

Die Bedeutung viraler Determinanten für die Pathogenität
muriner Leukämieviren
Dissertation
zur Erlangung des Doktorgrades
des Fachbereichs Biologie der
Universität Hamburg
vorgelegt von
Michaela Rodenburg
aus Hamburg
Hamburg 2003

Genehmigt vom
Fachbereich Biologie
der Universität Hamburg
auf Antrag von Herrn Professor Dr. Wolfram Ostertag
Weiterer Gutachter der Dissertation:
Herr Professor Dr. W.O. Abel
Tag der Disputation: 05 September 2003; 11 Uhr

Die vorliegende Arbeit wurde am Heinrich-Pette-Institut für Experimentelle Virologie
und Immunologie an der Universität Hamburg, Abteilung Zell- und Virusgenetik, in
der Zeit von September 1999 bis August 2003 angefertigt.
Herrn Professor Wolfram Ostertag danke ich für die Vergabe des Themas, die
Bereitstellung des Arbeitsplatzes und seinem Interesse am Werdegang dieser Arbeit.
Dr. Carol Stocking möchte ich an dieser Stelle für die optimale Betreuung und ihr
stets offenes Ohr für alle großen und kleinen Problemchen danken. Ein spezielles
Dankeschön gebührt Dr. Jürgen Löhler für seinen unermüdlichen und unersetzlichen
Einsatz an der „Mausfront“.
Allen Mitarbeitern der Abteilung 5 danke ich für ihre Unterstützung in allen fachlichen
und mentalen Fragen, ganz zu Schweigen vom tollen Arbeitsklima.
Ein dickes, fettes Dankeschön geht an meine Schwester Gaby und meine Freundin
Susu Rehaag für die Schulter zum Ausweinen, ein stets offenes Ohr und die
aufmunternden Worte. Darüber hinaus danke ich Susu für das superschnelle
Korrektulesen und die charmanten Kommentare.
„I thank the academy.......“

Inhaltsverzeichnis
Inhaltsverzeichnis
1. Zusammenfassung 1
2. Einleitung 3 2.1 Retroviren allgemein 3
2.2 Murine Leukämieviren 4
2.2.1 Genomorganisation 4
2.3 Retroviraler Entwicklungszyklus 6
2.3.1 Rezeptorbindung und Eintritt in die Wirtszelle 6
2.3.2 Reverse Transkription und Integration 10
2.3.3 Transkription, Translation und Assemblierung 12
2.4 Pathogenität muriner Leukämieviren 13
2.4.1 Replikationsinkompetente Retroviren 15
2.4.2 Replikationskompetente Retroviren 17
2.5 Verschiedene Faktoren beeinflussen den Typ der induzierten Krankheit 19
2.5.1 Virale Determinanten: LTR- und env-Sequenzen 19
2.5.2 Generation von rekombinanten Viren 20
2.5.3 Aktivierung von Genen durch Provirusintegrationen 21
2.5.3.1 Aktivierte Onkogene in T-Zell Leukämien 22
2.5.3.2 Aktivierte Onkogene in Erythroleukämien 24
3. Fragestellung dieser Arbeit 25
4. Material 26 4.1 Chemikalien und Enzyme 26
4.2 Bakterienstämme 26
4.3 Zelllinie 26
4.4 Medien 27
4.4.1 Bakterienkulturmedien 27
4.4.2 Zellkulturmedien 27
4.5 Verwendeter Mausstamm 28
4.6 Klonierte retrovirale Proviren 28
4.7 Oligonukleotide 28

Inhaltsverzeichnis
4.8 Größenstandards und Ladepuffer 29
4.8.1 λ-DNA/HindIII Marker für Southern Blot Analysen 29
4.8.2 λ-DNA/HindIII/EcoRI Marker für gelelektrophoretische Analysen 29
4.8.3 GeneRuler DNA Ladder Mix für gelelektrophoretische Analysen 29
4.8.4 10 x Yellow SubTM PCR-Additiv und Ladepuffer 29
4.9 DNA Sonden für Southern Hybridisierung 30
5. Methoden 31 5.1 Isolierung von DNA-Fragmenten 31
5.2 Dephosphorylierung des gespaltenen Vektors 31
5.3 Ligation 31
5.4 Screening nach positiven Klonen 32
5.5 Plasmidisolierung 33
5.6 Sequenzierung 33
5.7 Zellkultur 34
5.7.1 Virusproduktion 34
5.7.2 Transfektion mittels Elektroporation 34
5.7.3 Ernten des Virusüberstandes 34
5.7.4 Titerbestimmung 35
5.7.5 Infektion neonataler Mäuse 35
5.8 Analyse erkrankter Tiere 35
5.8.1 Überwachung und Autopsie der Mäuse 35
5.8.2 Analyse der Blutparameter 36
5.9 Durchführung von Blutausstrichen und Cytospins 37
5.9.1 Blutausstriche 37
5.9.2 Cytospin 37
5.9.3 AccustainTM Pappenheim-Färbung 37
5.10 FACS-Analysen 38
5.10.1 Vorbereitung der Organe 38
5.10.2 Vorbereitung der Zellen 40
5.10.3 FACS-Messung 41
5.11 Gewinnung genomischer DNA aus Organen 41
5.12 Restriktionsspaltung und gelelektrophoretische Auftrennung 42
genomischer DNA

Inhaltsverzeichnis
5.13 Southern Blotting 42
5.13.1 Hybridisierung 43
5.13.2 Waschen der Membranen 44
5.13.3 Herstellung und Markierung von Sonden 44
5.14 Vektorklonierung 45
5.14.1 Chimäre MuLVs mit einem Austausch der env-Sequenzen 45
5.14.2 Chimäre MuLVs mit einem Austausch der gag-Sequenzen 46
5.14.3 Chimäre MuLVs mit einem Austausch der NP-Sequenzen 46
5.14.4 Chimäre MuLVs mit einem Austausch der LTR-Sequenzen 48
6. Ergebnisse 50 6.1 Charakterisierung der von Moloney-, 4070A- und 10A1-MuLV induzierten
Krankheiten in NIH/Ola- Mäusen 50
6.1.1 Charakterisierung der phänotypischen Merkmale 50
6.1.1.1 Phänotyp von Moloney- bzw. 4070A-MuLV infizierten Tieren 50
6.1.1.2 Phänotyp von 10A1-MuLV infizierten Tieren 51
6.1.2 Weitere Charakterisierung der MuLV-induzierten Tumore 58
6.1.2.1 Moloney- und 4070A-MuLV induzierte Leukämien 59
6.1.2.2 10A1-MuLV induzierte Leukämien 60
6.1.2.3 Überprüfung des Differenzierungsgrades der B-Zellen 62
6.1.2.4 Kinetik des Krankheitsverlaufs 66
6.2 Charakterisierung der chimären Murinen Leukämieviren 67
6.2.1 Charakterisierung von MuLVs mit ausgetauschten env-Sequenzen 67
6.2.1.1 Phänotyp von Mo-10A1env-infizierten Tieren 68
6.2.1.2 Phänotyp von 10A1-4070env-infizierten Tieren 69
6.2.2 Charakterisierung von MuLVs mit einem LTR-Austausch 70
6.2.2.1 Phänotyp von Mo-10A1V-10A1LTR-infizierten Tieren 71
6.2.2.2 Phänotyp von 10A1V-MoLTR-infizierten Tieren 72
6.2.3 Charakterisierung von MuLVs mit einem Austausch von
gag- oder NP-Sequenzen 74
6.2.3.1 Phänotyp von 10A1V-MoNP-infizierten Tieren 75
6.2.3.2 Phänotyp von 10A1V-Mogag-infizierten Tieren 77
6.2.3.3 Phänotyp von Mo-10A1V-10A1NP-infizierten Tieren 78

Inhaltsverzeichnis
6.3 Aktivierung von Onkogenen 80
6.3.1 Klonalität der Tumore 80
6.3.2 Überprüfung der Tumore nach aktivierten Onkogenen 83
6.3.3 Untersuchung der von chimären MuLVs induzierten blastischen
Lymphome auf Insertionen im Fli-1-Gen 86
6.4 Vorkommen von MCF-Viren in MuLV infizierten Mäusen 86
6.4.1 Erhöhte MCFV-Frequenz in Mo-MuLV induzierten Tumoren 90
6.4.2 Niedrige MCFV-Frequenz in 10A1-MuLV induzierten Tumoren 91
6.4.3 Niedrige MCFV-Frequenz in Mo-10A1V-10A1NP-MuLV und
10A1V-MoNP-MuLV induzierten Tumoren 94
6.5 Messung des glykoGag-Expressionsniveaus auf MuLV infizierten Zellen 97
7. Diskussion 101 7.1 Pathogenität von Moloney-, 4070A- und 10A1-MuLV in NIH/Ola-Mäusen 101
7.2 Bedeutung viraler Determinanten für die Pathogenität 102
7.2.1 Einfluss der env-Sequenzen 102
7.2.2 Einfluss der LTR-Sequenzen 104
7.2.3 Einfluss der NP-Sequenzen 106
7.2.3.1 Einfluss des glykoGag-Expressionsniveaus auf die Pathogenität 109
7.2.4 Einfluss von MCF-Viren auf die Pathogenität 110
7.2.5 Aktivierung von Onkogenen 111
8. Appendix 114
9. Abkürzungsverzeichnis 116
10. Literaturverzeichnis 118

Zusammenfassung 1
1. Zusammenfassung
Murine Leukämieviren induzieren in infizierten Mäusen verschiedene
Leukämieformen. Die Leukämie wird durch die zufällige und stabile Integration der
Proviren in das Genom der infizierten Zellen induziert. Diese Integrationen können
zur Aktivierung zellulärer Gene führen. In vivo wird auf Provirusintegrationen
selektioniert, die der infizierten Zelle einen Wachstumsvorteil verschaffen. Dieser
rapide anwachsende Zellpool führt dann zur Tumorbildung. Die Charakterisierung
der Integrationsstellen hat zur Entdeckung von zahlreichen Onkogenen geführt, die
auch in der humanen Tumorentwicklung eine wichtige Rolle spielen.
In dieser Arbeit sollte untersucht werden, welche Sequenzen im Genom von murinen
Leukämieviren einen Einfluss auf die Art der induzierten Leukämie haben. Da das
Moloney-MuLV vorwiegend T-Zellen und das 10A1-MuLV im Gegensatz dazu
myeloblastische Zellen transformieren kann, wurden bestimmte Bereiche der
Genome durch die entsprechenden Sequenzen aus dem anderen MuLV ersetzt. Die
auf diese Weise hergestellten chimären Retroviren wurden dann im NIH/Ola-
Mausmodell getestet. In früheren Arbeiten wurde gezeigt, dass die „Long Terminal
Repeats“ (LTR) einen Einfluss auf die Zellspezifität eines MuLVs haben, da diese die
für die Transkription in bestimmten Zelltypen wichtigen Enhancer-Elemente
enthalten. In der vorliegenden Arbeit wurde dagegen gezeigt, dass es zwei Bereiche
außerhalb des LTRs gibt, die einen starken Einfluss auf die Art der induzierten
Leukämie haben. Für die Induktion von myeloblastischen Lymphomen sind vor allem
die 10A1 env-Sequenzen und die damit verbundene Rezeptornutzung von Pit1
wichtig. Die Aktivierung des Transkriptionsfaktors Fli-1 scheint ebenfalls für die
Transformation der Vorläuferzellen essentiell zu sein und einen Selektionsvorteil für
diese Zellen in vivo zu bedeuten. Des weiteren hat der stromabwärts vom LTR
gelegene Leader-Bereich einen stärkeren Einfluss auf die Art der induzierten
Leukämie als die LTR-Sequenzen. Der vom Moloney-MuLV stammende Leader-
Bereich ist in dem hier verwendeten Mausmodell eine stärkere Determinante für die
Induktion einer T-Zell-Leukämie als das thymotrope Moloney-LTR. Der
entsprechende vom 10A1-MuLV stammende Bereich scheint dagegen die
Entwicklung von blastischen Lymphomen zu ermöglichen, wenn dieser Einfluss nicht
durch das starke Moloney-LTR maskiert wird.

Zusammenfassung 2
In weiteren Untersuchungen sollte nun geklärt werden, wie der Leader-Bereich die
Induktion verschiedener Leukämieformen beeinflussen könnte. Diese Region
umfasst für den retroviralen Entwicklungszyklus wichtige regulatorische Elemente,
wie die Verpackungssequenz für die virale RNA (Ψ), die Primer-Bindungsstelle
(PBS), das Dimerisierungssignal (DLS) und eine interne Ribosomeneintrittsstelle
(IRES). Darüber hinaus liegt im Leader-Bereich auch das Initiations-Kodon für das
für die Virusstruktur nicht erforderliche glykoGag-Protein. Dieses Protein wird nach
der Glykosylierung und Prozessierung zur Zellmembran transportiert und konnte
auch in Viruspartikeln nachgewiesen werden. Die Funktion dieses Proteins im
retroviralen Entwicklungszyklus ist unbekannt, es konnte aber gezeigt werden, dass
es wichtig für die Virusausbreitung und Pathogenität in vivo ist. Analysen des
glykoGag-Expressionsniveaus in MuLV-infizierten SC-1 Zellen zeigten, dass die
Moloney Leader-Region zu einem niedrigen, die 10A1 Leader-Region dagegen zu
einem hohen glykoGag-Expressionslevel führte. Die Herkunft der den Leader-
Bereich flankierenden Sequenzen hatte dabei keine Auswirkungen auf die
Expression des Proteins. Das glykoGag-Expressionsniveau korreliert also mit der
Induktion bestimmter Leukämieformen. Das Expressionsniveau des glykoGag-
Proteins könnte also einen wichtigen Parameter für die Zellspezifität eines MuLVs
darstellen. Es kann jedoch nicht ausgeschlossen werden, dass andere Bereiche
innerhalb des Leaders, die die Virusreplikation in vivo beeinflussen, ebenfalls an der
Zellspezifität beteiligt sind. Die Ergebnisse der vorliegenden Arbeit zeigen, dass zwei
Bereiche außerhalb des LTRs durch die Virus-Wirts-Interaktion die Leukämie-
spezifität beeinflussen.

Einleitung 3
2. Einleitung
2.1 Retroviren allgemein
Retroviren sind weit verbreitet und konnten in Mollusken, Fischen, Reptilien, Vögeln
und Säugern nachgewiesen werden. Sie können sowohl horizontal (Infektion) als
auch vertikal (Keimbahn) übertragen werden. Die Viruspartikel sind von einer
Doppelmembran umgeben und haben einen Durchmesser von 80-100 nm. Das
Genom hat eine Größe von 7-12 kb und besteht aus zwei identischen, linearen (+)-
Strang RNA-Molekülen. Die genetische Struktur ist bei allen Retroviren gleich. Die in
drei Leserastern angeordneten Gene gag, pol und env werden von zwei redundanten
Enden („Long Terminal Repeats“, LTR) flankiert (Coffin et al., 1997). Einfache
Retroviren besitzen nur diese Gene. Sie werden aufgrund ihrer Partikelmorphologie
in B-, C- oder D-Typ Viren eingeteilt. Bei komplexen Retroviren wurden noch weitere
Leseraster gefunden, die für zusätzliche regulatorische Proteine kodieren. Diese
können zwischen den pol und env Genen und/oder zwischen dem env Gen und dem
3’-LTR lokalisiert sein. Die Retroviren wurden aufgrund der Morphologie, der
Genomorganisation und Unterschieden im Entwicklungszyklus in sieben Gattungen
unterteilt (Coffin et al., 1997).
Tab. 1: Die sieben Gattungen der Retroviren, ihre Genomorganisation und exemplarische Vertreter.
Retrovirus Gattung Genom Beispiel
Bezeichnung Bezeichnung nach ICTV
Vogel-C-Typ Viren α-Retroviren einfach aviäres Leukose Virus (ALV)
Säuger-B-Typ Viren β-Retroviren einfach Maus-Mamma-Tumorvirus (MMTV)
D-Typ Viren β-Retroviren einfach Mason-Pfitzer-Affen Virus (MPMV)
Säuger-C-Typ Viren γ-Retroviren einfach murine Leukämieviren (MuLV)
BLV-HTLV δ-Retroviren komplex humanes T-Zell-Leukämievirus-1 (HTLV-1)
ε-Retroviren komplex Walleye-Dermal-Sarkom-Virus (WDSV)
Spumaviren Spumaviren komplex Affen-Foamy-Virus (SFV)
Lentiviren Lentiviren komplex humanes Immundefizienz-Virus-1 (HIV-1)
ICTV: „International Committee on Taxomomy of Viruses“
Das erste Retrovirus (ALV, s. Tab. 1) wurde 1908 von Ellermann und Bang aus
Hühnern isoliert, die an Leukämie und Lymphomen erkrankt waren (Ellermann and

Einleitung 4
Bang, 1908). Rous isolierte 1911 aus Hühnern das Rous Sarkom Virus (Rous, 1911).
Das Virus HTLV-1 wurde 1977 entdeckt und ist das erste im Menschen
nachgewiesene Retrovirus mit einem onkogenen Potential (Takatsuki et al., 1977;
Uchiyama et al., 1977). Kurz danach wurde das Virus HIV-1 als Ursache für das
erworbene Immundefizienz Syndrom des Menschen entdeckt (Barre´-Sinoussi et al.,
1983; Gallo et al., 1984; Levy et al., 1984). Retroviren können darüber hinaus auch
als endogene Viren im Vertebratengenom vorliegen und werden wie andere Gene
über die Keimbahn weiter vererbt. Die Proviren können als vollständige oder
unvollständige Kopien vorliegen, die entweder mit den zellulären Genen abgelesen
werden oder als stumme Form integriert sind.
2.2 Murine Leukämieviren
Da murine Leukämieviren gut charakterisiert, sind werde ich mich in den folgenden,
allgemeinen Aussagen auf das Buch „Retroviruses“ von Coffin, Hughes und Varmus
(1997) beziehen. Spezifische bzw. neuere Ergebnisse sind durch Zitate belegt.
2.2.1 Genomorganisation
Murine Leukämieviren (MuLVs) gehören zu den Säuger-C-Typ bzw. γ-Retroviren und
besitzen die einfache Genomorganisation LTR-gag-pol-env-LTR (s. Abb. 2). Sie
können als exogene und/oder endogene Viren vorkommen (Gardner, 1978; Hoggan
et al., 1983; Jenkins et al., 1982; Stoye and Coffin, 1987). Die gag-Sequenzen
kodieren für die Gruppen-spezifischen Antigene: die Matrix- und Kapsidproteine (MA,
CA), sowie das Nukleokapsidprotein (NC). Das MA-Protein ist mit der
Lipiddoppelmembran des Viruspartikels assoziiert, während das CA-Protein den
viralen Core-Komplex bildet (s. Abb. 1). Das NC-Protein bindet die viralen RNA-
Moleküle. Das pol-Gen kodiert für die viralen Enzyme Protease (PR), Reverse
Transkriptase (RT), die eine zusätzliche RNaseH-Aktivität besitzt, und die Integrase
(IN). Die env-Sequenzen kodieren für die durch proteolytische Spaltung generierten
Proteine: das Oberflächenprotein (SU) und das Transmembranprotein (TM). Das
Transmembranprotein verankert das äußere SU-Protein in der Doppelmembran des
Viruspartikels.

Einleitung 5
MA
PR
CA
NCRNA
IN
RT
TM
SULipiddoppelmembran
Abb. 1: Schematische Darstellung eines Viruspartikels. MA: Matrixprotein; CA: Kapsidprotein; NC:
Nukleokapsid; PR: Protease; RT: reverse Transkriptase; IN: Integrase; SU: Oberflächenhüllprotein;
TM: Transmembran-Hüllprotein (Coffin et al., 1997).
Stromabwärts vom env-Gen vor dem 3’-LTR liegt der 15 Basenpaar große
Polypurintrakt (PPT), der während der reversen Transkription als Primer für die (+)-
Strang-DNA-Synthese dient. Die drei Leseraster werden von identischen LTR-
Sequenzen flankiert. Das LTR besteht aus der U3-Region, die die Promoter- und
Enhancer-Elemente umfasst. Es folgt die terminale Repeat-Sequenz (R), an der die
virale Transkription startet und die ein Polyadenylierungssignal mit einschließt. Den
Abschluss bildet die U5-Region, die ebenfalls eine regulatorische Funktion erfüllt.
Stromabwärts vom LTR liegt der sogenannte Leader-Bereich, der die
Verpackungssequenz für die virale RNA (Ψ), eine Primer-Bindungsstelle (PBS), das
Dimerisierungssignal (DLS) und nach (Berlioz and Darlix, 1995) eine interne
Ribosomeneintrittsstelle (IRES) umfasst.

Einleitung 6
pol envgag LTR LTR
Transkrip- start
PBS
R U5 U3 ATG (nt 619)
CTG (nt 355)
SD
Ψ+
IRESDLSPromotor Enhancer
Pr75glykoGag
Pr65gag
gag
PolyA PPTSASD
Ψ
INRTPRNC CA MA
TMSU
Abb. 2: Cis-aktive Elemente in murinen Leukämieviren: „Enhancer“ Verstärker der Transkription; DLS
Dimerisierungssignal; PBS Primer-Bindungsstelle; IRES interne Ribosomeneintrittsstelle; PPT
Polypurintrakt; PolyA Polyadenylierungssignal; SD Spleiß-Donor; SA Spleiß-Akzeptor; LTR „Long
Terminal Repeat“; Ψ Verpackungssignal; Ψ+ erweitertes Verpackungssignal. Virale Gene: gag; pol;
env; Virale Proteine: MA Matrixprotein; CA Kapsidprotein; NC Nukleokapsid; PR Protease; RT reverse
Transkriptase; IN Integrase; SU Oberflächenhüllprotein; TM Transmembran-Hüllprotein.
2.3 Retroviraler Entwicklungszyklus
2.3.1 Rezeptorbindung und Eintritt in die Wirtszelle
Die Bindung an den zellulären Rezeptor und der nachfolgende Eintritt des
Viruspartikels in die Wirtszelle wird über das virale Hüllprotein (Env-Protein) vermittelt
(s. Abb. 3). Das virale env-Gen kodiert für ein Polyprotein, welches von einer
zellulären Protease im Golgi-Apparat so gespalten wird, dass ein
Transmembranprotein (TM, p15E) und ein Oberflächenprotein (SU, gp70) entstehen,
wobei das TM-Protein die SU-Einheit in der Membran des Viruspartikels verankert.
Die Bindung des Env-Proteins an seinen zellulären Rezeptor vermittelt eine
Konformationsänderung im TM-Protein, was dann zur Membranfusion und zur
Aufnahme des viralen Core-Komplexes in die Wirtszelle führt. Die virale Protease
spaltet das p15E Protein zu p12E und p2E, ersteres wird auch als R-Peptid
bezeichnet (Januszeski et al., 1997). Erst die Freisetzung des R-Peptids ermöglicht
die Fusion des Viruspartikels mit der Wirtszellmembran (Rein et al., 1994). Die

Einleitung 7
viralen Proteine SU, TM und MA verbleiben wahrscheinlich in der zellulären
Membran. Das SU-Protein erkennt und bindet den zellulären Rezeptor und vermittelt
somit die Spezies- und Gewebespezifität eines Retroviruses (White, 1992).
Zellen, die mit einem Retrovirus infiziert sind, können nicht mehr durch Viren, die den
gleichen Rezeptor benutzen, überinfiziert bzw. superinfiziert werden. Diese Art der
Resistenz wird als Interferenz bezeichnet. Diese Zellen sind aber weiterhin
empfänglich für eine Infektion mit Retroviren, die einen anderen Rezeptor für den
Eintritt in die Wirtszelle benutzen. Bei den zellulären Rezeptoren, die von den
murinen Leukämieviren erkannt werden, handelt es sich fast ausnahmslos um
Transporterproteine, die 6-14 α-helikale Transmembrandomänen besitzen. Der
Transport von kleinen Molekülen, wie Aminosäuren oder anorganischem Phosphat,
ist an einen Ionengradienten gekoppelt. Bei den Ionen kann es sich um Natrium-
und/oder Wasserstoff-Ionen handeln. Diese werden dabei entweder in die gleiche
Richtung oder entgegengesetzt zum eigentlichen Substrat transportiert (Reizer et al.,
1994; Saier, 2000).
Im Env-Protein befinden sich drei Domänen, die zwischen den einzelnen
Virenstämmen stark variieren. Im aminoterminalen Bereich sind die variablen
Regionen A und B (VRA und VRB) lokalisiert, denen sich die prolinreiche
hypervariable Region (HVR) anschließt. Der carboxyterminale Bereich ist dagegen
zwischen den verschiedenen MuLVs weitgehend konserviert. Mutationen im env-Gen
stellen einen wichtigen Faktor für die Evolution endogener Viren und deren
Pathogenität dar. Die mutierten env-Sequenzen können zur Benutzung eines
anderen Rezeptors führen, was es den Viren ermöglicht, andere Spezies zu
infizieren. Die Hauptunterschiede zwischen den verschiedenen MuLVs liegen dabei
im 5’-env-Bereich, der für das SU-Protein kodiert. Die murinen Leukämieviren
werden aufgrund ihrer Rezeptorspezifität und Homologie in verschiedene
Interferenzgruppen unterteilt, wobei Viren, die aus der Wildmaus Mus musculus
isoliert wurden, am besten charakterisiert sind (s. Tab. 2).
Diese wurden in fünf Gruppen unterteilt: ökotrope, xenotrope, polytrope, amphotrope
und 10A1-Viren (Weiss, 1993). Darüber hinaus gibt es aber auch Viren, die aus
anderen Wildmausstämmen wie Mus dunni, Mus cervicolor und Mus caroli isoliert
werden konnten.

Einleitung 8
Tab. 2: Gruppen von murinen Leukämieviren eingeteilt aufgrund von Rezeptorspezifität und
Homologie (NZB: New Zealand Black-MuLV; MDEV: Mus dunni endogenes Virus).
Rezeptorspezifität Rezeptor Prototyp
Mus musculus
ökotrop mCat1 Moloney-MuLV
xenotrop XPR1* NZB-MuLV
polytrop XPR1 Moloney-MCF
amphotrop Pit2 4070A-MuLV
10A1 Pit1, Pit2 10A1-MuLV
Mus dunni
multitrop unbekannt MDEV
Mus cervicolor
CI unbekannt CERV CI
CII SMIT1 M813
Mus caroli
CI unbekannt CARO CI
*: xenotrope MuLVs können den murinen XPR1-Rezeptor nicht benutzen
Ökotrope MuLVs kommen sowohl als exogene als auch als endogene Viren vor
(Jenkins et al., 1982) und benutzen den kationischen Aminosäure Transporter Cat-1
als Rezeptor, wobei sie nur die CAT1-Proteine von Maus und Ratte erkennen
können (Albritton et al., 1989; Coffin et al., 1997). Die Rezeptornutzung von
ökotropen Viren wird durch die VRA vermittelt, der VRB kommt dabei nur eine
akzessorische Funktion zu (Battini et al., 1995; Battini et al., 1998; Battini et al.,
1996). Der aminoterminale env-Bereich von ökotropen Viren stammt entweder von
einem unbekannten Virus oder von zellulären Sequenzen, während die restlichen
env-Regionen xenotropen Ursprungs sind (Stoye and Coffin, 1987).
Polytrope und xenotrope Viren benutzen zwar beide das Glykoprotein XPR1 als
Rezeptor (Battini et al., 1999; Tailor et al., 1999; Yang et al., 1999), aber Mutationen
in dessen Aminosäuresequenz bestimmen, ob eine Spezies durch eines der beiden
Viren infizierbar ist oder nicht (Marin et al., 1999). Während der von xenotropen
MuLVs benutzbare XPR1-Rezeptor nicht von Mus musculus und von ihr
abstammenden Labormausstämmen, sondern nur von einigen Wildmausarten bzw.

Einleitung 9
von anderen Spezies exprimiert wird, können die polytropen Viren alle Mausstämme
und viele andere Spezies infizieren (Kozak, 1985). Xenotrope Viren kommen nur als
endogene Viren vor. Polytrope Viren sind durch Mutationen im xenotropen env-Gen
entstanden, wodurch sie ein breiteres Wirtsspektrum infizieren können (Marin et al.,
1999; Tomonaga and Coffin, 1999). Ein Gen im Rmc1-Lokus kodiert für den XPR1-
Rezeptor, Mutationen in diesem Lokus entscheiden darüber, ob das Protein von
polytropen oder xenotropen Viren benutzt werden kann (Battini et al., 1999; Hartley
et al., 1983; Tailor et al., 1999; Yang et al., 1999). Im Gegensatz zu ökotropen
MuLVs benötigen xenotrope und polytrope Viren neben der VRA und VRB, auch die
HVR für die Rezeptorerkennung (Battini et al., 1995; Battini et al., 1998; Battini et al.,
1996).
Amphotrope Viren benutzen den von vielen Spezies exprimierten Typ III Natrium
abhängigen Phosphatsymporter Pit2 (Kavanaugh et al., 1994; Miller and Miller, 1994;
Wilson et al., 1995). Amphotrope MuLVs benötigen ebenfalls nur die VRA und VRB
für die Erkennung ihres Rezeptors. Das 10A1-MuLV ist durch Rekombination
zwischen dem exogenen, amphotropen Virus 1504A und endogenen polytropen
Sequenzen entstanden, wobei die polytrope HVR in das amphotrope env-Gen
inseriert wurde (Ott et al., 1990; Rasheed et al., 1982). Durch die rekombinierten
env-Sequenzen ist das Virus in der Lage sowohl den Pit2-Rezeptor als auch den
verwandten Phosphatsymporter Pit1 zu nutzen (Miller and Miller, 1994; Wilson et al.,
1994). Die einzigartige Rezeptornutzung von Pit1 verleiht dem Virus ein breites
Wirtsspektrum und es stellt deshalb eine eigene Interferenzgruppe dar (Han et al.,
1997).
Murine Leukämieviren, die aus anderen Mausstämmen als Mus musculus isoliert
worden sind, wurden bisher noch nicht so ausführlich charakterisiert. Kürzlich konnte
in Mus dunni ein endogenes, multitropes Virus (MDEV) entdeckt werden, welches ein
breites Wirtsspektrum hat, dessen zellulärer Rezeptor unbekannt, aber nicht
identisch mit den schon bekannten Rezeptorklassen ist (Bonham et al., 1997). Das
aus Mus cervicolor isolierte Virus CERV CI zeigt ein mit den xenotropen MuLVs
vergleichbaren Wirtstropismus und scheint mit dem aus Mus caroli isolierten Virus
verwandt zu sein (Benveniste et al., 1977). Das aus Mus cervicolor isolierte M813-
MuLV ähnelt den ökotropen Viren, benutzt aber den Rezeptor SMIT1 für die Infektion
(Hein et al., 2003; Prassolov et al., 2001).

Einleitung 10
Aktivierung
Rezeptorbindung
Virusreifung
VirusknospungAssemblierung
Translation
Transkription
Membranfusion
Zellkernimport
Gagvon
Genen
glykoGag Integration RNA Export
Gag-Pol
Env-Prozessierung
Zerfall des Core-Komplexes
reverse Transkription
Abb. 3: Schematische Darstellung des retroviralen Entwicklungszyklus
2.3.2 Reverse Transkription und Integration Nach Bindung des zellulären Rezeptors und anschließender Membranfusion kommt
es zur Internalisierung des Nukleokapsids (s. Abb. 3). Dieser Prozess führt zu einer
Konformationsänderung, die den Eintritt von Nukleotiden in das Innere des Partikels
ermöglicht. Die beiden identischen RNA-Moleküle sind über ihre im Leader-Bereich
liegenden DLS-Sequenzen dimerisiert. Sie weisen bezüglich der Translation eine
positive Polarität auf. Die genomische RNA besitzt eine 5’-Kappenstruktur und ein 3’-
Polyadenylierungssignal wie es für eukaryontische mRNAs charakteristisch ist. Die
reverse Transkription der einzelsträngigen RNA findet im Zytoplasma der Wirtszelle
statt. Die Transkription des negativen DNA-Stranges beginnt an einer vom Virus
mitgeführten zellulären tRNA, die als Primer dient. Dieser Primer ist über homologe
Basenpaarung an die PBS der viralen RNA gebunden. Durch die Verlängerung des
3’-OH Endes des Primers werden die U5 und R-Sequenzen am 5’-Ende der

Einleitung 11
genomischen RNA transkribiert. Der bereits transkribierte Bereich der viralen RNA
wird durch die RNaseH-Funktion der reversen Transkriptase degradiert. Es kommt
nun zum ersten Strang-Transfer. Die neu erzeugte R-Region des negativen DNA-
Stranges bindet an die 3’ komplementäre R-Sequenz am 3-Ende des RNA-Genoms.
Das lineare RNA-Molekül wird dann in 5’-Richtung transkribiert, wobei bereits
transkribierte Bereiche degradiert werden. Die im 3’-Bereich des viralen Genoms
gelegene PPT-Region ist relativ resistent gegenüber des RNaseH-Abbaus und dient
als Primer für die Synthese des positiven DNA-Stranges. Die Synthese erfolgt in 3’-
Richtung bis zur PBS, die an diese gebundene tRNA wird degradiert. Dieses kurze
positive DNA-Fragment bindet an die komplementären Bereiche im negativen DNA
Strang und wird verlängert bis ein doppelsträngiges DNA-Molekül entsteht, das zwei
identische LTR-Bereiche an beiden Enden besitzt. Da die reverse Transkriptase
keine Fehlerkorrekturen durchführt, wie es von zellulären DNA-Polymerasen bekannt
ist, treten 10-4 Fehler pro inkorporierter Base auf. Das entspricht einer falschen Base
pro synthetisiertem Genom. Diese hohe Mutationsrate ist für die Anpassungs- und
Wandlungsfähigkeit der Retroviren verantwortlich. Wenn eine Zelle mit zwei
verschiedenen Viren infiziert ist, kann es zur Verpackung von zwei unterschiedlichen
RNA-Molekülen in einem Viruspartikel kommen. Darüber hinaus kann es während
der reversen Transkription zu Rekombinationen zwischen den beiden Nukleinsäure-
Molekülen kommen. Dies ist mit einem Selektionsvorteil verbunden, da es zu einer
erhöhten Variation führen kann (Hu und Temin, 1990).
Der Kapsidkomplex bleibt während des Transports zum und in den Zellkern erhalten.
Der Transfer in den Zellkern erfolgt während der Mitose, wenn die Membran des
Nukleus abgebaut wird. Die virale Integrase vermittelt die zufällige Integration der
proviralen DNA in das Genom der Wirtszelle. Allerdings scheinen aktiv transkribierte
Bereiche für die Inserierung etwas bevorzugt zu werden (Mooslehner et al., 1990;
Rohdewohld et al., 1987; Scherdin et al., 1990; Vijaya et al., 1986). Das HI-Provirus
inseriert präferentiell in kodierende Genbereiche (Schröder et al., 2002). Dabei
scheint der Präferenzgrad des Virus mit dem Expressionslevel des Gens zu
korrelieren. Darüber hinaus inserierte das HI-Virus vorwiegend in solchen Genen, die
nach einer HIV Infektion aktiviert sind.
Ein infizierendes Viruspartikel führt nur zur Integration eines Provirus in das
Wirtsgenom (Hu and Temin, 1990), welches dann stabil weitervererbt wird. Die

Einleitung 12
Integration des Provirus in zellulären Genen oder in deren regulatorische Sequenzen
kann zu einer Aktivierung oder Inaktivierung dieser Elemente führen, wobei die
Integration eines Provirus nicht unbedingt direkt zur Entartung der betroffenen Zelle
führen muss. Es sind meistens mehrere Integrationen in verschiedenen Genen
notwendig, wobei dann deren Zusammenwirken zum Auswachsen eines Tumors
führt. Es gibt natürlich auch Integrationen, die keinerlei Auswirkungen auf die
Regulation der betroffenen Zelle haben.
2.3.3 Transkription, Translation und Assemblierung
Die LTR-Sequenzen kodieren zwar für keine viralen Proteine, sind aber wichtig für
einige Stationen im retroviralen Replikationszyklus, wie Integration des Provirus in
die DNA der Zielzelle, sowie Initiation und Termination der Transkription. Die
Transkription beginnt erst wenn das Provirus in das Genom des Wirtes inseriert ist.
Das zelleigene Enzym RNA-Polymerase II synthetisiert die virale RNA. Die
Transkription startet an der 5’-R-Region und endet im 3’-LTR am Ende der dort
gelegenen R-Sequenz. Den regulatorischen Sequenzen in der U3-Region, Promotor-
und Enhancer-Elementen, wird eine besondere Bedeutung zugeschrieben, da diese
die Genexpression durch Rekrutierung zelleigener Transkriptionsfaktoren
beeinflussen. Das Vorhandensein dieser Transkriptionsfaktoren in bestimmten
Zelltypen oder bestimmten Differenzierungsstadien von Zellen könnte somit
Auswirkungen auf das Transkriptionsniveau spezifischer MuLVs haben.
Bei der Transkription wird gespleißte und ungespleißte RNA synthetisiert. Das
ungespleißte, genomische RNA-Transkript wird entweder über das Verpackungs-
signal Ψ in das Viruspartikel inseriert (Mann et al., 1983; Mann and Baltimore, 1985)
oder es werden von diesem die Gag- bzw. Gag-Pol-Polyproteine translatiert. Die
Initiation der Translation des Gag-Polyproteins (pr65Gag) erfolgt kappenunabhängig
an einer stromaufwärts vom AUG gelegenen IRES (Berlioz and Darlix, 1995). Die
kappenabhängige Translation startet an einem konservierten CUG stromaufwärts
vom Gag-Initiations-Kodon und führt zu einem aminoterminal verlängerten Gag-
Protein, dem glykoGag (pr75Gag) (Corbin et al., 1994). Dieses hat einen
modifizierten aminoterminalen Bereich, wird im endoplasmatischen Retikulum
glykosyliert (pr85glykoGag) und von einer zellulären Protease in zwei Hälften

Einleitung 13
gespalten. Die C-terminale Hälfte wird sekretiert, während der N-terminale Bereich in
der Plasmamembran lokalisiert bleibt. Beim N-terminalen, glykosylierten Teil handelt
es sich um ein Typ II Membranprotein (Ncyto, Cexo), d.h. der N-Terminus ist in der
Plasmamembran verankert und der C-Terminus ragt in den extrazellulären Raum
(Fujisawa et al., 1997). Möglicherweise interagiert glykoGag mit den Rezeptoren auf
Nachbarzellen und verstärkt so die Virusausbreitung von Zelle zu Zelle. Die
Translation des Gag-Pol-Polyproteins erfolgt durch Suppression eines Stop-Kodons.
Das Env-Polyprotein wird durch spleißen erzeugt. Der Spleiß-Donor ist im Leader-
Bereich lokalisiert, der Spleiß-Akzeptor liegt stromaufwärts von den env-Sequenzen.
Im endoplasmatischen Retikulum oligomerisiert das Vorläuferprotein zu Trimeren,
wird dann glykosyliert und das Signalpeptid entfernt. Im Golgi-Apparat wird das
Glykoprotein von einer zellulären Protease in das glykosylierte SU- und das nicht-
glykosylierte TM-Protein gespalten. Diese werden dann zur Plasmamembran
transportiert.
Der virale Kapsid-Komplex formt sich aus den dimerisierten RNA-Molekülen und den
Gag- und Gag-Pol-Polyprotein an der Zytoplasmamembran. Während des
Ausknospens erhält das Viruspartikel seine Doppelmembran mit den Env-Proteinen.
Die virale Protease prozessiert die Gag- und Gag-Pol-Polyproteine während und
nach der Virusfreisetzung. Wenn eine Zelle mit verschiedenen MuLVs infiziert ist,
können RNA-Moleküle von einem Virus, die Hüllproteine aber von einem anderen
Virus stammen. Dieser Vorgang wird als Pseudotypisierung bezeichnet.
2.4 Pathogenität muriner Leukämieviren
Retroviren induzieren sowohl in der freien Wildbahn als auch unter
Laborbedingungen eine Vielzahl von Erkrankungen. Dazu gehören Neoplasien,
Entzündungen, Immundefizienzsyndrome sowie degenerative Auswirkungen. Dabei
sind meistens das lymphohämatopoetische und/oder das Nervensystem betroffen.
Es können aber auch andere Organe und Gewebe beteiligt sein. Neben den viralen
Parametern, entscheidet der genetische Hintergrund der verwendeten Mäuse
darüber, welche Krankheitssymptome sich nach einer Virusinfektion manifestieren
können oder ob sich überhaupt eine chronische Infektion etablieren kann.

Einleitung 14
Die Resistenz gegenüber einer Infektion mit bestimmten Viren wird durch Wirtsgene
vermittelt, die auf unterschiedliche Weise in den retroviralen Entwicklungszyklus
eingreifen. Eine Möglichkeit ist die Modulation der Immunantwort. Es gibt Gene,
innerhalb und außerhalb der MHC-Region, die die Immunreaktion gegenüber einer
MuLV Infektion regulieren können. Eine weitere Möglichkeit eine Virusinfektion zu
verhindern, ist der Polymorphismus von bestimmten Rezeptorgenen in den
verschiedenen Spezies. Einige Mausstämme, die eine funktionelle Variante eines
bestimmten Rezeptors exprimieren, können nicht durch Viren infiziert werden, die
diesen modifizierten Rezeptor nicht binden können. Andere Rezeptor-vermittelte
Resistenzen basieren auf einem Interferenz-Mechanismus. Endogene env-
Sequenzen im Mausgenom können z.B. für ein Env-Protein kodieren, welches dann
an einen bestimmten, auf der Zellmembran lokalisierten, Rezeptor bindet. Der
Rezeptor ist dadurch blockiert und kann nicht mehr von einem bestimmten MuLV für
den Zelleintritt benutzt werden. Das Fv4-Gen kodiert für ein ökotropes Env-
Glykoprotein, welches die Bindung von exogenen, ökotropen Viren an ihren Rezeptor
inhibiert (Ikeda et al., 1985; Taylor et al., 2001). Analog dazu verhindert das
Genprodukt des Rmcf-Lokus die Bindung von polytropen MuLVs an den
entsprechenden Rezeptor (Jung et al., 2002; Ruscetti et al., 1981). Das Fv1-Gen
kodiert für ein Protein, welches dem viralen Kapsidprotein ähnelt, und wahrscheinlich
durch eine direkte Bindung an das virale Protein einen Präintegrationsblock
verursacht (Best et al., 1996; Bishop et al., 2001).
Es gibt aber auch Gene, die einen Mausstamm besonders sensitiv gegenüber der
Transformation durch bestimmte Viren machen. Das Fv2-Gen kodiert für eine
verkürzte Form der Stk Rezeptor-Tyrosinkinase, die wahrscheinlich mit dem
gp55/EpoR-Komplex assoziiert ist. Dies führt zu einer erhöhten Empfänglichkeit
gegenüber der Induktion von Erythroleukämien durch den Friend-Viruskomplex (FV-
Komplex) führt (Persons et al., 1999). Ein weiteres Gen Cdc25A, welches in der
Nähe von Fv2 lokalisiert ist, beeinflusst den Status des Zellzyklus von erythroiden
Vorläuferzellen und moduliert ebenfalls die Sensitivität der Tiere gegenüber einer FV-
Infektion, da dieser FV-Komplex keine ruhenden Zellen infizieren kann (Melkun et al.,
2002).
Die in den Mäusen induzierten Neoplasien lassen sich aufgrund der
Inkubationsphase grob in zwei Gruppen einteilen. Einige MuLVs induzieren nach
einer kurzen Latenzzeit akute Krankheitssymptome, während andere erst nach einer

Einleitung 15
langen Inkubationszeit klinische Symptome induzieren. Murine Leukämieviren
können auch zu chronischen Infektionen ohne die Entwicklung von Krankheits-
symptomen führen.
2.4.1 Replikationsinkompetente Retroviren
Bei diesen Retroviren handelt es sich um akut transformierende Viren, die nach einer
kurzen Latenzzeit maligne Erkrankungen induzieren. Diese Viren haben auf Kosten
eigener Sequenzen im gag- oder env-Bereich ein zelluläres Onkogen erworben und
sind deshalb meistens replikationsinkompetent. Sie benötigen für die Replikation ein
sogenanntes Helfer-Virus, das in derselben Zelle repliziert. Diese Viren spielen in der
Natur keine große Rolle, da die Erwerbung eines Onkogens zum einen sehr selten
vorkommt und zum anderen eine horizontale Ausbreitung sich durch die letalen
Auswirkungen einer Infektion in Grenzen hält. Auf der anderen Seite stellen sie für
die Erforschung der Zelltransformation ein wichtiges Hilfsmittel dar. Das virale
Onkogen (v-onc) ist meistens im gleichen Leseraster mit den anderen viralen Genen
inseriert, so dass bei der Translation Fusionsproteine entstehen. Der dominierende
Effekt der v-onc Gene basiert auf drei Eigenschaften. Das erworbene Onkogen
vermittelt den infizierten Zellen einen Selektionsvorteil. Durch die hohe Mutationsrate
in den Retroviren kann das onkogene Potential dieses Gens noch erhöht werden.
Zweitens kann v-onc mit Hilfe des Virus in Zellen eingebracht werden, in denen es
normalerweise nicht exprimiert wird oder die nicht die nötigen Faktoren besitzen, um
die Expression des Onkogens zu regulieren. Drittens sorgen die starken Promoter-
und Enhancer-Elemente im LTR für eine starke Expression in der infizierten Zelle.
Die von murinen Retroviren erworbenen v-onc Gene werden nach der Funktion des
zellulären Proto-Onkogens (c-onc) in verschiedene Klassen aufgeteilt (s.Tab. 3).

Einleitung 16
Tab. 3: Erworbene Onkogene und exemplarische Retrovirusvertreter (Coffin et al., 1997)
c-onc Funktion Onkogen Virus Art der Expression Tumor
Protein-Tyrosin-
Kinase abl Abelson-MuLV gag-abl-Fusion B-Zell-Lymphom
G-Protein Ha-ras Harvey-MSV
MHSVv-ras Sarkome, Erythroleukämien
maligne HistiozytoseSerin-Threonin-
Kinase mos Moloney-MSV env-mos-Fusion Fibrosarkom
Transkriptionsfaktor fos FBJ-MSV v-fos Osteosarkom
MSV: Maus-Sarkom-Virus; MHSV: Malignes-Histiozytose-Sarkom-Virus
Das Myeloproliferative-Sarkom Virus (MPSV) und das Moloney-Maus-Sarkom-Virus
(M-MSV) tragen beide das v-mos Onkogen. Während das MPSV in Mäusen eine
myeloproliferative Erkrankung und Fibrosarkome induziert, verursacht das M-MSV
ausschließlich Fibrosarkome. Dieser Unterschied in der Tumorspezifität wird nur
durch die Herkunft der LTR-Sequenzen bestimmt (Stocking et al., 1985). Darüber
hinaus ist jedes mos-Gen, unabhängig ob viralen oder zellulären Ursprungs, in der
Lage eine Myeloproliferation zu induzieren, wenn es im Kontext mit den richtigen
LTR-Sequenzen exprimiert wird. Auch für das v-ras tragende Maligne-Histiozytose-
Sarkom-Virus (MHSV) konnte gezeigt werden, dass dessen Fähigkeit in adulten
Mäusen Makrophagen zu transformieren durch seine LTR-Sequenzen bestimmt wird
(Friel et al., 1990; Franz et al., 1985; Löhler et al., 1987).
Das replikationsinkompetente Milzfokus-formende-Virus (SFFV, „Spleen-Focus-
Forming-Virus“) wirkt durch sein Env-Glykoprotein (gp55) ebenfalls akut
transformierend auf infizierte Zellen. Das deletierte, rekombinierte gp55 bindet an
den Erythropoetin-Rezeptor und stimuliert auf diese Weise die Proliferation
erythroider Vorläuferzellen, was zur Induktion einer Erythroleukämie führt (Li et al.,
1990; Li et al., 1995). Das SFFV bildet mit dem Friend-MuLV als Helfervirus den
sogenannten Friend-Viruskomplex (Ostertag et al., 1987).

Einleitung 17
2.4.2 Replikationskompetente Retroviren
Im Gegensatz zu den akut transformierenden Retroviren benötigen die
replikationskompetenten MuLVs kein virales Onkogen, um proliferative Erkrankungen
zu induzieren. Die Inkubationszeiten der verschiedenen Viren variiert sehr stark, von
einem Monat bis zu einem dreiviertel Jahr. Es treten vor allem Beeinträchtigungen
des hämatopoetischen Systems, der Knochen und des Zentralnervensystems auf.
Die Art der induzierten Krankheit hängt vom Virus, dem Infektionszeitpunkt, dem
Infektionsort und dem genetischen Hintergrund des Mausstammes ab (s. Tab. 4).
Rezeptorspezifität Virus induzierte Neoplasie
ökotrop Moloney-MuLV T-Zell-Leukämie
Friend-MuLV Erythroleukämie
Graffi-MuLV myeloische Leukämie
Gross-MuLV T-Zell-Leukämie
AKR-SL3-3-MuLV T-Zell-Leukämie
Cas-Br-E-MuLV myeloische Leukämie
nicht T-, nicht B-Zell-Leukämie
polytrop Moloney-MCF T-Zell-Leukämie
MCF-247-MuLV T-Zell-Leukämie
amphotrop 4070A myeloische Leukämie
nicht T-, nicht B-Zell-Leukämie
B-Zell-Leukämie
10A1 10A1-MuLV Stammzell-Leukämie
Tab. 4: Von replikationskompetenten MuLVs induzierte Leukämieformen, mit exemplarischen
Vertretern (Kozak and Ruscetti, 1992; Ostertag et al., 1980; Ott et al., 1994; Rasheed et al., 1982)
Die Entwicklung einer Neoplasie kann in eine präleukämische und eine leukämische
Phase eingeteilt werden. Nach der Infektion kann eine allgemeine Zellvermehrung
(Hyperplasie) in den lymphatischen Organen beobachtet werden, deren Integrität
aber erhalten bleiben (Fan, 1994). Dieser vermehrten Proliferation liegt
wahrscheinlich ein virusinduzierter Zytokin-Stimulus zugrunde (Brightman et al.,
1990). Die leukämische Phase zeichnet sich vor allem durch das klonale
Auswachsen einer transformierten Zelle aus. Die Deregulation von zelleigenen
Genen hat der Zelle einen Selektionsvorteil verschafft.

Einleitung 18
Die Maus ist zwar ein gutes Modell für Krebserkrankungen, aber es existieren auch
deutliche Unterschiede in der Entwicklung neoplastischer Erkrankungen des
lymphohämatopoetischen Systems zwischen Mensch und Maus. Der wichtigste
Unterschied betrifft den Ursprung der Neoplasien. Leukämien des Menschen
entstehen primär im Knochenmark, von wo aus die Ausschwemmung von
Tumorzellen in die Blutzirkulation erfolgt, was dann in seltenen Fällen zur
Tumorbildung in anderen Organen führen kann. In einigen Mausinzuchtstämmen
kann es spontan zur Entstehung von primär manifestierten Leukämien kommen, was
allerdings sehr selten beobachtet wird. Bei murinen Neoplasien, vor allem in
experimentell induzierten, treten dagegen häufiger solide Tumore im blutbildenden
System auf, wobei besonders die Milz (weiße und rote Pulpa), die Lymphknoten und
nur selten das Knochenmark betroffen sind. Bei Progression dieser soliden,
hämatopoetischen Tumore kommt es in ca. 30 % der Tiere zu einer sekundären
Ausschwemmung von Tumorzellen und der Entwicklung einer Leukämie. Diesen
unterschiedlichen Prozessen liegen wahrscheinlich die fundamentalen Unterschiede
im blutbildenden System der beiden Spezies zugrunde. Während die rote Milzpulpa
des erwachsenen Menschen nur zur Resorption beschädigter Erythrozyten dient, ist
diese in der adulten Maus immer noch ein hämatopoetisch wichtiges Gebilde. Das
Knochenmark einer ausgewachsenen Maus ist fast vollständig für die Hämatopoese
zuständig, während diese beim adulten Menschen nur noch in bestimmten, flachen
Knochenstrukturen stattfindet (z.B. Schädel, Wirbelkörper, Becken und Sternum). Die
aus der Maus gewonnenen Erkenntnisse können also nur bedingt auf den Menschen
übertragen werden, sind aber für die Erforschung von Mechanismen, die zu
Neoplasien führen sehr hilfreich, weil sich die Krebsentwicklung in den beiden
Spezies auf molekularer Ebene nur wenig unterscheidet.

Einleitung 19
2.5 Verschiedene Faktoren beeinflussen den Typ der induzierten Krankheit
Die Latenzzeit und die Art einer MuLV-induzierten Leukämie wird von mehreren
Faktoren beeinflusst:
1. viralen Determinanten
2. Generation von rekombinierten Viren
3. Aktivierung von Genen durch Provirusintegrationen
4. Mausstamm
2.5.1 Virale Determinanten: LTR- und env-Sequenzen
Die Enhancer-Elemente im LTR liegen meistens als zwei identische Kopien („Direct
Repeats“) vor und enthalten die Bindungsstellen für bestimmte Transkriptions-
faktoren, die in einer speziellen Reihenfolge und in einem bestimmten Abstand
zueinander angeordnet sind (DiFronzo and Holland, 1999; Fan et al., 1988; Golemis
et al., 1990). Die Anzahl und die Vollständigkeit dieser Direct Repeats beeinflusst die
Pathogenität eines MuLVs (Baum et al., 1997; Hanecak et al., 1988; Manley et al.,
1993; Speck et al., 1990; Wang et al., 1993). Im Allgemeinen haben Retroviren, die
nur eine Kopie der Enhancer-Elemente besitzen, die dann auch noch Mutationen in
einigen Bindungsstellen aufweisen kann (z.B. 10A1-, 4070A- und Friend-SFFV), eine
lange Latenzzeit. Hochpathogene Viren zeigen dagegen eine Tandem-Anordnung
der Enhancer-Elemente und haben eine kurze Inkubationszeit (DesGrosseilliers and
Jolicoeur, 1984; Holland et al., 1989). Das Mo-MuLV besitzt diese Tandem-
Anordnung in seinen LTR-Sequenzen und induziert T-Zell-Lymphome, während das
Friend-MuLV nur ein unvollständiges Enhancer-Element aufweist und ausschließlich
Erythroleukämien verursacht. Das Mo-MuLV induziert nach Einführung der Friend
Enhancer-Elemente vorwiegend Erythroleukämien anstatt T-Zell-Leukämien,
während das Friend-MuLV mit einem Moloney-LTR überwiegend T-Zell-Lymphome
anstatt Erythroleukämien verursacht (Chatis et al., 1983; Golemis et al., 1989).
Die Env-Proteine könnten durch ihre spezifische Rezeptorerkennung einen Einfluss
auf die infizierbaren Zellpopulationen haben und stellen möglicherweise einen
wichtigen Parameter für die Pathogenität eines MuLVs dar. Die Phosphatsymporter

Einleitung 20
Pit1 und Pit2 werden in fast allen Geweben exprimiert, wobei aber das Pit2
Expressionslevel im allgemeinen niedriger ist als das von Pit1 (Nielsen et al., 2001).
Dabei bestimmt die Menge an exprimiertem Rezeptor auf der Oberfläche bestimmter
Zellen über deren Empfänglichkeit gegenüber einer Virusinfektion (Tailor et al.,
2000). Pit1 wird vor allem im Knochenmark und auf hämatopoetischen Zellen
exprimiert (Kavanaugh et al., 1994). Der Austausch der env-Sequenzen eines Virus
hat gezeigt, dass deren Herkunft Auswirkungen auf die Art der induzierten Neoplasie
hatte (Ott et al., 1992). Darüber hinaus kann das nicht neuropathogene Moloney-
MuLV durch amphotrope env-Sequenzen in die Lage versetzt werden, das
Zentralnervensystem von infizierten Mäusen zu schädigen (Münk et al., 1997).
2.5.2 Generation von rekombinanten Viren
Durch die Rekombination von exogenen, ökotropen Viren mit endogenen Sequenzen
im Mausgenom entstehen MCF-Viren (Hartley et al., 1977). Da die chimären MCF-
Viren durch ihren rekombinierten Rezeptorerkennungsbereich in der Lage sind
andere Rezeptoren für den Eintritt in die Wirtszelle zu nutzen als das exogene Virus,
ist eine Superinfektion der Zellen möglich, was die Ausbreitung der Infektion
beschleunigt (Lavignon and Evans, 1996). Das ökotrope Friend-MuLV kann in
infizierten Mäusen nach einer kurzen Latenzzeit Erythroleukämien induzieren.
Mausstämme, die das Rmcf-1-Gen exprimieren, sind resistent gegenüber einer
Friend-MuLV Infektion. Da ihr Rezeptor blockiert ist, können die Zellen nicht durch
die nach der Infektion entstandenen Friend-MCF-Viren überinfiziert werden (Ruscetti
et al., 1981).
Die Superinfektion von infizierten Zellen durch MCF-Viren scheint also für die
Etablierung einer Krankheit wichtig zu sein. Eine Überinfektion führt zu weiteren
Provirusintegrationen, was die Wahrscheinlichkeit von zusätzlichen Genakti-
vierungen, die für eine vollständige Transformation der infizierten Zelle notwendig
sind, erhöht. Möglicherweise ermöglichen MCF-Viren eine Superinfektion durch
Pseudotypisierung, d.h. ökotrope genomische RNA wird in Viruspartikel mit
polytropen Hüllproteinen verpackt. Rekombinationen im LTR-Bereich von MCF-Viren
erhöhen deren Transkription in Thymozyten, was ebenfalls dafür spricht, dass diese
Rekombinanten einen wichtigen Beitrag zur Tumorentstehung leisten können
(Holland et al., 1989).

Einleitung 21
Die in der präleukämischen Phase beobachtete Hyperplasie von Moloney-MuLV
infizierten Mäusen wurde mit dem Auftreten von MCF-Viren in Zusammenhang
gebracht (Brightman et al., 1991). Es konnte gezeigt werden, dass das polytrope
Env-Protein (gp70) in der Lage ist, den zellulären Interleukin-2-Rezeptor zu binden
(Li and Baltimore, 1991). Diese Art der Interaktion ist vom SFFV gp55 bekannt, das
an den Erythropoetin-Rezeptor (EpoR) bindet. Diese Interaktion auf der
Zelloberfläche könnte einen Stimulus für die Proliferation der infizierten Zellen
induzieren (Fan, 1994; Li and Baltimore, 1991; Li et al., 1995).
Einen völlig anderen Einfluss von MCF-Viren schlagen (Yoshimura et al., 2001) vor.
Sie vermuten, dass die Bindung von gp70 an den zellulären Rezeptor keine
Proliferation, sondern Apoptose induziert. Weitere Integrationsereignisse durch
Superinfektion könnten dann eine Zelle aus der apoptotischen Krise retten. In wie
weit MCF-Viren für die Induktion einer T-Zell-Leukämie notwendig sind, kann nicht
beantwortet werden. Ratten besitzen zum Beispiel keine endogenen Sequenzen, die
zur Generierung von MCF-Viren beitragen könnten, trotzdem entwickeln diese Tiere
nach einer MuLV Infektion eine T-Zell-Leukämie.
2.5.3 Aktivierung von Genen durch Provirusintegrationen
Retrovirale Integrationen können durch verschiedene Mechanismen zur Aktivierung
von zellulären Genen beitragen (Coffin et al., 1997).
1. Promoter-Insertion: Die Aktivierung der zellulären Gene erfolgt über die viralen
Promoter im LTR. Diese Art der Genaktivierung wird vor allem in ALV
induzierten Tumoren beobachtet.
2. Enhancer-Insertion: Die Enhancer-Sequenzen des integrierten Provirus
erhöhen die Expression des zellulären Gens. Diese Aktivierung ist unabhängig
von der Orientierung des integrierten Virus. Darüber hinaus hat der
Integrationsort des Virus, 5’ oder 3’ vom Gen, keinen Einfluss auf die
Aktivierung. Dieser Mechanismus wird sehr häufig in MuLV induzierten
Tumoren beobachtet.

Einleitung 22
3. Fusionstranskripte: Die Integration eines Provirus in derselben
Transkriptionsrichtung wie die des zellulären Gens kann nach Veränderungen
im viralen 3’-LTR zur Bildung von Fusionstranskripten führen. Das Gen kann
durch die Bildung eines Fusionsproteins onkogenes Potential entwickeln.
4. Insertion in UTR-Regionen: Provirusintegrationen in 5’ oder 3’ gelegenen
untranslatierten, regulativen Sequenzen können diese zerstören, was zu einer
Deregulation des dazugehörigen Gens führt, so dass dieses ein onkogenes
Potential erwirbt.
Die Charakterisierung dieser Integrationsstellen hat zur Entdeckung von zahlreichen
Onkogenen geführt, die auch in der humanen Tumorentwicklung eine wichtige Rolle
spielen (Lund et al., 2002; Mikkers et al., 2002; Suzuki et al., 2002).
2.5.3.1 Aktivierte Onkogene in T-Zell-Leukämien
In 35-50 % der MuLV induzierten T-Zell-Lymphome ist eine provirale Integration in
das N-myc Gen zu beobachten gewesen (van Lohuizen et al., 1989). Wenn die
Insertion in einem kleinen Segment innerhalb des 3’ untranslatierten Bereichs (3’-
UTR) stattfindet, kommt es zur verstärkten Transkription einer verkürzten mRNA. Der
erhöhte Expressionslevel des N-myc Proteins führt zu einem fast vollständigen
Verschwinden des c-myc Genprodukts, so dass N-myc wahrscheinlich als
Suppressor für andere Mitglieder der myc-Familie fungiert (van Lohuizen et al.,
1989).
Das Pim-1 Gen kodiert für eine zytoplasmatische Serin-Threonin-Kinase, deren Rolle
in der Signaltransduktion noch nicht geklärt ist. Eine Aktivierung dieses Gens wurde
in 50 % der T-Zell-Leukämien beobachtet. Die Integration der Proviren erfolgt vor
allem in der 3’-UTR, seltener auch in der 5’-UTR, wobei es zu einer Akkumulation
von Wildtyp mRNA und Protein kommt, was eine stark transformierende Wirkung auf
die Zelle ausübt (Cuypers et al., 1984). In transgenen c-myc Mäusen konnte eine
Kooperation von c-myc und Pim-1 während der Onkogenese beobachtet und darüber
hinaus festgestellt werden, dass eine Integration in den Pim-2 Lokus die Integration
in Pim-1 ersetzen kann (van Lohuizen et al., 1991). Die hohe Aminosäuresequenz-
Homologie der beiden Proteine spricht auch für eine funktionelle Redundanz.

Einleitung 23
Eine Aktivierung des c-myc-Gens wird sehr häufig in T-Zell- und B-Zell-Lymphomen
beobachtet (Corcoran et al., 1984; Stanton et al., 1983). Die Rolle von c-myc in der
murinen und humanen Onkogenese ist unumstritten und es stellt eines der
bestuntersuchten Onkogene dar. Das Genprodukt von c-myc ist ein
Transkriptionsfaktor und wird vor allem während der Differenzierung von Zellen
exprimiert, die sich in einer bestimmten Phase des Zellzyklus befinden (Hann et al.,
1985; Kelly et al., 1983; Rabbitts et al., 1985). Die Expression dieses Proteins
unterliegt einer strengen Kontrolle. Darüber hinaus wird Myc durch die Bindung an
ein anderes Protein namens Max reguliert (Blackwood and Eisenmann, 1991;
Prendergast et al., 1991). Dieser Myc-Max-Kompex erhöht den Transkriptionslevel
von bestimmten Genen. Da Max in großer Menge in der Zelle vorkommt, kann eine
Myc Überexpression durch das virale LTR die Transformation einer infizierten Zelle
induzieren (Amati et al., 1993). In murinen Plasmazytomen und in humanen Burkitt
Lymphomen kann häufig eine Chromosomentranslokation beobachtet werden, die
das c-myc Gen unter die Kontrolle des Enhancers aus dem Immunoglobulingen-
Lokus stellt (Stanton et al., 1983). Dabei geht das erste Exon verloren, welches zwar
untranslatiert ist, aber wahrscheinlich regulatorische Sequenzen enthält. Die
Transkription startet an alternativen Stellen im ersten Intron. In MuLV induzierten T-
und B-Zell-Lymphomen kann eine Anhäufung von Provirusintegrationen 5’ von c-myc
beobachtet werden. Die Aktivierung erfolgt zum einen über die viralen Enhancer und
zum anderen eventuell über die Zerstörung von Suppressor-Bindungsstellen in
diesem Bereich (Corcoran et al., 1984).
Weitaus seltener konnte die Aktivierung von anderen Loci wie Vin-1 und Fis-1 in
MuLV induzierten T-Zell-Lymphomen beobachtet werden (Jonkers and Berns, 1996).
Vin-1 kodiert für Cyclin D2, welches zur großen Familie der Cyclin D Gene gehört,
die für den Übergang von der G1- zur S-Phase während des Zellzyklus mit
verantwortlich sind. Integrationen in Fis-1 scheinen die Expression von Cyclin D1 zu
beeinflussen. Diese Aktivierung scheint funktionell analog zu der in humanen B-
Zellen beobachteten BCL-1 Translokation zu sein, da diese auch zu einer erhöhten
Cyclin D1 Expression führt. Cyclin D1 transgene Mäuse haben gezeigt, dass eine
konstitutive Cyclin D1 Expression Lymphome induzieren kann und eine Kollaboration
mit c-myc vorliegt.

Einleitung 24
2.5.3.2 Aktivierte Onkogene in Erythroleukämien
Neben der oben schon erwähnten Aktivierung des EpoR durch das SFFV Env-
Protein gp55, kann dieser auch durch Provirusintegrationen aktiviert werden, was in
durch den Friend-Viruskomplex induzierten Erythroleukämien häufig zu beobachten
ist. Allerdings ist EpoR nicht das einzige Onkogen, dass in Erythroleukämien
involviert ist. Die meisten Tumore zeigen eine Aktivierung von Transkriptionsfaktoren
der ets-Familie. In SFFV induzierten Erythroleukämien weisen 95 % der Tumore eine
Insertion in dem stromaufwärts des Spi-1/PU.1 Gens gelegenen Bereichs auf. Das
Provirus integrierte dabei in 3’-5’ Orientierung, was wahrscheinlich zu einer
Aktivierung des Gens über die im 5’-LTR lokalisierten Enhancer-Regionen des Virus
geführt hatte (Moreau-Gachelin et al., 1989). Diese Provirus induzierte, anomale
Expression des Spi-1/PU.1 Proteins in erythroblastischen Vorläuferzellen führt zu
einem Block in der Differenzierung und zur Induktion einer Erythroleukämie. Spi-
1/PU.1 scheint darüber hinaus noch eine wichtige Schlüsselfunktion bei der
Differenzierung anderer Zellkompartimente einzunehmen. Ein hoher Spi-1/PU.1
Level induziert die Entwicklung von Makrophagen, ein niedriger Level hingegen die
Entwicklung von B-Zellen (DeKoter and Singh, 2000).
In 75-90 % der Friend-MuLV induzierten Tumore konnte eine Provirusintegration in
den Fli-1 Gen-Lokus beobachtet werden. Die Integrationen erfolgten dabei immer im
ersten Exon des Fli-1 Gens, wobei alle Proviren in 5’-3’ Orientierung, also in der
gleichen transkriptionellen Orientierung wie das Gen vorlagen (Ben-David et al.,
1991). Auch in 10A1-induzierten Tumoren wurde eine Insertion in den Fli-1 Lokus
beobachtet, wobei die Aktivierung des Gens durch Promoterinsertion erfolgte, da
eine 10A1/Fli-1-Fusions mRNA nachgewiesen wurde (Ott et al., 1994). In der Cap-
Struktur der Fli-1 mRNA wurden Bindungsstellen für die Transkriptionsfaktoren
Spi-1/PU.1 und GATA-1 nachgewiesen, so dass es sich um positive Regulatoren für
die Fli-1 Expression handeln könnte (Barbeau et al., 1999). Ein hoher GATA-1
Expressionslevel führt zu einer Differenzierung von Vorläuferzellen in die erythroide
Richtung. Ein hoher PU.1 Level zur Differenzierung in die myeloide Richtung. Die
beiden Proteine wirken also als antagonistische Regulatoren bei der Differenzierung
von Zellen (Orkin, 2000).

Einleitung 25
3. Fragestellung dieser Arbeit
Murine Leukämieviren (MuLV) induzieren in infizierten Mäusen verschiedene
Leukämieformen. In dieser Arbeit sollte untersucht werden, welche Bereiche im
Genom eines murinen Leukämievirus eine Determinante für dessen
Krankheitsspezifität darstellen. Das Moloney-Virus induziert vorwiegend T-Zell-
Leukämien, während das 10A1-Virus zur Entwicklung eine myeloblastischen
Leukämieform führt. Diese beiden MuLVs wurden daher als Basis für die
hergestellten chimären Retroviren benutzt. Bei den chimären Viruskonstrukten
wurden bestimmte Bereiche durch Sequenzen aus dem anderen MuLV ersetzt.
Diese sollten dann im NIH/Ola-Mausmodell getestet werden. Durch Analyse der
erkrankten Mäuse sollte der durch ein chimäres Virus induzierte Phänotyp
charakterisiert werden. Darüber hinaus sollten die induzierten Tumore auf
Provirusintegrationen in bekannten Onkogenen getestet werden. Die auf diese Weise
entdeckten Gen-Bereiche könnten zu einem größeren Verständnis für die
Entstehung von Krebserkrankungen betragen. Die viralen Determinanten, die die
Krankheitsspezifität beeinflussen konnten, sollten dann weiter untersucht werden, um
herauszufinden worin dieser Einfluss begründet sein könnte.

Material 26
4. Material
4.1 Chemikalien und Enzyme
Die verwendeten Enzyme wurden von den Firmen MBI Fermentas, Gibco BRL Life
Technologies und New England Biolabs bezogen. Die benutzten Chemikalien
stammen von den Firmen: Biochrom, Biomol, Difco-Laboratories, Geneo Bioproducts
GmbH, Merck, neoLab, Pharmacia, Roche, Serva, Sigma, Stratagene, Qiagen. Die
verwendeten Enzyme wie Restriktionsendonukleasen, Ligasen, Phosphatasen und
Polymerasen wurden gemäß der Herstellerangaben eingesetzt. Es wurden die
üblichen Standardqualitäten verwendet. Die α-[32P] markierten Radionukleotide
wurden von der Firma Hartmann Analytic bezogen.
4.2 Bakterienstämme
Für die Transformation und Amplifikation der hier verwendeten Plasmide wurden
folgende Escherichia coli Laborstämme verwendet:
XL-1 Blue Blau/Weiß-Selektion Stratagene
XL-10 Gold ultrakompetent, Blau/Weiß-Selektion Stratagene
CMK 603 Derivat des Stammes 600 (Appleyard, 1954)
Die Herstellung kompetenter Bakterien, deren Lagerung, Transformation und
anschließende Expansion erfolgte nach den allgemein üblichen Protokollen (Ausubel
et al., 2001; Sambrook et al., 1989).
4.3 Zelllinie
SC-1 ATCC Nr. CRL-1404: diese murine Fibroblastenzelllinie eignet
sich besonders für die Produktion von replikationskompetenten
murinen Leukämie Viren MuLVs; (Hartley and Rowe, 1975)

Material 27
4.4 Medien
4.4.1 Bakterienkulturmedien
Luria-Bertani (LB)-Medium, pH 7,4:
1 %0,5 %
5 %
(w/v)(w/v)(w/v)
Bacto-Trypton, Difco Hefeextrakt, Difco NaCl
LB-Agarplatte: LB-Medium + 1,5 % (w/v) Bacto-Agar, Difco
Medien für Flüssigkulturen und Agarplatten wurden 20 min bei 121°C autoklaviert.
Alle Zusätze, die nicht hitzestabil sind, wurden nach dem Abkühlen der Medien auf
ca. 40-50°C zugegeben. Für die Selektion transformierter Bakterien wurden die
abgekühlten Medien mit Ampicillin (Endkonzentration 100 ng/ml) versetzt.
4.4.2 Zellkulturmedien
Die Zellkulturmedien wurden mit fötalem Kälberserum (FCS, Sigma) versetzt. Zur
Inaktivierung der im Serum enthaltenen Komponenten des Komplementsystems
wurde dieses 30 min bei 56°C erwärmt.
Roswell Park Memorial Institute 1640 Medium (RPMI) von Gibco wurde für die
Organaufarbeitung supplementiert mit:
5 % FCS 2 % Penicillin/Streptomycin
0,005 % β-Mercaptoethanol
Minimal Essential Medium (MEM) von Sigma wurde für die Anzucht von SC-1 Zellen
supplementiert mit
10 % FCS 4 mM Glutamin 1 mM Natriumpyruvat
1 % Penicillin/Streptomycin

Material 28
4.5 Verwendeter Mausstamm
Für die intraperitoneale Infektion neonataler Mäuse mit replikationskompetenten
MuLVs wurde der Stamm Mus musculus NIH/OlaHsd (Jackson Laboratory, Bar
Harbor, USA) verwendet.
4.6 Klonierte retrovirale Proviren
Die in dieser Arbeit verwendeten proviralen Genome wurden in den Vektor pUC18
kloniert. Die Plasmide kodieren für replikationskompetente murine Leukämieviren.
Tab. 5: Dargestellt sind die Namen der im Plasmid enthaltenen Proviren, die laborinterne
Bezeichnung, der Virusklon und die dazugehörigen Referenzen.
Virus interne Nomenklatur Klon Referenz Moloney-MuLV R686 mov3 (Harbers et al., 1981) 10A1-MuLV R862 RRI (Ott et al., 1990) 4070A #434 4070A (Ott et al., 1990) SFFV-MCF #72 (Linemeyer et al., 1980)
4.7 Oligonukleotide Tab. 6: Oligonukleotide, deren Sequenz und ihr Verwendungszweck sind zusammengefasst
dargestellt (for: forward; rev: reverse).
Name Sequenz 5’→ 3’
Verwendung
MR6 (for) CGG AAT TCT TGT TGA GAA GG Amplifizierung Fli-1 Sonde MR7 (rev) GCT CAA AGC GAA TTC TGG GT Amplifizierung Fli-1 Sonde MR8 (for) CCC TAA GCC TCC GCC TCC Screening 10A1V-Mogag MR9 (rev) CCG GTC AGC AGA GTC CCC Screening 10A1V-Mogag MR14 (for) GCC CCC TCA ATA CCA GTT AC Screening 10A1-4070Aenv MR15 (rev) CAC ATT GTT CCG GCG GGT G Screening 10A1-4070Aenv A1 (for) CCG TAT GTC GGG TAT GGC TG Amplifizierung 4070Aenv
A2 (rev) GAC ACT TGG ACT TGT AG Amplifizierung 4070Aenv aus R654

Material 29
Alle in dieser Arbeit verwendeten Oligonukleotide wurden von der Firma Invitrogen
(Karlsruhe) hergestellt.
4.8 Größenstandards und Ladepuffer
4.8.1 λ-DNA/HindIII Marker für Southern Blot Analysen
Zur Herstellung dieses Größenstandards werden 2µg λ-DNA mit 20 U HindIII in
einem Volumen von 20 µl 1-1,5 h bei 37°C gespalten. Die vollständige Spaltung wird
mit einem Aliquot des Spaltungsansatzes gelelektrophoretisch überprüft.
Anschließend werden die verbliebenen 17 µl mit 30 µCi (≈ 1,11 MBq) α-[32P]-CTP,
einem Mix aus dGTP, dTTP, dATP und Klenow-Enzym in einem Volumen von 30 µl
radioaktiv markiert. Die Inkubationszeit beträgt 20 min bei Raumtemperatur. Zum
Stoppen der enzymatischen Reaktion und Erhöhung des Probenvolumens werden
5 µl 0,5 M EDTA und 45 µl 1x TE-Puffer zugesetzt. Zum Entfernen von nicht
eingebauten Nukleotiden wird der Reaktionsansatz mit Hilfe von Affinitäts-
Chromatografie-Säulchen (MobiSpin von MoBiTec) 2 min bei 3000 U/min
aufgereinigt. Der Erfolg der Markierung wird mit einem Aliquot im Szintillator
überprüft.
4.8.2 λ-DNA/HindIII/EcoRI Marker für gelelektrophoretische Analysen
Zur Herstellung dieses Größenstandards werden 50µg λ-DNA mit 50 U HindIII und
EcoRI in einem Volumen von 250 µl für 3 h bei 37°C gespalten. Die vollständige
Spaltung wird mit einem Aliquot des Spaltungsansatzes gelelektrophoretisch
überprüft. Wenn die DNA durchgespalten ist, wird eine Endkonzentration von
0,125 µg/µl eingestellt und 10 x Ladepuffer zugesetzt.
4.8.3 GeneRuler DNA Ladder Mix (MBI Fermentas) für gelelektrophoretische Analysen 4.8.4 10 x Yellow SubTMGeneo Bioproducts PCR-Additiv und Ladepuffer

Material 30
4.9 DNA Sonden für Southern Hybridisierung
Sonde Größe Herstellung Plasmid Bereich
Moenv* 1,14 kb BamHI/ClaI #522 Mo 3’-env 4070Aenv 0,6 kb A1/A2 PCR R654 VRA, VRB, HVR 10A1env 2,0 kb NotI/SalI R700 10A1 gesamtes envMCFenv 0,6 kb BamHI/EcoRI #72 MCF 5’-env Fli-1* 1,5 kb MR6/MR7 PCR gen. DNA Ex1a Ex1b In1 Pim-1* 0,9 kb BamHI #442 Ex5 In5 Ex6 c-myc* 2,6 kb XbaI #52 Ex2 In2 Ex3 Spi-1* 0,8 kb PstI #530 5’-UTR N-myc* 0,7 kb BamHI/SacI #528 Ex1 JH 3-4* 1,9 kb BamHI/EcoRI R438 Maus Ig JH-Cluster
Tab. 7: Die Auflistung zeigt die in dieser Arbeit verwendeten Sonden für die Hybridisierung. Für
weitere Informationen bezüglich einiger Sonden (*) siehe auch (Morita et al., 2000) für Moenv,
(Barbeau et al., 1996) für Fli-1, (Moreau-Gachelin et al., 1989) für Spi-1, (Stanton et al., 1983) für c-
myc, (van Lohuizen et al., 1989) für N-myc, (Cuypers et al., 1984) für Pim-1 und (Alt et al., 1984) für JH
3-4. (Ex: Exon; In: Intron; VRA, VRB: variable Region A, B; HVR: hypervariable Region)

Methoden 31
5. Methoden
5.1 Isolierung von DNA-Fragmenten
Die zu isolierenden DNA-Fragmente sind zuvor mittels PCR-Amplifikation oder
Restriktionsspaltung aus Plasmiden gewonnen worden. Nach der
gelelektrophoretischen Auftrennung und Ethidiumbromid-Färbung werden die
richtigen Fragmente auf einem UV-Transilluminator mit einem sauberen Skalpell aus
dem Gel ausgeschnitten und in ein Eppendorf-Reaktionsgefäß überführt. Die
anschließende Aufreinigung erfolgt mit Hilfe des Qiagen Extraktions-Kits gemäß der
Herstellerangaben. Die Konzentration der DNA-Fragmente wird gelelektrophoretisch
durch Vergleich der Bandenintensität mit einem Größenstandard abgeschätzt.
5.2 Dephosphorylierung des gespaltenen Vektors
Der linearisierte, aufgereinigte Vektor wird in einem Volumen von 40 µl unter Zugabe
von 4 µl 10 x B*-Puffer (MBI Fermentas) und 1 µl Shrimp Alkaline Phosphatase
(Boehringer Mannheim) für 1 h bei 37°C dephosphoryliert. Das Enzym wird
anschließend für 10 min bei 70°C inaktiviert. Der Vektor kann direkt für die
Ligationsreaktion eingesetzt werden. Die Konzentration der Vektor-DNA wird
gelelektrophoretisch durch Vergleich der Bandenintensität mit einem Größen-
standard abgeschätzt.
5.3 Ligation
Als Hintergrund-Kontrolle wird dem Ligationsansatz nur der dephosphorylierte Vektor
zugesetzt. Für die Ligation wird ein molares Vektor-/Insert-DNA Verhältnis von ca.
1:3 eingesetzt. Die Reaktion wird in einem Volumen von 15 µl unter Zugabe von 2 U
T4-Ligase (MBI Fermentas) und 0,1 Volumen 10 x T4-Ligasepuffer über Nacht bei
Raumtemperatur durchgeführt.

Methoden 32
5.4 Screening nach positiven Klonen
Um in möglichst kurzer Zeit eine große Anzahl von Klonen überprüfen zu können,
wurde die sogenannte „Dip-PCR-Methode“ angewendet. In einer Zellkultur 96-Loch
Platte werden je Vertiefung 20 µl H2O vorgelegt. Mit Hilfe von gelben Pipettenspitzen
wird je eine Bakterienkolonie von den Selektionsplatten in eine Vertiefung überführt.
In die 96-Loch PCR-Platte werden je Vertiefung 49 µl vom PCR-Mastermix pipettiert.
Ein PCR-Ansatz enthält pro Vertiefung folgende Mengen:
0,15 µl Taq-Polymerase (≈ 0,75 U, Gibco)
5,0 µl 10 x PCR-Mix (Gibco)
5,0 µl Yellow Sub (Geneo)
0,2 µl dNTP-Mix (≈ 0,04 mM each)
1,0 µl Primer 1 (15 pmol/µl)
1,0 µl Primer 2 (15 pmol/µl)
1,5 µl MgCl2 (≈ 1,5 mM, Gibco)
35,15 µl H2O
Die Gesamtmengen für den Mastermix hängt von der Anzahl der gepickten Klone ab.
Die Oligonukleotide werden entsprechend des Klonierungsvorhabens ausgewählt.
Die Bedingungen der anschließenden PCR-Reaktion hängen von den verwendeten
Primern und der zu erwartenden Fragmentgröße ab (*).
Grundprogramm:
1. 95°C, 3 min
2. 95°C, 15 sec
3. *°C, 15 sec
4. 72°C, *
5. 4°C, ∞
Schritte 2-4 werden 25 mal wiederholt.
Da die PCR-Ansätze mit dem Additiv Yellow Sub versetzt wurden, können Aliquots
direkt auf ein Agarosegel aufgetragen werden. In jede Vertiefung der 96-Loch
Zellkulturplatte mit den angeimpften Klonen werden 200 µl LB-Medium (s. 4.4.1),
supplementiert mit Ampicillin, gegeben und die Platte über Nacht bei 37°C
geschüttelt. Da die Nummer der Klone mit der Nummer des PCR-Ansatzes

Methoden 33
übereinstimmt, kann ein im Screening positiver Klon direkt aus der Zellkulturplatte für
eine Mini- oder Maxi-Präparation angeimpft werden.
5.5 Plasmidisolierung
Die Präparation von Plasmid-DNA aus Bakterien erfolgt nach dem Prinzip der
alkalischen Lyse (Birnboim and Doly, 1979; Ish-Horowicz and Burke, 1981). Bei der
Präparation im kleinen Maßstab erhält man eine DNA Menge von max. 20 µg, im
großen Maßstab von max. 500 µg. Die isolierte DNA wird mit Hilfe von
Restriktionsspaltung und Sequenzierung überprüft.
5.6 Sequenzierung
DNA wird nach der Dideoxy-Kettenabbruch-Methode nach (Sanger et al., 1977) mit
Hilfe von Fluoreszenz-markierten Nukleotiden sequenziert. Die Reaktion wird als
Cycle-Sequencing durchgeführt. In einem Ansatz von 20 µl werden 800 ng DNA ,
4 µl BigDye-Puffer (Applied Biosystems), 1,5 µl Primer (15 pmol/µl) und die
entsprechende Menge Wasser vermischt.
Die Ansätze werden auf Eis zusammenpipettiert und im Thermocycler nach
folgendem Programm inkubiert:
1. 96°C, 1 min
2. 96°C, 30 sec
3. 50°C, 15 sec
4. 60°C, 4 min
5. 4°C, ∞
Schritte 2-4 werden 25 mal wiederholt. Der gesamte Sequenzieransatz wird mit 50 µl
100 %-igem Ethanol gefällt, 30 min bei 13.000 rpm anzentrifugiert und mit 70 %
Ethanol gewaschen. Dann wird ein weiteres Mal zentrifugiert, der Überstand entfernt
und das Pellet in der Lyophylle getrocknet. Die gelelektrophoretische Analyse findet
im Sequenzierservicelabor (Institut für Zellbiochemie und klinische Neurobiologie,
Hamburg) statt.

Methoden 34
5.7 Zellkultur
Die SC-1 Zellen werden subkonfluent in MEM-Medium (s. 4.4.2) in Standardbrut-
schränken gehalten. Die Kultivierung erfolgt in Gegenwart von 5 % CO2 (v/v) bei
37°C und 95 % Luftfeuchtigkeit. Das Wechseln des Mediums und die Verdünnung
der Zellen erfolgt im 2-2-3-Tage Rhythmus. Dazu werden die konfluenten,
adhärenten Zellen durch Zugabe von Trypsin vom Boden der Zellkulturflasche gelöst.
Durch das Aufnehmen der gelösten Zellen in FCS-haltigem Medium wird das Trypsin
inaktiviert.
5.7.1 Virusproduktion
Die Virusproduktion erfolgt durch Transfektion der Plasmid-DNA in die murine
Fibroblastenzelllinie SC-1. Da die in der Zellkultur eingesetzten Plasmide für
replikationskompetente Retroviren kodieren, sind Verpackungszelllinien nicht nötig.
Nach der Infektion gelangen die Viruspartikel durch Ausknospung in den
Mediumüberstand und können geerntet werden.
5.7.2 Transfektion mittels Elektroporation
Die Zellen werden vom Boden gelöst und in Medium resuspendiert. Die Suspension
wird 5 min bei 1000 rpm abzentrifugiert und das Pellet so in Medium resuspendiert,
dass 400 µl 2 x 106 Zellen entsprechen. Für die Elektroporation werden 400 µl Zellen
und 10 µg Plasmid-DNA eingesetzt. Es werden folgende Parameter eingestellt: 4 mm
Elektrodenabstand, 1050 µF Feldstärke bei 260 V. Die Zellen werden nach der
Elektroporation in 5 ml Medium aufgenommen und sehr dünn ausgesät. Da in dieser
Kultur die Virusverbreitung stattfinden soll, werden die Zellen für mindestens 14 Tage
inkubiert.
5.7.3 Ernten des Virusüberstandes
Der Mediumüberstand enthält nach zwei Wochen eine hohe Konzentration an
infektiösen Partikeln. Der Überstand wird abgenommen, filtriert (Ausschlussgröße

Methoden 35
0,22 µm), um Kontaminationen mit Kulturzellen zu vermeiden, in Cryo-Röhrchen
aliquotiert und in der Gasphase des Stickstoff-Tankes gelagert.
5.7.4 Titerbestimmung
Der geerntete Virusüberstand wird 1:5 seriell verdünnt. SC-1 Zellen werden in 24-
Loch Zellkulturplatten dünn ausgesät (3 x 103 Zellen/Vertiefung) und mit dem
verdünnten Virusüberstand vermischt. Die Platten werden bei 37°C bis zur Konfluenz
kultiviert. Die Zellen werden mit 5 mM EDTA abgelöst (s. 5.10.2), gewaschen, mit FC-
Block und α-glykoGag-Cy5 gefärbt und im FACS-Calibur (Becton Dickinson)
vermessen. Der Titer ergibt sich aus der letzten seriellen Verdünnungsstufe, in der
noch α-glykoGag-Cy5 detektierbar gewesen ist, multipliziert mit der Anfangsver-
dünnung (Volumen des Virusüberstandes zu Volumen der SC-1 Zellen im Medium).
Es wird auf diese Weise der Titer der infektiösen Partikel ermittelt, es handelt sich
nicht um eine quantitative Virusbestimmung.
5.7.5 Infektion neonataler Mäuse
Neugeborene werden innerhalb von 48 h intraperitoneal mit replikationskompetentem
MuLV-Überstand infiziert. In Abhängigkeit vom Virustiter (1-3 106 CFU/ml) werden
50-100 µl injiziert. Da das Virus 10A1-MoLTR in Vorversuchen eine starke Neigung zu
neurodegenerativen Erkrankungen gezeigt hatte, wurden die Babys erst 5-8 Tage
nach der Geburt infiziert (Münk et al., 1997), was das Auftreten von spongiformen
Degenerationserscheinungen deutlich reduziert.
5.8 Analyse erkrankter Tiere
5.8.1 Überwachung und Autopsie der Mäuse
Die Tiere werden dreimal in der Woche genauestens auf das Auftreten etwaiger
Krankheitssymptome, wie vergrößerte Organe (Lymphknoten, Milz und Leber),
Apathie, buckelige Haltung, Hecheln und steifbeiniger Gang, untersucht. Darüber
hinaus muss auch auf Anzeichen einer neurodegenerativen Erkrankung wie Tremor
und Ataxie geachtet werden. Einem Tier mit eindeutigen Krankheitssymptomen wird

Methoden 36
Blut entnommen, es wird dann geopfert und eine Autopsie durchgeführt. Zur
Erleichterung der Blutabnahme, wird die Maus ein paar Minuten unter einer
Rotlichtlampe erwärmt, mit Hilfe eines Kapillarblutröhrchens ca. 50-100 µl Blut aus
der angeschnittenen Schwanzvene entnommen. Das Gefäß enthält zur
Gerinnungshemmung EDTA. Das Tier wird anschließend durch das Einatmen der
Ether-Gasphase betäubt und durch Genickbruch getötet. Die Maus wird auf der OP-
Unterlage fixiert und durch Besprühen mit Ethanol semisterilisiert. Als erstes wird
dann mit einem doppelten Y-Schnitt die Dermis eröffnet, so dass der Hals, die
Achseln, die Lenden und das Peritoneum frei liegen. Jetzt können die zervikalen,
axialen und peripheren Lymphknoten untersucht werden. Anschließend wird die
Bauchhöhle eröffnet und das Sternum durchtrennt. Auf diese Weise können alle
inneren Organe in Augenschein genommen werden, insbesondere die mediastinalen
Lymphknoten, die Milz; die Leber und der Thymus mit seinen assoziierten
Lymphknoten. Die vergrößerten Organe werden entnommen und gewogen. Für die
Präparation von genomischer DNA bzw. RNA wird ein Teil des Organs in flüssigem
Stickstoff schockgefroren. Für eine FACS-Analyse wird ein Stück des Organs in 5 ml
RPMI (s. 4.4.2) aufgenommen. Die Kadaver der getöteten Tiere werden zentral
gelagert und entsorgt. Die in Stickstoff fixierten Organe werden bis zu ihrer
Weiterverarbeitung bei –70°C aufbewahrt.
5.8.2 Analyse der Blutparameter
Die Blutparameter werden mit Hilfe des Coulter MaxM Blutanalysegerätes ermittelt,
von besonderem Interesse sind vor allem der Hämatokrit und die Leukozyten-
konzentration. Dazu werden 50 µl Mausblut mit 100 µl Isoton III-Lösung (Coulter) in
einem Eppendorfgefäß vermischt. Die Verdünnung muss später bei der
Datenauswertung mit einbezogen werden. Die Probenaufnahme erfolgt im
Sekundärmodus des Gerätes. Das Hochfahren und Spülen des MaxM wird gemäß
der Herstellerangaben durchgeführt.

Methoden 37
5.9 Durchführung von Blutausstrichen und Cytospins
5.9.1 Blutausstriche
Zur morphologischen Auswertung des entnommenen Blutes werden pro Tier zwei
Blutausstriche mit jeweils ca. 2,5 µl Blut hergestellt. Das Blut wird auf die schmale
Seite des Objektträgers pipettiert und mit einem im 45° Grad Winkel gehaltenen
zweiten Objektträger entlang der Längsachse gleichmäßig ausgestrichen. Die
Ausstriche sollten über Nacht trocknen.
5.9.2 Cytospin
Phosphat-Puffer-System (PBS):
0,14 M NaCl 3 mM KCl 1 mM NaH2PO42 mM KH2PO4
Die vereinzelten Zellen aus den Organen für die FACS-Analyse werden in einer
Konzentration von 0,48 x 106 Zellen pro 600 µl 1 x PBS aufgenommen. Es werden
pro Organ zwei Cytospins erstellt. Auf jeden Objektträger werden 200 µl der
Zellsuspension (entspricht 0,16 x 106 Zellen) mittels Zentrifugation (5 min 600 rpm)
aufgebracht. Die Färbung erfolgt wie bei den Blutausstrichen nach der Lufttrocknung.
5.9.3 AccustainTM Pappenheim-Färbung (Sigma):
0,25 %(w/v) May-Grünwald in Methanol
0,4 % (w/v) modifiziertes Giemsa in gepuffertem Methanol (pH 6,8)
Die Objektträger werden 5 min in der May-Grünwald-Lösung gefärbt und
anschließend mit Aqua dest. gründlich gewaschen bzw. neutralisiert. Der zweite
Färbeschritt erfolgt in 1:20 verdünnter Giemsa-Lösung für 20 min. Danach folgt ein
weiterer Waschschritt. Die Präparate werden getrocknet und können dann mit Eukitt
eingedeckelt werden. Die Auswertung erfolgt am Mikroskop. Detaillierte Photos
wurden freundlicherweise von Dr. Jürgen Löhler hergestellt.

Methoden 38
5.10 FACS-Analysen
5.10.1 Vorbereitung der Organe
10 x Erythrozytenlysepuffer: 1,7 M NH4Cl; 0,1 M KHCO3, 1M EDTA, pH 7,3
Die in der RPMI-Lösung (s. 4.4.2) aufgenommenen Organe werden innerhalb von
24 h weiterverarbeitet. Die Vereinzelung der Zellen erfolgt mit Hilfe eines eng-
maschigen Metallsiebes, durch das das Organ mit dem Stempel einer Spritze
gepresst wird. Das Sieb wird mit weiteren 5 ml RPMI-Medium gespült, so dass sich
die vereinzelten Zellen in einem Volumen von 10 ml Medium befinden. Die Zellen
werden 5 min bei 1200 rpm abzentrifugiert und in 10 ml 1 x PBS resuspendiert. Zur
Bestimmung der Zellzahl werden 10 µl Zellsuspension mit 90 µl 1 x Erythrozyten-
Lyse-Puffer vermischt und unter dem Mikroskop in einer Neubauer Zählkammer
gezählt. Die ermittelte Zahl muss mit dem Kammerfaktor (104) und dem
Verdünnungsfaktor (10) multipliziert werden.
Für die Antikörperfärbung werden pro Färbeansatz 1 x 106 Zellen in einem Volumen
von 200-300 µl 1 x PBS eingesetzt. Bei sehr geringen Zellzahlen sollte das Volumen
durch erneutes Zentrifugieren (5 min bei 2000 rpm, Eppendorf Tischzentrifuge) auf
300 µl eingeschränkt werden. Für die Analyse der Organe wurden folgende
Antikörper-Kombinationen (Pharmingen) eingesetzt:

Methoden 39
Ansätze FITC-konjugierte Ak
PE-konjugierte Ak PI-Zugabe Verwendung
1 - - Nein Zellen pur, Eigenfluoreszenz
2 CD45 (LCA) - Nein FITC Kompensation 3 - CD45 (LCA) Nein PE Kompensation 4 - - Ja PI Kompensation 5 rat IgG2α rat IgG2α Ja Isotypkontrolle 6 CD4 CD8 Ja T-Zellen 7 CD90 CD3e Ja T-Zellen 8 CD19 B220 Ja B-Zellen 9 CD79b B220 Ja B-Zellen 10 CD34 CD117 Ja Stammzellen 11 IgM Gr-1 Ja B-Zellen/Granulozyten 12 Sca-1 Ter119 Ja Stammzellen/Erythrozyten
Tab. 8: Auf 1 x 106 Zellen werden 0,6 µl FITC-konjugierter und 1,2 µl PE-konjugierter Antikörper
eingesetzt. Zusätzlich zu den aufgelisteten Antikörperkombinationen wurde zeitweise auch anti-
glykoGag Cy5 gekoppelter Antikörper eingesetzt (1,0 µl/1 x 106 Zellen). Dieser erkennt das virale, auf
der Zelloberfläche lokalisierte glykoGag-Protein.
Zu allen Ansätzen (außer 1 und 4) wird je 1,0 µl FC-Block (α-FCγR- Antikörper)
gegeben und diese anschließend für 15 min ohne Licht bei 4°C inkubiert. Dieses
Reagenz blockiert unspezifische Bindungsmöglichkeiten, so dass diese in der
nachfolgenden spezifischen Färbung nicht mehr zur Verfügung stehen. Anschließend
werden die FITC- bzw. PE-konjugierten Antikörper zugegeben und die Ansätze 30
min (oder über Nacht) bei 4°C ohne Lichteinwirkung inkubiert. Es wird dann zweimal
gewaschen, indem die Ansätze mit je 1 ml 1 x PBS versetzt, auf dem Vortexer
gemischt und dann 5 min bei 2000 rpm (Tischzentrifuge) abzentrifugiert werden.
Nach dem letzten Waschschritt wird das Zellpellet in 700 µl 1 x PBS resuspendiert.
Um apoptotische Zellen in der FACS-Messung ausschließen zu können, werden die
Ansätze 4 (PI-Kompensation) und 5-12 mit je 7 µl Propidiumiodid-Lösung (0,1 mg/ml
in H2O, Endkonzentration 10 µg/ml), versetzt. Die Proben werden am FACS-Calibur
(Becton Dickinson) vermessen.
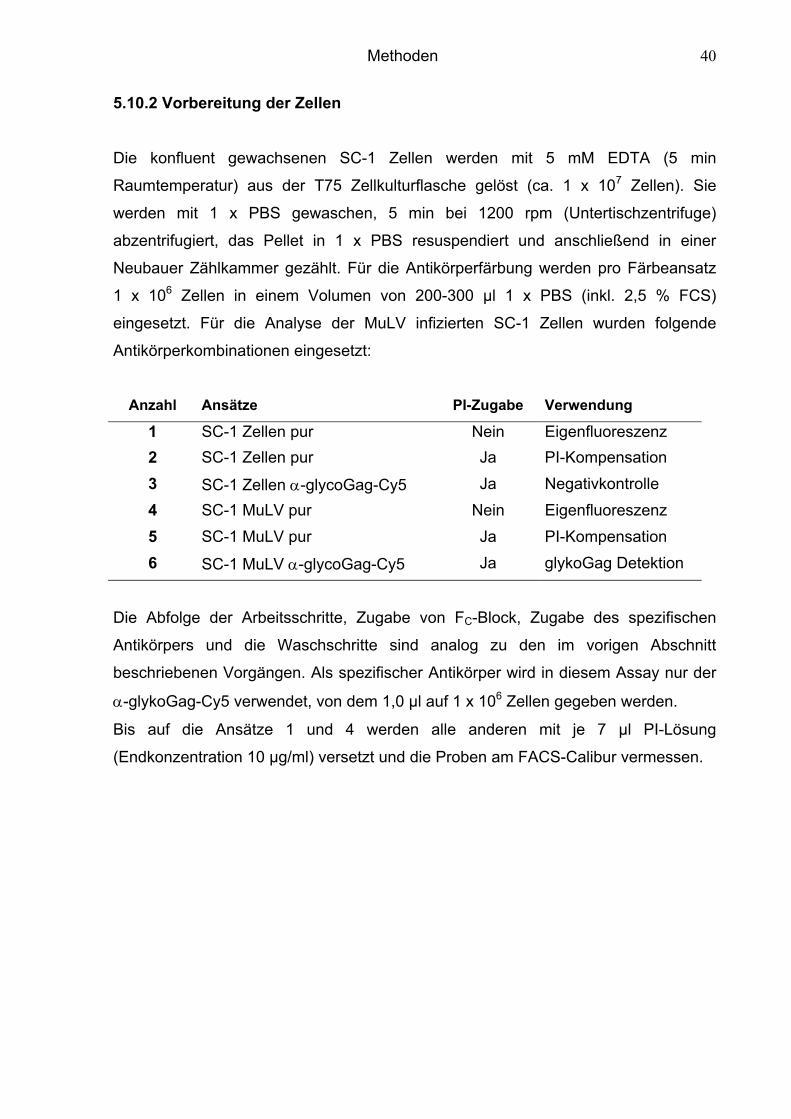
Methoden 40
5.10.2 Vorbereitung der Zellen
Die konfluent gewachsenen SC-1 Zellen werden mit 5 mM EDTA (5 min
Raumtemperatur) aus der T75 Zellkulturflasche gelöst (ca. 1 x 107 Zellen). Sie
werden mit 1 x PBS gewaschen, 5 min bei 1200 rpm (Untertischzentrifuge)
abzentrifugiert, das Pellet in 1 x PBS resuspendiert und anschließend in einer
Neubauer Zählkammer gezählt. Für die Antikörperfärbung werden pro Färbeansatz
1 x 106 Zellen in einem Volumen von 200-300 µl 1 x PBS (inkl. 2,5 % FCS)
eingesetzt. Für die Analyse der MuLV infizierten SC-1 Zellen wurden folgende
Antikörperkombinationen eingesetzt:
Anzahl Ansätze PI-Zugabe Verwendung
1 SC-1 Zellen pur Nein Eigenfluoreszenz 2 SC-1 Zellen pur Ja PI-Kompensation 3 SC-1 Zellen α-glycoGag-Cy5 Ja Negativkontrolle 4 SC-1 MuLV pur Nein Eigenfluoreszenz 5 SC-1 MuLV pur Ja PI-Kompensation 6 SC-1 MuLV α-glycoGag-Cy5 Ja glykoGag Detektion
Die Abfolge der Arbeitsschritte, Zugabe von FC-Block, Zugabe des spezifischen
Antikörpers und die Waschschritte sind analog zu den im vorigen Abschnitt
beschriebenen Vorgängen. Als spezifischer Antikörper wird in diesem Assay nur der
α-glykoGag-Cy5 verwendet, von dem 1,0 µl auf 1 x 106 Zellen gegeben werden.
Bis auf die Ansätze 1 und 4 werden alle anderen mit je 7 µl PI-Lösung
(Endkonzentration 10 µg/ml) versetzt und die Proben am FACS-Calibur vermessen.

Methoden 41
5.10.3 FACS-Messung
Das Vermessen der Proben erfolgt am FACS-Calibur (Becton Dickinson). Die
Datenaufnahme und die anschließende Auswertung werden mit Hilfe der Cellquest
Software (Becton Dickinson) durchgeführt. Als erstes wird die Autofluoreszenz der
ungefärbten Zellen so eingestellt, dass diese nicht als falsch Positive in die Messung
eingehen. Anschließend werden die einzelnen Fluoreszenzen gemessen und so weit
kompensiert, dass sie nicht in die anderen Kanäle einstrahlen können: FITC gegen
den PE-Kanal, PE gegen den FITC- und PI-Kanal, PI gegen den PE-Kanal sowie
APC gegen den PE-Kanal und umgekehrt. Nachdem diese Parameter eingestellt
sind, können die Proben vermessen werden.
5.11 Gewinnung genomischer DNA aus Organen
1 x TE (10/10): 10 mM Tris-HCl 1 x TE/SDS: 10mM Tris-HCl
10 mM EDTA, pH 7,5 10 m M EDTA
1 % SDS, pH 7,5
1 x TE (10/1): 10 mM Tri-HCl
1 mM EDTA, pH 7,5
Die genomische DNA wird aus den bei –70°C gelagerten Tumoren extrahiert. Dazu
werden die Organe im gefrorenen Zustand in einen Homogenisator gegeben und in
einem Volumen von 2-4 ml TE-Lösung (10/10), je nach Größe des Organstücks,
homogenisiert. Die Zellsuspension wird dann mit dem gleichen Volumen TE/SDS-
Lösung vermischt, mit 20 µg Proteinase K pro ml Lösung versetzt, gut geschüttelt
und bei 37°C über Nacht im Wasserbad inkubiert.
Zur Entfernung von Proteinen wird die Nukleinsäurelösung einer Phenol/Chloroform-
Extraktion unterzogen. Dazu wird die Lösung zweimal mit einem Volumen Phenol
versetzt, 15 min auf dem Schüttler langsam geschüttelt und 10 min bei 2000 rpm
zentrifugiert. Die obere Phase wird in ein neues Gefäß überführt. Zur Entfernung von
Phenolresten wird die Lösung zweimal mit einem Volumen Chloroform extrahiert,
analog zur Phenolextraktion.

Methoden 42
Zur Fällung der DNA wird 0,1 Volumen 5 M NaCl-Lösung zugegeben, gemischt und
vorsichtig mit 2,5 Volumen eisgekühltem 100 %-igen Ethanol überschichtet. Die
genomische DNA kondensiert zu gut sichtbaren Fäden, die mit einem Häkchen
(vorne angeschmolzene Pasteurpipette) aufgewickelt und in 70 % Ethanol
gewaschen werden, um etwaige Salzreste zu entfernen. Die aufgespulte DNA wird
kurz an der Luft getrocknet und in einem Volumen von 200-500 µl TE (10/1)
aufgenommen. Die Konzentration und Reinheit der Nukleinsäuren wird mit Hilfe
eines UV-Spektralphotometers durch Messung der Absorption bei 260 nm und 280
nm bestimmt. Die Konzentration wird durch weiteres Verdünnen mit TE auf ca.
1 µg/µl eingestellt.
5.12 Restriktionsspaltung und gelelektrophoretische Auftrennung genomischer DNA
1 x TAE-Lösung: 40 mM Tri-HCl, 10 mM EDTA, pH 7,6
20 mM Essigsäure
Für die Southern Blot Analyse werden 10 µg genomische DNA mit den
entsprechenden Restriktionsendonukleasen (35 U) in einem Volumen von 50 µl für
5 h bei der entsprechenden Temperatur gespalten. Nach ca. 1 h werden die Ansätze
durch Auf- und Abpipettieren gründlich gemischt. Die gespaltenen DNA-Ansätze
werden mit Ladepuffer versetzt und in einem 0,8 %-igen (w/v) 1 x TAE-Agarosegel
aufgetrennt. Als Größenstandard wird radioaktiv markierte (ca. 2 x 105 cpm) HindIII
gespaltene λ-DNA verwendet. Die Elektrophorese findet bei 15 V über Nacht statt.
5.13 Southern Blotting
Denaturierungslösung: 0,4 N NaOH
0,6 M NaCl
Neutralisierungslösung: 1,5 M NaCl
0,5 M Tris-HCl, pH 7,5
20 x SSC-Lösung: 3 M NaCl
0,4 M Na-Citrat

Methoden 43
Das Gel wird nach der Elektrophorese photographiert, ausgemessen, dann 30 min in
Denaturierungslösung und anschließend 30 min in Neutralisierungslösung
geschwenkt. Der Transfer der gespaltenen DNA auf eine Nylonmembran erfolgt
durch Ausnutzung von Kapillarkräften. Der Versuchsaufbau ist folgendermaßen: eine
Plastikwanne wird mit 1,5 l 10 x SSC Laufpuffer gefüllt (1:1 verdünntes 20 x SSC),
eine Glasscheibe wird als Brücke darüber gelegt, auf dieser werden zwei Dochte aus
Whatman-Papier so platziert, dass die Enden in den Laufpuffer hängen. Auf diese
angefeuchteten Dochte wird das Gel luftblasenfrei aufgelegt, es folgt die auf
Gelgröße zurechtgeschnittene, angefeuchtete Membran (Biodyne B Transfer
Membrane, PALL Europe Limited), dann zwei ebenfalls angefeuchtete Papierlagen.
Mehrere Lagen saugfähige Papierhandtücher, die mit einem Gewicht beschwert
werden, bilden den Abschluss. Der DNA Transfer findet über Nacht statt. Zur
Fixierung der DNA auf der Membran wird diese zweimal mit je 120 mJ im UV-
Stratalinker (Stratagene) vernetzt.
5.13.1 Hybridisierung
Hybridisierungslösung I: 1 M NaCl, 10% SDS, 10 % Dextransulfat
Hybridisierungslösung II: 0,5 M Formamid, 6 x SSC, 7,5 % Dextransulfat, 2,4 x
Denhardtslösung, 750 µg/ml Heringssperma-DNA
50 x Denhardts-Lösung: 1 % (w/v) Ficoll, 1 % (w/v) Polyvinylpyrrolidon, 1 % (w/v)
BSA (1 g/ml), ad 500 ml H2O
Die Membran wird in 10 ml Hybridisierungspuffer für mindestens 30 min
prähybridisiert. Welcher Puffer dabei zum Einsatz kommt, hängt von der
gewünschten Stringenz ab. Die Inkubation erfolgt entweder im Hybridisierungspuffer
II (mit Formamid) bei 55°C (hohe Stringenz) oder in Hybridisierungslösung I bei 65°C
(niedrige Stringenz). Hybridisierungslösung I wird vor der Inkubation 750 µg/ml
denaturierte Heringssperma-DNA zugesetzt. Die nach Anleitung (s. 5.13.3) markierte
Sonde wird vor der Zugabe zum Prähybridisierungspuffer 5 min bei 95°C denaturiert.
Die Hybridisierung findet über Nacht (ca. 12 h) statt.

Methoden 44
5.13.2 Waschen der Membranen
Die Hybridisierungslösung wird in den radioaktiven Flüssigabfall entsorgt.
Anschließend wird zweimal mit ca. 20 ml 2 x SSC (1:10 verdünntes 20 x SSC, frisch
hergestellt) für 5 min bei Raumtemperatur in dem Hybridisierungsröhrchen
gewaschen. Die Waschlösung wird ebenfalls in den radioaktiven Flüssigabfall
gegeben. Die nächsten Waschschritte finden in einer Plastikwanne statt. Es wird
zweimal mit ca. 250 ml Waschlösung II (0,1 x SSC, 0,1 % SDS, frisch hergestellt) für
1 h bei 65°C gewaschen. Die Membran wird kurz luftgetrocknet, in Folie
eingeschweißt und in einer Filmkassette fixiert. In der Dunkelkammer wird ein
Röntgenfilm (SuperRX Medical X-Ray, Fuji Photo Film GmbH) aufgelegt und die
Filmbox bei – 80°C bis zur Entwicklung des Filmes gelagert. Die Filmentwicklung
wird mit dem Fuji X-Ray Filmprocessor RG II durchgeführt.
5.13.3 Herstellung und Markierung von Sonden
Die für die Isolierung der in dieser Arbeit verwendeten Sonden wichtigen Parameter
sind in Tabelle 7 (s. 4.9) angegeben. Die Sonden werden durch Vergleich mit dem
Größenstandard auf eine Konzentration von 10 ng/µl eingestellt. Die radioaktive
Markierung erfolgt mit dem DecaLabelTM DNA Labeling Kit von MBI Fermentas
entsprechend den Herstellerangaben. Nach dem Einbau von 50 µCi (≈ 1,85 MBq) α-
[32P]-CTP wird der Reaktionsansatz mit TE (10/1) auf 100 µl aufgefüllt, die nicht
eingebauten Nukleotide entfernt und der Ansatz am Szintillator vermessen (s 4.8.1).
Es sollten mindestens 1,5 x 107 cpm für 10 ml Hybridisierungspuffer eingesetzt
werden.

Methoden 45
5.14 Vektorklonierung
NheI PstI
SalINheI
envpolgag
ClaI SphIDraIIIBsrGI PvuI
U5 R U3 U5R U3
Abb. 4: Schematische Skizze eines murinen Leukämievirus (MuLV) mit den für die Herstellung der
verschiedenen Chimären relevanten Restriktionsschnittstellen. Die strukturellen Gene gag, pol und
env werden von den LTR-Bereichen (U3, R, U5) flankiert.
Die Proviren liegen im Plasmid pUC18 in der Reihenfolge LTR-gag-pol-env-LTR vor.
Alle Moloney-Sequenzen stammen aus dem Plasmid R686, welches für das Moloney
Provirus kodiert (s. 4.6). Alle 10A1-Sequenzen stammen aus dem Vektor R862,
welches für das 10A1 Provirus kodiert (s. 4.6). Alle amphotropen Sequenzen
stammen aus dem Plasmid #434, welches für das 4070A Provirus kodiert (s. 4.6).
Der Vektor R602 kodiert nur für das 10A1-LTR, das Plasmid R603 nur für das
Moloney-LTR.
5.14.1 Chimäre MuLVs mit einem Austausch der env-Sequenzen
Für die Klonierung des Konstrukts Mo-10A1env wurden über die Schnittstellen SalI
und ClaI 10A1 Sequenzen in das Moloney-Rückgrat inseriert. Die vom 10A1-MuLV
stammende Region umfasst neben dem kompletten env- (2000 bp) auch den 3’ pol-
Bereich (1900 bp). Zur Herstellung der Chimäre 10A1-4070env wurden vom 4070A-
MuLV stammende Sequenzen über die Schnittstellenkombination SphI/ClaI in das
10A1-Rückgrat eingefügt. Dieser Bereich umfasst das gesamte env-Gen (2000 bp)
und einen kleinen Bereich der 3’ pol-Sequenzen (500 bp).

Methoden 46
Tab. 9: Zusammenfassung der für die Klonierung wichtigen Daten der beiden MuLVs mit einem
Austausch der env-Sequenzen.
Chimäre VektorrückgratHerkunft der env-
Sequenzen Enzym-
Kombination Interne
Nomenklatur
Mo-10A1env Moloney-MuLV 10A1-MuLV SalI/ClaI #427
10A1V-4070env 10A1-MuLV 4070A-MuLV SphI/ClaI R888
5.14.2 Chimäre MuLVs mit einem Austausch der gag-Sequenzen
Zur Herstellung der Konstrukts 10A1V-Mogag (R884) wurde die vom Moloney-MuLV
stammende gag-Sequenz über die Schnittstellenkombination BsrGI und DraIII in das
10A1-Rückgrat eingefügt.
5.14.3 Chimäre MuLVs mit einem Austausch der NP-Sequenzen
Die Abbildung 5 zeigt die Klonierungsstrategie für die Herstellung des Vektors
10A1V-MoNP (R633). Das Plasmid R686 kodiert für das Moloney-Provirus, der Vektor
R862 für das 10A1-Provirus. Beide werden mit den Restriktionsendonukleasen NheI
und PstI gespalten. Das Moloney-Fragment (NP) enthält das 5’-LTR und den Leader-
Bereich. Das 10A1 Fragment beinhaltet die strukturellen Gene gag, pol und env. Das
3’-LTR wird vom Plasmid R602 bereitgestellt, welches nur für das 10A1-LTR kodiert.
Die beiden isolierten Fragmente werden in den NheI gespaltenen Vektor R602
kloniert. Das auf diese Weise entstandene Provirusintermediat enthält ein Moloney
5’-LTR und ein 10A1 3’-LTR. Nach Transfektion der SC-1 Zellen und Durchlaufen
des retroviralen Replikationszyklus weist das neue Virus 10A1V-MoNP vom Anfang
des R-Bereichs bis zur PstI Schnittstelle eine Insertion von Moloney-Sequenzen auf.
Für die Einführung des 10A1 NP-Bereiches in das Moloney-Provirus wird umgekehrt
verfahren. Statt des Plasmids R602 wird der Vektor R603 verwendet, der für das
Moloney-LTR kodiert. Auf diese Weise wurde auch das Konstrukt Mo-10A1V-10A1NP
(R662) hergestellt.

Methoden 47
PstI NheI U3 R U5
Mo NP-Fragment
PstI NheI
U3 R U5
10
1. Die für die Proviren kodie
Enzymen NheI und PstI
anschließend isoliert.
+PstI NheI
U3 R U5
NheIU
2. Die isolierten Fragmen
welches nur das 10A1-LTR
PstI NheI U3 R U5
3. Das auf diese Weise klo
Erst nach Durchlaufen des
vor.
R686
NheI U5 U3 R pUC18
R862
NheI U5 U3 R pUC18
A1-Fragment
renden Plasmide R686 (Moloney) und R862 (10A1) werden mit den
gespalten. Die mit den Klammern markierten Fragmente werden
NheIPstI
U5 3 R pUC18
te werden in das mit NheI gespaltene Plasmid R602 kloniert,
enthält.
NheI U5 U3 R pUC18
nierte Provirus besitzt ein 5‘ Moloney-LTR und ein 3‘ 10A1-LTR.
retroviralen Entwicklungszyklus liegt das Konstrukt 10A1V-MoNP

Methoden 48
NheI U5 RU3 Wirtsgenom
PstI NheI U5 R U3
Abb. 5: Schematische Darstellung der Klonierungsstrategie für den Austausch von NP-Sequenzen
am Beispiel des Konstrukts 10A1V-MoNP.
5.14.4 Chimäre MuLVs mit einem Austausch der LTR-Sequenzen
Die Abbildung 6 zeigt die Klonierungsstrategie für die Herstellung des Vektors
10A1V-MoLTR (R606). Das Plasmid R862, das für das 10A1-Provirus kodiert, wird mit
dem Enzym NheI gespalten. Das 10A1-Fragment enthält das 5’-LTR, den Leader-
Bereich und die strukturellen Gene gag, pol und env. Das 3’-LTR wird vom Plasmid
R603 bereitgestellt, welches nur für das Moloney-LTR kodiert. Das isolierte Fragment
wird in den NheI gespaltenen Vektor R603 kloniert. Das auf diese Weise
entstandene Provirusintermediat enthält ein 10A1 5’-LTR und ein Moloney 3’-LTR.
Nach Transfektion der SC-1 Zellen und Durchlaufen des retroviralen
Replikationszyklus besitzt das neue Virus 10A1V-MoLTR nur den U3-Bereich vom
Moloney-Virus in einem 10A1-Rückgrat. Für die Einführung des 10A1 U3-Bereiches
in das Moloney-Provirus wird umgekehrt verfahren. Statt des Plasmids R603 wird der
Vektor R602 verwendet, der für das 10A1-LTR kodiert. Auf diese Weise wurde auch
das Konstrukt Mo-10A1V-10A1LTR (R661) hergestellt.

Methoden 49
R862
NheI R U5 U3 pUC18
NheI U5 R U3
10A1-Fragment
1. Das Plasmid R862, das für das 10A1-Provirus kodiert, wird mit dem Enzym NheI
gespalten. Das durch die Klammer markierte Fragment wird anschließend isoliert.
NheI R U5U3 pUC18
NheIU3
NheI U5 R U3
2. Das isolierte Fragment wird in das mit NheI gespaltene Plasmid R603 kloniert, welches nur
das Mo-LTR enthält.
NheI
R U5U3 pUC18
NheI U5 R U3
3. Das auf diese Weise klonierte Provirus besitzt ein 5‘ 10A1-LTR und ein 3‘ Moloney-LTR.
Erst nach Durchlaufen des retroviralen Entwicklungszyklus liegt das Konstrukt 10A1V-
MoLTR vor.
NheI R U5U3 Wirtsgenom
NheI U5 R U3
Abb. 6: Schematische Darstellung der Klonierungsstrategie für den Austausch von LTR-Sequenzen
am Beispiel des Konstrukts 10A1V-MoLTR.

Ergebnisse 50
6. Ergebnisse
6.1 Charakterisierung der von Moloney-, 4070A- und 10A1-MuLV induzierten Krankheiten in NIH/Ola- Mäusen
Um herauszufinden, welche Leukämieformen Moloney-, 4070A- und 10A1-MuLVs im
genetischen Hintergrund des NIH/OlaHsd-Inzuchtstammes induzieren, wurden die
Mäuse intraperitoneal mit 50-100 µl Virusüberstand infiziert. Die Virustiter lagen bei
infektiösen Partikelzahlen von 5,8 x 105 bis 3 x 106 pro ml. Die Infektion erfolgte
entweder innerhalb von 24-48 Stunden oder 5-8 Tage nach der Geburt (s. 5.7.5). Die
Tiere wurden dreimal in der Woche sorgfältig auf das Auftreten von
Krankheitssymptomen untersucht.
6.1.1 Charakterisierung der phänotypischen Veränderungen
6.1.1.1 Phänotyp von Moloney- bzw. 4070A-MuLV infizierten Tieren
Das Mo-MuLV induzierte nach einer mittleren Latenzzeit von 95 Tagen in allen
infizierten Tieren (n = 10) Tumore. Diese bildeten sich bei den meisten Mäusen in der
Milz bei einigen auch im Thymus, was zu einer starken Vergrößerung dieser Organe
führte. Das durchschnittliche Organgewicht lag bei 1,5 g (± 0,6), was einer
Gewichtszunahme um das 4,5-fache entspricht. Die Leber war nur selten, die
Lymphknoten dagegen fast immer betroffen. Der durchschnittliche Hämatokrit der
infizierten Tiere war nur geringfügig niedriger als normal, 36,1 %
(± 5,9) gegenüber Normalwerten von 38-50 %. Das 4070A-Virus induzierte dagegen
nach einer mittleren Latenzzeit von 217 Tagen in allen Mäusen (n = 14) Lymphome,
bei denen die Milz meistens nur leicht vergrößert war, mit durchschnittlichen
Organgewichten von 0,7 g (± 0,3), was dem doppelten der Normalgröße entspricht.
Die Lymphknoten waren immer betroffen und dabei meistens stark vergrößert. Der
durchschnittliche Hämatokrit der erkrankten Tiere war leicht vermindert gegenüber
dem uninfizierter Tiere, 31,3 % (± 5,6) versus 38-50%. Nur ein Tier zeigte einen
abweichenden Befund, neben den Lymphknoten waren auch Leber und Milz stark
vergrößert.
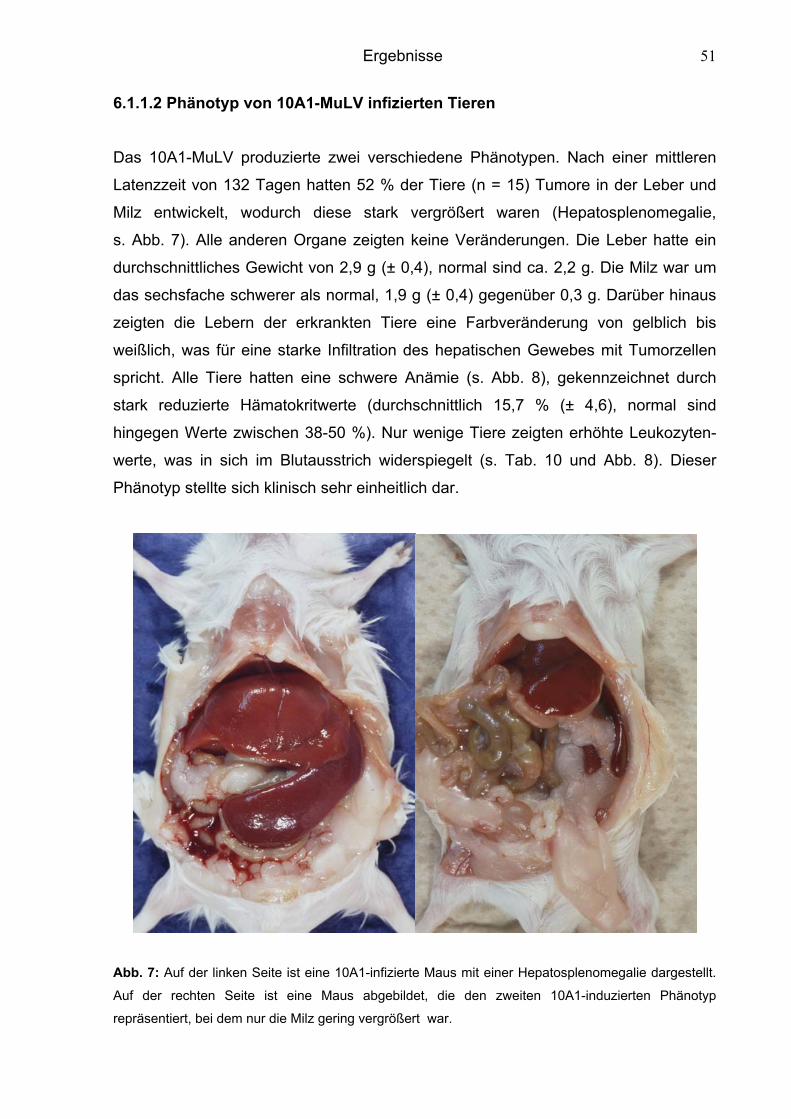
Ergebnisse 51
6.1.1.2 Phänotyp von 10A1-MuLV infizierten Tieren
Das 10A1-MuLV produzierte zwei verschiedene Phänotypen. Nach einer mittleren
Latenzzeit von 132 Tagen hatten 52 % der Tiere (n = 15) Tumore in der Leber und
Milz entwickelt, wodurch diese stark vergrößert waren (Hepatosplenomegalie,
s. Abb. 7). Alle anderen Organe zeigten keine Veränderungen. Die Leber hatte ein
durchschnittliches Gewicht von 2,9 g (± 0,4), normal sind ca. 2,2 g. Die Milz war um
das sechsfache schwerer als normal, 1,9 g (± 0,4) gegenüber 0,3 g. Darüber hinaus
zeigten die Lebern der erkrankten Tiere eine Farbveränderung von gelblich bis
weißlich, was für eine starke Infiltration des hepatischen Gewebes mit Tumorzellen
spricht. Alle Tiere hatten eine schwere Anämie (s. Abb. 8), gekennzeichnet durch
stark reduzierte Hämatokritwerte (durchschnittlich 15,7 % (± 4,6), normal sind
hingegen Werte zwischen 38-50 %). Nur wenige Tiere zeigten erhöhte Leukozyten-
werte, was in sich im Blutausstrich widerspiegelt (s. Tab. 10 und Abb. 8). Dieser
Phänotyp stellte sich klinisch sehr einheitlich dar.
Abb. 7: Auf der linken Seite ist eine 10A1-infizierte Maus mit einer Hepatosplenomegalie dargestellt.
Auf der rechten Seite ist eine Maus abgebildet, die den zweiten 10A1-induzierten Phänotyp
repräsentiert, bei dem nur die Milz gering vergrößert war.

Ergebnisse 52
Tab. 10: Relevante Befunde erkrankter Mäuse mit 10A1-induzierter früher Krankheitsmanifestation.
Das normale Milzgewicht liegt bei ca. 0,34 g, das normale Gewicht einer Leber bei ca. 2,4 g. (N:
normal; n.a.: nicht analysiert; Hkt: Hämatokrit; Leu: Leukozytenzahl).
Maus- Nr.
nach [d]
Milz [g]
Leber[g]
andere Organe
Hkt [%]
Leuk [106/ml]
#1824 145 >2,0 >3,0 normal 14 1,9 #2137 194 1,36 >2,5 normal 10,4 n.a. #2363 110 1,53 2,79 normal 16,2 <100 #2369 183 2,38 2,88 normal 16 53,2 #2375 144 1,99 2,68 normal 12 10,4 #2438 159 2,25 2,95 normal 14,4 181,2 #2442 116 1,86 2,98 normal 12,4 n.a. #2443 116 2,53 2,77 normal 12,6 11,8 #2453 170 1,76 3,62 normal 21,9 71,7 #2456 129 1,71 3,01 normal 15,6 9 #2457 129 1,3 N normal 26,7 11,4 #2461 159 1,65 N normal 23,4 n.a. #2463 106 2,6 3,6 normal 13,2 25 #2464 130 2,65 2,48 normal 13,8 7,2 #2465 149 2,03 3,06 normal 11,4 5,1
Es folgte dann, nach einer mittleren Latenzzeit von 164 Tagen, ein weiterer 10A1-
induzierter Phänotyp, bei dem 41 % der Mäuse (n = 14) Veränderungen an
verschiedenen Organen zeigten. Die Leber war in der Mehrzahl der analysierten
Tiere unverändert. Die Beteiligung der Milz war sehr unterschiedlich: bei einigen
Mäusen war eine starke, bei den übrigen dagegen nur eine leichte Vergrößerung zu
beobachten (s. Abb. 7). Auch die Beteiligung der Lymphknoten war sehr
uneinheitlich, bei einigen Tieren waren diese stark vergrößert, meistens war aber
keine Veränderung feststellbar. Bei allen Tieren konnte eine leichte Anämie
diagnostiziert werden, mit einem durchschnittlichen Hämatokrit von 31,9 % (± 9,3).
Nur wenige Tiere hatten erhöhte Leukozytenwerte, so dass in den Blutausstrichen
auch nur wenige Tumorzellen zu erkennen waren (s. Abb. 9). Morphologisch war
dieser Phänotyp deutlich heterogener als der uniforme Phänotyp der ersten
Krankheitswelle (s. Tab. 11).

Ergebnisse 53
Tab. 11: Zusammenstellung aller relevanten Daten der erkrankten Mäuse aus der 10A1-induzierten
späten Krankheitswelle. (N: normal; n.a.: nicht analysiert; Hkt: Hämatokrit; Leu: Leukozytenzahl)
Maus- Nr.
nach [d]
Milz [g]
Leber[g]
Andere Organe
Hkt [%]
Leuk [106/ml]
#1821 138 > 1,0 N Lymphknoten 12 33 #1822 138 > 1,5 > 2,5 Lymphknoten 32 10 #1823 145 > 0,4 N Lymphknoten 48 36 #1826 188 N N Lymphknoten 36 5,2 #2129 224 N N Lymphknoten n.a. n.a. #2135 180 N N Lymphknoten n.a. n.a. #2139 180 0,76 N Lymphknoten n.a. n.a. #2367 119 0,83 N Lymphknoten 36 5,1 #2432 161 0,79 N Lymphknoten 23,4 26,4 #2446 194 0,95 N normal 31,2 3,6 #2450 166 0,85 N Lymphknoten 36,3 18,3 #2454 201 N N normal 32,4 2,7
Um die Unterschiede zwischen den beiden 10A1-induzierten Phänotypen noch
genauer zu bestimmen, wurden histologische Untersuchungen durchgeführt. Dazu
wurden Blutausstriche und Organschnitte gefärbt (s. 5.9) und mikroskopisch
ausgewertet. Die Organschnitte und die histologischen Analysen wurden
freundlicherweise von Dr. Jürgen Löhler (Arbeitsgruppe Molekulare Pathologie des
HPI) durchgeführt.

Ergebnisse 54
Abb. 8: Blutausstrich einer 10A1-infizierten Maus mit dem frühen Phänotyp. An der Verformung der
Eryhtrozyten ist die starke Anämie zu erkennen (Akanthozytose). Die Erythrozyten sind darüber
hinaus polychromatisch. Des weiteren sind Proerythroblasten, basophile Makroblasten, Normoblasten
und ein Granulozyt zu erkennen (Vergrößerung x 880, Pappenheim-Färbung).
Abb. 9: Blutausstrich einer 10A1-infizierten Maus mit dem zweiten Phänotyp. Es sind drei
Lymphoblasten zu erkennen. Die Erythrozyten sind polychromatisch (Vergrößerung x 880, Pappen-
heim-Färbung).

Ergebnisse 55
Mäuse, die zur ersten Krankheitswelle gehörten, zeigten vermehrt Tumorzellinfiltrate
in der Leber, der Lunge und dem Knochenmark. Die normale Architektur der Milz war
durch das Tumorwachstum fast vollständig zerstört, wobei die weiße Pulpa durch die
stark expandierte und alterierte rote Pulpa weitgehend verdrängt wurde (s. Abb. 10).
Die Ausweitung der roten Pulpa war auf die massenhaft auftretenden Tumorzellen
zurückzuführen. Die Struktur des Knochenmarks wurde durch die Tumorzellinfiltrate
aufgelöst und die Erythro- und Myelopoese waren stark reduziert. An dieser Stelle
sollte darauf hingewiesen werden, dass die Erythro- und Myelopoese in der Maus
außer im Knochenmark auch in der roten Milzpulpa stattfindet. Die Leber zeigte eine
extreme Invasion von Tumorzellen, was zu einer Vergrößerung und Verfärbung
derselben führte (s. Abb. 12). Bei den Tumorzellen handelte es sich um
Vorläuferzellen der erythroiden, myeloiden und lymphoiden Abstammungslinie, was
für die Virus bedingte Transformation einer Stammzell-ähnlichen Zelle spricht. Diese
Leukämieform wird deshalb nachfolgend als blastische Leukämie bezeichnet. Die
Blutausstriche zeigten nur wenige normale, reife Erythrozyten, die teilweise starke
Verformungen aufwiesen (Akanthozytose) und myeloblastische sowie lympho-
blastische Zellen (s. Abb. 8). Des weiteren konnten verschiedene Reifungsstadien
der roten Blutzellreihe detektiert werden. Einige Erythrozyten hatten noch Zellkerne
(Normoblasten), was Zeichen einer gestörten Erythropoese ist. Die Blockierung
undifferenzierter, erythroider Zellen kann zu einer Verarmung an reifen Erythrozyten
führen, was die schwere Anämie der Tiere erklären würde. Die Mäuse erkrankten an
einer blastischen Leukämieform, bei der das erythroide Kompartiment stark
beeinträchtigt wurde.
Die Tiere der zweiten Krankheitsphase zeigten nur eine geringe, lokal begrenzte
Infiltration der Leber (s. Abb. 13). Die Gewebsarchitektur des Knochenmarks und der
Lymphknoten war weitgehend zerstört und deren Umgebung durch auswandernde
Tumorzellen stark infiltriert. In diesem Fall stellten die Tumorzellen lymphoide Zellen
verschiedener Reifungsgrade dar. Die weiße Pulpa der Milz zeigte eine stark
expandierte B-Zellzone, die teilweise die T-Zellzone verdrängt hatte (s. Abb. 11). Die
lymphoiden Tumorzellen infiltrierten auch die rote Pulpa. Daneben war eine
verstärkte Erythropoese zu beobachten. In den Blutausstrichen konnte eine erhöhte
Anzahl an lymphoiden Zellen, Lymphoblasten und Lymphozyten (Leukozytose)
beobachtet werden (s. Abb. 9).

Ergebnisse 56
Abb. 10: Schnitt durch die Milz einer 10A1-infizierten Maus aus der ersten Krankheitswelle.
Erythroblastische Tumorzellen besiedeln die rote Milzpulpa. Die normale Hämatopoese der roten
Pulpa ist vollständig verschwunden (Vergrößerung x 560, Hämotoxilin-Eosin-Fäbung).
wP
wP
L
E
Abb. 11: Schnitt durch die Milz einer 10A1-infizierten Maus aus der späten Krankheitswelle. Die weiße
Milzpulpa ist durch lymphoblastische Tumorzellen expandiert (wP). Bei „L“ sind residuale normale
Lymphozyten zu erkennen. Am rechen Bildrand ist eine hyperplastische Erythropoese zu erkennen (E,
Vergrößerung x 350, Hämotoxilin-Eosin-Fäbung).

Ergebnisse 57
Abb. 12: Schnitt durch die Leber einer 10A1-infizierten Maus aus der ersten Krankheitswelle. Die
Lebersinusoide sind stark erweitert durch infiltrierende hämatopoetische Tumorzellen. Die
Tumorzellen handelt es sich vorwiegend um unreife erythroide Zellformen. Es sind aber auch einige
myeloblastische und lymphoblastische Zellen vorhanden. Die Hepatozyten zeigen eine
mikrovesikuläre, fettige Degeneration (Vergrößerung x 560, Perjodsäure-Schiff-Reaktion).
Abb. 13: Das Bild zeigt ein leukämisches Infiltrat (Metastase) in einem Portalfeld der Leber. In der
Lymphomabsiedelung sind zahlreiche Mitosen und Apoptosen der neoplastischen Zellen zu erkennen.
Das an den Tumor angrenzende Lebergewebe ist intakt (Vergrößerung x 350, Giemsa-Färbung).

Ergebnisse 58
Die beiden Phänotypen waren mikroskopisch sehr unterschiedlich. Neben den
unterschiedlichen Tumorzellarten liegt der Hauptunterschied in einem
unterschiedlichen Verteilungsmuster des Gewebsprozesses in der Milz. Der
blastische Phänotyp beeinträchtigte vor allem die rote Pulpa, während die
Neoplasien der lymphoiden Zellen vor allem die weiße Pulpa betreffen. Dies könnte
unterschiedliche pathogenetische Mechanismen im Krankheitsverlauf reflektieren
(s. 6.1.2.4).
6.1.2 Weitere Charakterisierung der MuLV-induzierten Tumore
Bei der FACS-Analyse macht man sich die Tatsache zunutze, dass die verschie-
denen hämatopoetischen Zelltypen während und nach der Differenzierung ganz
bestimmte Proteinmoleküle auf der Oberfläche exprimieren, die von den
entsprechenden, monoklonalen Antikörpern erkannt werden. Da diese Antikörper an
fluoreszierende Farbstoffe gekoppelt sind, können die auf diese Weise markierten
Zellen mittels des FACS-Gerätes quantitativ erfasst werden. Zellen können
gleichzeitig mit mehreren Antikörpern gefärbt werden, wenn diese an andere
Farbstoffe gekoppelt sind, wie z.B. FITC (grün, x-Achse) und PE (rot, y-Achse).
Zellen, an die keiner der beiden Antikörper gebunden hat, befinden sich im linken
unteren Quadranten. Zellen, die nur positiv für den FITC-assoziierten Antikörper sind,
befinden sich im rechten, unteren Quadranten. Zellen, die dagegen nur vom PE-
gekoppelten Antikörper erkannt werden, befinden sich im linken oberen Quadranten
(s. Abb.14). Im rechten oberen Quadranten befinden sich nur Zellen, die doppelt
positiv für die beiden verwendeten Antikörper sind (s. Abb. 14). Bei der
Isotypkontrolle handelt es sich um die Negativkontrolle. Diese wird verwendet, damit
sich die ungefärbten Zellen nach der Justierung im linken unteren Quadranten
befinden und dann nicht als falsch positive gezählt werden können (s. Abb. 14). Im
Forward-Scatter (FSC) wird die Zellgröße und im Sideward-Scatter (SSC) die
Granularität der Zellen bestimmt. Es werden nur die Zellen ausgewählt, die eine
einheitliche Population bilden (s. Abb. 14). Auf diese Weise werden Zelltrümmer und
Erythrozyten nicht mit in die Messung einbezogen.

Ergebnisse 59
6.1.2.1 Moloney- und 4070A-MuLV induzierte Leukämien
Alle vom Moloney-Virus induzierten Tumore waren in der FACS-Analyse doppelt
positiv für die T-Zell-Marker CD90 und CD3 (s. Abb. 14). Im Gegensatz dazu waren
nur einige Lymphome doppelt positiv für die T-Zell-Marker CD4 und CD8, andere
dagegen nur positiv für einen der beiden Antikörper. Bei den doppelt positiven
Tumorzellen handelte es sich um unreife T-Zellen. T-Helfer-Zellen sind nur CD4
positiv, zytotoxische T-Zellen sind nur CD8 positiv. Die Tumore waren alle negativ für
die Oberflächenmarker von Zellen anderer Abstammungslinien (s. Abb. 14). Das
Moloney-MuLV hatte in allen infizierten NIH/Ola-Mäusen T-Zell-Lymphome/Leukämie
induziert, bei der entweder reife oder unreife T-Zellen betroffen waren.
R1
100
CD
4
CD19
B22
0
Isotypktr. FITC 100100
IIsot
ypkt
r. PE
10
4
Sca-
1
100
CD
3e
104
100
104
100
104
100
104
CD8 100 104 100 104 CD34 104
CD90 100 104
SSC
-Hei
ght
0 10
00
1000 00 1000 10FSC-H 0
Abb. 14: FACS-Analyse der Mo-MuLV infizierten Maus #2355, die an einer T-Zell-
Leukämie/Lymphom erkrankt war (Organ: Thymus). Die x- bzw. y-Achsen geben die
Fluoreszenzintensität der gefärbten Zellen an, die Einteilung ist logarithmisch.
In der FACS-Analyse waren 93 % (n = 13) der vom 4070A-MuLV induzierten Tumore
doppelt positiv für die B-Zell-Marker CD19 und B220. Es handelte sich bei den
Tumorzellen also um B-Zellen. Zellen aus anderen Abstammungslinien konnten nicht
detektiert werden. Die Maus, die andere morphologische Merkmale als die anderen

Ergebnisse 60
Tiere gezeigt hatte, lieferte in der FACS-Analyse das gleiche Bild wie die vom
Moloney-MuLV induzierten Tumore. Das Tier hatte eine T-Zell-Leukämie/Lymphom
(s. Abb. 15).
R1
SSC
-Hei
ght
0 10
00
FSC-H 1000 0
B22
0
CD
4 10
0 10
4
104
Sca-
1
104
CD34 104
CD
3e
100
104
Isot
ypkt
r. PE
Isotypktr. FITC
100
104
100 104 100 CD90 104
104 100 100
CD19 100 104 100100
CD8
Abb. 15: FACS-Analyse der 4070A-MuLV-infizierten Maus #2324, die an einer B-Zell-
Leukämie/Lymphom erkrankt war (Organ: Lymphknoten).
6.1.2.2 10A1-MuLV induzierte Leukämien
Die Charakterisierung der blastischen Leukämieform mittels FACS-Analyse stellte
sich schwierig dar, weil sich diese Tumore durch ein Fehlen von typischen
Zellmarkern auszeichneten. Die Tumore waren weder positiv für die B-Zell-Marker
(CD19, B220), noch für die T-Zell-Marker (CD90, CD3, CD4 und CD8). Darüber
hinaus konnten auch die Antikörper CD34 und CD117, die frühe Stammzellen
erkennen, nicht nachgewiesen werden (s. Abb. 16). Die Lymphome waren ebenfalls
negativ für die myeloiden Zellmarker CD11b und Gr-1 (nicht dargestellt).
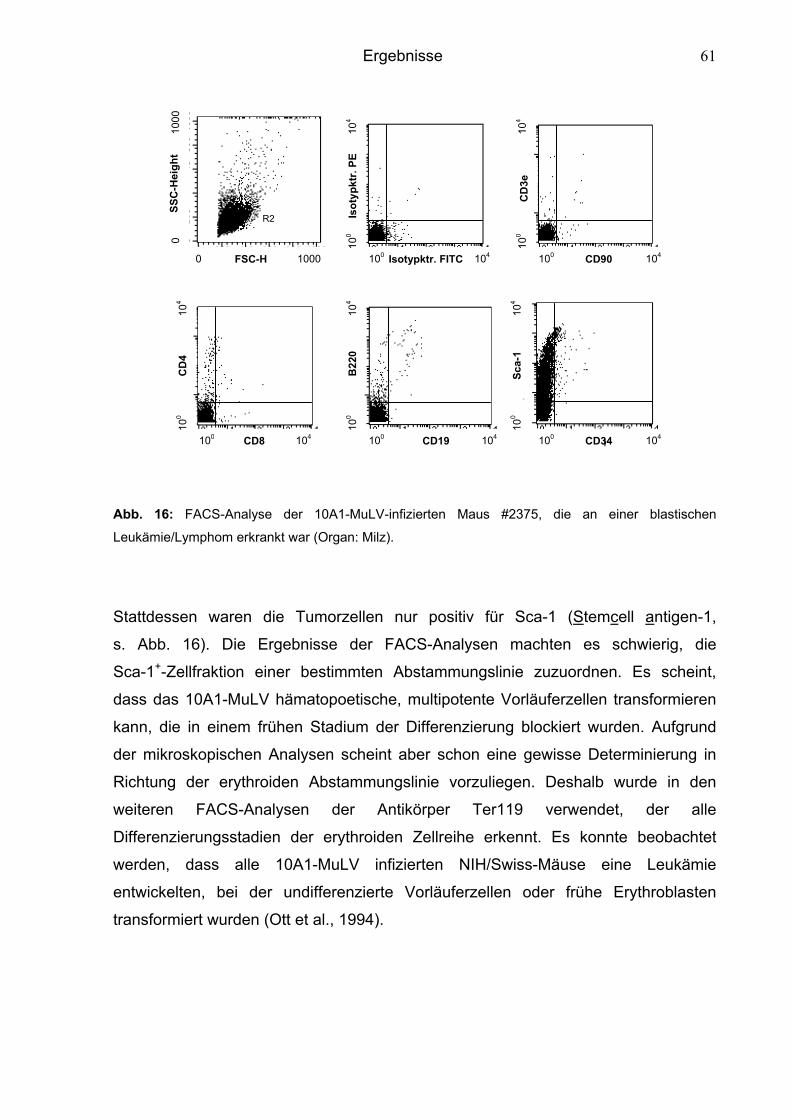
Ergebnisse 61
R2
Isotypktr. FITC
CD34
Sca-
1
CD
4
CD19
B22
0
CD
3e
Isot
ypkt
r. PE
SS
C-H
eigh
t 0
1000
100
104
100
104
100
104
100
104
100
104
FSC-H 0 1000
100 100 104 CD8 100 104 104
100 104 CD90 010 410
Abb. 16: FACS-Analyse der 10A1-MuLV-infizierten Maus #2375, die an einer blastischen
Leukämie/Lymphom erkrankt war (Organ: Milz).
Stattdessen waren die Tumorzellen nur positiv für Sca-1 (Stemcell antigen-1,
s. Abb. 16). Die Ergebnisse der FACS-Analysen machten es schwierig, die
Sca-1+-Zellfraktion einer bestimmten Abstammungslinie zuzuordnen. Es scheint,
dass das 10A1-MuLV hämatopoetische, multipotente Vorläuferzellen transformieren
kann, die in einem frühen Stadium der Differenzierung blockiert wurden. Aufgrund
der mikroskopischen Analysen scheint aber schon eine gewisse Determinierung in
Richtung der erythroiden Abstammungslinie vorzuliegen. Deshalb wurde in den
weiteren FACS-Analysen der Antikörper Ter119 verwendet, der alle
Differenzierungsstadien der erythroiden Zellreihe erkennt. Es konnte beobachtet
werden, dass alle 10A1-MuLV infizierten NIH/Swiss-Mäuse eine Leukämie
entwickelten, bei der undifferenzierte Vorläuferzellen oder frühe Erythroblasten
transformiert wurden (Ott et al., 1994).
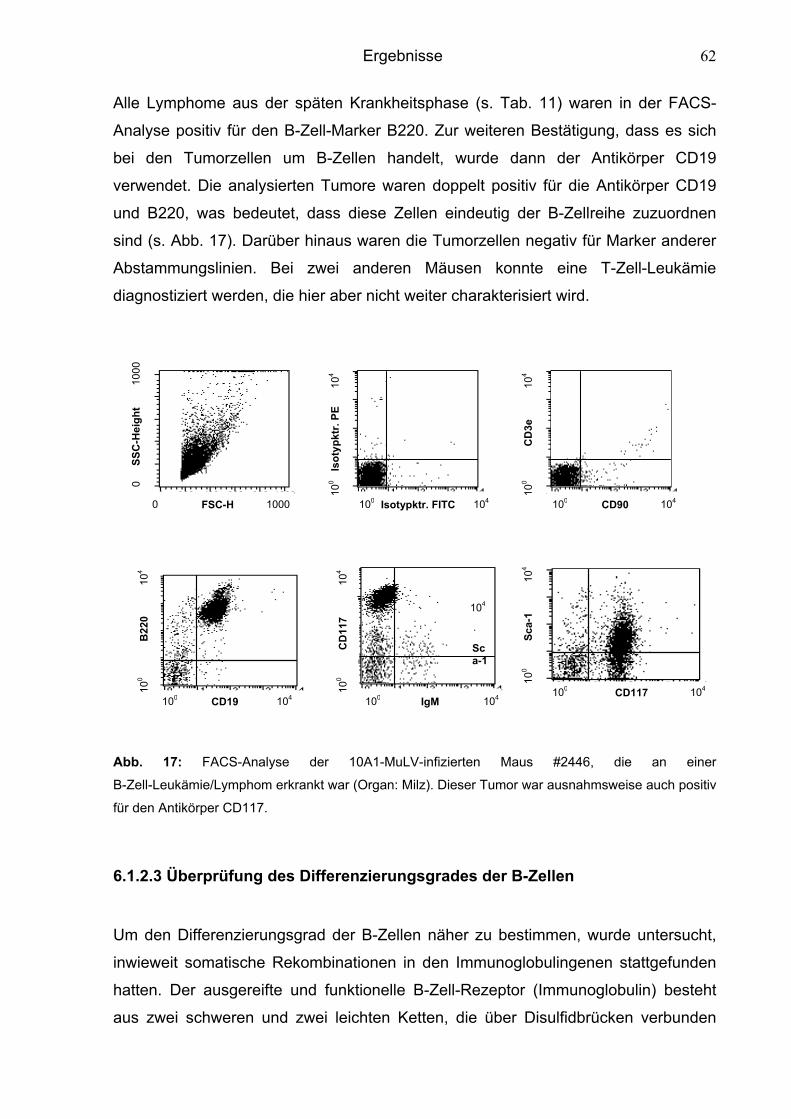
Ergebnisse 62
Alle Lymphome aus der späten Krankheitsphase (s. Tab. 11) waren in der FACS-
Analyse positiv für den B-Zell-Marker B220. Zur weiteren Bestätigung, dass es sich
bei den Tumorzellen um B-Zellen handelt, wurde dann der Antikörper CD19
verwendet. Die analysierten Tumore waren doppelt positiv für die Antikörper CD19
und B220, was bedeutet, dass diese Zellen eindeutig der B-Zellreihe zuzuordnen
sind (s. Abb. 17). Darüber hinaus waren die Tumorzellen negativ für Marker anderer
Abstammungslinien. Bei zwei anderen Mäusen konnte eine T-Zell-Leukämie
diagnostiziert werden, die hier aber nicht weiter charakterisiert wird.
R1
104
100
100
Sca-
1
100
100
104100 u000 uCD117
IgM 100 104
100
100
CD
117
100
B22
0
104
100 CD19
104
100
104
FSC-H
CD
3e
100 0 1000 100CD90 104
104
100
100
Isot
ypkt
r. PE
10
4 10
0
1041000 1000 1000 Isotypktr. FITC 0
SSC
-Hei
ght
0 10
00
Sca-1
104
Abb. 17: FACS-Analyse der 10A1-MuLV-infizierten Maus #2446, die an einer
B-Zell-Leukämie/Lymphom erkrankt war (Organ: Milz). Dieser Tumor war ausnahmsweise auch positiv
für den Antikörper CD117.
6.1.2.3 Überprüfung des Differenzierungsgrades der B-Zellen
Um den Differenzierungsgrad der B-Zellen näher zu bestimmen, wurde untersucht,
inwieweit somatische Rekombinationen in den Immunoglobulingenen stattgefunden
hatten. Der ausgereifte und funktionelle B-Zell-Rezeptor (Immunoglobulin) besteht
aus zwei schweren und zwei leichten Ketten, die über Disulfidbrücken verbunden

Ergebnisse 63
sind. Die schwere Kette besteht aus vier Domänen: einer variablen Domäne (VH), die
für die Antigenerkennung wichtig ist und drei konstanten Domänen (CH1-3). Der VH-
Bereich wird von drei verschiedenen genetischen Clustern kodiert: den
Variabilitätsgenen (V), den Diversitätsgenen (D) und den Verbindungsgenen (J). Die
somatische Rekombination beginnt mit dem Zusammenfügen jeweils eines D- und
eines J-Fragments aus den entsprechenden Clustern, erst dann folgt eines der V-
Fragmente (s. Abb. 18).
prä-B-Rezeptor
Umstrukturier
Umstrukturierungen beginne
große prä-B-Zelle
VH-DJH Rekombination findet statt
kein funktionelles Protein wird exprimiert
frühe pro-B-Zelle
ZellenProteineGene
Abb. 18: In frühen pro-B-Zellen beg
schwere Kette mit dem Zusammenfüg
Gen-Clustern. Es wird aber noch ke
großen prä-B-Zellen ist die Rekombina
surrogate leichte Kette
ungen in der leichten Kette werden beendet
reife B-Zelle
unreife B-Zelle
n in der leichten Kette und stoppen in der schweren Kette
innt die Rekombination in den Imm
en eines D- und eines J-Fragments
in funktionelles Protein auf der Ze
tion der schweren Kette abgeschloss
IgM
unoglobulingenen für die
aus den entsprechenden
lloberfläche exprimiert. In
en und Rekombination in
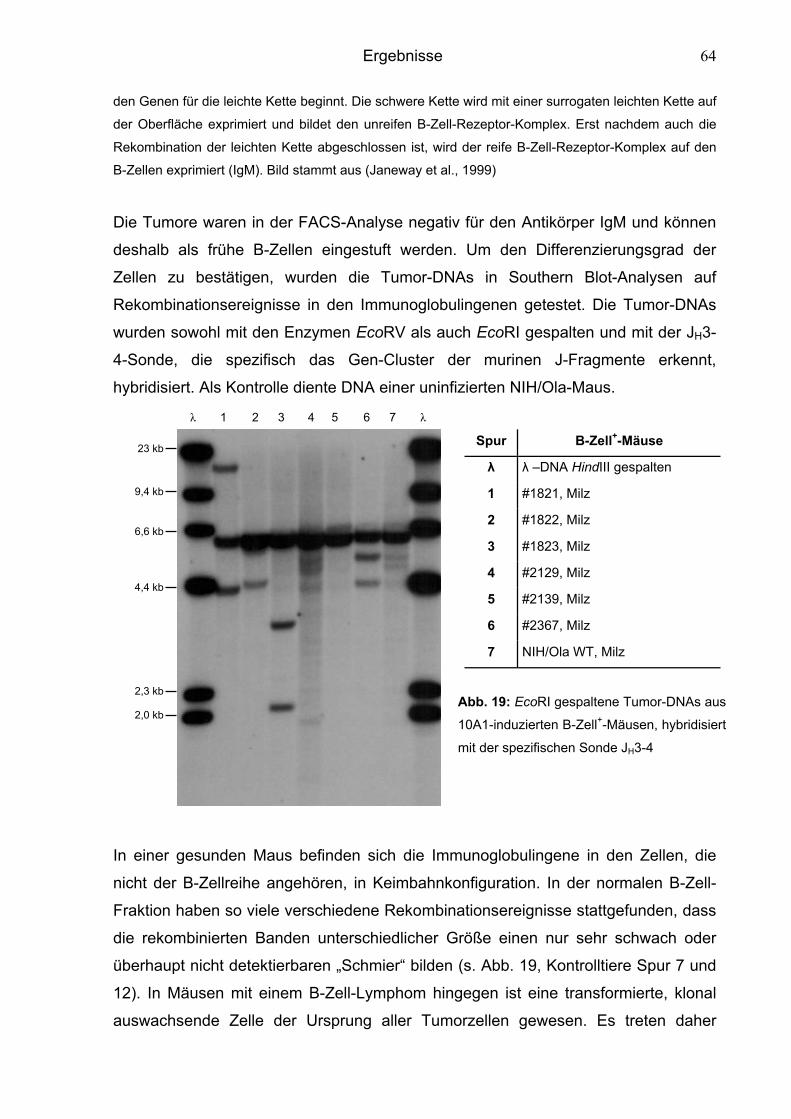
Ergebnisse 64
den Genen für die leichte Kette beginnt. Die schwere Kette wird mit einer surrogaten leichten Kette auf
der Oberfläche exprimiert und bildet den unreifen B-Zell-Rezeptor-Komplex. Erst nachdem auch die
Rekombination der leichten Kette abgeschlossen ist, wird der reife B-Zell-Rezeptor-Komplex auf den
B-Zellen exprimiert (IgM). Bild stammt aus (Janeway et al., 1999)
Die Tumore waren in der FACS-Analyse negativ für den Antikörper IgM und können
deshalb als frühe B-Zellen eingestuft werden. Um den Differenzierungsgrad der
Zellen zu bestätigen, wurden die Tumor-DNAs in Southern Blot-Analysen auf
Rekombinationsereignisse in den Immunoglobulingenen getestet. Die Tumor-DNAs
wurden sowohl mit den Enzymen EcoRV als auch EcoRI gespalten und mit der JH3-
4-Sonde, die spezifisch das Gen-Cluster der murinen J-Fragmente erkennt,
hybridisiert. Als Kontrolle diente DNA einer uninfizierten NIH/Ola-Maus.
λ 1 λ2 3 4 5 6 7
Spur B-Zell+-Mäuse
λ λ –DNA HindIII gespalten
1 #1821, Milz
2 #1822, Milz
3 #1823, Milz
4 #2129, Milz
5 #2139, Milz
6 #2367, Milz
7 NIH/Ola WT, Milz
2,3 kb
9,4 kb
2,0 kb
4,4 kb
6,6 kb
23 kb
Abb. 19: EcoRI gespaltene Tumor-DNAs aus
10A1-induzierten B-Zell+-Mäusen, hybridisiert
mit der spezifischen Sonde JH3-4
In einer gesunden Maus befinden sich die Immunoglobulingene in den Zellen, die
nicht der B-Zellreihe angehören, in Keimbahnkonfiguration. In der normalen B-Zell-
Fraktion haben so viele verschiedene Rekombinationsereignisse stattgefunden, dass
die rekombinierten Banden unterschiedlicher Größe einen nur sehr schwach oder
überhaupt nicht detektierbaren „Schmier“ bilden (s. Abb. 19, Kontrolltiere Spur 7 und
12). In Mäusen mit einem B-Zell-Lymphom hingegen ist eine transformierte, klonal
auswachsende Zelle der Ursprung aller Tumorzellen gewesen. Es treten daher
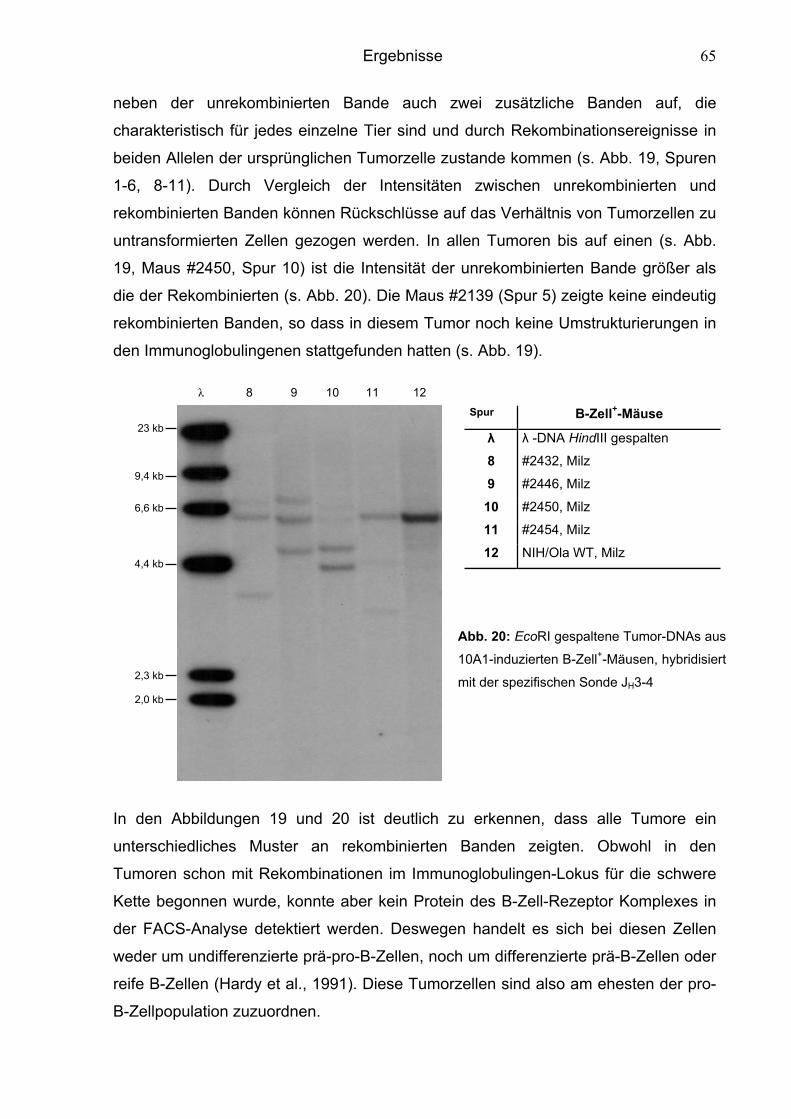
Ergebnisse 65
neben der unrekombinierten Bande auch zwei zusätzliche Banden auf, die
charakteristisch für jedes einzelne Tier sind und durch Rekombinationsereignisse in
beiden Allelen der ursprünglichen Tumorzelle zustande kommen (s. Abb. 19, Spuren
1-6, 8-11). Durch Vergleich der Intensitäten zwischen unrekombinierten und
rekombinierten Banden können Rückschlüsse auf das Verhältnis von Tumorzellen zu
untransformierten Zellen gezogen werden. In allen Tumoren bis auf einen (s. Abb.
19, Maus #2450, Spur 10) ist die Intensität der unrekombinierten Bande größer als
die der Rekombinierten (s. Abb. 20). Die Maus #2139 (Spur 5) zeigte keine eindeutig
rekombinierten Banden, so dass in diesem Tumor noch keine Umstrukturierungen in
den Immunoglobulingenen stattgefunden hatten (s. Abb. 19).
λ 8 9 10 11 12
Spur B-Zell+-Mäuse
λ λ -DNA HindIII gespalten
8 #2432, Milz
9 #2446, Milz
10 #2450, Milz
11 #2454, Milz
12 NIH/Ola WT, Milz
2,3 kb
9,4 kb
2,0 kb
4,4 kb
6,6 kb
23 kb
Abb. 20: EcoRI gespaltene Tumor-DNAs aus
10A1-induzierten B-Zell+-Mäusen, hybridisiert
mit der spezifischen Sonde JH3-4
In den Abbildungen 19 und 20 ist deutlich zu erkennen, dass alle Tumore ein
unterschiedliches Muster an rekombinierten Banden zeigten. Obwohl in den
Tumoren schon mit Rekombinationen im Immunoglobulingen-Lokus für die schwere
Kette begonnen wurde, konnte aber kein Protein des B-Zell-Rezeptor Komplexes in
der FACS-Analyse detektiert werden. Deswegen handelt es sich bei diesen Zellen
weder um undifferenzierte prä-pro-B-Zellen, noch um differenzierte prä-B-Zellen oder
reife B-Zellen (Hardy et al., 1991). Diese Tumorzellen sind also am ehesten der pro-
B-Zellpopulation zuzuordnen.

Ergebnisse 66
6.1.2.4 Kinetik des Krankheitsverlaufs
Um festzustellen, ob sich ein Unterschied zwischen den beiden 10A1-induzierten
Phänotypen bereits während der Latenzzeitphase nachweisen lässt, wurden jeweils
zwei Mäuse zu bestimmten Zeitpunkten nach der Infektion geopfert und sowohl
makroskopisch als auch mikroskopisch auf das Auftreten etwaiger Veränderungen
hin untersucht. Der Abstand zwischen den Beobachtungszeitpunkten betrug ein bis
zwei Wochen. Der gesamte Untersuchungszeitraum umfasste 15 Wochen, wobei mit
den Analysen zwei Wochen nach Infektion begonnen wurde.
Erste mikroskopische Veränderungen konnten sechs Wochen nach der Infektion in
den klinisch gesunden Tieren festgestellt werden. In der leicht vergrößerten Milz war
eine Vermehrung (Hyperplasie) von hämatopoetischen Zellen zu erkennen, was aber
die Architektur des Organs nicht beeinträchtigte. In der weißen Pulpa konnte eine
Vermehrung der B-Zellen, in der roten Pulpa eine Hyperplasie vor allem erythroider
Zellen und in geringerem Maße auch der myeloiden und megakaryozytischen Zellen
diagnostiziert werden. Auch in den Lymphknoten war eine beginnende
Zellproliferation nachweisbar. Nach acht Wochen zeigten die Lymphknoten und das
Knochenmark eine deutliche Hyperplasie.
Mikroskopisch ist bis zur neunten Woche nach Virusinokulation kein Unterschied
zwischen den beiden Phänotypen feststellbar. Die Auswirkungen der Virusinfektion
ist in diesem Zeitraum durch eine allgemeine Hyperplasie der roten Pulpa
gekennzeichnet. Das gleiche gilt übrigens auch für Moloney-MuLV infizierte Mäuse
(Daten nicht dargestellt). Erst nach diesem Zeitpunkt beginnt sich eine Manifestation
in die eine oder andere Richtung bemerkbar zu machen, wobei die Mäuse nach
Auftreten der ersten Anzeichen für einen blastischen Phänotyp sehr rasch moribund
werden, d.h. der Krankheitsverlauf ist in diesem Fall rasch propagiert. In der Leber
konnten ab der zehnten Woche Tumorzellinfiltrate nachgewiesen werden. Das
Auftreten von Infiltraten in der Leber, später auch in anderen Organen ist
charakteristisch für den blastischen Phänotyp, da beim B-Zell+-Phänotyp zu keinem
Zeitpunkt Infiltratbildungen nachgewiesen werden konnten, die die Leberarchitektur
beeinträchtigten (s. Abb. 12 und 13).

Ergebnisse 67
6.2 Charakterisierung der chimären murinen Leukämieviren NIH/Ola-Mäuse wurden mit den chimären Retroviren infiziert. Kranke Tiere wurden
geopfert und auf Veränderungen untersucht. Die entnommenen Tumore wurden
dann weiter analysiert.
6.2.1 Charakterisierung von MuLVs mit ausgetauschten env-Sequenzen
Um festzustellen inwieweit sich der Austausch von env-Sequenzen auf die
Pathogenität von murinen Leukämieviren auswirkt, wurden zwei chimäre Retroviren
hergestellt (s. Abb. 21). In das Moloney-MuLV wurden über die Schnittstellen SalI
und ClaI die 10A1 env-Sequenzen eingeführt. Das Virus 10A1V-4070Aenv trägt die
über die Schnittstellen SphI und ClaI inserierten amphotropen env-Sequenzen.
MuLVs infizieren Zellen über deren auf der Oberfläche exprimierten Rezeptoren.
Welchen Rezeptor ein Virus für den Eintritt in die Wirtszelle benutzt, wird durch
dessen env-Sequenzen determiniert. (Münk et al., 1998) haben gezeigt, dass die
Einführung eines amphotropen oder 10A1 env-Gens in das Moloney-MuLV, dieses
befähigt spongiforme Läsionen im Gehirn infizierter Mäuse zu verursachen. Der
Austausch von env-Sequenzen könnte also profunde Auswirkungen auf die Fähigkeit
eines MuLVs haben, bestimmte Spezies oder Gewebe zu infizieren und könnte damit
auch dessen Pathogenität beeinflussen. Es wurde deshalb in dieser Arbeit getestet,
inwieweit sich der Austausch von env-Sequenzen auf die Fähigkeiten vom Moloney-
und 10A1-MuLV auswirken, bestimmte Leukämieformen zu induzieren. Das 10A1-
Virus erkennt die beiden Phosphatsymporter Pit1 und Pit2, während das 4070A-
MuLV nur den Pit2-Rezeptor benutzt. Das Mo-MuLV erkennt den
Aminosäuretransporter mCat-1.

Ergebnisse 68
LTR Rezeptor: LTR envpolgag
10A1 ClaI SalI
4070A ClaI SphI
LTR LTR envpolgag
Mo-MuLV mCat1
Mo-101env-MuLV Pit1 + Pit2
Pit1 + Pit210A1-MuLV
Pit2 10A1-4070env-MuLV
Abb. 21: Genomorganisation und Rezeptornutzung von Moloney-MuLV, Mo-10A1env-MuLV, 10A1-
MuLV und 10A1-4070env. Das chimäre Mo-10A1env Virus trägt das env-Gen und den 3’-pol Bereich des
10A1MuLVs. Das Virus Konstrukt 10A1-4070env besitzt amphotropen Sequenzen von der SphI bis zur
ClaI Schnittstelle, was das env-Gen und einen kleinen Teil der 3’-pol Region umfasst.
6.2.1.1 Phänotyp von Mo-10A1env infizierten Tieren
Das chimäre Virus Mo-10A1env induzierte nach einer mittleren Latenzzeit von 128
Tagen in 93 % der infizierten Tiere (n = 15) eine T-Zell-Leukämie. Eine Maus hatte
eine B-Zell-Leukämie. Interessanterweise zeigten die meisten Tiere eine stark
vergrößerte Leber, was bei Moloney-MuLV infizierten Mäusen fast nie zu beobachten
gewesen war. Darüber hinaus hatten die erkrankten Tiere fast alle eine stark
vergrößerte Milz und die Lymphknoten waren ebenfalls fast immer betroffen (s. Tab.
12). Es wurden keine Blutanalysen von diesen Tieren durchgeführt.
In der FACS-Analyse waren die Tumore entweder nur CD90 oder CD90/CD3 positiv.
Die meisten Lymphome waren CD4/CD8 positiv, einige exprimierten aber nur CD4
oder CD8 auf der Oberfläche (s. Tab 12). Diese Tumore zeigten somit das gleiche
Bild wie die vom Moloney-MuLV induzierten Lymphome. Ein Tumor war positiv für
den B-Zell-Marker B220, der Antikörper anti-CD19 wurde nicht verwendet. Dieses
Virus-Konstrukt wurde bereits in NIH/Swiss Mäusen getestet (Ott et al., 1992). Die
Tiere zeigten ebenfalls den gleichen Phänotyp wie nach einer Infektion mit dem
Wildtyp Moloney-MuLV.
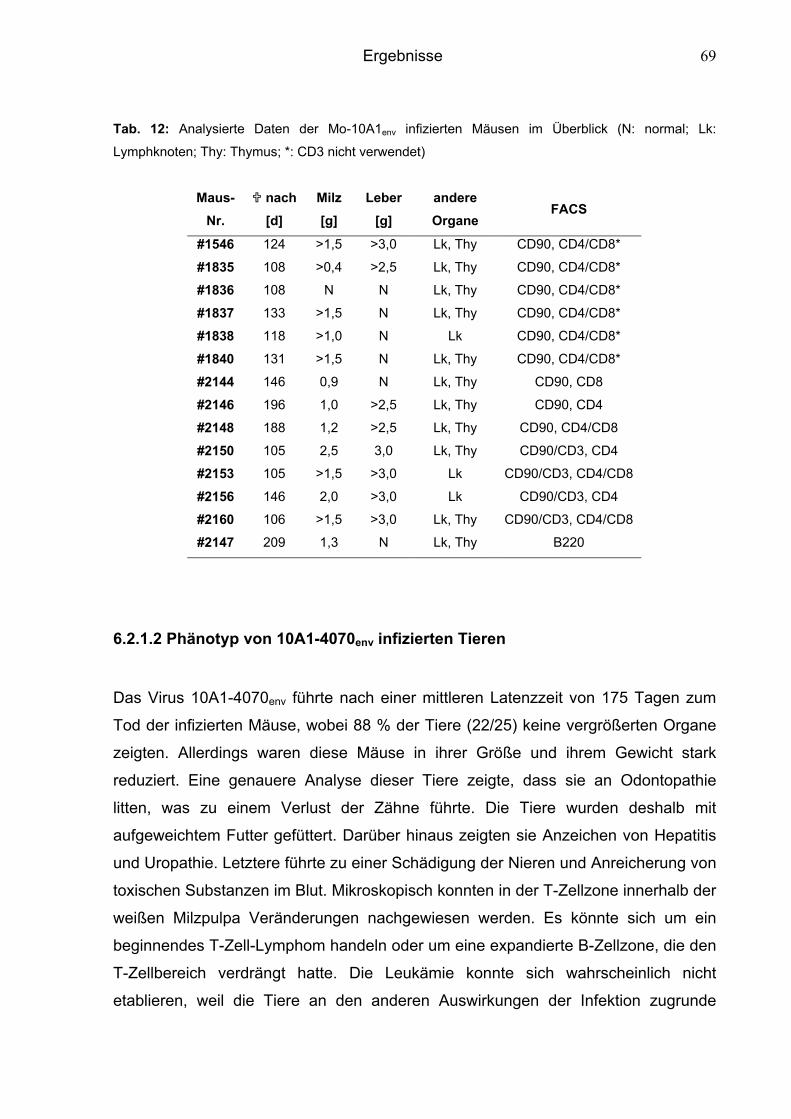
Ergebnisse 69
Tab. 12: Analysierte Daten der Mo-10A1env infizierten Mäusen im Überblick (N: normal; Lk:
Lymphknoten; Thy: Thymus; *: CD3 nicht verwendet)
Maus- Nr.
nach [d]
Milz [g]
Leber [g]
andere Organe
FACS
#1546 124 >1,5 >3,0 Lk, Thy CD90, CD4/CD8*
#1835 108 >0,4 >2,5 Lk, Thy CD90, CD4/CD8*
#1836 108 N N Lk, Thy CD90, CD4/CD8*
#1837 133 >1,5 N Lk, Thy CD90, CD4/CD8*
#1838 118 >1,0 N Lk CD90, CD4/CD8*
#1840 131 >1,5 N Lk, Thy CD90, CD4/CD8*
#2144 146 0,9 N Lk, Thy CD90, CD8
#2146 196 1,0 >2,5 Lk, Thy CD90, CD4
#2148 188 1,2 >2,5 Lk, Thy CD90, CD4/CD8
#2150 105 2,5 3,0 Lk, Thy CD90/CD3, CD4
#2153 105 >1,5 >3,0 Lk CD90/CD3, CD4/CD8
#2156 146 2,0 >3,0 Lk CD90/CD3, CD4
#2160 106 >1,5 >3,0 Lk, Thy CD90/CD3, CD4/CD8
#2147 209 1,3 N Lk, Thy B220
6.2.1.2 Phänotyp von 10A1-4070env infizierten Tieren
Das Virus 10A1-4070env führte nach einer mittleren Latenzzeit von 175 Tagen zum
Tod der infizierten Mäuse, wobei 88 % der Tiere (22/25) keine vergrößerten Organe
zeigten. Allerdings waren diese Mäuse in ihrer Größe und ihrem Gewicht stark
reduziert. Eine genauere Analyse dieser Tiere zeigte, dass sie an Odontopathie
litten, was zu einem Verlust der Zähne führte. Die Tiere wurden deshalb mit
aufgeweichtem Futter gefüttert. Darüber hinaus zeigten sie Anzeichen von Hepatitis
und Uropathie. Letztere führte zu einer Schädigung der Nieren und Anreicherung von
toxischen Substanzen im Blut. Mikroskopisch konnten in der T-Zellzone innerhalb der
weißen Milzpulpa Veränderungen nachgewiesen werden. Es könnte sich um ein
beginnendes T-Zell-Lymphom handeln oder um eine expandierte B-Zellzone, die den
T-Zellbereich verdrängt hatte. Die Leukämie konnte sich wahrscheinlich nicht
etablieren, weil die Tiere an den anderen Auswirkungen der Infektion zugrunde

Ergebnisse 70
gegangen sind. Drei Tiere zeigten eine leichte Vergrößerung der Milz und der
Lymphknoten und hatten eine B-Zell-Leukämie (positiv für CD19/B220).
Beim Virus 10A1-4070env hatte der Austausch der env-Sequenzen einen
dramatischen Einfluss auf dessen pathogenes Potential. Diese Abschwächung
könnte darin begründet sein, dass das chimäre MuLV nur noch den amphotropen
Rezeptor Pit2 für die Infektion benutzen konnte. Die einzigartige Rezeptornutzung
vom 10A1-MuLV (Pit1 und Pit2) scheint also wichtig für die Transformation von
Stammzellen zu sein. Pit1 wird vor allem im Knochenmark und auf hämato-
poetischen Zellen exprimiert (Kavanaugh et al., 1994), was erklären könnte, warum
das 10A1-MuLV in der Lage war, das Stammzell-Kompartiment zu transformieren.
Obwohl Mo-10A1env völlig andere zelluläre Rezeptoren erkennt als das Mo-MuLV,
scheint diese veränderte Rezeptorerkennung keinen nennenswerten Einfluss auf die
induzierte Leukämieform zu haben. Diese Ergebnisse legen den Schluss nahe, dass
die für die Induktion einer T-Zell-Leukämie verantwortlichen Sequenzen im Mo-MuLV
außerhalb des env-Bereiches liegen müssen.
6.2.2 Charakterisierung von MuLVs mit einem LTR-Austausch
Verschiedene Arbeitsgruppen haben gezeigt, dass die für eine Leukämie
verantwortlichen Bereiche wahrscheinlich in den Enhancer- und Promotersequenzen
der U3-Region des LTR lokalisiert sind. Mo-MuLV induziert T-Zell-Lymphome,
während das Friend-MuLV ausschließlich Erythroleukämien verursacht. Ein Friend-
Virus mit Moloney Enhancer-Elementen induzierte vorwiegend T-Zell-Leukämien,
während das Mo-MuLV nach Einführung der Friend Enhancer-Elemente vorwiegend
Erythroleukämien induzierte (Chatis et al., 1983; Golemis et al., 1989).
Um zu testen, inwieweit der Austausch von Enhancer-Elementen Auswirkungen auf
die Art der von 10A1 und Mo-MuLV induzierten Leukämie hat, wurden zwei
Rekombinanten hergestellt (s. Abb. 22). In das Mo-10A1env Virus, welches
ausschließlich T-Zell-Leukämien verursacht, wurde zusätzlich die 10A1 U3-Region
eingefügt. Das 10A1V-MoLTR entstand durch Insertion der Moloney U3-Region in das
10A1-MuLV.

Ergebnisse 71
gag pol env LTR LTR
env polgag LTR LTR
10A1 ClaI SalI
Mo-MuLV
Mo-10A1V-10A1LTR-MuLV
10A1-MuLV
10A1V-MoLTR-MuLV
Abb. 22: Genomorganisation von Moloney-MuLV, Mo-10A1V-10A1LTR-MuLV, 10A1-MuLV und
10A1V-MoLTR-MuLV. Das chimäre Mo-10A1V-10A1LTR Virus trägt den U3-Bereich des 10A1MuLVs.
Das Virus-Konstrukt 10A1V-MoLTR besitzt die vom Mo-MuLV stammende U3-Region.
6.2.2.1 Phänotyp von Mo-10A1V-10A1LTR infizierten Tieren
Das chimäre Virus Mo-10A1V-10A1LTR induzierte nach einer mittleren Latenzzeit von
124 Tagen in 94 % der infizierten Tiere (n = 17) eine T-Zell-Leukämie (s. Tab 13). Ein
Tier zeigte einen blastischen Phänotyp. Die 16 Lymphome waren ausschließlich
positiv für die T-Zell-Marker. Die Tiere hatten alle normale Hämatokritwerte. Die
Leber war nur bei der Hälfte der Mäuse vergrößert. Im Gegensatz dazu zeigten alle
Tiere eine Splenomegalie mit durchschnittlichen Organgewichten von 1,3 g (± 0,5).
Auch der Thymus und die Lymphknoten waren fast immer vergrößert. Eine Maus
hatte eine schwere Anämie und zeigte sich auch morphologisch so, wie es vom
10A1-induzierten Sca-1+-Phänotyp bekannt ist. Der Tumor war nur positiv für den
Antikörper anti-Sca-1.
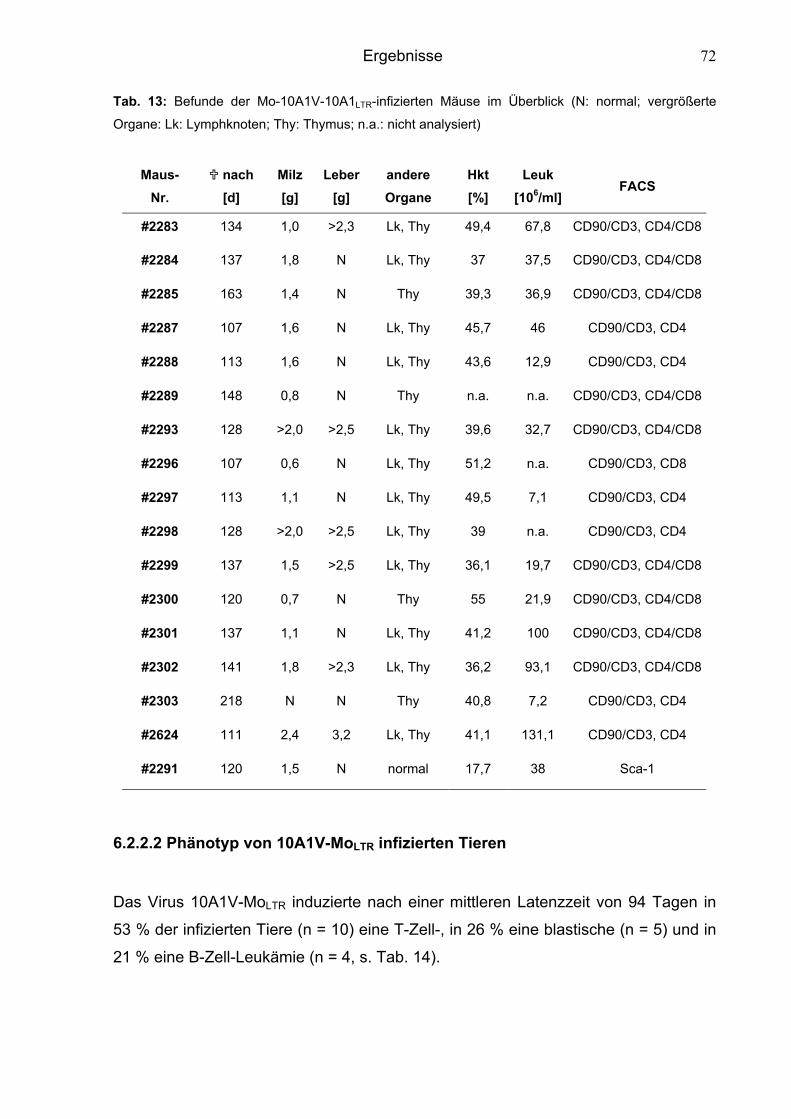
Ergebnisse 72
Tab. 13: Befunde der Mo-10A1V-10A1LTR-infizierten Mäuse im Überblick (N: normal; vergrößerte
Organe: Lk: Lymphknoten; Thy: Thymus; n.a.: nicht analysiert)
Maus-
Nr. nach [d]
Milz [g]
Leber [g]
andere Organe
Hkt [%]
Leuk [106/ml]
FACS
#2283 134 1,0 >2,3 Lk, Thy 49,4 67,8 CD90/CD3, CD4/CD8
#2284 137 1,8 N Lk, Thy 37 37,5 CD90/CD3, CD4/CD8
#2285 163 1,4 N Thy 39,3 36,9 CD90/CD3, CD4/CD8
#2287 107 1,6 N Lk, Thy 45,7 46 CD90/CD3, CD4
#2288 113 1,6 N Lk, Thy 43,6 12,9 CD90/CD3, CD4
#2289 148 0,8 N Thy n.a. n.a. CD90/CD3, CD4/CD8
#2293 128 >2,0 >2,5 Lk, Thy 39,6 32,7 CD90/CD3, CD4/CD8
#2296 107 0,6 N Lk, Thy 51,2 n.a. CD90/CD3, CD8
#2297 113 1,1 N Lk, Thy 49,5 7,1 CD90/CD3, CD4
#2298 128 >2,0 >2,5 Lk, Thy 39 n.a. CD90/CD3, CD4
#2299 137 1,5 >2,5 Lk, Thy 36,1 19,7 CD90/CD3, CD4/CD8
#2300 120 0,7 N Thy 55 21,9 CD90/CD3, CD4/CD8
#2301 137 1,1 N Lk, Thy 41,2 100 CD90/CD3, CD4/CD8
#2302 141 1,8 >2,3 Lk, Thy 36,2 93,1 CD90/CD3, CD4/CD8
#2303 218 N N Thy 40,8 7,2 CD90/CD3, CD4
#2624 111 2,4 3,2 Lk, Thy 41,1 131,1 CD90/CD3, CD4
#2291 120 1,5 N normal 17,7 38 Sca-1
6.2.2.2 Phänotyp von 10A1V-MoLTR infizierten Tieren
Das Virus 10A1V-MoLTR induzierte nach einer mittleren Latenzzeit von 94 Tagen in
53 % der infizierten Tiere (n = 10) eine T-Zell-, in 26 % eine blastische (n = 5) und in
21 % eine B-Zell-Leukämie (n = 4, s. Tab. 14).

Ergebnisse 73
Tab. 14: Befunde von den 10A1V-MoLTR-induzierten Lymphomen im Überblick (N: normal; vergrößerte
Organe: Lk: Lymphknoten; Thy: Thymus; n.a.: nicht analysiert)
Maus-
Nr. nach [d]
Milz [g]
Leber [g]
andere Organe
Hkt [%]
Leuk [106/ml]
FACS
#2537 89 2,4 3,8 Lk 42,6 265 CD90/CD3, CD4
#2539 108 1,5 2,6 Lk, Thy 39 2,6 CD90/CD3, CD4/CD8
#2541 124 1,3 N Lk, Thy 31,2 43,5 CD90/CD3, CD4/CD8
#2543 83 0,6 N Thy n.a. n.a. CD90/CD3, CD4/CD8
#2546 89 1,3 3,9 Lk, Thy 36 62,4 CD90/CD3, CD4/CD8
#2549 108 2,2 3,0 Lk 39,3 99,3 CD90/CD3, CD4
#2550 90 2,3 4,2 Lk, Thy 39 n.a. CD90/CD3, CD4
#2551 94 1,8 2,8 Lk, Thy 43,2 50,4 CD90/CD3, CD8
#2555 130 1,3 N Lk, Thy 46,5 21,3 CD90/CD3, CD4/CD8
#2558 83 3,5 5,1 Lk n.a. n.a. CD90/CD3, CD4
#2542 80 2,6 3,3 normal n.a. n.a. Sca-1, Sca-1/Ter119
#2545 77 1,8 2,5 normal n.a. n.a. Sca-1/ Ter119
#2547 90 2,3 2,9 normal 16,2 8,1 Sca-1/ Ter119
#2553 94 2,2 2,4 normal 18 18,3 Sca-1, Sca-1/Ter119
#2559 104 2,6 3,1 normal n.a. n.a. Sca-1, Sca-1/Ter119
#2538 94 0,6 1,3 normal 43,2 6,6 CD19/B220
#2548 104 1,0 1,8 Lk n.a. n.a. CD19/B220
#2554 103 1,2 2,0 Lk 49,5 n.a. CD19/B220
#2556 117 0,6 1,8 normal 41,4 11,4 CD19/B220
Die Mäuse mit T-Zell-Lymphomen hatten alle eine vergrößerte Milz. Bei einigen
Tieren war die Leber nicht betroffen, bei den anderen war diese stark vergrößert
(Gewicht über 2,2 g). Der Thymus und die Lymphknoten waren fast immer verändert.
Der Hämatokrit war fast immer normal. In FACS-Analysen zeigten sich die T-Zell-
Tumore ausschließlich positiv für die typischen Marker (s. Tab. 14). Der T-Zell+-
Phänotyp stellte sich wie der vom Mo-MuLV induzierte dar: Bei den 10A1V-MoLTR

Ergebnisse 74
infizierten Tieren konnte allerdings eine stärkere Beteiligung der Milz beobachtet
werden.
Alle Mäuse mit einem blastischen Phänotyp hatten eine stark vergrößerte Milz. Die
Leber zeigte immer starke Farbveränderungen (Tumorzellinfiltrate), auch wenn das
Organ nicht vergrößert war. Die anderen Organe waren nicht beteiligt. Die
analysierten Hämatokritwerte waren stark vermindert. Die Tumore zeigten in der
FACS-Analyse eine Sca-1 und eine Sca-1/Ter 119 positive Zellpopulation. Der durch
das Virus 10A1V-MoLTR induzierte Phänotyp entspricht dem vom Wildtyp 10A1-
MuLV.
Die Tiere, die an einer B-Zell-Leukämie erkrankt waren, hatten eine um das Doppelte
vergrößerte Milz. Die Leber und der Thymus waren nicht betroffen. Auch die
Lymphknoten zeigten nur eine geringe Beteiligung. Die Hämatokritwerte waren
normal. Die Tumore waren in der FACS-Analyse doppelt positiv für die B-Zell-Marker
CD19/B220. Es scheint sich auch bei diesen Tumorzellen, um unreife B-Zellen zu
handeln.
Erstaunlicherweise hatte der Verlust der Moloney Enhancer-Elemente keinen
Einfluss auf die Fähigkeit der Rekombinante Mo-10A1V-10A1LTR T-Zell-Leukämien
zu induzieren, so dass es Sequenzen außerhalb des LTR geben muss, die diesen
Verlust ausgleichen können. Diese Hypothese wird durch das 10A1V-MoLTR-Virus
untermauert, da die Einführung der starken Moloney Enhancer-Elemente nur in etwa
50 % der Tiere zu einer T-Zell-Leukämie führte. Das Mo-LTR scheint somit zwar
ausreichend, aber nicht unbedingt notwendig für das Auftreten von T-Zell-
Lymphomen zu sein.
6.2.3 Charakterisierung von MuLVs mit einem Austausch von gag- oder NP-Sequenzen
Da die bisherigen Untersuchungen gezeigt hatten, dass die 10A1 env-Sequenzen in
einem Moloney-Rückgrat bzw. die Moloney LTR-Sequenzen in einem 10A1-Rückgrat
entweder keine Auswirkungen zeigten bzw. nicht ausreichend für die Induktion einer
T-Zell-Leukämie waren, wurden noch weitere chimäre MuLVs hergestellt (s. Abb. 23
und 24). Im stromaufwärts vom strukturellen gag-Gen gelegenen Bereich (Leader)
sind regulatorische Elemente lokalisiert, die für den Entwicklungszyklus eines MuLVs

Ergebnisse 75
sehr wichtig sind, wie das Dimerisierungssignal, die Primer-Bindungsstelle, die
interne Ribosomeneintrittsstelle und das Verpackungssignal. Das erweiterte
Verpackungssignal überlappt mit den gag-Sequenzen. Im Leader-Bereich ist auch
das Initiations-Kodon für das N-terminal verlängerte glykoGag-Protein lokalisiert.
Dieses Protein beeinflusst auf bisher unbekannte Weise die Pathogenität von
murinen Leukämieviren. Um zu testen, inwieweit der Austausch der Leader- bzw.
NP-Sequenzen Auswirkungen auf die Art der vom 10A1-MuLV induzierten Leukämie
hat, wurde die Moloney NP-Region in das 10A1-Virus eingeführt. Das chimäre Virus
wurde als 10A1V-MoNP bezeichnet.
LTR envpolgag LTR
PstI NheI
10A1-MuLV
10A1V-MoNP-MuLV
Abb. 23: Genomorganisation von 10A1-MuLV und 10A1V-MoNP-MuLV. Das Virus Konstrukt 10A1V-
MoNP besitzt die vom Mo-MuLV stammende NP-Region. Für die genaue Klonierungsstrategie der NP-
Konstrukte s. 3.14.3.
6.2.3.1 Phänotyp von 10A1V-MoNP infizierten Tieren
Das chimäre Virus 10A1V-MoNP induzierte nach einer mittleren Latenzzeit von 102
Tagen in 74 % der infizierten Tiere (17/23) eine T-Zell- und in 22 % eine blastische
(5/23) Leukämie. Nur eine Maus hatte eine B-Zell-Leukämie (s. Tab 15).
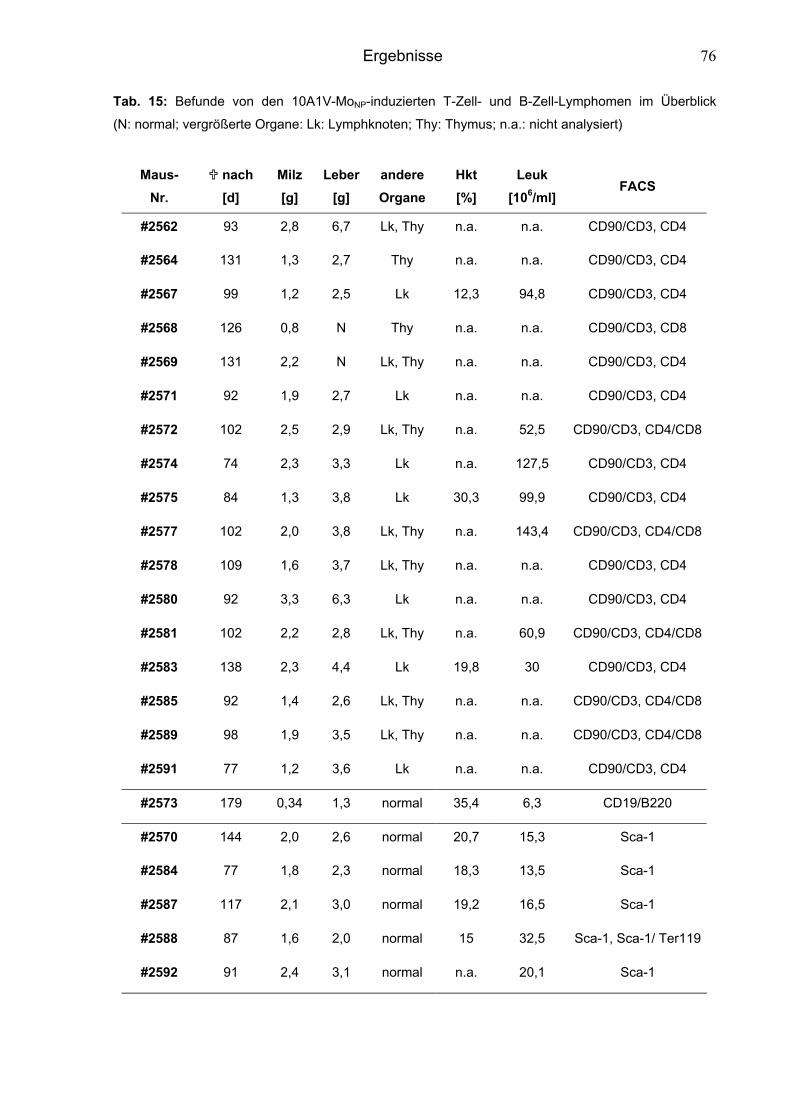
Ergebnisse 76
Tab. 15: Befunde von den 10A1V-MoNP-induzierten T-Zell- und B-Zell-Lymphomen im Überblick
(N: normal; vergrößerte Organe: Lk: Lymphknoten; Thy: Thymus; n.a.: nicht analysiert)
Maus-
Nr. nach [d]
Milz [g]
Leber [g]
andere Organe
Hkt [%]
Leuk [106/ml]
FACS
#2562 93 2,8 6,7 Lk, Thy n.a. n.a. CD90/CD3, CD4
#2564 131 1,3 2,7 Thy n.a. n.a. CD90/CD3, CD4
#2567 99 1,2 2,5 Lk 12,3 94,8 CD90/CD3, CD4
#2568 126 0,8 N Thy n.a. n.a. CD90/CD3, CD8
#2569 131 2,2 N Lk, Thy n.a. n.a. CD90/CD3, CD4
#2571 92 1,9 2,7 Lk n.a. n.a. CD90/CD3, CD4
#2572 102 2,5 2,9 Lk, Thy n.a. 52,5 CD90/CD3, CD4/CD8
#2574 74 2,3 3,3 Lk n.a. 127,5 CD90/CD3, CD4
#2575 84 1,3 3,8 Lk 30,3 99,9 CD90/CD3, CD4
#2577 102 2,0 3,8 Lk, Thy n.a. 143,4 CD90/CD3, CD4/CD8
#2578 109 1,6 3,7 Lk, Thy n.a. n.a. CD90/CD3, CD4
#2580 92 3,3 6,3 Lk n.a. n.a. CD90/CD3, CD4
#2581 102 2,2 2,8 Lk, Thy n.a. 60,9 CD90/CD3, CD4/CD8
#2583 138 2,3 4,4 Lk 19,8 30 CD90/CD3, CD4
#2585 92 1,4 2,6 Lk, Thy n.a. n.a. CD90/CD3, CD4/CD8
#2589 98 1,9 3,5 Lk, Thy n.a. n.a. CD90/CD3, CD4/CD8
#2591 77 1,2 3,6 Lk n.a. n.a. CD90/CD3, CD4
#2573 179 0,34 1,3 normal 35,4 6,3 CD19/B220
#2570 144 2,0 2,6 normal 20,7 15,3 Sca-1
#2584 77 1,8 2,3 normal 18,3 13,5 Sca-1
#2587 117 2,1 3,0 normal 19,2 16,5 Sca-1
#2588 87 1,6 2,0 normal 15 32,5 Sca-1, Sca-1/ Ter119
#2592 91 2,4 3,1 normal n.a. 20,1 Sca-1

Ergebnisse 77
Mäuse mit einer T-Zell-Leukämie hatten eine vergrößerte Leber. Das Milzgewicht war
um ca. das fünffache gegenüber normal erhöht. Der Thymus und die Lymphknoten
waren fast immer vergrößert. Die wenigen analysierten Blutproben zeigten einen
verminderten Hämatokrit. Die Tumore waren ausschließlich positiv für die typischen
T-Zell-Marker (s. Tab. 15). Die T-Zell+-Tumore entsprechen dem Mo-MuLV
induzierten Phänotyp. Die Maus, mit dem B-Zell-Phänotyp, hatte keine veränderten
Organe. Der Hämatokrit war leicht vermindert. Der Tumor war doppelt positiv für die
Antikörper anti-CD19 und anti-B220.
Mäuse mit einer blastischen Leukämie hatten alle eine stark vergrößerte Milz. Die
Leber zeigte immer starke Farbveränderungen (Tumorzellinfiltrate), auch wenn das
Organ nicht vergrößert war. Die anderen Organe zeigten keine Beteiligung. Die
analysierten Hämatokritwerte waren stark vermindert. Die Tumore zeigten in der
FACS-Analyse verschiedene Zellpopulationen, die entweder nur Sca-1 positiv oder
doppelt positiv für Sca-1/Ter 119 gewesen sind. Der hier beobachtete Phänotyp
entspricht dem vom Wildtyp 10A1-MuLV-induzierten blastischen Tumoren.
6.2.3.2 Phänotyp von 10A1V-Mogag infizierten Tieren
Um zu testen ob der Austausch der gag-Sequenzen ebenfalls Auswirkungen auf die
Art der vom 10A1-MuLV induzierten Leukämien hat, wurden Moloney gag-
Sequenzen in das 10A1-Virus eingeführt (s. Abb. 24). Das Konstrukt wurde als
10A1V-Mogag bezeichnet. LTR envpolgag LTR
10A1-MuLV
BsrGI Mo DraIII
10A1V-Mogag-MuLV
Abb. 24: Genomorganisation von 10A1-MuLV und 10A1V-Mogag-MuLV. Die Moloney gag-Sequenzen
wurden über die Schnittstellen BsrGI/DraIII in das 10A1-Rückgrat eingefügt.
Die Rekombinante 10A1V-Mogag induzierte nach einer mittleren Latenzzeit von 169
Tagen in 42 % der infizierten Tiere eine blastische (n = 8), in 37 % eine B-Zell-
(n = 7) und in 21 % eine T-Zell-Leukämie (n = 4). Die Befunde der analysierten Tiere
sind ausführlich im Appendix dargestellt (s. Tab 19).

Ergebnisse 78
Mäuse, die an einer blastischen Leukämie erkrankt waren, hatten die schon
beschriebenen typischen Merkmale: Hepatosplenomegalie und starke Anämie. Die
analysierten Tumore zeigten in der FACS-Analyse entweder nur eine Sca-1 positive
oder Sca-1/Ter 119 positive Zellpopulation. Einige Tiere wurden nicht mittels FACS-
Analysen getestet, da ihr morphologischer Phänotyp eindeutig gewesen ist. Der
Phänotyp war analog zu der vom 10A1-MuLV induzierten blastischen Leukämie.
Die meisten Tiere mit einer B-Zell-Leukämie hatten eine unauffällige Leber und eine
nur leicht vergrößerte Milz. Der Thymus und die Lymphknoten waren fast nie
beteiligt. Die Hämatokritwerte schwankten zwischen normal und sehr niedrig. Alle
Tumore waren in der FACS-Analyse doppelt positiv für die Antikörper anti-CD19 und
anti-B220. Um den Differenzierungsgrad der B-Zellen zu bestimmen, wurden die
Antikörper CD79β und IgM verwendet. Der Antikörper CD79β erkennt ein Protein,
das auf der Oberfläche mit dem B-Zell-Rezeptor ko-exprimiert wird, so dass der
sogenannte B-Zell-Rezeptor Komplex entsteht. CD79β wird sowohl auf unreifen als
auch reifen B-Zellen exprimiert. Der Marker IgM erkennt hingegen nur reife B-Zellen.
Die vollständige Auflistung der routinemäßig getesteten Antikörper ist im
Methodenteil aufgeführt (s. 5.10.1). Drei Tumore zeigten zusätzlich noch eine
Subpopulation von Zellen, die auch die reifen B-Zell-Marker CD79β und IgM
exprimierten, so dass noch reife B-Zellen vorhanden waren. Die Tumore zeigten
damit einen etwas anderen Phänotyp als die vom 10A1-Wildtyp induzierten B-Zell-
Lymphome.
Alle Mäuse mit einer T-Zell-Leukämie hatte Lymphome, die sich wie die vom Mo-
MuLV induzierten Tumore darstellten.
6.2.3.3 Phänotyp von Mo-10A1V-10A1NP infizierten Tieren
Um zu testen, ob der Austausch von Leader-Sequenzen ebenfalls Auswirkungen auf
die Art der vom Mo-MuLV induzierten Leukämien hat, wurden in das Mo-10A1env-
Virus, welches fast ausschließlich T-Zell-Leukämien induziert, die 10A1 NP-Region
eingefügt (s. Abb. 25).

Ergebnisse 79
Pr65gagPr75gag
TR LTR CA NC
10A1SalIΨ MA
LClaI
Mo-101env-MuLV
NheI PstI 10A1 ClaI SalI Mo-10A1V-10A1NP-MuLV
Abb. 25: Genomorganisation von Mo-10A1env-MuLV und Mo-10A1V-10A1NP-MuLV. Das chimäre Mo-
10A1V-10A1NP Virus trägt den NP-Bereich des 10A1MuLVs. Für die genaue Klonierungsstrategie der
NP-Konstrukte s. 3.14.3. MA, Matrix-; CA, Kapsid-; NC, Nukleokapsidprotein; Pr65gag, strukturelles
Gag-Polyprotein; Pr75gag, nicht-strukturelles Gag-Polyprotein; Ψ, Verpackungssignal
Das Virus Mo-10A1V-10A1NP induzierte nach einer mittleren Latenzzeit von 106
Tagen in 88 % der infizierten Tiere (n = 17) eine T-Zell-Leukämie. Zwei Mäuse
zeigten einen anderen Phänotyp, ein Tier hatte eine B-Zell-Leukämie, die andere
eine blastische Leukämie. Die Befunde der analysierten Tiere sind ausführlich im
Appendix dargestellt (s. Tab 20).
Die Leber von Mäusen mit einer T-Zell-Leukämie war unterschiedlich stark betroffen.
Das durchschnittliche Milzgewicht lag bei 1,6 g (± 0,7), was dem fünffachen des
normalen Gewichts entspricht. Auch beim Thymus variierte das Erscheinungsbild von
unauffällig bis sehr stark vergrößert. Die Lymphknoten zeigten nur bei 50 % der Tiere
eine Veränderung. Die Hämatokritwerte waren nur leicht vermindert gegenüber den
Normalwerten. Der B-Zell-Phänotyp wies keine Veränderungen der Leber oder der
Lymphknoten auf, die Milz und der Thymus waren allerdings leicht vergrößert. Der
Hämatokrit war ebenfalls normal. Der blastische Phänotyp stellte sich morphologisch
wie schon beschrieben dar, mit Hepatosplenomegalie, keine Beteiligung anderer
Organe und stark vermindertem Hämatokrit. Die T-Zell-Lymphome waren in den
FACS-Analysen ausschließlich positiv für die typischen T-Zell-Marker (CD4 und CD8
wurden nicht analysiert). Ein Lymphom exprimierte nur die B-Zell-Marker, während
das blastische Lymphom positiv für den Antikörper anti-Sca-1 gewesen ist.
Erstaunlicherweise war das Virus 10A1V-MoNP in der Lage in 74 % der Mäuse eine
T-Zell-Leukämie zu induzieren. D.h. der Moloney NP-Bereich scheint einen stärkeren
Einfluss auf die Induktion einer T-Zell-Leukämie zu haben als das Mo-LTR (74 %
gegenüber 53 %). Die Auswirkungen des 10A1 NP-Bereichs im Moloney-MuLV

Ergebnisse 80
waren nicht so deutlich, da sie durch die Gegenwart des Mo-LTR und der Moloney
gag-Sequenzen überdeckt wurden. Die beiden Chimären Mo-10A1V-10A1 und Mo-
10A1V-10A1NP
LTR induzierten in fast gleichem Maße T-Zell-Lymphome in den
infizierten Mäusen, 88 % gegenüber 94%. Interessanterweise scheint sich die
Gegenwart von verschiedenen Moloney-Sequenzen akkumulativ auf die Fähigkeit, T-
Zell-Lymphome zu induzieren, auszuwirken. Das Mo-gag induzierte in 21 %, das Mo-
LTR in 53 % der Tiere eine T-Zell-Leukämie. Das Virus Mo-10A1V-10A1NP trägt
beide Bereiche und induzierte in 88 % der Mäuse ein T-Zell-Lymphom. Im
Gegensatz dazu war der Mo-NP Bereich allein in der Lage in 74 % der Tiere ein T-
Zell-Lymphom zu induzieren. Es scheint, als ob mehrere Faktoren im Moloney-
Genom an der Induktion einer T-Zell-Leukämie beteiligt sind, was auch für die
Induktion von blastischen Lymphomen durch das 10A1-MuLV zutrifft.
6.3 Aktivierung von Onkogenen 6.3.1 Klonalität der Tumore
In einem klonalen Tumor haben bestimmte retrovirale Integrationsereignisse eine
infizierte Zelle soweit entartet, dass diese einen Selektionsvorteil hat und es zum
raschen Auswachsen eines Lymphoms kommt. Die Transformation findet durch die
Insertion der Proviren in für die Zelle funktionell wichtigen Genabschnitten statt, was
dieser einen Wachstumsvorteil verschafft. Die hierdurch bedingte virale Aktivierung
oder Inaktivierung von Wirtsgenen führt letztendlich zur Ausbildung eines malignen
Tumors. Dieser besteht dann aus den sich rasant vermehrenden, identischen
Nachkommen einer transformierten Zelle. Oligo- bzw. polyklonale Tumorgewebe
bestehen aus mehreren parallel ausgewachsenen Einzeltumoren. Es haben wichtige
Integrationsereignisse in verschiedenen Zellen stattgefunden, die dann langsam zu
einem gemischten Lymphom ausgewachsen sind.
Die 10A1-infizierten Mäuse erkrankten in zwei Schüben. Um zu testen in wie weit
sich die frühen und späten Lymphome in ihrem Klonalitätsgrad unterscheiden,
wurden die Tumor-DNAs mit dem Enzym BglII gespalten, das außerhalb des von der
10A1-env Sonde detektierbaren Bereichs des Provirus schneidet. Abhängig von der
Lage der zweiten Schnittstelle im Wirtsgenom, und somit vom Integrationsort, werden

Ergebnisse 81
Banden unterschiedlicher Größe erzeugt. Als Negativkontrollen wurden die DNAs
von nicht infizierten SC-1 Zellen (s. Abb. 26, Spur 9) bzw. einer nicht infizierten
NIH/Ola-Maus (s. Abb. 26, Spur 11) verwendet. Als Positivkontrolle dienten mit 10A1-
MuLV infizierte SC-1 Zellen (s. Abb. 26, Spur 10). Die Schärfe der Banden und deren
Intensität lassen Rückschlüsse auf die Häufigkeit bestimmter Integrationsereignisse
zu, d.h. je intensiver eine Bande ist, desto größer ist die Wahrscheinlichkeit, dass
diese Integrationen in allen Tumorzellen stattgefunden haben.
2,0 kb
6,6 kb
4,4 kb
2,3 kb
9,4 kb
8 λ
23 kb
9 10 11 12 13 14 λ λ 15 16 λ 2 1 3 4 5 76
4,4 kb
2,3 kb 2,0 kb
23 kb
9,4 kb
6,6 kb
Spur Sca-1+-Mäuse Spur Sca-1+-Mäuse
λ λ -DNA HindIII gespalten λ λ -DNA HindIII gespalten
1 #1824, Milz 9 SC-1 Zellen
2 #2137, Milz 10 10A1 infizierte SC-1 Zellen
3 #2363, Milz 11 NIH/Ola WT, Milz
4 #2369, Milz 12 #2457, Milz
5 #2375, Milz 13 #2461, Milz
6 #2438, Milz 14 #2463, Milz
7 #2443, Milz 15 #2464, Milz
8 #2456, Milz 16 #2465, Milz
Abb. 26: BglII gespaltene Tumor-DNAs aus 10A1-induzierten Sca-1+-Mäusen, hybridisiert mit der
10A1-env spezifischen Sonde. Spur 9 enthält sehr wenig DNA.
Die 10A1-induzierten Sca-1+-Tumore (Spuren 1-8, 12-16) hatten eine mittlere
Latenzzeit von 132 Tagen und zeigten klar abgrenzbare Banden ähnlicher Intensität
(s. Abb. 26). Von den Tieren #2442 und #2453 konnte keine Tumor-DNA gewonnen
werden, deshalb wurden sie nicht weiter charakterisiert. Es traten ca. ein bis drei
Integrationen pro Tumor auf, so dass diese als klonal eingestuft werden können. Im
Gegensatz dazu zeigt die DNA der 10A1-infizierten SC-1 Zellen keine scharf

Ergebnisse 82
umrissenen Banden, sondern nur einen diffusen Schmier, was deutlich macht, dass
diese Massenkultur der Zellen polyklonal ist. Die Banden, die in den Spuren 11
(uninfizierte Maus) und Spur 9 (uninfizierte SC-1 Zellen) zu erkennen sind,
entsprechen endogenen Sequenzen, die auch von der 10A1-env Sonde erkannt
wurden (s. Abb. 26).
2,0 kb2,3 kb
9,4 kb
6,6 kb
4,4 kb
23 kb8 9 10 11 12 13 14λλ5 6 72 3 4 λ 1
23 kb
6,6 kb
4,4 kb
2,0 kb
9,4 kb
2,3 kb
Spur B-Zell+-Mäuse Spur B-Zell+-Mäuse
λ λ -DNA HindIII gespalten λ λ -DNA HindIII gespalten
1 #1821, Milz 8 #2432, Milz
2 #1822, Milz 9 #2446, Milz
3 #1823, Milz 10 #2450, Milz
4 #2129, Milz 11 #2454, Milz
5 #2139, Milz 12 SC-1 Zellen
6 #2367, Milz 13 10A1 infizierte SC-1 Zellen
7 NIH/Ola WT, Milz 14 NIH/Ola WT, Milz
Abb. 27: BglII gespaltene Tumor-DNAs aus 10A1-induzierten B-Zell+-Mäusen, hybridisiert mit der
10A1-env spezifischen Sonde. Spur 2 enthält wenig DNA.
Die Banden, die in den Spuren 7 und 14 (uninfizierte Maus) und Spur 12 (uninfizierte
SC-1 Zellen) zu erkennen sind, entsprechen endogenen Sequenzen, die auch von
der 10A1-env Sonde erkannt wurden (s. Abb. 27). Von den Tieren #1826 und #2135
gab es keine Tumor-DNA, deshalb wurden diese nachfolgend nicht weiter
charakterisiert. Der B-Zell+-Phänotyp stellte sich auch in dieser Analyse sehr
uneinheitlich dar. Die meisten Tumore zeigten neben den scharfen Banden auch
einen starken Hintergrund (Spuren 1-6, 8-11), so dass sie als vorwiegend polyklonal

Ergebnisse 83
eingestuft werden können. Diese Einstufung spiegelt sich auch in der relativ langen
Latenzzeit von 164 Tagen wieder (s. Abb. 27).
Der homogene Phänotyp der Sca-1+-Mäuse und die kurze Latenzzeit sind also auf
einige wenige Provirusintegrationen in den klonalen Tumoren zurückzuführen. Der
sehr heterogene Phänotyp der B-Zell+-Mäuse und die verlängerte Latenzzeit lassen
sich auf das langsame Auswachsen unterschiedlicher, unabhängig transformierter
Zellpopulationen zurückführen.
6.3.2 Überprüfung der Tumore nach aktivierten Onkogenen
Es wurden Gene getestet, die in Erythroleukämien, und T- und B-Zell-Lymphomen
häufig durch die Insertion von Proviren aktiviert werden, darunter der
Transkriptionsfaktor Spi-1 (auch als PU.1 bezeichnet), der wie Fli-1 zur ets-Familie
gehört, sowie die Proto-Onkogene Pim-1, c-myc und N-myc (Jonkers and Berns,
1996).
In SFFV (Spleen Focus Forming Virus) induzierte Erythroleukämien weisen 95 % der
Tumore eine Insertion im stromaufwärts vom Spi-1 Gen gelegenen Bereich auf
(Moreau-Gachelin et al., 1989). In allen 10A1-induzierten blastischen Tumoren wurde
eine Insertion in den Fli-1 Lokus nachgewiesen (Ott et al., 1994). Eine Aktivierung
des Pim-1 Gens wurde in 50 % der T-Zell-Leukämien gefunden (Cuypers et al.,
1984). Der Transkriptionsfaktor c-myc ist sowohl in T-Zell-, als auch in B-Zell-
Lymphomen aktiviert, was eine wichtige Rolle in der Lymphomgenese impliziert
(Corcoran et al., 1984; Stanton et al., 1983). In 35-50 % der MuLV-induzierten T-Zell-
Lymphome ist eine provirale Integration in das N-myc Gen zu beobachten gewesen
(van Lohuizen et al., 1989).
Um zu überprüfen, ob eine Integration im Fli-1 Gen vorlag, wurden die Sca-1+ Tumor-
DNAs mit dem Enzym BamHI gespalten und mit der genomischen Fli-1-Sonde
hybridisiert. Die Sonde erkennt den Bereich, der das alternative Exon 1 und das
Intron 1 umfasst. Diese Region soll wie von Ott et al. gezeigt, alle proviralen
Integrationsstellen beinhalten. Als Kontrolle wurde die DNA einer nicht infizierten
NIH/Ola-Maus (s. Abb. 28, Spuren 10 und 15) verwendet.

Ergebnisse 84
λ9 10
Wildtyp Fli-1 Allel
Fli-1 Allel mit Insertion
876
6,6 kb
4,4 kb
2,3 kb
2,0 kb
λ 2 1 3 4 5
14 15
Wildtyp Fli-1 Allel
Fli-1 Allel mit Insertion
6,6 kb
4,4 kb
2,3 kb
2,0 kb
λ 11 12 13
Spur Sca-1+-Mäuse Spur Sca-1+-Mäuse
λ λ -DNA HindIII gespalten 8 #2456, Milz
1 #1824, Milz 9 #2457, Milz
2 #2137, Milz 10 NIH/Ola WT, Milz
3 #2363, Milz 11 #2461, Milz
4 #2369, Milz 12 #2463, Milz
5 #2375, Milz 13 #2464, Milz
6 #2438, Milz 14 #2465, Milz
7 #2443, Milz 15 NIH/Ola WT, Milz
Abb. 28: BamHI gespaltene Tumor-DNAs aus 10A1-induzierten Sca-1+-Mäusen, hybridisiert mit der
Fli-1 spezifischen Sonde

Ergebnisse 85
Wie die Abbildung 28 zeigt, weisen alle Sca-1+-Tumore eine Provirusintegration in
einem der beiden Fli-1 Allele auf (Spuren 1-9, 11-14). Die Insertion scheint darüber
hinaus, wie oben beschrieben, in einem bestimmten Bereich stattgefunden zu haben,
da alle Tumore bis auf einen (#2464, Spur 13) eine Insertionsbande der gleichen
Größe (4,4 kb) zeigen.
Die Tumore der beiden 10A1-induzierten Phänotypen wurden mit Hilfe von Southern
Blot-Analysen auf häufig vorkommende provirale Integrationsstellen untersucht.
Die Tumor-DNAs wurden mit den Enzymen EcoRV bzw. EcoRI gespalten. Die
Hybridisierung erfolgte mit den spezifischen Sonden für Spi-1/PU.1, c-myc, N-myc
und Pim-1, die alle den jeweiligen Bereich erkennen, in dem die meisten Provirus-
insertionen vorkommen.
Tab. 16: Zusammenfassung der Ergebnisse des Onkogenscreenings für die beiden 10A1-induzierten
Phänotypen (n.a. = nicht analysiert)
Sonde Sca-1+-Tumore B-Zell+-Tumore detektierter Bereich
Fli-1 13/13 1/10 3,4 kb inkl. Ex1a Ex1b In1
Spi-1 0/13 n.a. 3,6-18 kb inkl. 5’-UTR
N-myc n.a. 0/10 10-23 kb inkl. Ex1
c-myc 0/13 1/10 22 kb inkl. Ex2 In2 Ex3
Pim-1 2/13 1/10 11-22 kb inkl. Ex5 In5 Ex6
Alle Mäuse, die eine blastische Leukämie hatten, zeigten eine Integration in das Fli-1
Gen (s. Tab 16). In keinem der analysierten Sca-1+-Tumore konnte eine provirale
Integration in die Gene, die für Spi-1/PU.1 oder C-myc kodieren, festgestellt werden.
Zwei Tumore waren positiv für eine Integration im Pim-1 Gen, allerdings weist die
Insertionsbande im Vergleich zum Wildtyp-Allel eine geringere Intensität auf, so dass
wahrscheinlich nicht alle Tumorzellen eine Integration in diesem Lokus hatten (nicht
dargestellt). Einer der B-Zell+-Tumore hatte eine Integration in einem der Fli-1 Allele,
wobei die Bande wie in den Sca-1+ Tumor-DNAs, eine Größe von 4,4 kb zeigte.
Keiner der analysierten Tumore zeigte eine Aktivierung des N-myc Gens. Eine Maus
hatte jeweils eine Insertion in den Genen c-myc Pim-1, eine Kombination, die häufig
in T-Zell- und B-Zell-Lymphomen vorkommt (Cuypers et al., 1984).

Ergebnisse 86
6.3.3 Untersuchung der von chimären MuLVs induzierten blastischen Lymphome auf Insertionen im Fli-1-Gen
Virus-Konstrukte, die eine blastische Leukämie induzieren konnten, wurden ebenfalls
auf Integrationen in dem Fli-1 Gen-Lokus überprüft. Zehn 10A1V-MoLTR induzierte
Tumore, die einen der drei beschriebenen Phänotypen hatten, wurden analysiert. Die
getesteten Sca-1+-Tumore (4/4) zeigten eine Provirusinsertion im Fli-1 Gen, wobei
diese in der gleichen Region stattgefunden hatten, wie dies schon beim 10A1-MuLV
beschrieben wurde (Größe der rekombinierten Bande 4,4,kb). Weder die
analysierten T-Zell-Lymphome (0/6) noch der untersuchte B-Zell-Tumor hatten eine
provirale Insertion in diesem Gen-Lokus.
Neun 10A1V-MoNP induzierte Tumore, die entweder einen T-Zell- oder einen
blastischen Phänotyp hatten, wurden ebenfalls auf Integrationen in dem Fli-1 Gen-
Lokus überprüft. In den Sca-1+-Tumoren (3/3) konnte eine Provirusinsertion im Fli-1
Gen detektiert werden. Die Insertionsbande hatte dabei eine Größe von 4,4 kb, wie
es auch beim 10A1-MuLV beobachtet worden ist. Von den getesteten T-Zell-
Lymphomen zeigte keiner der Tumore (0/6) eine Fli-1 Integration.
Es wurden acht 10A1V-Mogag induzierte Sca-1+-Tumore auf Integrationen im Fli-1
Gen überprüft. Alle Tumore hatten eine Provirusintegration im Fli-1 Gen-Lokus (8/8).
Erstaunlicherweise scheinen die Integrationen aber in 7/8 Tumoren in einer anderen
Region stattgefunden zu haben, da hier eine Insertionsbande von ca. 6,6 kb
detektiert werden konnte. Nur einer der Tumore zeigte die typische Insertionsbande
von 4,4 kb.
6.4 Vorkommen von MCF-Viren in MuLV infizierten Mäusen
Replikationskompetente MCF-Viren entstehen durch Rekombinationsereignisse
zwischen dem exogenen (inokulierten) MuLV und endogenen polytropen Sequenzen
in vivo. Diese unvollständigen endogenen Viren liegen als vielfache Kopien im
gesamten Mausgenom vor. Die Rekombinationen finden meistens in dem Bereich
zwischen den 3’-pol und den 3’-env Sequenzen statt (Rekombinante Typ I). Darüber
hinaus können zusätzlich Rekombinationen zwischen den 3’-LTR Regionen
stattfinden, die allerdings weitaus seltener auftreten (Rekombinante Typ II). Die neu

Ergebnisse 87
entstandenen Rekombinanten vom Typ I weisen chimäre env-Sequenzen auf, bei
denen der 5’ Bereich von den polytropen endogenen Viren und der 3’ Bereich vom
exogenen MuLV stammt. Rekombinanten vom Typ II weisen neben dem
rekombinierten env-Bereich auch ein 3’-LTR auf, das von den endogenen Viren
stammt (Stoye and Coffin, 1987). Es wird postuliert, dass die Entstehung von MCF-
Viren ein kritischer Schritt für die Induktion einer T-Zell-Leukämie ist (Brightman et
al., 1991). Es wurde bereits gezeigt, dass bei einer Mo-MuLV Infektion eine große
Anzahl an Rekombinationsereignissen in vivo stattfinden (Lavignon and Evans,
1996).
Ob diese Rekombinantenbildung einen entscheidenden Einfluss auf die Art der
induzierten Leukämieform oder auf den Krankheitsverlauf haben kann, wurde in
dieser Arbeit getestet. Der Assay macht sich die Tatsache zunutze, dass bestimmte
Restriktionsschnittstellen im pol/env-Bereich verschiedener MuLVs unterschiedlich
verteilt sind bzw. gar nicht vorkommen (Belli et al., 1995). In polytropen env-
Sequenzen kommt zwar eine BamHI Schnittstelle vor, es fehlen aber die
Schnittstellen für die Enzyme HindIII und ClaI. Das ökotrope-env weist dagegen in
diesem Bereich eine BamHI und eine ClaI Schnittstelle auf, während die 10A1
pol/env-Region zwei HindIII und eine ClaI Schnittstelle besitzt. Da sich die Herkunft
des NP-Bereichs entscheidend auf die Induktion einer T-Zell-Leukämie auswirkte,
wurden in diesem Assay die Viren Mo-10A1V-10A1NP und 10A1V-MoNP auf ihr
MCFV-Generierungspotential getestet. Da aber postuliert wird, dass auch die
Herkunft der gag-Sequenzen einen Einfluss auf die Entstehung von MCF-Viren hat,
wurden zusätzlich die MuLVs Moloney und 10A1 untersucht. Dazu wurden die
Tumor-DNAs aus den MuLV infizierten Mäusen mit den Enzymen gespalten, deren
Schnittstellen im jeweiligen pol/env-Bereich vorkommen, anschließend
gelelektrophoretisch aufgetrennt, mittels Southern Blotting auf einer Membran
immobilisiert und anschließend mit den entsprechenden env-Sonden hybridisiert.

Ergebnisse 88
BamHI
env3‘-pol
ClaIBamHI
env3‘-pol
1,1 kb
Mo-env Sonde
2 kb
>2 kb
polytropes-env
Rekombination Typ II
ökotropes-env
Rekombination Typ I
Integration Wildtyp-Virus
Abb. 29: Schematische Darstellung der polytropen (oben) und der ökotropen pol/env-Sequenzen
(unten) und deren Schnittstellenverteilung. X gibt die Bereiche an, zwischen denen Rekombinationen
stattfinden können.
Die Wildtyp-Integration eines Virus mit ökotropen-env ergibt eine Virusbande bei 1,1
kb. Bei einer Typ I bzw. Typ II Rekombination treten zusätzliche Banden auf, die aber
größer als die Wildtyp-Virusbande sind. Durch Rekombinationen vom Typ I fällt die
BamHI (X), bei Typ II Rekombinationen auch zusätzlich die ClaI Schnittstelle weg
(X). Je nach Ereignis, Integration oder Rekombination, treten verschiedene
Fragmentgrößen auf (s. Abb. 29).

Ergebnisse 89
HindIII ClaI
3‘-pol
2,7 kb
10A1-env Sonde
1,9 kb
HindIII
env
0,8 kb
10A1-env
Integration Wildtyp-Virus
Rekombination Typ I Rekombination Typ II
>2,7 kb
BamHI
polytropes-env 3‘-pol env
BamHI ClaI
env
0,7 kb1,2 kb
10A1-env Sonde
1,9 kb
BamHI
3‘-pol
>1,9 kb
10A1-env
Integration Wildtyp-Virus
Rekombination Typ I Rekombination Typ II
Abb. 30: Schematische Darstellung der 10A1 (oben und unten) und der polytropen pol/env-
Sequenzen (Mitte) und die Verteilung der entsprechenden Restriktionsenzymschnittstellen. X gibt die
Bereiche an, zwischen denen Rekombinationen stattfinden können. Oben sind die resultierenden
Bande angegeben, wenn die 10A1 Tumor-DNAs mit HindIII und ClaI gespalten wurden. Unten sind
die resultierenden Banden angegeben, wenn die Enzymkombination BamHI und ClaI verwendet
wurde.
Stammen die env-Sequenzen von 10A1, können nach einer Integration jeweils zwei
Wildtyp-Virusbanden detektiert werden (ca. 0,8 und 1,9 kb). Durch ein
Rekombinationsereignis verschwinden die internen Schnittstellen und die
resultierenden Fragmente sind größer. Durch Rekombinationen vom Typ I fällt die

Ergebnisse 90
BamHI bzw. HindIII (X), bei Typ II Rekombinationen auch zusätzlich die ClaI
Schnittstelle weg (X, s. Abb. 30).
6.4.1 Erhöhte MCFV-Frequenz in Mo-MuLV induzierten Tumoren
Die DNA aus fünf Mo-MuLV induzierten Tumoren wurde mit den Enzymen
BamHI/ClaI gespalten und mit der Mo-env Sonde hybridisiert (s. Abb. 31). Die
infizierten Tiere gehörten verschiedenen Mausstämmen an, NIH/Ola und C57Bl.
Deshalb wurden als Kontrollen sowohl die DNA einer uninfizierten NIH/Ola-Maus
(Spur 1), als auch die einer C57Bl-Maus (Spur 4) verwendet. Bei dem Tier C9
handelte es sich um eine C57Bl-Maus, die anderen vier Tiere (#2207-2346) gehörten
zum NIH/Ola-Stamm.
0,5 kb
9,4 kb
4,4 kb
2,0 kb
6,6 kb
2,3 kb
WT Moloney
R Typ II
R Typ I
23 kb 2 4 7 6 1 3 5 λ λ
Spur Mo-MuLV inf.-Mäuse
λ λ –DNA HindIII
1 NIH/Ola WT, Milz
2 #2346, Thymus
3 #2353, Thymus
4 C57Bl WT, Milz
5 C9, TLN
6 #2205, TLN
7 #2207, TLN
Abb. 31: Fünf Mo-MuLV induzierte Tumor-DNAs wurden mit der Mo-env spezifischen Sonde auf
interne Rekombinationsereignisse getestet..
Wenn man nun die Bandenmuster der Kontroll-DNAs mit denen der Tumor-DNAs
vergleicht, fallen drei Dinge ins Auge (s. Abb. 31). Erstens zeigen alle DNAs,
unabhängig von ihrer Herkunft, oberhalb von ca. 2,7 kb ein nahezu einheitliches
Bandenmuster, was dafür spricht, dass die Mo-env Sonde auch mit endogenen
Sequenzen hybridisiert. Darüber hinaus sind die endogenen Sequenzen innerhalb
verschiedener Mausstämme relativ einheitlich verteilt. Zweitens zeigen die Tumor-
DNAs alle ein 1,1 kb Fragment, welches das integrierte Wildtyp Moloney-Virus

Ergebnisse 91
repräsentiert und in den Kontrollen nicht vorkommt. Drittens stellen die Banden in
dem Bereich zwischen der Virusbande und den endogenen Sequenzen die
verschiedenen Rekombinationsereignisse dar. Dabei scheint es sich bei den
Rekombinationen sowohl um Typ I als auch um Typ II Rekombinanten zu handeln.
Die Typ I Rekombinationen werden durch eine sehr intensive Bande auf Höhe von
ca. 2,0 kb repräsentiert. Die darüber liegenden Banden weisen auf Typ II
Rekombinanten hin, da durch den Wegfall der ClaI Schnittstelle ein größeres
Fragment von der Sonde erkannt wurde. Im Bereich der endogenen Sequenzen sind
nur bei Maus #2205 (Spur 6) Unterschiede zu erkennen. Bei einem Vergleich der
Bandenintensitäten zwischen Wildtyp und rekombinierten Virus ist auffallend, dass
rekombinierte Viren in etwa ebenso häufig wie Wildtyp-Viren auftreten. Setzt man die
Bandenintensität der unrekombinierten Virusbande (1,1 kb) gleich 1, so liegt das
Verhältnis von unrekombinierten zu rekombinierten Viren in den Tumoren zwischen
eins und drei, d.h. auf eine Mo-MuLV Integration kommen 1-3 MCF-Viren.
6.4.2 Niedrige MCFV-Frequenz in 10A1-MuLV induzierten Tumoren
Es wurden die DNAs von 13 Sca-1+- (s. Abb. 32) und 10 B-Zell+-Tumoren (s. Abb.
33) auf das Auftreten von MCF-Viren hin getestet. Die DNA von einer nicht infizierten
NIH/Ola-Maus und von nicht infizierten SC-1 Zellen diente als Negativkontrolle. Als
Positivkontrolle wurde DNA von 10A1 infizierten SC-1 Zellen verwendet. Da es sich
hier um 10A1 pol/env-Sequenzen handelt, wurden die DNAs mit der
Enzymkombination HindIII/ClaI gespalten und mit der 10A1-env Sonde hybridisiert.
Die Fragmente der Größe 0,8 kb und 1,9 kb repräsentieren das Wildtyp 10A1-MuLV.

Ergebnisse 92
WT 10A1
WT 10A1
R Typ I
23 kb
9,4 kb 6,6 kb
4,4 kb
2,3 kb 2,0 kb
0,5 kb
λ 1 λ 2 3 4 5 6 7 8 9 10 13 15 16 11 12 14 17 23 kb
9,4 kb
4,4 kb
2,3 kb
0,5 kb
6,6 kb
2,0 kb
Spur Sca-1+-Mäuse Spur Sca-1+-Mäuse
λ λ -DNA HindIII gespalten λ λ -DNA HindIII gespalten
1 #2456, Milz 10 #1824, Milz
2 #2457, Milz 11 #2137, Milz
3 #2461, Milz 12 #2363, Milz
4 #2463, Milz 13 #2369, Milz
5 #2464, Milz 14 #2375, Milz
6 #2465, Milz 15 #2488, Milz
7 NIH/Ola WT, Milz 16 #2443, Milz
8 SC-1 Zellen 17 NIH/Ola WT, Milz
9 10A1 infizierte SC-1 Zellen
Abb. 32: Dreizehn 10A1-MuLV induzierte Tumor-DNAs wurden mit der 10A1-env spezifischen Sonde
auf interne Rekombinationsereignisse getestet.
Beim Vergleich der Bandenmuster von Sca-1+ Tumor-DNAs mit denen von Mo-MuLV
sieht man, dass es deutlich weniger MCF-Viren gibt (s. Abb. 32). Es konnten keine
Typ II Rekombinanten detektiert werden (> 2,7 kb). Darüber hinaus ist das Verhältnis
von unrekombinierten zu rekombinierten Viren deutlich kleiner als bei Moloney, d.h.
auf eine 10A1-MuLV Integration kommen ca. 0,1 MCF-Viren. Da diese Tumore klonal
sind, treten also MCF-Viren nur in einer kleinen Zellpopulation des Tumors auf, so
dass eine Beteiligung an der Entartung sehr unwahrscheinlich ist.
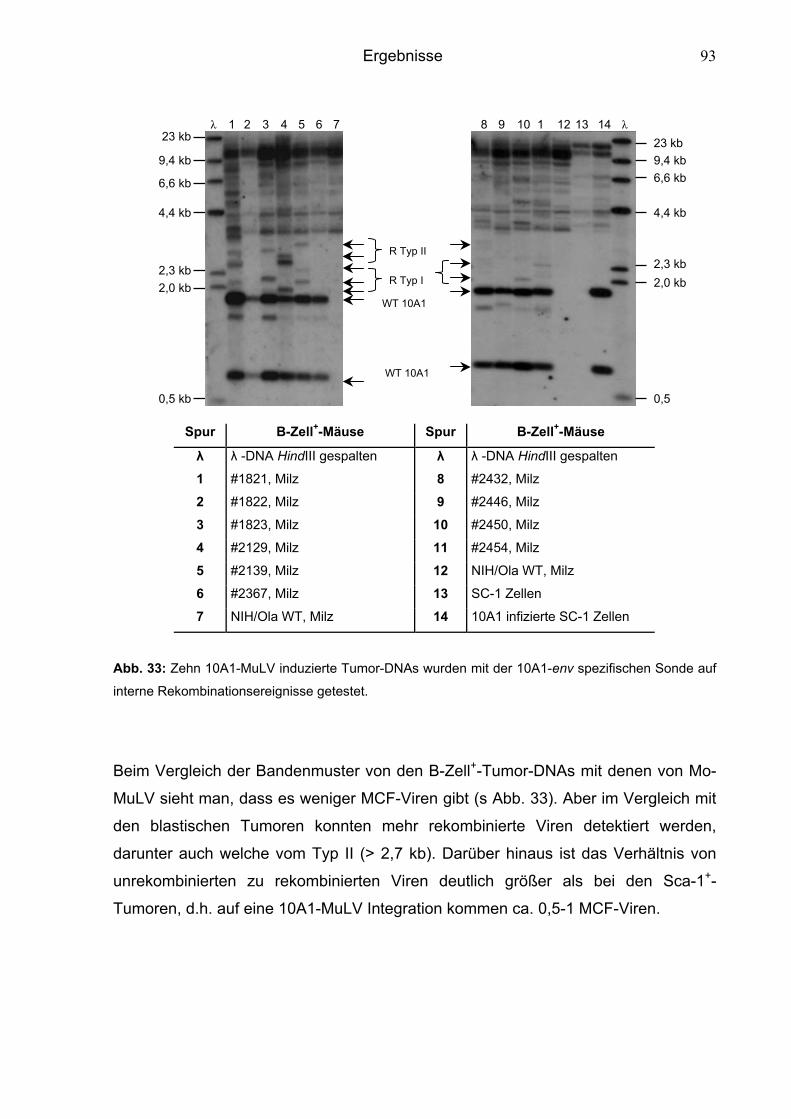
Ergebnisse 93
76 2 3 4 5 1λ
0,5 kb
2,0 kb 2,3 kb
4,4 kb
6,6 kb
9,4 kb
23 kb 14 12 13 11098 λ
R Typ II
R Typ I
WT 10A1
WT 10A1
23 kb
9,4 kb6,6 kb
4,4 kb
2,3 kb 2,0 kb
0,5
Spur B-Zell+-Mäuse Spur B-Zell+-Mäuse
λ λ -DNA HindIII gespalten λ λ -DNA HindIII gespalten
1 #1821, Milz 8 #2432, Milz
2 #1822, Milz 9 #2446, Milz
3 #1823, Milz 10 #2450, Milz
4 #2129, Milz 11 #2454, Milz
5 #2139, Milz 12 NIH/Ola WT, Milz
6 #2367, Milz 13 SC-1 Zellen
7 NIH/Ola WT, Milz 14 10A1 infizierte SC-1 Zellen
Abb. 33: Zehn 10A1-MuLV induzierte Tumor-DNAs wurden mit der 10A1-env spezifischen Sonde auf
interne Rekombinationsereignisse getestet.
Beim Vergleich der Bandenmuster von den B-Zell+-Tumor-DNAs mit denen von Mo-
MuLV sieht man, dass es weniger MCF-Viren gibt (s Abb. 33). Aber im Vergleich mit
den blastischen Tumoren konnten mehr rekombinierte Viren detektiert werden,
darunter auch welche vom Typ II (> 2,7 kb). Darüber hinaus ist das Verhältnis von
unrekombinierten zu rekombinierten Viren deutlich größer als bei den Sca-1+-
Tumoren, d.h. auf eine 10A1-MuLV Integration kommen ca. 0,5-1 MCF-Viren.

Ergebnisse 94
6.4.3 Niedrige MCFV-Frequenz in Mo-10A1V-10A1NP-MuLV und 10A1V-MoNP-MuLV induzierten Tumoren
Es wurden die DNAs von zehn Mo-10A1V-10A1NP- bzw. zehn 10A1V-MoNP-
induzierten Tumoren auf das Auftreten von MCF-Viren hin getestet. Bei allen Mo-
10A1V-10A1NP-induzierten Tumoren handelte es sich um T-Zell-Lymphome (s. Abb.
34). Bei den 10A1V-MoNP-induzierten Tumoren handelte es sich um sechs T-Zell-
und vier blastische Lymphome (s. Abb. 35). Die DNA von einer nicht infizierten
NIH/Ola-Maus diente als Negativkontrolle. Die DNAs wurden mit der
Enzymkombination BamHI/ClaI gespalten und mit der 10A1-env Sonde hybridisiert.
Die Fragmente der Größe 0,7 kb und 1,2 kb repräsentieren das Wildtyp Mo-10A1V-
10ANP- bzw. das 10A1V-MoNP-MuLV.
Spur Mo-10A1V-10A1NP
inf.-Mäuseλ λ –DNA HindIII 1 #2489, Milz
2 #2491, Milz
3 #2496, Milz
4 #2500, Milz
5 #2505, Milz
6 #2506, Milz
7 #2514, Milz
8 #2517, Milz
9 #2520, Milz
10 #2521, Milz
11 NIH/Ola WT, Milz
R Typ II
WT Mo-10A1V-10A1NP
WT Mo-10A1V-10a1NP
0,5 kb
2,0 kb 2,3 kb
4,4 kb
6,6 kb 9,4 kb 23 kb
2 3 4 7 8 9 10 111 5 6 λλ
Abb. 34: Zehn Mo-10A1V-10A1NP-MuLV induzierte Tumor-DNAs wurden mit der 10A1-env
spezifischen Sonde auf interne Rekombinationsereignisse getestet.
Beim Vergleich der Bandenmuster von den T-Zell+-Tumor-DNAs von beiden
Chimären mit denen von Mo-MuLV sieht man, dass es deutlich weniger MCF-Viren
gibt (vergleiche Abb. 31 mit 34 und 35). Diese Lymphome sind am ehesten mit den
10A1-induzierten blastischen Tumoren zu vergleichen (s. Abb. 32). Bei den
detektierten MCF-Viren des Mo-10A1V-10A1NP-MuLVs handelte es sich

Ergebnisse 95
ausschließlich um Typ II Rekombinanten (> 1,9 kb), während beim 10A1V-MoNP
neben den Typ II auch Typ I Rekombinanten zu erkennen sind. Beim Vergleich der
Bandenmuster von den 10A1V-MoNP-induzierten T-Zell+-Tumor-DNAs (s. Abb. 35)
mit den Sca-1+ (s. Abb. 32) sieht man auch hier, dass die blastischen Lymphome so
gut wie keine MCF-Viren hatten. Die detektierbaren MCF-Viren sind Typ I
Rekombinanten. Die Bandenintensität der MCF-Viren ist in allen hier untersuchten
Tumoren sehr gering im Vergleich zur Wildtyp-Bande. Bei beiden Chimären ist das
Verhältnis von unrekombinierten zu rekombinierten Viren so wie bei den 10A1-
induzierten Sca-1+-Tumoren, d.h. auf eine Mo-10A1V-10A1NP- bzw. 10A1V-MoNP-
MuLV Integration kommen ca. 0,1 MCF-Viren. Die Beteiligung dieser wenigen
rekombinierten Viren ist eher unwahrscheinlich. Da aber die nicht ganz optimale
Enzymkombination BamHI/ClaI benutzt wurde, konnten weiter 5’ gelegene
Rekombinationen nicht nachgewiesen werden, so dass die Anzahl der MCF-Viren
pro Wildtyp Integration höher als geschätzt sein könnte.
Spur 10A1V-MoNP-MuLV
inf.-Mäuseλ λ –DNA HindIII T-Zell+ 1 #2577, Milz T-Zell+
2 #2585, Milz T-Zell+
3 #2589, Milz T-Zell+
4 #2562, Milz T-Zell+
5 #2574, Milz T-Zell+
6 #2575, Milz T-Zell+
7 #2584, Milz Sca-1+
8 #2588, Milz Sca-1+
9 #2592, Milz Sca-1+
10 NIH/Ola WT, Milz -
R Typ II
WT 10A1V-MoNP0,5 kb
2,3 kb 2,0 kb
4,4 kb
6,6 kb 9,4 kb
23 kb
R Typ I
WT 10A1V-MoNP
λ 1 2 3 4 5 6 7 8 9 10
Abb. 35: Neun 10A1V-MoNP-MuLV induzierte Tumor-DNAs wurden mit der 10A1-env spezifischen
Sonde auf interne Rekombinationsereignisse getestet. Sechs Tumore waren T-Zell Lymphome, drei
waren blastische Lymphome.

Ergebnisse 96
Tab. 17: Überblick der in den Lymphomen von MuLV infizierten Mäusen ermittelten MCFV-Frequenz
im Zusammenhang mit der vorwiegend induzierten Leukämieform der verschiedenen Viren.
MuLVs Leukämietyp MCFV-Frequenz
Moloney T-Zell 1-3
10A1 blastisch 0,1
10A1 B-Zell 0,5-1
Mo-10A1V-10A1NP T-Zell 0,1
10A1V-MoNP T-Zell/blastisch 0,1
Das Moloney-MuLV zeigte sich in diesem Assay so wie es schon allgemein
beschrieben wurde. In allen Lymphomen konnte eine Vielzahl von MCF-Viren
detektiert werden, die in allen Tumorzellen vorzukommen scheinen. Diese in vivo
Rekombinationen scheinen wichtig für die Pathogenität dieses Virus zu sein. In
10A1-induzierten Tumoren konnten weniger MCF-Viren detektiert werden. Die
beiden Phänotypen zeigten auch in diesem Versuch ein unterschiedliches Bild
(s. Tab. 17). Eine Beteiligung der Rekombinanten an der Bildung einer blastischen
Leukämie ist sehr unwahrscheinlich, da nur eine kleine Subpopulation der
Tumorzellen Rekombinationsereignisse aufwies. In B-Zell+-Lymphomen konnten
mehr MCF-Viren detektiert werden. Da es sich hier um mehrere parallel
auswachsende Tumore handelte, haben wohl in einigen Zellklonen Rekombinationen
stattgefunden. Inwieweit diese für die Induktion einer B-Zell-Leukämie wichtig sind,
lässt sich nicht beantworten. Die Herkunft des NP-Bereichs scheint keinen Einfluss
auf die Häufigkeit von Rekombinationsereignissen zu haben. Da die detektierten
MCF-Viren in beiden NP-Konstrukten nur in einer Subpopulation der Tumorzellen
aufgetreten sind, scheint es zweifelhaft, dass diese an der Ausbildung einer T-Zell-
Leukämie beteiligt gewesen sind. Das Dogma, das MCF-Viren eine Voraussetzung
für die Induktion einer T-Zell-Leukämie sind, kann aufgrund dieser Ergebnisse nicht
bestätigt werden. Beim Konstrukt 10A1V-MoLTR, das in über 50 % der infizierten
Mäuse eine T-Zell-Leukämie induzierte, konnten überhaupt keine MCF-Viren
nachgewiesen werden (Daten nicht dargestellt).
Darüber hinaus lässt sich auch kein Zusammenhang mit dem Krankheitsverlauf
nachweisen, da das Auftreten von MCF-Viren nicht mit den Latenzzeiten der
verschiedenen MuLVs korrelierte. Die Viren Mo-MuLV und 10A1V-MoLTR haben zwar

Ergebnisse 97
eine vergleichbare mittlere Latenzzeit, 95 bzw. 94 Tage, aber ersterer hat eine hohe
und letzterer gar keine Neigung zur Rekombination. Obwohl die Herkunft des NP-
Bereichs, wie oben beschrieben, eine deutliche Auswirkung auf die Art der
induzierten Leukämie hat, scheint dieser Einfluss nicht in Zusammenhang mit dem
Vorkommen von MCF-Viren zu stehen. Diese Region muss also durch andere
Parameter das Krankheitsbild in vivo beeinflussen können.
6.5 Messung des glykoGag-Expressionsniveaus auf MuLV infizierten Zellen Das aminoterminal verlängerte glykoGag-Protein ist Bestandteil des Viruspartikels
(Fujisawa et al., 2001) und scheint einen wesentlichen Beitrag zur Pathogenese zu
leisten (Portis et al., 1990; Portis et al., 1996; Sitbon et al., 1986). Das Protein ist in
allen MuLVs konserviert und ist wichtig für dessen Ausbreitung in vivo (Corbin et al.,
1994). Es konnten auf der Oberfläche von infizierten Mauszellen bei einem
neuroinvasiven MuLV nur eine geringe Menge, bei einer nicht mehr neuroinvasiven
Mutante dagegen eine große Menge des glykoGag-Proteins detektieren (Fujisawa et
al., 1998). Es könnte also ein Zusammenhang zwischen der Menge an exprimiertem
glykoGag-Protein und der Pathogenität bestehen.
Um zu testen, ob das Expressionsniveau des glykoGag-Proteins in unserem Fall
auch eine Rolle bei der Pathogenität spielt, wurden SC-1 Zellen mit einigen aus dem
vorigen Kapitel vorgestellten MuLVs infiziert, mit dem Antikörper anti-glykoGag
gefärbt und anschließend mittels FACS-Analyse vermessen (s. 5.10.2). Der
Antikörper anti-glykoGag erkennt ein Epitop im C-terminalen Bereich des
glykosylierten Gag-Proteins. Das Protein wird auf der Zelloberfläche exprimiert,
wobei das Epitop in den extrazellulären Raum ragt. Es wurden von jedem Konstrukt
drei unabhängige Messungen durchgeführt und von den gemessenen
Fluoreszenzwerten (FW) der Mittelwert und die Standardabweichung ermittelt. Da
sich der NP-Bereich vom Mo-MuLV als eine wichtige Determinante für die Induktion
einer T-Zell-Leukämie präsentierte, wurden für diese Analyse Virusrekombinanten
ausgewählt, die entweder einen vom 10A1 oder Moloney stammenden NP-Bereich
aufwiesen (Mo-10A1V-10A1NP, 10A1V-MoNP). Allerdings könnte auch die Herkunft
der die NP-Region flankierenden Bereiche einen Einfluss haben, deshalb wurden
auch die chimären Viren 10A1V-Mogag, Mo-10A1V-10A1LTR und Mo-10A1env getestet

Ergebnisse 98
(s. Abb. 36). Da das 10A1-MuLV keine T-Zell-Leukämien induzierte, fungierte es als
eine Art Referenz.
Mo-10A1env infiziert
Mo-10A1V-10A1LTR infiziert
10A1V-MoNP infiziert
10A1V-Mogag infiziert
Mo-10A1V-10A1NP infiziert
10A1 infiziert
uninfiziert
Zellz
ahl
anti-glykoGag
blaa
60
0
blaa
60
0
blaa
60
0
blaa
60
0
blaa
60
0
blaa
60
0
blaa
60
0
100 104Cy5glyko
100 104Cy5glyko
100 104Cy5glyko
100 104Cy5glyko
100 104Cy5glyko
100 104Cy5glyko
Cy5glyko 104100
Abb. 36: Vergleich der FACS-Messungen von MuLV infizierten und mit dem Antikörper glykoGag-Cy5
gefärbten SC-1 Zellen. Konstrukte, die einen 10A1 NP-Bereich tragen sind in rot, Konstrukte mit
einem Moloney NP-Bereich in gelb dargestellt. Als Kontrolle dienten uninfizierte, glykoGag-Cy5
gefärbte SC-1 Zellen (schwarze Kurve).

Ergebnisse 99
Die Mittelwerte aus den drei unabhängigen FACS-Messungen von Viren mit einer
NP-Region vom 10A1- bzw. Moloney-MuLV wurden noch einmal gemittelt und das
Verhältnis (10A1:Mo) errechnet. Der NP-Bereich vom 10A1 vermittelte unabhängig
von der Herkunft der flankierenden Sequenzen eine um das 5,9-fach höhere
glykoGag-Expression als der Moloney NP-Bereich (s. Tab. 18).
Virus-Konstrukt NP-Bereich FW [MW] SD vorw. Leukämie
10A1 10A1 119 ± 43 blastisch/B-Zell
Mo-10A1V-10A1NP 10A1 108 ± 35 T-Zell
10A1V-Mogag 10A1 69 ± 14 blastisch/B-Zell
10A1V-MoNP Mo 16 ± 2.4 T-Zell
Mo-10A1V-10A1LTR Mo 22 ± 4 T-Zell
Mo-10A1env Mo 12 ± 4 T-Zell
Tab. 18: Zusammenfassung der im FACS ermittelten durchschnittlichen Fluoreszenzwerte (FW) mit
Standardabweichung (SD) der MuLV infizierten SC-1 Zellen. Die drei Mittelwerte von den Konstrukten
mit einem vom 10A1- bzw. Moloney-MuLV stammenden NP-Bereich wurden nochmals gemittelt und
das Verhältnis zwischen den Expressionsniveaus von 10A1- und Moloney NP-Bereich (10A1:Mo)
berechnet.
Darüber hinaus wurde das Expressionsniveau des glykosylierten Gag-Proteins in
Milzen von Mäusen zwei bzw. sechs Wochen nach Infektion ermittelt. Die Tiere
waren infiziert mit den MuLVs: 10A1, Mo-10A1V-10A1NP, Mo-10A1V-10A1LTR und
Mo-10A1env. Pro Virusrekombinante wurden zwei Tiere analysiert. Die gemessenen
Fluoreszenzwerte aus beiden Messungen wurden gemittelt und die
Standardabweichung bestimmt. Von allen Mittelwerten der Konstrukte mit einem vom
10A1- bzw. Moloney-stammenden NP-Bereich wurden ebenfalls die Mittelwerte
berechnet und das Verhältnis (10A1:Mo) ermittelt.
Der NP-Bereich vom 10A1 vermittelte in zwei Wochen alten Mäusen eine um das
doppelte höhere glykoGag-Expression als der Moloney NP-Bereich. Sechs Wochen
nach der Infektion war die Proteinexpression in Gegenwart eines vom 10A1-
stammenden NP-Bereichs um das 2,4-fache höher als das des Moloney NP-
Bereichs. Auch in vivo hatte die Herkunft der flankierenden Sequenzen keinen
Einfluss.

Ergebnisse 100
Das Expressionsniveau korrelierte sowohl in vitro als auch in vivo mit der Herkunft
des NP-Bereiches und zwar unabhängig von der Abstammung der flankierenden
Sequenzen, wobei aber die Korrelation in vivo nicht so stark war wie in vitro. Dabei
scheint das hohe glykoGag-Expressionslevel, das der 10A1-Leader vermittelt, mit
der Induktion des blastischen Phänotyps zu korrelieren, wenn dieser Einfluss nicht
durch das thymotrope Moloney-LTR überdeckt wird, wie das beim Konstrukt Mo-
10A1V-10A1NP der Fall ist. Die MuLVs mit einer vom Moloney-stammenden NP-
Region induzierten vorwiegend T-Zell-Lymphome (10A1V-MoNP 74%; Mo-10A1V-
10A1LTR 94 %; Mo-10A1env 93 %).

Diskussion 101
7. Diskussion
In dieser Arbeit konnte erstmals gezeigt werden, dass neben den Enhancer-
Elementen des LTR, auch der sich direkt stromabwärts anschließende Leader-
Bereich einen Einfluss auf die Art der induzierten Leukämie hat. Des weiteren scheint
der vom Moloney-MuLV stammende Leader-Bereich eine stärkere Determinante für
die Induktion einer T-Zell-Leukämie zu sein als das lymphotrope Moloney-LTR.
Ferner konnte gezeigt werden, dass die Generierung von MCF-Viren keine
Voraussetzung für die Induktion einer T-Zell-Leukämie ist. Die hier präsentierten
Daten zeigen darüber hinaus, dass die vom 10A1-MuLV stammenden env-
Sequenzen, und die damit verbundene Rezeptornutzung, eine wichtige Determinante
für die Proliferation des Myelo/Erythroblasten-Kompartiments darstellen. Wobei die
Transformation dieser Zellen in direktem Zusammenhang mit der Provirus
induzierten Aktivierung des Fli-1-Gens steht.
7.1 Pathogenität von Moloney-, 4070A- und 10A1-MuLV in NIH/Ola-Mäusen
Das Ziel dieser Arbeit war, die Sequenzbereiche im Genom des Moloney- bzw.
10A1-MuLVs einzugrenzen, die an der Induzierung einer bestimmten Leukämieform
beteiligt sind. Da aber sowohl der Mausstamm, der Infektionszeitpunkt als auch der
Infektionsort (z.B. intraperitoneal) Auswirkungen auf die Art der induzierten
Leukämieform in infizierten Mäusen haben, war es wichtig die Retroviren, auf denen
die chimären MuLVs basieren, in dem hier verwendeten Mausmodell zu testen. Das
Moloney-MuLV induzierte im NIH/Ola-Mausstamm nach einer mittleren Latenzzeit
von 95 Tagen ausschließlich T-Zell-Lymphome/Leukämien, wobei es sich bei den
betroffenen Lymphozyten um verschiedene Differenzierungsstadien innerhalb der T-
Zelllinie handelte. Mit dem 10A1-MuLV infizierte NIH/Ola-Mäuse zeigten nach einer
mittleren Latenzzeit von 132 Tagen eine blastische und nach 164 Tagen eine pro-B-
Zell-Leukämie. Das 4070A-MuLV induzierte nach 217 Tagen überwiegend B-Zell-
Leukämien. In Moloney-infizierten NIH/Swiss-Mäusen wurden nach einer Latenzzeit
von 125 Tagen ebenfalls T-Zell-Leukämien/Lymphomen unterschiedlicher Differen-
zierungsstadien induziert (Ott et al., 1992). Alle NIH/Swiss-Mäuse, die dem 10A1-
MuLV infiziert wurden, erkrankten ebenfalls an einer blastischen Leukämie (Ott et al.,

Diskussion 102
1994). Das 4070A-MuLV induzierte in NIH/Swiss-Mäusen nach 278 Tagen unreife T-
Zell-Lymphome (Ott et al., 1992). Eine andere Arbeitsgruppe diagnostizierte in 10A1-
bzw. 4070A-MuLV infizierten NIH/Swiss-Mäusen entweder primitive B-Zell-
Lymphome oder eine Leukämieform, bei der die Milz nicht betroffen war (Rasheed et
al., 1982). Die hier beobachteten Unterschiede in den Latenzzeiten und der Art der
induzierten Krankheit lassen sich wahrscheinlich auf den genetischen Hintergrund
des verwendeten Mausstammes zurückführen, da alle Mäuse innerhalb der ersten
48 h, intraperitoneal mit vergleichbaren Virustitern infiziert worden sind. Alle in dieser
Arbeit präsentierten Ergebnisse beziehen sich deshalb ausschließlich auf das hier
verwendete Mausmodell.
7.2 Bedeutung viraler Determinanten für die Pathogenität
7.2.1 Einfluss der env-Sequenzen
Der Austausch der env-Sequenzen wirkte sich bei den beiden Chimären Mo-10A1env
und 10A1-4070env ganz unterschiedlich aus. Während die 10A1 env-Sequenzen im
Moloney-MuLV, dessen Fähigkeit T-Zell-Lymphome zu induzieren nicht
beeinträchtigten, veränderte sich das onkogene Potential des 10A1-MuLVs durch die
Einführung der amphotropen env-Sequenzen dramatisch.
Die Chimäre Mo-10A1env war in der Lage, statt des ökotropen Rezeptors mCat-1, die
Phosphatsymporter Pit1 und Pit2 für die Bindung an die Wirtszelle zu nutzten. Der
einzige Unterschied zum Wildtyp Moloney-Virus war aber die vermehrte
Transformation von frühen, CD4/CD8 doppelt positiven T-Zellen, was auch schon
(Ott et al., 1992) in NIH/Swiss-Mäusen beobachtet hatten. Bei den Mo-10A1env-
induzierten Tumoren waren 9/14 CD4/CD8 doppelt positiv, während nur 3/7
Moloney-induzierten Lymphomen diesen Phänotyp zeigten. Da aber die Anzahl der
analysierten Tiere nicht besonders groß war, sollte diese Beobachtung nicht
überbewertet werden. Die Spezifität des Moloney-MuLVs hängt also nicht von der
Nutzung des Cat-1 Rezeptors ab. Der Austausch der env-Sequenzen hatte
wahrscheinlich keinen Einfluss auf die Spezifität der Chimäre, weil auch die
Phosphatsymporter von hämatopoetischen Zellen exprimiert werden. Die Tatsache,
dass das Virus Mo-10A1env vermehrt frühe T-Zellen transformierte, könnte mit der
unterschiedlichen Rezeptornutzung dieser Chimäre zusammenhängen. Eine andere

Diskussion 103
Möglichkeit wäre, dass die Gegenwart der 10A1-Sequenzen zur Aktivierung von
anderen Onkogenen beigetragen hat.
Die Einführung der amphotropen env-Sequenzen in das 10A1-MuLV veränderte die
Spezifität dieses Virus dramatisch. Das chimäre 10A1-4070env-MuLV konnte nur
noch den Phosphatsymporter Pit2 benutzen und war nicht mehr in der Lage eine
blastische Leukämie zu induzieren. Stattdessen zeigten die Mäuse nach einer
mittleren Latenzzeit von 175 Tagen eine starke Schädigung in bestimmten Geweben
bzw. Organen, die bei den anderen, hier untersuchten MuLVs, nicht beobachtet
werden konnte. Da sich diese Krankheitssymptome sehr spät manifestierten, hätten
Tiere mit einer blastischen Leukämie detektiert werden können, da diese
Leukämieform in 10A1-infizierten Mäusen eine deutlich kürzere Inkubationszeit von
132 Tagen hatte. Die Nutzung des Pit1-Rezeptors ist also eine wichtige
Determinante für die Pathogenität des 10A1-Virus, da dessen Nutzung für die
Transformation von Vorläuferzellen essentiell zu sein scheint.
Die beiden Phosphatsymporter Pit1 und Pit2 werden von hämatopoetischen Zellen
exprimiert. Während Pit1 ein hohes Expressionslevel im Knochenmark zeigt, ist Pit2
vor allem auf Herzzellen zu finden (Kavanaugh et al., 1994). Weitere
Untersuchungen haben gezeigt, dass hämatopoetische Zellen, auch Stammzellen,
sich effektiver über den Pit-1- als über den Pit2-Rezeptor infizieren lassen (Bauer et
al., 1995; Sabatino et al., 1997; von Kalle et al., 1994). Es wurde bereits gezeigt,
dass das 10A1-Virus murine Zellen vermehrt über den Pit1-Rezeptor infiziert,
während es bei der Infektion von humanen Zellen vorwiegend den Pit2-Rezeptor
benutzt, weil der humane Pit1-Rezeptor nur sehr ineffizient genutzt werden kann
(Miller, 1996). Die Möglichkeit zelluläre Rezeptoren für die Infektion zu nutzen, hängt
nicht nur von deren Verfügbarkeit auf bestimmten Zellen ab, sondern auch von ihrem
Expressionsniveau (Tailor et al., 2000) und der Art der Protein-Prozessierung. Die
veränderte Glykosylierung in bestimmten Geweben kann dazu führen, dass ein
MuLV seinen Rezeptor entweder nicht mehr erkennen oder ihn zwar binden, aber
nicht mehr für den Zelleintritt nutzen kann (Eiden et al., 1994; Marin et al., 2000;
Wang et al., 1996). Die vermehrte Expression von Pit1 im Knochenmark und die
bevorzugte Nutzung von Pit1 könnte eine mögliche Erklärung dafür sein, warum das
10A1-MuLV in der Lage ist, Vorläuferzellen zu transformieren. Eine andere
Möglichkeit ist, dass es nicht die Bindung des Env-Proteins, gp70, an den Pit1-
Rezeptor ist, die eine Proliferation des Stammzell- bzw. Vorläuferzellpools bewirkt,

Diskussion 104
sondern die Stimulation eines Zytokin-Rezeptors. Dieser Stimulus könnte bewirken,
dass die Zellen in ihrem Vorläuferstadium verharren und eine weitere Differenzierung
unterbunden wird. Das polytrope Env-Protein ist in der Lage ist, den zellulären
Interleukin-2-Rezeptor zu binden (Li and Baltimore, 1991). Diese Art der Interaktion
ist vom SFFV gp55 bekannt, das an den Erythropoetin-Rezeptor (EpoR) bindet und
dadurch die Proliferation des erythroiden Zellpools vermittelt (Li et al., 1995). Bisher
konnte eine derartige Wirkungsweise des gp70 allerdings nicht nachgewiesen
werden.
7.2.2 Einfluss der LTR-Sequenzen
Obwohl in der Chimäre Mo-10A1V-10A1LTR das lymphotrope Moloney-LTR fehlte,
induzierte es in fast allen Mäusen T-Zell-Lymphome. Während das chimäre 10A1V-
MoLTR nur in etwa der Hälfte der infizierten Tiere T-Zell-Lymphome induzieren
konnte. Die restlichen Mäuse entwickelten blastische oder B-Zell-Lymphome. Die
10A1-Sequenzen können scheinbar, gegen die Wirkung des Mo-LTR, noch immer
zur Transformation von Vorläuferzellen beitragen. Dieser Bereich liegt außerhalb der
env-Sequenzen, denn diese konnten sich in der Chimäre Mo-10A1env nicht gegen die
T-Zell-Determinanten durchsetzen. Auch bei den Latenzzeiten zeigten sich deutliche
Unterschiede zwischen den beiden Chimären. Während die Latenzzeit des 10A1V-
MoLTR-MuLV (94 Tage) mit der des Wildtyp Moloney-Virus (95 Tage) vergleichbar
war, hatte das chimäre Virus Mo-10A1V-10A1LTR eine deutlich längere
Inkubationszeit von 124 Tagen. Da bei allen diesen Viren, bis auf das Moloney-
MuLV, nur wenige oder keine MCF-Viren auftraten, können diese nicht für die
Unterschiede in den Latenzzeiten verantwortlich gemacht werden. Möglicherweise
beeinflusst die Herkunft der Enhancer-Elemente den Zeitraum, in dem sich die
Krankheit manifestiert.
Bei einem Vergleich der LTR-Sequenzen von Moloney-, 10A1- und 4070A-MuLV
kann man feststellen, dass sich die Enhancer-Elemente der drei Viren durchaus
ähneln (s. Abb. 37). Wobei aber zu beachten ist, dass das Moloney-MuLV zwei
identische Kopien der Enhancer-Elemente besitzt, während das 10A1- und das
4070A-MuLV nur jeweils eine Kopie besitzen. Die Bindung der Transkriptionsfaktoren
CBF und eines Ets-Proteins an die Core- bzw. LVt-Bindungstelle im Moloney-
Enhancer erhöht dessen Transkription in T-Zellen (Manley et al., 1993).

Diskussion 105
Comparison of LTR sequence
Mo-MuLV
4070A
10A1
F-SFFVp
d.r.d.r.
TCAAGGTCAGGAACAGATGGAACAGCTGAATATGGGCCAAACAGGATATCTGTGGTAAGCAGTTCCTGCCCCGGCTCAGGGCCAAGAACAGATGG
TCAAGGTCAGGAACAG ACAGCTGAA CCAAACAGGATATCTGTGGTAAGCAGTT CCCCGG GGCCAAGAACAGATGGTCCCCAGAT G CA CCCT
TCAAGGTCAGGAACAG ACAGCTGAA CCAAACAGGATATCTGTGGTAAGCAGTT CCCCGG GGCCAAGAACAGATGGTCCCCAGAT G CA CCC
GCAGTT CCCCGG GCCAAGAACAGATGGTCCCCAGA G CA CTTCAAG A A C GGCCAAACAGGATATCT T AGCAGTT C CCCGG GGCCAAGAACAGATGG
GAACAGCTGAATATGGGCCAAACAGGATATCTGTGGTAAGCAGTTCCTGCCCCGGCTCAGGGCCAAGAACAGATGGTCCCCAGATGCGGTCCAGCCCT
Sp1 C/EBP
Lvt /Ets
Lvt /Ets
NF-1 NF-1LVa C/EBPor CBF
MCREF-1 MCREF-1 FVcMCREF-1
A A GGG C G C C G G C
A A GGG C G C C G G C
C G C C G T G C CCG C GG AC A AG T A TG G GG GC C C G
-AA
-AA
TG-AA
GTT
GTT
CGTG G T T A
T
T
TT
G
G
GG G
C
C
CCC
G
G
GAG
AT
AT
AT
C
C
C
A
A
A
-
Abb. 37: Vergleich der U3-Sequenzen der LTR-Bereiche von verschiedenen MuLVs. Basenpaare, die
sich von der Moloney-MuLV Sequenz (oben) unterscheiden sind in rot dargestellt. Die bekannten
Bindungsstellen von verschiedenen Transkriptionsfaktoren sind gekennzeichnet (Baum et al., 1997;
Golemis et al., 1990; Manley et al., 1993).
Diese Transkriptionsfaktoren kommen nicht exklusiv in T-Lymphozyten vor, werden
dort aber verstärkt exprimiert. Mutationen in diesen Bindungsstellen im Moloney-LTR
verschieben dessen Krankheitsspezifität in Richtung Erythroleukämien (Speck et al.,
1990). Obwohl auch das 10A1- bzw. 4070A-LTR NF-1, LVt bzw. Core-
Bindungsstellen besitzt, fehlt ihnen, wie auch dem F-SFFV, die stromabwärts-
gelegene LVt-Bindungsstelle, stattdessen haben die drei Viren Bindungsstellen für
MCREF-1 und FVc (Manley et al., 1993). Wahrscheinlich ist die Transkription des
10A1-LTR in T-Lymphozyten nicht so effektiv wie die des Moloney-LTR, weil es zum
einen nur eine Kopie der Enhancer-Elemente besitzt und zum anderen diese nicht
absolut identisch mit den Moloney Enhancer-Elementen sind. Allerdings scheint das
10A1-LTR auch für die Transkription in hämatopoetischen Vorläuferzellen nicht
optimal geeignet, weil ihm die dafür wichtige Sp1-Bindungsstelle fehlt, wie sie in den SFFV Enhancer-Elementen vorkommt (Baum et al., 1997; Grez et al., 1991). Die
LTR-Sequenzen des Onkogen-tragenden Myeloproliferativen-Sarkom-Virus (MPSV)
beeinflussen dessen Krankheitsspezifität (Stocking et al., 1985). Allgemein scheinen
die 10A1 LTR-Sequenzen weder besonders gut, noch besonders schlecht für die
Transkription in verschiedenen Zellkompartimenten zu sein. Während das Moloney-

Diskussion 106
LTR für die Transkription in T-Zellen hochspezialisiert ist, können das 4070A- bzw.
das 10A1-LTR, deren Enhancer-Sequenzen sehr homolog sind, in verschiedenen
Zellarten replizieren. Sie sind unspezifischer, aber dafür auch ineffektiver.
Möglicherweise ist die verlängerte Inkubationszeit des 10A1-MuLVs im Vergleich
zum Moloney-MuLV auf die Mittelmäßigkeit seiner Enhancer-Elemente
zurückzuführen. Das Moloney-LTR ist also durchaus wichtig für die Transformation
von T-Zellen, aber wie die hier präsentierten Daten zeigen, keinesfalls ausreichend.
7.2.3 Einfluss der NP-Sequenzen
Die Bereiche, die neben dem LTR eine Auswirkung auf die Art der induzierten
Krankheit haben könnten, sind die gag- bzw. NP-Sequenzen. Das 10A1V-Mogag-
MuLV induzierte in 21 % der infizierten Mäuse T-Zell-Lymphome, so dass der Beitrag
dieses Bereichs nur als gering einzustufen ist. In den verbliebenen 80 % der Mäuse
induzierte die Chimäre dagegen blastische bzw. pro-B-Zell-Lymphome, so dass es
nahezu mit dem Wildtyp 10A1-Virus vergleichbar war. Auch die Latenzzeit dieses
Virus entsprach nahezu der des 10A1-MuLVs, 169 Tage gegenüber 155 Tagen. Die
Ergebnisse mit der Chimäre 10A1V-Mogag deuten daraufhin, dass sich neben den
10A1 env-Sequenzen, auch der Leader-Bereich des 10A1-MuLVs positiv auf die
Transformation von Vorläuferzellen auszuwirken scheint, wenn dessen Einfluss nicht
durch die Gegenwart des starken Moloney-LTR überdeckt wird. Interessanterweise
führte der Moloney NP- bzw. Leader-Bereich im chimären Virus 10A1V-MoNP in über
70 % der infizierten Mäuse zur Entwicklung von T-Zell-Lymphomen. Die mittlere
Latenzzeit betrug dabei nur 102 Tage und ist damit nur unwesentlich länger als die
des Wildtyp Moloney-Virus (95 Tage). Der Leader-Bereich enthält eine Reihe
wichtiger Kontrollelemente des retroviralen Entwicklungszyklus, wie die
Verpackungssequenz für die virale RNA (Ψ), die Primer-Bindungsstelle (PBS), das
Dimerisierungssignal (DLS) und eine interne Ribosomeneintrittsstelle (IRES).
Außerdem enthält der NP-Bereich das Start-Kodon für die mRNA des glykoGag-
Proteins. Dieses Protein ist für Retroviren wichtig, auch wenn seine Funktion nach
wie vor unbekannt ist.
Ein Vergleich zwischen der Moloney und der 10A1 NP-Region zeigt 49 bp
Unterschiede (s. Abb. 38). Es kann bisher nur spekuliert werden, welche Mutationen

Diskussion 107
im Moloney-NP ausschlaggebend für die T-Zell-Spezifität des Virus sind und welche
Mechanismen diesem Einfluss zu Grunde liegen könnten.
R
Mo-MuLV GCGCCAGTCCTCCGATTGACTGAGTCGCCCGGGTACCCGTGTATCCAATAAACCCTCTTG10A1-MuLV GCGCCAGTCCTCCGATAGACTGAGTCGCCCGGGTACCCGTGTATCCAATAAACCCTCTTG
Mo-MuLV CAGTTGCATCCGACTTGTGGTCTCGCTGTTCCTTGGGAGGGTCTCCTCTGAGTGATTGAC10A1-MuLV CTGTTGCATCCGACGAGTGGTCTCGCTGTTCCTTGGGAGGGTCTCCTCGGAGTGATTGAC
Mo-MuLV TACCCGTCAGCGGGGGTCTTTCATTTGGGGGCTCGTCCGGGATCGGGAGACCCCTGCCCA10A1-MuLV TACCCGTCA_CGGGGGTCTTTCATTTGGGGGCTCGTCCGGGATTTTGAGACCCCTGCCCA
Mo-MuLV GGGACCACCGACCCACCACCGGGAGGTAAGCTGGCCAGCAACTTATCTGTGTCTGTCCGA10A1-MuLV GGGACCACCGACCCACTACCGGGAGGTAAGCTGGCCAGCAACTGATCCGTGTCTGTCCGA
Mo-MuLV TTGTCTAGTGTCTATGACTGATTTTATGCGCCTGCGTCGGTACTAGTTAGCTAACTAGCT10A1-MuLV TTGTCCTGTGTCTATGACTGATTTTATGCGCCTGCGTCTGTATTAGTTGGCCGACTAGCT
Mo-MuLV CTGTATCTGGCGGACCCGTGGTGGAACTGACGAGTTCGGAACACCCGGCCGCAACCCTGG10A1-MuLV CTGTATCTGGCGGACCCGTGGTGGAGCTGACGAGTTCGGAACACCCGACCGCAACCCTGG
Mo-MuLV GAGACGTCCCAGGGACTTCGGGGGCCGTTTTTGTGGCCCGACCTGAGTCCAAAAATCCCG10A1-MuLV GAGACGTCCCAGGGACTTCGGGGGCCGTTTTTGTGGCCCGACCCGAGTCCAAAAGTCCCG
Mo-MuLV ATCGTTTTGGACTCTTTGGTGCACCCCCCTTAGAGGAGGGATATGTGGTTCTGGTAGGAG10A1-MuLV ATCGTTTTGGACTCTTTGGCGCACCCCCCTTAGAGGAGGGGTACGTGATTCTGGTAGGAG
Mo-MuLV ACGAGAACCTAAAACAGTTCCCGCCTCCGTCTGAATTTTTGCTTTCGGTTTGGGACCGAA10A1-MuLV ACGGGGACCTGAAACCGTTCCCGCCTCCGTCTGAGTTTTTGCTTTCGGTTTGGAGCCGAA
Mo-MuLV GCCGCGCCGCGCGTCTTGTCTGCTGCAGCATCGTTCTGTGTTGTCTCTGTCTGACTGTGT10A1-MuLV GCCGCGCCGCGCGTCTTGTCTGCTGCAGCATTGTTCTGTGTTGTCTCTGTTTGGCTGTTT
Mo-MuLV TTCTGTATTTGTCTGAGAATATGGGCCAGACTGTTACCACTCCCTTAAGTTTGACCTTAG10A1-MuLV TTCTGTATTGGTCTGAAAATATGGGCCAGACTGTTACCACACCCTTAAGTTTGACCTTAG
Mo-MuLV GTCACTGGAAAGATGTCGAGCGGATCGCTCACAACCAGTCGGTAGATGTCAAGAAGAGAC10A1-MuLV ATCATTGGAAAGATGTCGAGCGTACCGCTCACAACCAATCGGTGGATGTCAAGAAGAGAC
Mo-MuLV GTTGGGTTACCTTCTGCTCTGCAGpppppppppppppppppppppppppppppppppppp10A1-MuLV GCTGGGTCACCTTCTGCTCTGCAGpppppppppppppppppppppppppppppppppppp
Ende LTR
U5
Abb. 38: Ein Sequenzvergleich zwischen dem vom Moloney- und dem 10A1-MuLV stammenden
Leader-Bereich zeigt einen Unterschied von 49 bp. Mutationen in der 10A1-Sequenz sind als rote
Buchstaben dargestellt. Es sind der Anfang des R- und des U5-Bereichs angegeben, sowie das Ende
des LTR. Das Start-Kodon des glykoGag ist ein CTG, welches grün unterlegt ist. Das Start-Kodon für
das strukturelle Gag-Protein (ATG) ist rötlich unterlegt. Die im Moloney-Leader lokalisierte Ikaros-
Bindungsstelle ist gelb unterlegt.
Eine Möglichkeit ist das Vorhandensein einer alternativen Speiß-Stelle im Moloney
Leader-Bereich, die die Aktivierung von transformierend wirkenden Genen in T-
Zellen ermöglicht. (Audit et al., 1999) haben gezeigt, dass drei Punktmutationen im
strukturellen gag-Gen von Mo-MuLV, zu einem breiteren Spektrum an induzierten
Leukämien führten. Es handelte sich um sogenannte synonyme Mutationen, die die

Diskussion 108
Aminosäuresequenz des Kapsid-Proteins nicht veränderten. Neben T-Zell-
Lymphomen traten auch Erythro- und myelomonozytische Leukämien in den
infizierten Mäusen auf. Die Mutationen hatten keine Auswirkungen auf die
Inkubationszeit oder die Ausbreitung der Virusmutante in vivo. Dieselbe
Arbeitsgruppe hat später gezeigt, dass durch diese Mutationen eine stark
konservierte, alternative Spleiß-Donorstelle (SD’) zerstört worden ist, was zu einer
verminderten Replikation der Mutante in vitro führte. Diese Spleißstelle scheint für
die Herstellung eines subgenomischen RNA-Transkripts (4,4 kb) verantwortlich zu
sein, was für simple Retroviren sehr ungewöhnlich ist (De´jardin et al., 2000). Da
aber die Moloney gag-Sequenzen auch die alternative Speißstelle vom Moloney-
Virus enthalten, kann die Menge des subgenomischen RNA-Transkripts keinen
Einfluss auf die vom 10A1V-Mogag-induzierte Krankheitsverteilung haben. Außerdem
haben die hier präsentierten Daten gezeigt, dass die Herkunft des gag-Bereichs die
Art der induzierten Lymphome nur gering beeinflussen kann.
Die Moloney Leader-Region erweist sich vielleicht auch deshalb als eine wichtige
Determinante für die Induktion von T-Zell-Lymphomen in dem hier verwendeten
Mausmodell, weil in diesem Bereich unbekannte regulatorische Sequenzen oder
Bindungsstellen für bisher unbekannte Faktoren lokalisiert sind. Im Moloney-Leader
konnte von mir eine Bindungsstelle für den T-Zell-spezifischen Transkriptionsfaktor
Ikaros nachgewiesen werden, die in der 10A1-Sequenz mutiert ist. Die Arbeiten von
(Reuss et al., 2001) haben gezeigt, dass amphotrope Viren ohne Enhancer-
Elemente in der Lage sind, in humanen Zellen zu replizieren, so dass es Bereiche
außerhalb der U3-Region geben muss, die die Enhancer-Elemente im LTR ersetzen
können.
Neuere Untersuchungen von (Wolff et al., 2003) mit einer Moloney-Chimäre mit
amphotropen LTR-Sequenzen zeigten, dass dieses Virus in 23 % der infizierten
FVB- und 46 % der Balb/c-Mäuse, T-Zell-Lymphome induzieren konnte, die anderen
Tiere erkrankten an myeloiden Tumoren. Da myeloide Leukämien nach der Infektion
mit dem Mo-MuLV in diesen Mausstämmen nicht beobachtet werden konnten,
schlägt die Arbeitsgruppe eine Kollaboration zwischen den amphotropen und den
Moloney-Sequenzen als möglichen Mechanismus vor. Da die Mausstämme FVB und
Balb/c durch die Expression des Fv1-Lokus resistent gegenüber dem amphotropen
Virus sind, ist es schwer zu sagen, ob die induzierten T-Zell-Lymphome nur auf die
Gegenwart der Moloney-Sequenzen zurückzuführen sind. Der Moloney Leader-

Diskussion 109
Bereich scheint in diesen Mausmodellen nicht so effektiv zu sein, wie in dem von uns
verwendeten. Der genetische Hintergrund der infizierten Mäuse, hat wie bereits
erwähnt, einen starken Einfluss auf die Art der induzierten Krankheit. Nichts desto
trotz belegt auch die Arbeit von Wollf et al., dass es Sequenzen außerhalb des LTR
geben muss, die die induzierte Leukämieform beeinflussen können.
7.2.3.1 Einfluss des glykoGag-Expressionsniveaus auf die Pathogenität
Eine andere Möglichkeit, die wir genauer untersucht haben, ist eine Beeinflussung
der Krankheitsspezifität durch das glykoGag-Protein, dessen Start-Kodon im NP-
Bereich lokalisiert ist. Dazu wurden die hier verwendeten Viren bezüglich ihrer
glykoGag-Expression in SC-1 Zellen untersucht. Dabei stellte sich, wie bereits
erwähnt, heraus, dass die Moloney Leader-Region zu einem niedrigen, die 10A1
Leader-Region dagegen zu einem hohen glykoGag-Expressionsniveau sowohl in
vitro als auch in vivo führte. Die Herkunft der den Leader-Bereich flankierenden
Sequenzen hatte dabei keine Auswirkungen auf die Expression. Die Gegenwart des
Moloney NP-Bereichs scheint sich günstig auf die Induktion von T-Zell-Lymphomen
auszuwirken. Der entsprechende vom 10A1-MuLV-stammende Bereich scheint
dagegen die Entwicklung von blastischen Lymphomen zu ermöglichen, wenn dieser
Einfluss nicht durch das stark thymotrope Moloney-LTR maskiert wird, wie in der
Chimäre Mo-10A1V-10A1NP. Das glykoGag-Expressionsniveau korreliert also mit der
Induktion bestimmter Leukämieformen. Die Funktion des glykoGag-Proteins im
retroviralen Entwicklungszyklus ist unbekannt, es konnte aber bisher gezeigt werden,
dass dieses Protein wichtig für die Virusausbreitung und Pathogenität in vivo ist
(Corbin et al., 1994; Fujisawa et al., 1997). Mutationen im Leader-Bereich des
chimären Cas-Br-E-/Friend-MuLV führten zu einem hohen glykoGag-
Expressionslevel, welches sich inhibierend auf dessen Neuroinvasivität auswirkte
(Fujisawa et al., 1998). Im Gegensatz dazu haben (Münk et al., 2003) gezeigt, dass
ein hohes glykoGag-Expressionsniveau mit der Neuropathogenität von chimären
Moloney-MuLVs korreliert. Da das glykoGag-Protein in die Viruspartikel inkorporiert
wird (Fujisawa et al., 2001), könnte die Menge des eingebauten Proteins eine Rolle
bei der Infektiosität von Viren spielen. Möglicherweise kann das membranständige
glykoGag-Protein bestimmte Ko-Rezeptoren auf Nachbarzellen erkennen und
binden, was die Virusausbreitung in bestimmten Geweben begünstigt. Alternativ

Diskussion 110
könnte die Interaktion zur Freisetzung von Wachstums-Faktoren führen, die
entweder eine Differenzierung oder Proliferation von bestimmten Zellen bewirken.
7.2.4 Einfluss von MCF-Viren auf die Pathogenität
Da postuliert wird, dass MCF-Viren durch die Bereitstellung von rekombinierten env-
und/oder rekombinierten LTR-Sequenzen für die rasche Krankheitsentwicklung in
MuLV-infizierten Mäusen verantwortlich sind (Brightman et al., 1991; Hartley et al.,
1977; Lavignon and Evans, 1996; Ruscetti et al., 1981), wurden die hier induzierten
Tumore auf das Vorkommen von MCF-Viren untersucht. Interessanterweise traten in
den Mo-10A1env-MuLV-induzierten Tumoren nur wenige MCF-Viren auf, was
eventuell auf die Gegenwart der 10A1 env-Sequenzen zurückzuführen war. Die
Möglichkeit der dualen Rezeptornutzung scheint wohl das Auftreten von MCF-Viren
für die Induktion einer T-Zell-Leukämie/Lymphom unnötig zu machen. Das
verminderte Vorkommen von MCF-Viren könnte die verlängerte Inkubationszeit von
128 Tagen, im Vergleich zu 95 Tagen beim Moloney-MuLV, erklären. Auch in den
durch die anderen Chimären induzierten Lymphomen konnten, unabhängig von der
Krankheit, nur wenige Rekombinationsereignisse nachgewiesen werden, die dann
auch nicht in allen Tumorzellen vorlagen. Da auch in klonal ausgewachsenen,
blastischen Lymphomen nur in einigen Zellen MCF-Viren detektiert werden konnten,
ist deren Einfluss auf die Krankheitsverteilung eher unwahrscheinlich. Wenn diese
Viren durch ihre rekombinierten env-Sequenzen zu einer Superinfektion der Zellen
geführt und/oder durch ihre modifizierten Enhancer-Elemente einen
Transkriptionsvorteil in bestimmten Zellen gehabt hätten, wäre anzunehmen
gewesen, dass diese in dem gesamten klonalen Zellpool zu finden gewesen wären.
(Lavignon et al., 1997) hatten festgestellt, dass die Herkunft der gag-Sequenzen, den
Typ der generierten MCFVs beeinflussen kann. Die hier präsentierten Ergebnisse
lassen keinen Zusammenhang zwischen der Herkunft bestimmter Gen-Bereiche,
außer der vom 10A1-MuLV stammenden pol/env-Sequenz, und der Generierung von
MCF-Viren erkennen. Die duale Rezeptornutzung des 10A1-MuLVs ist auf die
Rekombination von amphotropen env-Sequenzen mit endogenen, MCF-ähnlichen
Sequenzen zurückzuführen (Ott et al., 1990). Vielleicht lässt die Gegenwart dieser
MCF-ähnlichen Sequenzen im 10A1-env nur bedingt Rekombinationen mit
endogenen Sequenzen zu. Obwohl dies eigentlich unwahrscheinlich ist, da durch die

Diskussion 111
hohe Sequenzhomologie eine verstärkte Rekombination zu erwarten gewesen wäre.
Tumore, die durch MuLVs induziert wurden, die keine 10A1 env-Sequenzen tragen,
wie das Moloney-, das 10A1-4070env- und das 4070A-Virus, zeigten eine hohe
MCFV-Frequenz in allen Tumorzellen. Möglicherweise erleichtern oder stimulieren
amphotrope bzw. ökotrope env-Sequenzen die MCFV-Generation, weil keine große
Homologie zu den endogenen Sequenzen vorhanden ist. Die Rezeptoren mCat-1
und Pit2 sind vielleicht nicht optimal für die Infektion lymphoider Zellen geeignet, weil
sie ein niedrigeres Expressionslevel haben oder durch Modifikationen nicht so
effizient genutzt werden können, so dass in vivo auf Viren selektioniert wird, die
durch modifizierte env-Sequenzen den polytropen Rezeptor nutzen können. In
Moloney-MuLV induzierten Tumoren konnten darüber hinaus auch viele Typ II
Rekombinanten detektiert werden, so dass wohl auch auf die Bereitstellung
rekombinierter LTR-Sequenzen selektioniert wurde.
7.2.5 Aktivierung von Onkogenen
Die 10A1-induzierten Tumore wurden auf das Vorkommen von häufig aktivierten
Onkogenen untersucht. Von den pro-B-Zell-Lymphomen zeigte nur je ein Tumor
(1/10) Provirusintegrationen in den Genen c-myc und Pim-1, die in T-Zell- und B-Zell-
Lymphomen häufig aktiviert sind. Da nur in wenigen Lymphomen Integrationen in
den bekannten Onkogenen festgestellt werden konnten, ist ein entscheidender
Beitrag dieser Insertionen zur malignen Entartung des pro-B-Zellpools eher
unwahrscheinlich. In diesen Tumoren haben verschiedene, zusammenwirkende
Integrationsereignisse in funktionellen Genen zur malignen Transformation in
mehreren Zellen geführt. Diese Zellpopulationen sind dann parallel ausgewachsen
und bildeten einen polyklonalen Tumor. Diese Polyklonalität spiegelt sich in der
relativ langen Latenzzeit von 164 Tagen wieder. Die Integrationen haben entweder in
anderen Bereichen der bekannten Loci stattgefunden oder es wurden bisher
unbekannte Onkogene aktiviert.
Alle 10A1-induzierten, blastischen Tumore waren positiv für Integrationen in den
Fli-1-Lokus, Provirusinsertionen in anderen, für Erythroleukämien typischen
Onkogenen, wie z.B. Spi-1/PU.1, konnten nicht detektiert werden. Da in nur 2/13
Tumoren Integrationen in das Pim-1-Gen detektiert werden konnten, ist eine
Auswirkung dieser Aktivierung auf den sehr einheitlichen Phänotyp der erkrankten

Diskussion 112
Tiere eher unwahrscheinlich. In NIH/Swiss-Mäusen konnten in allen 10A1-
induzierten, blastischen Lymphomen Integrationen im Fli-1-Lokus identifiziert und
darüber hinaus eine 10A1/Fli-1-Fusions mRNA nachgewiesen werden, was für eine
Aktivierung des Gens durch Promoterinsertion spricht (Ott et al., 1994). Alle
Integrationen fanden stromaufwärts vom Fli-1 Initiations-Kodon in der gleichen
transkriptionellen Orientierung wie die des Gens statt (s. Abb. 39). Auch in fast 70 %
der Cas-Br-E-MuLV induzierten blastischen Lymphome konnten Integrationen in dem
von Ott beschriebenen Bereich im Fli-1 Gen in der gleichen transkriptionellen
Orientierung nachgewiesen werden (Bergeron et al., 1991).
+1
Fli-1 Sonde
E E BB
Exon 9Exon 1Exon 1b
Abb. 39: Schematische Darstellung des Fli-1-Lokus mit den relevanten Schnittstellen und den
bevorzugten Integrationsstellen. Die transkriptionelle Orientierung der Proviren ist durch die Richtung
des Pfeils dargestellt. Die Lage der Fli-1-Sonde ist mit einem Doppelpfeil angegeben. B: BamHI; E:
EcoRI; +1: Transkriptionsstart.
In den hier analysierten Tumoren zeigten 12/13 Tieren ebenfalls Integrationen in
diesem Bereich (s. Abb. 39). Nur in einem Tumor konnte eine weiter stromaufwärts
gelegene Integration nachgewiesen werden. Alle blastischen Lymphome, die durch
die verschiedenen, hier getesteten chimären MuLVs induziert wurden, zeigten
ebenfalls eine Integration in den Fli-1-Lokus. Wobei die meisten Tumore eine
Insertion im gleichen Bereich wie in den 10A1-induzierten Lymphomen hatten,
nämlich stromaufwärts vom Fli-1 Initiations-Kodon in der gleichen transkriptionellen
Orientierung (s. Abb. 39). Interessanterweise konnten in den durch das 10A1V-Mogag
induzierten, blastischen Lymphomen Integrationen in einem anderen Bereich des Fli-
1-Lokus nachgewiesen werden, so wie es auch einer der 10A1-induzierten Tumore
zeigte. Die Integrationen haben weiter stromaufwärts in den regulatorischen

Diskussion 113
Sequenzen des Fli-1 Gens in umgekehrter Orientierung stattgefunden (s. Abb. 39).
Wahrscheinlich hatte die Integration in diesem Bereich einen Selektionsvorteil für die
infizierten Zellen. Möglicherweise hatten die Moloney gag-Sequenzen einen Einfluss
auf die Richtung der Provirusintegration. Auch in 95 % der Friend-MuLV induzierten
Leukämien konnten Integrationen stromaufwärts und in umgekehrter
transkriptioneller Orientierung zum Fli-1 Gen nachgewiesen werden (Ben-David et
al., 1991). Die Aktivierung des Fli-1-Gens scheint somit für die Transformation der
Vorläuferzellen essentiell zu sein, was durch die Beobachtung, dass diese Tumore
klonal sind, noch untermauert wird. Die Aktivierung dieses Gen-Lokus führt zu einer
schnellen Proliferation von Vorläuferzellen und dann zum Tod der infizierten Tiere.
Interessanterweise war auch ein B-Zell-Tumor positiv für eine Integration in den Fli-1-
Lokus, wobei das Provirus in den gleichen Bereich inseriert hatte, wie das auch in
den blastischen Tumoren der Fall gewesen ist. Möglicherweise hatte diese
Integration zu einem späteren Zeitpunkt oder in einer anderen Zellpopulation
stattgefunden, so dass eine Aktivierung dieses Lokus nicht mehr zur Proliferation im
Myelo/Erythroblasten-Kompartiment oder nicht zur Transformation der infizierten
Zelle führte. Ob die Gegenwart bestimmter 10A1-Bereiche die Proviren präferentiell
in den Fli-1-Lokus inserieren lässt, kann ohne weitere Untersuchungen nur spekuliert
werden. Wahrscheinlich hat dieser Lokus in Vorläuferzellen eine offene
Chromatinstruktur, die eine Provirusintegration erst ermöglicht. Es wurde bereits
gezeigt, dass das HI-Virus bevorzugt in aktive Gene inseriert, vor allem in solche, die
nach einer HIV-Infektion aktiviert sind (Schröder et al., 2002).

Appendix 114
8. Appendix Tab. 19: Befunde von den 10A1V-Mogag-induzierten Lymphomen im Überblick (N: normal; Lk:
Lymphknoten; Thy: Thymus; n.a.: nicht analysiert, *:Tumore zeigten auch eine anti-CD79ß und anti-
IgM positive Subpopulation).
Maus- Nr.
nach [d]
Milz [g]
Leber [g]
andere Organe
Hkt [%]
Leuk [106/ml]
FACS
#2662 86 2,6 3,2 normal 15,3 n.a. Sca-1/Ter119
#2663 113 2 2,5 normal n.a. n.a. Sca-1
#2669 104 2,2 3,2 normal 17,7 14,4 n.a.
#2671 121 2,6 3,5 normal 22,2 137,4 n.a.
#2680 138 2,2 4,7 normal n.a. n.a. n.a.
#2682 79 2,3 2,7 normal 15 41,1 Sca-1
#2749 83 2,1 3,4 normal n.a. n.a. n.a.
#2752 134 3,7 3,9 normal n.a. n.a. n.a.
#2665 205 0,4 N normal 39,2 3,6 CD19/B220*
#2673 199 0,5 N normal 20,1 29,7 CD19/B220
#2678 210 0,5 N normal 38,1 3 CD19/B220*
#2679 222 0,5 N Lk 34,5 n.a. CD19/B220*
#2684 222 0,8 N Lk 20,4 15,9 CD19/B220
#2750 125 1,0 N Lk, Thy 33,6 13,8 CD19/B220
#2753 169 3,1 3,2 normal 17,1 n.a. CD19/B220
#2664 176 >0,4 >2,3 Lk, Thy n.a. n.a. CD90/CD3, CD4
#2677 210 1,1 2,4 Thy 36,3 24,9 CD90/CD3, CD4/CD8
#2683 187 2,7 5,8 Lk, Thy 50,7 8,4 CD90/CD3, CD4/CD8
#2748 116 1,2 2,0 Lk, Thy 39,3 n.a. CD90/CD3, CD4/CD8

Appendix 115
Tab. 20: Befunde von Mo-10A1V-10A1NP-infizierten Mäusen im Überblick (N: normal; Lk:
Lymphknoten; Thy: Thymus; n.a.: nicht analysiert)
Maus-
Nr. nach [d]
Milz [g]
Leber [g]
andere OrganeHkt [%]
Leuk [106/ml]
FACS
#2489 76 2,0 4,2 Lk 38,2 n.a. CD90/CD3
#2491 76 1,9 3,3 Lk, Thy 32,7 57,6 CD90/CD3
#2495 83 1,6 3,0 Lk, Thy 25,4 39,3 CD90/CD3
#2496 85 0,9 N Thy 41,1 58,5 CD90/CD3
#2497 118 1,5 3,2 Lk 44,1 126,3 CD90/CD3
#2500 94 1,2 N Lk, Thy n.a. n.a. CD90/CD3
#2505 114 0,5 N Thy 38,7 10,5 CD90/CD3
#2506 93 1,9 2,7 Lk, Thy 36,9 48,9 CD90/CD3
#2508 125 1,5 2,8 Lk 39 38,4 CD90/CD3
#2514 113 2,1 2,5 Lk, Thy 45,9 n.a. CD90/CD3
#2516 94 2,7 >2,5 Lk, Thy 17,3 n.a. CD90/CD3
#2517 97 1,7 3,1 Lk, Thy 38,4 412 CD90/CD3
#2519 160 0,3 N Thy 43,5 5,4 CD90/CD3
#2520 107 2,7 >2,3 Lk, Thy 18,3 n.a. CD90/CD3
#2521 111 1,2 4,5 Lk, Thy 45 37,5 CD90/CD3
#2494 119 0,7 N Thy 40,2 13,2 CD19/B220
#2499 83 1,6 2,6 normal 17,4 47,7 Sca-1

Abkürzungsverzeichnis 116
9. Abkürzungsverzeichnis
Abb. Abbildung Ak Antikörper bp Basenpaar CA Kapsidprotein c-onc zelluläres Proto-Onkogen cpm engl. Counts per minute, Ereignisse pro Minute d engl. Day, Tag DLS Dimerisierungssignal DNA engl. Deoxyribonucleic acid, Desoxyribonukleinsäure EC embryonale Karzinomzellen env Gen für das retrovirale Hüllprotein EpoR Erythropoetin-Rezeptor ES embryonale Stammzellen FACS engl. Fluorescence Activated Cell Sorter, Fluoreszenz-aktivierter
Zellsortierer FCS engl. Fetal Calf Serum, fötales Kälber Serum gag Gen für die retroviralen Kapsidproteine glykoGag Gen für das glykosylierte Gag-Protein gp55 Glykoprotein des SFFV gp70 Glykoprotein muriner Leukämieviren h engl. Hour, Stunde HIV Humanes Immundefizienz Virus Hkt Hämatokrit HTLV-1 humanes T-Zell-Leukämievirus-1 HVR hypervariable Region IN retrovirales Enzym: Integrase IRES interne Ribosomeneintrittsstelle kb 1000 Basenpaare LB Luria-Bertani Leu Leukozytenzahl Lk Lymphknoten LTR engl. Long Terminal Repeat, lange terminale Wiederholung MA Matrixprotein MCFV engl. Mink-Cell-Focus-Forming-Virus, Nerz-Zellfokus-bildenes Virus MDEV Mus dunni endogenes Virus MEM Minimal Essentielles Medium MESV Murines embryonales Stammzell Virus MHC engl. Major Histocompatibility Complex, Haupt Histokompatibilitäts-
Komplex ml Milliliter MPSV murines Proliferatives Sarkom Virus mRNA engl. Messenger-RNA, Boten-RNA MSV murines Sarkom Virus MuLV murines -Leukämie Virus N normal NC Nukleokapsidprotein nt Nukleotid

Abkürzungsverzeichnis 117
NZB New Zealand Black OD Optische Dichte PBS Primerbindungsstelle pol Gen für die retroviralen Enzyme PPT Polypurintrakt PR retrovirales Enzym: Protease R Redundante Region RNA engl. Ribonucleic acid, Ribonukleinsäure rpm engl. Round per minute, Umdrehungen pro Minute RPMI Roswell Park Memorial Institute 1640 Medium RSV Rous Sarkom Virus RT retrovirales Enzym: reverse Transkriptase SA Spleiß-Akzeptor SD Spleiß-Donor SFFV engl. Spleen-Focus-forming-Virus, Milzfokus-bildenes Virus SU Oberflächenhüllprotein Tab Tabelle TAE Tris-Azetat-EDTA-Puffer Thy Thymus TM Transmembranes Hüllprotein tRNA Transfer RNA U3 engl. Unique, einzigartige 3’-Region U5 engl. Unique, einzigartige 5’-Region UTR Untranslatierte Region v-onc virales Onkogen VRA variable Region A VRB variable Region B

Literaturverzeichnis 118
10. Literaturverzeichnis
Albritton, L. M., Tseng, L., Scadden, D., and Cunningham, J. M. (1989). A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptability to virus infection., Cell 57, 659-666.
Alt, F. W., Yancopoulos, G. D., Blackwell, T. K., Wood, C., Thomas, E., Boss,
M., Coffman, R., Rosenberg, N., Tonegawa, S., and Baltimore, D. (1984). Ordered rearrangements of immunoglobulin heavy chain variable region segments., The EMBO Journal 3, 1209-1219.
Amati, B., Brooks, M. W., Levy, N., Littlewood, T. D., Evans, G. I., and Land, H.
J. (1993). Oncogenic activity of the C-Myc protein requires dimerization with Max., Cell 72, 233-245.
Appleyard, R. K. (1954). Segregation of new lysogenic types during growth of a
doubly lysogenic strain derived from Escherichia coli K12., Genetics 39, 440-452.
Audit, M., De´jardin, J. R., Hohl, B., Sidobre, C., Hope, T. J., Mougel, M. n., and
Sitbon, M. (1999). Introduction of a cis-Acting mutation in the capsid-coding gene of Moloney murine leukemia virus extends Its leukemogenic properties., Journal of Virology 73, 10472-10479.
Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith,
J. A., and Struhl, K. (2001). Current protocols in molecular biology., John Wiley and Sons, Inc.).
Barbeau, B., Barat, C., Bergeron, D., and Rassart, E. (1999). The GATA-1 and
Spi-1 transcriptional factors bind to a GATA/EBS dual element in the Fli-1 exon 1., Oncogene 18, 5535-5545.
Barbeau, B., Bergeron, D., Beaulieu, M., Nadjem, Z., and Rassart, E. (1996).
Characterization of the human and mouse Fli-1 promoter regions., Biochimica et Biophysica Acta 1307, 220-232.
Barre´-Sinoussi, F., Chermann, J. C., Rey, F., Nugeyre, M. T., Chamaret, S.,
Gruest, J., Dauguet, C., Axler-Blin, C., Ve´zinet-Brun, F., Rouzioux, C., et al. (1983). Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune defiency syndrome (AIDS)., SCIENCE 220, 868-871.
Battini, J.-L., Danos, O., and Heard, J. M. (1995). Receptor-Binding Domain of
Murine leukemia virus envelope glycoproteins., Journal of Virology 69, 713-719.
Battini, J.-L., Danos, O., and Heard, J. M. (1998). Definition of a 14-Amino-Acid
Peptide essential for the interaction between the murine leukemia virus amphotropic envelope glycoprotein and its receptor.,

Literaturverzeichnis 119
Journal of Virology 72, 428-435. Battini, J.-L., Rasko, J. E., and Miller, A. D. (1999). A human cell-surface
receptor for xenotropic and polytropic murine leukemia viruses: Possible role in G protein-coupled signal transduction., Proc Natl Acad Sci USA 96, 1385-1390.
Battini, J.-L., Rodrigues, P., Müller, R., Danos, O., and Heard, J. M. (1996).
Receptor-binding properties of a purified fragment of the 4070A amphotropic murine leukemia virus envelope glycoprotein., Journal of Virology 70, 4387-4393.
Bauer, T. J., Jr., Miller, A. D., and Hickstein, D. D. (1995). Improved transfer of
the leukocyte integrin CD18 subunit into hematopoietic cell lines by using retroviral vectors having gibbon ape leukemia virus envelope., BLOOD 86, 2379-2387.
Baum, C., Katsuhiko, I., Meyer, J., Laker, C., Yoshiaki, I., and Ostertag, W.
(1997). The potent enhancer activity of the polycythemic strain of spleen focus-forming virus in hematopoietic cells is governed by a binding site for Sp1 in the upstream control region and by a unique enhancer core motif, creating an exclusive target for PEBP/CBF., Journal of Virology 71, 6323-6331.
Belli, B., Patel, A., and Fan, H. (1995). Recombinant Mink Cell Focus-Inducing
Virus and long terminal repeat alterations accompany the increased leukemogenicity of the Mo+PyF101 variant of Moloney murine leukemia virus after Intraperitoneal Inoculation., Journal of Virology 69, 1037-1043.
Ben-David, Y., Giddens, E. B., Letwin, K., and Bernstein, A. (1991).
Erythroleukemia induction by Friend murine leukemia virus: insertional activation of a new member of the ets gene family, Fli-1, closely linked to c-ets-1., Genes & Development 5, 908-918.
Benveniste, R., Callahani, R., Sherr, C., Chapman, V., and Todaro, G. (1977).
Two distinct endogenous type C viruses isolated from the Asian rodent Mus cervicolor: Conservation of viral gene sequences in related rodent species., Journal of Virology 21, 849-862.
Bergeron, D., Poliquin, L., Kozak, A. C., and Rassart, E. (1991). Identification of
a common viral integration region in Cas-Br-E murine leukemia virus-induced non-T-, non-B-cell lymphomas., Journal of Virology 65, 7-15.
Berlioz, C., and Darlix, J.-L. (1995). An internal ribosomal entry mechanism
promotes translation of murine leukemia virus gag polyprotein precursors., Journal of Virology 69, 2214-2222.
Best, S., Tissier, P. L., Towers, G., and Stoye, J. P. (1996). Positional cloning of
the mouse retrovirus restriction gene Fv1., Nature 382, 826-829. Birnboim, H. C., and Doly, C. (1979). A rapid alkaline extraction procedure for
screening recombinant plasmid DNA., Nucl Acids Res 7, 1513-1523.

Literaturverzeichnis 120
Bishop, K. N., Bock, M., Towers, G., and Stoye, J. P. (2001). Identification of
the regions of Fv1 necessary for murine leukemia virus restriction., Journal of Virology 75, 5182-5188.
Blackwood, E. M., and Eisenmann, R. N. (1991). Max: A helix-loop-helix zipper
protein that forms a sequence-specific DNA-binding complex with Myc., SCIENCE 251, 1211-1217.
Bonham, L., Wolgamot, G., and Miller, A. D. (1997). Molecular cloning of Mus
dunni endogenous virus: an unusual retrovirus in a new murine viral interference Ggoup with a wide host range., Journal of Virology 71, 4663-4670.
Brightman, B. K., Davis, B. R., and Fan, H. (1990). Preleukemic hematopoietic
hyperplasia induced by Moloney murine leukemia virus is an indirect consequence of viral infection., Journal of Virology 64, 4582-4584.
Brightman, B. K., Rein, A., Trepp, D. J., and Fan, H. (1991). An enhancer
variant of Moloney murine leukemia virus defective in leukemogenesis does not generate detectable mink cell focus-inducing virus in vivo., Proc Natl Acad Sci USA 88, 2264-2268.
Chatis, A. P., Holland, A. C., Hartley, J. W., Rowe, W. P., and Hopkins, N.
(1983). Role for the 3' end of the genome in determining disease specificity of Friend and Moloney murine leukemia viruses., Proc Natl Acad Sci USA 80, 4408-4411.
Coffin, J. M., Hughes, S. H., and Varmus, H. E. (1997). Retroviruses.
(New York, Cold Spring Harbor Laboratory Press). Corbin, A., Prats, A. C., Darlix, J.-L., and Sitbon, M. (1994). A nonstructural
gag-encoded glycoprotein precursor is necessary for efficient spreading and pathogenesis of murine leukemia viruses., Journal of Virology 68, 3857-3867.
Corcoran, L. M., Adams, J. M., Dunn, A. R., and Cory, S. (1984). Murine T
lymphomas in which the cellular myc oncogene has been activated by retroviral insertion., Cell 37, 113-122.
Cuypers, H. T., Selten, G., Quint, W., Zijlstra, M., Maandag, E. R., Boelens, W.,
van Wezenbeek, P., Melief, C., and Berns, A. (1984). Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region., Cell 37, 141-150.
De´jardin, J. R., Bompard-Mare´chal, G., Audit, M., Hope, T. J., Sitbon, M., and
Mougel, M. N. (2000). A novel subgenomic murine leukemia virus RNA transcript results from alternative splicing., Journal of Virology 74, 3709-3714.

Literaturverzeichnis 121
DeKoter, R. P., and Singh, H. (2000). regulation of B lymphocyte and macrophage development by graded expression of PU.1., SCIENCE 288, 1439-1441.
DesGrosseilliers, L., and Jolicoeur, P. (1984). The tandem direct repeats within
the long terminal repeat of murine leukemia viruses are the primary determinant of their leukomogenic potential., Journal of Virology 52, 945-952.
DiFronzo, N. L., and Holland, A. C. (1999). Sequence-specific and/or
stereospecific constraints of the U3 enhancer elements of MCF 247-W are important for pathogenecity., Journal of Virology 73, 234-241.
Eiden, M. V., Farrell, K. B., and Wilson, C. A. (1994). Glycosylation-dependent
inactivation or ecotropic murine leukemia virus receptor., Journal of Virology 68, 626-631.
Ellermann, V., and Bang, O. (1908). Experimentelle Leukämie bei Hühnern.,
Zentralalbl. Bakteriol. Parasitenkd. Infectionskr. Hyg. Abt. Orig. 46, 595-609.
Fan, H. (1994). Retroviruses and their role in cancer. In The Retroviridae, J. A.
Levy, ed. (New York, Plenum Press), pp. 313-362. Fan, H., Chute, H., Chao, E., and Pattengale, P. K. (1988). Leukemogenicity of
Moloney murine leukemia viruses carrying polyoma enhancer sequences in the long terminal repeat is dependent on the nature of the inserted Polyoma sequences., Virology 166, 58-65.
Friel, J., Hughes, D., Pragnell, I., Stocking, C., Laker, C., Nowock, J., Ostertag,
W., and Padua, R. A. (1990). The malignant histiocytosis sarcoma virus, a recombinant of Harvey murine sarcoma virus and Friend mink cell focus-forming virus, has acquired myeloid transformation specificity by alterations in the long terminal repeat., Journal of Virology 64, 369-378.
Fujisawa, R., McAtee, F. J., Favara, C., Hayes, S. F., and Portis, J. L. (2001).
N-terminal cleavage fagment of glycosylated Gag is incorporated into murine oncornavirus particles., Journal of Virology 75, 11239-11243.
Fujisawa, R., McAtee, F. J., Wehrly, K., and Portis, J. L. (1998). The
neuroinvasiveness of a murine retrovirus is influenced by a dileucine-containing sequence in the cytoplasmic tail of glycosylated Gag., Journal of Virology 72, 5619-5625.
Fujisawa, R., McAtee, F. J., Zirbel, J. H., and Portis, J. L. (1997).
Characterization of glycosylated Gag expressed by a neurovirulent murine leukemia virus: Identification of differences in processing in vitro and in vivo., Journal of Virology 71, 5355-5360.

Literaturverzeichnis 122
Gallo, R. C., Salahuddin, S. Z., Popovic, M., Shearer, G. M., Kaplan, M., Haynes, B. F., Palker, T. J., Redfield, R., Oleske, J., Safai, B., et al. (1984). Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS., SCIENCE 224, 500-503.
Gardner, M. B. (1978). Type C viruses of wild mice: Characterization and
natural history of amphotropic, ecotropic, and xenotropic MuLV., CTMI, 215-269.
Golemis, E., Li, Y., Fredrickson, T. N., Hartley, J. W., and Hopkins, N. (1989).
Distinct segments within the Eehancer region collaborate to specify the type of leukemia induced by nondefective Friend and Moloney viruses., Journal of Virology 63, 328-337.
Golemis, E., Speck, N. A., and Hopkins, N. (1990). Alignment of U3 region
sequences of mammalian Type C viruses: Identification of highly conserved motifs and implications for enhancer design., Journal of Virology 64, 534-542.
Grez, M., Zornig, M., Nowock, J., and Ziegler, M. (1991). A single point mutation
activates the Moloney murine leukemia virus long terminal repeat in embryonal stem cells., Journal of Virology 65, 4691-4698.
Han, J.-Y., Cannon, P. M., Lai, K.-M., Zhao, Y., Eiden, M. V., and Anderson, W.
F. (1997). Identification of envelope protein residues required for the expanded host range of 10A1 murine leukemia virus., Journal of Virology 71, 8103-8108.
Hanecak, R., Pattengale, P. K., and Fan, H. (1988). Addition or substitution of
simian virus 40 enhancer sequences into the Moloney murine leukemia virus (M-MuLV) long terminal repeat yields infectious M-MuLV with altered biological properties., Journal of Virology 62, 2427-2436.
Hann, S. R., Thompson, C. B., and Eisenmann, R. N. (1985). C-myc onkogenic
protein synthesis is dependent of the cell cycle in human and avian cells., Nature 314, 366-369.
Harbers, K., Schnieke, A., Stuhlmann, H., Jähner, D., and Jaenisch, R. (1981).
DNA methylation and gene expression: Endogenous retroviral genome becomes infectious after molecular cloning., Proc Natl Acad Sci USA 78, 7609-7613.
Hardy, R. R., Carmack, C. E., Shinton, S. A., Kemp, J. D., and Hayakawa, K.
(1991). Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow., J Exp Med 173, 1213-1225.
Hartley, J. W., and Rowe, W. P. (1975). Clonal cell lines from feral mouse
embryo which lack host-range restrictions for murine leukemia viruses., Virology 65, 128-134.

Literaturverzeichnis 123
Hartley, J. W., Wolford, N. K., Old, L. J., and Rowe, W. P. (1977). A new class of murine leukemia virus associated with development of spontaneous lymphomas., Proc Natl Acad Sci USA 74, 789-792.
Hartley, J. W., Yetter, R. A., and Morse, H. C., III (1983). A mouse gene on
chromosome 5 that restricts infectivity of mink cell focus-forming recombinant murine leukemia viruses., J Exp Med 158, 16-24.
Hein, S., Prassolov, V., Zhang, Y., Ivanov, D., Löhler, J., Ross, S. R., and
Stocking, C. (2003). Sodium-dependent myo-inositol transporter 1 (SMIT1) is a cellular receptor for Mus cervicolor M813 murine leukemia virus., Journal of Virology 77, 1-7.
Hoggan, M. D., Buckler, C. E., Sears, J. F., Rowe, W. P., and Martin, M. A.
(1983). Organization and stability of endogenous xenotropic murine leukemia virus proviral DNA in mouse genomes., Journal of Virology 45, 473-477
. Holland, A. C., Thomas, C. Y., Chattopadhyay, S. K., Koehne, C., and
O'Donnell, P. V. (1989). Influence of enhancer sequences on thymotropism and leukemogenicity of Mink Cell Focus-Forming Virus., Journal of Virology 63, 1284-1292.
Hu, W. S., and Temin, H. M. (1990). Genetic consequence of packaging two
RNA genomes in one retroviral particle: Pseudodiploidy and higher rate of genetic recombination., Proc Natl Acad Sci USA 87, 1556-1560.
Ikeda, H., Laiget, F., Martin, M. A., and Repaske, R. (1985). Characterization of
a molecularly cloned retroviral sequence associated with Fv-4 resistance., Journal of Virology 55, 768-777.
Ish-Horowicz, D., and Burke, J. F. (1981). Rapid and efficient cosmid cloning.,
Nucl Acids Res 9, 2989-2998. Janeway, C. A., Travers, P., Walport, M., and Capra, J. D. (1999).
Immunobiology. The immunesystem in health and disease., Fourth Edition edn, Elsevier Science Ltd/ Garland Publishing).
Januszeski, M. M., Cannon, P. M., Chen, D., Rozenberg, Y., and Anderson, W.
F. (1997). Functional analysis of the cytoplasmic tail of Moloney murine leukemia vrus envelope protein., Journal of Virology 71, 3613-3619.
Jenkins, N. A., Copeland, N. G., Taylor, B. A., and Lee, B. K. (1982).
Organization, distribution, and stability of endogenous ecotropic murine leukemia virus DNA sequences in chromosomes of Mus musculus., Journal of Virology 43, 26-36.
Jonkers, J., and Berns, A. (1996). Retroviral insertional mutagenesis as a
strategy to identify cancer genes., Biochimica et Biophysica Acta 1287, 29-57.

Literaturverzeichnis 124
Jung, Y. T., Lyu, M. S., Buckler-White, A., and Kozak, A. C. (2002). Characterization of a polytropic murine leukemia virus proviral sequence associated with the virus resistence gene Rmcf of DBA/2 mice., Journal of Virology 76, 8218-8224.
Kavanaugh, M. P., Miller, D. G., Zhang, W., Law, W., Kozak, S. L., Kabat, D.,
and Miller, A. D. (1994). Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters., Proc Natl Acad Sci USA 91, 7071-7075.
Kelly, K., Cochran, B. H., Stiles, C. D., and Leder, P. (1983). Cell-specific
regulation of the c-myc gene by lymphocyte mitogens and platelet-derived-growth-factor., Cell 35, 603-610.
Kozak, A. C. (1985). Susceptibility of wild mouse cells to exogenous infection
with xenotropic leukemia viruses: Control by a single dominant locus on Chromosome 1., Journal of Virology 55, 690-695.
Kozak, A. C., and Ruscetti, S. K. (1992). Retroviruses in rodents. in the
retroviridae, J. A. Levy, ed. (New York, Plenum Press), pp. 405-481. Lavignon, M., and Evans, L. (1996). A multistep process of leukemogenesis in
Moloney murine leukemia virus-infected mice that is modulated by retroviral pseudotyping and interference., Journal of Virology 70, 3852-3862.
Lavignon, M., Richardson, J., and Evans, L. H. (1997). A small region of the
ecotropic murine leukemia virus (MuLV) gag gene profoundly influences the types of polytropic MuLVs generated in mice., Journal of Virology, 8923-8927.
Levy, J. A., Hoffmann, A. D., Kramer, S. M., A., L. J., Shimabukuro, J. M., and
Oshiro, L. S. (1984). Recovery of AIDS-associated retrovirus from patients with AIDS or AIDS-related conditions and from clinically healthy individuals., J Infect Dis 152, 734-738.
Li, J.-P., and Baltimore, D. (1991). Mechanism of leukomogenesis induced by
mink cell focus-forming murine leukemia viruses., Journal of Virology 65, 2408-2414.
Li, J.-P., D'Andrea, A. D., Lodish, H. F., and Baltimore, D. (1990). Activation of
cell growth by binding of Friend spleen focus-forming virus gp55 glycoprotein to the erythropoietin receptor., Nature 343, 762-764.
Li, J.-P., Hu, H.-O., Niu, Q.-T., and Fang, C. (1995). Cell surface activation of
the erythropoietin receptor by Friend spleen focus-forming virus gp55., Journal of Virology 69, 1714-1719.

Literaturverzeichnis 125
Linemeyer, D. L., Ruscetti, S. K., Menke, J. G., and Scolnick, E. M. (1980). Recovery of biologically active spleen focus-forming virus from molecularly cloned spleen focus-forming virus-pBR32 circular DNA by cotransfection with infectious type C retroviral DNA., Journal of Virology 35, 710-721.
Lund, A. H., Turner, G., Trubetskoy, A., Verhoeven, E., Wientjens, E., Hulsman,
D., Russell, R., DePinho, R. A., Lenz, J., and van Lohuizen, M. (2002). Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice., Nature Genetics 32, 160-165.
Manley, N. R., O'Connell, M., Sun, W., Speck, N. A., and Hopkins, N. (1993).
Two factors that bind to highly conserved sequences in mammalian type C retroviral enhancers., Journal of Virology 67, 1967--1975.
Mann, R., and Baltimore, D. (1985). Varying the position of a retrovirus
packaging sequence results in the encapsidation of both the unspliced and spliced RNAs., Journal of Virology 54, 401-407.
Mann, R., Mulligan, R. C., and Baltimore, D. (1983). Construction of a retrovirus
packaging mutant and its use to produce helper-free defective retrovirus., Cell 33, 153-159.
Marin, M., Tailor, C. S., Nouri, A., Kozak, S. L., and Kabat, D. (1999).
Polymorphisms of the cell surface receptor control mouse susceptibilities to xenotropic and polytropic leukemia viruses., Journal of Virology 73, 9362-9368.
Marin, M., Tailor, C. S., Nouri, A., Kozak, S. L., and Kabat, D. (2000). Sodium
dependent neutral amino acid transporter type 1 is an auxilliary receptor for baboon endogenous retrovirus., Journal of Virology 74, 8085-8093.
Melkun, E., Pilione, M., and Paulson, R. F. (2002). A naturally occurring point
substitution in Cdc25A, and not Fv2/Stk, is associated with altered cell-cycle status of early erythroid progenitor cells., BLOOD 100, 3804-3811.
Mikkers, H., Allen, J., Knipscheer, P., Romeyn, L., Hart, A., Vink, E., and Berns,
A. (2002). High-throughput retroviral tagging to identify components of specific signaling pathways in cancer., Nature Genetics 32, 153-159.
Miller, A. D. (1996). Cell-surface receptors for retroviruses and implications for
gene transfer., Proc Natl Acad Sci USA 93, 11407-11413. Miller, D. G., and Miller, A. D. (1994). A familiy of retroviruses that utilize related
phosphate transporters for cell entry., Journal of Virology 68, 8270-8276. Mooslehner, K., Karls, U., and Harbers, K. (1990). Retroviral integration sites in
transgenic mov mice frequently map to the vicinity of transcribed DNA regions., Journal of Virology 64, 3056-3058.

Literaturverzeichnis 126
Moreau-Gachelin, F., Ray, D., Mattei, M.-G. v., Tambourin, P., and Tavitian, A. (1989). The putative oncogene Spi-1: murine chromosomal localization and transcriptional activation in murine acute erythroleukemias., Oncogene 4, 1449-1456.
Morita, S., Kojima, T., and Kitamura, T. (2000). Plat-E: an efficient and stable
system for transient packaging of retroviruses., Gene Therapy 7, 1063-1066.
Münk, C., Löhler, J., Prassolov, V., Just, U., Stockschläger, M., and Stocking,
C. (1997). Amphotropic murine leukemia viruses induce spongiform encephalomyelopathy., Proc Natl Acad Sci USA 94, 5837-5842.
Münk, C., Prassolov, V., Rodenburg, M., Kalinin, V., Löhler, J., and Stocking, C.
(2003). 10A1-MuLV but not the related amphotropic 4070A MuLV is highly neurovirulent: importance of sequences upstream of the structural Gag coding region., Virology 309.
Münk, C., Thomsen, S., Stocking, C., and Löhler, J. (1998). Murine leukemia
virus recombinants that use phosphate transporters for cell entry induce similar spongiform encephalomyelopahties in newborn mice., Virology 252, 318-323.
Nielsen, L. B., Pedersen, F. S., and Pedersen, L. (2001). Expression of type III
sodium-dependent phosphate transporters/retroviral receptors mRNAs during osteoblast differentiation., Bone 28, 160-166.
Orkin, S. H. (2000). Diversification of haematopoietic stem cells to specific
lineages., Nature Reviews Genetics 1, 57-64. Ostertag, W., Stocking, C., Johnson, G. R., Kluge, N., Kollek, R., Franz, T., and
Hess, N. (1987). Transforming genes and target cells of murine spleen focus-forming viruses., Advanced Cancer Research 48, 193-355.
Ostertag, W., Vehmeyer, K., Fagg, B., Pragnell, I., Paetz, W., Le Bousse, M. C.,
Smadja-Joffe, F., Klein, B., Jasmin, C., and Eisen, H. (1980). Myeloproliferative virus, a cloned murine sarcoma virus with spleen focus-forming properties in adult mice., Journal of Virology 33, 573-582.
Ott, D. E., Friedrich, R., and Rein, A. (1990). Sequence analysis of amphotropic
and 10A1 murine leukemia viruses: Close relationship to mink cell focus-inducing viruses., Journal of Virology 64, 757-766.
Ott, D. E., Keller, J., and Rein, A. (1994). 10A1 MuLV induces a murine
leukemia that expresses hematopoietic stem cell markers by a mechanism that includes fli-1 integration., Virology 205, 563-568.
Ott, D. E., Keller, J., Sill, K., and Rein, A. (1992). Phenotypes of murine
leukemia virus-induced tumors: Influence of 3' viral coding sequences., Journal of Virology 66, 6107-6116.

Literaturverzeichnis 127
Persons, D. A., Paulson, R. F., Loyd, M. R., Herley, M. T., Bodner, S. M., Bernstein, A., Correll, P. H., and Ney, P. A. (1999). Fv2 encodes a truncated form of the Stk receptor tyrosine kinase., Nature Genetics 23, 159-165.
Portis, J. L., Czub, S., Garon, C. F., and McAtee, F. J. (1990).
Neurodegenerative disease induced by the wild mouse ecotropic retrovirus is markedly accelerated by the long terminal repeat and gag-pol sequences from nondefective Friend murine leukemia virus., Journal of Virology 64, 1648-1656.
Portis, J. L., Fujisawa, R., and McAtee, F. J. (1996). The glycosylated Gag-
protein of MuLV is a determinant of neuroinvasiveness: Analysis of second site revertants of mutant MuLV lacking expression of this protein., Virology 226, 384-392.
Prassolov, V., Hein, S., Ziegler, M., Ivanov, D., Münk, C., Löhler, J., and
Stocking, C. (2001). Mus cervicolor murine leukemia virus isolate M813 belongs to a unique receptor interference group., Journal of Virology 75, 4490-4498.
Prendergast, G. C., Lawe, D., and Ziff, E. B. (1991). Association of Myn, the
murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and Ras cotransformation., Cell 65, 395-407.
Rabbitts, P. H., Watson, J. W., Lamond, A., Forster, A., Stinson, M. A., Evan,
G., Fischer, W., Atherton, E., Shepard, M. A., and Rabbitts, T. H. (1985). Metabolism of c-myc gene products: c-myc mRNA and protein expression in the cell cycle., The EMBO Journal 4, 2009-2015.
Rasheed, S., Pal, B. K., and Gardner, M. B. (1982). Characterization of a highly
oncogenic murine leukemia virus from wild mice., Int J Cancer 29, 345-350.
Rein, A., Mirro, J., Haynes, J. G., Ernst, S. M., and Nagashima, K. (1994).
Function of the cytoplasmic domain of a retroviral transmembrane protein: p15E-p2E cleavage activates the membrane fusion capability of the murine leukemia virus Env protein., Journal of Virology 68, 1773-1781.
Reizer, J., Reizer, A., and Saier, M., Jr. (1994). A functional superfamily of
sodium/solute symporters., Biochimica et Biophysica Acta 1197, 133-166.
Reuss, F. U., Berdel, B., Ploss, M., and Heber, R. (2001). Replication of
enhancer-deficient amphotropic murine leukemia virus in human cells., Proc Natl Acad Sci USA 98, 10898-10903.

Literaturverzeichnis 128
Rohdewohld, H., Weiher, H., Reik, W., Jaenisch, R., and Breindl, M. (1987). Retrovirus integration and chromatin structure: Moloney murine leukemia proviral integration sites map near DNAse I-hypersensitive sites., Journal of Virology 61, 336-343.
Rous, P. (1911). A sarcoma of the fowl transmissible by an agent separable
from the tumor cells., J Exp Med 13, 397-411. Ruscetti, S. K., Davis, L., Feild, J., and Oliff, A. (1981). Friend murine leukemia
virus-induced leukemia is associated with the formation of mink cell focus-inducing viruses and is blocked in mice expressing endogenous mink cell focus-inducing xenotropic viral envelope genes., J Exp Med 154, 907-920.
Sabatino, D. E., Do, B. Q., Pyle, L. C., Seidel, N. E., Girard, L. J., Spratt, S. K.,
Orlic, D., and Bodine, D. M. (1997). Amphotropic or gibbon ape leukemia virus retrovirus binding and transduction correlates with the level of receptor mRNA in human hematopoietic cell lines., BLOOD 23, 422-433.
Saier, M., Jr. (2000). A functional-phylogenetic classification system for
transmembrane solute transporters., Microbiol Mol Bio Rev 64, 354-411. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular cloning, A
laboratory manual., 2nd Edition edn, Cold Spring Harbor Laboratory Press.
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-
terminating inhibitors., Proc Natl Acad Sci USA 74, 5463-5467. Scherdin, U., Rhodes, K., and Breindl, M. (1990). Transcriptionally active
genome regions are preferred targets for retroviral integration., Journal of Virology 64, 907-912
Schröder, A. R. W., Shinn, P., Chen, H., Berry, C., Ecker, J. R., and Bushman,
F. (2002). HIV-1 integration in the human genome favors active genes and local hotspots., Cell 110, 521-529.
Sitbon, M., Evans, L., Nishio, J., Wehrly, K., and Chesebro, B. (1986). Analysis
of two strains of Friend murine leukemia viruses differing in ability to induce early splenomegaly: lack of relationship with generation of recombinant mink cell focus-forming viruses., Journal of Virology 57, 389-393.
Speck, N. A., Renjifo, B., Golemis, E., Fredrickson, T. N., Hartley, J. W., and
Hopkins, N. (1990). Mutations of the core or adjacent LVb elements of the Moloney murine leukemia virus enhancer alters disease specificity., Genes & Development 4, 233-242.
Stanton, L. W., Watt, R., and Marcu, K. B. (1983). Translocation, breakage and
truncated transcripts of c-myc oncogene in murine plasmacytomas., Nature 303, 401-406.

Literaturverzeichnis 129
Stocking, C., Koller, R., Bergholz, U., and Ostertag, W. (1985). Long terminal repeat sequences impart hematopoietic transformation properties to the myeloproliferative sarcoma virus., Proc Natl Acad Sci USA 82, 5746-5750.
Stoye, J. P., and Coffin, J. M. (1987). The four classes of endogenous murine
leukemia virus: Structural relationships and potential for recombination., Journal of Virology 61, 2659-2669.
Suzuki, T., Shen, H., Akagi, K., Morse, H. C., III, Malley, J. D., Naiman, D. Q.,
Jenkins, N. A., and Copeland, N. G. (2002). New genes involved in cancer identified by retroviral tagging., Nature Genetics 32, 166-173.
Tailor, C. S., Nouri, A., and Kabat, D. (2000). Cellular and species resistance to
murine amphotropic, gibbon ape, and feline subgroup C leukemia viruses is strongly influenced by receptor expression levels and by receptor masking mechanisms., Journal of Virology 74, 9797-9801.
Tailor, C. S., Nouri, A., Lee, C. G., Kozak, A. C., and Kabat, D. (1999). Cloning
and characterization of a cell surface receptor for xenotropic and polytropic murine leukemia viruses., Proc Natl Acad Sci USA 96.
Takatsuki, K., Uchiyama, J., Sagawa, K., and Yodoi, J. (1977). Adult T-cell
leukemia in Japan. In Topics of hematology, S. e. a. Senos, ed. (Amsterdam, Excerpta Medica), pp. 73-77.
Taylor, G. M., Gao, Y., and Sanders, D. A. (2001). Fv-4: Identification of the
defect in Env and the mechanism of resistance to ecotropic murine leukemia virus., Journal of Virology 75, 11244-11248.
Tomonaga, K., and Coffin, J. M. (1999). Structures of endogenous nonecotropic
murine leukemia virus (MLV) long terminal repeats in wild mice: Implication for evolution of MLVs., Journal of Virology 73, 4327-4340.
Uchiyama, J., Yodoi, J., Sagawa, K., Takatsuki, K., and Uchino, H. (1977). Adult
T-cell leukemia: Clinical and hematologic features of 16 cases., BLOOD 50, 481-492.
van Lohuizen, M., Breuer, M., and Berns, A. (1989). N-myc is frequently
activated by proviral insertion in MuLV-induced T cell lymphomas., The EMBO Journal 8, 133-136.
van Lohuizen, M., Verbeek, S., Scheijen, B., Wientjens, E., van der Gulden, H.,
and Berns, A. (1991). Identification of cooperating oncogenes in Eµ-myc transgenic mice by provirus tagging., Cell 65, 737-752.
Vijaya, S., Steffen, D. L., and Robinson, H. L. (1986). Acceptor sites for
retroviral integrations map near DNAse I-hypersensitive sites in chromatin., Journal of Virology 60, 683-692.

Literaturverzeichnis 130
von Kalle, C., Kiem, H. P., Goehle, S., Darovsky, B., Heimfeld, S., Torok-Storb, B., Storb, R., and Schuening, F. G. (1994). Increased gene transfer into human hematopoietic progenitor cells by extended in vitro exposure to a pseudotyped retroviral vector., BLOOD 84, 2890-2897.
Wang, H., Klamo, E., Kuhmann, S. E., Kozak, S. L., Kavanaugh, M. P., and
Kabat, D. (1996). Modulation of ecotropic murine retroviruses by N-linked glycosylation of the cell surface receptor/amino acid transporter., Journal of Virology 70, 6884-6891.
Wang, S., Wang, Q., Crute, B. E., Melnikowa, I. N., Keller, S. R., and Speck, N.
A. (1993). Cloning and charcterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor., Molecular and Cellular Biology 13, 3324-3339.
Weiss, R. A. (1993). Cellular receptors and viral glycoproteins involved in
retrovirus entry. In the retroviridae, J. A. Levy, ed. (New York, Plenum Press), pp. 1-108.
White, J. M. (1992). Membrane fusion., SCIENCE 258, 917-924. Wilson, C. A., Eiden, M. V., Anderson, W. B., Csaba, L., and Olah, Z. (1995).
The dual-function hamster receptor for amphotropic murine leukemia Vvrus (MuLV), 10A1 MuLV, and gibbon ape leukemia virus is a phosphate symporter., Journal of Virology 69, 534-537.
Wilson, C. A., Farrell, K. B., and Eiden, M. V. (1994). Properties of a unique
form of the murine amphotropic leukemia virus receptor expressed on hamster cells., Journal of Virology 68, 7697-7703.
Wolff, L., Koller, R., Hu, X., and Anver, M. R. (2003). A Moloney murine
leukemia virus-based retrovirus with 4070A long terminal repeat sequences induces a high incidence of myeloid as well as lymphoid neoplasms., Journal of Virology 77, 4965-4971.
Yang, Y.-L., Guo, L., Xu, S., Holland, A. C., Kitamura, T., Hunter, K., and
Cunningham, J. M. (1999). Receptors for polytropic and xenotropic mouse leukaemia viruses encoded by a single gene at Rmc1., Nature Genetics 21, 216-219.
Yoshimura, F. K., Wang, T., and Nanua, S. (2001). Mink cell focus-forming
murine leukemia virus killing of mink cells involves apoptosis and superinfection., Journal of Virology 75, 6007-6015.







