


Neuartige „Arduengo“-Carben homologe Verbindungen mit den Elementen der Gruppe 14 und 15:
Untersuchungen zu Struktur und Reaktivität ungesättigter
N-Heterocyclen der Elemente Germanium, Zinn, Arsen und Antimon
Dissertation
zur Erlangung des Doktorgrades (Dr. rer. nat.)
der Mathematisch-Naturwissenschaftlichen Fakultät der
Rheinischen- Friedrichs-Wilhelms-Universität Bonn
vorgelegt von
Diplom-Chemiker
Timo Gans-Eichler
aus Neuwied
Bonn 2004
N2 Sn1
Sn1´
N5
C4 C3


Die vorliegende Arbeit wurde in der Zeit von September 2000 bis September
2003 am Institut für Anorganische Chemie der Universität Bonn unter Leitung
von Herrn Prof. Dr. D. Gudat angefertigt.
Angefertigt mit Genehmigung der Mathematisch-Naturwissenschaftlichen
Fakultät der Rheinischen- Friedrichs-Wilhelms-Universität Bonn
1. Referent: Prof. Dr. D. Gudat
2. Referent: Prof. Dr. E. Niecke
Tag der mündlichen Prüfung:


Danksagung:
Meinem akademischen Lehrer, Herrn Prof. Dr. D. Gudat, danke ich herzlich für seine
vielfältige Unterstützung und ständiges Interesse an der vorliegenden Arbeit.
Herrn Prof. Dr. E. Niecke danke ich besonders für die Übernahme des Koreferates und
die Aufnahme in seinen Arbeitskreis während der Zeit von März 2003 bis Januar 2004,
Frau Dahme für die Aufnahme mehrerer IR-Spektren,
Herrn Dr. Jörg Daniels für die Messung und Lösung einer Kristallstruktur,
Herrn Dr. Martin Nieger für die Messung und Lösung vieler Kristallstrukturen,
Herrn Prof. Dr. D. Gudat für die Messung der 2D-Spektren und quantenchemischen
Rechnungen,
Frau Nicole Heyne für die Messung der 13C-Spektren am AC-200,
Frau Karin Prochnicki für die 119Sn-Sondermessungen,
Herrn Dr. Wilfried Hoffbauer für die Messung der CP/MAS-Spektren,
Herrn Dr. Jörn Schmedt auf der Günne für die Herzfeld-Berger-Analyse,
Herrn Dr. Jürgen Tirre für die Aufnahme vieler MS-Spektren,
Herrn Dr. Lewall für die CV-Messungen,
Herrn Dr. U. Kessler und Herrn S. Schlüter für die Messung der vielen Ramanproben
sowie der Pulveraufnahmen,
Herrn Dr. M. Koch für die Messung der IR-Proben und Herrn Dr. R. Weißbarth für die
EDX-Messungen,
meinen Laborkollegen Dr. A. Haghverdi, Dr. S. Häp, G. Schröder und Dr.L. Szarvas für
theoretische und praktische Tips und insbesondere Herrn Zoltan Bajko für lustige Zeiten
in Würzburg und Bad Kösen sowie Herrn Sebastian Burck und Herrn Corvin Volkholz für
angenehme Zusammenarbeit im Labor und Biologenpraktika,
den ACF-Praktikanten für engagierte Mitarbeit und Interesse an dieser Arbeit.


Abkürzungen und Symbole
2D zweidimensional Ad Adamantyl
B3LYP Becke-3-Lee-Yang-Parr Bipy Bipyridyl Cp Cyclopentadienyl CV Cyclische Voltammetrie δ Chem. Verschiebung d Dublett DBN Diazabicyclononen Dipp 2,6-Diisopropylphenyl DFT Dichtefunktionaltheorie DMAP 4-Dimetylaminopyridin DMF Dimethylformamid E0 Standardpotential EPa anodischer Peakstrom EPk kathodischer Peakstrom Ether Diethylether EXSY exchange spectroscopy gs gradient selected HMBC Heteronuclear Multiple Bond Correlation HMQC Heteronuclear Multiple Quantum Coherence HOMO highest occupied molecular orbital IR Infrarot nJ Kopplungskonstante über n Bindungen LUMO lowest unoccupied molecular orbital M Multiplett Mes Mesityl NBO Natural Bond Orbital NOESY nuclear overhauser enhancement and
exchange spectroscopy OTf Triflat s Singulett SCE saturated calomel electrode t Triplett TMS Trimethylsilyl TMEDA Tetramethylethylendiamin Verb. Verbindung


Inhaltsverzeichnis
1. Einleitung 1
2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14 6
2.1. Einführung 6
2.1.1 Struktur und elektronischer Zustand von Carbenen und höheren Homologen 6
a) Struktur und elektronischer Zustand von subvalenten Verbindungen
EH2 mit Elementen der Gruppe 14 6
b) Stabilisierung von EH2 durch Einführung geeigneter Substituenten 7
c) Struktur und elektronischer Zustand in „Arduengo“-Carbenen und
höheren Homologen 8
2.1.2 Die Darstellung N-heterocyclischer Silylene, Germylene und Stannylene 10
a) Darstellung von N-heterocyclischen Carbenen 10
b) Darstellung von N-heterocyclischen Silylenen 10
c) Darstellung von N-heterocyclischen Germylenen 11
d) Darstellung von Benzo-2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-ylidenen 12
2.1.3 Reaktionsverhalten von N-heterocyclischen Carbenen und höheren Analoga 13
a) Das Reaktionsverhalten von N-heterocyclischer Carbenen 13
b) Das Reaktionsverhalten von N-heterocyclischer Silylenen 14
c) Das Reaktionsverhalten von N-heterocyclischer Germylenen 17
2.2 Eigene Untersuchungen 18
2.2.1 Synthesemethoden 18
a) Darstellung von Diazastannol-2-ylidenen: Methoden, Darstellung und
Untersuchung der Vorstufen, Reaktionsmechanismen 18
b) Versuche zur Darstellung von Diazaplumbol-2-ylidenen 22
c) Darstellung von 4,5-Dimethyl-2,3-Dihydro-1-H-[1,3,2]-diazagermol-2-ylidenen 24

Inhaltsverzeichnis
2.2.2 NMR-Spektroskopische Untersuchung an Diazastannol-2-ylidenen 25
2.2.3 Röntgenstrukturanalytische Untersuchungen 29
2.2.4 Elektrochemische Untersuchungen an Diazastannol-2-ylidenen 31
2.2.5 Reaktionsverhalten der Diazastannol-2-ylidene 36
a) Untersuchungen zur Stabilität von Diazastannol-2-ylidenen 36
b) Untersuchungen zum Verhalten von Diazastannol-2-ylidenen als Lewis-Base 37
c) Untersuchungen zum Lewis-sauren Verhalten von Diazastannol-2-ylidenen 37
d) Untersuchungen zum Reaktionsmechanismus alkylsubstituierter α-Amino
-aldimine mit Sn[N(TMS2)]2 42
e) Reaktion der Diazastannol-2-ylidenen mit Chalkogenen und einem
Triphosphinin 45
2.2.6 Untersuchungen zum Austausch von Zinn- und Germanium-Atomen zwischen
Diazastannol-, Diazagermol-2-ylidenen und Diazabutadienen 47
a) Kinetische Untersuchungen an Diazastannol-2-ylidenen 48
b) Quantenchemische Studien an Diazastannol-2-ylidenen 52
c) Untersuchung von Germanium-Transferreaktionen an Diazagermol-2-ylidenen 56
3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen 58
3.1 Einführung 58
3.1.1 Diazastannacyclopentene 58
3.1.2 Synthesemethoden 59
3.2 Eigene Untersuchungen 60
3.2.1 Versuche zur Darstellung von Diazastannacyclopentenen 60
3.2.2 Reaktionsverhalten 62
4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15 66
4.1 Einführung 66
4.1.1 „Arduengo“-Carbenanaloga mit dem Element Phosphor:
Darstellung von 1,3,2-Diazaphospholenium-Kationen 66

Inhaltsverzeichnis
4.1.2 „Arduengo“-Carbenanaloga mit den Elementen Arsen und Antimon:
1,3,2-Diazarsolenium-Kationen und 1,3,2-Diazastibolenium-Kationen 68
4.1.3 Struktur und elektronischer Zustand in Phosphenium Kationen und 1,3,2-Diaza-
phospholenen 70
a) Grenzorbitalbetrachtung an den acyclischen Molekülen +PH2 und [+P(NH2)]2 70
b) Struktur und elektronischer Zustand von 1,3,2-Diazaphospholenium-Kationen 71
c) Struktur und elektronischer Zustand von 1,3,2-Diazaphospholenen 73
4.1.4 Das Reaktionsverhalten von 1,3,2-Diazaphospholenen, Phospheniumkationen und
Analoga mit den Elementen Arsen Antimon und Bismut 74
4.2 Eigene Untersuchungen 78
4.2.1 Synthesemethoden für 1,3,2-Diazaarsolene und -Stibolene 79
4.2.2 Röntgenstrukturanalytische Untersuchungen 81
a) Kristallstrukturen der Diazastibol(arsol)ene und Diazastibol(arsol)enium-
Kationen 81
b) Vergleichende Diskussion der Kristallstrukturen von kationischen N-heterocyclischen
Carbenanaloga mit den Elementen Phosphor, Arsen und Antimon 89
4.2.3 NMR-spektroskopische Untersuchungen 92
4.2.4 Schwingungsspektroskopische Untersuchungen 94
4.2.5 Untersuchungen zur Dissoziation der As-Cl-Bindung im 1,3,2-Diazarsolen 12c[Cl] 99
4.2.6 Reaktionsverhalten der 1,3,2-Diazastibolene, 1,3,2-Diazaarsolene bzw.
1,3,2-Diazastibolenium- und 1,3,2-Diazarsolenium-Kationen 101
a) Thermische Stabilität 101
b) Reaktionsverhalten gegenüber Oxidationsmitteln 102
c) Koordinationschemische Untersuchungen 107
5. Zusammenfassung der Ergebnisse und Ausblick 114
6. Experimenteller Teil 118

Inhaltsverzeichnis
6.1 Allgemeines 118
6.1.1 Arbeitsbedingungen 118
6.1.2 verwendete Chemikalien 118
6.1.3 Analytik 118
6.2 Chemische Umsetzungen und analytische Untersuchungen 122
7. Literaturverzeichnis 159
8. Kristallographischer Anhang 167
fotographische Aufnahmen
Lebenslauf
Publikationen

1. Einleitung
1
1. Einleitung
Carbene sind als subvalente Verbindungen des zweiwertigen Kohlenstoffs zu betrachten.
Abb. 1.1: Unterschiedliche Verteilung der beiden nichtbindenden Elektronen in Carbenen
Prinzipiell können sich die beiden nichtbindenden Elektronen gepaart mit
unterschiedlichem Spin in einem Orbital (Abb. 1.1, a) oder ungepaart mit gleichem Spin in
zwei Orbitalen (Abb. 1.1, b) aufhalten.
Bereits im 19. Jahrhundert wurden erste Untersuchungen zur Carbenchemie von J.B.
Dumas (1835)[1] und A. Geuther (1862)[2] unternommen. Der Grundgedanke dieser sehr
frühen Arbeiten war die Möglichkeit aus vierwertigen Kohlenstoffverbindungen durch
Elimination von z. B. HCl zweiwertige Kohlenstoffverbindungen zu erzeugen.
Abb. 1.2: Darstellung des Dichlorcarbens CCl2 nach Geuter[2]
In der Tat gelingt die Darstellung des Dichlorcarbens wie in Abb. 1.2 beschrieben. Da
allerdings Dichlorcarben sehr schnell Folgereaktionen eingeht, kann es nur als instabile
Zwischenverbindung gesehen werden.
Im darauf folgenden Jahrhundert versuchten auch H. Scheibler (1926)[3] und M. Schmeisser
(1960)[4] ohne Erfolg die Existenz eines „stabilen“ Carbens nachzuweisen. Es setzte sich
daraufhin die Erkenntnis durch, dass es sich bei Carbenen um höchst reaktive
Zwischenstufen handeln muss, die in situ aus geeigneten Vorstufen dargestellt und sofort in
Folgereaktionen abgefangen werden. (Abb. 1.3) Trotz ihrer offensichtlichen Instabilität
erlangten Carbene als Intermediate in der klassischen organischen Synthese beträchtliche
C
ClC l H
ClKOEt
-KCl, EtOH
C:
ClCl
Folgereaktionen
C C(a) (b)

1. Einleitung
2
Bedeutung. Als Beispiel sei das Auftreten von Dichlorcarben in der Reimer-Tiemann-
Reaktion genannt [5]. Auch in der moderneren organische Chemie sind Carbene wichtige
Synthesebausteine [6,7,8,9].
R2CH-XB-
R2C N Nhν [R2C:] + N2
CN
N[R2C:] + N2
hν
[R2C:] + BH + X-
aus Diazoalkanen Photolyse
aus Diazirinen Photolyse
basenkatalysierteα-Eliminierung von HX
Abb. 1.3: Allgemeine Methoden zur Carbenerzeugung[10,11,12]
H.-W. Wanzlick, ein Schüler von H. Scheibler, verfolgte in den frühen 60er Jahren die Idee
der Isolierung eines stabilen Carbens weiter[13,14,15]. Nachdem allerdings viele seiner
Veröffentlichungen zu diesem Thema widerlegt werden konnten[16,17,18], sprach vorerst
niemand mehr von stabilen Carbenen.
Erst 1991 gelang es Arduengo und seinen Mitarbeitern das 1,3-Diadamantyl-2,3-dihydro-1-
H-imidazol-2-yliden 1a (Abb.1.4) als bis 240°C stabile Substanz zu isolieren und durch
eine Röntgenstrukturanalyse zweifelsfrei als N-heterocyclisches Carben zu
identifizieren[19].Im Verlauf weiterer Forschungen konnten auch Carbene mit den sterisch
weniger anspruchsvollen tert-Butyl- (1b) und Mesityl-Resten (1c) dargestellt werden.
Abb. 1.4: „Arduengo“-Carben 1a
N
N
C:
1
R
R
R = Ad (a) R = tBu (b) R = Mes (c)
1. Einleitung
3
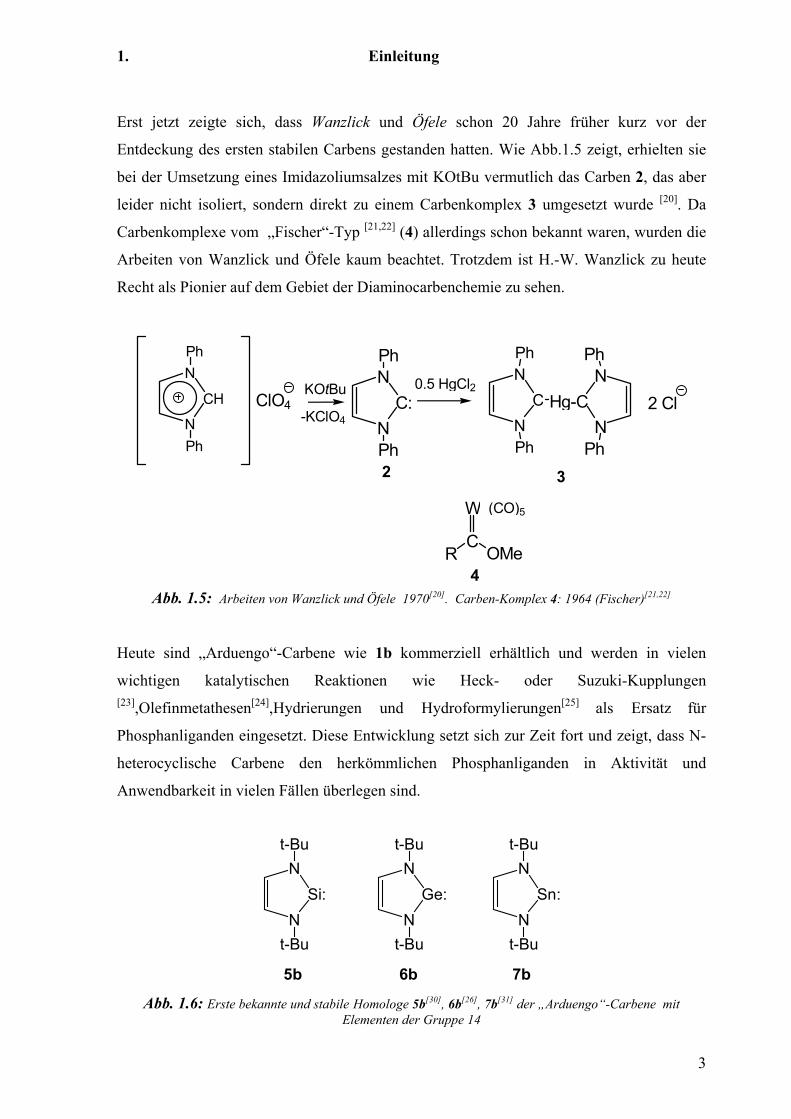
Erst jetzt zeigte sich, dass Wanzlick und Öfele schon 20 Jahre früher kurz vor der
Entdeckung des ersten stabilen Carbens gestanden hatten. Wie Abb.1.5 zeigt, erhielten sie
bei der Umsetzung eines Imidazoliumsalzes mit KOtBu vermutlich das Carben 2, das aber
leider nicht isoliert, sondern direkt zu einem Carbenkomplex 3 umgesetzt wurde [20]. Da
Carbenkomplexe vom „Fischer“-Typ [21,22] (4) allerdings schon bekannt waren, wurden die
Arbeiten von Wanzlick und Öfele kaum beachtet. Trotzdem ist H.-W. Wanzlick zu heute
Recht als Pionier auf dem Gebiet der Diaminocarbenchemie zu sehen.
Abb. 1.5: Arbeiten von Wanzlick und Öfele 1970[20]. Carben-Komplex 4: 1964 (Fischer)[21,22]
Heute sind „Arduengo“-Carbene wie 1b kommerziell erhältlich und werden in vielen
wichtigen katalytischen Reaktionen wie Heck- oder Suzuki-Kupplungen [23],Olefinmetathesen[24],Hydrierungen und Hydroformylierungen[25] als Ersatz für
Phosphanliganden eingesetzt. Diese Entwicklung setzt sich zur Zeit fort und zeigt, dass N-
heterocyclische Carbene den herkömmlichen Phosphanliganden in Aktivität und
Anwendbarkeit in vielen Fällen überlegen sind.
N
NSi:
t-Bu
t-Bu
N
NGe:
t-Bu
t-Bu
N
NSn:
t-Bu
t-Bu
5b 6b 7b Abb. 1.6: Erste bekannte und stabile Homologe 5b[30], 6b[26], 7b[31] der „Arduengo“-Carbene mit
Elementen der Gruppe 14
N
N
CH
Ph
Ph
3
ClO4
W
2 Cl0.5 HgCl2KOtBu
N
NC
Ph
Ph
-KClO4
2
4
N
NHg-C
Ph
Ph
N
NC:
Ph
Ph
(CO)5
COMeR

1. Einleitung
4
Die Entdeckung stabiler „Arduengo“-Carbens musste auch die Frage aufwerfen, ob höhere
Homologe, insbesondere Silylene, eine vergleichbare Stabilität aufweisen können. Die
Synthese der Germaniumverbindung 6b 1992 war wenig überraschend [26], da acyclische
Diaminogermylene, Diaminostannylene und -plumbylene im Gegensatz zu den
entsprechenden zu jener Zeit nicht bekannten Carbenen und Silylenen schon lange als
stabile Verbindungen galten [27,28,29]. Aber bereits 3 Jahre nach der Entdeckung des
„Arduengo“-Carbens konnte auch erstmals ein stabiles Silylen 5a von Denk et. al isoliert
werden [30]. Im Gegensatz zu 5b und 6b ist allerdings das Stannylen 7b, welches in
Vorarbeiten zur vorliegenden Dissertation isoliert, aber bislang strukturell nur unzureichend
untersucht werden konnte, relativ instabil [31].
Stabile Carbenanaloge Verbindungen lassen sich nicht nur mit Elementen der Gruppe 14
realisieren, sondern sind auch durch isoelektronischen Ersatz des zweiwertigen
Kohlenstoffatoms durch ein Element der benachbarten Gruppen 13 und 15 erhältlich. In der
Literatur sind bislang neben den N-heterocyclischen Carbenen 1, Silylenen 5 und
Germylenen 6 Verbindungen mit E = N+ (10)[32], P+ (11)[33], As+ (12)[34], Ga- (9)[35] bekannt.
Abb. 1.7: Bekannte Analoga N-heterocyclisher Carbene. Verbindungen, die in dieser Arbeit untersucht werden, sind grau unterlegt
Ziel der vorliegenden Arbeit ist die Untersuchung der Chemie höherer Homologer der
„Arduengo“-Carbene mit den Elementen Germanium (6), Zinn (7), Blei (8), Arsen (12),
Antimon (13) und Bismut (14). Hierzu müssen zunächst geeignete Synthesemöglichkeiten
für die noch völlig unbekannten Carbenanaloga mit den Elementen Blei, Antimon und
Bismut gefunden werden und bereits vorhandene Synthesemethoden für Carbenanaloga mit
den Elementen Zinn und Germanium optimiert werden.
E = C (1) E = N (10)
E = Si (5) E = P (11)
E = Ga (9) E = Ge (6) E = As (12)
E = Sn (7) E = Sb (13)
E = Pb (8) E = Bi (14)
N
NE(+)
R
R
N
NE (-)
R
R
N
NE
R
R

1. Einleitung
5
Weiter sollen insbesondere anhand von kristallographischen und spektroskopischen Daten
Aussagen über Bindungsverhältnisse in Carbenanaloga mit verschiedenen Elementen der
Gruppe 14 und 15 gemacht werden und diese miteinander verglichen sowie unter
Zuhilfenahme computerchemischer Methoden interpretiert werden.
Als Endziel soll versucht werden, ein umfassenderes Bild von Chemie, Struktur und
elektronischen Eigenschaften der höheren Analoga der „Arduengo“-Carbene zu gewinnen
und Gemeinsamkeiten und Unterschiede in isoelektronischen und subvalenten
Verbindungen mit unterschiedlichen Elementen herauszuarbeiten.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
6
2. Höhere Homologe der „Arduengo“-Carbene
2.1 Einführung
2.1.1 Struktur und elektronischer Zustand von Carbenen und höheren Homologen
a) Struktur und elektronischer Zustand von subvalenten Verbindungen EH2 mit
Elementen der Gruppe 14
Subvalente mit Verbindungen EH2 mit zweiwertigen Gruppe 14-Elemente bezeichnet man als
Carbene ( E = C ), Silylene ( E = Si ), Germylene ( E = Ge ), Stannylene ( E = Sn ) und
Plumbylene ( E = Pb ).
E
H
H b 1 a 1
E
H
H b1 (LUMO)a1 ( HOM O)
S i n g u l e t t - Z u stand (S) Triplett-Zustan d ( T ) Abb. 2.1: Frontorbitale und Besetzung mit Elektronen in subvalenten Verbindungen mit einem Gruppe 14-Element: a1-Orbital in der Molekülebene, b1-Orbital senkrecht zur Molekülebene
Die beiden nichtbindenden Elektronen am Element E können sich spingepaart im a1-Orbital
(HOMO) befinden oder mit gleichem Spin sowohl das a1- und b1-Orbital besetzten. Im
ersten Fall resultiert ein Singulett-Zustand (S) und im zweiten Fall ein Triplett-Zustand (T) (s.
Abb. 2.1). Für die Festsetzung des Grundzustands von EH2 ist die Energiedifferenz zwischen
dem a1- und dem b1-Orbital entscheidend. Je höher der Energieunterschied ∆E(a1-b1), desto
eher wird der Singulett-Zustand als Grundzustand bevorzugt. Bei höheren Elementen
derselben Periode, bei sinkender Elektronennegativität und bei Verringerung des
Bindungswinkels H-E-H, wird ∆E(a1-b1), zunehmend größer[36]. Als Folge bevorzugt das
Methylen CH2 einen Triplett-Grundzustand und die homologen Moleküle Silylen (SiH2) und
Germylen (GeH2) einen Singulett-Grundzustand [37](s. Tab. 2.1).

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
7
Tab. 2.1: Bindungswinkel und Singlett-Triplett-Anregungsenergien von EH2 [37]
b) Stabilisierung von EH2 durch Einführung geeigneter Substituenten
Eine Möglichkeit zur Darstellung isolierbarer Carbene, Silylene, Germylene und Stannylene
ist die Stabilisierung des Singulett-Zustands durch Herbeiführen einer hohen Energiedifferenz
∆E(a1-b1). Dieses ist zum einen damit zu begründen, dass Triplett-Carbene aufgrund der
Möglichkeit zur Ausbildung einer stabilen C=C-Doppelbindung eine große Neigung zur
Dimerisierung haben. Zum anderen impliziert eine hohe Energiedifferenz ∆E(a1-b1) eine
Herabsetzung der Reaktivität gegenüber Elektrophilen und Nucleophilen.
Wie jüngst gezeugt werden konnte, lässt sich die Neigung von Triplett-Carbenen zur
Dimersierung durch Einführung sterisch äußerst anspruchsvoller Reste soweit zurückdrängen,
dass ein persistentes Triplett-Carben 15 mit einer Halbwertszeit von bis zu einer Woche
erzeugt werden konnte [38].
C
15 Abb. 2.2: Persistentes Triplett-Carben 15; Halbwertszeit 1 Woche
Zur Stabilisierung des Singulett-Zustandes eines Carbens gegenüber Reaktionen mit
Nucleophilen und Elektrophilen kann man zwei Effekte nutzbar machen [39].
CH2 SiH2 GeH2
∆E(S T) [kJ/mol] -38 +88 +96
∠ H-E-H (S) 102° 92.8°
∠ H-E-H (T) 134° 118.5

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
8
Stabilisierung durch Polarisierung der σ-Bindung (-I-Effekt)
Ersetzt man z.B. die Wasserstoffe eines EH2 Moleküls durch ein elektronegativeres Element
wie z. B. Fluor, bleibt das LUMO nahezu unverändert und das HOMO wird stabilisiert. Die
Folge ist eine Vergrößerung von ∆ΕHOMO-LUMO und eine Bevorzugung des Singulett-
Zustandes. CF2 ist im Grundzustand daher ein Singulett-Carben.
Stabilisierung durch π−Donorwechselwirkung (+M-Effekt)
Wird ein Wasserstoffatom durch eine Gruppe ersetzt, die über ein freies Elektronenpaar
verfügt, das mit dem b1-Orbital (LUMO) am Element E in Wechselwirkung treten kann, wird
dieses durch Mesomerie destabilisiert. Diese Wechselwirkung führt ebenfalls zu einer
Vergrößerung von ∆HOMO-LUMO und damit wiederum zu einer Stabilisierung des Singulett-
Zustandes.
EH
Rb1
a1
Abb. 2.3: π-Wechselwirkung eines freien Elektronenpaars im Substituenten R mit dem b1-Orbital
Da Aminogruppen sowohl als +M als auch als –I-Substituenten einzustufen sind, bewirken
sie gleichzeitig eine mesomere Destabilisierung des LUMO und eine Absenkung des HOMO
durch induktive Effekte. Daher ist die erfolgreiche Darstellung der stabilen und isolierbaren
Singulett-Carbene (1), Silylene (5) bzw. Germylene (6) einer Kombination beider Effekte zu
verdanken, die man zusammengefasst auch als synergetischen „push–pull“-Effekt bezeichnen
kann [40,41].
C) Struktur und elektronischer Zustand in „Arduengo“-Carbenen und höheren
Homologen
N-heterocyclische Carbene des Typs 1 und ihre Homologen besitzen formal 6 π−Elektronen
im Fünfring. Daher besteht die Möglichkeit zur Ausbildung eines aromatischen π-Systems,
und in diesem Sinn wurde z. b. das Silylen 5b formal als Aromat mit einem delokalisierten

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
9
6π−Elektronensystem beschrieben [30]. In der Fachwelt wird der elektronische Charakter
solcher Systeme jedoch kontrovers diskutiert.
Arduengo et. al. begründen auf der Basis der Ergebnisse von DFT-Rechnungen an
perdeuterierten Tetramethyl-2,3-dihydro-1H-imidazol-2-ylidenen die Stabilisierung N-
heterocyclischer Carbene lediglich in einer Polarisierung der C-N-σ-Bindung zum
elektronegativeren Stickstoff [42]. Die Synthesen offenkettiger Germylene, Stannylene und
Plumbylene vor über 30 Jahren[27], der gesättigten cyclischen Carbene 17 (1995)[43] und 18
(1996)[44] und die Synthese eines acyclischen Carbens 19 durch (1996)[45] lassen tatsächlich
den Schluss zu, dass Aromazität nicht den entscheidenden Beitrag zur Stabilität der
ungesättigten Heterocyclen 1, 5 und 6 leistet.
Abb. 2.4: stabile Carbenanaloga sind nicht ausschließlich auf ungesättigte cyclische Systeme beschränkt
Zu einem differenzierten Ergebnis kommen Boehme und Frenking[46] sowie Heineman, Thiel,
Herrmann, Schwarz und Apeloig[47,48] aufgrund von computerchemischen Studien. Anhand
von NBO-Analysen und Berechnungen isodesmischer Reaktionsenergien deuten sie eine
stärker ausgeprägte π-Elektronendelokalisierung und höhere thermodynamische Stabilität von
ungesättigten N-heterocyclischen Carbenen und deren höheren Homologen im Gegensatz zu
gesättigten Verbindungen an. Insgesamt nehmen sowohl die π-Delokalisierung als auch die
thermodynamische Stabilität in Richtung C > Si, Ge ab. Die geringere Stabilität gesättigter
gegenüber ungesättigten Heterocyclen wird besonders am Silylen 18 deutlich, welches als
chemisch instabiler als 5b beschrieben wird und unter geeigneten Bedingungen im Gegensatz
zu diesem in Si-N-Bindungen insertieren kann [49].
Die zusätzliche Stabilisierung ungesättigter N-heterocyclischer Carbene und ihrer Homologen
wird durch die Mitwirkung der C=C Bindung an der bereits erwähnten mesomeren
Destabilisierung des Carben-HOMOs erklärt, die damit im Sinne einer 6π (C2N2) => π(E)
Wechselwirkung [50] zwischen dem HOMO eines Diazabutadien-Dianion-Fragments und dem
N
N
Si:
18
N
N
C:
17
(Pri)2N
(Pri)2N
C:
19
Mes
Mes
t-Bu
t-Bu

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
10
leeren b1(π)-Orbital des Elementes Ebeschrieben werden kann. Für die Bindungslängen im
Heteroyclus folgt aus diesem Modell eine Verkürzung der E-N-und C-N-Bindungen sowie
eine Verlängerung der C=C-Bindung gegenüber einem isolierten Diazabutadien-Dianion.
Abb. 2.5: qualitative Beschreibung der 6π (C2N2) => π (E) Wechselwirkung
In Summa kann der Stand der Diskussion so zusammengefasst werden, dass die Polarisierung
der E-N-σ-Bindung in Kombination mit einer mesomeren π-E-N-Wechselwirkung zur
Stabilisierung vieler subvalenter Diamide der Elemente C, Si, Ge, Sn, Pb qualitativ
ausreichend ist. Die Doppelbindung in N-heterocyclischen Carbenen (1), Silylenen (5) und
Germylenen (6) trägt quantitativ zur weiteren Stabilisierung bei.
2.1.2 Die Darstellung N-heterocyclischer Carbene, Silylene, Germylene und
Stannylene
a) Darstellung von N-heterocyclischen Carbenen
Die Darstellung der N-heterocyclischen Carbene bzw. 2,3-Dihydro-1-H-imidazol-2-ylidene
wird in diesem Abschnitt nicht behandelt. Die Methoden [51], die zur Darstellung dieser
Verbindungen angewendet werden, unterscheiden sich prinzipiell von den
Darstellungsmethoden ihrer höheren Homologen und sind daher für die vorliegende Arbeit
nicht relevant.
b) Darstellung von N-heterocyclischen Silylenen
Das N-heterocyclische Silylen bzw. 2,3-Dihydro-1-H-[1,3,2]-diazasilol-2-yliden (kurz
Diazasilol-2-yliden) 5b wurde von Denk et. al. durch Reduktion des 2-Dichloro-1,3-diaza-2-
silacyclopent-4-ens 22b mit Kalium dargestellt [30]. Das Diazasilacyclopenten 22b wurde
erstmals von tom Dieck durch Reaktion des Diazabutadiens 20b mit 2 Äquivalenten Lithium
und anschließender Metathese mit SiCl4 erhalten [52]. Eine alternative, von Karsch et. al.
N
N EN
EN

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
11
entwickelte Methode ist die Umsetzung des Diazabutadiens mit HSiCl3 in Gegenwart von
DABCO [53].
Abb. 2.6: Darstellung der des Diazasilacyclopentens 22b[52,53] und des Diazasilolylidens 5b nach Denk et.
al.[30]
b) Darstellung von N-heterocyclischen Germylenen
Das N-heterocyclische Germylen bzw. 2,3-Dihydro-1-H-[1,3,2]-diazagermol-2-yliden 6b
(kurz Diazagermol-2-yliden) wurde erstmals von Herrmann et. al. durch Metathese des
metallierten Diazabutadiens 21b mit GeCl2*Dioxan dargestellt [26].
Abb. 2.7: Darstellung des Germylens 6b nach Herrmann et. al.
Diese Methode konnte später von Heinicke et. al. erfolgreich auf die Darstellung von Pyrido-
2,3-Dihydro-1-H-[1,3,2]-diazagermol-2-ylidenen angewendet werden [64].
NGe:
N
t-Bu
t-Bu
N
N
t-Bu
t-Bu
N
N
t-Bu
t-Bu
2 - 2Li+2 Li
THF
GeCl2*Dioxan
-2 LiCl
6b 20b 21b
NSi:
N
t-Bu
t-B u
5b
N
N
t-B u
t-Bu
N
N
t-B u
t-B u
2 - 2Li+
NSi
N
t-Bu
t-Bu
22b
Cl
Cl
2 Li
THF
SiCl4-2 LiCl -KCl
2 K
HSiC l3 /DABCOCH2 Cl2 / -78°C
20b 21b

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
12
N
N
N G e :
n p
n p N
N H
N H
n p
n p
GeCl2
- 2LiClN
N Li
N Li
np
np
n -BuLi
np = neopentyl 2 3 Abb. 2.8 : Darstellung des Pyrido-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-ylidens 23 nach [64]
c) Darstellung von Benzo-2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-ylidenen
2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-ylidene und 2,3-Dihydro-1-H-[1,3,2]-diazaplumbol-
2-ylidene sind bislang in der Literatur bis auf die Benzo-2,3-dihydro-1-H-[1,3,2]-
diazastannol-2-ylidene 24d,e der Arbeitsgruppe von Lappert und Gehrhus nicht beschrieben [54].
Abb. 2.9 : Darstellung von Benzo-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-ylidenen durch Transaminierungs-
oder Salzeliminierungsreaktionen
Die Darstellung dieser Benzo-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-ylidene gelingt zum
einen durch Umsetzung von lithierten 1,2-Phenylendiaminen mit Zinn(II)chlorid oder
alternativ durch Transaminierung eines 1,2-Phenylendiamins mit Zinn(II)bis-
[bis(trimethylsilyl)]amid.
Sn[N(SiMe3)2]2
N
NS n:
R
R 24
NH
NH
R
R - 2 HN(SiMe3)2
SnCl2
- 2 LiCl
n-BuLi
CH2t-Bu ( d ) SiMe2tBu ( e )
NLi
NLi
R
R

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
13
2.1.3 Reaktionsverhalten von N-heterocyclischen Carbenen und höheren Analoga
a) Das Reaktionsverhalten N-heterocyclischer Carbene
Singulett-Carbene und ihre Analoga haben ein nichtbindendes Elektronenpaar und ein
energetisch hochliegendes, unbesetztes p-Orbital, was sie theoretisch befähigt sowohl als
Nucleophile als auch als Elektrophile zu reagieren. Als Präzedenzfall für eine derartige
ambidente Reaktivität kann das Beispiel der „Fischer-Carbenkomplexe“ 4 aufgeführt werden,
die schon seit über 30 Jahren bekannt sind. Die M-C Bindung kann hier durch Überlagerung
einer dativen C→M-„σ-Hinbindung“ und einer dativen M→C-„π-Rückbindung“ beschrieben
werden. Die σ−Hinbindung resultiert aus einem Ladungstransfer vom HOMO ( σ-lone pair )
des Carbens in ein leeres d-Orbital am Metall. Die π−Rückbindung resultiert aus einem
Elektronentransfer aus einem besetzten d-Orbital in das LUMO des Carbens.
Abb. 2.10: Schematische Darstellung der M-C-Bindung in Carben-Metallkomplexen
Wird das LUMO im Carbenfragment durch intramolekulare π-Donorwechselwirkungen
destabilisiert, wie es von N-heterocyclischen Carbenen bekannt ist, nimmt der elektrophile
Charakter des Carbens ab und die M→L-Rückbindung wird immer unwichtiger. So wirken
N-heterocyclische Carbene wie in Beispiel 25 vorwiegend als nucleophile 2e-Donoren [55,56],
die nur schwache M→C-Rückbindungen eingehen [57,58]. Aus den gleichen Gründen, die
gegen M→C-Rückbindungen in Übergangsmetallkomplexen sprechen, wird auch
Koordination einer Lewis-Base an das N-heterocyclische Carben energetisch ungünstiger. N-
heterocyclische Carbene reagieren im Gegenteil eher selbst als Lewis-Base gegenüber Lewis-
Säuren wie bei der Bildung von 26 [59,60].
M C
R
R

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
14
Abb. 2.11: Übergangsmetallkomplexe nucleophiler N-heterocyclischer Carbene
Das 1,3,4,5-Tetramethyl-imidazol-2-yliden 27 reagiert mit 2,4,6-tri-tert-Butyl-1,3,5-
triphosphabenzen unter Verkleinerung des P3C3t-Bu3-6-Ringes zum 1,2,4-Triphosphol-
Derivat 28. Diese Reaktion ist das erste Beispiel in der organischen Chemie in der
Verengung eines aromatischen 6-Ringes zu einem 5-Ring erfolgt [61].
NC :
N
M e
M e
2 7 2 8
NC
N
Me
Me
C t - B u
P P P
t - B u
t - B u
P P
P t-But - B u
t-Bu
+
Abb. 2.12: Reaktion des 1,3,4,5-tetramethyl-imidazol-2-ylidenes mit 2,4,6-tri-tert-Butyl-1,3,5-triphospha-benzen
b) Das Reaktionsverhalten von N-heterocyclischen Silylenen
Seit der Entdeckung des N-heterocyclischen Silylens 5b wurde das Reaktionsverhalten dieser
Verbindungsklasse eingehend untersucht. Analog zu den N-heterocyclischen Carbenen bildet
auch 5b Übergangsmetallkomplexe wie 29[49] und 30[62]. Die Silylen-Liganden sind dabei wie
Carbene als nucleophile 2e-Donatoren mit geringer Tendenz zu einer M→C π−Rückbindung
zu verstehen[62].
PdI
I
N C
N
H3C
CH3
N
CN
CH3
H3 C
25
NC
N
R
R
SnCl2
26

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
15
Abb. 2.13: Übergangsmetallkomplexe nucleophiler N-heterocyclischer Silylene
Das Silylen 5b reagiert mit Sauerstoff unter Öffnung des Ringes im wesentlichen zu
Siliziumdioxid und Diazabutadien[63].Die Reaktion mit Schwefel verläuft unter Erhalt des
Ringes durch Addition des Schwefels an das Silziumatom und anschließender Dimerisierung
zu 31 [63]. Ein Überschuss an Schwefel führt jedoch zur Freisetzung von Diazabutadien 20b
und laut den Autoren auch hier zu höheren Aggregaten 32.
Abb. 2.14: Reaktionen des Silylens 5b mit Chalkogenen
NSi
N
t-Bu
t-Bu
29
MN
SiN
t-Bu
t-Bu
OC CO
COOC
NSi
Nt-Bu t-Bu
Ni NSi
N
t-Bu
t-Bu
OCOC
30M = Cr, Mo,W
NSi:
N
t-Bu
t-B u
N
N
t-Bu
t-Bu
NSi
N
t-Bu
t-Bu
+ SiO2+ O2
S-78°C
1 eq S 8 RT
NS i
N
t-Bu
t-BuS
S
NSi
N
t-B u
t-Bu315b
20b
Überschuss S 8
NSi
N
t-Bu
t-BuN
N
t-Bu
t-Bu32
S
SSi
SSi
S
nN
N
t-Bu
t-Bu
20b
+

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
16
Das 1H-NMR-Spektrum von 32 zeigt eine große Anzahl von breiten sowie schwachen 1H-
NMR-Signale im Vinyl-Bereich [63], die von den Autoren als Beleg für eine polymere
Struktur angeführt wurden. Als weitere wichtige Reaktionstypen sind [1+4] Cycloadditionen
und Insertionsreaktionen zu nennen. So reagiert 5b mit Wasser durch Insertion in die HO-
Bindung und anschließender Kondensation zu 33[63]. Das Diazabutadien reagiert 20b mit 5b
mittels oxidativer Addition an das Siliziumatom zu 34b[63].
Abb. 2.15: Reaktionen von 5b mit Wasser und Diazabutadien
Das erstmals von der Arbeitsgruppe Lappert und Gehrhus dargestellte benzanellierte Silylen
36 [64] reagiert wie 5b mit 2,4,6-tri-tert-Butyl-1,3,5-triphosphabenzen über eine [1+4]
Cycloaddition zu den Addukten 35 [65] bzw. 37 [66].
N S i :
N
t -B u
t -B u N N t-Bu t - B u
5 b P P
P
S i
NS i :
N
C H 2 t - B u
CH 2 t - B u
3 6
3 5
N
N
C H 2 t - B u
C H 2 t - B u
P
PP
S i
3 7
P P
P t-But - Bu
t-Bu
P P
P t-But - Bu
t-Bu
+
+
Abb. 2.16: Rektionen von 5b und 36 mit einem Triphosphabenzen
N S i:
N
t-B u
t-Bu NSi
N
t-Bu
t-Bu
NSi
N
t-Bu
t-Bu
5b
H2O
Benzen RTH N
S iN
t-Bu
t-B uO
H
N
N
t-Bu
t-BuN Nt-Bu t-Bu
33
34b
20b

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
17
c) Das Reaktionsverhalten von N-heterocyclischen Germylenen
Die Chemie der Diazagermol-2-ylidene 6 ist im Gegensatz zu den Carbenen und Silylenen
bislang wenig untersucht worden. Von der Arbeitsgruppe Heinicke wurde vor kurzem die
erste Komplexverbindung 38 eines Diazagermol-2-ylidens 6d entdeckt [67].
Abb. 2.17 : Reaktion von 6d zu ersten Komplexverbindung eines Diazagermol-2-ylidenes
Anhand der Röntgenstrukturanalyse und der IR-Daten des Komplexes 38 konnten Hinweise
auf signifikante π-Rückbindungsanteile zwischen dem Molybdän-Atom und dem
Germanium-Atom im Diazagermol-2-yliden-Liganden gefunden werden. Im Gegensatz zu
Silylenen und Carbenen scheinen Diazagermol-2-ylidene neben nucleophilen somit auch
stärker ausgeprägte elektrophile Eigenschaften zu besitzen. Die Autoren erklären das
anwachsendene π-Akzeptorverhalten mit einer vom Carben zum Germylen abnehmenden π-
Delokalisation und einer daraus resultierenden geringeren π-Elektronendichte am
Germanium-Atom.
N
N Ge
np
np
38
2
COC OOC
CON
NGenp
np N N Ge
np
np
CONCEtOC
CONCEtOC
Mo+ Mo
6d np = neopentyl = CH2t-Bu (d)

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
18
2.2 Eigene Untersuchungen
2.2.1 Synthesemethoden
a) Darstellung von Diazastannol-2-ylidenen: Methoden, Darstellung und Untersuchung
der Vorstufen, Reaktionsmechanismen
In Vorarbeiten zur vorliegenden Arbeit konnte gezeigt werden, dass aufgrund des Auftretens
einer unerwünschten Reduktion des SnCl2 zu Zinn ein 2,3-Dihydro-1-H-[1,3,2]-diazastannol-
2-yliden 7b nicht analog zu Abb. 2.7 oder Abb.2.8 durch Salzeliminierungsreaktionen
darstellbar war [31]. Die Zielverbindung 7b konnte aber durch eine Transaminierungsreaktion
von Zinn(II)bis[bis(trimethylsilyl)]amid mit dem α-Aminoaldimin-LiCl-komplex 39b[LiCl],
erhalten werden.
Abb. 2.18: Darstellung des 2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-yliden 7b durch Transaminierung
Da das aus dem Komplex 39b[LiCl] freigesetzte (2-tert-Butylimino-ethyl)-tert-butylamin
39b zur Synthese des Diazastannol-2-ylidens 7b geeignet war, wurde versucht, die N-Aryl-
Diazabutadiene 20c,f sowie die C-alkylierten Diazabutadiene 41c,f, zur Darstellung
entsprechender α-Aminoaldiminen 39 einzusetzen (s. Abb. 2.18 ). Die Metallierung des 1,4-
Bis(2,4,6-Trimethylphenyl)diazabutadiens 20c bzw. 1,4-Bis(2,6-diisopropylphenyl)-
diazabutadiens 20f mit Lithium und die anschließende Umsetzung der Dilithiumsalze mit 2
Äquivalenten Ethanol liefert vermutlich zunächst Ethendiamine 40, die sich anschließend in
einem Keto-Enol anlogen Tautomerisierungschritt in die α-Aminoaldimine 39 umwandeln.
2 NEt3HCl Sn[N(S i M e 3 ) 2 ] 2 N
N
t - B u
t - B u
N L i
N L i
t - B u
t - B u
N
NH
t-Bu
LiCl- 2 NEt3
t-Bu
2 , 5 e q Li
- 2 HN( S i M e 3 ) 2
N
NSn
t Bu
t -Bu39b[LiCl] 7b2 0 b 2 1 b

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
19
Abb. 2.19: Darstellung von α-Aminoaldiminen 39 und Butendiaminen 43
Die Metallierung des 1,4-Bis(2,4,6-Trimethylphenyl)diazabutadiens 41c bzw. des 1,4-
Bis(2,6-diisopropylphenyl)-diazabutadiens 41f mit Lithium und anschließende Umsetzung
der Dilithiumsalze 42c,f mit 2 Äquivalenten Ethanol führt hingegen überraschenderweise
ohne weitere Tautomerisierung zu den Butendiaminen 43c,f. Das Dilithiumsalz 42g reagiert
mit Ethanol wiederum unter Tautomerisierung zum α-Aminoaldimin 44g. Eine
Literaturrecherche ergab, dass stabile N,N’-Bis(Alkyl)-but-2-en-2,3-diamine wie 43f bislang
völlig unbekannt sind.
Vom N,N`-Bis-(2,6-diisopropylphenyl)but-2-en-2,3-diamin 43f konnten aus Hexanlösungen
bei -30°C Kristalle erhalten werden, die für eine röntgenstrukturanalytische Untersuchung
geeignet waren. Das Ergebnis dieser Untersuchung zeigt dass die Doppelbindung eine Z-
Konfiguration besitzt. Die N(1) und N(2)-Atome weisen eine pyramidale Geometrie auf. Die
an den Stickstoffen gebundenen 2,6-Diisopropylphenyl-Gruppen stehen wie aus den beiden
C-N-C-C-Torsionswinkeln von 102.66° und 114.28° ersichtlich ist annähernd senkrecht zur
C2N2-Ebene.
N
N
R
R
N Li
N Li
R
R
N
NH
R
R
2,5 eqLi
39
NH
NH
R
R
2 EtOH
-2 EtOLi
40
N
N
R
R
NLi
NLi
R
R
2,5 eq LiNH
NH
R
R
2 EtOH
-2 EtOLi
43
R = Dipp ( f ) R = Mes (c )
R = Dipp (f )R = Mes (c)
N
NH
CH2H2NMe2
CH2H2NMe2 44g
20 21
41 42
R = CH2 C H 2 N M e 2 ( g )

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
20
C2 C1
N2 N1
C17
C18
C5
C6
Abb. 2.20: Struktur von 43f im Festkörper
Tab. 2.1: Ausgewählte Strukturparameter von 43f
Die C=C-Bindungslängen ähneln reinen Doppelbindungen (132.2 bis 132.6 pm [68]), während
die C-N-Bindungen gegenüber isolierten C-N-Einfachbindungen (145.7-148.7 pm [68])
verkürzt sind. Dieser Bindungslängenausgleich lässt sich durch eine geringfügige
Delokalisierung der π-Elektronen im isolierten π(C2N2)-Fragment erklären.
Das Butendiamin 43f reagiert mit Zinn(II)bis[bis(trimethylsilyl)]amid zum thermisch labilen
4,5-Dimethyl-2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-yliden 45f, dessen Konstitution durch
NMR-spektroskopische Untersuchungen belegt werden konnte. Eine Isolierung des Produktes
gelang nicht.
Bindungslängen [ Å ] Bindungs-/Torsionswinkel [°]
N2-C2 1.4283(17) C2-N2-C17 118.64(11)
N1-C1 1.4326(17) C2-N2-H2 109.0(9)
C1-C2 1.3390(18) C17-N2-H2 112.2(10)
C2-N2-C17-C18 102.66(15)
C1-N1-C5-C6 -114.28(15)
2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
21
Abb. 2.21: Erzeugung des 4,5-Dimethyl -2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-ylidens 45f.
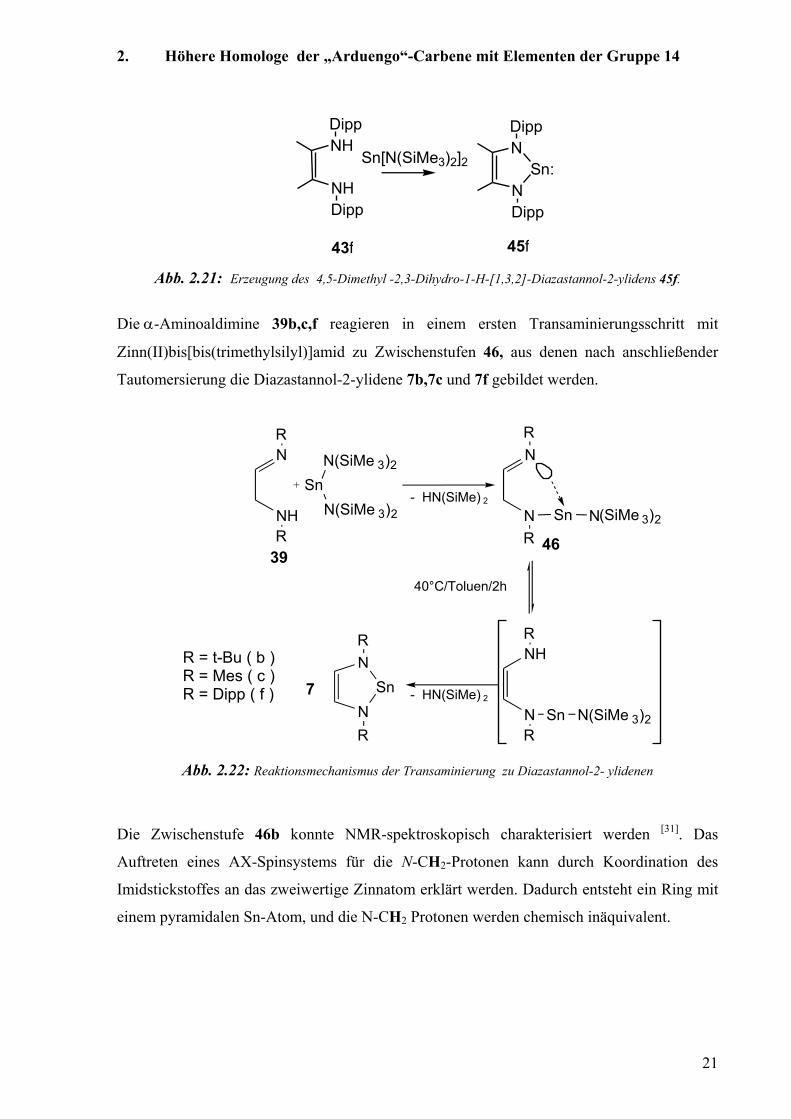
Die α-Aminoaldimine 39b,c,f reagieren in einem ersten Transaminierungsschritt mit
Zinn(II)bis[bis(trimethylsilyl)]amid zu Zwischenstufen 46, aus denen nach anschließender
Tautomersierung die Diazastannol-2-ylidene 7b,7c und 7f gebildet werden.
Abb. 2.22: Reaktionsmechanismus der Transaminierung zu Diazastannol-2- ylidenen
Die Zwischenstufe 46b konnte NMR-spektroskopisch charakterisiert werden [31]. Das
Auftreten eines AX-Spinsystems für die N-CH2-Protonen kann durch Koordination des
Imidstickstoffes an das zweiwertige Zinnatom erklärt werden. Dadurch entsteht ein Ring mit
einem pyramidalen Sn-Atom, und die N-CH2 Protonen werden chemisch inäquivalent.
NH
NH
Dipp
Dipp
N
N
Dipp
Dipp
Sn:
43f 45f
Sn[N(SiMe3)2]2
N
NH
R
R
N(SiMe 3)2
Sn
N(SiMe 3)2
NH
NR
R
Sn N(SiMe 3 ) 2
+
N
N
R
RSn N (SiMe 3 )2
N
NSn
R
R
- HN(SiMe) 2
40°C/Toluen/2h
- HN(SiMe) 2
46 39
7
R = t-Bu ( b ) R = Mes ( c ) R = Dipp ( f )

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
22
Tab. 2.2: Ausgewählte NMR-Daten von 46b ( C6D6)
Die gegenüber Zinn(II)amiden wie z.B. Zinn(II)bis[bis(trimethyl-silyl)]amid (δ119Sn = 759)
auftretende große Hochfeldverschiebung des 119Sn-NMR-Signals von 46b kann als Folge
dieser Koordination erklärt werden, da eine Koordination des Imidstickstoffes am Zinn zu
einer Destabilisierung des LUMO und so zu einer Verkleinerung des paramagnetischen
Abschirmungsterms σpara führen sollte. In Abschnitt 2.2.2 wird dieser Effekt noch einmal
besprochen.
b) Versuche zur Darstellung von Diazaplumbol-2-ylidenen
Die Reaktion der Dilithium-Salze 21b und 21c mit Blei(II)chlorid verläuft in Analogie zur
Reaktion mit Zinn(II)chlorid im Sinn einer Redoxreaktion zu elementarem Blei und den
Diazabutadienen 20b und 20c. Reaktionen der α-Aminoaldimine 39b,c,f mit
Blei(II)bis[bis(trimethylsilyl)]amid führten ebenso ausschließlich zu den entsprechenden
Diazabutadienen und elementarem Blei. Die Reaktion verläuft bei -40 °C über die
Zwischenstufen 47b,c,f, von denen 47b und 47c spektroskopisch charakterisiert werden
konnten. Die Bildung von Diazabutadienen und elementarem Blei lässt sich aus einer
schnellen cheletropen Zerfallsreaktion der möglicherweise als Intermediate entstandenen
Diazaplumbol-2-ylidene 8b,c,f erklären, die allerdings spektroskopisch nicht nachwiesen
werden konnten.
δ 1H(N=CH) 7.43 ppm
δ 1H(N-CH2) 4.52 ; 3.68 ppm
δ 119Sn 118.4 ppm
46b
N
NSn
t-Bu
t-Bu
N H´
H´
HSi(Me)3
Si(Me)3

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
23
Abb. 2.23: Die Transaminierungsreaktion von 39b,c,f mit Blei(II)bis[bis(trimethylsilyl)]amid. Temperaturerhöhung von -40°C auf RT führt zur Zersetzung der möglicherweise
intermediär entstandenen Diazaplumbol-2-ylidene 8
Die spektroskopisch untersuchten Intermediate 47b,c ähneln konstitutionell vermutlich den
im Verlauf der Untersuchung von Transaminierungsreaktionen mit Zinn(II)bis-
[bis(trimethylsilyl)]amid gefundenen Zinnverbindungen 46. Obwohl die deutlich erhöhte
Abschirmung der 207Pb-NMR-Signale der Intermediate 47b,c im Vergleich zu bekannten
acyclischen Diaminoplumbylenen[69] ein deutliches Indiz für eine intramolekulare
Koordination des freien Stickstoff-Elektronenpaars ist, spricht das Auftreten eines einzigen 1H-NMR-Signals für die (CH2)-Protonen in 47b,c jedoch für eine im Vergleich zum Zinn
höhere kinetische Labilität dieser koordinativen Wechselwirkung.
Tab. 2.3: Ausgewählte NMR-Daten von 47b,c in C6D6
47b 47c
δ 1H(CH) 8.08 7.80
δ 1H(CH2) 4.52 5.34
δ 207Pb 2590 2800
N
NH R
R
N(SiMe 3)2
Pb N(SiMe 3)2
NH
NR
R
Pb N(SiMe 3 ) 2
+ -HN( SiMe 3)2
N
N
R
RPb N(SiMe 3 ) 2
N
N Pb
R
R
N
N R
R
+ Pb-HN( SiMe 3)2
47 b,c,f 39 b,c,f
20 b,c,f 8b,c,f

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
24
c) Darstellung von 4,5-Dimethyl-2,3-Dihydro-1-H-[1,3,2]-diazagermol-2-ylidenen
C-methylierte Diazagermol-2-ylidene sind im Gegensatz zu 2,3-Dihydro-1-H-[1,3,2]-
diazagermol-2-ylidenen 6 bislang unbekannt. Das 4,5-Dimethyl-2,3-Dihydro-1-H-[1,3,2]-
diazagermol-2-yliden 48c wurde durch eine Transaminierungsreaktionen über das
Butendiamin 43c, welches durch Protonierung lithiierter Diazabutadiene 43c entsteht,
dargestellt und nach Isolierung durch spektroskopische und analytische Untersuchungen
charakterisiert.
Abb. 2.24: Darstellung des 4,5-Dimethyl-2,3-dihydro-1-H-[1,3,2]-diazagermol-2-ylidens 48c
N
N
Mes
Mes
2 - 2Li+2 Li
THFN
N
Mes
Mes
NH
NH
Mes
Mes
2EtOH-2LiOEt
N
N
Mes
Mes
Ge:Ge[N(SiMe3)2]2
41c 43c 48c42c

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
25
2.2.2 NMR-Spektroskopische Untersuchung an Diazastannol-2-ylidenen
Die Konstitution der Diazastannol-2-ylidene 7b, 7c, 7f, 45f wurde anhand von 1H, 13C, 119Sn
(aus 1H-detektierten 119Sn-HMQC-NMR-Spektren) und 15N NMR-Daten (aus 1H,15N gs-
HMBC-Spektren) aufgeklärt. Eine Aufstellung aller Daten befindet sich im experimentellen
Teil.
Ein Vergleich von 1H, 13C und 15N-NMR-Daten des Diazastannol-2-ylidens 7b mit denen des
Silylens 5b, Germylens 6b und Carbens 1a zeigt eine zunehmende Entschirmung der Signale
aller Ringatome mit zunehmender Ordnungszahl des zweifach koordinierten Elements
(Tab.2.4). Ein ähnlicher Schweratomeffekt wird auch bei den höheren Homologen des
Pyridins wie in Arsa-, Stiba- oder Bismabenzen beobachtet [70].
Tab. 2.4: Ausgewählte NMR-Daten von Stannylenen, Germylenen[26], Silylenen[30] und Carbenen[19]
Die für Diazastannol-2-ylidene 7b - 45f gefundenen 119Sn-NMR-Signale liegen zwischen 237
ppm und 260 ppm (Tab.2.5). Eine ähnliche 119Sn-NMR-Verschiebung wurde für das bereits
bekannte Benzo-2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-yliden 24d beobachtet [54].
Auffällig ist die relativ starke Abschirmung der 119Sn-Kerne der Diazastannol-2-ylidene 7b -
45f und 24d im Vergleich zu dem acyclischen SnII-Amid (Sn(N(SiCH3)2)2)[71], dem
cyclischen Bisaminostannylen 49[71] und dem N-silyl-substituierten Benzo-2,3-Dihydro-1-
H-[1,3,2]-diazastannol-2-yliden 24e (s. Tab. 2.5).
δ[ppm]
1a
5b
6b
7b
δ 1H
(H-C=C) 6.91 6.75 7.05 7.44
δ 13C
(H-C=C) 113.9 120 125.1 128.7
δ 15N -160.5 -170 -135 -119.9
t-Bu
N
N Sn
t-BuN
NSi
t-Bu
t-Bu
N
NC
Ad
Ad
N
NGe
t-Bu
t-Bu

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
26
Tab. 2.5: 119Sn- NMR-Daten von Diazastannol-2-ylidenen in C6D6
Eine mögliche Erklärung für diesen ungewöhnlichen Effekt konnte aus der Auswertung eines
unter „slow-spinning“-Bedingungen aufgenommenen 119Sn-CP/MAS-Spektrums von festem
7c erhalten werden (Abb. 2.25).
Abb. 2.25: 119Sn-CP/MAS-Spektrum von festem 7c
7b
7c
7f
45f δ 119Sn [ppm] 237 259 260 256
δ 119Sn [ppm] 269 456 759 639
49
t-Bu N
Me 2Si
N t-Bu
Sn
N
N Sn
t-Bu
t-Bu
N
NSn
Mes
Mes
N
NSn
Dipp
Dipp
N
NSn
Dipp
Dipp
NSn
NSiMe2tBu
SiMe2tBu 24eN
SnNCH2tBu
CH2tBu 24d Sn
N(SiCH3)2
N(SiCH3)2

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
27
Die Auswertung der Seitenbandenintensitäten, die nach der Herzfeld-Berger-Methode
durchgeführt wurde [72], liefert die in Tabelle 2.6 aufgeführten Hauptwerte δii des
Abschirmungstensors.
119Sn δ11 [ppm] δ22 [ppm] δ33 [ppm]
7c
960 -65 -251
29Si δ11 [ppm] δ22 [ppm] δ33 [ppm]
5b
284.9 -16.1 -43.6
13C δ11 [ppm] δ22 [ppm] δ33 [ppm]
1a 104 9 -184
Tab. 2.6: Aus CP/MAS-Spektren ermittelte Hauptwerte der anisotropen 119Sn, 29Si, 13C -Abschirmungstensoren von 7c, 5b[73] und 27[74]
Der Hauptwert δ11 von 7c ist gegenüber den Hauptwerten δ22 und δ33 stark entschirmt. Die
Hauptwerte δ11 des Silylens 5b[73] und des Carbens 27[74] zeigen ebenfalls eine starke
Entschirmung gegenüber den Hauptwerten δ22 und δ33. Die Aufnahme eines FK-NMR-
Spektrums einer pulverförmigen Probe des Diazastannol-2-ylidens lässt zwar die Ermittlung
der Beträge der Tensorhauptwerte δii zu, erlaubt aber keine Bestimmung der Ausrichtung der
Hauptachsen. Diese kann allerdings häufig durch Vergleich der experimentellen Daten mit
quantenchemischen Rechnungen abgeleitet werden. Die Durchführung entsprechender DFT-
Berechnungen an dem Diazasilol 5b ergab dabei, dass die zur am wenigsten abgeschirmte
Komponente δ11 zugehörige Hauptachse 1 parallel zur N-N-Verbindungsachse orientiert ist
und die zum am stärksten abgeschirmten Hauptwert δ33 gehörige Hauptachse 3 senkrecht auf
der Fünfringebene steht [73]. Eine analoge Ausrichtung des Hauptachsensystems wurde für das
Carben 27 gefunden [74].
N
N Si
t-Bu
t-Bu
N
NSn
Mes
Mes
N C :
N
M e
M e

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
28
N
NE
3
2
1
δ 33
δ 22δ 11
E = C, Si, Sn Abb. 2.26: Lage der Hauptachsen des Abschirmungstensors δii in N-heterocyclischen Carbenen [74],
Silylenen [73] und Stannylenen
Angesichts dieser Befunde sollte die Hauptachse 1 im Diazastannol-2-yliden 7c ebenfalls
parallel zur N-N-Verbindungsachse und die Hauptachse 3 senkrecht zur Fünfringebene
orientiert sein. Die Lage der Hauptachsen konnten durch computerchemische Rechnungen
bestätigt werden [50].
Die starke Entschirmung des Hauptwerts δ11 in den drei Homologen 27, 5b, 7c lässt sich
gleichermaßen damit erklären, dass der magnetisch erlaubte n π* Übergang einen großen
Beitrag zum paramagnetischen Abschirmungsterm σpara liefert und in der senkrecht zur Achse
der beteiligten n- und π*-Orbitale stehenden Raumrichtung eine starke Entschirmung
induziert [75]. Eine niedrige Anregungsenergie des n π* Übergangs ∆En π* wie z.B. in
Sn(N(SiCH3)2)2 impliziert einen großen paramagnetischen Beitrag σpara zur Abschirmungs-
konstante σ und damit insgesamt eine starke Entschirmung des 119Sn-NMR-Signals. Im
Gegensatz dazu führt eine Vergrößerung von ∆En π* zu einer Abschwächung des
paramagnetischen Beitrages σpara und damit zu einer Abschirmung des 119Sn-NMR-Signals.
In den Diazastannol-2-ylidenen 7b-45f kann diese relative Erhöhung der Anregungsenergie
als direkte Folge der Destabilisierung des LUMOs durch die 6π (C2N2) =>π (E)
Elektronendelokalisation interpretiert werden.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
29
2.2.3 Röntgenstrukturanalytische Untersuchungen
Eine Einkristallstrukturanalyse von 7b zeigt, dass im Kristall monomere Diazastannol-2-
ylidenen-Einheiten vorliegen. Die gewonnenen Strukturdaten sind jedoch von geringer Güte
und für eine eingehende Betrachtung der Bindungsparameter nicht geeignet. Ein besserer
Datensatz, der eine weitergehende Interpretation erlaubt, wurde von 7c erhalten. Auch hier
liegen isolierte Moleküle ohne intermolekulare Kontakte innerhalb der Summe der Van der
Waals-Radien vor. Demgegenüber liegen im Benzo-2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-
yliden 24d intermolekulare (η6-C6H4)…Sn-Kontakte [54] und im 1,3,2,4λ2-Diazasilastannetidin
49 Sn N-Kontakte [76] vor.
Das Sn-Atom von 7c liegt nicht in der Ringebene, sondern wird in zwei Positionen mit einer
relativen Besetzungswahrscheinlichkeit von 60:40 („Split“-Lage) oberhalb und unterhalb der
Ringebene gefunden. Dieser Befund kann als Folge des Vorhandenseins zweier übereinander
gelagerter, fehlgeordneter Ringe mit einer flachen envelope-Konformation interpretiert
werden.
Abb. 2.27: Struktur von 7c im Festkörper. Das Sn-Atom ist über zwei Lagen mit einer relativen
Besetzung von Sn1: Sn1´ = 60:40 fehlgeordnet.
Tab. 2.7: Ausgewählte Bindungsparameter des Diazastannol-2-ylidens 7c
Bindungslängen [ Å ] Bindungswinkel [°]
Sn1-N2 2.084 (3) C3-N2 1.378 (5) N2-Sn1-N5 77.63 (13)
Sn1-N5 2.102 (3) C4-N5 1.378 (5)
C3-C4 1.356 (6)
N2 Sn1
Sn1´
N5
C4 C3

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
30
Die C=C-Doppelbindung in 7c ist gegenüber isolierten C=C-Doppelbindungen (132.2 bis
132.6 pm) [68] etwas verlängert und die C-N-Einfachbindung sind gegenüber isolierten C-N-
Einfachbindungen deutlich (145.7-148.7 pm) [68] verkürzt.
Die C-C- und C-N-Bindungen in 7c sind unter Berücksichtigung der Standardabweichungen
vergleichbar groß wie im Diazagermol-2-yliden 6b [26]. Im Vergleich zu dem Butendiamin
43f wird in 7c und 6b ein stärker ausgeprägter Bindungslängenausgleich gefunden, aus dem
eine effektivere Delokalisierung der 6π−Elektronen gefolgert werden kann, die durch eine
Wechselwirkung der 6π−Elektronen im π(C2N2)-Fragment von 7c und 6b mit dem Element E
im Sinne einer 6π (C2N2) => π(E) Wechselwirkung (s. Abb. 2.5) erklärt werden kann.
Bindung Bindungslänge [Å]
E-N 1.753 (5) 1.856 (1) 2.084 (3) 2.102 (3)
2.051 (5) 2.067 (5)
N-C 1.400 (9) 1.384 (1) 1.378 (5) 1.378 (5)
1.382 (8) 1.376 (7)
1.4283 (17) 1.4326 (17)
C-C 1.347(21) 1.364 (1) 1.356 (6) 1.433 (9) 1.346 (6)
Winkel Winkel [°] N-E-N 90.5 (10) 84.8 (1) 77.63 (13) 78.5 (2)
Tab. 2.8: Vergleich ausgewählter Strukturdaten von Silylenen, Germylenen und Stannylenen 5b [30], 6b [26], 7c, 24d [54] und des Diaminobutens 43f
Die Sn-Abstände in 7c sind vergleichbar mit durchschnittlichen Sn-N-Bindungslängen in
Sn(II)amiden (209 pm) [77,78], aber etwas länger als typische Sn-N-Bindungen in Sn(IV)-
Verbindungen (204.7- 206.6 pm) [79,80,81] und die Sn-N-Bindungen im Benzo-2,3-Dihydro-1-
H-[1,3,2]-diazastannol-2-yliden 24d [54]. Die längeren Sn-N-Abstände in 7c gegenüber 24d
können die Folge einer weniger effektiveren π−Elektronendelokalisation im (N-Sn-N)-
Fragment von 7c sein. Dieses könnte aus einer im Vergleich mit dem C6H4-Fragment von
24d stärkeren π−Wechselwirkung innerhalb des Diaminoethenfragments von 7c resultieren.
N
NSi
t-Bu
t-Bu 5b N
SnNCH2tBu
CH2tBu 24d
N
NGe
t-Bu
t-Bu6b NH
NH
dipp
43f
dippN
NSn
Mes
Mes7c

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
31
2.2.4 Elektrochemische Untersuchungen an Diazastannol-2-ylidenen
Aus Berechnungen der freien Reaktionsenthalpie der in Abb. 2.28 dargestellten Reduktionen
von 1, 5 (B3LYP6-31G*-Niveau) bzw. 6 (B3LYP6-31G**-Niveau)und Vergleich der Werte
mit dem experimentell bestimmten Standardreduktionspotential des Redoxpaars 10/10. von -
1.84 V (gegen SCE) wurden von Heinicke et. al [82] für die Reduktion des Carbens 1 und der
Carbenanaloga 5 bzw. 6 stark negative Standardpotentiale von -4.41 V (1/1. –), -2.97 V (5/5. –)
und -2.88 V (6/6. –)abgeschätzt.
N
NC:
N
NSi:
N
NGe:
N
NC:
N
NSi:
N
NGe:
e-
1 5 6
1.- 5.- 6.-
N
NN
N
NN
e-e-e-
H
H
H
H
H
H
H
H
HH
H
H
H
H
H H
10
10.
-1.8
4 V
gege
n SC
EEx
perim
ente
ll
Abb. 2.28: Experimentell untersuchte Redoxreaktion 10/10.; Redoxpaare 1/1. – , 5/5. –, 6/6. –
Cyclovoltammetrische und chemische Untersuchungen [83] (Abb. 2.29) am Carben 27
zeigten, dass die Übertragung eines Elektrons vom divalenten Kohlenstoffatom zum
[Ru(bpy)3]2+ unter Bildung eines instabilen Radikalkations möglich ist. Eine Reduktion des
Carbenzentrums war demgegenüber in weiteren cyclovoltammetrische Untersuchungen selbst
bei stark negativen Potentialen nicht möglich [84]. Diese experimentellen Arbeiten bekräftigen
daher die durch Heinicke et. al vorgenommene Abschätzung eines stark negativen
Standardpotentials von -4.41 V für 1/1. – .

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
32
Abb. 2.29: Das Carben 27 reduziert [Ru(bpy)3]2+ zu [Ru(bpy)3]0 bzw. [Ru(bpy)3]+
Die Reduktion von Silylenen sollte unter Berücksichtigung des abgeschätzten
Standardpotentials von -2.97 V für 5/5. – gegenüber der Reduktion von Carbenen begünstigt
sein. Die Reduktion des Silylens 5b mit Alkalimetallen führt jedoch zum Diazabutadien 20b
und zur „Spiro“-Verbindung 34b (s. Abb. 2.15), die aus einer Folgereaktion des Silylens 5b
mit dem Diazabutadien 20b gebildet werden kann [85]. Aus cyclovoltammetrischen und
chemischen Untersuchungen am Silylen 18 konnte demgegenüber die reduktive
Dimerisierung zum Dianion 50 belegt werden, das durch Hydrolyse zum entsprechenden
Disilan abgefangen wurde [85,86].
Abb. 2.30: Reduktion des Silylens 18 zu einem Dianion 50; Abfangreaktion mit H2O
Erste erfolgreiche Versuche zur Reduktion ungesättigter N-heterocyclischer Carbenderivate
zu Radikalanionen wurden durch cyclovoltammetrische Untersuchungen am Diazagermol-2-
yliden 6d von Heinicke et. al durchgeführt [82]. Die bei einem Standardpotential von E0 = -2.75
V beobachtete Reduktion erwies sich jedoch nur bei hoher Vorschubgeschwindigkeit von 5
V/s-1 als reversibel, was für eine kurze Lebensdauer des gebildeten Radikalanions spricht.
Das gefundene Potential E0 = 2.75 V gegen SCE wurde als übereinstimmend mit den obigen
theoretischen Rechnungen betrachtet.
NC:
N
M e
Me
27
[Ru(bpy)3]2++[Ru(bpy)3]0
[Ru(bpy)3]+ NC
N
Me
Me
+
Zersetzung
O2
[Ru(bpy)3]2+
NSi
N
t-Bu
t-Bu
NSi
N
t-Bu
t-Bu
NSi :
N
t-Bu
t-Bu
18
NaK2
THF
50
2M+
2-
THF NSi
N
t-Bu
t-Bu
NSi
N
t-Bu
t-BuH2O
H
H

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
33
Um das Redoxverhalten der Diazastannol-2-ylidene zu untersuchen, wurden im Rahmen
dieser Arbeit cyclovoltammetrische Untersuchungen mit dem sterisch am stärksten
abgeschirmten Diazastannol-2-yliden 7f durchgeführt.
Abb. 2.31: Elektrochemisches Verhalten ; Potentiale E0 der Redoxpaare 7f/7f.+ und 7f/ 7f.-
Bei der Oxidation einer Lösung von 7f in CH2Cl2 (Vorschubgeschwindigkeit 200mV/s-1) im
Potentialbereich von 0 bis 620 mV zeigt die CV-Kurve eine Stromspitze EPa von 352 mV,
der bei Potentialumkehr eine zweite Stromspitze EPk bei 294 mV für die entsprechende
Reduktion entspricht. Die Potentialdifferenz EPa-EPk von 58 mV und das Verhältnis der
Peakströme IPk/IPa von 1.09 sprechen für das Vorliegen reversibler Redoxreaktionen zwischen
7f und einem in der Zeitskala des Experimentes stabilen Radikalkation 7f.+·. Aus dem
Mittelwert der Peakpotentiale EPa = 352 mV und EPk = 294 mV erhält man für das
Redoxpaar 7f/7f+· ein Standardpotential E0 von 0.32 V.
352 mV
294 mV
E0 = 0.32V
Abb. 2.32: Cyclovoltammogramm der Oxidation von 7c; Potentialverlauf 0 mV 620 mV 0 mV, Vorschubgeschwindigkeit 0.2 V/s-1
Bei Versuchen zur elektrochemischen Reduktion von 7f wurde im Potentialbereich zwischen
-1.2 und -1.8 V eine Stromspitze EPk bei 1532 mV beobachtet, die eine Reduktion von 7f
anzeigt und bei niedriger Vorschubgeschwindigkeit von 2 V/s-1 irreversibel ist.
N
NSn
Dipp
Dipp
N
NSn
Dipp
Dipp
N
NSn
Dipp
Dipp
+ e-- e-
7f 7f .-7f .+
E0 = 0.32 V E0 = - 1.57 V

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
34
-1532 mV
Abb. 2.33: Cyclovoltammogramm von 7f im Potentialbereich zwischen -1.2 und -1.8 V; Vorschubgeschwindigkeit 2 V/s-1
Wird die elektrochemische Reduktion jedoch mit einer höheren Vorschubgeschwindigkeit
von 10 V/s-1 durchgeführt, zeigt die CV-Kurve neben der Stromspitze für den
Reduktionsvorgang (EPk = -1626 mV) bei Potentialumkehr eine zweite anodische Stromspitze
bei einem Potential EPa = -1532 mV. Aus der Differenz der Peakpotentiale und dem
Verhältnis der Peakströme IPk/IPa von 1.02lässt sich ebenfalls auf einen reversiblen
Redoxprozess schließen, dessen Halbzellenpotential aus dem Mittelwert von EPa und EPk zu
E0 = - 1.57 V berechnet werden kann.
-1532 mV
-1626 mV E0 = -1.57 V
Abb. 2.34: Cyclovoltammogramm der Reduktion von 7f im Potentialbereich von -1.2 bis -1.8 V mit einer Vorschubgeschwindigkeit von 10 V/s-
In Analogie zu den Arbeiten von Heinicke et. al können diese Ergebnisse dahin gehend
interpretiert werden, dass vermutlich gleichfalls eine Reduktion des Stannylens 7f zum
Radikalanion 7f.- erfolgt, das ebenfalls nur kurze Zeit stabil ist und sich rasch zersetzt.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
35
Ein Vergleich der cyclovoltametrischen Studien [82] an 7f und 6d zeigt, dass das Stannylen 7f
sowohl leichter oxidierbar als auch leichter reduzierbar als das Germylen 6d ist Diese
Befunde können so interpretiert werden, dass in 7f einerseits das HOMO energetisch höher
und andererseits das LUMO energetisch niedriger liegt als im Germylen 6d, so dass die
Energiedifferenz ∆EHOMO-LUMO von 6d zu 7f insgesamt abnimmt.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
36
2.2.5 Reaktionsverhalten der Diazastannol-2-ylidene
a) Untersuchungen zur Stabilität von Diazastannol-2-ylidenen
Diazastannol-2-ylidene erweisen sich als äußerst empfindlich gegenüber Luft und in
geringerem Maße gegenüber Feuchtigkeit. Besonders hervorzuheben ist die Thermolabilität
der Diazastannol-2-ylidene 7b und 7c in Lösung, die innerhalb von wenigen Stunden bei
Raumtemperatur in einer cheletropen Reaktion zu Diazabutadien und tetragonalen Zinn
zerfallen. Im Gegensatz dazu schmelzen hochreine, feste Proben von 7c erst bei 105°C ohne
nachweisbare Zersetzung. Die N-(2,6-Diispropylphenyl)-substituierten Derivate 7f und 45f
erweisen sich vermutlich als Folge der deutlich höheren sterischen Abschirmung des
Zinnatoms durch die sterisch anspruchsvollen N-Aryl-Reste als wesentlich stabiler als 7b und
7c. 45f zersetzt sich bei 120°C rasch zu Diazabutadien und Zinn, während der analoge Zerfall
von 7f erst oberhalb von 130°C langsam einsetzt.
Abb. 2.35: Zersetzungsreaktion der Diazastannol-2-ylidene 7b,7c bei 20°C in Lösung
Durch Kontrollreaktionen, die zur Aufklärung der Ursachen der deutlich unterschiedlichen
Thermostabilität der einzelnen Stannylene durchgeführt wurden, konnte belegt werden, dass
Zusätze von SnO2 oder LiCl zu einer Beschleunigung des chelotropen Zerfalles von
Diazastannol-2-ylidenen führen können. So wurde gefunden, dass sich in einer Lösung von
7b in C6D6 nach 90 min bei 60°C nur 30% des Diazastannol-2-ylidens zersetzt hatte, während
nach Zusatz von LiCl zu einer Probe derselben Lösung unter sonst gleichen Bedingungen
eine Zersetzung von 41% beobachtet wurde. Bemerkenswert ist in diesem Zusammenhang
allerdings, dass während der über eine Transaminierungsreaktion bei 40°C laufende Synthese
von 7b ungeachtet der Anwesenheit von LiCl auch nach Reaktionszeiten von 90 min nur
wenig Zersetzungsprodukte gebildet wurden. Als Schluss aus den angestellten
Beobachtungen kann festgehalten werden, dass die niedrigere Stabilität von Diazastannol-2-
ylidenen in Lösung durch einen katalytischen Einfluss von Verunreinigungen auf die
N
NSn
R
R
N
NR
R
+ Sn (0)
20°C/ Einige Stunden
R = t-Bu (b) Mes (c)
7 20

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
37
cheletrope Zerfallsreaktion erklärt werden könnte. Umgekehrt wird möglicherweise eine
Stabilisierung der Diazastannol-2-ylidene durch die Anwesenheit einer Lewis-Base wie
Hexamethyldisilazan erreicht, die durch Bildung von Lewis-Säure/Base-Addukten entweder
mit LiCl oder mit dem Diazastannol-2-yliden (Abb.2.36) die Katalyse der
Zersetzungsreaktion inhibitieren könnte.
Abb. 2.36: mögliche Stabilisierung eines -2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-ylidens mit
Hexamethyldisilazan
b) Untersuchungen zum Verhalten von Diazastannol-2-ylidenen als Lewis-Base
2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-ylidene reagieren im Gegensatz zu den analogen
nucleophilen Carbenen und Silylenen [62] nicht mit Lewis-Säuren oder mit [Ni(CO)4],
[W(CO)5(C8H14)] oder [Pt(C2H4)(PPh3)2] zu entsprechenden Komplexverbindungen und
können daher eher als Verbindungen mit äußerst schwach ausgeprägtem nucleophilen
Charakter angesehen werden.
c) Untersuchungen zum Lewis-sauren Verhalten von Diazastannol-2-ylidenen
Die Annahme, dass Diazastannol-2-ylidene elektrophilen Charakters besitzen könnten, wird
durch die Tatsache nahe gelegt, dass die Bildung stabiler Addukte mit Lewis-Basen für
andere Aminostannylene gut bekannt ist. Um diese Hypothese experimentell zu überprüfen,
wurde eine Lösung von 7c mit einem Überschuß an DMAP versetzt und NMR-
spektroskopisch untersucht.
N
NSn
Mes
Mes
N NMe2+N
NSn
Mes
Mes
N NMe2
7c DMAP7c DMAP
Abb. 2.37: Lewis-Säure/Base-Addukt des Diazastannol-2-ylidens 7c mit DMAP
Sn[N(SiMe3)2]2N
NH
t-Bu
LiCl
t-Bu
N
NSn
t-Bu
t-Bu39b 7b
HN(SiMe3)2
40°C/ 90 min

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
38
In Anwesenheit der Lewis-Base erfährt das 119Sn-NMR-Signal von 7c eine Verschiebung
von ∆δ = -26 ppm. Diese Verschiebung kann aus einer Anhebung des LUMO ( π* ) erklärt
werden, die durch intermolekulare Wechselwirkung des Zinnatoms mit dem freien
Elektronenpaar des DMAP induziert wird. Eine ähnliche Adduktbildung konnte auch bei der
Reaktion eines benzanellierten Diazastannol-2-ylidens mit TMEDA [54] oder bei der
Dimerisierung gesättigter N-heterocyclischer Stannylene über intermolekulare N Sn-
Wechselwirkungen beobachtet werden [87].
Da die Isolierung eines stabilen Adduktes 7c DMAP nicht gelang, wurden weitere Versuche
zur Darstellung isolierbarer Addukte mit bifunktionellen Donoren (Chelateffekt) bzw.
intramolekular donorstabilisierten Diazastannol-2-ylidens unternommen.
Die Untersuchung der Umsetzung des Diazastannol-2-ylidens 7c mit dem Diaza-
butadien 20b als Lewis-Base ergab, dass die Reaktion überraschenderweise weder unter
Bildung eines Donor -Addukts 7c 20b noch unter oxidativer Addition des Diazabutadiens
an 7c zur Spiroverbindung 51 verlief, sondern das Diazastannol-2-yliden 7b und das
Diazabutadien 20c lieferte.
Abb. 2.38: Reaktion des Diazastannol-2-ylidens 7c mit Diazabutadien 20b
NSn:
N
Mes
MesN
SnN
Mes
Mes7c
N
N
Mes
MesN
N
t-B u
t-Bu
+
NSn
N
Mes
MesN
N
t-Bu
t-Bu
NSn:
N
t-Bu
t-Bu
7b
N
N
Mes
Mes
+
51
7c 20b
20b
20c

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
39
Formal wurde bei dieser Reaktion ein Zinnatom vom Diazastannol-2-yliden 7c auf das
Diazabutadien 20b übertragen. Um diese Atomübertragungsreaktion zu verifizieren und ihre
Anwendungsbreite auszuloten, wurden gezielt weitere Austauschreaktionen durchgeführt
(Abb.2.39).
Abb. 2.39: Zinntransferreaktionen von Diazastannol-2-ylidenen und Diazbutadienen
In den Reaktionen I-V konnte die Bildung eines neuen Diazastannol-2-ylidendurch Reaktion
des Diazabutadiens mit dem vorgelegten Diazastannol-2-yliden 1H-NMR-spektroskopisch
nachgewiesen und das Produkt durch Vergleich der Spektren mit denen authentischer
Vergleichsproben zweifelsfrei identifiziert werden. Versuche zur Isolierung wurden nicht
vorgenommen. Im Unterschied dazu wurden in den Reaktionen VI bis IX keine neuen 2,3-
Dihydro-1-H-[1,3,2]-diazastannol-2-ylidene gebildet, so dass der Schluss nahe liegt, dass die
Methylgruppen in 4,5-Position des Diazastannol-2-ylidenringes einen destabilisierenden
Effekt ausüben.
Die Möglichkeit zur Bildung eines stabilen intramolekularen Lewis-Säure/Base-Addukts
eines Diazastannol-2-ylidens wurde anhand der versuchten Darstellung von 44g (Abb. 2.40)
ausgelotet. Die geplante Synthese sollte analog zu Abb. 2.19 in einer Transaminierungs-
reaktion ausgehend von einem geeigneten α-Aminoaldimin geführt werden.
Reaktion R R´ R´´
I tBu Mes H
II Mes tBu H
III Mes Dipp H
IV Dipp Mes H
V Dipp tBu H
VI tBu Dipp Me VII Mes Mes Me
VIII Dipp CH2CH2N(Me)2 Me IX Mes CH2CH2N(Me)2 Me
N
N
R´
R´
N
N S n
R
R
+ N
N
R
R
N
NS n
R´
R´
+ R´ ́
R´ ́
´´R
´´R

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
40
Abb. 2.40: intramolekular basenstabilisiertes Diazastannol-2-yliden
Die sofortige Entstehung von elementarem Zinn bei Reaktionsbeginn deutete allerdings auf
einen unerwarteten Verlauf der Reaktion hin.
Abb. 2.41: Geplante Transaminierungsreaktion von 44g und Sn[N(SiMe3)2]2
Ein ersten Anhaltspunkt zum Verständnis der tatsächlich ablaufenden Vorgänge lieferte ein 119Sn, 1H-HMQC-Spektrum der Reaktionslösung, das zwei Kreuzsignale mit 119Sn-
Verschiebungen von -179.7 ppm und -262.7 ppm zeigt (Abb. 2.42) und auf Entstehung
zweier neuer Produkte hinweist .
Abb. 2.42: Ausschnitt aus einem 1H,119Sn-HMQC- Spektrum der Reaktionslösung von 44g mit Sn[N(SiMe3)2]2
N
N
Sn
NMe2
NMe2
44g
Sn[N(SiMe3)2]2N
NHCH2 CH2 NMe2
- 2 HN(SiMe3)2
N
NSn
CH2 CH2 NMe2
43g 44g
CH 2 CH2 NMe2
CH 2 CH2 NMe2
(ppm) 1.80 1.60 1.40
160
200
240
280
(ppm)

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
41
Aus weiter gehenden gradientenselektierten (gs) 1H-NOESY, 1H,13C-HMQC- und 1H,13C-
HMBC-NMR-Spektren ließ sich folgern, dass wahrscheinlich keines der Produkte die
Konstitution eines Heterocyclus 44g aufweist, sondern vielmehr CH3-CR=NR- und
H2C=CR2- Strukturfragmente besitzt, die mit einer cyclischen Struktur unvereinbar sind.
Unter Berücksichtigung aller verfügbaren Informationen wurde für das Produkt mit δ119Sn =
-262.7 ppm die in Abb.2.43 dargestellte Konstitution eines symmetrisch substituierten
acyclischen Stannylens 52 postuliert.
Tab. 2.9: ausgewählte NMR-Daten der Reaktionsprodukte aus der Reaktion von 44g mit Sn[N(SiMe3)2]2
N
NR
N
NSn
RR
52
NMe2
Abb. 2.43: Vorgeschlagene Konstitution eines der aus 44g und Sn[N(SiMe3)2]2 gebildeten Reaktionsprodukte
Die starke Abschirmung der 119Sn-Resonanz von 52 im Vergleich zu Sn[N(SiMe3)2]2 ist mit
einer Erhöhung der Koordinationszahl des Zinnatoms als Folge der Koordination einer
NMe2-Gruppe und eines C=N-Fragmentes vereinbar. Ähnliche 119Sn-Verschiebungen (-284.8
ppm) treten auch in Doppelkubanen [Sn7(2-NR)8] auf, in denen intramolekular
δ119Sn [ppm] δ1H [ppm] δ13C [ppm]
CH3-CR=NR 1.76 5J(1H,119/117Sn) = 7.5 Hz CH3-CR=NR 14.1
R2C=CH2 4.43 4J(1H,119/117Sn) = 12.8 Hz
3.92 4J(1H,119/117Sn) = 10.5 Hz
R2C=CH2 84.7
R2C=CH2 154.73 -179.9
CH3-CR=NR 170.9
CH3-CR=NR 1.63 5J(1H,119/117Sn) = 9 Hz CH3-CR=NR 11.5
R2C=CH2 4.39 4J(1H,119/117Sn) = 12.4 Hz 4.00 4J(1H,119/117Sn) = 8.5 Hz
R2C=CH2 84.1
R2C=CH2 154.70 -262.7
CH3-CR=NR 171.1

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
42
donorstabilisierte, vierfach N-koordinierte Sn(II)-Atome vorliegen [88]. Für das zweite
Produkt (mit δ119Sn = -179.9) könnte ebenfalls die Konstitution eines donorstabilisierten
acyclischen Stannylens postuliert werden, allerdings bleibt die genaue Molekülstruktur sowie
die Zusammensetzung der Koordinationssphäre des Zinnamtoms in diesem Fall noch unklar.
d) Untersuchungen zum Reaktionsmechanismus alkylsubstituierter α-Aminoaldimine
mit Sn[N(SiMe3)2]2
Eingehendere Untersuchungen der Reaktion von 44g und Sn[N(SiMe3)2]2 zeigten, dass
Einsatz gleicher Mengen von α-Aminoaldimin 44g und Sn[N(SiMe3)2]2 zum vollständigen
Verbrauch des Sn(II)amids führten, wohingegen etwa 50% nicht umgesetztes α-
Aminoaldimin in der Reaktionslösung zurückblieb. Zugabe des Diazabutadiens 20c zu dieser
Reaktionslösung ergab keinen Hinweis auf eine Sn-Tranferreaktion unter Bildung von 7c. Die
Bildung dieses Produktes konnte jedoch überraschenderweise nachgewiesen werden,
nachdem diese Lösung mit überschüssigem Sn[N(SiMe3)2]2 versetzt wurde, um das restliche
α-Aminoaldimin 44g vollständig abreagieren zu lassen.
Da 7c nicht durch direkte Reaktion von 20c und Sn[N(SiMe3)2]2 entsteht, wie auch
unabhängig festgestellt wurde, kommt für seine Bildung nur ein Weg unter Übertragung eines
Sn-Atomes von einem weiteren, durch Reaktion von 44g und Sn[N(SiMe3)2]2 gebildeten
Intermediats, in Frage. Hierbei könnte es sich um das unsymmetrische Sn(II)amid
Sn(H)N(SiMe3)2 handeln, das durch β-Eliminierung aus einem als Primärprodukt zu
postulierendem instabilen Stannylen 53 gebildet werden müsste (Abb. 2.44, Reaktion (I)).
Die weitere Umsetzung mit dem Diazabutadien 20c zu HN(SiMe3)2 und 7c könnte dann
entweder unter [4+2]-Cycloaddition zum 2-Hydro-2-bis(trimethylsilyl)amino-1,3-diaza-2-
stannacyclopent-4-en 55 und anschließender reduktiver Eliminierung, oder alternativ unter
Addition an die C=N-Doppelbindung zur Zwischenstufe 56 und nochmaliger
Tautomerisierung und cyclisierender Amineliminierung, verlaufen (Reaktion (II), Abb. 2.44).
Das Auftreten eines Hydrido-Stannylens HSnR als instabiles Intermediat wird durch die
kürzlich gelungene Darstellung eines stabilen Derivates dieses Typs durch Power et. al.
gestützt [89], das in Lösung und im Festkörper als Hydrid-verbrücktes Dimer [(µ-H)SnR]2 (R
= 2,6-Trip2H3C6) vorliegt. Eine [4+2]-Cycloaddition eines solchen Intermediats entspricht
weiterhin bekannten Abfangreaktionen kurzlebiger Stannylene SnR2 mit elektronenreichen
Systemen wie 1,2-Diketonen [90] oder Diazabutadienen [91], auf die in Kapitel 3 noch einmal
eingegangen wird.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
43
Sn[N(SiMe3)2]2N
NHR
- HN(SiMe3)2
R = CH2CH2NMe2
RN
NR
R
Sn N(SiMe3)2
HN
N
R
R
- HN(SiMe3)2
N
NH
R
R
1/2 Sn[N(SiMe3)2]2
- HN(SiMe3)2
N
N
R
R
N
N
R
R
Sn
SnH N(SiMe3)2 Sn(0) +
HN(SiMe3)2
SnH N(SiMe3)2
N
N
Mes
Mes
N
NSn
H
N(SiMe3)2
Mes
Mes - HN(SiMe3)2 N
NSn
Mes
Mes
7c5520c
+
+
53 40g
54 52
N
N
SnN(SiMe3)2
Mes
MesN
N
SnN(SiMe3)2
Mes
Mes
H
Reaktion I
Reaktion II
N
N
R
R
40g
Reaktion III
56
Abb. 2.44: Möglicher Mechanismus zur Bildung der Stannylene 52 und 7c in der Reaktion von 44g mit
Sn[N(SiMe3)2].
In Abwesenheit des Diazabutadiens 20c zersetzt sich HSnN(SiMe3)2 offensichtlich in einer α-
Eliminierung zu Zinn und HN(SiMe3)2. Die Entstehung des acyclischen Stannylens 52 kann
ausgehend vom intermediär entstandenem Diazabutadien 40g durch Tautomerisierung zu 54
und anschließender Transaminierung mit Sn[N(SiMe3)2]2 erklärt werden (Abb 2.44, Reaktion
III). Dieser letzte Schritt konnte durch eine Kontrollreaktion bestätigt werden, in der das
Diazabutadien 40g direkt mit Sn[N(SiMe3)2]2 zu 52 reagierte.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
44
Interessant ist im Zusammenhang mit den obigen Beobachtungen die bereits früher
untersuchte Reaktion (Abb. 2.45) von Sn[N(SiMe3)2]2 mit (1-Ethyl-2-tert-butylimino-ethyl)-
tert-butyl-amin 56, das durch das Diazabutadien 20b verunreinigt war [31].
Abb. 2.45: 4-Alkyl-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-ylidene sind nicht durch Transaminierungsreaktionen zugänglich
Als Hauptprodukt dieser Umsetzung wurden das offenkettige Stannylen 58 und das
Diazastannol-2-yliden 7b nachgewiesen , während das 4-Ethyl-2,3-dihydro-1-H-[1,3,2]-
diazastannol-2-yliden 58 nicht gebildet wurde. Die Entstehung der unerwarteten Produkte 7b
und 57 kann nach dem in Abb. 2.44 dargestellten Reaktionsmechanismus durch primäre
Reaktion von 57 mit Sn[N(SiMe3)2]2 zu HSnN(SiMe3)2 und einem unsymmetrischen, zu 53
analogen, 2-Ethyl-Diazadien erklärt werden, deren Umsetzung mit 20b bzw. Sn[N(SiMe3)2]2
dann die beobachteten Endprodukte liefert.
Zusammengefasst ergibt sich aus den in diesem und im Abschnitt 2.2.1 dargelegten
Erkenntnissen das in Abb. 2.46 veranschaulichte Bild der Transaminierungsreaktionen von c-
Alkyl- und c-H-substituierten α-Aminoaldiminen I bzw. Butendiaminen VI. c-Alkyl- wie c-
H-substituierte α-Aminoaldimine I bilden nach dem ersten Transaminierungsschritt eine
Zwischenstufe II, die entweder unter von Abspaltung von HN(SiMe3)2 zu III oder unter
Tautomerisierung zu V und letztendlich zum Diazastannol-2-yliden VI reagieren kann.
Sn[N(SiMe3)2]2N
NHt-Bu
- HN(SiMe3)2
t-Bu
E t
N
Nt-Bu
t-Bu
SnNTMS2
N
N
t-Bu
t-Bu
N
NSn
t-Bu
t-Bu
57 5820b 7bCH3
Et
S n[N(SiMe3)2]2 - 2 H N (S iMe3)2
N
NSn
t-Bu
t-Bu
59
+ +

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
45
Sn[N(SiMe3)2]2N
NHR
- HN(SiMe3)2
R
X
N
NR
R
XSn
NTMS2Sn
H NTMS2N
N
R
RX
- HN(SiMe3)2
N
NSn
R
R
NH
NR
R
XSn
NTMS2X
NH
NHR
R
X
Sn[N(SiMe3)2]2
- HN(SiMe3)2
X = H, Me , EtX = H
X = CH3
X X X
I II III
IV V VI
X = Me, Et+
Abb. 2.46: Aufgrund der schnelleren Geschwindigkeit der β-Eliminierung gegenüber dem Tauto- merieschritt bilden c-Alkyl- substituierte α-Aminoaldimine keine Diazastannol-2-ylidene.
Während die H-Verschiebung für C-H-substituierte Edukte bevorzugt ist, verläuft bei C-
Alkyl-substituierten α-Aminoaldiminen offenbar die β-Eliminierung schneller. Die Bildung
von 4,5-Dialkyl-diazastannol-2-ylidenen wie 45f (Typ IV) gelingt nur, wenn als Edukte
Butendiamine vom Typ VI zur Verfügung stehen, die eine zweifache Transaminierung ohne
intermediär erfolgende Tautomerisierung ermöglichen.
e) Reaktion der Diazastannol-2-ylidene mit Chalkogenen und einem Triphosphinin
Die Diazastannol-2-ylidene 7c und 7f reagieren im Unterschied zu ihren leichteren
Homologen [61,65] mit 2,4,6-tri-tert-Butyl-1,3,5-triphospha-benzen weder in einer [4+1]-
Cycloaddition noch in einer Insertionsreaktion. Sie reagieren allerdings schnell mit Sauerstoff
und Schwefel und langsam mit rotem Selen zu Diazabutadien und Zinnchalkogeniden. Eine
oxidative Addition eines Chalkogen-Atoms unter Ausbildung einer Sn(IV)-Verbindung 60
konnte nicht beobachtet werden.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
46
Abb. 2.47: Reaktionen von Diazastannol-2-ylidenen 7c und 7f mit Chalkogenen
Auch wenn Sn(IV)-Verbindungen mit Sn=X-Doppelbindungen bekannt sind [92], zeigen die
Ergebnisse von DFT-Rechnungen an Formaldehyd und seinen schweren Homologen, dass
nur im Formaldehyd energetisch gleichwertige σ-und π-Bindungen zwischen dem
Kohlenstoff- und dem Sauerstoffatom ausgebildet werden [93]. Im Thio- und
Selenoformaldehyd sowie in allen Verbindungen mit Doppelbindungen zu höheren
Homologen des Kohlenstoffs nimmt die Stabilität der π-Bindungen gegenüber den σ-
Bindungen deutlich ab. Die berechneten Sn-O-σ-Bindungen sind dementsprechend etwa um
einen Faktor 3 stärker als die Sn-O-π-Bindungen. Sn-S und Sn-Se-σ-Bindungen sind etwa
zweimal so stark wie die entsprechenden π-Bindungen.
X=O X=S X=Se X=Te
H2C=X σ π
93.6 95.3
73.0 54.6
65.1 43.2
57.5 32.0
H2Si=X σ π
119.7 58.5
81.6 47.0
73.7 40.7
63.2 32.9
H2Ge=X σ π
101.5 45.9
74.1 41.1
67.8 36.3
59.1 30.3
H2Sn=X σ π
94.8 32.8
69.3 33.5
64.3 30.6
56.4 26.3
H2Pb=X σ π
80.9 29.0
60.9 30.0
57.0 27.8
50.3 24.4
Tab. 2.10: berechnete (B3LYP-Niveau) σ und π -Bindungenergien (kcal mol-1) in H2C=X (nach [93])
N S n:
N
R
R N
N
R
R
NSn
N
R
R
E
E = O,S ,Se
+E
+ SnO2, S nS, S nSe2
60
7c, 7f

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
47
Die Berechnungen sind prinzipiell vereinbar mit der klassischen Doppelbindungsregel [94], die
eine Schwächung der π-Bindungen in schwereren Elementen aufgrund geringerer
Überlappungsintegrale ihrer p-Orbitale vorhersagt. Im Licht dieser Resultate dürfte ein
Zerfall eventuell intermediär auftretender Diazastannol-2-yliden-chalcogenide in
Zinnchalkogenid und Diazabutadien daher aufgrund der Bevorzugung stabiler σ-Bindungen
thermodynamisch begünstigt sein. Eine kinetische Reaktionskontrolle, die die Bildung von
Produkten des Typs 60 erlauben sollte, scheint selbst bei Anwesenheit der sterisch
anspruchsvollen Dipp-Reste nicht gegeben zu sein.
2.2.6 Untersuchungen zum Austausch von Zinn- und Germanium-Atomen zwischen
Carbenanaloga und Diazabutadienen
Im Abschnitt 2.2.5 wurde der Austausch von Zinnatomen zwischen Diazastannol-2-ylidenen
und Diazabutadienen beschrieben. Im folgenden Abschnitt sollen Aussagen über den
Reaktionsechanismus getroffen werden und folgende Fragen beantwortet werden:
Verläuft der Transfer eines Zinnatomes über einen direkten, bimolekularen Austausch
zwischen Diazastannol-2-ylidenen und Diazabutadienen gemäß Abb. 2.48, oder sind
eventuell Zwischenstufen beteiligt?
- Erfolgt der Austausch eines Zinnatomes reversibel ?
- Können Aussagen über die Geschwindigkeit des Austausches und ihre
Temperaturabhänigkeit gemacht werden?
- Können daraus die Aktivierungsparameter EA bzw. ∆S und ∆H bestimmt werden ?
Abb. 2.48: Dynamisches Gleichgewicht zwischen Diazabutadien 20c und Diazastannol-2-yliden 7c
N
N
Mes
Mes
N
NS n
Mes
Mes
+N
N
Mes
Mes
N
NSn
Mes
Mes
+k1
k-1
A B A B

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
48
a) Kinetische Untersuchungen an Diazastannol-2-ylidenen
1H -NMR-Spektren von Mischungen eines Diazastannols wie z.b. 7c mit dem entsprechenden
Diazabutadien 20c zeigen getrennte Signale beider Verbindungen (vgl. 2.49). 1H-EXSY-
Spektren solcher Mischungen zeigen jedoch bereits im Temperaturbereich von 0°C bis 30° C
das Auftauchen intensiver Austauschsignale, die den reversiblen, direkten Ausstauch eines
Zinnatoms zwischen beiden Molekülen ohne Bildung weiterer detektierbarer Zwischenstufen
belegt.
Abb. 2.49: Ausschnitt aus einem gradientenselektiven 1H-EXSY-NMR-Spektrum (300 MHz, C6D6, 30°C, Mischzeit 0.5 s). Die mit * bzw. + markierten Signale werden den NCH- und den C6H2Me3-Wasserstoffatomen im Diazadien 20c (R = Mes) bzw. im Heterocyclus 7c zugeordnet (R = Mes).
EXSY (Exchange Spectroscopy) ist eine Methode zur Untersuchung dynamischer
Gleichgewichte, in denen die Umwandlungsgeschwindigkeit im Bereich weiniger Sekunden
liegt, jedoch nicht so hoch ist, dass dynamisch induzierte Linienverbreiterungen oder
Koaleszensphänomene beobachtet werden können [95]. Daraus ergibt sich ein Zeitfenster für
k ~ 101 -105s-1[75]. Die auch als 2D-Austauschspektroskopie bekannte Methode verwendet
dieselbe Pulsfolge wie das NOESY-Experiment. In der Pulssequenz wird zunächst durch
einen 90°-Puls ein Kern A angeregt und transversale Magnetisierung erzeugt, die sich in der
* * + +
8.0 7.0
8.0
7.0
*

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
49
Evolutionszeit t1 mit der chem. Verschiebung δ(A) entwickelt. Durch einen zweiten 90°-Puls
wird diese Magnetisierung wieder in longitudinale Magnetisierung umgewandelt, die
während einer anschließenden Wartezeit tmix als Folge der Wirkung von NOE-
Wechselwirkungen oder eines chemischen Austausches auf einen anderen Kern B übertragen
werden kann. Diese transferierte longitudinale Magnetisierung kann durch einen dritten Puls
wieder in transversale Magnetisierung umgewandelt und detektiert werden. Wird eine Reihe
von Experimenten mit inkrementierter Evolutionszeit t1 durchgeführt so erhält man nach
zweimaliger Fourier-Transformation eine 2D-Matrix, in der ein Magnetisierungstransfer
zwischen zwei Kernen A nach B durch entsprechende Kreuzpeaks angezeigt wird. Zwischen
Korrelationen durch chem. Austausch und NOE-Transfer kann in der Regel durch die
Signalphase unterschieden werden: Austauschpeaks haben die gleiche Phase wie die
Diagonalsignale, während NOESY-Kreuzpeaks ein Signal mit negativer Phase geben.
Die Ermittlung der Geschwindigkeitskonstanten kAus der in Abb. 2.48 dargestellten Sn-
Austauschreaktion kann im durch Integration der Austauschsignale in EXSY-Spektren
erfolgen, gelingt aber noch einfacher durch 1D-Magnetisierungstransferexperimente nach der
Methode von Forsen und Hoffmann [96]. Dabei wird das Signal eines ausgewählten Kerns A
durch einen selektiven RF-Puls gesättigt oder invertiert und das NMR-Spektrum nach einer
Wartezeit t detektiert. Findet während dieser Zeit ein dynamischer Austausch zwischen dem
vorher angeregten Kern A und einem weiteren Kern B statt, so führt dieser Prozess zu einer
zeitabhängigen Modulation der Intensitäten beider Signale, die durch die Länge der Wartezeit
t, die Relaxationszeiten T1A und T1B sowie die pseudo-unimolekulare
Austauschgeschwindigkeit kobs = k1/[B] = k-1/[A] bestimmt wird und durch die so genannten
McConnell-Gleichungen [97] beschrieben werden kann:
MB(t) = MB(∞) .[ exp(-kobs.t) + T1
A/kobs]
MA(t) = [MA(0) – MA(∞)] .[exp(-kobs·t) + 1/k.T1B]
MA,B(0) = Magnetisierung bei t = 0
MA,B(∞) = Gleichgewichtsmagnetisierung
kobs = pseudo-unimolekulare Geschwindigkeitskonstante
T1 = longitudinale Relaxationszeit
Wird eine Reihe von Spektren mit variabler Wartezeit t aufgenommen, können die Parameter
kobs, T1A und T1B durch Fit der erhaltenen Signalintensitäten an eine durch Lösung der

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
50
McConnell-Gleichung erhaltenen Funktion M(t) erhalten werden. Die
Temperaturabhängigkeit der Geschwindigkeitskonstante kobs der Sn-Austauschreaktion
zwischen 7c und 20c wurde auf diesem Weg durch Aufnahme von Inversions-Recovery-
Experimenten bei verschiedenen Temperaturen zwischen 258 und 293 K ermittelt [98].
Temp. [K]
kobs [1/s]
Fehler [1/s]
258 2,68 0,13 263 4,03 0,20 268 5,17 0,52 273 6,89 1,38 278 9,27 1,85 283 13,0 5,22 288 15,1 3,02 293 19,2 5,76
Tab. 2.11: Temperaturabhängigkeit der Geschwindigkeitskonstante kobs
Zur Auswertung der Messwerte (s. Tab. 2.11) wurden die Größen ln(kobs) bzw. ln(kobs/T)
gegen die reziproke Temperatur aufgetragen 1/T und nach dem Modell von Arrhenius die
Aktivierungsenergie EA bzw. nach dem Ansatz von Eyring die Reaktionsentropie ∆S und
Reaktionsenthalpie ∆H durch Fit der experimentellen Daten an die Bestimmungsgleichungen
ln kobs = EA/RT + ln A (Abb. 2.50) bzw. ln (kobs/T) = 23.76-(∆H/R)*1/T + ∆S/R (Abb. 2.51,
mit R = 8.31 J*K-1) bestimmt.
Eyring-Modell
-5-4,5
-4-3,5
-3-2,5
-2-1,5
-1
0,0033 0,0034 0,0035 0,0036 0,0037 0,0038 0,0039 0,0041/T [1/K]
ln (k
/T) [
1/s*
K]
Abb. 2.50: Ermittlung von ∆H und ∆S der Sn-Austauschreaktion auf Basis der Eryring-Gleichung
∆S = -49.8 J/(K*mol) ∆H = 32.9 kJ/mol

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
51
Arrhenius-Modell
7
7,5
8
8,5
9
9,5
10
10,5
11
0,0033 0,0034 0,0035 0,0036 0,0037 0,0038 0,0039 0,0041/T [1/K]
ln k
[mol
/s]
Abb. 2.51: Ermittlung der Aktivierungsenergie ΕΑ auf Basis der Arrhenius-Gleichung
Zusätzlich wurde eine Verdünnungsreihe angesetzt, um den vermuteten bimolekularen
Mechanismus der in Abb. 2.48 dargestellten Sn-Transferreaktionen zu belegen. Hierzu wurde
davon ausgegangen, dass bei Annahme eines Geschwindigkeitsgesetzes 2. Ordnung für eine
bei einer bestimmten Temperatur im Gleichgewicht befindliche Lösung die Beziehung
[7c].[20c].k1 = const. erfüllt sein muss. Unter Berücksichtigung von k1 = kobs/[20c] folgt
daraus ebenfalls [7c].kobs = const.; d.h. das Produkt aus der NMR-spektroskopisch
beobachtbaren Zerfallsgeschwindigkeit und der Konzentration der zerfallenden Spezies sollte
unabhängig von der Größe der Konzentration im Idealfall eine Konstante darstellen. Erfolgt
ein bimolekularer Mechanismus des Sn-Transfers so ist der Quotient aus k(obs)/[20c] bei
verschiedenen Konzentrationen des Diazabutadiens [20c] im Idealfall konstant.
Erwartungsgemäß beobachtete man bei Verdünnung der Lösung eine Verminderung der
observierten Geschwindigkeitskonstante k(Obs.), da diese von der Konzentration des Diaza-
butadiens [20c] nach k(obs) = [20c] . kAus abhängen sollte.
ΕΑ = 32.0 kJ/mol

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
52
Konzentrationsabhängigkeit
0
5
10
15
20
25
30
0 0,05 0,1 0,15 0,2[mol/l] Diazadien
k(ob
s.)/[
20]
Abb. 2.52: Quotienten aus k(obs)/[20c] in Abhängigkeit der Konzentration [20c]
In Übereinstimmung mit dieser Annahme ergaben NMR-Untersuchungen an unterschiedlich
konzentrierten Lösungen von 20c/7c, dass mit zunehmender Verdünnung eine Abnahme der
Geschwindigkeitskonstante kobs zu beobachten war, Der experimentell erhaltene Quotient aus
k(obs) und [20c] kann unter Berücksichtigung der relativ hohen Standardabweichung als
konstant betrachtet werden (Abb.2.52) die erzielte Genauigkeit der einzelnen
Konzentrationsbestimmungen wird durch Signalüberlappungen, durch das Auftreten geringen
Mengen unlöslicher Zersetzungsprodukte verursachte unsymmetrische Signalformen und das
z.T. schlechte Signal/Rausch-Verhältnis ungünstig beinflusst). Das Ergebnis dieses
Experiments stützt damit die Annahme, dass die Sn-Transferreaktion wie vermutet einem
Geschwindigkeitgesetz 2. Ordnung gehorcht.
b) Quantenchemische Studien an Diazastannol-2-ylidenen
Um ein besseres Verständnis für die elektronischen Eigenschaften und die ungewöhnliche
Reaktivität der Diazastannol-2-ylidene zu erlangen, wurden DFT-Rechnungen an den
Modellverbindungen I-IV durchgeführt [99] und die Ergebnisse mit Hilfe von NBO-Analysen [100] interpretiert.

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
53
N
NE
H
H
N
NSn
Ar = 2,6-Me2C6H3
C I-HSi II-HGe III-HSn IV-H
N
N
N
N
H
H
VI-H VI-ArIV-Ar
Abb. 2.53: Modellverbindungen I-IV
Die NBO-Analyse der Kohn-Sham-Orbitale von I-H - IV-H ergibt einen hohen s-Charakter
(IV-H: 88%) für das als freies Elektronenpaar am Zinnatom identifizierte LMO und
impliziert damit einen geringen nucleophilen Charakter des Diazastannol-2-ylidens. Die
Besetzung des pπ(E)-Orbitals (0.57) in IV-H ist weitaus geringer als in ideal delokalisierten
Fünfringen mit 6π-Elektronen (pπ(E) = 1.2), aber immer noch höher als im Germylen III-H
bzw. im Silylen II-H (s. Tab. 2.13). Dies deutet auf einen stärkeren Beitrag an
π−Stabilisierung am Zinnatom hin und ist entgegengesetzt zum generell abnehmenden
Trend zur Ausbildung von π−Bindungen in höheren Perioden [101]. Dass dieser Trend nicht
allgemein gültig ist, konnte auch durch Untersuchungen von Grützmacher et. al an den
Kationen ([C3-n(XH)n] X= O, S, Se, Te ) gezeigt werden, die in der Reihenfolge S < Se < Te
eine zunehmende π−Konjugation zwischen den Chalcogenatomen und dem zentralen
Kohlenstoffatom aufweisen [102].
NBO-Populationsanalyse
E C (I-H) Si (II-H) Ge (III-H) Sn (IV-H)
Besetzung des pπ(E) –Orbitals
n[pπ(E)] 0.62 0.48 0.53 0.57
Wiborg-Bond-Indices (E-N)
WBI 1.27 0.79 0.77 0.70
Beitrag in % von E zum σ(E-N)-LMO
32.6 16.8 14.5 11.8
Tab. 2.13: Ausgewählte Resultate der NBO-Polulationsanalyse von Carbenen, Siylenen, Gemylenen und Stannylenen [103]

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
54
Ein Vergleich der Wiberg-Bindungsindices (WBI) und der Atombeiträge zu den lokalisierten
σ(E-N)-Orbitalen zeigt, dass die Bindungsordnung der E-N-Bindung vom Carben I-H zum
Stannylen IV-H kontinuierlich abnimmt und gleichzeitig die Polarität der σ-Bindungen steigt.
Dieser Effekt kann durch eine zunehmend stärkere Betonung der Grenzstrukur IV´ (Abb.
2.54) beschrieben werden. Die Analyse von auf DFT-Rechnungen basierenden
Elektronendichte-Konturliniendiagramme für II und III ermöglichten Arduengo et. al. schon
1996 den Vorschlag analoger Grenzstrukturen II´ und III´ für die entsprechenden Silylene
und Germylene [104]. Die dadurch implizierte Beschreibung der Heterocyclen als
„chelatisierte“ Metallatome dürfte insbesondere im Falle der Diazastannol-2-ylidene 7b bzw.
7c dem chelotropen Zerfall in Zinn und Diazabutadien unmittelbar förderlich sein.
Abb. 2.54: Grenzstrukur „chelatisierte“ Metalle
Experimentell zugängliche Verbindungen, für deren Beschreibung den Grenzstrukturen II´-
IV´ hohes Gewicht zukommt, finden sich auch in den durch Phenantrolinkomplexen 61 mit
einem formal nullwertigen Sn-Atom [105,104]. Analoge Germanium und Bleiderivate sind
ebenfalls bekannt.
Abb. 2.55: Phenanthrolin-E(0)-Komplexe 61
N N
E
Cr(CO)5(CO)5Cr
E = Sn Ge, Pb61
N
N
t-Bu
t-Bu
E
I-IV´
C I/ I´
Si II/ II´
Ge III/ III´
Sn IV/V´
N
N
E
t-Bu
t-Bu I-IV

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
55
Da die zuvor geschilderten experimentellen Untersuchungen den Schluss nahe legen, dass der
Transfer eines Sn-Atomes auf ein Diazabutadien über einen assoziativen Mechanismus
verläuft, wurden mit DFT-Methoden weiterhin die Energieprofile einer assoziativen Reaktion
von I-H - IV-H mit dem Diazabutadien VI berechnet [99].
Im Fall der Reaktion von IV mit VI (auf B3LYP/SDD-Niveau mit relativistischen
Pseudopotentialen am Zinn) wurden zwei stationäre Zustände auf der Energiehyperfläche
lokalisiert, von denen VIII-H einem lokalen Minimum (∆E + zpe = + 1.6 kcal mol-1) und
VII-H einem Übergangszustand (∆E + zpe = + 5.6 kcal mol-1) zwischen VIII-H und den
getrennten Molekülen zugeordnet werden kann.
Abb. 2.56: MOLDEN-Darstellung der Molekülstrukturen und berechnete Energieprofile für die Sn-Transferreaktion zwischen Diazabutadien und Diazastannol-2-ylidenen
Die Spiroverbindung VIII-H entspricht dem Produkt einer oxidativen Addition des
Diazabutadiens an IV und weist eine ψ−trigonal-bipyramidale Geometrie auf, die strukturell
eher SnII-Amidaten ähnelt [106]. Die oxidative Addition des Diazabutadiens an das Silylen II-
H, die zu einer D2d-symetrischen Si(IV)-Spiroverbindung führt, ist dagegen stark exotherm
(∆E + zpe = - 56.5 kcal mol-1). Die starke Abweichung von der tetraedischen Koordination
am Zinnatom in VIII-H kann auf die spitzen endocyclischen N-Sn-N Bindungswinkel und
die gleichzeitige Aufweitung der exocyclischen N-Sn-N Bindungswinkel zurückgeführt
werden. Die Einführung aromatischer Reste destabilisiert VIII-2,6-Me2C6H3 weiter, so dass
das vorherige lokale Minimum nun zu einem Übergangszustand der
Zinnübertragungsreaktion wird (∆E + zpe = 31.9 kcal mol-1) [103].
b) R = 2,6-Me2C6H3
0
10
20
30
40
IV + VI VII VIII VII VI + IV Reaction Coordinate
∆E + zpe kcal mol-1
a) R = H

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
56
d) Untersuchung von Germanium-Transferreaktionen an Diazagermol-2-ylidenen
Die theoretische Untersuchungen an den Carben-, Silylen-, Germylen- und Stannylen-
Modellverbindungen I-IV im vorigen Abschnitt) legt eine in der Reihe I-IV abnehmende
Stabilität der E-N-σ-Bindungen nahe. Auf der Basis dieser Ergebnisse erscheint für
Umsetzungen von Diazadienen mit Diazagermol-2-ylidenen sowohl eine Atom-
Transferreaktion als auch alternative eine oxidative Addition im Bereich des Möglichen. In
diesem Abschnitt soll daher der Frage nachgegangen werden, auf welche Weise
Diazagermol-2-ylidene mit Diazabutadienen reagieren können.
Als erste Umsetzungen wurden die Reaktionen der Diazagermol-2-ylidene 6c und 6b mit den
Diazabutadienen 20b und 20c unersucht (I und II, Abb. 2.57). NMR-spektroskopische
Untersuchungen an bis auf 80°C erwärmten Reaktionslösungen ergaben jedoch weder
Anhaltspunkte für die Bildung von Austauschprodukten noch von Additionsprodukten. Ein
zusätzlich aufgenommenes 1H-EXSY-Spektrum der Reaktionslösung II zeigte auch bei hoher
Temperatur keinerlei auf der NMR-Zeitskala nachweisbaren Austauschphänomene zwischen
Diazagermol-2-ylidenen und Diazabutadienen.
Abb. 2.57: Germaniumtranferreaktionen von Diazagermol-2-ylidenen
In Abschnitt 2.25 a) wurde der im Vergleich zu 7f deutlich leichter erfolgende cheletrope
Zerfall des Diazastannol-2-yliden 45f erwähnt und im Abschnitt 2.25 b) wurde festgestellt,
dass 4,5-Dimethyl-diazastannol-2-ylidene nicht durch Sn-Transfer erzeugt werden können.
Da beide Befunde auf eine Schwächung der Sn-Nσ-Bindungen durch einen destabilisierenden
Effekt der Methylgruppen hin deuten, könnte in 4,5-Dimethyl-diazagermol-2-ylidenen ein
analoger Effekt zur Schwächung der Ge-Nσ-Bindungen führen und dadurch den Transfer
eines Germaniumatoms auf Diazabutadiene gleichfalls erleichtern. Die NMR-
Reaktion R R´
I tBu Mes
II Mes tBu
N
N
R´
R´
N
NGe
R
R
+N
N
R
R
N
NGe
R´
R´
+

2. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 14
57
spektroskopische Untersuchung einer Reaktionslösung des 4,5-Dimethyl-2,3-dihydro-1-H-
[1,3,2]-diazagermol-2-ylidens 47c und des Diazabutadiens 20b (III, Abb. 2.58) zeigt in der
Tat, dass nach 8h bei 70°C durch Germanium–Transfer Diazagermol-2-yliden 6b entstanden
ist, das durch Vergleich der 1H, 13C und 15N NMR-Daten mit Literaturdaten zweifelsfrei
identifiziert werden konnte. Auch im Fall der Reaktion IV konnte das Diazagermol-2-yliden
6c durch Germanium–Transfer erzeugt und 1H-NMR-spektroskopisch anhand einer
authentischen Vergleichsprobe identifiziert werden. Kontrollreaktionen der Ddiazagermol-2-
ylidene 6c und 6b mit 20c führten nicht zu den erwarteten 4,5-Dimethyl-2,3-dihydro-1-H-
[1,3,2]-diazagermol-2-yliden 47c und 47b. Ebenso zeigten NOESY-Spektren, die von
Mischungen des Diazagermol-2-ylidens 47c mit Diazabutadien 20b aufgenommen wurden
keine Austauschphänomene, was auf eine in Relation zur NMR-Zeitskala zu geringere
Reaktionsgeschwindigkeit zurück geführt werden kann.
Abb. 2.58: Germaniumtranferreaktionen von 4,5-Dimethyl-2,3-dihydro-1-H[1,3,2]-diazagermol-2-ylidenen
Im Unterschied zu den geschilderten Sn-Transferreaktionen verlaufen die Reaktionen III und
IV demnach nicht in umgekehrter Richtung. Es konnte nicht abschließend geklärt werden, ob
diese Beobachtung durch eine Irreversibilität der Ge-Transferreaktion begründet ist oder
durch eine quantitative Verschiebung des Gleichgewichts auf die Seite eines der Produkte zu
erklären ist. Die im Vergleich zur analogen Übertragung eines Zinnatoms auffällig
langsamere Geschwindigkeit der Ge-Transferreaktionen III und IV kann dagegen als Folge
der im Gegensatz zu Sn-Nσ-Bindungen stärkeren Ge-Nσ-Bindungen in den Diazagermol-2-
ylidenen gedeutet werden und stützt die theoretischen Überlegungen aus Abschnitt 2.2.6 b).
Reaktion R R´
III Mes tBu
IV Mes Mes
N
N
R
R
N
NGe
R´
R´
+N
N
R´
R´
N
NGe
R
R
+70°C/ 8h

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
58
3. Untersuchungen zum Reaktionsverhalten von Diazastanna-
cyclopentenen
3.1 Einführung
3.1.1 Diazastannacyclopentene
Ungesättigte N-heterocyclische Verbindungen, die Zinn in der Oxidationstufe IV enthalten,
wurden erstmals durch eine [4+1]-Cycloaddition eines Stannylens mit Dizabutadien
dargestellt und spektroskopisch sowie strukturell untersucht. Das instabile nur intermediär
auftretende Stannylen konnte durch thermische oder photochemische Behandlung eines
Cyclotristannans 62 erzeugt werden [91].
Abb. 3.1: Die photochemische Behandlung eines Cyclotristannans führt zur Abspaltung eines Stannylens (SnR2), das mit Diazabutadien in einer [4+1] Cycloaddition weiter zum Diazastannacyclopenten reagiert.
Über eine eventuelle Reversibilität dieser [4+1]-Cycloaddition wurde weder im Hinblick
auf die Zinn(IV)verbindungen 63 noch auf homologe Germanium(IV) oder
Siliziumverbindungen (IV) berichtet. Die Ergebnisse aus Kapitel 2 lassen jedoch vermuten,
dass ein Zerfall der Zinn(IV)-verbindungen in Diazabutadien und SnR2 möglich sein
könnte. In diesem Kapitel soll daher untersucht werden, ob diese Vermutung zutrifft, und
ob es möglich ist, SnR2-Fragmente von Diazastannacyclopentenen direkt auf
Diazabutadiene zu übertragen. Des weiteren sollen NMR-spektroskopische Vergleiche
zwischen Diazastannacyclopentenen und Diazastannol-2-ylidenen gezogen werden.
R2S n
R2S nSnR2
hν, ∆:S nR2 + R2Sn=SnR2
NR´´RNN
N
SnR2
R´
R ́R = Dipp, R´= 2,6-Me2C6H362
63

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
59
3.1.2 Synthesemethoden
Vierwertige Silizium-Stickstoffverbindungen wie die Diazasilacyclopentene 64 können
durch Salzeliminierungsreaktionen aus lithiierten Diazabutadienen und Dialkyldichlor-
silizium(IV)-Verbindungen dargestellt werden. Solche Verbindungen werden in der
Literatur nicht als instabile Verbindungen beschrieben [52,53].
Abb. 3.2: Methoden zur Darstellung von Diazagermacyclopentenen (R = tBu)
Die Erzeugung von Diazagermacyclopentenen 65 wurde von Neumann et. al. durch [4+2]
Cycloaddition von in situ erzeugtem Ge(Me)2 mit Diazabutadienen oder durch
Salzeliminierungsreaktionen analog zur Darstellung der Silziumverbindung beschrieben [107]. In beiden Fällen konnte das Produkt offensichtlich nicht rein isoliert werden, da
dieSalzeliminierungsreaktion nur unvollständig abläuft und das entstehende Produkt
äußerst empfindlich gegenüber Sauerstoff und Feuchtigkeit ist und sich selbst in
ausgeheizten Glasgeräten unter Schutzgas zu Diazabutadien und (Me2GeO)n zersetzt.
Abb. 3.3: Reaktion der Diazagermacyclopentene 65 mit Wasser und Sauerstoff
Diazastannacyclopentene wurden durch die in Abb.3.1 beschriebene Reaktion von Neuman
et. al. dargestellt. Im folgenden Abschnitt sollen alternative Synthesemethoden untersucht
werden.
N
N
R
R
N
N
R
R
2 - 2Li+
NE
N
R
RMe
Me
2 Li Me2ECl2
-2 LiCl
+ :Ge(Me)2
E = Si(64),Ge(65)
N Ge N
R
RMe
Me
O2 ; H2O; ∆
N
N
R
R
+ (Me2GeO)
65

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
60
3.2 Eigene Untersuchungen
3.2.1 Versuche zur Darstellung von Diazastannacyclopentenen
Eigene Versuche zeigten, dass Diazastannacyclopentene vom Typ 63 nicht nur durch
Salzelimierung, sondern auch alternativ durch Reaktion eines α-Aminoaldimins 39c mit
Bis(diamino)-Zinn(IV)-dialkylen wie Me2Sn(NEt2)2 erzeugt werden können. Da in beiden
Fällen, insbesondere aber bei der Darstellung von 63b, als Nebenprodukte Diazabutadiene
entstehen, die eine Aufreinigung der Verbindungen erschweren oder sogar ganz
verhindern, konnten die Reaktionsprodukte 63b und 63c nicht isoliert werden. Die besten
Ergebnisse wurden durch Reaktion der Dilithiumsalze 21 mit Me2SnCl2 erzielt. Lösungen
von 63b enthielten zu einem geringen Prozentsatz als weiteres Nebenprodukt das 1,3-di-
tert-butyl-2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-yliden 7b. Eine Erklärung für dessen
Entstehung steht bislang noch aus.
Abb. 3.4: Methoden zur Darstellung der Diazastannacyclopentene 62
Die Produkte der Salzeliminierungsreaktionen wurden in der Reaktionslösung NMR-
spektroskopisch identifiziert. Die Konstitutionsaufklärung von 63b und 63c beruht
wesentlich auf der Beobachtung charakteristischer Korrelationen zwischen den
olefinischen Protonen im Fünfring und dem Zinnatom in zweidimensionalen
gradientenselektiven 1H-119Sn-NMR-. Die 119Sn-NMR-Verschiebung von 63b (66.6 ppm)
N
N
R
R
N
N
R
R
2 - 2Li +2 Li Me2SnCl2
-2 LiCl NSn
N
R
RMe
Me
63b,a
N
NH
Mes
Mes
R = t-Bu(b), Mes(c)
39c
Me2Sn(NEt2)2
- 2 NEt2

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
61
und 63c (67.7 ppm) liegen in dem für Diaminodialkylzinn(IV)-Verbindungen typischen
Spektralbereich [108].
Abb. 3 .5: 2D- gradientenselektive 1H-119Sn-NMR-Experimente zur Konstitutionsaufklärung der Zinn(IV)verbindungen 63b (links) und 63c (rechts)
Die Abschirmung des 119Sn-NMR-Signals der Diazastannacyclopentene gegenüber 2,3-
Dihydro-1-H-[1,3,2]-Diazastannol-2-ylidenen ist durch das Fehlen eines energetisch
tiefliegenden n π* Übergangs und dem daraus resultierenden deutlich kleineren
paramagnetischen Beitrag σpara zu erklären. Das Ausbleiben nennenswerter π(N-Sn)-
Wechselwirkungen in den Diazastanna(IV)cyclopentenen wird auch durch den Vergleich
der 15N-NMR-Daten von 63b (δ15N = -304.4) mit denen des Butendiamins 43c (δ15N = -
305.8) einerseits und der 2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-ylidene 7b und 7c
( δ15N = -111.9, -135.7 ppm) andererseits angedeutet.
240
160
80
0
(ppm)
160
-80
320
240
80
0
(ppm) 5.00 4.00 3.00 2.00 1.00 0.00
-80
(ppm) 6.00 5.00 4.00 3.00 2.00 1.00 0.00
(ppm)

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
62
3.2.2 Reaktionsverhalten
Direkte Transferreaktionen eines SnMe2-Fragmentes zwischen einem Diazabutadien und
einem Diazastannacyclopentenen können nicht festgestellt werden.
Diazastannacyclopentene 63c und 63b zersetzen sich allerdings gemäß der in Abb. 3.6
dargestellten Reaktion schon bei Raumtemperatur teilweise und bei längerer
Temperaturerhöhung auf 60°C vollständig zu Diazabutadien und Oligostannanen (SnMe2)n.
Abb. 3.6: Zersetzung der Diazastannacyclopentene zu Polystannanen
Die eindeutige Identifizierung der formulierten Thermolyseprodukte gelang durch NMR-
spektroskopischs Untersuchungen.
(a)
Abb. 3.7: 1H,119Sn-gsHMQC-NMR: 1H-entkoppelt
Während die Bildung von Diazabutadien bereits durch 1H-NMR-Spektren eindeutig belegt
wurde, konnte die Entstehung von Oligostannanen (SnMe2)n aus dem Auftreten mehrerer
Kreuzsignale in 1H,119Sn-gsHMQC-Spektren (s. Abb. 3.7) gefolgert werden. Der oligomere
Charakter wurde für das Hauptprodukt, das ein Signal mit δ119Sn = -233.3 ppm liefert,
NSn
N
R
RMe ∆
Me N
N
R
R
+ (Me2Sn)n
(ppm) 0.70 0.60 0.50 0.40 0.30
-216
-224
-232
-240
-248
(ppm)
63

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
63
anhand des Auftretens von 117Sn-Satelliten (Abb. 3.8, (b)) sowie von mehreren verschieden
großen 1H,119Sn-Kopplungen in einem gekoppelten 1H,119Sn-gsHMQC-Spektrum (Abb.
3.8, (c)) bewiesen. Alle gefundenen NMR-Daten sind in Tabelle 3.1 zusammengestellt.
(b) (c)
Abb. 3.8: 1H,119Sn-gsHMQC-NMR: 1H-entkoppelt( links) ; 1H-gekoppelt ( rechts )
Tab. 3.1: NMR-Daten der Produkte der Thermolyse aus 1H,119Sn-gsHMQC und 1H- DOSY NMR-Spektren
Produkt δ1H [ppm]
δ119Sn [ppm]
nJ(119Sn,117Sn) [Hz]
nJ(119Sn,1H) [Hz]
lg(D)
1 0.49 -247.2 3J = 18,7 2J = 42.8 -9.05
2 0.51 -243.0 3J = 19,6 2J = 42.0 -9.07
3 0.62 -233.3 1J = 905 2J = 253 3J = 19,6 2J = 41.8 -9.21
4 0.53 -212.6 3J = 19,1 2J = 42.6 -9.12
(Hz) 220 200 180 160 140
25200
25600
26000
26400
26800
(Hz)
(ppm)
(ppm) 0.60 0.40 0.20
-216
-224
-232
-240
-248

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
64
Abb. 3.9: 1H-DOSY-Spektrum der Thermolyseprodukte der Reaktion von ....
Unterschiedliche Molekulargewichte der einzelnen Produkte werden weiter durch die in
einem 1H-DOSY-Spektrum [109] (Abb. 3.9) der Mischung beobachtete Verteilung der
Diffusionskoeffizienten Dj nahe gelegt. Ein hoher Diffusionskoeffizient bedeutet dabei eine
höhere Beweglichkeit des Moleküls in der Lösung. Größere Moleküle besitzen einen
kleineren Diffusionskoeffizienten als kleinere Moleküle. Den kleinsten
Diffusionskoeffizient besitzt das als Komponente 3 bezeichnete Produkt, das anhand des
Vergleichs der NMR-Daten mit Literaturwerten als Hexamer (SnMe2)6 (δ119Sn = -231.0,
δ1H = 0.63, 1J(119Sn,117Sn) = 941 Hz, 2J(119Sn,117Sn) = 275 Hz) [110] identifiziert wurde. Die
Komponenen 4 und 1 bzw. 2 sollten aufgrund ihrer größeren Diffusionskoeffizienten
kleinere Molekulargewichte haben und wurden daher Cyclostannanen mit kleineren
Ringen zugeordnet. Komponente 4 konnte durch Vergleich der NMR-Daten mit den
Literaturwerten (δ119Sn = -241.4, δ1H = 0.53)[110] als Pentamer (SnMe2)5 identifiziert
werden. Die bevorzugte Bildung von cyclischen Stannanen mit mittlerer Ringgröße bei der
Zersetzung von 63b,c ist im Einklang mit Resultaten früherer Untersuchungen [110,111] an
Oligomerem bzw. Polymeren (SnR2)n. Dabei wurde gefunden, dass das kristalline
Hexamer (SnMe2)6 bei Temperaturerhöhung leicht eine Zersetzungsreaktion unter
Übertragung von Me2Sn-Fragmenten auf andere Ringe eingeht.
-8.96
-9.04
-9.12
-9.20
-9.28
-9.36
lg( Dj)
ppm 0.64 0.56 0.48 0.40

3. Untersuchungen zum Reaktionsverhalten von Diazastannacyclopentenen
65
Dadurch kann sich die Anzahl größerer und kleinerer Ringe auf Kosten von (SnMe2)6
erhöhen [112]. Kettenförmige Polystannane H(SnR2)nH sind andererseits instabiler als
Cyclostannane und zersetzen sich photochemisch hauptsächlich zu den cyclischen Penta-
oder Hexameren [110,113]. Cyclische Polystannane entstehen nicht nur aus langen
kettenförmigen Polystannanen, die gezielt synthetisiert werden müssen [114,115], sondern
auch aus Verbindungen die ein SnR2-Fragment unter Bestrahlung oder Erwärmung
eliminieren können [116].
SnMe2
H
SnMe2
H
<-75°CCH3 SnMe2
1/n (Me2Sn)2 Abb. 3.10: Eliminierung eines Stannylens SnMe2 aus Dimethylbenzylstannan
Im Licht dieser Befunde erscheint es nahe liegend, dass die als Hauptprodukte der
Thermolyse von Diazacyclopentenen bevorzugt entstandenen Cyclostannane (SnMe2)6 und
(SnMe2)5 die thermodynamisch am meisten begünstigten Produkte darstellen.

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
66
4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der
Gruppe 15
4.1 Einführung
4.1.1 „Arduengo“-Carbenanaloga mit dem Element Phosphor: Darstellung von
1,3,2-Diazaphospholenium-Kationen
Carbenanaloge Moleküle mit den Elementen der Gruppe 15 erhält man durch den
formalen Austausch des C-Atoms gegen ein positiv geladenes Pnicogenatom. Auf diese
Weise entspricht dem Carben :CH2 .das isolelektronische Phospheniumkation +PH2.
Stabile cyclische Phosphenium-Kationen 66i, welche dem „Arduengo“-Carben analog
sind, wurden erstmals 1988 von Pudovik et. al. aus dem 1,3,2-Diazaphospholen 67i[Cl]
durch Halogenid-Abstraktion mit Lewis-Säuren dargestellt [117].
Abb. 4.1: Darstellung des 1,3,2-Diazaphospholenium-Kations 66i nach Pudovik et al. [117]
Alternativ dazu wurden die 1,3,2-Diazaphospholenium-Kationen 11 durch Umsetzung
der 1,3,2-Diazaphospholene 11b[Cl] oder 11c[Cl] mit TMSOTf erhalten [118].
Abb. 4.2: Darstellung von 1,3,2-Diazaphospholenium-Kationen 11 mit TMSOTf [118]
R = tBu (b), Mes (c)
NP
N
R
R
NP
N
R
R
ClTMSOTf
OTf
11Cl 11[OTf]
-TMSCl
NP
N
c-Hex
c-HexCl
N P
N
c-Hex
c-Hex C l
ClSbCl5 SbCl6
67i[Cl] 66i[SbCl6]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
67
Die als Vorstufen für die Kationen benötigten 1,3,2-Diazaphospholene 67[Cl] wurden
entweder durch Umsetzung von 1,4-Diazabutadienen mit PCl3 in Gegenwart von
Triethylamin [117,118] oder durch Methathese von 2,2-dichloro-1,2,dihydro-1,3,2-
diazasilolen 22 mit PCl3 erhalten [119]. Im ersten Fall wird gleichzeitig ein Cl-Substituent
am C-4-Atom des Fünfringes eingeführt.
Abb. 4.3: Darstellung von 1,3,2-Diazaphospholenen nach Pudovic et. al. [117] oder Denk et. al. [119]
Nach einer weiteren Vorschrift von Cowley et. al. soll das C-H-substituierte 1,3,2-
Diazaphospholen 11b[Cl] wie auch das entsprechende Diazaarsolen auch durch
Salzeliminierung aus dem Diazabutadien-dilithiumsalz 21b und PCl3 zugänglich sein [34].
Abb. 4.4: Methode zur Darstellung von 1,3,2-Diazaphospholenen und -arsolenen nach Cowley et. al. [34]
NP
N
t-Bu
t-Bu
Cl PCl3
N
N
t-Bu
t-Bu
2 2 Li
11b[Cl] 21b
NP
N
R
R
ClPCl3
NSi
N
R
R
ClCl
NP
N
R
RC l
Cl
R = Mes (c), tBu(b), c-Hex(i)
N
N
R
R
PCl3 / NEt3
-NEt3HCl
20 67[Cl]
R = Mes (c), tBu(b)
22 11[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
68
4.1.2 „Arduengo“-Carbenanaloga mit den Elementen Arsen und Antimon: 1,3,2-
Diazarsolenium-Kationen und 1,3,2-Diazastibolenium-Kationen
1,3,2-Diazarsolenium-Kationen und 1,3,2-Diazastibolenium-Kationen wurden als höhere
Homologe der „Arduengo“-Carben-analogen 1,3,2-Diazaphospholenium-Kationen im
Gegensatz zu diesen bisher kaum erforscht.
RN
NR
Sb
13
RN
NR
Bi
14
RN
NR
As
12
Abb. 4.5: 1,3,2-Diazastibolenium- arsolenium und bismutenium- Kationen
Ein erstes 1,3,2-Diazarsolenium-Salz 12b[GeCl5-] wurde in einer Metathesereaktion,
deren Mechanismus nicht aufgeklärt werden konnte, durch Umsatz eines 1,3,2λ2-
Diazagermanols mit AsCl3 erhalten und als Gemisch mit dem 1,3,2-Diazarsolen 12b[Cl]
isoliert. Die Autoren berichten über eine röntgenstrukturanalytische Untersuchung eines
Mischkristalls beider Verbindungen, die jedoch ohne zufriedenstellende Ergebnisse blieb [34]. Eine genaue Untersuchung der Bindungstruktur in 1,3,2-Diazarsolenium-Kationen
und 1,3,2-Diazarsolen steht daher noch aus.
Abb. 4.6: Reaktion eines Diazagermanols mit AsCl3 zu einem Gemisch aus dem 1,3,2-Diazarsolenium-Salz 12b[GeCl5] und dem 1,3,2-Diazaarsolen 12b[Cl]
N
NAs
tBu
tBu
GeCl5
12b[GeCl5]
N
NGe
tBu
tB u
22 AsCl3
N
NA s-C l
tBu
tB u
+
6b 12b[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
69
Bekannt sind weiterhin die gesättigten 1,3-Diaza-2-arsolidenium-Kationen 69 [120,121] und
70 [122] sowie einige 2-Chloro-2-arsa-1,3,2-diazarsolidine 68 [34,122], die im Fall von 68j [122] und 68b [34] röntgenstrukturanalytisch untersucht werden konnten.
Abb. 4.7: Literaturbekannte Arsenium-Kationen
„Arduengo“-Carben analoge Bismutkationen sind bislang unbekannt. Cyclische neutrale
und ionische 1,3,2,4λ2-Diazasilaphosphetidine, -arsetitine, -stibetidine und -bismutetidine
71E, 72E wurden jedoch in der Arbeitsgruppe von Veith eingehend untersucht [123,124].
Abb. 4.8: Cyclische kationische und neutrale 1,3,2,4λ2-Diazasilaphosphetidine, -arsetitine, -stibetidine und -bismutetidine
NMe2S i
NE-Cl
tBu
tB u
E = P, As, Sb, Bi
NMe2Si
NE:
tBu
tBu
AlCl4
71E 72E
N
N As-Cl
R
R
N
NAs
R
R
N
NAs
Me
Me
GaCl4
2
OTf
R = tB u(b), Me(j) R = Et(k), Me(j)
6 8 69 70

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
70
4.1.3 Struktur und elektronischer Zustand von Phosphenium-Kationen und 1,3,2-
Diazaphospholenen
a) Grenzorbitalbetrachtung an den acyclischen Kationen +PH2 und [+P(NH2)]2
Die Stammsysteme der Phospheniumkationen und Carbene, +PH2 und CH2, ähneln sich
im Aufbau ihrer Frontorbitale. Beide besitzen als HOMO ein „lone-pair“ (a1) und ein
darauf senkrecht sitzendes unbesetztes p-Orbital als LUMO (b1).
PH
H
:
b1
a1
Abb. 4.9: Frontorbitale des Stammsystems +PH2
+PH2 kann daher als sowohl isoelektronischer als auch isolobaler Verwandter des
Methylens CH2 bezeichnet werden. Hieraus lässt sich die Annahme rechtfertigen, dass
Carbene und Phospeniumkationen im Prinzip vergleichbares Reaktionsverhalten
aufweisen sollten.
NH2
PNH2
1b1(π1)
a1(n)
a2 (π2)
2b1(π3)
Abb. 4.10: Frontorbitalsequenz von [P(NH2)2]+

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
71
Im einfachsten Diaminophosphenium-Ion [P(NH2)2]+ zeigt sich im Gegensatz zu den
Stammverbindungen der Carbene und Phospheniumkationen eine abweichende Abfolge
der Frontorbitale [125]. Die Bindungssituation in [P(NH2)2]+ (Abb.4.10) kann am besten im
Sinn einer heteroallylischen 4-Elektronen-3-Zentren-Bindung beschrieben werden, deren
Frontorbitale eher einem Allyl-Anion als einem Carben ähneln [126]. Durch eine induktive
Destabilisierung des a1-Orbitals bei Einführung von Alkylgruppen könnte eine Kreuzung
der 1b1- und a1-Orbitale eine Carben-ähnliche Frontorbitalsequenz wiederherstellen [127].
b) Struktur und elektronischer Zustand in 1,3,2-Diazaphospholenium-Kationen
Rechnungen auf MP2/6-31+g(d)-Niveau prognostizieren im Falle des cyclischen 1,3,2-
Diazaphospholenium-Kations 11 sowohl für das LUMO als auch HOMO einen b1(π)-
Charakter.
Abb. 4.11: Frontorbitalsequenz von 11
Damit ergibt sich für die „Arduengo“-Carben analogen 1,3,2-Diazaphospholenium-
Kationen eine ähnliche Frontorbitalsequenz wie in [P(NH2)2]+ [125].Die Analyse von
Photoelektronenspektren zeigte, dass in „Arduengo“-Carbenen die erste
Ionisierungsbande einer Ionisation aus dem "lone-pair“ zugeordnet werden kann, das
demzufolge unter Annahme der Gültigkeit von Koopmans Theorem [128] das HOMO sein
sollte. In den Silylenen 5 und Germylenen 6 wird das "lone-pair“- Orbital dem HOMO-1
zugeordnet. Das HOMO ist hier wie im [P(NH2)2]+ ein Orbital mit b1-Symetrie, das einer
π− -Kombination der freien Elektronenpaare am Stickstoff entspricht [104]. Heineman et.
al. zeigten hingegen, dass Hartree-Fock-Rechnungen für N-heterocyclische Carbene und
Silylene ebenfalls ein b1-Orbital als HOMO voraussagen [47]
N
NP
N
NP
b1(π*)
b1(π3)
N H
P
H N
11

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
72
Die Abweichung zwischen experimentell gefundenen Ionisierungsenergien und
theoretisch berechneten Orbitalenergien für die Carbene resultiert offenbar aus einer
Verletzung von Koopmans Theorem. Dies wird durch weitere Rechnungen auf
PUMP4/6-31G(d) oder CCSD(T)6-31G(d)-Niveau von Heineman et. al. bestätigt [48].
Diese Rechnungen, die die Vernachlässigung der Elektronenkorrelation in den Hartree-
Fock-Rechnungen kompensieren können, zeigen eine gute Übereinstimmung mit den
experimentell gefundenen Ionisierungsbanden und sagen in der Tat für die Carbene und
die Silylene als HOMO ein b1(π)-Orbital voraus.1,3,2-Diazaphospholenium-Kationen
scheinen daher in ihren Frontorbitalen den „Arduengo“-Carbenen und deren höheren
Homologen zu ähneln.Wie in isoelektronischen N-heterocyclischen Carbenen und deren
höheren Homologen (s. S.10) kann auch in ungesättigten 1,3,2-Diazaphosphenium-
Kationen eine energetische Stabilisierung im Sinne einer 6π (C2N2) π (E+)-
Wechselwirkung eintreten. Berechnete isodesmische Methatese-Reaktionen, bei denen
das 6π-Elektronen-System aus einer 4π-X2E-Einheit und einem Ethylen-Fragment
aufgebaut wird, zeigen das die berechneten Delokalisierungsenegien für die „Arduengo“-
Carben analogen Spezies ungefähr halb so groß sind wie für ein isoelektronisches
Phospholid-Anion, dessen aromatischer Charakter unstrittig ist. Dadurch kann gezeigt
werden, dass der durch eine Aromatisierung bedingte Stabilitätszuwachs in N-
heterocyclischen Carbenen und 1,3,2-Diazaphospholenium-Kationen im Vergleich zu
den stärkeren Effekten der N E π-Wechselwirkung und E-N-Bindungspolarität in
acyclischen oder gesättigten cyclischen Systemen eher gering ist [125].
XE
X+
XE
X+ + ∆E
NH
C
HN
NH
Si
HN
NH
P
HN
NH
P
HN
∆E[kcal/mol]: -25,7 -24,2 -28,8 -58,8 Abb. 4.12: isodesmische Reaktionen zur Beschreibung der Wechselwirkung zwischen einer 4π-X2E- Einheit und einem Ethylen-Fragment als Modell für die aromatische Stabilisierung in Analoga von „Arduengo“-Carbenen und Phospholiden

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
73
c) Struktur und elektronischer Zustand von 1,3,2-Diazaphospholenen
Die elektronische Situation in 1,3,2-Diazaphospholenen ähnelt der von 1,3,2-
Diazaphosphenium-Kationen insoweit, als dass in beiden Fällen eine Wechselwirkung
eines 6π(C2Ν2)-Fragments mit einem am Phosphoratom lokalisierten Orbital formuliert
werden kann. In 1,3,2-Diazaphospholenen entspricht dies einer Hyperkonjugation des
6π(C2N2 )-Fragments mit dem σ*-Orbital der P-Cl Bindung [118]. Das σ*(P-Cl)-Orbital
wird auf diese Weise mit Elektronen populiert, wodurch eine Verlängerung der P-Cl-
Bindung und Verkürzung der P-N-Bindung induziert wird. Als weitere Folge sollte in
Analogie zur 6π (C2N2) => π (E) Wechselwirkung ebenfalls eine Verlängerung der C-C-
und eine Verkürzung der C-N-Bindungen im Heterocyclus resultieren.
Abb. 4.13: Modell der 6π(C2N2) σ*(P-Cl)-Hyperkonjugation in 1,3,2-Diazaphospholenen nach [118]
Die röntgenstrukturanalytische Untersuchung von Verbindung 66i[Cl] weist in der Tat
eine ungewöhnlich lange P-Cl-Bindung auf (241.6 pm), die auf eine Wechselwirkung der
lone-pairs der Stickstoffatome [117] mit dem σ*-Orbital der P-Cl Bindung zurückgeführt
werden kann. Das Diazaphospholen 11b[Cl], deren Strukturdaten 1999 publiziert
wurden, zeigt eine noch stärkere Verlängerung der P-Cl–Bindung (275.9 pm). Die
Autoren weisen der P-Cl –Bindung in 11b[Cl] daher einen überwiegenden ionischen
Charakter zu und beschreiben 11b[Cl] somit als aromatisch stabilisiertes
Phospheniumkation [33]. Es konnte jedoch durch Leitfähigkeitsmessungen eindeutig
gezeigt werden, dass 11b[Cl] über eine kovalente P-Cl Bindung verfügt [118]. Weitere
vergleichende röntgenstrukturanalytische Untersuchungen an N-Aryl-Diazaphospholenen
zeigen wesentlich kürzere P-Cl Bindungen (224.3 pm [129] und 232.4 pm [118]), so dass
Diazaphospholene eine große Variation hinsichtlich ihrer P-Cl Bindungen aufzuweisen
scheinen. Die außergewöhnlich lange P-Cl-Bindung in 11b[Cl] ist daher eher auf
N
N
P
Cl
N
P
N
c-Hex
c-Hex
ClCl
67i[Cl]
N
P
N
tB u
tBu
Cl
11b[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
74
induktive Substituenteneffekte an den Stickstoffatomen als auf π-Konjugationseffekte
zurückzuführen. Die Ausbildung eines cyclisch delokalsierten 6π-System und eine
spontane Ionisation der P-Cl-Bindung im 1,3,2-Diazaphospholene 11b[Cl] kann aufgrund
dieser Erkenntnisse letztendlich nicht bestätigt werden [118]. Dennoch ist die Ähnlichkeit
von 1,3,2-Diazaphospholenen und 1,3,2-Diazaphosphenium-Kationen Als Folge von π-
(Hyper)konjugationseffekten nicht zu übersehen und wird insbesondere durch teilweise
analoge Reaktivitäten deutlich [118].
3.1.4 Das Reaktionsverhalten von 1,3,2-Diazaphospholenen, Phospheniumkationen
und Analoga mit den Elementen Arsen Antimon und Bismut
1,3,2-Diazaphospholenium-Kationen und ihre höheren Homologen können wie N-
heterocyclische Carbene prinzipiell sowohl elektrophil über ihr hoch liegendes LUMO
als auch nucleophil als Lewis-Basen über das „lone-pair“ reagieren. In Kapitel 2 wurde
bereits angesprochen, dass N-heterocyclische Carbene ausschließlich als Lewis-Basen
reagieren und einen nucleophilen Charakter aufweisen. Für andere bekannte
Phosphenium-Kationen und Arsenium-Kationen dominiert dagegen im Allgemeinen das
Lewis-saure Reaktionsverhalten [130,131,122]. Acyclische Diaminoposphenium-Kationen
sind dabei stärkere Lewis-Säuren sind als acyclische Diaminoarsenium-Kationen,
während in cyclischen Systemen der Lewis-saure Charakter der Diaminoposphenium-
Kationen abnimmt und ähnlich groß wird wie in cyclischen Diaminoarsenium-Kationen [122,120]. Die im vorherigen Abschnitt angesprochenen Variationen der Frontorbitale in
Phospheniumkationen scheinen damit auf das Reaktionsverhalten keinen Einfluss zu
haben, da dieses im wesentlichen durch das LUMO bestimmt wird und die Abfolge der
weiteren Grenzorbitale somit nur von geringerer Bedeutung ist [125].
Sowohl 1,3,2-Diazaphospholenium-Kationen als auch N-heterocyclische Carbene
reagieren als Liganden in Übergangsmetallkomplexen [132]. 1,3,2-Diazaphospholenium-
Kationen können durch Kombination ihres „lone-pairs“ mit einem Orbital passender
Symmetrie am Metall nicht nur eine dative L M-σ-Bindung, sondern durch
Kombination des LUMO mit einem gefüllten Metall-d-Orbital zusätzlich eine M L-π-
Rückbindung ausbilden.

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
75
Abb. 4.14: Das Reaktionsverhalten der Kationen ist elektrophil und wird durch ihr tiefliegendes LUMObestimmt.
Im Gegensatz zu den in Kapitel 2 beschriebenen Carbenen ist die M L π-Rückbindung
für die Stabilität eines Phospholenium-Komplexes 73 essentiell, da hierdurch der durch
die positive Ladung im Liganden bedingte Elektronenmangel ausgeglichen werden kann [133]. Die Hinbindung spielt aufgrund der verringerten Nucleophilie der
Phospheniumkationen nur eine untergeordnete Rolle. Trotz der gleichen Tendenz
nucleophiler Carbene und elektrophiler Phospheniumkationen, als 2e-Liganden
Übergangsmetallkomplexe zu bilden, werden auch hier grundsätzliche Unterschiede
sichtbar.
Da in Carben-Metallkomplexen die dative L M-σ-Bindung und in Phosphenium-
kationen-Metallkomplexen die M L-π-Rückbindung dominiert, können weitere
Liganden am jeweiligen Metallatom die Stabilität des gesamten Komplexes auf
unterschiedliche Weise beeinflussen [133,134].
Kationische 1,3,2-Diazaphospholenium-Komplexe sollten instabiler werden, je
mehr π−Akzeptoren am Metall gebunden sind und je stärker deren π-Acidität ist.
Nucleophile Carbenkomplexe sollten durch die gleichen Effekte nicht destabilisiert
sondern stabilisiert werden.
Die Erhöhung der Anzahl von reinen σ-Donorliganden (z.b. Aminen) führt zu
einem Anstieg der Elektronendichte am Metall. Dadurch steigt die Stabilität der
1,3,2-Diazaphospholeniumkomplexe und es sinkt im Gegensatz dazu die Stabilität
nucleophiler Carben-Komplexe.
NP
N
Mes
MesCl
MOC
N
CO
N
M = W ,Mo
NP :
N
Mes
MesC l
CO
:B
73

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
76
Für kationische 1,3,2-Diazaphosphenium Komplexe gilt weiterhin [133]:
Cyclische Phospholenium-Kationen bilden stabilere Komplexe als acyclische
Phospholenium-Kationen
Komplexe mit Mo und W sind stabiler als solche mit Cr.
Das 1,3,2-Diazaphospholen 67c[Cl] bildet trotz seiner erwiesenermaßen kovalenten P-Cl-
Bindung analog zu den 1,3,2-Diazaphospholenium-Kationen kationische Übergangs-
metallkomplexe 73, die unter Substitution von CO durch Chlorid zu den entsprechenden
neutralen Komplexverbindungen 74 weiterreagieren können. Die Gesamtreaktion
entspricht einer Metall-Insertion in eine P-Cl-Bindung [132].
Abb. 4.15: Reaktionen des 1,3,2-Diazaphospholens 67c[Cl] mit [(MeCN)M(CO)3bpy]
Ebenfalls neutrale Komplexverbindungen 75, die röntgenstrukturanalytisch
charakterisiert werden konnten, erhält man durch Reaktion des 1,3,2-Diazaphospholens
11c[Cl] mit TlCo(CO)4 unter TlCl–Elimination [135].
NP
N
Mes
MesCl
MOC
N
CO
N
M = W ,Mo
NP
N
Mes
MesCl
C l CO [( MeCN)M(CO)3bpy]
-MeCN
Cl
NP
N
Mes
MesCl
MOC
N
CO
NC l
-CO
73
74
67c[C l]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
77
Abb. 4.16: Reaktion des 1,3,2-Diazaphospholens 11c[Cl] mit TlCo(CO)4
Die von Veith et. al. beschriebenen cyclischen 1,3-Di-tert-butyl-2,2-dimethyl-4-chloro-
1,3,2,4λ2-diazasilastibetidine und bismutetidine 71Sb bzw. 71Bi reagieren analog zu den
1,3,2-Diazaphospholenen zwar mit anionischen Carbonylkomplexen zu neutralen
Komplexverbindungen 76Sb und 76Bi [136], die allerdings als metallsubstituierte
dreiwertige Sb- bzw. Bi-Verbindungen mit einer reinen M-E-σ-Bindung zu beschreiben
sind. Das freie Elektronenpaar am Element E nimmt nicht an der Wechselwirkung mit
dem Metall teil, und es resultiert damit eine pyramidale Geometrie am Element E.
Analoge „Metallophosphane“ wurden vor allem von Malisch et. al. als Vorstufen zu
Pospheniumkomplexen mit planar koordinierten Phosphoratomen beschrieben [137].
Abb. 4.17: Reaktion cyclischer Diaminoelementchlorverbindungen mit anionischen Carbonylkomplexen
NMe2 Si
NE-Cl
tBu
tBu
E = Sb, B i
-NaCl
M = Fe, n = 2M = Mo, n = 3M = W, n = 3
NMe2Si
NE -
tBu
tBu
N aM(CO)nCpM(CO)nCp
71E 76E
NP
N
Mes
Mes
C l+TlCo(CO)4
NP
N
Mes
Mes
Co
CO
C O
CO- TlCl
11c[Cl] 75

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
78
4.2 Eigene Untersuchungen
In den folgenden Kapiteln sollen die im vorigen Abschnitt beschriebenen Erkenntnisse
dazu herangezogen werden, auch die Bindungsverhältnisse in den höheren Homologen
der 1,3,2-Diazaphospholenium-Kationen und 1,3,2-Diazaphospholenen zu verstehen.
Insbesondere soll anhand von röntgenstrukturanalytischen, schwingungs-
spektroskopischen und NMR-spektroskopischen Studien sowie Untersuchungen zur
Koordinationschemie ein Einblick in die π-Elektronenstruktur von 1,3,2-
Diazastibolenium und -arsolenium-Kationen bzw. 1,3,2-Diazastibolen und -arsolen
gewährt werden. Es soll dabei im Einzelnen geklärt werden, welchen Einfluss das
Gruppe-15-Element in den Kationen bzw. die Atome in der E–X Bindung in den
Halogenverbindungen auf die Delokalisation im 6π-Elektronensystem bzw. die
6π(C2Ν2) σ*(E-X)-Hyperkonjugation in den höheren Homologen der 1,3,2-
Diazaphospholene ausüben. Um solche Aussagen zu treffen, bedarf es
Referenzsubstanzen, in denen π-Delokalisation nur im π(C2N2)-Fragment auftritt.
Verbindungen, die diese Voraussetzung erfüllen, sind die in Abb. 4.18 aufgeführten
Diaminoethylene bzw. 2-λ4-1,3,2-Diazasilole.
Abb. 4.18: Verbindungen in denen π-Delokalisation auf den π(C2N2)-Bereich beschränkt sein sollte.
NSi(CH3)2
N
c-Hex
c-Hex
NSi
N
dipp
dipp
22f
Cl
Cl
64i
NH
NH
dipp
dipp
43f

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
79
4.2.1 Synthesemethoden für 1,3,2-Diazaarsolene und -Stibolene
1,3,2- Diazastibolene 13[Cl] sind trotz tiefer Temperaturen und sorgfältiger Reaktions-
führung nicht durch Salzeliminierungsreaktionen mit SbCl3 zugänglich [138].Eine einfache
Methode zur Darstellung von sowohl 1,3,2-Diazarsolenen 12[Cl] als auch –stibolenen
13[Cl] ist die Reaktion der in Kapitel 2 beschriebenen α-Aminoaldimine mit AsCl3 bzw.
SbCl3 oder mit ClSb(NMe2)2 bzw. ClSb(N(SiMe3)2)2 . 1,3,2-Diazarsolene 12b,c[Cl] und
das 1,3,2- Diazastibolen 13b[Cl] erhält man dabei durch Reaktion der α-Aminoaldimine
mit AsCl3 bzw. SbCl3 und zwei Äquivalenten NEt3 glatt und in relativ guten Ausbeuten
von 40-60%.
Abb. 4.19: Methoden zur Darstellung von 1,3,2-Diazaphospholenen , 1,3,2-Diazarsolenen und 1,3,2- Diazastibolenen
Das 1,3,2- Diazastibolen 12c[Cl] konnte auch durch eine Transaminierungsreaktionen
aus ClSb(NMe2)2 dargestellt und mit 57% Ausbeute isoliert werden. Das Diaminoethan
43f reagiert mit ClSb(NMe2)2 ebenfalls zum 4,5-Dimethyl-substituierten Diazastibolen
13f[Cl] Eine Isolierung des 1H-NMR-spektroskopisch identifizierten Produktes aus
den Reaktionsgemischen gelang jedoch nicht. Das 2-Bromo-1,3,2-diazastibolen
NE
N
R
R
ClNH
N
R
R
ECl3 / 2 NEt3 - 2 NEt3HCl
NH
NH
Dipp
Dipp
13f[Cl]N
Sb-Cl N
Dipp
Dipp
ClSb(NTMS2)2
- 2HNTMS2
ClSb(NMe2)2
- 2HNMe2
ClSb(NMe2)2
- 2HNMe2
12[Cl] E = As; R = tBu(b),Mes(c) 13[Cl] E = Sb; R = tBu(b) 13[Cl] E = Sb; R = tBu(b)
13[Cl] E = Sb; R = Mes(c)
43f

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
80
13b[Br] lässt sich analog durch Reaktion des α-Aminoaldimin-Komplexes 39[LiBr] mit
SbBr3darstellen.
NSb-Br
N
tBu
tBu
N
N
tBu
tBu
LiBrSbBr3 / 2 NEt3
-NEt3HBr NSb
N
tBu
tBu
IN
Sb-ClN
tBu
tBu
+TMSI
-TMSCl
39[LiBr] 13b[Br] 13b[I]13b[Cl]
Abb. 4.20: Darstellung des 2-Bromo-1,3,2-diazastibolens 13b[Br]
Umsetzung des Chlor-1,3,2- Diazastibolen 13b[Cl] mit Trimethylsilyliodid führt zu den
entsprechenden 2-Iodo-1,3,2- Diazastibolen 13b[I].
Reaktionen von BiCl3 mit α-Aminoaldiminen ergaben schwarze Reaktionslösungen, in
denen 1H-NMR-spektroskopisch Diazabutadiene als einzige Reaktionsprodukte
nachgewiesen werden konnten.
Die Darstellung von Salzen mit 1,3,2-Diazastibolenium und 1,3,2-Diazarsolenium-
Kationen erfolgte analog durch Zugabe stöchiometrischer Mengen von TMSOTf oder
Lewis-Säuren wie AlCl3, GaCl3 oder SbCl3 zu Lösungen der 2-Chloro-1,3,2-
diazastibolene oder –arsolene 12[Cl] oder 13[Cl] in Ether oder Methylenchlorid-Ether-
Mischungen. Die zunächst in amorpher Form ausfallenden Feststoffe wurden aus
Acetonitril/Ether umkristallisiert.
Abb. 4.21: Methoden zur Darstellung von 1,3,2-Diazastibolenium und -arsolenium-Salzen
NSb-Cl
N
t-Bu
t-Bu
NSb
N
t-Bu
t-BuECl3Ether ECl4
E = Al (13[AlCl4]), Ga (13[GaCl4]), Sb (13[SbCl4])
NE
N
R
R
ClN
EN
R
R
Ether
TMSOTf-TMSCl O Tf
R = tBu(b) ; E = Sb (13b[O Tf]), As (12b[OTf])
R = Mes(c) ; E = Sb (13c[O Tf]), As (12c[OTf])
13b[Cl] 13b[ECl4]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
81
a
bc
ClSbNCH
4.2.2 Röntgenstrukturanalytische Untersuchungen
a) Kristallstrukturen der Diazastibol(arsol)ene und Diazastibol(arsol)enium-Kationen
Strukturdaten konnten vom 2-Chloro-1,3-di-tert-butyl-1,3,2-diazastibolen 13b[Cl] und
vom 2-Bromo-1,3-di-tert-butyl-1,3,2-diazastibolen 13b[Br] erhalten werden. 1,3,2-
diazastibolen 13b[Cl] liegt im Festkörper in Form isolierter Moleküle vor, die relativ
schwache intermolekulare Sb…Cl-Kontakte (386.2 pm) aufweisen. Sekundäre Sb…Cl-
Kontakte unterhalb der Summe der Van der Waals Radien von Chlor und Antimon (400
pm) werden in der Literatur von 334 pm bis 397 pm diskutiert [139,140,123, 141].
Abb. 4.22: Elementarzelle von 13b[Cl]: Orthorombisches Kristallsystem; Raumgruppe Pbca (Nr.62)
Die C2N2Sb-Einheit im 1,3,2-Diazastibolen 13b[Cl] ist planar aufgebaut und besitzt
keine „envelope“-Konformation wie der Ring im homologen tert-Butyl-1,3,2-
diazaphospholen 11b[Cl], sondern entspricht eher der Struktur des 1,3,2-Diaza-
phospholens 11c[Cl].
Die Stickstoffatome sind planar und das Antimonatom ist pyramidal koordiniert. Die Sb-
Cl Bindung steht im Winkel von etwa 99.8° zur C2H2N2Sb-Ebene. Die Sb-Cl Bindung
in 13b[Cl] ist mit 264.60(15) pm ca 13% länger als in SbCl3 in der Gasphase (233
pm)[68] sowie ca 12% länger als die mittlere Sb-Cl-Bindungslänge in festem SbCl3 (235.9
pm)[140]. Gegenüber dem cyclischen 1,3-Di-tert-butyl-2,2-dimethyl-4-chloro-1,3,2,4λ2-
diazasila-stibetidin 71Sb (Sb-Cl 247.2 (3) pm) wird eine Verlängerung der Sb-Cl
Bindung von ca 7% beobachtet[123].

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
82
Cl1
C1N1 C2
N2
Sb1
a
bc
Br Sb N CH
Abb. 4.23: Struktur des 1,3,2-Diazastibolens 13b[Cl] im Festkörper und ausgewählte Bindungs parameter
Die endocyclischen Bindungsabstände im 1,3,2- Diazastibolen 13b[Cl] zeigen eine Ver-
längerung der C-C-Doppelbindung gegenüber Standardwerten von 132.2 bis 132.6 pm [68]
für eine isolierte C-C-Doppelbindung, während die endocyclischen C-N-Bindungen
kürzer als isolierte C-N-Einfachbindungen (145.7-148.7 pm) [68] sind. Die Sb-N-
Bindungen in 13b[Cl] sind in Relation zu den im Tris(2,4,6-tri-tert-
butylphenylamino)stiban (Sb-N 204.1-206.4 pm)[142] bzw. 1,2,3,4-Tetramethyl-1,3,2,4-
λ2-diazasiladistibetidin (Sb-N 204.3-205.1 pm)[143} auftretenden Sb-N-Abständen
signifikant verkürzt.
Das 2-Bromo-1,3-di-tert-butyl-1,3,2-Diazastibolen 13b[Br] ist das erste Diamino-
antimon(III)bromid, das bislang röntgenstrukturanalytisch untersucht wurde [144].
Abb. 4.24:. Elementarzelle von 13b[Br]: Orthorombisches Kristallsystem; Raumgruppe Pbca (Nr.62)
Bindungslängen [Å]
Cl1-Sb1 2.6460 (15) Sb1-N1 1.998 (4) Sb1-N2 2.000 (4) N1-C1 1.407 (6) N2-C2 1.391 (6) C1-C2 1.346 (6)
Bindungswinkel [°]
N1-Sb1-N2 81.49 (16) N2-Sb1-Cl1 99.00 (12) N1-Sb1-Cl1 100.23(12)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
83
C2 C1N2
Br1
N1
Sb1
Kristalle von 13b[Br] sind isomorph zu denen von 13b[Cl] und enthalten ebenfalls
isolierte Moleküle, die relativ schwache intermolekulare Sb…Br-Kontakte (386.2 pm)
unterhalb Summe der Van der Waals Radien (415 pm) aufweisen. In der Literatur werden
Abstände von 351 pm bis 396 pm diskutiert [145,146,147].
Der Fünfring im 2-Bromo-1,3-di-tert-butyl-1,3,2- Diazastibolen 13b[Br] ist wie der im
Diazastibolen 13b[Cl] planar aufgebaut, wobei die Sb-Br-Bindung einen Winkel von
etwa 99° mit der C2H2Sb-Einheit bildet.
Die Sb-Br-Bindung ist mit 294.33(4) pm um ca 18 % länger als die mittlere Sb-Br-
Bindungslänge in gasförmigem SbBr3 (250 pm)[68] und um ca 17 % länger als die in
festem SbBr3 (252 [148] bzw. 250 pm [147]).
Abb. 4.25: Struktur des 1,3,2-Diazastibolens 13b[Br] im Festkörper und ausgewählte Bindungs- parameter Die C-C-Doppelbindung und die C-N-Bindungen unterscheiden sich kaum von den
entsprechenden Bindungen in 13b[Cl] und sind damit gleichfalls länger als isolierte C-C-
Doppelbindungen bzw. kürzer als isolierte C-N-Einfachbindungen.
Neben 13b[Cl] und 13b[Br] konnten auch vom 2-Chloro-1,3-di-tert-butyl-1,3,2-
diazarsolen 12b[Cl] Strukturdaten erhalten werden. Auch in dieser Verbindung liegen im
Festkörper isolierte Moleküle vor, die im Gegensatz zu den 1,3,2- Diazastibolenen
13b[Cl] und 13b[Br] jedoch keine intermolekularen Kontakte unterhalb der Summe der
Van der Waals-Radien aufweisen.
Bindungslängen [Å]
Br1-Sb1 2.9433 (4) Sb1-N1 1.998 (2) Sb1-N2 1.994 (3) N1-C1 1.386 (4) N2-C2 1.391 (4) C1-C2 1.346 (4)
Bindungswinkel [°]
N1-Sb1-N2 81.28 (10) N2-Sb1-Br1 97.95 (8) N1-Sb1-Br1 98.56 (7)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
84
C3C4
N2
Cl1
N5As1
a
bc
AsCl NCH
Abb. 4.26:. Elementarzelle von 12b[Cl]: Orthorombisches Kristallsystem; Raumgruppe Pbca (Nr.62)
Die C2N2As-Einheit in 12b[Cl] ist planar aufgebaut und besitzt wie 13b[Cl] und 13b[Br]
keine „envelope“-Konformation. Im 1,3,2-Diazarsolenring sind die Stickstoffe planar
koordiniert (Winkelsumme N(5) = 358.8°, N(2) = 359°) und das Arsen-Atom pyramidal
umgeben (Winkelsumme As(1) = 292.1°). Die As-Cl Bindung steht im Winkel von
104.8° zur Fünfringebene.
Abb. 4.27: Struktur und ausgewählte Bindungsparameter des 1,3,2-Diazarsolenes 12b[Cl] im Festkörper Die As-Cl Bindung in 12b[Cl] ist mit 2.6527(4) um ca 23% länger als die in AsCl3 in der
Gasphase (216 pm)[68] und um ca 13% länger als diejenige im 1,3-Di-tert-butyl-2,2-
dimethyl-4-chloro-1,3,2,4λ2-diazasila-arsetidin 70As ( 234.5 (1) pm )[123].
Bindungslängen [Å]
As1-Cl1 2.6527(4) As1-N5 1.8045(12) As1-N2 1.8057(12) N2-C3 1.382(2) C4-N5 1.3831(19) C3-C4 1.348(2)
Bindungswinkel [°] N5-As1-N2 86.03(5) N5-As1-Cl 102.28(4)
N2-As1-Cl1 103.92(4)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
85
O
F
S
F
OC
O
Sb1
F
N5N2
C4C3
Ein Bindungslängenausgleich der endocyclischen C-C- und C-N-Bindungen gegenüber
isolierten C-C-Doppelbindungen bzw. C-N-Einfachbindungen ist auch in diesem Fall
beobachtbar. Die As-N-Bindungen in 12c[Cl] sind zudem signifikant kürzer als in
acyclischen Trisaminoarsinen (185.3 -186.6 pm ) [149].
Neben den beschriebenen Halogenverbindungen konnten auch die Salze 13b[OTf] und
13b[SbCl4] röntgenstrukturanalytisch untersucht werden. Ebenso wie die Fünfringe der
1,3,2-Diazastibolene sind auch die der 1,3,2-Diazastibolenium-Kationen planar
aufgebaut.
Abb. 4.28: Struktur und ausgewählte Bindungsparameter von Anion und Kation im 1,3,2-
Diazastibolenium-Triflat 13b[OTf]
Im Salz 13b[SbCl4] sind die SbCl4--Anionen analog zur Struktur von [n-Prop4N][SbCl4]
über Cl-Brücken zu dimeren Einheiten mit pentakoordinierten Antimonatomen verknüpft [141]. Das Sb(1)-Atom des Kations weist vier intermolekulare Sb(1)…Cl-Kontakte
zwischen 343.26 und 380.57 pm sowie einen weiteren längeren Kontakt von 394.35 pm
zu Cl-Atomen in den Anionen auf. Unter Vernachlässigung des längsten Kontaktes ergibt
sich für das Sb(1)-Atom die Koordinationszahl 6. Die Länge des verbrückenden
Sb(2)…Cl(1a)-Kontaktes im Sb2Cl82--Anion von 13b[SbCl4] liegt mit 305.38 pm deutlich
unter der Summe der Van der Waals Radien und ist vergleichbar groß wie im
[Prop4N][SbCl4] (311(3) ppm).
Bindungslängen [Å]
Sb1-N2 2.0177 (19) Sb1-N5 2.0202 (19) N2-C3 1.354 (3) N2-C2 1.349 (3) C3-C4 1.364 (4)
Bindungswinkel [°] N1-Sb1-N2 79.38(8)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
86
Abb. 4.29: Struktur und ausgewählte Bindungsparameter des Salzes 13b[SbCl4] im Festkörper; Das Cl(2)-Atom mit dem Sb(1)…Cl(2) Kontakt von 380.57 pm ) sowie der Sb(1)…Cl(4)- Kontakt (394.35 pm) sind nicht eingezeichnet
Der dem Sb(2)…Cl(1a)-Kontakt gegenüberliegende Sb(2)-Cl(3)-Abstand (242.1 pm) ist
relativ zu den Sb(2)-Cl(2)- und Sb(2)-Cl(1)-Abständen verkürzt. Im [Prop4N][SbCl4] ist
die Bindungsituation vergleichbar und wird von den Autoren dadurch erklärt, dass trans-
ständige Sb-Cl-Bindungen als Dreizentren-Vierelektronen-bindungen anzusehen sind, in
der es zu einer Konkurrenz der Chlor-Atome um das zentrale p-Orbital am Antimon
kommt. Eine Schwächung einer Sb-Cl-Bindung führt daher zu einer Verkürzung der
trans-ständigen Sb-Cl-Bindung. Der Sb(2)-Cl(1)-Abstand (269.94 pm) ist größer als der
Sb(2)-Cl(2)-Abstand (251.73). Dieses könnte durch den relativ kurzen Kontakt des Sb(1)-
Bindungslängen [Å]
Sb1-N2 2.023 (2) Sb2-Cl3 2.4210(7) Sb1-Cl1 3.4326(7) Sb1-N1 2.025 (2) Sb2-Cl1 2.6994(7) Sb1-Cl2a 3.6927(7) N1-C1 1.356 (3) Sb2-Cl2 2.5173(7) Sb1-Cl4a 3.7188(7) N2-C2 1.353 (3) Sb2-Cl1a 3.0538(7) Sb1-Cl2 3.8057(8) C1-C2 1.364 (4) Sb1-Cl4 3.9435(7)
Bindungswinkel /Torsionswinkel [°]
N1-Sb1-N2 79.71 (9) C1-N1-Sb1-Cl1 114.53(7)
Sb1
Cl1a
N1C1
C2
N2
Cl1
Sb2 Cl2Sb3
Cl4
Cl4a
Cl2a Sb2a
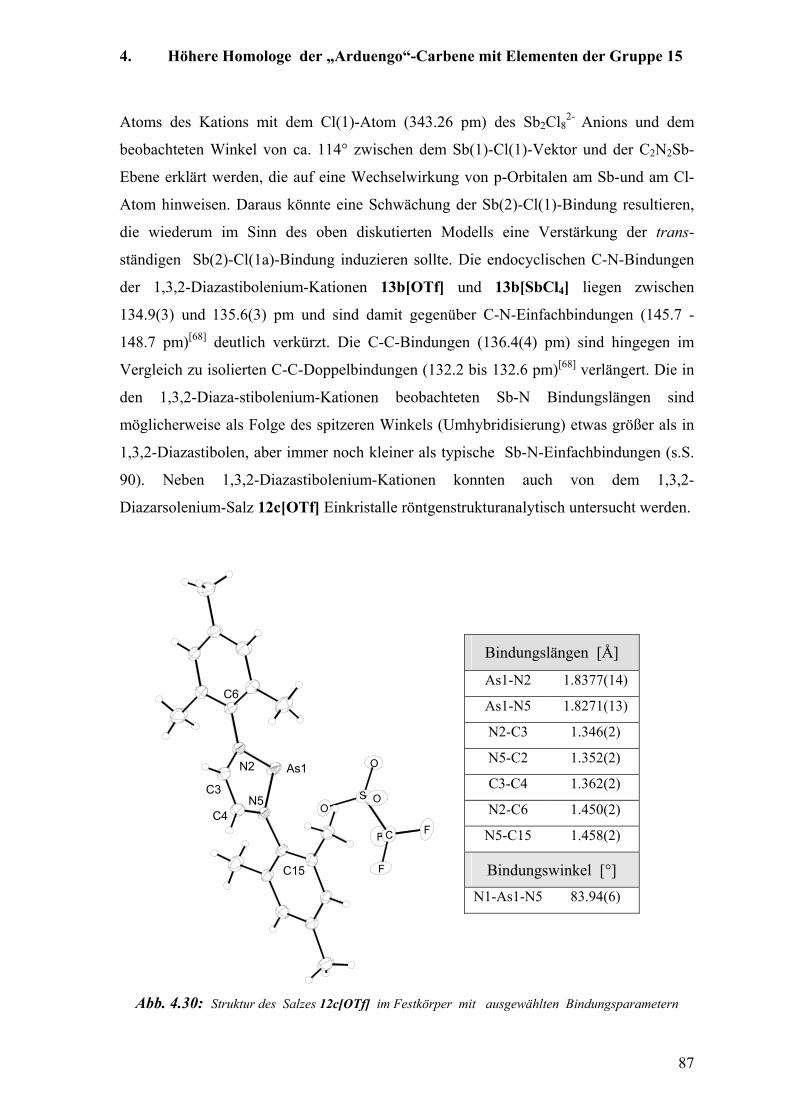
4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
87
F C F
O
F
OS
As1
O
C15
N5
N2
C6
C4
C3
Atoms des Kations mit dem Cl(1)-Atom (343.26 pm) des Sb2Cl82- Anions und dem
beobachteten Winkel von ca. 114° zwischen dem Sb(1)-Cl(1)-Vektor und der C2N2Sb-
Ebene erklärt werden, die auf eine Wechselwirkung von p-Orbitalen am Sb-und am Cl-
Atom hinweisen. Daraus könnte eine Schwächung der Sb(2)-Cl(1)-Bindung resultieren,
die wiederum im Sinn des oben diskutierten Modells eine Verstärkung der trans-
ständigen Sb(2)-Cl(1a)-Bindung induzieren sollte. Die endocyclischen C-N-Bindungen
der 1,3,2-Diazastibolenium-Kationen 13b[OTf] und 13b[SbCl4] liegen zwischen
134.9(3) und 135.6(3) pm und sind damit gegenüber C-N-Einfachbindungen (145.7 -
148.7 pm)[68] deutlich verkürzt. Die C-C-Bindungen (136.4(4) pm) sind hingegen im
Vergleich zu isolierten C-C-Doppelbindungen (132.2 bis 132.6 pm)[68] verlängert. Die in
den 1,3,2-Diaza-stibolenium-Kationen beobachteten Sb-N Bindungslängen sind
möglicherweise als Folge des spitzeren Winkels (Umhybridisierung) etwas größer als in
1,3,2-Diazastibolen, aber immer noch kleiner als typische Sb-N-Einfachbindungen (s.S.
90). Neben 1,3,2-Diazastibolenium-Kationen konnten auch von dem 1,3,2-
Diazarsolenium-Salz 12c[OTf] Einkristalle röntgenstrukturanalytisch untersucht werden.
Abb. 4.30: Struktur des Salzes 12c[OTf] im Festkörper mit ausgewählten Bindungsparametern
Bindungslängen [Å]
As1-N2 1.8377(14)
As1-N5 1.8271(13)
N2-C3 1.346(2)
N5-C2 1.352(2)
C3-C4 1.362(2)
N2-C6 1.450(2)
N5-C15 1.458(2)
Bindungswinkel [°] N1-As1-N5 83.94(6)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
88
Der Fünfring im 1,3,2-Diazarsolenium-Kation von 12c[OTf] ist planar aufgebaut und
zeigt bezüglich der endocyclischen C=C und C-N-Bindungen einen
Bindungslängenausgleich, welcher vergleichbar mit homologen 1,3,2-Diazastibolenium-
Kationen ist. Die exocyclischen C-N-Bindungen in 12c[OTf] sind reine
Einfachbindungen, da sich aufgrund der orthogonalen Stellung der Mesitylreste zur
Ringebene eine π−Delokalisation zwischen diesen Resten und dem Fünfring aus
geometrischen Gründen ausschließen lässt. Die As-N-Bindungen in 12c[OTf] sind etwas
kürzer als in acyclischen Trisaminoarsinen wie im Tris(morpholino)arsin[149]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
89
b) Vergleichende Diskussion der Kristallstrukturen von kationischen N-heterocyclischen
Carbenanaloga mit den Elementen Phosphor, Arsen und Antimon
In allen Verbindungen, die im vorliegenden Abschnitt 4.2.2 a) vorgestellt wurden,
beobachtet man einen endocyclischen C-N/C-C- sowie E-N-Bindungslängenausgleich,
der entweder durch eine cyclische Delokalisierung im formalen 6π−Elektronensystem der
Kationen oder alternativ durch π(C2N2) σ*(E-X)-Hyperkonjugation in dem Neutral-
verbindungen erklärt werden kann.
Der Einfluss dieser Effekte muss in Relation zu dem Bindungslängenausgleich diskutiert
werden, der in Verbindungen mit isolierten π(C2N2)-Fragmenten wie z.B. 43f, 22f und
64i vorliegt.
Tab. 4.1: Vergleich ausgewählter Bindungsparameter von 43f , 22f[53] ,64i[150], 11b[BF4][151]
Die Sb-N- bzw. As-N-Bindungen in den 1,3,2-Diazastibolenen 13b[Cl] (199.8/200.0 pm)
und 13b[Br] (199.4/199.8 pm) bzw. im 1,3,2-Diazarsolen 12b[Cl] (180.45/180.57 pm)
sind gegenüber typischen Sb-N-Bindungen in 1,2,3,4-Tetramethyl-1,3,2,4λ2-diazasila-
Bindungslängen [Å]
C-N 1.4283 (17) 1.4326 (17)
C-N 1.416 (2) 1.416 (2)
C-N 1.410 (4) 1.417 (4)
N-C 1.3912 (12) 1.3913 (12)
N-C 1.368 (2) 1.370 (2)
C-C 1.3390 (18)
C-C 1.328 (3)
C-C 1.317 (4)
C-C 1.3485 (14)
C-C 1.353 (2)
N-C 1.407 (6) 1.391 (6)
N- C 1.386 (4) 1.391 (4)
N-C 1.382 (2)
1.3831 (19)
N-C 1.346 (2) 1.352 (2)
N-C 1.356 (3) 1.353 (3)
N-C 1.354 (3) 1.349 (3)
C-C 1.346 (6)
C-C 1.346 (4)
C-C 1.348 (2)
C-C 1.362 (2)
C-C 1.364 (4)
C-C 1.364 (4)
Sb-N 1.998(4) 2.000 (4)
Sb-N 1.998(2) 1.994 (3)
As-N 1.8045(12) 1.8057(12)
As-N 1.8377(14) 1.8271(13)
Sb-N 2.023 (2) 2.025 (2)
Sb-N 2.0177 (19) 2.0202 (19)
NH
NH
dipp
4 3 f
dipp
NSi
N
dipp
22f
Cl
Cl
dipp
NSi(CH3)2
N
c-Hex
c-Hex
64i
NP-Cl
N
tBu
tBu
11b[Cl]
NP+BF4
-N
tBu
tBu
11b[BF4]
NAs-Cl
N
tBu
tBu
12b[Cl]
NSb -Cl
N
tBu
tBu
13b[Cl]
NSb+
N
tBu
tBu
13b[SbCl4]
SbCl4-
NSb -Br
N
tBu
tBu
13b[Br]
NSb +
N
tBu
tBu
13b[OTf]
OTf-
NAs+
N
Mes
Mes
12b[OTf]
OTf-

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
90
distibetidin [143] und Tris(2,4,6-tri-tert-butylphenylamino)stiban [142] bzw. der As-N-
Bindung in Tris(morpholino)arsin [149] verkürzt. Diese Bindungsverkürzung kann durch
die in Abschnitt 4.13 c) beschriebene 6π(C2N2 ) σ*(E-X)-Hyperkonjugation erklärt
werden.
Abb. 3.31: Verbindungen mit typischen Sb-N bzw. As-N- Bindungen
Die Verlängerung der Sb-X-Bindungen in den 1,3,2-Diazastibolenen 13b[Br] und
13b[Cl] gegenüber denen in SbBr3, SbCl3. und im 1,3-Di-tert-butyl-2,2-dimethyl-4-
chloro-1,3,2,4λ2-diazasila-stibetidin 71Sb sowie die Verlängerung der As-Cl-Bindung im
1,3,2-Diazarsolen 12b[Cl] gegenüber gasförmigem AsCl3. und 1,3-Di-tert-butyl-2,2-
dimethyl-4-chloro-1,3,2,4λ2-diazasila-arsetidin 71As kann ebenfalls durch 6π(C2N2 )
σ*(E-X)-Hyperkonjugation erklärt werden.
In Relation zu den E-X-Bindungen in gasförmigem EX3 ist die Verlängerung der As-Cl-
Bindung im 1,3,2-Diazarsolen 12b[Cl] (23 %) ausgeprägter als die Verlängerung der Sb-
Br-Bindung im 1,3,2-Diazastibolen 13b[Br] (18%). Die Verlängerung der Sb-Cl-
Bindung im 1,3,2-Diazastibolen 13b[Cl] ist am wenigsten ausgeprägt (13%).
Ein überwiegend ionischer Bindungscharakter der As-Cl-Bindung im 1,3,2-Diazarsolen
12b[Cl] kann ausgeschlossen werden, da selbst die extrem lange P-Cl Bindung
(Verlängerung um ca 34% gegenüber P-Cl-Standardbindungslängen von etwa 205 pm)
im 1,3,2-Diazaphospholen 11b[Cl] durch Leitfähigkeitsmessungen als kovalent
eingestuft werden konnte. Möglicherweise ist die außergewöhnlich lange As-Cl-Bindung
im 2-Chloro-1,3-di-tert-butyl-1,3,2-Diazarsolen 12b[Cl] und im 2-Chloro-1,3-di-tert-
N MeS b
NSbMe
Me
Me
SbNHMes*
Mes* HN
NHMes*As
N
NNO
O
O
1,2 ,3,4-Te tramethy l-1,3,2 ,4 λ-dia zasila-distib etidin
Tris(2 ,4 ,6-tri-tert-butylphenylamino)stiba n
Tris(morpholino)arsin
Sb-N: 204.1-206.4 pmSb-N 204.3-205.1 pm 185.3-186.6 pm

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
91
butyl-1,3,2-Diazaphospholen 11b[Cl] auf sterische Wechselwirkungen des Halogens mit
den Substituenten an den Stickstoffatomen zurückzuführen.
Die endocyclischen Bindungen in den 1,3,2-Diazastibolenen 13b[Cl] und 13b[Br] sind
ähnlich und zeigen einen Bindungslängenausgleich zwischen C-N-Bindungen und der C-
C-Doppelbindung. Ein Vergleich zwischen den 1,3,2- Diazastibolenen 13b[Br] und
13b[Cl] mit dem 1,3,2-Diazarsolen 12b[Cl] sowie dem 1,3,2-Diazaphospholen 11b[Cl]
zeigt keine nennenswert unterschiedlichen C-N-Bindungen und C-C-Doppelbindungen.
In Relation zu den Verbindungen 43f, 22f und 64i mit einem isolierten π(C2N2)-Fragment
wird jedoch eine geringfügige Verlängerung von C-C-Doppelbindungen und Verkürzung
der C-N-Bindungen beobachtet. Daraus könnte eine Delokalisierung der 6π-Elektronen
durch 6π(C2N2 ) σ*(E-X)-Hyperkonjugation über das (C2N2)-Fragment hinaus gefolgert
werden. Ein quantitativer Vergleich des Ausmaßes der Hyperkonjugation in 1,3,2-
Diazastibolenen, -arsolenen und -phospholenen ist jedoch in Anbetracht der geringen
Differenzen der experimentell gefundenen C=C- bzw. C-N-Bindungslängen in 13b[Br],
12b[Cl] und 11b[Cl], die nur wenig größer als die geschätzten Standardabweichungen
sind, nicht möglich.
Die C-N-Einfachbindungen und C-C-Doppelbindungen in den 1,3,2-Diazastibolenium
und -arsolenium-Kationen von 13b[SbCl4], 13b[OTf] und 12c[OTf] sind ähnlich lang
und zeigen im Vergleich zu den isolierten π(C2N2)-Fragmenten der Verbindungen 43f,
24c und 64i einen noch stärker ausgeprägten Bindungslängenausgleich als die vorher
diskutierten Halogenverbindungen. Dieser Befund steht im Einklang mit strukturellen
Untersuchung an analogen 1,3,2-Diazaphospholenen und 1,3,2-Diazaphospholenium-
Kationen [151]. Die Verkürzung der C-N-Bindungen von 11b[BF4] im Vergleich zu 43f,
22c und 64i ist demgegenüber etwas geringer. Man kann daher davon ausgehen, dass in
den 1,3,2-Diazastibolenium-Kationen 13b[SbCl4] und 13b[OTf] und im 1,3,2-
Diazarsolenium-Kation 12c[OTf] eine ähnlich starke oder noch effektivere 6π(C2N2)
=> p(E) Wechselwirkung als im 1,3,2-Diazaphospholenium-Kation 11b[BF4] existiert.

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
92
4.2.3 NMR-spektroskopische Untersuchungen
Alle bislang vorgestellten 1,3,2-Diazarsolene und 1,3,2-Diazastibolene sowie 1,3,2-
Diazarsolenium und 1,3,2-Diazastibolenium-Kationen wurden NMR-spektroskopisch
untersucht. In Tabelle 4.2 sind die 1H,13C,15N chemischen Verschiebungen der Atome im
Fünfring aufgeführt.
δ[ppm]
11b[Cl]
12b[Cl]
13b[Cl]
11b[OTf]
12b[OTf]
13b[OTf]
δ1H (H-C=C) 6.19a[151] 6.36a 6.60a 8.17b[151] 8.44d 8.62c, 8.72d
δ13C (H-C=C) 118.6a[151] 118.0a 119.5a 132.9b[151] 134.6d 138.5c
δ15N -206.0a -208.9a -99.6d -84.6d
δ[ppm]
11c[Cl]
12c[Cl]
13b[Cl]
11c[OTf]
12c[OTf]
13c[OTf]
δ1H (H-C=C) 5.85a 6.01a 6.37a 8.24b 8.21c 8.30c 8.41d
δ13C (H-C=C) 120.2a 120.2a 124.7a 131.5b 133.1d 132.5d
δ15N -235.7a -219.5a - 116.9d -99.6d
Tab. 4.2: ausgewählte NMR-Daten der Salze und Neutralverbindungen a) C6D6, b) CDCl3 c) CD2Cl2, d) CD3CN
Die Entschirmung der CH-Protonen und Stickstoffatome nimmt für strukturell
vergleichbare Verbindungen in der Regel mit steigender Ordnungszahl des Gruppe-15-
Elements zu. Eine Ausnahme wird lediglich für die Neutralverbindungen 11b[Cl],
12b[Cl], 13b[Cl] und die Kationen 11c, 12c, 13c beobachtet, deren wo die 13C und 15N
bzw. 1H und 13C Verschiebungen keine einheitlichen Trends zeigen. Ein formaler
Austausch des Halogens in 1,3,2-Diazastibolenen macht sich NMR-spektroskopisch
kaum bemerkbar (Tab.4.3).
NP-Cl
N
tBu
tB u
NP-Cl
N
Mes
Me s
NSb+
N
Mes
MesOTf-
NAs+
N
Mes
Me sOTf-
NSb-Cl
N
Mes
Mes
NAs-Cl
N
Mes
Mes
NP+
N
Mes
MesOTf-
NSb+
N
tBu
tBuOTf-
NP+
N
tBu
tBuOTf-
NAs-Cl
N
tBu
tBu
NSb-Cl
N
tBu
tBu
NAs+
N
tBu
tBuOTf-

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
93
δ[ppm]
13b[Br]
13b[I]
13b[AlCl4]
13b[GaCl4]
13b[SbCl4]
13b[OTf]
δ1H (H-C=C) 6.62a 6.60a 8.57c 8.49d 8.57d 8.72d
δ13C (H-C=C) 122.4a 122.4a 137.9c 136.2d 136.9d 138.5c
δ15N -91.8d -102.2 d -94.5d -84.6d
Tab. 4.3: ausgewählte NMR-Daten a) C6D6, b) CDCl3, c) CD2Cl2, d) CD3CN
Die 1,3,2-Diazastibolenium und -arsolenium-Salze weisen insgesamt eine starke
Entschirmung der 15N-Kerne gegenüber den Neutralverbindungen und dem
Diaminobuten 43f (δ15N = -305.8) auf, die auf die Einbindung der Stickstoff „lone-pairs“
in ein delokalisiertes π−Elektronensystem in den Kationen zurückgeführt werden könnte.
Ähnliche Entschirmungen sind auch für Phospheniumkationen (δ15N = -217.1 bis -181.6
ppm ) bekannt[152].
Die zusätzliche Zunahme der Entschirmung in den 1,3,2-Diazarsolenium und 1,3,2-
Diazastibolenium-Kationen 12b,c bzw. 13b,c gegenüber den Phosphenium-Kationen
11b,c könnte auf eine zunehmende Verkleinerung des HOMO-LUMO-Abstandes als
Folge des Einbaus zunehmend schwerer werdender Gruppe-15-Elemente und die damit
einhergehende Zunahme des paramagnetischen Beitrages zur magnetischen Abschirmung
zu erklären sein. Interessant ist, dass die 13C- wie auch die 15N-NMR-Daten des 1,3,2-
Diazaphospholenium-Kations 13b eine deutliche Abhängigkeit vom vorhandenen Anion
aufweist. Dies kann im Sinn des Vorliegens von Kontaktionenpaaren gedeutet werden.
NSb +
N
tB u
tBuOTf-
NSb+
N
tBu
tBuSbCl4-
NSb+
N
tBu
tBuGaCl4-
NSb+
N
tBu
tBuAlCl4-
NSb-I
N
tBu
tBu
NSb-Br
N
tBu
tBu

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
94
4.2.4 Schwingungsspektroskopische Untersuchungen
Um neben röntgenstrukturanalytischen Aussagen noch mehr Informationen über
Bindungszustände in 1,3,2-Diazarsolenium und -stibolenium-Kationen sowie 1,3,2-
Diazastibolenen und 1,3,2-Diazarsolenen zu erhalten, wurden systematische
Untersuchungen der Raman und Infrarotspektren durchgeführt.
Tab. 4.4: Wellenzahlen (in cm-1) für charakteristische Schwingungen der Kationen und Anionen in den Salzen 13b[SbCl4], 13b[GaCl4], 13b[AlCl4] und 13b[OTf . Werte in Klammern wurden der jeweiligen Literatur entnommen [153]. a) [ n-Bu4N][SbCl4] [154]; b) [n-Prop4N][SbCl4] [141]
Schwingungen der Kationen ν(Kation)
13b[GaCl4] 13b[AlCl4] 13b[SbCl4] 13b[OTf] Raman [cm-1]
IR [cm-1]
Raman [cm-1]
IR [cm-1]
Raman [cm-1]
IR [cm-1]
Raman [cm-1]
IR [cm-1]
3103 3104 3104 3098 3096 2981 2982 2989 2973 1508 1509 1509 1509 1513 1512 1512 1512 1224 1224 1225 1221 1224 1220 1223 1036 1205 1205
1192 1191 1191 1038 1025
950 951 951 954 770 771 774 722 721 722
678 679 679 679 668 684 563 563 564 569
517 518 515 391 391 391 393 287 287 290
Schwingungen der Anionen ν(Anion)
GaCl4- AlCl4
- SbCl4-
Raman [cm-1]
IR [cm-1]
Raman [cm-1]
IR [cm-1]
Raman [cm-1]
IR [cm-1]
344 (343) 348
(348) 343
(347)a
(342)b
343 (342)a
(343)b
123,116 (120) 121,130
(119) 292
(280)a (304)b
290 ( 290)a
(294)b
377, 365 (370) 370 479
(498) 491
(490)
243 (256)a
(248)b
241 (260)a
(-)b
150 (153) 150 165
(182) 182,177
(180)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
95
Die Schwingungsbanden der Anionen GaCl4- und AlCl4
- der Verbindungen 13b[GaCl4]
und 13b[AlCl4] konnten leicht durch Vergleich mit Literaturdaten zugeordnet werden [153]. Der Habitus der Spektren deutet auf hohe Symmetrie hin und impliziert die
Abwesenheit signifikanter intermolekularer Wechselwirkungen der Anionen GaCl4- und
AlCl4-. Die Zuordnung der Schwingungen des Cl-verbrückten dimeren Anions im Salz
13b[SbCl4] gelingt durch Vergleich mit den Schwingungsspektren von [n-Bu4N][SbCl4]
und [n-Prop4N][SbCl4], deren Anionen im Kristallverband ebenfalls diskrete Dimere
Sb2Cl82- bilden [154,141].
Die schwingungsspektroskopische Untersuchung der 1,3,2-Diazastibolenium–Salze
13b[SbCl4], 13b[GaCl4], 13b[AlCl4] und 13b[OTf] zeigt, dass die jeweiligen IR- und
Raman-Banden der Kationen in den unterschiedlichen Salzen nicht signifikant
voneinander abweichen und ein Einfluss des Anions auf die Bindungsstruktur des
Kations somit ausgeschlossen werden kann.
Die IR- und Raman-Banden um 1510 cm-1 liegen im Erwartungsbereich für eine C=C-
Valenzschwingung und wurden daher der Valenzschwingung der Doppelbindung in den
1,3,2-Diazastibolenium–Salzen zugeordnet. Ein Vergleich dieser Schwingungsbanden
mit isolierten C-C-Doppelbindungen zeigt im Falle der Salze eine zunehmend höhere
Intensität und Rotverschiebung. Das spricht für die Ausbildung eines konjugierten π-
Systems im Fünfring. Die Zuordnung und Auswertung der weiteren Schwingungsbanden
wird nicht diskutiert, da es sich hierbei überwiegend um Gerüstschwingungen handelt,
die nicht exakt einzelnen funktionellen Gruppen zugeordnet werden können.
Die IR- und Raman-spektroskopische Untersuchung an den 1,3,2-Diazastibolenen
13b[Cl], 13b[Br] und 13b[I], dem 1,3,2-Diazarsolen 12b[Cl] sowie dem Salz 12b[OTf]
zeigen, dass wie bei den entsprechenden 1,3,2-Diazaphospholenen [151] die beobachteten
Schwingungsfrequenzen deutliche Unterschiede untereinander als auch gegenüber den
νC=C-Schwingungen in den Salzen 11b[OTf] [129], 12b[OTf] und 13[OTf] aufweisen.
Dieses kann als deutlicher Hinweis auf einen Einfluss des kovalent gebundenen
Halogenatoms auf die Bindungsverhältnisse im Fünfring angesehen werden. Die für die
Diskussion dieses Einflusses wichtigen νC=C-Schwingungsfrequenzen der 1,3,2-
Diazaphospholene 11b[x] (x = F, Cl, Br, I) und 1,3,2-Diazastibolene 13b[X] (X = Cl, Br,
I ) sind in Tab.4.6 zusammen mit den Daten der Referenzverbindungen 64i, 22f und
43f,c aufgelistet.

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
96
11b[F]
11b[Cl]
11b[Br]
11b[I]
12b[Cl]
13b[Cl]
13b[Br]
13b[I]
Raman [cm-1]
ν(C=C)
-
1559[118]
1551[118]
1551[129]
1556
1569
1558
1549
IR [cm-1]
ν(C=C)
1625[118]
1560[118]
1552[118]
-
1557
1568
1558
-
11b[OTf]
12b[OTf]
13b[OTf]
13b[AlCl4] 13b[GaCl4]
13b[SbCl4]
Raman [cm-1]
ν(C=C) 1547[129] 1529 1512
1508
1509 1513 - 1643 (43f)
IR [cm-1] ν(C=C)
- 1527 1512 1509 1512 1628 1610 1650 (43c)
1642 (43f)
Tab. 4.5 : Wellenzahlen in cm-1 der C=C-Valenzschwingungen in Diazaphospholen, diazarsolen, diaza-stibolen -Derivaten
Für 13b[Cl] wurde zusätzlich eine im Raman-Spektrum auftretende Bande bei 150 cm-1
der Sb-Cl-Valenzschwingung zugeordnet. Diese Bande zeigt gegenüber der
entsprechenden Absorption im acyclischen ClSb(NMe2)2, (308 cm-1)[155] eine deutliche
Rotverschiebung von νSb-Cl, die auf eine erhebliche Schwächung der kovalenten
Bindungsanteile in 13b[Cl] hinweist. Eine Zuordnung der Sb-Br bzw. Sb-I-
Valenzschwingung in 13b[Br] bzw. 13b[I], die aufgrund der höheren reduzierten Massen
zu noch kleineren Wellenzahlen verschoben sein sollten, gelang nicht.
Die beobachteten C=C-Valenzschwingungen aller Kationen und Neutralverbindungen
sind gegenüber den Werten der Valenzschwingungen in den Butendiaminen 43f,c (1642,
1650 cm-1) sowie den Diazasilacyclopentenen 64i bzw. 22b (1610, 1628 cm-
1)[156,157,158,150] zu niedrigen niedrigeren Wellenzahlen verschoben. Diese Verschiebung
ist in den Salzen 11b[OTf], 12b[OTf] und 13[OTf] (1512-1547 cm-1) stärker ausgeprägt
als in den entsprechenden Chlorderivaten 11b[Cl], 12b[Cl] und 13[Cl] (1556-1569 cm-1).
NSb-I
N
tBu
tBu
NSb -Br
N
tBu
tBu
NSb -Cl
N
tBu
tBu
NP-
N
tBu
tBu
BrN
PN
tBu
tBu
IN
As-ClN
tBu
tBu
NSb+
N
tBu
tBuSbCl4-
NP+
N
tBu
tBu OTf-
NAs+
N
tBu
tBuOTf-
NSb+
N
tBu
tBuECl4-
NSb+
N
tBu
tBuOTf-
22b
NSi
N
tBu
tBu Cl
Cl
c-Hex
NSi
N
c-Hex
CH3
CH3
64i
NH
NH
R
RR =Mes(c) = Dipp(f)
43
NP
N
tBu
tBu
FN
P-N
tBu
tBu
Cl

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
97
In den Verbindungen 13b[X] (X = Cl, Br, I ) führt der Austausch eines leichteren gegen
ein schwereres Halogen zu einer deutlichen Rotverschiebung von νC=C, während für die
Salze 11b[OTf], 12b[OTf] und 13[OTf] und für die Chlorderivate bei Ersatz eines
leichteren gegen ein schwereres Gruppe-15-Element einander gegenläufige Trends
beobachtet werden:
Die Frequenz der C=C-Valenzschwingung fällt bei den Kationen und steigt bei den
Chlorderivaten mit der Ordnungszahl des Gruppe-15-Elements. Da die Richtung und
Größenordnung der beobachteten Änderungen von νC=C in den untersuchten
Verbindungen nicht zufrieden stellend allein durch die Zunahme der reduzierten Masse
bei Ersatz leichterer gegen schwerere Atome zu erklären ist, erscheint es nahe liegend,
dass die Änderung der Kraftkonstanten kC=C eine wichtige Rolle spielen muss. Die
signifikante Verschiebung der C=C-Valenzschwingung zu niedrigeren Wellenzahlen in
1,3,2-Diazastibolenen, -arsolenen und phospholenen können unter dieser Prämisse
dahingehend interpretiert werden, dass hier kein lokalisiertes π(C2N2)-Fragment mehr
vorliegt, sondern dass eine Hyperkonjugation des 6π(C2N2)-Fragments mit einem
antibindenden σ*(E-X)-Orbital eine deutliche Schwächung der C=C-Bindung herbeiführt.
Diese Schwächung der C=C-Bindung korreliert innerhalb der Reihe der 1,3,2-
diazastibolene 13b[Cl], 13b[Br], 13b[I] und der 1,3,2-Diazaphospholene 11b[F] bis
11b[I] mit abnehmender E-X Bindungsstärke und zunehmender Ladungskapazität und
Polarisierbarkeit der Halogene (Iod > Brom > Chlor > Fluor). Dies impliziert einen von
13b[Cl] nach 13b[I] zunehmenden ionischen Charakter der Sb-X-Bindung. Das Fluor-
1,3,2-diazaphospholen 11b[F] belegt diesen Trend in eindrucksvoller Weise in
gegenläufiger Richtung: die C=C-Valenzschwingung zeigt mit 1625 cm-1 die höchste
Wellenzahl aller untersuchten C=C-Bindungen, was im Einklang mit
röntgenstrukturanalytischen Untersuchungen auf einen hohen kovalenten P-F-
Bindungscharakter hindeutet [118]. Die hier beschriebenen Untersuchungen legen nahe,
dass das Ausmaß der π(C2N2) σ*(E-X)-Hyperkonjugation im Chlor-1,3,2-Diazastibolen
13b[Cl] weniger stark als im Chlor-1,3,2-Diazaphospholen 11b[Cl] bzw. Chlor-1,3,2-
Diazarsolen 12b[Cl] ausgeprägt ist, beim Ersatz des Cl-Atoms durch ein Br- oder I-Atom
aber wieder zunimmt. Die gleichen Werte für νC=C im Iod-1,3,2-Diazaphospholen 11b[I]
und Iod-1,3,2-Diazastibolen 13b[I] deuten darauf hin, dass die Hyperkonjugation in
beiden Verbindungen etwa gleich wirksam sein dürfte.

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
98
Die noch stärker ausgeprägte Rotverschiebung von νC=C in 1,3,2-Diazarsolenium-
Kationen und insbesondere in 1,3,2-Diazastibolenium-Kationen können gleichermaßen
als Folge einer Konjugation des 6π(C2N2)-Fragments mit dem freien b1(π)-Orbital am
Element E im Sinne einer 6π (C2N2) (E)Wechselwirkung erklärt werden. Die
beschriebenen schwingungsspektroskopischen Untersuchungen legen hier nahe, dass in
den Kationen die Effektivität der 6π(C2N2)-Elektronen-Delokalisation beim Übergang
von 1,3,2-Diazaphospholenium- zu 1,3,2-Diazarsolenium-Kationen und weiter zu 1,3,2-
Diazastibolenium-Kationen zunehmen sollte.
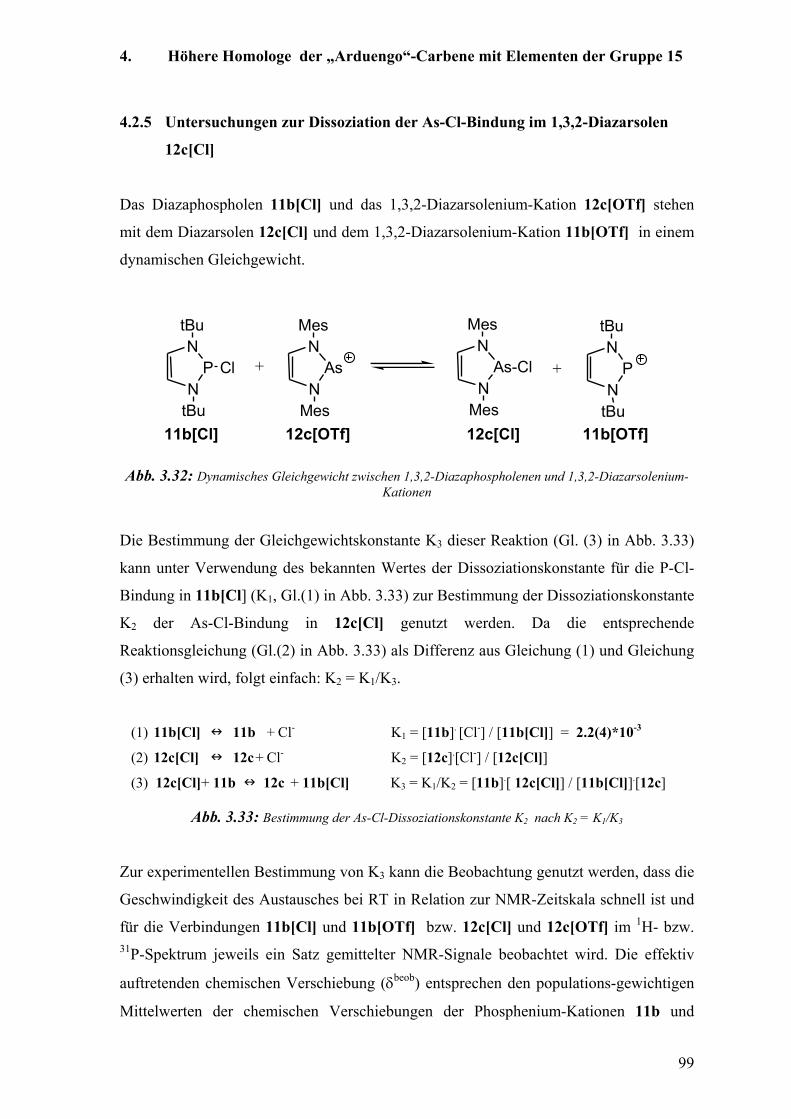
4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
99
4.2.5 Untersuchungen zur Dissoziation der As-Cl-Bindung im 1,3,2-Diazarsolen
12c[Cl]
Das Diazaphospholen 11b[Cl] und das 1,3,2-Diazarsolenium-Kation 12c[OTf] stehen
mit dem Diazarsolen 12c[Cl] und dem 1,3,2-Diazarsolenium-Kation 11b[OTf] in einem
dynamischen Gleichgewicht.
Abb. 3.32: Dynamisches Gleichgewicht zwischen 1,3,2-Diazaphospholenen und 1,3,2-Diazarsolenium-Kationen
Die Bestimmung der Gleichgewichtskonstante K3 dieser Reaktion (Gl. (3) in Abb. 3.33)
kann unter Verwendung des bekannten Wertes der Dissoziationskonstante für die P-Cl-
Bindung in 11b[Cl] (K1, Gl.(1) in Abb. 3.33) zur Bestimmung der Dissoziationskonstante
K2 der As-Cl-Bindung in 12c[Cl] genutzt werden. Da die entsprechende
Reaktionsgleichung (Gl.(2) in Abb. 3.33) als Differenz aus Gleichung (1) und Gleichung
(3) erhalten wird, folgt einfach: K2 = K1/K3.
(1) 11b[Cl] 11b + Cl- K1 = [11b]. [Cl-] / [11b[Cl]] = 2.2(4)*10-3
(2) 12c[Cl] 12c + Cl- K2 = [12c].[Cl-] / [12c[Cl]]
(3) 12c[Cl]+ 11b 12c + 11b[Cl] K3 = K1/K2 = [11b].[ 12c[Cl]] / [11b[Cl]].[12c]
Abb. 3.33: Bestimmung der As-Cl-Dissoziationskonstante K2 nach K2 = K1/K3
Zur experimentellen Bestimmung von K3 kann die Beobachtung genutzt werden, dass die
Geschwindigkeit des Austausches bei RT in Relation zur NMR-Zeitskala schnell ist und
für die Verbindungen 11b[Cl] und 11b[OTf] bzw. 12c[Cl] und 12c[OTf] im 1H- bzw. 31P-Spektrum jeweils ein Satz gemittelter NMR-Signale beobachtet wird. Die effektiv
auftretenden chemischen Verschiebung (δbeob) entsprechen den populations-gewichtigen
Mittelwerten der chemischen Verschiebungen der Phosphenium-Kationen 11b und
NP
N
tBu
tB u
ClN
As-ClN
Mes
Mes
N As
N
Mes
Mes
NP
N
tB u
tBu
+ +
11b[Cl] 12c[OTf] 12c[Cl] 11b[OTf]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
100
Phospholenen 11b[Cl] (4) bzw. der Arsolenium-Kationen 12c und Arsolenen 12c[Cl]
(5) und können durch die Gleichungen (4) und (5) ausgedrückt werden.
(4) δbeob(4) = δi (11b[Cl]) . Pi(11b[Cl]) + δi(11b) . Pi(11b)
(5) δbeob(5) = δi (12c[Cl]) . Pi(12c[Cl]) + δi(12c) . Pi(12c)
.
Durch 1H und 31P-spektroskopische Untersuchungen der Gleichgewichtsmischung aus
dem 1,3,2-Diazarsolenium-Salz 12b[OTf] und dem 1,3,2-Diazaphospholen 11b[Cl] kann
die Gleichgewichtskonstante K3 aus den aus Gleichung (4) zugänglichen
Gleichgewichtskonzentrationen errechnet werden. Die gemittelte Verschiebung δbeob(4)
wird experimentell aus Reaktionslösungen 31P-NMR-spektroskopisch bestimmt. Die
gemittelte Verschiebung δbeob(5) wird aus derselben Reaktionslösung 1H-spektroskopisch
bestimmt. Die Verschiebungen δi können ebenfalls experimentell bestimmt werden oder
aus der Literatur übernommen werden.
Man findet für die Dissoziationskonstante K2 der AsCl-Bindung in 12b[Cl] den Wert
0.12(2)*10-3 mol/l, welcher geringer ist als die Dissoziationskonstante K1 = 2.2(4)*10-3
mol/l der PCl-Bindung in 11b[Cl]. Daraus folgt, dass die PCl-Bindung im 1,3,2-
Diazaphospholen 11b[Cl] in Lösung eine höhere Tendenz zu Dissoziation aufweist als
die AsCl-Bindung im 1,3,2-Diazarsolen 12c[Cl]. Die AsCl-Bindung im 1,3,2-
Diazarsolen 12b[Cl] hat daher offensichtlich einen stärker kovalenten Charakter als die
PCl-Bindung im 1,3,2-Diazaphospholen 11b[Cl].

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
101
4.2.6 Reaktionsverhalten der 1,3,2-Diazastibolene, 1,3,2-Diazaarsolene bzw. 1,3,2-
Diazastibolenium- und 1,3,2-Diazarsolenium-Kationen
a) Thermische Stabilität
1,3,2-Diazastibolene sind den 2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-ylidenen sehr
ähnlich hinsichtlich ihrer ausgeprägten Thermolabilität. Orangefarbenes 1,3,2-
Diazastibolen 13c[Cl] färbt sich als Feststoff ab 90°C rasch schwarz. In Lösung tritt diese
Schwarzfärbung in wenigen Stunden bereits ab Temperaturen über 60°C ein.
Abb. 4.34: Zersetzung der 1,3,2-Diazastibolene über 60°C. Diazabutadien und metallisches Antimon konnten nachgewiesen werden.
In den schwarzen Lösungen konnte nach vollständiger Zersetzung quantitativ die Bildung
von Diazabutadien nachgewiesen werden. Die Schwarzfärbung resultiert aus fein
verteiltem metallischen Antimon, welches durch quantitatives EDX nachgewiesen
werden konnte. Vermutlich entsteht zuerst in einer cheletropen Reaktion eine Verbindung
der Zusammensetzung (SbCl)x, welche sofort zu Antimon (0) und Antimon(III)chlorid
disproportioniert.
N Sb-Cl
N
R
R
N
N
R
R
+ 6 SbCl60°C
6 6 2 SbCl3 + 4 Sb(0)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
102
b) Reaktionsverhalten gegenüber Oxidationsmitteln
Gibt man zu einer Etherlösung des 1,3,2-Diazastibolens 13b[Cl] ein halbes Äquivalent
Antimon(V)chlorid, so bildet sich nicht das Salz 13b[SbCl6], sondern 13b[SbCl4].
Hierbei reduziert der im Überschuss vorhandene Fünfring 13b[Cl] Antimon(V)chlorid zu
Antimon(III)chlorid und wird selbst zu Diazabutadien umgewandelt. Das entstehende
Antimon(III)chlorid reagiert mit noch vorhandenem Fünfring 13b[Cl] zum Salz
13b[SbCl4] weiter, welches durch Vergleich mit Infrarot- und Ramanspektren einer
identischen Probe identifiziert wurde. Die Reaktion äquimolarer Mengen 13b[Cl] mit
Antimon(V)chlorid verläuft vollständig im Sinn einer Redoxreaktion zu
Antimon(III)chlorid und Diazabutadien, welches in Form eines kristallinen 2:1 Addukts
(s. u.) isoliert werden konnte.
Abb. 4.35: Oxidation von 13b[Cl] zu SbCl3 und Weiterreaktion zum 1,3,2-Diazastibolenium-Kation
13b[SbCl4]
Die Reaktion von 13b[Cl] mit einem halben Moläquivalent Phosphor(V)chlorid verläuft
ähnlich unter Reduktion des Phosphor(V)chlorids zu Phosphor(III)chlorid und Oxidation
des 1,3,2-Diazastibolens 13b[Cl] zu Diazabutadien und Antimon(III)chlorid.
Anschließend erfolgt eine Methathesereaktion des verbliebenen 1,3,2-Diazastibolens
13b[Cl] mit Phosphor(III)chlorid zum 1,3,2-Diazaphospholen 11b[Cl], welches mit dem
gebildeten Antimon(III)chlorid zum 1,3,2-Diazaphospholenium-Salz 11b[SbCl4] weiter
reagiert.
NSb
N
t-Bu
t-Bu
Cl
NSb
N
t-Bu
t-Bu
+ SbCl5S bCl4
N
N
tBu
t-Bu
+ 2 SbCl3
SbCl 6
NS b
N
t-B u
t-Bu
13b[SbCl4]
13b[SbCl6]
13b[Cl] + SbCl5
+13b[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
103
Abb. 4.36: Oxidation von 13b[Cl] zu PCl3 und Weiterreaktion zum 1,3,2-Diazaphospholenium-Salz
11b[SbCl4]
Die Existenz des 1,3,2-Diazaphospholenium-Kations in 11b[SbCl4] wurde durch 1H, 31P-
2D-NMR-Spektroskopie nachgewiesen. Das Anion SbCl4- konnte durch IR- und Raman-
spektroskopische Untersuchungen identifiziert werden.
Im Unterschied zu den beschriebenen Reaktionen verläuft die Umsetzung von 13b[Cl]
mit Aluminium(III)chlorid ausschließlich im Sinn einer Lewis-Säure/Base-Reaktion zum
Salz 13b[AlCl4]. NMR-spektroskopische Untersuchungen von CD2Cl2-Lösungen dieses
Produktes in eingeschmolzenen Glasröhrchen zeigen auch nach 1 Woche Aufbewahrung
bei Raumtemperatur kaum Zersetzungsprodukte. Unter Luftzutritt erweisen sich die
Lösungen allerdings als instabil. NMR-Messungen weisen auf eine rasche Zersetzung des
Heterocycluses unter Bildung von Diazabutadien hin.
Eine analoge oxidative Zersetzung wurde auch an Lösungen von 13b[Cl] beobachtet.
Die Oxidationsprodukte fallen aus den Lösungen in der Regel als amorphe weiße
Niederschläge aus. Aus Lösungen des Salzes 13b[AlCl4] konnten zusätzlich gelbe
Kristalle, die röntgenstrukturanalytisch als 2:1 Adukkt aus Diazabutadien und
Antimon(III)chlorid 77 identifiziert ( s. Abb. 4.49 ) wurden, erhalten werden. Auch wenn
die Sb-haltigen Produkte der Oxidationsreaktionen nicht identifiziert wurden, ist
anzunehmen, dass das 1,3,2-Diazastibolen 13b[Cl] mit Sauerstoff zu Diazabutadien und
vermutlich SbOCl reagiert.
NSb-Cl
N
t-Bu
t-Bu
NSb
N
t-Bu
t-Bu
+PCl 5 S bCl4 PCl3 +N
N
t-Bu
t-Bu
SbCl3 +
P Cl6
NP
N
t-B u
t-B u
+ PCl5
13b[PCl6]
11b[SbCl4] 13b[Cl]
NP-Cl
N
t-Bu
t-Bu
11b[Cl]
+SbCl3
+ 13b[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
104
Abb. 4.37: Mit Sauerstoff reagiert das 1,3,2-Diazastibolen 13b[Cl] vermutlich zu Diazabutadien und
SbOCl
Das Salz 13b[AlCl4], welches im Gleichgewicht mit 1,3,2-Diazastibolen 13b[Cl] und
AlCl3 steht könnte auf diese Weise mit Sauerstoff zu SbOCl, Antimon(III)chlorid und
Diazabutadien reagieren. Das Addukt 77 könnte sich anschließend durch Assoziation
von Diazabutadien und Antimon(III)chlorid bilden.
Abb. 4.38: Sauerstoff oxidiert Kation 13b[AlCl4] ; Diazabutadien komplexiert mit gelöstem SbCl3 zu 77
77 liegt im Festkörper als dimeres Addukt eines Diazabutadiens mit zwei pyramidal
aufgebauten SbCl3-Molekülen vor. Das Sb(1)-Atom befindet sich in einer Ebene, die von
den Stickstoffatomen N(1)/N(2) des Diazabutadiens und den Chloratomen Cl(2)/Cl(3)
aufgespannt wird. Die Sb(1)-Cl(1)-Bindung steht annähernd senkrecht auf dieser
Ebene.Die Sb(1)-N-Abstände in 77 liegen deutlich unter der Summe der Van der Waals-
Radien (370 pm), aber über der Summe der Kovalenzradien beider Elemente (215 pm).
NSb-Cl
N
t-Bu
t-Bu
AlCl4
N
N
t-Bu
t-Bu
NSb
N
t-Bu
t-B u
+ 4 AlCl3 + 4 SbOCl
+ O2
2SbCl3
2
N
N
t-Bu
t-B u
13b[AlCl4]
77
+ 4 AlCl3
+ 4 SbCl3
+ 4 AlOCl
N
N
t-Bu
t-B u
4 4 4
* 4 + 4 Al OC lN
N
t-Bu
t-B u
2
13b[Cl]
NSb-Cl
N
t-Bu
t-Bu
N
N
t-Bu
t-Bu
+ SbOCl + 1/2 O2
13b[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
105
Das Sb(1)-Atom besitzt einen zusätzlichen intermolekularen Sb(1)…Cl(4)-Kontakt
(336.33(6) pm ), der deutlich unter der Summe der Van der Waals-Radien liegt (400
pm). Das Sb(2)-Atom zeigt neben den drei Sb-Cl-Bindungen mit Abständen von 238.41-
259.62 pm drei längere, intermolekulare Sb…Cl-Kontakte von 304.08-333.36 pm. Beide
Sb-Atome befinden sich damit insgesamt in einer verzerrt oktaedrischen
Koordinationssphäre.
Abb. 4.39: Struktur SbCl3-Diazabutadien Adduktes 77 im Festkörper und ausgewählte Bindungsparameter
Bindungslängen [Å]
Sb1-N2 2.418 (2) Sb1-Cl1 2.3841(6) Sb2-Cl4 2.4062 (7)
Sb1-N1 2.460 (2) Sb1-Cl2 2.5464 (7) Sb2-Cl5 2.4116 (7)
N1-C1 1.278 (3) Sb1-Cl3 2.5962 (7) Sb2-Cl6 2.3804 (7)
C1-C2 1.472 (3) Sb1-Cl4 3.3633 (6) Sb2-Cl3 3.0408 (7)
Sb2-Cl2 3.1963 (7)
Bindungswinkel [°]
Cl1-Sb1-Cl2 87.05 (2) Cl4-Sb2-Cl5 87.38 (2) Cl1-Sb1-Cl3 86.05 (2)
Cl2-Sb1-Cl3 86.55 (2) Cl6-Sb2-Cl5 91.98 (3) Cl6-Sb2-Cl4 93.23 (3)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
106
Bei näherer Betrachtung der Geometrie stellt man am N-koordinierten Sb(1)-Atom von
77 eine Abnahme der Sb-Cl-Bindungswinkel gegenüber reinem SbCl3 fest. Man findet
zudem für die Sb(1)-Cl-Bindungen in trans-Position zu den koordinierenden Stickstoffen
einen mittleren Wert von 257.13 pm, der deutlich von der senkrecht dazu stehenden
Sb(1)-Cl(1)-Bindung (238.41(6) pm) sowie von den mittleren Sb-Cl-Bindungslängen in
reinem SbCl3 ( 235.9 pm ) abweicht [140]. Die beobachtete Sb-Cl-Bindungsaufweitung
kann durch die Wechselwirkung der freien Elektronenpaare am Stickstoff mit σ*-(SbCl)-
Orbitalen des Sb(1)Cl3-Moleküls erklärt werden.
Addukte des SbCl3 mit S- und N- Donoren wie z.B. Anilin [159] sind in der Literatur
zahlreich beschrieben[160]. Addukte von SbCl3 mit chelatisierenden N-Donoren wie
Diazabutadienen sind dagegen bislang kaum strukturell untersucht worden. Kurze Sb-N-
Kontakte (228.1 pm) werden in einem Addukt 78 aus Bipyridin und SbCl3, gefunden [139].
Abb. 4.40: Durch Bipyridin chelatisiertes SbCl3
78
In 78 existieren intermolekulare Sb…Cl-Kontakte, die zu einer Verbrückung der
Bipyridin-SbCl3-Einheiten in Form einer Kette führt. Daraus resultiert für das
Antimonatom eine verzerrt oktaedrische Koordination. Die Abstände der Sb…Cl-
Kontakte in 78 (334 pm) sind mit denen in 77 vergleichbar groß. Die Sb-N-Kontakte in
77 sind zwar erheblich länger als in 78, aber dennoch kürzer als in Addukten des nicht
chelatisierenden Anilins mit SbCl3 (253 pm).

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
107
c) Koordinationschemische Untersuchungen
Die Koordinationschemie der 1,3,2-Diazastibolenium-Kationen und 1,3,2-Diazastibolene
sowie analoger Arsenverbindungen ist im Gegensatz zu der von 1,3,2-
Diazaphospholenium -Kationen und 1,3,2-Diazaphospholenen bislang nicht untersucht
worden.
Obwohl Arbeiten von Nakazawa [134] zeigen, dass ein 16VE M(CO)3bipy-Fragment (M =
Mo, W) gut zur Stabilisierung von Phosphenium-Komplexen geeignet ist, reagieren tert-
butyl-substituierte 1,3,2-Diazaphospholene offenbar aus sterischen Gründen nicht mit
Übergangsmetallkomplexen wie [W(MeCN)(CO)3bipy] zu Diazaphospholenium-
Komplexen[151].Entsprechende Reaktionen mit N-Aryl-1,3,2-Diazaphospholenen und
1,3,2-Diazaphospholenium-Kationen konnten dagegen erfolgreich durchgeführt werden [151]. Versuche zur Darstellung der analogen Diazastibolenium- bzw. Diazarsolenium-
Komplexe ergaben überraschenderweise, dass weder tert-butyl- noch aromatisch
substituierte 1,3,2-Diazastibolene und 1,3,2-Diazarsolene noch deren Kationen mit
[W(MeCN)(CO)3bipy] oder anderen neutralen Metallkomplexvorstufen wie z.B.
([W(CO)5(C8H14)] oder [Pt(C2H4)(PPh3)2] stabile kationische Komplexe bilden. Um
neutrale Komplexverbindungen aus 1,3,2-Diazastibolenen und 1,3,2-Diazarsolenen bzw.
1,3,2-Diazarsolenium-Kationen zu erhalten, wurden diese mit anionischen
Carbonylkomplexen wie TlCo(CO)4 umgesetzt.
Abb. 4.41: Chemische Umsetzungen von 1,3,2-Diazaarsolenen und 1,3,2-Diazastibolenium bzw. 1,3,2-Diazarsolenium-Kationen mit TlCo(CO)4
NE
N
Mes
Mes
C l
NAs
N
Mes
Mes
OTf
+Tl Co(CO)4 -TlC l N
EN
Mes
Mes
Co COCO
CO
CO
+TlCo(CO)4
-Tl OTf
NE
N
Mes
Mes
Co COCO
CO
-CO
-70°C
79 E = As80 E = Sb
81 E = As82 E = Sb
13c[OTf]
E = As 12c[Cl] E = Sb 13c[Cl]

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
108
Die Reaktion des 1,3,2-Diazarsolens 12c[Cl] mit TlCo(CO)4 verläuft glatt unter TlCl-
Eliminierung und anschließender Abspaltung eines CO-Moleküls über das Intermediat 79
zur dunkelroten kristallinen Komplexverbindung 81, welche bei RT kurzzeitig stabil ist
und sich bei längerer Lagerung oberhalb von -30°C zersetzt. Die Konstitution des
Komplexes 81 konnte durch IR- und NMR-Spektroskopie in Lösung nachgewiesen
werden. Die Umsetzung des 1,3,2-Diazastibolens 13c[Cl] mit TlCo(CO)4 führt nach
kurzer Zeit zur Entfärbung der ursprünglich roten Reaktionslösung und anschließend bei
Erwärmung auf Temperaturen oberhalb von -70°C zur Ausfällung eines schwarzen
Materials. Das nur kurzzeitig stabile Reaktionsprodukt lässt sich nicht isolieren. Die
Bildung eines Tricarbonylstiboleniumkomplexes 82 kann jedoch durch IR- und 2D-
NMR-spektroskopische Untersuchungen an den Reaktionslösungen nahe gelegt werden.
Im 1H-NMR-Spektrum von 81 wird für die Signale der Protonen an den C5/C4-
Kohlenstoffatomen im Diazarsolen-Ring eine geringe Entschirmung im Vergleich zu
12c[Cl] und eine deutliche Hochfeldverschiebung im Vergleich zu 12c[OTf] gefunden.
Ein ähnliches NMR-spektroskopisches Verhalten wurde auch bei homologen
Phospheniumkomplexen 75 beobachtet [151]. Der 1,3,2-Diazastibolen-Ligand im Komplex
82 zeigt erwartungsgemäß eine Entschirmung der 1H-NMR-Signale gegenüber 13c[Cl].
Vom Produkt der Reaktion des 1,3,2-Diazarsolens 12c[Cl] mit Tl[Co(CO)4] konnten aus
Acetonitril bei -30°C Kristalle erhalten werden, die für eine Röntgenstrukturanalyse
geeignet waren. Überraschenderweise ergab die Röntgenstrukturanalyse des ersten
untersuchten Kristalls, dass es sich bei der untersuchten Probe um den als Vorstufe von
81 auftretenden Tetracarbonylkomplex 79 handelte. Es stellte sich im Verlauf weiterer
Untersuchungen heraus, dass dieses Produkt als geringfügige Verunreinigung im Produkt
vorhanden war Kristallisationsansatzes anzusehen war. Eine gezielte Separation von 79
gelang nicht. Daher war eine Isolierung sowie IR- bzw. NMR-spektroskopische
Untersuchung des Komplexes 79 nicht möglich. Die Röntgenstrukturanalyse eines
weiterern Kristalls aus dem Kristallisationsansatz bestätigte die vorgeschlagene
Konstitution eines monomeren Komplexes 81 mit einem tetraedisch koordinierten
Cobalt-Atom und einem annähernd trigonal-planar koordinierten Arsen-Atom. Da
aufgrund der mangelhaften Kristallqualität keine zufrieden stellende Verfeinerung der
Strukturdaten gelang, muss auf eine weitere Diskussion von Bindungsparametern an
dieser Stelle verzichtet werden.

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
109
O1B C1B
As1
O1CC1C
Co1
N5
C1A
O1A
N2
C4
C1D
C3
O1D
AsCo
Abb. 4.42 : Struktur des Komplexes 81 im Festkörper
Der Diazarsolenring im Tetracabonylarsolenium-Komplex 79 ist annähernd planar
aufgebaut. Das Arsenatom weist eine pyramidale Umgebung auf und das Kobaltatom ist
verzerrt trigonal- bipyramidal koordiniert. Der Co-As-Abstand in 79 ist gegenüber den
entsprechenden Abständen in bekannten Co-AsPh3-Komplexen (228.9(1)[161] - 229.49(2) [162] pm) deutlich verlängert. Die drei in der Ebene liegenden CO-Liganden besitzen in
etwa gleich lange Co-C- und C-O-Bindungen. Der trans zum As-Liganden ständige CO-
Ligand zeigt demgegenüber einen etwas längeren Co-C- und einen geringfügig kleineren
C-O-Abstand.
Abb. 4.43: Molekülstruktur des Komplexes 79 im Festkörper

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
110
Tab. 4.6: Ausgewählte Bindungsparameter im Komplex 79
Die C=C und C-N-Bindungen im Diazarsol-Liganden von 79 sind denen im freien 1,3,2-
Diazarsolen 12b[Cl] (C-N 138.2(2), 138.31(19) pm; C-C 134.8(2) pm) vergleichbar. Die
As-N-Bindungen in 79 sind dagegen im Vergleich zu 12b[Cl] (1.8045(12), 1.8057(12)
pm) merklich aufgeweitet. Im Vergleich zum 1,3,2-Diazarsolenium-Kation von
12c[OTf] (C-N 134.6(2), 135.2(2) pm; C-C 136.2(2) pm; As-N 1.8377(14), 1.8271(13)
pm) weist 79 deutlich kürzere C-N-Bindungen und längere C=C- und As-N-Bindungen
auf.
Die Molekülstruktur und Bindungslängen des Diazarsolenium-Liganden in 79 ähneln
insgesamt eher einem 1,3,2-Diazarsolen als einem 1,3,2-Diazarsolenium-Kation, wobei
die Bindungsaufweitung der As-N-Bindung in 79 durch einen geringen π-
Rückbindungsanteil der As-Co-Bindung erklärt werden kann. Zusammengenommen
weisen die strukturchemischen Befunde darauf hin, dass der Arsoleniumligand als π-
Akzeptorligand mit einer im Vergleich zu Kohlenmonoxid stärker ausgeprägten π-
Acidität anzusehen ist. Eine Untermauerung dieser Annahme durch aussagekräftige IR-
spektroskopische Daten gelang bislang nicht, da Komplex 79 nicht in ausreichenden
Mengen rein isoliert werden konnte.
Die IR-Spektren des Tricarbonylkomplexes 81 in Hexanlösungen zeigen zwei
Absorptionsbanden von CO-Valenzschwingungen bei 2021 und 1962 cm-1, während in
Spektren von festen, als Nujolverreibungen vermessenen Proben insgesamt fünf CO-
Schwingungsbanden etwa gleicher Intensität beobachtbar sind. Zwei Banden entsprechen
denen des Lösungsspektrums und können durch eine partielle Löslichkeit des Komplexes
Bindungslängen [Å] Bindungs-/Torsionswinkel [°] As1-Co1 2.7263 (4) C(1A)-O(1A) 1.128 (3) N5-As1-N2 85.40 (8)
As1-N2 1.8353 (17) C(1B)-O(1B) 1.144 (3) N5-As1-Co1 111.59 (6)
As1-N5 1.8459 (19) C(1C)-O(1C) 1.153(3) N2-As1-Co1 103.15 (6)
C1-C2 1.330(3) C(1D)-O(1D) 1.149(3) C(1A)-Co1-As1 178.07 (9)
N2-C3 1.394 (3) Co1-C(1A) 1.822 (3) Co1-As1-N2-C3 105.63 (14)
N5-C4 1.396 (3) Co1-C(1B) 1.789 (3) C3-N2-C6-C11 121.5 (2)
N5-C15 1.445 (3) Co1-C(1C) 1.796 (3) C4-N5-C15-C20 106.3 (3)
N2-C6 1.432 (3) Co1-C(1D) 1.790 (3) As1-N2-C3-C4 4.6 (3)

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
111
in Nujol erklärt werden. Das Auftauchen der drei zusätzlichen Banden (ν = 2004, 1934,
1928 cm-1) kann unter der Maßgabe erklärt werden, dass die Konstitution des Komplexes
81 in Lösung und im Festkörper unterschiedlich ist.
Ausgehend von der röntgenstrukturanalytisch belegten Festkörperstruktur von 81 können
die drei dem Festkörper zugeschriebenen Banden den drei Valenzschwingungen des
Co(CO)3-Fragments zugeordnet werden. Da für Tricarbonylkomplexe mit lokaler C3v-
Symmetrie nur zwei IR-aktive Normalmoden (A1 + E) zu erwarten sind, wird die
beobachtete zusätzliche Aufspaltung als Folge einer Symmetrieerniedrigung gedeutet, die
wahrscheinlich auf die π-Wechselwirkung zwischen dem Metall und dem
Diazaarsoleniumliganden zurück geht. Das Auftreten einer unterschiedlichen
Konstitution in Lösung könnte damit zu erklären sein, dass eine Dimerisation zu einem
Produkt (81)2 stattfindet[94] (Abb.4.44). Vergleichbare, in Lösung dimere
Komplexverbindungen 83 werden durch Reaktion von AsPh3 mit Co2(CO)8 gebildet
(Abb.4.44) [163,162].
Abb. 4.44: Monomere und vermutliche dimere Struktur von 81
Für 83 werden in Lösung zwei CO-Schwingungen gefunden, die auf D3h oder D3d
Symmetrie hinweisen. Röntgenstrukturanalytisch wurde für 83 D3d-Symmetrie gefunden
in der die CO-Liganden wie vermutet in gestaffelter Position zueinander stehen.
N
NAs
Mes
Mes
CoCO
COCO
Co
OC CO
CO
Co
OC C O
CON
NAs
Mes
MesN
NAs
Mes
Mes
2
81 (81)2 Lösung (Hex an)Festkörper (N ujol )
Co
OC CO
CO
Co
OC C O
CO
A sPh3Ph3As
83
ν(CO)= 2023, 1964 c m-1ν(CO)= 2004, 1934, 1928 cm-1
Lösung (H ex an)
ν(CO)= 1999, 1980 cm-1

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
112
Das Vorliegen des Arsoleniumkomplexes 81 als Monomer im Festkörper sowie als
Dimer in Lösungen wird durch analoges Verhalten homologer Phospheniumkomplexe
75 bekräftigt, die in Nujolverreibungen ein ähnliches IR-Bandenmuster ( ν = 2025,
2005, 1957, 1934 cm-1 ) zeigen[135]. Röntgenstrukturanalytische Untersuchungen belegen
für 75 das Vorliegen monomerer Moleküle im Festkörper [135]. Der monomeren Form mit
lokaler C3v-Symmetrie können die beiden IR-Banden bei 2005 und 1934 cm-1 zugeordnet
werden, während die zusätzlichen IR-Banden bei 2025 und 1957 cm-1 wie im Fall des
Arsoleniumkomplexes einem Dimeren (75)2 zugeordnet werden können.
Wie in den Arsolenium- und Phospholenium-Komplexen 81 und 75 [135] beobachtet man
auch in Hexanlösungen des vermuteten, kurzzeitig stabilen Komplexes 82 zwei intensive
Banden bei 2062 und 1977 cm-1 und zwei weitere Banden geringerer Intensität bei 2002
und 1942 cm-1. Vergleicht man diese Daten mit denen der Komplexe 81 und 75, kann das
intensive Bandenpaar 2062, 1977 cm-1 einem Dimeren (82)2 und das weniger intensive
Bandenpaar 2002, 1942 cm-1 einem monomeren Komplex 82 zugeordnet werden.
Offenbar existieren in Hexanlösungen beide Spezies nebeneinander im Gleichgewicht.
Der Vergleich der IR-Daten (Tab. 4.7) aller homologen Komplexverbindungen zeigt,
dass sich die Phospholenium und Arsoleniumkomplexe 75 und 81 in ihren CO-
Schwingungen nur unwesentlich unterscheiden.
IR
(82)2
(81)2
(75)2
ν(CO) 2002a 2062a 2004b 2023a,b 2005b[135] 2025b[135]
[cm-1] 1942a 1977a 1934,1928b 1964a,b 1934b[135] 1957b[135]
Tab. 4.7: Vergleich der ν(CO)-Schwingungen der monomeren und dimeren Komplexe a) Lösung in Hexan b) Nujolverreibung
Der Stiboleniumkomplex 82 zeigt hingegen eine Verschiebung der Wellenzahlen der C-
O-Schwingungen zu höheren Werten. Daraus lässt sich auf eine verstärkte π-
Rückbindung vom Metall zum Antimon und eine daraus resultierende Stärkung der C-O-
Bindung durch anwachsende π-Bindungsanteile schließen. Offenbar sind 1,3,2-
Diazastibolenium-Kationen, die formal als Liganden im Co(CO)3-Komplex fungieren,
N
NSb
Mes
Mes
CoCO
COCO
82
N
NAs
Mes
Mes
CoCO
COCO
81
N
NP
Mes
Mes
CoCO
COCO
75

4. Höhere Homologe der „Arduengo“-Carbene mit Elementen der Gruppe 15
113
elektrophiler als entsprechende 1,3,2-Diazaphospholenium-und 1,3,2-Diazarsolenium-
Kationen.

5. Zusammenfassung der Ergebnisse und Ausblick
114
5. Zusammenfassung der Ergebnisse und Ausblick
Im Rahmen der vorliegenden Arbeit konnten neuartige höhere Homologe des „Arduengo“-
Carbenes mit den Elementen Zinn und Antimon dargestellt und hinsichtlich ihrer
strukturellen und spektroskopischen Eigenschaften untersucht werden. Bereits bestehende
Synthesemethoden zur Darstellung isoelektronischer Phosphor- und Arsenverbindungen
wurden durch Verwendung von α-Aminoaldiminen bzw. Butendiaminen als Edukte
entscheidend verbessert. Zudem konnte mit 43f erstmals ein Z-N,N’-Bis(Alkyl)-but-2-en-
2,3-diamin dargestellt und strukturell charakterisiert werden.
Die Transaminierungsreaktionen einiger α-Aminoaldimine mit Pb(N(SiCH3)2)2 wurden
NMR-spektroskopisch verfolgt. Obwohl 2,3-Dihydro-1-H-[1,3,2]-Diazaplumbol-2-ylidene
letztendlich nicht direkt nachweisbar oder isolierbar waren, konnten dennoch kurzzeitig
stabile Vorstufen nachgewiesen werden, die sich möglicherweise direkt in Diazaplumbol-2-
ylidene umwandeln können. Die in Kapitel 2 erarbeiteten Möglichkeiten zur Stabilisierung
thermolabile Diazastannol-2-ylidene durch sperrige Reste an den Stickstoffen sind offenbar
zur Stabilisierung von Diazaplumbol-2-ylidenen nicht ausreichend.
Transaminierungsreaktionen C-Alkyl-substituierter α-Aminoaldimine mit Sn(N(SiCH3)2)2
führen, wie die Untersuchung der Reaktionsmechanismen zeigte, nicht zu Diazastannol-2-
ylidenen sondern zu acyclischen Stannylenen. Die Synthese intramolekular stabilisierter
Diazastannol-2-ylidene ausgehend von C-Alkyl-substituierten α-Aminoaldimine gelang
daher nicht. Zur Synthese dieser Zielverbindungen müssten wahrscheinlich entsprechende
CH-substituierte α-Aminoaldimine eingesetzt werden.
Überraschende Ergebnisse ergaben Untersuchungen zum Reaktionsverhalten der
dargestellten Diazastannol-2-ylidene. Ihre teilweise geringe thermische Stabilität und ihr in
mancher Hinsicht abweichendes Reaktionsverhalten gegenüber Imidazol-2-ylidenen,
Diazasilolen und Diazagermol-2-ylidenen wurde insbesondere an bislang unbekannten
Zinntransferreaktionen und chelotropen Zerfallsreaktionen deutlich gemacht.
N
N
R´
R´
N
NSn
R
R
+N
N
R
R
N
NSn
R´
R´
+N
NSn
R
R
N
N
R
R
+ Sn(0)

5. Zusammenfassung der Ergebnisse und Ausblick
115
Nach den Ergebnissen von quantenchemischen Rechnungen kann dieses ungewöhnliche
Reaktionsverhalten auf zunehmende π-Elektronendelokalisation und gleichzeitige
Polarisierung der σ−Sn-N-Bindungen zurückgeführt werden. Die von Arduengo et. al.
vorgeschlagenen Grenzstrukturen der Diazasilol-2-ylidene und Diazagermol-2-ylidene als
„chelatisierte“ Metalle [104] scheinen sich somit auch in der elektronischen Struktur der
homologen Diazastannol-2-ylidene widerzuspiegeln.
Ein schwach elektrophiler Charakter der Diazastannol-2-ylidene konnte durch Reaktion mit
4-Dimethylaminopyridin belegt werden. Ein ähnliches Verhalten zeigen auch Diazagermol-
2-ylidene, die im Vergleich zu Carbenen und Diazasilol-2-ylidenen als Liganden ein
erhöhtes π-Akzeptorverhalten in Molybdän-Komplexen zeigen[67]. Eine weitere Ähnlichkeit
zwischen Diazagermol-2-ylidenen und Diazastannol-2-ylidenen zeigt sich auch an der
Reaktion des Diazagermol-2-ylidens 48 mit dem Diazabutadien 20b, in der erstmals der
Transfer eines Germaniumatoms zwischen zwei Diazabutadienfragmenten beobachtet
werden konnte. Da die untersuchte, nicht entartete Germanium-Transferreaktion viel
langsamer als der analoge Zinn-Transfer verläuft und das Gleichgewicht praktisch
vollständig auf einer Seite liegt, konnte die Reversibilität der Reaktion bisher nicht
experimentell nachgewiesen werden. Ein solcher Nachweis könnte jedoch durch
Einführung einer Isotopenmarkierung bei der Reaktion des 4,5-Dimethyl-2,3-Dihydro-1-H-
[1,3,2]-Diazagermol-2-ylidens mit einem entsprechenden perdeuterierten Diazabutadien
geführt werden.
N
N
R
R
E
E = Si, Ge, Sn
N
N
R
R
N
NGe
R
R
+N
N
R
R
N
NGe
R
R
+CD3
CD3
D3C
D3C

5. Zusammenfassung der Ergebnisse und Ausblick
116
In Kapitel 3 wurde außerdem nachgewiesen, dass der für Sn(II)-Verbindungen beobachtete
cheletrope Zerfall auch bei Diazastannacyclopentenen mit Sn(IV) auftritt. Die geringe
Stabiltät dieser Verbindungen ist erstaunlich und verhinderte ihre vollständige
Charakterisierung. Die Eliminierung des reaktiven Stannylens Sn(CH3)2 konnte durch
Nachweis der daraus entstandenen cyclischen Polystannane (Me2Sn)x belegt werden.
Die isoelektronischen kationischen Analoga der „Arduengo“-Carbene mit den Elementen
Arsen und Antimon sowie die entsprechenden neutralen Chloro-Arsen- bzw. Chloro-
Antimonverbindungen konnten in dieser Arbeit intensiv untersucht werden. Die
Darstellung analoger Verbindungen mit Bismut blieben bislang ohne Erfolg und könnten
das Ziel künftiger Untersuchungen sein.
Anhand röntgenstrukturanalytischer und schwingungsspektroskopischer Untersuchungen an
1,3,2-Diazastibolenium-Kationen und 1,3,2-Diazarsolenium-Kationen gelang der
Nachweis, dass π-Delokalisation in 1,3,2-Diazastibolenium-Kationen eine gewichtige Rolle
spielt und von 1,3,2-Diazaphospholenium-Kationen zu den höheren Homologen in der
Reihenfolge P < As < Sb zunimmt. Dieser Trend steht im Widerspruch zur abnehmenden
Tendenz der Elemente in höheren Perioden π-Bindungen auszubilden, stimmt allerdings mit
den Beobachtungen überein, die an Diazastannol-2-ylidenen gemacht wurden. Durch
röntgenstrukturanalytische und schwingungsspektroskopische Untersuchungen war es
möglich, in 1,3,2-Diazastibolenen Hyperkonjugation der π(C2N2)-Einheit mit dem σ*-
Orbital der Sb-Cl-Bindung nachzuweisen, die hier allerdings etwas weniger ausgeprägt ist
als in Chlor-1,3,2-Diazaphospholenen und Chlor-1,3,2-Diazarsolenen.
1,3,2-Diazastibolene wie auch 1,3,2-Diazastibolenium-Kationen sind bedingt durch ihre
Oxophilie nur äußerst schwer handhabbar. Das 1,3,2-Diazastibolenium-Kation 11b[AlCl4]
reagiert mit Sauerstoff zu einem Addukt aus Antimon(III)chlorid und Diazabutadien. 1,3,2-
Diazastibolenium-Kationen 13b[Cl] reagieren zudem mit Phosphor(V)chlorid oder
Antimon(V)chlorid unter Oxidation des Ringsystems zu Diazabutadien und Folgeprodukten
wie dem 1,3,2-Diazaphospholenium-Kation 11b[SbCl4] oder dem 1,3,2-Diazastibolenium-
Kation 13b[SbCl4]. 1,3,2-Diazastibolene sind wie 2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-
ylidene thermolabil. Der Transfer eines SbCl-Fragmentes zwischen 1,3,2-Diazastibolenen
und Diazabutadienen konnte bislang nicht beobachtet werden. Denkbar wäre jedoch analog
zum Transfer eines Germaniumatoms auch ein Austausch eines SbCl-Fragmentes zwischen
4,5-Dimethyl-1,3,2-Diazastibolenen und Diazabutadienen.

5. Zusammenfassung der Ergebnisse und Ausblick
117
1,3,2-Diazastibolenium-Kationen und 1,3,2-Diazarsolenium-Kationen sowie die
entsprechenden Neutralverbindungen reagieren überraschenderweise nicht wie 1,3,2-
Diazaphospholenium-Kationen mit elektronenreichen Übergangsmetallkomplexen. Dieses
kann auf den schwächer ausgeprägten σ-Donor-Charakter des freien Elektronenpaares als
Folge eines höheren s-Charakters sowie möglicherweise zusätzlich auf die erhöhte π-
Elektronendichte (= geringere π-Akzeptorfähigkeit) am Gruppe-15-Element zurückgeführt
werden. 1,3,2-Diazastibolenium-Kationen und 1,3,2-Diazarsolenium-Kationen fungieren
jedoch als Liganden in Komplexen mit einem Metallfragment wie Co(CO)4-. oder Co(CO)3
-
, von denen der 1,3,2-Diazarsolenium-Co(CO)3-Komplex isoliert werden konnte. Eine
vollständige röntgenstrukturanalytische Untersuchung dieses Komplexes steht aber noch
aus. Demgegenüber gelang die röntgenstrukturanalytische Untersuchung des 1,3,2-
Diazarsolenium-Co(CO)4-Komplexes, der als Vorstufe zum 1,3,2-Diazarsolenium-
Co(CO)3-Komplex auftritt. Ein 1,3,2-Diazastibolenium-Co(CO)3-Komplex konnte aufgrund
mangelnder thermischen Stabilität bislang nicht isoliert werden.
IR-Untersuchungen und Vergleich der CO-Schwingungen der homologen Verbindungen
mit P, As und Sb deuten auf einen etwas stärker ausgeprägten elektrophilen Charakter des
1,3,2-Diazastibolenium-Liganden in Co(CO)3-Komplexen hin. Dieses steht im Widerspruch
zu den aus schwingungsspektroskopischen Untersuchungen erhaltenen, die eine erhöhte
Elektronendichte im b1(π)-Orbital des 1,3,2-Diazastibolenium-Kations implizieren. Die
Frage inwieweit ein trotz der ausgeprägten Konjugation des 6π (C2N2)-Fragmentes mit dem
b1(π)-Orbital im 1,3,2-Diazastibolenium-Kation tiefer liegendes LUMO auch den stärkeren
elektrophilen Charakter hervorrufen kann, könnte durch quantenchemische Rechnungen
beantwortet werden.
N
N
R
R
N
NSb-Cl
R´
R´
+N
N
R´
R´
N
NSb-Cl
R
R
+

6. Experimenteller Teil
118
6. Experimenteller Teil 6.1 Allgemeines
6.1.1 Arbeitsbedingungen
Alle Reaktionen wurden aufgrund der Oxidations-und Hydrolyseempfindlichkeit der
verwendeten Substanzen in ausgeheizten Glasapparaturen oder in einer Glove-Box unter
Ausschluß von Feuchtigkeit und Sauerstoff in einer Argon-Schutzgasatmosphäre
durchgeführt. Die verwendeten Lösemittel wurden nach den üblichen Verfahren gereinigt
und getrocknet.
6.1.2 Chemikalien
Folgende Chemikalien wurden nach Literaturangaben dargestellt:
1-tert-Butylimino-2-tert-butylamino-ethan-LiBr-Komplex (39b[LiCl])[31]
1,2-Dimethyl-1,2-(2,4,6-trimethylphenyl)-diazabutadien[164]
1,2-Dimethyl-1,2-(2,6-diisopropylphenyl)-diazabutadien[165]
1,3-Di-tert-butyl-diazabutadien[156]
Zinn(II)bis[bis(trimethylsilyl)]amid][27]
Germanium (II)bis[bis(trimethylsilyl)]amid [27]
Blei(II)bis[bis(trimethylsilyl)]amid[27]
[W(bipy)(CO)3(MeCN)][134]
Thaliumtetracabonylcobaltat(-I)[138]
Alle übrigen Substanzen wurden im Handel käuflich erworben, von Kollegen freundlich
überlassen oder aus Laborbeständen übernommen.
6.1.3 Analytik
Kernresonanzspektren
1 H, 13C, 15N, 31P, 119Sn,207Pb,27Al NMR-Spektren in Lösung wurden auf AMX 300 oder
AC-200 Spektrometern der Firma Bruker gemessen. Die 119Sn und Pb207-NMR-Daten

6. Experimenteller Teil
119
wurden aus 1H-detektierten HMQC-Experimenten und die 15N-NMR-Daten aus 15N-
gsHMBC-Experimenten ermittelt.
Zur Strukturaufklärung einiger Verbindungen wurden (1H,31P)-HMQC, (1H, 13C)-HMQC
und (1H, 13C)-HMBC-Spektren aufgenommen.
1H-NMR: 300 MHz, ext. Standard: Tetramethylsilan (TMS); δ =0
Ξ = 100.000000 MHz 13C-NMR: 75.47 MHz, ext. Standard: Tetramethylsilan (TMS); δ =0
Ξ = 25.145020 MHz
119Sn-NMR: 148.7 MHz, ext. Standard: Tetramethylzinn ( SnMe4) ; δ =0
Ξ = 37.290632 MHz
15N-NMR: 30.42 MHz, ext. Standard: MeNO2; δ =0
Ξ = 10.136767 MHz
31P-NMR: 121.5 MHz, ext. Standard: 85% H3PO4 ; δ =0
Ξ = 40.480742 MHz 207Pb-NMR: 62.9 MHz , ext. Standard: PbMe4 ; δ =0
Ξ =20.920599 MHz 27Al-NMR: 78.2 Mhz ext. Standard: Al(NO3)3 ; δ =0
Ξ =26.056859 MHz
Alle NMR-Verschiebungswerte werden auf der δ-Skala angegeben. Dabei werden
Spinmultiplizitäten wie folgt angegeben: s-Singulett, d-Dublett, t-Triplett, q-Quartett, m-
Multiplett
IR-Spektren
Nicolet 550 FT
Bruker FT-IR IFS 100
Bruker Vektor 22
Messung in KBr-Küvetten (in Lösung oder als Nujolverreibung) im Bereich von 4000-500
cm-1 oder als PE-Pressling im Bereich von 500-100 cm-1 bzw. als KBr-Pressling im
Bereich von 4000-500 cm-1.

6. Experimenteller Teil
120
Raman-Spekten
Bruker RFS 100 ( Die Pulverförmigen Proben wurden in Glaskapillaren eingeschmolzen;
Messungen bei -80°C )
Massenspektren
VG Instruments VG 12-250 (Direkteinlasssystem )
Concept 1H der Firma Cratos Analytical Ltd., Manchester, GB.
Die Massenspektren wurden unter Elektronenstoßionisation mit 12-16eV gemessen
Die angegebenen Molmassen beziehen sich auf das jeweils häufigste Isotop eines Elements.
Pulverdiffraktogramme
Stadi P der Firma STOE ( Cu Kα-Strahlung, Ge-Monochromator) Röntgenstrukturanalyse
Verwendet wurde ein Diffraktometer vom Typ Nonius Kappa CCD (Mo Kα-Strahlung) ,
Die Berechnug der Strukturparameter wurden mit den Programmpaketen SHELXS,
SHELXL und SHEXTL-PLUS durch Herrn Dr. M Nieger durchgeführt
Elementaranalysen
Die quantitativen C,H,N-Analysen wurden auf einem CHNO-Rapid Gerät der Firma
Heraeus im Institut für Organische Chemie und Biochemie, Abt. Mikroanalyse, der
Universität Bonn durchgeführt.
Schmelzpunktbestimmung
Die Bestimmung erfolgte in abgeschmolzenen Kapillaren mit einem Gerät der Firma Büchi
Flawil (Schweiz). Die Schmelzpunkte wurden nicht korrigiert.

6. Experimenteller Teil
121
CV-Aufnahmen
Die Aufnahmen wurden auf einem Potentiostaten der Fa. Autolab aufgenommen.
Referenzelektrode: 0.004M [(Me5C5)2Fe+]/ [(Me5C5)2Fe] in 0.2M Bu4NPF6 /CH3CN–
Lösung/Pt-Draht; Arbeitselekrode: Glassy Carbon Electrode, 2mm ø; Gegenelektrode: Pt-
Draht, Brückenelektrolyt: 0.2M (Bu)4NPF6 / CH3CN, Leitsalz: 0.2M (Bu)4NPF6 / CH3CN
Elektronenmikroskopie (REM, EDX, TEM)
Rasterelektronenmikroskop DSM 940 (Fa. Zeiss)
EDX-System zu Elementanalytik (HPGe-Detektor, Voyager, Fa. Noran). Die Abbilder
werden mit einer CCD-Kamera (1024 x 1024 Pixel) aufgenommen. Elektronentransparente
Proben werden erhalten, indem die Substanzen als fein verriebene Pulver auf Kupfer-Netze
gegeben wurden.
Im Beugungsmodus ergeben kristalline Proben ein charakteristisches Beugungsbild. Durch
Elektronenstreuexperimente an amorphen Proben lässt sich durch Messung der
Streuintensität, Ermittlung der Interferenzfunktion i(Q) und Fouriertransformation die
Paarverteilungsfunktion (PDF) erhalten.

6. Experimenteller Teil
122
6.2 Chemische Umsetzungen und analytische Untersuchungen
1,2-Dimethyl-1,2- (N,N-dimethylethylendiamin)-diazabutadien (42g)
Eine Lösung aus 230 mmol (19.78 g) 2,3 Butandion und 460 mmol (40.48 g) N,N-Di-
methylethylendiamin wurde in 150 ml Benzen unter Rühren zum Sieden erhitzt. Das bei der
Reaktion frei werdende Wasser wurde durch einen Wasserabscheider abgetrennt.
Nach 3 Stunden Reaktion wurde das Lösemittel unter verminderten Druck entfernt.
Der Rückstand wurde unter Hochvakuum fraktioniert abdestilliert. Man erhielt eine rötliche
Flüssigkeit. Ausbeute: 39 g (75%)
1H-NMR (C6D6) 2.00 [s, 6 H N=CCH3], 2.15 [s, 12H N(CH3)2], 2.63 [t, 2H 3J( 1H,1H)
= 7Hz CH2], 3.43 [t, 2H 3J( 1H,1H) = 7Hz CH2]
13C-NMR (C6D6) 11.1 [CH3], 44.7 [N(CH3
)2], 50.4 [CH2], 59.4 [CH2], 166.7 [N=C]
MS: (EI, 16eV, 100°C) m/z(%) = 226(20) [M+], 225(20) [M+-H], 211(5) [M+-CH3],
181(15) [M+-HNMe2], 168(20) [M+-CH2NMe2], 112(2)
[M+- 2 CH2NMe2] 170(34) [M+-CH2NMe2]
IR: (KBr/Film) 1028(s) ; 1024(s) ; 1098(s) ; 1152(s) ; 1180(w) ; 1267(m) ; 1358(m);
1480(s) ; 1637(s) ; 1699(w) ; 2767(s) ; 2817(s) ; 2860(s) ; 2943(s) ;
2971(s) cm-1
N
N
CH2CH2NMe2
CH2CH2NMe2

6. Experimenteller Teil
123
Allgemeine Vorschrift für Diazabutadien-Dilithiumsalze 21b,c,f,g und 42c,f,g
Zu 200 mol einer Lösung des entsprechenden Diazabutadiens in 150 ml THF wurden 500
mmol Lithiumspäne gegeben. Die Mischung wurde 2 Tage bei RT unter sorgfältigem
Ausschluss von Sauerstoff gerührt. Das überschüssige Lithium wurde über eine G-2-Fritte
abfiltriert. Anschließend wurde das Lösemittel entfernt und der feste Rückstand mit Hexan
aufgenommen und filtriert. Im Filterrückstand blieb festes gelbes bis ockerfarbenes Produkt
zurück. Bedingt durch die extreme Luftempfindlichkeit der Substanzen wurden keine
Versuche zur Charakterisierung des Rohproduktes unternommen und dieses für weitere
Reaktionen eingesetzt.
(2-Dimethylamino-ethyl)-[2-(2-dimethylamino-ethylimino)-1-methyl-propyl]amin
(44g)
100 mmol (24 g) Dilithiumsalz 42g wurde in 200 ml THF gelöst und die Lösung in einen
250 ml Tropftrichter auf einem 500 ml Dreihalskolben, umgefüllt. In den Dreihalskolben
wurden 100 ml THF vorgelegt und etwas Dilithiumsalzlösung zugetropft. Zu dieser Lösung
wurde über ein Septum mit einer Spritze wasserfreies Ethanol gegeben, bis sich die
Reaktionslösung gerade gelb färbt. Dieser Vorgang wird solange wiederholt bis 200 mmol
(9.2 g) Ethanol verbraucht wurden und die Reaktionslösung farblos ist.
Anschließend wurde das Lösemittel entfernt und der Rückstand unter Hochvakuum
fraktioniert destilliert. Man erhielt eine schwach gelbliche viskose Flüssigkeit.
Ausbeute: 9g (40%)
N
NH
CH2CH2NMe2
CH2CH2NMe2

6. Experimenteller Teil
124
1H-NMR (C6D6) 1.08 [d, 3H 3J( 1H,1H) = 6.8 Hz CHCH3], 1.53 [s, 3H N=CCH3], 2.05
[s, 6H N(CH3)2], 2.16 [s, 6H N(CH3)2], 2.30 [m, 2H NHCH2CH2N],
2.57 [m, 2H C=NCH2CH2N], 2.65 [t, 2H, 3J( 1H,1H) = 7.0 Hz
C=NCH2CH2N], 3.31 [m, 1H CHCH3], 3.35 [m, 2H NHCH2CH2N]
13C-NMR (C6D6) 20.0 [CHCH3], 13.4 [N=CCH3], 45.8 [N(CH3)2], 46.1 [NHCH2
CH2N] 46.3 [N(CH3)2], 50.7 [NHCH2CH2N], 60.2 [C=NCH2CH2N]
62.3 [C=NCH2CH2N], 63.2 [CHCH3], 172.1 [N=C]
MS: (EI, 16eV, 250°C) m/z(%) = 229(5) [M++1], 228(5) [M+], 226(5) [M+-2H], 225(5)
[M+-3H], 182(5) [M+-H –HNMe], 181(4)
[M+-2H –HNMe]
IR: (KBr/Film) 1042(s) ; 1097(m) ; 1142(m) ; 1180(m) ; 1242(w) ; 1268(m); 1360(m)
; 1457(s) ; 1662(w) ; 2765(s) ; 2815(s) ; 2855(s) ; 2941(s) ; 2970(s) ;
3313(w) ; 3445(w) cm-1
(2-tert-Butylimino-ethyl)-tert-butylamin-LiBr-Komplex (39[LiBr])
Eine Lösung von 180 mmol (30.24 g) 1,3-Di-tert-butyl-diazabutadien 20b in 150 ml THF
wurde mit 450 mmol (3.15 g) Lithiumstücken versetzt. Nach 2 Tagen Rühren wurde die
Reaktionslösung filtriert und der Filterrückstand in einen Tropftricher auf einem 500 ml
Dreihalskolben, der mit 100 ml THF gefüllt ist, überführt. Unter Rühren wurde eine kleine
Menge der Lösung des Diazabutadien-Dilithiumsalzes 21b in den Reaktionskolben getropft
und so lange trockenes Trietylaminhydrochlorid hinzugegeben bis sich die Reaktionslösung
gerade entfärbt. Der Vorgang wurde solange wiederholt, bis 360 mmol
N
NH
tBu
tBu
11/2 LiBr

6. Experimenteller Teil
125
Trietylaminhydrochlorid verbraucht waren. Nach Beendigung der Reaktion wurde das
Lösemittel im Vakuum entfernt, der Rückstand mit Hexan versetzt und abfiltriert. Der
Filterrückstand wurde mit Methylenchlorid aufgenommen und filtriert um überschüssiges
LiBr zu entfernen. Aus dem Filtrat wurde nach Entfernung des Lösemittels im Vakuum
ein weiß-grauer Feststoff isoliert. Ausbeute: 17 g
Smp.: 135°C
Elementaranalyse: Berechnet für (C10H20N2LiBr): C 40.07 H 7.35 N 9.35 %
Gefunden: C 40.88 H 8.17 N 9.13 %
1H-NMR (CDCl3): 7. 70 [t, 1H, 3JHH = 1.53 Hz N=CH], 3.50 [d, 2H 3JHH = 1.53 Hz, CH-
CH2], 1.29 [s, 9H C(CH3) 3], 1.20 [s, 9H C(CH3) 3]
13C-NMR (CDCl3) 27.9 [C(CH3)3], 28.7 [C(CH3)3], 45.4 [C(CH3)3], 50.8 [C(CH3)3], 57.1
[CH-CH2], 159.7 [N=CH]
[2-(1,3,5-Trimethylphenyl)imino-ethyl]-(1,3,5-trimethylphenyl)amin (39c)
100 mmol (30.6 g) Dilthiumsalz 21b wurden in 150-200 ml THF gelöst und in einen 250
ml Tropftrichter, auf einem 500 ml Dreihalskolben umgefüllt. Im Dreihalskolben wurden
50 ml THF vorgelegt und etwas Dilithiumsalzlösung zugeben. Zu dieser Lösung im
Dreihalskolben wurde über ein Septum mit einer Spritze wasserfreies Ethanol gegeben bis
sich die Reaktionslösung gerade gelb färbt. Dieser Vorgang wurde solange wiederholt bis
200 mmol (9,2 g) Ethanol verbraucht waren .
Anschließend wurde das Lösemittel entfernt, der gelbe pastöse Rückstand in 100 ml Hexan
aufgenommen und das Lithiumethanolat über eine G-4 Fritte abfiltriert. Bei der
N
NH
Mes
Mes

6. Experimenteller Teil
126
Aufarbeitung ist darauf zu achten, dass das Produkt temperaturempfindlich ist und nach
einigen Stunden bei Raumtemperatur eine Zersetzungsreaktion eingeht. Zur Reinigung des
Rohproduktes zu erhalten wurde die Hexanlösung auf 50 ml eingeengt und über Nacht bei –
2°C gelagert. Dabei kristallisierte ein gelber, faseriger Feststoff aus. Durch nochmaliges
Einengen der Mutterlauge ließ sich weiteres Produkt auskristallisieren.
Ausbeute: 12 g ( 41% )
Um die Ausbeute zu erhöhen genügte es, die Hexanlösung bis zu Trockene einzuengen und
den Rückstand gut zu trocknen. Man erhielt das Produkt als einen pasteusen hellgelben bis
weißen Feststoff. Ausbeute: 18 g ( 62 % )
Smp.: 48°C
1H-NMR (C6D6) 2.01 [s, 6H CH3], 2.16 [s, 3H CH3], 2.19 [s, 3H CH3], 2.27 [s, 6H
CH3], 3.62 [dd, 2H, 3J(H,H) = 5.2, 2.2 Hz, N-CH2], 4.73 (t, 1H, 3J(H,H) = 5.2, NH], 6.78 [s, 2H m-H], 6.79 [s, 2H m-H], 7.22 (t, 1H, 3J(H,H) = 2.2 Hz, N=CH]
13C-NMR (C6D6) 164,0 [N=CH], 148,9 [i-C], 145,0 [i-C], 133,2 [p-C], 130.7 [p-C],
130,1 [m-C], 129,4 [m-C], 128,9 [o-C], 127,1 [o-C], 53,7 [N-CH2],
21.0 [p-CH3], 20,9 [p-CH3], 19,3 [o-CH3], 18,5 [o-CH3].
MS (EI, 12eV) m/z(%) = 294 (89) [M+], 277 (42) [M+-CH3,-2H],
IR (KBr): 3350 (NH), 1666 (C=N) cm-1

6. Experimenteller Teil
127
[2-(2,6-diisopropylphenyl )imino-ethyl]-(2,6-diisopropylphenyl)amin (39f)
60 mmol (23.4) Dilthiumsalz 21f wurden in 200 ml THF gelöst und in einen 250 ml
Tropftrichter, auf einem 500 ml Dreihalskolben umgefüllt.
Die Reaktion und Aufarbeitung des Produktes wurde auf die gleiche Weise durchgeführt
wie bei der Darstellung von 39b beschrieben.
Man erhielt durch Kristallisation aus einer Hexanlösung über Nacht bei –2°C einen gelben,
kristallinen Feststoff Ausbeute: 13 g ( 57 % )
Smp.: 91°C
1H-NMR (CDCl3) 1.32 [d, 12H 3J = 6.9 Hz CH(CH3) 2], 1.22 [d, 12H 3J = 6.9 Hz
CH(CH3)2], 3.48 [m, 2H, 3J = 6.9 Hz CH(CH3) 2)], 2.99 [m, 2H, 3J =
6.9 Hz CH(CH3)2], 4.10 [dd, 2H, 3J(H,H) = 5.2, 2.1 Hz, N-CH2], 4.71
[t, 1H, 3J(H,H) = 5.2, NH], 7.17 [m, 3 H aromat-H], 7.15 [m, 3 H
aromat-H], 7.85 [t, 1H, 3J(H,H) = 2.1 Hz, N=CH]
13C-NMR (CDCl3) 23.9 [CH(CH3)2], 24.5 [CH(CH3)2], 28.2 [CH(CH3)2], 28.4 [CH(CH3)
2], 56.0 [N-CH2], 123.4 [m-C], 128.3 [p-C], 124.0 [m-C], 124.7 [p-C],
134.0 [o-C], 142.0 [o-C], 144.3 [i-C], 148.6 [i-C],
163.4 [N=CH]
MS (EI, 16eV) m/z(%) = 378 (56) [M+], 334(4) [M+-C3H7], 333 (16) [M+-C3H7 -H]
N
NH
Dipp
Dipp

6. Experimenteller Teil
128
N,N´-Di-(2,6-diisopropylphenyl)-but-2-en-2,3-diamin (43f)
48 mmol (20.0 g) Dilthiumsalz 42f wurde in 150 ml THF gelöst und in einen 250 ml
Tropftrichter, auf einem 500 ml Dreihalskolben umgefüllt.
Die Reaktion und Aufarbeitung des Produktes wurde auf die gleiche Weise durchgeführt
wie bei der Darstellung von 39b beschrieben.
Man erhielt durch Kristallisation aus einer Hexanlösung über Nacht bei –2°C einen
farblosen bis gelblichen, kristallinen Feststoff, der getrocknet wurde. Aus der Mutterlauge
ließ sich weiteres Produkt auskristallisieren. Ausbeute: 8 g ( 41 % )
Smp.: 85°C
1H-NMR (C6D6) 7.17 [s, 6H, aromat-H), 4.84 [s, 2H, NH), 3.41 [m, 4H, 3J = 6.54 Hz
CH(CH3)2), 1.58 [s, 6H, =C-CH3,), 1.29 [d, 12H 3J = 6.54 Hz
CH(CH3)
MS (EI, 16eV) m/z(%) = 406 (4) [M+], 362 (5) [M+-C3H7], 362 (19) [M+-C3H7 -H]
Raman: (Pulver) 276 ; 441 ; 576 ; 698 ; 1044 ; 1110 ; 1159 ; 1176 ; 1265 ; 1390 ; 1445 ;
1591 ;1643; 1667 ; 2864 ; 2922 ; 2962 ; 3064 cm-1
IR: (CH2Cl2) 796(m) ; 818(m) ; 934(m) ;1043(m) 1058(m) ; 1118(s) ; 1160(w) ;
1184(m) ; 1239(w) ; 1329(m) ; 1365(s) ; 1384(s) ; 1464(s) ; 1589(m)
; 1642(s) ; 1667(s) ; 2870(s) ; 2927(s) ; 3305(m) ; 3356(m) cm-1
NH
NH
Dipp
Dipp
CH3
CH3

6. Experimenteller Teil
129
N,N´-Di-(2,4,6-trimethylphenyl)-but-2-en-2,3-diamin (43c)
57 mmol (24.74g) Dilthiumsalz wurden in 150 ml THF gelöst und in einen 250 ml
Tropftrichter auf einem 500 ml Dreihalskolben umgefüllt. Die Reaktion und Aufarbeitung
des Produktes wurde auf die gleiche Weise durchgeführt wie bei der Darstellung von 39b
beschrieben.
Man erhielt durch Kristallisation aus einer Hexanlösung über Nacht bei –30°C einen
gelblichen kristallinen Feststoff, der getrocknet wurde. Ausbeute: 6 g ( 31 % )
Smp.: 80°C
1H-NMR (C6D6) 6.80 [s, 4H m-H], 4.53 [s, 2H, NH], 2.18 [s, 6H p-CH3], 2.18 [s, 12H
o-CH3], 1.50 [s, 6H 4,5 CH3]
13C-NMR (C6D6) 14.6 [-CH3] 18.8 [o-CH3], 21.1 [p- CH3], 132.9 [ o-C], 132,5 [i-C],
139.9 [p-C], 126.9 [4,5C], 129.7 [m-C] 15N-NMR (C6D6) -305.8
MS (EI, 16eV) m/z(%) = 322 (25) [M+], 305 (41) [M+-2H, - CH3], 162 (100) [M+-
MesCN-H]
IR: (CH2Cl2) 1621(w) ; 1638(m) ; 1650(w) ; 1666(s)
NH
NH
Mes
Mes

6. Experimenteller Teil
130
1,3-Di-tert-butyl-2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-yliden (7b)
4.7 mmol ( 1.0 g ) 39[LiCl] wurde in 30 ml Toluen gelöst. Nach Zugabe von 3.8 mmol
(1.65 g) Zinn(II)bis[bis(trimethylsilyl)]amid wurde die Lösung bei 40°C zwei Stunden
gerührt. Anschließend wurde Toluen im Vakuum entfernt und der Rückstand mit Hexan
aufgenommen. Es wurde filtriert und das Filtrat bis zur Trockene eingeengt. Man erhielt
einen dunkelroten, pastösen Feststoff. Durch Umkristallisation konnte ein roter kristalliner
Feststoff gewonnen werden. 0.27 g ( 20% )
Smp.: 40°C
1H-NMR (C6D6) 7.44 [s, 2H, 3J( 119Sn, 1H) = 8 Hz, CH], 1.41 (s, 18H C(CH3)3)
13C-NMR (C6D6): 33.9 [C(CH3) 3], 56.7 [C(CH3) 3], 125.5 [4,5C]
119Sn-NMR (C6D6): 237.0
MS: (EI, 16eV) m/z(%) = 288 (4) [M+]
N
NSn
tBu
tBu

6. Experimenteller Teil
131
1,3-Dimesityl-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-yliden (7c)
N
NSn
Mes
Mes
Zu 6.8 mmol (2 g) 39c wurden 6.8 mmol ( 2.99 g) Zinn(II)bis[bis(trimethylsilyl)]amid
gelöst in 15 ml Toluen gegeben.
Die Mischung wurde nach Zugabe weniger Tropfen Hexamethyldisilazan 90 min bei genau
40°C gerührt. Die Reaktionslösung wurde danach sofort filtriert oder abdekantiert und mit
Hexan unterschichtet. Bei –30°C fielen nach wenigen Stunden dunkelrote bis schwarze
Kristalle aus, welche getrocknet und in der Glove-Box in einen sauberen Kolben umgefüllt
wurden. Das Produkt kann dann über Wochen bei -30° C unter Schutzgas aufbewahrt
werden. Ausbeute: 1.1 g (40% )
Smp.: 105° C
Elementaranalyse: Berechnet für (C20H24N2Sn): C 58.43 H 5.88 N 6.81
Gefunden: C 56.22 H 5.81 N 6.92
1H-NMR (C6D6) 6.88 [s, 2H, 3J( 119Sn, 1H) = 8Hz, CH], 6.89 [s, 4H m-H], 2.22 [s, 6H
p-CH3], 2.27 [s, 12H o- CH3]
13C-NMR (C6D6) 18.5 [o-CH3], 20.9 [p- CH3], 128.7 [4,5C], 129.2 [m-C], 133.3 [p-C],
134.3 [o-C], 146.1 [i-C]
15N-NMR (C6D6) -135.7
119Sn-NMR (C6D6) 259
MS: (EI, 16eV) m/z(%) = 412(30) [M+], 277(100) [M+ - Sn -CH3]

6. Experimenteller Teil
132
1,3-Di(2,6-diisopropylphenyl) -2,3-dihydro-1-H-[1,3,2]-diazastannol-2-yliden (7f)
9 mmol (3.4 g) 39f wurden mit einer Lösung von 9 mmol Zinn(II)-
bis[bis(trimethylsilyl)]amid in 50 ml Hexan und wenigen Tropfen Hexamethyldisilazan
versetzt. Es wurde 4h bei genau 60°C gerührt. Die warme Reaktionslösung wurde danach
sofort filtriert und etwas eingeengt. Bei –30°C fielen über Nacht lange orangefarbene
Nadeln aus, welche getrocknet wurden. Ausbeute: 3.1g (68% )
Smp. Zersetzung bei 130°
Elementaranalyse: Berechnet für (C26H38N2Sn): C 63.07 H 7.28 N 5.66
Gefunden: C 62.22 H 7.70 N 5.43
1H-NMR (C6D6) 7.04 [s, 2H, 3J( 119Sn, 1H) = 9.2Hz CH], 7.19 [s, 6H aromat-H], 3.31
[m, 4H, 3J = 7 Hz CH(CH3) 2], 1.21 [d, 12H 3J = 7 Hz CH(CH3)2],
1.19 [d, 12H 3J = 7 Hz CH(CH3) 2]
13C-NMR (C6D6) 144.9 [ o-C], 144,7 [i-C], 130.3 [p-C], 126.9 [4,5C], 123.7 [m-C), 28.4
[CH(CH3) 2], 26.16 [CH(CH3) 2], 25.1[CH(CH3) 2]
119Sn-NMR (C6D6) 260.1
MS: (EI, 16eV) m/z(%) = 492(6) [M+], 378(12) [M+ - Sn]
15N-NMR (C6D6): − 111.9
N
NSn
Dipp
Dipp

6. Experimenteller Teil
133
1,3-Di(2,6-diisopropylphenyl)-4,5-dimethyl-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-
yliden (45f)
2.2 mmol (900 mg) 43f wurden mit einer Lösung von 2.2 mmol (966 mg)
Zinn(II)bis[bis(trimethylsilyl)]amid in 15 ml Toluen versetzt. Es wurde bei 120°C gerührt.
Nach 90 min fiel elementares Zinn aus und die Reaktion wurde beendet. Das Zinn wurde
abfiltriert und das Lösemittel unter vermindertem Druck abdestilliert. Das Rohprodukt ließ
sich nicht weiter aufreinigen und wurde ausschließlich NMR-spektroskopisch untersucht.
Ausbeute durch Integration des 1H-NMR-Spektrums: 43f (36%); Diazabutadien 41f (13%)
1H-NMR (C6D6) 7.18 [s, 6H, aromat-H], 3.12 [m, 4H, 3J = 6.91 Hz CH(CH3) 2) ] 1.95
[s, 6H, 4,5 CH3], 1.22 [d, 12H 3J = 6.91 Hz CH(CH3) 2], 1.13
[d, 12H 3J = 7 Hz CH(CH3) 2 , 3J( 119Sn, 1H) = 6.3 Hz]
119Sn-NMR (C6D6) 256
Umsetzung von 1,3-Dimesityl-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-yliden mit DMAP
Eine Lösung von 0.1 mmol 7c in 0.5 ml C6D6 wurde mit DMAP im Überschuss versetzt
und NMR-spektroskopisch untersucht. Für die 1H-Signale des freien und des koordinierten
DMAP wurden aufgrund des schnellen chemischen Austausches mit dem Diazastannol-2-
yliden Mittelwertsignale gefunden.
1H-NMR (C6D6) 8.38 [dd 2H, 3J(1H, 1H) = 9.6 Hz , H-aromat ], 6.83 [s, 2H, 3J( 119Sn,
1H) = 9.4 Hz, CH], 6.90 [s, 4H m-H], 6.09 [dd 2H, 3J(1H, 1H) = 9.6
Hz , H-aromat ], 2.21[s, 6H p-CH3], 2.27 [s, 12H o- CH3], 2.24 [s, 6H
CH3] 119Sn-NMR (C6D6) 236
N
NSn
Dipp
Dipp

6. Experimenteller Teil
134
a) Umsetzung von 43g mit Zinn(II)bis[bis(trimethylsilyl)]amid
Eine Lösung von 1 mmol (440 mg) Zinn(II)bis[bis(trimethylsilyl)]amid in 5 ml Hexan
wurde mit 1 mmol (230 mg) 43g versetzt und bei 40-50°C gerührt. Nach 30 min fiel
elementares Zinn aus der roten Reaktionslösung aus. Nach weiteren 3h Rühren bei 40-50°C
wurde der Niederschlag abfiltriert und das Lösemittel des Filtrates unter vermindertem
Druck abdestilliert.
Man erhielt ein dunkelrotes Öl, das NMR-spektroskopisch untersucht wurde. Das 119Sn-
und 1H-NMR-Spektrum zeigte 3 Produkte von denen zwei als 52 und 43g identifiziert
wurden.
als 52 idenfiziertes Produkt mit 119Sn = -262.7 ppm:
1H-NMR (C6D6) 1.63 [s, 3H, 5J(1H,119/117Sn) = 9 Hz , CH3-C=N], 2.03 [s, 6H, NMe2],
2.11 [s, 6H, NMe2], 4.00 [s, 1H, 4J(1H,119/117Sn) = 8.5 Hz, C=CH2 ]
4.39 [s, 1H, 4J(1H,119/117Sn) = 12.4 Hz , C=CH2 ]
13C-NMR (C6D6) 11.5 [CH3-C=N], 43.21 [ N-CH3], 43.58 [ N-CH3], 84.1 [C=CH2 ] ,
154.7 [C=CH2 ], 171.1 [CH3-C=N]
119Sn-NMR (C6D6) -262.7
unbekanntes Produkt mit 119Sn = -179.9 ppm
1H-NMR (C6D6) 1.76 [s, 3H, 5J(1H,119/117Sn) = 7.5 Hz , CH3-C=N], 1.99 [s, 6H, NMe2],
2.06 [s, 6H, NMe2], 3.92 [s, 1H, 4J(1H,119/117Sn) = 10.5 Hz, C=CH2 ]
4.43 [s, 1H, 4J(1H,119/117Sn) = 12.8 Hz , C=CH2 ]
13C-NMR (C6D6) 14.1 [CH3-C=N], 43.78 [ N-CH3], 44.45 [ N-CH3], 84.7 [C=CH2 ] ,
154.73 [C=CH2 ], 170.9 [CH3-C=N]
119Sn-NMR (C6D6) -179.9

6. Experimenteller Teil
135
b) Umsetzung von 44g mit Zinn(II)bis[bis(trimethylsilyl)]amid
Eine Lösung von 0.8 mmol (350 mg) Zinn(II)bis[bis(trimethylsilyl)]amid in 10 ml Hexan
wurde mit 0.8 mmol (230 mg) 44g versetzt und 8h bei 60°C gerührt. Das Lösemittel wurde
unter vermindertem Druck abdestilliert. Man erhielt ein dunkelrotes Öl das NMR-
spektroskopisch untersucht wurde. Es wurde neben den noch nicht abreagierten Edukten
44g und Zinn(II)bis[bis(trimethylsilyl)]amid ein Produkt mit δ119Sn = -262.7 ppm
nachgewiesen, das durch Vergleich mit den 1H und 119Sn-NMR-Daten aus a) als Stannylen
52 identifiziert werden konnte.
Thermolyse des 1,3-Dimesityl-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-ylidens (7c)
Eine Lösung von 0.5 mmol (200 mg) 7c in Toluen wurde 1h auf 60°C erwärmt. Das aus der
Lösung ausfallende kristalline Feststoff wurde abfiltriert mit Toluen gewaschen und
getrocknet. Der Feststoff wurde anhand eines Pulverdiffraktogrammes als tetragonales Zinn
identifiziert. Im Filtrat konnte NMR-spektroskopisch als einziges Produkt das
Diazabutadien 20c nachgewiesen werden
Allgemeine Vorschrift zur Reaktion von 2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-
ylidenen 7b, 7c, 7f mit den Diazabutadienen 20f , 20b, 20c
Eine Lösung von 0.2 mmol des Diazastannol-2-ylidens in 0.5 ml C6D6 wurde mit 0.2 mmol
Diazabutadien versetzt. Die Reaktionslösung wurde in ein NMR-Röhrchen überführt und
ca. 1h auf 40° erwärmt. Anschließend wurde die Lösung 1H-NMR-spektroskopisch
untersucht. Die bei der Zinntransferreaktion entstehenden Austauschprodukte konnten
anhand von authentischen Vergleichsproben identifiziert werden.

6. Experimenteller Teil
136
Allgemeine Vorschrift zur Reaktion des 2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-ylidens
7f mit Schwefel und Selen
Eine Lösung von 0.6 mmol (300 mg) 7f in 5 ml Toluen wurde mit 0.6 mmol Schwefel bzw.
Selen versetzt. Anschließend wurde die Reaktionslösung 90 min auf 40°C erwärmt. Die
aus-fallenden Feststoffe wurden abfiltriert und getrocknet. Durch quantitative EDX-
Elementanalytik wurden die Feststoffe als Zinn(II)sulide bzw. Zinn(IV)selenide
identifiziert. Im Filtrat konnte NMR-spektroskopisch als einziges Produkt das
Diazabutadien 20f nachgewiesen werden.
EDX-Elementanalytik ( in Atom %): ber. für SnS: Sn (50) S (50)
gef.: Sn (48.7) S(51.3)
Ber. SnSe2: Sn (33.3) S (66.7) gef.: Sn (31.5) S(68.5)
Reaktion des 2,3-Dihydro-1-H-[1,3,2]-diazastannol-2-ylidens 7c mit Sauerstoff
In eine Lösung von 1 mmol (410 mg) 7c in 5ml Toluen wurde langsam so lange über
H2SO4 getrocknete Luft eingeleitet bis sich die rote Lösung gelb färbt. Der ausfallende
weiße Niederschlag wurde mit Toluen gewaschen und getrocknet. Durch quantitative EDX-
Elementanalytik wurde der Feststoff als Zinn(IV)oxid identifiziert. Im Filtrat konnte NMR-
spektroskopisch als einziges Produkt das Diazabutadien 20c nachgewiesen werden
EDX-Elementanalytik ( in Atom %): ber. für SnO2: Sn (33.3) O (66.7) gef.: Sn (28.6) O (71.4)

6. Experimenteller Teil
137
1,3- Dimesityl-2,3-Dihydro-1-H-[1,3,2]-Diazagermol-2-yliden (6c)
Zu einer Lösung von 2.6 mmol GeCl2*Dixoan in 20 ml THF wurde langsam bei 0°C eine
Lösung von 2.6 mmol (800 mg) des Dilithiumsalzes 21c in 20 ml THF getropft. Nach
Beendigung der Zugabe wurde das Lösemittel unter verminderten Druck abdestilliert und
der Rückstand mit 15 ml Hexan aufgenommen. Das LiCl wurde über eine G4-Ftitte
abfiltiert. Aus dem Filtrat fielen bei -30°C über Nacht ockerfarbene Kristalle aus. Die
Kristalle wurden getrocknet. Ausbeute: 0.75g ( 76% )
1H-NMR (C6D6) 6.56 [s, 2H CH], 6.85 [s, 4H m-H], 2.17 [s, 6H p-CH3], 2.22 [s, 12H
o- CH3]
1,3-Dimesityl-4,5-Dimethyl-2,3-dihydro-1-H-[1,3,2]-diazagermol-2-yliden (48c)
Eine Lösung von 6 mmol Germanium(II)bis[bis(trimethylsilyl)]amid (2.35 g) und 6 mmol
(1.93 g) 43c in 20 ml Hexan wurde 180 min bei 70°C gerührt. Nach Beendigung der
Reaktion die Lösung auf ihr halbes Volumen eingeengt und der Ansatz bei -30°C stehen
gelassen. Es bildeten sich gelbe Kristalle, die getrocknet wurden. Ausbeute: 1g ( 41% )
Smp: 60°C
N
NGe
Mes
Mes
N
N
Ge
Mes
Mes

6. Experimenteller Teil
138
Elementaranalyse: Berechnet für (C22H28N2Ge): C 67.23 H 7.18 N 7.13
Gefunden: C 66.07 H 7.01 N 7.39
1H-NMR (C6D6) 6.87 [s, 4H m-H], 2.18 [s, 6H p-CH3], 2.13 [s, 12H o- CH3], 1.72 [s,
6H, 4,5 CH3]
13C-NMR (C6D6) 17.4 [o-CH3], 19.6 [p- CH3], 125.5 [4,5C], 127.7 [m-C], 139.7 [p-C],
133.5 [o-C], 133.8 [i-C]
15N-NMR ( C6D6) -157.7
MS: (EI, 12eV) m/z(%) = 394(56) [M+], 305 (45) [M+ - Ge -CH3]
Allgemeine Vorschrift zur Reaktion des 1,3-Dimesityl-4,5-Dimethyl-2,3-Dihydro-1-H-
[1,3,2]-Diazagermol-2-ylidens (48c) mit den Diazabutadienen 20b und 20c
Eine Lösung von 0.2 mmol des betreffenden Diazagermol-2-ylidens in 0.5 ml C6D6 wurde
mit 0.2 mmol Diazabutadien versetzt. Die Reaktionslösung wurde in ein NMR-Röhrchen
überführt und ca. 8h auf 70° erwärmt. Anschließend wurde die Lösung 1H-NMR-
spektroskopisch untersucht. Die bei der Germaniumtransferreaktion entstehenden
Austauschprodukte wurden anhand von authentischen Vergleichsproben bzw. im Falle des
Diazagermol-2-ylidens 6b durch Vergleich mit Literaturdaten identifiziert.

6. Experimenteller Teil
139
Cyclovoltammetrische-Messungen an
1,3-Di(2,6-diisopropylphenyl)-2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-yliden (7f)
Die cyclovoltammetrischen-Messungen wurden mit einer 3mM (0.02g/10ml) Lösung von
7f in CH2Cl2 mit verschiedenen Vorschubgeschwindigkeiten aufgenommen.
ν (mV/s)
Epa(mV) Epk (mV)
δEp (mV)
Ipa (µA) Ipk (µA) Ipk/Ipa E ½ Epa+ Epk/2 (mV)
200 352 294 58 23.2-1.8 = 22.4
-24.4 1.09 323
400 355 295 60 29.3-2.3= 27
-30.2 1.12 325
600 364 299 65 31.5-1.2 = 30.4
-34.35 1.13 331
800 364 299 65 55.6-2.4 = 53.2
-60.1 1.13 331
1000 356 291 65 58.8-2.8 = 56
-64.4 1.15 323.5
2000 -1543 -1640 97.5 -30.3+3.1 = -27
24 0.89 -1591
10000 -1532 -1626 84.5- -211+17.9
= -193
196.8 1.02 -15744
50000 -1532 -1650 118 -270+15 = -255
280 1.1 -1591
Untersuchung der Kinetik der Sn-Austauschreaktion zwischen 20c und 7c
a) Untersuchung der Temperaturabhängigkeit der Reaktion:
A+ B B + A // A = 7c B = 20c
Die Geschwindikeit des Austausches zwischen 20c und 7c wurde an einer Lösung von
88.16 mg 7c in 0.5 ml d8-THF durch Inversions-Recovery-Experimente bei verschiedenen
Temperaturen ermittelt.
6. Experimenteller Teil
140
Aus den Spektren wurden folgende Werte der pseudo-unimolekularen
Geschwindigkeitskonstante kobs erhalten (Tab.). Die Auswertung mit Hilfe der Ansätze von
Arrhenius und Eyring ist im Text beschrieben und ergab folgende Resultate:
Temp. [K]
∆T [K]
k(pseudo 1.Ord.) [mol/s]
∆k(pseudo 1.Ord.) [mol/s]
258 0,5 1,2 0,05 263 0,5 1,7 0,05 268 0,5 2,2 0,1 273 0,5 3.0 0,2 278 0,5 4.0 0,2 283 0,5 5,6 0,4 288 0,5 6,5 0,2 293 0,5 8,3 0,3
b) Konzentrationsabhängigkeit kAus
Eine Lösung von 89 mg 7c und 13 mg 20c in 0.5 ml d8-THF wurde jeweils dreimal im
Verhältnis 1: 2 verdünnt. In den einzelnen Lösungen wurden durch Inversions-Recovery-
Experimente die Austauschgeschwindigkeiten bstimmt.
Konz. [20c]
[mol/L] Konz. 7c [mol/L]
K(obs.) [mol/s]
K(obs.) /[20c] [1/s]
0,16 0,43 2,96 18,8 0,08 0,21 1,51 20.0 0,04 0,12 0,82 19.0 0,03 0,08 0,49 15,9
Reaktion von 21c mit Dimethylzinn(IV)dichlorid
Zu einer Lösung von 5 mmol (1.1 g) Dimethylzinn(IV)dichlorid in 20 ml Toluen wurde bei
-70°C eine Lösung aus 5 mmol (1.53 g) des Lithiumsalzes 21c in 30 ml Toluen getropft.
Das ausfallende LiCl wurde abfiltriert und das Lösemittel unter verminderten Druck
abdestilliert. Das Rohprodukt wurde anschließend NMR-spektroskopisch untersucht. 1H-NMR-spektroskopisch wurden 85% 1,3-Dimesityl-2,2-bis(dimethyl)-1,3-diaza-2-
stannacyclopent-4-en (63c), 10% Diazabutadien 20c und 5 % Polystannane (Me2Sn)n
nachgewiesen. Eine Isolierung von 63c gelang nicht.

6. Experimenteller Teil
141
NMR-Daten von 63c:
1H-NMR (C6D6) 5.68 [s, 2H, 3J( 117/119Sn, 1H) = 65 Hz, CH), 6.86 [s, 4H m-H],
2.18[s, 6H p-CH3], 2.36 [s, 12H o-CH3], 0.22 [s, 6H, 2J(117/119Sn, 1H)
= Hz, Sn-CH3]
13C-NMR (C6D6) -1.6 [Sn-CH3],18.0 [o-CH3], 17.0 [p- CH3], 119.0 [2J( 117/119Sn, 13H) =
32 Hz 4,5C], 128.3 [m-C], 131.4 [p-C],134.0 [o-C], 144.0 [i-C]
119Sn-NMR (C6D6) 66.6
15N-NMR (C6D6) -304.4
Reaktion von 21b mit Dimethylzinn(IV)dichlorid
Zu einer Lösung von 3 mmol (660 mg) Dimethylzinn(IV)dichlorid in 15 ml Toluen wurde
bei -70°C eine Lösung aus 3 mmol (920 mg) Lithiumsalz 21b in 15 ml Toluen getropft. Das
ausfallende LiCl wurde abfiltriert und das Lösemittel unter verminderten Druck
abdestilliert. Das Rohprodukt wurde anschließend NMR-spektroskopisch untersucht. 1H-NMR-spektroskopisch wurden 12.6% 2,3-Di-tert-butyl-2,2-bis(dimethyl)-1,3-diaza-2-
stannacyclopent-4-en (63b), 42.7% des Diazabutadiens 20b, 41% Polystannane (Me2Sn)n
und 3% 2,3-Dihydro-1-H-[1,3,2]-Diazastannol-2-yliden 7b nachgewiesen. Eine Isolierung
von 63b gelang nicht.
NMR-Daten von 63b:
1H-NMR (C6D6) 5.86 [ s, 2H, 3J( 117/119Sn, 1H] = 72 Hz, CH], 1.16 [s, 18H,-CH3], 0.37 [s, 6H; Sn-CH3]
119Sn-NMR (C6D6): 67.7

6. Experimenteller Teil
142
2-Chloro-1,3-di-tert-butyl-1,3,2-diaza-stibolen (13b[Cl])
16.6 mmol ( 3.5 g ) 39[LiCl] und 36.6 mmol (3.7 g) Triethylamin wurden in 20 ml Toluen
gelöst. Zu dieser Lösung wurde unter Rühren 18.2 mmol (4.15 g ) SbCl3 gelöst in 10 ml
Toluen zugetropft. Die entstehende rote bis schwarze Reaktionslösung wurde 45 min bei
50°C gerührt. Anschließend wurde das Lösemittel entfernt, der Rückstand mit 20 ml Hexan
aufgenommen und filtriert. Das orange-rote Filtrat wurde eingeengt und über Nacht bei –
30°C stehengelassen. Die entstehenden orange-roten Kristalle wurden durch Abdekantieren
der Mutterlauge isoliert und getrocknet. Ausbeute: 2.2 g ( 41 %)
Smp: 90° C (Zersetzung)
Elementaranalyse: Berechnet für (C10H20N2SbCl): C 36.9 H 6.15 N 8.62
Gefunden: C 37.0 H 6.53 N 8.45
1H-NMR (C6D6) 1.25 [s, 18H C(CH3) 3 ], 6.6 [s, 2H CH]
13C-NMR (C6D6) 119.5 [ 4,5C], 55.2 [C(CH3) 3], 30.8 [C(CH3) 3]
15N-NMR (C6D6) -208.9
MS: (EI, 16eV, 150°C) m/z(%) = 324(85) [M+], 289(85) [M+-Cl], 232(50) [M+-Cl –C4H9]
Raman: (Pulver) 150 ; 299 ; 396 ; 569 ; 690 ; 968 ; 1220 ; 1262 ; 1569 ; 2868 ; 2905 ;
2972 ; 3058 cm-1
N
NSb-Cl
tBu
tBu

6. Experimenteller Teil
143
IR: (KBr) 3047 (w) ; 2955(ss) ; 1562(w) ; 1458(s) ; 1386(s) ; 1386(ss) ; 1255(ss)
; 1145(m) ; 1058(m) ; 964(m) ; 829(m) ; 686(s) ; 484 (s) cm-1
IR: (Nujol) 3058 CH (w) ; 1568 C=C (w) cm-1
1,3-Di-tert-butyl-2-iodo-1,3,2-diaza-stibolen (13b[I])
Zu einer Lösung von 2 mmol (650 mg) 13b[Cl] in 20 ml THF wurde unter Rühren bei 0°C
2 mmol (400 mg) Trimethylsilyliodid getropft. Die Mischung wurde 90 min bei RT gerührt,
im Vakuum auf ein Volumen von ca. 10 ml eingeengt und über Nacht bei –30°C
stehengelassen. Die entstandenen dunkelroten Kristalle wurden isoliert und getrocknet.
Ausbeute 0.3g ( 36% )
Smp: 90° C (Zersetzung)
Elementaranalyse: Berechnet für (C10H20N2SbI ): C 28.81 H 4.83 N 6.72
Gefunden: C 28.79 H 5.15 N 6.60
1H-NMR (C6D6) 1.18 [s, 18H C(CH3) 3 ], 6.60 [s, 2H CH]
13C-NMR (C6D6) 122.4 [ 4,5C], 56.3 [C(CH3) 3], 29.4 [C(CH3) 3]
MS: (EI, 16eV, 150°C) m/z(%) = 416(9) [M+], 289(100) [M+-I]
IR: (KBr) 3063(w) ; 3028(w) ; 2952(s) ; 2850(m) ; 1539(s) ; 1458( s) ; 1387
(m) 1358(ss) ; 1254(m) ; 1195(ss) ; 1160(s) ; 1057(m) ; 1026(w) ;
1013(s) ; 810(m) ; 710(s) ; 686(s) cm-1
N
NSb-I
tBu
tBu

6. Experimenteller Teil
144
Raman: (Pulver) 129 ; 158 ; 287 ; 570 ; 691 ; 970 ; 1042 ; 1202 ; 1223 ;
1262 ; 1549 ; 2862 ; 2903; 2968 ; 3042 cm-1
2-Bromo-1,3-di-tert-butyl-1,3,2-diaza-stibolen (13b[Br])
1.17 mmol (300 mg) 39b[LiBr] und 2.7 mmol (270 mg) Triethylamin wurden in 10 ml
Toluen gelöst. Zu dieser Lösung wurden unter Rühren 1.3 mmol (470 mg) SbBr3 gelöst in
10 ml Toluen getropft. Die entstehende rote bis schwarze Reaktionslösung wurde 60 min
bei 50°C gerührt. Anschließend wurde das Lösemittel entfernt und der Rückstand mit 15 ml
Hexan und 5 ml Toluen aufgenommen und filtriert. Das orange-rote Filtrat wurde eingeengt
und über Nacht bei –30°C stehengelassen. Die entstandenen roten Kristalle wurden isoliert
und getrocknet. Ausbeute: 0.2 g ( 46 % )
Smp: 90° C (Zersetzung)
Elementaranalyse: Berechnet für (C10H20N2SbBr): C 32.47 H 5.45 N 7.58
Gefunden: C 33.52 H 5.75 N 7.56
1H-NMR ( C6D6 ) 1.22 [s, 18H C(CH3) 3 ], 6.62 [s, 2H CH]
13C-NMR (C6D6) 122.4 [4,5C], 57.3 [C(CH3) 3], 31.9 [C(CH3) 3]
MS: (EI, 16eV, 150°C) m/z(%) = 386(4) [M+], 289(100) [M+-Br], 232(30) [M+-Cl –C4H9]
Raman: (Pulver) 157 ; 570 ; 973 ; 1163; 1205 ; 1266 ; 1559 ; 1862 ; 2905 ; 2971; 3048
cm-1
N
NSb-Br
tBu
tBu

6. Experimenteller Teil
145
IR: (KBr) 2955(ss) ; 2850(m) ; 1595(w) ; 1546(w) ; 1497(s) ; 1488(s) ; 1313(m) ;
1210(ss) ; 1165(m) ; 1057(m) ; 1028(w) ; 704(s) ; 686(s) ; 487 (m) cm-1
IR: (Nujol) 3044 CH (w) ; 1558 C=C (w) cm-1
2-Chloro-1,3-dimesityl-1,3,2-diaza-stibolen (13c[Cl])
Eine Lösung aus 20 mmol ( 5.88 g) 39c in 20ml Toluen wurde auf –78°C abgekühlt und
mit einer Lösung von 20 mmol (4.91 g) Bis(Dimethylamino)antimon(III)chlorid in 20 ml
Toluen versetzt. Die Reaktionslösung wurde langsam auf etwa –40 °C erwärmt. Das hierbei
entstehende Dimethylamin wurde über einen Blasenzähler abgeleitet. Nachdem sich die
Reaktionslösung bei weiterem Erwärmen rot färbte, wurde etwa 60 min bis 90 min bei 40°C
gerührt, bis eine Schwarzfärbung der Reaktionslösung eintrat. Die Reaktionslösung wurde
filtriert und stark eingeengt, mit 15 ml Diethylether überschichtet und über 1-2 Tage bei –
30°C gelagert. Es bildete sich ein orange bis ockerfarbener Feststoff, der von der
überstehenden Lösung durch Abdekantieren befreit und getrocknet wurde.
Ausbeute: 4.2 g ( 47 % )
Smp: 80° C (Zersetzung)
1H-NMR (C6D6) 6.79 [s, 4H, m-H], 6.38 [s, 2H CH], 2.29 [s, 12H o-CH3], 2.13 [s, 6H
p-CH3]
13C-NMR (C6D6) 19.5 [o-CH3], 17.6 [ p-CH3], 124.8 [4,5C], 128.2 [m-C], 140.1 [p-C],
134.0 [o-C], 134.8 [i-C]
N
NSb-Cl
Mes
Mes

6. Experimenteller Teil
146
15N-NMR (C6D6) -219.5
MS: (EI) m/z (%) = 448(20) [M+], 413(16) [M+-Cl], 277(100) [M+-Cl –Sb –CH3]
2-Chloro-1,3-dimesityl-1,3,2-diazarsolen (13c[Cl])
Eine Lösung aus 20 mmol (5.88 g) 39c in 100 ml Toluen wurde auf 10°C abgekühlt und
mit 24 mmol (2.42 g) Triethylamin versetzt. Zu dieser Lösung wurde langsam 22 mmol
(3.99 g) Arsentrichlorid getropft. Die Reaktionslösung wurde auf etwa 60°C erwärmt und 3
Stunden gerührt. Die Reaktionslösung wurde filtriert und auf ein Volumen von ca 50 ml
eingeengt und mit 30 Diethylether überschichtet. Der ausfallende gelbe oder ockerfarbene
Feststoff wurde abfiltriert und getrocknet. Ausbeute: 3.5 g ( 44 % )
Smp.: 200°C
1H-NMR (C6D6) 6.72 [s, 4H, m-H], 6.01 [s, 2H CH], 2.32 [s, 12H o-CH3], 2.08 [s, 6H
p-CH3]
13C-NMR (C6D6) 17.7 [o-CH3], 19.5 [p-CH3], 120.2 [4,5C], 128.6 [m-C], 136.0 [p-C],
134.8 [o-C], 135.7 [i-C]
15N-NMR (C6D6 ) -235.8
MS: (EI) m/z (%) = 402 (20) [M+], 367(16) [M+-Cl], 277(100) [M+-Cl –As –CH3]
N
NAs-Cl
Mes
Mes

6. Experimenteller Teil
147
2-Chloro-1,3-di-tert-butyl-1,3,2-diazarsolen (12b[Cl])
Eine Lösung aus 18 mmol (3.83 g) 38[LiCl] in 100 ml Toluen wurde auf 10°C abgekühlt
und mit 39 mmol (3.94 g) Triethylamin versetzt. Zu dieser Lösung wurde langsam 21 mmol
(3.81 g) Arsentrichlorid getropft. Die Reaktionslösung wurde auf etwa 60°C erwärmt und 3
Stunden gerührt. Nach Abkühlen auf Raumtemperatur wurde filtriert und das Filtat auf ein
Volumen von ca. 50 ml eingeengt. Anschließend wurde die eingeengte Lösung mit 30 ml
Diethylether überschichtet. Der ausfallende weiße Feststoff wurde abfiltriert und
getrocknet. Ausbeute: 2.8 g ( 56 % )
Smp.: 155°C
1H-NMR (C6D6) 1.29 [s, 18H C(CH3) 3 ], 6.36 [s, 2H CH]
13C-NMR (C6D6) 118.0 [ 4,5C], 55.7 [C(CH3) 3], 29.3 [C(CH3) 3]
15N-NMR (C6D6) -206.0
MS: (EI) m/z (%) = 278(40) [M+], 243(100) [M+-Cl]
Raman: (Pulver) 559 ; 984 ; 1163; 1195 ; 1267 ; 1556 ; 2909 ; 2974; 3066 cm-1
IR: (Nujol) 3041 CH (w) ; 1556 C=C (w) cm-1
N
N
As-Cl
tBu
tBu

6. Experimenteller Teil
148
1,3-Di-tert-butyl-1,3,2-diaza-stibolenium-aluminiumtetrachlorid (13b[AlCl4])
Zu 1.55 mmol (500 mg) 13b[Cl] gelöst in 20 ml Diethylether wurde eine Suspension von
1.55 mmol (210mg) AlCl3 in 10 ml Diethylether getropft. Es bildete sich schnell ein gelber
Niederschlag, der nach 1 Stunde Rühren abfiltriert und mit Diethylether gewaschen wurde.
Der Filterrückstand wurde getrocknet und lieferte ein gelbes Pulver als Rohprodukt.
Umkristallisation des Rohprodukts aus Acetonitril und Diethylether ergab reines Produkt in
Form gelber Kristalle. Ausbeute: 0,5g ( 73% )
Smp.: 180° C (Zersetzung)
1H-NMR (CD2Cl2) 1.78 [s, 18H C(CH3) 3 ], 8.57 [s, 2H CH ]
13C-NMR (CD2Cl2) 137.9 [ 4,5C ], 62.4 [ C(CH3) 3 ], 32.0 [ C(CH3) 3 ]
15N-NMR (CD3CN) -91.8
27Al-NMR (CD3CN) 105.2
IR: (Nujol) 491(s) ; 518(w) ; 678(w) ; 722(w) ; 1191(m) ; 1205(m) ; 1509(m) ; 3089
(w); 3104(w) cm-1
(Polyethylen) 491(s) ; 377(m) ; 182(m) ; 177(m) cm-1
Raman: (Pulver) 121 ; 130 ; 165 ; 188 ; 233 ; 287 ; 347 ; 391; 479 ; 563 ; 679 ; 796 ;
923 ; 951 ; 1038 ; 1136 ; 1225 ; 1442 ; 1472 ; 1509 ; 2910 ; 2982 ;
3104 cm-1
N
NSb
tBu
tBu
AlCl4

6. Experimenteller Teil
149
1,3-Di-tert-butyl-1,3,2-diaza-stibolenium-galliumtetrachlorid (13b[AlCl4])
Zu 1 mmol (330 mg) 13b[Cl] gelöst in 10 ml Diethylether wurde eine Suspension von 1
mmol (180 mg) GaCl3 suspendiert in 10 ml Diethylether getropft. Es bildete sich schnell ein
gelber Niederschlag, der nach 1 Stunde Rühren abfiltriert, mit Diethylether gewaschen und
getrocknet wurde. Umkristallisation des Rohprodukts aus Acetonitril und Diethylether
ergab reines Produkt in Form gelber Kristalle. Ausbeute: 0,35 g ( 70% )
Smp.: 190° C (Zersetzung)
Elementaranalyse: Berechnet für (C10H20N2SbGaCl4): C 23.95 H 4.02 N 5.59
Gefunden: C 23.97 H 4.04 N 5.47
1H-NMR (CD3CN) 1.70 [s, 18H C(CH3) 3], 8.49 [s, 2H CH ]
13C-NMR (CD3CN) 136.2 [4,5C], 62.5 [C(CH3) 3], 31.5 [ C(CH3) 3]
15N-NMR (CD3CN) -102.2
IR: (Nujol) 518(w) ; 678(w) ; 722(w) ; 770(m) ; 1205(m) ; 1509(m) ; 3089(m) ; 3103
(m) cm-1
(Polyethylen) 478 ; 430 ; 375 ; 286 ; 188 ; 150 cm-1
Raman: (Pulver) 123 ; 150 ; 188 ; 233 ; 287 ; 344; 369; 375 ; 391 ; 563 ; 678 ; 795 ; 922
; 950 ; 1036 ; 1135 ; 1224 ; 1444 ; 1471 ; 1508 ; 2909 ; 2981 ; 3103
cm-1
N
N
Sb
tBu
tBu
GaCl4

6. Experimenteller Teil
150
1,3-Di-tert-butyl-1,3,2-diaza-stibolenium-tetrachloroantimonat (13b[SbCl4])
Zu 2 mmol (650 mg) 13b[Cl] gelöst in 15 ml Diethylether wurde eine Lösung von 2 mmol
(460 mg) SbCl3 in 10 ml Diethylether getropft. Es bildete sich schnell ein gelber
Niederschlag, der nach 1 Stunde Rühren abfiltriert, mit Diethylether gewaschen und
getrocknet wurde. Umkristallisation des Rohprodukts aus Acetonitril und Diethylether
ergab reines Produkt in Form gelber Kristalle. Ausbeute: 0,56 g ( 51% )
Smp.: 130° C (Zersetzung)
1H-NMR (CD3CN) 1.71 [s, 18H C(CH3) 3 ], 8.57 [s, 2H CH ]
13C-NMR (CD3CN) 136.9 [ 4,5C ], 62.7[ C(CH3) 3 ], 31.5 [ C(CH3) 3 ]
15N-NMR (CD3CN) - 94.5
IR: (Nujol) 515(w) ; 668(w) ; 722 (w) ; 774(w) ; 1191(m) ; 1224(m) ; 1512 (w) ; 3096
(w) cm-1
(Polyethylen) 343(m) ; 290(m) ; 241(w) cm-1
Raman: (Pulver) 187 ; 235 ; 343 ; 292 ; 243 ; 564 ; 951 ; 1513 ; 2973 ; 3098 cm-1
N
NSb
tBu
tBu
SbCl4

6. Experimenteller Teil
151
1,3-Di-tert-butyl-1,3,2-diaza-stibolenium-triflat (13b[OTf])
Zu einer Lösung von 3.5 mmol ( 1.14 g ) 13b[Cl] in 20 ml Diethylether wurden 3.5 mmol
(780 mg) Trimethylsilyltriflat getropft. Es bildete sich schnell ein gelber Niederschlag, der
nach 1 Stunde Rühren abfiltriert, mit Diethylether gewaschen und getrocknet wurde.
Umkristallisation des Rohprodukts aus Acetonitril und Diethylether ergab reines Produkt in
Form gelber Kristalle. Ausbeute: 1.1 g ( 72% )
Smp.: 180° C (Zersetzung)
1H-NMR (CD2Cl2) 1.78 [s, 18H C(CH3) 3 ], 8.62 [s, 2H CH ]
13C-NMR (CD2Cl2 ) 138.5 [ 4,5C ], 63.4 [ C(CH3) 3 ], 33.3 [ C(CH3) 3 ]
15N-NMR (CD3CN ) -84.49
IR: ( Nujol ) 515(m) ; 639(s) ; 1147(m) ; 1195(w) ; 1223(m) ; 1512(w) ; 3083(vw) cm-1
(Polyethylen) 193(m); 208(m) ; 295(m) ; 346(m) ; 379(s) ; 448(w) cm-1
Raman: (Pulver) 168 ; 194 ; 290 ; 310 ; 346 ; 393 ; 569 ; 684 ; 754 ; 800 ; 954 ;1025 ;
1139 ; 1220 ; 1476 ; 1512 cm-1
N
NSb
tBu
tBu
OTf
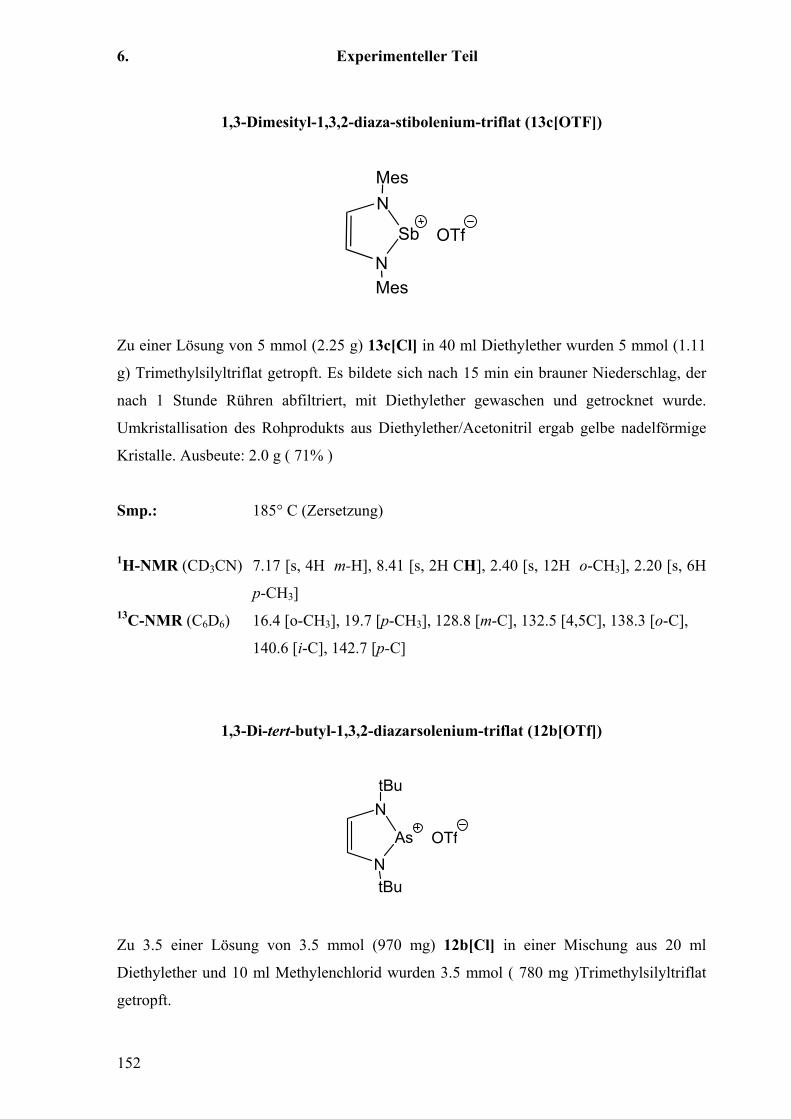
6. Experimenteller Teil
152
1,3-Dimesityl-1,3,2-diaza-stibolenium-triflat (13c[OTF])
Zu einer Lösung von 5 mmol (2.25 g) 13c[Cl] in 40 ml Diethylether wurden 5 mmol (1.11
g) Trimethylsilyltriflat getropft. Es bildete sich nach 15 min ein brauner Niederschlag, der
nach 1 Stunde Rühren abfiltriert, mit Diethylether gewaschen und getrocknet wurde.
Umkristallisation des Rohprodukts aus Diethylether/Acetonitril ergab gelbe nadelförmige
Kristalle. Ausbeute: 2.0 g ( 71% )
Smp.: 185° C (Zersetzung)
1H-NMR (CD3CN) 7.17 [s, 4H m-H], 8.41 [s, 2H CH], 2.40 [s, 12H o-CH3], 2.20 [s, 6H
p-CH3] 13C-NMR (C6D6) 16.4 [o-CH3], 19.7 [p-CH3], 128.8 [m-C], 132.5 [4,5C], 138.3 [o-C],
140.6 [i-C], 142.7 [p-C]
1,3-Di-tert-butyl-1,3,2-diazarsolenium-triflat (12b[OTf])
Zu 3.5 einer Lösung von 3.5 mmol (970 mg) 12b[Cl] in einer Mischung aus 20 ml
Diethylether und 10 ml Methylenchlorid wurden 3.5 mmol ( 780 mg )Trimethylsilyltriflat
getropft.
N
NSb
Mes
Mes
OTf
N
NAs
tBu
tBu
OTf

6. Experimenteller Teil
153
Es bildete sich schnell ein weißer flockiger Niederschlag, der nach 1 Stunde Rühren
abfiltriert und mit Diethylether gewaschen wurde. Der Filterrückstand wurde getrocknet
und lieferte ein weißes flockiges Pulver, das nicht weiter gereinigt wurde.
Ausbeute: 1.0 g ( 73% )
Smp.: 160°C
1H-NMR (CD3CN) 1.76 [s, 18H C(CH3) 3 ], 8.44 [s, 2H CH ]
13C-NMR (CD3CN) 134.6 [ 4,5C ], 62.9 [ C(CH3) 3 ], 30.6 [ C(CH3) 3 ]
15N-NMR (CD3CN) -99.6
IR: ( Nujol ) 515(m) ; 637(s) ; 722(m); 1138(m) ; 1526(w) ; 3097(w) cm-1
Raman: (Pulver) 3134 ; 3099 ; 2989 ; 2915 ; 1529 ; 1479 ; 1260 ; 1237 ; 1151 ; 1029 ;
986 ; 932 ; 809 ; 754 ; 719 ; 606 ; 415 ; 347 ; 248 cm-1
1,3-Dimesityl-1,3,2-diazarsolenium-triflat (12c[OTf])
Zu einer Lösung von 2.5 mmol (1.0 g) 12c[Cl] einer Mischung aus 20 ml Diethylether und
10 ml Methylenchlorid wurden 2.5 mmol (560 mg) Trimethylsilyltriflat getropft.
Es bildete sich schnell ein rosa bis weißfarbener Niederschlag der nach 1 Stunde Rühren
abfiltriert und mit Diethylether gewaschen wurde. Der Filterrückstand wurde getrocknet
und lieferte ein rosafarbenes flockiges Pulver als Rohprodukt. Umkristallisation aus
Diethylether/Acetonitril ergibt farblose rechteckige Kristalle. Ausbeute: 1.2 g ( 93% )
N
N
As
Mes
Mes
OTf

6. Experimenteller Teil
154
Smp: 185°C
1H-NMR (CD2Cl2) 7.17 [s, 4H, m-H], 8.21 [s, 2H CH], 2.43 [s, 12H o-CH3], 2.23 [s, 6H
p-CH3] 13C-NMR (CD3CN) 16.4 [o-CH3], 19.8 [p-CH3], 129.3 [m-C], 133.1 [4,5C], 139.5 [o-C],
135.9 [i-C], 140,1 [p-C]
15N-NMR (CD3CN) -116.9
Raman: (Pulver) 165 ; 224 ; 248 ; 311 ; 347 ; 415 ; 503 ; 571 ; 606 ; 719 ; 754 ; 809 ; 932
; 986 ; 1029 ; 1054 ; 1151 ; 1214 ; 1226 ; 1237 ; 1260 ; 1450 ; 1479 ;
1529 ; 2731 ; 2796 ; 2915 ; 2989 ; 3099 ; 3134 cm-1
Reaktion des 2-Chloro-1,3-Di-tert-butyl-1,3,2-diaza-stibolens 13b[Cl] mit SbCl5
Zu einer Lösung von 1.5 mmol (490 mg) 13b[Cl] in 20 ml Ether wurde langsam bei -40°C
eine Lösung von 1.5 mmol (450 mg) SbCl5 in 15 ml Ether getropft. Es fiel sofort ein gelber
Niederschlag aus. Sobald eine grünliche Färbung des Niederschlags eintritt, wurde die
Reaktion beendet und der Niederschlag abfiltiert und getrocknet. Im Filtrat wurde NMR-
spektroskopisch das Diazabutadien 20b nachgewiesen. Der Feststoff wurde Raman- und
NMR-spektroskopisch untersucht und durch Vergleich mit einer authentischen Probe als
1,3-Di-tert-butyl-1,3,2-diaza-stibolenium-tetrachloroantimonat 13b[SbCl4] identifiziert.
Reaktion des 2-Chloro-1,3-Di-tert-butyl-1,3,2-diaza-stibolens 13b[Cl] mit PCl5
N
NP
tBu
tBu
SbCl4

6. Experimenteller Teil
155
Zu einer Lösung von 1.3 mmol (420 mg) 13b[Cl] in 15 ml Ether wurde langsam bei 0°C
eine Lösung von 1.3 mmol (270 mg) PCl5 in 10 ml Ether getropft. Nach wenigen Minuten
fiel ein grünlicher Niederschlag aus, der abfiltriert und getrocknet wurde. Im Filtrat wurden
NMR-spektroskopisch das Diazabutadien 20b und PCl3 nachgewiesen. Der Feststoff wurde
durch IR- und NMR-spektroskopische Untersuchungen untersucht und wurde als 1,3-Di-
tert-butyl-1,3,2-diaza-phospholenium-tetrachloroantimonat 11b[SbCl4] identifiziert.
NMR- und IR-Daten von 11b[SbCl4]:
1H-NMR (CD3CN) 1.75 [d, 4JPH = 1.68 Hz, 18H C(CH3) 3 ], 8.14 [s, 2H CH ]
31P-NMR (CD3CN) 204.7
IR: (Polyethylen) 340(m) ; 294(m) cm-1
1,3-Di-tert-butyl-diazabutadien-Antimon(III)chlorid-Addukt 77
N
N
t-Bu
t-Bu
2SbCl3
2
*
Eine Lösung von 1 mmol (460 mg) 13b[AlCl4] in 10 ml CH2Cl2 wurde in einen Kolben
überführt, in den langsam Sauerstoff eindiffundieren kann. Der Kolben wurde einige Tage
bei -30°C gelagert. Es bildete sich ein amorpher weißer Niederschlag und gelbe Kristalle. In
der Reaktionslösung konnte NMR-spektroskopisch das Diazabutadien 20b nachgewiesen
werden. Der weiße Niederschlag wurde nicht näher analysiert. Durch eine
röntgenstrukturanalytische Untersuchung konnten die gelben Kristalle als Addukt von 1,3-
Di-tert-butyl-diazabutadien mit Antimon(III)chlorid identifiziert werden.

6. Experimenteller Teil
156
Tricarbonyl-1,3-Dimesityl-1,3,2-diazarsoleniumcobaltat(-1) 81
Zu einer Lösung von 2 mmol (800 mg) 12c[Cl] in 10 ml Acetonitril wurde eine Lösung von
2 mmol (750 mg) TlCo(CO)4 in 10 ml Acetonitril getropft. Das aus der Reaktionslösung
ausfallende TlCl wurde abfiltriert und das Filtrat eingeengt. Bei -30 °C kristallisierte das
Produkt über Nacht in Form dunkelroter Kristalle. Ausbeute: 0.4 g (42%). Die isolierten
Kristalle von 81 waren mit einigen Kristallen von 79 verunreinigt. Eine optische
Separierung dieser Kristalle war nicht möglich.
Smp.: 60° C
Elementaranalyse: Berechnet für (C23H24N2O3AsCo): C 54.12 H 4.7 N 5.49
Gefunden: C 53.62 H 4.79 N 5.6
1H-NMR (C6D6) 6.73 [s, 4H, m-H], 6.31 [s, 2H CH], 2.042 [s, 12H o-CH3], 2.034 [s,
6H p-CH3]
13C-NMR (C6D6) 18.0 [o-CH3], 21.3 [p-CH3], 127.2 [4,5C], 130.1 [m-C], 134.9 [o-C],
135.9 [i-C], 139.1, [p-C], 208.8 [C=O]
IR: (Nujol) 2021, 2004, 1962, 1934, 1928 cm-1
IR: (Hexan) 2023, 1964 cm-1
N
N
As
Mes
Mes
Co
CO
CO
CO

6. Experimenteller Teil
157
Tricarbonyl-1,3-Dimesityl-1,3,2-diazastiboleniumcobaltat(-1) 82
Eine Lösung von 0.1 mmol (55 mg) 13c[Cl] in 1 ml d8-Toluen bzw. 1 ml Hexan wurde auf
-70°C abgekühlt und mit 0.1 mmol (38 mg) TlCo(CO)4 versetzt. Die Reaktionslösung
färbte sich bei – 40 °C erst Rot und nach einer halben Stunde bei RT schwarz. Das Produkt
wurde in der d8-Toluen-Reaktionslösung NMR-spektroskopisch bzw. in der Hexan-
Reaktionslösung durch IR-Spektroskopie nachgewiesen. Eine Isolierung des Produktes war
nicht möglich.
1H-NMR (C7D8) 6.73 [s, 4H, m-H], 6.23 [s, 2H CH], 2.16 [s, 12H o-CH3], 2.36 [s, 6H
p-CH3]
13C-NMR (C7D8) 19.0 [o-CH3], 20.4 [p-CH3], 126.4 [4,5C], 129.0 [m-C], 129.6 [o-C],
135.0 [i-C], 141.0, [p-C]
IR: (Hexan) 2002, 1942 cm-1
N
NSb
Mes
Mes
Co
CO
CO
CO

6. Experimenteller Teil
158
Bestimmung der Dissoziationskonstanten KAs(Dis) der As-Cl-Bindung
Durch Einwaage von 97.15 mg 12c[OTf] bzw. 51,87 mg 11b[Cl] und Verdünnen auf 2.76
ml bzw. 1.78 ml wurden zwei Stammlösungen hergestellt. Durch Mischen dieser
Stammlösungen im Verhältnis von 1 : 4, 2 : 3 und 3 : 2 wurden insgesamt drei
Reaktionslösungen hergestellt, in denen die gemittelten chem. Verschiebungen δ31P von
11b[Cl]/11b[OTf] bestimmt wurden. Unter Berücksichtigung der Werte von δ31P = 205.5
ppm für reines 11b[OTf] bzw. 170.5 ppm für reines 11b[Cl] wurde nach der Formel δbeob
= δi (11b[Cl]) . Pi(11b[Cl]) + δi(11b[OTf]) . Pi(11b[OTf]) der Molenbruch von 11b[OTf]
und 11b[Cl] im Gleichgewicht berechnet. Unter Ausnutzung der Beziehung K3 = 11b[OTf].
12c[Cl]/11b[Cl].12c[OTf] konnte dann die Gleichgewichtskonstante K3 berechnet
werden. Die Gleichgewichtskonstante K2 für die As-Cl-Dissoziation wurde nach K3 =K1/K2
mit dem bekannten Wert K3 für die P-Cl-Dissoziation[151] berechnet.
Verbindung 12c[OTf] 11b[Cl] M (g/mol) 516.17 234.71
Einwaage (mg) 97,15 51,87
Vol (ml) 2,76 1,78
Konz (mg/ml) 35,20 29,14
Konz (mmol/ml) 0,068 0,124
Probe Ars1 Probe Ars2 Probe Ars3 12c[OTf] 11b[Cl] 12c[OTf] 11b[Cl] 12c[OTf] 11b[Cl]
Vol (ml) 0,1 0,4 0,2 0,3 0,3 0,2
Einwaage (mg) 3,52 11,66 7,04 8,74 10,56 5,83
Stoffmenge (µmol) 6,82 49,66 13,64 37,25 20,46 24,83
Stoffmenge (rel) 1,00 7,28 1,00 2,73 1,00 1,21
Integral (1H-NMR) 1 2,83 1 1,38 1 0,78 δ P
11b[OTf] δ P+ 205,5 ppm
11b[Cl] δ PCl 170,5 ppm δ Pbeob 182,5 ppm 193,5 ppm 201,6 ppm
K3=K1/K2 = 11b[OTf].12c[Cl]/ 11b[Cl].12c[OTf] 17,0367893 18,6620429 18,008006
KAsCl(Dis)=K2
0,12913231 *10-3 mol/l
0,11789266 *10-3 mol/l
0,12216788 *10-3 mol/l
Mittelwert Standardabw Resultat 17,9019462 0,81730441 18(2)
0,12306429 0,00567319
KAs(Dis) = 0,12(2)*10-3 mol/l

7. Literaturverzeichnis
159
[1] J. B. Dumas und E. Peligot, Ann. chem. Phys. 1835, 58, 5
[2] A. Geuther, Ann. Chem. Pharm. 1862, 123, 21
[3] H. Scheibler, Chem. Ber. 1926, 59,
[4] M.Schmeisser, H. Schröter, Angew. Chemie 1960, 72, 349
[5] K. Reimer, Ber. Dtsch. Chem. Ges. 1876, 9, 423
[6] G. A. Russel, D.G. Hendry, J. Org. Chem. 1963, 28, 1933
[7] W. M. Jones, J.W. Wilson, Jr. Und R.G. Bergman, J. Am. Chem. Soc. 1973, 85, 3796
[8] L.A. Paquette, S.E. Wilson R. P. Henzel , G. H. Allen, Jr., J. Am. Chem. Soc. 1972,94,
7762
[9] Z. Majerski, Z. Hamersak, R. Sarac-Arneri, J. Org. Chem. 1988, 53, 5053
[10] W. J. Baron, M. R. Decamp, M .E. Hendrick, M. Jones, R. H. Levin, M. B., Sohn, in
Carbenes Wiley, New York 1973, 1
[11] H.M. Frey, Adv. Photochem. 1966, 4, 225
[12] Kirmse, Carbene Chemistry, AcademicPress, New York, 1971
[13] H.-W. Wanzlick, E. Schikora, Angew. Chemie 1960, 72, 494
[14] H.-W. Wanzlick, H.-J. Kleiner, Angew. Chemie 1963, 75, 1204
[15] H.-W. Wanzlick, Angew. Chemie 1962, 74, 129
[16] H. Quast, S. Hünig, Angew. Chemie 1964, 76, 989
[17] H. Quast, S. Hünig, Chem. Ber. 1966, 99, 2017
[18] D. M. Lemal, R. A. Lovald und K.I. Kawano, J. Am. Chem. Soc. 1964, 86, 2518
[19] A. J. Arduengo, Richard L. Harlow, M. Kline, J. Am. Chem. Soc. 1991,
113, 362-363
[20] H. J. Schönherr, H.-W. Wanzlick, Chem. Ber. 1970, 103, 1037
[21] E.O Fischer, A. Maasböl, Angew. Chemie 1964, 76, 645
[22] E.O Fischer, A. Maasböl, Angew. Chemie 1974, 86, 651
[23] W. A. Hermann, M. Elison, J. Fischer, C. Köcher, G. R. J. Artus, Angew. Chemie
1995, 107, 2602
[24] T. Weskamp, W. C. Schattenmann, M. Spiegler, W. A. Hermann, Angew. Chemie
1998, 110, 2631
T. Weskamp, F. J. Kohl, W. A. Hermann, J. Organomet. Chem. 1999, 582, 362
T. Weskamp, V. P. W. Böhm, W. A. Hermann, J. Organomet. Chem. 2000, 600, 12

7. Literaturverzeichnis
160
[25] H. M. Lee, D. C. Smith, Z. He, E. D. Stevens, C. S. Yi, S. P. Nolan, Organometallics
2001, 20, 794
[26] W.A.Hermann, M. Denk, J. Behm, W. Scherer, F.R. Klingan, H. Bock, B. Soluki, M.
Wagner, Angew. Chemie 1992, 104, 1489
[27] M. Gynane, David H. Harris, Michael F. Lappert, Phillip P. Power, Pierre Riviere,
Monique Riviere-Baudet, J. Chem. Soc. Dalton Trans. 1977, 2004
[28] M. Veith, Angew. Chem. Internat. Edn. 1975, 14, 263
[29] B. Wrackmeyer, C. Stader, K. Horchler, Hong Zhou, Inorg. Chim. Acta 1990, 176,
205
[30] M. Denk, R. Lennon, R. Hayashi, R. West; A.V. Belyakov, H.P. Verne, A. Haaland,
M. Wagner, N. Metzler, J. Am. Chem. Soc. 1994, 116, 2691-2692
[31] T. Gans-Eichler, Diplomarbeit Universität Bonn, 2000
[32] G. Boche, P. Andrews, K. Harms, M. Marsh, K. S. Rangappa, M. Schimeczeck, C.
Willeke, J. Am. Chem. Soc. 1996, 118, 4925-4930
[33] M. K. Denk, Shilipi Gupta, A. J. Lough, Eur. J. Inorg. Chem. 1999, 41
[34] C. J. Carmalt, V. Lomeli, B. G. McBurnett, A. H. Cowley, Chem Commun. 1997,
2095
[35] E. Schmidt, A. Jockisch, H. Schmidbaur, J. Am. Chem. Soc. 1999, 121, 9758
[36] T.A. Albright, J.K. Burdett, M. Whangbo, Orbital Interactions in Chemistry, Wiley,
New York , 1985
[37] Hollemann, Wiberg, Lehrbuch der Anorganischen Chemie, 101. Auflage, de Gruyter
[38] H. Tomoika, E. Iwamoto und K. Harai, J. Am. Chem. Soc. 2003, 125, 14664
[39] R. Gleiter, R. Hoffmann, J. Am. Chem.,1968 , 90, 1475
[40] a) S. S. Krishnamurthy, Curr. Sci. 1991, 60, 629 b) E. A. Carter, W. A. Goddard, J.
Phys. Chem.,1986 , 90, 998
[41] W. A. Herrmann und C. Köcher, Angew. Chemie 1997, 109, 2256
[42] D. A. Dixon., A. J. Arduengo , J. Phys. Chem. 1991, 95, 4180
[43] A.J. Arduengo, J. R. Görlich und W. J. Marschall, J. Am. Chem. Soc.
1995, 117, 11027-11028
[44] M. Haaf, T. A. Schmedake, R. West, Acc. Chem Res 2000, 33, 704
R. West, M. Denk, Pure & Appl. Chem. 1996, Vol. 68. No. 4, pp. 785
[45] R. W. Alder, P. R. Allen, M. Murray, A. G. Orpen, Angew. Chemie 1996, 108, 1211
[46] C.Boehme und G. Frenking, J. Am. Chem. Soc. 1996, 118, 2039-2046

7. Literaturverzeichnis
161
[47] C. Heinemann, W.A. Herrmann, W. Thiel, J. Organometal. Chem. 1994, 475, 73
[48] C. Heinemann, T. Müller, Y. Apeloig, H. Schwarz, J. Am. Chem. Soc. 1996, 118,
2023-2038
[49] T. A. Schmedake, M. Haaf, Y. Apeloig, T. Müller, S. Bukalov, R. West, J. Am.
Chem. Soc. 1999, 121, 9479-9480
[50] D. Gudat, pers. Mitteilung
[51] a) W. A. Herrmann, C. Köcher, L. J. Gooßen, G. R. J. Artus, Chem. Eur. J. 1996, 2,
162 b) W. A. Herrmann, F. J. Kohl, J. Schwarz, in Synthetic Methods of
Organometallic and Inorganic Chemistry , Vol 9, Enke, Stuttgart, 2000, s84
c) N. Kuhn , T. Kratz, Synthesis, 1992, 561 d) H. A. Craig, J. R. Goerlich, W. J.
Marshall, M . Unverzagt, Tetrahedron 1999, 55, 14523 e) A. J. Arduengo III (E.I. Du
Pont de Nemours & Company), US 5.077.414 A2, 1992
[52] H. tom Dieck, , M. Svoboda T. Greiser, Z. Naturforsch. 36b 1981, 823
[53] H. H. Karsch, P. A. Schlüter, F. Bienlein, M. Herker, E. Witt, A. Salek, Z. Anorg.
Allg. Chem. 1998 624, 295
[54] H. Braunschweig, B. Gerhus, P. B. Hitchcock, M. F. Lappert, Z.
anorg. allg. Chem. 1995, 621, 1922
[55] K.Öfele, J. Organometal. Chem. 1968, 12, 42
[56] E.D. Fischer, A. Maasböl, Angew. Chemie 1994, 106, 2253
[57] M. Regitz, Angew. Chemie 1996, 108, 791
[58] D. Gudat , Eur. J. Inorg. Chem. 1998, 1087-1094
[59] a) N. Kuhn, T. Kratz, D. Bläser, R. Boese, Chem. Ber. 1995, 128, 245
b) A. Schäfer, M. Weidenbruch, W. Saak, S. Pohl, J. Chem. Soc., Chem. Commun.
1995, 1157
[60] A.J. Arduengo, V. R. Dias, F. Davidson, R. L. Harlow, M. Kline, J. Organometal.
Chem. 1993, 462, 13
[61] S. B. Clendering, P. B. Hitchcock, J. F. Nixon, L. Nyulaszi J. Chem. Soc.,Chem.
Commun. 2000, 1305-1306
[62] M. K. Denk, R. K. Hayashi, R. West, J. Chem. Soc., Chem. Com. 1994, 33
[63] M. Haaf, A. Schmiedl, T. A. Schmedake, D. R. Powell, A. J. Millevolte, M. Denk, R.
West, J. Am. Chem. Soc. 1998, 120, 12714
[64] Gehrhus B., M. F. Lappert, J. Heinicke, R. Boese, D. Blaeser, J. Chem. Commun.
1999, 24, 2451

7. Literaturverzeichnis
162
[65] M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland,
M. Wagner, N. Metzler, J. Am. Chem. Soc. 1994, 116, 2691
[66] S. B. Clendering, B. Gehrhus, P. B. Hitchcock, J. F. Nixon, J. Chem. Soc.,Chem.
Commun. 1995, 1931
[67] O. Kühl, P. Lönnecke, J. Heinicke, Inorg. Chem 2003, 42, 2836
[68] a) R. T. Sanderson. Chemical Bonds and Bond Energy, Academic Press, New York
San Francisco London 1976, Second Edition
b) A. J. C. Wilson, Int. Tables for Crystallography Vol. C, Kluwer Academic
Publishers Dordrecht, Boston, London
[69] C. Stader, B. Wrackmeyer, Z. Naturforsch. 42b 1987, 1515
[70] a) Arthur J. Ashe, Robert R. Sharp und John W. Tolan, J. Am. Chem. Soc. 1976, 5451
b) Arthur J. Ashe, Top. Curr. Chem., 105, 1982, 125
[71] B. Wrackmeyer J. Schiller, Z. Naturforsch. 47b 1992, 662
[72] Die Tensoranalyse wurde von Herrn Dr. Jörn Schmedt auf der Günne nach der
Methode von J. Herzfeld und A. E. Berger durchgeführt. J .Herzfeld und A. E. Berger,
J. Chem. Phys. 1980, 73, 6021
[73] West R. Buffy J. Haaf M. Mueller T. Gerhus B. Lappert M. F. Apeloig Y., J. Am.
Chem. Soc. 1998, 120, 1639
[74] A. J. Arduengo, D. A. Dixon. K. K. Kumashiro, C. Lee, W. P. Power, K. W. Zilm, J.
Am. Soc. 1994, 116, 6361-6367
[75] H.Günther, NMR-Spektroskopie, Georg Thieme, New York . Stuttgart
[76] M. Veith, Z. Naturforschung, 1978, 33b, 7
[77] T. Fjeldberg, H. Hope, M. F. Lappert, P. P. Power und A. J.
Thorne, J. Chem. Soc., Chem. Commun.,1983, 639
[78] M. F. Lappert, P. Power, A. Sanger, R. Srivastava “ Metal and Metalloid Amides”,
Horwood-Wiley: Chichester, England, 1978
[79] H. Hohmeister, H. Wessel, P. Lobinger, H. W. Roesky, P. Muller, H. G. Schmidt, M.
Noltemeyer, J. Magull, J. Flourine Chem. 2003, 120, p59
[80] M. Rannenberg, J. Weidlein, Z. Naturforsch. 1991, 46b, 459
[81] D. Hänsgen, J.Organomet. Chem. 1989, 37, 373
[82] L. Pause, M. Robert, J. Heinicke, O. Kühl, J. Chem. Soc., Perkin Trans. 2001, 1383
[83] D. Enders, K. Breuer, G. Raabe, J. Simonet, A. Ghanimi, H. B. Stegmann, H. Teles
[84] Y. Zhang and M. A. Fox, Acta Chem. Scand. 1999, 53, 857

7. Literaturverzeichnis
163
[85] R. West, T.A. Schmedake, M. Haaf, J. Becker, T. Mueller, Chemistry Letters 2001,
68
[86] M. Haaf , T.A. Schmedake, B. J. Paradise, R. West, Can. J. Chem 2000, 78, 1528
[87] B. Wrackmeyer, C.Stader, K. Horchler, H. Zhou, Inorg. Chim. Acta 1990, 176, 205-
214
[88] D. R. Armstrong, F. Benevelli, A. D. Bond, N. Feeder, E. Harron, A. D. Hopkins, M.
McPartlin, D. Moncrieff, D. Saez, E. A. Quadrelli, A. D. Woods. D . S. Wright, Inorg.
Chem. 1991, 30, 3390
[89] B. E. Eichler, P. P. Power, J. Am. Chem. Soc. 2000, 122, 8785
[90] K. Hilner, W. P. Neumann, Tetrahedron Lett. 1986, 27, 5347
[91] M. Weidenbruch, M. Stürmann, H. Killian, S. Pohl, W. Saak, Chem. Ber. 1997, 130,
735-738
[92] M. Saito, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc. 1997, 119, 11124
[93] H. Suzuki, N. Tokitoh, R. Okazaki, Shigeru Nagase , M. Goto J. Am. Chem. Soc.
1998, 120, 11096
[94] Ch. Elschenbroich, A. Salzer, Organometallchemie, 3. Auflage, Teubner, 1993
[95] G. Binsch, H. Kessler, Angew. Chem. 1980, 92, 445
[96] S., Forsen, R. A Hoffmann. J. Chem. Phys. 1963, 39, 2892
[97] H. M. McConnell, J. Chem. Phys., 1962, 847
[98] Die Inversions-Recovery-Experimente wurden von Herrn Prof. Dr. D. Gudat
durchgeführt und ausgewertet.
[99] Die quantenchemischen Rechnungen wurden von Herrn Prof. Dr. D. Gudat am Inst.
für Anorg. Chemie der Universität Bonn durchgeführt
[100] NBO 5.0 E. D. Glendening, J. K. Badenhoop, A.E. Reed, J. E. Carpenter, J. A.
Bohman, C. M. Morales, F. Weinhold, Theroetical chemistry Institute, University
Wisconsin, Madison, WI, 2001; http://www.chem.-wisc.edu/~nbo5.
[101] W. Kutzelnigg, Angew. Chem. Internat. Ed. Eng. 1984, 23, 272
[102] D. Ohlmann, C.M. Marchand, H. Grützmacher, G.S. Chen, D. Framer, R. Glaser, A.
Currao, R. Nesper, H. Pritzkow, Angew. Chem. 1996, 108, 317.
[103] T. Gans-Eichler, D. Gudat, M. Nieger, Angew. Chem. 2002, 114, 11, 1966
[104] A. J. Arduengo, H. Bock, H. Chen, M. Denk, D. A. Dixon, J. C.
Green, Wolfgang A. Herrmann, Nancy L. Jones, M. Wagner und R.West,
J. Am. Chem. Soc. 1994, 116, 6641

7. Literaturverzeichnis
164
[105] P. Kircher, G. Huttner, K. Heinze, B. Schiemenz, L. Zsolani, M. Büchner, A. Driess,
Eur. J. Inorg. Chem. 1998, 703
[106] a) Y. Zhou, D. S. Richeson, J. Am. Chem. Soc. 1996, 118, 10850
b) S.R. Foley, Y. Zhou, G. P. A. Yap, D. S. Richeson, Inorg. Chem. 2000, 30, 924
[107] K.Bootz, W. P. Neumann, Tetrahedron Lett. 1989, 30(48), 6669
[108] C.Stader, B.Wrackmeyer, Z. Naturforschung 1988, 43b, 707
[109] G. Morris H. Barjat, J. Magn. Reson. Ser. B 1995, 108, 170
[110] B. Watta, W. P. Neuman, J. Sauer, Organometallics 1985, 11, 1955
[111] T. Imori, T. Don Tilley, J. Am. Chem. Soc. 1995, 117(40), 9931
[112] B. Watta, W. P. Neuman, J. Sauer, J. Organometallics 1985, 4 1955
[113] T. Imori, V. Lu, H. Cai , T. Don Tilley, J. Chem. Soc.,Chem. Commun. 1993, 1607
[114] W. K. Zou, L. N. Yang, Polym. Prepr.(Am. Chem. Soc., Div. Polym. Chem.) 1992,
33, 188
[115] S. J. Holder, R. G. Jones, R. E. Benfield , M. J. Went, Polymer 1996, 37, 3477
[116] P. Bleckmann, H. Maly, R. Minkwitz, W. P. Neumann, B. Watta, Tetrahedron Lett.
1982, 32(45), 4655
[117] A. M. Kibardin, I. A. Litvinov, V. A. Naumov, T. Truchkov, T. V. Gryaznova,, B.
Mikhailov, A. N. Pudovik, Dokl. Akad. Nauk SSSR 1988, 298(2), 369
[118] D. Gudat, A. Haghverdi, H. Hupfer, M. Nieger, J. Eur. Chem. 2000, 6 (18), 3414
[119] M. K. Denk, Shilipi Gupta, R. Ramachandran, Tetrahedron Letters 1996, 9025
[120] C. Payastre, Y. Madaule, J. G. Wolf, Heteroatom Chemistry 1992, 3(2), 157
[121] C. Payastre, Y. Madaule, J. G. Wolf, Tetrahedron Letters 1990, 1145
[122] N. Burford, C. Macdonald, T. M.. Parks, Gang Wu, B. Borecka, W. Kwiatkowski, T.
S. Cameron, Can. J. Chem 1996. 74, 2209
[123] M. Veith, B. Bertsch, Z. anorg. Allg. Chem. 1988, 557, 7
[124] M. Veith, B. Bertsch, V. Huch, Z. anorg. Allg. Chem. 1988, 559, 73
[125] D. Gudat, Eur. J. Inorg. Chem. 1998, 1087
[126] W.W. Schoeller, T. Busch, Chem. Ber. 1990,123, 971
[127] W. Kutzelnigg, Angew. Chem. Internat. Ed. Eng. 1994 , 23, 272
[128] T.Koopmans, Physica 1934, 1, 104
[129] S. Burck, Diplomarbeit, Bonn 2002
[130] C. W. Schultz, R. W. Parry, Inorg. Chem. 1976, 15, 3042
[131] R. Reed, R. Reau, F. Dahan, G. Bertrand, Angew. Chem. 1993, 105, 464

7. Literaturverzeichnis
165
[132] D. Gudat, A. Haghverdi, M. Nieger, J. Organometallic Chemistry 2001, 627
[133] D. Gudat, Coordination Chem. Rev. 1997, 163 ,71
[134] Y. Yamaguchi, H. Nakazawa, T. Itoh, K. Miyoshi, Bull. Chem. Soc . Jpn. 1996, 69,
983
[135] S. Burck, unveröffentlichte Ergebnisse
[136] M. Veith, V. Huch, W. Malisch, Organometallics 1990, 9, 1798-1802
[137] a) R. Maisch, M. Barth, W. Malisch, J. Organomet. Chem. 1984, 260, C35-C39.
b) R. Maisch, E. Ott. W. Buchner, W. Malisch, J. Organomet. Chem. 1985, 286,
C31-C35
[138] A. Haghverdi, persönliche Miteilung
[139] A. Lipka, H. Wunderlich, Z. Naturforschung 1980, 35b, 1548
[140] A. Lipka, Acta Crystallogr. 1979, B35, 3020-3022
[141] U. Ensinger, W. Schwarz, A. Schmidt, Z. Naturforschung 1982, 37b, 1584
[142] N. Burford, L. B. Macdonald, K .N. Robertson, T. Cameron, Inorg. Chem 1996, 35,
4013-4016
[143] B. Ross, J. Belz, M. Nieger, Chem. Ber. 1990, 975-978
[144] Ergebnis einer CCDC-Datenbankrecherche
[145] H. Schmidbaur, J. M. Wallis, R. Nowak, B. Huber ,G. Müller, Chem. Ber. 1987,
120, 1837-1843
[146] A. Lipka, D. Mootz, Z. Naturforschung 1982, 37b, 694.698
[147] D. W. Cushen, R. Hulme, J. Chem. Soc. 1964, 4162
[148] A. F. Wells, Structural Inorganic Chemistry 2 Auflage, S. 458, Oxford 1950
[149] C. Romming, J. Songstad, Acta Chim. Scand. 1980, A34, 365
[150] M. Weidenbruch, A. Lesch, J. Organometallic Chemistry 1991, 407, 31
[151] A. Haghverdi, Dissertation, Bonn 2000
[152] B. Weidenbruch, J. Schiller, Z. Naturforsch. 1992, 47b, 662
[153] G. L. Carlson, Spectrochimica Acta 1963, 19, 1291
[154] G. Y. Ahlijah, M. Goldstein, J. Chem. Soc. A 1970, 326
[155] U. Ensinger, A. Schmidt, Z. anorg. allg. Chem. 1984, 514, 137
[156] H. tom Dieck, M. Zettlitzer, Chem. Ber. 1987, 120, 795-801
[157] H. tom Dieck, B. Bruder, K. Franz, Chem. Ber. 1983, 116, 136-145
[158] H. tom Dieck, M. Zettlitzer, L. Stutz, Z. Naturforschung 1986, 41b, 1230
[159] a) R. Hulme, D. Mullen, J. C. Scruton, Acta Crystallogr. 1969, A25, 171

7. Literaturverzeichnis
166
b) R. Hulme, J. C. Scruton, J. Chem. Soc. A 1968, 2448
[160] a) H. Schiff, C. R. Acad. Sci. 1863, 56, 1059 ; Ber., 1901, 34, 804
b) G. Kiel, R. Engler, Chem. Ber. 1974, 107, 3444
c) W. Lindemann, R. Wögerbauer, P. Berger, Z. anorg. allg. Chem. 1977, 437, 155
[161] C. Loubser, J. L. M. Dillen , S. Lotz, Polyhedron 1991, 10, 2535-2539
[162] P. Macchi, D. M. Proserpio A. Sironi, J. Am. Chem. Soc. 1998, 120, 13429-13435
[163] P. Macchi, L. Garlaschelli, S. Martinengo, A. Sironi, Inorg. Chem. 1998, 37, 6263-
6268
[164] H. tom Dieck, Z. Naturforsch. 36b 1981, 814-831
[165] H. tom Dieck, Z. Naturforsch. 36b 1981, 814-831

8. Kristallographischer Anhang
167
Kristallographische Daten der Verbindungen
7c, 13b[Cl], 13b[Br], 13b[OTf], 13b[SbCl4], 43f, 77, 12c[OTf],
12b[Cl], 79

8. Kristallographischer Anhang
168
Table 1. Crystal data and structure refinement for
1,3-Dimesityl-2,3-dihydro-1-H-[1,3,2]-diazastannol-2-yliden (7c)
Identification code gudat47m Empirical formula C20 H24 N2 Sn Formula weight 411.10 Temperature 123(2) K Wavelength 0.71073 A (MoKa) Crystal system, space group Triclinic, P-1 (No.2) Unit cell dimensions a = 8.1325(5) A alpha = 92.083(3) deg. b = 10.3532(7) A beta = 104.517(3) deg. c = 12.4569(9) A gamma = 111.763(3) deg. Volume 933.18(11) A^3 Z, Calculated density 2, 1.463 Mg/m^3 Absorption coefficient 1.370 mm^-1 F(000) 416 Crystal size 0.20 x 0.08 x 0.03 mm Diffractometer Nonius KappaCCD Theta range for data collection 2.92 to 25.00 deg. Limiting indices -9<=h<=9, -12<=k<=12, -14<=l<=14 Reflections collected / unique 10158 / 3224 [R(int) = 0.0573] Completeness to theta = 25.00 98.0 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.8678 and 0.7594
N2 Sn1
Sn1´
N5
C4 C3

8. Kristallographischer Anhang
169
Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 3224 / 14 / 214 Goodness-of-fit on F^2 1.003 Final R indices [I>2sigma(I)] R1 = 0.0444, wR2 = 0.0974 R indices (all data) R1 = 0.0732, wR2 = 0.1068 Largest diff. peak and hole 1.458 and -1.260 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat47. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ Sn(1) 1872(2) 5064(1) 4353(1) 23(1) a) N(2) 3445(5) 7220(4) 4595(3) 29(1) C(3) 2909(7) 7875(5) 3714(4) 29(1) C(4) 1598(7) 7068(5) 2792(4) 31(1) N(5) 914(5) 5632(4) 2794(3) 30(1) Sn(1') 2343(3) 5004(1) 4189(1) 25(1) b) C(21) 5008(6) 7984(5) 5525(4) 25(1) C(22) 6801(7) 8381(5) 5409(4) 27(1) C(23) 8291(7) 9019(5) 6344(4) 30(1) C(24) 8088(7) 9281(5) 7392(4) 30(1) C(25) 6322(7) 8917(5) 7478(4) 29(1) C(26) 4765(7) 8287(5) 6565(4) 26(1) C(27) 2860(7) 7984(6) 6689(5) 40(1) C(28) 9773(8) 9954(6) 8401(5) 48(2) C(29) 7080(8) 8110(6) 4281(4) 40(1) C(51) -529(6) 4725(5) 1841(4) 23(1) C(52) -150(6) 3833(5) 1148(4) 26(1) C(53) -1605(7) 2891(5) 292(4) 28(1) C(54) -3396(7) 2816(5) 85(4) 29(1) C(55) -3708(7) 3733(5) 760(4) 31(1) C(56) -2316(7) 4684(5) 1642(4) 28(1) C(57) -2780(8) 5598(6) 2394(5) 46(2) C(58) -4951(7) 1773(6) -872(4) 44(2) C(59) 1779(7) 3902(6) 1307(4) 39(1) ________________________________________________________________ a) s.o.f.= 0.60(1); b) s.o.f.= 0.40(1)

8. Kristallographischer Anhang
170
Table 3. Bond lengths [A] and angles [deg] for gudat47. _____________________________________________________________ Sn(1)-N(2) 2.086(3) Sn(1)-N(5) 2.095(4) N(2)-C(3) 1.373(6) N(2)-C(21) 1.426(6) N(2)-Sn(1') 2.118(4) C(3)-C(4) 1.346(6) C(4)-N(5) 1.382(6) N(5)-C(51) 1.439(5) N(5)-Sn(1') 2.103(4) C(21)-C(26) 1.400(6) C(21)-C(22) 1.408(7) C(22)-C(23) 1.380(7) C(22)-C(29) 1.510(7) C(23)-C(24) 1.386(7) C(24)-C(25) 1.377(7) C(24)-C(28) 1.520(6) C(25)-C(26) 1.391(6) C(26)-C(27) 1.512(7) C(51)-C(56) 1.396(7) C(51)-C(52) 1.408(7) C(52)-C(53) 1.387(6) C(52)-C(59) 1.505(7) C(53)-C(54) 1.387(7) C(54)-C(55) 1.378(7) C(54)-C(58) 1.525(6) C(55)-C(56) 1.389(6) C(56)-C(57) 1.514(7) N(2)-Sn(1)-N(5) 77.70(14) C(3)-N(2)-C(21) 120.8(4) C(3)-N(2)-Sn(1) 113.3(3) C(21)-N(2)-Sn(1) 125.8(3) C(3)-N(2)-Sn(1') 113.0(3) C(21)-N(2)-Sn(1') 123.4(3) C(4)-C(3)-N(2) 118.0(4) C(3)-C(4)-N(5) 117.2(4) C(4)-N(5)-C(51) 119.4(4) C(4)-N(5)-Sn(1) 113.0(3) C(51)-N(5)-Sn(1) 126.8(3) C(4)-N(5)-Sn(1') 113.7(3) C(51)-N(5)-Sn(1') 126.6(3) N(5)-Sn(1')-N(2) 76.83(15) C(26)-C(21)-C(22) 120.0(4) C(26)-C(21)-N(2) 120.2(4) C(22)-C(21)-N(2) 119.8(4) C(23)-C(22)-C(21) 118.7(4) C(23)-C(22)-C(29) 120.9(5) C(21)-C(22)-C(29) 120.5(4) C(22)-C(23)-C(24) 122.5(5) C(25)-C(24)-C(23) 117.8(4) C(25)-C(24)-C(28) 121.8(5) C(23)-C(24)-C(28) 120.5(5) C(24)-C(25)-C(26) 122.5(4) C(25)-C(26)-C(21) 118.5(4) C(25)-C(26)-C(27) 120.5(4) C(21)-C(26)-C(27) 121.0(4) C(56)-C(51)-C(52) 120.3(4)

8. Kristallographischer Anhang
171
C(56)-C(51)-N(5) 119.8(4) C(52)-C(51)-N(5) 119.7(4) C(53)-C(52)-C(51) 118.2(5) C(53)-C(52)-C(59) 120.2(4) C(51)-C(52)-C(59) 121.5(4) C(54)-C(53)-C(52) 122.6(5) C(55)-C(54)-C(53) 117.7(4) C(55)-C(54)-C(58) 121.5(5) C(53)-C(54)-C(58) 120.8(5) C(54)-C(55)-C(56) 122.5(5) C(55)-C(56)-C(51) 118.7(4) C(55)-C(56)-C(57) 119.7(5) C(51)-C(56)-C(57) 121.6(4) _____________________________________________________________ Table 4. Torsion angles [deg] for gudat47. ________________________________________________________________ N(5)-Sn(1)-N(2)-C(3) 7.2(3) N(5)-Sn(1)-N(2)-C(21) -168.9(4) N(5)-Sn(1)-N(2)-Sn(1') -84.6(2) C(21)-N(2)-C(3)-C(4) 170.6(4) Sn(1)-N(2)-C(3)-C(4) -5.7(6) Sn(1')-N(2)-C(3)-C(4) 9.1(6) N(2)-C(3)-C(4)-N(5) -1.2(7) C(3)-C(4)-N(5)-C(51) 178.2(4) C(3)-C(4)-N(5)-Sn(1) 7.5(6) C(3)-C(4)-N(5)-Sn(1') -7.3(6) N(2)-Sn(1)-N(5)-C(4) -7.8(4) N(2)-Sn(1)-N(5)-C(51) -177.7(4) N(2)-Sn(1)-N(5)-Sn(1') 88.06(19) C(4)-N(5)-Sn(1')-N(2) 9.0(4) C(51)-N(5)-Sn(1')-N(2) -177.0(4) Sn(1)-N(5)-Sn(1')-N(2) -81.03(19) C(3)-N(2)-Sn(1')-N(5) -9.6(4) C(21)-N(2)-Sn(1')-N(5) -170.6(4) Sn(1)-N(2)-Sn(1')-N(5) 84.4(2) C(3)-N(2)-C(21)-C(26) 108.7(5) Sn(1)-N(2)-C(21)-C(26) -75.4(6) Sn(1')-N(2)-C(21)-C(26) -91.6(5) C(3)-N(2)-C(21)-C(22) -73.9(6) Sn(1)-N(2)-C(21)-C(22) 101.9(5) Sn(1')-N(2)-C(21)-C(22) 85.7(5) C(26)-C(21)-C(22)-C(23) 2.5(7) N(2)-C(21)-C(22)-C(23) -174.8(4) C(26)-C(21)-C(22)-C(29) -177.6(5) N(2)-C(21)-C(22)-C(29) 5.0(7) C(21)-C(22)-C(23)-C(24) 0.0(7) C(29)-C(22)-C(23)-C(24) -179.8(5) C(22)-C(23)-C(24)-C(25) -1.8(7) C(22)-C(23)-C(24)-C(28) 178.5(5) C(23)-C(24)-C(25)-C(26) 1.0(7) C(28)-C(24)-C(25)-C(26) -179.3(5) C(24)-C(25)-C(26)-C(21) 1.5(7) C(24)-C(25)-C(26)-C(27) -176.4(5) C(22)-C(21)-C(26)-C(25) -3.2(7) N(2)-C(21)-C(26)-C(25) 174.1(4) C(22)-C(21)-C(26)-C(27) 174.7(5) N(2)-C(21)-C(26)-C(27) -8.0(7) C(4)-N(5)-C(51)-C(56) -71.7(6) Sn(1)-N(5)-C(51)-C(56) 97.6(5) Sn(1')-N(5)-C(51)-C(56) 114.6(5)

8. Kristallographischer Anhang
172
C(4)-N(5)-C(51)-C(52) 111.5(5) Sn(1)-N(5)-C(51)-C(52) -79.2(5) Sn(1')-N(5)-C(51)-C(52) -62.2(5) C(56)-C(51)-C(52)-C(53) -2.0(7) N(5)-C(51)-C(52)-C(53) 174.8(4) C(56)-C(51)-C(52)-C(59) 177.0(4) N(5)-C(51)-C(52)-C(59) -6.2(7) C(51)-C(52)-C(53)-C(54) 1.2(7) C(59)-C(52)-C(53)-C(54) -177.8(5) C(52)-C(53)-C(54)-C(55) 0.6(7) C(52)-C(53)-C(54)-C(58) 179.5(5) C(53)-C(54)-C(55)-C(56) -1.8(7) C(58)-C(54)-C(55)-C(56) 179.4(5) C(54)-C(55)-C(56)-C(51) 1.0(7) C(54)-C(55)-C(56)-C(57) -176.2(5) C(52)-C(51)-C(56)-C(55) 1.0(7) N(5)-C(51)-C(56)-C(55) -175.8(4) C(52)-C(51)-C(56)-C(57) 178.1(5) N(5)-C(51)-C(56)-C(57) 1.3(7) ________________________________________________________________
Table 1. Crystal data and structure refinement for 2-Chloro-1,3-Di-tert-butyl-1,3-diazastibolen (13b[Cl])

8. Kristallographischer Anhang
173
Identification code gudat53m Empirical formula C10 H20 Cl N2 Sb Formula weight 325.48 Temperature 123(2) K Wavelength 0.71073 A (MoKa) Crystal system, space group Orthorhombic, Pbca (No.61) Unit cell dimensions a = 11.5242(5) A alpha = 90 deg. b = 10.9946(4) A beta = 90 deg. c = 21.0980(9) A gamma = 90 deg. Volume 2673.20(19) A^3 Z, Calculated density 8, 1.617 Mg/m^3 Absorption coefficient 2.234 mm^-1 F(000) 1296 Crystal size 0.50 x 0.20 x 0.03 mm Diffractometer Nonius KappaCCD Theta range for data collection 2.62 to 25.00 deg. Limiting indices -13<=h<=10, -12<=k<=13, -25<=l<=25 Reflections collected / unique 13862 / 2344 [R(int) = 0.1043] Completeness to theta = 25.00 99.8 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.5204 and 0.3830 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 2344 / 0 / 127 Goodness-of-fit on F^2 0.933 Final R indices [I>2sigma(I)] R1 = 0.0375, wR2 = 0.0826 R indices (all data) R1 = 0.0627, wR2 = 0.0911 Largest diff. peak and hole 1.123 and -0.970 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat53. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________

8. Kristallographischer Anhang
174
Cl(1) 5999(1) 3917(1) 1486(1) 40(1) Sb(1) 7363(1) 5853(1) 1507(1) 28(1) N(1) 6792(3) 6666(4) 2294(2) 25(1) N(2) 6287(3) 7055(4) 1105(2) 28(1) C(1) 5907(4) 7516(5) 2171(2) 28(1) C(2) 5669(4) 7715(5) 1555(2) 27(1) C(11) 7040(4) 6257(5) 2949(2) 26(1) C(12) 7250(4) 7363(5) 3376(2) 32(1) C(13) 6016(4) 5513(5) 3198(2) 35(1) C(14) 8146(4) 5488(5) 2932(2) 32(1) C(21) 5988(4) 7149(5) 426(2) 30(1) C(22) 6783(5) 6295(5) 61(3) 38(1) C(23) 4728(5) 6758(6) 325(3) 44(2) C(24) 6185(6) 8457(5) 201(3) 47(2) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat53. _____________________________________________________________ Cl(1)-Sb(1) 2.6460(15) Sb(1)-N(1) 1.998(4) Sb(1)-N(2) 2.000(4) Sb(1)-Cl(1)#1 3.8620(15) N(1)-C(1) 1.407(6) N(1)-C(11) 1.481(6) N(2)-C(2) 1.391(6) N(2)-C(21) 1.478(6) C(1)-C(2) 1.346(6) C(11)-C(13) 1.530(7) C(11)-C(14) 1.531(7) C(11)-C(12) 1.533(7) C(21)-C(22) 1.522(7) C(21)-C(23) 1.528(7) C(21)-C(24) 1.531(7) N(1)-Sb(1)-N(2) 81.49(16) N(1)-Sb(1)-Cl(1) 100.23(12) N(2)-Sb(1)-Cl(1) 99.00(12) N(1)-Sb(1)-Cl(1)#1 77.28(12) N(2)-Sb(1)-Cl(1)#1 73.84(12) Cl(1)-Sb(1)-Cl(1)#1 172.64(3) C(1)-N(1)-C(11) 120.9(4) C(1)-N(1)-Sb(1) 112.5(3) C(11)-N(1)-Sb(1) 125.2(3) C(2)-N(2)-C(21) 120.4(4) C(2)-N(2)-Sb(1) 111.9(3) C(21)-N(2)-Sb(1) 127.0(3) C(2)-C(1)-N(1) 115.7(4) C(1)-C(2)-N(2) 118.0(4) N(1)-C(11)-C(13) 109.5(4) N(1)-C(11)-C(14) 107.8(4) C(13)-C(11)-C(14) 110.8(5) N(1)-C(11)-C(12) 109.8(4) C(13)-C(11)-C(12) 110.1(4) C(14)-C(11)-C(12) 108.7(4) N(2)-C(21)-C(22) 107.8(4) N(2)-C(21)-C(23) 109.7(4) C(22)-C(21)-C(23) 109.1(5) N(2)-C(21)-C(24) 109.4(4) C(22)-C(21)-C(24) 109.5(4) C(23)-C(21)-C(24) 111.2(5) _____________________________________________________________

8. Kristallographischer Anhang
175
Symmetry transformations used to generate equivalent atoms: #1 -x+3/2,y+1/2,z Table 4. Torsion angles [deg] for gudat53. ________________________________________________________________ N(2)-Sb(1)-N(1)-C(1) 5.6(3) Cl(1)-Sb(1)-N(1)-C(1) -92.1(3) Cl(1)#1-Sb(1)-N(1)-C(1) 80.8(3) N(2)-Sb(1)-N(1)-C(11) 172.0(4) Cl(1)-Sb(1)-N(1)-C(11) 74.4(4) Cl(1)#1-Sb(1)-N(1)-C(11) -112.7(4) N(1)-Sb(1)-N(2)-C(2) -4.8(3) Cl(1)-Sb(1)-N(2)-C(2) 94.3(3) Cl(1)#1-Sb(1)-N(2)-C(2) -84.0(3) N(1)-Sb(1)-N(2)-C(21) -175.0(4) Cl(1)-Sb(1)-N(2)-C(21) -75.9(4) Cl(1)#1-Sb(1)-N(2)-C(21) 105.8(4) C(11)-N(1)-C(1)-C(2) -172.6(4) Sb(1)-N(1)-C(1)-C(2) -5.5(5) N(1)-C(1)-C(2)-N(2) 1.5(7) C(21)-N(2)-C(2)-C(1) 174.2(4) Sb(1)-N(2)-C(2)-C(1) 3.3(6) C(1)-N(1)-C(11)-C(13) 67.3(6) Sb(1)-N(1)-C(11)-C(13) -98.1(5) C(1)-N(1)-C(11)-C(14) -172.0(4) Sb(1)-N(1)-C(11)-C(14) 22.6(6) C(1)-N(1)-C(11)-C(12) -53.7(6) Sb(1)-N(1)-C(11)-C(12) 140.9(3) C(2)-N(2)-C(21)-C(22) -178.1(4) Sb(1)-N(2)-C(21)-C(22) -8.6(6) C(2)-N(2)-C(21)-C(23) -59.3(6) Sb(1)-N(2)-C(21)-C(23) 110.1(4) C(2)-N(2)-C(21)-C(24) 62.9(6) Sb(1)-N(2)-C(21)-C(24) -127.6(4) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+3/2,y+1/2,z Table 5. Hydrogen bonds for gudat53 [A and deg.]. _________________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(1)-H(1)...Cl(1)#2 0.95 2.96 3.902(5) 171.9 C(23)-H(23C)...Cl(1)#3 0.98 3.04 3.982(6) 162.7 _________________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+3/2,y+1/2,z #2 -x+1,y+1/2,-z+1/2 #3 -x+1,-y+1,-z
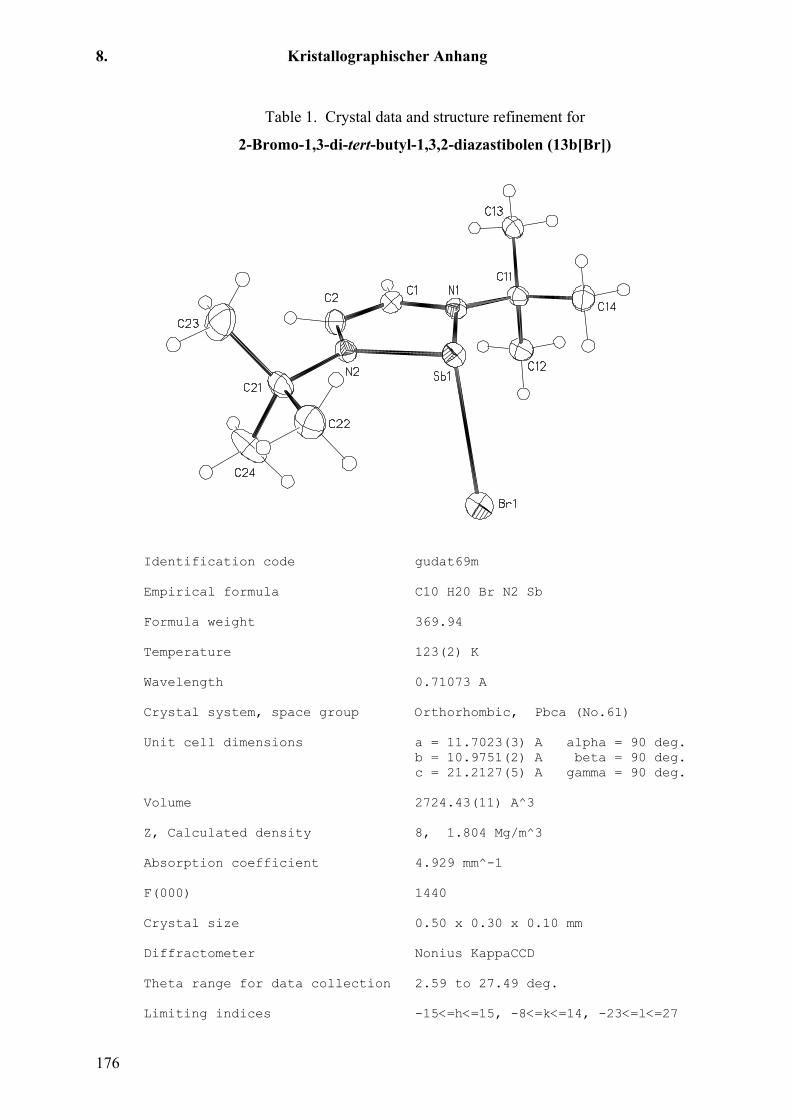
8. Kristallographischer Anhang
176
Table 1. Crystal data and structure refinement for
2-Bromo-1,3-di-tert-butyl-1,3,2-diazastibolen (13b[Br]) Identification code gudat69m Empirical formula C10 H20 Br N2 Sb Formula weight 369.94 Temperature 123(2) K Wavelength 0.71073 A Crystal system, space group Orthorhombic, Pbca (No.61) Unit cell dimensions a = 11.7023(3) A alpha = 90 deg. b = 10.9751(2) A beta = 90 deg. c = 21.2127(5) A gamma = 90 deg. Volume 2724.43(11) A^3 Z, Calculated density 8, 1.804 Mg/m^3 Absorption coefficient 4.929 mm^-1 F(000) 1440 Crystal size 0.50 x 0.30 x 0.10 mm Diffractometer Nonius KappaCCD Theta range for data collection 2.59 to 27.49 deg. Limiting indices -15<=h<=15, -8<=k<=14, -23<=l<=27

8. Kristallographischer Anhang
177
Reflections collected / unique 12347 / 3094 [R(int) = 0.0677] Completeness to theta = 27.49 98.9 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.29127 and 0.13256 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 3094 / 0 / 127 Goodness-of-fit on F^2 0.977 Final R indices [I>2sigma(I)] R1 = 0.0305, wR2 = 0.0669 R indices (all data) R1 = 0.0442, wR2 = 0.0701 Largest diff. peak and hole 0.646 and -1.782 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat69. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ Br(1) 994(1) 3738(1) 3534(1) 25(1) Sb(1) 2402(1) 5960(1) 3510(1) 18(1) N(1) 1790(2) 6697(2) 2718(1) 17(1) N(2) 1267(2) 7096(2) 3891(1) 16(1) C(1) 922(3) 7528(3) 2833(1) 17(1) C(2) 655(3) 7745(3) 3440(1) 19(1) C(11) 2072(3) 6285(3) 2071(1) 17(1) C(12) 1074(3) 5526(3) 1816(2) 23(1) C(13) 2282(3) 7389(3) 1645(2) 23(1) C(14) 3156(3) 5514(3) 2106(2) 23(1) C(21) 955(3) 7193(3) 4570(1) 22(1) C(22) 1727(3) 6332(3) 4939(2) 29(1) C(23) 1130(4) 8497(3) 4795(2) 38(1) C(24) -289(3) 6792(4) 4652(2) 37(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat69. _____________________________________________________________ Br(1)-Sb(1) 2.9433(4) Sb(1)-N(2) 1.994(3) Sb(1)-N(1) 1.998(2) Sb(1)-Br(1)#1 3.5813(5) N(1)-C(1) 1.386(4) N(1)-C(11) 1.483(4) N(2)-C(2) 1.391(4) N(2)-C(21) 1.489(4) C(1)-C(2) 1.346(4) C(11)-C(14) 1.527(4) C(11)-C(13) 1.531(5) C(11)-C(12) 1.533(4)

8. Kristallographischer Anhang
178
C(21)-C(23) 1.523(5) C(21)-C(22) 1.523(5) C(21)-C(24) 1.530(4) N(2)-Sb(1)-N(1) 81.28(10) N(2)-Sb(1)-Br(1) 97.95(8) N(1)-Sb(1)-Br(1) 98.56(7) N(2)-Sb(1)-Br(1)#1 79.11(8) N(1)-Sb(1)-Br(1)#1 81.63(7) Br(1)-Sb(1)-Br(1)#1 177.007(11) C(1)-N(1)-C(11) 121.7(2) C(1)-N(1)-Sb(1) 112.44(19) C(11)-N(1)-Sb(1) 125.2(2) C(2)-N(2)-C(21) 120.2(3) C(2)-N(2)-Sb(1) 112.56(19) C(21)-N(2)-Sb(1) 126.8(2) C(2)-C(1)-N(1) 117.0(3) C(1)-C(2)-N(2) 116.7(3) N(1)-C(11)-C(14) 108.0(2) N(1)-C(11)-C(13) 110.0(3) C(14)-C(11)-C(13) 109.5(3) N(1)-C(11)-C(12) 108.8(3) C(14)-C(11)-C(12) 110.4(3) C(13)-C(11)-C(12) 110.2(3) N(2)-C(21)-C(23) 109.7(3) N(2)-C(21)-C(22) 107.9(3) C(23)-C(21)-C(22) 110.0(3) N(2)-C(21)-C(24) 108.9(3) C(23)-C(21)-C(24) 111.2(3) C(22)-C(21)-C(24) 109.1(3) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,y+1/2,z Table 4. Torsion angles [deg] for gudat69. ________________________________________________________________ N(2)-Sb(1)-N(1)-C(1) -2.1(2) Br(1)-Sb(1)-N(1)-C(1) 94.7(2) Br(1)#1-Sb(1)-N(1)-C(1) -82.3(2) N(2)-Sb(1)-N(1)-C(11) -172.6(3) Br(1)-Sb(1)-N(1)-C(11) -75.8(2) Br(1)#1-Sb(1)-N(1)-C(11) 107.2(2) N(1)-Sb(1)-N(2)-C(2) 1.6(2) Br(1)-Sb(1)-N(2)-C(2) -95.9(2) Br(1)#1-Sb(1)-N(2)-C(2) 84.7(2) N(1)-Sb(1)-N(2)-C(21) 174.6(3) Br(1)-Sb(1)-N(2)-C(21) 77.0(2) Br(1)#1-Sb(1)-N(2)-C(21) -102.4(2) C(11)-N(1)-C(1)-C(2) 173.2(3) Sb(1)-N(1)-C(1)-C(2) 2.3(4) N(1)-C(1)-C(2)-N(2) -1.0(4) C(21)-N(2)-C(2)-C(1) -174.3(3) Sb(1)-N(2)-C(2)-C(1) -0.8(4) C(1)-N(1)-C(11)-C(14) 172.7(3) Sb(1)-N(1)-C(11)-C(14) -17.7(4) C(1)-N(1)-C(11)-C(13) 53.3(4) Sb(1)-N(1)-C(11)-C(13) -137.1(2) C(1)-N(1)-C(11)-C(12) -67.5(4) Sb(1)-N(1)-C(11)-C(12) 102.2(3) C(2)-N(2)-C(21)-C(23) -63.3(4)

8. Kristallographischer Anhang
179
Sb(1)-N(2)-C(21)-C(23) 124.2(3) C(2)-N(2)-C(21)-C(22) 176.9(3) Sb(1)-N(2)-C(21)-C(22) 4.4(4) C(2)-N(2)-C(21)-C(24) 58.5(4) Sb(1)-N(2)-C(21)-C(24) -113.9(3) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,y+1/2,z Table 5. Hydrogen bonds for gudat69 [A and deg.]. ____________________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(1)-H(1)...Br(1)#2 0.95 2.96 3.898(3) 168.4 C(24)-H(24B)...Br(1)#3 0.98 3.02 3.979(3) 166.8 ____________________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,y+1/2,z #2 -x,y+1/2,-z+1/2 #3 -x,-y+1,-z+1
Table 1. Crystal data and structure refinement for
1,3-Di-tert-butyl-1,3,2-diazastibolenium-triflat (13b[OTf)
I

8. Kristallographischer Anhang
180
Identification code gudat60m Empirical formula C12 H22 Cl2 F3 N2 O3 S Sb Formula weight 524.03 Temperature 123(2) K Wavelength 0.71073 A / MoKa Crystal system, space group Monoclinic, P2(1)/c No.14 Unit cell dimensions a = 11.0192(1) A alpha = 90 deg. b = 10.3184(1) A beta = 104.138(1) deg. c = 18.6936(2) A gamma = 90 deg. Volume 2061.09(4) A^3 Z, Calculated density 4, 1.689 Mg/m^3 Absorption coefficient 1.737 mm^-1 F(000) 1040 Crystal size 0.35 x 0.25 x 0.20 mm Diffractometer Nonius KappaCCD Theta range for data collection 3.14 to 25.00 deg. Limiting indices -13<=h<=13, -12<=k<=12, -22<=l<=22 Reflections collected / unique 32263 / 3603 [R(int) = 0.0744] Completeness to theta = 25.00 99.7 % Absorption correction Empirical Max. and min. transmission 0.8459 and 0.4978 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 3603 / 0 / 217 Goodness-of-fit on F^2 1.045 Final R indices [I>2sigma(I)] R1 = 0.0234, wR2 = 0.0595 R indices (all data) R1 = 0.0259, wR2 = 0.0606 Largest diff. peak and hole 1.304 and -0.861 e.A^-3

8. Kristallographischer Anhang
181
Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat60m. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ Sb(1) 6750(1) 5889(1) 5760(1) 18(1) N(2) 8287(2) 6622(2) 5503(1) 17(1) C(3) 9374(2) 6260(2) 5974(1) 22(1) C(4) 9276(2) 5454(2) 6535(1) 24(1) N(5) 8111(2) 5106(2) 6570(1) 20(1) C(6) 8289(2) 7484(2) 4858(1) 21(1) C(7) 9026(3) 8716(3) 5138(2) 30(1) C(8) 6934(2) 7835(2) 4486(1) 25(1) C(9) 8879(3) 6741(3) 4323(1) 29(1) C(10) 7900(2) 4193(2) 7153(1) 24(1) C(11) 8524(3) 2904(2) 7075(2) 30(1) C(12) 6496(3) 4005(3) 7039(2) 37(1) C(13) 8462(3) 4798(3) 7909(2) 39(1) S(1) 3550(1) 6518(1) 6186(1) 20(1) O(1) 3796(2) 5323(2) 5845(1) 27(1) O(2) 4636(2) 7084(2) 6671(1) 30(1) O(3) 2432(2) 6520(2) 6455(1) 32(1) C(1) 3169(2) 7644(3) 5411(2) 31(1) F(1) 4084(2) 7701(2) 5059(1) 43(1) F(2) 2134(2) 7273(2) 4916(1) 49(1) F(3) 2978(2) 8834(2) 5630(1) 52(1) C(1S) 6569(3) 9436(3) 7060(2) 39(1) Cl(1S) 7988(1) 8582(1) 7136(1) 47(1) Cl(2S) 6095(1) 10176(1) 6185(1) 62(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat60m. _____________________________________________________________ Sb(1)-N(2) 2.0177(19) Sb(1)-N(5) 2.0202(19) N(2)-C(3) 1.354(3) N(2)-C(6) 1.499(3) C(3)-C(4) 1.364(4) C(4)-N(5) 1.349(3) N(5)-C(10) 1.502(3) C(6)-C(9) 1.527(3) C(6)-C(8) 1.528(3) C(6)-C(7) 1.530(3) C(10)-C(12) 1.521(4) C(10)-C(11) 1.521(4) C(10)-C(13) 1.530(4) S(1)-O(2) 1.4378(18) S(1)-O(3) 1.4400(18) S(1)-O(1) 1.4436(18) S(1)-C(1) 1.825(3) C(1)-F(3) 1.328(3) C(1)-F(1) 1.333(3) C(1)-F(2) 1.336(3) C(1S)-Cl(2S) 1.765(3) C(1S)-Cl(1S) 1.770(3)

8. Kristallographischer Anhang
182
N(2)-Sb(1)-N(5) 79.38(8) C(3)-N(2)-C(6) 120.79(19) C(3)-N(2)-Sb(1) 113.72(15) C(6)-N(2)-Sb(1) 125.49(14) N(2)-C(3)-C(4) 116.4(2) N(5)-C(4)-C(3) 117.0(2) C(4)-N(5)-C(10) 121.3(2) C(4)-N(5)-Sb(1) 113.49(16) C(10)-N(5)-Sb(1) 125.18(15) N(2)-C(6)-C(9) 108.28(19) N(2)-C(6)-C(8) 108.27(19) C(9)-C(6)-C(8) 110.2(2) N(2)-C(6)-C(7) 108.87(19) C(9)-C(6)-C(7) 111.4(2) C(8)-C(6)-C(7) 109.7(2) N(5)-C(10)-C(12) 108.1(2) N(5)-C(10)-C(11) 109.0(2) C(12)-C(10)-C(11) 110.1(2) N(5)-C(10)-C(13) 108.4(2) C(12)-C(10)-C(13) 110.4(2) C(11)-C(10)-C(13) 110.8(2) O(2)-S(1)-O(3) 115.51(11) O(2)-S(1)-O(1) 114.11(11) O(3)-S(1)-O(1) 115.10(11) O(2)-S(1)-C(1) 103.72(12) O(3)-S(1)-C(1) 103.05(12) O(1)-S(1)-C(1) 102.99(12) F(3)-C(1)-F(1) 107.9(2) F(3)-C(1)-F(2) 107.8(2) F(1)-C(1)-F(2) 107.0(2) F(3)-C(1)-S(1) 111.52(19) F(1)-C(1)-S(1) 111.38(18) F(2)-C(1)-S(1) 111.03(19) Cl(2S)-C(1S)-Cl(1S) 110.21(17) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: Table 4. Torsion angles [deg] for gudat60m. ________________________________________________________________ N(5)-Sb(1)-N(2)-C(3) -0.80(16) N(5)-Sb(1)-N(2)-C(6) 178.94(18) C(6)-N(2)-C(3)-C(4) -178.8(2) Sb(1)-N(2)-C(3)-C(4) 1.0(3) N(2)-C(3)-C(4)-N(5) -0.5(3) C(3)-C(4)-N(5)-C(10) 178.3(2) C(3)-C(4)-N(5)-Sb(1) -0.2(3) N(2)-Sb(1)-N(5)-C(4) 0.52(17) N(2)-Sb(1)-N(5)-C(10) -177.83(19) C(3)-N(2)-C(6)-C(9) 66.6(3) Sb(1)-N(2)-C(6)-C(9) -113.09(19) C(3)-N(2)-C(6)-C(8) -173.9(2) Sb(1)-N(2)-C(6)-C(8) 6.4(3) C(3)-N(2)-C(6)-C(7) -54.7(3) Sb(1)-N(2)-C(6)-C(7) 125.60(19) C(4)-N(5)-C(10)-C(12) 180.0(2) Sb(1)-N(5)-C(10)-C(12) -1.8(3) C(4)-N(5)-C(10)-C(11) -60.4(3)

8. Kristallographischer Anhang
183
Sb(1)-N(5)-C(10)-C(11) 117.8(2) C(4)-N(5)-C(10)-C(13) 60.2(3) Sb(1)-N(5)-C(10)-C(13) -121.5(2) O(2)-S(1)-C(1)-F(3) -57.3(2) O(3)-S(1)-C(1)-F(3) 63.4(2) O(1)-S(1)-C(1)-F(3) -176.53(19) O(2)-S(1)-C(1)-F(1) 63.3(2) O(3)-S(1)-C(1)-F(1) -175.96(19) O(1)-S(1)-C(1)-F(1) -55.9(2) O(2)-S(1)-C(1)-F(2) -177.59(18) O(3)-S(1)-C(1)-F(2) -56.8(2) O(1)-S(1)-C(1)-F(2) 63.2(2) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: Table 5. Hydrogen bonds for gudat60m [A and deg.]. _______________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(1S)-H(1S1)...O(2) 0.99 2.32 3.193(4) 147.1 C(4)-H(4)...Cl(1S)#1 0.95 2.99 3.916(3) 166.4 C(11)-H(11A)...Cl(1S)#1 0.98 2.89 3.830(3) 160.5 C(9)-H(9B)...F(2)#2 0.98 2.68 3.528(3) 144.5 C(11)-H(11B)...F(2)#3 0.98 2.74 3.619(3) 149.2 C(13)-H(13B)...F(3)#4 0.98 2.76 3.617(4) 146.8 C(8)-H(8A)...O(1)#3 0.98 2.47 3.377(3) 154.2 C(9)-H(9A)...O(1)#3 0.98 2.75 3.588(3) 144.3 C(12)-H(12B)...O(2)#4 0.98 2.62 3.573(3) 165.4 C(3)-H(3)...O(3)#2 0.95 2.46 3.279(3) 144.7 C(11)-H(11C)...O(3)#4 0.98 2.56 3.481(3) 157.2 C(1S)-H(1S2)...O(3)#5 0.99 2.51 3.472(4) 164.1 _______________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+2,y-1/2,-z+3/2 #2 x+1,y,z #3 -x+1,-y+1,-z+1 #4 -x+1,y-1/2,-z+3/2 #5 -x+1,y+1/2,-z+3/2

8. Kristallographischer Anhang
184
Table 1. Crystal data and structure refinement for
1,3-Di-tert-butyl-1,3,2-diazastibolenium-tetrachloroantimonat (13b[SbCl4]) Identification code gudat62m Empirical formula C10 H20 Cl4 N2 Sb2 Formula weight 553.58 Temperature 123(2) K Wavelength 0.71073 A Crystal system, space group Monoclinic, P2(1)/n (No.14) Unit cell dimensions a = 10.1078(1) A alpha = 90 deg. b = 19.5200(2) A beta = 114.408(1) deg. c = 10.2340(1) A gamma = 90 deg. Volume 1838.75(3) A^3 Z, Calculated density 4, 2.000 Mg/m^3 Absorption coefficient 3.506 mm^-1 F(000) 1056 Crystal size 0.60 x 0.40 x 0.30 mm Diffractometer Nonius KappaCCD Theta range for data collection 2.38 to 25.00 deg. Limiting indices -12<=h<=12, -23<=k<=23, -12<=l<=12 Reflections collected / unique 16986 / 3213 [R(int) = 0.0408]

8. Kristallographischer Anhang
185
Completeness to theta = 25.00 99.7 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.3297 and 0.2458 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 3213 / 0 / 163 Goodness-of-fit on F^2 1.214 Final R indices [I>2sigma(I)] R1 = 0.0180, wR2 = 0.0416 R indices (all data) R1 = 0.0192, wR2 = 0.0419 Largest diff. peak and hole 0.556 and -0.438 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat62. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ Sb(1) 8304(1) 971(1) 3416(1) 17(1) N(1) 8318(2) 1993(1) 3762(2) 16(1) C(1) 8289(3) 2369(1) 2637(3) 22(1) C(2) 8214(3) 2021(1) 1458(3) 21(1) N(2) 8159(2) 1329(1) 1506(2) 17(1) C(11) 8215(3) 2328(1) 5034(3) 18(1) C(12) 8438(3) 1787(2) 6171(3) 26(1) C(13) 9371(4) 2884(2) 5621(3) 35(1) C(14) 6698(3) 2639(2) 4548(3) 37(1) C(21) 7997(3) 908(1) 227(3) 20(1) C(22) 7833(3) 159(2) 571(3) 25(1) C(23) 9354(3) 1004(2) -51(3) 26(1) C(24) 6637(3) 1132(2) -1067(3) 27(1) Sb(2) 3567(1) 789(1) 5036(1) 17(1) Cl(1) 5006(1) 523(1) 3382(1) 28(1) Cl(2) 2075(1) 907(1) 6476(1) 27(1) Cl(3) 2362(1) 1753(1) 3524(1) 29(1) Cl(4) 1789(1) 21(1) 3521(1) 21(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat62. _____________________________________________________________ Sb(1)-N(2) 2.023(2) Sb(1)-N(1) 2.025(2) Sb(1)-Cl(1) 3.4326(7) Sb(1)-Cl(2)#1 3.6927(7) Sb(1)-Cl(4)#1 3.7188(7) Sb(1)-Cl(2)#2 3.8057(8) Sb(1)-Cl(4)#2 3.9435(7) N(1)-C(1) 1.356(3) N(1)-C(11) 1.499(3) C(1)-C(2) 1.360(4)

8. Kristallographischer Anhang
186
C(2)-N(2) 1.353(3) N(2)-C(21) 1.497(3) C(11)-C(12) 1.519(4) C(11)-C(13) 1.524(4) C(11)-C(14) 1.528(4) C(21)-C(23) 1.523(4) C(21)-C(24) 1.526(4) C(21)-C(22) 1.528(4) Sb(2)-Cl(4) 2.3624(6) Sb(2)-Cl(3) 2.4210(7) Sb(2)-Cl(2) 2.5173(7) Sb(2)-Cl(1) 2.6994(7) Sb(2)-Cl(1)#1 3.0538(7) Cl(1)-Sb(2)#1 3.0538(7) N(2)-Sb(1)-N(1) 79.71(9) N(2)-Sb(1)-Cl(1) 113.31(6) N(1)-Sb(1)-Cl(1) 100.92(6) N(2)-Sb(1)-Cl(2)#1 113.88(6) N(1)-Sb(1)-Cl(2)#1 165.04(6) Cl(1)-Sb(1)-Cl(2)#1 68.592(17) N(2)-Sb(1)-Cl(4)#1 167.76(6) N(1)-Sb(1)-Cl(4)#1 111.47(6) Cl(1)-Sb(1)-Cl(4)#1 60.865(15) Cl(2)#1-Sb(1)-Cl(4)#1 54.466(15) N(2)-Sb(1)-Cl(2)#2 117.03(6) N(1)-Sb(1)-Cl(2)#2 87.48(6) Cl(1)-Sb(1)-Cl(2)#2 129.660(16) Cl(2)#1-Sb(1)-Cl(2)#2 91.365(16) Cl(4)#1-Sb(1)-Cl(2)#2 69.815(14) N(2)-Sb(1)-Cl(4)#2 84.03(6) N(1)-Sb(1)-Cl(4)#2 121.13(6) Cl(1)-Sb(1)-Cl(4)#2 137.161(16) Cl(2)#1-Sb(1)-Cl(4)#2 68.570(15) Cl(4)#1-Sb(1)-Cl(4)#2 93.773(14) Cl(2)#2-Sb(1)-Cl(4)#2 51.874(14) C(1)-N(1)-C(11) 121.1(2) C(1)-N(1)-Sb(1) 112.90(17) C(11)-N(1)-Sb(1) 125.74(16) N(1)-C(1)-C(2) 117.1(2) N(2)-C(2)-C(1) 117.1(2) C(2)-N(2)-C(21) 120.5(2) C(2)-N(2)-Sb(1) 113.09(17) C(21)-N(2)-Sb(1) 126.42(17) N(1)-C(11)-C(12) 108.6(2) N(1)-C(11)-C(13) 109.6(2) C(12)-C(11)-C(13) 110.3(2) N(1)-C(11)-C(14) 108.1(2) C(12)-C(11)-C(14) 109.7(2) C(13)-C(11)-C(14) 110.4(3) N(2)-C(21)-C(23) 108.3(2) N(2)-C(21)-C(24) 109.6(2) C(23)-C(21)-C(24) 111.1(2) N(2)-C(21)-C(22) 107.8(2) C(23)-C(21)-C(22) 110.7(2) C(24)-C(21)-C(22) 109.2(2) Cl(4)-Sb(2)-Cl(3) 91.61(2) Cl(4)-Sb(2)-Cl(2) 88.00(2) Cl(3)-Sb(2)-Cl(2) 92.16(2) Cl(4)-Sb(2)-Cl(1) 85.80(2) Cl(3)-Sb(2)-Cl(1) 90.43(2) Cl(2)-Sb(2)-Cl(1) 173.35(2) Cl(4)-Sb(2)-Cl(1)#1 83.14(2)

8. Kristallographischer Anhang
187
Cl(3)-Sb(2)-Cl(1)#1 173.26(2) Cl(2)-Sb(2)-Cl(1)#1 91.88(2) Cl(1)-Sb(2)-Cl(1)#1 84.98(2) Sb(2)-Cl(1)-Sb(2)#1 95.02(2) Sb(2)-Cl(1)-Sb(1) 135.82(3) Sb(2)#1-Cl(1)-Sb(1) 87.962(17) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1,-y,-z+1 #2 x+1,y,z Table 4. Torsion angles [deg] for gudat62. ________________________________________________________________ N(2)-Sb(1)-N(1)-C(1) -2.54(17) Cl(1)-Sb(1)-N(1)-C(1) -114.53(17) Cl(2)#1-Sb(1)-N(1)-C(1) -158.63(17) Cl(4)#1-Sb(1)-N(1)-C(1) -177.34(16) Cl(2)#2-Sb(1)-N(1)-C(1) 115.52(17) Cl(4)#2-Sb(1)-N(1)-C(1) 73.98(18) N(2)-Sb(1)-N(1)-C(11) 171.3(2) Cl(1)-Sb(1)-N(1)-C(11) 59.35(19) Cl(2)#1-Sb(1)-N(1)-C(11) 15.2(4) Cl(4)#1-Sb(1)-N(1)-C(11) -3.5(2) Cl(2)#2-Sb(1)-N(1)-C(11) -70.61(18) Cl(4)#2-Sb(1)-N(1)-C(11) -112.14(17) C(11)-N(1)-C(1)-C(2) -172.4(2) Sb(1)-N(1)-C(1)-C(2) 1.8(3) N(1)-C(1)-C(2)-N(2) 0.7(4) C(1)-C(2)-N(2)-C(21) 176.8(2) C(1)-C(2)-N(2)-Sb(1) -2.9(3) N(1)-Sb(1)-N(2)-C(2) 2.90(18) Cl(1)-Sb(1)-N(2)-C(2) 100.45(17) Cl(2)#1-Sb(1)-N(2)-C(2) 176.34(16) Cl(4)#1-Sb(1)-N(2)-C(2) 159.5(2) Cl(2)#2-Sb(1)-N(2)-C(2) -78.89(18) Cl(4)#2-Sb(1)-N(2)-C(2) -120.28(17) N(1)-Sb(1)-N(2)-C(21) -176.8(2) Cl(1)-Sb(1)-N(2)-C(21) -79.2(2) Cl(2)#1-Sb(1)-N(2)-C(21) -3.3(2) Cl(4)#1-Sb(1)-N(2)-C(21) -20.2(4) Cl(2)#2-Sb(1)-N(2)-C(21) 101.45(19) Cl(4)#2-Sb(1)-N(2)-C(21) 60.06(19) C(1)-N(1)-C(11)-C(12) -174.0(2) Sb(1)-N(1)-C(11)-C(12) 12.5(3) C(1)-N(1)-C(11)-C(13) -53.5(3) Sb(1)-N(1)-C(11)-C(13) 133.1(2) C(1)-N(1)-C(11)-C(14) 67.0(3) Sb(1)-N(1)-C(11)-C(14) -106.5(2) C(2)-N(2)-C(21)-C(23) 65.2(3) Sb(1)-N(2)-C(21)-C(23) -115.2(2) C(2)-N(2)-C(21)-C(24) -56.2(3) Sb(1)-N(2)-C(21)-C(24) 123.5(2) C(2)-N(2)-C(21)-C(22) -175.0(2) Sb(1)-N(2)-C(21)-C(22) 4.6(3) Cl(4)-Sb(2)-Cl(1)-Sb(2)#1 -83.46(2) Cl(3)-Sb(2)-Cl(1)-Sb(2)#1 -175.05(2) Cl(2)-Sb(2)-Cl(1)-Sb(2)#1 -62.0(2) Cl(1)#1-Sb(2)-Cl(1)-Sb(2)#1 0.0 Cl(4)-Sb(2)-Cl(1)-Sb(1) -175.70(4) Cl(3)-Sb(2)-Cl(1)-Sb(1) 92.71(4) Cl(2)-Sb(2)-Cl(1)-Sb(1) -154.26(19)

8. Kristallographischer Anhang
188
Cl(1)#1-Sb(2)-Cl(1)-Sb(1) -92.24(4) N(2)-Sb(1)-Cl(1)-Sb(2) -130.20(7) N(1)-Sb(1)-Cl(1)-Sb(2) -46.80(7) Cl(2)#1-Sb(1)-Cl(1)-Sb(2) 122.07(4) Cl(4)#1-Sb(1)-Cl(1)-Sb(2) 61.82(3) Cl(2)#2-Sb(1)-Cl(1)-Sb(2) 49.04(4) Cl(4)#2-Sb(1)-Cl(1)-Sb(2) 122.46(3) N(2)-Sb(1)-Cl(1)-Sb(2)#1 134.70(7) N(1)-Sb(1)-Cl(1)-Sb(2)#1 -141.90(6) Cl(2)#1-Sb(1)-Cl(1)-Sb(2)#1 26.973(16) Cl(4)#1-Sb(1)-Cl(1)-Sb(2)#1 -33.282(14) Cl(2)#2-Sb(1)-Cl(1)-Sb(2)#1 -46.06(3) Cl(4)#2-Sb(1)-Cl(1)-Sb(2)#1 27.36(3) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1,-y,-z+1 #2 x+1,y,z Table 5. Hydrogen bonds for gudat62 [A and deg.]. ___________________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(1)-H(1)...Cl(2)#3 0.95 2.76 3.610(3) 148.8 C(12)-H(12A)...Cl(2)#2 0.98 3.01 3.951(3) 162.2 C(2)-H(2)...Cl(3)#3 0.95 2.77 3.655(3) 156.0 C(12)-H(12B)...Cl(4)#1 0.98 2.90 3.558(3) 125.2 C(22)-H(22A)...Cl(4)#2 0.98 2.97 3.899(3) 158.2 C(23)-H(23C)...Cl(4)#4 0.98 2.86 3.815(3) 165.1 ___________________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1,-y,-z+1 #2 x+1,y,z #3 x+1/2,-y+1/2,z-1/2 #4 -x+1,-y,-z Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat53. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ Cl(1) 5999(1) 3917(1) 1486(1) 40(1) Sb(1) 7363(1) 5853(1) 1507(1) 28(1) N(1) 6792(3) 6666(4) 2294(2) 25(1) N(2) 6287(3) 7055(4) 1105(2) 28(1) C(1) 5907(4) 7516(5) 2171(2) 28(1) C(2) 5669(4) 7715(5) 1555(2) 27(1) C(11) 7040(4) 6257(5) 2949(2) 26(1) C(12) 7250(4) 7363(5) 3376(2) 32(1) C(13) 6016(4) 5513(5) 3198(2) 35(1) C(14) 8146(4) 5488(5) 2932(2) 32(1) C(21) 5988(4) 7149(5) 426(2) 30(1) C(22) 6783(5) 6295(5) 61(3) 38(1) C(23) 4728(5) 6758(6) 325(3) 44(2) C(24) 6185(6) 8457(5) 201(3) 47(2) ________________________________________________________________

8. Kristallographischer Anhang
189
Table 3. Bond lengths [A] and angles [deg] for gudat53. _____________________________________________________________ Cl(1)-Sb(1) 2.6460(15) Sb(1)-N(1) 1.998(4) Sb(1)-N(2) 2.000(4) Sb(1)-Cl(1)#1 3.8620(15) N(1)-C(1) 1.407(6) N(1)-C(11) 1.481(6) N(2)-C(2) 1.391(6) N(2)-C(21) 1.478(6) C(1)-C(2) 1.346(6) C(11)-C(13) 1.530(7) C(11)-C(14) 1.531(7) C(11)-C(12) 1.533(7) C(21)-C(22) 1.522(7) C(21)-C(23) 1.528(7) C(21)-C(24) 1.531(7) N(1)-Sb(1)-N(2) 81.49(16) N(1)-Sb(1)-Cl(1) 100.23(12) N(2)-Sb(1)-Cl(1) 99.00(12) N(1)-Sb(1)-Cl(1)#1 77.28(12) N(2)-Sb(1)-Cl(1)#1 73.84(12) Cl(1)-Sb(1)-Cl(1)#1 172.64(3) C(1)-N(1)-C(11) 120.9(4) C(1)-N(1)-Sb(1) 112.5(3) C(11)-N(1)-Sb(1) 125.2(3) C(2)-N(2)-C(21) 120.4(4) C(2)-N(2)-Sb(1) 111.9(3) C(21)-N(2)-Sb(1) 127.0(3) C(2)-C(1)-N(1) 115.7(4) C(1)-C(2)-N(2) 118.0(4) N(1)-C(11)-C(13) 109.5(4) N(1)-C(11)-C(14) 107.8(4) C(13)-C(11)-C(14) 110.8(5) N(1)-C(11)-C(12) 109.8(4) C(13)-C(11)-C(12) 110.1(4) C(14)-C(11)-C(12) 108.7(4) N(2)-C(21)-C(22) 107.8(4) N(2)-C(21)-C(23) 109.7(4) C(22)-C(21)-C(23) 109.1(5) N(2)-C(21)-C(24) 109.4(4) C(22)-C(21)-C(24) 109.5(4) C(23)-C(21)-C(24) 111.2(5) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+3/2,y+1/2,z Table 4. Torsion angles [deg] for gudat53. ________________________________________________________________ N(2)-Sb(1)-N(1)-C(1) 5.6(3) Cl(1)-Sb(1)-N(1)-C(1) -92.1(3) Cl(1)#1-Sb(1)-N(1)-C(1) 80.8(3) N(2)-Sb(1)-N(1)-C(11) 172.0(4) Cl(1)-Sb(1)-N(1)-C(11) 74.4(4) Cl(1)#1-Sb(1)-N(1)-C(11) -112.7(4) N(1)-Sb(1)-N(2)-C(2) -4.8(3) Cl(1)-Sb(1)-N(2)-C(2) 94.3(3)

8. Kristallographischer Anhang
190
Cl(1)#1-Sb(1)-N(2)-C(2) -84.0(3) N(1)-Sb(1)-N(2)-C(21) -175.0(4) Cl(1)-Sb(1)-N(2)-C(21) -75.9(4) Cl(1)#1-Sb(1)-N(2)-C(21) 105.8(4) C(11)-N(1)-C(1)-C(2) -172.6(4) Sb(1)-N(1)-C(1)-C(2) -5.5(5) N(1)-C(1)-C(2)-N(2) 1.5(7) C(21)-N(2)-C(2)-C(1) 174.2(4) Sb(1)-N(2)-C(2)-C(1) 3.3(6) C(1)-N(1)-C(11)-C(13) 67.3(6) Sb(1)-N(1)-C(11)-C(13) -98.1(5) C(1)-N(1)-C(11)-C(14) -172.0(4) Sb(1)-N(1)-C(11)-C(14) 22.6(6) C(1)-N(1)-C(11)-C(12) -53.7(6) Sb(1)-N(1)-C(11)-C(12) 140.9(3) C(2)-N(2)-C(21)-C(22) -178.1(4) Sb(1)-N(2)-C(21)-C(22) -8.6(6) C(2)-N(2)-C(21)-C(23) -59.3(6) Sb(1)-N(2)-C(21)-C(23) 110.1(4) C(2)-N(2)-C(21)-C(24) 62.9(6) Sb(1)-N(2)-C(21)-C(24) -127.6(4) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+3/2,y+1/2,z Table 5. Hydrogen bonds for gudat53 [A and deg.]. _______________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(1)-H(1)...Cl(1)#2 0.95 2.96 3.902(5) 171.9 C(23)-H(23C)...Cl(1)#3 0.98 3.04 3.982(6) 162.7 _______________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+3/2,y+1/2,z #2 -x+1,y+1/2,-z+1/2 #3 -x+1,-y+1,-z
Table 1. Crystal data and structure refinement for
1,2-Dimethyl-1,2-(2,6-diisopropylphenyl)-Diaminoethan (43f)

8. Kristallographischer Anhang
191
Identification code gudat65 Empirical formula C28 H42 N2 Formula weight 406.64 Temperature 123(2) K Wavelength 0.71073 A Crystal system, space group Monoclinic, P2(1)/n (No.14) Unit cell dimensions a = 16.1735(3) A alpha = 90 deg. b = 9.0446(1) A beta = 106.403(1) deg. c = 18.4566(3) A gamma = 90 deg. Volume 2590.00(7) A^3 Z, Calculated density 4, 1.043 Mg/m^3 Absorption coefficient 0.060 mm^-1 F(000) 896 Crystal size 0.30 x 0.25 x 0.20 mm Diffractometer Nonius KappaCCD Theta range for data collection 2.70 to 25.00 deg. Limiting indices -19<=h<=19, -10<=k<=10, -21<=l<=21 Reflections collected / unique 45384 / 4547 [R(int) = 0.0579] Completeness to theta = 25.00 99.9 % Absorption correction None Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 4547 / 0 / 279 Goodness-of-fit on F^2 1.004 Final R indices [I>2sigma(I)] R1 = 0.0417, wR2 = 0.1053 R indices (all data) R1 = 0.0666, wR2 = 0.1145 Largest diff. peak and hole 0.215 and -0.182 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat65. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.

8. Kristallographischer Anhang
192
________________________________________________________________ x y z U(eq) ________________________________________________________________ N(1) 3802(1) 4761(1) 2036(1) 29(1) H(1) 3694(10) 4963(16) 2463(9) 44 N(2) 5261(1) 5720(1) 3029(1) 29(1) H(2) 5008(10) 4815(18) 2998(8) 44 C(1) 4244(1) 5976(1) 1815(1) 25(1) C(2) 4975(1) 6432(1) 2310(1) 25(1) C(3) 3822(1) 6683(2) 1067(1) 32(1) C(4) 5504(1) 7734(2) 2198(1) 35(1) C(5) 3116(1) 3994(2) 1517(1) 27(1) C(6) 2279(1) 4074(2) 1604(1) 30(1) C(7) 1638(1) 3200(2) 1140(1) 37(1) C(8) 1795(1) 2329(2) 580(1) 40(1) C(9) 2604(1) 2312(2) 478(1) 36(1) C(10) 3283(1) 3115(2) 945(1) 30(1) C(11) 4182(1) 2948(2) 849(1) 37(1) C(12) 4538(1) 1408(2) 1113(1) 51(1) C(13) 4203(1) 3211(2) 34(1) 51(1) C(14) 2075(1) 5089(2) 2191(1) 33(1) C(15) 2205(1) 4292(2) 2942(1) 48(1) C(16) 1171(1) 5757(2) 1933(1) 50(1) C(17) 6175(1) 5663(2) 3403(1) 27(1) C(18) 6516(1) 6646(2) 4001(1) 30(1) C(19) 7396(1) 6593(2) 4357(1) 36(1) C(20) 7921(1) 5601(2) 4132(1) 38(1) C(21) 7577(1) 4638(2) 3549(1) 38(1) C(22) 6700(1) 4645(2) 3172(1) 32(1) C(23) 6341(1) 3557(2) 2531(1) 40(1) C(24) 6636(1) 3947(2) 1841(1) 59(1) C(25) 6563(2) 1967(2) 2782(1) 70(1) C(26) 5938(1) 7697(2) 4277(1) 38(1) C(27) 5664(1) 6977(2) 4916(1) 58(1) C(28) 6340(1) 9207(2) 4512(1) 64(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat65. _____________________________________________________________ N(1)-C(5) 1.4247(17) N(1)-C(1) 1.4326(17) N(1)-H(1) 0.872(16) N(2)-C(2) 1.4283(17) N(2)-C(17) 1.4457(17) N(2)-H(2) 0.910(16) C(1)-C(2) 1.3390(18) C(1)-C(3) 1.4997(18) C(2)-C(4) 1.5043(19) C(5)-C(10) 1.4072(19) C(5)-C(6) 1.4092(19) C(6)-C(7) 1.390(2) C(6)-C(14) 1.525(2) C(7)-C(8) 1.380(2) C(8)-C(9) 1.374(2) C(9)-C(10) 1.394(2) C(10)-C(11) 1.520(2) C(11)-C(13) 1.533(2) C(11)-C(12) 1.534(2) C(14)-C(15) 1.524(2)

8. Kristallographischer Anhang
193
C(14)-C(16) 1.529(2) C(17)-C(22) 1.398(2) C(17)-C(18) 1.4028(19) C(18)-C(19) 1.390(2) C(18)-C(26) 1.518(2) C(19)-C(20) 1.378(2) C(20)-C(21) 1.374(2) C(21)-C(22) 1.393(2) C(22)-C(23) 1.522(2) C(23)-C(24) 1.522(2) C(23)-C(25) 1.523(2) C(26)-C(27) 1.518(2) C(26)-C(28) 1.522(2) C(5)-N(1)-C(1) 122.60(11) C(5)-N(1)-H(1) 113.1(10) C(1)-N(1)-H(1) 109.8(10) C(2)-N(2)-C(17) 118.64(11) C(2)-N(2)-H(2) 109.0(9) C(17)-N(2)-H(2) 112.2(10) C(2)-C(1)-N(1) 117.26(11) C(2)-C(1)-C(3) 125.57(12) N(1)-C(1)-C(3) 117.07(11) C(1)-C(2)-N(2) 119.10(12) C(1)-C(2)-C(4) 125.08(12) N(2)-C(2)-C(4) 115.66(12) C(10)-C(5)-C(6) 120.63(13) C(10)-C(5)-N(1) 120.04(12) C(6)-C(5)-N(1) 119.27(12) C(7)-C(6)-C(5) 118.28(13) C(7)-C(6)-C(14) 120.45(13) C(5)-C(6)-C(14) 121.27(12) C(8)-C(7)-C(6) 121.54(14) C(9)-C(8)-C(7) 119.58(14) C(8)-C(9)-C(10) 121.60(14) C(9)-C(10)-C(5) 118.24(13) C(9)-C(10)-C(11) 119.17(13) C(5)-C(10)-C(11) 122.50(13) C(10)-C(11)-C(13) 112.78(13) C(10)-C(11)-C(12) 109.78(12) C(13)-C(11)-C(12) 110.02(13) C(15)-C(14)-C(6) 111.13(12) C(15)-C(14)-C(16) 110.13(13) C(6)-C(14)-C(16) 113.38(12) C(22)-C(17)-C(18) 121.33(13) C(22)-C(17)-N(2) 120.33(12) C(18)-C(17)-N(2) 118.35(12) C(19)-C(18)-C(17) 118.15(13) C(19)-C(18)-C(26) 120.54(13) C(17)-C(18)-C(26) 121.25(12) C(20)-C(19)-C(18) 121.11(14) C(21)-C(20)-C(19) 120.07(14) C(20)-C(21)-C(22) 121.20(14) C(21)-C(22)-C(17) 118.13(13) C(21)-C(22)-C(23) 119.75(13) C(17)-C(22)-C(23) 122.11(13) C(22)-C(23)-C(24) 111.10(13) C(22)-C(23)-C(25) 111.65(13) C(24)-C(23)-C(25) 111.77(15) C(18)-C(26)-C(27) 109.55(13) C(18)-C(26)-C(28) 113.81(14) C(27)-C(26)-C(28) 111.00(14) _____________________________________________________________

8. Kristallographischer Anhang
194
Table 4. Torsion angles [deg] for gudat65. ________________________________________________________________ C(5)-N(1)-C(1)-C(2) -167.82(12) C(5)-N(1)-C(1)-C(3) 15.55(19) N(1)-C(1)-C(2)-N(2) -1.53(19) C(3)-C(1)-C(2)-N(2) 174.78(12) N(1)-C(1)-C(2)-C(4) -176.69(13) C(3)-C(1)-C(2)-C(4) -0.4(2) C(17)-N(2)-C(2)-C(1) 150.77(13) C(17)-N(2)-C(2)-C(4) -33.62(18) C(1)-N(1)-C(5)-C(10) 68.45(18) C(1)-N(1)-C(5)-C(6) -114.28(15) C(10)-C(5)-C(6)-C(7) 3.7(2) N(1)-C(5)-C(6)-C(7) -173.53(12) C(10)-C(5)-C(6)-C(14) -176.45(12) N(1)-C(5)-C(6)-C(14) 6.30(19) C(5)-C(6)-C(7)-C(8) -3.5(2) C(14)-C(6)-C(7)-C(8) 176.70(14) C(6)-C(7)-C(8)-C(9) 0.5(2) C(7)-C(8)-C(9)-C(10) 2.3(2) C(8)-C(9)-C(10)-C(5) -2.0(2) C(8)-C(9)-C(10)-C(11) 174.68(14) C(6)-C(5)-C(10)-C(9) -1.1(2) N(1)-C(5)-C(10)-C(9) 176.17(12) C(6)-C(5)-C(10)-C(11) -177.64(13) N(1)-C(5)-C(10)-C(11) -0.4(2) C(9)-C(10)-C(11)-C(13) 53.14(18) C(5)-C(10)-C(11)-C(13) -130.31(15) C(9)-C(10)-C(11)-C(12) -69.93(17) C(5)-C(10)-C(11)-C(12) 106.63(16) C(7)-C(6)-C(14)-C(15) 89.72(17) C(5)-C(6)-C(14)-C(15) -90.10(16) C(7)-C(6)-C(14)-C(16) -34.97(19) C(5)-C(6)-C(14)-C(16) 145.21(14) C(2)-N(2)-C(17)-C(22) -77.57(16) C(2)-N(2)-C(17)-C(18) 102.66(15) C(22)-C(17)-C(18)-C(19) 0.6(2) N(2)-C(17)-C(18)-C(19) -179.67(12) C(22)-C(17)-C(18)-C(26) -176.85(12) N(2)-C(17)-C(18)-C(26) 2.92(19) C(17)-C(18)-C(19)-C(20) -0.2(2) C(26)-C(18)-C(19)-C(20) 177.27(13) C(18)-C(19)-C(20)-C(21) -0.3(2) C(19)-C(20)-C(21)-C(22) 0.4(2) C(20)-C(21)-C(22)-C(17) 0.0(2) C(20)-C(21)-C(22)-C(23) -179.71(13) C(18)-C(17)-C(22)-C(21) -0.5(2) N(2)-C(17)-C(22)-C(21) 179.74(12) C(18)-C(17)-C(22)-C(23) 179.23(12) N(2)-C(17)-C(22)-C(23) -0.5(2) C(21)-C(22)-C(23)-C(24) -70.17(18) C(17)-C(22)-C(23)-C(24) 110.10(16) C(21)-C(22)-C(23)-C(25) 55.35(19) C(17)-C(22)-C(23)-C(25) -124.37(16) C(19)-C(18)-C(26)-C(27) -85.21(17) C(17)-C(18)-C(26)-C(27) 92.14(16) C(19)-C(18)-C(26)-C(28) 39.7(2) C(17)-C(18)-C(26)-C(28) -142.96(14) ________________________________________________________________

8. Kristallographischer Anhang
195
Table 1. Crystal data and structure refinement
(tBuNC=CNCtBu*2SbCl3)2 77
Identification code gudat57m Empirical formula C10 H20 Cl6 N2 Sb2 Formula weight 624.48 Temperature 123(2) K Wavelength 0.71073 A (MoKa) Crystal system, space group Monoclinic, C2/c (No.15) Unit cell dimensions a = 12.0617(1) A alpha = 90 deg. b = 17.4255(2) A beta = 102.081(1) deg. c = 19.3913(2) A gamma = 90 deg. Volume 3985.42(7) A^3 Z, Calculated density 8, 2.082 Mg/m^3 Absorption coefficient 3.508 mm^-1 F(000) 2384 Crystal size 0.15 x 0.10 x 0.05 mm Diffractometer Nonius KappaCCD Theta range for data collection 2.72 to 25.00 deg. Limiting indices -14<=h<=14, -20<=k<=20, -23<=l<=23

8. Kristallographischer Anhang
196
Reflections collected / unique 34881 / 3512 [R(int) = 0.0705] Completeness to theta = 25.00 99.8 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.7920 and 0.5753 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 3512 / 0 / 181 Goodness-of-fit on F^2 0.979 Final R indices [I>2sigma(I)] R1 = 0.0182, wR2 = 0.0391 R indices (all data) R1 = 0.0261, wR2 = 0.0406 Largest diff. peak and hole 0.414 and -0.541 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat57. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ Sb(1) 5302(1) 2347(1) 6042(1) 18(1) Cl(1) 5597(1) 2237(1) 4868(1) 25(1) Cl(2) 3865(1) 3394(1) 5580(1) 26(1) Cl(3) 3652(1) 1379(1) 5657(1) 28(1) N(1) 6954(2) 1486(1) 6271(1) 16(1) C(11) 6908(2) 629(1) 6370(1) 20(1) C(12) 8079(2) 262(2) 6563(1) 26(1) C(13) 6261(2) 518(2) 6969(1) 25(1) C(14) 6238(2) 283(2) 5686(1) 24(1) C(1) 7887(2) 1856(2) 6427(1) 19(1) C(2) 7923(2) 2698(1) 6378(1) 17(1) N(2) 7023(2) 3095(1) 6180(1) 18(1) C(21) 7076(2) 3961(2) 6159(1) 21(1) C(22) 6396(2) 4237(2) 6703(1) 26(1) C(23) 6515(2) 4199(2) 5408(1) 28(1) C(24) 8276(2) 4273(2) 6344(1) 26(1) Sb(2) 1962(1) 2443(1) 6136(1) 19(1) Cl(4) 3501(1) 2422(1) 7151(1) 27(1) Cl(5) 1028(1) 3340(1) 6772(1) 31(1) Cl(6) 1032(1) 1379(1) 6538(1) 34(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat57. _____________________________________________________________ Sb(1)-Cl(1) 2.3841(6) Sb(1)-N(2) 2.418(2) Sb(1)-N(1) 2.460(2) Sb(1)-Cl(2) 2.5464(7) Sb(1)-Cl(3) 2.5962(7) Sb(1)-Cl(4) 3.3633(6)

8. Kristallographischer Anhang
197
Cl(1)-Sb(2)#1 3.3336(7) Cl(2)-Sb(2) 3.1963(7) Cl(3)-Sb(2) 3.0408(7) N(1)-C(1) 1.278(3) N(1)-C(11) 1.508(3) C(11)-C(12) 1.524(4) C(11)-C(14) 1.526(3) C(11)-C(13) 1.541(3) C(1)-C(2) 1.472(3) C(2)-N(2) 1.277(3) N(2)-C(21) 1.510(3) C(21)-C(24) 1.517(4) C(21)-C(23) 1.529(3) C(21)-C(22) 1.542(3) Sb(2)-Cl(6) 2.3804(7) Sb(2)-Cl(4) 2.4061(7) Sb(2)-Cl(5) 2.4116(7) Sb(2)-Cl(1)#1 3.3336(7) Cl(1)-Sb(1)-N(2) 81.54(5) Cl(1)-Sb(1)-N(1) 81.15(5) N(2)-Sb(1)-N(1) 70.43(7) Cl(1)-Sb(1)-Cl(2) 87.05(2) N(2)-Sb(1)-Cl(2) 99.10(6) N(1)-Sb(1)-Cl(2) 165.19(5) Cl(1)-Sb(1)-Cl(3) 86.08(2) N(2)-Sb(1)-Cl(3) 166.07(5) N(1)-Sb(1)-Cl(3) 101.44(5) Cl(2)-Sb(1)-Cl(3) 86.55(2) Cl(1)-Sb(1)-Cl(4) 149.17(2) N(2)-Sb(1)-Cl(4) 124.25(5) N(1)-Sb(1)-Cl(4) 121.02(4) Cl(2)-Sb(1)-Cl(4) 73.372(19) Cl(3)-Sb(1)-Cl(4) 69.522(19) Sb(1)-Cl(1)-Sb(2)#1 103.85(2) Sb(1)-Cl(2)-Sb(2) 89.654(19) Sb(1)-Cl(3)-Sb(2) 92.23(2) C(1)-N(1)-C(11) 121.6(2) C(1)-N(1)-Sb(1) 112.00(17) C(11)-N(1)-Sb(1) 125.28(16) N(1)-C(11)-C(12) 113.0(2) N(1)-C(11)-C(14) 107.9(2) C(12)-C(11)-C(14) 110.0(2) N(1)-C(11)-C(13) 104.93(19) C(12)-C(11)-C(13) 110.6(2) C(14)-C(11)-C(13) 110.3(2) N(1)-C(1)-C(2) 121.6(3) N(2)-C(2)-C(1) 121.7(2) C(2)-N(2)-C(21) 120.8(2) C(2)-N(2)-Sb(1) 113.39(17) C(21)-N(2)-Sb(1) 125.13(16) N(2)-C(21)-C(24) 113.3(2) N(2)-C(21)-C(23) 106.6(2) C(24)-C(21)-C(23) 109.7(2) N(2)-C(21)-C(22) 105.13(19) C(24)-C(21)-C(22) 110.1(2) C(23)-C(21)-C(22) 112.0(2) Cl(6)-Sb(2)-Cl(4) 93.23(3) Cl(6)-Sb(2)-Cl(5) 91.98(3) Cl(4)-Sb(2)-Cl(5) 87.38(2) Cl(6)-Sb(2)-Cl(3) 91.01(2) Cl(4)-Sb(2)-Cl(3) 77.80(2) Cl(5)-Sb(2)-Cl(3) 165.02(2)

8. Kristallographischer Anhang
198
Cl(6)-Sb(2)-Cl(2) 159.25(2) Cl(4)-Sb(2)-Cl(2) 78.33(2) Cl(5)-Sb(2)-Cl(2) 106.36(2) Cl(3)-Sb(2)-Cl(2) 68.787(18) Cl(6)-Sb(2)-Cl(1)#1 83.07(2) Cl(4)-Sb(2)-Cl(1)#1 160.34(2) Cl(5)-Sb(2)-Cl(1)#1 73.51(2) Cl(3)-Sb(2)-Cl(1)#1 121.435(18) Cl(2)-Sb(2)-Cl(1)#1 111.029(17) Sb(2)-Cl(4)-Sb(1) 88.232(19) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,-y+1/2,-z+1 Table 4. Anisotropic displacement parameters (A^2 x 10^3) for gudat57. The anisotropic displacement factor exponent takes the form: -2 pi^2 [ h^2 a*^2 U11 + ... + 2 h k a* b* U12 ] ______________________________________________________________________ U11 U22 U33 U23 U13 U12 ______________________________________________________________________ Sb(1) 20(1) 19(1) 13(1) -1(1) 3(1) 1(1) Cl(1) 28(1) 34(1) 13(1) -2(1) 4(1) 4(1) Cl(2) 26(1) 25(1) 26(1) 3(1) 3(1) 5(1) Cl(3) 25(1) 24(1) 33(1) -6(1) 2(1) -2(1) N(1) 22(1) 15(1) 12(1) 0(1) 5(1) 2(1) C(11) 28(2) 15(2) 16(1) 1(1) 5(1) 0(1) C(12) 33(2) 17(2) 25(2) 1(1) 2(1) 3(1) C(13) 34(2) 20(2) 21(2) 1(1) 6(1) -2(1) C(14) 33(2) 18(2) 21(2) -3(1) 4(1) -3(1) C(1) 22(2) 21(2) 14(1) -1(1) 3(1) 4(1) C(2) 19(2) 23(2) 11(1) -4(1) 4(1) -5(1) N(2) 24(1) 19(1) 10(1) -1(1) 4(1) -2(1) C(21) 31(2) 12(1) 19(2) 1(1) 3(1) 0(1) C(22) 36(2) 17(2) 27(2) -4(1) 8(1) 3(1) C(23) 37(2) 23(2) 22(2) 7(1) 5(1) -2(1) C(24) 34(2) 17(2) 27(2) 1(1) 6(1) -3(1) Sb(2) 21(1) 20(1) 16(1) 0(1) 2(1) 0(1) Cl(4) 24(1) 38(1) 17(1) 2(1) 2(1) 2(1) Cl(5) 31(1) 30(1) 29(1) -12(1) 1(1) 5(1) Cl(6) 32(1) 23(1) 46(1) 11(1) 4(1) -4(1) _________________________________________________________________ Table 5. Hydrogen coordinates ( x 10^4) and isotropic displacement parameters (A^2 x 10^3) for gudat57. ________________________________________________________________ x y z U(eq) ________________________________________________________________ H(12A) 8507 493 6998 38 H(12B) 8483 346 6181 38 H(12C) 7998 -291 6634 38 H(13A) 6701 741 7405 37 H(13B) 6148 -31 7039 37 H(13C) 5523 774 6844 37 H(14A) 5491 527 5562 36 H(14B) 6144 -270 5749 36

8. Kristallographischer Anhang
199
H(14C) 6648 369 5306 36 H(1) 8575 1581 6577 23 H(2) 8634 2953 6498 21 H(22A) 5623 4032 6579 40 H(22B) 6368 4799 6701 40 H(22C) 6765 4056 7173 40 H(23A) 5742 3994 5289 41 H(23B) 6955 3996 5077 41 H(23C) 6490 4761 5376 41 H(24A) 8631 4120 6826 39 H(24B) 8256 4834 6310 39 H(24C) 8715 4066 6014 39 ________________________________________________________________ Table 6. Torsion angles [deg] for gudat57. ________________________________________________________________ N(2)-Sb(1)-Cl(1)-Sb(2)#1 -134.92(6) N(1)-Sb(1)-Cl(1)-Sb(2)#1 153.72(5) Cl(2)-Sb(1)-Cl(1)-Sb(2)#1 -35.26(2) Cl(3)-Sb(1)-Cl(1)-Sb(2)#1 51.48(2) Cl(4)-Sb(1)-Cl(1)-Sb(2)#1 14.53(5) Cl(1)-Sb(1)-Cl(2)-Sb(2) 122.81(2) N(2)-Sb(1)-Cl(2)-Sb(2) -156.25(4) N(1)-Sb(1)-Cl(2)-Sb(2) 159.9(2) Cl(3)-Sb(1)-Cl(2)-Sb(2) 36.561(19) Cl(4)-Sb(1)-Cl(2)-Sb(2) -33.087(16) Cl(1)-Sb(1)-Cl(3)-Sb(2) -126.07(2) N(2)-Sb(1)-Cl(3)-Sb(2) -153.3(2) N(1)-Sb(1)-Cl(3)-Sb(2) 153.79(4) Cl(2)-Sb(1)-Cl(3)-Sb(2) -38.80(2) Cl(4)-Sb(1)-Cl(3)-Sb(2) 34.731(16) Cl(1)-Sb(1)-N(1)-C(1) 91.47(15) N(2)-Sb(1)-N(1)-C(1) 7.36(15) Cl(2)-Sb(1)-N(1)-C(1) 53.9(3) Cl(3)-Sb(1)-N(1)-C(1) 175.61(15) Cl(4)-Sb(1)-N(1)-C(1) -111.54(15) Cl(1)-Sb(1)-N(1)-C(11) -100.53(17) N(2)-Sb(1)-N(1)-C(11) 175.36(18) Cl(2)-Sb(1)-N(1)-C(11) -138.09(18) Cl(3)-Sb(1)-N(1)-C(11) -16.39(17) Cl(4)-Sb(1)-N(1)-C(11) 56.47(18) C(1)-N(1)-C(11)-C(12) -8.4(3) Sb(1)-N(1)-C(11)-C(12) -175.32(15) C(1)-N(1)-C(11)-C(14) -130.3(2) Sb(1)-N(1)-C(11)-C(14) 62.8(2) C(1)-N(1)-C(11)-C(13) 112.1(2) Sb(1)-N(1)-C(11)-C(13) -54.8(3) C(11)-N(1)-C(1)-C(2) -175.57(19) Sb(1)-N(1)-C(1)-C(2) -7.1(3) N(1)-C(1)-C(2)-N(2) 0.1(3) C(1)-C(2)-N(2)-C(21) 177.88(19) C(1)-C(2)-N(2)-Sb(1) 7.1(3) Cl(1)-Sb(1)-N(2)-C(2) -90.92(16) N(1)-Sb(1)-N(2)-C(2) -7.37(15) Cl(2)-Sb(1)-N(2)-C(2) -176.53(15) Cl(3)-Sb(1)-N(2)-C(2) -63.4(3) Cl(4)-Sb(1)-N(2)-C(2) 107.45(15)

8. Kristallographischer Anhang
200
Cl(1)-Sb(1)-N(2)-C(21) 98.73(17) N(1)-Sb(1)-N(2)-C(21) -177.71(18) Cl(2)-Sb(1)-N(2)-C(21) 13.12(17) Cl(3)-Sb(1)-N(2)-C(21) 126.3(2) Cl(4)-Sb(1)-N(2)-C(21) -62.90(18) C(2)-N(2)-C(21)-C(24) 3.8(3) Sb(1)-N(2)-C(21)-C(24) 173.45(15) C(2)-N(2)-C(21)-C(23) 124.5(2) Sb(1)-N(2)-C(21)-C(23) -65.8(2) C(2)-N(2)-C(21)-C(22) -116.5(2) Sb(1)-N(2)-C(21)-C(22) 53.2(3) Sb(1)-Cl(3)-Sb(2)-Cl(6) -142.84(3) Sb(1)-Cl(3)-Sb(2)-Cl(4) -49.76(2) Sb(1)-Cl(3)-Sb(2)-Cl(5) -41.31(9) Sb(1)-Cl(3)-Sb(2)-Cl(2) 32.310(19) Sb(1)-Cl(3)-Sb(2)-Cl(1)#1 134.660(19) Sb(1)-Cl(2)-Sb(2)-Cl(6) -19.19(7) Sb(1)-Cl(2)-Sb(2)-Cl(4) 48.30(2) Sb(1)-Cl(2)-Sb(2)-Cl(5) 132.03(2) Sb(1)-Cl(2)-Sb(2)-Cl(3) -32.994(19) Sb(1)-Cl(2)-Sb(2)-Cl(1)#1 -149.747(18) Cl(6)-Sb(2)-Cl(4)-Sb(1) 126.43(2) Cl(5)-Sb(2)-Cl(4)-Sb(1) -141.73(2) Cl(3)-Sb(2)-Cl(4)-Sb(1) 36.089(16) Cl(2)-Sb(2)-Cl(4)-Sb(1) -34.438(15) Cl(1)#1-Sb(2)-Cl(4)-Sb(1) -155.17(5) Cl(1)-Sb(1)-Cl(4)-Sb(2) -6.23(5) N(2)-Sb(1)-Cl(4)-Sb(2) 136.30(6) N(1)-Sb(1)-Cl(4)-Sb(2) -137.33(6) Cl(2)-Sb(1)-Cl(4)-Sb(2) 46.51(2) Cl(3)-Sb(1)-Cl(4)-Sb(2) -46.04(2) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,-y+1/2,-z+1 Table 7. Hydrogen bonds for gudat57 [A and deg.]. ____________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(1)-H(1)...Cl(6)#2 0.95 3.00 3.844(3) 148.8 C(2)-H(2)...Cl(5)#2 0.95 2.90 3.829(3) 164.9 C(22)-H(22B)...Cl(6)#3 0.98 2.79 3.764(3) 172.0 C(13)-H(13B)...Cl(5)#4 0.98 2.88 3.820(3) 160.0 C(24)-H(24A)...Cl(5)#5 0.98 2.99 3.926(3) 160.1 ____________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,-y+1/2,-z+1 #2 x+1,y,z #3 x+1/2,y+1/2,z #4 x+1/2,y-1/2,z #5 -x+1,y,-z+3/2

8. Kristallographischer Anhang
201
Table 1. Crystal data and structure refinement for
1,3-Dimesityl-1,3,2-diazarsolenium-triflat (12c[OTf])
Identification code gudat78m Empirical formula C23 H29 As F3 N2 O3.50 S C20 H24 As N2 - CF3SO3 - 1/2 Et2O Formula weight 553.46 Temperature 123(2) K Wavelength 0.71073 A Crystal system, space group Monoclinic, C2/c (No.15) Unit cell dimensions a = 25.0588(3) A alpha = 90 deg. b = 15.8785(3) A beta = 132.657(1) deg. c = 17.2022(3) A gamma = 90 deg. Volume 5033.74(14) A^3 Z, Calculated density 8, 1.461 Mg/m^3 Absorption coefficient 1.485 mm^-1 F(000) 2280 Crystal size 0.70 x 0.60 x 0.50 mm Diffractometer Nonius KappaCCD

8. Kristallographischer Anhang
202
Theta range for data collection 2.92 to 27.48 deg. Limiting indices -32<=h<=32, -20<=k<=20, -22<=l<=22 Reflections collected / unique 32626 / 5599 [R(int) = 0.0431] Completeness to theta = 27.48 97.0 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.48133 and 0.46155 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 5599 / 15 / 306 Goodness-of-fit on F^2 0.959 Final R indices [I>2sigma(I)] R1 = 0.0282, wR2 = 0.0644 R indices (all data) R1 = 0.0437, wR2 = 0.0673 Largest diff. peak and hole 0.331 and -0.456 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat78. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ S(1) 3666(1) 2577(1) 9092(1) 23(1) O(1) 4162(1) 2969(1) 9047(1) 32(1) O(2) 3256(1) 1884(1) 8356(1) 34(1) O(3) 3253(1) 3136(1) 9160(1) 38(1) C(1) 4255(1) 2031(1) 10353(2) 31(1) F(1) 4686(1) 1481(1) 10415(1) 42(1) F(2) 3873(1) 1604(1) 10502(1) 46(1) F(3) 4687(1) 2568(1) 11159(1) 52(1) As(1) 2117(1) 2361(1) 6386(1) 25(1) N(2) 1246(1) 2633(1) 5070(1) 21(1) C(3) 658(1) 2359(1) 4877(1) 25(1) C(4) 826(1) 1926(1) 5702(1) 24(1) N(5) 1545(1) 1860(1) 6556(1) 19(1) C(6) 1181(1) 3182(1) 4337(1) 20(1) C(7) 1186(1) 2834(1) 3592(1) 23(1) C(8) 1104(1) 3385(1) 2892(1) 24(1) C(9) 1012(1) 4242(1) 2906(1) 25(1) C(10) 1013(1) 4558(1) 3663(1) 30(1) C(11) 1097(1) 4043(1) 4385(1) 26(1) C(12) 1091(1) 4401(1) 5198(2) 38(1) C(13) 928(1) 4826(1) 2134(2) 37(1) C(14) 1280(1) 1903(1) 3555(2) 41(1) C(15) 1766(1) 1453(1) 7497(1) 19(1) C(16) 1742(1) 1930(1) 8152(1) 20(1) C(17) 1886(1) 1512(1) 8991(1) 22(1) C(18) 2046(1) 656(1) 9170(1) 22(1) C(19) 2085(1) 217(1) 8509(1) 25(1) C(20) 1947(1) 602(1) 7662(1) 22(1)

8. Kristallographischer Anhang
203
C(21) 1999(1) 110(1) 6967(2) 33(1) C(22) 2147(1) 207(1) 10036(2) 32(1) C(23) 1558(1) 2852(1) 7970(1) 25(1) C(2E) -133(4) 108(5) 992(5) 43(2) s.o.f.= 0.50 C(1E) 5(5) -410(4) 1814(6) 31(2) s.o.f.= 0.50 O(1E) 73(3) 68(1) 2567(4) 28(1) s.o.f.= 0.50 C(3E) 93(5) -467(5) 3255(6) 38(3) s.o.f.= 0.50 C(4E) -7(4) 142(5) 3857(6) 50(3) s.o.f.= 0.50 ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat78. _____________________________________________________________ S(1)-O(3) 1.4267(13) S(1)-O(1) 1.4365(12) S(1)-O(2) 1.4511(14) S(1)-C(1) 1.816(2) O(2)-As(1) 2.6636(13) C(1)-F(2) 1.332(2) C(1)-F(1) 1.336(2) C(1)-F(3) 1.337(2) As(1)-N(5) 1.8271(13) As(1)-N(2) 1.8377(14) N(2)-C(3) 1.346(2) N(2)-C(6) 1.450(2) C(3)-C(4) 1.362(2) C(4)-N(5) 1.352(2) N(5)-C(15) 1.458(2) C(6)-C(11) 1.394(3) C(6)-C(7) 1.402(2) C(7)-C(8) 1.386(2) C(7)-C(14) 1.505(3) C(8)-C(9) 1.383(3) C(9)-C(10) 1.395(2) C(9)-C(13) 1.511(2) C(10)-C(11) 1.380(2) C(11)-C(12) 1.520(2) C(15)-C(20) 1.393(2) C(15)-C(16) 1.393(2) C(16)-C(17) 1.397(2) C(16)-C(23) 1.502(3) C(17)-C(18) 1.391(3) C(18)-C(19) 1.391(2) C(18)-C(22) 1.511(2) C(19)-C(20) 1.391(2) C(20)-C(21) 1.506(2) C(2E)-C(1E) 1.461(7) C(1E)-O(1E) 1.408(6) O(1E)-C(3E) 1.431(7) C(3E)-C(4E) 1.556(7) O(3)-S(1)-O(1) 115.87(9) O(3)-S(1)-O(2) 114.77(8) O(1)-S(1)-O(2) 114.06(8) O(3)-S(1)-C(1) 104.25(8) O(1)-S(1)-C(1) 103.84(8) O(2)-S(1)-C(1) 101.68(9) S(1)-O(2)-As(1) 114.19(8) F(2)-C(1)-F(1) 107.49(17) F(2)-C(1)-F(3) 108.00(15) F(1)-C(1)-F(3) 106.90(16) F(2)-C(1)-S(1) 111.40(13)

8. Kristallographischer Anhang
204
F(1)-C(1)-S(1) 111.38(13) F(3)-C(1)-S(1) 111.45(14) N(5)-As(1)-N(2) 83.94(6) N(5)-As(1)-O(2) 87.35(5) N(2)-As(1)-O(2) 171.21(5) C(3)-N(2)-C(6) 121.47(14) C(3)-N(2)-As(1) 114.56(12) C(6)-N(2)-As(1) 123.58(10) N(2)-C(3)-C(4) 113.12(15) N(5)-C(4)-C(3) 114.49(15) C(4)-N(5)-C(15) 117.65(13) C(4)-N(5)-As(1) 113.88(11) C(15)-N(5)-As(1) 128.37(10) C(11)-C(6)-C(7) 122.49(16) C(11)-C(6)-N(2) 118.03(15) C(7)-C(6)-N(2) 119.46(16) C(8)-C(7)-C(6) 117.10(17) C(8)-C(7)-C(14) 121.26(16) C(6)-C(7)-C(14) 121.64(16) C(9)-C(8)-C(7) 122.43(16) C(8)-C(9)-C(10) 118.26(16) C(8)-C(9)-C(13) 121.04(16) C(10)-C(9)-C(13) 120.69(18) C(11)-C(10)-C(9) 122.08(18) C(10)-C(11)-C(6) 117.65(16) C(10)-C(11)-C(12) 121.13(18) C(6)-C(11)-C(12) 121.22(17) C(20)-C(15)-C(16) 123.32(15) C(20)-C(15)-N(5) 118.85(14) C(16)-C(15)-N(5) 117.71(16) C(15)-C(16)-C(17) 117.06(17) C(15)-C(16)-C(23) 122.41(15) C(17)-C(16)-C(23) 120.52(15) C(18)-C(17)-C(16) 121.84(16) C(17)-C(18)-C(19) 118.56(15) C(17)-C(18)-C(22) 120.64(16) C(19)-C(18)-C(22) 120.76(17) C(18)-C(19)-C(20) 122.07(17) C(19)-C(20)-C(15) 117.10(15) C(19)-C(20)-C(21) 120.76(17) C(15)-C(20)-C(21) 122.15(15) O(1E)-C(1E)-C(2E) 112.7(6) C(1E)-O(1E)-C(3E) 110.8(2) O(1E)-C(3E)-C(4E) 104.6(5) _____________________________________________________________ Table 4. Torsion angles [deg] for gudat78. ________________________________________________________________ O(3)-S(1)-O(2)-As(1) 56.58(9) O(1)-S(1)-O(2)-As(1) -80.48(9) C(1)-S(1)-O(2)-As(1) 168.43(7) O(3)-S(1)-C(1)-F(2) 60.49(15) O(1)-S(1)-C(1)-F(2) -177.76(13) O(2)-S(1)-C(1)-F(2) -59.10(15) O(3)-S(1)-C(1)-F(1) -179.51(13) O(1)-S(1)-C(1)-F(1) -57.76(15) O(2)-S(1)-C(1)-F(1) 60.90(15) O(3)-S(1)-C(1)-F(3) -60.21(16) O(1)-S(1)-C(1)-F(3) 61.53(15) O(2)-S(1)-C(1)-F(3) -179.81(13) S(1)-O(2)-As(1)-N(5) -114.45(8)

8. Kristallographischer Anhang
205
S(1)-O(2)-As(1)-N(2) -106.3(4) N(5)-As(1)-N(2)-C(3) -0.02(13) O(2)-As(1)-N(2)-C(3) -8.2(5) N(5)-As(1)-N(2)-C(6) 172.81(14) O(2)-As(1)-N(2)-C(6) 164.7(3) C(6)-N(2)-C(3)-C(4) -173.29(16) As(1)-N(2)-C(3)-C(4) -0.3(2) N(2)-C(3)-C(4)-N(5) 0.6(2) C(3)-C(4)-N(5)-C(15) 176.00(16) C(3)-C(4)-N(5)-As(1) -0.6(2) N(2)-As(1)-N(5)-C(4) 0.33(13) O(2)-As(1)-N(5)-C(4) 179.09(13) N(2)-As(1)-N(5)-C(15) -175.81(15) O(2)-As(1)-N(5)-C(15) 2.95(14) C(3)-N(2)-C(6)-C(11) 82.5(2) As(1)-N(2)-C(6)-C(11) -89.89(17) C(3)-N(2)-C(6)-C(7) -95.9(2) As(1)-N(2)-C(6)-C(7) 91.77(17) C(11)-C(6)-C(7)-C(8) 0.1(2) N(2)-C(6)-C(7)-C(8) 178.39(14) C(11)-C(6)-C(7)-C(14) 179.85(17) N(2)-C(6)-C(7)-C(14) -1.9(2) C(6)-C(7)-C(8)-C(9) -0.7(2) C(14)-C(7)-C(8)-C(9) 179.63(17) C(7)-C(8)-C(9)-C(10) 0.8(3) C(7)-C(8)-C(9)-C(13) 179.71(16) C(8)-C(9)-C(10)-C(11) -0.4(3) C(13)-C(9)-C(10)-C(11) -179.33(16) C(9)-C(10)-C(11)-C(6) -0.1(3) C(9)-C(10)-C(11)-C(12) -179.56(17) C(7)-C(6)-C(11)-C(10) 0.2(3) N(2)-C(6)-C(11)-C(10) -178.07(14) C(7)-C(6)-C(11)-C(12) 179.71(16) N(2)-C(6)-C(11)-C(12) 1.4(2) C(4)-N(5)-C(15)-C(20) 96.17(19) As(1)-N(5)-C(15)-C(20) -87.82(18) C(4)-N(5)-C(15)-C(16) -80.0(2) As(1)-N(5)-C(15)-C(16) 96.05(16) C(20)-C(15)-C(16)-C(17) -2.1(2) N(5)-C(15)-C(16)-C(17) 173.86(13) C(20)-C(15)-C(16)-C(23) 178.83(15) N(5)-C(15)-C(16)-C(23) -5.2(2) C(15)-C(16)-C(17)-C(18) -0.1(2) C(23)-C(16)-C(17)-C(18) 179.04(15) C(16)-C(17)-C(18)-C(19) 2.0(2) C(16)-C(17)-C(18)-C(22) -175.59(15) C(17)-C(18)-C(19)-C(20) -1.9(3) C(22)-C(18)-C(19)-C(20) 175.68(15) C(18)-C(19)-C(20)-C(15) -0.1(2) C(18)-C(19)-C(20)-C(21) 179.54(16) C(16)-C(15)-C(20)-C(19) 2.2(2) N(5)-C(15)-C(20)-C(19) -173.73(13) C(16)-C(15)-C(20)-C(21) -177.47(16) N(5)-C(15)-C(20)-C(21) 6.6(2) C(2E)-C(1E)-O(1E)-C(3E) 170.9(5) C(1E)-O(1E)-C(3E)-C(4E) -167.7(5) ________________________________________________________________

8. Kristallographischer Anhang
206
Table 5. Hydrogen bonds for gudat78 [A and deg.]. ____________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(3)-H(3)...O(1)#1 0.95 2.43 3.018(2) 119.9 C(4)-H(4)...O(1)#1 0.95 2.54 3.072(2) 115.8 C(23)-H(23C)...O(3) 0.98 2.53 3.263(2) 131.3 ____________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 x-1/2,-y+1/2,z-1/2 Table 1. Crystal data and structure refinement for 2-Chloro-1,3-di-tert-butyl-1,3,2-diazarsolen (12b[Cl])
Identification code gudat83m Empirical formula C10 H20 As Cl N2 Formula weight 278.65 Temperature 123(2) K Wavelength 0.71073 A Crystal system, space group Orthorhombic, Pbca (No.61) Unit cell dimensions a = 11.6481(2) A alpha = 90 deg. b = 12.0818(2) A beta = 90 deg. c = 18.7968(3) A gamma = 90 deg. Volume 2645.27(8) A^3 Z, Calculated density 8, 1.399 Mg/m^3 Absorption coefficient 2.741 mm^-1 F(000) 1152 Crystal size 0.50 x 0.25 x 0.15 mm

8. Kristallographischer Anhang
207
Diffractometer Nonius KappaCCD Theta range for data collection 3.26 to 27.47 deg. Limiting indices -15<=h<=15, -15<=k<=15, -24<=l<=24 Reflections collected / unique 22019 / 2978 [R(int) = 0.0426] Completeness to theta = 27.47 98.3 % Absorption correction Empirical from multile refl. Max. and min. transmission 0.46567 and 0.38736 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 2978 / 0 / 127 Goodness-of-fit on F^2 1.043 Final R indices [I>2sigma(I)] R1 = 0.0223, wR2 = 0.0583 R indices (all data) R1 = 0.0280, wR2 = 0.0596 Largest diff. peak and hole 0.285 and -0.643 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat83. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ As(1) 5109(1) 6192(1) 915(1) 18(1) Cl(1) 3565(1) 4773(1) 1410(1) 32(1) N(2) 4282(1) 7419(1) 720(1) 21(1) C(3) 4299(1) 8183(1) 1268(1) 24(1) C(4) 5004(1) 7906(1) 1808(1) 23(1) N(5) 5556(1) 6904(1) 1712(1) 19(1) C(6) 3471(1) 7511(1) 105(1) 26(1) C(7) 2255(1) 7266(2) 370(1) 33(1) C(8) 3817(2) 6646(2) -448(1) 36(1) C(9) 3558(2) 8668(2) -209(1) 41(1) C(10) 6263(1) 6386(1) 2286(1) 21(1) C(11) 5511(2) 6224(1) 2943(1) 30(1) C(12) 7279(1) 7141(1) 2454(1) 29(1) C(13) 6698(1) 5267(1) 2025(1) 26(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat83. _____________________________________________________________ As(1)-N(5) 1.8045(12) As(1)-N(2) 1.8057(12) As(1)-Cl(1) 2.6527(4) N(2)-C(3) 1.382(2) N(2)-C(6) 1.4975(19)

8. Kristallographischer Anhang
208
C(3)-C(4) 1.348(2) C(4)-N(5) 1.3831(19) N(5)-C(10) 1.4943(19) C(6)-C(9) 1.520(2) C(6)-C(8) 1.528(2) C(6)-C(7) 1.529(2) C(10)-C(13) 1.525(2) C(10)-C(12) 1.527(2) C(10)-C(11) 1.527(2) N(5)-As(1)-N(2) 86.03(5) N(5)-As(1)-Cl(1) 102.28(4) N(2)-As(1)-Cl(1) 103.92(4) C(3)-N(2)-C(6) 122.30(12) C(3)-N(2)-As(1) 112.92(10) C(6)-N(2)-As(1) 123.65(10) C(4)-C(3)-N(2) 113.85(14) C(3)-C(4)-N(5) 113.69(14) C(4)-N(5)-C(10) 121.88(12) C(4)-N(5)-As(1) 113.06(10) C(10)-N(5)-As(1) 124.00(9) N(2)-C(6)-C(9) 108.99(14) N(2)-C(6)-C(8) 107.91(12) C(9)-C(6)-C(8) 110.29(14) N(2)-C(6)-C(7) 108.60(12) C(9)-C(6)-C(7) 111.49(14) C(8)-C(6)-C(7) 109.48(14) N(5)-C(10)-C(13) 108.73(12) N(5)-C(10)-C(12) 109.07(12) C(13)-C(10)-C(12) 109.82(13) N(5)-C(10)-C(11) 108.75(12) C(13)-C(10)-C(11) 109.72(13) C(12)-C(10)-C(11) 110.71(13) _____________________________________________________________ Table 4. Torsion angles [deg] for gudat83. ________________________________________________________________
N(5)-As(1)-N(2)-C(3) -6.17(11) Cl(1)-As(1)-N(2)-C(3) 95.51(10) N(5)-As(1)-N(2)-C(6) -174.22(12) l(1)-As(1)-N(2)-C(6) -72.54(12) C(6)-N(2)-C(3)-C(4) 173.74(14) As(1)-N(2)-C(3)-C(4) 5.51(17) N(2)-C(3)-C(4)-N(5) -1.0(2) C(3)-C(4)-N(5)-C(10) -172.57(13) C(3)-C(4)-N(5)-As(1) -3.90(17) N(2)-As(1)-N(5)-C(4) 5.61(11) Cl(1)-As(1)-N(5)-C(4) -97.78(10) N(2)-As(1)-N(5)-C(10) 174.01(12) Cl(1)-As(1)-N(5)-C(10) 70.61(11) C(3)-N(2)-C(6)-C(9) 51.38(19) As(1)-N(2)-C(6)-C(9) -141.66(12) C(3)-N(2)-C(6)-C(8) 171.15(14) As(1)-N(2)-C(6)-C(8) -21.88(18) C(3)-N(2)-C(6)-C(7) -70.26(18) As(1)-N(2)-C(6)-C(7) 96.70(14) C(4)-N(5)-C(10)-C(13) 177.12(13) As(1)-N(5)-C(10)-C(13) 9.71(17) C(4)-N(5)-C(10)-C(12) -63.15(18) As(1)-N(5)-C(10)-C(12) 129.44(11) C(4)-N(5)-C(10)-C(11) 57.69(17)

8. Kristallographischer Anhang
209
As(1)-N(5)-C(10)-C(11) -109.72(13) ________________________________________________________________
Table 5. Hydrogen bonds for gudat83 [A and deg.]. _________________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(7)-H(7C)...Cl(1)#1 0.98 2.79 3.7290(17) 161.8 C(12)-H(12B)...Cl(1)#2 0.98 3.00 3.8720(17) 149.3 C(13)-H(13C)...Cl(1)#2 0.98 2.78 3.7076(16) 157.8 _________________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1/2,y+1/2,z #2 x+1/2,y,-z+1/2
Table 1. Crystal data and structure refinement for 79
Identification code gudat70_m Empirical formula C24 H24 As Co N2 O4 Formula weight 538.30 Temperature 123(2) K Wavelength 0.71073 A Crystal system, space group Triclinic, P-1 (No.2) Unit cell dimensions a = 10.6396(3) A alpha = 112.236(2) deg. b = 10.7829(3) A beta = 96.521(2) deg. c = 11.2066(4) A gamma = 93.104(2) deg. Volume 1175.76(6) A^3 Z, Calculated density 2, 1.521 Mg/m^3

8. Kristallographischer Anhang
210
Absorption coefficient 2.159 mm^-1 F(000) 548 Crystal size 0.30 x 0.20 x 0.10 mm Theta range for data collection 2.97 to 27.48 deg. Limiting indices -13<=h<=13, -13<=k<=13, -14<=l<=14 Reflections collected / unique 22309 / 5252 [R(int) = 0.0736] Completeness to theta = 27.48 97.5 % Absorption correction Empirical from multiple refl. Max. and min. transmission 0.6988 and 0.5668 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 5252 / 0 / 295 Goodness-of-fit on F^2 0.907 Final R indices [I>2sigma(I)] R1 = 0.0322, wR2 = 0.0584 R indices (all data) R1 = 0.0592, wR2 = 0.0626 Largest diff. peak and hole 0.561 and -0.503 e.A^-3 Table 2. Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for gudat70m. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________ x y z U(eq) ________________________________________________________________ As(1) 4135(1) 2968(1) 3508(1) 20(1) Co(1) 1866(1) 2467(1) 1923(1) 25(1) C(1A) 353(2) 2071(3) 847(3) 35(1) O(1A) -575(2) 1818(2) 167(2) 50(1) C(1B) 2371(2) 4075(3) 1920(3) 29(1) O(1B) 2672(2) 5110(2) 1920(2) 43(1) C(1C) 1242(2) 2565(2) 3376(3) 28(1) O(1C) 761(2) 2651(2) 4271(2) 37(1) C(1D) 2610(2) 1038(3) 981(3) 27(1) O(1D) 3059(2) 121(2) 333(2) 36(1) N(2) 4189(2) 1464(2) 3915(2) 20(1) C(3) 5034(2) 597(2) 3258(2) 23(1) C(4) 5698(2) 1058(2) 2548(2) 24(1) N(5) 5433(2) 2321(2) 2574(2) 21(1) C(6) 3419(2) 1172(2) 4764(2) 20(1) C(7) 2550(2) -4(2) 4295(2) 23(1) C(8) 1817(2) -220(3) 5161(3) 27(1) C(9) 1913(2) 664(3) 6454(3) 26(1) C(10) 2782(2) 1797(2) 6882(2) 25(1) C(11) 3542(2) 2077(2) 6061(2) 21(1) C(12) 4510(2) 3308(2) 6617(2) 27(1) C(13) 1126(2) 373(3) 7383(3) 37(1)

8. Kristallographischer Anhang
211
C(14) 2400(2) -1024(3) 2906(3) 33(1) C(15) 6374(2) 3172(2) 2320(2) 21(1) C(16) 7433(2) 3792(2) 3283(3) 26(1) C(17) 8346(2) 4586(2) 3020(3) 29(1) C(18) 8231(2) 4800(3) 1872(3) 31(1) C(19) 7185(2) 4170(2) 947(3) 29(1) C(20) 6243(2) 3352(2) 1146(3) 26(1) C(21) 5133(2) 2645(3) 87(3) 34(1) C(22) 9253(2) 5686(3) 1632(3) 44(1) C(23) 7578(2) 3630(3) 4560(3) 32(1) ________________________________________________________________ Table 3. Bond lengths [A] and angles [deg] for gudat70m. _____________________________________________________________ As(1)-N(5) 1.8353(17) As(1)-N(2) 1.8459(19) As(1)-Co(1) 2.7263(4) Co(1)-C(1B) 1.789(3) Co(1)-C(1D) 1.790(3) Co(1)-C(1C) 1.796(3) Co(1)-C(1A) 1.822(3) C(1A)-O(1A) 1.128(3) C(1B)-O(1B) 1.144(3) C(1C)-O(1C) 1.153(3) C(1D)-O(1D) 1.149(3) N(2)-C(3) 1.394(3) N(2)-C(6) 1.432(3) C(3)-C(4) 1.330(3) C(4)-N(5) 1.396(3) N(5)-C(15) 1.445(3) C(6)-C(11) 1.397(3) C(6)-C(7) 1.413(3) C(7)-C(8) 1.389(3) C(7)-C(14) 1.512(3) C(8)-C(9) 1.389(3) C(9)-C(10) 1.382(3) C(9)-C(13) 1.517(3) C(10)-C(11) 1.395(3) C(11)-C(12) 1.513(3) C(15)-C(20) 1.394(3) C(15)-C(16) 1.406(3) C(16)-C(17) 1.390(3) C(16)-C(23) 1.498(3) C(17)-C(18) 1.384(4) C(18)-C(19) 1.378(3) C(18)-C(22) 1.521(3) C(19)-C(20) 1.392(3) C(19)-H(19) 0.9500 C(20)-C(21) 1.511(3) N(5)-As(1)-N(2) 85.40(8) N(5)-As(1)-Co(1) 111.59(6) N(2)-As(1)-Co(1) 103.15(6) C(1B)-Co(1)-C(1D) 118.28(11) C(1B)-Co(1)-C(1C) 113.64(11) C(1D)-Co(1)-C(1C) 121.92(11) C(1B)-Co(1)-C(1A) 100.13(12) C(1D)-Co(1)-C(1A) 97.37(11) C(1C)-Co(1)-C(1A) 97.51(11) C(1B)-Co(1)-As(1) 81.59(8) C(1D)-Co(1)-As(1) 80.99(8)

8. Kristallographischer Anhang
212
C(1C)-Co(1)-As(1) 82.54(7) C(1A)-Co(1)-As(1) 178.07(9) O(1A)-C(1A)-Co(1) 179.1(3) O(1B)-C(1B)-Co(1) 178.8(2) O(1C)-C(1C)-Co(1) 175.1(2) O(1D)-C(1D)-Co(1) 177.3(2) C(3)-N(2)-C(6) 122.94(19) C(3)-N(2)-As(1) 112.42(15) C(6)-N(2)-As(1) 124.59(14) C(4)-C(3)-N(2) 114.4(2) C(3)-C(4)-N(5) 115.1(2) C(4)-N(5)-C(15) 120.48(17) C(4)-N(5)-As(1) 112.36(15) C(15)-N(5)-As(1) 123.43(15) C(11)-C(6)-C(7) 120.9(2) C(11)-C(6)-N(2) 118.6(2) C(7)-C(6)-N(2) 120.6(2) C(8)-C(7)-C(6) 117.8(2) C(8)-C(7)-C(14) 119.4(2) C(6)-C(7)-C(14) 122.8(2) C(7)-C(8)-C(9) 122.6(2) C(10)-C(9)-C(8) 117.9(2) C(10)-C(9)-C(13) 120.8(2) C(8)-C(9)-C(13) 121.2(2) C(9)-C(10)-C(11) 122.3(2) C(10)-C(11)-C(6) 118.5(2) C(10)-C(11)-C(12) 119.1(2) C(6)-C(11)-C(12) 122.4(2) C(20)-C(15)-C(16) 121.2(2) C(20)-C(15)-N(5) 120.8(2) C(16)-C(15)-N(5) 118.0(2) C(17)-C(16)-C(15) 117.6(2) C(17)-C(16)-C(23) 120.5(2) C(15)-C(16)-C(23) 121.9(2) C(18)-C(17)-C(16) 122.5(2) C(19)-C(18)-C(17) 118.3(2) C(19)-C(18)-C(22) 121.1(3) C(17)-C(18)-C(22) 120.5(2) C(18)-C(19)-C(20) 122.0(3) C(19)-C(20)-C(15) 118.4(2) C(19)-C(20)-C(21) 120.2(2) C(15)-C(20)-C(21) 121.4(2) _____________________________________________________________ Symmetry transformations used to generate equivalent atoms: ________________________________________________________________ Table 4. Torsion angles [deg] for gudat70m. ________________________________________________________________ N(5)-As(1)-Co(1)-C(1B) -90.00(10) N(2)-As(1)-Co(1)-C(1B) 179.77(10) N(5)-As(1)-Co(1)-C(1D) 30.55(10) N(2)-As(1)-Co(1)-C(1D) -59.68(10) N(5)-As(1)-Co(1)-C(1C) 154.65(10) N(2)-As(1)-Co(1)-C(1C) 64.42(10) N(5)-As(1)-Co(1)-C(1A) 63(3) N(2)-As(1)-Co(1)-C(1A) -27(3) C(1B)-Co(1)-C(1A)-O(1A) 87(16) C(1D)-Co(1)-C(1A)-O(1A) -33(17) C(1C)-Co(1)-C(1A)-O(1A) -157(16)

8. Kristallographischer Anhang
213
As(1)-Co(1)-C(1A)-O(1A) -65(18) C(1D)-Co(1)-C(1B)-O(1B) 147(11) C(1C)-Co(1)-C(1B)-O(1B) -60(11) C(1A)-Co(1)-C(1B)-O(1B) 43(11) As(1)-Co(1)-C(1B)-O(1B) -138(11) C(1B)-Co(1)-C(1C)-O(1C) 79(3) C(1D)-Co(1)-C(1C)-O(1C) -129(3) C(1A)-Co(1)-C(1C)-O(1C) -26(3) As(1)-Co(1)-C(1C)-O(1C) 156(3) C(1B)-Co(1)-C(1D)-O(1D) -76(5) C(1C)-Co(1)-C(1D)-O(1D) 133(5) C(1A)-Co(1)-C(1D)-O(1D) 29(5) As(1)-Co(1)-C(1D)-O(1D) -152(5) N(5)-As(1)-N(2)-C(3) -5.49(15) Co(1)-As(1)-N(2)-C(3) 105.63(14) N(5)-As(1)-N(2)-C(6) 177.05(19) Co(1)-As(1)-N(2)-C(6) -71.83(18) C(6)-N(2)-C(3)-C(4) -177.9(2) As(1)-N(2)-C(3)-C(4) 4.6(3) N(2)-C(3)-C(4)-N(5) -0.3(3) C(3)-C(4)-N(5)-C(15) 154.9(2) C(3)-C(4)-N(5)-As(1) -4.1(3) N(2)-As(1)-N(5)-C(4) 5.29(16) Co(1)-As(1)-N(5)-C(4) -97.04(15) N(2)-As(1)-N(5)-C(15) -152.95(19) Co(1)-As(1)-N(5)-C(15) 104.72(18) C(3)-N(2)-C(6)-C(11) 121.5(2) As(1)-N(2)-C(6)-C(11) -61.3(3) C(3)-N(2)-C(6)-C(7) -58.8(3) As(1)-N(2)-C(6)-C(7) 118.4(2) C(11)-C(6)-C(7)-C(8) 0.6(3) N(2)-C(6)-C(7)-C(8) -179.1(2) C(11)-C(6)-C(7)-C(14) -178.9(2) N(2)-C(6)-C(7)-C(14) 1.4(3) C(6)-C(7)-C(8)-C(9) -0.2(3) C(14)-C(7)-C(8)-C(9) 179.3(2) C(7)-C(8)-C(9)-C(10) -0.4(3) C(7)-C(8)-C(9)-C(13) -178.1(2) C(8)-C(9)-C(10)-C(11) 0.7(3) C(13)-C(9)-C(10)-C(11) 178.4(2) C(9)-C(10)-C(11)-C(6) -0.3(3) C(9)-C(10)-C(11)-C(12) -177.5(2) C(7)-C(6)-C(11)-C(10) -0.3(3) N(2)-C(6)-C(11)-C(10) 179.4(2) C(7)-C(6)-C(11)-C(12) 176.7(2) N(2)-C(6)-C(11)-C(12) -3.6(3) C(4)-N(5)-C(15)-C(20) 106.3(3) As(1)-N(5)-C(15)-C(20) -97.1(2) C(4)-N(5)-C(15)-C(16) -72.5(3) As(1)-N(5)-C(15)-C(16) 84.0(2) C(20)-C(15)-C(16)-C(17) -0.2(3) N(5)-C(15)-C(16)-C(17) 178.7(2) C(20)-C(15)-C(16)-C(23) 178.9(2) N(5)-C(15)-C(16)-C(23) -2.2(3) C(15)-C(16)-C(17)-C(18) 1.2(4) C(23)-C(16)-C(17)-C(18) -177.8(2) C(16)-C(17)-C(18)-C(19) -1.7(4) C(16)-C(17)-C(18)-C(22) 179.6(2) C(17)-C(18)-C(19)-C(20) 1.0(4) C(22)-C(18)-C(19)-C(20) 179.7(2) C(18)-C(19)-C(20)-C(15) 0.0(4) C(18)-C(19)-C(20)-C(21) -178.0(2) C(16)-C(15)-C(20)-C(19) -0.4(4)

8. Kristallographischer Anhang
214
N(5)-C(15)-C(20)-C(19) -179.3(2) C(16)-C(15)-C(20)-C(21) 177.6(2) N(5)-C(15)-C(20)-C(21) -1.3(3) ________________________________________________________________ Symmetry transformations used to generate equivalent atoms: Table 5. Hydrogen bonds for gudat70m [A and deg.]. ____________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(19)-H(19)...O(1B)#1 0.95 2.66 3.596(3) 169.0 C(4)-H(4)...O(1D)#2 0.95 2.67 3.441(3) 138.2 ____________________________________________________________________ Symmetry transformations used to generate equivalent atoms: #1 -x+1,-y+1,-z #2 -x+1,-y,-z

Fotographische Aufnahmen
Verbindung 7c
Verbindung 13b[Cl]
Verbindung 39c
Verbindung 39b[LiCl]


Lebenslauf
Lebenslauf
Zur Person
Name: Timo Gans-Eichler
Geburtsdatum: 29.11.1973 in Neuwied
Konfession: evangelisch
Familienstand: ledig
Schulausbildung
1980-1984 Grundschule Anhausen
1984-1993 Besuch des staatl. Rhein-Wied-Gymnasiums in Neuwied mit Abschluß der allgemeinen Hochschulreife am 18.6.1993
Zivildienst
1993-1994 15 Monate Zivildienst in der Behindertenwerkstatt Engers bei der Josephs Gesellschaft Köln
Hochschulausbildung
14.10.1994 Beginn des Hochschulstudium an der Unversität Bonn
Fachrichtung: Chemie
Oktober 1996 Erlangung des Vordiploms in Chemie
August 1999 Beginn der Diplomarbeit bei Prof. Dr. D. Gudat am Institut für Anorganische Chemie der Universität Bonn mit dem Thema: „Versuche zur Darstellung eines 1,3,2-Diazastannolidens“
1.9. 2000 Erlangung des Grades Diplom-Chemiker mit dem Nebenfach Biochemie
2000-2004 Dissertation bei Prof. Dr. D. Gudat in Bonn
Berufstätigkeit
01.10.97- 30.11.97 studentische Hilfskraft am Agrikulturchemischen Institut in Bonn
01.04.98-30.06.98 studentische Hilfskraft am Agrikulturchemischen Institut in Bonn
02.08.00-31.08.00 studentische Hilfskraft am Anorganisch-chemischen Institut/Bonn


01.09.00-31.01.03 wissenschaftliche Hilfskraft am Anorganisch-chemischen Institut in Bonn
01.02.03-31.03.03 wissenschaftlicher Mitarbeiter am Anorganisch-chemischen Institut in Bonn
01.04.03-31.01.04 wissenschaftliche Hilfskraft am Anorganisch-chemischen Institut in Bonn
Fortbildungen
2000 Teilnahme an der International Conference on Inorganic Ring Systems in Saarbrücken
2001 Posterpräsentation auf der GDCh Jahrestagung Chemie in Würzburg
2002 Vortrag auf dem Regional Seminar of Ph.D.-Students on Organo-metallic and Coordination Chemistry in Bad Kösen mit dem Titel:
„On the Heaviest Homologues of „Arduengo“ Carbenes with Tin and Lead”
Publikationen:
T. Gans-Eichler, D. Gudat, M. Nieger, Angew. Chem. 2002, 114, 11, 1966
D. Gudat, A.Haghverdi, T. Gans-Eichler, M. Nieger, Posphorous, Sulfur, and Silicon, 2002,
1177,1637
Timo Gans-Eichler

Erklärung Ich erkläre hiermit, dass ich die vorliegende Arbeit selbstständig und ohne Benutzung anderer als der angegbenen Hilfsmittel angefertigt habe ; die aus aus fremden Werken wörtlich oder sinngemäß übernommenen Gedanken sind unter Angabe der Quellen gekennzeichnet. Ich versichere, dass ich bisher keine Arbeit mit gleichem oder ähnlichem Thema bei einer Prüfungsbehörde oder anderen Hochschule vorgelegt habe. Bonn den 1.3.2004 …………………………………………………….. Ort, Datum Timo Gans-Eichler








