatenbanken
• z.B.– Pfam
– ClusTr
• Eigene Suche mit Sequenzvergleichsprogrammen
• z.B.– BLAST
– FastA
Beziehung zwischen Proteinen
Ortholog = gemeinsamer Ursprung, Evolution der SpeziesParalog = Gendublikation, Evolution in einer SpeziesAnaloge = Koevolution
Nahe Homologe
Entfernte Homologe
Proteom -58-
weit entfernte Homologe Analoge
Annu Rev Biochem, 2005, 74, 867

Alignment, Identity, Homology, Similarity
• Alignment: The process of lining up two or more sequences to achieve maximal levels of identity (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology.
• Conservation: Changes at a specific position of an amino acid (or less commonly, DNA) sequence that preserve the physico-chemical properties of the original residue
Proteom -59-
the original residue.
• Homology: Similarity attributed to descent from a common ancestor.
• Identity: The extent to which two (nucleotide or amino acid) sequences are invariant.
• Similarity: The extent to which nucleotide or protein sequences are related. The extent of similarity between two sequences can be based on percent sequence identity and/or conservation. In BLAST similarity refers to a positive matrix score.
Die Suchprinzipien in Sequenzdatenbanken –wie finde ich eine ähnliche Sequenz (DNA/Protein)?
logische Algorithmen
Smith-Waterman Algorithmusmathematisch vollständige Suchel iefert exakt nur die gegebenen Parameterlangsam
heuristische Algorithmen
BLAST ("Basic Local Alignment and Search Tool")neuere Varianten BLASTP für Proteine in Protein Datenbanken
BLASTX für DNA in Protein DatenbankenBLASTN für DNA in DNA Datenbanken
Proteom -60-
BLASTN für DNA in DNA Datenbanken
PSI BLAST ("Position Specific Iterated BLAST)Weiterentwicklung von BLAST, erlaubt Lücken,erstellt intermediäres Profil (eine Art Konsensus-Sequenz)
FASTAPrinzip Punkt-Grafikschnell (ca. 10 bis 100 mal schneller als exakte Suche)beste Übereinstimmung wird durch Parameter festgelegt
Profil gestützte Algorithmen
Hidden Markov Modelle (HMM)aus paarweisen Sequenzabgleichen wird ein Profil erstellt mit Positions-spezifischer Informationmeist Suche mit bereits vorhandenen HMMErstellung eigener HMM möglich
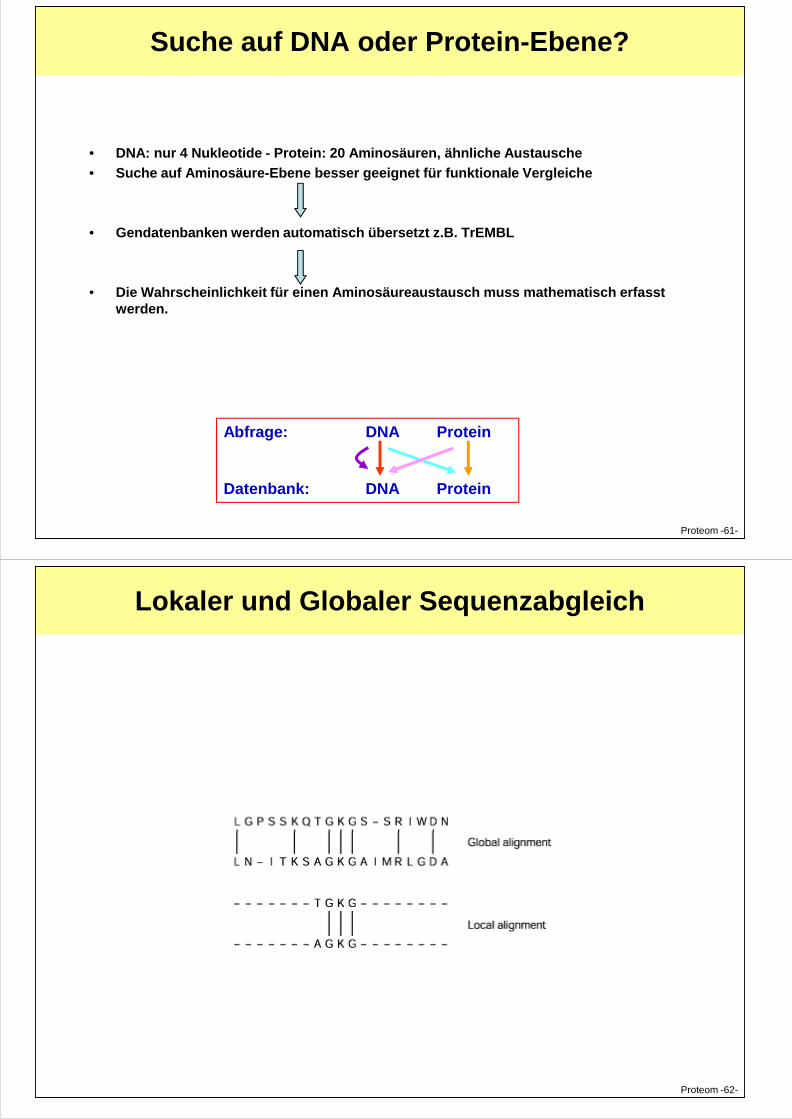
Suche auf DNA oder Protein-Ebene?
• DNA: nur 4 Nukleotide - Protein: 20 Aminosäuren, ähnliche Austausche
• Suche auf Aminosäure-Ebene besser geeignet für funktionale Vergleiche
• Gendatenbanken werden automatisch übersetzt z.B. TrEMBL
Proteom -61-
• Die Wahrscheinlichkeit für einen Aminosäureaustausch muss mathematisch erfasst werden.
Abfrage: DNA Protein
Datenbank: DNA Protein
Lokaler und Globaler Sequenzabgleich
Proteom -62-
NCBI BLAST
Proteom -63-Altschul et al., J Mol Biol 1990 215:403-10
BLAST auf der EBI Webseite
Proteom -64-
http://www.ebi.ac.uk/blast2/

BLAST Ausgabe
Score ESequences producing significant alignments: (bits) Value
gi|208343|gb|AAA72563.1| epidermal growth factor >gi|208520... 186 1e-46 gi|46242544|gb|AAS83395.1| epidermal growth factor [Homo sa... 186 1e-46 gi|4503491|ref|NP_001954.1| epidermal growth factor (beta-u... 186 1e-46 gi|55623060|ref|XP_517395.1| PREDICTED: epidermal growth fa... 186 1e-46 gi|11225524|gb|AAG33031.1| PR-S/epidermal growth factor fus... 186 1e-46 gi|207908|gb|AAA72814.1| epidermal growth factor 186 1e-46 gi|40288425|gb|AAR84237.1| truncated epidermal growth facto... 186 1e-46 gi|16975129|pdb|1JL9|B Chain B, Crystal Structure Of Human ... 180 8e-45 gi|37927454|pdb|1P9J|A Chain A, Solution Structure And Dyna... 169 2e-41 gi|938287|emb|CAA42102.1| epidermal growth factor [Sus scro... 145 3e-34
Proteom -65-
...insgesamt 117 Treffer
Ein Treffer als Beispielgi|47522862|ref|NP_999185.1| epidermal growth factor [Sus scrofa]
gi|13324649|gb|AAK18830.1| epidermal growth factor precursor [Sus scrofa]
gi|55977733|sp|Q00968|EGF_PIG Pro-epidermal growth factor precursor (EGF)
Length = 1214
Score = 145 bits (335), Expect = 3e-34
Identities = 45/53 (84%), Positives = 48/53 (90%)
Query: 1 NSDSECPLSHDGYCLHDGVCMYIEALDKYACNCVVGYIGERCQYRDLKWWELR 53
NS SECP SHDGYCLH GVCMYIEA+D YACNCV GY+GERCQ+RDLKWWELR
Sbjct: 970 NSYSECPPSHDGYCLHGGVCMYIEAVDSYACNCVFGYVGERCQHRDLKWWELR 1022
Der E-Wert (expectation value)
• Der Treffer einer Suchanfrage ist gut, wenn die Wahrscheinlichkeit ihn zufällig zu finden gering ist. Diese Eigenschaft wird durch den E-Wert beschrieben.
• Der E-Wert hängt von dem Suchalgorithmus und der Größe der Datenbank ab.
• Je kleiner der E-Wert desto besser.
Proteom -66-
• Durch Probieren lassen sich für eine Abfrage sinnvolle E-Werte ermitteln.
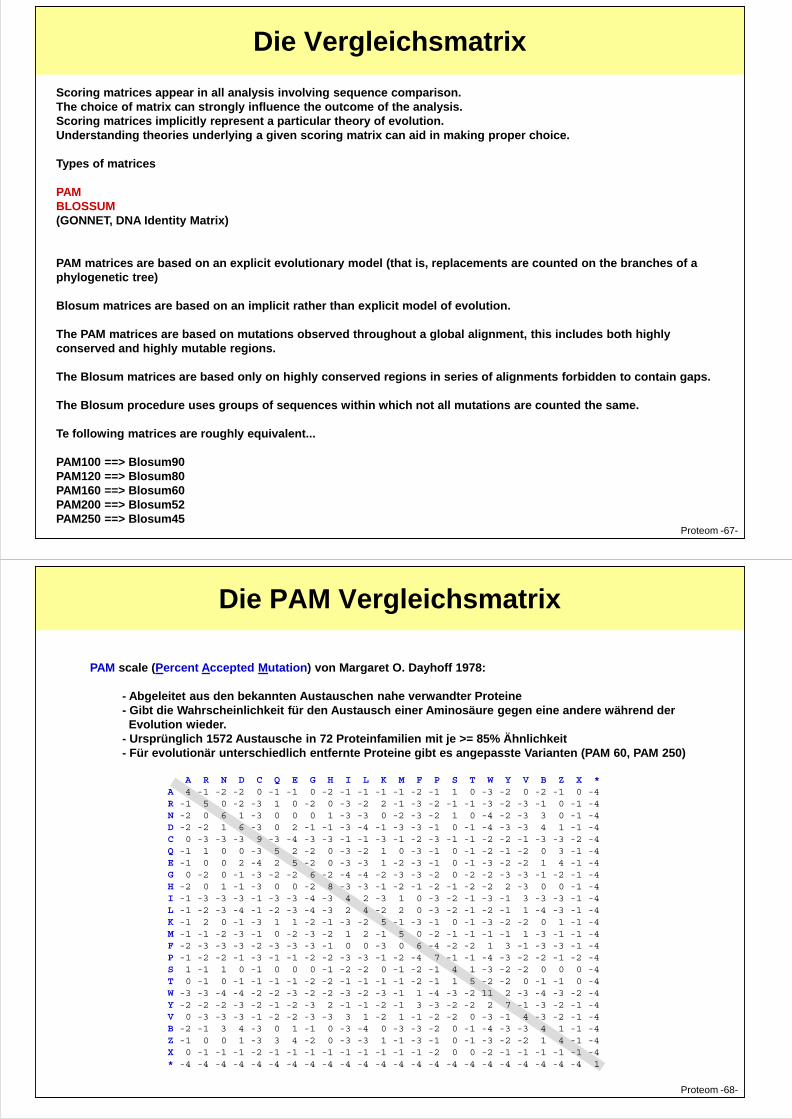
Die Vergleichsmatrix
Scoring matrices appear in all analysis involving sequence comparison. The choice of matrix can strongly influence the outcome of the analysis. Scoring matrices implicitly represent a particular theory of evolution. Understanding theories underlying a given scoring matrix can aid in making proper choice.
Types of matrices
PAM BLOSSUM (GONNET, DNA Identity Matrix)
PAM matrices are based on an explicit evolutionary model (that is, replacements are counted on the branches of a h l i )
Proteom -67-
phylogenetic tree)
Blosum matrices are based on an implicit rather than explicit model of evolution.
The PAM matrices are based on mutations observed throughout a global alignment, this includes both highly conserved and highly mutable regions.
The Blosum matrices are based only on highly conserved regions in series of alignments forbidden to contain gaps.
The Blosum procedure uses groups of sequences within which not all mutations are counted the same.
Te following matrices are roughly equivalent...
PAM100 ==> Blosum90 PAM120 ==> Blosum80 PAM160 ==> Blosum60 PAM200 ==> Blosum52 PAM250 ==> Blosum45
A R N D C Q E G H I L K M F P S T W Y V B Z X *A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4
Die PAM Vergleichsmatrix
PAM scale (Percent Accepted Mutation) von Margaret O. Dayhoff 1978:
- Abgeleitet aus den bekannten Austauschen nahe verwandter Proteine - Gibt die Wahrscheinlichkeit für den Austausch einer Aminosäure gegen eine andere während derEvolution wieder.
- Ursprünglich 1572 Austausche in 72 Proteinfamilien mit je >= 85% Ähnlichkeit- Für evolutionär unterschiedlich entfernte Proteine gibt es angepasste Varianten (PAM 60, PAM 250)
Proteom -68-
C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4* -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1

Die BLOSUM Vergleichsmatrix
BLOSUM (BLOcks Amino Acid SUbstitution Matrices) von Henikoff und Henikoff 1992
• Basiert auf den Austauschen in 2000 konservierten Aminosäuremustern aus 500 Proteinfamilien
• Je nach Ähnlichkeit gibt es angepasste Matrices z.B. Blosum60 für 60% identische Proteine.
• Die Werte der Matrix sind Logarithmen zur Basis 2 der Wahrscheinlichkeitsverhältnisse aus der Wahrscheinlichkeit einer richtigen Übereinstimmung geteilt durch die Wahrscheinlichkeit einer zufälligen Übereinstimmung, die mit dem Faktor zwei multipliziert wurden .
Beispiel: Sequenz A: Val Cys Asp AlaSequenz B: Val Met Glu Gly
Proteom -69-
C 9S -1 4T -1 1 4P -3 -1 1 7 BLOSUM62 Substitution MatrixA 0 1 -1 -1 4G -3 0 1 -2 0 6N -3 1 0 -2 -2 0 6D -3 0 1 -1 -2 -1 1 6E -4 0 0 -1 -1 -2 0 2 5Q -3 0 0 -1 -1 -2 0 0 2 5H -3 -1 0 -2 -2 -2 1 1 0 0 8R -3 -1 -1 -2 -1 -2 0 -2 0 1 0 5K -3 0 0 -1 -1 -2 0 -1 1 1 -1 2 5M -1 -1 -1 -2 -1 -3 -2 -3 -2 0 -2 -1 -1 5I -1 -2 -2 -3 -1 -4 -3 -3 -3 -3 -3 -3 -3 1 4L -1 -2 -2 -3 -1 -4 -3 -4 -3 -2 -3 -2 -2 2 2 4V -1 -2 -2 -2 0 -3 -3 -3 -2 -2 -3 -3 -2 1 3 1 4F -2 -2 -2 -4 -2 -3 -3 -3 -3 -3 -1 -3 -3 0 0 0 -1 6Y -2 -2 -2 -3 -2 -3 -2 -3 -2 -1 2 -2 -2 -1 -1 -1 -1 3 7W -2 -3 -3 -4 -3 -2 -4 -4 -3 -2 -2 -3 -3 -1 -3 -2 -3 1 2 11
C S T P A G N D E Q H R K M I L V F Y W
BLOSUM62 matrix value: 4 -1 2 0
Total score for alignment: 4 - 1 + 2 + 0 = 5
d.h. die Übereinstimmung ist 2(5/2) = 5,7 mal wahrscheinlicher als eine zufällige Übereinstimmung
FastA – Prinzip des Algorithmus
Sequence A
1. Ein Suchalgorithmus findet Identitäten größer einer minimalen Länge.
Sequence A
2. Eine Punktzahl wird berechnet und die 10 Besten erhalten
Proteom -70-
Sequence A
3. Übereinstimmungen, die nicht zum Treffer gehören, werden eliminiert.
4. Durch dynamische Programmierung wird der Abgleich optimiert.
Sequence A
W. R. Pearson and D. J. Lipman (1988) PNAS 85:2444- 2448, W. R. Pearson (1990) Methods in Enzymology 183:63- 98).

Sequenzabgleich Darstellung
Proteom -71-
Sequenzabgleich mit ClustalW: EGF
Proteom -72-

Suche nach der Struktur von einem Protein
Proteom -73-
Protein Data Bank PDB
www.pdb.org
Proteom -74-

Klassifizierung von Proteinenz.B. CATH
Proteom -75-
http://cathwww.biochem.ucl.ac.uk/
Klassifizierung von Proteinenz.B. SCOP
Proteom -76-
http://scop.mrc-lmb.cam.ac.uk/scop/

Vector Alignment Search Tool VAST
Protein structure neighbors in Entrez are determined by direct
http://www.ncbi.nlm.nih.gov/Structure/VAST/vast.shtml
Proteom -77-
NCBI's structure database is called MMDB (Molecular Modeling DataBase), and it is a subset of three-dimensional structures obtained from the Protein Data Bank (PDB), excluding theoretical models.
Protein structure neighbors in Entrez are determined by direct comparison of 3-dimensional protein structures with the VAST algorithm. Each of the more than 87,804 domains in MMDB is compared to every other one. From the MMDB Structure summary pages, retrieved via Entrez, structure neighbors are available for protein chains and individual structural domains. If you already know a PDB/MMDB-Id you can try this at once
VAST 3D Strukturabgleich1IVO:C (=EGF) ausgewähle Ergebnisse RMSD <=1A
10 20 30 40....*....|....*....|....*....|....*....|....*..
1IVO_C 5 ECPLSHDGYCLHDGVCMYIEA---LDKYACNCVVGYIGERCQYRDLK 481IVO_D 5 ECPLSHDGYCLHDGVCMYIEA---LDKYACNCVVGYIGERCQYRDLK 481XDT_R 38 dpclrkykdfcihgECKYVKE---LRAPSCICHPGYHGERCHGLS-- 791J9C_L 1 --dqcasSPCQNGGSCKdql-----qsyICFCLPAFegRNCETHKdd 40
Proteom -78-
1JL9_A 5 ecplshdGYCLHDGVCMYIEA---LDKYACNCVVGYIGERCQYRDlk 481DAN_L 46 dgdqcasSPCQNGGSCKDql-----qsYICFCLPAFEGRNCETHKdd 871FSB 1 -tascqdmscskqGECLEtig-----nytCSCYPGFYGPECEYVre- 401AUT_L 13 plehpcaslccghGTCIxgi-----gsFSCDCRSGWEGRFCQREVsf 541QFK_L 1 ---qcasSPCQNGGSCKDql-----qsYICFCLPAFEGRNCETHKDd 391O5D_L 46 dgdqcaSSPCQNGGSCKDql-----qsYICFCLPAFEGRNCETHKdd 871HAE 5 kcaekekTFCVNGGECFMvkdlsnpsRYLCKCQPGFTGARCTENVpm 511MOX_D 6 ndcpdshtqfcfhGTCRFLVQ---EDKPACVCHSGYVGARCEHADLL 491NQL_B 5 ecplshdgyCLHDGVCMYIEA---LDKYACNCVVGYIGERCQYRDLK 481FAK_L 46 dgdqcasSPCQNGGSCKDql-----qsYICFCLPAFEGRNCETHKdd 871HAF 5 kcaekekTFCVNGGECFMvkdlsnpsRYLCKCQPGFTGARCTENVpm 511FFM_A 2 dgdqcassPCQNGGSCKDQl-----QSYICFClpAFEGRNCETHKDd 43
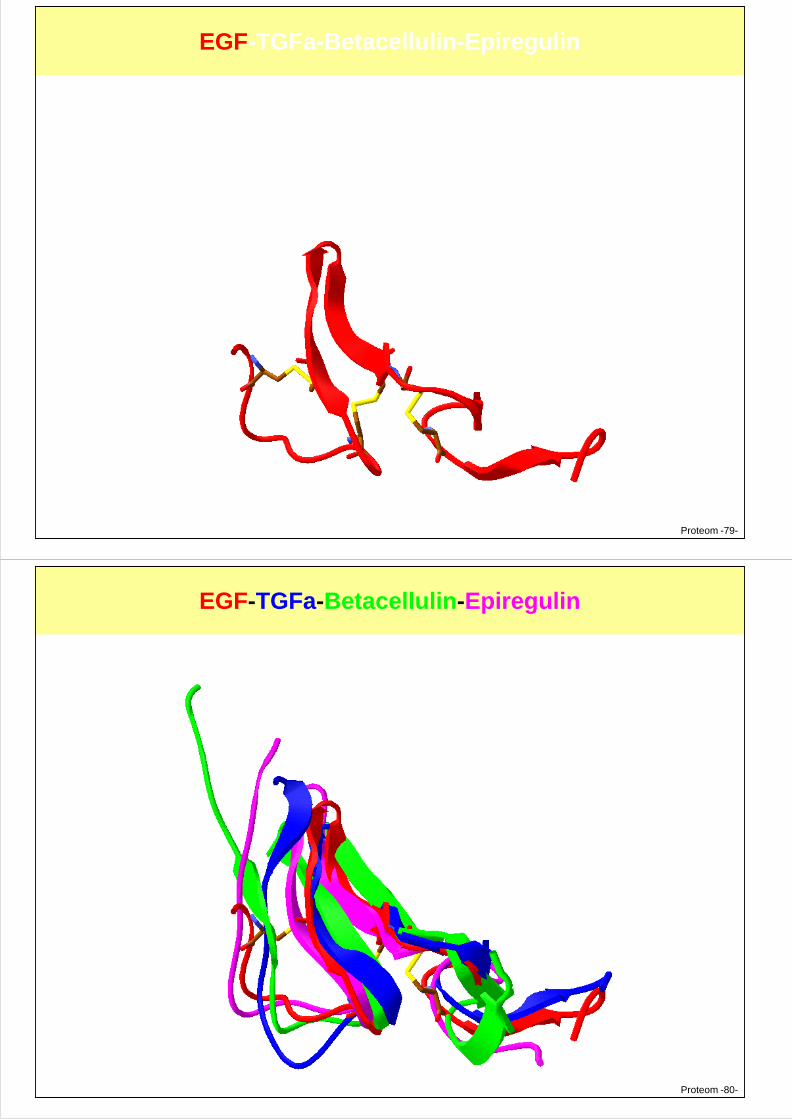
EGF-TGFa-Betacellulin-Epiregulin
Proteom -79-
EGF-TGFa-Betacellulin-Epiregulin
Proteom -80-

Gene Ontology Nomenklaturdatenbank
Current Ontologies:The three organizing principles of GO are molecular function, biological process and cellular component.
Proteom -81-
Gene Ontology of EGF-R signaling pathway
Proteom -82-
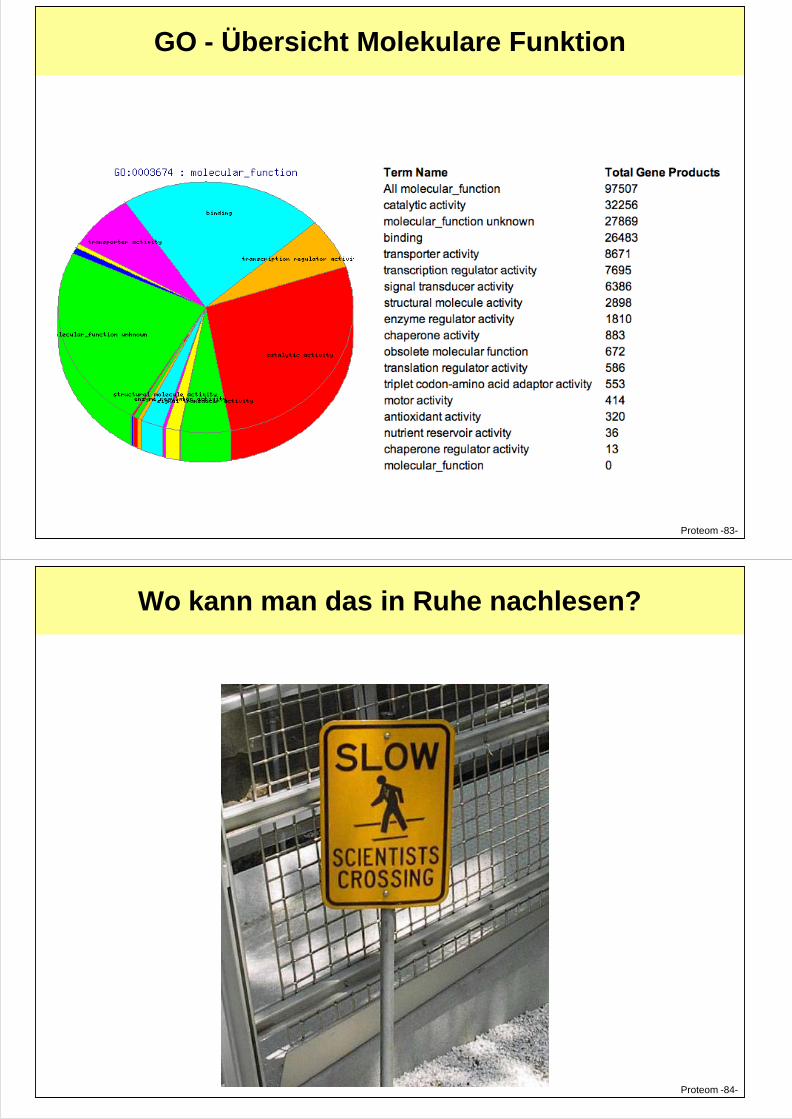
GO - Übersicht Molekulare Funktion
Proteom -83-
Wo kann man das in Ruhe nachlesen?
Proteom -84-
National Center for Biotechnological Information NCBI
http://www.ncbi.nlm.nih.gov/Education/index.html
Proteom -85-
EBI -Datenbank Einführung
Ensembl
Proteom -86-







