

Aus dem Institut für Pathologie
der Ruhr-Universität Bochum
an den Berufsgenossenschaftlichen Universitätskliniken Bergmannsheil
Direktorin: Prof. Dr. med. A. Tannapfel
Epitheloides Sarkom, Klarzellsarkom und
Synovialsarkom:
Morphologische Befunde und klinisch-pathologische Korrelationen an 167 Tumoren
Inaugural-Dissertation
zur Erlangung des Doktorgrades der Medizin
einer Hohen Medizinischen Fakultät
der Ruhr-Universität Bochum
vorgelegt von
Denise Rosenberger
aus Bielefeld
2007

Dekan: Prof. Dr. med. G. Muhr
Referent: Prof. Dr. med. C. Kuhnen
Koreferent: Pro. Dr. med. Voigtmann
Tag der mündlichen Prüfung: 24.6.2008

3
1 EINLEITUNG.......................................................................................... 9
1.1 Allgemeines............................................................................................................................. 9 1.1.2 Definition der Weichgewebstumoren ....................................................................................... 9 1.1.3 Ätiologie und Prävalenz ......................................................................................................... 10 1.1.4 Lokalisation der Tumoren ...................................................................................................... 11 1.1.5 Tumorgrößen .......................................................................................................................... 11 1.1.6 Alter und Geschlechtsverteilung ............................................................................................ 11 1.1.7 Krankheitssymptome.............................................................................................................. 12
1.2 Klinisch-pathologische Parameter...................................................................................... 12 1.2.1 Tumorgrading und Stadieneinteilung, prognostische Parameter ............................................ 12 1.2.2 Therapieprinzipien der Weichgewebstumore ......................................................................... 17
1.3 Tumorentitäten im Speziellen: Epitheloides Sarkom, Klarzellsarkom und
Synovialsarkom .................................................................................................................................. 18 1.3.1 Epitheloides Sarkom............................................................................................................... 18 1.3.2 Klarzellsarkom ....................................................................................................................... 21 1.3.4 Synovialsarkom...................................................................................................................... 25
1.4 Immunhistochemie............................................................................................................... 30 1.4.1 MIB-1 ..................................................................................................................................... 30 1.4.2 Epidermal Growth Factor Receptor (EGFR) .......................................................................... 30
1.5 Zielsetzung und Fragestellung ............................................................................................ 35
2 MATERIAL UND METHODEN ............................................................. 36
2.1 Material ................................................................................................................................ 36
2.2 Untersuchungsmethoden..................................................................................................... 36 2.2.1 Datenerhebung........................................................................................................................ 36 2.2.2 Konventionelle Histologie...................................................................................................... 37 2.2.3 Immunhistochemie ................................................................................................................. 37 2.2.4 Auswertung ............................................................................................................................ 39
3 ERGEBNISSE ...................................................................................... 40
3.1 Geschlechtsverteilung bei den Primärtumoren................................................................. 40
3.2 Altersverteilung und Geschlechtsverteilung der Tumore ................................................ 40

4
3.2.1 Alters- und Geschlechtsverteilung: Epitheloides Sarkom ...................................................... 40 3.2.2 Alters- und Geschlechtsverteilung: Klarzellsarkom ............................................................... 40 3.2.3 Alters- und Geschlechtsverteilung: Synovialsarkom.............................................................. 41
3.3 Tumorgrößen ....................................................................................................................... 41 3.3.1 Tumorgrößen: Epitheloides Sarkom....................................................................................... 41 3.3.2 Tumorgrößen: Klarzellsarkome.............................................................................................. 41 3.3.3 Tumorgrößen: Synovialsarkom .............................................................................................. 42
3.4 Lokalisation der Tumoren .................................................................................................. 43 3.4.1 Epitheloides Sarkom............................................................................................................... 43 3.4.2 Klarzellsarkom ....................................................................................................................... 44 3.4.3 Synovialsarkom................................................................................................................. 45
3.5 Malignitätsgrad.................................................................................................................... 46 3.5.1 Grading: Epitheloides Sarkom................................................................................................ 46 3.5.2 Grading: Klarzellsarkom ........................................................................................................ 46 3.5.3 Grading: Synovialsarkom....................................................................................................... 47
3.6 Krankheitsverlauf / Lokalrezidive und Metastasen.......................................................... 47 3.6.1 Epitheloides Sarkom............................................................................................................... 47 3.6.2 Klarzellsarkom ....................................................................................................................... 47 3.6.3 Synovialsarkom...................................................................................................................... 48
3.7 Histologisches Erscheinungsbild......................................................................................... 49 3.7.1 Histologisches Erscheinungsbild : Epitheloides Sarkom........................................................ 49 3.7.2 Histologisches Erscheinungsbild: Klarzellsarkom ................................................................. 54 3.7.3 Histologisches Erscheinungsbild: Synovialsarkom................................................................ 58
3.8 MIB-1 Expression ................................................................................................................ 63 3.8.1 MiB-1 Expression in Abhängigkeit von der Tumorgröße ...................................................... 63 3.8.2 MiB-1 Expression in Korrelation zum Tumorgrading............................................................ 63 3.8.3 Tab: 2 Ergebnistabelle: Epitheloides Sarkom......................................................................... 65 3.8.4 Tab 3: Ergebnistabelle: Klarzellsarkom ................................................................................ 66
3.9 EGF-R-Expression............................................................................................................... 67 3.9.1 Epitheloides Sarkom............................................................................................................... 67 3.9.2 Klarzellsarkom ....................................................................................................................... 67 3.9.3 Synovialsarkom...................................................................................................................... 67
4 DISKUSSION ....................................................................................... 70
4.1 Geschlechtsverteilung.......................................................................................................... 70

5
4.1.1 Epitheloides Sarkom............................................................................................................... 70 4.1.2 Klarzellsarkom ....................................................................................................................... 70 4.1.3 Synovialsarkom...................................................................................................................... 70
4.2 Altersverteilung.................................................................................................................... 71 4.2.1 Epitheloides Sarkom............................................................................................................... 71 4.2.2 Klarzellsarkom ....................................................................................................................... 71 4.2.3 Synovialsarkom...................................................................................................................... 72
4.3 Tumorgrößen ....................................................................................................................... 72 4.3.1 Epitheloides Sarkom............................................................................................................... 72 4.3.2 Klarzellsarkom ....................................................................................................................... 72 4.3.3 Synovialsarkom...................................................................................................................... 73
4.4 Lokalisation der Tumoren .................................................................................................. 73 4.4.1 Epitheloides Sarkom............................................................................................................... 73 4.4.2 Klarzellsarkom ....................................................................................................................... 74 4.4.3 Synovialsarkom...................................................................................................................... 74
4.5 Histopathologisches Erscheinungsbild, Subtypisierung und Grading ............................ 74 4.5.1 Epitheloides Sarkom............................................................................................................... 74 4.5.2 Klarzellsarkom ....................................................................................................................... 75 4.5.3 Synovialsarkom...................................................................................................................... 75
4.6 Krankheitsverlauf / Lokalrezidive und Metastasen.......................................................... 76 4.6.1 Epitheloides Sarkom............................................................................................................... 77 4.6.2 Klarzellsarkom ....................................................................................................................... 77 4.6.3 Synovialsarkom...................................................................................................................... 78 4.6.4 MiB-1-Expression .................................................................................................................. 79
4.7 EGF-R-Expression in Synovialsarkomen und in epitheloiden Sarkomen ...................... 81
5 ZUSAMMENFASSUNG........................................................................ 82
6 LITERATUR ......................................................................................... 86
DANKSAGUNG LEBENSLAUF

6
Abkürzungsverzeichnis
APAAP: alkalische Phosphatase Anti-Alkalische Phosphatase Bcl-2: menschliches Proto-Oncogen, verhindert Zelltod durch
Apoptose CD-34: Cluster of determination Nr. 34 CK: Zytokeratin EGF-R: Epithelial growth factor-receptor EMA: Epitheliales Membranantigen FISH: Fluoreszenz in situ Hybridisierung FNCLCC: Fèderation Nationale des Centres de Lutte Contre le
Cancer HE: Hämatoxylin-Eosin HMB45: Human Melanoma Black 45 Antikörper
HPF: High Power Field (Hochvergrößerte Mikroskop-Felder,
bezogen auf 40er Objektiv) KI-67: Kiel, Antigen Nr. 67 MiB-1: Made in Borstel, Antikörper Nr. 1 MPNST: Maligner peripherer Nervenscheidentumor NK1C3: monoklonaler Antikörper, der u.a.Melanome markiert
S-100: Kalzium bindendes Protein mit niedrigem
Molekulargewicht
UICC: Union Internationale Contre le Cancer

7
Tabellenverzeichnis
Tabelle 1: EGF-R-Expressionsrate unterschiedlicher Tumore S.34
Tabelle 2: Ergebnisse epitheloide Sarkome S.65
Tabelle 3: Ergebnisse Klarzellsarkome S.66

8
Abbildungsverzeichnis: Abb. 1: EGF-R-Signaltransduktionsweg S. 33
Abb. 2: Geschlechtsverteilung bei den Primärtumoren S. 40
Abb. 3: Tumorlokalisation des epitheloiden Sarkoms S. 43
Abb. 4: Tumorlokalisation des Klarzellsarkoms S. 44
Abb. 5: Tumorlokalisation des biphasischen
Synovialsarkoms S. 45
Abb. 6: Tumorlokalisation des monophasischen
Synovialsarkoms S. 45
Abb. 7: Histologie des epitheloiden Sarkoms (1) S. 50
Abb. 8: Histologie des epitheloiden Sarkoms (2) S. 51
Abb. 9: Histologie des epitheloiden Sarkoms (3) S. 53
Abb. 10: Makroskopisches Erscheinungsbild und
Histologie des Karzellsarkoms (1) S. 55
Abb. 11: Histologie des Klarzellsarkoms (2) S. 57
Abb. 12: Makroskopisches und radiologisches Erscheinungsbild
des Synovialsarkoms S. 60
Abb. 13: Histologie des Synovialsarkoms (1) S. 62
Abb. 14: EGF-R-Expression im Synovialsarkom S. 68
Abb. 15: EGF-R-Expression im Synovialsarkom und
im epitheloiden Sarkom S. 69

9
1 Einleitung
1.1 Allgemeines
Gegenstand der hier vorliegenden Arbeit sind 167 Weichgewebstumore,
davon 30 Klarzellsarkome, 32 epitheloide Sarkome und 105
Synovialsarkome. Zu den Weichgeweben zählen alle nicht-epithelialen
extraskelettalen Gewebe. Die in dieser Arbeit ausgewählten Tumore
zeichnen sich vor allem durch eine enge Assoziation zu Sehnen, Faszien
und Aponeurosen aus, und werden daher zusammengefasst und
vergleichend untersucht.
Das Klarzellsarkom als Entität wurde zuerst 1965 von Enzinger beschrieben
(Enzinger, 1965), das Epitheloidzellsarkom, oder epitheloides Sarkom, wurde
zunächst 1960 von Laskowski, und später im Detail von Enzinger
beschrieben.
Berichte über Synovialsarkome gab es bereits um die Jahrhundertwende
(Stuer et al.,1893), die Bezeichnung „Synovialsarkom“ wurde 1936 von Knox,
aufgrund der Ähnlichkeit zur Synovialis vorgeschlagen.
Die hier untersuchten Tumore entstammen dem „Register für
Gliedmaßentumoren“ des Institutes für Pathologie der Ruhr-Universität
Bochum an den Berufsgenossenschaftlichen Universitätskliniken
Bergmannsheil.
Dieses ist Ende der neunziger Jahre in Kooperation zwischen der Klinik für
Plastische Chirurgie und Schwerbrandverletzte und dem Institut für
Pathologie entstanden. Es umfasst bisher ca. 2056 maligne
Weichgewebstumore bei 1597 Patienten, welche über einen Zeitraum von 15
Jahren registriert worden sind.
1.1.2 Definition der Weichgewebstumoren
Unter diesem Begriff werden alle diejenigen Tumore verstanden, die nicht-
epithelial sind, sondern aus Zellen des extraskelettalen Gewebes,
einschliesslich des peripheren Nervengewebes, (ausgenommen die Tumore

10
des Makrophagen-, hämato-, und lymphopoetischen Systems und des
Stützgewebes) hervorgehen.
In der aktuellen WHO-Klassifikation werden mehr als 140 Subtypen
unterschieden, sie werden aufgrund ihrer Ähnlichkeit zu einem
mesenchymalen Muttergewebe klassifiziert, die unterschiedlichen
Tumorarten weisen dabei erhebliche Varianzen auf. Die histologische
Typisierung ist daher oft schwierig.
1.1.3 Ätiologie und Prävalenz
Es ist im Gegensatz zu den Karzinomen in der Regel keine klar definierte
Vorstufe im Sinne einer Präneoplasie bekannt (Ausnahme z. B. Neurofibrom/
MPNST bei Morbus Recklinghausen).
Ebenso lässt sich bislang kein spezifisches ätiologisches Geschehen
zuorden, eine Traumatische Genese, wie oft vermutet, konnte bisher nicht
bestätigt werden (Traeger et al., 2007).
Bei einigen Weichgewebstumoren wurden chromosomale Translokationen
nachgewiesen, sowie Tumorsuppressorgene (z.B. p53), deren Inaktivierung
an der Entstehung der Tumore beteiligt ist. Andere identifizierte Gene, z.B.
Ki-67, sind nicht nur an der Genese der Tumoren beteiligt, sondern haben
zudem Einfluß auf die Prognose (Hoos et al., 2000, Sreekantaiah, 1994)
Weichgewebstumoren können grundsätzlich in allen Altersstufen
vorkommen, manche Tumoren treten jedoch gehäuft in einer bestimmten
klinischen Konstellation auf. So treten die hier untersuchten Tumoren
vornehmlich im jungen Erwachsenenalter auf.
Insgesamt sind die Weichgewebstumoren mit einer Prävalenz von ca.1%
aller bösartigen Neubildungen im Erwachsenenalter seltene Neoplasien.
Bei Kindern machen die Weichgewebstumore ohne Einbeziehung der
Neuroblastome ca. 6% aller Neoplasien aus, und nehmen so in ihrer
Häufigkeit einen höheren Stellenwert ein.

11
1.1.4 Lokalisation der Tumoren
Es werden zentrale Weichteiltumoren, welche man z.B. im Mediastinum,
Retroperitoneum, Mesenterium oder der Orbita findet, von peripheren
Tumoren, die im Bereich des Kopfes, Rumpfes, der Extremitäten oder des
Halses auftreten unterschieden.
Das Klarzellsarkom tritt gehäuft an der unteren Extremität auf,
das Epitheloide Sarkom an der oberen Extremität,
das Synovialsarkom betrifft meistens die nähere Umgebung der großen
Gelenke, davon bevorzugt das Kniegelenk.
1.1.5 Tumorgrößen
Da die Tumore in der Regel für den Patient indolent sind, fallen sie erst ab
einer bestimmten Größe auf. Die Ausnahme bildet hierbei das
Synovialsarkom, welches häufig schmerzhaft ist.
Die in der Literatur beschriebenen Tumoren haben meistens eine Größe von
0,5 bis 10 cm, es wurden jedoch auch Tumoren von mehr als 20 cm
Durchmesser beschrieben.
Aus mehreren Studien geht hervor, dass eine Größe von mehr als 5 cm
(Russell et al., 1977, Pisters et al. 1996), oder mehr als 10 cm (Singer et al.,
1996), den Krankheitsverlauf negativ beeinflusst.
Als Prognosefaktoren sind die Tumorgrößen bei den hier untersuchten
Tumorentitäten nur bedingt geeignet, da das Klarzellsarkom, das epitheloide
Sarkom und das Synovialsarkom oft schon bei einer weitaus geringeren
Größe metastasieren.
1.1.6 Alter und Geschlechtsverteilung
Weichgewebstumoren können grundsätzlich bei Patienten beiderlei
Geschlechts und jeden Alters auftreten. Für manche Subtypen läßt sich ein
Altersgipfel und eine Häufung bei einem Geschlecht feststellen.
Die hier untersuchten Tumore sind Neoplasien, die vorwiegend bei
Erwachsenen jüngeren oder mittleren Alters angetroffen werden.

12
Der Altersgipfel beim Klarzellsarkom liegt bei ca. 30 Jahren (Eckardt J et al.,
1983), beim epitheloiden Sarkom zwischen 10 und 35 Jahren, und beim
Synovialsarkom zwischen 15 und 40 Jahren.
Das Verhältnis Männer:Frauen beträgt beim Klarzellsarkom etwa 1:1.5,
das epitheloide Sarkom und das Synovialsarkom betreffen etwas häufiger
das männliche Geschlecht (Verhältnis Männer : Frauen ca. 1.2:1).
1.1.7 Krankheitssymptome
Die peripheren Weichgewebstumoren fallen klinisch meist als indolente,
abgrenzbare Schwellung auf, welche an Größe zunimmt.
So klagt nur ein Drittel der Patienten mit einem Klarzellsarkom (Eckhardt et
al., 1983), ein Viertel der Patienten mit einem Epitheloidzellsarkom (Halling
et al., 1996) und die Hälfte der Patienten mit einem Synovialsarkom, über
Schmerzen. Manche Autoren beschrieben ein schmerzhafteres Verhalten
des biphasischen Typs im Vergleich zum monophasischen Typ (Hajdu et al.,
1977, Krall et al., 1981).
Die stammnah wachsenden Weichgewebstumore fallen oft erst anhand der
Metastasen oder durch Funktionsausfälle der betroffenen Organe auf.
Allgemeinsymptome, wie Unwohlsein, Gewichtsabnahme, Nachtschweiß etc.
(„B-Symptomatik“) werden in der Regel nicht beobachtet.
1.2 Klinisch-pathologische Parameter
In zahlreichen Untersuchungen wurde versucht, unterschiedliche klinische
und pathologische Parameter mit dem Krankheitsverlauf maligner
Erkrankungen zu korrelieren.
1.2.1 Tumorgrading und Stadieneinteilung, prognostische Parameter
Da zahlreiche Untersuchungen gezeigt haben, dass der Differenzierungsgrad
eines Tumors sich umgekehrt proportional zu seiner
Wachstumsgeschwindigkeit, der Strahlenempfindlichkeit und seiner

13
Bösartigkeit verhält (d.h. je undifferenzierter ein Tumor, desto bösartiger),
kann es prognostisch richtungsweisend sein, einen Tumor nach seinem
Differenzierungsgrad zu klassifizieren (Jensen et al., 1998, Markhede et al.
1982, Singer et al., 1994).
„Low grade“- (G1 und G2) unterscheiden sich in ihrer Prognose deutlich von
„high grade“-Tumoren: „Low grade“-Tumoren bilden in ca. 15%
Fernmetastasen, das Gesamtüberleben beträgt über 80%, „high grade“-
Tumoren bilden in über 50% der Fälle Fernmetastasen, das
Gesamtüberleben liegt bei ca. 50% (Lewis et al., 1999).
Bei der Graduierung von Sarkomen gilt:
Unterschiedlichste Parameter, wie zelluläre Atypie, infiltratives
Wachstumsmuster, Interzellularsubstanz, Entzündungsinfiltrat, insbesondere
aber Mitoserate und Ausmaß von Nekrosen werden beurteilt (Coindre et al.,
1998, Coindre et al. 2001).
Je geringer die Ähnlichkeit zum Muttergewebe, je höher die Mitosezahl und
je ausgedehnter die Tumornekrosen, desto höher der Malignitätsgrad.
Das Grading richtet sich nach dem jeweils niedrigsten Differenzierungsgrad
im Tumorgewebe, auch wenn diese Bereiche nur fokal ausgebildet sind, wie
man es oft bei Weichgewebstumoren findet.
Häufig ändert sich das Tumorbild auch erheblich bei den Rezidiven oder
nach einer Strahlen- oder Chemotherapie.
Die Tumorstadien werden nach der TNM-Klassifikation wie folgt eingeteilt:
T Primärtumor
Tx Primärtumor nicht beurteilbar
T0 kein Primärtumor
T1 Primärtumor < 5cm
A oberflächlicher Tumor, oberhalb der Faszie, ohne Infiltration derselben
B tiefer Tumor (retroperitoneale, mediastinale und
Beckenweichteilsarkome),unterhalb der Faszie, oder oberhalb der Faszie mit
Infiltration

14
T2 Primärtumor > 5cm
A oberflächlicher Tumor
B tiefer Tumor (s.o.)
N Lymphknotenbefall
Nx regionäre Lymphknoten nicht beurteibar
N0 Lymphknoten tumorfrei
N1 regionärer Lymphknotenbefall
M Metastasen
Mx Metastasierung nicht beurteilbar
M0 keine Fernmetastasen
M1 Fernmetastasen (auch Befall kontralateraler oder entfernter
Lymphknoten)
Die Stadieneinteilung der Weichgewebstumore erfolgt entsprechend AJCC
(American Joint Committee on Cancer, 1987):
Stadium GradingTNM Stadium
IA G1,2 T1a-b N0 M0
IB G1,2 T2a N0 M0
IIA G1,2 T2b N0 M0
IIB G3,4 T1a-b N0 M0
IIC G3,4 T2a N0 M0
III G3,4 T2b N0 M0
IV jedes G jedesT, jedes N, jedes M
Im Vergleich zu anderen Tumoren, für die die TNM-Klassifikation
aussagekräftig ist, neigen hochmaligne Weichgewebstumoren ungewöhnlich

15
stark zu Rezidiven und metastasieren außergewöhnlich schnell, nahezu
unabhängig von der Größe des Primärtumors. Spätmetastasen sind häufig.
Sehr wichtig im Hinblick auf die Prognose ist der Resektionsstatus des
Tumors, der R-Faktor. Bei positivem R-Faktor ist das Risiko für ein
Lokalrezidiv hoch, die Prognose somit schlecht (Pisters et al., 1996).
Lymphknotenmetastasen werden häufiger beim epitheloiden Sarkom als bei
den übrigen Sarkomen beobachtet (Chase et al., 1985). Insgesamt spielt bei
Sarkomen die lymphogene Metastasierung, im Vergleich zu Karzinomen,
eine untergeordnete Rolle.
Die in Europa am häufigsten angewandte Malignitätsgraduierung ist das von
Coindre und Guillou eingeführte, französische FNCLCC System (Coindre et
al., 2001).
Bei diesem System werden drei Parameter bestimmt: Die
Tumordifferenzierung, die Mitoserate und der Nekroseanteil.
Für jeden der untersuchten Parameter gibt es eine bestimmte Anzahl von
Punkten (Scores). Anhand der Gesamtpunktzahl wird in Grade eingeteilt.
Tumoren mit einer Gesamtpunktzahl von 0-3 werden danach als Grad 1
bezeichnet, eine Gesamtpunktzahl von 4-5 entspricht Grad 2 und eine
Gesamtpunktzahl von 6-8 entspricht nach diesem System Grad 3.
Für das Kriterium der Tumordifferenzierung gilt dabei: Einen Punkt erhalten
Tumoren, die dem Ursprungsgewebe nahezu entsprechen, zwei Punkte
erhalten Tumoren, bei denen eine eindeutige Diagnose möglich ist, und drei
Punkte erhalten biphasische und monophasische Synovialsarkome.
Die Mitoserate wird bestimmt, indem die Mitosen pro 10 HPF ausgezählt
werden (Gesichtsfeldgröße 0.174 mm). 0-9 Mitosen entsprechen dann einem
Punkt, 10-19 Mitosen zwei Punkten, und 20 und mehr Mitosen drei Punkten.
Die Nekroseareale werden prozentual bestimmt: 0 Punkte bedeutet, dass
keine Nekrosen zu finden sind, ein Punkt bedeutet bis zu 50% Nekrosen im
untersuchten Tumor und zwei Punkte über 50% Nekroseareale. Die
jeweiligen Punktzahlen des zu untersuchenden Tumors werden am Ende
addiert und es ergibt sich:

16
-Grad 1 für eine Gesamtsumme von 2 oder 3
-Grad 2 für eine Gesamtsumme von 4 oder 5
-Grad 3 für eine Summe von 6 bis 8 Punkten.
Diese Einteilung korreliert bei den meisten Tumoren streng mit der 5-Jahres-
Überlebensrate, ist also ein Kriterium für den klinischen Verlauf eines
Tumors.
Diese Methode des Gradings sollte nur bei Tumoren angewandt werden, die
noch nicht zytostatisch oder mittels Radiotherapie behandelt wurden, da die
Nekrosen nach einer Therapie nicht abschließend zu bewerten sind (Coindre
et al., 2001).
Auch das FNCLCC System des Gradings ist bei ca. 10% der
Weichgewebstumoren nicht aussagekräftig. Manche Tumoren sind nach
dieser Methode als hochmaligne anzusehen, sind jedoch im klinischen
Verhalten wenig maligne, andere wiederum verhalten sich höchstmaligne,
werden mittels Grading jedoch als weniger maligne klassifiziert
Beim epitheloiden Sarkom und bei Klarzellsarkomen werden bislang keine
unterschiedlichen Malignitätsgrade abgegrenzt, das Coindre-System wird
eingeschränkt verwendet, und bei malignen kindlichen Weichgewebstumoren
läßt sich aus dem Grading ebenfalls keine Prognose ableiten (Katenkamp
1999).
Bislang ist bei Sarkomen der Tumormalignitätsgrad unter Einbeziehung von
Tumorgröße, Infiltrationstiefe, chirurgischem Entfernungsabstand zum
gesunden Gewebe und histologischem Typ der wichtigste prognostische
Faktor (Russel et al., 1970).
Weitere Faktoren zur Beurteilung des Malignitätsgrades sind Tumorgröße
und Entzündungszellinfiltrate. Eine Tumorgröße von 5 cm (Russell et al.,
1977) oder 10 cm (Singer et al., 1996) gelten als Grenzwert für einen
schlechteren Krankheitsverlauf. Entzündungszellinfiltrate sind besonders in
prognostisch ungünstigen Tumoren zu finden. Eine prognostische Aussage
kann unter anderem durch den Mastzellgehalt eines Tumors getroffen
werden (Katenkamp und Hünerbein, 1992).

17
1.2.2 Therapieprinzipien der Weichgewebstumore
In der Vergangenheit wurden Weichgewebstumoren oft durch Amputation
einer Extremität oder durch verstümmelnde Eingriffe therapiert. Durch
interdisziplinäre Zusammenarbeit und multimodale Therapiestrategien hat
sich die chirurgische Vorgehensweise geändert (Steinau et al., 1996, Steinau
und Biemer., 1990). Besonders die Fortentwicklung plastisch-chirurgischer
Techniken in den letzten Jahren, macht die in ca. einem Viertel der Patienten
erforderiche plastische Rekonstruktion möglich. Der wichtigste
Prognosefaktor ist die in-sano Resektion, in der TNM-Klassifikation als R0
Resektion bezeichnet. Sie konnte in mehreren Studien als wichtigster Faktor
zur Verhinderung von Lokalrezidiven und zur Steigerung der Überlebensrate
ermittelt werden (Dickinson et al., 2006, Mandard et al., 1989, Markhede et
al., 1982, Pisters et al. 1996, Tunn et al., 2007).
Ein primär nicht operabler Tumor (bzw. keine sichere R0 Resektion möglich)
ohne Metastasen kann zunächst mit einer neoadjuvanten Therapie mit dem
Ziel der Tumorverkleinerung angegangen werden (Tunn et al.).
Auch bei kleineren Tumoren sollte vor einer Operation geprüft werden, ob
durch Vorschaltung ergänzender Therapien ein optimaleres operatives
Ergebnis erzielt werden kann.
Als Optionen dieser multimodalen Therapiestartegien stehen hier eine
systemische Chemotherapie mit und ohne Hyperthermiebehandlung und
Strahlentherapie, und eine Extremitätenperfusion zur Verfügung. Abhängig
vom Operationsergebnis erfolgt eine adjuvante Chemotherapie mit oder ohne
Radiotherapie.
Ein bereits metastasierter Tumor wird zunächst mit einer neoadjuvanten
Chemotherapie angegangen, bei Ansprechen der Therapie erfolgt die
Resektion des Tumors und der Metastasen, ggf. postoperativ eine weitere
Chemotherapie oder Radiotherapie. Bei Patienten mit diffuser
Metastasierung können eine palliative Chemotherapie und entsprechend des
Beschwerdebildes lokale Therapiemaßnahmen mit Extremitätenperfusion,
Resektion, Radiotherapie durchgeführt werden.

18
Insgesamt bewirkt eine Radiotherapie bei „high-grade“-Sarkomen eine
signifikante Verbesserung der Tumorkontrolle, im Gegensatz zur alleinigen
chirurgischen Resektion (Eilber et al., 2006).
Die wirksamsten derzeit verfügbaren Chemotherapeutika für
Weichgewebstumore sind Doxorubicin, und Ifosfamid mit Ansprechraten von
20-30%. Andere Chemotherapeutika mit ca. 10% Ansprechraten sind
Epirubicin, Actinomycin-D, Dacarbacin, Methotrexat, Cisplatin und
Gemcitabin (Berger et al., 2002). Neuere Chemotherapeutika werden in
Kombination angewendet (z.B. Adriamycin, Vinchristin, Adriablastin
Novatron), Langzeitergebnisse liegen dazu bisher nicht vor.
1.3 Tumorentitäten im Speziellen: Epitheloides Sarkom, Klarzellsarkom und Synovialsarkom
1.3.1 Epitheloides Sarkom
1.3.1.1 Allgemeines
Das epitheloide Sarkom entwickelt sich wahrscheinlich aus einer
undifferenzierten, mesenchymalen Stammzelle, diess wird durch
immunhistochemische Untersuchungen gestützt (Schmidt und Harms, 1987,
Enzinger et al., 1995, Mukai et al., 1984).
Es entwickelt sich typischerweise an den distalen Extremitäten (Bos et al.,
1988) und ist der häufigste maligne Weichgewebstumor der Hand und des
Handgelenkes.
In seltenen Fällen findet man Primärtumoren im Kopfbereich und im
Stammbereich, jedoch metastasiert dieses Sarkom häufig in die Kopfhaut.
1.3.1.2 Makroskopische Befunde
Es handelt sich um eine multinoduläre Geschwulst, die oberflächlich oder tief
lokalisiert sein kann. Faszien, Sehnen und das Subkutangewebe sind
meistens betroffen. Oberflächlich lokalisierte Tumoren können ulzerieren.

19
Je nach Subtyp können die Epitheloidzellsarkome rund bis oval sein, sie
können multipel oder solitär auftreten, eine Kapsel oder Pseudokapsel
aufweisen, aber auch kapsellos sein.
Die Schnittfläche kann von fleischartiger bis fibröser Konsistenz sein, und mit
mehr oder weniger ausgeprägten Nekrosen und Hämorrhagien durchsetzt
sein.
1.3.1.3 Mikroskopische Befunde
Das äußere Erscheinungsbild ist vielgestaltig. Die Tumoren ahmen offenbar
kein erkennbares Normalgewebe nach (Stiller et al., 1988).
Die Zellen sind groß, polygonal bis spindelig mit eosinophilem Zytoplasma.
Im Zentrum der Tumoren finden sich oft Nekroseareale und Kalzifikationen in
einer hyalinisierten Matrix. Um das Zentrum herum sind die Zellen
palisadenartig angeordnet ( Enzinger 1970).
Das Verhältnis von epitheloiden zu spindeligen Zellen variiert von Tumor zu
Tumor. Mitosen können immer nachgewiesen werden, in den meisten
Tumoren findet man außerdem ein entzündliches Infiltrat aus Lymphozyten,
Histiozyten und Plasmazellen.
1.3.1.4 Immunhistochemie
Alle Epitheloidzellsarkome sind Vimentin und Keratin positiv ( Chase et al.,
1984, Halling et al., 1985, Hazalbag et al., 1996, Manivel et al., 1987). 50%
der Tumorzellen sind CD-34 positiv, zu 85% reagieren sie ebenso mit EMA-
Antikörpern (Kuhnen et al., 1998, Miettinen et al., 1984/1983).
1.3.1.5 Prognose
Der Tumor neigt, wie das Klarzellsarkom, zu Rezidiven und Metastasen
(Evans et al., 1980).
Ca. 80% der Tumoren bilden Rezidive, in 50% der Fälle kommt es zu
Metastasen. Bevorzugte Metastasierungsorte sind die regionalen
Lymphknoten und die Lunge, seltener Haut, ZNS und Kopfhaut/-
weichgewebe.

20
Die hohe Metastasierungsrate läßt sich durch die Tendenz der Tumoren,
subklinische Satellitenknoten entlang von Sehnen, Faszien und
neurovaskulären Strukturen zu bilden, erklären.
Die Prognose wird entscheidend durch die Lokalisation des Primärtumors
beeinflußt. Dabei gilt: je peripherer der Primärtumor, desto günstiger die
Prognose. Das Vorkommen von Nekrosen und eine Tumorgröße über 5 cm
sind weitere Kriterien für einen ungünstigen Krankheitsverlauf.
Beim klassischen epitheloiden Sarkom versterben ca. 27-46% der Patienten
an den unmittelbaren Folgen der Erkrankung. Beim proximalen Typ des
Tumors versterben ca. 65% der Patienten (Chase et al., 1985, Hasegawa et
al.,1989, Guillou et al., 1998).
1.3.1.6 Therapie
Das epitheloide Sarkom muß mit ausreichendem Abstand im Gesunden
exzidiert werden. Verschiedene Chemotherapeutika und Radiotherapien
werden angewandt, zeigen jedoch nur wenig Wirkung (Chase et al., 1985,
Callister et al., 2001).
1.3.1.7 Differentialdiagnosen
• Synovialsarkom
Im Gegensatz zum Synovialsarkom weist das epitheloide Sarkom kein
granulomähnliches Wachstumsmuster auf. In ca. einem Drittel der Fälle
findet man Verkalkungen, und ein zumindest partiell
hämangioperizytomartiges Wachstum (Lombardi et al., 1984).
Die X;18 Translokation ist für Synovialsarkome spezifisch, der Nachweis ein
eindeutiges Unterscheidungskriterium zu anderen Tumoren.
Synovialsarkome sind CD-34 negativ und bcl-2 positiv (Turc-Carel et al.,
1986).
• Karzinom
Liegt eine Metastase vor, so hilft der Nachweis des Primärtumors. Ist der
Tumor örtlich entstanden, so helfen die Beziehungen zu vorbestehenden
epithelialen Strukturen in der Unterscheidung weiter.

21
Für das Vorliegen eines Karzinoms sprechen glanduläre oder
plattenepitheliale Differenzierungen, der Nachweis von CK7 und das Fehlen
von CD-34.
• Malignes Melanom
Die Zellen des malignen Melanoms sind durchgehend S-100 positiv und
meist NK1C3-positiv. Sie reagieren mit Melanin A und/oder HMB-45.
• Epitheloider MPNST
Das epitheloide MPNST kann CK- und EMA-positiv sein, und in 60-70% der
Tumoren liegt eine herdförmige S-100 Positivität vor.
• Epitheloides Leiomyosarkom
Es kann EMA- und/oder CK-positiv sein. Ein größerer Anteil der Tumorzellen
ist Aktin positiv, die Tumorzellen mit abgestumpften Enden weisen ein
eosinophiles Zytoplasma auf.
• Nekrobiotisches Granulom
Das Granulom weist mehr Entzündungszellen als das epitheloide Sarkom
auf. Man findet häufig Riesenzellen und Areale mit starker Gefäßproliferation.
Mitosen sind nicht, oder nur vereinzelt zu finden.
Das Granulom ist weder CK- noch EMA-positiv.
(Miettinen et al. 1983/1984)
1.3.2 Klarzellsarkom
1.3.2.1 Allgemeines
Das Klarzellsarkom ist neuroektodermalen Ursprunges und wird aufgrund
seiner Fähigkeit zur Melaninsynthese auch als Melanom des Weichgewebes
bezeichnet (Berman et al. 1975, Bourdeaux et al., 1978, Mackanzie et al.,
1974 ).

22
Im Gegensatz zum Melanom ist das Klarzellsarkom jedoch tiefer lokalisiert,
die Epidermis ist nicht einbezogen. Oft tritt es in topographischer Beziehung
zu Sehnen und Aponeurosen auf (Angervall et al., 1969, Alitale et al., 1977).
Prädilektionsstellen sind die Extremitäten, wobei statistisch die Füße
überwiegen.
Das Klarzellsarkom weist in der Regel eine t(12;22) Translokation auf
( Fletcher et al., 1992, Mrozek et al., 1993, Reeves et al., 1992, Rodriguez et
al., 1992, Zucman et al., 1993) , welche für diese Tumorart spezifisch zu sein
scheint, und ein wichtiges Diagnosekriterium ist (Stenman et al., 1992,
Peulve et al.,1991, Travis et al., 1992). Die Häufigkeit der Translokation wird
mit 70% angegeben.
1.3.2.2 Makroskopische Befunde
Klarzellsarkome wachsen meist in direkter Verbindung zu Sehnen oder
Aponeurosen.
Die Tumoren haben eine grau-weiße, homogene Schnittfläche von fester,
gummiartiger Konsistenz. Sie können lobuliert bis multilobulär wachsen, ca.
die Hälfte ist von einer Kapsel umgeben, zum Teil finden sich
Pigmentierungen.
1.3.2.3 Mikroskopische Befunde
Die klassischen histologischen Befunde beim Klarzellsarkom sind kleine
„Cluster“ von polygonalen bis spindeligen Zellen, welche von feinen, fibrösen
Septen unterteilt werden (Speleman et al., 1992).
Das Zytoplasma ist hell bis basophil, der Nukleus ovoid und vesikulär mit
einem prominenten, meistens eosinophilen Nukleolus. Der Anteil des
fibrösen Stromas ist variabel (Colome-Grimmer et al., 1996, Benson et al.,
1985, Enzinger et al., 1995).
Mitosen sind bei den meisten Klarzellsarkomen selten, meistens weniger als
5 Mitosen pro 10 hpf, es gibt jedoch auch Fälle mit bis zu 30 Mitosen pro 10
hpf. Nekrosen findet man bei ca. der Hälfte der Tumoren, ebenso
mehrkernige Riesenzellen in ca. 50% der Fälle (Mukai M et al., 1984, Bridge
JA et al., 1991). Im Elektronenmikroskop erkennt man eine bruchstückhafte

23
Basallamina, Glykogen und Melanosomen (Amr et al., 1984, Tsuneyoshi et
al., 1975, Fisher C., 1988, Kubo et al., 1969).
1.3.2.4 Immunhistochemie
Ca. 70% der Tumoren sind HMB-45 und Vimentin positiv und weisen
intrazelluläres Melanin auf (Bearman et al., 1975, Benson et al., 1985, Ekfors
et al., 1970, Raynor et al., 1979, Sara et al., 1990). Zytokeratin ist nicht
nachweisbar (Lukas et al., 1992, Deenik et al., 1999). Ebenso findet man
eine positive S100- und NK1C3-Reaktion. Oftmals, aber nicht obligat, gelingt
auch ein Melan-A-Nachweis (Kindblom et al., 1983, Boudreaux et al., 1978,
Hoffmann et al., 1973, Mechtersheimer et al., 1989).
1.3.2.5 Prognose
Die Prognose für Patienten mit einem Klarzellsarkom ist schlecht.
Der Tumor neigt zu Rezidiven und 50% der Patienten entwickeln im
Krankheitsverlauf Metastasen, besonders in Lunge, Lymphknoten und
Knochen.
Nach einem 5-Jährigen Krankheitsverlauf leben noch ca. zwei Drittel der
Patienten, nach 20 Jahren nur noch ca. 20%. Spätmetastasen sind nicht
selten.
Prognosefaktoren scheinen u.a. die Größe des Primärtumors und das
Vorliegen von Nekrosen und Mitosen zu sein (Marques et al., Lucas et al.,
1992, Sara et al., 1990, Choong et al., 1994). Wie bereits erwähnt, lässt sich
aus dem Grading der Klarzellsarkome keine Prognose ableiten (Katenkamp
1999).
1.3.2.6 Therapie
Die Therapie der Klarzellsarkome liegt in erster Linie in der chirurgischen
Intervention. Die Tumoren sollten je nach Dignität mit ausreichendem
Abstand exzidiert werden. Chemotherapie und Bestrahlungstherapie sind bis
heute wenig effektiv (Steinau et al. 1990, Deenik et al., 1999, Eckhardt et
al., 1983, Radstone et al., 1979).

24
1.3.2.7 Differentialdiagnosen
• Synovialsarkom
Beim Synovialsarkom liegt ein biphasisches Zellbild mit epithelialen und
spindeligen Arealen, oder ein fibrös-monophasisches Bild mit uniformen
Spindelzellen in gebogenen Faszikeln vor.
Es finden sich keine intrazellulären Melaninablagerungen. Die Zellen sind
herdförmig Zytokeratin- und/oder EMA positiv, Melanin A und HMB-45
negativ. S-100 kann nachweisbar sein. Im Zweifelsfall hilft der Nachweis der
für das Synovialsarkom spezifischen X;18-Translokation (Balasz et al., 1997,
Bridge et al., 1991).
• Epitheloides Sarkom
Das Epitheloide Sarkom wächst eher multinodulär, granulomähnlich und
weist im Zentrum oft Nekrosen auf.
Dieser Sarkomtyp kann insbesondere mit einer granulomatösen Entzündung
verwechselt werden.
Die Zellen sind epitheloid bis spindelig und sind positiv für Zytokeratin, EMA
und Vimentin.
• Malignes kutanes Melanom
Das Melanom liegt in der Regel oberflächlicher. Primär ist es in der Haut
älterer Menschen zu finden. Das Zellbild ist bunt, Mitosen sind häufiger als
beim Klarzellsarkom, man findet landkartenartige Nekrosen und nicht die für
das Klarzellsarkom typische t(12;22) Translokation (Sonobe et al., 1997,
Sonobe et al., 1999, Tsuneyoshi et al., 1975)
• Riesenzelltumor der Sehnenscheide
Das Zellbild ist bunt. Man findet histiozyten- und fibroblastenartige Zellen,
Schaumzellen und osteoklastenartige Riesenzellen.
Immunhistochemisch lassen sich keine melanozytäre Differenzierungen
nachweisen.
• Konventioneller Maligner peripherer Nervenscheidentumor (MPNST)

25
Man findet im Gegensatz zum Klarzellsarkom kein Melaninpigment, kein
intrazelluläres Glykogen, eine erhöhte Mitoserate und nur ein Teil der
Tumorzellen ist S-100 positiv.
Der konventionelle MPNST metastasiert in der Regel nicht in die
Lymphknoten.
1.3.4 Synovialsarkom
1.3.4.1 Allgemeines
Als Ursprungszelle des Synovialsarkoms vermutet man eine mesenchymale
Zelle, welche sich in eine epitheliale Zelle umdifferenziert oder eine
pluripotente Stammzelle.
Das Synovialsarkom macht beim Erwachsenen ca. 8-12% aller
Weichgewebstumoren aus, und ist somit der vierthäufigste maligne
Weichgewebstumor des Erwachsenen.
Es ist vorwiegend in den paraartikulären Regionen der Extremitäten
lokalisiert und wächst in Verbindung zu Sehnen, Bursae und Gelenkkapseln,
jedoch nicht intraartikulär. In ca. 10% der Fälle findet man auch
Synovialsarkome in Bereichen ohne Synovialstrukturen, wie z. B. in der
Zunge oder der Bauchwand oder Kopf/Hals-Bereich.
Die Bezeichnung „Synovialsarkom“ ist im klinischen Alltag fest verwurzelt,
jedoch irreführend, da der Tumor sich weder innerhalb der Synovialis
entwickelt, noch Strukturen aufweist, welche mit der normalen Synovialis
vergleichbar sind (Ghadially et al., 1987).
Ein wichtiges Unterscheidungskriterium zu anderen Tumoren ist die
reziproke X;18 Translokation. Ca. 90% aller Synovialsarkome weisen diese
Translokation auf (Turc-Carel et al., 1986, Balazs et al., 1997, Hirakawa et
al., 1995, Kawai et al., 1998).
1.3.4.2 Makroskopische Befunde
Die Tumoren wachsen langsam, und sind daher gut von der Umgebung
abgrenzbar. Sie können eine runde Form aufweisen oder multinodulär
wachsen, und sind häufig von einer weichen Pseudokapsel umgeben.

26
Die Schnittfläche der Synovialsarkome ist grau-weiß bis gelb, zystische
Formationen bis hin zu multizystischen Wachstumsformen sind zu finden.
Typisch sind die bereits im Röntgenbild sichtbaren Verkalkungen.
1.3.4.3 Mikroskopische Befunde
Histologisch unterscheidet man mehrere Subtypen von Synovialsarkomen
(Evans et al., 1980, Dardick et al., 1991, Lopes et al. 1994, Mickelson et al.,
1980):
1. Einen biphasischen Typ mit epithelialen karzinomartigen und
spindelzelligen sarkomartigen Arealen in unterschiedlicher
Zusammensetzung,
2. Einen monophasisch-fibrösen Typ mit nahezu ausschließlich
spindelzelligen Anteilen und
3. Einen monophasisch-epithelialen Typ mit nahezu ausschließlich
epithelialem Erscheinungsbild.
Weiterhin wird ein schlecht differenziertes Synovialsarkom mit rund- und
kleinzelligem, großzellig-epitheloidem oder high-grade spindelzelligem
Erscheinungsbild mit überwiegend Spindelzellen beschrieben (Dardick et al.,
1991, Machen et al., 1999, Nickelson et al., 1980).
Das klassische Synovialsarkom ist biphasisch und weist epitheliale,
karzinomähnliche und spindelzellige, sarkomähnliche Strukturen auf.
Der Tumor wächst zungenartig in das benachbarte Gewebe ein. Es kommen
Verkalkungen mit und ohne Ossifikation vor. Zelldichte Areale und Areale mit
reduzierter Zellzahl und einem wechselndem Anteil an kollagener
Interzellularsubstanz kommen vor (Mills et al., 1981). Die Zellen können
diffus, band-, oder plaqueartig angeordnet sein, myxoide Auflockerungen
kommen vor.
Die Synovialsarkome weisen oft Mastzellen auf und können eine dichtes
Gefäßnetz zeigen (hämangioperizytomartig). Synovialsarkome sind in der
Regel bcl-2-positiv und CD34-negativ, 90% der Tumoren weisen eine
reziproke X;18-Translokation auf.

27
1.3.4.4 Immunhistochemie
Normalerweise liegt eine Koexpression von Keratin und EMA vor. Die
Tumoren sind manchmal positiv für S-100 (Guillou et al., 1989, Lopes et al.
1994).
Vimentin ist besonders bei Tumoren mit einem hohen Anteil von
Spindelzellen positiv (Lopes et al., 1994). Die meisten bcl-2-positiven
Tumoren sind auch CD-34 negativ (Hirakawa et al., 1996, Lopes et al.,
1994).
Immunhistochemisch liegen keine großen Unterschiede zwischen
monophasischem und biphasischen Typ vor, jedoch zeigen ausschließlich
die soliden, großzelligen Areale der biphasischen Synovialsarkome eine
Expression von Keratin 13 und CEA (Lopes et al. 1994). Bei
elektronenmikroskopischen und immunhistochemischen Untersuchungen
stellte Ghadially fest, dass die Tumoren im Allgemeinen keine
synovialisähnlichen Strukturen aufweisen. Daher wird alternativ die
Bezeichnung „Karzinosarkom“, „Karzinom des Weichgewebes“
vorgeschlagen (Ghadially, 1987).
1.3.4.5 Prognose
Die Synovialsarkome neigen zu Rezidiven und Metastasen, späte
Metastasen sind nicht selten.
Ca. 50% der Patienten entwickeln Metastasen, besonders häufig betroffen
sind die regionalen Lymphknoten, Lunge und Knochenmark.
Ein prognostisch günstiger Faktor ist das Vorkommen von ausgedehnten
Verkalkungen, ein ungünstiger Faktor scheint das Auftreten von rhabdoiden
Zellen und das Vorliegen eines undifferenzierten Subtypes zu sein.
Die Prognose hängt ausschließlich vom Malignitätsgrad ab, weitere
Einflussfaktoren, wie Patientenalter, Tumorgröße und Lokalisation scheinen
nur wenig, oder keinen Einfluß auf den Krankheitsverlauf zu haben.
1.3.4.6 Therapie
Wie auch bei den anderen untersuchten Tumortypen ist die chirurgische
Therapie die wirksamste. Dabei ist auf einen ausreichenden

28
Sicherheitsabstand im Gesunden zu achten, da es sonst zu Rezidiven
kommt (radikale Resektion) (Rajpal et al., 1984).
Eine Chemotherapie mit Ifosfamid wird besonders beim schlecht
differenzierten, rundzelligen Subtyp angewandt.
1.3.4.7 Differentialdiagnosen
Monophasisch-fibröses Synovialsarkom:
• Fibrosarkom
Das Fibrosarkom weist langgestreckte Faszikel auf, die als
Fischgrätenmuster sichtbar sein können.
Im Gegensatz zum Synovialsarkom weist es mehr Mitosen auf, es fehlen
jedoch Mastzellen, Verkalkungen und ein hämangioperizytomartiges
Gefäßmuster.
Die Tumorzellen sind Vimentin positiv, CK und EMA sind nicht nachweisbar.
• Maligner peripherer Nervenscheidentumor (MPNST)
Der MPNST wächst in Assoziation mit einem großen Nerven. Eine
vorbestehende Neurofibromatose oder Reststrukturen eines Neurofibroms
sind hinweisend auf einen MPNST.
Im Synovialsarkom sind CK7 und CK19 nachweisbar, nicht jedoch im
MPNST, S-100 und CK kann in beiden Tumoren nachweisbar sein.
• Spindelzellkarzinom
Bei einem im tiefen Weichgewebe liegenden Tumor handelt es sich in den
meisten Fällen um eine Metastase. Es sind in der Regel stärkere
Unregelmäßigkeiten und zytologische Atypien als im monophasisch-fibrösen
Synovialsarkom zu finden.
Das zuverlässigste Diagnosekriterium ist der Nachweis der für
Synovialsarkome typischen X;18-Translokation.

29
Biphasisches Synovialsarkom:
• Glandulärer MPNST
Patienten mit einem glandulären MPNST sind fast immer an einer
Neurofibromatose erkrankt. Die Tumoren wachsen in direkter Verbindung zu
einem großen Nerven.
Im Gegensatz zum Synovialsarkom liegen keine Hyalinisierungen oder
Verkalkungen vor, es fehlen herdförmige CK-und/oder EMA positive
Bereiche.
Beim MPNST sind fokal S-100 positive Zellen zu finden.
• Karzinosarkom
Bei der Abgrenzung zu diesem Tumor helfen immunhistochemische
Untersuchungen nicht weiter. Es handelt sich meist um eine Metastase eines
nicht im Weichgewebe liegenden Tumors. Das wichtigste
Unterscheidungskriterium ist auch hier wieder die spezifische Translokation
X;18 des Synovialsarkoms.

30
1.4 Immunhistochemie
Immunhistochemische Untersuchungen sind ein unverzichtbarer Bestandteil
der diagnostischen Pathologie geworden.
Besonders bei einer so heterogenen Gruppe von Tumoren ist eine eindeutige
Zuordnung zur jeweiligen Entität, und somit eine eindeutige Diagnose ohne
Immunhistochemie nur schwer möglich.
1.4.1 MIB-1
Der Nachweis des MIB-1 Antikörpers, welcher gegen das Ki-67-Antigen
gerichtet ist, erlaubt eine Beurteilung der proliferativen Aktivität eines
Gewebes.
Das Ki-67-Antigen ist ein 395 kDa-Genprodukt, welches eng mit dem
Zellzyklus assoziiert ist.
Für die hohe Spezifität des Markers spricht, daß das Genprodukt in der G0-
Phase und in allen aktiven Phasen des Zellzyklus exprimiert wird.
Cattoretti und Mitarbeiter entwickelten den paraffingängigen MIB-1 Antikörper
gegen das Ki-67-Antigen (Cattoretti et al., 1992).
1.4.2 Epidermal Growth Factor Receptor (EGFR)
Der Epidermale Growth Factor Receptor (EGFR) ist ein transmembranöser
Rezeptor, und gehört wie die Herzeptin Rezeptoren HER2, HER3 und HER4
zur Gruppe der Tyrosinkinasen.
Bindet ein Ligand der EGF-Gruppe (Endothelial Growth Factor) an den
Rezeptor, wird eine Signalkaskade aktiviert, welche Gene aktiviert und den
Zellzyklus und Zelldifferenzierung beeinflusst (s. Abb.1).
EGFR spielt verschiedene Rollen bei der Tumorentstehung. Seine
Signalkaskaden nehmen Einfluß auf alle Schlüsselprozesse des
Tumorwachstums.
EGFR stimuliert die Zellteilung, reguliert die Apoptose und ein
Schnellvorgang des programmierten Zelltodes in der Embryogenese. Eine
Fehlsteuerung der Apoptose ist ein entscheidender Faktor des
Tumorwachstums. Weitere Tumorprozesse, welche durch EGFR beeinflusst

31
werden, sind die Angiogenese, Zelldifferenzierung, Zellmigration und
Metastasierung.
Bei der Tumorentstehung kommt es zu einer EGFR-Aktivierung aufgrund
einer EGFR-Genamplifikation, einer Genmutation oder durch eine
Wachstumsfaktorüberproduktion.
EGFR wird bei einer Vielzahl von Tumoren, nicht aber im angrenzenden
Normalgewebe gefunden. Unterschiedliche Tumoren zeigen eine
unterschiedliche EGFR-Expression. Ebenso gibt es Variationen der EGFR-
Expression innerhalb eines Tumors. Untersuchungen zeigen, dass die
EGFR-Expression mit dem Fortschreiten der Erkrankung zunimmt, es scheint
ein spätes Ereignis in der Tumorenstehung zu sein. Z.B. nimmt die EGFR-
Expression bei der Entstehung des Cervixkarzinoms von der Präneoplasie
stetig zu. Bei der Zervikalen Intraepithelialen Neoplasie (CIN) findet man eine
geringe EGFR-Expression, beim invasiven Cervixcarzinom eine hohe.
Einige Studien zeigen einen positiven Zusammenhang zwischen
Tumorgröße und EGFR-Expression, sowie zwischen
Lymphknotenmetastasen und EGFR-Expressionsrate. Zwischen dem Alter
der Patienten und der EGFR-Expressionsrate scheint ein negativer
Zusammenhang zu bestehen. Jüngere Patienten haben eine höhere EGFR-
Expressionsrate in ihren Tumoren. Bei einigen Tumorarten, z.B. Kopf-Hals-
Tumoren, Ovarialkarzimom, Blasen-und Ösophagus-Karzinom scheint eine
EGFR-Expression mit einer schlechteren Prognose vergesellschaftet zu sein
(Grandis et al., 1996). In anderen Tumoren, wie z.B. nicht kleinzelligem
Bronchialkarzinom erlaubt die EGFR-Expression keine Prognosevorhersage.
Einige Untersuchungen zeigten, dass eine hohe EGFR-Expression mit einem
geringen Ansprechen des Tumors auf Hormon-Chemo-und Radiotherapie
und somit wieder mit einer schlechteren Prognose einhergeht (Nicholson et
al., 1991).
Bisher gibt es mehrere verschiedene therapeutische Ansätze für eine EGFR-
Hemmung, z.B. Anti-EGFR-Antikörper, Tyrosinkinaseinhibitoren,
Oligonucleotide, EGFR/Liganden-spezifische Vaccine und mit radioaktiven
Isotopen oder Cytotoxinen armierte Antikörper.

32
In der vorliegenden Arbeit soll die EGFR-Expression bei seltenen
Weichgewebstumoren mit Assoziation zu Sehnen, Faszien und Aponeurosen
untersucht werden.
Insbesondere im Hinblick auf die schlechte Prognose der
Weichgewebstumoren, trotz multimodaler Therapieprinzipien, wären neue
Therapieansätze wünschenswert.

33
Abbildung 1:
Vereinfachte Darstellung des EGF-R-Signaltransduktionsweges für die Zellteilung aus: (Harari PM und Huang SM, 2000)

34
Tab. 1: Überblick über bereits auf EGFR-Expression untersuchte Tumoren
und ihre jeweilige Expressionsrate geben (Black et al., 1997).
Tumorart EGFR-Expressionsrate
Kopf-Hals-Tumore 80-100%
Nierenzellkarzinom 50-90%
Nicht-kleinzelliges Bronchialkarzinom 40-80%
Gliom 40-50%
Ovarialkarzinom 35-70%
Blasenkarzinom 31-48%
Pankreaskarzinom 30-50%
Colonkarzinom 25-77%
Mammakarzinom 14-91%

35
1.5 Zielsetzung und Fragestellung
Epitheloide Sarkome, Klarzellsarkome und Synovialsarkome sind seltene
maligne mesenchymale Tumore insbesondere mit Assoziation zu Sehnen,
Faszien und Aponeurosen.
In der neuen WHO-Klassifikation werden diese Tumoren als Neoplasien des
Weichgewebes mit „unklarer Differenzierungsrichtung“ eingeordnet, wobei
insbesondere für Synovialsarkome und epitheloide Sarkome eine epitheloide
Differenzierungsrichtung angegeben wird.
Ziel der hier vorliegenden Arbeit ist eine detaillierte Charakterisierung
hinsichtlich klinisch-pathologischer Parameter einschließlich der
Krankheitsverläufe bei einem Kollektiv von 135 Patienten.
Ein Grading von Klarzellsarkomen und epitheloiden Sarkomen nach der
Coindre-Klassifikation gilt bis zum heutigen Stand der Forschung bei
Weichgewebstumoren als eingeschränkt aussagekräftig. Somit ist eine
Prognose bezüglich des Krankheitsverlaufes im Einzellfall schwer möglich.
Insgesamt sollten folgende Fragestellungen untersucht werden:
1. Sind die erhobenen klinisch-pathologischen Parameter vergleichbar mit
anderen in der Literatur beschriebenen Kollektiven?
2. Ergibt sich eine Korrelation zwischen histologischen Parametern wie
Mitosen/Nekroseanteil und dem klinischen Verlauf?
3. Findet sich eine Korrelation zwischen Proliferationsindex (anhand der Ki-
67-Expression) und Tumorgröße/Lokalisation/Mitoseanzahl?
4. Ist ein Grading bei epitheloiden Sarkomen und Klarzellsarkomen möglich?
Ist eine Vorhersage des Krankheitsverlaufes möglich?
5. Ist eine EGF-R-Expression mit spezifischem Verteilungsmustern in
epitheloiden Sarkomen/Klarzellsarkomen /Synovialsarkomen zu belegen?

36
2 Material und Methoden
2.1 Material
In der vorliegenden Arbeit sind insgesamt 167 Weichgewebstumore von 135
Patienten untersucht worden, davon 30 Klarzellsarkome, 32 epitheloide
Sarkome und 105 Synovialsarkome.
Bei dem Kollektiv der Klarzellsarkome handelt es sich um 15 Primärtumore
und 15 Rezidivtumore bei 15 Patienten, bei den epitheloiden Sarkomen
liegen 24 Primärtumore und 8 Rezidivtumore bei insgesamt 24 Patienten vor.
Das Kollektiv der Synovialsarkome ist in 68 monophasische
Synovialsarkome und 37 Synovialsarkome vom biphasischen Typ unterteilt.
Es handelt sich dabei ausschließlich um Primärtumore.
Die Tumore wurden über einen Zeitraum von 15 Jahren (1991-2006) in der
Abteilung für Plastische Chirurgie und Schwerbrandverletzte der
Berufsgenossenschaftlichen Kliniken Bergmannsheil reseziert, die
histologischen Befunde am Institut für Pathologie der Ruhr-Universität
Bochum der Berufsgenossenschaftlichen Universitätsklinik Bergmannsheil
erhoben.
2.2 Untersuchungsmethoden
2.2.1 Datenerhebung
Es wurden klinisch-pathologische Daten erhoben zur Geschlechts- und
Altersverteilung, Tumorgröße und Tumorlokalisation sowie zu histologischen
Subtypen. Bei den epitheloiden Sarkomen und Klarzellsarkomen wurde eine
Bestimmung von Mitoserate und Nekroseausmaß, mit Erstellung eines
Malignitätsgrades für Weichgewebstumore anhand der Coindre-Klassifikation
durchgeführt
Informationen über Rezidive, Metastasen, krankheitsfreie Zeit,
Überlebenszeit und Therapie wurden unter anderem den Aufzeichnungen
und Krankenunterlagen der behandelnden Hausärzten und den jeweiligen
Krankenhäusern entnommen.

37
2.2.2 Konventionelle Histologie
Von den in Paraffin eingebetteten Tumoren wurden Schnittpräparate
angefertigt und mit Hilfe der HE-Färbung (Hämatoxilin-Eosin) nach
histologischem Standardprotokoll gefärbt.
2.2.3 Immunhistochemie
Im Rahmen der Untersuchung wurden Antikörper gegen folgende Antigene
verwendet:
Ki-67-Antigen (MIB1) und EGF-Rezeptor.
Färbemethode:
Die immunhistochemischen Färbungen wurden nach der APAAP-Methode
(alkalische Phosphatase-anti-alkalische Phosphatase) mit Hilfe der
entsprechenden Antikörper angefertigt.
3-4 μm dicke Schnittpräparate des in Formalin fixierten und in Paraffin
eingebetteten Gewebes wurden auf einen speziellen Kapillarobjektträger der
Firma DAKO aufgezogen. Anschliessend wurden die Präparate über Nacht
bei 37 Grad Celsius im Wärmeschrank inkubiert.
Die Schnittpräparate wurden in Xylol entparaffiniert und in einer
absteigenden Alkoholreihe (100%, 96%, 70%) rehydriert und in Tris-Puffer
(pH 7.2) gespült.
Die Präparate wurden mit den Primärantikörpern in deren jeweiligen
Gebrauchsverdünnung für 25 Minuten bei Raumtemperatur inkubiert. Die
Antikörperverdünnung erfolgte mit Hilfe eines Diluents der Firma Zytomed,
Der MiB-1 Antikörper wurde dabei auf 1:500 verdünnt.
Zwischen den einzelnen Inkubationen erfolgten mehrere Waschvorgänge mit
Puffern. Hierzu wurden Puffer aus einem Puffer-Kit der Firma DAKO (K5006)
benutzt.
Nach erneuter Spülung wurden die Präparate mit einem
Sekundärantikörper=Link (APPAP-Kit 5000, Firma DAKO, gebrauchsfertig)
25 Minuten lang bei Raumtemperatur inkubiert. Bei diesem

38
Brückenantikörper handelte es sich um ein Kaninchen-Anti-Maus-
Immunglobulin.
Es erfolgte ein weiterer Spülvorgang und danach eine weitere Inkubation mit
einem APAAP-Komplex (APAAP-Kit K5000, Firma DAKO, gebrauchsfertig)
für 25 Minuten bei Raumtemperatur. Der APAAP-Komplex bestand aus
monoklonalen Maus Ig1-Antikörpern, die spezifisch an die alkalische
Phosphatase aus der Kälberdarmmukosa gebunden waren.
Es erfolgte eine Wiederholung der Schritte 5 und 6 für jeweils 10 Minuten.
Zur Farbenentwicklung wurde ebenfalls das APAAP-Kit benutzt. Beim
Chromogen-Substrat handelte es sich um Neufuchsin. Die Inkubationszeit
betrug 4x5 Minuten, wobei zwischendurch Spülvorgänge mit Tris-Puffer
stattfanden.
Die Gegenfärbung erfolgte in Mayers-Hämatoxylin für 2 Minuten.
Anschließend wurden die Präparate in Wasser für 2 Minuten gebläut und
danach für weitere 2 Minuten in Tris-Puffer verbracht. Durch die aufsteigende
Alkoholreihe wurden die Präparate daraufhin dehydriert, bevor sie über Xylol
mit Eukitt oder Eindeckfolie (Eindeckautomat, Firma Vogel) abgedeckt
wurden.
Für den Nachweis einer EGFR-Expression wurde das DAKO EGFR
pharmDx Kit in folgenden Arbeitsschritten angewendet:
Applikation der Proteinkinase K und Inkubation für 5 Minuten
Applikation des Peroxidase-Blockes und Inkubation für 5 Minuten
Aufbringen des Primärantikörpers und Inkubation für 30 Minuten
Aufbringen des markierten Polymers HRP und Incubationfür 30 Minuten
Aufbringen des Substrat-Chromogens und Inkubation für 10 Minuten
Die Auswertung der nach den Prinzipien der konventionellen Histologie und
der Immunhistochemie gefärbten Präparate erfolgte mit einem
Lichtmikroskop (Axioskop, Carl-Zeiss, Jena). Die Abbildungen wurden mit
Hilfe einer CCD-Kamera (HV-C20 M, Hitachi Denshi Ltd., Japan) angefertigt

39
und mit Hilfe eines Bildbearbeitungsprogrammes (Diskus V4.20, Technisches
Büro Hilgers, Königswinter) am Computer bearbeitet.
2.2.4 Auswertung
Die Auswertung der histologischen Schnitte erfolgte mit Hilfe des
Lichtmikroskopes. Die Tumoren wurden dabei komplett durchgemustert,
repräsentative Areale ausgewählt und fotografiert.
Zur immunhistochemischen Auswertung wurden bei jedem Tumorschnitt
mindestens 15 hpfs (hpf = high power fields, hochvergrößertes Gesichtsfeld)
bei 250-facher Vergrößerung ausgezählt, sofern die Präparate ausreichend
groß waren, wurden 50hpfs bei 250-facher Vergrößerung ausgezählt. Bei der
Auszählung wurde zwischen Hotspots (Areale mit im Vergleich zur
Umgebung deutlich stärkerer Aktivität) und Non-Hotspots unterschieden. Bei
der Einteilung wurde ein Score zur Ki-67-Expression (Choong et
al.,1994/1995) verwendet. Dabei wurde das Verhältnis von markierten zu
nicht markierten Zellen berücksichtigt (siehe Ergebnistabellen).
40
3 Ergebnisse
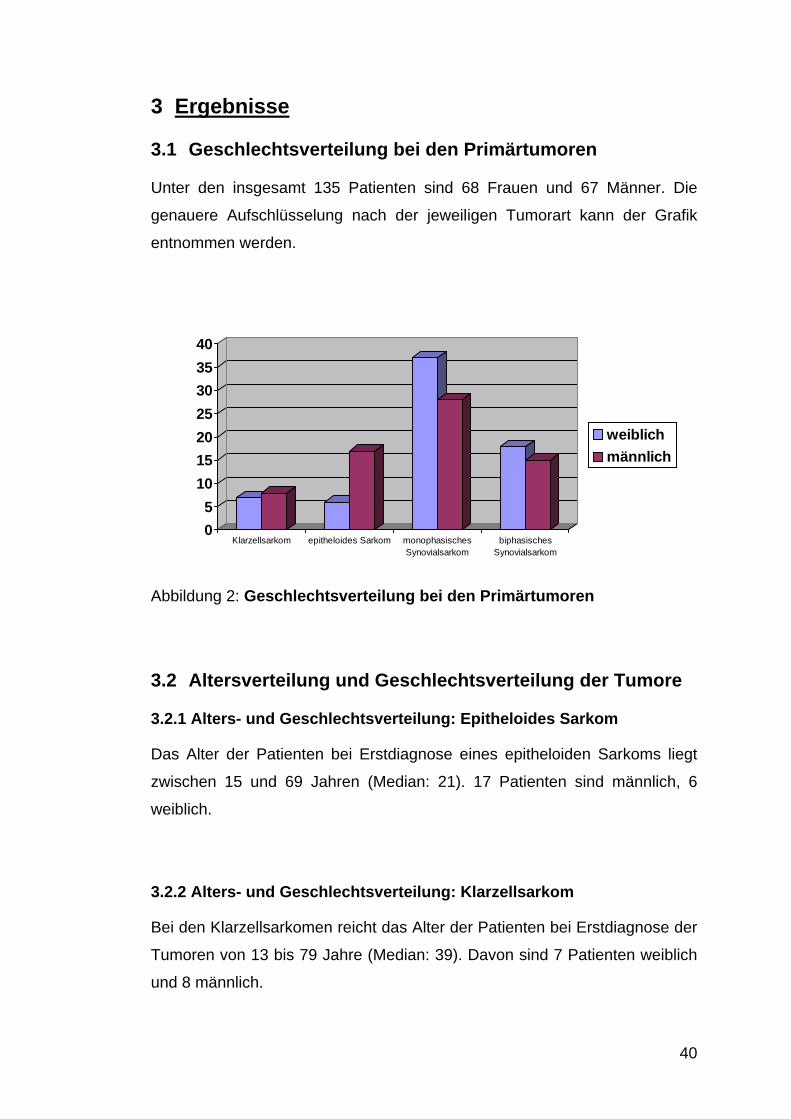
3.1 Geschlechtsverteilung bei den Primärtumoren
Unter den insgesamt 135 Patienten sind 68 Frauen und 67 Männer. Die
genauere Aufschlüsselung nach der jeweiligen Tumorart kann der Grafik
entnommen werden.
05
10152025303540
Klarzellsarkom epitheloides Sarkom monophasischesSynovialsarkom
biphasischesSynovialsarkom
weiblichmännlich
Abbildung 2: Geschlechtsverteilung bei den Primärtumoren
3.2 Altersverteilung und Geschlechtsverteilung der Tumore
3.2.1 Alters- und Geschlechtsverteilung: Epitheloides Sarkom
Das Alter der Patienten bei Erstdiagnose eines epitheloiden Sarkoms liegt
zwischen 15 und 69 Jahren (Median: 21). 17 Patienten sind männlich, 6
weiblich.
3.2.2 Alters- und Geschlechtsverteilung: Klarzellsarkom
Bei den Klarzellsarkomen reicht das Alter der Patienten bei Erstdiagnose der
Tumoren von 13 bis 79 Jahre (Median: 39). Davon sind 7 Patienten weiblich
und 8 männlich.

41
3.2.3 Alters- und Geschlechtsverteilung: Synovialsarkom
Bei der Erstdiagnose eines monophasischen Synovialsarkoms liegt das
Patientenalter zwischen 6 und 80 Jahre (Median: 36, Mittelwert: 39,6), es
handelt sich um 37 Frauen und 31 Männer. Bei der Erstdiagnose eines
biphasischen Synovialsarkoms hingegen reicht das Patientenalter von 17 bis
76 Jahre (Median: 38, Mittelwert: 39,8), davon sind 19 Patienten männlich
und 18 Patienten weiblich.
3.3 Tumorgrößen
Die vorliegenden Tumoren wurden anhand ihrer Größe in zwei Gruppen
eingeteilt. Als Grenzwert wurde eine Größe bis 5 cm für die 1. Gruppe
angenommen, da diese Größe in der Literatur als Grenzwert für einen
schlechteren Krankheitsverlauf gilt (Russel et al., 1977).
Bei 119 der untersuchten Tumoren ist eine genaue Tumorgröße bekannt,
hieraus ergibt sich folgende Einteilung:
3.3.1 Tumorgrößen: Epitheloides Sarkom
Bei den epitheloiden Sarkomen lag die Tumorgröße zwischen 1 cm und 26
cm, in einigen Fällen lagen nur Gewebeproben vor.
Median 4,25 cm und Mittelwert 6,6 cm.
Epitheloide Sarkome bis 5 cm Größe: 12/32
Epitheloide Sarkome über 5 cm Größe: 10/32
Bei 10 Tumoren lag keine Größenangabe vor.
3.3.2 Tumorgrößen: Klarzellsarkome
Bei den Klarzellsarkomen lag die Tumorgröße zwischen 1,2 cm und 7 cm, in
einigen Fällen lagen nur Gewebeproben vor.
Median 2,5cm und Mittelwert 2,8 cm.
Klarzellsarkome bis 5 cm Tumorgröße: 18/30
Klarzellsarkome über 5 cm Tumorgröße: 3/30
Bei 10 Tumoren lag keine Größenangabe vor.

42
3.3.3 Tumorgrößen: Synovialsarkom
Bei den Synovialsarkomen lag die Tumorgröße zwischen 1 cm und 17,9 cm,
in einigen Fällen lagen nur Gewebeproben vor.
Median der Tumorgrößen beim monophasisch-fibrösen Typ: 5,5 cm.
Median der Tumorgrößen beim biphasischen Typ: 5,5 cm.
Biphasische Synovialsarkome bis 5 cm: 10/37
Biphasische Synovialsarkome über 5 cm: 22/37
Bei 5 Tumoren lag keine Größenangabe vor.
Monophasische Synovialsarkome bis 5 cm: 23/68
Monophasische Synovialsarkome über 5 cm: 24/68
Bei 21 Tumoren lag keine Größenangabe vor.

43
3.4 Lokalisation der Tumoren
46 der insgesamt 167 Tumoren waren an der oberen Extremität zu finden,
101 an der unteren Extremität. Bei 20 Tumoren waren keine Angaben zur
Lokalisation vorhanden. Eine genauere Aufschlüsselung der Lokalisationen
der jeweiligen Tumorart sind den Schemata zu entnehmen:
3.4.1 Epitheloides Sarkom
Abbildung 2: Tumorlokalisation des epitheloiden Sarkoms

44
3.4.2 Klarzellsarkom
Abbildung 4: Tumorlokalisation des Klarzellsarkoms

45
3.4.3 Synovialsarkom
Abbildung 5: Abbildung 6:
Tumorlokalisation Tumorlokalisation
des monophasischen des biphasischen
Synovialsarkoms Synovialsarkoms

46
3.5 Malignitätsgrad
Für das Patientenkollektiv der Berufsgenossenschaftlichen
Kliniken Bergmannsheil ergaben sich folgende Verteilungen des
Malignitätsgrades:
3.5.1 Grading: Epitheloides Sarkom
Für insgesamt 32 epitheloide Sarkome konnte ein Grading durchgeführt
werden.
G1: 0/32
G2: 15/32
G3: 17/32
3.5.2 Grading: Klarzellsarkom
Für insgesamt 30 Klarzellsarkome konnte ein Grading durchgeführt werden.
G1: 2/30
G2: 19/30
G3: 9/30

47
3.5.3 Grading: Synovialsarkom
Insgesamt konnte bei 50 Synovialsarkomen ein Grading vorgenommen
werden:
Monophasisches Synovialsarkom:
G1: 0/29
G2: 18/29
G3: 11/29
Biphasisches Synovialsarkom:
G1: 1/21
G2: 13/21
G3: 7/21
3.6 Krankheitsverlauf / Lokalrezidive und Metastasen
3.6.1 Epitheloides Sarkom
Bei 10 von 20 Patienten mit einem epitheloiden Sarkom (= 50%) entwickelte
sich im Beobachtungszeitraum ein Rezidiv, frühestens nach 2 Monaten,
spätestens nach 49 Monaten.
Ein Patient entwickelte 5 Rezidive,
4 der Patienten entwickelten jeweils 2 Rezidive.
Bei 5 der Patienten des Kollektives (= 25%) entwickelten sich Metastasen,
frühestens nach einem Monat, spätestens nach 24 Monaten.
Bei 3 der Patienten kam es zuvor zu einem Rezidiv (= 15% des Kollektives).
3.6.2 Klarzellsarkom
Bei sechs von 13 Patienten mit einem Klarzellsarkom (= 46,2%) entwickelte
sich im Untersuchungszeitraum ein Rezidiv, frühestens 12 Monate,
spätestens 118 Monate nach der Erstdiagnose.
Bei einem Patienten entwickelten sich 7 Rezidive, zwei der Patienten
entwickelten jeweils 4 Rezidive, ein Patient entwickelte 2 Rezidive.

48
Bei 4 der 13 Patienten (= 30,8%) entwickelten sich Metastasen, frühestens
12 Monate nach der Erstdiagnose, spätestens nach 208 Monaten. Bei allen
Patienten mit einer Metastase kam es zuvor zu einem Lokalrezidiv.
3.6.3 Synovialsarkom
Insgesamt entwickelten 38 Patienten mit einem Synovialsarkom Metastasen.
Biphasische Synovialsarkome:
15 der Patienten mit einem biphasischen Synovialsarkom (=40,5%)
entwickelten im Untersuchungszeitraum ein Rezidiv, zwei Patienten hiervon
entwickelten 3 Rezidive, 3 Patienten jeweils zwei Rezidive.
Monophasische Synovialsarkome:
23 der Patienten mit einem monophasischen Synovialsarkom entwickelten im
Untersuchungszeitraum ein Rezidiv (=33,8%), davon entwickelten sich bei 6
Patienten jeweils zwei Rezidive, bei einem Patienten insgesamt 3 Rezidive.

49
3.7 Histologisches Erscheinungsbild
3.7.1 Histologisches Erscheinungsbild : Epitheloides Sarkom
Das histologische Bild der untersuchten epitheloiden Sarkome war
charakterisiert durch häufig in kleineren, nodulären Arealen gelagerte
epitheloide Tumorzellen mit teils in der Übersicht granulomartigem
Wachstumsmuster mit zentraler Nekrose (Abb. 7).
Die epitheloiden, oft polygonalen Tumorzellen ließen zumeist ein deutlich
eosinophiles Zytoplasma mit teils vesikulären, teils hyperchromatischen
Kernen erkennen (Abb. 8 und 9).
Neben epitheloiden fanden sich Übergänge in eher spindelige Tumorzellen.
Bei der großzelligen Variante, dem sogenannten großzelligen, „proximalen“
epitheloiden Sarkom fanden sich große, polygonale Tumorzellen mit
praktisch ausschließlich vesikulären Kernen und vermehrter Zelldissoziation,
ohne spindelige Anteile.
Immunhistochemisch zeigten die epitheloiden Sarkome eine Koexpression
von Keratin und/oder EMA und Vimentin (Abb. 8).

50
Abb. 7: Histologie epitheloides Sarkom (1)
a: granulomartige Tumorzellanordnung und zentrale Nekrose
b: epitheloide neben mehr plump-ovalären bis spindeligen Tumorzellen im
epitheloiden Sarkom
c: polygonale epitheloide Tumorzellen mit paranukleären eosinophilen, teils
rhabdoid-ähnlichen Zytoplasmastrukturen als Korrelat für
Intermediärfilamente
d: starke zytoplasmatische Keratinexpression der Tumorzellen
51
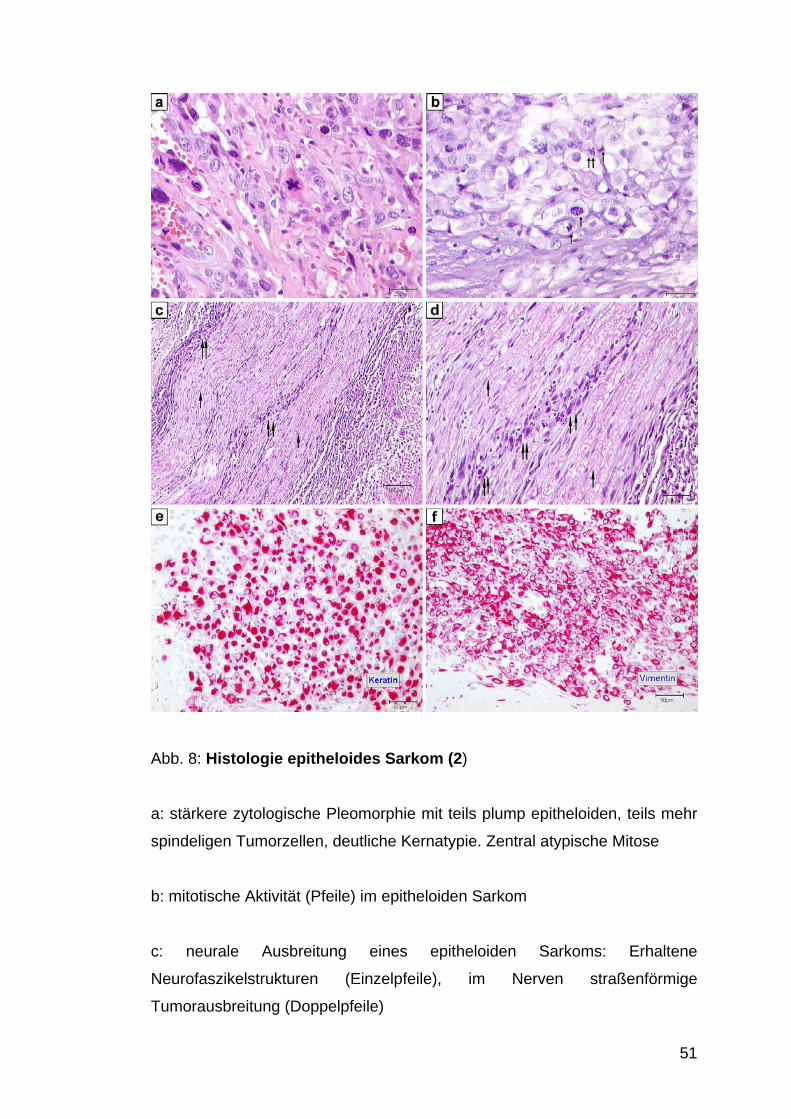
Abb. 8: Histologie epitheloides Sarkom (2)
a: stärkere zytologische Pleomorphie mit teils plump epitheloiden, teils mehr
spindeligen Tumorzellen, deutliche Kernatypie. Zentral atypische Mitose
b: mitotische Aktivität (Pfeile) im epitheloiden Sarkom
c: neurale Ausbreitung eines epitheloiden Sarkoms: Erhaltene
Neurofaszikelstrukturen (Einzelpfeile), im Nerven straßenförmige
Tumorausbreitung (Doppelpfeile)

52
d: stärkere Vergrößerung aus C: Straßenförmig angeordnete Tumorzellen
(Doppelpfeile) mit Ausbreitung in einem Nerven. Erhaltene Nervenfasern:
Doppelpfeile
e: starke Keratin-Expression
f: starke Vimentinexpression im epitheloiden Sarkom

53
Abb.9: Histologie epitheloides Sarkom (3)
a: Lymphknotenmetastase eines epitheloiden Sarkoms: Tumorinfiltrate
überwiegend im Randsinusbereich, zentral erhaltener sekundärer
Lymphfollikel
b: stärkere Vergrößerung aus a: Obere Bildhälfte mit originären Lymphozyten
des Lymphknotens, in der unteren Bildhälfte epitheloide Tumorzellen mit
vesikulären Kernen und prominenten Nukleolen
c: proximale (großzellige) Variante des epitheloiden Sarkoms: Große
Tumorzellen mit esinophilem Zytoplasma, vesikuläre Kerne und deutliche
Nukleolen
d: Epitheloides Sarkom mit mehr spindeligen Tumorzellen.
Hämosiderineinlagerungen (linke Bildhälfte) und Tumornekrose (rechte
Bildhälfte)

54
3.7.2 Histologisches Erscheinungsbild: Klarzellsarkom
Die untersuchten Klarzellsarkome waren durch häufig ballenartig gelagerte
Tumorzellkomplexe mit vesikulären Kernen und deutlichen Nukleolen, teils
ähnlich dem histologischen Erscheinungsbild der Melanome (Abb.10 und
11).
Es fanden sich häufig fibröse, septenartige Bindegewebszüge innerhalb des
Tumors, auch Riesenzellen waren nachweisbar. Immunhistochemisch war
eine Expression von S100 und HMB-45 (melanozytärer Marker) typisch
(Abb.13).
Makroskopisch imponiert ein weiß nodulärer Tumor mit weißer Schnittfläche
und Einblutungen (Abb. 10).

55
Abb. 10: Histologie Klarzellsarkom (1)
a. Makroskopisches Bild eines Klarzellsarkoms, Schnittfläche

56
b: typische Histologie des Klarzellsarkoms mit ballenförmig gelagerten
Tumorzellen mit relativ hellem Zytoplasma, vesikuläre Kerne mit prominenten
Nukleolen
c: ballenförmige Lagerung von Tumorzellen mit kleinen Bindegewebssepten,
mehrkernige Tumorzelle (rechter Bildrand)
d: starke zytoplasmatische und nukleäre S-100 Expression im Klarzellsarkom
e: starke zytoplasmatische Markierung mit dem Antikörper HMB-45
57
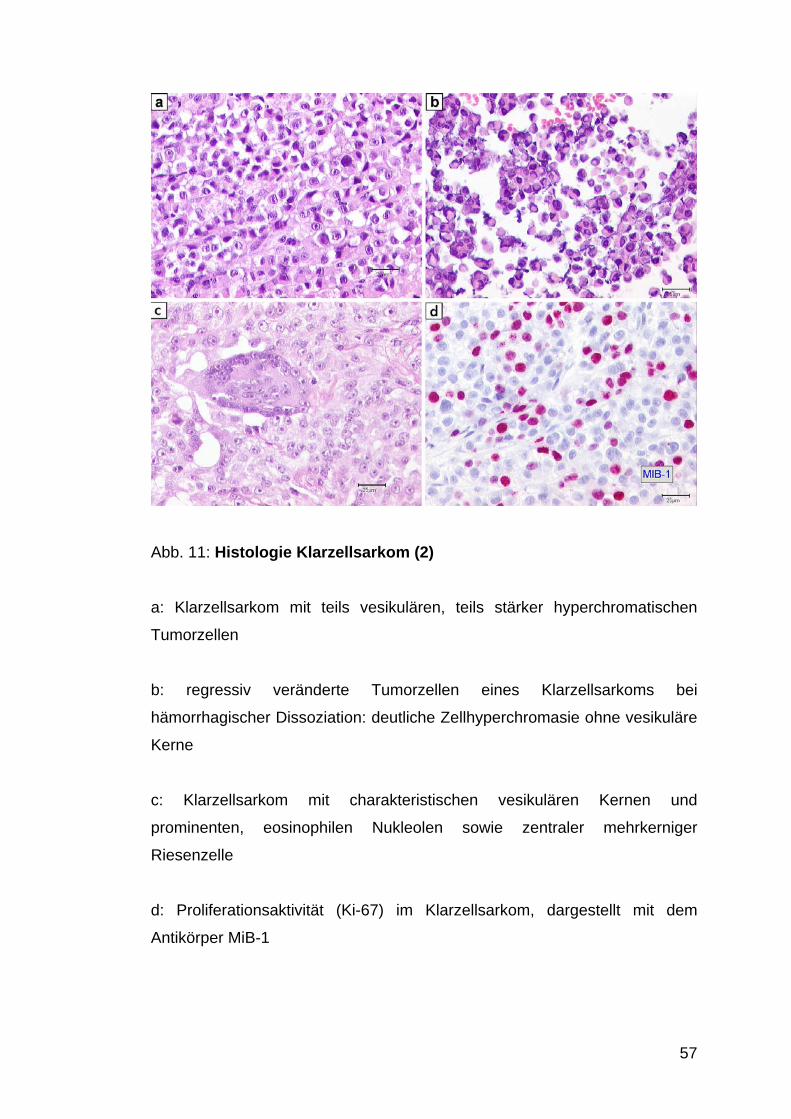
Abb. 11: Histologie Klarzellsarkom (2)
a: Klarzellsarkom mit teils vesikulären, teils stärker hyperchromatischen
Tumorzellen
b: regressiv veränderte Tumorzellen eines Klarzellsarkoms bei
hämorrhagischer Dissoziation: deutliche Zellhyperchromasie ohne vesikuläre
Kerne
c: Klarzellsarkom mit charakteristischen vesikulären Kernen und
prominenten, eosinophilen Nukleolen sowie zentraler mehrkerniger
Riesenzelle
d: Proliferationsaktivität (Ki-67) im Klarzellsarkom, dargestellt mit dem
Antikörper MiB-1

58
3.7.3 Histologisches Erscheinungsbild: Synovialsarkom
Im vorliegenden Kollektiv ließen sich 33,7% biphasische Synovialsarkome
von 63,4% monophasischen Synovialsarkomen unterscheiden.
In 2,9% lag der schlecht differenzierte (undifferentiated) Typ mit überwiegend
runden, kleinen Tumorzellen (dieser wurde bei deutlichem Überwiegen der
kleinzelligen Komponente angegeben) vor.
Der in der Literatur beschriebene rein epitheliale Subtyp mit großen,
epitheloiden Tumorzellen konnte in dem hier vorliegenden Untersuchungsgut
nicht identifiziert werden (Abb. 15).
Mikroskopische Ergebnisse monophasisches Synovialsarkom
Die untersuchten Tumoren wiesen zum Teil ein sehr unterschiedliches
Erscheinungsbild auf. Es fanden sich große, solide Tumoren mit großen
zystischen Strukturen oder ohne Zysten, aber auch zerklüftete Tumoren
unterschiedlicher Größe mit nekrotischen Arealen aller Größen mit und ohne
Einblutungen. Manche Tumoren bestanden aus mehreren ballenartig
zusammengelagerten Tumornestern, mit oder ohne Bindegewebe. Zwei der
untersuchten Tumoren wiesen eine palisadenartige Anordnung der Zellen
auf, ähnlich dem Bild eines Nervenscheidentumors. Zwei weitere der
Tumoren wiesen ein für Synovialsarkome untypisches pflanzenzellartiges
Erscheinungsbild der Zellen auf.
Bei den Tumoren fielen jeweils epithelartige, runde Zellen, sowie
Spindelzellen mit unterschiedlichem Verteilungsmuster auf. In fast allen
Tumoren überwog die spindelzellige Variante, diese war von wenigen
Rundzellen durchsetzt. In neun der untersuchten Tumoren waren deutlich
mehr rundzellige Anteile zu finden, 3 dieser Tumoren waren dem
rundzelligen, schlecht differenzierten Synovialsarkomtyp zuzuordnen.
Die MIB-1-Expression fiel unterschiedlich stark aus. Es traten Tumoren mit
einer sehr hohen Expression von über 25%, jedoch auch Tumoren ohne eine
MIB-1 Expression auf.
Bei einigen Tumoren waren Hotspotareale deutlich zu erkennen, bei anderen
fand sich eine gleichmäßige Verteilung der MIB-1-Expression.

59
Wie die MIB-1-Expression, fiel auch die Keratin und EMA-Expression sehr
unterschiedlich aus. Es fanden sich Tumoren ohne jegliche Keratin-und
EMA-Expression, aber auch Tumoren mit starker Expression eines oder
beider immunhistochemischen Färbungen. Insgesamt fiel eine stärkere EMA-
Expressionsrate in den spindelzelligen Anteilen auf, die Keratinexpression
war insgesamt stärker in den rundzelligen Anteilen.
Mikroskopische Ergebnisse biphasisches Synovialsarkom
Das Erscheinungsbild der untersuchten biphasischen Synovialsarkome war
wie bei dem der monophasischen Synovialsarkome sehr unterschiedlich. Es
traten solide Tumoren mit einer deutlichen Trennung der beiden
Zellkomponenten auf: Nester von epithelartigen, runden Zellen, durch
Spindelzellzüge voneinander abgegrenzt, aber auch weniger deutliche
Muster. Einige Tumoren waren durch Bindegewebe zum gesunden Gewebe
deutlich abgegrenzt. Einige wuchsen diffus in das gesunde Gewebe ein, das
Muster der Gefäßverteilung erschien häufig hämangioperizytomartig. Im
Gegensatz zu den monophasischen Synovialsarkomen fanden sich fast
keine zystische Strukturen, dafür häufig Verkalkungen. Einige Tumoren
wiesen Nekrosen mit und ohne Einblutungen auf, einige
Entzündungsreaktionen.
Die Keratin und EMA-Expression fiel unterschiedlich stark, insgesamt jedoch
in den rundzelligen Arealen deutlich stärker aus. Konnten im Tumor die
Zellkomponenten deutlich in epitheliale und fibrös/spindelzellige Areale
abgegrenzt werden, ließ sich dort auch ein Verteilungsmuster bezüglich der
MIB-1-Expression erkennen: In diesen Tumoren war die MIB-1-
Expressionsrate in den epithelialen Arealen deutlich stärker. Die
spindelzelligen Areale wiesen eine geringere oder keine MIB-1-Expression
auf (Abb. 15).
Makroskopisch imponiert jeweils ein bräunlich/beiger Tumor mit weißlichen
bis hellgelben stippchenartigen Herden (Abb. 12).

60
Abb.12: Makroskopisches und radiologisches Erscheinungsbild des Synovialsarkoms
A: Synovialsarkom mit Lokalisation in der Oberschenkelmuskulatur:
bräunlich-beiger Tumor mit weißlichen bis hellgelben, stippchenartigen
Herden der Schnittfläche, korrespondierend zu kleinen Verkalkungen

61
B: Korrespondierendes konventionelles Röntgenbild des Tumors aus A:
Verkalkungen im Weichteilmantel des Oberschenkels
C: Kernspintomogramm des Tumors aus A und B
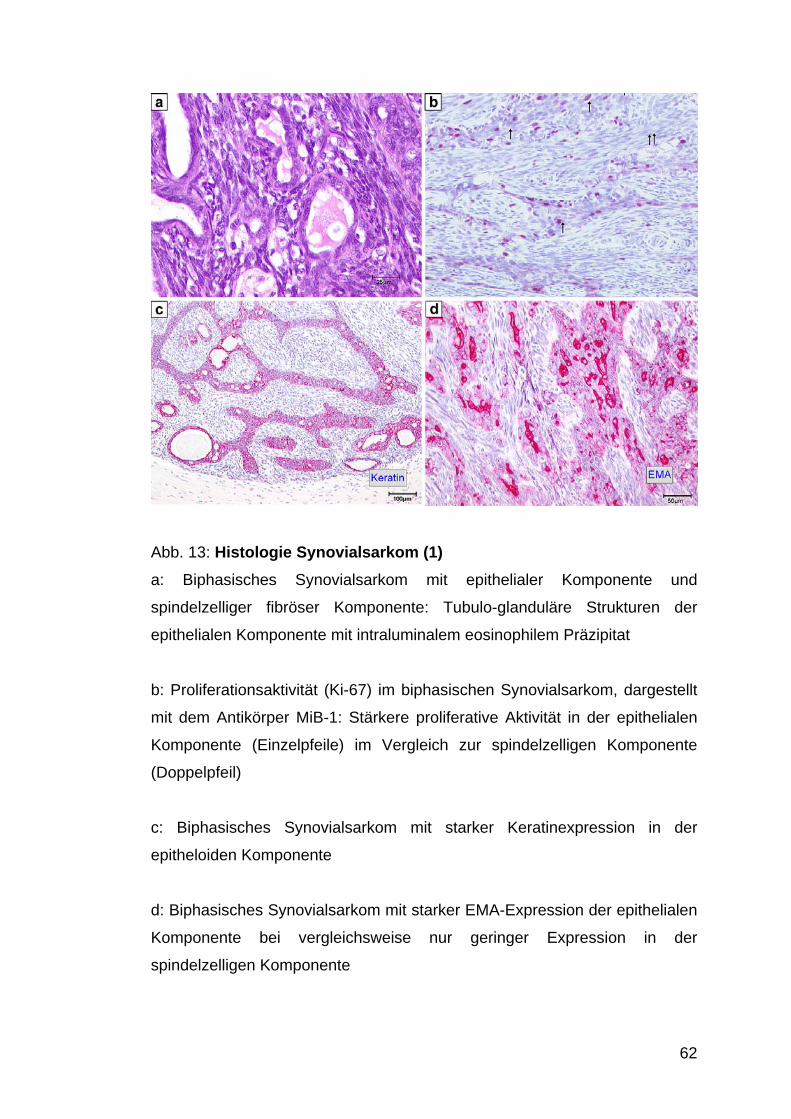
62
Abb. 13: Histologie Synovialsarkom (1) a: Biphasisches Synovialsarkom mit epithelialer Komponente und
spindelzelliger fibröser Komponente: Tubulo-glanduläre Strukturen der
epithelialen Komponente mit intraluminalem eosinophilem Präzipitat
b: Proliferationsaktivität (Ki-67) im biphasischen Synovialsarkom, dargestellt
mit dem Antikörper MiB-1: Stärkere proliferative Aktivität in der epithelialen
Komponente (Einzelpfeile) im Vergleich zur spindelzelligen Komponente
(Doppelpfeil)
c: Biphasisches Synovialsarkom mit starker Keratinexpression in der
epitheloiden Komponente
d: Biphasisches Synovialsarkom mit starker EMA-Expression der epithelialen
Komponente bei vergleichsweise nur geringer Expression in der
spindelzelligen Komponente

63
3.8 MIB-1 Expression
In der vorliegenden Arbeit wurden 30 Klarzellsarkome und 32 Epitheloide
Sarkome auf MiB-1-Expression untersucht. In lediglich vier Fällen (davon 3
Klarzellsarkome und ein Epitheloides Sarkom) zeigte sich keine MiB-1
Expression. Die meisten der vorliegenden Tumoren (48 Tumoren= 77,4%)
wiesen eine MiB-1 Expressionsrate >10% und <25% auf.
3.8.1 MiB-1 Expression in Abhängigkeit von der Tumorgröße
Zwischen Tumorgröße und MiB-1 Expression konnte kein Zusammenhang
festgestellt werden.
3.8.2 MiB-1 Expression in Korrelation zum Tumorgrading
Zwischen dem Tumorgrading und der MiB-1 Expressionsrate bestand in der
vorliegenden Untersuchung eine positive Korrelation.
Höher maligne Tumoren wiesen dabei eine höhere MiB-1-Expressionsrate
auf als niedriger maligne. Aufgrund der niedrigen Fallzahl war keine
statistische Aussage möglich.
Folgende Verteilungen der MiB-1-Expression in Abhängigkeit vom Grading
ergeben sich:
Klarzellsarkome G1 (2 Tumoren):
Weder in Hotspot-Bereichen noch in Non-Hotspotbereichen ist eine MiB-1-
Expression vorhanden.
Klarzellsarkome G2 (19 Tumore):
<10% MiB-1 positive Zellen : 1 Tumor = 5,3%
>10%<25% MiB-1 positive Zellen: 16 Tumore = 84,2%
>25% MIB-1 positive Zellen: 2 Tumore = 10,5%

64
Klarzellsarkome G3 (9 Tumore):
<10% MiB-1 positive Zellen: 0 Tumore
>10%<25% MiB-1 positive Zellen : 6 Tumore= 67%
>25% MiB-1 positive Zellen: 3 Tumore= 33%
Unter den epitheloiden Sarkomen ist kein Tumor mit einem Grading von 1
vorhanden.
epitheloides Sarkom G2 (14 Tumore):
<10% MiB-1 positive Zellen: 5 Tumore = 35,7%
>10%<25% MiB-1 positive Zellen: 6 Tumore = 42,9%
>25% MiB-1 positive Zellen: 3 Tumore = 21,4%
epitheloides Sarkom G3 (16 Tumore):
<10% MiB-1 positive Zellen: 0 Tumore
>10%<25% MiB-positive Zellen: 3 Tumore = 18,75%
>25% MiB-positive Zellen: 13 Tumore = 81,3%
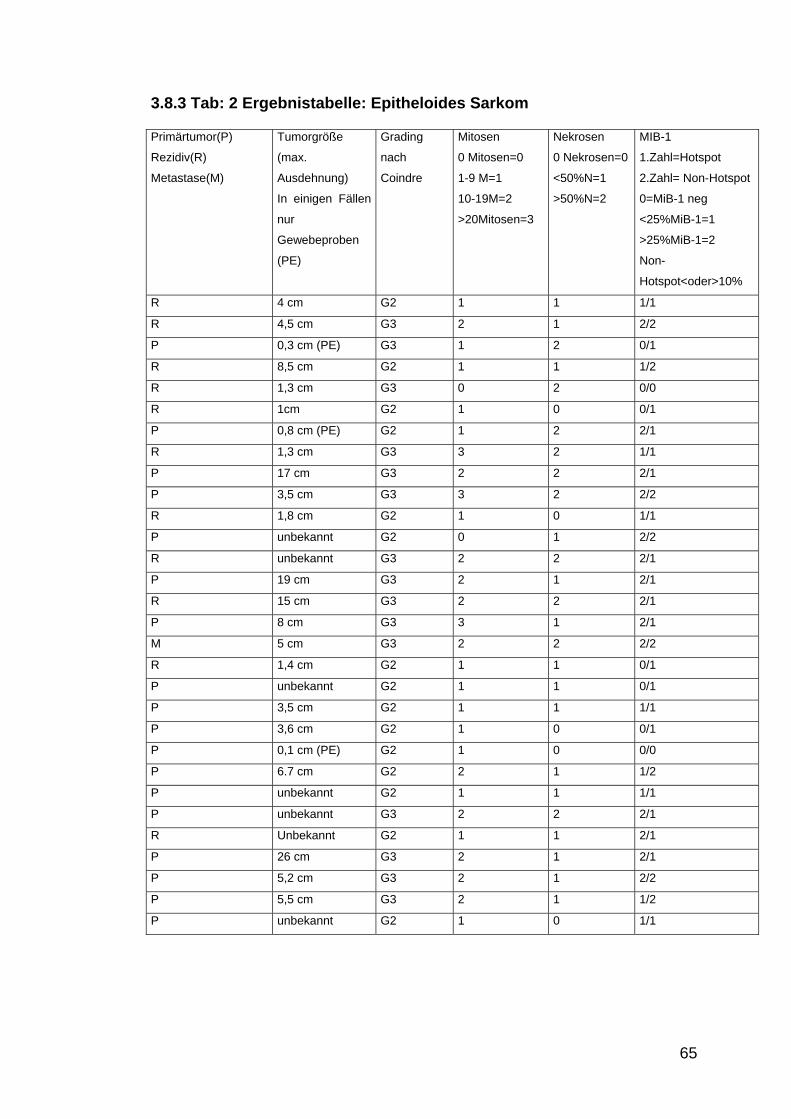
65
3.8.3 Tab: 2 Ergebnistabelle: Epitheloides Sarkom
Primärtumor(P)
Rezidiv(R)
Metastase(M)
Tumorgröße
(max.
Ausdehnung)
In einigen Fällen
nur
Gewebeproben
(PE)
Grading
nach
Coindre
Mitosen
0 Mitosen=0
1-9 M=1
10-19M=2
>20Mitosen=3
Nekrosen
0 Nekrosen=0
<50%N=1
>50%N=2
MIB-1
1.Zahl=Hotspot
2.Zahl= Non-Hotspot
0=MiB-1 neg
<25%MiB-1=1
>25%MiB-1=2
Non-
Hotspot<oder>10%
R 4 cm G2 1 1 1/1
R 4,5 cm G3 2 1 2/2
P 0,3 cm (PE) G3 1 2 0/1
R 8,5 cm G2 1 1 1/2
R 1,3 cm G3 0 2 0/0
R 1cm G2 1 0 0/1
P 0,8 cm (PE) G2 1 2 2/1
R 1,3 cm G3 3 2 1/1
P 17 cm G3 2 2 2/1
P 3,5 cm G3 3 2 2/2
R 1,8 cm G2 1 0 1/1
P unbekannt G2 0 1 2/2
R unbekannt G3 2 2 2/1
P 19 cm G3 2 1 2/1
R 15 cm G3 2 2 2/1
P 8 cm G3 3 1 2/1
M 5 cm G3 2 2 2/2
R 1,4 cm G2 1 1 0/1
P unbekannt G2 1 1 0/1
P 3,5 cm G2 1 1 1/1
P 3,6 cm G2 1 0 0/1
P 0,1 cm (PE) G2 1 0 0/0
P 6.7 cm G2 2 1 1/2
P unbekannt G2 1 1 1/1
P unbekannt G3 2 2 2/1
R Unbekannt G2 1 1 2/1
P 26 cm G3 2 1 2/1
P 5,2 cm G3 2 1 2/2
P 5,5 cm G3 2 1 1/2
P unbekannt G2 1 0 1/1
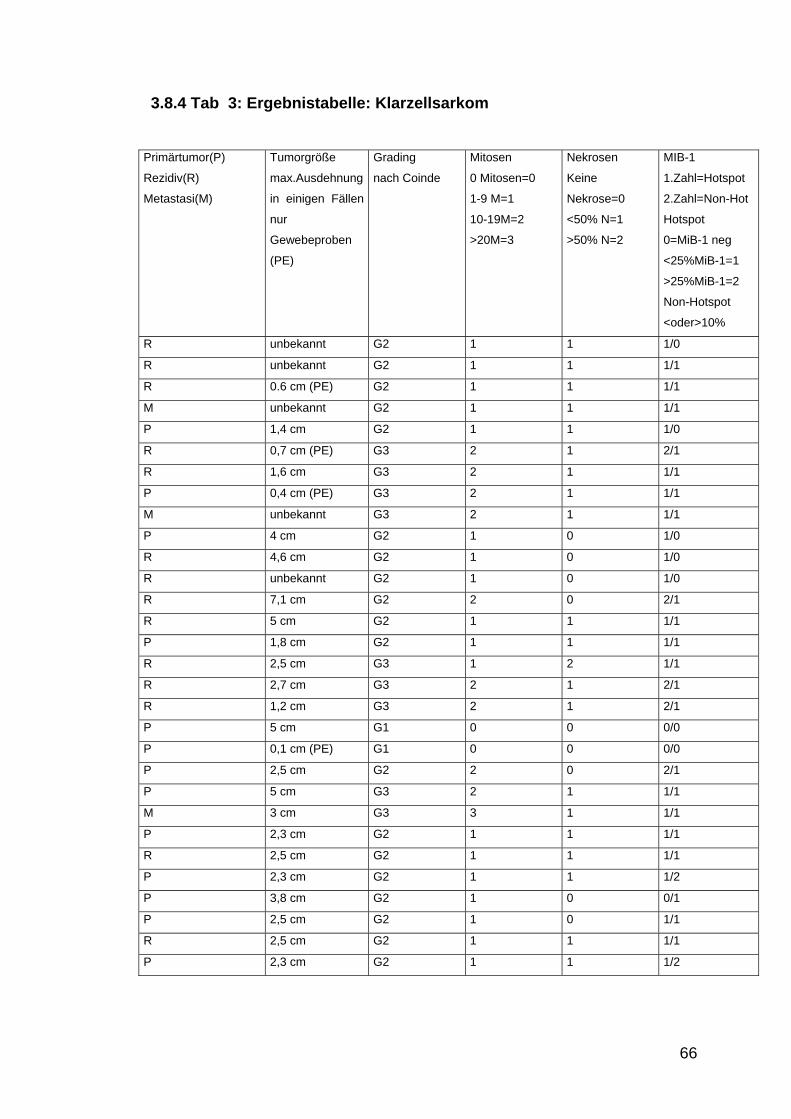
66
3.8.4 Tab 3: Ergebnistabelle: Klarzellsarkom
Primärtumor(P)
Rezidiv(R)
Metastasi(M)
Tumorgröße
max.Ausdehnung
in einigen Fällen
nur
Gewebeproben
(PE)
Grading
nach Coinde
Mitosen
0 Mitosen=0
1-9 M=1
10-19M=2
>20M=3
Nekrosen
Keine
Nekrose=0
<50% N=1
>50% N=2
MIB-1
1.Zahl=Hotspot
2.Zahl=Non-Hot
Hotspot
0=MiB-1 neg
<25%MiB-1=1
>25%MiB-1=2
Non-Hotspot
<oder>10%
R unbekannt G2 1 1 1/0
R unbekannt G2 1 1 1/1
R 0.6 cm (PE) G2 1 1 1/1
M unbekannt G2 1 1 1/1
P 1,4 cm G2 1 1 1/0
R 0,7 cm (PE) G3 2 1 2/1
R 1,6 cm G3 2 1 1/1
P 0,4 cm (PE) G3 2 1 1/1
M unbekannt G3 2 1 1/1
P 4 cm G2 1 0 1/0
R 4,6 cm G2 1 0 1/0
R unbekannt G2 1 0 1/0
R 7,1 cm G2 2 0 2/1
R 5 cm G2 1 1 1/1
P 1,8 cm G2 1 1 1/1
R 2,5 cm G3 1 2 1/1
R 2,7 cm G3 2 1 2/1
R 1,2 cm G3 2 1 2/1
P 5 cm G1 0 0 0/0
P 0,1 cm (PE) G1 0 0 0/0
P 2,5 cm G2 2 0 2/1
P 5 cm G3 2 1 1/1
M 3 cm G3 3 1 1/1
P 2,3 cm G2 1 1 1/1
R 2,5 cm G2 1 1 1/1
P 2,3 cm G2 1 1 1/2
P 3,8 cm G2 1 0 0/1
P 2,5 cm G2 1 0 1/1
R 2,5 cm G2 1 1 1/1
P 2,3 cm G2 1 1 1/2

67
3.9 EGF-R-Expression
3.9.1 Epitheloides Sarkom
Es wurden exemplarisch 2 epitheloide Sarkome auf eine EGF-R-Expression
untersucht. Beide Tumoren zeigten eine deutliche EGF-R-Expression (Abb.
15).
3.9.2 Klarzellsarkom
4 Klarzellsarkome wurden auf eine EGF-R-Expression untersucht. Keiner der
Tumoren zeigte eine EGF-R-Expression (Abb. 14 und 15).
3.9.3 Synovialsarkom
22 Synovialsarkome wurden auf eine EGF-R-Expression untersucht, davon 9
monophasisch-fibröse und 13 biphasische Synovialsarkome.
96% der untersuchten Synovialsarkome wiesen eine EGF-R-Expression auf.
In 59% der Tumoren war eine starke immunhistochemische EGF-R-
Expression mit membranöser Lokalisation nachzuweisen.
Auffällig war insbesondere das Verteilungsmuster der EGF-R Expression in
biphasischen Synovialsarkomen:
In nahezu allen Fällen fand sich eine starke Immunreaktivität in der fibrösen
Komponente, wobei in der epitheloid differenzierten Komponente weitgehend
keine EGF-R-Expression zu erkennen war.
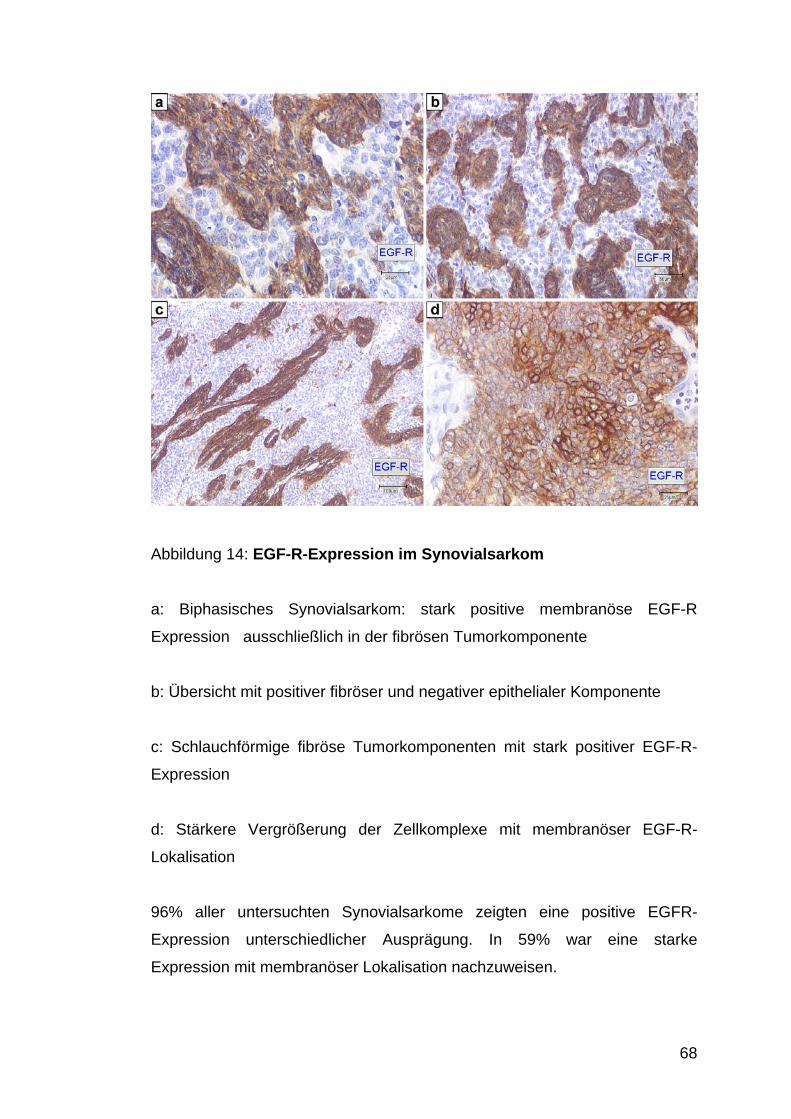
68
Abbildung 14: EGF-R-Expression im Synovialsarkom
a: Biphasisches Synovialsarkom: stark positive membranöse EGF-R
Expression ausschließlich in der fibrösen Tumorkomponente
b: Übersicht mit positiver fibröser und negativer epithelialer Komponente
c: Schlauchförmige fibröse Tumorkomponenten mit stark positiver EGF-R-
Expression
d: Stärkere Vergrößerung der Zellkomplexe mit membranöser EGF-R-
Lokalisation
96% aller untersuchten Synovialsarkome zeigten eine positive EGFR-
Expression unterschiedlicher Ausprägung. In 59% war eine starke
Expression mit membranöser Lokalisation nachzuweisen.
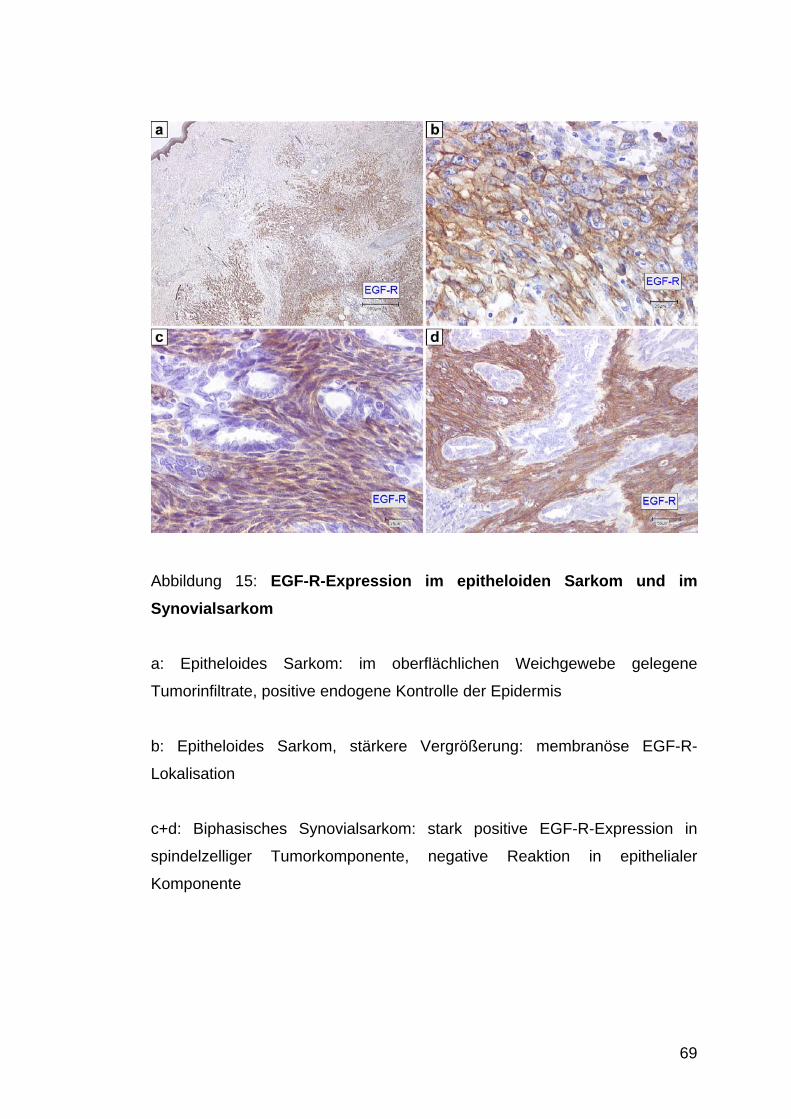
69
Abbildung 15: EGF-R-Expression im epitheloiden Sarkom und im Synovialsarkom
a: Epitheloides Sarkom: im oberflächlichen Weichgewebe gelegene
Tumorinfiltrate, positive endogene Kontrolle der Epidermis
b: Epitheloides Sarkom, stärkere Vergrößerung: membranöse EGF-R-
Lokalisation
c+d: Biphasisches Synovialsarkom: stark positive EGF-R-Expression in
spindelzelliger Tumorkomponente, negative Reaktion in epithelialer
Komponente

70
4 Diskussion
4.1 Geschlechtsverteilung
4.1.1 Epitheloides Sarkom
Bezüglich des Geschlechts scheint laut Literatur keine eindeutige
Bevorzugung vorzuliegen: Die Geschlechtsverteilungen der an einem
epitheloiden Sarkom erkrankten Personen variiert von Kollektiv zu Kollektiv
(Callister et al., 2001). In dem hier untersuchten Kollektiv sind hingegen
deutlich mehr männliche Personen als weibliche erkrankt: 18 Männer : 6
Frauen.
4.1.2 Klarzellsarkom
Die Geschlechtsverteilung der an einem Klarzellsarkom erkrankten Personen
variiert von Kollektiv zu Kollektiv, jedoch wird in einigen Kollektiven eine
Bevorzugung des weiblichen Geschlechts angegeben (s.u.).
In dem hier vorliegenden Kollektiv der Klarzellsarkome läßt sich keine
Geschlechtsbevorzugung ableiten: Es erkrankten 7 Frauen und 8 Männer.
4.1.3 Synovialsarkom
An dem Subtyp des monophasischen Synovialsarkomes erkrankten im
Bergmannsheilkollektiv 37 Frauen und 31 Männer.
An den biphasischen Synovialsarkomen erkrankten 18 Frauen und 19
Männer, somit liegt keine Geschlechtsbevorzugung vor.
Gleiche Verhältnisse findet man bei den in der Literatur vorhandenen
Kollektiven: Machen et al. untersuchten insgesamt 34 Tumoren, davon 15
Synovialsarkome bei weiblichen Patienten und 19 Synovialsarkome bei
Männern (Machen et al., 1999), bei Folpe et al. und Singer et al. überwog
das weibliche Geschlecht (Folpe et al., 1998, Singer et al., 1996).

71
4.2 Altersverteilung
4.2.1 Epitheloides Sarkom
Übereinstimmend mit der Literatur liegt in dem hier untersuchten Kollektiv der
Epitheloiden Sarkome eine große Altersspanne bei Erstdiagnose vor (15-57
Jahre), im Vergleich zu anderen Kollektiven liegt jedoch eine geringere
Alterspanne vor: Callister et al. : 3-72 Jahre (Callister et al., 2001), Bos et al.:
8-80 Jahre (Bos et al., 1988). Das Erkrankungsmaximum liegt in allen
Kollektiven im jungen Erwachsenenalter.
4.2.2 Klarzellsarkom
Ähnliche Verhältnisse liegen bei den Klarzellsarkomen vor.
Eckard et al. untersuchten 27 Klarzellsarkome bei 18 Frauen und 9 Männern
von 9-57 Jahren und einem Durchschnittsalter von 26 Jahren bei den Frauen
und 33 Jahren bei Männern. Deenik et al. untersuchten 30 Tumore bei 16
Männern und 14 Frauen von 9-68 Jahren (Deenik et al., 1999), das
Durchschnittsalter betrug bei seinem Kollektiv 30,8 Jahre. Lucas et al.
untersuchten 35 Tumore an der Mayo-Klinik (Lucas et al. 1992), in seinem
Kollektiv überwiegt das weibliche Geschlecht gegenüber dem männlichen
(1.5:1), das Durchschnittsalter liegt bei 30 Jahren, die Altersspanne reicht
von 10-64 Jahre.
In dem größten bisher untersuchten Kollektiv von Klarzellsarkomen des
Armed Forces Institute of Pathology wurden 141 Klarzellsarkome bei 66
Männern und 75 Frauen untersucht (Chung et al., 1983). Das Alter reichte
von 7-83 Jahren, das Durchschnittsalter lag bei 27 Jahren.
In der hier vorliegenden Arbeit wurden 30 Klarzellsarkome bei 15 Patienten,
davon 7 Frauen und 8 Männer untersucht, eine Geschlechtsbevorzugung
lässt sich somit für das Bergmannsheilkollektiv nicht feststellen. Die
Altersspanne lag zwischen 13 und 79 Jahren, mit einem Median von 39
Jahren und ist somit vergleichbar zu den anderen Patientenkollektiven.
Zito et al. beschrieben einen Zusammenhang zwischen Patientenalter und
Prognose der Erkrankung (Zito et al., 1984), andere Autoren fanden diesen
Zusammenhang nicht (El-Naggar et al., 1990, Brodsky et al., 1992). Auch in
dem hier vorliegendem Kollektiv konnte dieses nicht bestätigt werden.

72
4.2.3 Synovialsarkom
Bei den monophasischen Synovialsarkomen lag das Patientenalter bei
Erstdiagnose zwischen 6 und 80 Jahren, das Durchschnittsalter lag bei 39,6
Jahren.
Bei den biphasischen Synovialsarkomen lag das Alter der Patienten bei
Erstdiagnose zwischen 17 und 75 Jahren, das Duchschnittsalter lag bei 38
Jahren.
Machen et al. unterschieden nicht zwischen monophasischem und
biphasischem Typ (Machen et. al., 1999). Das Durchschnittsalter des von
Ihnen untersuchten Kollektives lag bei 36 Jahren, die Altersspanne reichte
ähnlich wie bei dem hier vorliegendem Kollektiv von 11-82 Jahren.
4.3 Tumorgrößen
Ebenso wie die in der Literatur untersuchten Kollektive, weist auch das hier
untersuchte Kollektiv eine große Variabilität der Tumorgrößen auf.
4.3.1 Epitheloides Sarkom
Das kleinste epitheloide Sarkom des Bergmannsheil Kollektives weist einen
Durchmesser von 1 cm auf, das größte epitheloide Sarkom 26 cm.
In den bisher in der Literatur vorhandenen Untersuchungen waren keine
genauen Tumorgrößen angegeben, lediglich Größenkategorien von unter
5 cm und über 5 cm, oder es handelte sich um einzelne Fallbeschreibungen.
4.3.2 Klarzellsarkom
Bei den Klarzellsarkomen finden sich ähnliche Tumorgrößen: 1,2 cm
Durchmesser weist das kleinste untersuchte Klarzellsarkom auf, 13 cm das
größte Klarzellsarkom des hier untersuchten Patientenkollektives. Im
Vergleich dazu fanden sich in dem von Deenik et al. untersuchten Kollektiv
Tumorgrößen von 2-14,4 cm (Deenik et al., 1998), im Kollektiv von Lucas et
al. fanden sich Tumorgrößen von 1-14,5 cm (Lucas et al., 1992).

73
4.3.3 Synovialsarkom
Bei den untersuchten monophasischen Synovialsarkomen ist der kleinste
Tumordurchmesser 0,8 cm, der größte 17,9 cm, bei den biphasischen
Synovialsarkomen 1 cm und 17 cm. Das kleinste von Machen et al.
untersuchte Synovialsarkom hat einen Durchmesser von 1,2 cm, das größte
16 cm, wobei keine Unterteilung zwischen monophasischem und
biphasischem Synovialsarkom vorgenommen wurde (Machen et al., 1999).
Der Median der Tumorgröße lag bei 6 cm.
4.4 Lokalisation der Tumoren
Übereinstimmend mit der Literatur finden sich die in dieser Arbeit
untersuchten Weichgewebstumore überwiegend an den Extremitäten.
4.4.1 Epitheloides Sarkom
In der Literatur findet man unterschiedliche Angaben zur Lokalisation der
epitheloiden Sarkome.
Übereinstimmend mit Bos et al. (Bos et al., 1988) überwiegt bei dem hier
untersuchten Kollektiv der epitheloiden Sarkome als Lokalisation die obere
Extremität, hier finden sich 75% der Tumoren.
Im Vergleich zu den anderen Kollektiven findet sich im Bergmannsheil-
Kollektiv jedoch kein Tumor an den Fingern.
Auffällig ist ein Tumor („proximaler Typ“) fern der Extremitäten, im
Damm/Leistenbereich. Für den proximalen Typ des epitheloiden Sarkoms ist
diese Loklaisation charakteristisch.
Im Gegensatz zum Bergmannsheil-Kollektiv und dem Mayoklinik-Kollektiv
traten im Kollektiv von Callister (Callister et al., 2001) die epitheloiden
Sarkome in gleicher Häufigkeit im Bereich der oberen und unteren Extremität
auf, jedoch fanden sich ebenso häufig Tumoren im Bereich des Rumpfes.

74
4.4.2 Klarzellsarkom
Bei den Klarzellsarkomen des Bergmannsheilkollektives finden sich 75% der
Tumoren an der unteren Extremität, 25% an der oberen Extremität, keiner
der Tumoren ist an anderen Lokalisationen zu finden. Im Vergleich dazu
waren im Kollektiv von Lucas et al. 90% der Tumoren an den Extremitäten zu
finden am häufigsten am Fuß, jedoch auch zwei Tumoren am Stamm und ein
Tumor in der Kopf-Hals-Region (Lucas et al., 1992).
Ähnliche Ergebnisse lagen auch im Patientenkollektiv von Chung et al. vor:
97% der Tumoren waren an den Extremitäten lokalisiert, 75% davon an der
unteren Extremität, lediglich vier der 141 Tumoren fanden sich am
Körperstamm (Chung et al., 1983).
4.4.3 Synovialsarkom
Bei den Synovialsarkomen des hier vorliegenden Kollektives überwiegt wie
auch in den meisten der in der Literatur vorliegenden Kollektiven eindeutig
eine Lokalisation an den unteren Extremitäten (Machen et al.,1999, Dardick
et al.,2001), insbesondere an den Oberschenkeln:
41 monophasische Synovialsarkome fanden sich an der unteren Extremität,
19 an der oberen Extremität,
24 biphasische Synovialsarkome fanden sich an der unteren Extremität, 8
monophasische Synovialsarkome an der oberen Extremität.
4.5 Histopathologisches Erscheinungsbild, Subtypisierung und Grading
4.5.1 Epitheloides Sarkom
Das Tumorgrading bei Weichgewebstumoren und insbesondere der hier
untersuchten Tumoren wird in der Literatur unterschiedlich diskutiert.
Bos et al. konnten für die von ihnen untersuchten epitheloiden Sarkome kein
Grading vornehmen, da die Tumoren untereinander kaum histologische
Abweichungen zeigten. Bei Bos et al. hatte die Anzahl von Mitosen keine
Korrelation zu Rezidiven oder dem Gesamtüberleben, lediglich der Anteil der

75
Nekroseareale ergab eine negative Korrelation zur 10-Jahres Überlebensrate
und dem krankheitsfreien Intervall. Bei Bos et al. waren in 70% der Tumoren
männlicher Patienten, aber nur in 35% der Tumoren weiblicher Patienten
nekrotische Areale zu finden (Bos et al., 1988).
4.5.2 Klarzellsarkom
Die von Deenik untersuchten Klarzellsarkome wiesen fast alle Mitosen auf,
jedoch oft in geringer Zahl (Deenik et al., 1999). Lukas et al. untersuchten
1992 Patienten der Mayo Klinik mit einem Klarzellsarkom (Lukas et al.,
1992). Übereinstimmend mit Deenik et al. lagen bei diesen Tumoren wenig
mitotische Aktivitäten vor (unter 5 pro hpf), jedoch gab es auch wenige
Tumoren mit hoher mitotischer Aktivität (30 pro hpf). Bei Lukas et al. fanden
sich nur in 30% der Fälle nekrotische Areale, ausgedehnte Nekroseareale
nur in wenigen Einzelfällen. Tumorgröße und Nekroseareale korrelierten
auch hier mit einer schlechteren Prognose.
Chung et al. fanden im Kollektiv der Klarzellsarkome des Armed Forces
Institute of Pathology ähnliche Ergebnisse (Chung et al., 1978). Es zeigte
sich in den meisten Tumoren eine geringe mitotische Aktivität, jedoch gab es
auch in diesem Kollektiv Tumoren mit 23 oder mehr Mitosen pro hpf. Über
Nekrosen oder die Prognose wurden keine Aussagen getroffen.
Die untersuchten Klarzellsarkome des eigenen Kolektives wiesen bis auf 3
Tumore alle Mitosen auf. Bei 10 der untersuchten Tumoren konnten keine
Nekroseareale gefunden werden, 5 der untersuchten Tumoren wiesen eine
Nekroserate von über 25% auf, 2 Tumoren sogar über 50%.
4.5.3 Synovialsarkom
Im hier untersuchten Kollektiv fand sich ein Anteil von 65% des
monophasischen Types und 35% des biphasischen, schlecht differenzierten
Types. Der Anteil des schlecht-differenzierten Typs erscheint relativ hoch,
insbesondere bei Fehlen des rein epithelialen Subtyps im eigenen Kollektiv.
Dieses ist unter anderem darauf zurückzuführen, dass auch teils biphasische
Tumoren mit einem hohen Anteil einer schlecht differenzierten Komponente
dem schlecht-differenzierten Typ zugeordnet wurden.

76
Sicherlich ist zu diskutieren, ob womöglich ein Teil des schlecht
differenzierten rundzelligen Typs in früheren Studien der Literatur dem rein
epithelialen Typ entsprach.
Insgasamt lässt sich feststellen, das der histologische Typ der
Metastasen/Rezidive mit dem Primärtumor übereinstimmen. In der Literatur
wurde ein Sonderfall beschrieben, dabei lag als Primarius ein
monophasisches Synovialsarkom vor, dieses bildete jedoch biphasische
Lungenmetastasen (Lotz et al., 1993). Solche Besonderheiten waren in dem
hier vorliegendem Kollektiv nicht zu finden.
4.6 Krankheitsverlauf / Lokalrezidive und Metastasen
Die Prognose aller drei untersuchten Tumortypen ist insgesamt schlecht. Die
meisten Patienten entwickelten im Untersuchungszeitraum ein Rezidiv:
46,2% der Patienten mit einem Klarzellsarkom, 50% der Patienten mit einem
epitheloiden Sarkom, 41,7% der Patienten mit einem biphasischen
Synovialsarkom und 36,5% der Patienten mit einem monophasischen
Synovialsarkom. Einige der Patienten entwickelten mehrere Rezidive.
30,8% der Patienten mit einem Klarzellsarkom und 25% der Patienten mit
einem epitheloiden Sarkom entwickelten im Untersuchungszeitraum eine
Metastase.
Es wurden unterschiedliche Prognosefaktoren, wie Tumorgröße, Anzahl der
Mitosen/MIB-1-Expression, Nekrosen und Grading untersucht.
Die in der Literatur als wichtigster Prognosefaktor angegebene Tumorgröße
(Chase et al., 1985, Halling et al., 1996, Montgomery et al., 1993) gibt auch
bei dem hier vorliegendem Kollektiv wichtige Hinweise auf den
Krankheitsverlauf.
In der Literatur wurde das Grading schon früh durch Russell et al. als
weiterer wichtiger Parameter für den Krankheitsverlauf beschrieben.
Insgesamt gilt es bei Weichgewebstumoren aufgrund des sehr schlechten
Kranheitsverlaufes auch bei einem niedrigen Grading als problematisch
(Hashimoto et al., 1992). Jedoch korreliert das Grading bei den hier
untersuchten Tumoren, wie auch bei den von Coindre et al. untersuchten

77
Weichgewebstumoren (Coindre et al., 2001) mit der Gesamtprognose.
Aufgrund der geringen Fallzahl war eine statistische Auswertung nicht
aussagekräftig, jedoch ist im Einzellfall ein schlechtes Grading mit einem
schlechten Krankheitsverlauf und häufiger Metastasenbildung verknüpft.
4.6.1 Epitheloides Sarkom
Bos et al. beschrieben eine schlechte Prognose bei epitheloiden Sarkomen
über 3 cm Ausdehnung (Bos et al., 1988). Bei den hier untersuchten
epitheloiden Sarkomen kam es bei Tumoren <2 cm in ihrer maximalen
Ausdehnung nicht zu einer Metastasen oder Rezidivbildung, bei einer Größe
über 2 cm hingegen in allen Fällen zu einer Rezidiv bzw. Metastasenbildung.
Ein weiterer in der Literatur beschriebener Prognosefaktor ist das Grading.
25% der Primärtumoren als G2 eingestuft entwickelten ein Rezidiv und 80%
der Primärtumoren als G3 eingestuft.
Das klassische Grading korreliert bei den meisten Tumoren deutlich mit der
5-Jahres Überlebensrate.
Auf die hier untersuchten Tumoren lässt sich dieses nicht ohne
Einschränkung übertragen.
Die Wahrscheinlichkeit, ein Rezidiv zu entwickeln, liegt auch bei G2-Tumoren
deutlich höher als bei anderen Tumoren.
4.6.2 Klarzellsarkom
Bei Lucas et al. wurde eine Tumorgröße unter 5 cm bei Klarzellsarkomen mit
einem günstigen Verlauf verbunden (Lucas et al., 1992). Eine Tumorgröße
größer 5 cm bedeutete für die Patienten in seinem Kollektiv einen
schlechteren Verlauf. Bei unserem Kollektiv lässt sich keine deutliche
Größengrenze für einen günstigen bzw. ungünstigen Verlauf erkennen. Der
größte Primärtumor, welcher in dem hier vorliegendem
Beobachtungszeitraum keine Metastase bildete, war 5 cm groß, der kleinste
Primärtumor, der eine Metastase bildete, war 1,4 cm in seiner größten
Ausdehnung.

78
Bezüglich des Gradings finden sich bei den hier untersuchten
Klarzellsarkomen ähnliche Ergebnisse wie auch schon bei den epitheloiden
Sarkomen:
Bei den hier vorliegenden Klarzellsarkomen kam es bei 100% der als G3
eingestuften Primärtumoren, bei 60% der als G2 eingestuften Primärtumoren
und bei 33,3% der als G1 eingestuften Primärtumoren zu einem Rezidiv.
Ebenso wie bei den epitheloiden Sarkomen lässt sich das klassische Grading
für die Prognose nicht uneingeschränkt anwenden, da auch bei einem
niedrigen Grading und somit zu erwartendem günstigeren Krankheitsverlauf
deutlich häufiger Rezidive und Metastasen zu erwarten sind, als bei anderen
Tumoren.
4.6.3 Synovialsarkom
Machen et al. beschrieben bei Synovialsarkomen eine schlechtere Prognose
bei Tumoren über 5 cm Ausdehnung, sie unterschieden dabei nicht zwischen
monophasischem oder biphasischen Typ (Machen et al., 1999).
Singer et al. fanden in dem von ihnen untersuchten Kollektiv ebenfalls eine
bessere Prognose bei Patienten mit einer Tumorgröße unter 5 cm (Singer et
al.,1996): Sie fanden eine 100%ige 10-Jahres Überlebensrate bei einer
Tumorgröße unter 5 cm, jedoch nur eine 32%ige 10-Jahres Überlebensrate
bei Tumorgrößen zwischen 5 und 10 cm.
In dem hier untersuchten Kollektiv ergab sich ebenso eine schlechtere
Prognose für Patienten mit Tumoren größerer Ausdehnung. Bei den
biphasischen Synovialsarkomen kam es bei 25% der Patienten mit einem
Tumor unter 5 cm Ausdehnung zu einem Rezidiv, bei 38,5% der Patienten
mit einem Tumor >5 cm und <10 cm Ausdehnung, 77,8% der Patienten mit
einem biphasischen Synovialsarkom >10 cm Ausdehnung entwickelten ein
Rezidiv. Zwei der Patienten des eigenen Kollektives mit einem biphasischen
Synovialsarkom >5 cm entwickelten im Untersuchungszeitraum eine
Metastase.
23,7% der Patienten mit einem monophasischen Synovialsarkom <5 cm
Ausdehnung, 70% der Patienten mit einem monophasischen Synovialsarkom
>5 cm und <10 cm Ausdehnung und 73% der Patienten mit einem

79
monophasischen Synovialsarkom >10 cm Ausdehnung entwickelten im
Untersuchungszeitraum ein Rezidiv.
Singer et al. beobachteten in ihren Untersuchungen einen deutlich
schlechteren Krankheitsverlauf bei einer Tumorgröße über 5cm, sowie bei
mehr als 10 Mitosen pro hpf, sie empfehlen daher bei Vorliegen beider
Kriterien zusätzliche Therapieoptionen neben chirurgischer Resektion
(Singer et al., 1996).
Einige Studien beschrieben einen schlechteren klinischen Verlauf bei
Synovialsarkomen mit distaler Lokalisation (Evans et al.,1993, Hajdu et
al.,1979, Rajpal et al.,1984). Wie hingegen bei Machen et al. lässt sich
dieses in dem hier vorliegenden Kollektiv der Synovialsarkome nicht
bestätigen (Machen et al., 1999).
Über eine 5-Jahres oder 10-Jahres Überlebensrate des eigenen Kollektves
kann in dieser Untersuchung keine Aussage getroffen werden, da bei
geringer Fallzahl nur wenige klinische Verlaufsdaten vorlagen.
4.6.4 MiB-1-Expression
Ein guter Prognosefaktor scheint in der hier vorliegenden Untersuchung die
MiB-1-Expressionsrate zu sein. Bereits Jensen et al. und Choong et al.
fanden eine Korrelation zwischen dem Grading der von ihnen untersuchten
Tumoren und der jeweiligen MiB-1-Expressionsrate (Jensen et al., 1996,
Choong et al., 1994).
Die hier untersuchten Tumore wiesen bis auf zwei Ausnahmen bei den
Klarzellsarkomen und einem Tumor bei den epitheloiden Sarkomen alle eine
zumindest fokale MiB-1 Expression auf. Ähnliche Verhältnisse fanden sich
bei Jensen et al., die in sämtlichen ihrer untersuchten Tumore eine MiB-1-
Expression fanden. Alle der hier vorliegenden G1 Tumoren wiesen eine MiB-
1-Expressionsrate unter 10% in Hotspot-Arealen auf, die drei Tumoren ohne
MiB-1-Expression waren alle G1 –Tumoren.
Ähnliche Verhältnisse fanden sich bei den G2 Tumoren, wobei jedoch 14,2%
der G2 Klarzellsarkome und 64,3% der G2 epitheloiden Sarkome eine MiB-1-
Expression von >10% und <25% aufwiesen. 33,3% der G3 Klarzellsarkome
und 80% der G3 epitheloiden Sarkome wiesen eine MiB-1 Expression >25%

80
auf. Für das Gesamtkollektiv der Klarzellsarkome lag eine MiB-1-Expression
von <10% in 69% der Fälle vor, bei den epitheloiden Sarkomen waren es
37% der Gesamtfälle. Bei Choong et al. fand sich in der Gruppe der G1 und
G2 Tumoren in 47% der Fälle eine MiB-1-Expression <10%, die Ergebnisse
sind jedoch nur eingeschränkt vergleichbar, da ein anderes Gradingschema
nach Angervall et al. benutzt wurde (Choong et al.1994).
Ein von Choong et al. beobachteter Zusammenhang zwischen Tumorgröße
und MiB-1-Expressionsrate fand sich bei der hier vorliegenden Untersuchung
nicht.

81
4.7 EGF-R-Expression in Synovialsarkomen und in epitheloiden Sarkomen
Es gibt bisher wenig Untersuchungen zu EGF-R-Expression in
Synovialsarkomen. Thomas et al. untersuchten insgesamt 38
Synovialsarkome bei 30 Patienten auf eine EGF-R-Expression. Sie fanden
bei 21 Tumoren eine EGF-R-Expression, dabei in den spindelzelligen
Arealen eine stärkere Expression als in den epitheloiden Arealen.
In den auf EGF-R-Expression untersuchten Synovialsarkomen des hier
vorliegenden Kollektives lässt sich kein Überwiegen einer Komponente
beobachten. Bei biphasischen Tumoren ist eine deutlich höhere Expression
in der spindelzelligen Komponente zu beobachten, bei anderen überwiegt
eine Expression in der epithelialen Komponente. Zusammenfassend läßt sich
jedoch festellen, dass die EGF-R-Expression immer membranös lokalisiert
ist.
Aufgrund der Untersuchungsergebnisse der Arbeit von Thomas et al. und der
hier vorliegenden Arbeit ergeben sich neue Therapieüberlegungen bei
Synovialsarkomen (Thomas et al., 2005).
Tyrosinkinaseinhibitoren könnten eine zusätzliche Therapieoption bei
Synovialsarkomen darstellen.

82
5 Zusammenfassung
In der vorliegenden Arbeit werden klinisch-pathologische Parameter an
insgesamt 167 Weichgewebstumoren des Registers für Gliedmaßentumoren
der Berufsgenossenschaftlichen Kliniken Bergmannsheil, Universitätsklinik
Bochum untersucht.
Das hier untersuchte Kollektiv stimmt in den untersuchten Aspekten im
wesentlichen mit bereits in der Literatur beschriebenen Kollektiven überein.
1. Epitheloides Sarkom:
Bei den Patienten mit einem epitheloiden Sarkom handelte es sich um 6
Frauen und 18 Männer, das Patientenalter lag zwischen 15 und 69 Jahren
mit einem Median von 21 Jahren.
Die Tumorgröße lag zwischen 0,3 und 26 cm,
die Tumoren waren gleich häufig an der oberen und der unteren Extremität
lokalisiert.
Eine Malignitätsgraduierung wurde orientierend anhand der Coindre-
Klassifikation vorgenommen, kein epitheloides Sarkom wies ein Grading von
G1 auf, 15 Tumoren G2, 17 Tumoren wiesen ein Grading von G3 auf.
Klinische Verläufe konnten für 20 Patienten erhoben werden.
Es konnte keine eindeutige Korrelation mit statistischer Signifikanz zwischen
dem Krankheitsverlauf und Mitosen/Nekrosen nachgewiesen werden.
Allerdings war eine Tendenz eines schlechteren Krankheitsverlaufes bei
hoher Mitoseanzahl oder hohem Nekroseanteil zu belegen.
Sämtliche epitheloiden Sarkome ließen eine deutliche EGF-R-Expression
erkennen.
2. Klarzellsarkom:
Bei den Patienten mit einem Klarzellsarkom handelte es sich um 7 Frauen
und 8 Männer, das Patientenalter lag zwischen 13 bis 79 Jahren mit einem
Median von 39 Jahren. Die Tumorgrößen lagen zwischen 0,4 cm und 13 cm,
bei der Lokalisation überwog die untere Extremität.
Ein orientierendes Grading nach Coindre ergab bei 2 Tumoren G1, bei 19
Tumoren G2, bei 9 Tumoren G3.
Klinische Verläufe konnten für 30 Patienten erhoben werden. Wie auch bei
den epitheloiden Sarkomen konnte keine Korrelation mit statistischer

83
Signifikanz zwischen hoher Mitosezahl und großem Nekroseanteil
nachgewiesen werden, jedoch ließ sich bei den Klarzellsarkomen mit hoher
Mitosezahl und/oder großem Nekroseanteil ein schlechterer
Krankheitsverlauf erkennen.
3. Synovialsarkom:
Abweichungen von anderen Kollektiven ergaben sich bei den
Synovialsarkomen. Das mikroskopische Erscheinungsbild der
Synovialsarkome des Bergmannsheilkollektives war sehr inhomogen. Neben
einem deutlich biphasischem und einem deutlich monophasischem
Synovialsarkomtyp fanden sich auch Mischformen mit weniger deutlichem
Verteilungsmuster.
Bei insgesamt 105 Synovialsarkomen ergab sich ein Anteil von 37
biphasischen und 68 monophasisch-fibrösen, schlecht differenzierten und
rundzelligen Synovialsarkomen. Der in der Literatur beschriebene rein
eptiheliale Subtyp war nicht eindeutig zu identifizieren.
Bei Tumoren mit deutlich biphasischen Arealen ließ sich ein bisher in der
Literatur nicht beschriebenes Verteilungsmuster der MIB-1-Expression
erkennen:
In der epithelialen Komponente war die MIB-1 Expression deutlich stärker als
in den übrigen Anteilen der Tumoren.
Insgesamt waren 55 Frauen und 50 Männer an einem Synovialsarkom
erkrankt.
Die Altersverteilung erstreckte sich von 6 bis 80 Jahren mit einem Median
von 36 Jahren und einem Mittelwert von 39,5 Jahren.
Als Lokalisation überwog bei den Synovialsarkomen die untere Extremität,
hier insbesondere die Oberschenkel: 46 monophasische Synovialsarkome
und 24 biphasische Synovialsarkome an den unteren Extremitäten, 20
monophasische Synovialsarkome und 8 biphasische Synovialsarkome an
den oberen Extremitäten.
Die Größe der Tumoren erstreckte sich von 0.3 cm bis 17.9 cm. Bei 50
Synovialsarkomen wurde ein Grading nach Coindre vorgenommen. Ein
biphasisches Synovialsarkom wies ein Grading von 1 auf, 31 Tumore wiesen
ein Grading von 3, 18 Tumore ein Grading von 2 auf.

84
Der Proliferationsmarker Ki-67 zeigte eine verstärkte Expression in den
epithelialen Anteilen gegenüber fibrösen Tumoranteilen. Von 22 auf eine
EGF-R-Expression untersuchten Synovialsarkomen zeigten 96% eine
deutliche Expression, in mehr als der Hälfte der Tumore zeigte sich eine
starke immunhistochemische Reaktion in membranöser Lokalisation. Ein
Verteilungsmuster zwischen spindelzelliger und epithelialer Komponente
fand sich bei den biphasischen Synovialsarkomen: In nahezu allen Fällen
war eine sehr starke Immunoreaktivität in der fibrösen (spindelzeligen)
Komponente vorhanden, die epitheloid differenzierte Komponente zeigte
überwiegend keine EGF-R-Expression (Abb.13). Aufgrund dieser Ergebnisse
könnten Tyrosinkinaseinhibitoren als zusätzliche Therapieoption, bei
insgesamt schlechter Prognose der Tumoren, diskutiert werden.
Fazit:
Die klinisch pathologischen Parameter des hier untersuchten Kollektives sind
vergleichbar mit anderen in der Literatur beschriebenen Kollektiven.
Eine eindeutige, statistische Korrelation zwischen histologischen Parametern
und klinischem Verlauf konnte bei den epitheloiden Sarkomen und den
Klarzellsarkomen aufgrund der geringen Fallzahl nicht nachgewiesen
werden, jedoch zeigten Tumoren mit hohem Nekrose- und/oder Mitoseanteil
ein aggressiveres Verhalten mit einem klinisch schlechterem Verlauf.
Es besteht keine Korrelation zwischen Proliferationsindex (MiB-1 Expression)
und der Tumorgröße und Lokalisation. Bei einer hohen Mitoserate war
jedoch auch die MiB-1-Expression hoch.
Es lässt sich ein spezifisches Muster der EGF-R-Expression bei
Synovialsarkomen belegen:
Alle untersuchten Synovialsarkome zeigten eine deutliche EGF-R-
Expression, immer in membranöser Lokalisation.
Alle epitheloiden Sarkome zeigten eine starke EGF-R-Expression, ohne
Verteilungsmuster.
Alle untersuchten Klarzellsarkome waren EGF-R negativ.

85
Ein Grading ist orientierend bei Synovialsarkomen, Klarzellsarkomen und
epitheloiden Sarkomen möglich.
Ein hohes Grading korreliert mit einem schlechteren Krankheitsverlauf,
jedoch zeigen auch Tumoren mit einem niedrigeren Grading oft aggressive
Verläufe.

86
6 Literatur
Alitalo, K., Paavolainen, P., Franssila, K., Ritsila, V. (1977). Clear-cell
sarcoma of tendons and aponeuroses. Report of two cases. Acta Orthop
Scand. 48, 241-244
Amr, S.S., Farah, G.R., Muhtaseb, H.M., Al-Hajj, H.A., Levene, A. (1984).
Clear cell sarcoma: Report of two cases with ultrastructural observations and
review of the literature. Clin Oncol. 10, 59-65
Angervall, L., Stener, B. Clear-cell sarcoma of tendons. (1969). A study of 4
cases. Acta Pathol Microbiol Scand. 77, 589-597
Azumi, N., Turner, R.R. (1983). Clear cell sarcoma of tendons and
aponeuroses: Electron microscopic findings suggesting Schwann cell
differentiation. Hum Pathol. 14, 1084-1089
Balazs, L., Parham, D., Shurtleff, S. (1997). Identification of the t(X;18) as a
diagnostic tool in the differential diagnosis of synovial sarcomas versus
peripheral nerve sheath tumors. Mod Pathol. 10, 8A
Bearman, R.M., Nof, J., Kempson, R.L. (1975) Clear cell sarcoma with
melanin pigment. Cancer 36, 977-984
Benson, J.D., Kraemer, B.B., Mackay, B. (1985). Malignant melanoma of soft
parts: An ultrastructural study of four cases. Ultrastructural Pathol. 8, 57-70
Berger, D.P., Engelhardt, R., Mertelsmann, R. (2002). Das Rote Buch.
Hämatologie und Internistische Onkologie. Ecomed Verlag
Black, R.J., Bray, F., Ferlay, J. (1997). Cancer incidence and mortality in the
European Union: cancer registry data and estimates of national incidence for
1990. Eur J Cancer. 33, 1075-1107

87
Boudreaux, D., Waisman, J. (1978). Clear cell sarcoma with melanogenesis.
Cancer 41, 1387-1394
Bos, G.D., Pritchard, D.J., Reiman, H.M., Dobyns, J.H., Ilstrup, D.M.,
Landon, G.C. (1988). Epithelioid sarcoma. J Bone and Joint Surg. 70A, 862-
870
Bridge, J.A., Borek, D.A., Neff, J.R., Huntrakoon, M. (1990). Chromosomal
abnormalities in clear cell sarcoma: implications for histogenesis. Am J Clin
Pathol. 93, 26-31
Bridge, J.A., Sreekantaiah, C., Neff, J.R., Sandberg, A.A. (1991). Cytogenetic
findings in clear cell sarcoma of tendons and aponeuroses. Malignant
melanoma of soft parts. Cancer Genet Cytogenet. 52, 101-106
Brodsky, J.T., Burt, M.E., Hajdu, S.I. (1992). Tendosynovial sarcoma.
Clinicopathologic features, treatment and prognosis. Cancer 70, 484-489
Callister, M.D., Ballo, M.T., Pisters, P.W.T., Patel, S.R., Feig, B.W., Pollock,
R.E., Benjamin, R.S., Zargas, G.K. (2001). Epithelioid sarcoma: Results of
conservative surgery and radiotherapy. Radiation Oncol. 51(2), 384-386
Chase, D.R., Enzinger, F.M. (1985). Epithelioid sarcoma: Diagnosis,
prognostic indicators, and treatment. Am J Surg Pathol. 9, 241-263
Chase, D.R., Enzinger, F.M., Weiss, S.W. (1984). Keratin in epithelioid
sarcoma: An immunohistochemical study. Am J Surg Pathol. 8, 435-441
Choong, P.F., Akerman, M., Willen, H., Andersson, C., Gustafson, P.,
Baldetorp, B., Ferno, M., Alvegard, T., Rydblom, A. (1994). Prognostic value
of Ki-67 expression in 182 soft-tissue sarcoma. Proliferation-a marker of
metastasis? APMIS. 102, 915-924

88
Choong, P.F., Akerman, M., Willen, H., Andersson, C., Gustafson, P.,
Alvegard, T., Rydholm, A. (1994). Expression of proliferating cell nuclear
antigen (PCNA) and Ki-67 in soft tissue sarcoma. Is prognostic significance
histiotype-specific ? APMIS. 103, 797-805
Chung, E.B., Enzinger, F.M. (1978). Clear cell sarcoma of tendons and
aponeuroses: Further observation. (Abstract) Lab Invest 38, 338
Chung, E.B., Enzinger, F.M. (1983). Malignant melanoma of soft parts. A
reassessment of clear cell sarcoma. Ann J Surg Pathol. 7, 405-413
Coindre, J.M. (1998). Histologic grading of adult soft tissue sarcomas. Verh.
Dtsch. Ges. Path. 82, 59-63
Coindre, J.M., Terrier, P., Guillou, L., Le Doussal, V., Collin, F., Ranchere,
D., Sastre, X., Vilain, M.O., Bonichon, F., Bui, B.G. (2001). Predictive value
of grade for metastasis development in the main histologic types in adult soft
tissue sarcomas. A study of 1240 patients from the French Federation of
Cancer Centers Sarcoma Group. Cancer. 91(10), 1914-1926
Colome-Grimmer, M.I., Evans, H.L. (1996). Metastasizing cellular
dermatofibroma. A report of two cases. Am J Surg Pathol. 20, 1361-1367
Dardick, I., Ramjohn, S., Thomas, M.J., Jeans, D., Hammar, S.P. (1991).
Synovial sarcoma interrelationship of the biphasic and monophasic subtypes.
Path Res Pract. 187, 871-885
Deenik, W., Mooi, W.J., Rutgers, E.J., Peterse, J.L., Hart, A.A., Kroon, B.B.
(1999). Clear cell sarcoma (malignant melanoma) of soft parts. A
clinicopathologic study of 30 cases. Cancer 86, 969-975
Dickinson, I.C., Whitwell, D.J., Battituta, D. (2006). Surgical margin and its
influence on survival in soft tissue sarcoma. J Surg 76 (3), 104-109.

89
Ducatman, B.S., Scheithauer, B.W., Piepgras, D.G., Reimann, H.M., Ilstrup,
D.M. (1986). Malignant peripheral nerve sheath tumors. A clinicopathologic
study of 120 cases. Cancer 57, 2006-2021
Dutra, F.R. (1970). Clear cell-sarcoma of tendons and aponeuroses. Three
additional cases. Cancer 25, 942-946
Eckard, J.J., Pritchard, D., Soule, E.H. (1983). Clear cell sarcoma. A study of
27 cases. Cancer 52, 1482-1488
Eilber, F.C., Tap, W.D., Nelson, S.D. (2006). Advances in chemotherapy for
patients with extremity soft-tissue sarcoma. Orthop Clin North Am. 37(1), 15-
22.
Ekfors, T.O., Rantakokko, V. (1970). Clear cell sarcoma of tendons and
aponeuroses: Malignant melanoma of soft tissues? A report of four cases.
Pathol Res Pract. 165, 422-428
El-Naggar, A.K., Ayala, A.G., Abdul-Karim, F.W. (1990). Synovial sarcoma. A
DNA flow cytometric study. Cancer 65, 2295-2300
El-Naggar, A.K., Ordonez, N.G., Sara, A., McLemore, D., Batsakis, J.G.
(1991). Clear cell sarcomas and metastatic soft tissue melanomas: a flow
cytometric comparison and prognostic implications. Cancer 67, 2173-2179
Enzinger, F.M. (1965). Clear-cell sarcoma of tendons and aponeuroses. An
analysis of 21 cases. Cancer. 18, 1163-1174
Enzinger, F.M. (1970). Epithelioid sarcoma: A sarcoma simulating a
granuloma or a carcinoma. Cancer 26, 1029-1041
Enzinger, F.M., Weiss, S.W. (1995). Epithelioid sarcoma: malignant tumors
of uncertain histogenesis. Enzinger/Weiss Soft Tissue Tumors, St. Louis,
MO, Mosby. 1074-1083

90
Erlandson, R.A., Woodruff, J.M. (1982). Peripheral nerve sheath tumors: An
electron microscopic study of 43 cases. Cancer 49, 273-287
Evans, H.L. (1980). Synovial sarcoma. A study of 23 biphasic on 17
probable monophasic examples. Pathol Ann. 15, 309-31
Evans, H.L., Baer, S.C. (1993). Epithelioid sarcoma: A clinicopathologic and
prognostic study of 26 cases. Semin Diagn Pathol. 10, 286-291
Fisher, C. (1988). Epithelioid sarcoma: The spectrum of ultrastructural
differentiation in seven immunohistochemically defined cases. Hum Pathol.
19, 265-275
Fletcher, J.A. (1992). Translocation (12;22)(q13-14;q12) is a nonrandom
aberration in soft-tissue clear-cell sarcoma. Genes Chromosomes Cancer.
5,184
Folpe, A.L., Schmidt, R.A., Chapman, D., Gown, A.M. (1998). Poorly
differentiated synovial sarcoma. Immunohistochemical distinction from
primitive neuroectodermal tumors and high-grade malignant peripheral nerve
sheath tumors. Am J Surg Pathol. 22, 673-682
Front, R.L., Truong, L.D. (1984). Melanotic Schwannoma of soft tissues.
Electron microscopic observations and review of the literature. Am J Surg
Pathol. 8, 129-138
Ghadially, F.N. (1987). Is synovial sarcoma a carcinosarcoma of connective
tissue? Ultrastruct Pathol. 11, 147-151
Grandis, J.R., Melham, M.F., Barnes, E.L. (1996). Quantitative
immunohistochemical analysis of transforming growth factor-alpha and
epidermal growth factor receptor in patients with squamous cell carcinoma of
the head and neck. Cancer 78, 1284-1292

91
Guillou, L., Coindre, J.M. (1998). How should we grade soft tissue sarcomas
and what are the limitations? Pathology 3, 105-110
Hajdu, S.I. (1979). Pathology of soft tissue tumors. Lea and Febinger,
Philadelphia. 204-210
Hajdu, S.I., Shiu, M.H., Fortner, J.G. (1977). Tendosynovial sarcoma. A
clinicopathologic study of 136 cases. Cancer 39, 1201-1217
Halling, A.C., Wollan, P.C., Pritchard, D.J., Vlasak, R., Nascimento, A.G.
Epithelioid sarcoma: a clinicopathologic review of 55 cases. (1996). Mayo Cin
Proc. 71, 636-642
Hasegawa, T., Hirose, T., Kudo, E., Hizawa, K. (1989). Clear cell sarcoma:
an immunohistochemical and ultrastructural study. Acta Pathol Jpn. 39, 321-
327
Hashimoto, H., Daimaru, Y., Takeshita, S., Tsuneyoshi, M., Enjoji, M. (1992).
Prognostic significance of histologic parameters of soft tissue sarcomas.
Cancer 79, 2816-22
Hazelbag, H.M., Mooi, W.J., Fleuren, G.J. (1996). Chain-specific keratin
profile of epithelioid soft-tissue sarcomas: an immunohistochemical study on
synovial sarcoma and epitheloid sarcoma. Appl Immunohistochem. 4, 176-
183
Hedley, D.W., Frielander, M.L., Taylor, I.W., Rugg, C.A., Musgrove, E.A.
(1983). Method for analysis of cellular DNA content of paraffin-embedded
pathological material using flow cytometry. J Histochem Cytochem. 31, 1333-
1335
Hicks, M.J., Saldivar, V.A., Chintagumpala, M.M., Horowitz, M.E., Cooley,
L.D., Barrish JP, Hawkins EP, Langston C. (1995). Malignant melanoma of

92
soft parts involving the head and neck region: Review of literature and case
report. Ultrastructural Pathol. 19, 395-400
Hoffmann, G.J., Cartner, D. (1973). Clear cell sarcoma of tendons and
aponeuroses with melanin. Arch Pathol. 95, 22-25
Hoos, A., Lewis, J., Brennan, M. (2000). Weichgewebssarkome-
Prognostische Faktoren und multimodale Therapie. Chirurg 71, 787-794
Huang SM, Harari PM. (2000). Modulation of of radiation response after
epidermal growth factor receptor bockade in squamous cell carcinomas:
inhibition of damage repair, cell cycle kinetics and tumor angiogenesis. Clin
Cancer Res 6, 2166-2174.
Jensen, V., Hoyer, M., Sorensen, F.B., Keller, J., Jensen, O.M. (1996). MIB-1
expression and iododeoxyuridine labelling in soft tissue sarcomas: an
immunohistochemical study including correlations with p53, bcl-2 and
histological characteristics. Histopathology 28, 437-444
Katenkamp, D. (1999). Aktuelles Referat Stand der Dinge bei den
Weichgewebstumoren. Path Res Pract. 2, 136-137
Katenkamp, D., Hünerbein, R. (1992). Untersuchungen zur prognostischen
Bedeutung von Entzündungszellen in malignen Weichgewebstumoren des
Menschen. Zentralbl Pathol. 138, 21-25
Katenkamp, D., Kosmehl, H. (1995). Heterogeneity in malignant soft tissue
tumors. In: Soft tissue tumors. Springer Verlag, Berlin Heidelberg New York.
123-151.
Kawai, A., Woodruff, J., Healey, J.H., Brennan, M.F., Antonescu, C.R.,
Ladanyi, M. (1998). SYT-SSX gene fusion as a determinant of morphology
and prognosis in synovial sarcoma. N Engl J of Medicine. 338, 153-160

93
Kindblom, L.G., Lodding, P., Angervall, L. (1983). Clear-cell sarcoma of
tendons and aponeuroses. An immunohistochemical and electron
microscopic analysis indicating neural crest origin. Virchows Arch. (Pathol
Anat) 401, 109-128
Krall, R.A., Kostianowski, M., Patchefsky, A.S. Synovial sarcoma. A clinical,
pathological, and ultrastructural study of 26 cases supporting the recognition
of a monophasic variant. (1981). Am J Surg Pathol. 5, 137-151
Kubo, T. (1969). Clear-cell sarcoma of patellar tendon studied by electron
microscopy. Cancer 24, 948-953
Kuhnen, C., Herter, P., Steinau, H.U., Müller, O., Müller, K.M. (1998).
Intrazelluläre Verteilung von ß-Catenin in Weichgewebstumoren. Verh Dtsch
Ges Path. 82, 322-326
Kuhnen, C., Lehnhardt, M., Tolnay, E., Muehlberger, T., Vogt, P.M., Müller,
K.M. (2000). Patterns of expression and secretion of vascular endothelial
growth factor in malignant soft-tissue tumours. J Cancer Res Clin Oncol. 126,
219-225
Kuhnen, C., Tolnay, E., Steinau, H.U., Voss, B., Müller, K.M. (1998).
Expression of c-Met receptor and hepatocyte growth factor/scatter factor in
synovial sarcoma and epithelioid sarcoma. Virchows Arch. 432, 337-342
Ladanyi, M., Bridge, J.A. (2000). Contribution of molecular genetic data to the
classification of sarcomas. Human Pathol. 31, 532-538
Lewis, J., Leung, D., Casper, E., Woodruff, J. (1999). Multifactorial analysis
of log-term follow-up (more than 5 years) of primary extremity sarcoma. Ann
Surg. 231, 655-663

94
Lombardi, L., Rilke, F. (1984). Ultrastructural similarities and differences of
synovial sarcoma, epithelioid sarcoma, and clear cell sarcoma of the tendons
and aponeuroses. Ultrastruct Pathol. 6, 209-219
Lopes, J.M., Bjerkenhagen, B., Holm, R., Bruland, Ö., Sobrinho-Simoes, M.,
Nesland, J.M. (1994). Immunohistochemical profile of synovial sarcoma with
emphasis on the epithelial-type differentiation. Path Res Pract. 190, 168-177
Lotz, S., Caselitz, J., Bullerdiek, J., Rieckhoff, K.U. (1993). Monophasisch
fibroses Synovialsarkom der Hand mit biphasisch differenzierten
Lungenmetastasen. Pathologe 14, 54-57
Lucas, D.R., Nascimento, A.G., Sim, F.H. (1992). Clear cell sarcoma of soft
tissues. Mayo Clinic experience with 35 cases. Am J Surg Pathol. 16, 1197-
1204
Machen, S.K., Easley, K.A., Goldblum, J.R. (1999). Synovial sarcoma of the
extremities. A clinicopathologic study including semi-quantitative analysis of
spindled, epithelial, and poorly differentiated areas. Am J Surg Pathol. 23,
268-275
Mackenzie, D.H. (1974). Clear cell sarcoma of tendon and aponeuroses with
melanin production. J Pathol. 114, 231-232
Mandard, A.M., Petiot, J.F., Marnay, (1989). Prognostic factors in soft-tissue
sarcomas. A multivariate analysis of 109 cases. Cancer 63, 1437-1451
Manivel, J.C., Wick, M.R., Dehner, L.P. (1987). Epithelioid sarcoma: An
immunohistochemical study. Am J Clin Pathol. 87, 319-326
Mechtersheimer, G., Tilgen, W., Klar, E., Möller, P. (1989). Clear cell
sarcoma of tendons and aponeuroses: Case presentation with special
reference to immunohistochemical findings. Hum Pathol. 20, 914-917

95
Meis, J.M., Enzinger, F.M., Martz, K.L., Neal, J.A. (1992). Malignant
peripheral nerve sheath tumors (malignant schwannomas) in children. Am J
Surg Pathol. 16, 694-707
Mickelson, M.R., Brown, G.A., Maynard, J.A., Cooper, R.R., Bonfiglio, M.
(1980). Synovial sarcoma. An electron microscopic study of monophasic and
biphasic forms. Cancer 45, 2109-2118
Miettinen, M., Lehto, V-P., Badley, R.A., Virtanen, I. (1983). Monophasic
synovial sarcoma of spindle-cell type: epithelial differentiation as revealed by
ultrastructural features, content of prekeratin and binding peanut agglutinin.
Virchows Arch. 44, 187-199
Miettinen, M., Virtanen, I. (1984). Synovial sarcoma- a misnomer. Am J
Pathol. 117, 18-25
Mills, S.E., Fechner, R.E., Bruns, D.E., Bruns, M.E., O´Hara, M.F. (1981).
Intermediate filaments in eosinophilic cells of epithelioid sarcoma. A light
microscopic, ultrastructural, and electrophoretic study. Am J Surg Pathol. 5,
195-202
Mirra, J.M., Kessler, S., Bhuta, S., Eckard, J. (1992). The fibroma-like variant
of epithelioid sarcoma. A fibrohistiocytic/myoid cell lesion often confused with
benign and malignant spindle cell tumors. Cancer 69, 1382-1395
Montgomery, E.A., Meis, J.M., Ramos, A.G., Frisman, D.M., Martz, K.L.
(1993). Clear cell sarcoma of tendons and aponeuroses. A clinicopathologic
study of 58 cases with analysis of prognostic factors. Int J Surg Pathol. 1, 89-
100
Mrozek, K., Karakousis, C.P., Perez-Mesa, C., Bloomfield, C.D. (1993).
Translocation t(12;22)(q13;q12.2-12.3) in a clear cell sarcoma of tendons and
aponeuroses. Genes Chromosomes and Cancer 6, 249-252

96
Mukai, M., Torikata, C., Risami, I. (1985). Cellular differentiation of epithelioid
sarcoma: An electron-microscopic, enzyme-histochemical, and
immunohistochemical study. Am J Pathol. 119, 44-56
Mukai, M., Torikata, C., Iri, C., Mikata, A., Kawai, T., Hanaoka, H., Yakumaru,
K., Kageyama, K. (1984). Histogenesis of clear cell sarcoma of tendons and
aponeuroses. An electron-microscopic, biochemical, enzyme histochemical
and immunohistochemical study. Am J Pathol. 114, 264-272
Nicholson, S., Richard, J., Sainsbury, C. (1991). Epidermal growth factor
receptor (EGF-R); results of a 6-year follow-up study in operable breast
cancer with emphasis on the node negative group. Br J Cancer. 63, 146-150
Peulve, P., Michot, C., Vannier, J.P., Tron, P., Hemet, J. (1991). Clear cell
sarcoma with t(12;22)(q13-14;q12). Genes, Chromosomes and Cancer. 3,
400-402
Pisters, P., Leung, D., Woodruff, J. (1996). Analysis of prognostic factors in
1041 patients with localized soft tissue sarcoma of the extremity. Clin Oncol.
14, 1679-1689
Quezado, M.M., Middelton, L.P., Bryant, B., Lane, K., Weiss, S., Merino, M.J.
(1998). Allelic loss on chromosome 22q in epithelioid sarcomas. Hum Pathol.
29, 604-608
Radstone, D.J., Revell, P.A., Mantell, B.S. (1979). Clear cell sarcoma of
tendons and aponeuroses treated with bleomycin and vincristine. Br J Radiol.
52, 238-241
Rajpal, S., Moore, R.H., Karakousis, C.P. (1984). Synovial sarcoma. A
review of treatment and survival in 52 patients. NY State J Med. 84, 17-19

97
Raynor, A.C., Vargas-Cortes, F., Alexander, R.W., Bingham, H.G. (1979).
Clear-cell sarcoma with melanin pigment: A possible soft-tissue variant of
malignant melanoma. J Bone Joint Surg. 61A , 276-280
Reeves, B.R., Fletcher, C.D.M., Gusterson, B.A. (1992). Translocation
t(12;22)(q13;q13) is a nonrandom rearrangement in clear cell sarcoma.
Cancer Genet Cytogenet. 64, 101-103
Reeves, B.R., Fusher, C., Smith, S. (1987). Ultrastructural,
immunocytochemical, and cytogenetic characterization of human epithelioid
sarcoma cell line (RM-HS1). J Nat Cancer Inst. 87, 7-11
Rodriguez, E., Sreekantaiah, C., Reuter, V.E., Motzer, R.J., Chaganti, R.S.K.
(1992). t(12;22)(q13;q13) and trisomy 8 are nonrandom aberrations in clear-
cell sarcoma. Cancer Genet Cytogenet. 64, 107-110
Russell, W.O., Cohen, J., Enzinger, F.M. (1977). A clinical and pathologic
staging system for soft-tissue sarcomas. Cancer 40, 1562-1570
Sara, A.S., Evans, H.L., Benjamin, R.S. (1990). Malignant melanoma of soft
parts (clear cell sarcoma): a study of 17 cases, with emphasis on prognostic
factors. Cancer 65, 367-374
Stenman, G., Kindblom, L.G., Angervall, L. (1992). Reciprocal translocation
t(12;22)(q13;q13) in clear cell sarcoma of tendons and aponeuroses. Genes
Chromosomes and Cancer. 4, 122-127
Singer, S., Baldini, E.H., Dimetri, G.D. (1996). Synovial sarcoma: Prognosis
significance of tumor size, margin of resection, and mitotic activity for
survival. J Clin Oncol. 14,1201-1208
Sonore, H., Furihata, M., Iwata, J. (1993). Morphological characterization of a
new human epithelioid sarcoma cell line, ES020488, in vitro and in vivo.
Virchows Archiv B Cell Pathol. 63, 219-225

98
Sonobe, H., Ohtsuki, Y. (1997). Involvement of 8q, 22q, and monosomy 21 in
an epithelioid sarcoma. Cancer Genet Cytogenet. 96, 178-180
Sonobe, H., Takeuchi, T., Shimizu, K., Iwata, J., Furihata, M., Ohtsuki, Y.
(1999). Further characterization of the human clear cell sarcoma line HS-MM
demonstrating a specific t(12;22)(q13;q12) translocation and hybrid
EWS/ATF-1 transcript. J Pathol. 187, 594-597
Speleman, F., Delatore, O., Peter, M., Hauben, E., Van Roy, N., Van Marck,
E. (1997). Malignant melanoma of the soft parts (clear-cell sarcoma):
confirmation of EWS and ATF-1 gene fusion caused by a t(12;22)
translocation. Mod Pathol. 10, 496-499
Speleman, F., Colpaert, C., Goovaerts, G., Leroy, J.G., Van Marck, E.
(1992). Malignant melanoma of soft parts. Further cytogenetic
characterization. Cancer Genet Cytogenet. 58, 176-179
Steinau, H.U., Biemer, E. (1990). Resektionsmethodik und funktionelle
Wiederherstellungschirurgie maligner Weichgewebstumoren der
Extremitäten. Langenb Arch Chir. 375, 239-245
Steinau, H.U., Büttemeyer, R., Vogt, P., Hussmann, J., Hebebrand, D.
(1996). Reconstruction and salvage of extremities. Kroll SS (Hrsg.)
Reconstructive plastic surgery for cancer. Mosby, St. Louis Baltimore Berlin,
S. 68-77
Stenman, G., Kindblom, L.G., Willems, J. (1990). A cell culture, chromosomal
and quantitative DNA analysis of a metastatic epithelioid sarcoma: Deletion
1p, a possible primary chromosomal abnormality in epithelioid sarcoma.
Cancer 65, 2013

99
Stiller, D., Holzhausen, H.J., Meyer, H. (1988). Histologische Diagnose und
ultrastrukturelle Zytologie des epitheloiden Sarkoms. Zentralbl allg Pathol
pathol Anat. 134, 467-477
Sreekantaiah, C., Landanyi, M., Rodriguez, E. (1994). Chromosomal
aberrations in soft tissue tumors. Relevance to diagnosis, classification, and
molecular mechanisms. Am J Pathol. 144, 1121-1134
Swanson, P.E., Wick, M.R. (1989). Clear cell sarcoma: an
immunohistochemical analysis of six cases and comparison with other
epithelioid neoplasms of soft tissue. Arch Pathol Lab Med. 113, 55-60
Takenoushi, T., Ito, K., Kazama, T., Ito, M. (1994). Establishment and
characterization of a clear-cell sarcoma (malignant melanoma of the soft
parts) cell line. Arch Dermatol Res. 286, 254-260
Thomas, D.G., Giordano, T.J., Sanders, D., Biermann, S., Sondak, V.K.,
Trent, J.C., Yu, D., Pollock, R.E., Baker, L. (2005). Expression of Receptor
Tyrosine Kinases Epidermal Growth Factor Receptor and HER-2/neu in
Synovial Sarcoma. Cancer 103, 830-838
Traeger,G., Ruchholz,S., Schütte, J., Nast-Kolb, D. (2004). Diagnostik und
Therapiestrategien bei Weichteilsarkomen. Unfallchirurg 107, 601-618
Travis, J.A., Bridge, J.A. (1992). Significance of both numerical and structural
chromosomal abnormalities in clear cell sarcoma. Cancer Genet Cytogenet.
64, 104-106
Tsuneyoshi, M. (1975). Distinction of melanoma of soft parts from clear-cell
sarcoma of soft parts from clear-cell sarcoma of tendons and aponeuroses. A
histopathological and electron microscopic study. Fukuoka Acta Med. 66,
666-681

100
Tsuneyoshi, M., Enjoji, M., Kubo, T. (1978). Clear cell sarcoma of tendon and
aponeuroses: A comparative study of 13 cases with a provisional
subgrounding into the melanotic and synovial types. Cancer 42, 243-252
Tunn, P.U., Dürr, H.R. (2007). Diagnostik und Therapie von
Weichgewebstumoren im Erwachsenenalter. arthritis+rheuma 27, 153-161.
Turc-Carel, C., Dal Cin, P., Limon, J., Li, F., Sandberg, A.A. (1986).
Translokation X;18 in synovial sarcoma. Cancer Genet Cytogenet. 23, 93
Zito, R. (1984). Synovial sarcoma: An Australian series of 48 cases.
Pathology 16, 45-52
Zucman, J., Delattre, O., Desmaze, C. (1993). EWS and ATF-1 gene fusion
induced by t(12;22) (q13; q12.2-12.3) translocation in malignant melanoma of
soft parts. Nature Genet. 6, 341-345

Danksagung
An erster Stelle bedanke ich mich bei meinem Doktorvater Prof. Dr. C.
Kuhnen für die Überlassung des Themas dieser Arbeit, seiner stetigen
Hilfsbereitschaft und Geduld mit mir.
Ich danke Frau C. Troske für die Unterstützung bei der Erstellung des
Bildmaterials dieser Arbeit, sowie Dr. T. Stark für die Hilfestellung bei
sämtlichen Computerproblemen und der Formatierung, ganz besonders
jedoch für die Unterstützung meiner bisherigen beruflichen Laufbahn.
Ich bedanke mich bei Frau Prof. Dr. Tannapfel für die Möglichkeit der
Promotion im Institut für Pathologie, sowie Herrn Prof. emer. Dr. K-M-Müller,
der mir vor Jahren durch seine Ausstrahlung und beeindruckenden
Vorlesungen die Pathologie nahegebracht hat, was mich letztendlich zu
dieser Arbeit geführt hat.
Mein letzter Dank geht an meine beste Freundin, Frau Dr. Marlene Helwing
und an meinen Kay, die mich uneingeschränkt unterstützt haben und meine
Stimmungsschwankungen ertragen haben.

Lebenslauf Persönliche Daten
Name: Denise Rosenberger Geburtsdatum: 9. Juli 1975 Geburtsort: Bielefeld
Schulausbildung
1986 – 1995 Evangelisch Stiftisches Gymnasium in Gütersloh
1995 Abitur
Studium
1995 – 1996 Studium der Applied Biosciences an der Glasgow Caledonian University in Schottland
10 / 1997 Beginn des Studiums der Humanmedizin an der Ruhr-
Universität Bochum 08 / 1999 Physikum 08 / 2000 Erster Abschnitt der Ärztlichen Prüfung 04 / 2003 Zweiter Abschnitt der Ärztlichen Prüfung 03 – 07 / 2003 Praktisches Jahr in der Chirurgischen Abteilung der
Augusta Krankenanstalten Bochum 07 – 11 / 2003 Praktisches Jahr in der Onkologischen und
Nephrologischen Abteilung der Augusta Krankenanstalten in Bochum
12 / 2003 -03/2004 Praktisches Jahres in der Klinik für Hals-Nasen-Ohrenheilkunde des St. Elisabeth Hospitales Bochum
05 / 2004 Dritter Abschnitt der Ärztlichen Prüfung

Famulaturen / weitere Tätigkeiten
07 / 1996 - 07 / 1997 Freiwilliges Soziales Jahr im Alten-und Pflegeheim Haus Ubbedissen in Bielefeld
09 / 2000 vierwöchige Unfallchirurgische Famulatur im
Klinikum Rosenheim 02 – 04 / 2001 Famulatur in der Geburtshilfe und in der Ambulanz
des Nkandla Hospital, Zululand in Südafrika 03 / 2002 vierwöchige Famulatur im Institut für Pathologie
an den Berufsgenossenschaftlichen Kliniken Bergmannsheil in Bochum
08 – 09 / 2002 sechwöchige Famulatur Innere Medizin/Ambulanz
im Lourdes Hospital in Ernakulam in Südindien 10 / 1997 – 03 / 2003 Studentische Hilfskraft im Institut für Pathologie an
den Berufsgenossenschaftlichen Kliniken Bergmannsheil in Bochum
06 / 1998 – 06 / 2004 Studentische Hilfskraft im Pflegedienst in der
Gefäßchirurgischen Abteilung des St. Josef Hospital in Bochum
Ärztliche Tätigkeit
Seit August 2004 Assistenzärztin in der Abteilung für Hals-Nasen-Ohrenheilkunde, Kopf- und Halschirurgie der Ruhr-Universität Bochum am St. Elisabeth Hospital Bochum







