

Die Bedeutung Adenosin-abhängiger Signalkaskaden
in präklinischen Modellen
der systemischen Sklerose
Medizinische Klinik III
Klinik für Rheumatologie und Immunologie
Der Medizinischen Fakultät
der
Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
Dr. med. dent.
vorgelegt von
Florian Philip Layritz
aus
Erlangen

Als Dissertation genehmigt von der
Medizinischen Fakultät der Friedrich-Alexander-Universität
Erlangen-$ürnberg
Tag der mündlichen Prüfung:
04.04. 2016
Vorsitzender des Promotionsorgans: Prof. Dr. med. Dr. h.c. Jürgen Schüttler
Gutachter: Prof. Dr. med. Jörg Distler
Gutachter: Prof. Dr. med. univ. Georg Schett

Für meine Eltern

Gliederung
1. Zusammenfassung .................................................................................................. 1
2. Summary ................................................................................................................. 3
3. Einleitung ................................................................................................................ 5
3.1 Überblick über die Systemische Sklerose ........................................................... 5
3.1.1 Klinische Manifestation und Epidemiologie ............................................... 5
3.1.2 Pathogenese der systemischen Sklerose ...................................................... 6
3.2 Mausmodelle zur Erforschung der Systemischen Sklerose ................................ 8
3.2.1 Mausmodell der sklerodermiformen chronischen Graft-versus-Host Erkrankung . 8
3.2.2 Fra-2-Mausmodell ....................................................................................... 9
3.3 Das Adenosin-Rezeptor-System ........................................................................ 11
3.3.1 Die Adenosinrezeptoren: Aufbau und Funktion ........................................ 11
3.3.2 Der Adenosin-Metabolismus ..................................................................... 12
3.3.3 Störungen des Adenosin-Rezeptor-Systems in der Fibrose ....................... 13
4. Fragestellung ........................................................................................................ 15
5. Material und Methoden ....................................................................................... 16
5.1 cGvHD-Mausmodell ......................................................................................... 16
5.1.1 Hintergrundinformationen zum cGvHD-Mausmodell............................... 16
5.1.2 Klinische Verlaufskontrolle ....................................................................... 17
5.1.3 Analyse der Hautveränderung ................................................................... 18
5.1 3.1 Histologische Analyse ............................................................................. 18
1.) Hämatoxylin-Eosin-Färbung ...................................................................... 18
2.) Masson-Trichrom-Färbung ........................................................................ 18
5.1.3.2 Immunhistochemische Analyse der Myofibroblasten ................................. 19
5.1.3.3 Hydroxyprolin-Assay .............................................................................. 19
5.2 Fra-2-Mausmodell ............................................................................................ 21

5.2.1 Hintergrundinformationen zum Fra-2-Mausmodell .................................. 21
5.2.2 Analyse der Hautveränderung ................................................................... 21
5.2.2.1 Histologische Analyse: Sirius Red-Färbung .............................................. 22
5.2.2.2 Immunhistochemische Analyse der Myofibroblasten ................................. 22
5.2.2.3 Hydroxyprolin-Assay ............................................................................. 22
5.2.3 Analyse der Lungenveränderung ............................................................... 23
5.2.3.1 Histologische Analyse: Sirius Red-Färbung .............................................. 23
5.2.3.2 Immunhistochemische Analyse der Myofibroblasten ................................. 23
5.2.3.3 Hydroxyprolin-Assay ............................................................................. 23
5.2.4 Analyse der Lungenarterien mittels Hämatoxylin-Eosin-Färbung ............ 23
5.2.4.1 Analyse der Gefäßwanddicke .................................................................. 24
5.2.4.2 Quantifizierung der stark stenosierten Lungenarterien ............................... 24
5.3 Statistische Auswertung ........................................................................................... 24
6. Ergebnisse ............................................................................................................. 24
6.1 cGvHD-Mausmodell ......................................................................................... 25
6.1.1 Klinischer Verlauf...................................................................................... 25
1. Körpergewicht ............................................................................................... 25
2. Klinische Beurteilung ..................................................................................... 26
6.1.2 Histologische Veränderungen der Haut ..................................................... 27
6.1.2.1 Histologische Analyse ............................................................................. 27
1.) Hämatoxylin-Eosin-Färbung ...................................................................... 27
2.) Masson-Trichrom-Färbung ........................................................................ 28
6.1.2.2 α-SMA Immunhistologie ......................................................................... 30
6.1.2.2 Hydroxyprolin-Assay .............................................................................. 31
6.2 Fra-2-Mausmodell ............................................................................................ 33
6.2.1 Histologische Veränderungen der Haut ..................................................... 33
6.2.1.1 Histologische Analyse: Sirius Red-Färbung .............................................. 33
6.2.1.2 α-SMA Immunhistologie ......................................................................... 35

6.2.1.3 Hydroxyprolin-Assay .............................................................................. 36
6.2.2 Histologische Veränderungen der Lunge .................................................. 38
6.2.2.1 Histologische Analyse: Sirius Red-Färbung .............................................. 38
6.2.2.2 α-SMA Immunhistologie ......................................................................... 40
6.2.2.3 Hydroxyprolin-Assay .............................................................................. 41
6.2.3 Ergebnisse zur Umgestaltung der Lungenarterien ..................................... 42
6.2.3.1 Gefäßwanddicke: Hämatoxylin-Eosin-Färbung ......................................... 42
6.2.3.2 Stark stenosierte Lungenarterien: Hämatoxylin-Eosin-Färbung .................. 43
7. Diskussion ............................................................................................................. 45
8. Literaturverzeichnis ............................................................................................. 49
9. Abkürzungsverzeichnis........................................................................................ 57
10. Chemikalien/Materialien ................................................................................... 59
11. Danksagung ........................................................................................................ 61
12. Lebenslauf ........................................................................................................... 62

1
1. Zusammenfassung
Hintergrund und Ziele der Arbeit. Bei der systemischen Sklerose (SSc) handelt es
sich um eine autoimmun-vermittelte seltene Krankheit mit signifikant erhöhter Sterb-
lichkeit. Die Kennzeichen dieser Erkrankung sind u.a. der Verlust von Kapillaren,
die Umformung von Lungenarterien und eine Gewebefibrose aufgrund einer erhöh-
ten Kollagenproduktion pathologisch aktivierter Fibroblasten. Diese vermehrte Pro-
duktion von Kollagen beruht unter anderem auf einer durch Adenosin gesteuerten
Aktivierung des A2A-Rezeptors. Der Gewebespiegel von extrazellulärem Adenosin
wird hauptsächlich durch das Enzym Adenosin-Desaminase (ADA) gesteuert. Tier-
experimentelle Studien zeigten, dass Mäuse mit einem Mangel an ADA einen daraus
resultierenden Anstieg von endogenem Adenosin haben und eine Fibrose in der
Haut, der Lunge und anderen Organen entwickelten, welche durch Gabe eines A2A-
Rezeptor-Antagonisten verhindert werden konnte. Ziel dieser Arbeit war es, anhand
tierexperimenteller Mausmodelle die Wirkung rekombinanter Adenosin-Desaminase
(r-ADA) bei der Entstehung und der Therapie einer SSc zu beurteilen.
Methoden. Die Wirksamkeit r-ADA wurde anhand zweier verschiedener Mausmo-
delle untersucht. Zuerst wurde die Wirksamkeit r-ADA anhand eines Mausmodells
der sklerodermiformen chronischen Graft-versus-Host-Erkrankung (cGvHD) analy-
siert. Mittels allogener Stammzelltransplantation bei sich in minor histocompatibility
Antigenen unterscheidenden Mäusen wurden klinische und histopathologische
Symptome einer cGvHD induziert und die Wirksamkeit einer nach Transplantation
beginnenden Behandlung mit r-ADA hinsichtlich Krankheitsverlauf und Ausprä-
gungsgrad untersucht. Hierzu wurden Hautschnitte zur Bestimmung der Hautdicke
histologisch gefärbt, die Anzahl der Myofibroblasten wurde immunhistologisch un-
tersucht. Ein Hydroxyprolin-Assay diente zur Quantifizierung des Kollagengehaltes.
Der Krankheitsverlauf wurde anhand einer zuvor definierten klinischen Bewertung
zusammen mit dem Körpergewicht täglich ab Stammzelltransplantation erhoben. In
einem zweiten Modell wurde die Wirksamkeit von r-ADA bei Fos-related-antigen-2
(Fra-2) transgenen Mäusen untersucht. Fra-2 ist ein Transkriptionsfaktor der Aktiva-
tor-Protein-1 (AP-1)-Familie, dessen transgene Überexpression zu Haut- und Lun-
genfibrose und Gefäßveränderungen (Lungenarterienstenose sowie Gefäßwandverdi-
ckung) führt, welche charakteristische Symptome einer SSc sind. Die Wirksamkeit
der Therapie mit r-ADA wurde in der Haut und Lunge untersucht. Die analysierten
Punkte der Haut und der Lunge waren denen der cGvHD identisch. Zusätzlich er-

2
folgte eine Beurteilung der Lungenfibrose und der Veränderungen von Lungenarte-
rien mit Hilfe von histologischen Färbungen.
Ergebnisse und Beobachtungen. In beiden Modellen zeigte die Gabe von r-ADA
antifibrotische Effekte. Im Modell der cGvHD linderte diese sowohl den Krankheits-
verlauf als auch den Grad der Krankheitsausprägung. So zeigten mit r-ADA behan-
delte Mäuse eine geringere Hautdicke, eine geringere Anzahl an Myofibroblasten in
der Haut sowie einen niedrigeren Kollagenhalt. Bei den Fra-2 transgenen Mäusen be-
wirkte eine Therapie mit r-ADA eine Besserung der Haut und Lungenfibrose. Durch
Gabe von r-ADA konnte die Anzahl an Myofibroblasten und die Kollagenmenge in
Haut und Lunge gesenkt werden, was die Abnahme fibrotischer Lungenbereiche so-
wie der Hautdicke widerspiegelt. Der Umbau der Lungenarterien mit Verdickung der
Gefäßwand wurde ebenfalls reduziert. So zeigten sich durch die Therapie weniger
stark stenosierte Pulmonalarterien und dünnere Gefäßwände. Nebenwirkungen der
Therapie wurden in beiden Modellen nicht beobachtet.
Praktische Schlussfolgerungen. Die vorliegende Arbeit zeigt, dass in beiden Maus-
modellen der SSc die Gabe von r-ADA sowohl vaskuläre als auch fibrotische Verän-
derungen reduzieren kann. Diese Ergebnisse liefern weitere Hinweise auf die zentra-
le Bedeutung Adenosin-abhängiger Signalkaskaden in der SSc und zeigen, dass die
Gabe von r-ADA ein möglicher Behandlungsansatz sein könnte.

3
2. Summary
Objectives. Systemic sclerosis (SSc) is a systemic, autoimmune mediated, orphan
disease with significantly increased mortality. The hallmarks of the disease are vas-
cular changes such as loss of capillaries and remodeling of the pulmonary arteries
and tissue fibrosis due to increased release of collagen from pathological activated
fibroblasts. This increased collagen production is caused e.g. by adenosine triggered
activation of the A2A-Receptor. The tissue level of extracellular adenosine is mainly
controlled by the enzyme adenosine deaminase (ADA). Mice with deficiency in
ADA have increased levels of endogenous adenosine and develop systemic fibrotic
manifestations. These fibrotic changes were prevented by treatment with an A2A-
receptor antagonist. Aim of this study was, to evaluate the effect of exogenous re-
combinant ADA in murine models of SSc.
Methods. The efficiency of recombinant ADA was examined in two murine models.
The first model was a model of chronic-graft-versus-host-disease (cGvHD) induced
by allogenic bone marrow transplantation with a mismatch in minor histocompatibi-
lity antigens. The effects of treatment with recombinant adenosine deaminase were
analyzed with clinical, histological and biochemical readouts. Skin sections were
stained with Hematoxylin and eosin (HE) to quantify dermal thickening and stained
for α-smooth muscle actin (αSMA) to identify myofibroblasts. The collagen content
in skin was determined by hydroxyproline assays. Clinical changes were assessed
using an established score for cutaneous cGvHD. As a second model, we analyzed
Fos-related-antigen-2 (Fra-2) transgenic mice. Fra-2 is a transcription factor of the
Activator-Protein-1 (AP-1)-family. The transgenic overexpression of Fra-2 results in
fibrosis of the skin and lungs. In addition, Fra-2 transgenic mice develop SSc-like
vascular manifestations with pulmonary-arterial hypertension like changes. Fibrotic
changes were assessed by determination of dermal thickness (skin) and of the Sirius
Red stained area (lungs), by assessment of myofibroblast counts and by quantifica-
tion of the hydroxyproline content. Vascular changes were analyzed by assessment
of the average thickness of the vessel walls of pulmonary arteries and by counting
the number of highly narrowed vessels.
Results. In both models treatment with recombinant adenosine deaminase exerted
anti-fibrotic effects. In the cGvHD model, preventive treatment with recombinant
ADA ameliorated fibrotic manifestations with decreased dermal thickening, reduced
myofibroblast counts and lower hydroxyproline content. Treatment with recombinant

4
ADA also reduced the cutaneous cGvHD score. Treatment with recombinant ADA
also ameliorated dermal and pulmonary fibrosis in Fra-2 transgenic mice. Moreover,
pulmonary arterial remodeling was reduced. The average thickness of the vessel wall
in pulmonary arteries was reduced and less highly narrowed pulmonary arteries were
detected. Adverse events of the treatment were not observed in the two models.
Conclusions. The present study demonstrates that recombinant ADA ameliorates
both vascular and fibrotic changes in two murine models of SSc. These results fur-
ther highlight the role of adenosine signaling in the pathogenesis of systemic sclero-
sis and may provide first evidence that recombinant ADA may be a potential treat-
ment option for SSc.
5
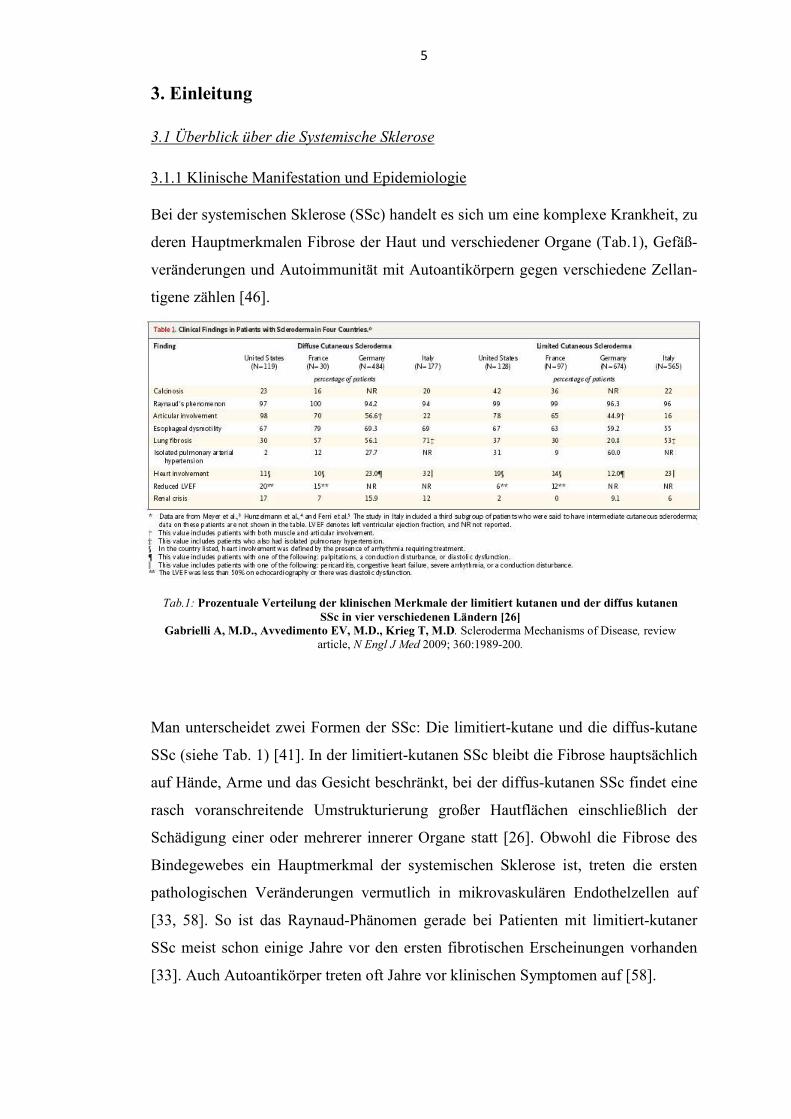
Tab.1: Prozentuale Verteilung der klinischen Merkmale der limitiert kutanen und der diffus kutanen
SSc in vier verschiedenen Ländern [26]
Gabrielli A, M.D., Avvedimento EV, M.D., Krieg T, M.D. Scleroderma Mechanisms of Disease, review article, . Engl J Med 2009; 360:1989-200.
3. Einleitung
3.1 Überblick über die Systemische Sklerose
3.1.1 Klinische Manifestation und Epidemiologie
Bei der systemischen Sklerose (SSc) handelt es sich um eine komplexe Krankheit, zu
deren Hauptmerkmalen Fibrose der Haut und verschiedener Organe (Tab.1), Gefäß-
veränderungen und Autoimmunität mit Autoantikörpern gegen verschiedene Zellan-
tigene zählen [46].
Man unterscheidet zwei Formen der SSc: Die limitiert-kutane und die diffus-kutane
SSc (siehe Tab. 1) [41]. In der limitiert-kutanen SSc bleibt die Fibrose hauptsächlich
auf Hände, Arme und das Gesicht beschränkt, bei der diffus-kutanen SSc findet eine
rasch voranschreitende Umstrukturierung großer Hautflächen einschließlich der
Schädigung einer oder mehrerer innerer Organe statt [26]. Obwohl die Fibrose des
Bindegewebes ein Hauptmerkmal der systemischen Sklerose ist, treten die ersten
pathologischen Veränderungen vermutlich in mikrovaskulären Endothelzellen auf
[33, 58]. So ist das Raynaud-Phänomen gerade bei Patienten mit limitiert-kutaner
SSc meist schon einige Jahre vor den ersten fibrotischen Erscheinungen vorhanden
[33]. Auch Autoantikörper treten oft Jahre vor klinischen Symptomen auf [58].

6
Ergebnisse epidemiologischer Studien zur Prävalenz schwanken zwischen 50-300
Fällen pro Million Einwohner und einer Inzidenz mit 2,3-22,8 neuen Erkrankungen
im Jahr pro Million Einwohner [8]. Frauen sind hierbei mit einem Verhältnis von
3-14:1 deutlich häufiger als Männer betroffen. Ebenso wird von einer leicht erhöhten
Anfälligkeit an SSc zu erkranken, bei Afroamerikanern berichtet [45, 55]. Aufgrund
familiärer Häufung der systemischen Sklerose, einer zusätzlich erhöhten Inzidenz
anderer Autoimmunkrankheiten bei Familienmitgliedern der an SSc erkrankten Pati-
enten, z.B. systemischer Lupus erythematodes, rheumatoide Arthritis und Sjögren-
Syndrom sowie unterschiedlicher Phänotypen in verschiedenen ethnischen Gruppen,
liegt es nahe, dass genetische Faktoren zu der Entstehung einer SSc beitragen [55,
18]. So hat man festgestellt, dass bei SSc-Patienten gehäuft Polymorphismen bei
Genen auftreten, die Immunantworten regulieren, wobei viele dieser Abweichungen
nur bei einzelnen Patienten-Kohorten beobachtet wurden und nur wenige dieser Po-
lymorphismen unabhängig bestätigt werden konnten [1]. Bestimmte Human Leu-
kocyte Antigen (HLA) Klasse II-Moleküle und damit verbundenen klinischen Er-
scheinungsbildern der systemischen Sklerose sowie spezielle Autoantikörper zeigen
demgegenüber eine hohe Evidenz [42]. Diese Erkenntnisse unterstützen die Ansicht,
dass die systemische Sklerose nicht als ein klar definiertes Krankheitsbild betrachtet
werden sollte, sondern vielmehr als ein Syndrom, welches viele Erscheinungsbilder
umfasst [26]. Weiter unterstützt wird diese Sichtweise durch die Tatsache, dass be-
stimmte Umweltgifte, z.B. Vinylchlorid oder Siliziumdioxid, klinische Erschei-
nungsbilder hervorrufen können, welche der systemischen Sklerose ähneln [48].
3.1.2 Pathogenese der systemischen Sklerose
Man unterscheidet bei der SSc zwischen frühen und späten pathologischen Gewebe-
veränderungen, wobei zu Beginn Gefäßschäden, vor allem an kleinen Gefäßen, ins-
besondere Arteriolen [51, 22], und entzündliche Veränderungen im Vordergrund
stehen. Die fortgeschrittene Krankheit zeichnet sich durch exzessive Fibrose, Zellun-
tergang und Atrophie aus [26].
Histopathologisch zeichnen sich die mikrovaskulären Veränderungen durch große
Lücken zwischen den Endothelzellen, dem Verlust der endothelialen Integrität, der
Vakuolisierung des Zellplasmas sowie vermehrter Apoptose von Endothelzellen aus
[27, 31]. Zudem kommt es zur Aufsplittung der Basallamina und es bilden sich peri-

7
vaskuläre Infiltrate aus mononukleären Entzündungszellen [21]. So finden sich peri-
vaskulär zu Beginn der Erkrankung vor allem T-Lymphozyten, Makrophagen und
Mastzellen [36, 59, 63]. Bei Hautläsionen findet man hauptsächlich CD4+-T-Zellen
[59], vornehmlich vom Typ 2 (Th2) [44, 59]. Dies ist in Übereinstimmung mit er-
höhten Th2 Zytokinen im Serum [28, 29]. Außerdem befinden sich auch CD20+ -B-
Zellen in den Hautläsionen [63]. Diese könnten durch die Sekretion von Interleukin-
6, TGF-β [14, 29, 63] sowie von Autoantikörpern Fibroblasten direkt aktivieren und
so zur Entstehung der Fibrose beitragen. Zusätzlich kommt es zur Obliteration
kleinster Gefäße und einem Verlust an Kapillaren [21, 22, 23, 27, 51].
Ein Kennzeichen der fortgeschrittenen SSc ist eine vermehrte Ansammlung von Be-
standteilen der extrazellulären Matrix (EZM) aufgrund einer Überproduktion von
Kollagen, v.a. Typ I und Glykoproteinen, wie z.B. Fibronektin und Fibrillin [15, 40].
Fibroblasten spielen bei der Produktion, der extrazellulären Ansammlung sowie bei
der Umformung von Kollagen und anderen EZM-Bestandteilen eine zentrale Rolle
[26]. Die Mechanismen der Kollagenüberproduktion durch die pathologisch aktivier-
ten Fibroblasten sind vielfältig [40]. Fibroblasten von SSc-Patienten scheinen endo-
gen über epigenetische Veränderungen aktiviert zu sein und behalten diesen aktivier-
ten Phänotyp auch in vitro [12]. Vor allem in frühen Krankheitsstadien scheint aber
die exogene Aktivierung zu dominieren. So sezernieren Endothelzellen, Epithelzel-
len und Leukozyten profibrotische Zytokine, wie z.B. Transforming-Growth-Factor-
β (TGF-β) und platelet-derived growth factor (PDGF), die die Kollagensynthese in
Fibroblasten steigern [60].
Fibroblasten von SSc-Patienten können sich unter dem Einfluss von TGF-β in Myo-
fibroblasten umwandeln [52]. Myofibroblasten sind spezialisierte kontraktile Fibro-
blasten, welche das Protein α-Smooth-Muscle-Actin (α-SMA) exprimieren und große
Mengen Kollagen sowie andere Bestandteile der extrazellulären Matrix produzieren
[60]. Während Myofibroblasten bei der normalen Wundheilung im Granulationsge-
webe nur vorübergehend auftreten und mittels programmierten Zelltods wieder ent-
fernt werden, persistieren sie in fibrotischen Erkrankungen und tragen zur Überpro-
duktion der extrazellulären Matrix bei [60].

8
3.2 Mausmodelle zur Erforschung der Systemischen Sklerose
Die komplexe Pathogenese der SSc ist nur zum geringen Teil verstanden. Viele Zu-
sammenhänge sind weiterhin rätselhaft [3]. Ein besseres Verständnis der Erkrankung
wäre jedoch für die Entwicklung neuer Therapien essentiell [3]. Eine wichtige Hilfe-
stellung könnten hierbei Tiermodelle geben [3]. Keines der vorhandenen Tiermodelle
kann jedoch alle Merkmale dieser komplexen Erkrankung widerspiegeln. Es lassen
sich mittels unterschiedlicher Tiermodelle einzelne Aspekte der Erkrankung abbilden
[3]. Diese Tiermodelle können in genetische und induzierbare Tiermodelle der SSc
eingeteilt werden [3]. In genetischen Modellen entwickeln sich Schlüsselmerkmale
der humanen SSc anhand spezifischer Änderung im Genom der Tiere, bei induzier-
baren Tiermodellen bewirken von außen zugeführte Agenzien oder Prozeduren einen
der SSc ähnlichen Phänotyp [3].
3.2.1 Mausmodell der sklerodermiformen chronischen Graft-versus-Host Erkrankung
Eine chronische Graft-versus-Host-Erkrankung (cGvHD) tritt bei 40-60% der Lang-
zeitüberlebenden nach einer Stammzelltransplantation auf [4]. Sie ist der Hauptfaktor
bezüglich der Langzeitprognose [3]. Es gibt zwei Hauptformen der cGvHD: Die zy-
totoxische und die sklerodermiforme cGvHD, wobei letztere die Fibrose hervorru-
fende und der SSc ähnelnde Variante ist und 10-15% der cGvHD Fälle verursacht
[61]. Aufgrund der im klinischen Erscheinungsbild großen Ähnlichkeit zwischen der
sklerodermiformen cGvHD und der diffus-kutanen SSc kam man zu der Hypothese,
dass die SSc eine Variante der cGvHD sein könnte [39], welche durch Mikrochimä-
rismus, das heißt durch das Überleben einiger körperfremder Zellen eines anderen
Individuums, im Körper entsteht [37, 47]. In dem von uns verwendeten Mausmodell
der sklerodermiformen cGvHD sind Spender- und Empfänger-Mäuse bezüglich der
major histocompatibility complexes (MHCs) identisch, aber in den minor histocom-
patibility Antigenen (MHA) verschieden [3]. Deshalb kann es zu einer „Fremderken-
nung“ des Empfängergewebes durch das transplantierte Immunsystem des Spenders
kommen, die sich als sklerodermiforme cGvHD mit Fibrose und Entzündung mani-
festiert [3]. In unserem Modell werden Splenozyten zusammen mit Knochenmarks-
zellen aus der Tibia und des Femur von B10.D2 Spender-Mäusen in die Schwanzve-
ne der Empfänger Balb/c-Maus, welche kurz zuvor subletal bestrahlt wurde, injiziert

9
[32]. Ungefähr zwei Wochen nach der Knochenmarktransplantation sind die be-
troffenen Empfänger-Gewebe von T-Zellen, Monozyten, Mastzellen und Leukozyten
infiltriert, was ortsständige Fibroblasten dazu stimuliert, große Mengen an extrazellu-
lärer Matrix freizusetzen [11, 32]. Ab der dritten Woche nach Transplantation entwi-
ckeln die Mäuse eine Hautfibrose mit Verlust an subkutanem Fettgewebe [9, 32].
Dieses Tiermodell stellt ein gut etabliertes und standardisiertes Mausmodell der SSc
dar [3].
3.2.2 Fra-2-Mausmodell
Es handelt sich hier um ein genetisch induziertes Tiermodell der SSc. Fra-2 ist ein
Transkriptionsfaktor, der zur Aktivator-Protein-1-Familie zählt [3]. Dieser ist an
Zellproliferation, Entzündung und Wundheilung mit beteiligt [62]. In Abb.1 ist die
Gensequenz zu sehen.
Abb.1: Gensequenz der Fra-2 Mäuse [4]
Beyer C, Schett G, Distler O, Distler JH: Animal models of systemic sclerosis: prospects and limitations. Arthritis Rheum. 2010; 62(10):2831-44
Das transgene Konstrukt der Fra-2 Maus besteht aus einem ubiquitär exprimierten
H2Kb Promotor, dem Fra-2 Genom, einem grün fluoreszierenden Reporter-Gen mit
interner Ribosomenbindungsstelle (IRES-EGFP) sowie einem Polyadenylierungs-
signal (PolyA) [3]. Die transgene Überexpression von Fra-2 führt zu generalisierter
Entzündung und Fibrose, vor allem in der Haut und der Lunge [54]. Zusätzlich zur
Entzündung und Fibrose entwickeln Fra-2 transgene Mäuse einen vaskulären Phäno-
typ, welcher sich unter anderem in der Lunge und der Haut manifestiert [3]. Dieser
lässt sich ab einem Alter von neun Wochen zuerst als Apoptose in mikrovaskulären
Endothelzellen mit konsekutiver Abnahme der Kapillardichte in der Haut nachwei-
sen [43]. Zusätzlich zum Verlust kleiner Blutgefäße entwickeln Fra-2 transgene
Mäuse eine proliferierende Gefäßerkrankung in der Lunge, ähnlich den histologi-
schen Veränderungen der pulmonal-arteriellen Hypertonie (PAH). Aufgrund der
Lungenfibrose und der PAH sterben Fra-2 transgene Mäuse in einem Alter von

10
16-20 Wochen an Herz-Lungen-Versagen [17]. Interessanterweise zeigt sich bei Pa-
tienten mit SSc eine erhöhte Expression von Fra-2 in Myofibroblasten, Endothelzel-
len sowie in glatten Muskelzellen, was darauf hindeutet, dass Fra-2 an der Pathoge-
nese der humanen SSc mitbeteiligt sein könnte [43, 54]. Des Weiteren konnte gezeigt
werden, dass profibrotische Mediatoren, wie TGF-β und PDGF, die Expression von
Fra-2 in Fibroblasten stimuliert [54]. Die Manifestationen im Fra-2-Modell entwi-
ckeln sich dabei in einem zeitlichen Ablauf, der dem systemischer Sklerose ähnelt
[3]. Das Fra-2-Modell kann damit auch die komplexen Interaktionen zwischen Fib-
rose und Vaskulopathie widerspiegeln [3].

11
3.3 Das Adenosin-Rezeptor-System
Da es Ziel dieser Arbeit ist, die Aktivierung des Adenosin-Rezeptors bei der Entste-
hung und Therapie der SSc zu bewerten, sind Grundkenntnisse zum Adenosin-
Rezeptor-System zum Verständnis nötig, welche im folgenden Kapitel dargestellt
werden.
3.3.1 Die Adenosinrezeptoren: Aufbau und Funktion
Die Adenosinrezeptoren gehören zur Familie der P1-Rezeptoren und sind Mitglied
der Rhodopsin ähnlichen G-Protein gekoppelten Rezeptorfamilie [6]. So besitzen die
Adenosin-Rezeptoren sieben Transmembran-Domänen, welche vor allem aus hydro-
phoben Aminosäuren bestehen, wobei man annimmt, dass jede der Transmembran-
domänen eine α-Helix aus 21-28 Aminosäuren darstellt [25, 53]. Die Transmem-
brandomänen sind mittels drei extrazellulärer und drei zytoplasmatischer, hydrophi-
ler Schleifen ungleicher Länge verbunden [53] (siehe Abb. 2). Man unterscheidet bei
den Adenosinrezeptoren vier unterschiedliche Vertreter, nämlich ADORA1 (A1R),
Abb.2: Aufbau der Adenosinrezeptoren [38]
Latta V, Cabiati M, Rocchicciolia S, Del Ry S, Morales MA. Review: The role of the adenosinergic system in lung fibrosis,
Pharmacological Research 2013; 76:182–189

12
ADORA2A (A2AR), ADORA2B (A2BR) und ADORA3 (A3R) [38]. Die A2-
Rezeptoren unterteilt man in hoch-affine A2A- und niedrig-affine A2B-Rezeptoren be-
züglich eines Anstiegs an zyklischem Adenosinmonophosphat (cAMP) [68]. So sind
die A1R- und A3R-Rezeptoren an inhibitorische G-Proteine gekoppelt, was zu einem
intrazellulären Abfall an cAMP führt. Im Gegensatz dazu sind A2-Rezeptoren an sti-
mulatorische G-Proteine gekoppelt und induzieren einen Anstieg der intrazellulären
cAMP-Konzentration [6, 10, 24]. Die durch Stimulation der verschiedenen Adeno-
sinrezeptoren erzielten Wirkungen sind sowohl innerhalb dieser als auch in den ver-
schiedenen Geweben sehr unterschiedlich [16]. So stimuliert die Aktivierung des
A2A-Rezeptors auf humanen dermalen Fibroblasten die Kollagenproduktion [7]. Auf-
grund dieser vielfältigen Wirkungen der Adenosinrezeptoren werden diese als poten-
tieller Angriffspunkt zur Therapie vieler verschiedener Erkrankungen in Betracht
gezogen [34].
3.3.2 Der Adenosin-Metabolismus
Adenosin ist ein Nukleosid, bestehend aus der Purinbase Adenin, welche über eine
β-N9-glykosidische Bindung mit einer Ribose verknüpft ist [38].
Abb.3: Schematische Darstellung des Adenosin-Metabolismus [38]
Latta V, Cabiati M, Rocchicciolia S, Del Ry S, Morales MA. Review: The role of the adenosinergic system in lung fibrosis, Pharmacological Research 2013; 76:182– 189
Abbildung 3 veranschaulicht den Adenosin-Metabolismus. Im Zytoplasma ist Ade-
nosin hauptsächlich in seinen phosphorylierten Formen Adenosinmonophosphat
(AMP), Adenosindiphosphat (ADP) und Adenosintriphosphat (ATP) vorhanden [38].
Adenosin wird intrazellulär über das Enzym Ecto-5‘-Nucleotidase (CD73) aus AMP

13
generiert [38]. Unter physiologischen Bedingungen wird die intrazelluläre Adenosin-
Konzentration mittels der Enzyme S-Adenosylhomo-cysteinase und S-Adenosyl-
homocystein-Hydrolase niedrig gehalten [2]. Als Antwort auf zellulären Stress und
Zellschaden wird ATP in den Extrazellularraum freigegeben und schnell anhand der
Enzyme Ectonucleoside triphosphate Diphosphohydrolase (CD39) zu AMP und an-
schließend durch Ecto-5‘-Nucleotidase (CD73) zu Adenosin umgesetzt [38]. Dieses
Adenosin kann entweder mit den vorausgehend erklärten Adenosinrezeptoren intera-
gieren [38] oder mittels passiver Diffusion anhand eines beidseitig funktionierenden
Adenosin-Transport-Systems (equilibrative nucleoside transporter (ENT)) wieder in
die Zelle gelangen [38]. Durch das Enzym Adenosin-Desaminase (ADA) kann so-
wohl extra- als auch intrazelluläres Adenosin zu Inosin (INO) desaminiert werden
[38]. Intrazelluläres Adenosin muss hierfür entweder durch das Adenosin-
Transporter-System nach außen sezerniert werden oder wieder zu ATP phosphory-
liert werden, wobei die Umwandlung von Adenosin zu AMP durch das Enzym Ade-
nosinkinase (ADK) stattfindet [11, 30, 57, 64]. Adenosin-Desaminase (ADA) ist
dagegen das bedeutendste Enzym zum Abbau von Adenosin [20].
3.3.3 Störungen des Adenosin-Rezeptor-Systems in der Fibrose
Neben den physiologischen Funktionen des Adenosin-Rezeptor-Systems sind auch
die Störungen dieses Systems zum Erkenntnisgewinn von Bedeutung. So hat man
festgestellt, dass eine Aktivierung der A2A-Rezeptoren in humanen Fibroblasten die
Kollagenproduktion steigert [50]. Ebenso konnte man zeigen, dass Mäuse, welche
einen Mangel an Adenosin-Desaminase aufwiesen und daraus resultierend erhöhte
Gewebespiegel an Adenosin bekamen, spontan Fibrose in der Haut und Lunge ent-
wickelten [16, 20]. Durch Gabe eines A2A-Rezeptor-Antagonisten, z.B. ZM-241385,
konnte man bei diesen Mäusen die Entwicklung der Fibrose verhindern [20]. Durch
Gabe von A2A-Rezeptor-Antagonisten oder durch Knockout des A2A-Rezeptors konn-
te zudem eine anhand von externer Bleomycin-Gabe induzierte Fibrose bei Mäusen
verhindert werden [20]. Mäuse, denen die Enzyme CD39 oder CD73 fehlen, sind
dagegen weitgehend resistent gegen eine Bleomycin-induzierte Hautfibrose [19].
Man stellte fest, dass bei ihnen im Vergleich zu nicht CD39/ CD73-defizienten Mäu-
sen niedrigere Gewebespiegel an profibrotischen Zytokinen, wie TGF-β und connec-
tive tissue growth factor (CTGF), vorhanden waren [19]. Aufgrund dieser Ergebnisse

14
kam man zu der Hypothese, dass extrazelluläres Adenosin, welches anhand der En-
zyme CD39 und CD73 erzeugt wird, die Fibrogenese der Haut unterstützt [19]. Die-
selbe Arbeitsgruppe zeigte, dass der A2A-Rezeptor möglicherweise an der Rekrutie-
rung peripherer im Blut zirkulierender Fibroblastenvorläufer, sog. Fibrozyten, zum
Ort der Fibrose beteiligt ist [35].

15
4. Fragestellung
Das Adenosin-Rezeptor-System scheint aufgrund der im letzten Kapitel beschriebe-
nen Erkenntnisse eine wichtige Rolle in der Pathogenese fibrotischer Erkrankungen
zu spielen. Deshalb ist es Ziel dieser Arbeit anhand tierexperimenteller Mausmodelle
zu untersuchen, ob die Gabe rekombinanter Adenosin-Desaminase einen positiven
Effekt bei der Entstehung und der Therapie der systemischen Sklerose haben könnte.
Rekombinante Adenosin-Desaminase ist zur Behandlung bei Kindern mit angebore-
nem Adenosin-Desaminase (ADA)-Mangel zugelassen und stünde damit auch zur
Behandlung der SSc zur Verfügung.

16
5. Material und Methoden
5.1 cGvHD Mausmodell
5.1.1 Hintergrundinformationen zum cGvHD Mausmodell
Die in dieser Arbeit beschriebene cGvHD wurde mittels einer allogenen Stammzel-
lentransplantation hervorgerufen. Spender- und Empfänger-Mäuse sind bezüglich der
major histocompatibility complexes (MHCs) aufeinander abgestimmt, aber unter-
scheiden sich in den minor histocompatibility Antigenen (MHA). Bei den allogenen
Spendermäusen handelt es sich um B10.D2 (H-2d)-Mäuse, welche von der Firma
Jackson Laboratory (Bar Harbour, ME) bestellt wurden. Von diesen wurden Kno-
chenmarkzellen und Splenozyten gewonnen und in die Schwanzvene der Empfänger
Balb/c (H-2d)-Mäuse injiziert. Die Balb/c (H-2d)-Mäuse wurden von der Firma Jan-
vier (Le Genest, St. Isle, France) bestellt. Alle Mäuse wurden unter pathogenfreien
Bedingungen besonders gepflegt. Sie erhielten sterile Pellet-Nahrung sowie steriles
Wasser und hatten einen normalen Tag-Nacht-Zyklus. Zur Knochenmarkzellgewin-
nung wurden die Tibia und der Femur der Spendermäuse unter sterilen Bedingungen
präpariert. Um die Knochenmarkzellen aus den präparierten Knochenkavitäten zu
spülen, verwendete man eine phosphatgepufferte Salzlösung (PBS) (Life technolo-
gies, Germany). Anschließend wurden die Knochenmarkzellen mittels 70 µm Nylon-
Netzen (BD Biosciences, Heidelberg, Germany) filtriert und es erfolgte eine Hämo-
lyse. Die erhaltenen Knochenmarkzellen wurden bis zur Transplantation in Eis gela-
gert.
Die Empfänger Balb/c (H-2d)-Mäuse waren zum Zeitpunkt der Knochenmarkzellen-
und Splenozytentransplantation acht Wochen alt. Sechs Stunden vor der Transplanta-
tion bekamen sie eine Ganzkörperbestrahlung mit einer Dosis von 700 Zentigray
(cGy). Zur Transplantation erhielten alle Empfänger Balb/c (H-2d)-Mäuse Knochen-
mark von entweder gleichen „syngenen“ Balb/c (H-2d) oder allogenen B10.D2 (H-
2d) Mäusen. Zur Transplantation wurden 5 × 106 Splenozyten und 1 × 106 Kno-
chenmarkzellen von den Spendermäusen in 0,2 ml PBS (Life technologies, Germa-
ny) resuspendiert und in die Schwanzvene injiziert. Die Behandlung der Knochen-
markempfängermäuse mit rekombinanter Adenosin-Desaminase begann zehn Tage
nach der Knochenmarktransplantation. 45 Tage nach der Knochenmarktransplantati-
on wurden die Mäuse getötet und Proben zur Analyse gewonnen. Um die Effekte der

17
Gabe rekombinanter Adenosin-Desaminase zu studieren, gab es vier Gruppen mit je
acht Mäusen.
• Die erste Gruppe war die syngene Kontrollgruppe. Hierbei erfolgte eine
syngene Transplantation der Splenozyten und Knochenmarkzellen Balb/c (H-
2d) �Balb/c (H-2d). Jede Woche wurden 100 µl 0,9% NaCl (Carl Roth,
Karlsruhe, Germany), welches als Lösungsmittel für rekombinante ADA
diente, intraperitoneal injiziert.
• Bei der zweiten Gruppe handelte es sich um eine allogene Gruppe, bei der
man eine allogene Knochenmarkzellen- und Splenozytentransplantation
B.10D2 (H-2d)� Balb/c (H-2d) durchführte. Jede Woche wurden hier ebenso
100 µl 0,9% NaCl intraperitoneal injiziert.
• Gruppe drei war Behandlungsgruppe 1: Es erfolgte eine allogene Knochen-
markzellen- und Splenozytentransplantation B.10D2 (H-2d)� Balb/c (H-2d).
Wöchentlich wurde rekombinante ADA gegeben, die hierzu in 0,9% NaCl
gelöst wurde und mit einer Konzentration von 0,5 mg/ml (0,5 Units = 0,5 U)
wurden 100 µl intraperitoneal injiziert.
• Gruppe vier war Behandlungsgruppe 2: Es erfolgte eine allogene Knochen-
markzellen- und Splenozytentransplantation B.10D2 (H-2d)� Balb/c (H-2d).
Auch hier wurde wöchentlich rekombinante ADA gegeben, die hierzu in
0,9% NaCl gelöst wurde und mit einer Konzentration von 5 mg/ml (5 Units =
5 U) injizierte man 100 µl intraperitoneal.
5.1.2 Klinische Verlaufskontrolle
Zur Beurteilung des klinischen Verlaufs und Ausprägungsgrades der Erkrankung
erfolgten nach der Transplantation täglich eine Bestimmung des Körpergewichtes der
Mäuse sowie die Einstufung in ein klinisches Bewertungssystem. Dieses unterteilte
man in Haarverlust sowie in das Erscheinungsbild der Ohren und des Schwanzes. Es
erfolgte eine Punktevergabe, wobei 0 Punkte bei gesundem Erscheinungsbild und 3
Punkte bei max. krankhaftem Erscheinungsbild gegeben wurden. Die Punktevergabe
bei Ohr und Schwanz erfolgte anhand optischer Beurteilung.

18
Der Haarverlust wurde zuvor graduiert in:
• Kein Haarverlust und gesundes Erscheinungsbild der Haut = 0 Punkte.
• Haut-Läsion mit Apoplezie ≤ 1cm2 = 1 Punkt.
• Haut-Läsion mit Apoplezie 1-2cm2 = 2 Punkte.
• Haut-Läsion mit Apoplezie ≥ 2cm2 = 3 Punkte.
Die einzelnen Punkte der unterschiedlichen Kategorien addierte man hierbei und es
konnte somit ein Maximalergebnis von 9 Punkten erreicht werden.
5.1.3 Analyse der Hautveränderung
5.1.3.1 Histologische Analyse
Um die Hautfibrose der Mäuse zu analysieren, wurden Hautareale herausgeschnitten,
in 4%igem Formalin (Carl Roth, Karlsruhe, Germany) für sechs Stunden fixiert und
in Paraffin eingebettet. 5 µm Schnitte wurden hergestellt.
1.) Hämatoxylin-Eosin-Färbung
Um die Hautdicke zu bestimmen, wurden die histologischen Schnitte mit Hämatoxy-
lin (Merk, Germany) und Eosin (Carl Roth, Karlsruhe, Germany) (HE) - wie be-
schrieben (Distler et al. 2007) - gefärbt [13] und mithilfe eines Nikon Eclipse 80i
Mikroskops (Nikon, Badhoevedorp, Niederlande) untersucht. Die Hautdicke wurde
mit 100-facher Vergrößerung bestimmt, indem die größte Distanz von der dermoepi-
dermalen Junktionszone bis zur Grenze zwischen Dermis und Subkutis an vier ver-
schiedenen Stellen je Präparat gemessen wurde (Distler et al. 2007) [13]. Die Aus-
wertung erfolgte verblindet.
2.) Masson-Trichrom-Färbung
Um die Kollagenfasern selektiv darzustellen, führten wir eine Trichrom-Färbung
nach Masson durch. Dazu wurden die Hautschnitte entparaffiniert und rehydriert,
bevor sie in Bouin’scher Lösung (Sigma-Aldrich, USA) bei 56°C für 15 Minuten
inkubiert wurden. Anschließend erfolgte die Färbung der Schnitte mittels Wei-
gert’scher Hämatoxylin-Eisenlack-Lösung (Sigma, Germany) und Biebrich-Schar-

19
lachrot-Säurefuchsin-Lösung (Sigma-Aldrich, USA). Im Anschluss an die Behand-
lung mit Phosphor-Wolframsäure (Carl Roth, Karlsruhe, Germany) und Molybdato-
phosphorsäure (Carl Roth, Karlsruhe, Germany) wurden die Schnitte mit Anilinblau
(Sigma-Aldrich, USA) für zwei Minuten bei Raumtemperatur inkubiert, danach die
Schnitte dehydriert und mit dem Einschlussmittel Roti®-Histokitt (Carl Roth, Karls-
ruhe, Germany) fixiert. Repräsentative Areale wurden durch das Nikon Eclipse 80i
Mikroskop (Nikon, Badhoevedorp, Niederlande) dargestellt. Die Auswertung erfolg-
te verblindet.
5.1.3.2 Immunhistochemische Analyse der Myofibroblasten
Zur Quantifizierung der Myofibroblasten führte man eine immunhistochemische
Darstellung des Proteines α-smooth muscle actin (α-SMA) durch. Myofibroblasten
sind durch die Produktion α-SMA charakterisiert.
Zunächst wurden die histologischen Schnitte deparaffinisiert und mit 5%igem Pfer-
deserum (Life technologies, Germany) und anschließend mit 3%igem H2O2 (Carl
Roth, Karlsruhe, Germany) inkubiert. α-SMA-positive Zellen konnte man durch In-
kubation mit monoklonalen anti-α-SMA-Antikörpern (clone 1A4, Sigma-Aldrich,
Steinheim, Germany) nachweisen. Als Sekundär-Antikörper wurden mit Meerrettich-
Peroxidase (Dako, Hamburg, Deutschland) verknüpfte Antikörper verwendet. Die
Expression von α-SMA machte man mittels DAB (3,3‘-Diaminobenzidin tetrahydro-
chloride) Peroxidase-Substrat-Lösung (Sigma-Aldrich, USA) sichtbar. Als Kontroll-
gruppe wurden monoklonale IgG Maus-Antikörper (Calbiochem, San Diego, CA,
USA) verwendet.
Zur Bestimmung der Anzahl der Myofibroblasten wurde ein Nikon Eclipse 80i Mik-
roskop (Nikon, Badhoevedorp, Niederlande) verwendet. Die Auswertung erfolgte
verblindet.
5.1.3.3 Hydroxyprolin-Assay
Man bestimmte den Kollagengehalt in den Hautproben mit Hilfe eines Hydroxypro-
lin-Assay. Hierfür wurden Stanzbiopsien mit einem Durchmesser von 3 mm herge-
stellt und in 6 M HCl (Carl Roth, Karlsruhe, Germany) bei 120°C für drei Stunden

20
verarbeitet. Danach wurde der pH-Wert mittels 6 M NaOH (Carl Roth, Karlsruhe,
Germany) auf einen pH-Wert von 6 eingestellt. Als Farbindikator wurde Lackmus-
papier (Carl Roth, Karlsruhe, Germany) verwendet. Anschließend wurde 0,06 M
Chloramin T (Sigma, Germany) den Proben zugegeben und man ließ sie für 20 Mi-
nuten bei Raumtemperatur inkubieren. Danach gab man 3,15 M Perchlorsäure
(Merck, Germany) und 20%igen para-Dimethylaminobenzaldehyd (Fluka, Germany)
hinzu und inkubierte die Proben für weitere 20 Minuten bei 60°C. Die Absorption
der Proben wurde bei einer Wellenlänge von 557 nm mit einem Spectra MAX 190
Mikroplatten-Spektralphotometer (Molecular Devices, Sunnyvale, CA, USA) be-
stimmt.

21
5.2 Fra-2-Mausmodell
5.2.1 Hintergrundinformationen zum Fra-2-Mausmodell
In diesem Modell führte, wie bereits in Abschnitt 3.2.2 ausführlich erläutert, die
transgene Überexpression von Fra-2 zu systemischer Fibrose, zu Mikroangiopathien
und zu einer proliferierenden Gefäßerkrankung in der Lunge, ähnlich den histologi-
schen Veränderungen der pulmonal-arteriellen Hypertonie (PAH). Aufgrund der
Lungenfibrose und der PAH starben die Fra-2 transgenen Mäuse in einem Alter von
16-20 Wochen an Herz-Lungen-Versagen. Die Manifestationen im Fra-2-Modell
entwickeln sich dabei in einem zeitlichen Ablauf, der dem der systemischen Sklerose
ähnelt. So zeigen sich die ersten mikrovaskulären Veränderungen in einem Alter von
neun Wochen vor Beginn der Fibrose. Deshalb begann die Behandlung mit rekombi-
nanter Adenosin-Desaminase in einem Alter von acht Wochen. Mit einem Alter von
16 Wochen wurden die Mäuse getötet und Proben zur Analyse der Lunge und Haut
gewonnen. Mäuse, welche eine Kopie des Transgens exprimieren, haben die Be-
zeichnung Fra2+/-. Die das Transgen nicht exprimierende Kontrollgruppe hat die
Bezeichnung Fra2-/-. In diesem Modell gab es vier Gruppen mit je sechs Mäusen.
• Gruppe eins war die das Transgen nicht exprimierende Kontrollgruppe
(Fra2-/-). Jede Woche wurden 100 µl 0,9% NaCl (Carl Roth, Karlsruhe,
Germany), welches als Lösungsmittel für rekombinante ADA diente, intrape-
ritoneal injiziert.
• Gruppe zwei waren Fra2+/- Mäuse, die mit dem Lösungsmittel NaCl behan-
delt wurden. Hierzu wurde ebenso jede Woche 100 µl 0,9% NaCl intraperi-
toneal injiziert.
• Gruppe drei war Fra2+/- Behandlungsgruppe 1. Wöchentlich wurde rekombi-
nante ADA gegeben, die hierzu in 0,9% NaCl gelöst wurde und mit einer
Konzentration von 0,5 mg/ml (0,5 Units = 0,5 U) wurden 100 µl intraperi-
toneal injiziert
• Gruppe vier war Fra2+/- Behandlungsgruppe 2. Auch hier wurde wöchentlich
rekombinante ADA gegeben, die hierzu in 0,9% NaCl gelöst wurde und mit

22
einer Konzentration von 5 mg/ml (5 Units = 5 U) injizierte man 100 µl intra-
peritoneal.
5.2.2 Analyse der Hautveränderung
Um die Hautfibrose der Mäuse zu analysieren, wurden Hautareale herausgeschnitten,
in 4%igem Formalin für sechs Stunden fixiert und in Paraffin eingebettet.
5.2.2.1 Histologische Analyse: Sirius Red-Färbung
Um die Hautdicke zu bestimmen, führte man eine Sirius Red-Färbung durch. Dazu
wurden die Hautschnitte entparaffiniert und rehydriert, bevor man sie mittels Wei-
gert’scher Hämatoxylin-Eisenlack-Lösung (Sigma, Germany) für acht Minuten färb-
te. Danach wurden die Schnitte für fünf Minuten mit Leitungswasser gewaschen.
Darauf folgte die Färbung mit Picro-Sirius Red (Sigma-Aldrich, USA) für eine Stun-
de. Abschließend wurden die Schnitte dehydriert und mit dem Einschlussmittel Ro-
ti®-Histokitt (Carl Roth, Karlsruhe, Germany) fixiert.
Mit Hilfe eines Nikon Eclipse 80i Mikroskops (Nikon, Badhoevedorp, Niederlande)
wurden die Hautschnitte im Dunkelfeld untersucht. Die Hautdickenbestimmung
wurde bei 100-facher Vergrößerung, identisch der im Punkt 5.1.3.1 beschriebenen
Methode, durchgeführt.
5.2.2.2 Immunhistochemische Analyse der Myofibroblasten
Wurde identisch Punkt 5.1.3.2 vorgenommen.
5.2.2.3 Hydroxyprolin-Assay
Wurde identisch Punkt 5.1.3.3 vorgenommen

23
5.2.3 Analyse der Lungenveränderung
Zur Analyse der Lunge wurde der linke Lungenflügel in 4%igem Formalin für acht
Stunden fixiert, in Paraffin eingebettet und anschließend stellte man 5 µm dicke
Schnitte her.
5.2.3.1 Histologische Analyse: Sirius Red-Färbung
Die Färbung erfolgte identisch der im Punkt 5.2.2.1 aufgeführten Beschreibung.
Zur histologischen Quantifizierung der Lungenfibrose wurden die aufgrund der Fib-
rose mit Sirius Red gefärbten Lungenbereiche ins Verhältnis zum Gesamtlungenge-
webe gesetzt. Hierfür wurde ein mit Digitalkamera und Bild-Analyse-Programm
OsteoMeasure (Osteometrics, Decatur, GA) ausgestattetes Zeiss Axioskop-2 Mikro-
skop (Carl Zeiss AG, Oberkochen, Germany) verwendet. Man schoss hierzu Bilder
von allen auf dem Präparat befindlichen Lungenabschnitten. Anschließend markierte
man mit Hilfe eines Eingabestiftes und des Programmes OsteoMeasure alle fibroti-
schen Lungenbereiche. Das Programm gab den prozentualen Anteil der markierten
Fläche - bezogen auf die Gesamtfläche eines Bildes - an. Dieser Vorgang wurde bei
jedem Bild des Präparates wiederholt und abschließend berechnete man den Gesamt-
durchschnitt an fibrotischem Lungengewebe, bezogen auf das ganze Präparat.
5.2.3.2 Immunhistochemische Analyse der Myofibroblasten
Wurde identisch Punkt 5.1.3.2 vorgenommen.
5.2.3.3 Hydroxyprolin-Assay
Wurde identisch Punkt 5.1.3.32 vorgenommen.
5.2.4 Analyse der Lungenarterien mittels HE-Färbung
Um die Veränderungen der Lungenarterien zu bestimmen, wurden die histologischen
Schnitte mit Hämatoxylin (Merk, Germany) und Eosin (Carl Roth, Karlsruhe, Ger-
many) - wie beschrieben (Distler et al. 2007) - gefärbt [13].

24
5.2.4.1 Analyse der Gefäßwanddicke
Unter Zuhilfenahme eines Nikon Eclipse 80i Mikroskops (Nikon, Badhoevedorp,
Niederlande) und des Programmes LC Micro berechnete man hierfür die Differenzen
zwischen innerem und äußerem Gefäßwanddurchmesser. Eine Gefäßwanddicke von
>10% des Gesamtgefäßdurchmessers wurde als pathologisch bewertet. Pro Gefäß
wurden drei Messungen vorgenommen und anschließend der Durchschnitt gebildet.
Pro Maus wurden hierbei mindestens fünf Gefäße vermessen.
5.2.4.2 Quantifizierung der stark stenosierten Lungenarterien
Unter Zuhilfenahme eines Nikon Eclipse 80i Mikroskops (Nikon, Badhoevedorp,
Niederlande) wurden die stark stenosierten Lungenarterien manuell gezählt. Als stark
stenosiert wurden hierbei Lungenarterien, deren Lumendurchmesser < 10% der Ge-
fäßwanddicke beträgt, gewertet. Pro Maus wurden mindestens drei Bilder geschos-
sen. Anschließend berechnete man bei jedem Bild den Anteil der stark stenosierten
Gefäße an allen auf dem Bild befindlichen Gefäßen und bildete im Anschluss den
Gesamtdurchschnitt an stark stenosierten Gefäßen pro Maus und pro Gruppe.
5.3 Statistische Auswertung
Alle Daten werden als Mittelwert ± Standardfehler dargestellt. Die Unterschiede
zwischen den Gruppen wurden durch nicht-parametrischen Mann-Whitney-U-Test
auf ihre statistische Signifikanz untersucht. Ein p-Wert von kleiner als 5% wurde als
statistisch signifikant erachtet.
* entspricht einem signifikanten p-Wert, ** entspricht einem sehr signifikanten und
*** einem hoch signifikanten p-Wert.
Zur Verarbeitung der statistischen Daten und der Erstellung der Graphen sowie Bal-
kendiagramme wurden die Programme Excel (Microsoft Corporation, USA) und
GraphPad PrismV (GraphPad Software, USA) verwendet.

25
Abb. 4: Veränderung des Körpergewichtes nach der Knochenmarkzellen- und Splenozyten-transplantation. Die aufgrund der allogenen Knochenmarkzellen- und Splenozytentransplantation verursachte sklerodermiforme cGvHD senkte das Durchschnittskörpergewicht der Mäuse. Bei der syngenen Transplantation kam es zu keinem Gewichtsverlust, sondern zu einer Gewichtszunahme. Die Therapie der cGvHD mit rekombinanter ADA bewirkte ein höheres Durchschnittsgewicht als mit dem Lösungsmittel NaCl.
6.Ergebnisse
6.1 cGvHD-Mausmodell
6.1.1 Klinischer Verlauf
1. Körpergewicht
Die durch allogene Knochenmarkzellen- und Splenozytentransplantation hervorgeru-
fene sklerodermiforme cGvHD verursachte ein signifikant niedrigeres Durchschnitts-
körpergewicht bei den mit dem Lösungsmittel behandelten Mäusen - verglichen mit
der syngenen Kontrollgruppe am 45. Tag nach der Transplantation („allogene Grup-
pe + .aCl“ 99 ± 5% gegenüber der „syngenen Kontrollgruppe + .aCl“ 122 ± 4%
(p = 0,0003); verglichen zum Durchschnittskörpergewicht am Transplantationstag).
Eine Behandlung mit rekombinanter ADA reduzierte den durch die allogene
Splenozyten- und Knochenmarkzellentransplantation verursachten Gewichtsverlust
und bewirkte eine signifikante Steigerung des Durchschnittskörpergewichtes. Hierbei
bewirkte die Gabe rekombinanter ADA mit der Konzentration 0,5 U bzw. 5 U eine
Steigerung des Durchschnittkörpergewichtes auf 111 ± 8% (p = 0,032) bzw. 106 ±
4% (p = 0,039), verglichen mit dem Durchschnittskörpergewicht der Mäuse am
Transplantationstag. (siehe Abb. 4)
26
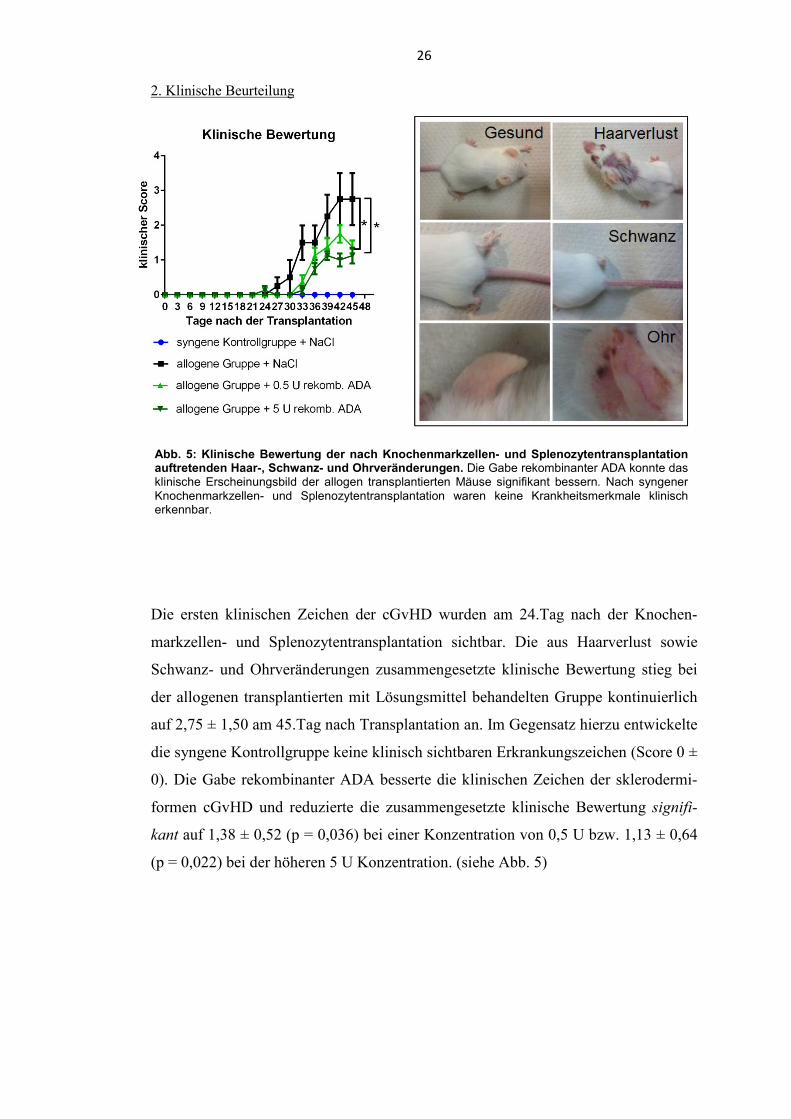
Abb. 5: Klinische Bewertung der nach Knochenmarkzellen- und Splenozytentransplantation auftretenden Haar-, Schwanz- und Ohrveränderungen. Die Gabe rekombinanter ADA konnte das klinische Erscheinungsbild der allogen transplantierten Mäuse signifikant bessern. Nach syngener Knochenmarkzellen- und Splenozytentransplantation waren keine Krankheitsmerkmale klinisch erkennbar.
2. Klinische Beurteilung
Die ersten klinischen Zeichen der cGvHD wurden am 24.Tag nach der Knochen-
markzellen- und Splenozytentransplantation sichtbar. Die aus Haarverlust sowie
Schwanz- und Ohrveränderungen zusammengesetzte klinische Bewertung stieg bei
der allogenen transplantierten mit Lösungsmittel behandelten Gruppe kontinuierlich
auf 2,75 ± 1,50 am 45.Tag nach Transplantation an. Im Gegensatz hierzu entwickelte
die syngene Kontrollgruppe keine klinisch sichtbaren Erkrankungszeichen (Score 0 ±
0). Die Gabe rekombinanter ADA besserte die klinischen Zeichen der sklerodermi-
formen cGvHD und reduzierte die zusammengesetzte klinische Bewertung signifi-
kant auf 1,38 ± 0,52 (p = 0,036) bei einer Konzentration von 0,5 U bzw. 1,13 ± 0,64
(p = 0,022) bei der höheren 5 U Konzentration. (siehe Abb. 5)

27
Abb. 6: Histologische Veranschaulichung der Hautveränderungen mit Hilfe der HE-Färbung. Deutliche Zunahme der Hautdicke bei der allogen transplantierten mit Lösungsmittel behandelten Gruppe. Die Gabe rekombinanter ADA konnte die Zunahme der Hautdicke verbessern. (100 fache Vergrößerung)
6.1.2 Histologische Veränderungen der Haut
6.1.2.1 Histologische Analyse
1.) Hämatoxylin-Eosin-Färbung
Die zur Veranschaulichung der Hautveränderungen durchgeführten HE-Färbungen
zeigen die typischen Merkmale der sklerodermiformen cGvHD. So führte die alloge-
ne Knochenmarkzellen- und Splenozytentransplantation bei der mit dem Lösungs-
mittel NaCl behandelten Gruppe zu einer Zunahme der Hautdicke und einem Verlust
an subkutanem Fettgewebe. Durch Therapie mit rekombinanter ADA konnte die Zu-
nahme der Hautdicke deutlich verringert werden. (siehe Abb. 6)
syngene Kontrollgruppe + NaCl allogene Gruppe + NaCl
allogene Gruppe + 5 U rekomb. ADA allogene Gruppe + 0.5 U rekomb. ADA

28
Abb. 7: Histologische Analyse der Hautdicke und selektive Kollagendarstellung mit Hilfe der Masson-Trichrom-Färbung. Starke Zunahme der Hautdicke und der blau dargestellten Kollagen-menge bei der allogen transplantierten mit Lösungsmittel behandelten Gruppe. Rekombinante ADA verminderte sowohl die Zunahme der Hautdicke als auch der Kollagenmenge. (100 fache Vergröße-rung)
2.) Masson-Trichrom-Färbung
Die zur Hautdickenbestimmung sowie zur selektiven Kollagendarstellung angefertig-
ten Masson-Trichrom-Färbungen zeigen die aufgrund der allogenen Knochenmark-
zellen- und Splenozytentransplantation hervorgerufene Hautfibrose. Diese zeigt sich
deutlich in Form einer Zunahme der Kollagenmenge (blau) und daraus resultierender
Vergrößerung der Hautdicke. Zudem erkennt man einen Verlust an subkutanem Fett-
gewebe. Die Gabe rekombinanter ADA verringerte die Kollagen- und Dickenzu-
nahme der Haut. (siehe Abb. 7)
allogene Gruppe + NaCl syngene Kontrollgruppe + NaCl
allogene Gruppe + 5 U rekomb. ADA allogene Gruppe + 0.5 U rekomb. ADA

29
Abb. 8: Statistische Auswertung der Hautdicke. Die statistische Auswertung der Hautdicke zeigt eine Zunahme der Hautdicke bei der allogen transplantierten mit NaCl behandelten Gruppe. Die Therapie mit rekombinanter ADA verringerte die Zunahme der Hautdicke.
Die folgenden statistischen Auswertungen werden in Form von Balkendiagrammen
dargestellt, welche mit GraphPad PrismV erstellt wurden.
Die allogen transplantierten mit dem Lösungsmittel NaCl behandelten Mäuse besa-
ßen eine 1,51 ± 0,1 -fach (p < 0,0001) hoch signifikant erhöhte Hautdicke im Ver-
gleich zur syngenen Kontrollgruppe. Die Behandlung mit rekombinanter ADA ver-
minderte die Zunahme der Hautdicke. Durch Behandlung mit einer Konzentration
von 0,5 U und 5 U war die Hautdicke auf das 1,29 ± 0,12 -fache (p < 0,0001) bzw.
1,14 ± 0,10 -fache (p < 0,0001) in beiden Fällen hoch signifikant im Vergleich zur
mit Lösungsmittel NaCl therapierten allogenen Gruppe verringert. (siehe Abb. 8)

30
Abb. 9: Immunhistologische Analyse der in der Haut befindlichen Myofibroblasten anhand der Darstellung des Proteins α-Smooth-Muscle-Actin. Myofibroblasten erkennt man anhand des Proteins α-SMA, welches braun angefärbt und charakteristisch für diese ist. Man sieht eine deutliche Zunahme der Myofibroblasten bei der allogenen mit Lösungsmittel behandelten Gruppe. Rekombi-nante ADA verringerte die Anzahl der in der Haut befindlichen Myofibroblasten deutlich. (200 fache Vergrößerung)
6.1.2.2 α-SMA Immunhistologie
Um die Anzahl der Myofibroblasten zu bestimmen, wurden Gewebeschnitte der
Mäuse immunhistochemisch für α-SMA braun angefärbt und die Anzahl der α-SMA-
positiven Fibroblasten ausgezählt. Die allogene Splenozyten- und Knochenmarkzel-
lentransplantation stimulierte die Differenzierung ortsständiger Fibroblasten zu Myo-
fibroblasten. Die Bilder veranschaulichen die deutliche Zunahme an Myofibroblasten
bei der allogen transplantierten mit Lösungsmittel behandelten Gruppe gegenüber der
syngenen Kontrollgruppe. Ebenso sieht man die Verringerung der Myofibroblasten-
anzahl bei den mit rekombinanter ADA therapierten Gruppen. (siehe Abb. 9)
syngene Kontrollgruppe + NaCl allogene Gruppe + NaCl
allogene Gruppe + 0.5 U rekomb. ADA allogene Gruppe + 5 U rekomb. ADA
31
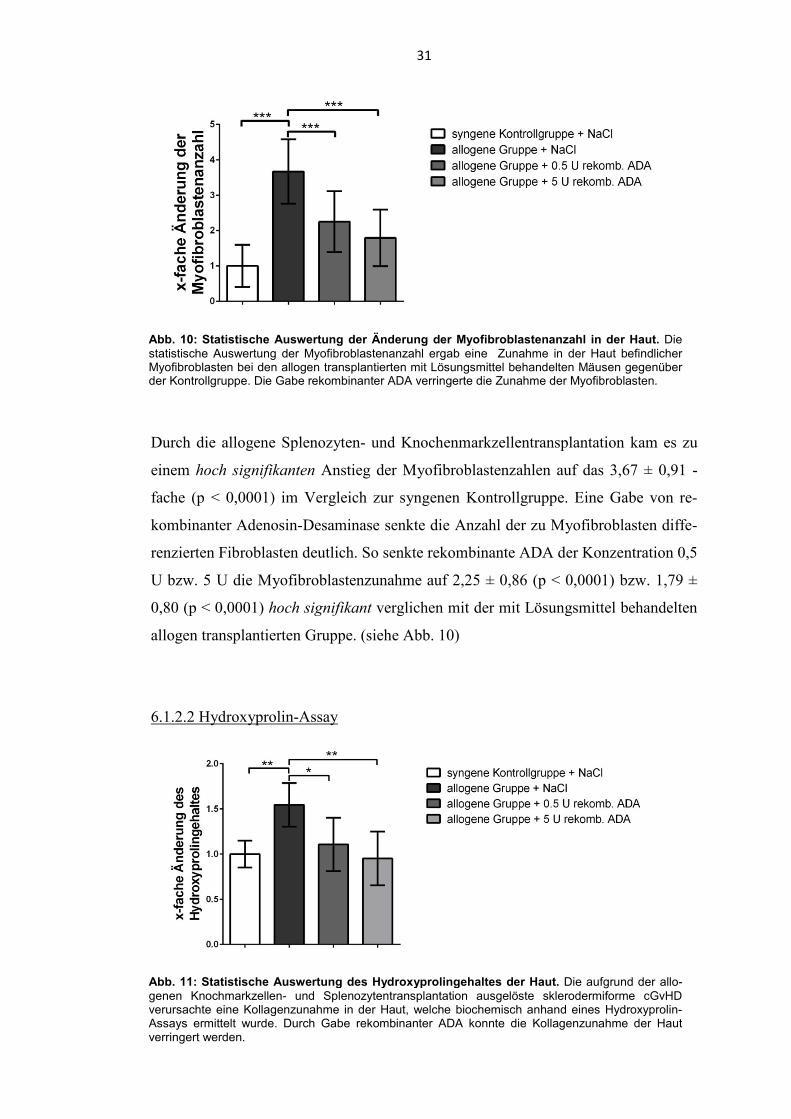
Abb. 10: Statistische Auswertung der Änderung der Myofibroblastenanzahl in der Haut. Die statistische Auswertung der Myofibroblastenanzahl ergab eine Zunahme in der Haut befindlicher Myofibroblasten bei den allogen transplantierten mit Lösungsmittel behandelten Mäusen gegenüber der Kontrollgruppe. Die Gabe rekombinanter ADA verringerte die Zunahme der Myofibroblasten.
Abb. 11: Statistische Auswertung des Hydroxyprolingehaltes der Haut. Die aufgrund der allo-genen Knochmarkzellen- und Splenozytentransplantation ausgelöste sklerodermiforme cGvHD verursachte eine Kollagenzunahme in der Haut, welche biochemisch anhand eines Hydroxyprolin-Assays ermittelt wurde. Durch Gabe rekombinanter ADA konnte die Kollagenzunahme der Haut verringert werden.
Durch die allogene Splenozyten- und Knochenmarkzellentransplantation kam es zu
einem hoch signifikanten Anstieg der Myofibroblastenzahlen auf das 3,67 ± 0,91 -
fache (p < 0,0001) im Vergleich zur syngenen Kontrollgruppe. Eine Gabe von re-
kombinanter Adenosin-Desaminase senkte die Anzahl der zu Myofibroblasten diffe-
renzierten Fibroblasten deutlich. So senkte rekombinante ADA der Konzentration 0,5
U bzw. 5 U die Myofibroblastenzunahme auf 2,25 ± 0,86 (p < 0,0001) bzw. 1,79 ±
0,80 (p < 0,0001) hoch signifikant verglichen mit der mit Lösungsmittel behandelten
allogen transplantierten Gruppe. (siehe Abb. 10)
6.1.2.2 Hydroxyprolin-Assay

32
Um den Kollagengehalt mit einer biochemischen Methode zu vergleichen, wurde der
Hydroxyprolin-Assay durchgeführt. Es zeigte sich eine sehr signifikante Zunahme
der Kollagenmenge bei der mit dem Lösungsmittel NaCl behandelten allogenen
Transplantationsgruppe auf das 1,55 ± 0,24 -fache (p = 0,009) im Vergleich zur
syngenen Kontrollgruppe. Die Gabe rekombinanter ADA konnte die erhöhte Kol-
lagenproduktion senken. So verringerte 0,5 U bzw. 5 U rekombinante Adenosin-
Desaminase den Kollagengehalt der Haut auf 1,11 ± 0,3 (p = 0,029) bzw. 5 U 0,95 ±
0,30 (p = 0,006) signifikant und sehr signifikant im Vergleich zur allogenen mit dem
Lösungsmittel NaCl behandelten Gruppe. (siehe Abb. 11)

33
Abb. 12: Histologische Analyse der Hautdicke mit Hilfe der Sirius Red-Färbung. Die das Fra-2 Gen exprimierenden Mäuse entwickelten eine Fibrose in der Haut mit deutlicher Zunahme der Haut-dicke. Eine Therapie mit rekombinanter ADA verringerte die Hautdickenzunahme. (100 fache Ver-größerung)
6.2 Fra-2-Mausmodell
6.2.1 Histologische Veränderungen der Haut
6.2.1.1 Histologische Analyse: Sirius Red-Färbung
Die zur Veranschaulichung der Hautfibrose und zur Hautdickenbestimmung angefer-
tigten Sirius Red-Färbungen, deren Bilder im Dunkelfeld geschossen wurden, zeigen
die aufgrund der transgenen Überexpression von Fra-2 ausgelöste Hautfibrose sehr
deutlich. Fra2-/- steht für die das Transgen nicht exprimierende Kontrollgruppe,
Fra2+/- kennzeichnet Tiere mit einer Kopie des Transgens. Durch Therapie mit re-
kombinanter ADA konnte die Zunahme der Hautdicke in beiden Konzentrationen
vermindert werden. (siehe Abb. 12)
Fra2-/- NaCl Fra2+/- NaCl
100X
Fra2+/- 0.5 U rekomb. ADA Fra2+/- 5 U rekomb ADA

34
Abb. 13: Statistische Auswertung der Hautdicke. Man erkennt eine starke Zunahme der Hautdi-cke bei den mit dem Lösungsmittel NaCl behandelten Fra2+/- Mäusen verglichen mit der Fra2-/- Kontrollgruppe. Eine Therapie mit rekombinanter ADA verringerte die Zunahme der Hautdicke deut-lich.
Die mit dem Lösungsmittel NaCl behandelten Fra2+/- Mäuse entwickelten eine star-
ke Hautfibrose mit einer 2,57 ± 0,75 -fach (p < 0,0001) hoch signifikant erhöhten
Hautdicke gegenüber der das Transgen nicht exprimierenden Kontrollgruppe. Die
Gabe rekombinanter ADA der Konzentrationen 0,5 U und 5 U konnte die Zunahme
der Hautdicke auf 1,44 ± 0,23 (p < 0,0001) bzw. 1,49 ± 0,16 (p < 0,0001) hoch signi-
fikant im Vergleich zur mit Lösungsmittel therapierten Fra2+/- Gruppe senken.
(siehe Abb. 13)
35
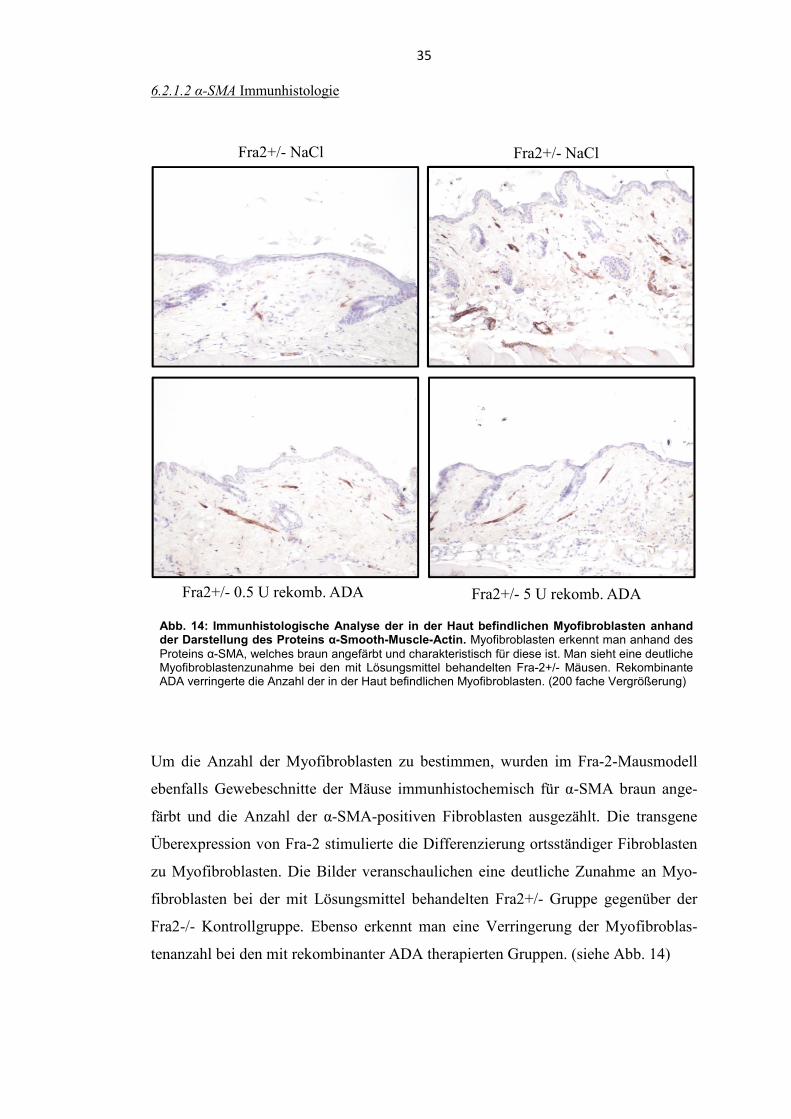
Abb. 14: Immunhistologische Analyse der in der Haut befindlichen Myofibroblasten anhand der Darstellung des Proteins α-Smooth-Muscle-Actin. Myofibroblasten erkennt man anhand des Proteins α-SMA, welches braun angefärbt und charakteristisch für diese ist. Man sieht eine deutliche Myofibroblastenzunahme bei den mit Lösungsmittel behandelten Fra-2+/- Mäusen. Rekombinante ADA verringerte die Anzahl der in der Haut befindlichen Myofibroblasten. (200 fache Vergrößerung)
6.2.1.2 α-SMA Immunhistologie
Um die Anzahl der Myofibroblasten zu bestimmen, wurden im Fra-2-Mausmodell
ebenfalls Gewebeschnitte der Mäuse immunhistochemisch für α-SMA braun ange-
färbt und die Anzahl der α-SMA-positiven Fibroblasten ausgezählt. Die transgene
Überexpression von Fra-2 stimulierte die Differenzierung ortsständiger Fibroblasten
zu Myofibroblasten. Die Bilder veranschaulichen eine deutliche Zunahme an Myo-
fibroblasten bei der mit Lösungsmittel behandelten Fra2+/- Gruppe gegenüber der
Fra2-/- Kontrollgruppe. Ebenso erkennt man eine Verringerung der Myofibroblas-
tenanzahl bei den mit rekombinanter ADA therapierten Gruppen. (siehe Abb. 14)
Fra2+/- NaCl
Fra2+/- 0.5 U rekomb. ADA Fra2+/- 5 U rekomb. ADA
Fra2+/- NaCl

36
Die statistische Auswertung der α-SMA Immunhistologie ergab eine hoch signifikan-
te Zunahme an Myofibroblasten bei den mit Lösungsmittel behandelten Fra2+/-
Mäusen auf das 2,75 ± 0,54 -fache (p < 0,0001) im Vergleich zur Fra2-/- Kontroll-
gruppe. Durch Therapie der transgenen Mäuse mit rekombinanter ADA konnte die
Anzahl der Myofibroblasten in beiden Konzentrationen deutlich gesenkt werden. So
verringerte rekombinante ADA der Konzentrationen 0,5 U bzw. 5 U die Zunahme
der Myofibroblastenanzahl mit 1,76 ± 0,60 (p = 0,0005) bzw. 1,56 ± 0,61 (p <
0,0001) hoch signifikant gegenüber den mit NaCl behandelten Fra2+/- Mäusen.
(siehe Abb.15)
6.2.1.3 Hydroxyprolin-Assay
Abb. 15: Statistische Auswertung der Änderung der Myofibroblastenanzahl in der Haut. Die statistische Auswertung der Myofibroblastenanzahl ergab eine starke Zunahme in der Haut befindli-cher Myofibroblasten bei den mit dem Lösungsmittel NaCl behandelten Fra2+/- Mäusen gegenüber der Fra2-/- Kontrollgruppe. Die Gabe rekombinanter ADA verringerte die Zunahme an Myofibroblas-ten.
Abb. 16: Statistische Auswertung des Hydroxyprolingehaltes der Haut. Die durch Überexpres-sion von Fra-2 ausgelöste Fibrose bewirkte eine starke Kollagenzunahme in der Haut der mit Lö-sungsmittel therapierten Fra2+/- Mäuse. Die Therapie mit rekombinanter ADA verringerte in beiden Konzentrationen die Kollagenmenge in der Haut.

37
Der mit Hilfe eines Hydroxyprolin-Assays quantifizierte Kollagengehalt der Haut
zeigt einen hoch signifikanten Anstieg bei den mit dem Lösungsmittel behandelten
Fra2+/- Mäusen auf 2,39 ± 0,47 -fach (p < 0,0001) verglichen mit der Fra2-/- Kon-
trollgruppe. Die Therapie mit rekombinanter ADA verringerte den Kollagengehalt
der transgenen Mäuse auf das 1,67 ± 0,60 -fache (p = 0,029) bei 0,5 U signifikant
bzw. auf das 1,16 ± 0,53 -fache (p < 0,0001) bei 5 U hoch signifikant gegenüber den
mit Lösungsmittel behandelten Fra2+/- Mäusen. (siehe Abb. 16)

38
6.2.2 Histologische Veränderungen der Lunge
6.2.2.1 Histologische Analyse: Sirius Red-Färbung
Zur Bestimmung der Lungenfibrose wurden Sirius Red-Färbungen angefertigt und
Bilder im Dunkelfeld gemacht. Zur histologischen Quantifizierung der Lungenfibro-
se wurden die aufgrund der Fibrose mit Sirius Red gefärbten Lungenbereiche ins
Verhältnis zum Gesamtlungengewebe gesetzt. An den dargestellten Bildern ist die
massive Lungenfibrose der transgenen Fra2+/- Mäuse im Gegensatz zur Fra2-/- Kon-
trollgruppe, welche keine Lungenfibrose entwickelte, sehr gut erkennbar. (siehe Abb.
17)
Fra2-/- NaCl Fra2+/- NaCl
Fra2+/- 0.5 U rekomb. ADA Fra2+/- 5 U rekomb. ADA
Abb. 17: Histologische Analyse der Lungenfibrose mit Hilfe der Sirius Red-Färbung. Die das Fra-2 Gen exprimierenden Mäuse entwickelten eine starke Lungenfibrose. Die fibrotisch veränderten Lungenbereiche färbten sich mit Sirius Red an. Eine Therapie mit rekombinanter ADA verringerte den prozentualen Anteil von fibrotisch verändertem Lungengewebe. (200 fache Vergrößerung)

39
Die mit Lösungsmittel Fra2+/- behandelten Mäuse entwickelten einen 5,94 ± 1,83 -
fach (p < 0,0001) hoch signifikant erhöhten Anteil an fibrotischem Lungengewebe
gegenüber den Fra2-/- Kontrollmäusen. Die Gabe der hohen Dosis rekombinanter
ADA konnte den Anteil an fibrotischem Lungengewebe signifikant im Vergleich zu
den Fra2+/- mit NaCl behandelten Mäusen senken. So verminderte eine Dosis von
0,5 U bzw. 5 U den fibrotischen Anteil auf das 3,71 ± 0,34 -fache bzw. 2,58 ± 0,72
-fache (p = 0,011) der Fra2-/- Kontrollgruppe. (siehe Abb. 18)
Abb. 18: Statistische Auswertung der Lungenfibrose. Man erkennt bei den mit dem Lösungsmit-tel NaCl behandelten Fra2+/- Mäusen eine starke Zunahme des Prozentsatzes fibrotischer Lungen-bereiche. Die Gabe rekombinanter ADA verringerte den Anteil fibrotisch veränderter Lungenberei-che.

40
Abb. 19: Immunhistologische Analyse der in der Lunge befindlichen Myofibroblasten anhand der Darstellung des Proteins α-Smooth-Muscle-Actin. Myofibroblasten erkennt man anhand des Proteins α-SMA, welches braun angefärbt und charakteristisch für diese ist. Man sieht eine Myo-fibroblastenzunahme bei den mit dem Lösungsmittel NaCl behandelten Fra-2+/- Mäusen. Rekombi-nante ADA verringerte die Anzahl der in der Lunge befindlichen Myofibroblasten deutlich. (200 fache Vergrößerung)
6.2.2.2 α-SMA Immunhistologie
Fra2-/- NaCl Fra2+/- NaCl
Fra2+/- 0.5 U rekomb. ADA Fra2+/- 5 U rekomb. ADA
Abb. 20: Statistische Auswertung der Änderung der Myofibroblastenanzahl in der Lunge. Die statistische Auswertung der Myofibroblastenanzahl ergab eine starke Zunahme in der Lunge befind-licher Myofibroblasten bei den mit Lösungsmittel behandelten Fra2+/- Mäusen gegenüber der Fra2-/- Kontrollgruppe. Die Gabe rekombinanter ADA verringerte die Zunahme an Myofibroblasten.

41
Abb. 21: Statistische Auswertung des Hydroxyprolingehaltes der Lunge. Die durch Überex-pression von Fra-2 ausgelöste Fibrose bewirkte eine Kollagenvermehrung in der Lunge der mit Lö-sungsmittel therapierten transgenen Mäuse. Die Therapie mit rekombinanter ADA verringerte in beiden Konzentrationen die Zunahme der Kollagenmenge in der Lunge.
Die mit dem Lösungsmittel NaCl behandelten Fra2+/- Mäuse besaßen eine 5,44 ±
0,22 -fach (p < 0,0001) hoch signifikant erhöhte Anzahl an Myofibroblasten gegen-
über der Fra2-/- Kontrollgruppe. Die Therapie mit rekombinanter ADA verringerte
die Myofibroblastenzunahme. Die Konzentrationen 0,5 U bzw. 5 U verringerten die-
se auf 4,25 ± 0,23 (p = 0,009) bzw. 3,28 ± 0,19 (p = 0,007) sehr signifikant vergli-
chen mit den mit NaCl therapierten Fra2+/- Mäusen. (siehe Abb. 20)
6.2.2.3 Hydroxyprolin-Assay
Die bei Fra2+/- Mäusen entstehende Lungenfibrose ist an einem erhöhten Kollagen-
gehalt in der Lunge erkennbar. Der Kollagengehalt der Lunge wurde biochemisch
mit Hilfe eines Hydroxyprolin-Assays gemessen. Die nur mit Lösungsmittel behan-
delten Fra2+/- Mäuse haben einen 1,61± 0,06 -fach (p = 0,007) sehr signifikant er-
höhten Hydroxyprolingehalt im Vergleich zur Fra2-/- Kontrollgruppe. Die Gabe re-
kombinanter ADA der Konzentration 0,5 U bzw. 5 U verminderte die Erhöhung des
Hydroxyprolingehalt mit 1,21 ± 0,29 (p = 0,028) signifikant bzw. 1,06 ± 0,31 (p =
0,008) sehr signifikant. (siehe Abb. 21)

42
Abb. 22: Analyse der Gefäßwanddicke mit Hilfe der HE-Färbung. Die dargestellten Bilder veran-schaulichen den vaskulären Phänotyp des Fra-2-Modells mit verdickten Gefäßwänden und perivas-kulären Infiltraten von Entzündungszellen. Mit dem Lösungsmittel NaCl therapierte Fra2+/- Mäuse entwickelten dicke Gefäßwände. Durch Therapie mit rekombinanter ADA entstanden dünnere Ge-fäßwände im Vergleich zu den mit NaCl therapierten Fra2+/- Mäusen. (200 fache Vergrößerung)
6.2.3 Ergebnisse zur Umgestaltung der Lungenarterien
6.2.3.1 Gefäßwanddicke: Hämatoxylin-Eosin-Färbung
.
Fra2-/- NaCl Fra2+/- NaCl
Fra2+/- 0.5 U rekomb. ADA Fra2+/- 5 U rekomb. ADA
Abb. 23: Statistische Auswertung der Gefäßwanddicke. Man sieht eine deutlich höhere Gefäß-wanddicke bei den mit NaCl therapierten Fra2+/- Mäusen im Vergleich zur Fra2-/- Kontrollgruppe. Fra2+/- transgene Mäuse, welche mit rekombinanter ADA therapiert wurden, haben geringere Ge-fäßwanddicken als die mit NaCl therapierten Fra2+/- Mäuse.

43
Abb. 24: Darstellung stark stenosierter Lungenarterien mit Hilfe der HE-Färbung. Neben den verdickten Gefäßwänden sind stark stenosierte Lungenarterien typisch für den vaskulären Phänotyp des Fra-2 Modelles. Man sieht, dass die das Transgen nicht exprimierende Fra2-/- Kontrollgruppe keine stark stenosierten Lungenarterien aufweist. Fra2+/- transgene Mäuse besitzen dagegen stark stenosierte Gefäße und diese sind auch nach Therapie mit rekombinanter ADA noch vorhanden. (200 fache Vergrößerung)
Der durchschnittliche Lungenarteriengefäßwanddurchmesser war bei den mit Lö-
sungsmittel NaCl behandelten Fra2+/- Mäusen mit 1,81 ± 0,31 -fach (p = 0,016) sehr
signifikant gegenüber der Kontrollgruppe erhöht. Eine Behandlung der transgenen
Fra2+/- Mäuse mit rekombinanter ADA bewirkte im Vergleich zu den mit NaCl be-
handelten Fra2+/- Mäusen sehr signifikant geringere Gefäßwanddicken. Hierbei
wurde bei einer Konzentration von 0,5 U bzw. 5 U die Gefäßwanddicke auf das 1,21
± 0,19 -fache (p = 0,006) bzw. 1,09 ± 0,04 -fache (p = 0,003) der Fra2-/- Kontroll-
gruppe verringert. (siehe Abb. 23)
6.2.3.2 Stenosierte Lungenarterien: Hämatoxylin-Eosin-Färbung
Fra2-/- NaCl Fra2+/- NaCl
Fra2+/- 5 U rekomb. ADA Fra2+/- 0.5 U rekomb. ADA
44
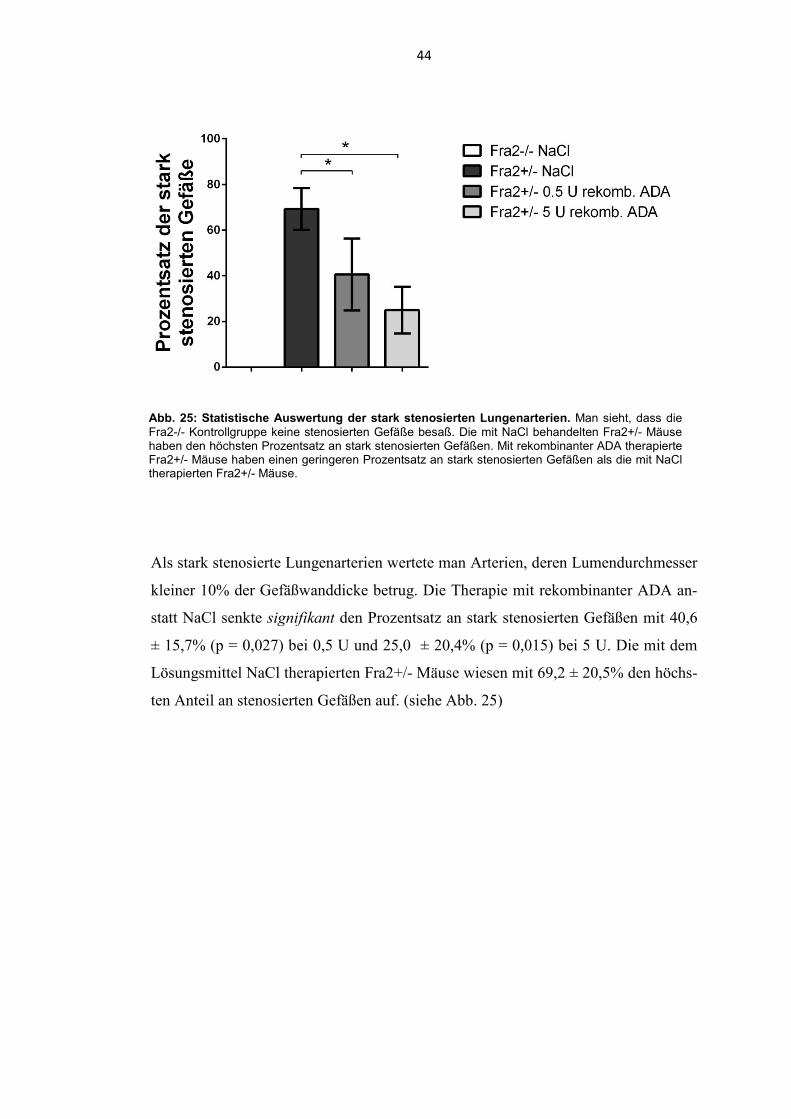
Abb. 25: Statistische Auswertung der stark stenosierten Lungenarterien. Man sieht, dass die Fra2-/- Kontrollgruppe keine stenosierten Gefäße besaß. Die mit NaCl behandelten Fra2+/- Mäuse haben den höchsten Prozentsatz an stark stenosierten Gefäßen. Mit rekombinanter ADA therapierte Fra2+/- Mäuse haben einen geringeren Prozentsatz an stark stenosierten Gefäßen als die mit NaCl therapierten Fra2+/- Mäuse.
Als stark stenosierte Lungenarterien wertete man Arterien, deren Lumendurchmesser
kleiner 10% der Gefäßwanddicke betrug. Die Therapie mit rekombinanter ADA an-
statt NaCl senkte signifikant den Prozentsatz an stark stenosierten Gefäßen mit 40,6
± 15,7% (p = 0,027) bei 0,5 U und 25,0 ± 20,4% (p = 0,015) bei 5 U. Die mit dem
Lösungsmittel NaCl therapierten Fra2+/- Mäuse wiesen mit 69,2 ± 20,5% den höchs-
ten Anteil an stenosierten Gefäßen auf. (siehe Abb. 25)

45
7. Diskussion
In der vorliegenden Arbeit konnten wir zeigen, dass die Behandlung mit rekombi-
nanter ADA antifibrotische Effekte in dem cGvHD und dem Fra-2 Modell der SSc
bewirkte. Zusätzlich reduzierte die Behandlung mit ADA die vaskulären Verände-
rungen im Fra-2 Modell.
Das sklerodermiforme cGvHD Mausmodell spiegelte die Schlüsselmerkmale Fibro-
se, Entzündung und Autoimmunität der SSc wider. Durch Gabe rekombinanter ADA
konnte das Durchschnittsgewicht gegenüber mit Lösungsmittel therapierten alloge-
nen Mäusen erhöht werden. Die Therapie mit rekombinanter ADA bewirkte eine
signifikante Besserung der klinischen Beurteilung.
Die histologischen, immunhistologischen und biochemischen Untersuchungen verifi-
zierten die bessere klinische Beurteilung der mit rekombinanter ADA behandelten
Mäuse. So zeigten diese Mäuse eine signifikant geringere Hautdicke und Kollagen-
menge in der Haut im Vergleich zu den allogenen mit Lösungsmittel behandelten
Mäusen. Dieselben Ergebnisse brachte die immunhistologische Analyse. Rekombi-
nante ADA konnte die Anzahl der differenzierten Myofibroblasten hoch signifikant
gegenüber den mit dem Lösungsmittel NaCl behandelten allogenen Mäusen senken.
Insgesamt bewirkte die Gabe rekombinante ADA im Modell der cGvHD eine Ab-
schwächung des Krankheitsverlaufes und eine Besserung der Krankheitssymptome.
Gleichzeitig ließen sich keine Nebenwirkungen feststellen.
Das Fra-2-Mausmodell entwickelte neben Entzündung und Fibrose auch noch einen
vaskulären Phänotyp. So entwickelten die Fra-2 transgenen Mäuse neben der Fibrose
zusätzlich Gefäßveränderungen, wie den Verlust kleinster Hautkapillaren, eine Ver-
dickung der Gefäßwand in Lungenarterien und die Zunahme von stark stenosierten
Lungenarterien.
Wir untersuchten in diesem Modell die Wirkung der rekombinanten ADA in der
Haut und in der Lunge. Die starke Hautfibrose der Fra2+/- transgenen Mäuse konnte
durch Therapie mit rekombinanter ADA gebessert werden. So verringerte sich die
Hautdicke, die Kollagenmenge und die Myofibroblastenanzahl bei den mit rekombi-
nanter ADA therapierten Mäusen signifikant gegenüber den mit Lösungsmittel be-
handelten Fra2+/- Mäusen. Bei der Untersuchung der Lunge bewirkte die Therapie
mit rekombinanter ADA ähnliche Ergebnisse. Mit rekombinanter ADA therapierte

46
Mäuse besaßen signifikant weniger fibrotische Lungenbereiche, weniger Myo-
fibroblasten und einen niedrigeren Kollagengehalt in den Lungen als mit NaCl thera-
pierte Fra2+/- Mäuse. Die Therapie mit rekombinanter ADA bewirkte signifikant
dünnere und weniger stark stenosierte Lungengefäße im Vergleich zur Therapie der
Fra2+/- Mäuse mit dem Lösungsmittel NaCl.
Zusammengefasst brachte die Gabe rekombinanter ADA auch in diesem Modell sehr
gute therapeutische Effekte, ohne dass Nebenwirkungen beobachtet werden konnten.
Unsere Arbeit liefert damit nicht nur Hinweise darauf, dass die Behandlung mit re-
kombinanter ADA ein potentieller Behandlungsansatz sein könnte, sondern unter-
streicht in Übereinstimmung mit vorherigen Arbeiten die Bedeutung des Adenosin-
Systems für die Pathogenese fibrotischer Erkrankungen. So zeigte die Arbeitsgruppe
um Fernández et al. [20], dass Mäuse mit einem Mangel an ADA und einen daraus
resultierend erhöhten Gewebespiegel an Adenosin spontan Fibrose in der Lunge und
Haut entwickelten. Diese Fibrosierung kann durch Gabe eines A2A-Rezeptor-
Antagonisten ZM-241385 verhindert werden [20]. Ebenso zeigten sie in einem
Mausmodell, dass eine aufgrund externer Bleomycin-Gabe hervorgerufene Fibrose
durch Blockade des A2A-Rezeptors oder Knockout desselben verhindert werden
konnte [20]. Sie stellten zudem fest, dass Mäuse mit einem Mangel an den Enzymen
CD39 oder CD73 keine Bleomycin induzierte Hautfibrose entwickelten und geringe-
re Gewebespiegel an profibrotischen Zytokinen aufwiesen [19]. Die Enzyme CD39
und CD73 sind für den extrazellulären Umbau von ATP zu Adenosin verantwortlich
[65]. Das Enzym Adenosin-Desaminase kann sowohl extra- als auch intrazelluläres
Adenosin zu Inosin (INO) desaminieren [38] und es ist das bedeutendste Enzym zum
Abbau von Adenosin [20].
Rekombinante Adenosin-Desaminase wird bereits seit 1986 als Therapiemöglichkeit
bei Kindern mit angeborenem Adenosin-Desaminase (ADA)-Mangel angewendet,
welche bei Nichtbehandlung ansonsten einen schweren kombinierten Immundefekt
entwickeln [5]. Bis heute wurde dieses rekombinante Enzym bei mehr als 150 Pati-
enten verwendet und gut vertragen [5]. Bei Menschen mit angeborenem Mangel an
ADA kommt es neben einer Adenosin- auch zu einer Desoxyadenosin (dAdo)-
Anhäufung. Desoxyadenosin wird zu Desoxadenosintriphosphat (dATP) umgebaut
[5]. Der daraus resultierend erhöhte dATP-Spiegel hat eine toxische Wirkung auf
Lymphozyten und behindert die Lymphozytenproliferation [5]. Rekombinante ADA
senkt den extrazellulären dATP-Spiegel [5].

47
Bei ADA-defizienten Mäusen konnte bewiesen werden, dass die Stoffwechselpro-
zesse, welche zu Fibrose führen, trennbar von denen sind, die den Immundefekt aus-
lösen. [5]. Blackburn et al. [4] stellte fest, dass bei Mäusen, die aufgrund ADA-
Mangel einen Immundefekt sowie eine Fibrose und Entzündung in der Lunge entwi-
ckelten, die Gabe rekombinanter ADA in niedriger Konzentration zwar die Lungen-
fibrose und Entzündung besserte, aber auf den Immundefekt keine Wirkung hatte [4].
Durch Gabe rekombinanter ADA in niedriger Konzentration konnten die erhöhten
Gewebespiegel an dATP in der Lunge gesenkt werden [4]. Die dATP-Spiegel im
Thymus und der Milz blieben allerdings weiterhin erhöht [4]. Durch Therapie mit
einer hohen Konzentration rekombinanter ADA konnte auch der Immundefekt ver-
bessert werden [4]. Diese Ergebnisse legten nahe, dass die Lungenfibrose und der
Immundefekt zwei voneinander trennbare Erscheinungsbilder darstellen, welche
durch einen unterschiedlichen Grad an Stoffwechselstörungen im Zusammenhang
mit ADA-Mangel entstehen [4].
Diese Ergebnisse verstärken, zusammen mit unseren Ergebnissen, dass rekombinante
ADA einen positiven Einfluss auf mehrere pathologische Stoffwechselprozesse ha-
ben könnte.
Neben den antifibrotischen und antiinflammatorischen Effekten rekombinanter
ADA, welche bereits Blackburn et al. [4] feststellten, konnten wir in unserer Arbeit
zusätzlich noch eine gefäßschützende Wirkung rekombinanter ADA im Fra-2-
Modell feststellen.
Rekombinante ADA wird mittlerweile seit knapp 30 Jahren in der Klinik verwendet.
Bis heute gibt es keine Berichte von toxischen Reaktionen oder Überempfindlich-
keitsreaktionen, welche durch rekombinante ADA ausgelöst wurden [5]. Somit
scheint es sich um ein sicheres Medikament mit großer klinischer Erfahrung zu han-
deln.
A2A-Rezeptorantagonisten, wie z.B. ZM-241385, bewirkten bei ADA-defizienten
Mäusen ebenfalls eine Besserung der Fibrose [16, 20]. Diese wirken eventuell auch
selektiver als rekombinante ADA. ZM-241385 befindet sich derzeit in klinischer
Erprobung, unter anderem bei der Therapie von Morbus Alzheimer und Morbus Par-
kinson [49]. Im Vergleich zu rekombinanter ADA fehlen jedoch Langzeitergebnisse
und Erfahrungen mit diesem Medikament.

48
Diese Arbeit untersuchte die Wirkung rekombinanter ADA zur Prävention SSc-
artiger Veränderungen in zwei etablierten Mausmodellen. Für die klinische Anwen-
dung wäre es jedoch ebenfalls interessant, ob eine Behandlung mit ADA auch bei
bereits manifesten Symptomen wirksam ist. Hierzu sollte in weiteren Studien die
Wirksamkeit von ADA auch im therapeutischen Einsatz bei bereits klinisch manifes-
ten Symptomen in präklinischen Modellen untersucht werden. Außerdem sollte wei-
ter analysiert werden, welche molekularen Mechanismen der gefäßprotektiven Wir-
kung zugrunde liegen.
Zusammenfassend konnten wir somit zeigen, dass rekombinante ADA in zwei ver-
schiedenen Mausmodellen der SSc sowohl vaskuläre als auch fibrotische Verände-
rungen reduzieren kann, ohne dass Nebenwirkungen durch die Therapie ausgelöst
wurden.

49
8. Literaturverzeichnis
1. Agarwal SK, Tan FK, Arnett FC. Genetics and genomic studies in sclero-
derma (systemic sclerosis). Rheum Dis Clin .orth Am. 2008; 34:17-40
2. Antonioli A, Fornai M, Colucci R, Ghisu $, Tuccori M, Del Tacca M, et
al. Pharmacological modulation of adenosine system: novel options for
treatment of inflammatory bowel diseases. Inflamm Bowel Dis. 2008;
14:566–74
3. Beyer C, Schett G, Distler O, Distler JH: Animal models of systemic scle-
rosis: prospects and limitations. Arthritis Rheum. 2010; 62(10):2831-44
4. Blackburn MR, Aldrich M, Volmer JB, et al. The use of enzyme therapy to
regulate the metabolic and phenotypic consequences of adenosine deaminase
deficiency in mice. Differential impact on pulmonary and immunologic ab-
normalities. J Biol Chem. 2000; 275(41):32114–32121
5. Booth C, Gaspar HB. Review: Pegademase bovine (PEG-ADA) for the
treatment of infants and children with severe combined immunodeficiency
(SCID). Biologics. 2009; 3:349-58
6. Burnstock G. Purine and pyrimidine receptors. Cell Mol Life Sci. 2007;
64:1471–83
7. Chan ES, Fernandez P, Merchant AA, Montesinos MC, Trzaska S, Desai
A, et al. Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role
in human dermal fibroblasts and in a murine model of scleroderma. Arthritis
Rheum. 2006; 54(8):2632–42
8. Chifflot H, Fautzi B, Sordet C, Chatelus E, Sibilia J. Incidence and preva-
lence of systemic sclerosis: a systematic literature review. Semin Arthritis
Rheum. 2008; 37:223-35

50
9. Claman H$, Jaffee BD, Huff JC, Clark RA. Chronic graft-versus-host dis-
ease as a model for scleroderma. II. Mast cell depletion with deposition of
immunoglobulins in the skin and fibrosis. Cell Immunol. 1985; 94:73–84
10. Del Ry S, Cabiati M, Lionetti V, Aquaro GD, Martino A, Mattii L, et al.
Pacing-induced regional differences in adenosine receptors mRNA expres-
sion in as wine model of dilated cardiomyopathy. PLoS O.E 2012;
7(10):e4701
11. Del Ry S, Cabiati M, Martino A, Simioniuc A, Morales M-A, Picano E.
Adenosine-receptor mRNA expression in normal and failing minipig hearts. J
Cardiovasc Pharmacol. 2011; 58:149–56
12. Derk CT, Jimenez SA. Systemic sclerosis: current views of its pathogenesis.
Autoimmun Rev. 2003; 2:181-91.
13. Distler JH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE,
Michel BA, Hauser T, Schett G, Gay S, Distler O. Imatinib mesylate
reduces production of extracellular matrix and prevents development of
experimental dermal fibrosis. Arthritis Rheum. 2007; 56:311-22.
14. Duncan MR, Berman B. Stimulation of collagen and glycosaminoglycan
production in cultured human adult dermal fibroblasts by recombinant human
interleukin 6. J Invest Dermatol. 1991; 97:686-92.
15. Eckes B, Mauch C, Hüppe G, Krieg T. Differential regulation of transcrip-
tion and transcript stability of pro-alpha1(I) collagen and fibronectin in acti-
vated fibroblasts derived from patients with systemic scleroderma. Biochem
J. 1996; 315:549-54.
16. Edwin S. L., Chan ES, Cronstein B$. Adenosine in fibrosis. Mod Rheu-
matol. 2010; 20(2): 114–122

51
17. Eferl R, Hasselblatt P, Rath M, Popper H, Zenz R, Komnenovic V, et al.
Development of pulmonary fibrosis through a pathway involving the tran-
scription factor Fra-2/AP-1. Proc .atl Acad Sci USA 2008; 105:10525–30
18. Englert H, Small-McMahon J, Chambers P, et al. Familial risk estimation
in systemic sclerosis. Aust .ZJ Med. 1999; 29:36-41
19. Fernández P, Perez-Aso M, Smith G, Wilder T, Trzaska S, Chiriboga L,
Franks A Jr, Robson SC, Cronstein B$, Chan ES. Extracellular genera-
tion of adenosine by the ectonucleotidases CD39 and CD73 promotes dermal
fibrosis. Am J Pathol. 2013; 183(6):1740-6
20. Fernández P, Trzaska S, Wilder T, Chiriboga L, Blackburn MR,
Cronstein B$, Chan ES. Pharmacological blockade of A2A receptors pre-
vents dermal fibrosis in a model of elevated tissue adenosine. Am J Pathol.
2008; 172(6):1675-82
21. Fleischmajer R, Perlish JS, West WP. Ultrastructure of cutaneous cellular
infiltrates in scleroderma. Arch Dermatol. 1977; 113:1661
22. Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad
Dermatol. 1980; 2:161-70
23. Fleming J$, $ash RA, McLoad DO, et al. Capillary regeneration in sclero-
derma: stem cell therapy reverses phenotype? PLoS One 2008; 3(1):e1452
24. Freedholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W.
Structureand function of adenosine receptors and their genes. .aunyn-
Schmiedeberg’s Arch Pharmacol. 2000; 362:364–74
25. Freedholm BB, Ljzerman AP, Jacobson KA, Klotz K$, Linden J. Inter-
national Union of Pharmacology. XXV. Nomenclature and classification of
adenosine receptors. Pharmacol Rev. 2001;53:527–52

52
26. Gabrielli A, M.D., Avvedimento EV, M.D., Krieg T, M.D. Scleroderma
Mechanisms of Disease, review article, . Engl J Med. 2009; 360:1989-2003
27. Harrison $K, Myers AR, Corrin B, et al. Structural features of interstitial
lung disease in systemic sclerosis. Am Rev Respir Dis. 1991; 144:706-13
28. Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels
of interleukin 4 (IL-4), IL 10, and IL-13 in patients with systemic sclerosis. J
Rheumato.l 1997; 24:328-32
29. Hasegawa M, Sato S, Fujimoto M, Ihn H, Kikuchi K, Takehara K. Serum
levels of interleukin 6 (IL-6), oncostatin M, soluble IL-6 receptor, and soluble
gp130 in patients with systemic sclerosis. J Rheumatol. 1998; 25:308-13
30. Hasko G, Cronstein B$. Adenosine: an endogenous regulator of innate im-
munity. Trends Immunol. 2004; 25:33–9
31. Hoskins LC, $orris HT, Gottlieb LS, Zamcheck $. Functional and mor-
phologic alterations of the gastrointestinal tract in progressive systemic scle-
rosis (scleroderma). Am J Med. 1962; 33:459-70
32. Jaffee BD, Claman H$. Chronic graft-versus-host disease (GVHD) as a
model for scleroderma. I. Description of model systems. Cell Immunol. 1983;
77:1–12
33. Kahaleh, M.D. Raynaud phenomenon and the vascular disease in SSc.
Curr. Opin. Rheumatol. 2004; 16:718–722
34. Karmouty-Quintana H, Xia Y, Blackburn MR. Adenosine signaling dur-
ing acuteand chronic disease states. J Mol Med. 2013; 91:173–81
35. Katebi M, Fernandez P, Chan ES, Cronstein B$. Adenosine A2A receptor
blockade or deletion diminishes fibrocyte accumulation in the skin in a mu-
rine model of scleroderma, bleomycin-induced fibrosis. Inflammation. 2008;
31(5):299-303

53
36. Kräling BM, Maul GG, Jimenez SA. Mononuclear cell infiltrates in clini-
cally involved skin from patients with systemic sclerosis of recent onset pre-
dominantly consist of monocytes/macrophages. Pathobiology 1995; 63:48-56
37. Lambert $C, Lo YM, Erickson TD, Tylee TS, Guthrie KA, Furst DE, et
al. Male microchimerism in healthy women and women with scleroderma:
cells or circulating DNA? A quantitative answer. Blood 2002; 100:2845–51
38. Latta V, Cabiati M, Rocchicciolia S, Del Ry S, Morales MA. Review: The
role of the adenosinergic system in lung fibrosis, Pharmacological Research
2013; 76:182– 189
39. Lawley TJ, Peck GL, Moutsopoulos HM, Gratwohl AA, Deisseroth AB.
Scleroderma, Sjögren-like syndrome, and chronic graft-versus-host disease.
Ann Intern Med. 1977; 87:707–9
40. LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in
vitro: a possible defect in the regulation or activation of the scleroderma fi-
broblast. J Clin Invest. 1974; 54:880-9
41. LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclero-
sis): classification, subsets and pathogenesis. J Rheumatol 1988; 15: 202-5
42. Loubière LS, Lambert $C, Madeleine MM, et al. HLA allelic variants en-
coding DR11 in diffuse and limited systemic sclerosis in Caucasian women.
Rheumatology (Oxford) 2005; 44:318-22
43. Maurer B, Busch $, Jungel A, Pileckyte M, Gay RE, Michel BA, et al.
Transcription factor fos-related antigen-2 induces progressive peripheral vas-
culopathy in mice closely resembling human systemic sclerosis. Circulation
2009; 120:2367–76

54
44. Mavalia C, Scaletti C, Rom agnani P, et al. Type 2 helper T-cell predomi-
nance and high CD30 expression in systemic sclerosis. Am J Pathol. 1997;
151:1751-1758
45. Mayes MD, Lacey JV, Beebe-Dimmer J, et al. Prevalence, incidence, sur-
vival, and disease characteristics of systemic sclerosis in a large US popula-
tion. Arthritis Rheum. 2003; 48:2246-55
46. Medsger TA. Systemic sclerosis (scleroderma): clinical aspects. In:
Koopman WJ. ed.: Arthritis and allied conditions: a textbook of rheumatol-
ogy. Philadelphia: Williams & Wilkins 1997:1433-65
47. $elson JL, Furst DE, Maloney S, Gooley T, Evans PC, Smith A, et al.
Microchimerism and HLA-compatible relationships of pregnancy in sclero-
derma. Lancet 1998; 351:559–62
48. $ietert PJ, Silver R M. Systemic sclerosis: environmental and occupational
risk factors. Curr Opin Rheumatol. 2000; 12:520-6
49. Oscar P Dall'lgna, Lisiane O Porciúncula, Diogo O Souza, Rodrigo A
Cunha, and Diogo R Lara :Neuroprotection by caffeine and adenosine A2A
receptor blockade of β-amyloid neurotoxicity Br J Pharmacol. 2003; 138(7):
1207–1209
50. Perez-Aso M, Fernandez P, Mediero A, Chan ES, Cronstein B$. Adeno-
sine 2A receptor promotes collagen production by human fibroblasts via
pathways involving cyclic AMP and AKT but independent of Smad2/3. FA-
SEB J. 2014; 28(2):802-12
51. Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P. Sequential
dermal microvascular and perivascular changes in the development of sclero-
derma. J Pathol. 1992; 166:255-63
52. Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham
DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a

55
convergence of pericytes and fibroblasts to a myofibroblast lineage in fibro-
sis. Arthritis Res Ther. 2005; 7: R1113-R1123
53. Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol
Rev. 1998; 50:413–92
54. Reich $, Maurer B, Akhmetshina A, Venalis P, Dees C, Zerr P, et al. The
transcription factor Fra-2 regulates the production of extracellular matrix in
systemic sclerosis. Arthritis Rheum. .2010; 62:280–90
55. Reveille JD. Ethnicity and race in systemic sclerosis: how it affects suscepti-
bility, severity, antibody genetics, and clinical manifestations. Curr Rheu-
matol Rep. 2003; 5:16 0 -7
56. Roumm AD, Whiteside TL, Medsger TA Jr, Rodnan GP. Lymphocytes in
the skin of patients with progressive systemic sclerosis: quantification, sub-
typing, and clinical correlations. Arthritis Rheum. 1984; 27:645-53
57. Sachdeva S, Gupta M. Adenosine and its receptors as therapeutic targets: an
overview. Saudi Pharmaceutical Journal 2013; 21:245-53
58. Sakkas, L.I. and Platsoucas, C.D. Is systemic sclerosis an antigen-driven T
cell disease? Arthritis Rheum. 2004; 50:1721–1733
59. Sakkas LI, Xu B, Artlett CM, Lu S, Jiminez SA, Platsoucas CD. Oligo-
clonal T cell expansion in the skin of patients with systemic sclerosis. J Im-
munol. 2002; 68:3649-59
60. Varga J, Abraham D Systemic sclerosis: a prototypic multisystem fibrotic
disorder. J Clin Invest. 2007; 117:557-567
61. Vogelsang GB, Lee L, Bensen-Kennedy DM. Pathogenesis and treatment of
graft-versus-host disease after bone marrow transplant. Annu Rev Med. 2003;
54:29–52

56
62. Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system.
Immunol Rev. 2005; 208:126–40
63. Whitfield ML, Finlay DR, Murray JI, et al. Systemic and cell type-specific
gene expression patterns in scleroderma skin. Proc .atl Acad Sci USA 2003;
100:12319-24
64. Zhou Y, Murthy J$, Zeng D, Belardinelli L, Blackburn MR. Alterations
in adeno-sine metabolism and signaling in patients with chronic obstructive
pulmonary disease and idiopathic pulmonary fibrosis. PLoS O.E 2010;
5(2):e9224
65. Zhou Y, Schneider DJ, Blackburn MR. Adenosine signaling and the regu-
lation of chronic lung disease. Pharmacol Ther. 2009; 123:105–16

57
9. Abkürzungsverzeichnis
ADA Adenosin-Desaminase
ADK Adenosinkinase
ADP Adenosindiphosphat
AMP Adenosinmonophosphat
AP-1 Aktivator-Protein-1
ATP Adenosintriphosphat
A1R Adenosin-Rezeptor-1
A2AR Adenosin-Rezeptor-2A
A2BR Adenosin-Rezeptor-2B
A3R Adenosin-Rezeptor-3
cAMP zyklisches Adenosinmonophosphat
CD39 Ectonucleoside triphosphate Diphosphohydrolase
CD73 Ecto-5‘-Nucleotidase
cGvHD chronische Graft-versus-Host-Erkrankung
cGy Zentigray
CTGF connective tissue growth factor
DAB 3,3’-Diaminobenzidin tetrahydrochloride
dAdo Desoxyadenosin
dATP Desoxadenosintriphosphat
ENT equilibrative nucleoside transporter
EZM Extrazelluläre Matrix
Fra-2 Fos-related-Antigen-2
HE Hämatoxylin und Eosin
HLA Human Leukocyte Antigen
INO Inosin
MHA minor histocompatibility Antigene

58
MHCs major histocompatibility complexes
PAH pulmonal-arterielle Hypertonie
PBS phosphatgepufferte Salzlösung
PDGF platelet-derived growth factor
r-ADA rekombinante Adenosin-Desaminase
rekomb. ADA rekombinante Adenosin-Desaminase
αSMA α-Smooth-Muscle-Actin
SSc Systemische Sklerose
TGF-β Transforming-Growth-Factor-β

59
10. Chemikalien/ Materialien
PBS Life technologies, Germany
70 µm Nylon-Netze BD Biosciences, Heidelberg, Germany
NaCl Carl Roth, Karlsruhe, Germany
4%iges Formalin Carl Roth, Karlsruhe, Germany
Hämatoxylin Merk, Germany
Eosin Carl Roth, Karlsruhe, Germany
Nikon Eclipse 80i Nikon, Badhoevedorp, Niederlande
Mikroskop
Bouin’scher Lösung Sigma-Aldrich, USA
Weigert’sche Hämatoxylin Sigma, Germany
-Eisenlack-Lösung
Biebrich-Scharlachrot- Sigma-Aldrich, USA
Säurefuchsin-Lösung
Phosphor-Wolframsäure Carl Roth, Karlsruhe, Germany
Molybdatophosphorsäure Carl Roth, Karlsruhe, Germany
Anilinblau Sigma-Aldrich, USA
Roti®-Histokitt Carl Roth, Karlsruhe, Germany
5%iges Pferdeserum Life technologies, Germany
3%iges H2O2 Carl Roth, Karlsruhe, Germany
monoklonaler anti- clone 1A4, Sigma-Aldrich, Steinheim, Germany
α-SMA-Antikörper
Meerrettich-Peroxidase Dako, Hamburg, Deutschland

60
DAB (3,3‘-Diaminobenzidin Sigma-Aldrich, USA
tetrahydrochloride)
Peroxidase-Substrat-Lösung Sigma-Aldrich, USA
monoklonaler IgG Maus Calbiochem, San Diego, CA, USA
Antikörper
6 M HCl Carl Roth, Karlsruhe, Germany
6 M NaOH Carl Roth, Karlsruhe, Germany
Lackmuspapier Carl Roth, Karlsruhe, Germany
0,06 M Chloramin T Sigma, Germany
3,15 M Perchlorsäure Merck, Germany
20%iger para-Dimethyl- Fluka, Germany
aminobenzaldehyd
Spectra MAX 190 Molecular Devices, Sunnyvale, CA, USA
Mikroplatten Spektral
Photometer
Picro-Sirius Red Sigma-Aldrich, USA
Zeiss Axioskop 2 Carl Zeiss AG, Oberkochen, Germany
Mikroskop
OsteoMeasure Osteometrics, Decatur, GA
GraphPad PrismV GraphPad Software, USA
Excel Microsoft Corporation, USA

61
Verwendete Mäuse
B10.D2 (H-2d) Mäuse Laboratory (Bar Harbour, ME)
Balb/c (H-2d) Mäuse Janvier (Le Genest, St. Isle, France)
Fra-2-Mäuse Arbeitsgruppe Prof. Erwin F. Wagner

62
11. Danksagung
Zuallererst möchte ich mich bei meinem Doktorvater, Herrn Prof. Dr. Jörg Distler,
für die Überlassung des Dissertationsthemas und die außerordentlich gute Betreuung
während der gesamten Arbeitszeit bedanken.
Mein ganz besonderer Dank geht an Frau Yun Zhang, ohne deren Unterstützung und
Rat bei Problemen das Gelingen dieser Arbeit sicher nicht möglich gewesen wäre.
Ebenso möchte ich mich bei Herrn PD Dr. Christian Beyer sehr bedanken, der stets
ein offenes Ohr hatte, wenn Probleme auftauchten.
Allen Mitarbeitern der AG Distler gilt ebenso großer Dank für das angenehme Ar-
beitsklima und der Hilfestellung bei Laborarbeiten.
Meinem Bruder Dr. Christian Layritz möchte ich für hilfreiche Ratschläge während
meines Studiums und bei der Dissertation sehr danken.
Schließlich möchte ich mich bei meinen Eltern bedanken, die mir stets hilfsbereit
zur Seite standen und mich in jeder erdenklichen Art und Weise während meines
gesamten Studiums unterstützten.

63
12. Lebenslauf
Persönliche Daten:
Name: Florian Philip Layritz
Geburtsdatum: 16.03.1989
Geburtsort: Erlangen
Adresse: Erlenstraße 40
90562 Kalchreuth
Familienstand: Ledig
Persönlicher Werdegang
Schulbildung:
09/1995 - 07/1999 Grundschule Kalchreuth
09/1999 - 07/2008 Gymnasium Eckental
Abschluss: Abitur
Zivildienst:
09/2008 - 05/2009 Bayerisches Rotes Kreuz Erlangen
Studium:
10/2009 - 06/2015 Studium der Zahnmedizin an der Fried-
rich-Alexander-Universität Erlangen-
Nürnberg
Studienabschluss: Staatsexamen
08/2010 Naturwissenschaftliche Vorprüfung
Gesamtnote:1
03/2012 Zahnärztliche Vorprüfung
Gesamtnote: 1
06/2015 Zahnärztliche Prüfung
Gesamtnote: 2
11/2012 - 11/2015 Promotionsstudien in der Medizinischen
Klinik III des Universitätsklinikums Er-
langen







