

BiokatalytischeSynthesenvon
ɛ–CaprolactonundchiralenAminen
Dissertation
zurErlangungdesDoktorgrades
derNaturwisenschaften
(Dr.rer.nat.)
vorgelegtder
FakultätfürChemiederUniversitätBielefeld
von
AnnaReimer
ausPretorija/Russland
Bielefeld,August2015


Inmemoriam
JAKOBREIMER


AlsDissertationvorgelegtderFakultätfürChemiederUniversitätBielefeld.
Erstberichterstatter: Prof.Dr.HaraldGröger
Zweitberichterstatter: Prof.Dr.NorbertSewald
Die vorliegende Arbeit wurde in der Organischen Chemie der Fakultät für Chemie der Universität
BielefeldunterderLeitungvonProf.Dr.HaraldGrögerimZeitraumvomApril2011bisAugust2015
erstellt.
AnnaReimer,BiokatalytischeSynthesenvonɛ–CaprolactonundchiralenAminen
©August2015


TeiledieserArbeitsindbereitsveröffentlicht:
Posterpräsentation:
AnnaReimer,SvenjaStaudt,UweBornscheuer,UlfMenyes,HaraldGröger,“EstablishingofGCanalytics
forenzymicallycatalysedBaeyer‐Villigerreaction”,7.CeBiTecSymposium,ZentrumfürInterdisziplinäre
Forschung(ZiF),UniversitätBielefeld,17.–19.Dezember2012.
Publikationen:
AnnaReimer,SeverinWedde,SvenjaStaudt,SandySchmidt,DanielaHöffer,WernerHummel,UdoKragl,
UweBornscheuer,HaraldGröger,ProcessDevelopmentThroughSolventEngineeringintheBiocatalytic
SynthesisoftheHeterocyclicBulkChemicalɛ–Caprolactone, J.Heterocyc.Chem.2015,eingereicht.
FlorianUthoff,AnnaReimer,AndreasLiese,HaraldGröger,EnantioselectiveSynthesisofChiralAmines
ThroughEnzymaticResolutionunderSolvent‐freeConditions,Adv.Synth.Catal.2015,eingereicht.


Danksagung
AndieserStellemöchteichalldenendanken,diedurchihrefachlicheundpersönlicheUnterstützung
zumGelingendieserArbeitbeigetragenhaben.
MeinbesondererDankgiltProf.Dr.HaraldGrögerfürdieBetreungmeinerDissertation.Insbesondere
dankeichfürdieBereitstellungderinteressantenThemenunddieHilfsbereitschaft,diekontinuierliche
FörderungunddiefachlichenGespräche.
IchdankeProf.Dr.NorbertSewaldfürdieÜbernahmedesZweitgutachtens.
DerDeutschenBundeststiftungUmwelt (DBU)danke ich fürdie finanzielleFörderungdesProjektes
„Nachhaltige,biokatalytischeOxidationsprozesse“.DerFirmaEnzymicalsAGundderArbeitsgruppevon
Prof.Dr.WernerHummelgiltmeinDankfürdiezurVerfügunggestelltenMonooxygenasenbzw.der
Alkohol‐Dehydrogenase.
EingroßerDankgehörtdenbeidengutenSeelenderArbeitsgruppe:ThomasGeislerundArjaGaestel.
BeidehabenmitIhrerunendlichfreundlichenArteineschöneAtmosphäregezaubert.Niemandkann
einen morgens fröhlicher begrüßen, mit einem Schmunzeln besser die Ruhe bewahren und eine
leckerereFeuerzangenbowlebastelnalsThomas.ArjadankeichfürdiezahlreichenPläuschchenund
Urlaubsimpressionenimmermalwiederzwischendurch.
Des Weiteren danke ich der MS‐Abteilung: Dr. Jens Spross, Sandra Heitkamp (Danke für die ESI‐
Messungen)undHeinz‐WernerPatruck,derbeiHilfe‐Rufen inComputerfragensofortzurStellewar.
Herrn PeterMester danke ich für die superschnellen NMR‐Messungen, die teilweise schon auf den
Serverhochgeladenwaren,bevormandenWegzurückinsLaborgeschaffthatte.FrauBrigitteMichel
dankeichfürdieDurchführungderElementaranalysen.
IchdankedemehemaligenAKMattayfürdiefreundlicheAufnahmeindiewunderbareWeltderOCI.
MeinDankgiltdenehemaligenundaktuellenMitgliedernderArbeitsgruppe:Dr.KaiAltenhöner*Daniel
Bakonyi–ischweiß,wodingGrillsteht,mingJung,unischfühlemicheingeladen!*TobiasBetke*Dr.
SebastianBringmann–dankefürdeinehumorvolleArt*Dr.JensEberhard–einwandelndesLehrbuch,
sprudelndvorIdeen*WilkoGreschner–einVirtouseanderKaffeemaschine*Dr.AnkeHummel–danke
fürdasKorrekturlesen!*Dr.FlorianMay*Dr.RichardMetzner–dankefürdasKorrekturlesenundnoch
vielesmehr(Fünnünününü!)*Dr.MichaelPeter*MatthiasPieper*PhilippRommelmann*Katharina
Tenbrink*Dr.OliverTosic*FlorianUthoff–denichumseineGelassenheitbeneide*SeverinWedde*
Dr.SebastianWiegmann–erkönntedieWeltmitStativstangenundAlufolieretten*sowiesämtlichen
KollegendesAKinErlangen.DankefürdienetteAtmosphäreimLabor,dievielenschönenausufernden
DiskussionenindenPausensowiedieMario‐Kart‐Abende,dieprofessionellenBrauerei‐Besichtigungen
undBoßel‐Hochleistungssport‐Ausflüge,PubQuiz‐Abende,Glühweinmarkt‐Besuchenundnoch vieles
mehr.EswareineschöneZeit!
Dr.SvenjaStaudtdankeichfürdiefreundlicheHilfestellungenundTippsundTricksfürdenEinstiegin
die Thematik. Ich danke Mathilde Brax und Jan‐Phlipp Broschinski für die synthetischen Arbeiten

während ihrerForschungspraktikaundsämtlichenAzubis (DankanEvelynSchulde fürdiehelfende
HandamKolben)undHiWis.DenDoktorandenderOCIIundOCIIIdankeichfürdengemeinsamen
ZeitvertreibwährendderPraktikumsbetreuungimSaal.
Meiner Büro‐Familie, Ramona Bringmann, Dr. Tina Reß und Marc Biermann, danke ich für die
fruchtbarenDiskussionenüberChemie,MSWordsowiedieWeltunddasLeben.„WennausKollegen
Freundewerden“odereinfach:ihrseidmirindiesenJahrensehransHerzgewachsen.Inbesonderem
MaßemöchteichmichbeiTinafürihreFreundschaftbedanken.EsgibtnochvieleschlechteFilmezu
schauen,vielWeinzutrinkenundunendlichvielzubereden–ichfreuemichdarauf!
MeinenFreundenundLiebenaußerhalbderUnidankeichfürihrVerständnis.Ichkonntemichimmer
aufihroffenesOhrundihrenUnterstützungverlassen,auchwennihnennichtimmersoklarwar,was
genauichdainderUnieigentlichsomache.
ZumSchlussdankeichdenfürmichkostbarstenMenschen:ichdankemeinemFreundRobertfürseine
geduldige moralische Unterstützung und seine Liebe. Mein Bruder Viktor danke ich für seine
Ermutigungen und dafür, dass er der beste Bruder ist. MeinerMutter Erna, diemir diese Chancen
ermöglichthat,dankeichaustiefstemHerzenfürdasinmichgesetzteVertrauen.Ohneeuchhätteich
diesenlangenWegnichtgehenkönnen.DANKE!

I
Inhalt
Inhalt....................................................................................................................................................................I
Abkürzungsverzeichnis..............................................................................................................................IX
1 Einleitung..........................................................................................................................................................1
2 MotivationundZielsetzung.........................................................................................................................3
2.1 EnzymkatalysierteOxidationvonCyclohexanol(1).....................................................................3
2.2 EnantioselektiveRacematspaltungvonAminen.............................................................................5
3 EnzymatischeOxidationmittelsMonooxygenasen.............................................................................9
3.1 ChemischeBAEYER‐VILLIGER‐Oxidation................................................................................................9
3.1.1 KlassischeBAEYER‐VILLIGER‐Oxidation.................................................................................................9
3.1.2 StandderWissenschaft:klassischeBAEYER‐VILLIGER‐Oxidation...........................................12
3.1.3 StereoselektiveBAEYER‐VILLIGER‐Oxidation....................................................................................14
3.2 EnzymkatalysierteSynthesen..............................................................................................................15
3.2.1 EnzymatischeBAEYER‐VILLIGER‐Oxidation.......................................................................................15
3.2.2 BAEYER‐VILLIGER‐Monooxygenasen.....................................................................................................16
3.2.2.1 Allgemeines...................................................................................................................................................16
3.2.2.2 StrukturundMechanismus...................................................................................................................18
3.2.3 Alkohol‐Dehydrogenasen.......................................................................................................................23
3.2.4 Cofaktor‐Regeneration............................................................................................................................26
3.3 EigeneErgebnisseundDiskussion.....................................................................................................29
3.3.1 Standardreaktion:DoppeloxidationvonCyclohexanol(1)zuɛ‐Caprolacton(3).........29
3.3.2 EtablierunganalytischerMethoden...................................................................................................30
3.3.2.1 UmsatzbestimmungmittelsNMR‐Analytik....................................................................................30
3.3.2.2 UmsatzbestimmungmittelsGaschromatographie......................................................................32
3.3.2.3 ErmittlungderRückgewinnungsrate................................................................................................33
3.3.2.4 VergleichderAnalytik‐MethodenanhandderStandard‐Reaktion.....................................36
3.3.2.5 AktivitätsbestimmungmittelsUV/Vis‐SpektroskopieundBRADFORD‐Test.....................37
3.3.2.6 OptimierungderStandardreaktion...................................................................................................39
3.3.3 UntersuchungenmittelsTitrations‐Apparatur.............................................................................43
3.3.4 VerwendungvonAdsorberharzen.....................................................................................................46

II
3.3.5 VariationderSubstrate...........................................................................................................................48
4 EnantioselektiveRacematspaltungvonAminen...............................................................................53
4.1 Lipasen............................................................................................................................................................53
4.1.1 Allgemeines..................................................................................................................................................53
4.1.2 StrukturundMechanismus...................................................................................................................54
4.1.3 EnantioselektivitätvonLipasen..........................................................................................................57
4.2 StandderWissenschaft:Aminsynthesen........................................................................................58
4.2.1 KlassischchemischeundchemokatalytischeSynthesen.........................................................58
4.2.2 EnzymkatalysierteSynthesen..............................................................................................................64
4.3 EigeneErgebnisseundDiskussion.....................................................................................................73
4.3.1 ReferenzenundAnalytik........................................................................................................................73
4.3.1.1 SynthesederReferenzverbindungen................................................................................................73
4.3.1.2 Analytik..........................................................................................................................................................75
4.3.2 VorversuchemitsubstituiertenBenzylaminenalsSubstrate................................................79
4.3.2.1 SynthesedesBenzylamids133...........................................................................................................79
4.3.3 EnzymatischeAcylierungsubstituierterBenzylamine.............................................................80
4.3.4 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)........................................85
4.3.4.1 Prozessoptimierung..................................................................................................................................86
4.3.4.1.1 EinflussderTemperaturundReaktionszeit..................................................................................86
4.3.4.1.2 VergleichmitbestehendenindustriellenProzessen..................................................................88
4.3.5 Enzymrecycling...........................................................................................................................................89
4.3.6 VerwendungvonalternativenLipasen............................................................................................90
4.3.6.1 Lipasen‐Screening.....................................................................................................................................90
4.3.6.2 VerwendungderLipasenvonC‐LECTAalsBiokatalysatoren..................................................93
4.3.7 VariationderAcyldonoren....................................................................................................................96
4.3.7.1 VergleichderAcyldonorenunterStandard‐Reaktionsbedingungen..................................97
4.3.7.2 VergleichderAcyldonorenbeiverringerterTemperatur.......................................................99
4.3.7.3 ParallelablaufendechemischeAcylierungvon1‐Phenylethylamin(rac‐4)................100
4.3.8 ReaktionskinetikenderRacematspaltungvon1‐Phenylethylamin(rac‐4)mit
unterschiedlichenAcyldonoren.......................................................................................................102
4.3.9 Substratbreite...........................................................................................................................................107
4.3.9.1 1‐Aminoindan(rac‐130).....................................................................................................................107

III
4.3.9.2 SekundäreAmine....................................................................................................................................112
4.3.10 ɛ–Caprolacton(3)alsAcyldonor......................................................................................................116
4.3.10.1 ChemischeAcylierungmitɛ–Caprolacton(3)............................................................................116
4.3.10.2 EnzymatischeAcylierungvonBenzylamin(134)mitɛ‐Caprolacton(3)......................118
4.3.10.3 EnzymatischeAcylierungvon1‐Phenylethylamin(rac‐4)mitɛ–Caprolacton
(3)...................................................................................................................................................................119
4.3.10.4 EnzymatischeAcylierungvonsekundärenAminenmitɛ‐Caprolacton(3)..................123
5 Zusammenfassung.....................................................................................................................................125
5.1 EnzymatischeOxidationvonCyclohexanol(1)mittelsCHMO...........................................125
5.2 EnzymatischeRacematspaltungchiralerAmine.......................................................................127
6 Summary.......................................................................................................................................................132
6.1 Enzymaticoxidationofcyclohexanol(1)bymeansofCHMO.............................................132
6.2 Enzymaticresolutionofchiralamines..........................................................................................134
7 ExperimentellerTeil.................................................................................................................................139
7.1 VerwendeteChemikalienundGeräte............................................................................................139
7.2 Synthesen,MethodenundspektroskopischeDaten................................................................142
7.2.1 EnzymatischeOxidationvonCyclohexanol(1)unterEinsatzvonCHMOund
LK‐ADH........................................................................................................................................................142
7.2.1.1 Standardreaktion:AllgemeineArbeitsvorschrift1(AAV1):Enzymatische
OxidationvonCyclohexanol(1)zuε‐Caprolacton(3)...........................................................142
7.2.1.2 Analytik........................................................................................................................................................143
7.2.1.2.1 Umsatzbestimmungmittels1H‐NMR‐Spektroskopie..............................................................143
7.2.1.2.2 StabilitätvonUrotropin(51)alsStandard.................................................................................143
7.2.1.2.3 BestimmungderRückgewinnungsratemittelsNMR..............................................................143
7.2.1.3 UmsatzbestimmungmittelsGC.........................................................................................................144
7.2.1.3.1 ErstellenderEichgerade......................................................................................................................144
7.2.1.3.2 BestimmungderDurchschnittsabweichungen..........................................................................144
7.2.1.3.3 BestimmungdesWiederfindungsratemittelsGC.....................................................................145
7.2.1.3.4 BestimmungderWiederfindungsrateinAnwesenheitvonLk‐ADH...............................146
7.2.1.3.5 EinflussderEnzympellet‐BildungaufdieRückgewinnungsrate......................................146
7.2.1.3.6 VergleichderAnalytikmethoden:GCvs.NMR...........................................................................147
7.2.1.4 Photometertests......................................................................................................................................148

IV
7.2.1.4.1 BestimmungderEnzymaktivitätderLk‐ADHgegenüber3,3,5‐Trimethyl‐
cyclohexanol(53)...................................................................................................................................148
7.2.1.4.2 AllgemeineArbeitsvorschrift2(AAV2):BestimmungderEnzymaktivitätder
CHMO............................................................................................................................................................149
7.2.1.4.3 BVMO‐ScreeningundBRADFORD‐TestzurOxidationvonCyclohexanol(2).................149
7.2.1.4.4 AktivitätsbestimmungderCHMOgegenüberIsophoron(52)...........................................150
7.2.1.5 OptimierungderStandard‐Reaktion..............................................................................................150
7.2.1.5.1 AllgemeineArbeitsvorschrift3(AAV3):Doppeloxidationzuɛ‐Caprolacton(3)
mitoptimierterAufarbeitung............................................................................................................150
7.2.1.5.2 VariationdesLösungsmittels............................................................................................................151
7.2.1.5.3 VariationderReaktionsbedingungen............................................................................................152
7.2.1.6 AllgemeineArbeitsvorschrift4(AAV4):UntersuchungderStabilitätderLk‐
ADHmittelsTitrations‐Apparatur..................................................................................................152
7.2.1.7 VariationdesSubstrates......................................................................................................................153
7.2.1.7.1 3,5,5‐Trimethylcyclohexanol(53)alsSubstrat.........................................................................153
7.2.1.7.2 Isophoron(52)alsSubstrat...............................................................................................................154
7.2.1.8 VerwendungvonAdsorberharzen..................................................................................................155
7.2.1.8.1 BestimmungderRückgewinnungsrateinAnwesenheitvonXAD‐7und
XAD‐1180...................................................................................................................................................155
7.2.1.8.2 DoppeloxidationvonCyclohexanol(1)inAnwesenheitvonXAD‐7................................155
7.2.2 EnantioselektiveRacematspaltungvonAminenmittelsLipasen.....................................157
7.2.2.1 AllgemeineArbeitsvorschrift5(AAV5):SynthesevonReferenzverbindungen........157
7.2.2.1.1 SynthesedesDL‐Ethyl‐3‐oxo‐3‐(1‐phenylethylamino)propanoats(rac‐10)..............157
7.2.2.1.2 Synthesedes2‐Phenoxy‐N‐(1‐phenylethyl)acetamids(rac‐121)....................................158
7.2.2.1.3 Synthesedes2‐Cyano‐N‐(1‐phenylethyl)‐acetamids(rac‐123).......................................158
7.2.2.1.4 SynthesedesN‐(1‐Phenylethyl)acetamids(rac‐7).................................................................159
7.2.2.1.5 Synthesedes2‐Phenyl‐N‐(1‐phenylethyl)acetamids(rac‐126).......................................160
7.2.2.1.6 Synthesedes2‐Methoxy‐N‐(1‐phenylethyl)acetamids(rac‐8).........................................161
7.2.2.1.7 SynthesedesEthyl‐3‐((2,3‐dihydro‐1H‐inden‐1‐yl)amino)‐3‐oxo‐propanoats
(rac‐129)....................................................................................................................................................161
7.2.2.2 Analytik.......................................................................................................................................................162
7.2.2.2.1 ValidierungderAufarbeitungvonAAV5mittelsNMR‐Analytik.......................................162

V
7.2.2.2.2 BestimmungderEnantiomerenüberschüssedurchchemischeAcylierungdes
resultierendenAmins(S)‐4................................................................................................................163
7.2.2.3 VorversuchezurenzymatischenAcylierungmitBenzylaminenalsSubstrate...........164
7.2.2.3.1 SynthesevonEthyl‐3‐(benzylamino)‐3‐oxopropanoat(133)............................................164
7.2.2.3.2 AnalysedesDiamidsN,N‘‐Dibenzylmalonamid(137)...........................................................165
7.2.2.3.3 AllgemeineArbeitsvorschrift6(AAV6):EnzymatischeAcylierung
substituierterBenzylamine................................................................................................................166
7.2.2.3.4 EnzymatischeSynthesevonEthyl‐3‐(benzylamino)‐3‐oxopropanoat(133).............166
7.2.2.3.5 EnzymatischeSynthesevonN‐(m‐Hydroxybenzyl)‐N‐ethylmalonamid(141)..........166
7.2.2.3.6 EnzymatischeSynthesevonN‐(m‐Bromobenzyl)‐N‐ethylmalonamid(145).............167
7.2.2.3.7 SynthesevonN‐(m‐Methoxybenzyl)‐N‐ethylmalonamid(143)........................................168
7.2.2.3.8 BestimmungdesVerlustsvonBenzylamin(134)undMalonsäurediethylester
(9)...................................................................................................................................................................168
7.2.2.3.9 AllgemeineArbeitsvorschrift7(AAV7):Kontrollederchemischen
ParallelreaktionderAcylierungverschiedenerBenzylamine............................................169
7.2.2.3.10 UntersuchungenzurDiamidbildungausgehendvon3‐Methoxybenzylamin
(139).............................................................................................................................................................170
7.2.2.3.11 LösungsmitteleffektderenzymatischenAcylierungsubstituierterBenzylamine.....170
7.2.2.4 EnzymatischenRacematspaltungvon1‐Phenylethylamin(rac‐4)..................................171
7.2.2.4.1 Standardreaktion:Racematspaltungvon1‐Phenylethylamin(rac‐4)mit
Malonsäurediethylester(9)................................................................................................................171
7.2.2.4.2 ProzessoptimierungderenzymatischenRacematspaltungvonrac‐4............................172
7.2.2.4.3 UntersuchungderParallelreaktionderAcylierungvon1‐Phenylethylamin
(rac‐4)..........................................................................................................................................................173
7.2.2.4.4 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)beigeringerer
Enzymbeladung.......................................................................................................................................173
7.2.2.4.5 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)beigeringerer
EnzymbeladunginEt2O........................................................................................................................174
7.2.2.5 Enzymrecycling........................................................................................................................................174
7.2.2.6 Lipasen‐ScreeningdurchAcylierungvonAminen...................................................................175
7.2.2.6.1 Lipasen‐ScreeningdurchAcylierungvonBenzylamin(134).............................................175
7.2.2.6.2 Lipasen‐ScreeningdurchAcylierungvon3‐Hydroxybenzylamin(138).......................176
7.2.2.6.3 Lipasen‐ScreeningdurchAcylierungvon1‐Phenylethylamin(rac‐4)...........................177

VI
7.2.2.7 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)mit
alternativenLipasenCAL‐BvonC‐LECTA......................................................................................178
7.2.2.7.1 AllgemeineArbeitsvorschrift9(AAV9):VergleichderAktivitätvonLipasen
vonC‐LECTAmittelsTitrationmitNaOH.......................................................................................178
7.2.2.7.2 BestimmungderEnzymaktivitätenderLipasenvonC‐LECTAmittels
enzymatischerSpaltungdesEssigesters5bei20°C..............................................................178
7.2.2.7.3 BestimmungderEnzymaktivitätenderLipasenmittelsenzymatischer
SpaltungdesEssigesters5bei60°C..............................................................................................179
7.2.2.7.4 EnzymatischeRacematspaltungvonrac‐4mittelsimmobilisierterCAL‐Bvon
C‐LECTA.........................................................................................................................................................180
7.2.2.7.5 EnzymatischeRacematspaltungvonrac‐4mittelslyophilisierterCAL‐Bvon
C‐LECTA.........................................................................................................................................................180
7.2.2.8 VariationderAcyldonoren.................................................................................................................181
7.2.2.8.1 EnzymatischeRacematspaltungvonrac‐4mitalternativenAcyldonoren...................181
7.2.2.8.2 ParallelablaufendechemischeAcylierungvonrac‐4mitunterschiedlichen
Acyldonoren..............................................................................................................................................182
7.2.2.8.3 KinetikderRacematspaltungvonrac‐4mitMalonsäurediethylester(9)....................183
7.2.2.8.4 KinetikderRacematspaltungvonrac‐4mitPhenoxyessigsäureethylester
(122).............................................................................................................................................................184
7.2.2.8.5 KinetikderRacematspaltungvonrac‐4mitCyanessigsäureethylester(124)...........185
7.2.2.8.6 KinetikderRacematspaltungvonrac‐4mitMethoxyessigsäureethylester
(111).............................................................................................................................................................185
7.2.2.9 1‐Aminoindan(rac‐130)alsSubstrat...........................................................................................186
7.2.2.9.1 AnalysedesDiamidsN1,N3‐bis(2,3‐dihydro‐1H‐inden‐1‐yl)malonamid(129).........186
7.2.2.9.2 BestimmungdesVerlustsvon1‐Aminoindan(rac‐130)währendder
Aufarbeitung.............................................................................................................................................187
7.2.2.9.3 Temperatur‐undZeiteffektderenzymatischenRacematspaltungvon
1‐Aminoindan(rac‐130).....................................................................................................................187
7.2.2.9.4 ParallelablaufendechemischeAcylierungvon1‐Aminoindan(rac‐130)....................188
7.2.2.10 SekundäreAminealsSubstrate........................................................................................................189
7.2.2.10.1 BestimmungdesVerlustsdersekundärenAmine149bzw.150.....................................189
7.2.2.10.2 SynthesevonEthyl‐3‐(2‐methylpiperidin‐1‐yl)‐3‐oxopropanoat(rac‐151)..............189
7.2.2.10.3 SynthesevonEthyl‐3‐(3‐methylpiperidin‐1‐yl)‐3‐oxopropanoat(rac‐152)..............190
7.2.2.10.4 EnzymatischeAcylierungvon3‐Methylpiperidin(rac‐150)..............................................190

VII
7.2.2.10.5 ProzessoptimierungderenzymatischenRacematspaltungvon
3‐Methylpiperidin(rac‐150).............................................................................................................191
7.2.2.10.6 ParallelablaufendechemischeAcylierungvonrac‐150.......................................................191
7.2.2.11 Verwendungvonɛ–Caprolacton(3)alsAcyldonor................................................................192
7.2.2.11.1 BestimmungderStabilitätvonɛ–Caprolacton(3)währendderReaktionund
Aufarbeitung..............................................................................................................................................192
7.2.2.11.2 EnzymatischeAcylierungvonBenzylamin(134)mitɛ‐Caprolacton(3)......................193
7.2.2.11.3 VariationderReaktionsbedingungenderenzymatischenAcylierungvon
Benzylamin(134)mitɛ‐Caprolacton(3).....................................................................................193
7.2.2.11.4 ParallelablaufendechemischeAcylierungvonBenzylamin(134)mit
ɛ‐Caprolacton(3)....................................................................................................................................194
7.2.2.11.5 Synthesevon6‐Hydroxy‐N‐(1‐phenylethyl)hexanamid(157)..........................................195
7.2.2.11.6 EnzymatischeAcylierungvon1‐Phenylethylamin(rac‐4)mit
ɛ‐Caprolacton(3)....................................................................................................................................195
7.2.2.11.7 TemperaturabhängigkeitderenzymatischenAcylierungvon
1‐Phenylethylamin(rac‐4)mitɛ–Caprolacton(3)..................................................................196
7.2.2.11.8 ParallelablaufendechemischeAcylierungvonrac‐4mitɛ‐Caprolacton(3)..............197
7.2.2.11.9 ReaktionskinetikderAcylierungvon1‐Phenylethylamin(rac‐4)mit
ɛ‐Caprolacton(3)....................................................................................................................................197
7.2.2.11.10 Synthesevon6‐Hydroxy‐1‐(3‐methylpiperidin‐1‐yl)hexan‐1‐on(155).......................198
7.2.2.11.11 EnzymatischeAcylierungensekundärerAminemitɛ‐Caprolacton(3).........................198
8 Literaturverzeichnis.................................................................................................................................201

VIII

IX
Abkürzungsverzeichnis
AAV AllgemeineArbeitsvorschrift
ADH Alkohol‐Dehydrogenase
AD‐H HPLC‐Säule,CHIRALPAK®
Amolysetris‐(3,5‐dimethylphenylcarbamat)
Äq. Äquivalent(e)
atm Atmosphäre,Druckeinheit
BVMO Baeyer‐Villiger‐Monooxygenase
br breit
c Umsatz(conversion)
CAL‐A LipaseAausCandidaantarctica
CAL‐B LipaseBausCandidaantarctica,Novozym435
CH Cyclohexan
CHMO Cyclohexan‐Monooxygenase
δ chemischeVerschiebungin[ppm]
d Dublett
d Küvettendickein[cm]
DBU DeutscheBundesstiftungUmwelt
DBU 1,8‐Diazabicyclo[5.4.0]undec‐7‐en
DC Dünnschichtchromatographie
DCM Dichlormethan
dd dupliziertesDublett
dest. destilliert
DKR dynamisch‐kinetischeRacematspaltung
ε ExtinktionskoeffizientfürNAD(P)Hin[mL.μmol‐1.cm‐1]
E.coli Escherichiacoli
EA Elementaranalyse
EC EinteilungderEnzymeinKlassen(enzymecommission)
EDTA Ethyldiamintetraacetat
ee Enantiomerenüberschuss(enantiomericexcess)
EI Elektronenstoß‐Ionisation
EtOAc Essigester
ESI Elektrospray‐Ionisation
Et Ethyl
etal. undandere(lat.etalii)
Et2O Diethylether
f Probenverdünnungsfaktor

X
FDA US‐Arzeimittelbehörde(USFoodandDrugAdministration)
FMN Flavinmononukleotid
GC Gaschromatographie
GDH Glucosedehydrogenase
ges. gesättigt
h Stunde(n)
HPLC Hochleistungsflüssigkeitschromatographie
(HighPerformanceLiquidChromatography)
Hz Hertz
i‐PrOH iso‐Propanol
IR Infrarot
i.Vak. imVakuum
J skalareKopplungskonstantein[Hz]
kDa Kilodalton
KM MICHAELIS‐Konstantein[mol/l]
KPi Kaliumphosphatpuffer
Lb‐ADH Alkohol‐DehydrogenaseausLactobacillusbrevis
Lk‐ADH Alkohol‐DehydrogenaseausLactobacilluskefir
M molar,Stoffmengenkonzentrationin[mol/l]
m Multiplett
m‐CPBA meta‐Chlorperbenzoesäure(meta‐chloroperoxybenzoicacid)
Me Methyl
mg Milligramm
MHz Megahertz
min Minute(n)
mL Milliliter
mm Millimeter
mM millimolar,Stoffmengenkonzentrationin[mmol/l]
mmol Millimol
MTBE Methyl‐tert‐butylether
MS Massenspektrometrie
m/z VerhältnisMassezuLadung
μL Mikroliter
NADH,NAD+ Nikotinsäureamid‐Adenin‐Dinukleotid
NADPH,NADP+ Nikotinsäureamid‐Adenin‐Dinukleotid‐Phosphat
n.b. nichtbestimmt
NEt3 Triethylamin
nm Nanometer
NMR KernmagnetischeResonanz(Nuclearmagneticresonance)
org. organisch

XI
PAMO Phenylaceton‐Monooxygenase
PBA Peroxybenzoesäure(peroxybenzoicacid)
PCL Poly‐ε‐caprolacton
PMP para‐Methoxyphenyl‐Schutzgruppe
ppm TeileproMillion(partspermillion)
prim primär
q Quartett
qui Quintett
R Substituent
rac Racemat
rel. relativ
rpm UmdrehungenproMinute(roundsperminute)
RT Raumtemperatur
s Singulett
Sdp. Siedepunkt
sec sekundär
Smp. Schmelzpunkt
spez. spezifisch
T Temperaturin[°C]
t Triplett
t Zeit
TBAI Tetrabutylammoniumiodid
td TripletteinesDubletts
tert Tertiär
THF Tetrahydrofuran
tR Retentionszeit
TMS Tetramethylsilan
TRIS Tris‐(hydroxymethyl)‐aminmethan
U Unitsin[µmolmin‐1]
U/mg Aktivität
U/mL volumetrischeAktivität
UV/Vis Ultraviolett/sichtbar(ultraviolett/visible)
v Volumen
Umrechnungsfaktor
Wellenzahlin[cm‐1]
vmax Maximalgeschwindigkeit
wässr. wässrig

XII

XIII
ˆfv|xÇvx? Åç Ätw? |á Åtwx âÑ Éy Å|áàt~xá?
uâà à{xç tÜx Å|áàt~xá ã{|v{ |à |á âáxyâÄ àÉ Åt~x? uxvtâáx à{xç Äxtw Ä|ààÄx uç Ä|ààÄx àÉ à{x àÜâà{AÊ
JULESVERNE,
AJourneytotheCenteroftheEarth

XIV

1 Einleitung
1
1 Einleitung
Die Biotechnologie hat sich zu einer der Schlüssel‐ und Zukunftstechnologien entwickelt. Die
Bezeichnung„weißeBiotechnologie“grenztdieindustrielleBiotechnologievonder„grünen“undder
„roten“Biotechnologieab,diesichmitPflanzenundLebensmittelbefassen,jedochgibtesmitbeiden
BereichenÜberschneidungen.[1]DieweißeBiotechnologiesetztdabeibiotechnologischeMethodenfür
industrielleVerfahrenzurHerstellungvonpharmazeutischenundchemischenProduktenwieAmino‐
säuren,Vitaminen,Antibiotika,Enzymen,MedikamentenundPolymerenein.Bereitsheutebeträgtder
AnteilvonProduktenundVerfahren,dieBiotechnologienutzen,inderchemischenIndustrieetwa5%.
EinAnstiegauf10–20%biszumJahr2025wirderwartet.[2]Etwa80%derWirkstoffeinderProdukt‐
pipeline der Pharmafirmen sind chiral und ein weiterer Anstieg ist sicher. Seit 1992 wird durch
RegularienderUSFoodandDrugAdministration (FDA)unddesEuropeanCommittee forProprietary
MedicinalProductsvorgeschrieben,dassdiephysiologischeWirkungjedesEnantiomerseinesPharma‐
wirkstoffseinzelnuntersuchtwerdenmuss.[4]
Die hervorragenden katalytischen Fähigkeiten von Enzymen zeigen sich in ihrer Vielfalt und Kom‐
plexität.EinGrundfürdasgroßePotentialvonBiokatalysatorenliegtauchdarin,dassdieunendliche
VielfaltderNaturerstansatzweiseentziffertist.MittedesJahres2015sind6759verschiedeneEnzyme
identifiziert und beschrieben worden, wobei etwa 25000 unterschiedliche Enzyme in der Natur
vermutetwerden.[5,6]DurchchemischeModifizierungen,zufälligeMutationenundProteinEngineering
wirdeinegroßeAnzahlanmodifiziertenEnzymenauchfürdieindustrielleAnwendungzurVerfügung
stehen. Zuden etabliertenAnwendungsfeldern vonEnzymen imBereichderweißenBiotechnologie
zählenvorallemdieAgro‐, Lebensmittel‐,Waschmittel‐undTextilchemiesowiedie chemischeund
pharmazeutischeIndustrie.VondenschonbekanntenEnzymenwerdenderzeitgeradeeinmaletwa130
industriellgenutzt.DieUnterteilungvonEnzymenerfolgtinsechsKlassen(EC,enzymecommission)in
AbhängigkeitvonihrenKatalyse‐EigenschafteninderNatur(Tabelle1).[7]
Tabelle1. UnterteilungderEnzymeinEC‐Klassen.[7,8]
Enzymklasse EC FunktioninMikroorganismen
Oxidoreduktasen 1 Reduktionbzw.Oxidation
Transferasen 2 TransfervonAldehyd‐,Keton‐,Acyl‐,Zucker‐,
Phosphoryl‐oderMethylgruppen
Hydrolasen 3 SpaltungoderBildungvonEstern,Amiden,Lactonen,
Lactamen,Epoxiden,Nitrilen,Anhydriden,Glycosiden
Lyasen 4 Addition/EliminierungvonkleinenMolekülen
Isomerasen 5 Isomerisierung,z.B.Racemisierung,Epimerisierung
Ligasen 6 BildungvonC‐C‐,C‐O‐,C‐S‐,C‐N‐Bindungen

1 Einleitung
2
Enzyme lassensichallerdings fürweitmehrReaktionenalsKatalysatoreinsetzenalsvonderNatur
vorgesehenist.BeimVergleichvonchemischenmitenzymatischenReaktionensindvieleVorteileder
Enzymkatalysezuerkennen.TypischerweiseverlaufenenzymkatalysierteReaktionenumeinenFaktor
108 bis 1012mal schneller verglichenmit den nicht‐katalysiertenReaktionen[8,9] und bis zu 107mal
schnelleralschemischkatalysierteReaktionenunterphysiologischenBedingungen.[10]
EnzymatischeUmsetzungenkönnenmeistbeimoderatenBedingungendurchgeführtwerden,während
chemischeReaktionenhäufigErhitzenzumRückflussoderaberHerunterkühlenaufniedrigeTempe‐
raturen erfordern. Metallkatalysatoren, die beispielsweise Platin oder Palladium enthalten, sind oft
teuer und aufwendig in derHerstellung. Im Gegensatz dazu sind vieleMikroorganismen im großen
MaßstabkultivierbarundfürzahlreicheEnzymestehenetablierteExpressionssystemezurVerfügung,
sodassBiokatalysatoren leichtundrelativgünstig ingroßemUmfangerhaltenwerdenkönnen.Aus‐
gehendvoneinemprochiralenkanneinchiralesZentrumerzeugtwerden.Auchkanndurchdiehohe
Chemo‐undRegioselektivitätdermeistenEnzymeaufaufwändigesSchützenundEntschützenvonähn‐
lichenfunktionellenGruppeninnerhalbeinesMolekülsverzichtetwerden.[6,11]
ZudenNachteilenderEnzymezählendieoftmäßigeStereoselektivität,dielimitierteSubstratbreiteund
dierelativgeringeStabilität,dasie in ihrerAktivitätansehrengeMilieubedingungengebundenund
schnelldenaturierbarsind,z.B.durchextremepH‐WerteoderTemperaturen.[12]DabeigibtesinHin‐
blickaufdieStabilitätauchUnterschiedeinnerhalbderBiokatalysatoren.SokönnenmancheEnzyme
lyophilisiertoderinGlycerin‐LösungenebensowieimmobilisiertaufTrägermaterialiendurchauslange
Lagerzeiten ohne größeren Aktivitätsverlust überstehen. DesWeiteren besteht dieMöglichkeit, das
immobilisierteEnzymnachderReaktionabzutrennenundeseinerweiterenUmsetzungzuzuführen.
Auchwird dadurch die Verwendung von organischen Lösungsmitteln als Reaktionsmedium ermög‐
licht.[13]
AlseinlimitierenderFaktorgaltlangediegeringeVerfügbarkeitvonEnzymeninbenutzerfreundlicher
Form.DurchheterogeneGenexpressionfremderGene innicht‐pathogenenundgutcharakterisierten
OrganismenwieEscherichiacoli (E.coli)oderderBäckerhefe (Sachharomycescerevisiae)kanndiese
Limitierungüberwundenwerden.DieHauptvorteileeiner rekombinantenExpressiongegenüberder
NutzungeinesWildtyp‐Enzymssind,dasseinhöheresExpressionsniveauerreichtwerdenkannunddie
WahrscheinlichkeitderkatabolenVerstoffwechslungdesgebildetenProduktsehergeringist.
Biotransformationen lassen sich in isoliertenEnzymenund in ganzenZellendurchführenundbeide
Biokatalysator‐FormenhabenihreVor‐undNachteile.BeiderVerwendungvonGanzzell‐Katalysatoren
entfallendieIsolationderEnzymesowiedieVerwendungeineszusätzlichenCofaktor‐Regenerations‐
systems.ZudenNachteilenzählendieniedrigeAktivitätundderUmgangmitPathogenen.Außerdem
sinddieseZell‐SystemeaufSubstratebeschränkt,diezumeinendieZellmembrandesMikroorganismus
passierenkönnen,zumanderensolltenimFalleinerFermentationwederSubstratnochProdukttoxisch
für den Organismus sein oder von diesem auf anderemWege umgesetzt werden.[11,14] Andererseits
treten bei isolierten Enzymen, im Gegensatz zu Ganzzell‐Katalysatoren, keine Massentransfer‐
Limitierungen und keinemöglicheNebenproduktbildung durch Übermetabolisierung des Produktes
auf.[15,16] Weitere Vorteile sind die einfachere Aufarbeitung und es kann mit höheren Substrat‐
konzentrationengearbeitetwerden.

2 MotivationundZielsetzung
3
2 MotivationundZielsetzung
Lactone dienen in der Industrie als wichtige Precursor für die Herstellung von unterschiedlichen
großtechnischenProdukten.[17]AusgehendvonCyclohexanol(1)sollindieserArbeitüberdieZwischen‐
stufe Cyclohexanon (2) die Synthese von ɛ–Caprolacton (3) mittels enzymkatalysierter Oxidation
erfolgen.FürdieDarstellungdesLactons3wirdzumeineneineAlkoholdehydrogenase(ADH)undzum
anderen eine Cyclohexanon‐Monooxygenase (CHMO) als Biokatalysator in einem Eintopf‐Prozess
eingesetzt(Abbildung1).
Die Synthese enantiomerenreiner Amine ist ein wichtiger Bestandteil bei der Entwicklung und
HerstellungneuartigerArzneimittelsowiebeiderOptimierungderSynthesewegebekannterpharma‐
zeutischerWirkstoffe.[18]FürdieDarstellungvon(S)‐AminenmithohenEnantiomerenüberschüssensoll
im Rahmen dieser Arbeit die kinetische Racematspaltung von racemischen Aminen mit Hilfe von
Lipasen als Biokatalysatoren untersucht werden. In Abbildung 1 sind die untersuchten Enzym‐
reaktionenaufgezeigt.
Abbildung1. BiokatalytischeReaktionen:DoppeloxidationvonCyclohexanol(1)zuε‐Caprolacton(3)undRacematspaltungvonAminen.
2.1 EnzymkatalysierteOxidationvonCyclohexanol(1)
LactonesindcyclischeEster,vondenenε–Caprolacton(3)dasbekanntesteunddasgrößtekommerziell
erhältlicheist.LactonemitkleinererRinggrößewieβ–Propio‐,γ‐Butyro‐undδ‐Valerolactondienenals
BausteinefürPolyesterfürunterschiedlicheAnwendungen.[19]2003lagdieweltweiteProduktionvon

2 MotivationundZielsetzung
4
ɛ‐Caprolacton(3)bei40.000‐60.000t/a.[20]Etwa50%desproduziertenLactons3wirddurchRing‐
öffnungspolymerisationzuPolycaprolacton(PCL)verarbeitet,einemaliphatischenPolyester,derbio‐
resorbierbar und nicht toxisch für Organismen ist.[17] PCL ist unter physiologischen Bedingungen
biologischleichtabbaubarundfindetgroßeVerwendungalsBiomaterial‐ImplantatoderalsMatrixfür
Drug‐Delivery‐SystemeundinderTherapiefürKnochenregeneration.[17,21]WeitereAnwendungfindet
ɛ‐Caprolacton 3 als Precursor für die Caprolactam‐Synthese, welches für die Nylon‐6‐Produktion
benötigt wird, sowie in der Synthese von Urethan‐Beschichtungen, Elastomeren, Lösungsmittel‐
VerdünnernundFasern.[22]
Industriell wird Caprolacton (3) im sogenannten UCC‐Prozess (Union Carbide Corporation) durch
OxidationvonCyclohexanon(2)mitPeressigsäurebei50°CundAtmosphärendruckhergestellt.Die
üblichen Selektivitäten liegen bei 90% bezogen auf Cyclohexanon (2) und 85‐90% bezüglich Per‐
essigsäure.[23]DieindustrielleSyntheseweisteinigeökonomischeundauchökologischeNachteileauf.
DurchdieVielzahlderbenötigtenSynthese‐undAufreinigungsschritteistdieAbfallproduktionbeiden
industriellenProzessenenormhoch.ZudembenötigtdieReaktionsführunghoheTemperaturensowie
Trägergase. Erst zu Beginn des Jahres 2014 hat BASF die Preise von Caprolactam und Adipinsäure
aufgrundgestiegenerRohstoffpreiseerhöht.[24]
AusgehendvondeneinleitendenArbeitenvonWILLETSundSTAUDTistdasHauptzieldieserArbeitdie
WegbereitungzurVergrößerungdesReaktionsmaßstabesderOxidationvonCyclohexanol(1)überdie
Zwischenstufe Cyclohexanon (2) zum Produkt ε‐Caprolacton (3) mit elementarem Sauerstoff als
alleinigesOxidationsmittel in einemEintopf‐Prozess (Abbildung2).[25,26]Dabei soll dasHauptaugen‐
merkdieserArbeitaufdemSolvenz‐EngineeringzurVermeidungderProduktinhibierungliegen.
Abbildung2. Enzymatische Doppeloxidation von Cyclohexanol (1) zu ɛ–Caprolacton (3) unterEinsatzvonLk‐ADHundCHMO.[26]
DieAlkoholdehydrogenaseausLactobacilluskefir (Lk‐ADH)unddieCHMOausAcinetobactercalcoa‐
ceticus(ECSMo‐1)wurdeninderLiteraturalseffektivsteBiokatalysatorenbeschriebenunddement‐
sprechendindieserArbeiteingesetzt.[25,26]AlseinzigesAbfallproduktdieserOxidationsreaktionwird
Wassererhalten.
An erster Stelle steht in dieser Arbeit die Etablierung einer stabilen Analytikmittels Gaschromato‐
graphie, um für die anschließenden enzymatischen Reaktionen die Umsätzemitmöglichst geringer
Schwankungsbreitezubestimmen.DesWeiterensollendieoptimalenBedingungenfürdieBiokatalyse

2 MotivationundZielsetzung
5
entwickeltwerden.ZielistdieEntwicklungeineseffizientenVerfahrensdurchVariationvonReaktions‐
zeit, Temperatur undWahl des Lösungsmittels. Auch die Verwendung von Adsorberharzen soll zur
insitu‐Produktentfernunguntersuchtwerden.DiezuoptimierendenParameterderStandard‐Reaktion
desCyclohexanols(1)zumLacton3sindinAbbildung3dargestellt.
Abbildung3. UntersuchungderenzymatischenDoppeloxidation.
2.2 EnantioselektiveRacematspaltungvonAminen
EnantiomerenreineAminesindwichtigeBausteine füreinegroßeAnzahlanArzneimittel,Agro‐und
anderen Spezialchemikalien. Untersuchungen zeigten, dass etwa 40 – 50% der pharmazeutischen
Wirkstoffe chiraleAmineenthalten.[18] InderLiteratur ist diekinetischeRacematspaltungvon race‐
mischenaliphatischenundaromatischenAminenmittelsLipaseninvielfältigerAusführungbeschrie‐
ben.DieLipaseBausCandidaantarctica(CAL‐B)wurdedabeialseffektivstesEnzymbeschriebenund
indieserArbeitverwendet.[27–29]DasamhäufigstenverwendeteracemischeAminzurRacematspaltung
istdasModelsubstrat1‐Phenylethylamin(rac‐4),welchesmitAcyldonorenwieEssigsäureethylester(5)
oderMethoxyessigsäurepropylester (6) inorganischenLösungsmittelnumgesetztwurde (Abbildung
4).[30,31]Zwarkönnendieerhaltenen(R)‐Amide,(R)‐7und(R)‐8,unddasresultierende(S)‐Amin(S)‐4
mitsehrhohenEnantiomerenüberschüssenerzieltwerden,jedochliegendieReaktionszeitenbeibiszu
60Stunden.DieReaktionsgeschwindigkeitdieserAcylierungenkannerhöhtwerden,wennAcyldonoren
wie6eingesetztwerden,dieinβ‐PositioneinHeteroatomtragen.DabeiwerdendiebestenErgebnisse
in Bezug auf Reaktionsgeschwindigkeit und Enantioselektivität mit Sauerstoff als Heteroatom
erhalten.[31]
Industriell findetdiekinetischeRacematspaltungvonAminenbereitsAnwendung.EinVerfahrenzur
AcylierungvonAminenwurdevonBASFSEpatentiert.[32]Seit2002wirddieenzymatischeRacemat‐
spaltungvonAminenimMaßstabvon>3500t/aproduziert.[4]

2 MotivationundZielsetzung
6
Abbildung4. EnzymatischeRacematspaltungvonrac‐4mitbekanntenAcyldonorenunterEinsatzvonCAL‐B.[30,31]
In Vorarbeiten von SIMON hat sich das Malonsäurediethylester (9) als effizientester Acyldonor
dargestellt.[33,34]DabeikonntedasAmid(R)‐10unterdengewähltenStandard‐Reaktionsbedingungen
von80°CReaktionstemperaturnachnur4.5StundenmitvollständigemUmsatznahezuenantiomeren‐
reinerhaltenwerden(Abbildung5).
Abbildung5. Benchmark‐ExperimentzurenzymatischenRacematspaltungvonrac‐4mitMalonsäurediethylester(9).[33,34]
EinleitendeVersuchevonUTHOFFzeigten,dassdieRacematspaltungvonrac‐4mitMalonsäurediethyl‐
ester (9) auch unter lösungsmittelfreien Bedingungen sehr gute Umsätze und Enantiomerenüber‐
schüssebeiniedrigerenReaktionstemperaturenliefert.[35]ImRahmendieserArbeitsolldieRacemat‐
spaltungmitMalonsäurediethylester(9)alsAcyldonorunterlösungsmittelfreienBedingungenweiter
etabliertwerden. Durch die Variation vonwichtigen Parametern, unter anderem vonReaktionszeit,
Temperatur,EnzymbeladungsowieSubstratkonzentration, istdasZieldieserArbeitdieOptimierung
der enzymkatalysierten kinetischen Racematspaltung hinsichtlich hoher Umsätze und Enantioselek‐
tivitäten. Da sich die CAL‐B durch eine Akzeptanz eines breiten Substratspektrums auszeichnet[4],
werdenimRahmendieserArbeitunterschiedlicheAminegetestet.ZusätzlichsollderAcyldonorvariiert
werden. Außerdem sollte der Prozess eine hohe Selektivität und Reaktionsgeschwindigkeit sowie
möglichst quantitative Umsätze aufweisen. Die Verwendung eines lösungsmittelfreien Reaktions‐
systemsisteingroßerSchritthinsichtlichder„grünen“Chemie,dadasorganischeLösungsmitteleinen

2 MotivationundZielsetzung
7
großen Teil der Abfallmenge einer kinetischen enzymatischen Racematspaltung ausmacht.[36] Die
meistenorganischenMedienhabenverschiedeneNachteile,unteranderemdieToxizitätfürdieUmwelt
sowiepotentielleExplosionsgefahraufgrundihrerFlüchtigkeitundEntflammbarkeit.[37]InderLiteratur
wurdevoneinerverringertenReaktionszeitderRacematspaltung inAbwesenheitvonLösungsmittel
berichtet, was aus ökonomischer Hinsicht vorteilhaft ist.[36] Die zu optimierenden Parameter der
kinetischenRacematspaltungwerdeninAbbildung6amBeispielderRacematspaltungdesAminsrac‐4
mitMalonester9alsStandard‐AcyldonorzumentsprechendenAmid(R)‐10dargestellt.
Abbildung6. UntersuchungderenzymatischenRacematspaltung.

8

3 EnzymatischeOxidationmittelsMonooxygenasen
9
3 EnzymatischeOxidationmittels
Monooxygenasen
3.1 ChemischeBAEYER‐VILLIGER‐Oxidation
3.1.1 KlassischeBAEYER‐VILLIGER‐Oxidation
VONBAEYERundVILLIGERberichtetenimJahre1899mitderRingöffnung„vonKetoneninderTerpen‐
gruppe“.[38]DabeiwurdensieaufBeobachtungenvonCAROaufmerksam,dereinJahrzuvordieOxidation
vonAnilin zuNitrosoaminmitdemstärksten zuderZeit bekanntenOxidationsmittel,H2SO5, durch‐
führte,welcheserauskonzentrierterSchwefelsäureundKalimpersulfatherstellte(Abbildung7).[39]
Abbildung7. SynthesedesCARO’schenReagenz(H2SO5).[39]
DiesesOxidationsmittelwandtenVONBAEYERundVILLIGERaufdieTerpen‐Derivateanund„fanden,dass
dasCARO’scheReagenzeinebensospezifischaufKetonewirkendesMittel ist,welchesgestattet,eine
ReihevonbisherunausführbarenAufgabenspielendleichtzulösen“.[38]Sokonntensieunteranderem
Menthon, Tetrahydrocarvon und Campher in die entsprechenden ε‐Caprolactone überführen
(Abbildung8).
Abbildung8. BAEYER‐VILLIGER‐OxidationvonMenthon(A),Tetrahydrocarvon(B)undCampher(C)mitdemCARO’schenReagenz.[38,40]

3 EnzymatischeOxidationmittelsMonooxygenasen
10
Sie schlussfolgerten, dass das hypothetisch gebildete Oxidationsprodukt „Syperoxid“, ähnlich einer
BECKMANN‐Umlagerung vonOximen, zu den Lactonen umgewandeltwird.[38] In ihrer Studie zurOxi‐
dation von 13‐ bis 17‐gliedrigen ringförmigen Ketonen mit dem CARO’schen Reagenz untersuchten
RUZICKA und STOLL im Jahre 1928 erstmals den Einfluss von Temperatur und Lösungsmittel.[41] Die
bestenErgebnisseerhieltensiebeiTemperaturenvon50–65°CinEisessig.DieBildungvonOligomeren
alsNebenproduktekonntebeiniedrigenTemperaturenvermiedenwerden.
Die ersten Vorschläge zum Ablauf der Reaktion reichten von dem von VON BAEYER und VILLIGER
vorgeschlagenenDioxiran‐Mechanismus[38]überdieBildungeinesPeroxid‐DimersalsNebenprodukt[42]
biszumWITTIG‐Mechanismus[43]mitderformalenÜbertragungeinesOH+‐TeilchensvoneinerPersäure
11 zumCarbonylsauerstoffdesKetons12.EinenweiterenAnsatz lieferteCRIEGEE1948,alsereinen
nucleophilen Angriff der Persäure 11 auf die Keto‐Funktion postulierte, wodurch das sogenannte
CRIEGEE‐Intermediat13gebildetwird.[44]ImJahre1953schließlichkonntenDOERINGundDORFMANdurch
IsotopenmarkierungsexperimentedieTheorievonCRIEGEEbestätigen,indemsie18O‐markiertesBenzo‐
phenon (12) einer Oxidationmit Persäure11 aussetzten und dadurch die Position desmarkierten
SauerstoffatomsimProdukt14bestimmenkonnten(Abbildung9).[45]
Abbildung9. Isotopenmarkierungsexperimente zur Aufklärung des Mechanismus der BAEYER‐VILLIGER‐Oxidation.[40] Dargestellt sind die nach den oben vorgeschlagenenMecha‐nismenzuerwartendenReaktionsprodukte.
Im Fall der Dioxiran‐Bildung (15) nach VON BAEYER und VILLIGER wurde eine statistische 50:50‐
Verteilung erwartet.[40] Sollte die Reaktion über das CRIEGEE‐Intermediat (13) laufen, somüsste das
markierteSauerstoffatomTeilderKeto‐Funktionsein.TatsächlichenthieltdieCarbonylfunktionzu93%
18O‐Sauerstoff.DasProduktdersäurekatalysiertenAdditioneinerPersäureaneineCarbonylfunktion
wirdallgemeinalsCRIEGEE‐Intermediatbezeichnet.DieseAdditionkannhierbeisowohlSäuren‐alsauch

3 EnzymatischeOxidationmittelsMonooxygenasen
11
Basen‐katalysiertablaufen.DieSäure‐EinheitfungiertdabeinachdemnucleophilenAngriffalsAbgangs‐
gruppe, während die Carbonylfunktion wieder hergestellt wird und ein Rest (RM) vom Carbonyl‐
KohlenstoffzumpositivpolarisiertenSauerstoffwandert,wodurchderEstererhaltenwird(Abbildung
10). Als klassische organische Persäuren dienen unter anderem m‐Chlorbenzoesäure (m‐CPBA),
TrifluorperessigsäuresowiePeroxybenzoesäure(PBA).
Abbildung10. ReaktionsmechanismusderBAEYER‐VILLIGER‐OxidationmiteinerPersäure(RM=wan‐dernderRest,RR=verbleibenderRest,RL=Abgangsgruppe).[46]
ImFallderOxidationeinesasymmetrischenKetonswandertamwahrscheinlichstendiejenigeGruppe,
diediepartiellepositiveLadungambestenstabilisierenkann.[40]SokanndieWanderungstendenzim
allgemeinenwiefolgtdefiniertwerden:tert‐Alkyl>Cyclohexyl>sec‐Alkyl>Benzyl>Phenyl>prim‐
Alkyl>Cyclopentyl,Cyclopropyl>Methyl.[46]SinddiebeidenRestegleichoderähnlich,dannwirdein
Gemisch aus beiden möglichen Estern erhalten. Sterische Ansprüche der Reste sowie sie Art der
verwendeten Persäure spielen ebenfalls eine entscheidende Rolle. Demnachmuss im CRIEGEE‐Inter‐
mediat ein nicht‐bindendes freies Elektronenpaar des Sauerstoffatoms sowie die O‐O‐Bindung anti‐
periplanarzumwanderndenRestRMstehen(Abbildung11).[47]
Abbildung11. Antiperiplanare Anordnung des wandernden Restes im CRIEGEE‐Intermediat (RM=wandernderRest,RR=verbleibenderRest,RL=Abgangsgruppe).[47]
TURNERbeobachtetebereits1950amBeispielderOxidationvonsowohlcis‐alsauchtrans‐1‐Acetyl‐2‐
Methylcyclohexan (17), dass dabei nur das jeweilige cis‐ bzw. trans‐Produkt (18) gebildet wird
(Abbildung12).[48]
Abbildung12. ErsterBeweisderErhaltungderKonfigurationdeswanderndenResteswährendderBAEYER‐VILLIGER‐Oxidation.[48]

3 EnzymatischeOxidationmittelsMonooxygenasen
12
BERSONundSUZUKIschlugen1959vor,dassdieWanderungdesRestesunddieCarboxylatabspaltung
konzertiert ablaufen und nannten dieses als Grund für die Stereoselektivität der Reaktion.[49] Die
BeobachtungenzurStereoselektivitätderBAEYER‐VILLIGER‐OxidationwurdenvonMISLOWundBRENNER
bestätigt,indemsiedasoptischaktive3‐Phenyl‐2‐butanon(20)oxidiertenunddabeieineRetentionvon
98% erhielten (Abbildung 13).[50] Die Retention der Stereoinformation macht die BAEYER‐VILLIGER‐
OxidationzueinemwichtigenWerkzeugfürdieasymmetrischeorganischeSynthese.
Abbildung13. Reaktionssequenz zur Bestätigung des Erhalts der Retention des wanderndenKohlenstoffatomsdesoptischaktivenKetons20.[50]
3.1.2 StandderWissenschaft:klassischeBAEYER‐VILLIGER‐Oxidation
ObwohlmehralseinJahrhundertseitEntdeckungderBAEYER‐VILLIGER‐Oxidationvergangenist,istein
Ende der Entwicklung derReaktion noch nicht abzusehen. In der Industrie findet die Synthese von
ɛ‐Caprolacton (3) großtechnische Anwendung. Dabei spielten in den letzten Jahrzehnten neben der
OptimierungderReaktionsbedingungenauchUmweltaspekteeineimmerwichtigereRolle.
ImAllgemeinenwerdendiebisherverwendetenPersäureninsituausdenentsprechendenSäurenund
Wasserstoffperoxid (H2O2, 90%) hergestellt,welches jedochwegen der hohen Explosionsgefahr aus
demHandelgenommenwurde.[51,52]Dasheuteerhältliche70%igeH2O2liefertjedochnurPersäurenmit
geringerer Konzentration. Die Verwendung von Persäuren erfordert den Einsatz geeigneter Kataly‐
satoren.KatalytischeVerfahrenhabengegenüberdenbisherangewendetenstöchiometrischenReak‐
tionenmehrereVorteile,wiedieVereinfachungderReaktionsbedingungenunddenEinsatzvonkosten‐
günstigerenReagenziensowieVerringerungderAbfallmenge.AllerdingsfälltalsNebenproduktWasser
an,waszurHydrolysederentstandenenEsterführenkann.VerwendeteKatalysatorenenthaltenunter
anderemKomplexederÜbergangsmetalleMo(VI)[53],Re(V)[54–56],Pt(II)[57],Ni(II)[51,52],Se[58]sowieTi‐
Silicate[59,60].EinBeispielfüreinemetallkatalysierteBAEYER‐VILLIGER‐OxidationistinAbbildung14dar‐
gestellt.

3 EnzymatischeOxidationmittelsMonooxygenasen
13
Abbildung14. VorgeschlagenerMechanismusderBAEYER‐VILLIGER‐OxidationdurcheinenRe‐Kata‐lysator(23)amBeispielvonCyclobutanon(25).[56]
Anstelle von Wasserstoffperoxid wird seit 1991 auch elementarer Sauerstoff in Kombination mit
BenzaldehydalsOxidationsmitteleingesetzt,welchesalsMUKAIYAMA‐Systembezeichnetwirdunddas
Keton selbst aktiviert.[61] Die Oxidation kann bei Raumtemperatur durch die Übergangsmetalle
Ni(II)[62,63],Cu(II)[63,64]oderFe2O3[65]katalysiertwerden.AllerdingsistderEinsatzsolcherKatalysatoren
vorwiegendaufdieOxidationvoncyclischenKetonenbeschränktundliefernteilweisenurgeringebis
mäßigeStoffumsätze,waszumTeildurchInaktivierungdesKatalysatorsdurchdenzurentsprechenden
Säure hydrolysierten Ester zurückzuführen ist. 1993wurde vonBOLM undKANEDA gezeigt, dass die
BAEYER‐VILLIGER‐OxidationmitdiesemSystemselbstinAbwesenheiteinesKatalysatorsnochmitguten
Umsätzenverläuft,wobei jedocheinÜberschuss anAldehyd sowiedieVerwendungvonchlorierten
Lösungsmittelnnotwendigist.[63,66]
AlternativeAnsätzewiebeispielsweisederEinsatzvonrecyclisierbarenmesoporösenMaterialien,z.B.
Fe‐MCM‐48[67], Fe‐Porphyrin‐Systemen[68] sowie die Oxidation mit Zinn‐ oder Titan‐haltigen β‐Zeo‐
lithen[60,69,70]undzahlreichenweiterenheterogenenZinn(IV)‐Spezies[71],Hydrocalcite[72],Heteropoly‐
oxometalate[73], organische Ionenaustauscherharze[74] sowie organische Acridinium‐Systeme[75],
konntenmit guten bis sehr guten Umsätzen durchgeführt werden. Um die Bildung vonWasser als
NebenproduktundsomitHydrolysedesgebildetenLactonszuvermeiden,wurdenBis(trimethylsilyl)‐
monoperoxidsulfat[76,77]sowie Peroxodisulfat‐Salze(S2O82‐)[78]alsOxidationsmittelanstellevonH2O2
verwendet,wassowohldieAusbeutealsauchdieReinheitdesProdukteserhöhte.2009wurdedieses
System in Kombination mit ionischen Flüssigkeiten noch weiter optimiert.[77] Auch mit anderen
KatalysatorenfandenionischeFlüssigkeitenbereitsAnwendung.[55,70]UmdenEinsatzvonhalogenierten
Lösungsmitteln zu vermeiden konnten erfolgreich auf Silica‐immobilisierten Persäuren in über‐
kritischen Kohlenstoffdioxid eingesetzt werden.[79] Im Jahr 2011 fand auch Oxon, das Triple‐Salz
2KHSO5.KHSO4.K2SO4, erneut Anwendung in Kombination mit einem Phasentransfer‐Katalysator,
welchesineinemZweiphasensystembereitsnach6Stundenbei40°CguteUmsätzelieferte.[80]
Es sind auchVorschläge veröffentlichtworden, Lactone direkt aus den entsprechenden sekundären
Alkoholenzusynthetisieren.2013verwendetenMENGetal.PerameisensäurealsOxidationsmittelund
konnten beispielsweise ɛ‐Caprolacton (3) nach 65MinutenReaktionszeitmit vollständigemUmsatz
erhalten.[81]AllerdingswirddabeidasSubstratineinerKonzentrationvon0.2MunddieSäureineinem
etwa20fachenÜberschusseingesetzt.DesWeiteren findenbeiderDoppeloxidationvonsekundären
AlkoholenionischenFlüssigkeitenundMikrowellen[82],Nafion‐H®[83]oderorganischeKatalysatoren[84]
Anwendung.JedochsindauchindiesenFällendieUmsätzebeilangenReaktionszeitennurmäßigund
die Verwendung vonPeroxo‐Verbindungen ist vomökologischen Standpunkt aus nachteilig imVer‐

3 EnzymatischeOxidationmittelsMonooxygenasen
14
gleich zur Enzymkatalyse, bei der eine Zugabe vonweiterenKatalysatoren nicht notwendig ist. Des
Weiteren dient elementarer Sauerstoff bei enzymkatalysierten Oxidationsreaktionen als Oxidations‐
mittel.
3.1.3 StereoselektiveBAEYER‐VILLIGER‐Oxidation
Katalysatorensolltenzusätzlichzu ihrenKatalyseeigenschaftenauchdieFähigkeitbesitzen,enantio‐
selektiveOxidationsreaktionenzukatalysieren.SoberichteteSTRUKULbereits1994voneinerOxidation
mit chiralen Pt(II)‐Komplexen, mit denen unterschiedliche 2‐substituierte cyclische Ketone zu den
entsprechendenLactonenmitee‐Wertenvonbiszu58%umgesetztwerdenkonnten(Abbildung15).[85]
Abbildung15. EnantioselektiveBAEYER‐VILLIGER‐OxidationmitchiralemKatalysator.[85]
SpäterkonntedurchdenEinsatzvonPt(II)‐oderCo(Salen)‐KomplexenundTensiden inWasserder
Enantiomerenüberschussauf90%gesteigertwerden.[86,87]InderheutigenZeitwerdenhoheUmsätze
mit mäßigen bis guten ee‐Werten durch Verwendung von chiralen enantiomerenreinen Organo‐
katalysatoren[88]oderchiralenMetallkomplexenderÜbergangsmetalleCo[87],Cu[89],Pd[90],Pt[91],Zr[92],
Hf[93]oderMg[94]undAl[95]erhalten.
AuspräperativerSichtscheintdennochdieVerwendungchiralerÜbergangsmetallkatalysatorenfürdie
enantioselektiveSynthesevonEsterninmittlererbishoheroptischerReinheitdieeinzigeaussichtsreise
Alternative zur Katalyse durch Enzyme zu sein.[51,52] Der Nachteil der meisten der genannten
Katalysator‐SystemeistjedochdiegeringeSubstratbreite,danurcyclischeKetoneumgesetztwerden
können.DesWeiterenmüssen sieaufwendig synthetisiertwerden, liefernniedrigeUmsätzeunddie
ReaktionenmüsseninorganischenLösungsmittelndurchgeführtwerden.

3 EnzymatischeOxidationmittelsMonooxygenasen
15
3.2 EnzymkatalysierteSynthesen
3.2.1 EnzymatischeBAEYER‐VILLIGER‐Oxidation
DerEinsatzvonEnzymenbietetimVergleichzudenobengenanntenorganischenKatalysatorenviele
VorteileinHinblickaufdieRegio‐,Chemo‐undEnantioselektivität.
1995beschriebROBERTSeinedurchdieLipaseBderCandidaantarctica (CAL‐B)katalysierteBAEYER‐
VILLIGER‐OxidationvoncyclischenKetonen.[96]DieCAL‐BisthierbeijedochnichtdirektanderOxidation
desKetonsbeteiligt,sondernkatalysiertdieBildungeinerPersäure,ausgehendvoneinerlangkettigen
Carbonsäure mit Wasserstoffperoxid. Die eingesetzten 2‐ und 3‐substituierten Cyclopentanone
und‐hexanonewurdensozuracemischenLactonenoxidiert.DiedabeierhaltenenAusbeutenlagenbei
57–73%undwarennurgeringfügigniedrigeralsdieaufklassischemWegerhaltenen.EineAusnahme
stellte jedoch 2‐Oxocyclohexanessigsäure (28) dar, bei der durch intramolekulare BAEYER‐VILLIGER‐
OxidationdasentsprechendeLacton29beieinemUmsatzvon54%einEnantiomerenüberschussvon
21% ee erhalten werden kann (Abbildung 16). Der Grund für die Enantioselektivität der Reaktion
scheintdieetwasschnellereAcylierungdesEnzymsdurcheinsderbeidenEnantiomerezusein,sodass
diePersäureimAnschlussdarandieintramolekulareReaktiondurchläuft.DielangenReaktionszeiten
vonetwaeinerWocheunddasaufwändigeEntfernenderentstandenenSäurebedürfenweitererOpti‐
mierung.
Abbildung16. EnantioselektiveBAEYER‐VILLIGER‐Oxidationvon2‐Oxocyclohexanessigsäure(28)katalysiertdurchLipaseBderCandidaAntarctica(CAL‐B).[96]
Ähnlichverläuftdie2007vonOLIVObeschriebeneOxidation,derzusätzlichdenKomplexUreahydrogen‐
peroxidinEssigester5einsetzte.[97]DerVorteilhierbeiwardieVermeidungderHydrolysedesLactons
durchAbwesenheitvonWasseralsNebenprodukt.EskonntensoLactonemitee‐Wertenvon20‐60%
undUmsätzevon20–50%nachetwazweiTagenerhaltenwerden.
EinenalternativenWeggingenGUIBÉ‐JAMPELetal.,aufbauendauffrühereArbeitenvonKIRK[98],mitder
autokatalytischenOxidationvonKetonen(Abbildung17).[99]DabeisollteWasserstoffperoxidmithilfe

3 EnzymatischeOxidationmittelsMonooxygenasen
16
vonCarboxyl‐SeitengruppendesEnzymsdieBAEYER‐VILLIGER‐Oxidationstarten.DasgebildeteLacton3
sollte eine CAL‐B‐katalysierte Perhydrolyse zur Peroxysäure 30 durchlaufen, die wiederum die
OxidationvonweiteremKeton2 katalysierenkann,wodurchein autokatalytischerProzess erhalten
wird. Der niedrige Umsatz von 20% konnte durch Ersatz des klassischen H2O2/H2O‐System durch
Ureaperoxidnach48StundenReaktionszeit65%Umsatzgesteigertwerden.ImFallvonsubstituiertem
CyclohexanonliegendieEnantiomerenüberschüssedererhaltenenLactoneauchhierbeinurzwischen
10und70%ee.
Abbildung17. Lipasenkatalysierte Perhydrolysemit anschließender chemischer BAEYER‐VILLIGER‐Oxidation mittels Autokatalyse am Beispiel von Cyclohexanon (2) nach GUIBÉ‐JAMPEL.[99]
3.2.2 BAEYER‐VILLIGER‐Monooxygenasen
3.2.2.1 Allgemeines
HöhereAusbeutenundhöhereEnantiomerenreinheitkönnendurchdenEinsatzvonBAEYER‐VILLIGER‐
Monooxygenasen (BVMOs, EC 1.13.14.x) erzielt werden.[100] Dadurch lässt sich im Gegensatz zu
chemischenoderLipasen‐katalysiertenReaktionenauchdieVerwendungvonWasserstoffperoxidoder
PersäurenalsOxidationsmittelundorganischeLösungsmittelvermeiden,dadieMonooxygenasenmit
molekularemSauerstoffinwässrigemMediumaktivsind.DesWeiterensindsieinderLage,nichtnur
cyclische, sondern auch aliphatische und aromatische Ketone in die entsprechenden Ester umzu‐
wandeln.DieEpoxidierungvonC=C‐Doppelbindungen[101]sowieunteranderemdieSulfoxidation[102],
Phosphoxidation[103], Aminoxidation[104], Boronsäurenoxidation[105] und Selenoxidation[106] können
ebenfallsdurchBVMOskatalysiertwerden.
Die BAEYER‐VILLIGER‐Monooxygenasen gehören zu einer Superfamilie, die drei Klassen von flavin‐
abhängigen Monooxygenasen umfassen: die sogenannten Flavin‐enthaltenden Monooxygenasen
(FMOs), die N‐hydroxylierenden Monooxygenasen (NMOs) und die BVMOs.[107] Monooxygenasen
werdenanhanddervonihnenbenötigtenCofaktorenunterteilt.MonooxygenasenvomTypIsindFlavin‐
abhängigundbenötigenNADPHalsElektronenquelle,währendEnzymedesTyps IIFMNundNADH

3 EnzymatischeOxidationmittelsMonooxygenasen
17
brauchen,hauptsächlichHeteroatomeoxidierenundeukaryotischemUrsprungssind.[108]DieTypenIII
und IV von Monooxygenasen sind kupferabhhängige bzw. eisenhaltige Enzyme.[109,110] BVMOs vom
TypOsindeinneuartigerBVMO‐Typ,dieebenfallsFADundNADPHbenötigen.[111]Strukturellunter‐
scheidensiesichjedochvondenBVMOsvomTypI,dasienichtdasfürBVMOscharakteristischekonser‐
vierteSequenzmotiv(vgl.Abschnitt3.2.2.2)besitzenundnur8%ÜbereinstimmungzurPAMOzeigen.
AllebisherkloniertenBVMOsgehörendemTypIan.
Die BVMOs aktivieren reduktiv Sauerstoff (O2) und bauen diesen in ein Substrat ein. Siewerden in
MonooxygenasenundDioxygenaseneingeteilt.[109,110]DabeiführenMonooxygenasen,zudenenauchdie
BVMOs zählen, ein Sauerstoffatom in das Substrat ein; das zweite Sauerstoffatom wird zu Wasser
reduziert(Abbildung18).DioxygenasenhingegeninserierenbeideAtomedeselementarenSauerstoffs
indasSubstrat.
Abbildung18. BAEYER‐VILLIGER‐OxidationeinesKetonskatalysiertdurchBVMO.[112]
Die ersten Anzeichen für eine enzymatische BAEYER‐VILLIGER‐Oxidation wurden von TURFITT 1948
währendseinenStudienzuSteroid‐SyntheseinPilzenentdeckt.[113]1953konntendieArbeitsgruppen
vonFRIEDundPETERSONinderSynthesevonTestololacton(32),ausgehendvonProgesteron(33),zwei
Reaktionsschrittenachweisen,diedurchdieMonooxygenaseausCyclindrocarponradicolakatalysiert
werden(Abbildung19).[114,115]
Abbildung19. SchematischerSynthesevonProgeston(33)zuTestololacton(32).[116]DiedurchBVMOkatalysiertenSchrittesindmitSternchengekennzeichnet.

3 EnzymatischeOxidationmittelsMonooxygenasen
18
DieersteVerknüpfungzwischenderLactonisierungvonSteroiden,derbiochemischenOxidationvon
CampherdurcheinBakteriumundder chemischenBAEYER‐VILLIGER‐Oxidation erstelltenPRAIRIEund
TALALY 1963.[117] Durchweiteremechanistische StudienwurdeNADPH als essenzieller Cofaktor für
dieseArtderOxygenaseidentifiziert.[118]
BAEYER‐VILLIGER‐Monooxygenasen tretenvorwiegend inMikroorganismenaufundsinddortankata‐
bolischenVorgängenbeteiligt,etwaindenPilzenCylindrocarpon[113–115],Exophiala[119],Xanthobacter[120]
und Aspergillus[121] und in den Bakterien der Gattungen Cormamonas (ehemals Pseudomonas)[122],
Nocardia[100],Rhoddococcus[123],Gordonia[124],Mycobacterium[125]undAcinetobacter[126].Aberauchbei
höherenOrganismensindMonooxygenasenanzutreffen,beispielsweisebeiderSynthesevonSteroiden
undIridoideninPflanzen[127]odervonToxineninSchalentieren[128].InBakterienscheintdienatürliche
RollederMonooxygenaseninderKatalysevoneinemodermehrerenwichtigenSchrittenimoxidativen
Katabolismuszuliegen,währenddieseEnzymeinPilzeneineschwacheRolleimWechselzwischendem
Primär‐unddemSekundärmetabolismusspielen.[129]
3.2.2.2 StrukturundMechanismus
WALSHetal. konnten1988anhandderOxidationvonCyclohexanon (2) zuɛ–Caprolacton (3)durch
CHMOausAcinetobactercalcoaceticusdenMechanismusderReaktionaufklären(Abbildung20).[109,110]
ImerstenSchrittdesMechanismuswirddasenzymgebundeneFlavin(34)durchdenCofaktorNADPH
reduziertundderEnzym‐Cofaktor‐Komplex35gebildet,welchermolekulareSauerstoffbindet.[130]Das
so entstandene C4a‐Peroxyflavin (36) greift, ähnlich wie die Persäure in der chemischen BAEYER‐
VILLIGER‐Oxidation, nucleophil andie CarbonylfunktiondesKetons2 an.[12]DieBildungdesCRIEGEE‐
Intermediats37istdergeschwindigkeitsbestimmendeSchritt.[130]BeideranschließendenUmlagerung
müssen die stereoelektronischenVoraussetzungender antiperiplanarenAnordnung derO‐O‐Einheit
sowiedassynchroneWanderndesRestes,wiebeidemPeroxid‐Mechanismus(Abbildung11,Abschnitt
3.1.1), stimmen. Das C4a‐Hydroxyflavin (38)wird hydrolysiert und das oxidierte Flavin39 für den
nächstenkatalytischenZyklusregeneriert.NADPH,welcheswährendeinesReaktionszyklusalserstes
amEnzymgebundenwirdundmitdemFlavin34reagiert,wirdbiszumletztenSchrittalsNADP+im
aktivenZentrumgehalten.

3 EnzymatischeOxidationmittelsMonooxygenasen
19
NH
N
NH
N
O
OR
OO
NH
N
NH
N
O
OR
OO
NADP
O
NH
N
NH
N
O
OR
HO
NADP
O
O
N
N
NH
N
O
OR
NADP
O
ON
N
NH
N
O
OR NADP
N
N
NH
N
O
OR
NH
N
NH
N
O
OR
NADP
O2
H2O
O
O
36C4a‐
Peroxyflavin
38C4a‐
Hydroxyflavin
H+
37
1 23
4
56a 4a7
89
9a10
10a
6
O
2
3 3
3
34
35
39
NADPH
NADP
CRIEGEE‐Intermediat
39
NADP
Abbildung20. MechanismusderenzymatischenBAEYER‐VILLIGER‐OxidationamBeispielvonCyclo‐hexanon(2)bezogenaufPAMO.[130]
WALSH et al. untersuchten 1982 die Fähigkeit der CHMO zur Oxidation von Heteroatom‐Verbin‐
dungen.[131] Der ambivalente Charakter des C4a‐Peroxyflavins 36 ist demnach der Grund für die
katalytischeAktivitätgegenüberelektronenreichen,z.B.Ketone,alsauchelektronenarmenSubstraten,
wie Sulfide (Abbildung 21).[116,132] So greift die Schwefelverbindung 40 die C4a‐Peroxy‐Spezies 36
nucleophilan,imUnterschiedzuderOxidationeinesKetons,beiderdasPeroxyflavin36alsNucleophil
agiert.

3 EnzymatischeOxidationmittelsMonooxygenasen
20
Abbildung21. AmbivalenteReaktivitätderC4a‐Peroxyflavins36gezeigtanderSulfid‐(links)undBAEYER‐VILLIGER‐Oxidation(rechts).[116,132]
BiochemischeStudienanBVMOssindaufgrundderenrelativgeringenAnzahl,dergeringenStabilität
und/oderderschwierigenAufreinigungsehrrar.[133]DieambestenuntersuchteMonooxygenaseistdie
Cyclohexanon‐Monooxygenase(CHMO,EC1.14.13.22)vomStammAcinetobactercalcoaceticusNCIMB
9871aufgrundihrerkatalytischenundkinetischenEigenschaften.[130]DasEnzymhateineMolekülmasse
vonetwa59kDaundbestehtauseinereinzelnenPolypeptidkette,dieeinMolgebundenesFADenthält,
welchesauchnachderAufreinigungnichtabgetrenntwird.[100] ImAcinetobacter‐Bakterium,welches
natürlich in mariner Umgebung vorkommt, ist es im biologischen Abbau von Cyclohexanol (1), zu
Adipinsäure (41) beteiligt, indem es Cyclohexanon (2) zu ε–Caprolacton (3) oxidiert (Abbildung
22).[112,134] Weitere Oxidation der Adipinsäure (41) durch β‐Oxidation liefert Acetyl‐ und Succinyl‐
CoenzymA.Cyclohexanol (1)selbst isteinAbbauproduktvonCyclohexan,welchesderCHMOsowie
weiterenMonooxygenasenalsKohlenstoffquelledientundzudenüber1000BestandteilenvonErdöl
gehört.[135]DieseCHMOakzeptierthundertevonunterschiedlichenKetonenundweistsomiteinaußer‐
gewöhnlich breites Substratspektrum in Kombination mit einer sehr hohen Regio‐ und Enantio‐
selektivitätauf.[136]
Abbildung22. KatabolischerAbbauvonCyclohexanol(1)zuAdipinsäure(41)imBakteriumAcinetobactersp.[134]
AufgrundderInstabilitätderCHMOwaresallerdingslangeZeitnichtmöglich,eineKristallstrukturdes
Enzymszuerhalten,dieeinenEinblick indie strukturellenDetailsdesEnzymsgebenwürden.2002

3 EnzymatischeOxidationmittelsMonooxygenasen
21
verglichenFRAAIJEetal.verschiedenesequenziertebakterielleBVMOsundstießendabeiaufeinkonser‐
viertesSequenzmotiv(FxGxxxHxxxW).[107]MitdiesenDatenkonntensiedieSuperfamiliederBVMOs
enthüllen.ZweiJahrespäterveröffentlichtedieselbeGruppeeineKristallstrukturvonderPhenylaceton‐
Monooxygenase(PAMO,EC1.14.13.92)ausThermobifidafusca,einemungewöhnlichstabilenundhitze‐
beständigenEnzym,dasjedochnureinensehrengenSubstratbereichhat(Abbildung23).[137]
Abbildung23. KristallstrukturvonPAMOausThermobifidafusca.[138]DieFAD‐Domäneistgrün,dieNADPH‐Domäneistblau,dasArg337istzentriertdargestellt.
PAMOisteinMonomermiteinerMolekülmassevon62kDa,welchesdieOxidationvonPhenylacetonzu
Phenylessigester katalysiert, undweist eineHomologie von 40.3% zur CHMO vonAcinetobacter sp.
auf.[116,137]DieKristallstrukturwurdemitgebundenemFAD,jedochinAbwesenheitvonCofaktorund
Substrat, aufgeklärt. Für PAMO wurde eine Multidomänen‐Struktur, bestehend aus einer FAD‐
bindendenundeinerNADPH‐bindendenDomäne,vorgeschlagen.DasaktiveZentrumbefindetsichin
direkterNachbarschaftzudenbeidenDomänen.DasFlavinisttiefinderFAD‐Domäneverborgenund
durchVANDERWAALS‐undWasserstoffbrückenbindungenfixiert.DiesesstimmtmitderBeobachtung
überein,dassFADnichtvomEnzymdissoziiert,währendNADP+zwischenderFAD‐undderNADPH‐
Domäne eingekeilt ist. Bemerkenswert ist, dass sich das Sequenzmotiv der BVMO nicht im aktiven
Zentrumbefindet, sondern ineinerSchleife,diediebeidenDomänenverbindet.DasaktiveZentrum
enthält zudem die Aminosäure Arg337, der die Funktion zugeschriebenwird, das negativ geladene
Flavin‐Peroxid‐Intermediat35durchWasserstoffbrückenbindungenzustabilisieren.
Arg337 ist außerdem nicht in einer einzelnen Konformation fixiert, sondern besitzt eine inhärente
Flexibilität.ImLaufedesKatalysezyklusnimmtArg337zweiKonformationenein:die„IN“‐Position,wie
es in der erhaltenenKristallstruktur beobachtetwurde, bei der das Flavin stabilisiertwird, unddie
„OUT“‐Position, inderderCofaktorangrenzendzumFlavinpositioniertwird.DieÜbertragungeines
Hydrid‐IonsvomNicotinamid‐RingzumFADfindetfolglicherstnacheinerKonformationsänderungim
Enzym statt. Mutagenese‐Studien zeigten, dass die Mutation des Arginin‐Restes zu Alanin zu einer

3 EnzymatischeOxidationmittelsMonooxygenasen
22
vollständigenInaktivierungdesEnzymsführt.[137,139]DieMutationbeeinflusstzwarnichtdieWechsel‐
wirkungendesEnzymsmitdemCofaktor, jedochschaltetesdieFähigkeitaus,Sauerstoff zubinden.
DieseszeigtdieBedeutungdesArginin‐ResteszurStabilisierungdesnegativgeladenenPeroxid‐Flavins
36.
MIRZAetal.löstenschließlicheineKristallstrukturderCyclohexanon‐MonooxygenaseausRhodococcus
sp.vomStammHI‐31(CHMORhodo.)inzweiverschiedenenZuständen,deroffenenunddergeschlossenen
Form,mit FAD undNADP+ auf (Abbildung 24).[140] Diese CHMO ist ein stabiles,monomeres Enzym,
welches ebenfalls FAD‐ und NADPH‐Domänen besitzt und zu 55% identisch mit der CHMO aus
Acinetobactersp.ist.WerdennunbeideKristallstrukturenderCHMORhodo.übereinandergelegt,soergibt
sichein„äquivalentesAtom‐Vektordiagramm“,dasdieVerschiebungderAtompositionenillustriert.[140]
WährenddieFAD‐Domäneunverändertbleibt,rotiertdieNADPH‐unddieHelix‐Domäneumetwa5°
bzw. 3°, was zu einer Verschiebung um 5Å bzw. 2Å von Resten führt, dieweiter entfernt von der
Rotationsachse sind. Durch diese Rotation wird der Cofaktor seitlich näher zum Flavin geschoben,
sodassderbenötigteHydridtransferstattfindenkann.
Abbildung24. KristallstrukturderCHMOvonRhodococcus sp.HI‐31 (links); dieFAD‐Domäne istbeige,dieNADPH‐Domäneistblau,dieLinker‐Regionistgrün,dieflexibleSchleifeistorange,dieHelix‐Domäneistbraun,dasBVMO‐Sequenzmotivistgelbdargestellt.[140]ÄquivalentesVektordiagrammderselbenCHMO(rechts).
Obwohlschonlangebekanntist,dassdieCHMOvonAcinetobactersp.vieleKetonemitguterRegio‐und
Stereoselektivität umsetzen kann,warwenig über ihre Chemoselektivität bekannt.Die Chemoselek‐
tivität ist „die bevorzugte Reaktion eines chemischen Reagenz‘mit einem von zwei odermehreren
unterschiedlichenfunktionellenGruppen“.[141]VoreinigenJahrenwurdebeschrieben,dassdieOxida‐
tionbevorzugtanderCarbonyl‐Funktionstattfindet,wenneineweitereoxidierbareFunktion,beispiels‐
weiseeinAlkenoderThioether,enthaltenist.WALSHschlugvor,dassα,β‐ungesättigteKetonenichtvon
derCHMOumgesetztwerdenkönnen.[109,110]DieseTheoriewurdevonOTTOLINAbestätigt,derzeigte,
dassbeiderchemoselektivenOxidationdesracemischenDiketonsrac‐44dasLacton(S)‐45mit35%
UmsatzinenantiomerenreinerFormerhaltenwurde(Abbildung25).[142]

3 EnzymatischeOxidationmittelsMonooxygenasen
23
Abbildung25. ChemoselektiveOxidationeinesracemischenDiketonsrac‐44mitCHMOnachOTTOLINA.[142]
ImFalleeineszweifachsubstituiertenCyclohexanon‐RingsnimmtderEnergieunterschiedimmerweiter
ab,jeähnlichersichdiebeidenRestesind.DadurchverschwimmtderEnergieunterschiedderbeiden
möglichenCRIEGEE‐IntermediateundbeideEnantiomerewerdengebildet.[136]
Wieschonbeschriebenwurde,könnenauchheterocyclischeVerbindungenwieThioether(40)durch
Monooxygenasenoxidiertwerden.BefindetsichjedocheineKeto‐FunktioninderselbenVerbindung,so
findeteineOxidationausschließlichdortstatt(Abbildung26).SelbstnachVerlängerungderReaktions‐
zeitkonntekeineOxidationdesHeteroatomsfestgestelltwerden.DiesesErgebnisistbemerkenswert,
danachorganisch‐chemischemProtokollmitm‐CPBAdasHeteroatombevorzugtoxidiertwird.[143]
Abbildung26. CHMO‐katalysierteBAEYER‐VILLIGER‐OxidationvonThioether(40)[109,110](links)undvonheterocyclischemKeton[143](rechts).
3.2.3 Alkohol‐Dehydrogenasen
Dehydrogenasen gehören allgemein zur Klasse der Oxidoreduktasen und haben in der Natur die
Funktion, stereospezifisch prochirale Cabonylverbindungen zu reduzieren.[144] Zur biokatalytischen
Darstellung von sekundären Alkoholen aus Carbonylverbindungen als wichtige Bausteine für die
pharmazeutische Industrie werden in großem Umfang Alkohol‐Dehydrogenasen (ADH) eingesetzt.
Diese treten ineinerVielzahlvon lebendenOrganismenauf, indenensie fürdieEntgiftungundden
Metabolismus von Ethanol und anderen Alkoholen zuständig sind.[145] Diese ADHs sind NAD(P)‐
abhängigundkatalysierendenirreversiblenTransfervonHydridionenvonNAD(P)HaufdieCarbonyl‐
verbindung.
ZudenbekanntestenAlkohol‐Dehydrogenasengehörenunteranderemdiejenigen,dieausHefe,Pferde‐
leberoderThermoanaerobicumbrockiiisoliertwurden.[144]Dieseweisenjedocheinegeringethermische
StabilitätundEnantioselektivitätsowieeinenteilweiseengenSubstratbereichauf,sodassSubstratemit
größeren Seitenketten, beispielsweise Acetophenon, nicht umgesetzt werden können. Verschiedene
Alkohol‐Dehydrogenasenkönnen,inAnalogiezuPRELOGSRegel,einHydrid‐IonvomNAD(P)Hentweder

3 EnzymatischeOxidationmittelsMonooxygenasen
24
aufdiesi‐oderdiere‐SeiteeinesKetonsübertragen.DarausresultierteinAlkoholmit(R)‐bzw.(S)‐Kon‐
figurationmitindenmeistenFällenhoherEnantioselektivität.[145,146]DerGroßteilderbekanntenADHs
gehört zu den (S)‐selektiven Dehydrogenasen, wie beispielsweise Rhodococcus erythropolis und
Theormoanaerobicumsp.[144,147]DiezuBeginnder1990erJahreentdeckten(R)‐ADHsgehörenzuden
kurzkettigenDehydrogenasen,zudenendieADHausLactobacilluskefir(Lk‐ADH,EC1.1.1.2)zählt.[144]
DieLk‐ADHisteinHomotetramermit251AminosäurenundeinemMolekulargewichtvon26.6kDapro
Untereinheit.[148]AufgrundderstrukturellenÄhnlichkeit(92.1%Identität)zuderADHausLactobacillus
brevis(Lb‐ADH)kanndessenMechanismusaufdieLk‐ADHübertragenwerden.DieAminosäurender
Lk‐ADHwerdenanalogzudenenderLb‐ADHnummeriert.DerkatalytischeMechanismusumfassteine
hochkonservierteTetradebestehendausSer142,Tyr155,Lys159undAsn113.DerTyrosin‐Restnimmt
dabeidiezentralekatalytischeSäurebeiderReduktioneinerCarbonylverbindungein.Erwirdwährend
desReaktionsverlaufsdeprotoniertundmussdurchdaspositivgeladeneLys159stabilisiertwerden.
Ser142hatdieAufgabe,dasSubstratzufixierenundzusätzlichdieLadungdesTyrosin‐Restesauszu‐
gleichen,sodasseinProtonvonzweiBindungspartnerngeteiltwird.DerkonservierteAsn113‐Restsoll
zusammenmitder2‘‐HydroxygruppedesCofaktorsundeinemgleichzeitigamLysin‐undAsparagin‐
RestgebundenemWassermolekül(Wat122)einProtonen‐Übertragungssystembilden.[149]FILLINGetal.
postulierten ein erweitertesProtonen‐Übertragungssystem,welcheseineGruppevonverschiedenen
Wassermolekülen umfasst, die eine hydrophobe Höhle ausfüllen, welche mit Carbonyl‐Sauerstoff‐
atomenbedecktist(Abbildung27).[150]
Abbildung27. NahaufnahmederwichtigstenRestedeserweitertenProtonen‐Übertragungssystemsder Lb‐ADH.[149] Die katalytische Tetrade ist inmagenta undWasserstoffbrücken‐bindungensindgestricheltdargestellt.
DieLk‐ADHenthältzweiMg2+‐Ionen,dieeherstrukturellealskatalytischeEigenschaftenbesitzen,dasie
jeeinekoordinativeBindungzueinemMonomerundelektrostatischeWechselwirkungenausbilden.
Durch Zugabe des Komplexbildners Ethyldiamintetraacetat (EDTA) wurde die Inaktivierung des
Enzymsgezeigt,dasodiequarternäreStrukturdesEnzymsnichtmehrstabilisiertwerdenkann.Durch
ZugabevonMg+‐IonenkanndessenAktivitätzuetwaeinemDrittelwiederregeneriertwerden.[148]Die
Bedeutung der Anwesenheit von zusätzlichen Mg2+‐Ionen in der Reaktionsmischung macht sich im

3 EnzymatischeOxidationmittelsMonooxygenasen
25
Anstieg der Aktivität der Lk‐ADH bemerkbar, was bedeutet, dass ein Teil der Ionen vom Enzym
dissoziiert.[151]
DavielederSubstratenichtwasserlöslichsind,kanneineZugabevonorganischemLösungsmittelzur
Reaktionsmischungsinnvollsein.LösungsmittelmitlogP‐Werten<3.5könnenjedochsowohldiespezi‐
fischeAktivitätalsauchdieStabilitätdesisoliertenEnzymsverringern.[152]DiehöchsteAktivitätweist
dieLk‐ADHbei50°Cauf,währenddaspH‐OptimumbeipH7fürdieReduktionundbeipH8fürdie
Oxidationliegt.[151]
HUMMELetal.setzten1990erfolgreichdasprochiraleSubstratAcetophenon(46)zu(R)‐Phenylethanol
((R)‐47)mitder(R)‐spezifischenLk‐ADHmiteinerAktivitätvon558U/mLum(Abbildung28).[151]Die
Lk‐ADH akzeptiert ein sehr breites Spektrum an Substraten wie aliphatische, offenkettige Ketone,
2‐oder3‐KetoesterundcyclischeKetonemithoherspezifischerAktivität(100–500U/mg).[153]
Abbildung28. SchemaderLk‐ADH‐katalysiertenOxidations‐sowieReduktionsreaktionen.[151]
AuchwenndieReduktionprochiralerKetonezuchiralenAlkoholenderwichtigsteAnwendungsbereich
derLk‐ADHist,kanndieOxidationsreaktionebenfallsgenutztwerden.SowirddieLk‐ADHbereitszur
enzymatischenRacematspaltungvonracemischenAlkoholen,beispielsweisevon(R)‐und(S)‐Phenyl‐
ethanol, eingesetzt, bei der nur der (R)‐Alkohol zum entsprechenden Keton oxidiert wird.[153] Im
RahmendieserArbeitsolldieLk‐ADHdieOxidationvonCyclohexanol(1)zuCyclohexanon(2)mitdem
kostengünstigeneCofaktorNADP+katalysieren(Abbildung29).DieLk‐ADHbesitztfürdieOxidationdes
Alkohols1einegeringereAktivitätalsfürdieReduktioneinesKetons2,sodassdieWahrscheinlichkeit
derRückreaktionsehrgroßist.AllerdingskanndasGleichgewichtzugunstenderOxidationverschoben
werden,wenndasgebildeteKeton2durchdieCHMOdirektweiterumgesetztwird(vergl.Abschnitt
2.1).
Abbildung29. EnzymatischeOxidationdurchLk‐ADHamBeispielvonCyclohexanol(1)zuCyclo‐hexanon(2).
AufgrundderCofaktor‐AbhängigkeitderLk‐ADHistauchindiesemFalldieAnwesenheiteinesRegene‐
rationssystemsnotwendig.AufverschiedeneSystemewirdinKapitel3.2.4nähereingegangen.

3 EnzymatischeOxidationmittelsMonooxygenasen
26
3.2.4 Cofaktor‐Regeneration
DieAnwendungvonBVMOsimindustriellenMaßstabistnochnichtvollständigentwickelt.Einesder
größtenHindernisse istdieAbhängigkeitderLk‐ADHsowiederMonooxygenasenvondemCofaktor
NADP+bzw.NADPH,welches instöchiometrischenMengenzurReaktionsführungeingesetztwerden
muss. Aus diesem Grund wurden Systeme zur in situ‐Cofaktor‐Regenerationssysteme entwickelt.
GenerellwerdenzweiunterschiedlicheStrategienzurCofaktor‐Regenerationangewandt(Abbildung
30).[14,154] Zum einen die substratgekoppelte Cofaktor‐Regeneration, die auf einen Alkohol als Co‐
substrat,meist iso‐Propanol,basiert,der inderRückreaktionvonderselbenAlkohol‐Dehydrogenase
zumKeton oxidiertwird und somit den Cofaktor regeneriert. Durch Zugabe eines Überschusses an
CosubstratkanndasGleichgewichtzurProduktseiteverschobenwerden.
Abbildung30. Strategienzursubstrat‐undenzymgekoppeltenCofaktor‐Regeneration.[154]
EinanderesKonzeptnutztdasenzymgekoppeltesRegenerationssystem.DabeiwirdderEinsatzeines
zweiten Enzyms notwendig, welches ein Cosubstrat unter Rückbildung des Cofaktors umsetzt. Die
Reaktionistirreversibel,wodurchdasGleichgewichtebenfallszurProduktseiteverschobenwird.Ein
gutesRegenerationssystemsolltepreiswertsein,keineNebenprodukteproduzierenundnichtmitden
katalytischen Eigenschaften des Enzyms wechselwirken. Klassisch werden als Redox‐Enzym Dehy‐
drogenasen, beispielsweise die Glucose‐ (GDH)[155] und Glucose‐6‐phosphat‐Dehydrogenase
(G6PDH)[131,156] sowiedieFormiat‐[157]oderPhosphit‐Dehydrogenase[158,159] eingesetzt.DieDehydro‐
genasenkönnendabeisowohlalsisoliertesEnzym[158,159]alsauchalsFusionsprotein[160]oderGanzzell‐
biokatalysator[14,161] zusammenmitderBVMOzurReaktionsmischunggegebenwerden.Ganzzellbio‐
katalysatoren, die neben dem rekombinanten produzierenden Enzym ein Cofaktor‐regenerierendes
Enzym coexpremiert enthalten, besitzen weitere Vorteile wie die leichte Handhabbarkeit von Bio‐
transformationen durch Verzicht auf Isolierung und Reinigung von Enzymen sowie die zellinterne
Cofaktor‐Regeneration.GleichzeitigschließensiedasVorhandenseinkonkurrierenderEnzyme,wiees
beiWildtypzellenderFallist,aus.ZubeachtenistsowohlbeiisoliertenEnzymenalsauchbeiGanzzell‐
Systemen die Kompatibilität der beiden Enzymreaktionen, da Inhibierungsreaktionen durch unter‐
schiedlicheReaktandenauftretenkönnen.

3 EnzymatischeOxidationmittelsMonooxygenasen
27
DiefürdieCofaktor‐RegenerationhäufiggenutzteGDHoxidiertdasCo‐SubstratD‐Glucose(48‐a)und
regeneriert dadurch den Cofaktor NAD(P)+ (Abbildung 31).[155] Durch die spontane Hydrolyse des
entstandenen Gluconolactons (49‐a) zur Gluconsäure (50‐a) ist die Reaktion irreversibel und ver‐
schiebtdasGleichgewichtzurProduktseite.DieGDHistgünstig,kommerziellerhältlichundstabilerals
diemeistenzurRegenerationverwendetenEnzyme.WeitereVorteilederGDHsinddieAkzeptanzvon
NAD+sowievonNADP+undihrehohespezifischeAktivität.
DieG6PDHistähnlicheffizientundwirdebenfallsvielfachverwendet.[16,131,156]SieoxidiertdenZucker
Glukose‐6‐Phosphat (48‐b, G6P) zu Glukonolacton (49‐b), welches spontan zu Glukonsäure (50‐b)
hydrolysiert.EinengroßenNachteildiesesSystemsstellendiehohenKostenfürdasG6P(48‐b)dar.Die
G6PDH wurde bereits in Ganzzellsystemen zusätzlich zur BVMO überexprimiert.[16] Die erhöhte
Verfügbarkeit von NADPH für die Oxidation führt zu einer Verbesserung der Produktion von
ε‐Caprolacton(3).
Abbildung31. CofaktorregenerationmitdemD‐Glucose(48‐a)/GDH‐unddemG6P(48‐b)/G6PDH‐System.[131,155,156]
DieVerwendungderFormiat‐Dehydrogenase(FDH)isteineweiterebekannteMethodeundliefertCO2
alsNebenprodukt.[157]Dadiesesflüchtigist,erfolgtdadurcheineVerschiebungdesGleichgewichtesder
ReaktionzurProduktseite.NachteilediesesSystemssinddiegeringespezifischeAktivitätderFDHsowie
dieAbhängigkeitvonNADHalsCofaktor.
Die Phosphit‐Dehydrogenase (PtxD) katalysiert die Oxidation von Phosphit zu Phosphatmit gleich‐
zeitiger Reduktion vonNAD+ zuNADH (Abbildung 32).[158,159] Anstelle von PtxD kann eineAlkohol‐
Dehydrogenase(ADH)zusammenmitiPrOHzugefügtwerden,umNADP+wiederzuregenerieren.[162]
Dabei wird Aceton gebildet, welches flüchtig ist und das Gleichgewicht zu Gunsten des Produktes
verschiebt.

3 EnzymatischeOxidationmittelsMonooxygenasen
28
Abbildung32. SchematischeDarstellungfürdenEinsatzvonPhosphit‐Dehydrogenase(PtxD)[158,159](links)undAlkohol‐Dehydrogenase(ADH)[162](rechts)zurCofaktor‐Regeneration.
EineeleganteLösungfürdieCofaktor‐RegenerationstelltenWILLETSetal.vor,indemsieeineCHMOmit
einerisoliertenADHkoppelten(Abbildung33).[163,164]DieADHoxidiertdenAlkoholalsSubstratzum
entsprechendenKeton und reduziertwährenddessendasNADP+ zuNADPH.Anschließendwirddas
Keton durch die Monooxygenase zum Lacton umgewandelt und NADPH verbraucht. Diese Art der
Cofaktor‐RegenerationsollauchindieserArbeitAnwendungfinden.EinweitererVorteildieserReak‐
tionsführungistderEinsatzvonnursehrgeringenMengenderCofaktorenNADP+bzw.NADPH(700€
bzw.2700€/g).[165]
Abbildung33. SchematischeDarstellungdessubstratgekoppeltenRegenerationssystemsausBVMOundADH.[163,164]
NebendenbishererwähntenMethodenzurCofaktor‐RegenerationwurdeninneuererZeitauchphoto‐
chemischeVerfahren beschrieben. Bei dieserArt derCofaktor‐Regeneration dient Ethyldiamintetra‐
acetat(EDTA)alsElektronendonor,welchesunterLichteinwirkungzerfällt.[166]DiesesSystemistaller‐
dingswenigeffizient.DanebengibtesAnsätzefürelektrochemischeVerfahren,diezurZeitjedochnoch
nichtausgereiftsind.[167]

3 EnzymatischeOxidationmittelsMonooxygenasen
29
3.3 EigeneErgebnisseundDiskussion
DieindiesemTeilderArbeitverwendeteCyclohexanon‐MonooxygenaseausAcinetobactercalcoaceticus
NCIMB9871(CHMO,EC1.14.13.22,HandelsnameECSMO‐1vonENZYMICALSAG)wurdeausschließlich
alsRohextrakteingesetzt.DieverwendetenChargenderAlkohol‐DehydrogenaseausLactobacilluskefir
(Lk‐ADH,EC1.1.1.2,DSM20587)wurdenebenfallsalsRohextrakteingesetztundinGlycerinineiner
1:1‐Mischung(v/v)aufbewahrt.DieBearbeitungdiesesForschungsprojektesfandimRahmendesvon
der„DeutschenBundesstiftungUmwelt“(DBU)gefördertenProjektsstatt.
3.3.1 Standardreaktion:DoppeloxidationvonCyclohexanol(1)zuɛ‐Caprolacton(3)
Bei der in der Arbeitsgruppe bereits etablierte Doppeloxidation wird Cyclohexanol(1) durch den
BiokatalysatorLk‐ADHinsituzuCyclohexanon(2)unddiesesanschließenddurchdieCyclohexanon‐
Monooxygenasesofortweiterzuɛ‐Caprolacton(3)oxidiert(Abbildung34).[26]DieEnzymaktivitätender
der Lk‐ADHwerden zuvor auf Acetophenon und die der CHMOauf Cyclohexanon (2) als Standard‐
Reagenzbezogen.
Abbildung34. Standard‐ReaktionderDoppeloxidationvonCyclohexanol(1).[26]
BeidieserReaktionbenötigtdieCHMOdieoxidierteCofaktorformNADP+,welchedurchdieLk‐ADHals
Cofaktor‐RegenerationssystemwiederzurreduziertenFormNADPHumgewandeltwird.STAUDTerhielt
denhöchstenproduktbezogenenUmsatz von>97%bei einer Substratkonzentrationvon20mMbei
Raumtemperatur und 24 Stunden Reaktionszeit.[26] Nach aktuellemKenntnisstand ist diese Doppel‐
oxidationdiebisdatoerstebiokatalytischeSynthesevonɛ‐Caprolacton(3)ausgehendvonCyclohexanol
(1).[26,168]

3 EnzymatischeOxidationmittelsMonooxygenasen
30
3.3.2 EtablierunganalytischerMethoden
Vor Beginn der synthetischen Arbeitenmusste zunächst eine schnelle und robuste Analytik für die
enzymatischeBAEYER‐VILLIGER‐OxidationzurqualitativenundquantitativenBestimmungderUmsätze
erarbeitetwerden.HierzuwurdezumeineneineNMR‐AnalytikinKombinationmitUrotropin(51)als
externerStandardetabliert.ZumanderenwurdeeineUmsatzbestimmungmittelsGaschromatographie
mittels Eichgeraden validiert sowie die Wiederfindungsraten unter verschiedenen Bedingungen
bestimmt.
3.3.2.1 UmsatzbestimmungmittelsNMR‐Analytik
Zur Bestimmung der Umsätzewurde im Anschluss an die Reaktion extraktiv in EPPENDORF‐Gefäßen
aufgearbeitet.NachEntfernendesorganischenLösungsmittelsuntervermindertemDruckwurdeder
vollständigeReaktionsansatzinNMR‐Röhrchenüberführt.Urotropin(51)dientedabeiinFormeiner
frisch angesetzten Stammlösung (0.17 M in CDCl3, 0.1 mL) als externer Standard. Mittels 1H‐NMR‐
SpektroskopiewurdedasVerhältnisvonProdukt3zumStandard51anhanddererhaltenenIntegrale
ermittelt (Abbildung 35). Das im Spektrum vorhandene Ethylacetat wurde bei der Versuchsdurch‐
führungimRahmendesSolvenz‐ScreeningszugegebenundistfürdieBestimmungdesUmsatzesnicht
vonBedeutung.
Abbildung35. Umsatzbestimmungmittels1H‐NMR‐SpektroskopiemitUrotropin(51)alsexternemStandard.

3 EnzymatischeOxidationmittelsMonooxygenasen
31
ImvorliegendenBeispielwurdederUmsatzwiefolgtanhandderIntegralebestimmt(Gleichung1):
Gleichung1
Des Weiteren kann der produktbezogene Umsatz am selben Beispiel nach folgender Gleichung 2
ermitteltwerden:
Gleichung2
BeidemVergleichderbeidenBestimmungsmethodenfälltjedochauf,dassesgroßeUnterschiedeinden
mithilfe der Gleichungen 1 und 2 ermittelten Umsätzen gibt. Dieses kann mit der Flüchtigkeit der
Komponenten Cyclohexanol (1) und Cyclohexanon (2) während der extraktiven Aufarbeitung mit
anschließendemEntfernendesLösungsmittelsbeivermindertemDruckbegründetwerden,dainAnwe‐
senheiteinesexternenStandardsnurdasVerhältnisderKomponentenuntereinanderbestimmtwerden
kann und nicht der quantitative Umsatz. Es wurde somit, soweit nicht anders angegeben, auf die
BestimmungdesUmsatzesmittelsexternemStandard50zurückgegriffen.
DieStabilitätdesStandardswurdeanschließendermittelt.DafürwurdenUrotropin (51)undCyclo‐
hexanol(1)eingewogenunddasVerhältnisbeiderKomponentensofortunderneutnach24Stunden
LagerzeitbeiRaumtemperaturineinemgeschlossenenGefäßmittels1H‐NMR‐Spektroskopiebestimmt
(Tabelle2).LautEinwaage liegtdasVerhältnisbei1 :0.80 (51/1),nach24Stundenwirddasselbe
Verhältnis beider Komponenten erhalten. Urotropin (51) ist folglich optimal zur Verwendung als
externerStandardgeeignet.
Tabelle2. VerhältnisvonUrotropin(51)undCyclohexanol(1)nachEinwaageundnach24StundenLagerzeitbeiRaumtemperatur.
EintragVerhältnis51 /1
Einwaage
Verhältnis51/1
nach24h
1 1:0.80 1:0.80
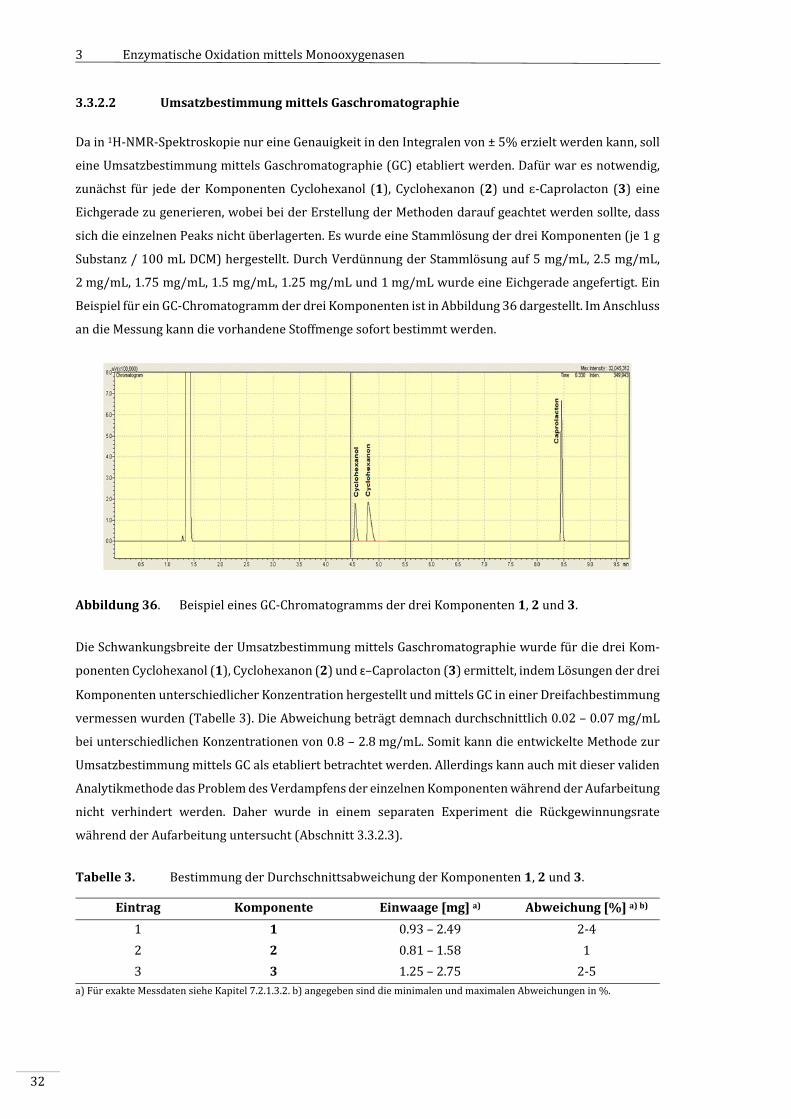
3 EnzymatischeOxidationmittelsMonooxygenasen
32
3.3.2.2 UmsatzbestimmungmittelsGaschromatographie
Dain1H‐NMR‐SpektroskopienureineGenauigkeitindenIntegralenvon±5%erzieltwerdenkann,soll
eineUmsatzbestimmungmittelsGaschromatographie(GC)etabliertwerden.Dafürwaresnotwendig,
zunächst für jedederKomponentenCyclohexanol (1), Cyclohexanon (2)undɛ‐Caprolacton (3) eine
Eichgeradezugenerieren,wobeibeiderErstellungderMethodendaraufgeachtetwerdensollte,dass
sichdieeinzelnenPeaksnichtüberlagerten.EswurdeeineStammlösungderdreiKomponenten(je1g
Substanz/100mLDCM)hergestellt.DurchVerdünnungderStammlösungauf5mg/mL,2.5mg/mL,
2mg/mL,1.75mg/mL,1.5mg/mL,1.25mg/mLund1mg/mLwurdeeineEichgeradeangefertigt.Ein
BeispielfüreinGC‐ChromatogrammderdreiKomponentenistinAbbildung36dargestellt.ImAnschluss
andieMessungkanndievorhandeneStoffmengesofortbestimmtwerden.
Abbildung36. BeispieleinesGC‐ChromatogrammsderdreiKomponenten1,2und3.
DieSchwankungsbreitederUmsatzbestimmungmittelsGaschromatographiewurdefürdiedreiKom‐
ponentenCyclohexanol(1),Cyclohexanon(2)undɛ–Caprolacton(3)ermittelt,indemLösungenderdrei
KomponentenunterschiedlicherKonzentrationhergestelltundmittelsGCineinerDreifachbestimmung
vermessenwurden(Tabelle3).DieAbweichungbeträgtdemnachdurchschnittlich0.02–0.07mg/mL
beiunterschiedlichenKonzentrationenvon0.8–2.8mg/mL.SomitkanndieentwickelteMethodezur
UmsatzbestimmungmittelsGCalsetabliertbetrachtetwerden.Allerdingskannauchmitdieservaliden
AnalytikmethodedasProblemdesVerdampfensdereinzelnenKomponentenwährendderAufarbeitung
nicht verhindert werden. Daher wurde in einem separaten Experiment die Rückgewinnungsrate
währendderAufarbeitunguntersucht(Abschnitt3.3.2.3).
Tabelle3. BestimmungderDurchschnittsabweichungderKomponenten1,2und3.
Eintrag Komponente Einwaage[mg]a) Abweichung[%]a)b)
1 1 0.93–2.49 2‐4
2 2 0.81–1.58 1
3 3 1.25–2.75 2‐5a)FürexakteMessdatensieheKapitel7.2.1.3.2.b)angegebensinddieminimalenundmaximalenAbweichungenin%.

3 EnzymatischeOxidationmittelsMonooxygenasen
33
3.3.2.3 ErmittlungderRückgewinnungsrate
UmdieRückgewinnungsratenvondenKomponenten2und3zuerhalten,wurdedieAufarbeitungder
ReaktionsimuliertunddieRückgewinnungsrateabhängigvonderjeweiligenMethodeaufzweiWegen
ermittelt(Abbildung37).
Abbildung37. SchematischeDarstellungderAufarbeitungsmethodenfürA)dieNMR‐SpektroskopieundB)dieGC.
ZurBestimmungderRückgewinnungsratemittelsNMR‐SpektroskopiewurdenCyclohexanon(2)bzw.
ε‐Caprolacton (3, je 40 mM) in KPi‐Puffer (5 mL) über einen Zeitraum von 24Stunden bei Raum‐
temperatur gerührt. ImAnschlusswurde die jeweilige Lösung extraktiv aufgearbeitet, das Lösungs‐
mittelentferntunddergesamteKolbeninhalt ineinNMR‐Röhrchenüberführt.Mithilfedesexternen
Standards50wurdendieWiederfindungsratenmittelsNMR‐Spektroskopieermittelt,diebeisehrguten
91 – 98% lagen. Dieses entspricht einem Verlust von 9% bei Cyclohexanon (2) und von 2% beim
ɛ‐Caprolacton(3)(Tabelle4).
Tabelle4. RückgewinnungsratederSubstanzen2und3nachsimulierterAufarbeitungnach24hRührenbeiRT.
Eintrag Substanz Rückgewinnungsrate[%] *
1 2 91
2 3 98*ermitteltmittelsNMRmitUrotropin(51)alsStandard.
DieRückgewinnungsratewurdeparallel dazumitderGC‐Methodebestimmt,wobei dasAugenmerk
dabei auf die unterschiedlichenRückgewinnungsratenbei unterschiedlichenKonzentrationen gelegt
wurde.DafürwurdendieKomponenten1,2und3 inverschiedenenKonzentrationen(20–80mM)
eingewogenund für24Stunden inKPi‐Puffer (5mL)gerührt. ImAnschlussandieextraktiveAufar‐
beitungwirddasLösungsmittelentfernt,derKolbeninhaltineinMaßkolbenüberführtunddieserzum
Eichstrichaufgefüllt(Abbildung37,B).DarauswerdendreiProbenentnommenundmittelsFünffach‐
bestimmungmittelsGCvermessen.DieErgebnissesindinAbbildung38aufgezeigt.

3 EnzymatischeOxidationmittelsMonooxygenasen
34
OH
1
O
2
O
O
3
Abbildung38. RückgewinnungsratederKomponenten1,2und3beiunterschiedlichenKonzentrationen,ermitteltmittelsGC.
MitzunehmenderKonzentrationsteigtdieRückgewinnungsratevon71–80%bei0.1mMaufhervor‐
ragende90–97%beieinerKonzentrationvon0.4mMan.Bemerkenswert istdabei,dassdieRück‐
gewinnungsratenbeiallenKonzentrationenfürCyclohexanol(1)amniedrigstenundfürdasLacton3
amhöchstensind.AuchdiesesErgebnisistmitderFlüchtigkeitderSubstanzenzubegründen,dabei
höherenKonzentrationeninsgesamteingeringererAnteildesSubstratesnotwendigist,umdieAtmo‐
sphäre im Reaktionsgefäß zu sättigen, und somit der Verlust geringer ist. DesWeiteren spielt der
VerteilungskoeffizientdereinzelnenSubstanzenwährendAufarbeitungeinewichtigeRolle.DerlogP‐
Wert ist charakteristisch für jede einzelne Verbindung und somit auch die Effizienz des jeweiligen
Extraktions‐VorgangsauseinerwässrigenPhase.
Die zunächst aus vorhergehenden Arbeiten[26] übernommene Extraktionsmethode in EPPENDORF‐
Gefäßenwurdeoptimiert.AnfangswurdedieoberewässrigePhaseinneueEPPENDORF‐Gefäßeüberführt
unddasEnzym‐PelletmitderorganischenPhase(DCM)zurückgelassen,sodassindennächstenExtrak‐
tionsschrittennurdiewässrigePhaseweiterextrahiertwerdenkonnte(Abbildung39).Beiderneuen
ExtraktionsmethodewirddieuntereorganischePhaseausdemEPPENDORF‐GefäßineinSammelgefäß
gegeben und die zurückbleibende wässrige Phase, die noch das Enzym‐Pellet enthält, erneut mit
Lösungsmittelversetzt.DurchdieseÄnderungwirdeinehöhereRückgewinnungsrateerzielt,danoch
weitereSubstanzausdemPelleterhaltenwerdenkann.SomitkonntediegesamteinderBiotransfor‐
mation eingesetzte Stoffmenge quantitativ zurückerhaltenwerden,während bei der bisherigenAuf‐
arbeitungsmethodenuretwa84%derStoffmengenachgewiesenwerdenkonnten.
0102030405060708090100
20 40 80
71
8490
75
8895
8088
97
Rückgew
innungsrate[%]
c[mM]
Cyclohexanol(1) Cyclohexanon(2) ɛ‐Caprolacton(3)(2)(1) (3)
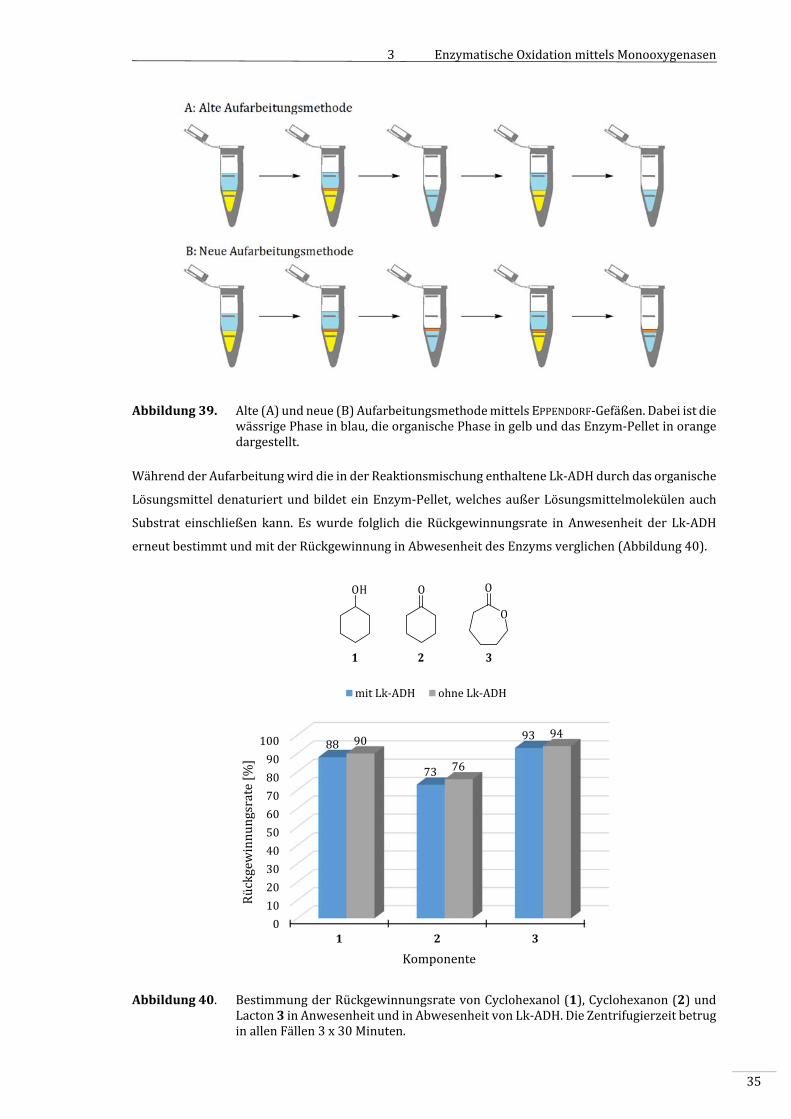
3 EnzymatischeOxidationmittelsMonooxygenasen
35
Abbildung39. Alte(A)undneue(B)AufarbeitungsmethodemittelsEPPENDORF‐Gefäßen.DabeiistdiewässrigePhaseinblau,dieorganischePhaseingelbunddasEnzym‐Pelletinorangedargestellt.
WährendderAufarbeitungwirddieinderReaktionsmischungenthalteneLk‐ADHdurchdasorganische
LösungsmitteldenaturiertundbildeteinEnzym‐Pellet,welchesaußerLösungsmittelmolekülenauch
Substrat einschließen kann. Eswurde folglich die Rückgewinnungsrate in Anwesenheit der Lk‐ADH
erneutbestimmtundmitderRückgewinnunginAbwesenheitdesEnzymsverglichen(Abbildung40).
OH
1
O
2
O
O
3
Abbildung40. BestimmungderRückgewinnungsratevonCyclohexanol(1),Cyclohexanon(2)undLacton3inAnwesenheitundinAbwesenheitvonLk‐ADH.DieZentrifugierzeitbetruginallenFällen3x30Minuten.
0
10
20
30
40
50
60
70
80
90
100
1 2 3
88
73
9390
76
94
Rückgew
innungsrate[%]
Komponente
mitLk‐ADH ohneLk‐ADH
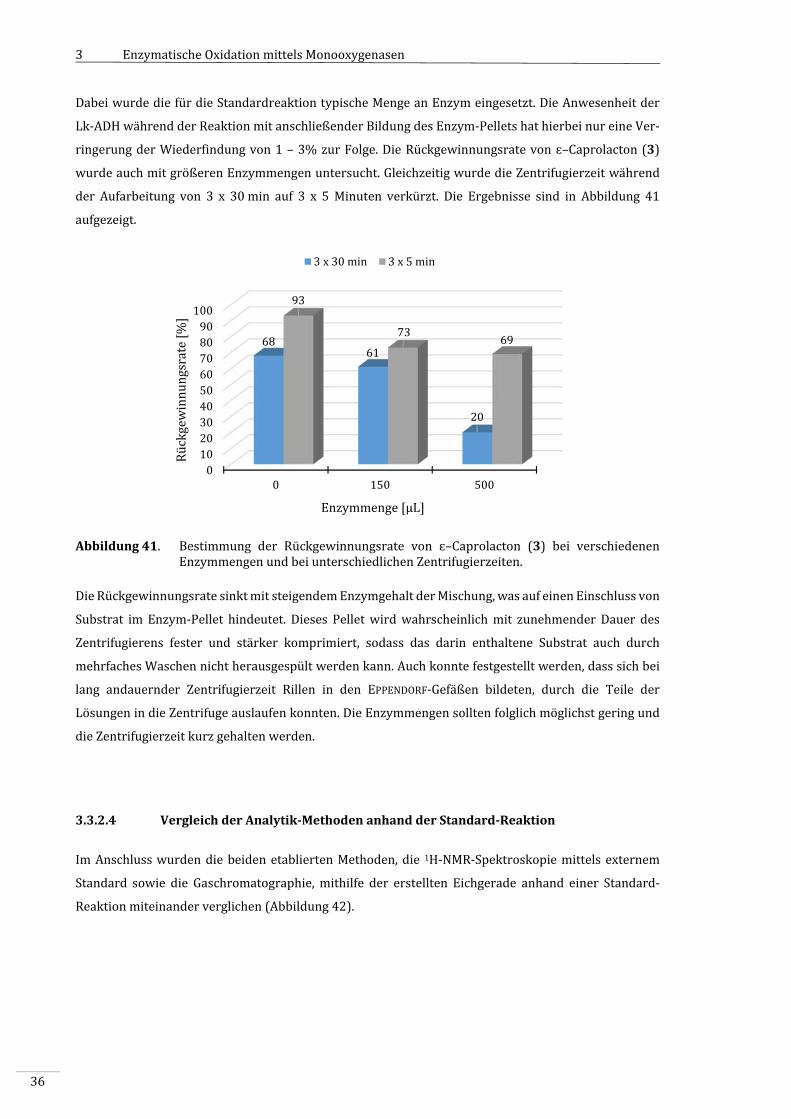
3 EnzymatischeOxidationmittelsMonooxygenasen
36
DabeiwurdediefürdieStandardreaktiontypischeMengeanEnzymeingesetzt.DieAnwesenheitder
Lk‐ADHwährendderReaktionmitanschließenderBildungdesEnzym‐PelletshathierbeinureineVer‐
ringerungderWiederfindungvon1–3%zurFolge.DieRückgewinnungsratevonɛ–Caprolacton(3)
wurdeauchmitgrößerenEnzymmengenuntersucht.GleichzeitigwurdedieZentrifugierzeitwährend
der Aufarbeitung von 3 x 30min auf 3 x 5Minuten verkürzt. Die Ergebnisse sind in Abbildung 41
aufgezeigt.
Abbildung41. Bestimmung der Rückgewinnungsrate von ɛ–Caprolacton (3) bei verschiedenenEnzymmengenundbeiunterschiedlichenZentrifugierzeiten.
DieRückgewinnungsratesinktmitsteigendemEnzymgehaltderMischung,wasaufeinenEinschlussvon
Substrat imEnzym‐Pellet hindeutet. Dieses Pelletwirdwahrscheinlichmit zunehmenderDauer des
Zentrifugierens fester und stärker komprimiert, sodass das darin enthaltene Substrat auch durch
mehrfachesWaschennichtherausgespültwerdenkann.Auchkonntefestgestelltwerden,dasssichbei
lang andauernder Zentrifugierzeit Rillen in den EPPENDORF‐Gefäßen bildeten, durch die Teile der
LösungenindieZentrifugeauslaufenkonnten.DieEnzymmengensolltenfolglichmöglichstgeringund
dieZentrifugierzeitkurzgehaltenwerden.
3.3.2.4 VergleichderAnalytik‐MethodenanhandderStandard‐Reaktion
ImAnschlusswurdendiebeidenetabliertenMethoden,die 1H‐NMR‐Spektroskopiemittelsexternem
Standard sowie die Gaschromatographie,mithilfe der erstellten Eichgerade anhand einer Standard‐
Reaktionmiteinanderverglichen(Abbildung42).
0102030405060708090100
0 150 500
6861
20
93
7369
Rückgew
innungsrate[%]
Enzymmenge[µL]
3x30min 3x5min
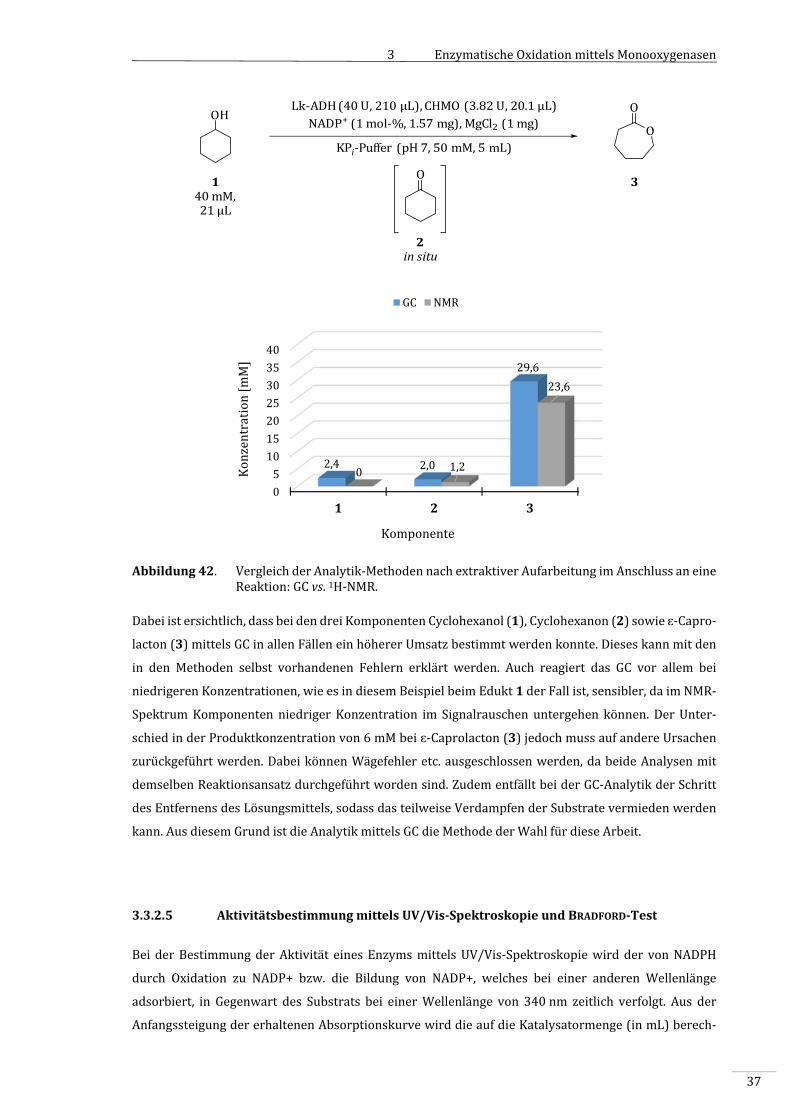
3 EnzymatischeOxidationmittelsMonooxygenasen
37
OH
140mM,21µL
O
2insitu
O
O
3
Lk‐ADH(40U,210µL),CHMO(3.82U,20.1µL)NADP+ (1mol‐%,1.57mg),MgCl2 (1mg)
KPi‐Puffer (pH7,50mM,5mL)
Abbildung42. VergleichderAnalytik‐MethodennachextraktiverAufarbeitungimAnschlussaneineReaktion:GCvs.1H‐NMR.
Dabeiistersichtlich,dassbeidendreiKomponentenCyclohexanol(1),Cyclohexanon(2)sowieɛ‐Capro‐
lacton(3)mittelsGCinallenFälleneinhöhererUmsatzbestimmtwerdenkonnte.Dieseskannmitden
in den Methoden selbst vorhandenen Fehlern erklärt werden. Auch reagiert das GC vor allem bei
niedrigerenKonzentrationen,wieesindiesemBeispielbeimEdukt1derFallist,sensibler,daimNMR‐
SpektrumKomponenten niedrigerKonzentration imSignalrauschen untergehen können.DerUnter‐
schiedinderProduktkonzentrationvon6mMbeiɛ‐Caprolacton(3)jedochmussaufandereUrsachen
zurückgeführtwerden.DabeikönnenWägefehleretc.ausgeschlossenwerden,dabeideAnalysenmit
demselbenReaktionsansatzdurchgeführtwordensind.ZudementfälltbeiderGC‐AnalytikderSchritt
desEntfernensdesLösungsmittels,sodassdasteilweiseVerdampfenderSubstratevermiedenwerden
kann.AusdiesemGrundistdieAnalytikmittelsGCdieMethodederWahlfürdieseArbeit.
3.3.2.5 AktivitätsbestimmungmittelsUV/Vis‐SpektroskopieundBRADFORD‐Test
BeiderBestimmungderAktivität einesEnzymsmittelsUV/Vis‐Spektroskopiewirdder vonNADPH
durch Oxidation zu NADP+ bzw. die Bildung von NADP+, welches bei einer anderen Wellenlänge
adsorbiert, in Gegenwart des Substrats bei einerWellenlänge von 340nm zeitlich verfolgt. Aus der
AnfangssteigungdererhaltenenAbsorptionskurvewirddieaufdieKatalysatormenge(inmL)berech‐
0
5
10
15
20
25
30
35
40
1 2 3
2,4 2,0
29,6
0 1,2
23,6
Konzentration[m
M]
Komponente
GC NMR

3 EnzymatischeOxidationmittelsMonooxygenasen
38
neteAktivitätbestimmtundmittelsGleichung4(Kapitel7.2.1.4.1)berechnet.DadieReagenziennach
demZusammengebengutgeschütteltwerdenmüssen,startetdieMessungmiteinigenSekundenVerzö‐
gerung.AusdiesemGrundistzuerwarten,dassdieermittelteAktivitätetwasgeringeralsdietatsäch‐
licheausfällt. InsolchenFällen isteswichtig,dieEnzymlösungentsprechendzuverdünnen,umeine
LinearitätderMessgeradenderAnfangssteigungzugewährleisten.
Tabelle5. BVMO‐ScreeningzurOxidationvonCyclohexanon(2).
Eintrag BVMOAktivität
[U/mLRohextrakt]Aktivität
[U/mgProtein]RelativeAktivität[%]
1 ECSMo‐01 7.38 3.00 100
2 ECSMo‐03 0.50 0.20 7
3 ECSMo‐05 0.14 0.05 2
DieAktivitätderCHMOwurdeinGegenwartvonCyclohexanon(2,1mM)alsSubstratbestimmt,indem
derVerbrauchdesCofaktorsNADPHzuNADP+beobachtetwird.Diesebeträgtdemnach0.082U/mL.
MithilfeeinesEnzymscreeningsmitdenvonENZYMICALSAGzurVerfügunggestelltenMonooxygenasen
ECS Mo‐03, einer Phenylaceton‐Monooxygenase (PAMO), sowie ECS Mo‐05, einer p‐Hydroxyaceto‐
phenon‐Monooxygenase(HAPMO),mitCyclohexanon(2)alsStandardsubstratsolltendieAktivitäten
mit der von CHMO (ECSMo‐01) verglichenwerden. Da alle drei BVMOs in flüssiger Form erhalten
wurden,kannkeindirekterVergleichdermittelsUV/Vis‐SpektroskopieerhaltenenAktivitäten(U/mL)
erfolgen. Durch einen BRADFORD‐Test wurde im Anschluss der Proteingehalt der einzelnen BVMOs
bestimmtunddarausdieAktivitätpromgProtein(U/mg)ermittelt.DieerhalteneAktivitätderCHMO
(ECSMo‐01)beträgt3.00U/mgundwurdegleich100%gesetzt.ImVergleichdazubeträgtdierelative
AktivitätderECSMo‐03undderECSMo‐05nur7%bzw.2%(Tabelle5).
DadasSubstratspektrumderenzymatischenDoppeloxidationdurchdieCHMOerweitertwerdensoll,
wurdedieEnzymaktivitätgegenüberIsophoron(52,1mM)alsmöglichesweiteresSubstratbestimmt.
DieerhalteneAktivitätwerdenaufdiedesCyclohexanons(2)alsReferenzsubstanzbezogen,welches
gleich100%gesetztwird,umeinerelativeAussageüberdieEnzymaktivitättreffenzukönnen(Tabelle
6).DabeikonnteeineAktivitätderCHMOgegenüberIsophoron(52)vonnur0.003U/mgimVergleich
zurAktivitätgegenüberCyclohexanon(2)von0.101U/mLnachgewiesenwerden.DierelativeAktivität
derCHMOgegenüberIsophoron(52)beträgtdemnachnur3%,wasunterdengegebenenReaktions‐
bedingungeneineschwierigeReaktionsführungist.

3 EnzymatischeOxidationmittelsMonooxygenasen
39
Tabelle6. BestimmungderAktivitätvonCHMOgegenüberIsophoron(52).
Eintrag Substrat Aktivität[U/mg] RelativeAktivität[%]
1 2 0.101 100
2 51 0.003 3
Die Aktivität der Lk‐ADH wurde gegenüber dem Standard‐Substrat 1‐Phenylethanol (47, 10 mM)
bestimmtundgleich100%relativeAktivitätgesetzt(Tabelle8).AnschließendwurdensowohlCyclo‐
hexanol(1,10mM)alsauch3,3,5‐Trimethylcyclohexanol(52,10mM)aufgleicheWeisevermessen.
Cyclohexanol(1)besitztdabeieinerelativeAktivitätvon153%(33.0U/mL),wirdalsodemnachvon
derLk‐ADHsehrgutalsSubstratakzeptiert.DieLk‐ADHweistgegenüberdemAlkohol53jedochkeine
Aktivitätauf.EventuellistdieReaktionszeitvon3MinutenindiesemFallnichtausreichend,sodassbei
einerverlängertenReaktionsdauervonbeispielsweise24StundentrotzdemProduktgebildetwerden
kann.
Tabelle7. BestimmungderAktivitätderLk‐ADHgegenüber3,3,5‐Trimethylcyclohexanol(53)imVergleichzu1‐Phenylethanol(47)undCyclohexanol(1).
Eintrag Substrat Aktivität[U/mL] RelativeAktivität[%]
1 47 21.5 100
2 1 33.0 153
3 53 0 0
3.3.2.6 OptimierungderStandardreaktion
EsistinderLiteraturbereitsbekannt,dassbeieinerKonzentrationdesLactons3höherals60mMeine
Inhibierung der CHMO auftritt.[26] Um Produktinhibierungen zu vermeiden, wurden in den letzten
Jahren verschiedene Methoden zur in situ‐Produktentfernung entwickelt, darunter der Einsatz von
Adsorberharzen[169,170],worauf imKapitel 3.3.4 eingegangenwird, sowiedieVerwendungvonZwei‐
phasen‐Systemen.[171–173]HydrophobeLösungsmittel,alsosolchemithohenlogP‐Werten,besitzenim
GegensatzzuhydrophilenSolventiennichtdieFähigkeit,dasamEnzymgebundeneessentielleWasser,
welchesalsSubstrathüllefungiert,zuentfernen,waszueinerVerringerungderEnzymaktivitätführen

3 EnzymatischeOxidationmittelsMonooxygenasen
40
würde.[174]IneinemzweiphasigenSystembefindensichdasEnzymunddiehydrophilenKomponenten
inderwässrigenPhase,währenddiehydrophobenAnteileinderorganischenPhasevorhandensind.In
der Literatur wurde beschrieben, dass für die BAEYER‐VILLIGER‐Oxidation von benzolgebundenen
Ketonen durch Katalyse durch die Monooxygenasen PAMO und HAPMO höhere Umsätze durch
VerwendungvonverschiedenenorganischenLösungsmittelnineiner5%(v/v)Konzentrationerzielt
werden können.[112] Der Einsatz eines zweiphasigenReaktionsmediums kann auch dazu dienen, die
CHMOzustabilisieren,wennmithohenSubstratkonzentrationengearbeitetwerdensoll.Zudemistauch
bekannt, dass die getesteten Monooxygenasen unterschiedliche organische Lösungsmittel tolerie‐
ren.[172,173]
AusgehendvomStandard‐VersuchausKapitel3.3.2.4wurdezunächstderLösungsmitteleffektunter‐
sucht.Dafür sollte sowohldasorganischeLösungsmittel als auchdieKonzentrationvariiertwerden
(Abbildung 43). Die Bestimmung der Stoffmenge ist bei den gezeigten Reaktionen auf zweiWegen
durchgeführt worden. In den meisten Fällen ist die Stoffmenge mittels 1H‐NMR‐Spektroskopie mit
Urotropin (51) als externer Standard bestimmtworden. Bei den Reaktionenmit 10% organischem
Lösungsmittel sowie mit Methylcyclohexan‐gesättigtem Puffer wurden die Stoffmengen mittels GC‐
Methodeermittelt.DerUmsatzdesBenchmark‐VersuchsinPuffer(vgl.Kapitel3.3.2.4)wurdeebenfalls
mtitelsGCbestimmtunddientzumVergleich.Dabei istder inKapitel 3.3.2.3bestimmteVerlustder
Substanzenmitbiszu10%proKomponentebeidenhierverwendetengeringenKonzentrationenzu
berücksichtigen.
Zunächst ist ein großer Unterschied der Reaktionen in Puffer und dest. Wasser zu erkennen. Der
geringereUmsatz inWasser istmit einermöglichenVeränderungdespH‐Wertesdesungepufferten
Systemszuerklären,diedurchhydrolytischeSpaltungdesgebildetenLactons3auftretenkann.
Allgemeinkonntegezeigtwerden,dassdieenzymatischeBAEYER‐VILLIGER‐OxidationauchinAnwesen‐
heit eines organischen Lösungsmittels stattfindet. Zwar liefert dieOxidation in einem zweiphasigen
SystemallgemeingeringereUmsätzealsinreinemPuffer,jedochwirddiesernegativeEffektteilweise
durch eine bessere Löslichkeit des Lactons 3 in dem Lösungsmittel‐Gemisch aufgehoben. Ähnliche
Ergebnisse beschrieben DE GONZALO et al. bei der BVMO‐katalysierten Oxidation von orga‐nischen
SulfideninZweiphasen‐Systemen.[173]BiokatalytischeReaktionenlaufenimAllgemeinenlang‐samabin
LösungsmittelnmitlogP‐Werten<2,mäßigbeilogP‐Wertenzwischen2und4undschnellmithohen
logP‐Wertenvon>4,wasmitdenjeweiligenFähigkeitendesLösungsmittelszusammenhängt,dasdas
EnzymumgebendeessentielleWasserzuentfernen.[175]DerhöchsteUmsatzvon87%konntemit10%
Isooctan (v/v) erzielt werden. Dieses ist nicht weiter überraschend, da Isooctan von den hier ver‐
wendetenLösungsmittelndenhöchstenlogP‐Wertvon4.54besitzt.[176]
DieUmsätzemitjeweils10%(v/v)Methylcyclohexan(logP=3.88)[177]undToluol(logP=2.73)[177]sind
geringer und folgen dem Trend.[177] n‐Heptan hättemit einem logP‐Wert von 4.40[176] jedoch einen
höhererUmsatzähnlichdemvonIsooctanliefernsollen.MäßigeUmsätzevon35–49%wurdenmit
Lösungsmittel‐gesättigtemKPi‐Puffer(pH7,50mM)erhalten.

3 EnzymatischeOxidationmittelsMonooxygenasen
41
OH
140mM,21µL
O
2insitu
O
O
3
Lk‐ADH(40U,210µL),CHMO(3.82U,20.1µL)NADP+ (1mol‐%,1.57mg),MgCl2 (1mg)
Lösungsmittel‐Gemisch, 5mL
Abbildung43. VariationdesLösungsmittelgemischesbeiderDoppeloxidationvonCyclohexanol(1).Angegeben ist der Umsatz der Komponenten2 und3 sowie die nicht umgesetzteStoffmenge von 1. Die Reaktion in PKi‐Puffer ist in Kapitel 3.3.2.4 im DetailbeschriebenunddienthierzumVergleichalsStandard‐Reaktion.
Gemischemit20bzw.40%(v/v)organischemLösungsmittelliefertenpraktischkeinLacton3.Inder
Literaturwurdebereitsbeschrieben,dassmitanderenMonooxygenasenkeineUmsätzeinAnwesenheit
0 5 10 15 20 25 30 35 40
KPi
dest.Wasser
MTBE‐ges.dest.Wasser
Methylcyclohexan‐ges.KPi
n‐Hexan‐ges.KPi
n‐Heptan‐ges.KPi
EtOAc‐ges.KPi
MTBE‐ges.KPi
KPi/n‐Heptan9:1
KPi/Isooctan9:1
KPi/Methylcyclohexan9:1
KPi/Toluol9:1
KPi/EtOAc6:4
KPi/MTBE6:4
KPi/MTBE8:2
Stoffmenge[mmol]
Lösungsm
ittel‐Gem
isch
Stoffmenge3 Stoffmenge2 Stoffmenge1ɛ‐Caprolacton(3)
i
i
i
i
i
i
i
i
i
i
i
i
n
n
n
i
Cyclohexanon(2) Cyclohexanol(1)

3 EnzymatischeOxidationmittelsMonooxygenasen
42
desEssigesters5(logP=0.68)erhaltenwerdenkonnten.[173,175]DiesesistauchhierderFall.FürdieLk‐
ADHscheintdieVerwendungdesEssigesters5alsLösungsmitteljedochkeinHinderniszusein,dennes
konntenbiszu6%desCylcohexanons(2)alsZwischenstufenachgewiesenwerden.Wahrscheinlichlief
dieOxidationvonCyclohexanol(1)zumKeton2soweitab,bisderhinzugegebeneCofaktorvollständig
zuNADPHreduziertwordenistunddieReaktionstagnierte.Esistauchbekannt,dassdieLk‐ADHihre
spezifischeAktivitätundStabilität inAnwesenheitvonorganischenLösungsmittelnmit logP‐Werten
<3.5einbüßt.[153]
Zusammenfassend kann geschlussfolgert werden, dass die Zugabe von 10% Isooctan (v/v), einem
LösungsmittelmithohemlogP‐Wert,zurReaktionsmischungimmernochzuhervorragendenUmsätzen
biszu87%führt.DadieAnsätzestetsnach24Stundenbeendetwordensind,könntehiereineVerlän‐
gerung der Reaktionszeit zu einer quantitativen Reaktion führen. Des Weiteren sollte untersucht
werden, inwiefernunterdiesenBedingungen eineProduktinhibierungauftritt,wennbeiKonzentra‐
tionenvonüber80mMgearbeitetwird.
AnschließendwurdederStandard‐VersuchbeiunterschiedlichenBedingungendurchgeführt.Sowurde
dieMengedesCofaktors,dieMengederLk‐ADHsowiedieReaktionstemperaturvariiert(Tabelle8).
Tabelle8. VariationderReaktionsbedingungenderOxidationvonCyclohexanol(1).
EintragNADP+[mol‐%]
Lk‐ADH
[U]
T
[°C]
Umsatz3
[%]b)
Umsatz2
[%]b)
1a) 1 40 RT 97 3
2 2 40 RT 97 0
3 10 40 RT 61 11
4 1 40 30 48 5
2 1 13 RT 55 3a)Standard‐Reaktion;b)Umsatzbestimmtmittels1H‐NMR‐Spektroskopie,angegebenistderproduktbezogeneUmsatz.
EinevermehrteZugabevonNADP+zurReaktionsmischunghatdemnachkeinenpositivenEinflussauf
denUmsatz.BeieinerErhöhungderCofaktormengeauf2mol‐%wurdenebenfalls97%desLactons3
erhalten.DieZugabevon10mol‐%vonNADP+lieferteimVergleichzurStandard‐Reaktion(Eintrag1)
nurnochetwa2/3desUmsatzes(Eintrag2);dazupassendkonnten11%anCyclohexanon(2)ermittelt
werden.DieseTatsachesprichtdafür,dassdieOxidationvonCyclohexanol(1)zumKeton2durchdie
Lk‐ADHsehrviel schneller abläuftalsdieOxidationvon2 zumLacton3 durchdieMonooxygenase.
Diesesdeutetdaraufhin,dassbeiderverwendetenCofaktor‐KonzentrationderMICHAELIS‐Konstante,

3 EnzymatischeOxidationmittelsMonooxygenasen
43
der KM‐Wert, überschritten worden ist. Der KM‐Wert gibt diejenige Konzentration an, bei der die
ReaktionsgeschwindigkeitderhalbenMaximalgeschwindigkeitvmaxentspricht.BeiweitererErhöhung
derKonzentrationwirdschließlichvmaxangestrebtundeineSättigungtrittein,daalleaktivenZentren
derEnzymebesetztsind.Esistbekannt,dassNADP+festimEnzyminseinerreduziertenFormgebunden
ist und das sonst labile C4a‐Hydroperoxyflavin (36) der CHMO solange stabilisiert, bis ein Substrat
verfügbar ist.[178] Liegt nun in der Reaktionsmischung ein Überschuss an NADP+ im Vergleich zum
Substrat vor ([S] >KM), sokommt es in solchenFällen zu einer kompetitiven Inhibierungbezüglich
NADPH.DieseArtderHemmungdesEnzymskannjedochdurchErhöhungderSubstratkonzentration
wiederrückgängiggemachtwerden.
Die Erhöhung derTemperatur von 20 °C auf 30 °C führte lediglich zu 48%Umsatz (Eintrag 4). Die
Halbwertszeitder isoliertenCHMObeträgt lautLiteraturbei25°CundpH8.5,demOptimumseiner
katalytischeAktivität,etwaeinenTag[179],sodassdavonausgegangenwerdenkann,dassdiekatalytische
Aktivität der CHMO bei einer Reaktionstemperatur von 30 °C bereits rapide gesunken ist. Die
MonooxygenasewirdalsdaskritischereEnzymindiesemSystemangesehen,dadieLk‐ADHfür ihre
StabilitätinAnwesenheitvonhydrophobenMoleküleningroßenMolekülenbekanntist.[26]EinWeg,die
CHMOzustabilisieren,istdieImmobilisationaufeinemTräger.
DerEinflussderVerringerungderEnzymmengevonLk‐ADHaufdenReaktionsverlaufwurdeebenfalls
untersucht(Eintrag5),dafürdieReaktiondiezehnfacheMengeanUnitsderAlkoholdehydrogenase
verwendet imVergleichzurCHMOwird.EskonntendemnachauchmitnureinemDrittelderbisher
verwendetenMenge an Lk‐ADH ein immer noch guter Umsatz von 55% Lacton3 erhaltenwerden.
EventuellkönntehiereineVerlängerungderReaktionszeitzueinemvollständigenUmsatzführen,was
zuKosteneinsparungeninHinblickaufdenPreisderAlkohol‐Dehydrogenaseführenkann.
3.3.3 UntersuchungenmittelsTitrations‐Apparatur
Mithilfe der Titrino‐Apparatur kann der Umsatz einer Reaktion bestimmtwerden, indem D‐Glucose
(47‐a)/Glucose‐Dehydrogenase(GDH)beiderReaktion,indiesemFalldieReduktionvonAcetophenon
(46)durchdieLk‐ADH,alsCofaktor‐Regenerationssystemeingesetztwird(Abbildung44).Dabeiwird
D‐Glucose(48‐a)überdieZwischenstufeGluconolacton(49‐a)zuGluconsäure(50‐a)umgewandelt.In
derApparaturwirddieseSäuremitNatronlaugebekannterKonzentrationgegentitriert.NachBeenden
derReaktionkanndirektdieMengeanverbrauchterNaOHabgelesenunddarausderUmsatzermittelt
werden.

3 EnzymatischeOxidationmittelsMonooxygenasen
44
O
O
O
+
NADPH NADP+
GDH
ADH OH
O
O
+
D‐GlucoseGlucono‐lacton
Glucon‐säure Na‐Gluconat
NaOH
46 3(R)‐473
48‐a 49‐a 50‐a
Abbildung44. OxidationvonAcetophenon(46)durchLk‐ADHinAnwesenheitvonɛ‐Caprolacton(3)mitD‐Glucose(48‐a)/GDHalsCofaktor‐Regenerationssystem.
ZunächstsolltedieInhibierungderLk‐ADHdurchɛ–Caprolacton(3)inunterschiedlichenKonzentra‐
tionenübereinenZeitraumvon24Stundenuntersuchtwerden(Abbildung45).DafürwurdeAcetophe‐
non(4),welchesalsStandardsubstratfürdieLk‐ADHfungiert,zuPhenylethanol((R)‐47)reduziert.
Abbildung45. UntersuchungderInhibierungderLk‐ADHdurchɛ–Caprolacton(3).DieReaktionenbei0.25und1MLacton‐Konzentrationwurdenautomatischnach3.5hReaktionszeitbeendet.
DieProduktkonzentrationenwurdenimAnschlusszumeinenausgehendvonderverbrauchtenMenge
anNatronlaugewährendderReaktionermittelt.ZumanderenwurdendieProduktkonzentrationendes
gebildetenPhenylethanols(R)‐47imAnschlussandieextraktiveAufarbeitungmittels1H‐NMR‐Spektro‐
skopiemitUrotropin(51)alsexternemStandardbestimmt.Dabeiistzuerkennen,dassdieerhaltenen
KonzentrationendesAlkohols(R)‐47beietwa16mMkonstantbleiben.FolglichtrittkeineInhibierung
derLk‐ADHdurchdasLacton3auf.DiemittelsTitrino‐ApparaturberechnetenProduktkonzentrationen
desAlkohols(R)‐47sindbeigeringenKonzentrationendesɛ‐Caprolactons(3,0–10mM)zunächstzwar
erhöht,bleibenjedochkonstant.MitsteigenderKonzentrationdesLactons3jedochnimmtauchdievia
TitrinoermittelteProduktkonzentrationzu.DiesesliegtzumTeilanKonzentrationsschwankungender
verwendeten Natronlauge. Außerdem kann die Neutralisation von Enzymbestandteilen durch die
05101520253035404550
0 0.01 0.04 0.1 0.25 1
25 24
30
42
49 50
1916 16 15 16 14
Produktkonzentration[m
M]
ɛ‐Caprolacton(3)[M]
Titrino NMR

3 EnzymatischeOxidationmittelsMonooxygenasen
45
Natronlauge erfolgen. Zudem kann davon ausgegangen werden, dass hierbei eine hydrolytische
SpaltungdeszugegebenenLactons3zurentsprechendenSäure53stattfindet,dieebenfallsmitNaOH
neutralisiertwird(Abbildung46).DiemittelsTitrino‐ApparaturbestimmtenProduktkonzentrationen
könnensomitnichtalsqualitativeWerteangesehenwerden.
O
O
OH
OHO
3 54
hydrolytischeSpaltung
Abbildung46. HydrolytischeSpaltungvonɛ–Caprolacton(3)zu6‐Hydroxyhexansäure(54).
DieReaktionwurdeinD2OmitanschließenderNMR‐spektroskopischerUntersuchungderwässrigen
Phasedurchgeführt.Mittels1H‐NMR‐SpektroskopiekonntedienahezuvollständigeRückgewinnungdes
eingesetzten Lactons 3 durch Verwendung des externen Standards Urotropin (51) nachgewiesen
werden.IndenSpektrenkonntenzwardiegebildetenwasserlöslichenSalzenichtbelegtwerden,jedoch
konntedieBildungvon6‐Hydroxyhexansäure(54)nachgewiesenwerden,welchedurchhydrolytische
SpaltungdesLactons3entstandenist.Abbildung47zeigteinNMR‐SpektrumeinerBiotransformation,
inderdieHydrolysedesLactons3zurSäure54stattgefundenhat.SosindcharakteristischeSignaleim
aliphatischenBereichdesSpektrums,beispielsweisebei2.16und1.53ppm,vorhanden.DasVerhältnis
desgebildetenLactons3zurentsprechendenSäure54beträgtindiesemFall1.0:2.2.
Abbildung47. NRM‐Spektrumvonɛ‐Caprolacton(3,links)undBeispieleinesNMR‐SpektrumsderHydrolysedesLactons3zurSäure54(rechts).
DadieReaktionsgefäßederTitrino‐ApparaturennurmitParafilmverschlossenwerdenkonnten,istes
nichtunwahrscheinlich,dassüberdenReaktionszeitraumvon24StundeneinTeilderSubstrateauf‐
grundderhohenFlüchtigkeitverdampftist.DiemittelsNMR‐SpektroskopieerhaltenenProduktkonzen‐
trationendesAlkohols (R)‐47 bleibenkonstantbeietwa16mM.DavorallembeidenAnsätzenmit
hohenKonzentrationendesLactons3dasGesamtvolumenanSubstratzunimmt,könnensichFehler

3 EnzymatischeOxidationmittelsMonooxygenasen
46
durchInhomogenitätderProduktmischungenergeben.Insgesamtlässtsichsagen,dassauchbeihohen
Konzentrationenanɛ–Caprolacton(3)keineProduktinhibierungderLk‐ADHstattfindet.DiesesEnzym
istdemnachfürdieDoppeloxidationvonCyclohexanol(1)übereinenZeitraumvon24Stundensehrgut
geeignet.
3.3.4 VerwendungvonAdsorberharzen
DieerstenVersuche, eineProduktinhibierungwährendderReaktiondurchVerwendungvonAdsor‐
berharzenzuvermeiden,wurdenbereits1997vonELILILLYANDCOMPANYveröffentlicht.[169]DasHarz
wirdzurReaktionsmischungzugegebenundkanndabeialsfestesorganischesLösungsmittelangesehen
werden.Die treibendeKraft hinter derAdsorption ist dieWechselwirkung zwischendenunpolaren
Regionen des zu adsorbierenden Moleküls und der polaren Oberfläche des Harzes. So können viel
höhere Substrat‐ und Produktkonzentrationen erreicht werden, während das Enzym nur geringen
Konzentrationenausgesetztist.
DieindieserArbeitgetestetenHarzeXAD‐7undXAD‐1180sindeinfacheporöse,nicht‐funktionalisierte
Kunststoff‐KügelchenmitgroßenOberflächen,dieausunterschiedlichenPolymerenhergestelltwurden
undsomitverschiedenePartikel‐undPorengrößenbesitzen.XAD‐7 isteinPolyacrylat‐Harzundnur
mäßigpolarundsolltesomitsowohldasSubstratCyclohexanol(1)alsauchdasProdukt3gutbinden.
XAD‐1180isteinunpolares,porösesmakrovernetztesaromatischesPolystyren‐Divinylbenzol‐Polymer.
DieHarzebesitzeneinedurchschnittlichePorengrößevon400bzw.450Å.
ZunächstsolltedieRückgewinnungsratederSubstanzenbeibeidenHarzenXAD‐7undXAD‐1180durch
Simulation der Aufarbeitung untersucht werden, indem das Substrat 1 bzw. das Lacton 3 über
20Stunden bei Raumtemperatur in Puffer zusammen mit dem jeweiligen Adsorberharz gerührt.
Anschließend wird zum einen das Harz abfiltriert und mehrmals mit organischen Lösungsmitteln
gewaschen,zumanderendieorganischePhaseextraktivaufgearbeitet(Tabelle9).DieAufnahmefähig‐
keitderbeidenHarzeXAD‐7undXAD‐1180fürdieSubstrateCyclohexanol(1)undɛ–Caprolacton(3)
liegen abhängig von Substrat, Lösungsmittel und Harz nur bei 2 –20%.Nach der extraktiven
Aufarbeitungkönnennur46bzw.74%vonCyclohexanol(1)bzw.ɛ‐Caprolacton(3)inAnwesenheitvon
XAD‐7 zurückgewonnen werden. Die Verwendung von MTBE anstelle von Dichlormethan als
Extraktionsmittel jedoch führt zu einer drastischen Abnahme der Rückgewinnungs‐rate auf 30%.
EventuellkanndieserUnterschiedmitdenlogP‐Werten(DCM:1.25,MTBE:0.94)begründetwerden,da
dasProdukt3(logP=1.25)[180]alsauchderAlkohol1(logP=1.23)[177]inDCMbesserlöslichist.Die
höchsteRückgewinnungsratevomSubstrat1von89%wurdeinAnwesenheitvon100mgXAD‐1180
mitDCMalsExtraktionsmittelerhalten.

3 EnzymatischeOxidationmittelsMonooxygenasen
47
Tabelle9. RückgewinnungsratederSubstanzen1bzw.3inAnwesenheitderAdsorberharzeXAD‐7bzw.XAD‐1180.
Eintrag Substanz Harz/[mg] SolvenzRückgewinnungsrate[%]
Harz insgesamt
1 1 XAD‐7/100 DCM 2 46
2 3 XAD‐7/100 DCM 8 68
3 3 XAD‐7/200 DCM 8 74
4 3 XAD‐7/100 MTBE 4 30
5 1 XAD‐1180/100 DCM 20 89
6 1 XAD‐1180/200 DCM 10 41
Da der Fokus bei der Doppeloxidation von Cyclohexanol (1) vor allem bei der Vermeidung der
ProduktinhibierungdurchdasLacton3liegt,wurdendieinAnsatz3verwendetenBedingungenfürdie
imAnschlussdurchgeführteReaktionaus‐gewählt(Abbildung48).
Abbildung48. DoppeloxidationvonCyclohexanol(1)inAnwesenheitvonXAD‐7.
DasHarzXAD‐7wurdezusammenmitdemCyclohexanol(1)sowiedenrestlichenKomponentenfür
24Stunden gerührt. Der dadurch erhaltene Umsatz beträgt 24% bezüglich des Urotropins (51) als
externerStandard(produktbezogenerUmsatz:48%).DieinsgesamtzurückgewonneneStoffmengealler
dreiKomponentenlagnachderAufarbeitungbeinur50%.DadieReaktionnurzurHälfteablief,istes
wahrscheinlich, dass ein Teil des Cyclohexanols (1) gleich zu Beginn im Harz durch Wasserstoff‐
brückenbindungenundVANDERWAALS‐KräftegebundenwurdeundfürdieReaktiondadurchnichtmehr
zurVerfügungstand.
Eswurde indieserArbeiterstmalsgezeigt,dassdieVerwendungvonHarzenbeiderenzymatischen
DoppeloxidationvonCyclohexanol(1)zumLacton3 istmöglichistundmit48%bereitseinenguten
produktbezogenenUmsatzliefert,derjedochnochweitererOptimierungbedarf.DesWeiterenkönnten
weitereAdsorberharze getestetwerden,derenPorengröße sowiePolaritätnochbesser aufdashier

3 EnzymatischeOxidationmittelsMonooxygenasen
48
erhalteneProdukt3 zugeschnitten ist.DieVerwendungvonLösungsmittelmithöheren logP‐Werten
(>1.25)könntezueinererhöhtenAusbeutewährendderAufarbeitungführen.
3.3.5 VariationderSubstrate
Alkylsubstituierte Caprolactone sind von großem Interesse für die Synthese von entsprechenden
Polymeren.DieCopolymerisationvonɛ‐Caprolacton (3)mitTrimethylcaprolactonenwurdevonder
FirmaDAICELpatentiert.[22]DurchUmsetzungderdarausentstandenenPolyolemitDiisocyanatewerden
Polyurethane erhalten, die sich durch exzellente Hydrolysebeständigkeit sowie durch hohe mecha‐
nischeStärkeundStabilitätgegenüberhohenTemperaturenundFeuchtigkeitauszeichnen.
Bei der BAEYER‐VILLIGER‐Oxidation von 3,5,5‐Trimethylcyclohexanon (55) entsteht auf chemischem
WegeeineracemischeMischungausdenRegioisomeren3,3,5‐und3,5,5,‐Trimethyl‐ɛ‐caprolacton(56
bzw. 57) (Abbildung 49). In diesem Fall werden vier Produkte gebildet, da es nur einen geringen
energetischenUnterschied der beidenwandernden Kohlenstoffatome gibt.[181] Die Anwesenheit von
Alkylsubstituenten macht sich auch in der längeren Reaktionszeit im Vergleich zur Oxidation von
Cyclohexanon(2)bemerkbar;sosinktdieReaktionsgeschwindigkeitmitsteigenderAnzahlanSubsti‐
tuentenebenfalls.AuchdieLängedesAlkylrestes in3‐PositionhateinenEinflussaufdieGeschwin‐
digkeit.
Abbildung49. ProduktederchemischenundenzymatischenBAEYER‐VILLIGER‐OxidationdesKetonsrac‐55.
IndieserArbeitsollteaußerdemdieMöglichkeitderbiokatalytischenDarstellungdesentsprechenden
Lactonsausgehendvon3,5,5‐Trimethylcyclohexanol(53)mithilfederLk‐ADHundderCHMOunter‐
suchtwerden(Abbildung50).
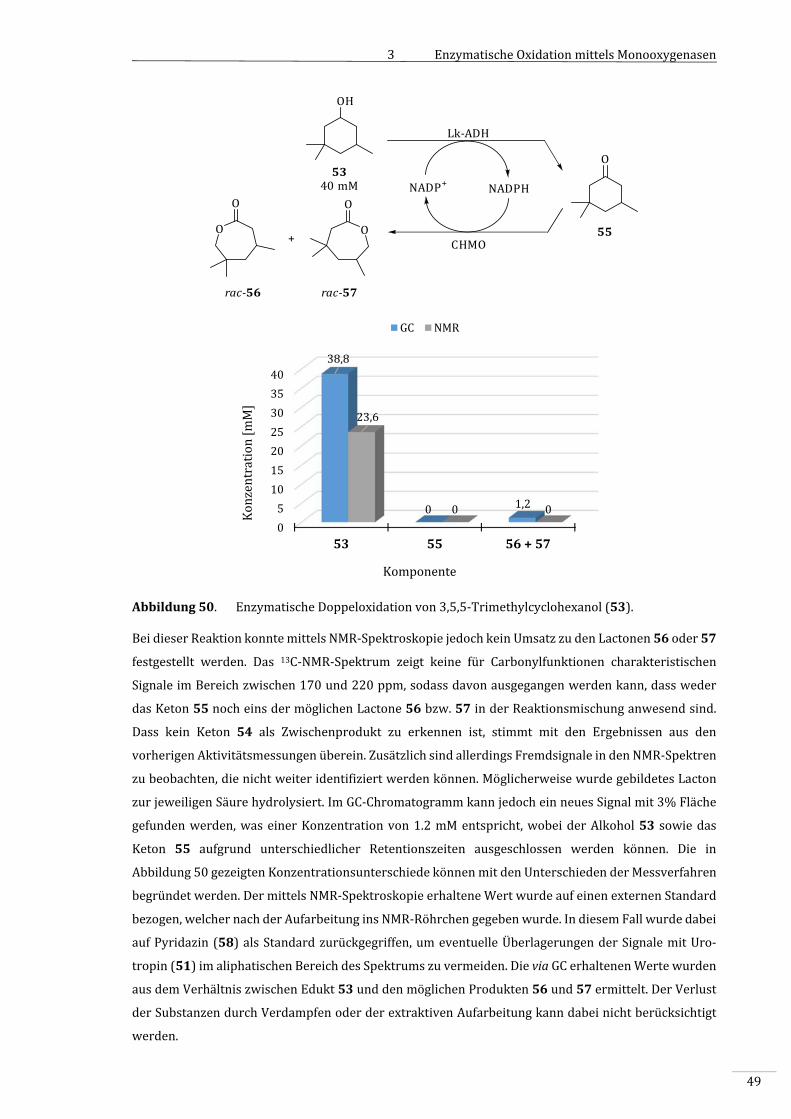
3 EnzymatischeOxidationmittelsMonooxygenasen
49
Lk‐ADH
NADP+ NADPH
OH
O
O
5340mM
rac‐57
O
55O
O
+
rac‐56
CHMO
Abbildung50. EnzymatischeDoppeloxidationvon3,5,5‐Trimethylcyclohexanol(53).
BeidieserReaktionkonntemittelsNMR‐SpektroskopiejedochkeinUmsatzzudenLactonen56oder57
festgestellt werden. Das 13C‐NMR‐Spektrum zeigt keine für Carbonylfunktionen charakteristischen
SignaleimBereichzwischen170und220ppm,sodassdavonausgegangenwerdenkann,dassweder
dasKeton55nocheinsdermöglichenLactone56bzw.57inderReaktionsmischunganwesendsind.
Dass kein Keton 54 als Zwischenprodukt zu erkennen ist, stimmt mit den Ergebnissen aus den
vorherigenAktivitätsmessungenüberein.ZusätzlichsindallerdingsFremdsignaleindenNMR‐Spektren
zubeobachten,dienichtweiteridentifiziertwerdenkönnen.MöglicherweisewurdegebildetesLacton
zurjeweiligenSäurehydrolysiert.ImGC‐ChromatogrammkannjedocheinneuesSignalmit3%Fläche
gefundenwerden,waseinerKonzentrationvon1.2mMentspricht,wobeiderAlkohol53 sowiedas
Keton 55 aufgrund unterschiedlicher Retentionszeiten ausgeschlossen werden können. Die in
Abbildung50gezeigtenKonzentrationsunterschiedekönnenmitdenUnterschiedenderMessverfahren
begründetwerden.DermittelsNMR‐SpektroskopieerhalteneWertwurdeaufeinenexternenStandard
bezogen,welchernachderAufarbeitunginsNMR‐Röhrchengegebenwurde.IndiesemFallwurdedabei
aufPyridazin(58)alsStandardzurückgegriffen,umeventuelleÜberlagerungenderSignalemitUro‐
tropin(51)imaliphatischenBereichdesSpektrumszuvermeiden.DieviaGCerhaltenenWertewurden
ausdemVerhältniszwischenEdukt53unddenmöglichenProdukten56und57ermittelt.DerVerlust
derSubstanzendurchVerdampfenoderderextraktivenAufarbeitungkanndabeinichtberücksichtigt
werden.
0
5
10
15
20
25
30
35
40
53 55 56+57
38,8
0 1,2
23,6
0 0Konzentration[m
M]
Komponente
GC NMR
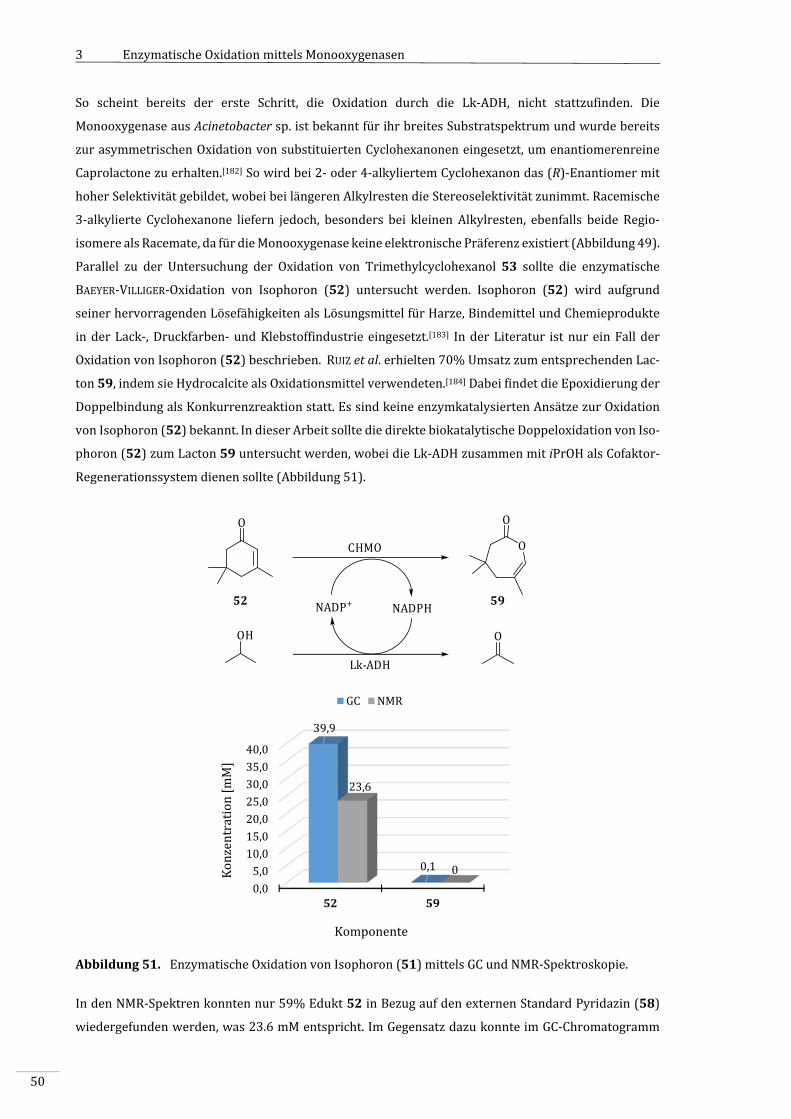
3 EnzymatischeOxidationmittelsMonooxygenasen
50
So scheint bereits der erste Schritt, die Oxidation durch die Lk‐ADH, nicht stattzufinden. Die
MonooxygenaseausAcinetobactersp.istbekanntfürihrbreitesSubstratspektrumundwurdebereits
zurasymmetrischenOxidationvonsubstituiertenCyclohexanoneneingesetzt,umenantiomerenreine
Caprolactonezuerhalten.[182]Sowirdbei2‐oder4‐alkyliertemCyclohexanondas(R)‐Enantiomermit
hoherSelektivitätgebildet,wobeibeilängerenAlkylrestendieStereoselektivitätzunimmt.Racemische
3‐alkylierte Cyclohexanone liefern jedoch, besonders bei kleinen Alkylresten, ebenfalls beide Regio‐
isomerealsRacemate,dafürdieMonooxygenasekeineelektronischePräferenzexistiert(Abbildung49).
Parallel zu der Untersuchung der Oxidation von Trimethylcyclohexanol 53 sollte die enzymatische
BAEYER‐VILLIGER‐Oxidation von Isophoron (52) untersucht werden. Isophoron (52) wird aufgrund
seinerhervorragendenLösefähigkeitenalsLösungsmittelfürHarze,BindemittelundChemieprodukte
inderLack‐,Druckfarben‐undKlebstoffindustrie eingesetzt.[183] InderLiteratur istnureinFallder
OxidationvonIsophoron(52)beschrieben.RUIZetal.erhielten70%UmsatzzumentsprechendenLac‐
ton59,indemsieHydrocalcitealsOxidationsmittelverwendeten.[184]DabeifindetdieEpoxidierungder
DoppelbindungalsKonkurrenzreaktionstatt.EssindkeineenzymkatalysiertenAnsätzezurOxidation
vonIsophoron(52)bekannt.IndieserArbeitsolltediedirektebiokatalytischeDoppeloxidationvonIso‐
phoron(52)zumLacton59untersuchtwerden,wobeidieLk‐ADHzusammenmitiPrOHalsCofaktor‐
Regenerationssystemdienensollte(Abbildung51).
Lk‐ADH
NADP+ NADPH
O
O
59
O
52
OH O
CHMO
Abbildung51. EnzymatischeOxidationvonIsophoron(51)mittelsGCundNMR‐Spektroskopie.
IndenNMR‐Spektrenkonntennur59%Edukt52inBezugaufdenexternenStandardPyridazin(58)
wiedergefundenwerden,was23.6mMentspricht.ImGegensatzdazukonnteimGC‐Chromatogramm
0,05,010,015,020,025,030,035,040,0
52 59
39,9
0,1
23,6
0Konzentration[m
M]
Komponente
GC NMR

3 EnzymatischeOxidationmittelsMonooxygenasen
51
der Reaktionsmischung ein neues Signal beobachtet werden, was etwa 0.3% Fläche und 0.12 mM
entspricht. Auch in diesem Fall ist der Unterschied zwischen den erhaltenen Konzentrationen des
Ketons52sehrgroßundaufunterschiedlicheMessver‐fahrenzurückzuführen.EbensowieinAbbildung
50 wurden die via GC erhaltenenWerte durch das Verhältnis zwischen Edukt 52 und Produkt59
bestimmt.AuchindiesemFallwerdenVerlustederSubstanzenwährendderReaktionsführungsowie
Aufarbeitungnichtnäherbetrachtet.
Zwarscheintes,alskönnedieBVMOnahezualleSubstrateumsetzen,diedasaktiveZentrumerreichen,
jedochsind2‐Cyclohexenon‐DerivateAusnahmen,dienichtodernursehrlangsamvonderCHMOunter
denhiergewähltenReaktionsbedingungenumgesetztwerdenkönnen.[109,110]

52

4 EnantioselektiveRacematspaltungvonAminen
53
4 EnantioselektiveRacematspaltungvon
Aminen
4.1 Lipasen
4.1.1 Allgemeines
Lipasen (Triacylglycerol‐Hydrolasen, EC 3.1.1.3) sind in der Natur allgegenwärtig und werden in
verschiedenenPflanzen,TierenundMikroorganismenexprimiert.[185]LipasenmikrobiellenUrsprungs,
hauptsächlichausBakterienundPilzen,repräsentierendieamhäufigstenverwendeteKlasseanEnzy‐
meninbiotechnologischenAnwendungenundinderorganischenChemie.SiefindenEinsatzineinem
breiten Spektrum industrieller Anwendungen, wie beispielsweise in der Lebensmittel‐Technologie,
Waschmittelundchemischen Industriesowie inderbiomedizinischenForschung.[185,186]Diemeisten
Lipasenwerden vonBakterien produziert,wodurch sich deren großtechnischeHerstellung viel ein‐
fachergestaltetunddiesedadurchvonbesonderemkommerziellem Interessesind. IhreEntdeckung
wurdebereits1901vonEIJKMANbeschrieben.[187]
ObwohlbereitseinebeträchtlicheAnzahlanStämmenzurProduktionvonLipasenverwendetwurden,
sind Candida sp., Pseudomonas sp. und Rhizopus sp. die wichtigsten Quellen.[186] Die natürlichen
SubstratederLipasensindlangkettigeTriacylglycerolevomTyp60,dieeinesehrgeringeLöslichkeitin
Wasserbesitzen(Abbildung52).[185]DurchHydrolysewerdenGlycerol(61)sowiedreiÄquivalenteder
entsprechendenFettsäure62erhalten.
Abbildung52. LipasenkatalysierteHydrolyseundSyntheseeinesTriacylglycerols60.[185]
Neben den lipolytischen, d.h. fettspaltenden Eigenschaften, besitzen die Lipasen noch eine ester‐
spaltende Aktivität.[185] Sie katalysieren neben der Hydrolyse und Umesterung außerdem die Alko‐
holyse, Acidolyse, Veresterung und Aminolyse.[186] Die Vorteile der Verwendung von Lipasen sind
vielfältig.DurchbereitsaufgelösteKristallstrukturenkanndaskatalytischePotentialvonLipasendurch

4 EnantioselektiveRacematspaltungvonAminen
54
molekularbiologische Ansätze wie Protein engineering oder gerichtete Evolution weiter verbessert
werden.[188]LipasenbenötigenkeineteurenCofaktorenundsindchemoselektiv.Siebesitzeneinehohe
Substratbreite und sind hochspezifische Katalysatoren in Hinblick auf Regio‐ und Enantioselekti‐
vität.[189]AufgrunddieserEigenschaftensindLipasendieambreitestenangewendeteGruppevonBio‐
katalysatoren,wasauchdurchdiejährlichdurchschnittlich>1000VeröffentlichungenzudemEnzym
deutlichwird.[190]
LipasenbesitzenzudemdieFähigkeit, zwischendenEnantiomereneines racemischenGemisches zu
unterscheiden, was zu einem Einsatz von Lipasen zur kinetischen Racemattrennung führte. Solche
enantiomerenreinenoder–angereichertenKomponentengewinnen inderSynthesevonpharmazeu‐
tischen,synthetischorganischenundnatürlichenProduktensowieAgrarerzeugnissenimmermehran
Bedeutung.[185]
4.1.2 StrukturundMechanismus
BRADYetal. publizierten1990die ersteKristallstruktur einerLipase ausdemPilzMucormiehei.[191]
Gleichzeitig wurde die Struktur der Lipase aus humaner Pankreas von WINKLER veröffentlicht.[192]
SeitdemwurdennocheinigeweitereLipasenbakteriellenUrsprungsmittelsRöntgenstrukturaufgelöst.
AlledieseLipasenvariierenzwarstarkinderGröße(20–60kDa),habenjedochtrotzrelativgeringer
sequenziellerÜbereinstimmungenalle eineähnlichedreidimensionaleGesamtstrukturmitder soge‐
nanntenα/β‐Hydrolase‐Falte,diealsallgemeinesFaltmuster identifiziertwerdenkonnteundhaupt‐
sächlichausβ‐Faltblätternbestehtundvonα–Helicesflankiertwerden.[187]Solcheineα/β‐Hydrolase‐
FalteisttypischfürEnzyme,diekeineoffensichtlicheSequenzähnlichkeitaufweisen,jedochvoneinem
gemeinsamenVorfahrenabstammen.[187]
Die Bestimmung der dreidimensionalen Struktur von unterschiedlichen Lipasen bestätigt deren
KlassifikationalsSerin‐Hydrolasen.[186]DasaktiveZentrumderLipasenenthälteinekatalytischeTriade
bestehend aus Ser‐His‐Asp‐Resten, wobei sich der Serin‐Rest in einem β‐εSer‐α‐Motiv im aktiven
Zentrumbefindet.DiesesMotivbestehtauseinemβ‐Faltblatt,einerSchleifemitSerininderε‐Position
undeinerα–Helix.DerunveränderlicheersteundletzteGlycin‐RestinderConsensus‐Sequenz(Gly‐x‐
Ser‐x‐Gly)diesesMotivsliegeninhelicalerKonformationvor,dieaufgrunddersterischenAnsprüche
diesesMotivskonserviertsind.[187]
Ein unübliches und interessantes Merkmal der Struktur der Lipasen ist, dass das aktive Zentrum
vollständig unter einer deckelartigen Struktur, bestehend aus ein oder zwei α–Helices, verdeckt ist.
DurcheineBewegungdesDeckelswirddasaktiveZentrumfürdasSubstratzugänglich.Diesestruk‐
turelleÄnderungdesProteinswirdbegleitet voneinerweiterenBewegung in einernahegelegenen
Schleife,wodurchdiesogenannteOxyanion‐Höhlefreigelegtwird.EineweitereFolgedieserKonforma‐
tionsänderungenistdieerheblicheVergrößerungderhydrophobenOberflächedesEnzyms,wasinder
Lipid‐Oberflächen‐ErkennungeineRollespielt.[187]
DasaktiveZentrumbestehtausdreiResten:einemSerin‐undeinemCarboxylat‐Rest,diebeidewasser‐
stoffverbrücktzueinemHistidin‐Restsind(Abbildung53).BeidemCarboxylat‐Restkannessichum

4 EnantioselektiveRacematspaltungvonAminen
55
einen Aspartat‐ oder Glutamat‐Rest handeln. Während der Hydrolyse einer Ester‐Bindung findet
zunächsteinnucleophilerAngriffdesSauerstoffsdesSerin‐RestesaufdasCarbonyl‐Kohlenstoffatom
desEstersstatt,waszuderBildungdestetraedrischenIntermediatesführt(Schritt1).Dieseswirddurch
Wasserstoffbrückenbindungen zu Resten in der Oxyanion‐Höhle stabilisiert. Der Imidazol‐Ring des
Histidinswirdprotoniertunddadurchpositiv geladenunderhöht somitdieNucleophiliederSerin‐
Hydroxygruppe.DiesepositiveLadungwirddurchdienegativeLadungdesSäure‐Restes stabilisiert
(Schritt 2). Das tetraedrische Intermediat wird durch zweiWasserstoffbrückenbindungen zu Amin‐
FunktionenvonResten,die sich inderOxyanion‐Höhlebefinden, stabilisiert.Abschließendwirdder
Alkohol freigesetzt,welcherdanndenAcyl‐Enzym‐Komplexverlässt (Schritt3).Durcheinennucleo‐
philenAngriffeinesHydroxy‐IonswirddieFettsäurefreigesetztunddasEnzymregeneriert(Schritt4).
Abbildung53. DetaillierterMechanismusderHydrolyseeinerEster‐BindungdurcheineLipase.[187]Wasserstoffbrückenbindungenwerdengestricheltdargestellt.
LipasenbesitzendieFähigkeit,anderWasser/Öl‐Grenzflächezuagieren;diesesunterscheidetsievon
denEsterasen.[186]DasKonzeptderPhasengrenzen‐Aktivierung(‘interfacialactivation‘)ergibtsichaus
derAbhängigkeitderkatalytischenAktivitätderLipasenvondemAggregatzustanddesSubstrates,da
lipolytischeSubstrateineinemGleichgewichtzwischenMonomeren,MicellenundemulgiertemZustand
vorliegenkönnen.DieAktivitätderLipasenwirddurchunlöslicheSubstrate,dieeineEmulsionbilden,
erhöht.[193] Diese Erhöhung der Aktivierung wird ausgelöst durch strukturelle Veränderungen am
aktiven Zentrum.Während der Abwesenheit einerWasser/Öl‐Grenzflächewird das aktive Zentrum

4 EnantioselektiveRacematspaltungvonAminen
56
durcheinenDeckel„geschlossen“.InAnwesenheitvonhydrophobenSubstanzenjedochöffnensichder
DeckelunddasaktiveZentrumundeswirdeinegroßehydrophobeOberflächeexponiert.
AusderHefeCandidaantarcticakonntenzweiLipasen,AundBgenannt,isoliertwerden.[194,195]Wievom
Namenersichtlichist,wurdederStammursprünglichinderAntarktismitdemZielisoliert,Enzymemit
extremen Eigenschaften zu finden.[195] Beide Lipasen unterscheiden sich stark. Die Lipase A ist ein
unspezifisches,thermostabilesundcalciumabhängigesEnzym.DieindieserArbeitverwendeteLipaseB
(CAL‐B)istetwaswenigerthermostabilundnichtcalciumabhängig.ObwohlderoptimalepH‐Bereich
beipH7liegt, istes imwässrigenMediumimBereichvonpH3.5–9 nochstabil.DieCAL‐Bhatein
Molekulargewicht von 33.3kDa und besteht aus 317 Aminosäuren (Abbildung 54). Die Consensus‐
sequenz (Gly‐x‐Ser‐x‐Gly) ist in der CAL‐B nicht vorhanden, da das erste Glycin durch Threonin
ausgetauschtwurde (Thr‐x‐Ser‐x‐Gly). Dadurch ergibt sich eine leicht veränderte Konformation des
Faltblattesundderα–HeliximVergleichzuanderenLipasen.AuchindieserLipaseisteinekurzeα‐Helix
desEnzymsdafürzuständig,alsDeckelzurKontrolledesaktivenZentrumszufungieren.Andersalsdie
meistenLipasenzeigtdieCAL‐BkeineGrenzflächen‐Aktivierung.DieCAL‐Bistnichtsoeffektivbeider
HydrolysevonTriacylglycolenwieandereLipasen,reagiertjedochsehrstereospezifischbeiHydrolysen
oderchemischenSynthesen.[98]
Abbildung54. KristallstruktureinerCAL‐B‐Mutante(links).DiestrukturellenWassermolekülesindalsrotePunktedargestellt(rechts).[11]
DieCAL‐B(Novozym435)istaufeinemmacroporösenPolymer‐Trägerimmobilisiert,deraufMethyl‐
undButylmethacrylesterbasiertunddurchDivinylbenzolquervernetztist.[36]DasImmobilisatbesitzt
einenProteingehaltvonetwa5%(w/w)deslyophilisiertenPulvers,wovonnurca.40%denGehaltder
Lipasedarstellen.[196]

4 EnantioselektiveRacematspaltungvonAminen
57
4.1.3 EnantioselektivitätvonLipasen
ObwohldiegrobeStrukturunddiekatalytischeTriadederLipasensehrähnlichsindgibtesunterihnen
einengroßenUnterschiedinderSubstratspezifitätundderenAusmaß.[197]EineallgemeineRegelfürdie
Vorhersage der Enantioselektivität wurde von KAZLAUSKAS aufgestellt.[198] Die Regel basiert auf der
GrößederSubstituentenamStereozentrum.MittelsKristallstrukturdesEnzymkomplexesderLipase
ausCandida rugosa wurdemit dem schnell reagierenden (R)‐Ester eines sekundären Alkohols eine
WasserstoffbrückenbindungvomkatalytischenHistidin‐RestzumSauerstoffatomdesEstersgefunden.
DieseWasserstoffbrückenbindungfehltimKomplexdesEnzymszumlangsamreagierenden(S)‐Enan‐
tiomer.Generell gilt zudem,dassdieStereoselektivitätbeiunterschiedlichgroßenSubstituentenam
chiralenKohlenstoffatomhöheristalsbeiSubstituentengleicherGröße.[11,199]
Abbildung55. Schnellreagierendes(a)undlangsamreagierendes(b)EnantiomerindemModelldesaktivenZentrums,abgeleitetvonderKAZLAUSKAS‐Regel.[199]
TriacylglyceridekönnenanallendreiEsterfunktionenoderspezifischnuraneineroderzweiPositionen
gespaltenwerden.LipasenzeigenauchverschiedeneSelektivitätenabhängigvonderKettenlängeder
Ester.[11,199]

4 EnantioselektiveRacematspaltungvonAminen
58
4.2 StandderWissenschaft:Aminsynthesen
Für die pharmazeutische, agrochemische und feinchemische Industrie ist die Synthese von enantio‐
merenreinen Aminen einwichtiger Bereich der organischen Chemie. Schätzungen zufolge enthalten
40‐50% aller optisch aktiver Wirkstoffe chirale Amine.[18] Zu den klassischen Synthesewegen von
chiralenAminengehörendieasymmetrischeHydrierung[200,201], Aminierung[18],Alkylierung[202]oder
Hydrosilylierung[203]vonIminen,oderdieHydrierungvonEnamiden.[204,205]Außerdemfindenbiokata‐
lytischeAnsätzewiederEinsatzvonTransaminasen[206,207]oderdiekinetischeRacematspaltungvon
racemischen Ausgangsverbindungen[31,208] zunehmend Anwendung. Einige Beispiele sind in den
folgendenAbschnittennäherbeschrieben.
4.2.1 KlassischchemischeundchemokatalytischeSynthesen
Die katalytische asymmetrische Addition von Organometall‐Reagenzien an Imine ist ein wichtiger
Syntheseweg zu optisch aktiven Aminen.[202] Die asymmetrische Addition an C=N‐Bindungen wird
generelldurchVerwendungvonchiralenAuxiliarenoderchiralenLigandenerreicht.Diesesbringtden
Vorteil einer einfachen Trennung der diastereomeren Produkte vor der Abspaltung des chiralen
Auxiliars mit sich. In den letzten Jahrzehnten fand die Addition von Organometall‐Reagenzien in
AnwesenheiteineschiralenLigandenvermehrtAnwendung.DieersteSynthesedieserArtberichtete
TOMIOKA.[202,209]DabeiwirddasgeschützteAmin63mitdemchiralenAminoether‐Liganden(S)‐64um‐
gesetzt(Abbildung56).
Abbildung56. AsymmetrischeAlkylierungdesgeschütztenImins63.[202,209]
Das entstehende Amin (R)‐65 kann mit einen nahezu vollständigen Umsatz von 98% und guten
ee‐Werten von75%erhaltenwerden,wobei derEinsatz von2.6Äquivalenten an (S)‐64 notwendig
waren.Selbstmit 5mol‐%desLiganden(S)‐64wirdimmernochenantiomerenangereichertesAmin
(R)‐65 mit 40%ee beobachtet. Der ee‐Wert kann bei ähnlichen Aminen durch Verwendung des
Bidentat‐LigandenBisoxazalinauf91%gesteigertwerden.[210]AuchDialkylzink‐Verbindungenkönnen
in Anwesenheit von chiralen Liganden zu sehr guten Enantiomerenüberschüssen führen.[202,211] In
neuerer Zeit zeigten auchAllylstannaneundAllylsilane guteErgebnisse.[212] Aufgrundder extremen

4 EnantioselektiveRacematspaltungvonAminen
59
Reaktionsbedingungen (‐100°C) sind diese Organometall‐katalysierten Reaktionen aus industrieller
Sichtschwierigzurealisieren.
EinederinteressantestenReaktionenderorganischenChemieistlautdemGreenChemistryInstituteof
AmericanChemicalSocietydieasymmetrischereduktiveAminierungvonprochiralenCarbonylverbin‐
dungen.[18]DabeigibteszweiMöglichkeitenderAminsynthese:diedirekteunddieindirektereduktive
Aminierung(Abbildung57).BeiderindirektenAminierungwirddieCarbonylverbindung66miteinem
AminzueinemImin66oderEnaminalsZwischenproduktumgesetzt,welchesisoliertundanschließend
enantioselektivweiterzumchiralenAmin68reduziertwird.EineLimitierungbestehtallerdingsinder
StabilitätdeszuisolierendenZwischenproduktes67.DiedirekteAminierungumgehtdieIsolationdes
Zwischenproduktes,indemdieCarbonyl‐Komponente66direktzumchiralenAmin68reduziertwird.
AlshomogeneKatalysatorendienenPt,Pd,NioderRhmitchiralenP‐Liganden.Veränderungeninder
StrukturderDiphosphin‐LigandensowiedersterischenundelektronischenFaktorenmachensichin
drastischen Abweichungen in Reaktivität und Stereokontrolle bemerkbar. Die Aminierung mit Am‐
moniakalsStickstoff‐DonoristimmernocheinungelöstesProblem.AufdiebiokatalytischeAminierung
wirdinKapitel4.2.2eingegangen.
Abbildung57. IndirekteunddirektereduktiveAminierungvonCarbonylverbindungen66.[18]
Die direkte asymmetrische Aminierung findet in der industriellen Herstellung von (S)‐Metolachlor
((S)‐69)derFirmaSYNGENTA(ehemalsNOVARTIS)Anwendung(Abbildung58).[213,214](S)‐69istderaktive
BestandteilinDUAL®,einemderwichtigstenHerbizide.DurchdiechiraleAchse(Atropisomerie)sowie
das stereogeneZentrumergeben sich für (S)‐69 vierStereoisomere.DieseMischungwird seit 1976
durch Pt‐Katalyse in Anwesenheit von Schwefelsäure mit >20000 t/a produziert. Nachdem 1982
erkanntwurde,dass95%derAktivitätvondenbeiden(1‘S)‐Diastereomerenausgeht,konnte1997eine
enantiomerenangereicherte Form (Handelsname DUAL MAGNUM®) der beiden (1‘S)‐Diastereomere
(90% ee)mit >10000 t/a in den Handel gebracht werden. Dieses führte zu einer Reduzierung der
Umweltbelastungumetwa40%imVergleichzurracemischenMischung.FürdieSynthesedes(S)‐69
wird das sterisch gehinderte Anilin70mitMethoxyaceton (71) umgesetzt. Der Schlüsselschritt der
SynthesevonMetolachlor((S)‐69)istdieIridium‐katalysierteenantioselektiveHydrierungdesImins
72.DeroptimierteProzessverläuftbei50°Cund80barWasserstoff‐Druck.DerKatalysatorwirdinsitu
aus [Ir(COD)Cl]2 unddemFerrocenyl‐Diphosphin‐LigandenXyliPhos (73) generiert. InAnwesenheit
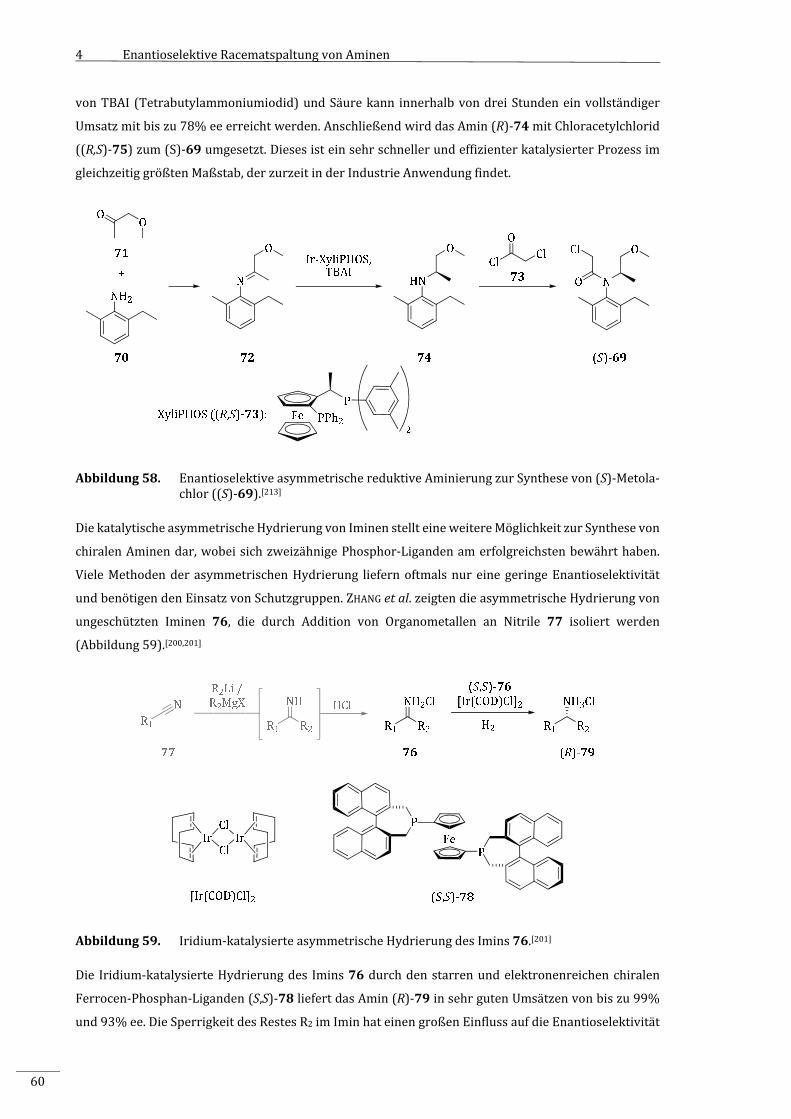
4 EnantioselektiveRacematspaltungvonAminen
60
vonTBAI(Tetrabutylammoniumiodid)undSäurekann innerhalbvondreiStundeneinvollständiger
Umsatzmitbiszu78%eeerreichtwerden.AnschließendwirddasAmin(R)‐74mitChloracetylchlorid
((R,S)‐75)zum(S)‐69umgesetzt.DiesesisteinsehrschnellerundeffizienterkatalysierterProzessim
gleichzeitiggrößtenMaßstab,derzurzeitinderIndustrieAnwendungfindet.
Abbildung58. EnantioselektiveasymmetrischereduktiveAminierungzurSynthesevon(S)‐Metola‐chlor((S)‐69).[213]
DiekatalytischeasymmetrischeHydrierungvonIminenstellteineweitereMöglichkeitzurSynthesevon
chiralenAminendar,wobeisichzweizähnigePhosphor‐Ligandenamerfolgreichstenbewährthaben.
VieleMethodenderasymmetrischenHydrierung liefernoftmalsnureinegeringeEnantioselektivität
undbenötigendenEinsatzvonSchutzgruppen.ZHANGetal.zeigtendieasymmetrischeHydrierungvon
ungeschützten Iminen 76, die durch Addition von Organometallen an Nitrile 77 isoliert werden
(Abbildung59).[200,201]
Abbildung59. Iridium‐katalysierteasymmetrischeHydrierungdesImins76.[201]
Die Iridium‐katalysierteHydrierungdes Imins76durchdenstarrenundelektronenreichenchiralen
Ferrocen‐Phosphan‐Liganden(S,S)‐78liefertdasAmin(R)‐79insehrgutenUmsätzenvonbiszu99%
und93%ee.DieSperrigkeitdesRestesR2imIminhateinengroßenEinflussaufdieEnantioselektivität

4 EnantioselektiveRacematspaltungvonAminen
61
derHydrierung.Miteinertert‐Butyl‐GruppeanstelleeinerMethyl‐Gruppesinktderee‐Wertvon99%
auf80%.TrotzdersehrgutenErgebnisseergibtsicheinNachteilinderVerwendungvonKetoiminen
76 als Substrate, dadieseoftmals nur inMischungen vonE/Z‐IsomerenundKeto‐Enol‐Tautomeren
erhaltenwerdenkönnen.
DiereduktiveAminierungvonCarbonylverbindungenzuAminendurchAmeisensäureunterHydrid‐
Übertragung ist als LEUCKART‐WALLACH‐Reaktion bekannt.[215] KADYROV et al. beschrieben 2003 die
asymmetrische reduktive Aminierung von Acetophenon (46) mittels Ruthenium‐Katalyse (80) und
AmmoniumformatalsHydrid‐Donor(Abbildung60).[216]SäurenbeschleunigendieReduktion, jedoch
habensieeinennegativenEinflussaufdieasymmetrischeInduktion.DerEinsatzvonAmmoniakführt
zueinerErhöhungderSelektivität,aberauchzueinerVerringerungderReaktivität.Bei5 ‐10Äqui‐
valentenHCOONH4inNH3/Methanol(15–25%)undTemperaturenvon60–85°Cwurdendiebesten
Enantioselektivitätenerhalten.DasProduktwurdealsfreiesAmin(rac‐4)undinN‐formulierterForm
(81)alsHauptproduktdieserReduktionerhalten.NachHydrolysevon81wirddasAmin(rac‐4)mit
hoher Enantioselektivität (86 – 98% ee) erzielt,wenn ein aromatischesKeton als Edukt verwendet
worden ist. Mit aliphatischen Ketonen konnten unter den gegebenen Bedingungen nur Spuren des
entsprechendenAminsbeobachtetwerden.
Abbildung60. Asymmetrische Aminierung von Acetophenon (46) mittels Ruthenium‐Katalyse(80).[216]
EbenfallsinteressantistdiedreistufigeSyntheseoptischaktiverAmidevonBURKetal.durchenantio‐
selektivekatalytischeHydrierungvonEnamidenmitRhI/DuPhos (S,S)‐82 (Abbildung61).[204,205]Die
EnamidewiederumsinddurchReduktionderOximemitEisenzugänglich.AusgehendvomPinakolon‐
Oxim83wirddasnahezuenantiomerenreineAmid(R)‐85überdenZwischenschrittdesEnamins84
erhalten,wobeinur0.1mol‐%desKatalysatorsbenötigtwird.DasAmid(R)‐85kannanschließendzum
primärenAmin(R)‐86hydrolysiertwerden.NachteilediesesProzessessinddieaufwändigeSynthese
der Enamin‐Vorstufe83, da diese durch Addition von Alkyl‐Grignard an Nitrile hergestelltwerden,
sowiediegeringeAusbeutevon20‐40%desEnamids84.
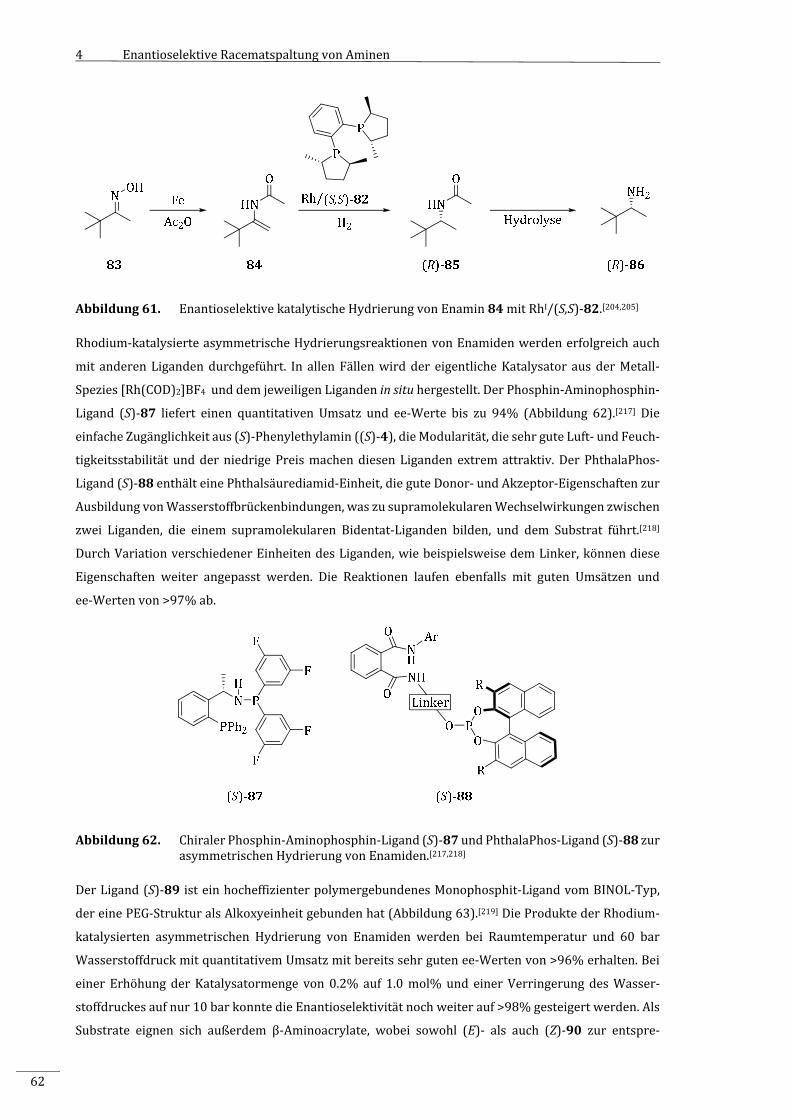
4 EnantioselektiveRacematspaltungvonAminen
62
Abbildung61. EnantioselektivekatalytischeHydrierungvonEnamin84mitRhI/(S,S)‐82.[204,205]
Rhodium‐katalysierteasymmetrischeHydrierungsreaktionenvonEnamidenwerdenerfolgreichauch
mit anderenLigandendurchgeführt. In allen Fällenwirdder eigentlicheKatalysator ausderMetall‐
Spezies[Rh(COD)2]BF4unddemjeweiligenLigandeninsituhergestellt.DerPhosphin‐Aminophosphin‐
Ligand (S)‐87 liefert einen quantitativen Umsatz und ee‐Werte bis zu 94% (Abbildung 62).[217] Die
einfacheZugänglichkeitaus(S)‐Phenylethylamin((S)‐4),dieModularität,diesehrguteLuft‐undFeuch‐
tigkeitsstabilitätundderniedrigePreismachendiesenLigandenextremattraktiv.DerPhthalaPhos‐
Ligand(S)‐88enthälteinePhthalsäurediamid‐Einheit,dieguteDonor‐undAkzeptor‐Eigenschaftenzur
AusbildungvonWasserstoffbrückenbindungen,waszusupramolekularenWechselwirkungenzwischen
zwei Liganden, die einem supramolekularen Bidentat‐Liganden bilden, und dem Substrat führt.[218]
DurchVariationverschiedenerEinheitendesLiganden,wiebeispielsweisedemLinker,könnendiese
Eigenschaften weiter angepasst werden. Die Reaktionen laufen ebenfalls mit guten Umsätzen und
ee‐Wertenvon>97%ab.
Abbildung62. ChiralerPhosphin‐Aminophosphin‐Ligand(S)‐87undPhthalaPhos‐Ligand(S)‐88zurasymmetrischenHydrierungvonEnamiden.[217,218]
DerLigand(S)‐89 isteinhocheffizienterpolymergebundenesMonophosphit‐LigandvomBINOL‐Typ,
dereinePEG‐StrukturalsAlkoxyeinheitgebundenhat(Abbildung63).[219]DieProduktederRhodium‐
katalysierten asymmetrischen Hydrierung von Enamiden werden bei Raumtemperatur und 60 bar
WasserstoffdruckmitquantitativemUmsatzmitbereitssehrgutenee‐Wertenvon>96%erhalten.Bei
einer ErhöhungderKatalysatormenge von 0.2%auf 1.0mol%und einerVerringerungdesWasser‐
stoffdruckesaufnur10barkonntedieEnantioselektivitätnochweiterauf>98%gesteigertwerden.Als
Substrate eignen sich außerdem β‐Aminoacrylate, wobei sowohl (E)‐ als auch (Z)‐90 zur entspre‐

4 EnantioselektiveRacematspaltungvonAminen
63
chendenAminosäure91umgesetztwerdenkönnen.Hierbeiweist(E)‐90einehöhereAktivitätalsdas
(Z)‐Isomer(94%bzw.73%Umsatz)auf, jedochbleibendieEnantiomerenüberschüssemit96%bzw.
97%bei einerKatalysatorbeladungvon0.1mol%etwagleich. SperrigereSubstituentenwiedie iso‐
Propyl‐GruppeführenzuoptimiertenErgebnissen,allerdingswirddabeieineUmkehrderSelektivität
von(S)zu(R)beobachtet.WeitereVorteilevon(S)‐89sinddieleichteZugänglichkeit,Luftstabilität,ein‐
facheAbtrennungsowiedieRecyclisierbarkeitdesKatalysator‐Systems.
Abbildung63. AsymmetrischeRh‐katalysierteHydrierungvonEnamiden90(links)mitdemMono‐phosphit‐Liganden(S)‐89(rechts).[219]
DieMöglichkeiten zur chemischenasymmetrischenSynthese chiralerAmine sind sehr vielfältigund
liefern indenmeistenFällensehrguteUmsätzesowiehoheEnantiomerenreinheiten.DieSynthesen
verlaufenjedochoftmehrstufigunderfordernOrganometall‐KatalysatorenundLiganden,diekosten‐
intensivsindundaufwendighergestelltwerdenmüssen.AuchkönnendieReaktionsbedingungennicht
direktaufandereSubstrateübertragenwerden,sodassjederSchrittindividuelloptimiertwerdenmuss.

4 EnantioselektiveRacematspaltungvonAminen
64
4.2.2 EnzymkatalysierteSynthesen
ZurbiokatalytischenSynthesevon chiralenAminengibt esunterschiedlicheAnsätzeausgehendvon
prochiralen Carbonylverbindungen oder von racemischen Aminen bzw. Amiden wie die Transami‐
nierung, die Deracemerisierung und die kinetische sowie dynamisch kinetische Racematspaltung
(Abbildung64).
Abbildung64. MethodenzurbiokatalytischenSynthesevonchiralenAminen.[220]
SeitBRAUNSTEIN1939erstmalsvondemTransfereinerAminofunktionzwischeneinerDonor‐Amino‐
säure und einer α–Ketosäure als Akzeptor durch lebendes Gewebe tierischenUrsprungs berichtete,
zähltdieEnzymklassederTransaminasezudenambestenuntersuchtePyridoxalphosphat‐abhängigen
Enzymen.[221]EinvonCELGENEentwickeltesindustriellesVerfahrenzurbiokatalytischenSynthesevon
chiralen Aminen nutzt die Transaminierung. Dabei katalysieren Transaminasen die asymmetrische
ÜbertragungeinerAminofunktionaufeineCarbonylverbindungwieKeton46odereineα‐Ketosäure,
wobei Pyridoxalphosphat 92 als Cofaktor dient, in dem die Aminofunktion sowie das Reduktions‐
äquivalentgespeichertwerden(Abbildung65,A).[207,222]Dasowohl(S)‐alsauch(R)‐selektiveTrans‐
aminasenentwickeltwurden,sindbeideEnantiomerederAminezugänglich.[208]EinprochiralesKeton
46 übernimmt dabei vom Amin‐Donor (S)‐93 eine Aminofunktion. Enantiomerenreines (S)‐4 und
AcetonentstehenmiteinerAusbeutevon>90%.DesWeiterenkanndieReaktionauchimSinneeiner
Racematspaltung geführt werden (Abbildung 65, B). Dabei wird das (S)‐4 in das entsprechende
Acetophenon(46)umgewandelt.AlsAminogruppenakzeptorwerdenAldehydewie94verwendet,aus
demdasAmin95entsteht.DasAmin(R)‐4wirdenantiomerenreinmitmaximal50%Umsatzerhalten.
AllerdingserweistsichdieTrennungderProduktmischungenausderRacematspaltungalsaufwendig.
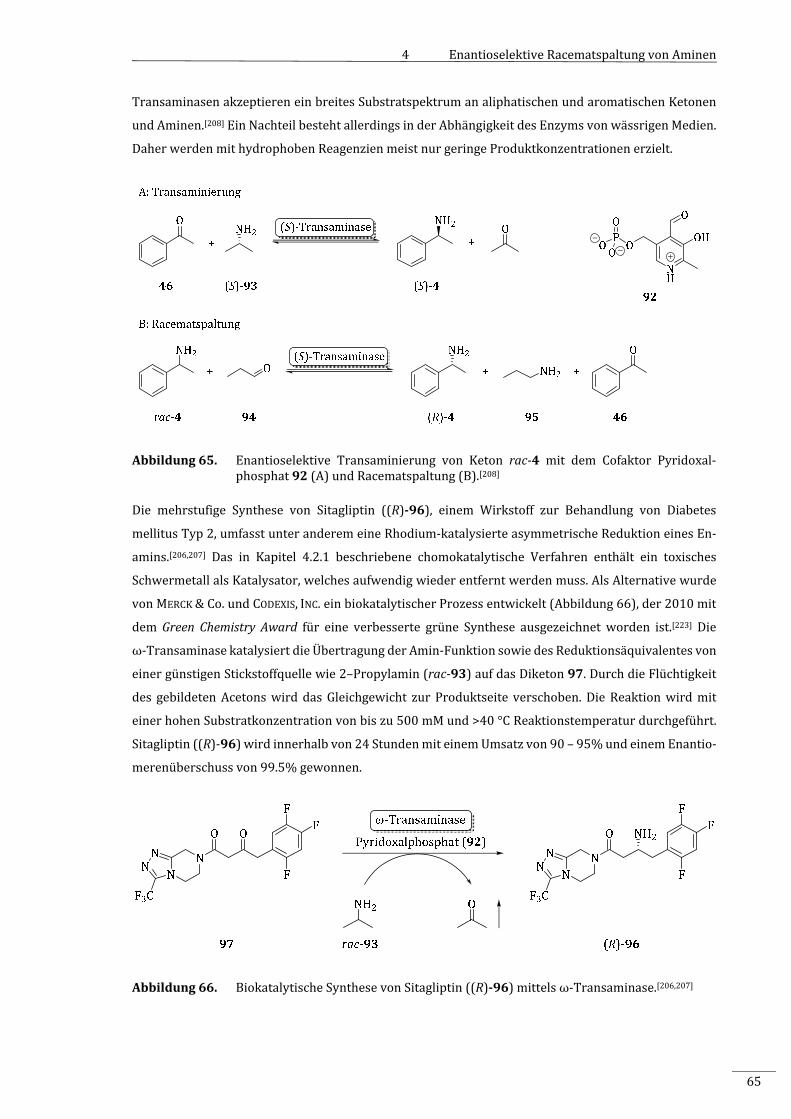
4 EnantioselektiveRacematspaltungvonAminen
65
TransaminasenakzeptiereneinbreitesSubstratspektrumanaliphatischenundaromatischenKetonen
undAminen.[208]EinNachteilbestehtallerdingsinderAbhängigkeitdesEnzymsvonwässrigenMedien.
DaherwerdenmithydrophobenReagenzienmeistnurgeringeProduktkonzentrationenerzielt.
Abbildung65. Enantioselektive Transaminierung von Keton rac‐4mit dem Cofaktor Pyridoxal‐phosphat92(A)undRacematspaltung(B).[208]
Die mehrstufige Synthese von Sitagliptin ((R)‐96), einem Wirkstoff zur Behandlung von Diabetes
mellitusTyp2,umfasstunteranderemeineRhodium‐katalysierteasymmetrischeReduktioneinesEn‐
amins.[206,207] Das in Kapitel 4.2.1 beschriebene chomokatalytische Verfahren enthält ein toxisches
SchwermetallalsKatalysator,welchesaufwendigwiederentferntwerdenmuss.AlsAlternativewurde
vonMERCK&Co.undCODEXIS,INC.einbiokatalytischerProzessentwickelt(Abbildung66),der2010mit
demGreen ChemistryAward für eine verbesserte grüne Synthese ausgezeichnetworden ist.[223] Die
ω‐TransaminasekatalysiertdieÜbertragungderAmin‐FunktionsowiedesReduktionsäquivalentesvon
einergünstigenStickstoffquellewie2–Propylamin(rac‐93)aufdasDiketon97.DurchdieFlüchtigkeit
des gebildetenAcetonswirddasGleichgewicht zurProduktseite verschoben.DieReaktionwirdmit
einerhohenSubstratkonzentrationvonbiszu500mMund>40°CReaktionstemperaturdurchgeführt.
Sitagliptin((R)‐96)wirdinnerhalbvon24StundenmiteinemUmsatzvon90–95%undeinemEnantio‐
merenüberschussvon99.5%gewonnen.
Abbildung66. BiokatalytischeSynthesevonSitagliptin((R)‐96)mittelsω‐Transaminase.[206,207]

4 EnantioselektiveRacematspaltungvonAminen
66
Die Deracemerisierung von racemischen Aminen, wie α–Aminosäuren, wurde kürzlich durch die
KupplungeinerenantioselektivenOxidationmittelseinerD‐spezifischenAminoxidaseundeinerche‐
mischen,nicht‐selektivenReduktiondurchgeführt(Abbildung67).[224]DieserProzessführtsomitzur
Bildung von L‐98 ausgehend vom prochiralen Imin99. Da chirale Amine jedoch nicht von Amino‐
säureoxidasenoxidiertwerdenkönnen,wurdeninneuererZeiteineAminoxidaseausAspergillusniger
als aktiverBiokatalysator identifiziert.[225] Somitkannnuneinbreites Substratspektruman chiralen
Aminenmit guterEnantioselektivitätumgesetztwerden. InallenbekanntenFällen istdiesesEnzym
(S)‐selektiv.AllerdingssindfürAnwendungeningroßemMaßstabnochOptimierungeninHinblickauf
Selektivität,AktivitätundStabilitätnotwendig.
Abbildung67. DeracemerisierungeinerD‐α‐AminosäureD‐98mittelseinerAminoxidase.[224]
ImgroßenMaßstabwerdenLipasenzurbiokatalytischenSynthesevonchiralenAminenherangezogen.
Nach einer Empfehlung von IUPAC ist eine kinetische Racematspaltung wie folgt definiert: „Das
ErreichenvoneinerteilweisenodervollständigenTrennungaufgrundvonunterschiedlichenReaktions‐
geschwindigkeitenderEnantiomereeinesRacematesmiteinemchiralenAgens(Reagenz,Katalysator,
Lösungsmittel, etc).“[226] Im einfachsten Fall einer Racematspaltung regieren die Enantiomere des
SubstratsmiteinemchiralenAgensoderKatalysatorüberzweidiastereomereÜbergangszustände.Die
EnergiendieserÜbergangszuständebestimmendieGeschwindigkeitskonstantenfürdieUmsetzungdes
langsamunddesschnellerreagierendenEnantiomersunddamitdieProduktverteilung.[37]Zwarwurden
vermehrtchemischeKatalysatorenzurRacematspaltunggetestet,allerdingskonntendieErgebnisse,die
mitvielenBiokatalysatorenerzieltwurden,nichtübertroffenwerden.[208]
Anfangder90erJahrenwurdenbeiSHELLersteArbeitenzurenantioselektivenHydrolyseracemischer
AmidemittelsArthrobactersp.durchgeführt,wobeisichdieVerwendungvonganzenZellensowiedie
sehrlangenReaktionszeitenalsnachteiligerwiesen.[227]Mitteder90erJahrewurdenvonderBAYERAG
MethodenzurselektivenHydrolysevonracemischenAcetamidenwie100mitLipaseBausCandida
antarctica (CAL‐B) als Biokatalysator ausgearbeitet (Abbildung 68).[228] Die CAL‐B hat sich, in Form
eines Immobilisats auf einemAcrylamid‐Träger (Novozym435), in einerReihevonArbeiten alsdie
geeignetsteLipaseerwiesen.[27–29]AufgrundunbefriedigenderRaum‐Zeit‐Ausbeuten,43%Umsatznach
sieben Tagen Reaktionszeit, wurde dieser Prozess trotz sehr guter Enantiomerenüberschüssen von
>99.5%technischnichtangewendet.[208]

4 EnantioselektiveRacematspaltungvonAminen
67
Abbildung68. SelektiveHydrolysedesracemischenAcetamidsrac‐100nachBAYERAG.[228]
Nach einemvonBASFSEpatentiertenVerfahrender enantioselektivenAcylierung, der sogenannten
kinetischen Racematspaltung, von Amin rac‐4 wird in der ersten Stufe nahezu enantiomerenreines
Amin(S)‐4unddasentsprechendeAmid(R)‐102mit>99%eeerhalten.[208]EineBesonderheitstelltder
induktiveEffektderMethoxygruppedar,diedieReaktivitätanderCarbonylfunktionaktiviert.Durch
MolecularModellingeinesÜbergangszustandeskonnteeineWasserstoffbrückenbindungzwischendem
β‐Sauerstoffatom des Esters103 und der Aminofunktion von rac‐4 ermittelt werden.[229,230] Wahr‐
scheinlichstabilisierendieseWechselwirkungsowieinduktiveEffekteundsterischeGründedenÜber‐
gangszustandunderhöhendadurchdieReaktionsgeschwindigkeit.
NachAbtrennungvon(R)‐102durchDestillationoderExtraktionwirddasebenfallsenantiomerenreine
(R)‐4 durch basische Hydrolyse freigesetzt. Die Substratbreite ermöglicht die Racematspaltung von
vielenstrukturellsehrunterschiedlichenAminen.WeitereVorteilediesesProzessessinddieMöglich‐
keit zur Rückführung der unerwünschten Enantiomere durch Racemisierung[231–233] und die Rück‐
gewinnung des Acylierungsmittels[234]. Seit Anfang 2002 produziert BASF SEmit diesem Verfahren
optischaktiveAminemit>3500t/aundistsomitderweltweitführendeAnbieterchiralerAmine.[208]
Abbildung69. Enantioselektive Amidbildung ausgehend von rac‐4 nach BASF SE.[208] BasischeHydrolysevon(R)‐102ergibt(R)‐4.
Bei der Umsetzung von cyclischen Aminoalkoholen[235] und Diaminen (wie (±)‐104)[236,237] wird
ebenfalls CAL‐B als Biokatalysator verwendet. Im Fall des Diamins (±)‐104 können mit Dimethyl‐
Malonester (105) als Acyldonor zwei sequenzielle Acylierungsreaktionen durchgeführt werden
(Abbildung70).[236,237]DabeiwirdzunächstdurchMonoacylierungdasoptischangereicherteMonoamid

4 EnantioselektiveRacematspaltungvonAminen
68
(R,R)‐106mit 66%Enantiomerenüberschuss unddas nicht umgesetzteAmin (S,S)‐104mit 83%ee
erhalten.DerzweiteAcylierungsschrittistenantioselektiv,wodurchdasDiamid(R,R)‐107mit>99%ee
erzieltwerden.DieCAL‐BzeigtfolglichinbeidenSchrittendiegleichePräferenzinBezugaufdas(R,R)‐
Enantiomer.DiesesErgebnisstehtinEinklangmitderRegelvonKAZLAUSKAS,dadasmonoacylierteAmid
(R,R)‐106 einengrößerensterischenUnterschied indenSubstituentenaufweistalsdas freieDiamin
(±)‐104.[28]
Abbildung70. Lipasen‐katalysierte doppelte Acylierung des Diamins (±)‐104 mit Malonsäure‐dimethylester(105)alsAcyldonor.[236,237]
Die Optimierung der katalytischen Aktivität und Selektivität der CAL‐B kann durch Variation des
LösungsmittelssowiedesAcyldonorserreichtwerden.DaAminesehrguteNucleophilesind,können
hochreaktiveAcyldonorenbereitsdurchspontanenicht‐enzymatischenucleophileSubstitutionzurace‐
mischenAmidenführen.[31]GILetal.zeigtendieAkzeptanzeinesbreitenSubstratspektrumsderCAL‐B
amBeispielderAcylierungvonrac‐108(Abbildung71).[31]Sowohlkurz‐alsauchlangkettigeCarbon‐
säurenundEster,wieLaurinsäure(109)unddessenEster110,wurdendabeialsAcyldonorengetestet,
wobeidieEnantioselektivitätsowiedieReaktionsgeschwindigkeitmitsteigenderKettenlängezunimmt.
AllerdingskanndieserEinflussnichtalsallgemeinerTrendfürLipasenangesehenwerden.Währendmit
simplenEstern,dasbekanntestedavon istEssigester5,nurbiszu91%eeerhaltenwerdenkönnen,
liefertMethoxyessigsäureethylester(111)innur1.5Stunden>99.5%ee,dasAmid(R)‐8allerdingsnur
mit77.4%ee.

4 EnantioselektiveRacematspaltungvonAminen
69
Abbildung71. EnzymatischeAcylierungvonrac‐108mitunterschiedlichenAcyldonoren.[31]
Ein großerNachteil der kinetischenRacematspaltung ist die LimitierungdesUmsatzes aufmaximal
50%.AusdiesemGrund istdieRacemisierungeinwichtigesHilfsmittel,das inKombinationmitder
biokatalytischenkinetischenRacematspaltungzueinemUmsatzvontheoretisch100%undeinemeffi‐
zienterenGebrauchdes gesamtenRacemates führenkann. EinmöglicherMechanismusderRacemi‐
sierungbestehtauseinerKombinationvonDehydrierungdeschiralenAminsundeinerRe‐Additionvon
WasserstoffandasImin.[231]DesWeiterenistdiebasenkatalysierteRacemisierungdesresultierenden
chiralenAminsmöglich.[238]EinSchlüsselproblemderdynamisch‐kinetischenRacematspaltung(DKR)
istes,Bedingungenzufinden,unterdenenderBio‐undderChemokatalysatoreffizientarbeitenkönnen.
WenndaserwünschteEnantiomerdas(R)‐Produktist,dannistbeidenReaktionsgeschwindigkeitender
einzelnenSchrittefolgendeReihenfolgevorausgesetzt:krac>kR>kS(Abbildung72).[239]
Abbildung72. Schemaderdynamisch‐kinetischenRacematspaltung(DKR).[239]
REETZundSCHIMOSSEKbeschrieben1996dieerstedynamisch‐kinetischeRacematspaltungmitPalladium
aufAktivkohlealsKatalysator,wobeinachfünfTageReaktionszeiteinUmsatzvon64%zumAmidund
ein Enantiomerenüberschuss von 99% erreicht werden konnte.[240] Weitere Ansätze verwendeten
Pd‐Partikel immobilisiert auf Trägern wie BaSO4[241] oder einen Palladium‐Nanokatalysator einge‐
schlosseninAluminiumhydroxid.[242]BÄCKVALLetal.kombiniertendieCAL‐BschließlichineinerZwei‐
stufen‐SynthesemitdemRuthenium‐Katalysator116,derinähnlicherFormbereitserfolgreichbeider
RacemisierungvonAlkoholenVerwendungfindet(Abbildung73).[231]AusgehendvondenAminenrac‐4
und rac‐117werdendieAmide (R)‐7 und (R)‐118nahezuenantiomerenreinmit>98% eeundmit
Umsätzenvon69%bzw.66%erhalten.DieAnwesenheitvonelektronenschiebendenGruppenerhöht

4 EnantioselektiveRacematspaltungvonAminen
70
dieGeschwindigkeitderRacemisierung.AllerdingserfordertdieseMethodeeineextraktiveTrennung
des(S)‐AminsvomProduktfürdieanschließendeRacemisierung.IndenletztenJahrenwurdenweitere
Katalysatorenentwickelt,beispielsweisedurchModifizierungenvon116[232,233]oderdurchEinsatzvon
MetallenwieIridium[239]oderdemThiyl‐Radikal[243],diehöhereReaktionsgeschwindigkeitenundSelek‐
tivitätenaufweisen.
Abbildung73. DKRausgehendvonrac‐4undrac‐117mitdemRuthenium‐Katalysator116.[231]
InneuererZeitgibtesauchVersuche,dieRacemisiserungbiokatalytischdurchEinsatzvonRacemasen
anstellevonChemokatalysatorendurchzuführen.InderNaturgibteswegenderMehrzahlanstereo‐
spezifischenProzessen nur eine kleineAnzahl anRacemasen.Die ambesten untersuchteMandelat‐
RacemaseausPseudomonasputidaATCC12633istaufgrundderStabilität,demrelativbreitenSubstrat‐
spektrumsowiederCofaktor‐UnabhängigkeiteinattraktiverBiokatalysatorfürRacemisierungenvon
Hydroxy‐Carbonsäuren.[244] Die geringe Aktivität gegenüber nicht‐natürlichen Substraten konnte in
diesemFallbereitsdurchMutationenoptimiertwerden.DieAnwendungdermeistenRacemasen ist
jedochaufAminosäurenundderenDerivatebeschränkt.[238]Dafür jedeSubstratklassediegeeignete
Racemase gefunden und verbessert werden muss steckt die generelle Anwendung von Racemasen
jedochnochindenAnfängen.[11]
TrotzseinesVorteilsalsnicht‐toxischesundnatürlichesLösungsmittel istWasser fürvieleunpolare
organische Substrate ein schlechtes Lösungsmittel. Oft finden Nebenreaktionen statt und die Rück‐
gewinnungderProduktegestaltetsichschwierig.SogehörtdieimVergleichzuWassererhöhteLöslich‐
keitvonunpolarenSubstraten,sowiedieVermeidungderHydrolysedesgebildetenProdukteszuden
Vorteilen der Verwendung von Enzymen in organischen Lösungsmitteln.[245] Dennoch ist die kata‐
lytische Aktivität der Enzyme in organischen Lösungsmitteln häufig um einige Größenordnungen
geringeralsinwässrigenSystemen.DieseslässtsichteilweiseaufdieLimitierungderDiffusionsowie
ÄnderungenderFlexibilitätundDestabilisierungdesEnzymszurückführen.AuchbeiEnzymen,diein
organischenMedienaktivsind,isteinegeringeMengeanWassernotwendig.GenerellsindzweiArten
von Wassermolekülen im Enzym vorhanden. Die Wassermoleküle in der „inneren Klasse“ sind im
InnerendesEnzymsgebundenundspieleneinewichtigeRolleindessenTertiär‐Struktur.DieWasser‐
moleküleder„Kontakt‐Klasse“sindnurschwachanderOberflächedesEnzymsgebundenundkönnen
beigetrocknetenEnzymendreiGew.%odermehrausmachen.[246]Diesekönnenschnelldurchandere
Moleküleausgetauschtwerden,waseinegroßeFlexibilitätdesEnzymszurFolgehat.UnpolareLösungs‐
mitteltauschennureinenTeildieserWassermoleküleausundführensomitzuverringerterFlexibilität,
während polare Lösungsmittel stärker an der Oberfläche binden. Die Verwendung von organischen

4 EnantioselektiveRacematspaltungvonAminen
71
LösungsmittelnistausmehrerenGründennachteiligfürdenEinsatzinderIndustrie.[247]Siesindmeist
starkflüchtigundderenVerwendungbedarfkostenintensivenNachbehandlungenwieAbdampfenund
Recycling.DesWeiterenführtderEinsatzvonLösungsmitteldurchdiehoheVerdünnungzurVerringe‐
rungderRaum‐Zeit‐Ausbeute.DemnachwäreeinlösungsmittelfreierProzesssowohlausökologischer
undökonomischerPerspektivevongroßemVorteil.
Lösungsmittelfreie Systeme sind hauptsächlich bei der biokatalytischen Acylierung von Alkoholen
bekannt.[248,249]Fürdielipasenkatalysierte,lösungsmittelfreieRacematspaltungvonAminen,indenen
derAcyldonornichtalsLösungsmitteldient,sindstrengeReaktionsbedingungensowieerhöhteTempe‐
raturenundverminderterDrucknotwendig.SobeschriebenKATOetal.,aufbauendaufden früheren
ArbeitenvonHULT, eineder ersten enantioselektiven lipasenkatalysierteAcylierungvonAminrac‐4
durch Reaktion mit Carbonsäuren 119 und 109 in einem lösungsmittelfreien System (Abbildung
74).[250,251]DasGleichgewichtderReaktionwirdzugunstenderBildungderAmide(R)‐120verlagert,
indem das als Nebenprodukt gebildeteWasser kontinuierlich unter niedrigemDruck entferntwird.
AufgrundderViskositätderReaktionsmischungmusstebei55°Cgearbeitetwerden,umeineeffiziente
Durchmischung des Systems zu ermöglichen. Die Acylierung liefert Enantiomerenüberschüsse von
>97%fürdasAmid(R)‐120‐abzw.>99%für(R)‐120‐bbeimäßigenUmsätzenvon32bzw.45%.Kurze
ZeitspäterberichteteGUPTA,dasseinvollständigerUmsatzdurchzusätzlichesErhitzenderReaktions‐
mischungauf90°Cerreichtwerdenkann.[252]AllerdingswerdenhierbeikeineAngabenzurStabilität
desEnzymsgemacht.
Abbildung74. LösungsmittelfreiebiokatalytischeAcylierungvonrac‐4.[250]
KANERVAetal.schließlichführtendielösungsmittelfreieRacematspaltungunterschiedlicherAminemit
äquimolarerMenge an aktiviertenMethoxyesternmittels CAL‐B bei Raumtemperatur durch.[36] Alle
(S)‐Amineund (R)‐AmidekonntenmithoherEnantiomerenreinheit von≥95%erhaltenwerden.Die
größtenUnterschiedezwischenReaktioneninGegenwartoderAbwesenheitvonorganischenLösungs‐
mittelnliegeninderbenötigtenZeit,um50%Umsatzzuerreichen,sowieinderEnzymeffizienz,dastatt
100mgEnzymprommolSubstratindiesemFallnurnoch25mgEnzymbenötigtwurden.Außerdem
konnte die Reaktionstemperatur auf 23 °C gesenkt werden ohne Verluste in den Umsätzen oder
ee‐Wertenzubeobachten.
In der Arbeitsgruppe von GRÖGER wurde Malonsäurediethylester (9) als effizienter Acyldonor in
n‐HeptanalsorganischesLösungsmitteleingesetzt.[33,34]DabeiwurdedasAmid(R)‐10bei80°CReak‐
tionstemperaturnach4.5StundenmitvollständigemUmsatznahezuenantiomerenreinerhalten.[33,34]

4 EnantioselektiveRacematspaltungvonAminen
72
InderselbenArbeitsgruppewurdeanschließendderMalonester9erfolgreichunterlösungsmittelfreien
Bedingungeneingesetzt.SokonnteineinemgrößeremMaßstabvon50mmoldasAmid(R)‐10beinur
60°Cmit50%Umsatzund99%eeerzieltwerden(Abbildung75).[35]
Abbildung75. EnzymatischeRacematspaltungvonrac‐4mitMalonsäurediethylester(9).[35]
DieVerwendungeineslösungsmittelfreienReaktionssystemsisteingroßerSchrittinHinblickaufGreen
Chemistry,dadasLösungsmitteleinengroßenTeildesAbfallsderenzymatischenkinetischenRacemat‐
spaltungausmacht.LimitierungenderlösungsmittelfreienRacematspaltungsindEnzymtoleranzenund
Selektivitäten sowie langsame Reaktionsraten in äquimolaren Reaktionsmischungen. Zudem muss
gewährleistetsein,dassdieSubstrateunterdenReaktionsbedingungenflüssigoderineinanderlöslich
sind.[4]

4 EnantioselektiveRacematspaltungvonAminen
73
4.3 EigeneErgebnisseundDiskussion
DieindieserArbeitverwendeteLipaseBausCandidaantarctica(CAL‐B)wurde,sofernnichtanders
erwähnt,ausschließlichinimmobilisierterForm(Novozym435)eingesetzt.DiesewirdimFolgenden
nuralsCAL‐Bbezeichnet.
4.3.1 ReferenzenundAnalytik
Im Folgenden wurde die enantioselektive Racematspaltung an racemischen Aminen durch die
(R)‐spezifischeCAL‐Buntersucht.UmdieEnantiomerenüberschüssederdarausresultierendenAmide
mittels HPLC bestimmen zu könnenwurden zunächst die entsprechenden Racemate chemisch syn‐
thetisiert. Diese dienten als Referenz zur Identifikation der Signale der Enantiomere im Chromato‐
grammsowiealsVergleichfürdie1H‐und13C‐NMR‐Spektren.
4.3.1.1 SynthesederReferenzverbindungen
DiechemischeAcylierungerfolgteimFallderAmiderac‐10undrac‐121ausgehendvonPhenylethyl‐
aminrac‐4mitdenAcyldonorenMalonsäurediethylester(9)undPhenoxyessigsäureethylester(122)
unterErhitzenzumRückflussfür4bzw.5Stunden(Abbildung76).[253]
Abbildung76. SynthesederReferenzverbindungenrac‐10,rac‐121undrac‐123.[253,254]
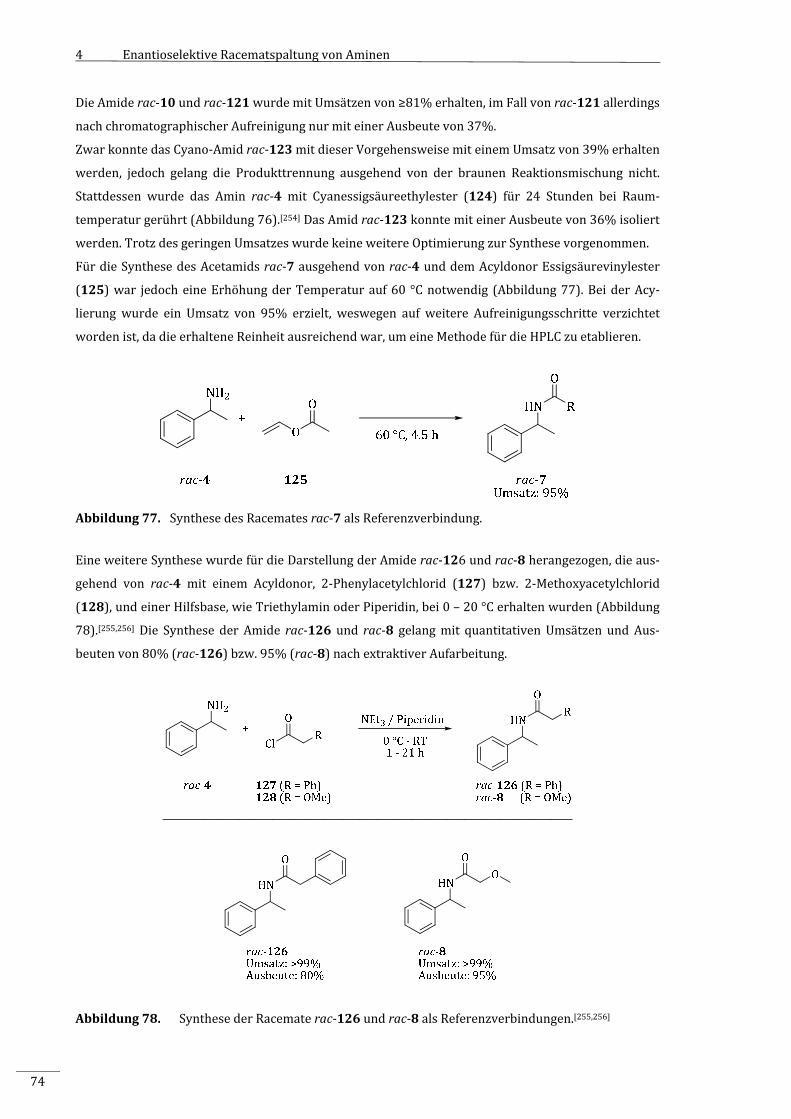
4 EnantioselektiveRacematspaltungvonAminen
74
DieAmiderac‐10undrac‐121wurdemitUmsätzenvon≥81%erhalten,imFallvonrac‐121allerdings
nachchromatographischerAufreinigungnurmiteinerAusbeutevon37%.
ZwarkonntedasCyano‐Amidrac‐123mitdieserVorgehensweisemiteinemUmsatzvon39%erhalten
werden, jedoch gelang die Produkttrennung ausgehend von der braunen Reaktionsmischung nicht.
Stattdessen wurde das Amin rac‐4 mit Cyanessigsäureethylester (124) für 24 Stunden bei Raum‐
temperaturgerührt(Abbildung76).[254]DasAmidrac‐123konntemiteinerAusbeutevon36%isoliert
werden.TrotzdesgeringenUmsatzeswurdekeineweitereOptimierungzurSynthesevorgenommen.
FürdieSynthesedesAcetamidsrac‐7ausgehendvonrac‐4unddemAcyldonorEssigsäurevinylester
(125)war jedocheineErhöhungderTemperaturauf60 °Cnotwendig (Abbildung77).BeiderAcy‐
lierung wurde ein Umsatz von 95% erzielt, weswegen auf weitere Aufreinigungsschritte verzichtet
wordenist,dadieerhalteneReinheitausreichendwar,umeineMethodefürdieHPLCzuetablieren.
Abbildung77. SynthesedesRacematesrac‐7alsReferenzverbindung.
EineweitereSynthesewurdefürdieDarstellungderAmiderac‐126undrac‐8herangezogen,dieaus‐
gehend von rac‐4 mit einem Acyldonor, 2‐Phenylacetylchlorid (127) bzw. 2‐Methoxyacetylchlorid
(128),undeinerHilfsbase,wieTriethylaminoderPiperidin,bei0–20°Cerhaltenwurden(Abbildung
78).[255,256] Die Synthese der Amide rac‐126 und rac‐8 gelangmit quantitativenUmsätzen undAus‐
beutenvon80%(rac‐126)bzw.95%(rac‐8)nachextraktiverAufarbeitung.
Abbildung78. SynthesederRacematerac‐126undrac‐8alsReferenzverbindungen.[255,256]

4 EnantioselektiveRacematspaltungvonAminen
75
Die Synthese des Aminoindan‐Amids rac‐129 benötigteweit längere Reaktionszeiten als die bisher
beschriebenen Synthesen. Das Aminoindan rac‐130 wurde zusammen mit dem Malonester 9 und
1,8‐Diazabicyclo[5.4.0]undec‐7‐en(DBU)inDCMfürdreiTagebei40°Cgerührt(Abbildung79).[257]Das
Amidrac‐129konntemiteinerAusbeutevon17% isoliertwerden.ZusätzlichzumProduktrac‐129
konntedurchsäulenchromatographischeAufreinigungdasentsprechendeDiamid131alsNebenpro‐
duktmit9%Ausbeuteisoliertwerden.
Abbildung79. SynthesedesAminoindan‐Amidsrac‐129.[257]
4.3.1.2 Analytik
Um eine genaue Umsatzbestimmung mittels 1H‐NMR‐Spektroskopie etablieren zu können, wurde
zunächstdieAufarbeitungvalidiert.Dafürwurdeeine1:1‐MischungausMalonsäurediethylester(9)und
rac‐4 eingewogen und das genaue Verhältnis mittels 1H‐NMR‐Spektroskopie bestimmt. Nach an‐
schließender Aufarbeitung analog der Allgemeinen Arbeitsvorschrift 5 (AAV 6, Abschnitt 7.2.2.3.3)
erfolgteerneuteineBestimmungdesVerhältnissesmittels1H‐NMR‐Spektroskopie.Diedabeierhaltenen
AbweichungenlagennachdreifacherBestimmungzwischen1–4%undsomitmitdurchschnittlich2%
im akzeptablen Bereich. Im Rahmen von Messunsicherheit kann dieser Wert toleriert werden. Es
konntenauchkeineProdukteeinerHydrolyse‐Reaktionbeobachtetwerden.
Die produktbezogene Bestimmung des Umsatzes mithilfe der 1H‐NMR‐Spektroskopie erfolge nach
Gleichung 2. In Abbildung 80 ist beispielhaft ein Spektrum einer Acylierung bei 60°C Reaktions‐
temperaturund4.5StundenReaktionszeitgezeigt.AnhandderambestenisoliertenIntegraledesAmids
(R)‐10 bei5.15ppmunddesMalonats9 bei3.36ppmkonnte indiesemFall einproduktbezogener
Umsatzvon42%ermitteltwerden.DadurchVariationderSignaleSchwankungenderUmsätzemitbis
zu2%auftretenkönnen,wurdenfürdieUmsatzbestimmungimmeraufdiegenanntenSignalezurück‐
gegriffen.
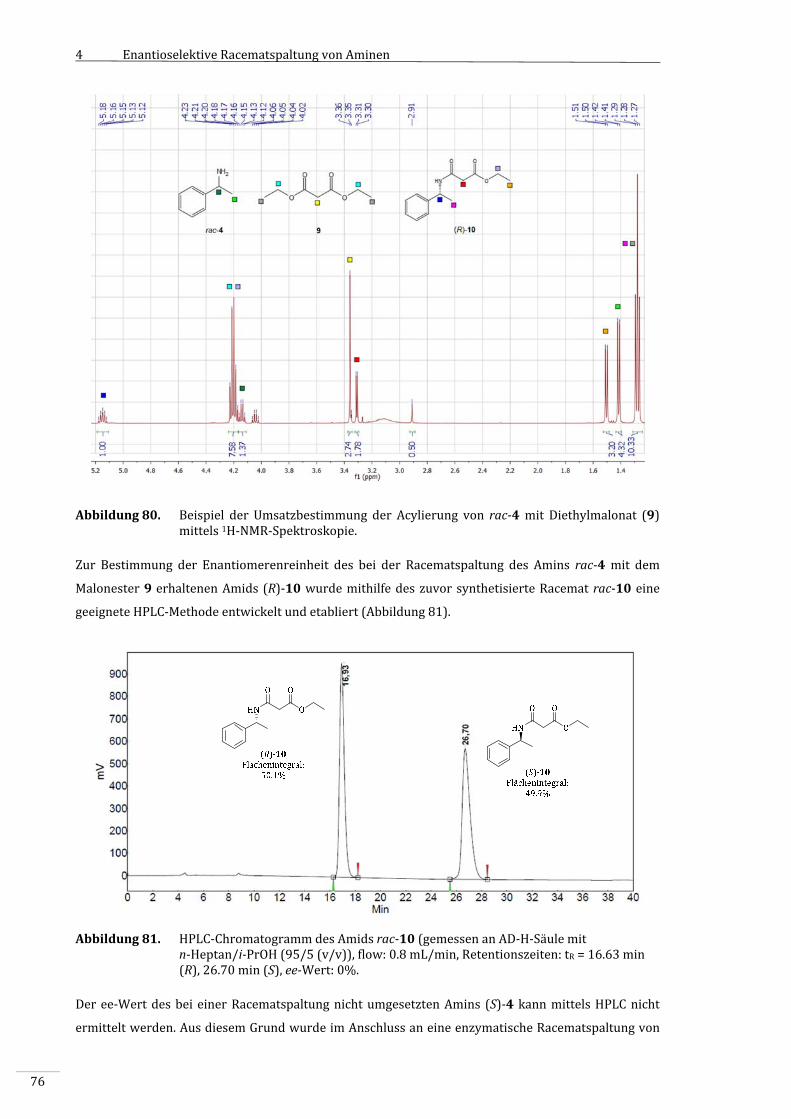
4 EnantioselektiveRacematspaltungvonAminen
76
Abbildung80. Beispiel der Umsatzbestimmung der Acylierung von rac‐4mit Diethylmalonat (9)mittels1H‐NMR‐Spektroskopie.
Zur Bestimmung der Enantiomerenreinheit des bei der Racematspaltung des Amins rac‐4 mit dem
Malonester9erhaltenenAmids(R)‐10wurdemithilfedeszuvorsynthetisierteRacematrac‐10eine
geeigneteHPLC‐Methodeentwickeltundetabliert(Abbildung81).
Abbildung81. HPLC‐ChromatogrammdesAmidsrac‐10(gemessenanAD‐H‐Säulemitn‐Heptan/i‐PrOH(95/5(v/v)),flow:0.8mL/min,Retentionszeiten:tR=16.63min(R),26.70min(S),ee‐Wert:0%.
Deree‐WertdesbeieinerRacematspaltungnichtumgesetztenAmins (S)‐4 kannmittelsHPLCnicht
ermitteltwerden.AusdiesemGrundwurdeimAnschlussaneineenzymatischeRacematspaltungvon

4 EnantioselektiveRacematspaltungvonAminen
77
rac‐4diegesamteReaktionsmischungohnevorherigeAufarbeitungmitAcetanhydrid(132)umgesetzt,
sodasseineAcylierungdesnicht‐umgesetztenAmins(S)‐4erfolgenkann(Abbildung82).DasAcetamid
(S)‐7 wurde dabei quantitativ erhalten. Mithilfe des zuvor dargestellten Racemates rac‐7 konnte
ebenfallseineHPLC‐Analytiketabliertwerdenundderee‐WertdesAmins(S)‐7somitermitteltwerden.
Dieserbeträgt<0.2%ee.
Abbildung82. ChemischeAcylierungdesresultierendenAmins(S)‐4.DieBerechnungdesee‐Wertesvon(S)‐4erfolgtemithilfedesProgramms„Enantioselectivity“durchAngabedesUm‐satzeszumAmid(R)‐10unddessendetektiertenee‐Wertes.[258]
Derermittelte ee‐WertdesAmins (S)‐4 von88%stimmtemitdemee‐Wert,dermittelsUmsatzder
enzymatischenAcylierungunddenee‐WertendeserhaltenenAmids(R)‐4berechnetwurden,bisauf
2%Differenzsehrgutüberein,sodassdieEnantiomerenüberschüssederSubstrateimFolgendennur
berechnet worden sind. Die Berechnung der Selektivität sowie der E‐Werte kannmithilfe des Pro‐
gramms„Enantioselectivity“derUniversitätGrazerfolgen(Abbildung83).[258]
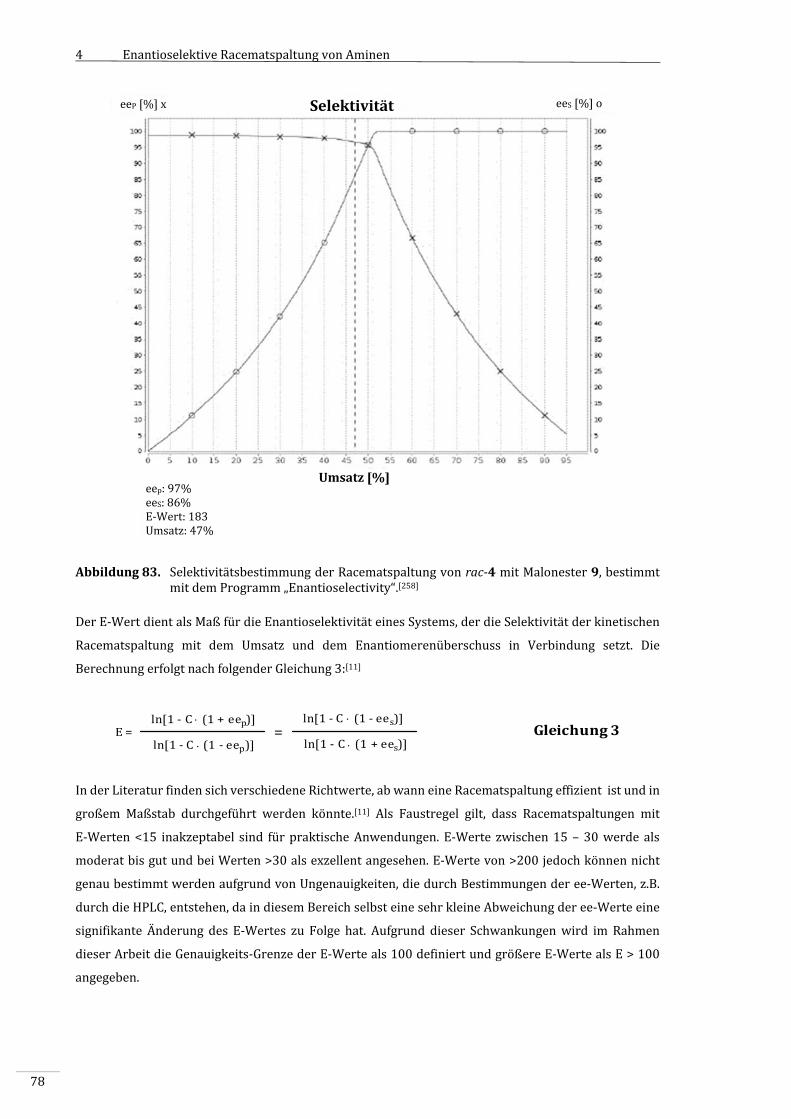
4 EnantioselektiveRacematspaltungvonAminen
78
Abbildung83. SelektivitätsbestimmungderRacematspaltungvonrac‐4mitMalonester9,bestimmt
mitdemProgramm„Enantioselectivity“.[258]
DerE‐WertdientalsMaßfürdieEnantioselektivitäteinesSystems,derdieSelektivitätderkinetischen
Racematspaltung mit dem Umsatz und dem Enantiomerenüberschuss in Verbindung setzt. Die
BerechnungerfolgtnachfolgenderGleichung3:[11]
E=ln[1‐C (1+eep)]
ln[1‐C (1‐eep)]=
ln[1‐C (1‐ees)]
ln[1‐C (1+ees)]Gleichung3
InderLiteraturfindensichverschiedeneRichtwerte,abwanneineRacematspaltungeffizientistundin
großem Maßstab durchgeführt werden könnte.[11] Als Faustregel gilt, dass Racematspaltungen mit
E‐Werten<15 inakzeptabel sind fürpraktischeAnwendungen.E‐Werte zwischen15–30werdeals
moderatbisgutundbeiWerten>30alsexzellentangesehen.E‐Wertevon>200jedochkönnennicht
genaubestimmtwerdenaufgrundvonUngenauigkeiten,diedurchBestimmungenderee‐Werten,z.B.
durchdieHPLC,entstehen,daindiesemBereichselbsteinesehrkleineAbweichungderee‐Werteeine
signifikante Änderung des E‐Wertes zu Folge hat. Aufgrund dieser Schwankungenwird imRahmen
dieserArbeitdieGenauigkeits‐GrenzederE‐Werteals100definiertundgrößereE‐WertealsE>100
angegeben.
eeS[%]oeeP[%]x
eep:97%eeS:86%E‐Wert:183Umsatz:47%
Selektivität
Umsatz[%]

4 EnantioselektiveRacematspaltungvonAminen
79
4.3.2 VorversuchemitsubstituiertenBenzylaminenalsSubstrate
4.3.2.1 SynthesedesBenzylamids133
ZurdetailliertenEtablierungvonMethodenderAnalytikundUmsatzbestimmungsolltezunächstein
Modelsubstrat zum Einsatz kommen, das keine Stereoinformation trägt. Dafür wurde sowohl das
einfachstearomatischeAmin,Benzylamin(134),alsauchsubstituierteDerivateausgewählt.Inmeta‐
PositionsubstituiertearomatischeAminedienenalsAusgangsstoffefürdieSynthesepharmazeutischer
Wirkstoffe. Beispiele dafür sind Rivastigmin (135)[259,260] oder das Calcimimeticim136[261], die zur
Behandlung der Alzheimerschen Krankheit bzw. zur Therapie von Hyperparathyroidismus, einer
RegulationsstörungderNebenschilddrüse,eingesetztwerden(Abbildung84).
Abbildung84. Beispiele für pharmazeutische Wirkstoffe mit meta‐substituierten aromatischenAminen:Rivastigmin(135)[259,260]unddasCalcimimeticum136[261].
FüreinevollständigeAnalytikwurdedieseAcylierungsreaktionzunächstchemisch,d.h.beimErhitzen
zumRückfluss für3Stunden,durchgeführt(Abbildung85).[253]DabeikonntenebendemBenzylamid
133,dasmit48%Ausbeuteisoliertwurde,dieBildungdesentsprechendenDiamids137beobachtet
werden. Nach säulenchromatographischer Aufreinigung wurde das Diamid 137 mit 20% Ausbeute
isoliert.DieBildungdiesesNebenproduktes137kanndurchschrittweiseZugabedesAcyldonorszur
Reaktionsmischungunterdrücktwerden.
Abbildung85. ChemischeAcylierungvonBenzylamin(134)mitMalonsäurediethylester(9).[253]
ImAnschlusswurdederVerlustderSubstanzen134und9währendderAufarbeitungbestimmt,indem
diese eingewogen und bei unterschiedlichen Drücken bei einer Badtemperatur von 40 °C gerührt
wurden.DiejeweiligenVerlustewurdenimAnschlussmittelsAuswaageermittelt(Abbildung86).Ein
großerVerlustanbeidenSubstanzen134und9währendderAufarbeitungmachtdiegenaueBestim‐
mungeinesproduktbezogenenUmsatzesunmöglich.DerVerlustderbeidenKomponenten134und9

4 EnantioselektiveRacematspaltungvonAminen
80
istabhängigvondenDampfdrücken(0.6bzw.1.3hPa)derKomponenten.[262]SomitkannderVerlust
desAmins134beimErhöhendesDruckesvon230auf550mbarbeieinerRührdauervon120Minuten
von69%aufnurnoch12%gesenktwerden.DainderRegelaberRührzeitenvonunter30Minutenfür
dieAufarbeitungausreichenkanndermittelsAuswaageermittelteVerlustvon<3%fürbeideKompo‐
nentenvernachlässigtwerden.DerproduktbezogeneUmsatzwird,sofernnichtandersangegeben,in
dieserArbeitaufdenAcyldonorMalonsäurediethylester(9)bezogen.
Abbildung86. VerlustderSubstanzen134und9währendderAufarbeitung.
4.3.3 EnzymatischeAcylierungsubstituierterBenzylamine
ZurUntersuchungdesEinflusseseinesSubstituenteninmeta‐Positionwurdenunterschiedlichsubsti‐
tuierteBenzylamine134und138–140mitMalonsäurediethylester(9)undCAL‐BalsBiokatalysator
lösungsmittelfreiumgesetzt (Abbildung87).BeiallenAcylierungsreaktionenwurden,mitAusnahme
desm‐Hydroxybenzylamins(138),nahezuvollständigeUmsätzeerzielt.DasAmin138istimGegensatz
zudenMethoxy‐undBromo‐substituiertenBenzylaminen139und140einkristallinerFeststoff,der
sichnurzueinemgewissenAnteil imMalonester9 löst.EineVerlängerungderReaktionszeitwürde
hierbeizueinemhöherenUmsatzführen.AllerdingskonntebeiderAcylierungdesHydroxyamins138,
welche nicht in lösungsmittelfreier Umgebung durchgeführt werden kann, neben der Bildung des
Produktes 141 auch die Bildung des entsprechenden Diamids 142 mit einem Umsatz von 17%
beobachtetwerden.DajeweilsnureinTeildesm‐Hydroxybenzylamins(138)gelöstinderReaktions‐
mischungvorliegtunddasentstandeneAmid141mitdemMalonester9gutmischbarist,istdieWahr‐
scheinlichkeiteinerzweitenAcylierungsehrgroß.
DieBildungdesentsprechendenDiamidskonntebeidenAcylierungsreaktionenderAmine134bzw.
139und140mit9bzw.5%nachgewiesenwerden.DieAnwesenheiteineselektronenziehendenSubsti‐
tuenteninmeta‐PositionscheintdemnacheinenpositivenEinflussaufdenUmsatzzuhaben.Alleindie
schlechteLöslichkeitdesHydroxyamins138istfürdessensehrniedrigenUmsatzverantwortlich.Durch
0102030405060708090100
0 0,5 1 1,5 2
Verlust[%
]
Zeit[h]
112,230mbar 112,550mbar 9,230mbar 9,550mbar 134,230mbar 134,550mbar 9,230mbar 9,550mbar
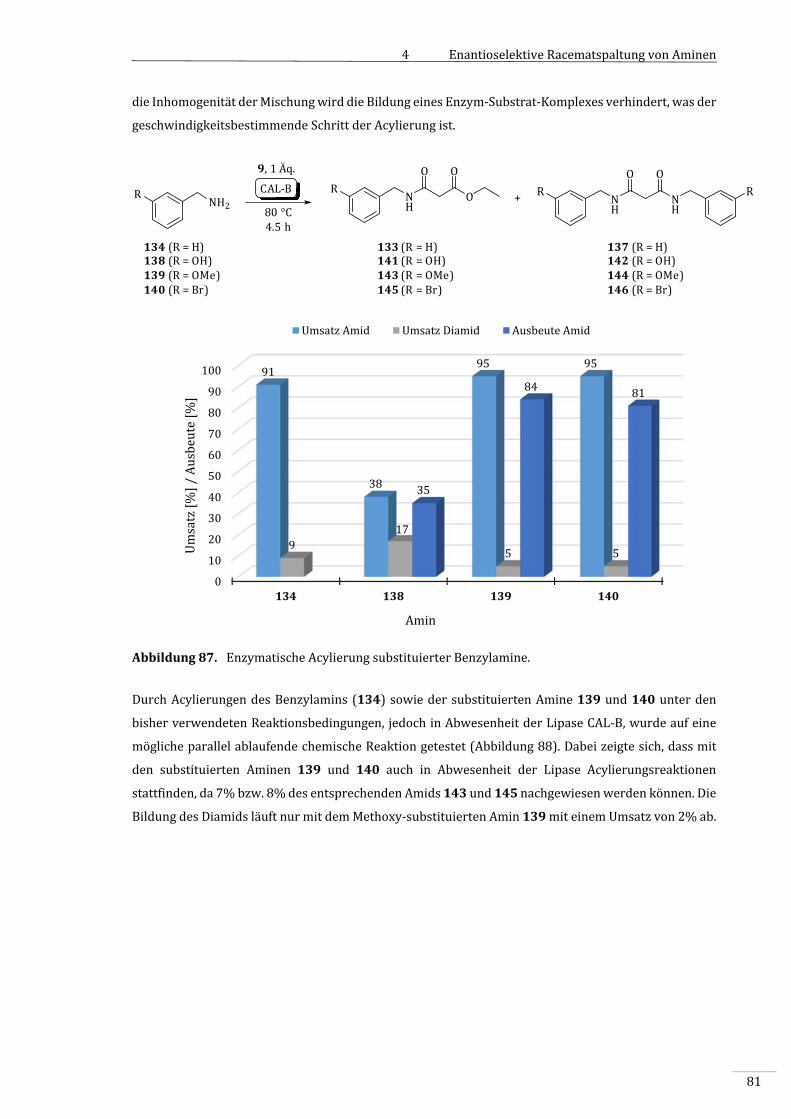
4 EnantioselektiveRacematspaltungvonAminen
81
dieInhomogenitätderMischungwirddieBildungeinesEnzym‐Substrat‐Komplexesverhindert,wasder
geschwindigkeitsbestimmendeSchrittderAcylierungist.
NH2CAL‐B
NH
O
O
O
R R
134 (R=H)138 (R=OH)139 (R=OMe)140 (R=Br)
9,1Äq.
NH
O
NH
OR R+
80°C4.5h
133 (R=H)141 (R=OH)143 (R=OMe)145 (R=Br)
137 (R=H)142 (R=OH)144 (R=OMe)146 (R=Br)
Abbildung87. EnzymatischeAcylierungsubstituierterBenzylamine.
DurchAcylierungendesBenzylamins(134)sowiedersubstituiertenAmine139und140unterden
bisherverwendetenReaktionsbedingungen,jedochinAbwesenheitderLipaseCAL‐B,wurdeaufeine
möglicheparallelablaufendechemischeReaktiongetestet(Abbildung88).Dabeizeigtesich,dassmit
den substituierten Aminen 139 und 140 auch in Abwesenheit der Lipase Acylierungsreaktionen
stattfinden,da7%bzw.8%desentsprechendenAmids143und145nachgewiesenwerdenkönnen.Die
BildungdesDiamidsläuftnurmitdemMethoxy‐substituiertenAmin139miteinemUmsatzvon2%ab.
0
10
20
30
40
50
60
70
80
90
100
134 138 139 140
91
38
95 95
917
5 5
35
84 81
Umsatz[%
]/Ausbeute[%]
Amin
UmsatzAmid UmsatzDiamid AusbeuteAmid
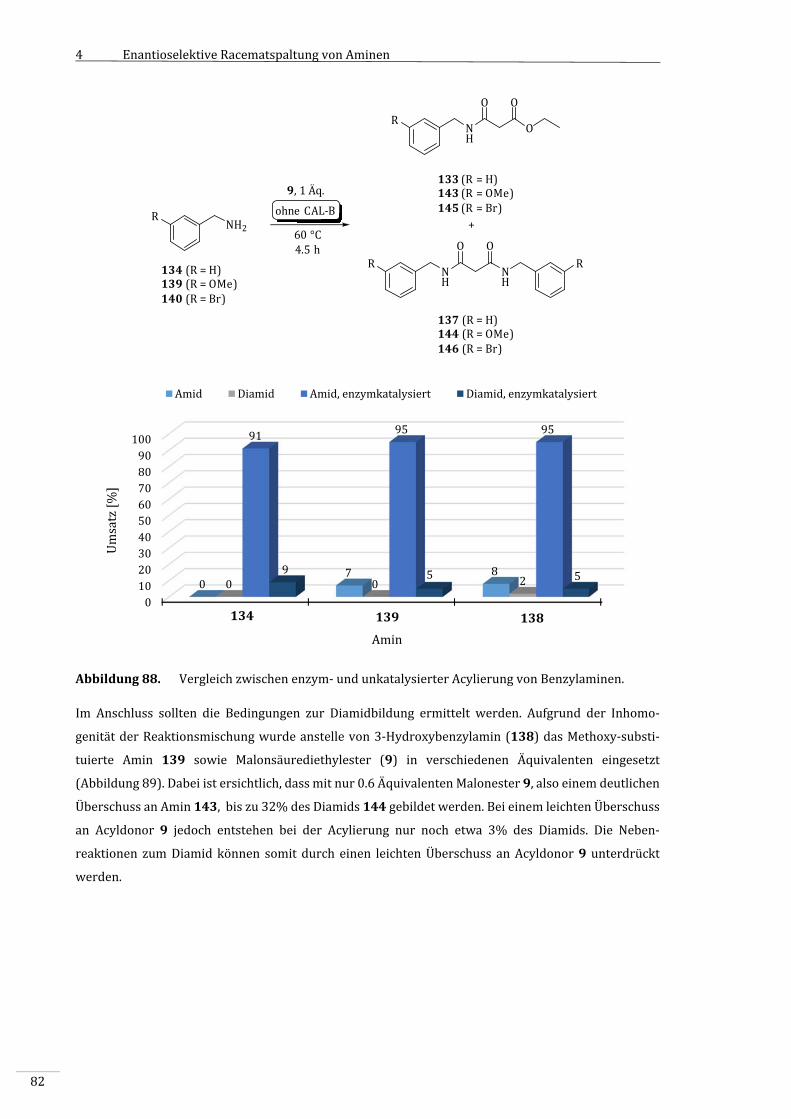
4 EnantioselektiveRacematspaltungvonAminen
82
NH2ohneCAL‐B
NH
O
O
O
R
R
134 (R=H)139 (R=OMe)140 (R=Br)
9,1Äq.
NH
O
NH
O
R R
+60°C4.5h
133 (R=H)143 (R=OMe)145 (R=Br)
137 (R=H)144 (R=OMe)146 (R=Br)
Abbildung88. Vergleichzwischenenzym‐undunkatalysierterAcylierungvonBenzylaminen.
Im Anschluss sollten die Bedingungen zur Diamidbildung ermittelt werden. Aufgrund der Inhomo‐
genitätderReaktionsmischungwurdeanstellevon3‐Hydroxybenzylamin(138)dasMethoxy‐substi‐
tuierte Amin 139 sowie Malonsäurediethylester (9) in verschiedenen Äquivalenten eingesetzt
(Abbildung89).Dabeiistersichtlich,dassmitnur0.6ÄquivalentenMalonester9,alsoeinemdeutlichen
ÜberschussanAmin143,biszu32%desDiamids144gebildetwerden.BeieinemleichtenÜberschuss
an Acyldonor 9 jedoch entstehen bei der Acylierung nur noch etwa 3% des Diamids. Die Neben‐
reaktionen zumDiamidkönnen somitdurch einen leichtenÜberschuss anAcyldonor9 unterdrückt
werden.
0102030405060708090100
130 133 134
07 8
0 0 2
91 95 95
9 5 5
Umsatz[%
]
Amin
Amid[%] Diamid[%] Amid,enzymkatalysiert[%] Diamid,enzymkatalysiert[%]
133 137 138134 139 138
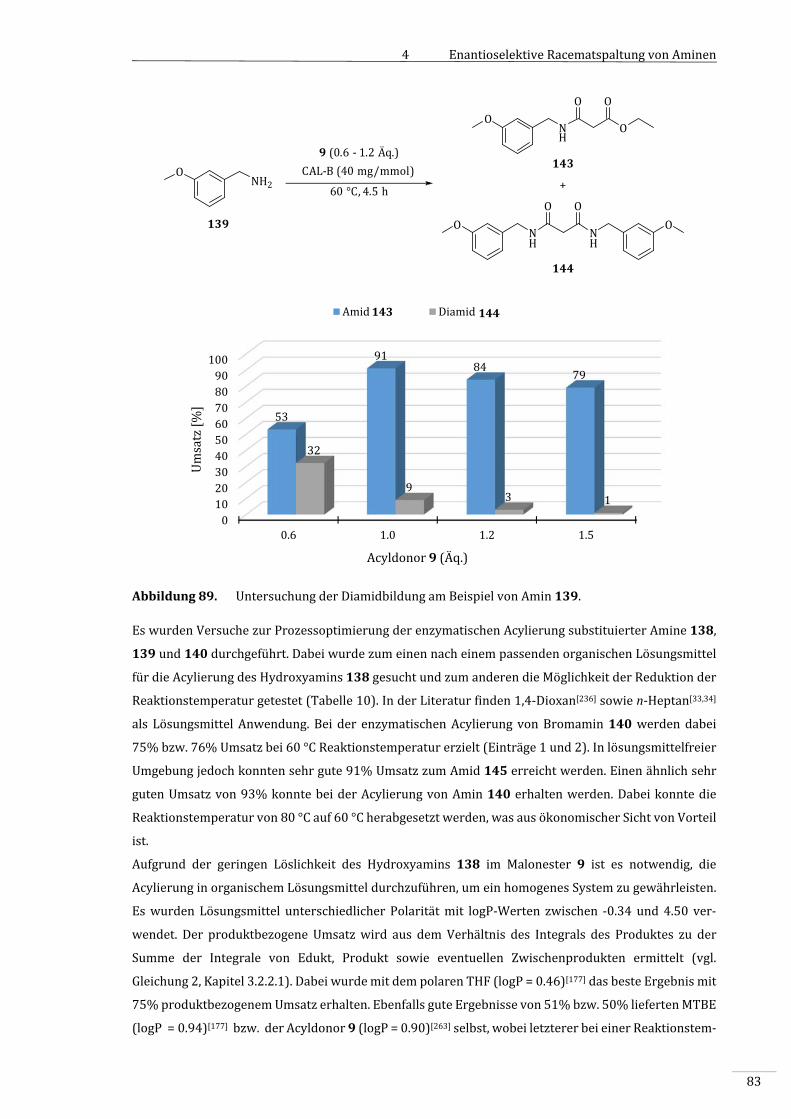
4 EnantioselektiveRacematspaltungvonAminen
83
NH2
NH
O
O
O
O
O
143
NH
O
NH
O
O O
144
+60°C,4.5h
139
9 (0.6‐1.2Äq.)
CAL‐B(40mg/mmol)
Abbildung89. UntersuchungderDiamidbildungamBeispielvonAmin139.
EswurdenVersuchezurProzessoptimierungderenzymatischenAcylierungsubstituierterAmine138,
139und140durchgeführt.DabeiwurdezumeinennacheinempassendenorganischenLösungsmittel
fürdieAcylierungdesHydroxyamins138gesuchtundzumanderendieMöglichkeitderReduktionder
Reaktionstemperaturgetestet(Tabelle10).InderLiteraturfinden1,4‐Dioxan[236]sowien‐Heptan[33,34]
als LösungsmittelAnwendung.Bei der enzymatischenAcylierungvonBromamin140werdendabei
75%bzw.76%Umsatzbei60°CReaktionstemperaturerzielt(Einträge1und2).Inlösungsmittelfreier
Umgebungjedochkonntensehrgute91%UmsatzzumAmid145erreichtwerden.Einenähnlichsehr
gutenUmsatzvon93%konntebeiderAcylierungvonAmin140erhaltenwerden.Dabeikonntedie
Reaktionstemperaturvon80°Cauf60°Cherabgesetztwerden,wasausökonomischerSichtvonVorteil
ist.
Aufgrund der geringen Löslichkeit des Hydroxyamins 138 im Malonester 9 ist es notwendig, die
AcylierunginorganischemLösungsmitteldurchzuführen,umeinhomogenesSystemzugewährleisten.
Eswurden Lösungsmittel unterschiedlicher Polaritätmit logP‐Werten zwischen ‐0.34 und4.50 ver‐
wendet. Der produktbezogene Umsatzwird aus dem Verhältnis des Integrals des Produktes zu der
Summe der Integrale von Edukt, Produkt sowie eventuellen Zwischenprodukten ermittelt (vgl.
Gleichung2,Kapitel3.2.2.1).DabeiwurdemitdempolarenTHF(logP=0.46)[177]dasbesteErgebnismit
75%produktbezogenemUmsatzerhalten.EbenfallsguteErgebnissevon51%bzw.50%liefertenMTBE
(logP=0.94)[177]bzw.derAcyldonor9(logP=0.90)[263]selbst,wobeiletztererbeieinerReaktionstem‐
0102030405060708090100
0.6 1.0 1.2 1.5
53
9184
79
32
93 1
Umsatz[%
]
Acyldonor9 (Äq.)
Amid121[%] Diamid122[%]143 144

4 EnantioselektiveRacematspaltungvonAminen
84
peraturvonnur60°Ceingesetztwordenist.BeideVariantenbietenVorteile,denndasMTBEistauf‐
grundseineshohenDampfdruckeseinfachausdemReaktionsgemischzuentfernen,währendMalon‐
säurediethylester(9)nachDestillationeinerweiterenAcylierungsreaktionzugeführtwerdenkönnte.
EineweitereBesonderheitzeigtsichbeiderVerwendungvonDiethylether(logP=0.89)[177]alsLösungs‐
mittel,dadasHerabsenkenderTemperaturvon60°CaufRaumtemperaturmitgleichzeitigerVerlän‐
gerungderReaktionsdauerauf24StundendengleichenUmsatzvon30%liefert.DiesesstehtinEinklang
mitBeobachtungenvonDITRICH,dermitMethoxyacetat6alsAcyldonormitCAL‐BinDiethyletherbei
Raumtemperaturüber24StundenReaktionszeitdasentsprechendeAmidenantiomerenreinmitvoll‐
ständigem Umsatz erzielen konnte.[30] Dieses zeigt deutlich die hohe Aktivität der CAL‐B in einem
breitenTemperaturbereich.
Tabelle10. VersuchezurProzessoptimierungderenzymatischenAcylierungmeta‐substituierterBenzylamine.
Eintrag Amin T[°C] t[h] Solvens logP Umsatz[%]
1 140 60 4.5 1,4‐Dioxan ‐1.10a) 75
2 140 60 4.5 n‐Heptan 4.50 b) 76
3 140 60 4.5 ‐ ‐ 91
4 139 60 4.5 ‐ ‐ 93
5 138 80 4.5 MTBE 0.94b) 51
6 138 80 4.5 THF 0.46b) 75
7 138 80 4.5 2‐Me‐THF 1.00c) 70
8 138 80 4.5 MeCN ‐0.34b) 43
9 138 80 4.5 i‐PrOH 0.28b) 22
10 138 60 4.5 n‐Heptan 4.50b) 45
11 138 60 4.5 EtOAc 0.73b) 41
12 138 60 4.5 Malonester 9(100Äq.) 0.90 c) 50
13 138 60 4.5 Et2O 0.89b) 30
14d) 138 RT 24 Et2O 0.89b) 30
15 138 RT 24 EtOH ‐0.24a) 5Datenentnommenaus:a)LAANEetal.[175];b)SANGSTERetal.[177];c)BestimmtdurchAdvancedChemistryDevelopment(ACD/Labs)SoftwareV11.02[263];d)Zugabevon1.5ÄquivalenteMalonsäurediethylester(9).

4 EnantioselektiveRacematspaltungvonAminen
85
4.3.4 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)
In der Literatur ist die enzymatischeRacematspaltung von aliphatischen und aromatischenAminen
durchunterschiedlicheAcyldonoreninAnwesenheitvonorganischenLösungsmittelnbereitsvielfach
beschrieben worden. Häufig finden dabei Acyldonoren wie langkettige Säuren sowie deren Ester
Anwendung,dieallerdingssehrlangeReaktionszeitenfüreinenvollständigenUmsatzbenötigen(vgl.
Kapitel4.2.2).[31]WeiterehäufigverwendeteAcyldonorensindEssigester5oderMethoxyesterwie6
bzw.111(Abbildung90).[30,31]Zwarwerdendiedabeierhaltenen(R)‐AmidemitsehrhohenEnantio‐
merenüberschüssenerhalten,allerdingsliegendieReaktionszeitenauchhierbeinochbei15bzw.60
Stunden.DurchEinsatzeinesHeteroatomsinβ‐PositiondesAcyldonorskanndieReaktionsgeschwin‐
digkeiterhöhtwerden,wobeidiebestenErgebnissemitO‐Substituentenerhaltenwerden.[31]
DieRacematspaltungracemischerAmineunterlösungsmittelfreienBedingungenfindeterstseitBeginn
diesesJahrhundertsBeachtung.[36,250–252]ZudenerstenenantioselektivenlipasenkatalysiertenAcylie‐
rungendesPhenylethylaminsrac‐4inAbwesenheitvonorganischenLösungsmittelnzählendieReak‐
tionenmitCarbonsäuren(119oder109)[250,252]oderMethoxyestern(6oder111)[36]alsAcyldonoren.
ImFallderCarbonsäurenistzurGleichgewichtseinstellungeinverringerterDrucknotwendig,während
mitMethoxyesternbereitsbeiRaumtemperaturgearbeitetwerdenkann.Methoxyestersinddurchdas
Heteroatominβ–PositionaktivierteAcyldonoren.
Abbildung90. EnzymatischeRacematspaltungvonrac‐4mitunterschiedlichenAcyldonoreninAbwesenheitvonorganischenLösungsmitteln.[36,250]
Der indieserArbeitsgruppebereits erfolgreich etablierteAcyldonorMalonsäurediethylester (9) soll
unterlösungsmittelfreienBedingungenalsStandard‐Acyldonordienen.[33–35]Ausgehendvondenvoran‐
gegangenArbeitensolltesielösungsmittelfreiAcylierungvonrac‐4weiteroptimiertundinHinblickauf
weiteremöglicheAcyldonorenuntersuchtwerden,DerWeiterensollenweiteremöglicheLipasenauf
ihre Fähigkeit zur solvenzfreien Acylierung von rac‐4 getestet und die Kinetiken dieser Reaktionen
geprüftwerden.
(111)
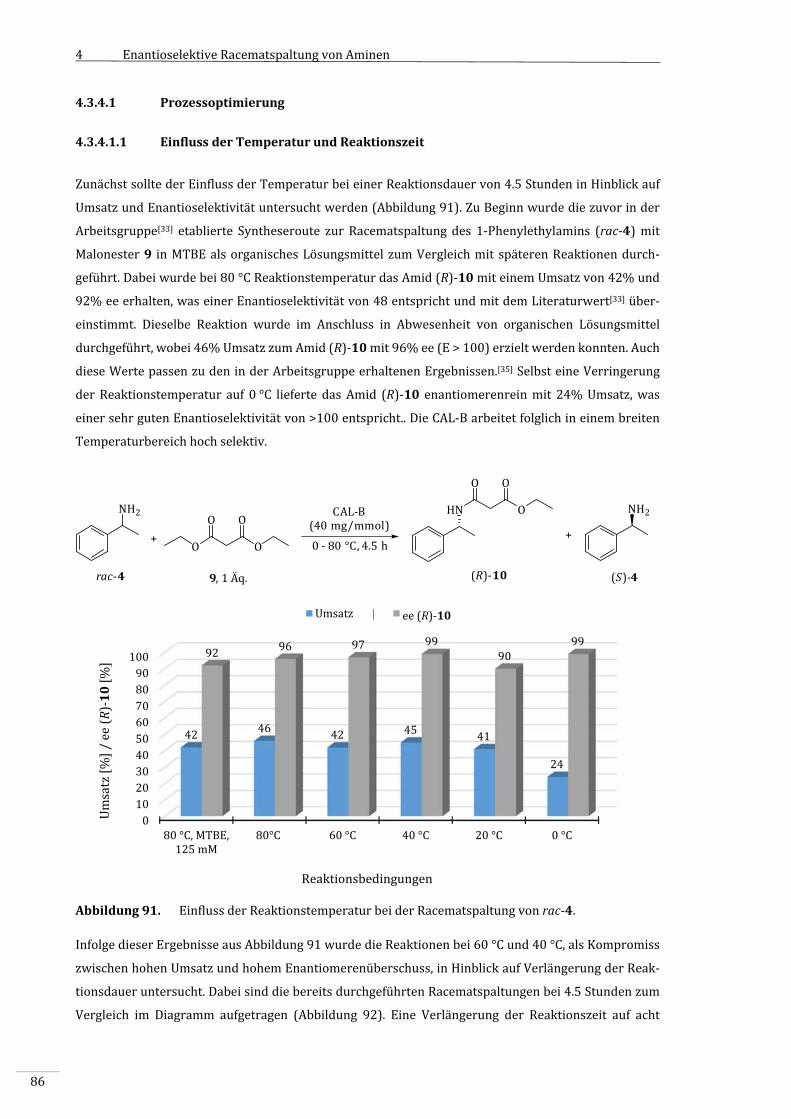
4 EnantioselektiveRacematspaltungvonAminen
86
4.3.4.1 Prozessoptimierung
4.3.4.1.1 EinflussderTemperaturundReaktionszeit
ZunächstsolltederEinflussderTemperaturbeieinerReaktionsdauervon4.5StundeninHinblickauf
UmsatzundEnantioselektivitätuntersuchtwerden(Abbildung91).ZuBeginnwurdediezuvorinder
Arbeitsgruppe[33] etablierte Syntheseroute zur Racematspaltung des 1‐Phenylethylamins (rac‐4)mit
Malonester9 inMTBEalsorganischesLösungsmittelzumVergleichmitspäterenReaktionendurch‐
geführt.Dabeiwurdebei80°CReaktionstemperaturdasAmid(R)‐10miteinemUmsatzvon42%und
92%eeerhalten,waseinerEnantioselektivitätvon48entsprichtundmitdemLiteraturwert[33]über‐
einstimmt. Dieselbe Reaktion wurde im Anschluss in Abwesenheit von organischen Lösungsmittel
durchgeführt,wobei46%UmsatzzumAmid(R)‐10mit96%ee(E>100)erzieltwerdenkonnten.Auch
dieseWertepassenzudeninderArbeitsgruppeerhaltenenErgebnissen.[35]SelbsteineVerringerung
derReaktionstemperatur auf 0°C lieferte dasAmid (R)‐10 enantiomerenreinmit 24%Umsatz,was
einersehrgutenEnantioselektivitätvon>100entspricht..DieCAL‐Barbeitetfolglichineinembreiten
Temperaturbereichhochselektiv.
NH2
O
O
O
O
+
CAL‐B(40mg/mmol)
0‐80°C,4.5h
9,1Äq.
HN
O O
O NH2
+
rac‐4 (S)‐4(R)‐10
Abbildung91. EinflussderReaktionstemperaturbeiderRacematspaltungvonrac‐4.
InfolgedieserErgebnisseausAbbildung91wurdedieReaktionenbei60°Cund40°C,alsKompromiss
zwischenhohenUmsatzundhohemEnantiomerenüberschuss,inHinblickaufVerlängerungderReak‐
tionsdaueruntersucht.DabeisinddiebereitsdurchgeführtenRacematspaltungenbei4.5Stundenzum
Vergleich im Diagramm aufgetragen (Abbildung 92). Eine Verlängerung der Reaktionszeit auf acht
0102030405060708090100
80°C,MTBE,125mM
80°C 60°C 40°C 20°C 0°C
42 46 42 45 41
24
92 96 97 9990
99
Umsatz[%
]/ee(R)‐10[%]
Reaktionsbedingungen
Umsatz[%] ee(R)‐10[%]ee(R)‐10
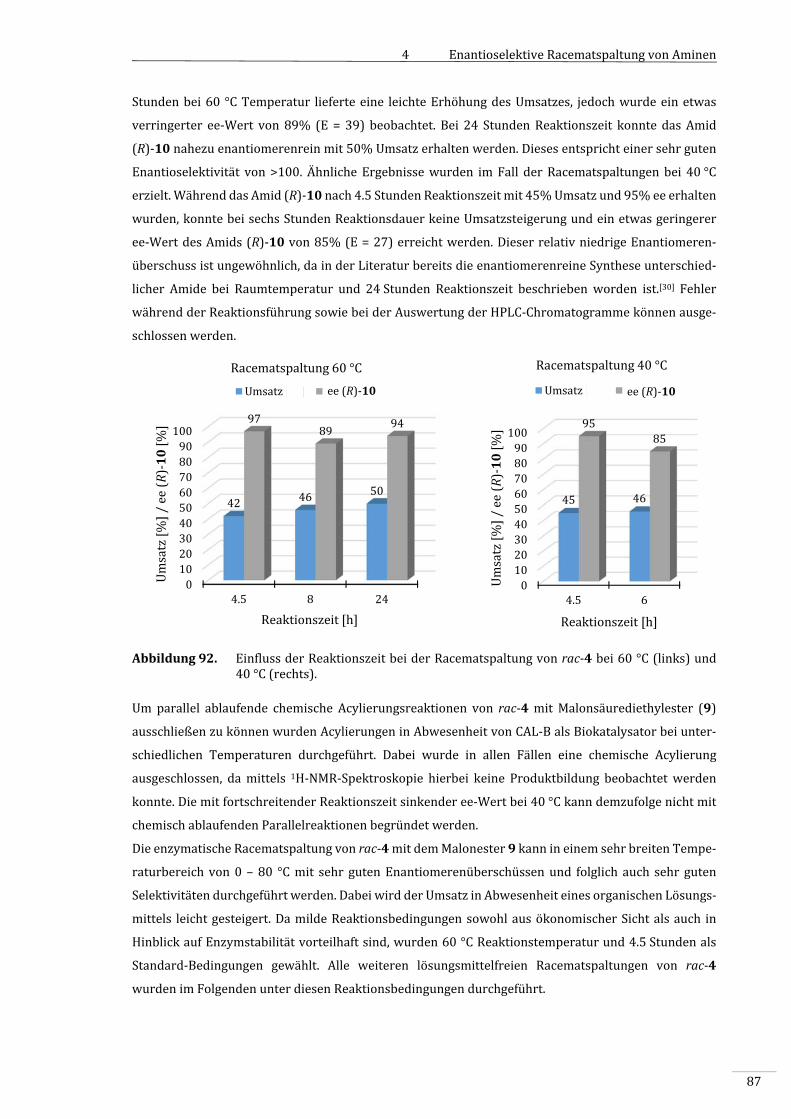
4 EnantioselektiveRacematspaltungvonAminen
87
Stundenbei60 °CTemperatur lieferteeine leichteErhöhungdesUmsatzes, jedochwurdeeinetwas
verringerter ee‐Wert von 89% (E = 39) beobachtet. Bei 24 StundenReaktionszeit konnte dasAmid
(R)‐10nahezuenantiomerenreinmit50%Umsatzerhaltenwerden.Diesesentsprichteinersehrguten
Enantioselektivität von>100.Ähnliche Ergebnissewurden imFall derRacematspaltungenbei 40°C
erzielt.WährenddasAmid(R)‐10nach4.5StundenReaktionszeitmit45%Umsatzund95%eeerhalten
wurden,konntebeisechsStundenReaktionsdauerkeineUmsatzsteigerungundeinetwasgeringerer
ee‐WertdesAmids(R)‐10von85%(E=27)erreichtwerden.DieserrelativniedrigeEnantiomeren‐
überschussistungewöhnlich,dainderLiteraturbereitsdieenantiomerenreineSyntheseunterschied‐
licher Amide bei Raumtemperatur und 24Stunden Reaktionszeit beschrieben worden ist.[30] Fehler
währendderReaktionsführungsowiebeiderAuswertungderHPLC‐Chromatogrammekönnenausge‐
schlossenwerden.
Abbildung92. EinflussderReaktionszeitbeiderRacematspaltungvonrac‐4bei60°C(links)und40°C(rechts).
Um parallel ablaufende chemische Acylierungsreaktionen von rac‐4mitMalonsäurediethylester (9)
ausschließenzukönnenwurdenAcylierungeninAbwesenheitvonCAL‐BalsBiokatalysatorbeiunter‐
schiedlichen Temperaturen durchgeführt. Dabei wurde in allen Fällen eine chemische Acylierung
ausgeschlossen, damittels 1H‐NMR‐Spektroskopie hierbei keine Produktbildung beobachtetwerden
konnte.DiemitfortschreitenderReaktionszeitsinkenderee‐Wertbei40°Ckanndemzufolgenichtmit
chemischablaufendenParallelreaktionenbegründetwerden.
DieenzymatischeRacematspaltungvonrac‐4mitdemMalonester9kannineinemsehrbreitenTempe‐
raturbereichvon0 – 80 °Cmit sehr gutenEnantiomerenüberschüssenund folglich auch sehr guten
Selektivitätendurchgeführtwerden.DabeiwirdderUmsatzinAbwesenheiteinesorganischenLösungs‐
mittels leichtgesteigert.DamildeReaktionsbedingungensowohlausökonomischerSichtalsauch in
HinblickaufEnzymstabilitätvorteilhaftsind,wurden60°CReaktionstemperaturund4.5Stundenals
Standard‐Bedingungen gewählt. Alle weiteren lösungsmittelfreien Racematspaltungen von rac‐4
wurdenimFolgendenunterdiesenReaktionsbedingungendurchgeführt.
0102030405060708090100
4.5 8 24
42 46 50
9789
94
Umsatz[%
]/ee(R)‐10[%]
Reaktionszeit[h]
Racematspaltung60°C
Umsatz[%] ee[%]
0102030405060708090100
4.5 6
45 46
9585
Umsatz[%
]/ee(R)‐10[%]
Reaktionszeit[h]
Racematspaltung40°C
Umsatz[%] ee[%]ee(R)‐10 ee(R)‐10
Reaktionszeit[h]

4 EnantioselektiveRacematspaltungvonAminen
88
4.3.4.1.2 VergleichmitbestehendenindustriellenProzessen
NachdemdieoptimaleReaktionstemperaturauf60°CunddieReaktionsdauerauf4.5Stundenfestgelegt
wordensind,wurdedielösungsmittelfreieenzymatischeRacematspaltungdesAminsrac‐4mitMalon‐
ester9alsAcyldonormitbestehendenindustriellenProzessenverglichen(Tabelle11,Eintrag1).Nach
einerPatentanmeldungvonBASFSEwirdeinaromatischesracemischesAminmitMethoxyester111
(1.5Äq.) und2mg/mmolCAL‐Bbei20 °Cumgesetztunddas entsprechendeAmid (R)‐8mit hoher
Enantiomerenreinheit erhalten.[264] Die Übertragung dieser Vorschrift auf die Racematspaltung mit
Malonsäurediethylester(9)alsAcyldonorliefertejedochnur25%Umsatzmit95%ee(Eintrag2).Der
bestimmteguteE‐Wertvon53liegtjedochdeutlichunterdenunterStandard‐Bedingungenerhaltenen
Wert. Der Unterschied in den Enantioselektivitäten von >100 und 53 kannmit der geringeren ver‐
wendeten Enzymmenge sowie niedrigeren Reaktionstemperatur und die dadurch etwas geringere
AktivitätderLipasebegründetwerden.DesWeiterenistderMethoxyester104alsAcyldonorstärker
aktiviertalsdasMalonat9.DurchVerlängerungderReaktionszeitkönnteauchindiesemFalleinvoll‐
ständigerUmsatzunddadurcheinehöhereEnantioselektivitäterreichtwerden.
Tabelle11. VergleichmitbestehendenindustriellenProzessen.
Eintrag Ester(Äq.)
CAL‐B[mg/mmol]
SolvensTemperatur
[°C]
Umsatz
[%]
eeP[%]
eeS
[%]a)E
1b)c) 9(1) 40 ‐ 60 42 96 70 >100
2d) 9(1) 2 ‐ 20 25 95 n.b. 53
3d) 9(1.5) 10 Et2O 20 48 95 88 >100
4d) 111(1.5) 10 Et2O 20 52 95 99 >100a)Bestimmtmittels„Enantioselectivity“[258];b)DarstellungdiesesVersucheserfolgtebereitsinAbbildung91undisthierzumVergleichaufgeführt;c)Reaktionszeit:4.5h;d)Reaktionszeit:24h;n.b.=nichtbestimmt.
DITRICHetal.setztenrac‐4mitMethoxyessigsäurepropylester(6)beiRaumtemperaturinDiethylether
um, wobei 10mg/mmol CAL‐B verwendet wurden.[30] Dabei wird das entsprechende Amid nach
24StundenReaktionszeitmit46%Umsatzenantiomerenrein(E>100)erhalten.BeiderReproduktion
dieserReaktionmitdemAcyldonor104konnten52%UmsatzzumAmid(R)‐8sowiesehrgute95%ee
(E>100)erreichtwerden(Eintrag4).DurchErsatzdesAcyldonors111durchdenMalonester9wurde
unter den gleichen Bedingungen ebenfalls ein sehr guter Umsatz von 48% erzielt (Eintrag 3). Der
EnantiomerenüberschusswurdefürdasAmid(R)‐10auf95%eebestimmt,wasebenfallseinerEnan‐
tioselektivitätvon>100entspricht.ZwarwirdnachdieserVorschriftnureinViertelderindieserArbeit
eingesetztenEnzymmengevon40mg/mmolbeiRaumtemperaturverwendet,allerdingsistderEinsatz

4 EnantioselektiveRacematspaltungvonAminen
89
vonDiethyletheralsLösungsmittelnotwendig.DesWeiterenwirdfüreinenvollständigenUmsatzzum
(R)‐AmideineReaktionszeitvon24Stundenbenötigt,wasausökonomischerSichtunvorteilhaftist.
BeiderRacematspaltungvonrac‐4unterdenindieserArbeitgewähltenlösungsmittelfreienReaktions‐
bedingungenmitdemMalonester9alsAcyldonorkonnteeinesehrguteEnantioselektivitätvon>100
erreichtwerden,dieinderselbenGrößenordnungderbestehendenindustriellenProzesseliegt.Durch
Up‐ScalingzugrößerenMaßstäbenkönntedieAufarbeitungdannmittelsDestillationerfolgen,sodass
kein organisches Lösungsmittel für extraktive Aufarbeitung mehr benötigt würde. Der im Rahmen
dieser Arbeit gewählte Syntheseweg stellt folglich eine sehr gute Alternative zu den bestehenden
industriellenProzessendar.
4.3.5 Enzymrecycling
Im Rahmen eines Enzymrecyclings wurde die Aktivität und die Stabilität der CAL‐B aus Candida
antarcticagetestetwerden.DurchFiltration,WaschenundTrocknenwardieRückgewinnungderLipase
imAnschlussaneineReaktionsehreinfachinderHandhabung.DerVerlustderCAL‐BbeiderReinigung
sowiedurchmechanischeBelastungwährenddesReaktionsablaufswurdedurcheinenentsprechend
größeren Reaktionsaufbau gering gehalten. In vorhergehenden Versuchen von SIMON konnte bei
starkemmechanischen Rühren ein starker Enzymabrieb beobachtet werden, der durch Rührenmit
<200rpmverringertwerdenkonnte.[33]DasEnzymrecyclingwurdeunterStandardbedingungendurch‐
geführt,indemdasracemischeAminrac‐4zusammenmitdemMalonester9unddieCAL‐Bbei60°C
gerührtwird(Abbildung93).
DieAuswertungderErgebnissedeutetaufeinestarkeAbnahmederAktivitätundauchderStabilitätder
CAL‐BwährendderZyklisierungsversuchedurchInaktivierunghin.SokonnteschonimzweitenZyklus
des Recyclings nur noch 30% Umsatz anstelle der 43% des ersten Zyklus erreicht werden. In den
folgendenZyklen(Zyklus3und4)sankderUmsatzweiterauf25%bzw.20%Umsatz.ImfünftenZyklus
konnten24%UmsatznurdurchVerlängerungderReaktionszeitaufübersechsStundenerzieltwerden.
AuchwenneinstetigerAbfall inderAktivitätderCAL‐Bzuerkennenist,bleibtdieStereoselektivität
konstanthoch.InallenFällen,mitAusnahmedeszweitenZyklus(81%ee),konntenguteEnantiomeren‐
überschüssevon>88%undsomitE‐Wertevon>13erreichtwerden.
InderLiteratursowieineinerDoktorarbeitdiesesArbeitskreisessinderfolgreicheRecyclingprozesse
derCAL‐BbeiderRacematspaltungvonAmineninAnwesenheitvonLösungsmittelnsowieinlösungs‐
mittelfreierUmgebungbeschriebenworden.[33,36]DabeiwurdengleichbleibendhoheUmsätzemitsehr
gutenee‐WertenfürdieerstenZyklenerhalten,bevoreinleichterRückgangderUmsätzezubeobachten
war.DierapidesinkendenUmsätzeinderRecycling‐VersuchsreiheimRahmendieserArbeitinlösungs‐
mittelfreiem Milieu deuten auf eine verringerte Aktivität der CAL‐B hin. In der Tat wurde dieses
PhänomenauchvonderArbeitsgruppeumKANERVAbeiderlösungsmittelfreienRacematspaltungvon
rac‐4mitMethoxyester6 beobachtet.[36]DesWeiterenwurdebeobachtet,dassdieLipasebeimehr‐
facherAnwendungbeihöherenTemperaturenteilweisevonTrägermaterialabgelöstwird.[33]
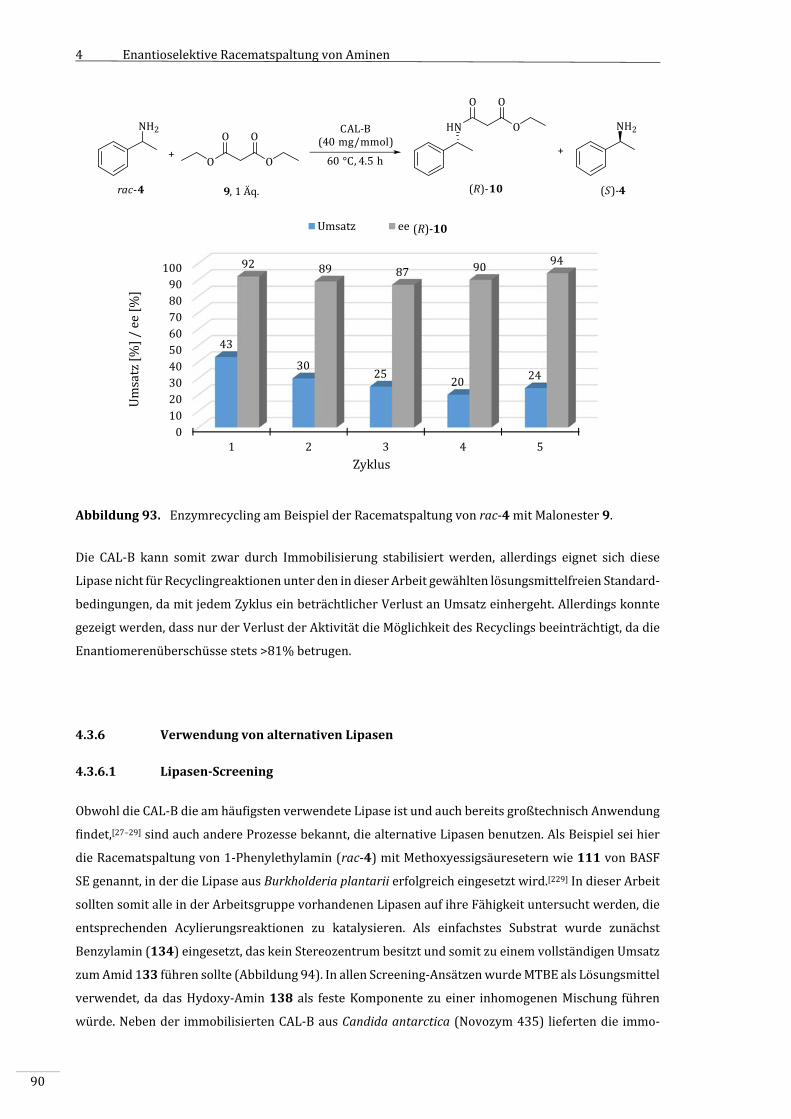
4 EnantioselektiveRacematspaltungvonAminen
90
NH2
O
O
O
O
+
CAL‐B(40mg/mmol)
60°C,4.5h
9,1Äq.
HN
O O
O NH2
+
rac‐4 (S)‐4(R)‐10
Abbildung93. EnzymrecyclingamBeispielderRacematspaltungvonrac‐4mitMalonester9.
Die CAL‐B kann somit zwar durch Immobilisierung stabilisiert werden, allerdings eignet sich diese
LipasenichtfürRecyclingreaktionenunterdenindieserArbeitgewähltenlösungsmittelfreienStandard‐
bedingungen,damitjedemZykluseinbeträchtlicherVerlustanUmsatzeinhergeht.Allerdingskonnte
gezeigtwerden,dassnurderVerlustderAktivitätdieMöglichkeitdesRecyclingsbeeinträchtigt,dadie
Enantiomerenüberschüssestets>81%betrugen.
4.3.6 VerwendungvonalternativenLipasen
4.3.6.1 Lipasen‐Screening
ObwohldieCAL‐BdieamhäufigstenverwendeteLipaseistundauchbereitsgroßtechnischAnwendung
findet,[27–29]sindauchandereProzessebekannt,diealternativeLipasenbenutzen.AlsBeispielseihier
dieRacematspaltungvon1‐Phenylethylamin(rac‐4)mitMethoxyessigsäureseternwie111vonBASF
SEgenannt,inderdieLipaseausBurkholderiaplantariierfolgreicheingesetztwird.[229]IndieserArbeit
solltensomitalleinderArbeitsgruppevorhandenenLipasenaufihreFähigkeituntersuchtwerden,die
entsprechenden Acylierungsreaktionen zu katalysieren. Als einfachstes Substrat wurde zunächst
Benzylamin(134)eingesetzt,daskeinStereozentrumbesitztundsomitzueinemvollständigenUmsatz
zumAmid133führensollte(Abbildung94).InallenScreening‐AnsätzenwurdeMTBEalsLösungsmittel
verwendet,dadasHydoxy‐Amin138 als festeKomponente zueiner inhomogenenMischung führen
würde.NebenderimmobilisiertenCAL‐BausCandidaantarctica(Novozym435)liefertendie immo‐
0102030405060708090100
1 2 3 4 5
43
3025
20 24
92 89 87 90 94
Umsatz[%
]/ee[%]
Zyklus
Umsatz[%] ee[%] (R)‐10

4 EnantioselektiveRacematspaltungvonAminen
91
bilisierteundlyophilisierteLipasenCAL‐B(beidevonC‐LECTA)einennahezuvollständigenUmsatzzum
Amid133(89%bzw.94%).WeitereLipasenführtenzuniedrigenUmsätzenimBereichvon15‐22%.
Beachtlichisthierbei,dassdieLipasenL5,GC4,R10,AY30sowieCE5undD20(allevonAMANO)alle
ungewöhnlichhoheUmsätze zumDiamid137 zeigten.Dieseskannals Indiz füreine sehr langsame
Acylierungsreaktion gesehenwerden, in der die zweite Acylierungmit größererWahrscheinlichkeit
abläuft als die gewünschte einfache Reaktion zumAmid133. Eventuell spielt die durch den ersten
AcylierungsschrittschonentstandeneNähedesAmids133zuraktivenSeitederLipaseeinewichtige
Rolledabei.
NH2
O
O
O
O NH
O
O
O
+60°C,3h
134 91Äq.
133
Lipase(40mg/mmol)
Abbildung94. Lipasen‐ScreeningmitBenzylamin(134)alsSubstrat.
0 10 20 30 40 50 60 70 80 90 100
CAL‐B(Novozym435)
Toyobo
Hogpancreas(Fluka)
L„Amano“5
Rhizopusniveus(Fluka)
L.P.L.„Amano“100S
GC„Amano“4
R„Amano“10
AY„Amano“30
NConc(Amano)
AP6(Amano)
PSimmobilisiertaufToyonite‐200‐P(Amano)
PSimmobilisiertaufDiatomite(Amano)
M‐AP10(Amano)
F‐AP15(Amano)
R10(Amano)
CE„Amano“5
D„Amano“20
AK„Amano“
AYS„Amano“
PS„Amano“SD
CAL‐Bimmobilisiert(c‐LEcta)
CAL‐Blyophilisiert(c‐LEcta)
Umsatz[%]
Lipase
UmsatzAmid113[%] UmsatzDimer114[%]
CE„AMANO“5
D„AMANO“20
AK„AMANO“AYS„AMANO“
PS„AMANO“SDCAL‐Bimmobilisiert(C‐LECTA)
CAL‐Blyophilisiert(C‐LECTA)
R10(AMANO)F‐AP15(AMANO)
M‐AP10(AMANO)
AP6(AMANO)
NConc(AMANO)AY„AMANO“30
R„AMANO“10
GC„AMANO“4L.P.L.„AMANO“100S
L„AMANO“5
Rhizopusniveus(FLUKA)
Hogpancreas(FLUKA)
PSimmob.aufToyonite‐200‐P(AMANO)
PSimmob.Diatomite(AMANO)
133 137
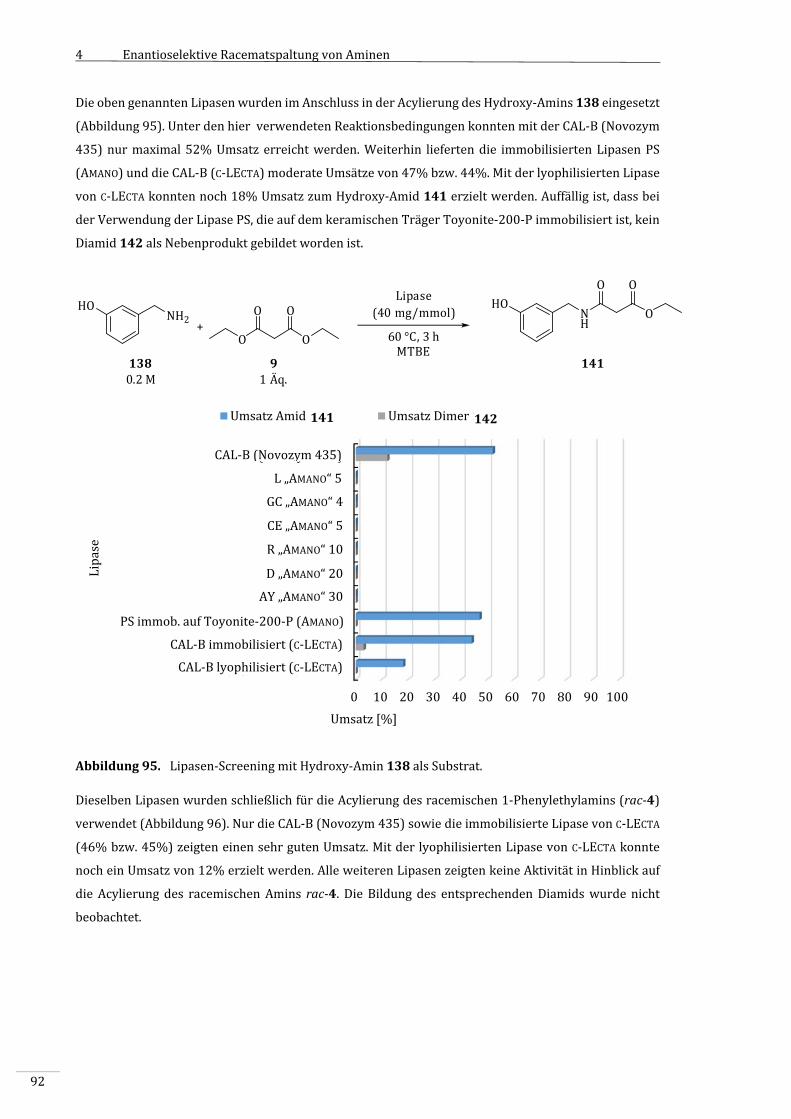
4 EnantioselektiveRacematspaltungvonAminen
92
DieobengenanntenLipasenwurdenimAnschlussinderAcylierungdesHydroxy‐Amins138eingesetzt
(Abbildung95).UnterdenhierverwendetenReaktionsbedingungenkonntenmitderCAL‐B(Novozym
435)nurmaximal52%Umsatzerreichtwerden.Weiterhin liefertendie immobilisiertenLipasenPS
(AMANO)unddieCAL‐B(C‐LECTA)moderateUmsätzevon47%bzw.44%.MitderlyophilisiertenLipase
vonC‐LECTAkonntennoch18%UmsatzzumHydroxy‐Amid141erzieltwerden.Auffälligist,dassbei
derVerwendungderLipasePS,dieaufdemkeramischenTrägerToyonite‐200‐Pimmobilisiertist,kein
Diamid142alsNebenproduktgebildetwordenist.
NH2
O
O
O
O NH
O
O
O
+
1380.2M
91Äq.
141
LipaseHO HO
(40mg/mmol)
60°C,3hMTBE
Abbildung95. Lipasen‐ScreeningmitHydroxy‐Amin138alsSubstrat.
DieselbenLipasenwurdenschließlichfürdieAcylierungdesracemischen1‐Phenylethylamins(rac‐4)
verwendet(Abbildung96).NurdieCAL‐B(Novozym435)sowiedieimmobilisierteLipasevonC‐LECTA
(46%bzw.45%)zeigteneinensehrgutenUmsatz.MitderlyophilisiertenLipasevonC‐LECTAkonnte
nocheinUmsatzvon12%erzieltwerden.AlleweiterenLipasenzeigtenkeineAktivitätinHinblickauf
dieAcylierungdes racemischenAmins rac‐4. DieBildungdes entsprechendenDiamidswurde nicht
beobachtet.
0 10 20 30 40 50 60 70 80 90 100
CAL‐B(Novozym435)
L„Amano“5
GC„Amano“4
CE„Amano“5
R„Amano“10
D„Amano“20
AY„Amano“30
PSimmobilisiertaufToyonite‐200‐P(Amano)
CAL‐Bimmobilisiert(c‐LEcta)
CAL‐Blyophilisiert(c‐LEcta)
Umsatz[%]
Lipase
UmsatzAmid118[%] UmsatzDimer119(%)
CAL‐Blyophilisiert(C‐LECTA)
CAL‐Bimmobilisiert(C‐LECTA)
PSimmob.aufToyonite‐200‐P(AMANO)
AY„AMANO“30
D„AMANO“20
R„AMANO“10
CE„AMANO“5
GC„AMANO“4
L„AMANO“5
141 142
CAL‐B(Novozym435)
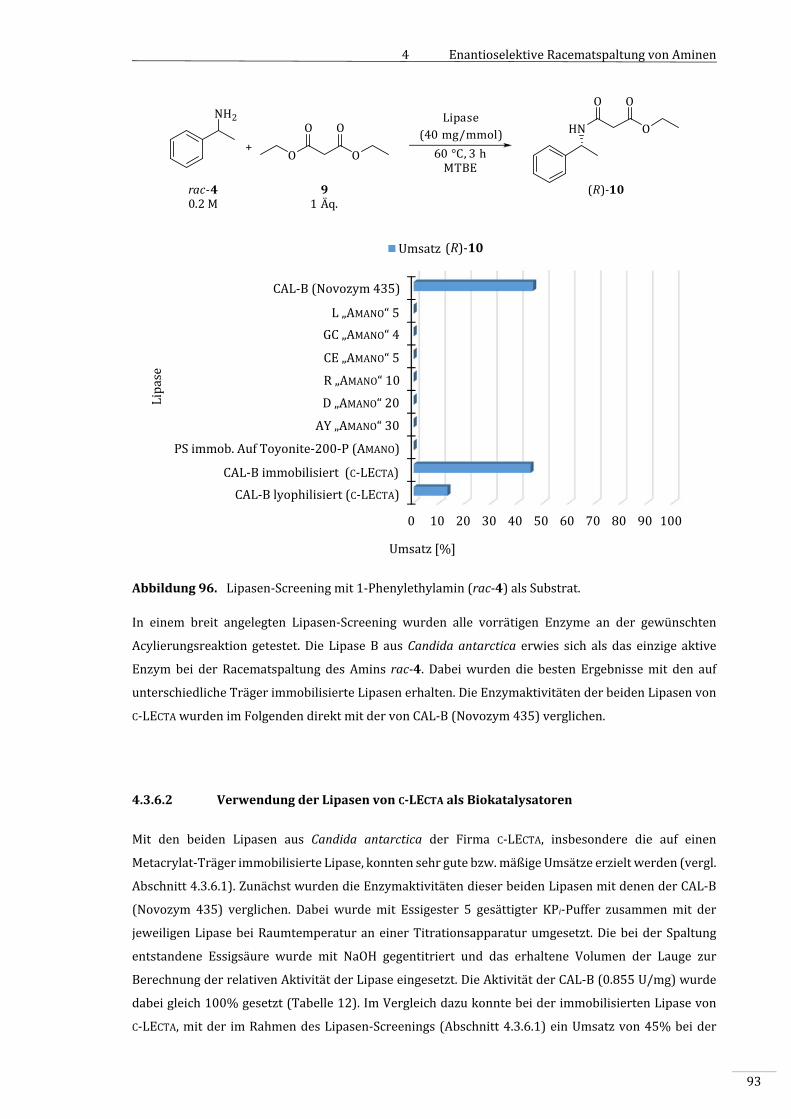
4 EnantioselektiveRacematspaltungvonAminen
93
O
O
O
O
+
(R)‐10rac‐40.2M
91Äq.
NH2HN
O
O
OLipase
(40mg/mmol)
60°C,3hMTBE
Abbildung96. Lipasen‐Screeningmit1‐Phenylethylamin(rac‐4)alsSubstrat.
In einem breit angelegten Lipasen‐Screening wurden alle vorrätigen Enzyme an der gewünschten
Acylierungsreaktion getestet. Die LipaseB ausCandidaantarctica erwies sich als das einzige aktive
Enzymbei der Racematspaltung des Amins rac‐4. Dabeiwurden die besten Ergebnissemit den auf
unterschiedlicheTrägerimmobilisierteLipasenerhalten.DieEnzymaktivitätenderbeidenLipasenvon
C‐LECTAwurdenimFolgendendirektmitdervonCAL‐B(Novozym435)verglichen.
4.3.6.2 VerwendungderLipasenvonC‐LECTAalsBiokatalysatoren
Mit den beiden Lipasen aus Candida antarctica der Firma C‐LECTA, insbesondere die auf einen
Metacrylat‐TrägerimmobilisierteLipase,konntensehrgutebzw.mäßigeUmsätzeerzieltwerden(vergl.
Abschnitt4.3.6.1).ZunächstwurdendieEnzymaktivitätendieserbeidenLipasenmitdenenderCAL‐B
(Novozym 435) verglichen. Dabei wurdemit Essigester 5 gesättigter KPi‐Puffer zusammenmit der
jeweiligenLipasebeiRaumtemperaturaneinerTitrationsapparaturumgesetzt.DiebeiderSpaltung
entstandene Essigsäure wurde mit NaOH gegentitriert und das erhaltene Volumen der Lauge zur
BerechnungderrelativenAktivitätderLipaseeingesetzt.DieAktivitätderCAL‐B(0.855U/mg)wurde
dabeigleich100%gesetzt(Tabelle12).ImVergleichdazukonntebeiderimmobilisiertenLipasevon
C‐LECTA,mitder imRahmendesLipasen‐Screenings(Abschnitt4.3.6.1)einUmsatzvon45%beider
0 10 20 30 40 50 60 70 80 90 100
CAL‐BCandidaantarctica(Novozym435)
L„Amano“5
GC„Amano“4
CE„Amano“5
R„Amano“10
D„Amano“20
AY„Amano“30
PSimmob.aufToyonite‐200‐P(Amano)
CAL‐Bimmob.(c‐LEcta)
CAL‐Blyo.(c‐LEcta)
Umsatz[%]
Lipase
Umsatz[%]
L„AMANO“5
GC„AMANO“4
CE„AMANO“5
R„AMANO“10
D„AMANO“20
AY„AMANO“30
PSimmob.AufToyonite‐200‐P(AMANO)
CAL‐Bimmobilisiert(C‐LECTA)
CAL‐Blyophilisiert(C‐LECTA)
CAL‐B(Novozym435)
(R)‐10

4 EnantioselektiveRacematspaltungvonAminen
94
Acylierungvonrac‐4erhaltenwurde,nureinerelativeAktivitätvonnur68%nachgewiesenwerden.
Von der lyophilisierte Lipase von C‐LECTA wurde nur 1/20 der Enzymmenge eingesetzt. Die dabei
ermittelterelativeAktivitätdieserLipasebetrug2000%.AufgrunddesniedrigenUmsatzesimRahmen
desLipasen‐ScreeningsistdiesesErgebnisüberraschend.
Tabelle12. EnzymkatalysierteSpaltungdesEssigesters5mitunterschiedlichenLipasenbei20°C.
Eintrag LipaseEnzym
[mg/mmol]
Steigung
[mL/sec]
Aktivität
[U/mg]
Rel.Aktivität
[%]
1 Novozym435 20 0.0057 0.855 100
2 C‐LECTAimmobilisiert 20 0.0039 0.585 68
3 C‐LECTAlyophilisiert 1 0.0057 17.100 2000
DieenzymatischeSpaltungdesEssigesters5durchdieCAL‐B(Novozym435)sowiedieimmobilisierte
und lyophilisierte Lipasen von C‐LECTA wurden im Anschluss bei 60 °C an der Titrationsapparatur
durchgeführt(Tabelle13).DabeiwurdeerneutdieAktivitätderCAL‐B(Novozym435)gleich100%
gesetzt.Diemittels immobilisierterLipase(C‐LECTA)erhaltenerelativeAktivitätgleichtmit69%den
WerteninTabelle12(Eintrag2).
Tabelle13. EnzymkatalysierteSpaltungdesEssigesters5mitunterschiedlichenLipasenbei60°C.
Eintrag LipaseEnzym
[mg/mmol]
Aktivität
[U/mg]
Rel.Aktivität
[%]
1 Novozym435 20 3.180 100
2 C‐LECTAimmobilisiert 20 2.190 69
3 C‐LECTAlyophilisiert 1 31.800 1000
Bei der lyophilisierten CAL‐B von C‐LECTA konnte eine relative Aktivität von 1000% nachgewiesen
werden. Dieses entspricht der Hälfte desWertes bei einer Temperatur von 20 °C und lässt darauf
schließen, dass die nicht‐immobilisierte CAL‐B bei höherenTemperaturen an Stabilität einbüßt.Die
erhöhteStabilitätvielerEnzymedurchImmobilisierungistinderLiteraturoftmalsbeschrieben.[13]Auch
in denProduktinformationsblätternderFirma C‐LECTAwirdder immobilisiertenCAL‐B einehöhere
Thermostabilitätzugewiesen.[265]

4 EnantioselektiveRacematspaltungvonAminen
95
DadiebeidenLipasenvonC‐LECTAhohebzw.sehrhoheAktivitäteninHinblickaufdieenzymatische
Esterspaltung aufweisen, sollte im Folgenden die enzymatische Racematspaltung von rac‐4 unter
Standardbedingungenuntersuchtwerden.DiejeweiligeEnzymmengewurdedafürentsprechendandie
ermittelterelativeAktivitätangepasst.
DieAcylierungvon1‐Phenylethylamin(rac‐4)mitderimmobilisiertenCAL‐BvonC‐LECTAwurdezum
einen lösungsmittelfreiundzumanderen inAnwesenheitvonMTBEalsLösungsmitteldurchgeführt
(Tabelle14).Zwar führtendabeibeideAcylierungsreaktionen zu einemvollständigenUmsatz, aller‐
dingskonntenurinAbwesenheitvonLösungsmitteln(Eintrag1)sehrguteee‐Wertevon96%(E=194)
erreichtwerden.ImGegensatzdazuwurdeninAnwesenheitvonMTBEnur84%eeundeinguterE‐Wert
von29erzielt (Eintrag2).Die immobilisierteLipasevonC‐LECTA stellt somit einweitereswichtiges
WerkzeugzurDarstellungenantiomerenreinerAmineinlösungsmittelfreierUmgebungdar.
Tabelle14. EnzymatischRacematspaltungvonrac‐4mittelsimmobilisierterCAL‐BvonC‐LECTA.
Eintrag MTBE[mL] Umsatz[%] eeP [%] eeS[%]a) E
1 0 52 96 96 194
2 0.5 49 84 81 29a)berechnetmit„Enantioselectivity“[258].
ParallelzuderenzymatischenRacematspaltungvonrac‐4mittelsimmobilisierterCAL‐BvonC‐LECTA
wurdedieTrennungderEnantiomeremithilfederlyophilisiertenCAL‐B,ebenfallsvonC‐LECTA,unter‐
sucht.AuchindiesemFallwurdedieEnzymmengeandieermittelterelativeAktivitätangepasst.
Die Racematspaltung von rac‐4 durch die lyophilisierte CAL‐B wurde zunächst unter Standard‐
bedingungeninAbwesenheitvonLösungsmittelndurchgeführt(Tabelle15,Eintrag1).Dabeiwurdenur
eingeringerUmsatzvon9%erzielt,jedochmiteinemgutenEnantiomerenüberschussvon81%(E=1).
Mitder fünffachenEnzymmengekonnte sowohlderUmsatzauf26%als auchder ee‐Wert auf91%
gesteigertwerden.DiesesentsprichteinerexzellentenEnantioselektivitätvon29.EineVerlängerung
derReaktionszeitauf24Stundenmit4mg/mmolCAL‐BliefertenureinenetwashöherenUmsatzvon
19%, jedoch war hierbei der Enantiomerenüberschuss mit 74% (E = 8) nicht zufriedenstellend
(Eintrag3). Eventuell hat in diesem Fall bei der langen Reaktionsdauer eine parallel chemische
Acylierungstattgefunden,sodassderee‐Wertdadurchsinkt.InteressanterweisewurdebeieinerReak‐
tionstemperaturvon20°Cbei24StundenReaktionsdauernahezudasidentischeErgebnisderReaktion
unterStandardbedingungenerhalten.AuchdieDurchführungderRacematspaltunginAnwesenheitvon
MTBEalsLösungsmittelliefertenur7%Umsatzmit87%ee(Eintrag5).DieZugabevonWasserzuder
ansonstenlösungsmittelfreienSyntheseführtenurzueinemUmsatzvon6%.AufeineBestimmungdes
EnantiomerenüberschussesistindiesemFallaufgrunddesniedrigenUmsatzesverzichtetworden.

4 EnantioselektiveRacematspaltungvonAminen
96
Tabelle15. EnzymkatalysierteRacematspaltungvonrac‐4mittelslyophilisierterCAL‐BvonC‐LECTA.
Eintrag CAL‐B[mg/mmol]
T[°C] t[h] Umsatz[%] ee[%] E
1 4 60 4.5 9 81 10
2 20 60 4.5 26 91 29
3 4 60 24 19 74 8
4 4 20 24 6 80 10
5a) 4 60 4.5 7 87 2
6b) 4 60 4.5 6 n.b. n.b.a)Zugabevon0.5mLMTBE;b)Zugabevon10µLdest.Wasser;n.b.=nichtbestimmt.
Insgesamt ist festzustellen, dass die enzymatischeRacematspaltung des Amins rac‐4 unter lösungs‐
mittelfreienBedingungentrotzdersehrhohenrelativenAktivitätderlyophilisiertenCAL‐BimVergleich
zudenbeidenimmobilisiertenLipasen(Novozym435unddieCAL‐BvonC‐LECTA)nichtzuzufrieden‐
stellendenErgebnissenführt.AlleindieErhöhungderEnzymmengeumdasFünffache(20mg/mmol
anstellevon4mg/mmolunterStandardbedingungen)lieferteeinenmäßigenUmsatzmithoherEnan‐
tiomerenreinheitundEnantioselektivitätvon29(Eintrag2).DieVerwendungeinerhohenEnzymmenge
istdemnachsinnvoll,umNebenreaktionenzumDiamidzuunterdrücken.AusökonomischerSichtist
dieseArtderReaktionsführungjedochnichtratsam.
4.3.7 VariationderAcyldonoren
BeiderRacematspaltungvonAminenistdieWahldesAcyldonorsvongroßerBedeutung.Hochreaktive
Acyldonoren, wie beispielsweise Enolester, können Carbonyl‐Verbindungen freisetzen, die dann
wiederummitderAminfunktionzueinemIminreagierenkönnten.[31]AufgrundderhohenNucleophilie
desAminsführendiesespontanen,nicht‐enzymatischenAcylierungsreaktionensomitzueinerverrin‐
gertenEnantioselektivität.SehrguteErgebnissewurdeninderLiteraturmitAcyldonorenerhalten,die
inβ–PositioneinHeteroatom,meistensSauerstoff,tragen,wieMethoxyessigsäureethyl‐oder–propyl‐
ester (111 bzw.6,Abbildung4).DieAnwesenheiteinesHeteroatomshateineerhöhteAktivitätdes
Carbonyl‐KohlenstoffszurFolge.[229]InderArbeitvonSIMONwurdeschließlichMalonsäurediethylester
(9) als sehr effizienter Acyldonor gefunden.[33,34] Aufgrund des sehr breiten Substratspektrums der
CAL‐BkanneineVielzahlanAcyldonorenverwendetwerden.EineAuswahlanAcyldonorenwurdebei
derenzymatischenRacematspaltungvonAminrac‐4unterlösungsmittelfreienBedingungeneingesetzt
unddieErgebnissederAcylierungmiteinanderundmitMalonsäurediethylester(9)alsStandard‐Acyl‐
donorverglichen.BeidenuntersuchtenAcyldonorenhandeltessichumPhenoxyessigsäureethylester

4 EnantioselektiveRacematspaltungvonAminen
97
(122), Phenylessigsäureethylester (147), Methoxyessigsäureethylester (111), Cyanessigsäureethyl‐
ester(124)undEssigsäurevinylester(125).DieausgewähltenAcyldonorensindinAbbildung97dar‐
gestellt.
Abbildung97. AlternativeAcyldonoren.
4.3.7.1 VergleichderAcyldonorenunterStandard‐Reaktionsbedingungen
Bei der enzymatischen Racematspaltung von 1‐Phenylethylamin (rac‐4) unter Standard‐Reaktions‐
bedingungenbei60°CReaktionstemperatursolltendieAktivitätenderAcyldonorenausAbschnitt4.3.5
untereinander verglichenwerden. Die Ergebnisse sind in Abbildung 98 graphisch dargestellt. Es ist
ersichtlich,dassbeiderAcylierungdesAminsrac‐4bereitsnach4.5StundenReaktionszeitmitdem
Standard‐AcyldonorMalonsäurediethylester(9)46%Umsatzsowiesehrgute98%eeerhaltenwurden.
DiesesentsprichteinemexzellentenE‐Wertvon>200.
DerAcyldonorPhenoxyessigester122lieferte51%Umsatzmit65%ee(E=9).EineVerringerungder
Reaktionstemperaturvon60°Cauf40°C lieferteeinähnlichesErgebnis,da49%UmsatzzumAmid
(R)‐121sowie67%ee(E=10)erreichtwordensind.TrotzdergutenUmsätzesinddieseWertefüreine
effizienteRacematspaltungnichtausreichend.
Der Cyanoester124 wurde in der Literatur bereits als Acyldonor für die Synthese von Zytostatica
beschrieben,jedochnurmittelsChemokatalyse.[266]BeiderenzymatischkatalysiertenAcylierungliefert
der Acyldonor124 einen guten Umsatz von 45%. Allerdings können dabei nur 10% ee beobachtet
werden,demnachisteineRacematspaltungauchmitdiesemDonornichterfolgreich.MitMethoxyessig‐
ester111konntennach4.5StundenReaktionszeitzwarnurmäßige25%UmsatzzumAmid(R)‐8erzielt
werden, jedoch lagderEnantiomerenüberschuss erfreulicherweise bei 92%.Dieses entspricht einer
sehrgutenEnantioselektivitätvon33.AllgemeinwerdenmitMethoxyessigesternbeiderAcylierungvon
AmineninderLiteraturvollständigeUmsätzeerhalten.[30]SosetztenDITRICHetal.dasAminrac‐4mit

4 EnantioselektiveRacematspaltungvonAminen
98
Methoxyessigsäurepropylester(6)mit46%UmsatzzumAmid(R)‐8enantiomerenrein(E>100)um.
DieseEnantioselektivitätenkönnenimRahmendieserArbeitundauchbeivorherigenAcetylierungs‐
reaktioneninorganischemLösungsmittelinnerhalbderArbeitsgruppe[33,34]nichtmehrerreichtwerden.
DerGrunddafürkannunterandereminderViskositätdeslösungsmittelfreienSystemsunddesdadurch
verminderten Transportprozesses innerhalb der Reaktionsmischung liegen. Es sollte im Anschluss
überprüftwerden, inwiefern eineVerlängerungderReaktionszeit eine SteigerungdesUmsatzes zur
Folgehat.
R O
OR'+
NH2NH R
O
rac‐4
CAL‐B(40 mg/mmol)
Acyldonor, 1 Äq.
+
NH2
(S)‐4
60 °C, 4.5 h
9 (R = CH2COOEt, R' = Et)122 (R = CH2COPh, R ' = Et)147 (R = CH2Ph, R' = Et)111 (R = CH2OMe. R' = Et)124 (R = CH2CN, R' = Et)125 (R = CH3, R' = CHCH2)
(R)‐10 (R = CH2COOEt)(R)‐121 (R = CH2COPh)(R)‐126 (R = CH2Ph)(R)‐8 (R = CH2OMe)(R)‐123 (R = CH2CN)(R)‐7 (R = CH3)
(R)‐Amid
Abbildung98. VergleichderalternativenAcyldonorenunterStandard‐Reaktionsbedingungen.
EnttäuschendeErgebnisseliefertendieRacematspaltungenvonrac‐4mitPhenylessigester147sowie
Vinylester 125. Mit dem Acyldonor 147 konnten nur 15% Umsatz zum Amid (R)‐126 beobachtet
werden.DieseskannmitdemfehlendenHeteroatominβ‐Positionunddiedadurchbedingtegeringere
AktivitätdesEsters147begründetwerden.[31]VermutlichwirktsichdieKettenlängedesDonors147
negativ auf die SelektivitätdesEnzymsaus.Der ee‐Wert dieserAcylierungsreaktionbeträgt für das
Amid(R)‐12653%miteinerinakzeptablenEnantioselektivitätvon4.DerAcyldonor147imdemnach
ungeeignetfüreineenzymatischeAcylierungdesAminsrac‐4.DieAcylierungmitdemVinylester125
liefertbeiebenfallsnur16%UmsatzzumAmid(R)‐7jedochnur10%ee.
0
10
20
30
40
50
60
70
80
90
100
9 122 147 111 121 125
4351
15
25
45
16
96
65
53
92
10 10Umsatz[%
]/ee[%]
Acyldonor
Umsatz[%] ee[%]

4 EnantioselektiveRacematspaltungvonAminen
99
UnterdengewähltenStandard‐ReaktionsbedingungenkonntefolglichmitMalonsäurediethylester(9)
alsAcyldonorder besteUmsatz unddie höchsteEnantiomerenreinheit von96%erzieltwerden. Im
AnschlussandieseVersuchsreihewurdendieAcylierungsreaktionenmitPhenoxy‐oderVinylessigester
(122bzw.125)beiverringerterTemperaturdurchgeführtunddieErgebnissemiteinanderverglichen.
4.3.7.2 VergleichderAcyldonorenbeiverringerterTemperatur
DiealternativenAcyldonorenwurdenauchbeiverringerterTemperaturvon40°CmitdemAminrac‐4
umgesetztunddieErgebnissemiteinanderverglichen.DasHerabsetzenderReaktionstemperaturkann
sich,besondersbei starkaktiviertenAcyldonorenwie122 und124, als vorteilhaft erweisen,dadie
Reaktionendannlangsamerundsomitselektiverablaufenkönnten.AlsStandard‐Acyldonordientehier‐
beiwiederMalonsäurediethylester(9).DieErgebnissesindinAbbildung99graphischdargestellt.
R O
O
R'+
NH2NH R
O
rac‐4
CAL‐B(40 mg/mmol)
Acyldonor, 1 Äq.
+
NH2
(S)‐4
40 °C, 4.5 h
9 (R = CH2COOEt, R' = Et)122 (R = CH2COPh, R ' = Et)147 (R = CH2Ph, R' = Et)111 (R = CH2OMe. R' = Et)124 (R = CH2CN, R' = Et)125 (R = CH3, R' = CHCH2)
(R)‐10 (R = CH2COOEt)(R)‐121 (R = CH2COPh)(R)‐126 (R = CH2Ph)(R)‐8 (R = CH2OMe)(R)‐123 (R = CH2CN)(R)‐7 (R = CH3)
(R)‐Amid
Abbildung99. VergleichderalternativenAcyldonorenbeiverringerterReaktionstemperatur.
0
10
20
30
40
50
60
70
80
90
100
9 122 147 111 124 125
34
49
4
3140
17
95
67
82
11
22
Umsatz[%
]/ee[%]
Acyldonor
Umsatz[%] ee[%]

4 EnantioselektiveRacematspaltungvonAminen
100
Der Malonester9 liefert bei 40 °C Reaktionstemperatur mit 34% Umsatz und sehr hoher Enantio‐
merenreinheitvon96%einensehrgutenE‐Wertvon80.DiesesistfüreineeffizienteRacematspaltung
einhervorragendesErgebnis.MitPhenoxyessigester122alsAcyldonorwurdenzwarsehrgute49%
Umsatz erzielt, jedoch konnten nur mäßige 67% ee beobachtet werden. Dieses entspricht einer
Enantioselektivitätvon10undist,ebensowiebei60°C,nichtzufriedenstellend.
Der Methoxyessigester 111 lieferte zwar einen leicht erhöhten Umsatz von 31% im Vergleich zur
Acylierungbei60°C,allerdingskonnteeinleichtesAbsinkendesee‐Wertesvon92%auf82%bei40°C
(E=14)detektiertwerden.AucheineweitereVerringerungderTemperaturauf20°Cführtenochzu
guten31%Umsatz,allerdingsnurmitmäßigen71%ee(E=8).DieniedrigenEnantiomerenüberschüsse
desAmids(R)‐8beiVerringerungderReaktionstemperaturaufnurnoch71%eeweichendeutlichvon
deninderLiteraturbeiRaumtemperaturerhaltenenWertenmitAcyldonor6ab(>99%ee,E>100).[30]
Eine Verringerung der Selektivität der CAL‐B bei niedrigeren Temperaturen kann demnach ausge‐
schlossenwerden.
UnveränderteErgebnisse (40%Umsatzmit11%eezumAmid(R)‐123) imVergleichzudenAcylie‐
rungsreaktionenbei60°CwurdenmitCyanoessigester124erhalten.UnterdenhierverwendetenReak‐
tionsbedingungenisteineRacematspaltungmitdiesemDonornichtmöglich.MitVinylessigester125
konntenauchbei40 °CReaktionstemperaturnur17%Umsatzerzieltwerden.FüreineguteDurch‐
mischungderdickflüssigenReaktionsmischungwarindiesemFalldieZugabevonMTBEalsLösungs‐
mittelnotwendig.Deree‐Wertbeträgthierbei22%.DiesesentsprichteinemE‐Wertvon2undistnicht
ausreichendfürdieRacematspaltung.EbenfallsnegativeErgebnissemit4%Umsatzwerdenmitdem
Phenylessigester147 als Acyldonor erhalten. Auf eine Bestimmung des Enantiomerenüberschusses
wurdeindiesemFallverzichtet.
EshatsichfolglichauchbeiverringerterReaktionstemperaturvon40°CMalonsäurediethylester(9)als
dergeeignetsteAcyldonorerwiesen,sowohlinHinblickaufdenUmsatzalsauchaufdieEnantiomeren‐
reinheitderRacematspaltung.MitdenAcyldonorenMalonester9,Phenoxyessigester122,Cyanessig‐
ester124sowiedemMethoxyessigester111wurdenimFolgendenKinetikenderRacematspaltungdes
Aminsrac‐4unterlösungsmittelfreienStandard‐Bedingungenuntersucht.
4.3.7.3 ParallelablaufendechemischeAcylierungvon1‐Phenylethylamin(rac‐4)
In den beiden vorangegangenen Abschnitten sind teilweise ungewöhnlich niedrige Enantiomeren‐
überschüsse erhaltenworden.Dieses sollte daraufhin näher untersuchtwerden, indemdie entspre‐
chendenAcylierungsreaktionenvonrac‐4 zwarunterStandard‐Bedingungen, jedoch inAbwesenheit
derLipaseCAL‐Bdurchgeführtwurden(Abbildung100).
SowohlmitdemCyanessigester124,alsauchinnochstärkeremMaßemitdemVinylessigester125,
findenunkatalysierteAcylierungsreaktionenstatt.DieseserklärtdiesehrniedrigenEnantiomerenüber‐
schüsse,dieindenAbschnitten4.3.7.1und4.3.7.2beschriebenwordensind.SokonnteninAbwesenheit
der CAL‐B bei 60 °C und 4.5StundenReaktionszeit bereits 33%Umsatz zum entsprechenden race‐
mischenAmidrac‐123mit3%ee(E=1)erhaltenwerden.DurchVerringerungderTemperaturauf

4 EnantioselektiveRacematspaltungvonAminen
101
40°CkanndiechemischeReaktionzueinemGroßteilunterdrücktwerden,danurnoch12%Umsatz
ermitteltwerden.
R O
OR'+
NH2HN R
O
rac‐4 Acyldonor, 1 Äq.
20 ‐ 60 °C4.5 h
9 (R = CH2COOEt, R' = Et)122 (R = CH2COPh, R ' = Et)147 (R = CH2Ph, R' = Et)111 (R = CH2OMe. R' = Et)124 (R = CH2CN, R' = Et)125 (R = CH3, R' = CHCH2)
rac‐10 (R = CH2COOEt)rac‐123 (R = CH2COPh)rac‐126 (R = CH2Ph)rac‐8 (R = CH2OMe)rac‐123 (R = CH2CN)rac‐7 (R = CH3)
Abbildung100. ParallelablaufendechemischeAcylierungvonrac‐4.
ImFalledesVinylessigesters125könnenbei60°CReaktionstemperaturbiszu93%desAmidsrac‐8
erreichtwerden.DieGeschwindigkeitderchemischenAcylierungkannwährendderReaktionsführung
mit und ohne Lipase beobachtet werden, da bereits nach wenigen Minuten eine Braunfärbung zu
erkennenist.Dieses istmiteinererhöhtenReaktivitätdesEsters125zubegründenunderklärtden
relativniedrigenUmsatzvon16%inAbschnitt4.3.7.1,derdurchenzymatischeAcylierungdesAmins
rac‐4 erhaltenwurde. Obwohl der Vinylester125 bereits erfolgreich zur Acylierung von Alkoholen
unter lösungsmittelfreien Bedingungen[248] eingesetzt worden ist, eignet er sich nicht zur Racemat‐
spaltungvonrac‐4.
Wird mit stark aktivierten Acyldonoren gearbeitet, so lässt sich die chemische Acylierung durch
VerringerungderReaktionstemperaturzueinemgewissenTeilvermeiden.Allerdingsmussinsolchen
Fällen die vorhandeneAktivität der CAL‐B bei entsprechendenTemperaturen bestimmtwerden, da
dieseihrTemperaturoptimumbei60–80°Chat.[195]
40°C60°C
0102030405060708090100
9 122 147 111 122 125
0 0 0 0
12
81
0 0 0 2
33
93
Umsatz[%
]
Acyldonor
40°C 60°C

4 EnantioselektiveRacematspaltungvonAminen
102
4.3.8 ReaktionskinetikenderRacematspaltungvon1‐Phenylethylamin(rac‐4)mit
unterschiedlichenAcyldonoren
Um die Effizient der unterschiedlichen Acyldonoren effektiv miteinander vergleichen zu können,
wurdendieAnfangsgeschwindigkeitenkinetischerRacematspaltungendieserAcyldonorenmiteinander
undmitMalonsäurediethylester(9)alsStandard‐Acyldonorverglichen.
DieReaktionskinetikenderAcylierungsreaktionendesAminsrac‐4wurdenunterdenStandard‐Reak‐
tionsbedingungen durchgeführt. Dabei wurden die Reaktionen nach der entsprechenden Zeit abge‐
brochenundaufgearbeitet,wasgenerellzukleinerenAbweichungendurchWägefehlernführenkann.
WeitereFehlerquellensinddieSchwankungsbreitenbeiderAuswertungder1H‐NMR‐Spektrensowie
derHPLC‐Chromatogramme.DerUmsatzwirdbeidiesenAcylierungsreaktioneninBezugaufdasAmin
rac‐4bestimmt,dadieFlüchtigkeitderAcyldonorennichtuntersuchtwordenist.
AlsVergleichfürdiealternativenAcyldonorendientedieReaktionskinetikderAcylierungvonrac‐4mit
Malonsäurediethylester(9)alsAcyldonorunterlösungsmittelfreienBedingungen(Abbildung101).Es
ist ein steterAnstiegdesUmsatzesmit der Zeit zu erkennen bis nach 24 Stunden ein vollständiger
Umsatzerreichtwird.DieEnantiomerenüberschüssefürdasAmid(R)‐10liegenbeidiesenAcylierungs‐
reaktionenimmer>94%ee.EineAusnahmestelltdabeiderReaktionmitachtStundenReaktionszeitan,
beidernur89%eebeobachtetwerdenkönnen.DieseAbweichungkannnichtaufFehlerwährendder
Reaktionsführungbegründetwerden.
Die Kinetik der Racematspaltung von rac‐4 mit Malonsäurediethylester (9) wurde in vorherigen
ArbeitenimArbeitskreis[33]bereitsuntersucht,allerdingsbeieinerReaktionstemperaturvon80°Cund
inAnwesenheit vonn‐Heptanals Lösungsmittel.Dabei konnte ebenfalls eine schnelleReaktionsrate
festgestelltwerden,wobeidervollständigeUmsatzbereitsnachdreiStundenerreichtwurde.Allerdings
warderEinsatzvon200mg/mmolCAL‐B,alsoderfünffachenMengedesimRahmendieserKinetik‐
studieverwendetenEnzyms,fürdieVersuchsreihenotwendig.
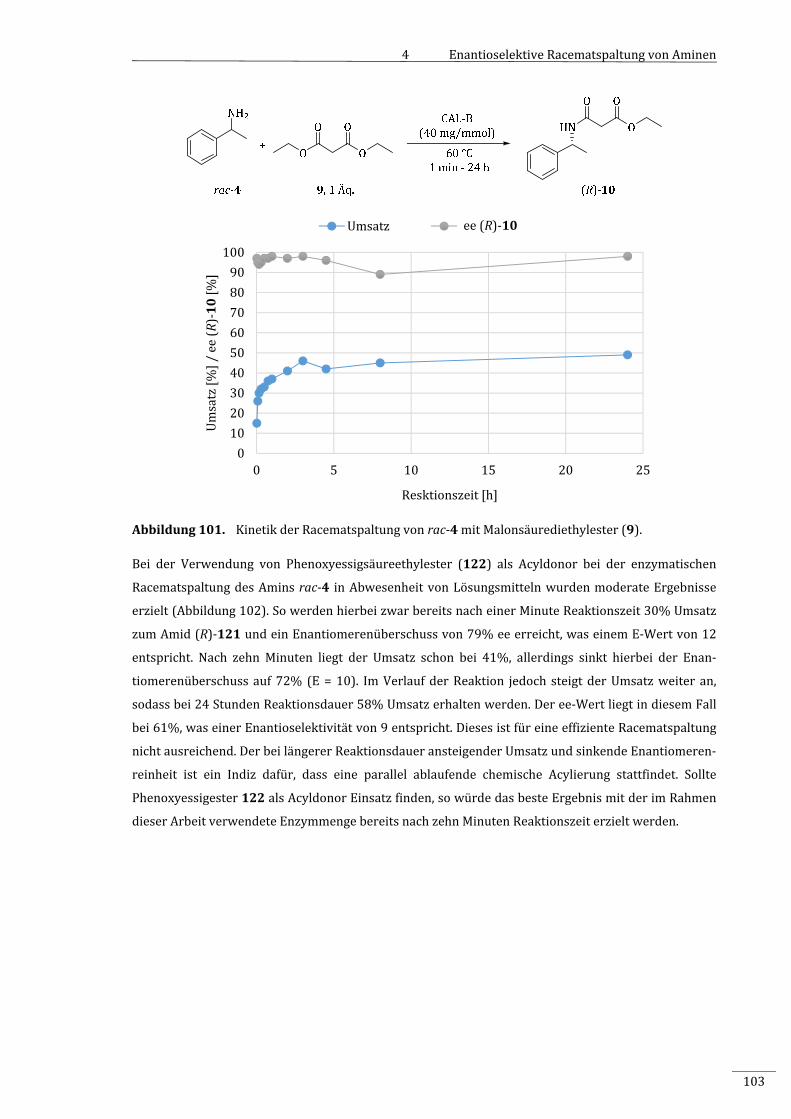
4 EnantioselektiveRacematspaltungvonAminen
103
Abbildung101. KinetikderRacematspaltungvonrac‐4mitMalonsäurediethylester(9).
Bei der Verwendung von Phenoxyessigsäureethylester (122) als Acyldonor bei der enzymatischen
RacematspaltungdesAminsrac‐4 inAbwesenheitvonLösungsmittelnwurdenmoderateErgebnisse
erzielt(Abbildung102).SowerdenhierbeizwarbereitsnacheinerMinuteReaktionszeit30%Umsatz
zumAmid(R)‐121undeinEnantiomerenüberschussvon79%eeerreicht,waseinemE‐Wertvon12
entspricht. Nach zehn Minuten liegt der Umsatz schon bei 41%, allerdings sinkt hierbei der Enan‐
tiomerenüberschussauf72% (E=10). ImVerlaufderReaktion jedoch steigtderUmsatzweiter an,
sodassbei24StundenReaktionsdauer58%Umsatzerhaltenwerden.Deree‐WertliegtindiesemFall
bei61%,waseinerEnantioselektivitätvon9entspricht.DiesesistfüreineeffizienteRacematspaltung
nichtausreichend.DerbeilängererReaktionsdaueransteigenderUmsatzundsinkendeEnantiomeren‐
reinheit ist ein Indiz dafür, dass eine parallel ablaufende chemische Acylierung stattfindet. Sollte
Phenoxyessigester122alsAcyldonorEinsatzfinden,sowürdedasbesteErgebnismitderimRahmen
dieserArbeitverwendeteEnzymmengebereitsnachzehnMinutenReaktionszeiterzieltwerden.
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25
Umsatz[%
]/ee(R)‐10[%]
Resktionszeit[h]
Umsatz[%] ee(R)‐10[%]ee(R)‐10
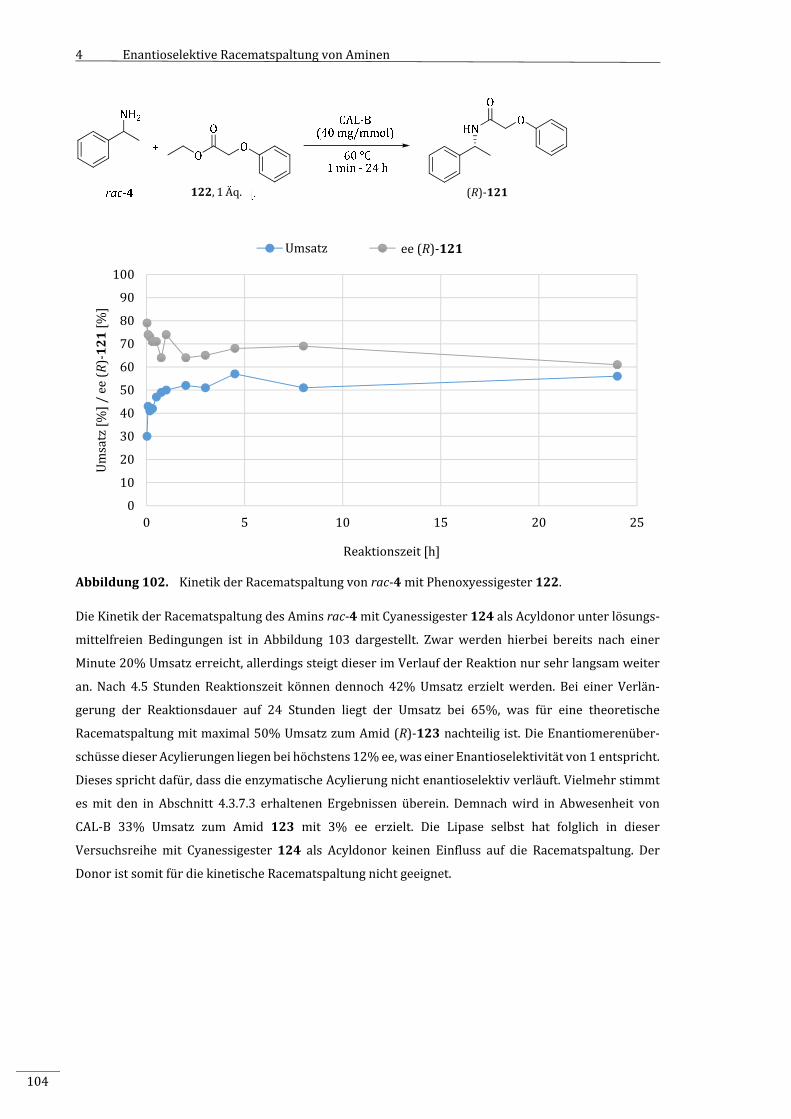
4 EnantioselektiveRacematspaltungvonAminen
104
Abbildung102. KinetikderRacematspaltungvonrac‐4mitPhenoxyessigester122.
DieKinetikderRacematspaltungdesAminsrac‐4mitCyanessigester124alsAcyldonorunterlösungs‐
mittelfreien Bedingungen ist in Abbildung 103 dargestellt. Zwar werden hierbei bereits nach einer
Minute20%Umsatzerreicht,allerdingssteigtdieserimVerlaufderReaktionnursehrlangsamweiter
an. Nach 4.5 Stunden Reaktionszeit können dennoch 42%Umsatz erzieltwerden. Bei einer Verlän‐
gerung der Reaktionsdauer auf 24 Stunden liegt der Umsatz bei 65%, was für eine theoretische
Racematspaltungmitmaximal50%UmsatzzumAmid(R)‐123nachteiligist.DieEnantiomerenüber‐
schüssedieserAcylierungenliegenbeihöchstens12%ee,waseinerEnantioselektivitätvon1entspricht.
Diesessprichtdafür,dassdieenzymatischeAcylierungnichtenantioselektivverläuft.Vielmehrstimmt
esmit den in Abschnitt4.3.7.3 erhaltenen Ergebnissen überein. Demnachwird in Abwesenheit von
CAL‐B 33% Umsatz zum Amid 123 mit 3% ee erzielt. Die Lipase selbst hat folglich in dieser
Versuchsreihe mit Cyanessigester124 als Acyldonor keinen Einfluss auf die Racematspaltung. Der
DonoristsomitfürdiekinetischeRacematspaltungnichtgeeignet.
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25
Umsatz[%
]/ee(R)‐121[%]
Reaktionszeit[h]
Umsatz[%] ee(R)‐117[%]ee(R)‐121
(R)‐121122,1Äq.
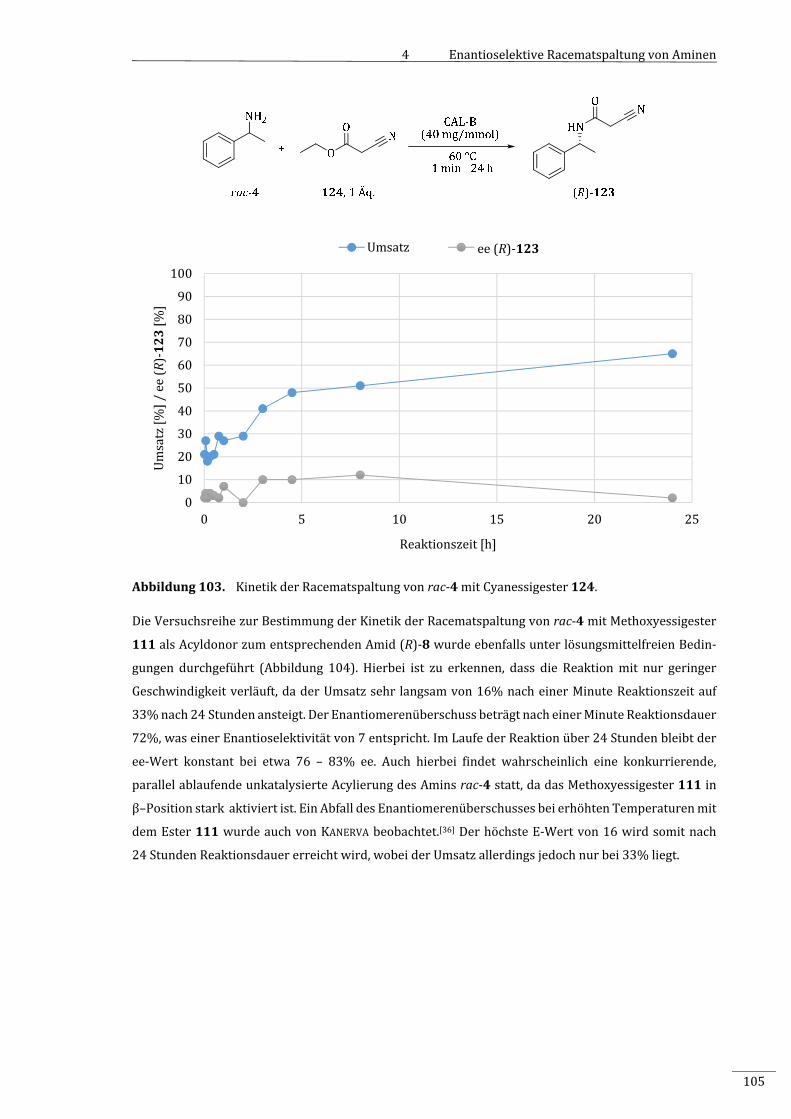
4 EnantioselektiveRacematspaltungvonAminen
105
Abbildung103. KinetikderRacematspaltungvonrac‐4mitCyanessigester124.
DieVersuchsreihezurBestimmungderKinetikderRacematspaltungvonrac‐4mitMethoxyessigester
111alsAcyldonorzumentsprechendenAmid(R)‐8wurdeebenfallsunterlösungsmittelfreienBedin‐
gungen durchgeführt (Abbildung 104). Hierbei ist zu erkennen, dass die Reaktionmit nur geringer
Geschwindigkeitverläuft,daderUmsatzsehr langsamvon16%nacheinerMinuteReaktionszeitauf
33%nach24Stundenansteigt.DerEnantiomerenüberschussbeträgtnacheinerMinuteReaktionsdauer
72%,waseinerEnantioselektivitätvon7entspricht.ImLaufederReaktionüber24Stundenbleibtder
ee‐Wert konstant bei etwa 76 – 83% ee. Auch hierbei findet wahrscheinlich eine konkurrierende,
parallelablaufendeunkatalysierteAcylierungdesAminsrac‐4statt,dadasMethoxyessigester111in
β–Positionstarkaktiviertist.EinAbfalldesEnantiomerenüberschussesbeierhöhtenTemperaturenmit
demEster111wurdeauchvonKANERVAbeobachtet.[36]DerhöchsteE‐Wertvon16wirdsomitnach
24StundenReaktionsdauererreichtwird,wobeiderUmsatzallerdingsjedochnurbei33%liegt.
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25
Umsatz[%
]/ee(R)‐123[%]
Reaktionszeit[h]
Umsatz[%] ee[%]ee(R)‐123
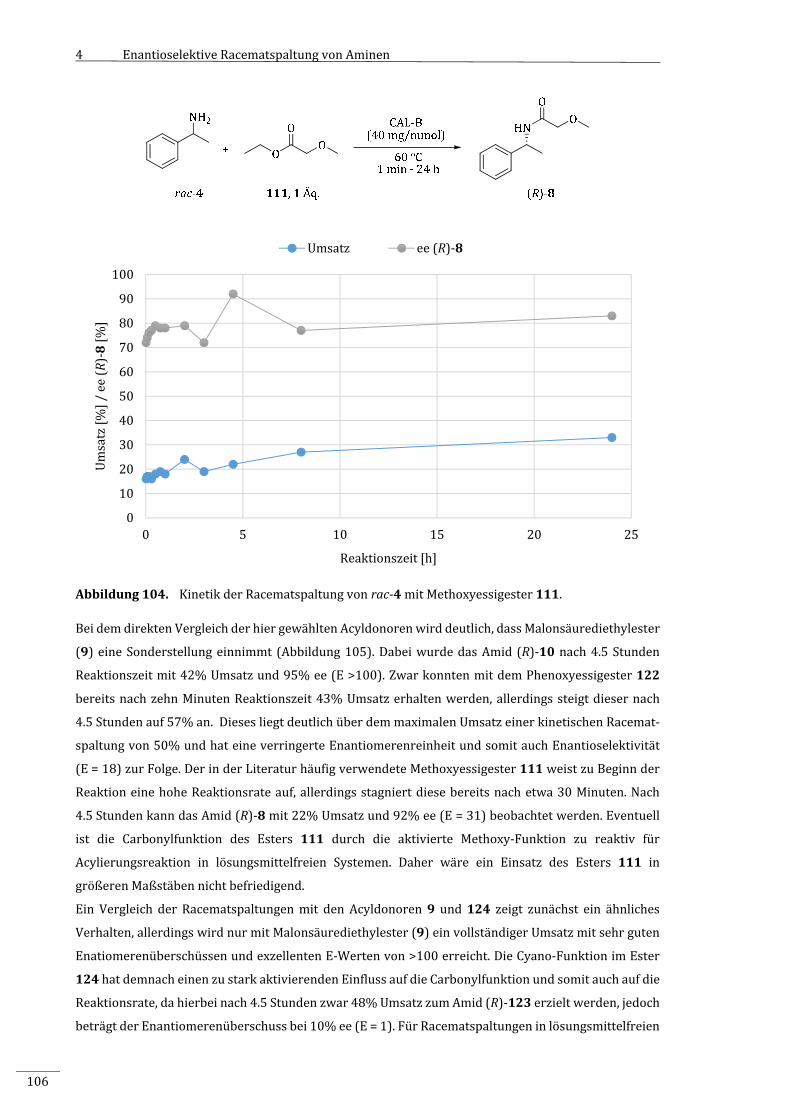
4 EnantioselektiveRacematspaltungvonAminen
106
Abbildung104. KinetikderRacematspaltungvonrac‐4mitMethoxyessigester111.
BeidemdirektenVergleichderhiergewähltenAcyldonorenwirddeutlich,dassMalonsäurediethylester
(9)eineSonderstellungeinnimmt(Abbildung105).DabeiwurdedasAmid (R)‐10nach4.5Stunden
Reaktionszeitmit42%Umsatzund95%ee(E>100).ZwarkonntenmitdemPhenoxyessigester122
bereitsnachzehnMinutenReaktionszeit43%Umsatzerhaltenwerden,allerdingssteigtdiesernach
4.5Stundenauf57%an.DiesesliegtdeutlichüberdemmaximalenUmsatzeinerkinetischenRacemat‐
spaltungvon50%undhateineverringerteEnantiomerenreinheitundsomitauchEnantioselektivität
(E=18)zurFolge.DerinderLiteraturhäufigverwendeteMethoxyessigester111weistzuBeginnder
ReaktioneinehoheReaktionsrateauf,allerdingsstagniertdiesebereitsnachetwa30Minuten.Nach
4.5StundenkanndasAmid(R)‐8mit22%Umsatzund92%ee(E=31)beobachtetwerden.Eventuell
ist die Carbonylfunktion des Esters 111 durch die aktivierte Methoxy‐Funktion zu reaktiv für
Acylierungsreaktion in lösungsmittelfreien Systemen. Daher wäre ein Einsatz des Esters 111 in
größerenMaßstäbennichtbefriedigend.
Ein Vergleich der Racematspaltungenmit denAcyldonoren9 und124 zeigt zunächst ein ähnliches
Verhalten,allerdingswirdnurmitMalonsäurediethylester(9)einvollständigerUmsatzmitsehrguten
EnatiomerenüberschüssenundexzellentenE‐Wertenvon>100erreicht.DieCyano‐FunktionimEster
124hatdemnacheinenzustarkaktivierendenEinflussaufdieCarbonylfunktionundsomitauchaufdie
Reaktionsrate,dahierbeinach4.5Stundenzwar48%UmsatzzumAmid(R)‐123erzieltwerden,jedoch
beträgtderEnantiomerenüberschussbei10%ee(E=1).FürRacematspaltungeninlösungsmittelfreien
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25
Umsatz[%
]/ee(R)‐8[%]
Reaktionszeit[h]
Umsatz[%] ee[%]ee(R)‐8
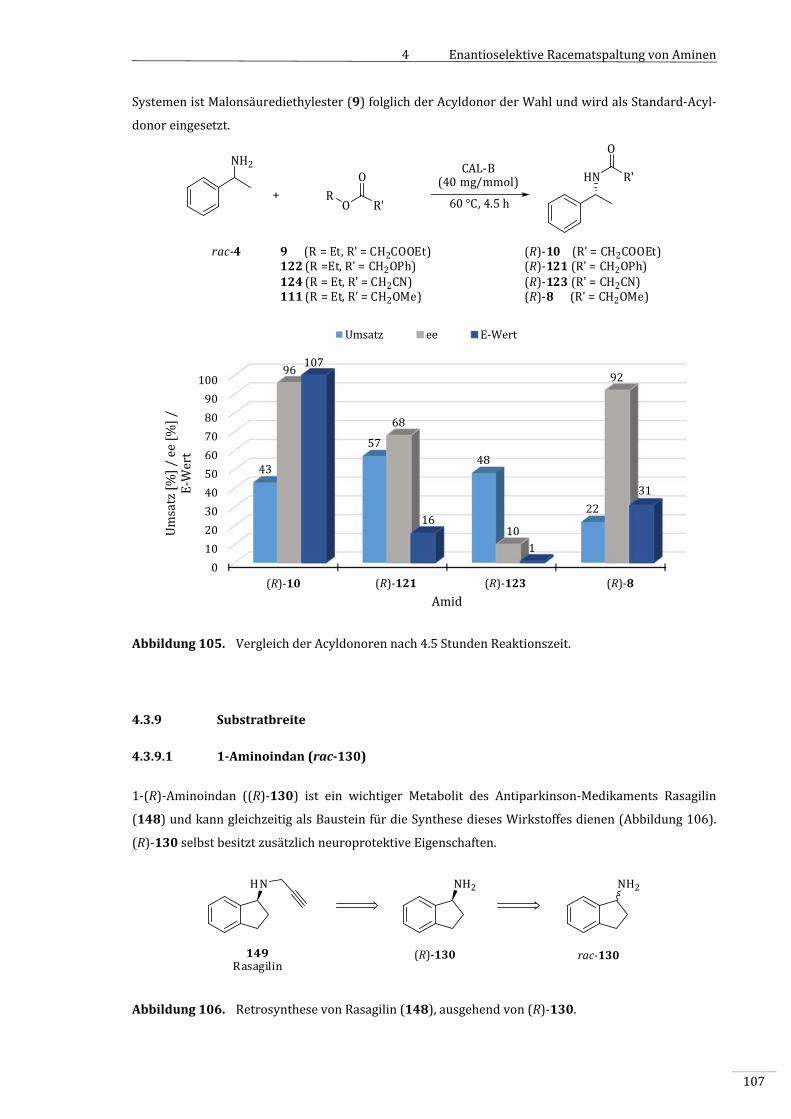
4 EnantioselektiveRacematspaltungvonAminen
107
SystemenistMalonsäurediethylester(9)folglichderAcyldonorderWahlundwirdalsStandard‐Acyl‐
donoreingesetzt.
RO
O R'+
NH2HN R'
O
rac‐4
CAL‐B(40 mg/mmol)
9 (R = Et, R' = CH2COOEt)122 (R =Et, R' = CH2OPh)124 (R = Et, R' = CH2CN)111 (R = Et, R' = CH2OMe)
(R)‐10 (R' = CH2COOEt)(R)‐121 (R' = CH2OPh)(R)‐123 (R' = CH2CN)(R)‐8 (R' = CH2OMe)
60 °C, 4.5 h
Abbildung105. VergleichderAcyldonorennach4.5StundenReaktionszeit.
4.3.9 Substratbreite
4.3.9.1 1‐Aminoindan(rac‐130)
1‐(R)‐Aminoindan ((R)‐130) ist ein wichtiger Metabolit des Antiparkinson‐Medikaments Rasagilin
(148)undkanngleichzeitigalsBausteinfürdieSynthesediesesWirkstoffesdienen(Abbildung106).
(R)‐130selbstbesitztzusätzlichneuroprotektiveEigenschaften.
NH2HN
149Rasagilin
(R)‐148
NH2
rac‐148
Abbildung106. RetrosynthesevonRasagilin(148),ausgehendvon(R)‐130.
0
10
20
30
40
50
60
70
80
90
100
(R)‐10 (R)‐121 (R)‐123 (R)‐8
43
5748
22
96
68
10
92107
16
1
31
Umsatz[%
]/ee[%]/
E‐Wert
Amid
Umsatz[%] ee[%] E‐Wert
(R)‐10 (R)‐121 (R)‐123 (R)‐8
(R)‐130 rac‐130
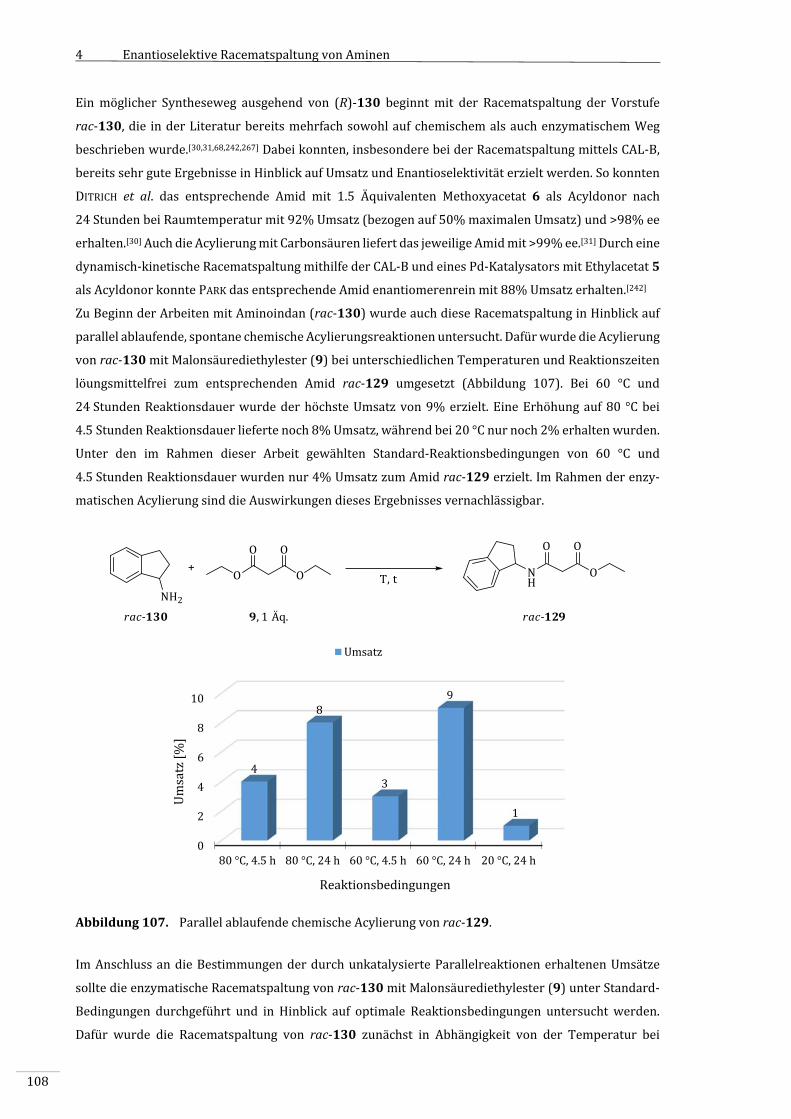
4 EnantioselektiveRacematspaltungvonAminen
108
Einmöglicher Syntheseweg ausgehend von (R)‐130 beginntmit der Racematspaltung der Vorstufe
rac‐130,die inderLiteraturbereitsmehrfachsowohlaufchemischemalsauchenzymatischemWeg
beschriebenwurde.[30,31,68,242,267]Dabeikonnten,insbesonderebeiderRacematspaltungmittelsCAL‐B,
bereitssehrguteErgebnisseinHinblickaufUmsatzundEnantioselektivitäterzieltwerden.Sokonnten
DITRICH et al. das entsprechende Amid mit 1.5 Äquivalenten Methoxyacetat 6 als Acyldonor nach
24StundenbeiRaumtemperaturmit92%Umsatz(bezogenauf50%maximalenUmsatz)und>98%ee
erhalten.[30]AuchdieAcylierungmitCarbonsäurenliefertdasjeweiligeAmidmit>99%ee.[31]Durcheine
dynamisch‐kinetischeRacematspaltungmithilfederCAL‐BundeinesPd‐KatalysatorsmitEthylacetat5
alsAcyldonorkonntePARKdasentsprechendeAmidenantiomerenreinmit88%Umsatzerhalten.[242]
ZuBeginnderArbeitenmitAminoindan(rac‐130)wurdeauchdieseRacematspaltunginHinblickauf
parallelablaufende,spontanechemischeAcylierungsreaktionenuntersucht.DafürwurdedieAcylierung
vonrac‐130mitMalonsäurediethylester(9)beiunterschiedlichenTemperaturenundReaktionszeiten
löungsmittelfrei zum entsprechenden Amid rac‐129 umgesetzt (Abbildung 107). Bei 60 °C und
24StundenReaktionsdauerwurdederhöchsteUmsatzvon9%erzielt.EineErhöhungauf80 °Cbei
4.5StundenReaktionsdauerliefertenoch8%Umsatz,währendbei20°Cnurnoch2%erhaltenwurden.
Unter den im Rahmen dieser Arbeit gewählten Standard‐Reaktionsbedingungen von 60 °C und
4.5StundenReaktionsdauerwurdennur4%UmsatzzumAmidrac‐129erzielt.ImRahmenderenzy‐
matischenAcylierungsinddieAuswirkungendiesesErgebnissesvernachlässigbar.
O
O
O
O+
9, 1 Äq.rac‐130
NH
O
O
O
NH2rac‐129
T, t
Abbildung107. ParallelablaufendechemischeAcylierungvonrac‐129.
ImAnschlussandieBestimmungenderdurchunkatalysierteParallelreaktionenerhaltenenUmsätze
solltedieenzymatischeRacematspaltungvonrac‐130mitMalonsäurediethylester(9)unterStandard‐
Bedingungen durchgeführt und in Hinblick auf optimale Reaktionsbedingungen untersuchtwerden.
Dafür wurde die Racematspaltung von rac‐130 zunächst in Abhängigkeit von der Temperatur bei
0
2
4
6
8
10
80°C,4.5h 80°C,24h 60°C,4.5h 60°C,24h 20°C,24h
4
8
3
9
1
Umsatz[%
]
Reaktionsbedingungen
Umsatz[%]

4 EnantioselektiveRacematspaltungvonAminen
109
4.5StundenReaktionsdaueruntersucht.DieErgebnisse sind inAbbildung108graphischdargestellt.
Dabeiistersichtlich,dassbeiderRacematspaltungkeinTrendderAbhängigkeitvonderTemperatur
hinsichtlich des Umsatzes vorliegt. So werden bei 80 °C Reaktionstemperatur 39% Umsatz erzielt,
währendbei20°Cnoch43%Umsatzerhaltenwerden.DieEnantiomerenüberschüsseliegendabeifür
dasAmidrac‐129imBereichvonnur73‐79%ee.DieRacematspaltungvonrac‐130mitdemMethoxy‐
ester6wurdevonBASFSEmithoherEnantiomerenreinheitsowohldesAmids(R)‐129alsauchdes
Amins(S)‐130durchgeführt.[30]KANERVAetal.führtendieAcylierung,ebenfallsmitdemMethoxyester
6,unterlösungsmittelfreienBedingungenbei23°CmithoherEnantioselektivitätdurch,wobeibereits
nacheinerStunde39%Umsatzerzieltwordensind.[36]BeidenindieserArbeitgewähltenReaktions‐
bedingungen spielt die Wahl des Acyldonors sowie die Abwesenheit von Lösungsmitteln eine ent‐
scheidendeRolle.
O
O
O
O+
9, 1 Äq.rac‐130
NH
O
O
O
NH2
(R)‐129
CAL‐B(40 mg/mmol)
T, 4.5 h
Abbildung108. Racematspaltungvonrac‐130inAbhängigkeitderTemperaturbei4.5h.
DieReaktionsdauerwurdeunterdensonstgleichbleibendenBedingungenauf24Stundenverlängert,
umdenEinflussderReaktionszeit zuüberprüfen (Abbildung109).Dabeiwirdbei 80 °CReaktions‐
temperatureinUmsatzvon42%erreicht,wasnureineleichteSteigerungzudemWertvon39%bei
4.5Stunden Reaktionszeit ist. Der Enantiomerenüberschuss für das Amid (R)‐129 ist mit 97% ee
(E>100)sehrgut.Allerdingswurdebereitsbeschrieben,dassbiszu8%desAmids(R)‐129bei80°C
und24StundenReaktionsdaueraufchemischemWegegebildetwird.DieEnantiomerenüberschüsse
derAcylierungennehmen jedochmitweitersinkenderTemperaturab,sodassdasAmid(R)‐129bei
40°Czwarnochmit50%Umsatzund90%ee,bei20°Cjedochnurnocheinee‐Wertvon54%detektiert
werdenkann.DieseniedrigeEnantioselektivität istüberraschend,dadieArbeitsgruppeumKANERVA
sehrguteErgebnissemitderCAL‐BinHinblickaufEnantioselektivitätbei23°CReaktionstemperatur
erhaltenhat.[36]
0102030405060708090100
80 60 20
3949
6673
7973
Umsatz[%
]/ee(R)‐129[%]
Reaktionstemperatur[°C]
Umsatz[%] ee[%]ee(R)‐129
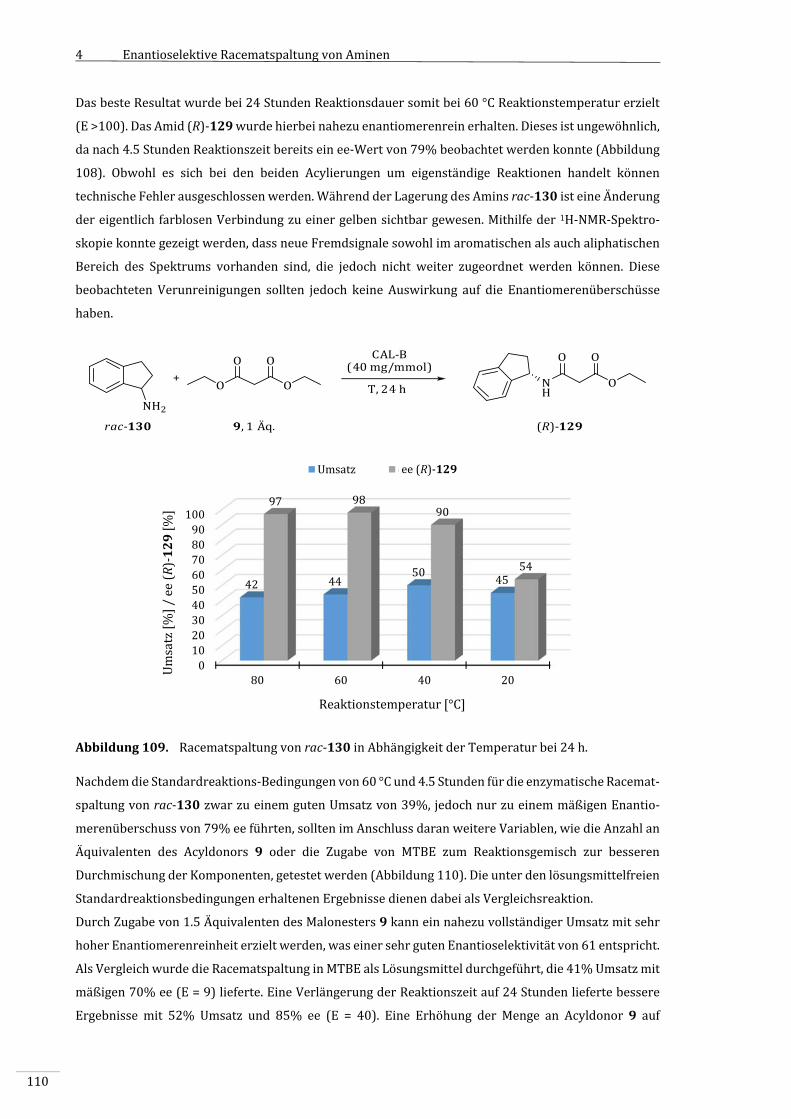
4 EnantioselektiveRacematspaltungvonAminen
110
DasbesteResultatwurdebei24StundenReaktionsdauersomitbei60°CReaktionstemperaturerzielt
(E>100).DasAmid(R)‐129wurdehierbeinahezuenantiomerenreinerhalten.Diesesistungewöhnlich,
danach4.5StundenReaktionszeitbereitseinee‐Wertvon79%beobachtetwerdenkonnte(Abbildung
108). Obwohl es sich bei den beiden Acylierungen um eigenständige Reaktionen handelt können
technischeFehlerausgeschlossenwerden.WährendderLagerungdesAminsrac‐130isteineÄnderung
dereigentlichfarblosenVerbindungzueinergelbensichtbargewesen.Mithilfeder1H‐NMR‐Spektro‐
skopiekonntegezeigtwerden,dassneueFremdsignalesowohlimaromatischenalsauchaliphatischen
Bereich des Spektrums vorhanden sind, die jedoch nicht weiter zugeordnet werden können. Diese
beobachteten Verunreinigungen sollten jedoch keine Auswirkung auf die Enantiomerenüberschüsse
haben.
O
O
O
O+
9, 1 Äq.rac‐130
NH
O
O
O
NH2
(R)‐129
CAL‐B(40 mg/mmol)
T, 24 h
Abbildung109. Racematspaltungvonrac‐130inAbhängigkeitderTemperaturbei24h.
NachdemdieStandardreaktions‐Bedingungenvon60°Cund4.5StundenfürdieenzymatischeRacemat‐
spaltungvonrac‐130zwarzueinemgutenUmsatzvon39%,jedochnurzueinemmäßigenEnantio‐
merenüberschussvon79%eeführten,solltenimAnschlussdaranweitereVariablen,wiedieAnzahlan
Äquivalenten des Acyldonors 9 oder die Zugabe von MTBE zum Reaktionsgemisch zur besseren
DurchmischungderKomponenten,getestetwerden(Abbildung110).Dieunterdenlösungsmittelfreien
StandardreaktionsbedingungenerhaltenenErgebnissedienendabeialsVergleichsreaktion.
DurchZugabevon1.5ÄquivalentendesMalonesters9kanneinnahezuvollständigerUmsatzmitsehr
hoherEnantiomerenreinheiterzieltwerden,waseinersehrgutenEnantioselektivitätvon61entspricht.
AlsVergleichwurdedieRacematspaltunginMTBEalsLösungsmitteldurchgeführt,die41%Umsatzmit
mäßigen70%ee(E=9)lieferte.EineVerlängerungderReaktionszeitauf24Stundenliefertebessere
Ergebnisse mit 52% Umsatz und 85% ee (E = 40). Eine Erhöhung der Menge an Acyldonor 9 auf
0102030405060708090100
80 60 40 20
42 4450
45
97 9890
54
Umsatz[%
]/ee(R)‐129[%]
Reaktionstemperatur[°C]
Umsatz[%] ee[%]ee(R)‐129
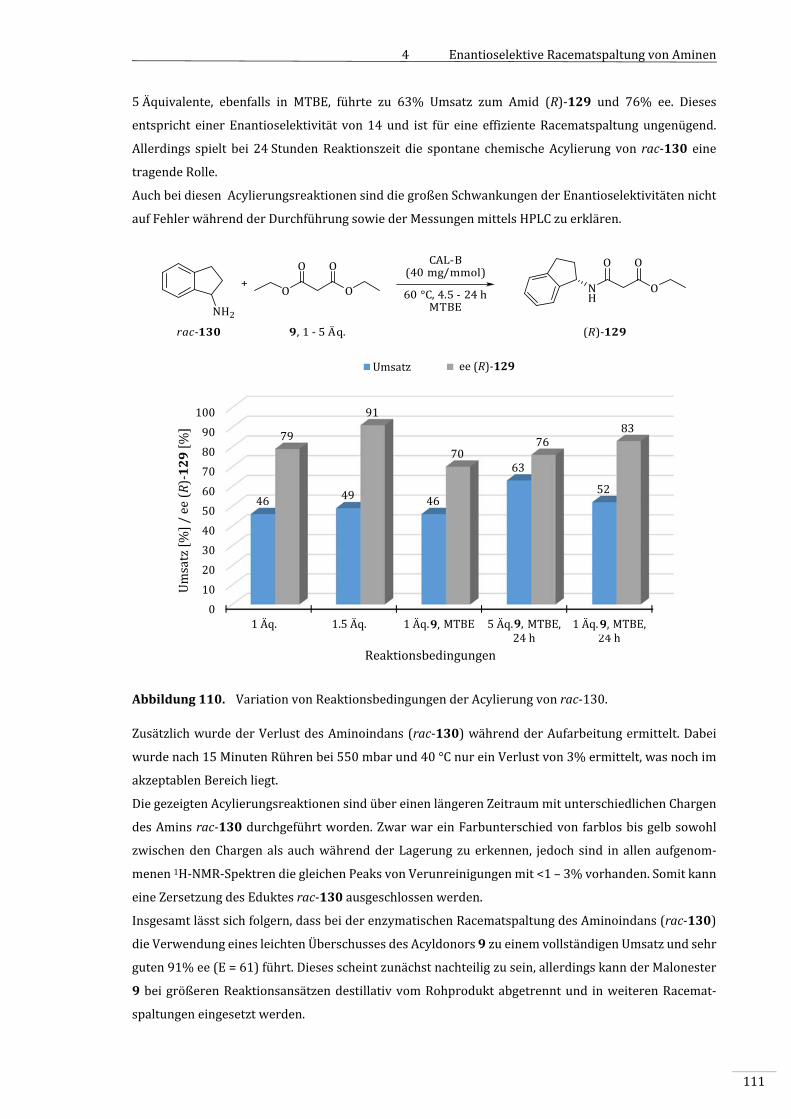
4 EnantioselektiveRacematspaltungvonAminen
111
5Äquivalente, ebenfalls in MTBE, führte zu 63% Umsatz zum Amid (R)‐129 und 76% ee. Dieses
entspricht einerEnantioselektivität von14und ist für eine effizienteRacematspaltungungenügend.
Allerdings spielt bei 24StundenReaktionszeit die spontane chemischeAcylierung von rac‐130 eine
tragendeRolle.
AuchbeidiesenAcylierungsreaktionensinddiegroßenSchwankungenderEnantioselektivitätennicht
aufFehlerwährendderDurchführungsowiederMessungenmittelsHPLCzuerklären.
O
O
O
O+
9, 1 ‐ 5 Äq.rac‐130
NH
O
O
O
NH2
(R)‐129
CAL‐B(40 mg/mmol)
60 °C, 4.5 ‐ 24 hMTBE
Abbildung110. VariationvonReaktionsbedingungenderAcylierungvonrac‐130.
ZusätzlichwurdederVerlustdesAminoindans(rac‐130)währendderAufarbeitungermittelt.Dabei
wurdenach15MinutenRührenbei550mbarund40°CnureinVerlustvon3%ermittelt,wasnochim
akzeptablenBereichliegt.
DiegezeigtenAcylierungsreaktionensindübereinenlängerenZeitraummitunterschiedlichenChargen
desAminsrac‐130durchgeführtworden.ZwarwareinFarbunterschiedvonfarblosbisgelbsowohl
zwischendenChargenals auchwährendderLagerung zu erkennen, jedoch sind inallen aufgenom‐
menen1H‐NMR‐SpektrendiegleichenPeaksvonVerunreinigungenmit<1–3%vorhanden.Somitkann
eineZersetzungdesEduktesrac‐130ausgeschlossenwerden.
Insgesamtlässtsichfolgern,dassbeiderenzymatischenRacematspaltungdesAminoindans(rac‐130)
dieVerwendungeinesleichtenÜberschussesdesAcyldonors9zueinemvollständigenUmsatzundsehr
guten91%ee(E=61)führt.Diesesscheintzunächstnachteiligzusein,allerdingskannderMalonester
9beigrößerenReaktionsansätzendestillativvomRohproduktabgetrenntund inweiterenRacemat‐
spaltungeneingesetztwerden.
0
10
20
30
40
50
60
70
80
90
100
1Äq.9 1.5Äq.9 1Äq.9,MTBE 5Äq.9,MTBE,24h
1Äq.9,MTBE,24h
46 49 46
63
52
79
91
7076
83
Umsatz[%
]/ee(R)‐129[%]
Reaktionsbedingungen
Umsatz[%] ee[%]
9, 9, 9,
ee(R)‐129

4 EnantioselektiveRacematspaltungvonAminen
112
4.3.9.2 SekundäreAmine
Enantiomerenreine Amine sind wichtige Bausteine für pharmazeutische Wirkstoffe und Agro‐
chemikalien.EssindjedochnurwenigeMethodenzurRacematspaltungsekundärerAminebekannt.Der
StandderWissenschaftistdabeiaufChromatographiemitchiralenstationärenPhasenoderklassische
RacematspaltungdurchBildung vonDiastereomerenpaarenbeschränkt. SekundäreAmine reagieren
aufgrundihrerstarkenNucleophilliemitdenAcyldonorenunterStandard‐Reaktionsbedingungen,wie
niedrige Temperaturen und Konzentrationen, meist nicht. Die Schwierigkeit bei der Acylierung
cyclischer sekundärer Amine liegtwahrscheinlich in der großen Anzahl an potentiellen Übergangs‐
zuständen sowie an denmöglichen Konformationen, die energetische sehr ähnlich sind.[268] Bei der
RacematspaltungfindendabeiklassischLipasenausCandidarugosa[239]sowiedieCAL‐AausCandida
antarctica[28,269]Anwendung.
In dieser Arbeit sollten zunächst die Referenzverbindungen der ausgehend von 2‐ bzw. 3‐Methyl‐
piperidin(149bzw.150)synthetisiertwerden,umeineMethodefürdieHPLCzufinden.ImAnschluss
daransolltedieenzymatischeAcylierungmitMalonsäurediethylester(9)untersuchtwerden.
Die chemische Acylierung erfolgt im Fall der sekundären Amine 149 und 150mit dem Acyldonor
Malonsäurediethylester(9)unterErhitzenzumRückflussfür3Stunden(Abbildung111).[253]DieAmide
rac‐151 und rac‐152 wurde mit Umsätzen von 56% bzw. 63% erhalten. Die isolierten Ausbeuten
betrugendabei41%fürrac‐151und42%fürrac‐152,wasfürdieAnalytikjedochausreichendwar.
Abbildung111. SynthesederRacematerac‐151undrac‐152alsReferenzverbindungen.[253]
AnschließendkonntefürdieAmideeinegeeigneteMethodefürdieHPLCetabliertwerden.InAbbildung
112isteincharakteristischesHPLC‐ChromatogrammamBeispielvonrac‐152dargestellt.Deree‐Wert
desRacematesrac‐144beträgt<0.2%.
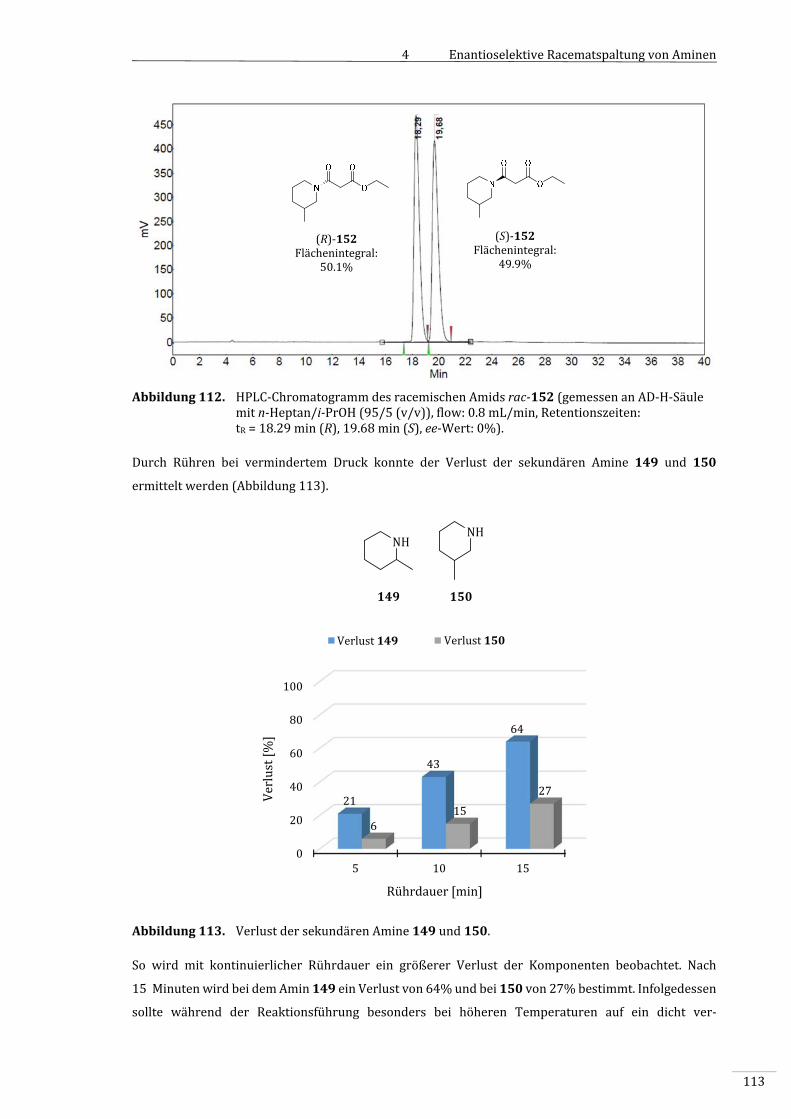
4 EnantioselektiveRacematspaltungvonAminen
113
Abbildung112. HPLC‐ChromatogrammdesracemischenAmidsrac‐152(gemessenanAD‐H‐Säulemitn‐Heptan/i‐PrOH(95/5(v/v)),flow:0.8mL/min,Retentionszeiten:tR=18.29min(R),19.68min(S),ee‐Wert:0%).
Durch Rühren bei vermindertem Druck konnte der Verlust der sekundären Amine 149 und 150
ermitteltwerden(Abbildung113).
NH
149
NH
150
Abbildung113. VerlustdersekundärenAmine149und150.
So wird mit kontinuierlicher Rührdauer ein größerer Verlust der Komponenten beobachtet. Nach
15MinutenwirdbeidemAmin149einVerlustvon64%undbei150von27%bestimmt.Infolgedessen
sollte während der Reaktionsführung besonders bei höheren Temperaturen auf ein dicht ver‐
0
20
40
60
80
100
5 10 15
21
43
64
615
27Verlust[%
]
Rührdauer[min]
Verlust142[%] Verlust138[%]Verlust149 Verlust150
(R)‐152Flächenintegral:
50.1%
(S)‐152Flächenintegral:
49.9%
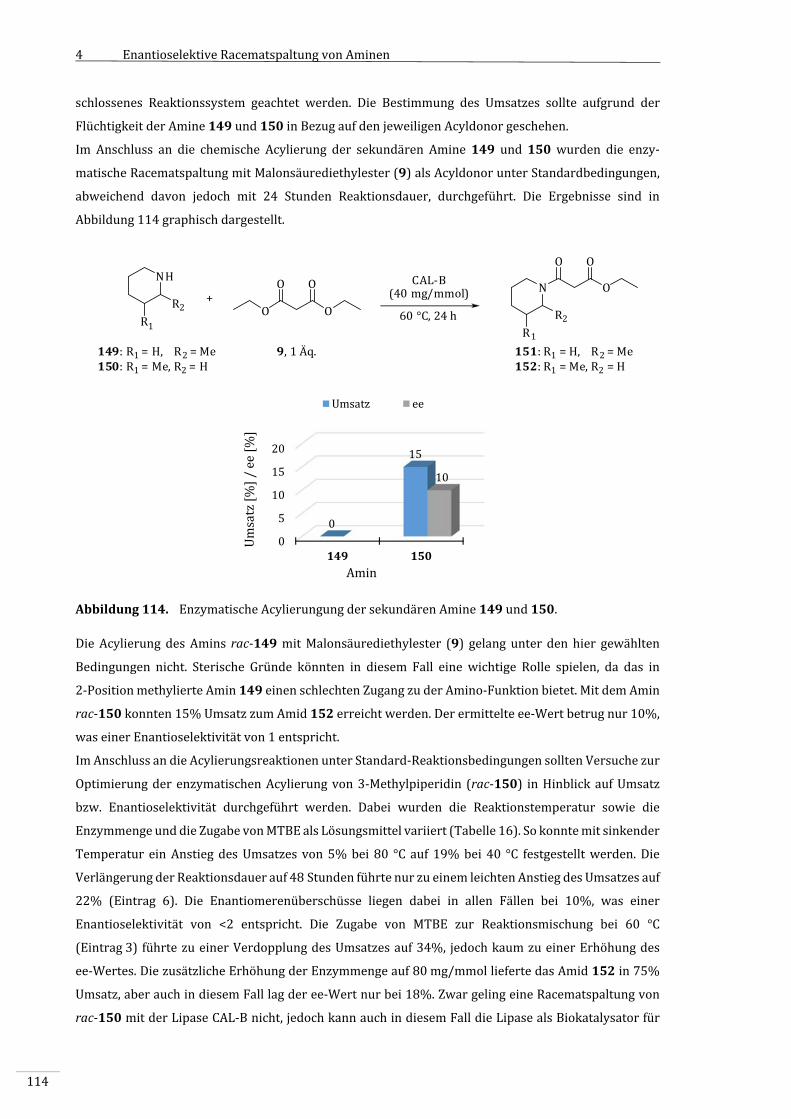
4 EnantioselektiveRacematspaltungvonAminen
114
schlossenes Reaktionssystem geachtet werden. Die Bestimmung des Umsatzes sollte aufgrund der
FlüchtigkeitderAmine149und150inBezugaufdenjeweiligenAcyldonorgeschehen.
Im Anschluss an die chemische Acylierung der sekundären Amine 149 und150 wurden die enzy‐
matischeRacematspaltungmitMalonsäurediethylester(9)alsAcyldonorunterStandardbedingungen,
abweichend davon jedoch mit 24 Stunden Reaktionsdauer, durchgeführt. Die Ergebnisse sind in
Abbildung114graphischdargestellt.
N
O O
O
O
O O
O+
CAL‐B(40 mg/mmol)
60 °C, 24 h
9, 1 Äq.
R1
NH
R2R1
R2
149: R1 = H, R2 = Me150: R1 = Me, R2 = H
151: R1 = H, R2 = Me152: R1 = Me, R2 = H
Abbildung114. EnzymatischeAcylierungungdersekundärenAmine149und150.
DieAcylierung desAmins rac‐149mitMalonsäurediethylester (9) gelang unter den hier gewählten
Bedingungen nicht. Sterische Gründe könnten in diesem Fall eine wichtige Rolle spielen, da das in
2‐PositionmethylierteAmin149einenschlechtenZugangzuderAmino‐Funktionbietet.MitdemAmin
rac‐150konnten15%UmsatzzumAmid152erreichtwerden.Derermittelteee‐Wertbetrugnur10%,
waseinerEnantioselektivitätvon1entspricht.
ImAnschlussandieAcylierungsreaktionenunterStandard‐ReaktionsbedingungensolltenVersuchezur
OptimierungderenzymatischenAcylierungvon3‐Methylpiperidin (rac‐150) inHinblickaufUmsatz
bzw. Enantioselektivität durchgeführt werden. Dabei wurden die Reaktionstemperatur sowie die
EnzymmengeunddieZugabevonMTBEalsLösungsmittelvariiert(Tabelle16).Sokonntemitsinkender
Temperatur einAnstiegdesUmsatzes von5%bei 80 °C auf 19%bei 40 °C festgestelltwerden.Die
VerlängerungderReaktionsdauerauf48StundenführtenurzueinemleichtenAnstiegdesUmsatzesauf
22% (Eintrag 6). Die Enantiomerenüberschüsse liegen dabei in allen Fällen bei 10%, was einer
Enantioselektivität von <2 entspricht. Die Zugabe von MTBE zur Reaktionsmischung bei 60 °C
(Eintrag3) führtezueinerVerdopplungdesUmsatzesauf34%, jedochkaumzueinerErhöhungdes
ee‐Wertes.DiezusätzlicheErhöhungderEnzymmengeauf80mg/mmolliefertedasAmid152in75%
Umsatz,aberauchindiesemFalllagderee‐Wertnurbei18%.ZwargelingeineRacematspaltungvon
rac‐150mitderLipaseCAL‐Bnicht,jedochkannauchindiesemFalldieLipasealsBiokatalysatorfür
0
5
10
15
20
149 150
0
15
10
Umsatz[%
]/ee[%]
Amin
Umsatz[%] ee[%]
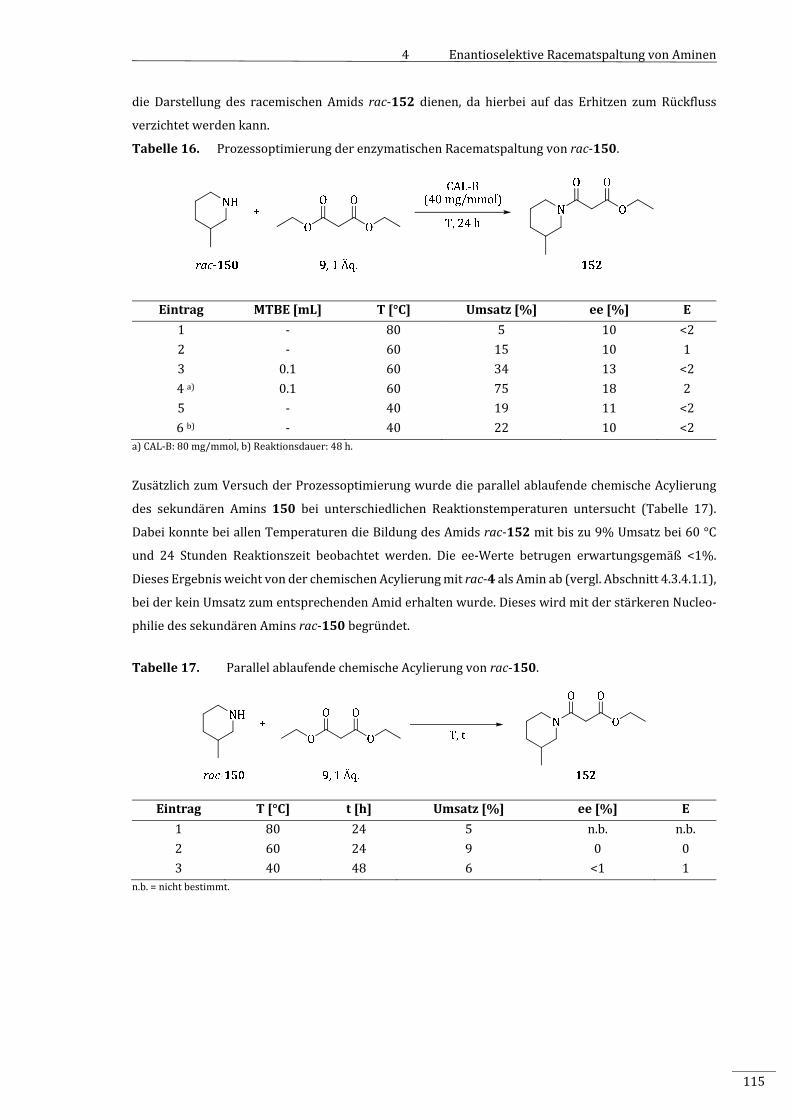
4 EnantioselektiveRacematspaltungvonAminen
115
die Darstellung des racemischen Amids rac‐152 dienen, da hierbei auf das Erhitzen zumRückfluss
verzichtetwerdenkann.
Tabelle16. ProzessoptimierungderenzymatischenRacematspaltungvonrac‐150.
Eintrag MTBE[mL] T[°C] Umsatz[%] ee[%] E
1 ‐ 80 5 10 <2
2 ‐ 60 15 10 1
3 0.1 60 34 13 <2
4a) 0.1 60 75 18 2
5 ‐ 40 19 11 <2
6b) ‐ 40 22 10 <2a)CAL‐B:80mg/mmol,b)Reaktionsdauer:48h.
ZusätzlichzumVersuchderProzessoptimierungwurdedieparallelablaufendechemischeAcylierung
des sekundären Amins 150 bei unterschiedlichen Reaktionstemperaturen untersucht (Tabelle 17).
DabeikonntebeiallenTemperaturendieBildungdesAmidsrac‐152mitbiszu9%Umsatzbei60°C
und 24 Stunden Reaktionszeit beobachtet werden. Die ee‐Werte betrugen erwartungsgemäß <1%.
DiesesErgebnisweichtvonderchemischenAcylierungmitrac‐4alsAminab(vergl.Abschnitt4.3.4.1.1),
beiderkeinUmsatzzumentsprechendenAmiderhaltenwurde.DieseswirdmitderstärkerenNucleo‐
philiedessekundärenAminsrac‐150begründet.
Tabelle17. ParallelablaufendechemischeAcylierungvonrac‐150.
Eintrag T[°C] t[h] Umsatz[%] ee[%] E
1 80 24 5 n.b. n.b.
2 60 24 9 0 0
3 40 48 6 <1 1n.b.=nichtbestimmt.

4 EnantioselektiveRacematspaltungvonAminen
116
4.3.10 ɛ–Caprolacton(3)alsAcyldonor
4.3.10.1 ChemischeAcylierungmitɛ–Caprolacton(3)
In dem ersten Teil dieser Arbeit (vergl. Abschnitt 3.3) wurde die enzymatische Herstellung von
ɛ‐Caprolacton (3) beschrieben. Im Folgendenwurde nun die Fähigkeit des Lactons3 zur Racemat‐
spaltung vonAminenwie 1‐Phenylethylamin (rac‐4) näher untersucht. Dafürwurden dieReferenz‐
verbindungenchemischhergestelltsowiedieStabilitätdesLactons3ermittelt.ImAnschlusswurdedie
enzymatischeAcylierungderAminedurchgeführtundderTemperatureffektuntersucht.
Als Vorbereitung der Acylierungsreaktionen wurde zunächst die Stabilität von ɛ–Caprolacton (3)
untersucht. Dazuwurde das Lacton3 in An‐ undAbwesenheit der CAL‐B bei 60 °C für 24 Stunden
gerührt(Tabelle18). ImFallderCAL‐BerfolgtzusätzlicheineAufarbeitungnachAAV6.DieAnsätze
wurdenmittels1H‐NMR‐Spektroskopieuntersucht.
Tabelle18. BestimmungderStabilitätvonɛ–Caprolacton(3).
Eintrag Bedingungen Verhältnis3/146
1 RührenohneCAL‐B 100:0
2RührenmitCAL‐Bund
AufarbeitungnachAAV61:77
So wird nach 24 Stunden Rühren in Abwesenheit der Lipase nur das reine Lacton 3 im 1H‐NMR‐
Spektrumerhalten.InAnwesenheitderCAL‐BmitanschließenderAufarbeitungmittelsFiltrationund
SpülenmitorganischenLösungsmittelnwerdenjedochnurnochSpurendesLactons3gefunden.Den
größtenAnteilderMischungmachtdieentsprechendeHydroxysäure54aus,diedurchdieSpaltungdes
Lactons3gebildetwird.FürdieSpaltungdesLactons3kanndiebereitsinSpurenvorhandeneSäure54
undzumanderendieLipaseselbstverantwortlichsein..FürdieReaktionsführungistdiesesjedochnicht
vonNachteil,dainderLiteraturCarbonsäurenbereitsmehrfacherfolgreichalsAcyldonoreneingesetzt
wordensind.[250,251]
DiechemischeAcylierungdesAminsrac‐4mitdemAcyldonorɛ‐Caprolacton(3)erfolgtunterErhitzen
zumRückflussfür4Stunden(Tabelle19,Eintrag1).[253]DasAmidrac‐147wurdedabeimit62%Umsatz
erhalten, was für die Analytik jedoch ausreichend war. Zusätzlich wurden auf gleichem Wege
AcylierungsreaktionendersekundärenAmine142und143durchgeführt(Eintrag2und3).Dieent‐
sprechendenAmidekonntendabeinurmitgeringenUmsätzenvon7bzw.17%erhaltenwerden.
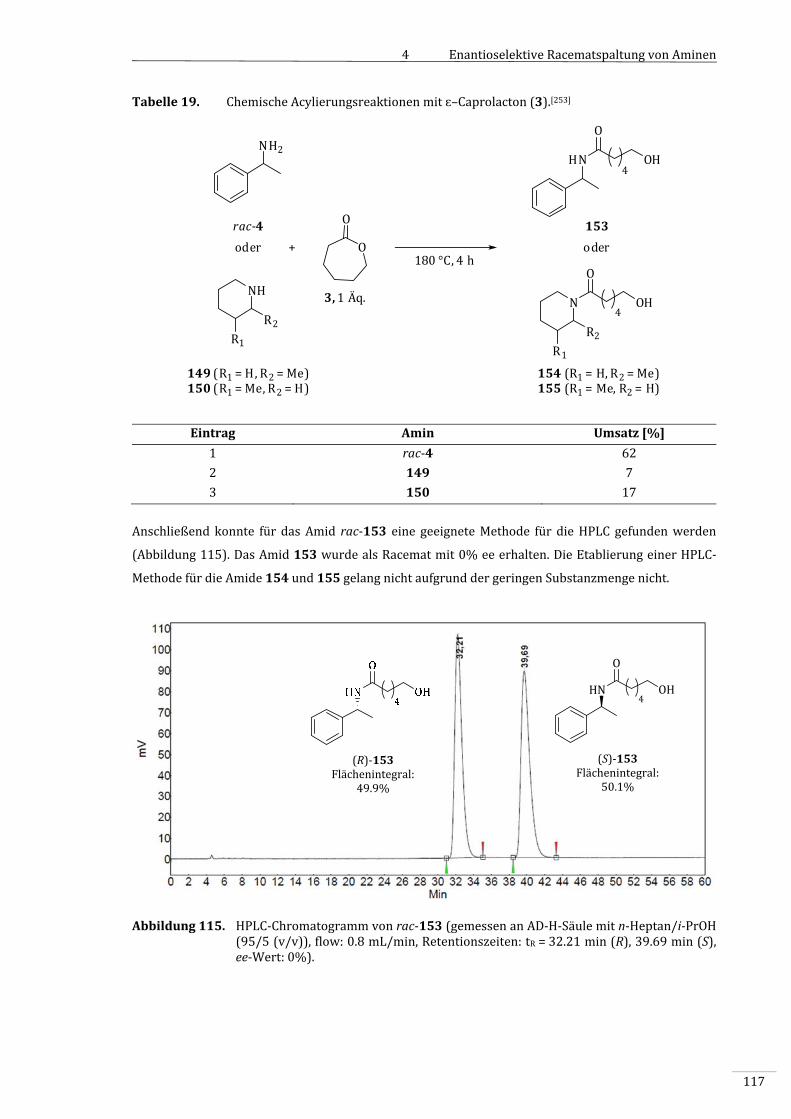
4 EnantioselektiveRacematspaltungvonAminen
117
Tabelle19. ChemischeAcylierungsreaktionenmitɛ–Caprolacton(3).[253]
180 °C, 4 h
rac‐4
3,1 Äq.
153
O
O
NH2OH
O
4HN
oder
NH
R1
149 (R1 = H, R2 = Me)150 (R1 = Me, R2 = H)
oder
N
R1
O
OH4
+
R2R2
154 (R1 = H, R2 = Me)155 (R1 = Me, R2 = H)
Eintrag Amin Umsatz[%]
1 rac‐4 62
2 149 7
3 150 17
Anschließend konnte für dasAmid rac‐153 eine geeigneteMethode für dieHPLC gefundenwerden
(Abbildung115).DasAmid153wurdealsRacematmit0%eeerhalten.DieEtablierungeinerHPLC‐
MethodefürdieAmide154und155gelangnichtaufgrunddergeringenSubstanzmengenicht.
Abbildung115. HPLC‐Chromatogrammvonrac‐153(gemessenanAD‐H‐Säulemitn‐Heptan/i‐PrOH(95/5(v/v)),flow:0.8mL/min,Retentionszeiten:tR=32.21min(R),39.69min(S),ee‐Wert:0%).
OH
O
4HN
(S)‐147Flächenintegral:
50.1%
(R)‐153Flächenintegral:
49.9%
(S)‐153Flächenintegral:
50.1%

4 EnantioselektiveRacematspaltungvonAminen
118
4.3.10.2 EnzymatischeAcylierungvonBenzylamin(134)mitɛ‐Caprolacton(3)
ImAnschlussandieBestimmungderStabilitätdesLactons3solltezunächstdessenAktivitätuntersucht
werden,bevorVersuchezurSelektivitätdurchgeführtwerden.DafürwurdedieseszunächstmitBenzyl‐
amin(134)alsModel‐SubstratunterStandardbedingungenenzymatischumgesetzt(Abbildung116).
DaessichbeidemAmin134nichtumeineracemischeVerbindunghandelt isthiervoneinemvoll‐
ständigen Umsatz auszugehen. Im 1H‐NMR‐Spektrum konnte allerdings nur ein Umsatz von 46%
bezogenaufdasAmin130nachgewiesenwerden.AuchindiesemFallistetwaeinDritteldesLactons3
zur Säure54 gespaltenworden.Wird der Umsatz zumAmid156 auf die Summeder Integrale des
Lactons3 und der Säure54 bezogen, so ergibt sich ein Umsatz von 44%.Während der Reaktions‐
führungundderAufarbeitungtrittdemnachkeinVerlustderKomponentenauf.
Abbildung116. EnzymatischeAcylierungvonBenzylamin(130)mitɛ‐Caprolacton(3).
Durch Variation der Temperatur sollte die enzymatische Acylierung in Hinblick auf eine mögliche
Umsatzsteigerung untersuchtwerden (Tabelle 20). Sowurden bei 80 °C Reaktionstemperatur 51%
UmsatzzumAmid156erreicht.DurchZugabevonMTBEalsorganischesLösungsmittelbei80°Ckonnte
eineUmsatzsteigerungauf65%erzieltwerden.Erfreulicherweisekonntenbereitsbei20°CReaktions‐
temperatur nach 4.5 Stunden 57% Umsatz erhalten werden. Diese Ergebnisse zeigen, dass eine
Erhöhung der Temperatur auf 80 °C keinen positiven Einfluss auf die enzymatische Acylierung des
Benzylamins134hat.
Tabelle20. Prozessoptimierung der enzymatischen Racematspaltung von Benzylamin (134) mitɛ‐Caprolacton(3).
Eintrag MTBE[mL] T[°C] t[h] Umsatz[%]
1 ‐ 80 4.5 51
2 0.5 80 4.5 65
3 ‐ RT 4.5 57
Die parallel ablaufende chemische Acylierung des Amins 134 mit dem Lacton 3 wurde bei unter‐
schiedlichen Temperaturen untersucht (Tabelle 21). Bei 80 °C Reaktionstemperatur können in Ab‐
wesenheitderLipasenoch30%UmsatzzumAmiderzieltwerden.MitsinkenderTemperatursinktder

4 EnantioselektiveRacematspaltungvonAminen
119
Umsatz, bis schließlich bei 0 °C nur noch 3% Amid156 gebildet werden. Durch Verlängerung der
Reaktionszeitauf24Stundenbei20°C(Eintrag4)werdennoch53%Umsatzerhalten.
Tabelle21. ParallelablaufendechemischeAcylierungvonBenzylamin(134)mitɛ–Caprolacton(3).
Eintrag T[°C] t[h] Umsatz[%]
1 80 4.5 30
2a) 60 4.5 69
3 RT 4.5 12
4 RT 24 53
5 0 4.5 3a)VerwendungeineraltenChargeanɛ–Caprolacton(3).
DurchVerwendungeinerälterenChargedesLactons3(Eintrag2),inderlaut1H‐NMR‐Spektroskopie
bereits17%desLactons3währendderLagerungzurentsprechendenSäure54gespaltenwordensind,
konnten bei 60 °C und 4.5 Stunden Reaktionszeit 69% Umsatz zum Amid 156 erhalten werden.
CarbonsäurenwerdenseltenalsAcyldonorenverwendet,daeinereversibleSalzbildungmitdemAmin
stattfindet.[250] Dieses unreaktive Salz ist nicht mehr für die Bildung des Acyl‐Enzym‐Komplexes
vorhanden.SomitstehtnureinTeilderSäurealsAcyldonorzurVerfügung,waszueinerverringerten
Reaktionsgeschwindigkeitführt.UnaktiviterteCarbonsäurenbenötigenfolglicheinKupplungsreagenz
zurAktivierungundhäufigdrastischeReaktionsbedingungen,ummiteinemAmineineAmid‐Bindung
zubildenunddieBildungdesentsprechendenSalzeszuvermeiden.[270]DieWahrscheinlihckeiteiner
Acylierungsreaktions durch die Säure54 als Acyldonor ist unter den verwendeten Reaktionsbedin‐
gungendemnach sehr gering.DurchdieWahl einermöglichstkurzenReaktionsdauerundniedriger
TemperaturkanndieunkatalysierteAcylierungdesAmins134somitunterdrücktwerden.Dascyclische
Lacton3 ist,eventuelldurchdieRingspannung,sehrstarkaktiviert,sodassbeidenfolgendenAcylie‐
rungendesracemischenAminsrac‐4aufeineVermeidungderchemischenAcylierunggeachtetwurde.
4.3.10.3 EnzymatischeAcylierungvon1‐Phenylethylamin(rac‐4)mitɛ–Caprolacton(3)
ZunächstwurdedieMöglichkeiteinerparallelzuderenzymatischenablaufendechemischeAcylierung
des Amins rac‐4 mit dem Lacton 3 untersucht. Dazu wurden die beiden Komponenten bei unter‐
schiedlichenTemperaturenfür4.5StundeninAbwesenheitderLipasegerührt.DieErgebnissesindin
Abbildung117graphischdargestellt.Sokannbei80°CnocheinUmsatzzumAmid157von10%erzielt
werden.MitsinkenderTemperaturnimmtauchderUmsatzab,bisbei0°CkeinAmid157mehr im
1H‐NMR‐Spektrumdetektiertwerdenkann.
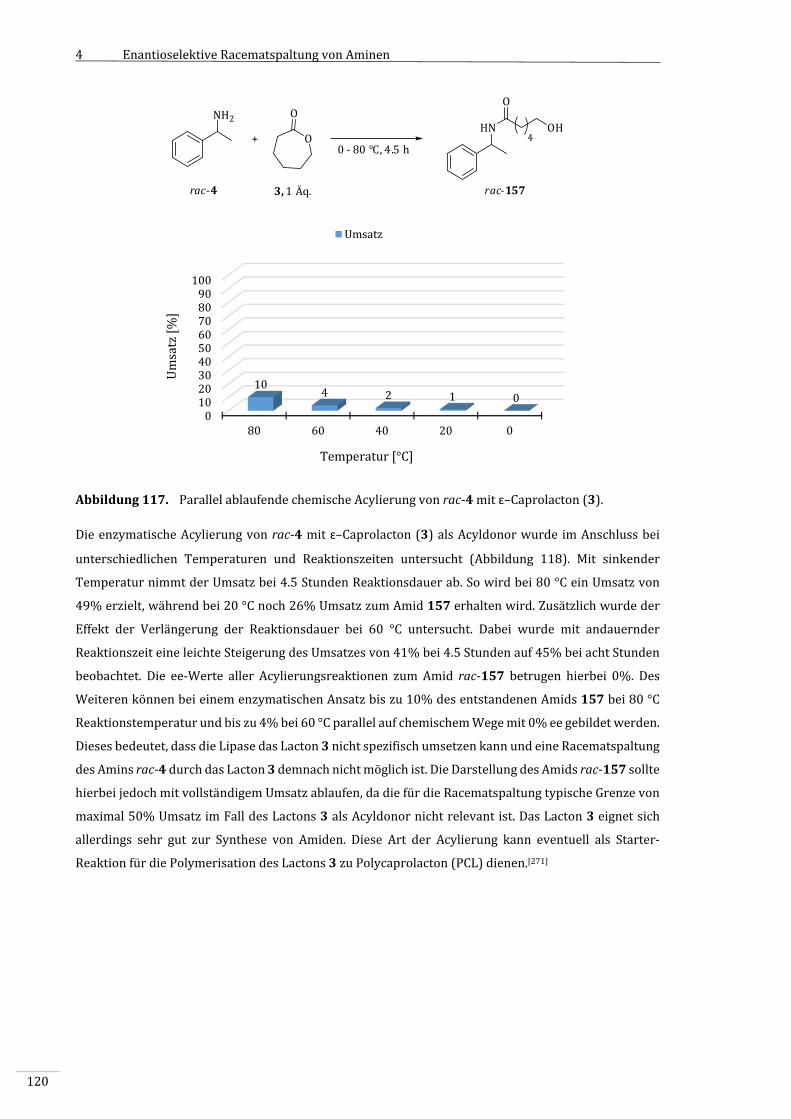
4 EnantioselektiveRacematspaltungvonAminen
120
+0 ‐ 80 °C, 4.5 h
rac‐4 3,1 Äq. rac‐157
O
ONH2OH
O
4HN
Abbildung117. ParallelablaufendechemischeAcylierungvonrac‐4mitɛ–Caprolacton(3).
DieenzymatischeAcylierungvonrac‐4mitɛ–Caprolacton(3)alsAcyldonorwurde imAnschlussbei
unterschiedlichen Temperaturen und Reaktionszeiten untersucht (Abbildung 118). Mit sinkender
TemperaturnimmtderUmsatzbei4.5StundenReaktionsdauerab.Sowirdbei80°CeinUmsatzvon
49%erzielt,währendbei20°Cnoch26%UmsatzzumAmid157erhaltenwird.Zusätzlichwurdeder
Effekt der Verlängerung der Reaktionsdauer bei 60 °C untersucht. Dabei wurde mit andauernder
ReaktionszeiteineleichteSteigerungdesUmsatzesvon41%bei4.5Stundenauf45%beiachtStunden
beobachtet. Die ee‐Werte aller Acylierungsreaktionen zum Amid rac‐157 betrugen hierbei 0%. Des
WeiterenkönnenbeieinemenzymatischenAnsatzbiszu10%desentstandenenAmids157bei80°C
Reaktionstemperaturundbiszu4%bei60°CparallelaufchemischemWegemit0%eegebildetwerden.
Diesesbedeutet,dassdieLipasedasLacton3nichtspezifischumsetzenkannundeineRacematspaltung
desAminsrac‐4durchdasLacton3demnachnichtmöglichist.DieDarstellungdesAmidsrac‐157sollte
hierbeijedochmitvollständigemUmsatzablaufen,dadiefürdieRacematspaltungtypischeGrenzevon
maximal50%UmsatzimFalldesLactons3alsAcyldonornichtrelevantist.DasLacton3eignetsich
allerdings sehr gut zur Synthese von Amiden. Diese Art der Acylierung kann eventuell als Starter‐
ReaktionfürdiePolymerisationdesLactons3zuPolycaprolacton(PCL)dienen.[271]
0102030405060708090100
80 60 40 20 0
104 2 1 0
Umsatz[%
]
Temperatur[°C]
Umsatz[%]
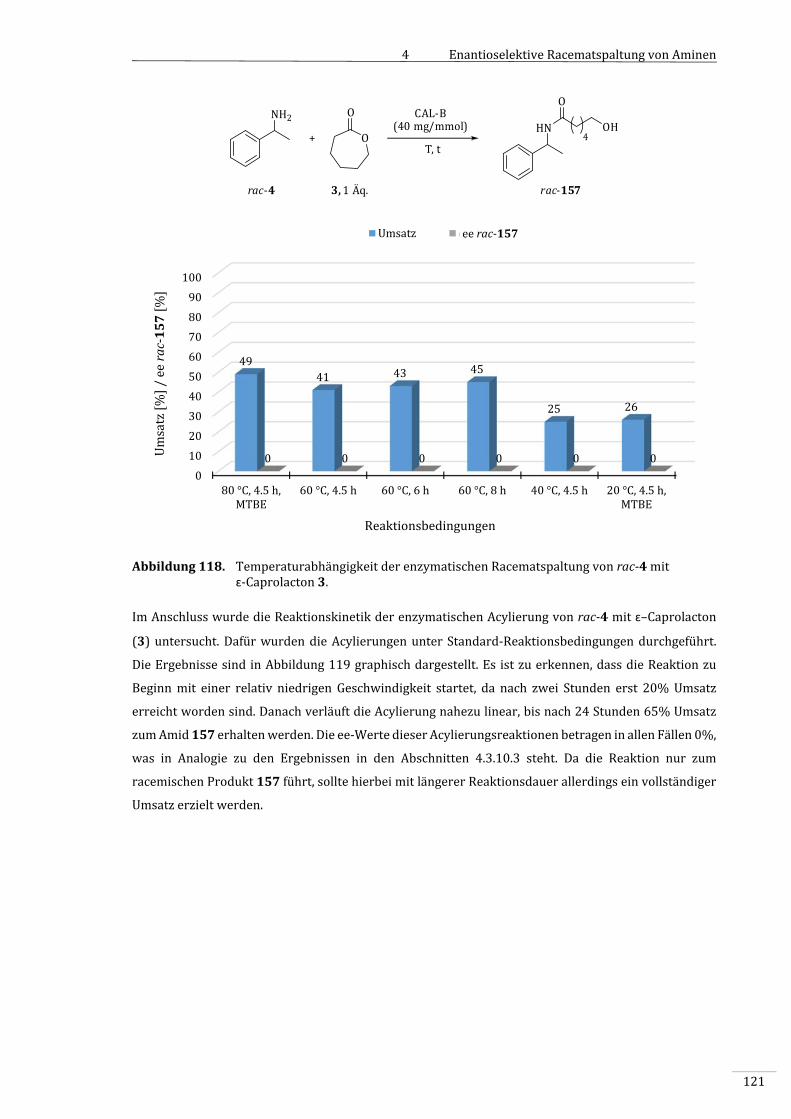
4 EnantioselektiveRacematspaltungvonAminen
121
+T, t
rac‐4 3,1 Äq. rac‐157
O
O CAL‐B(40 mg/mmol)
NH2OH
O
4HN
Abbildung118. TemperaturabhängigkeitderenzymatischenRacematspaltungvonrac‐4mitɛ‐Caprolacton3.
ImAnschlusswurdedieReaktionskinetikderenzymatischenAcylierungvonrac‐4mitɛ–Caprolacton
(3)untersucht.DafürwurdendieAcylierungenunterStandard‐Reaktionsbedingungendurchgeführt.
DieErgebnissesindinAbbildung119graphischdargestellt.Es istzuerkennen,dassdieReaktionzu
Beginnmit einer relativ niedrigen Geschwindigkeit startet, da nach zwei Stunden erst 20%Umsatz
erreichtwordensind.DanachverläuftdieAcylierungnahezulinear,bisnach24Stunden65%Umsatz
zumAmid157erhaltenwerden.Dieee‐WertedieserAcylierungsreaktionenbetrageninallenFällen0%,
was in Analogie zu den Ergebnissen in den Abschnitten 4.3.10.3 steht. Da die Reaktion nur zum
racemischenProdukt157führt,solltehierbeimitlängererReaktionsdauerallerdingseinvollständiger
Umsatzerzieltwerden.
0
10
20
30
40
50
60
70
80
90
100
80°C,4.5h,MTBE
60°C,4.5h 60°C,6h 60°C,8h 40°C,4.5h 20°C,4.5h,MTBE
4941 43 45
25 26
0 0 0 0 0 0Umsatz[%
]/eerac‐157[%]
Reaktionsbedingungen
Umsatz[%] ee[%]eerac‐157
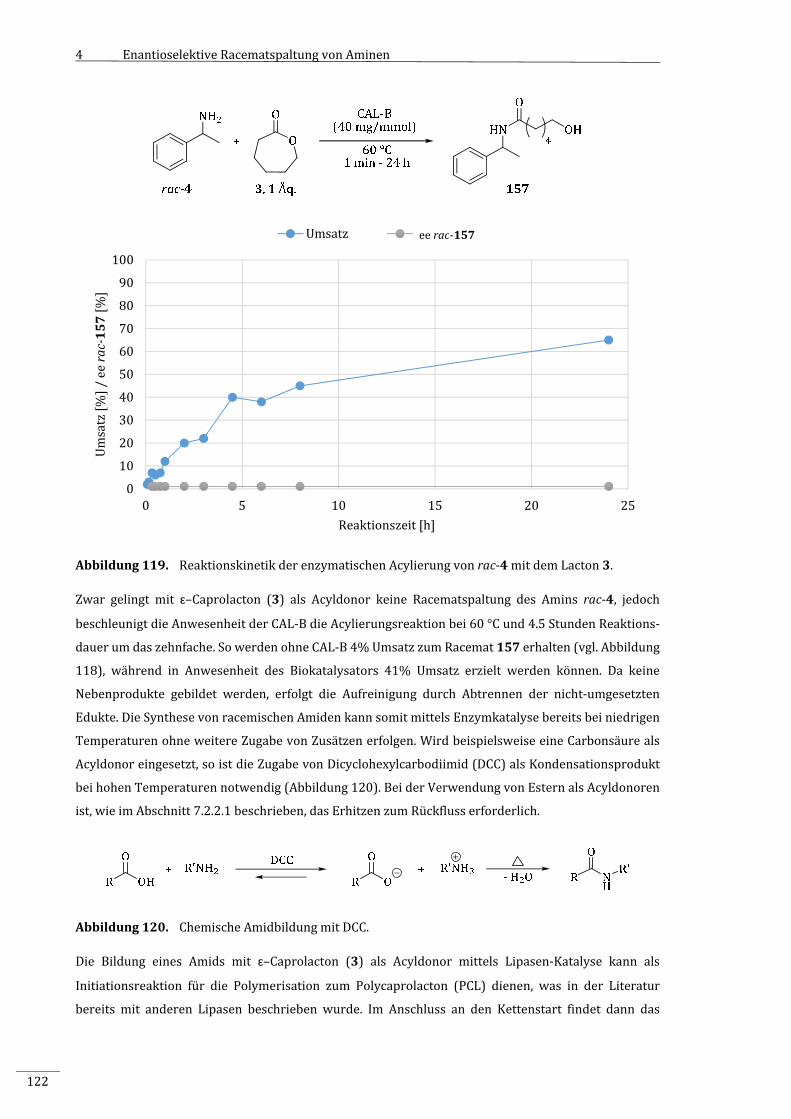
4 EnantioselektiveRacematspaltungvonAminen
122
Abbildung119. ReaktionskinetikderenzymatischenAcylierungvonrac‐4mitdemLacton3.
Zwar gelingt mit ɛ–Caprolacton (3) als Acyldonor keine Racematspaltung des Amins rac‐4, jedoch
beschleunigtdieAnwesenheitderCAL‐BdieAcylierungsreaktionbei60°Cund4.5StundenReaktions‐
dauerumdaszehnfache.SowerdenohneCAL‐B4%UmsatzzumRacemat157erhalten(vgl.Abbildung
118), während in Anwesenheit des Biokatalysators 41% Umsatz erzielt werden können. Da keine
Nebenprodukte gebildet werden, erfolgt die Aufreinigung durch Abtrennen der nicht‐umgesetzten
Edukte.DieSynthesevonracemischenAmidenkannsomitmittelsEnzymkatalysebereitsbeiniedrigen
TemperaturenohneweitereZugabevonZusätzenerfolgen.WirdbeispielsweiseeineCarbonsäureals
Acyldonoreingesetzt,soistdieZugabevonDicyclohexylcarbodiimid(DCC)alsKondensationsprodukt
beihohenTemperaturennotwendig(Abbildung120).BeiderVerwendungvonEsternalsAcyldonoren
ist,wieimAbschnitt7.2.2.1beschrieben,dasErhitzenzumRückflusserforderlich.
Abbildung120. ChemischeAmidbildungmitDCC.
Die Bildung eines Amids mit ɛ–Caprolacton (3) als Acyldonor mittels Lipasen‐Katalyse kann als
Initiationsreaktion für die Polymerisation zum Polycaprolacton (PCL) dienen, was in der Literatur
bereits mit anderen Lipasen beschrieben wurde. Im Anschluss an den Kettenstart findet dann das
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25
Umsatz[%
]/eerac‐157[%]
Reaktionszeit[h]
Umsatz[%] ee[%]eerac‐157

4 EnantioselektiveRacematspaltungvonAminen
123
KettenwachstumdurchringöffnendePolymerisationstatt,wasebenfallsdurchdieLipasekatalysiert
werdenkann.[271]
4.3.10.4 EnzymatischeAcylierungvonsekundärenAminenmitɛ‐Caprolacton(3)
Im Anschluss an die chemische Darstellung der Amide 154 bzw. 155 wurden die entsprechenden
sekundären Amine 149 und 150 enzymatisch unter Standard‐Reaktionsbedingungen umgesetzt
(Tabelle22). SokonntemitbeidenAminennur9%Umsatzzuden jeweiligenAmiden154und155
erzieltwerden.DieAcylierungsreaktionendersekundärenAmine149und150liefernunterlösungs‐
mittelfreienBedingungensomitungenügendeUmsätzevon7bzw.9%.DurchZugabevonMTBEzudem
Reaktionsgemisch(Eintrag3)zurbesserenDurchmischungderKomponentenkonnteeineSteigerung
desUmsatzesauf18%erreichtwerden.DieenzymatischeAcylierungderin2‐Positionsubstituierter
sekundärerAminegelingtdemnachnicht.
Tabelle22. EnzymatischeAcylierungdersekundärenAmine149und150mitɛ‐Caprolacton(3).
Eintrag Amin MTBE[mL] Umsatz[%]
1 149 ‐ 7
2 150 ‐ 9
3 150 0.1 18

124

5 Zusammenfassung
125
5 Zusammenfassung
ZielderArbeitwares,effizienteundnachhaltigeSyntheseroutenvonɛ–Caprolacton(3)undchiralen
Aminenzuetablieren.InnerhalbdieserArbeitkonntezumeinendieenzymatischeDoppeloxidationvon
Cyclohexanol(1)zumLacton3unterVerwendungderCHMOausAcinetobactercalcoaceticussowiedie
Lk‐ADHausLactobacilluskefirerfolgreichdurchgeführtwerden.DieSynthesewurdeinBezugaufihre
Effizienz optimiert. Zum anderen stand besonders die enzymatische Racematspaltung von chiralen
AminendurchdieVerwendungvonLipaseBausCandidaantarctica(CAL‐B)ineinemlösungsmittel‐
freienSystemimFokusderUntersuchungen.
5.1 EnzymatischeOxidationvonCyclohexanol(1)mittels
CHMO
DiezweistufigebiokatalytischeOxidationvonCyclohexanol(1)zuε–Caprolacton(3)mittelsADHaus
LactobacilluskefirsowieeinerCyclohexanon‐MonooxygenaseausAcinetobactercalcoaceticusüberdas
insitu‐gebildeteCyclohexanon(2)alsZwischenstufewurdeeingehenduntersucht.
EinleitendwurdenzweiMethodenzurzuverlässigenUmsatzbestimmungetabliert,dieVerwendungvon
Urotropin(51)alsexternerStandardfür1H‐NMR‐SpektroskopiesowiedieAnalytikmittelserstellter
EichgeradedurchGaschromatographie.BeimVergleichderbeidenMethodenhatsichgezeigt,dassdie
GC‐MethodehöhereUmsätzeundgeringereSchwankungsbreitenliefert,dadieseMethodebesonders
bei geringen Konzentrationen sensibler reagiert und der Schritt des Entfernens des Lösungsmittels
entfällt.AusdiesemGründenwardieAnalytikmittelsGCdieMethodederWahl.
AnschließendwurdendieRückgewinnungsratendereinzelnenSubstanzenbestimmt.Diesesindsowohl
von der eingesetzten Enzymmenge als auch von der Zentrifugierzeit während der Aufarbeitung
abhängig.Bei steigenderKonzentrationdesSubstratsCyclohexanol (1)wurdeeinAnstiegderRück‐
gewinnungsratevon71–80%auf90–97%beobachtet.ZusätzlichkonntediezuvorimArbeitskreis
verwendete Extraktionsmethodemittels EPPENDORF‐Gefäßen optimiert und dadurch der Verlust der
Substanzenverringertwerden.
ZunächstwurdediespezifischeAktivitätderhierverwendetenCHMObezüglichCyclohexanon(2)als
SubstratbestimmtundmitweiterenBVMOs,ECSMo‐03undECSMo‐05vonENZYMICALSAG,verglichen.
Dazuwurden zum einen die volumetrischen Aktivitätenmittels Photometertest bestimmt und zum
anderen BRADFORD‐Tests durchgeführt, um den jeweiligen Proteingehalt der Enzyme zu ermitteln.
Dadurch konnten die spezifischen Aktivitäten der drei Monooxygenasen berechnet werden. Die
spezifischeAktivitätderCHMOwurdegleich100%relativeAktivitätgesetzt.DiebeidenBVMOsECS
Mo‐03 und ECS Mo‐05 wiesen spezifische Aktivitäten von 0.2 U/mg bzw. 0.05 U/mg auf. Dieses

5 Zusammenfassung
126
entsprichtrelativenAktivitätenvon7%bzw.2%.FolglichsinddiebeidenMonooxygenasennichtfähig,
Cyclohexanon(2)effektivzuoxidieren.
DesWeiterenwurdedieAktivitätderEnzymegegenüberdenReferenz‐Substanzensowiegegenüber
weiterenSubstratenphotometrischbestimmt.DieCHMOwiesbezüglichCyclohexanon(2)eineAktivität
von0.082U/mL,wasgleich100%relativerAktivitätgesetztwurde.GegenüberdemSubstratIsophoron
(52)besitztdieCHMOeinerelativeAktivitätvonnur3%.SokonntebeiderbiokatalytischenOxidation
von Isophoron(52) mit dem D‐Glucose (48‐a)/GDH‐Regenerationssystem kein Umsatz festgestellt
werden. Unter den in dieser Arbeit gewählten Reaktionsbedingungen ist diese Aktivität nicht aus‐
reichendfüreineökonomischsinnvolleOxidation.
DieAktivitätderLk‐ADHwurdegegenüberdemStandard‐Substrat1‐Phenylethanol(47)bestimmtund
gleich 100% relative Aktivität gesetzt. Im Vergleich dazu wurde gegenüber Cyclohexanol (1) und
3,3,5‐Trimethylcyclohexanol(53)einerelativeAktivitätvon153%bzw.0%bestimmt.Währendsich
Cyclohexanol(1)demnachhervorragendalsSubstratfürdieOxidationdurchdieLk‐ADHeignet,liefert
die enzymatischeDoppeloxidationdesdreifachmethyliertenAlkohols53 unter den Standard‐Reak‐
tionsbedingungenkeinLacton56oder57.
ImAnschlusswurdedieStandard‐Reaktion inHinblickaufProduktinhibierungderCHMOoptimiert,
indemunterschiedlicheorganischeLösungsmittelineinemZweiphasensystemgetestetwurden.Dabei
zeigtesicheinGemischausKPi‐Pufferund10%Isooctan(v/v)alsbestesLösungsmittel.Dieseslieferte
einensehrgutenUmsatzvon87%(Abbildung121).AllgemeinkönnenmitLösungsmittelnmithöheren
logP‐WertenauchhöhereUmsätzeerzieltwerden.
Abbildung121. EnzymatischeDoppeloxidationvonCyclohexanol(1)mit10%(v/v)Isooctan.
AußerdemwurdensowohlderEinflussderMengenderLk‐ADHunddesCofaktorssowiederReaktions‐
temperatur untersucht. Bei der Erhöhungder Cofaktormenge von 1 auf 10mol‐% sinkt derUmsatz
dabei um einDrittel. Einen noch stärkeren negativen Einfluss haben die Verringerung der Lk‐ADH‐
Mengevon40auf13UsowiedieErhöhungderReaktionstemperaturvonRTauf30°C.
Eskonntenachgewiesenwerden,dassselbstbei sehrhohenProduktkonzentrationenvon1Mkeine
ProduktinhibierungderLk‐ADHauftritt.AllerdingswurdemittelsTitrino‐Apparaturdiehydrolytische
SpaltungdesLactons3zurentsprechendenSäure54beobachtet.
Dieinsitu‐ProduktentfernungmittelsAdsorberharzengelangmitdenfürdieseArbeitgewähltenHarzen
nicht.AuchdieRückgewinnungsratederSubstanzenausdenHarzendurchextraktiveAufarbeitungist
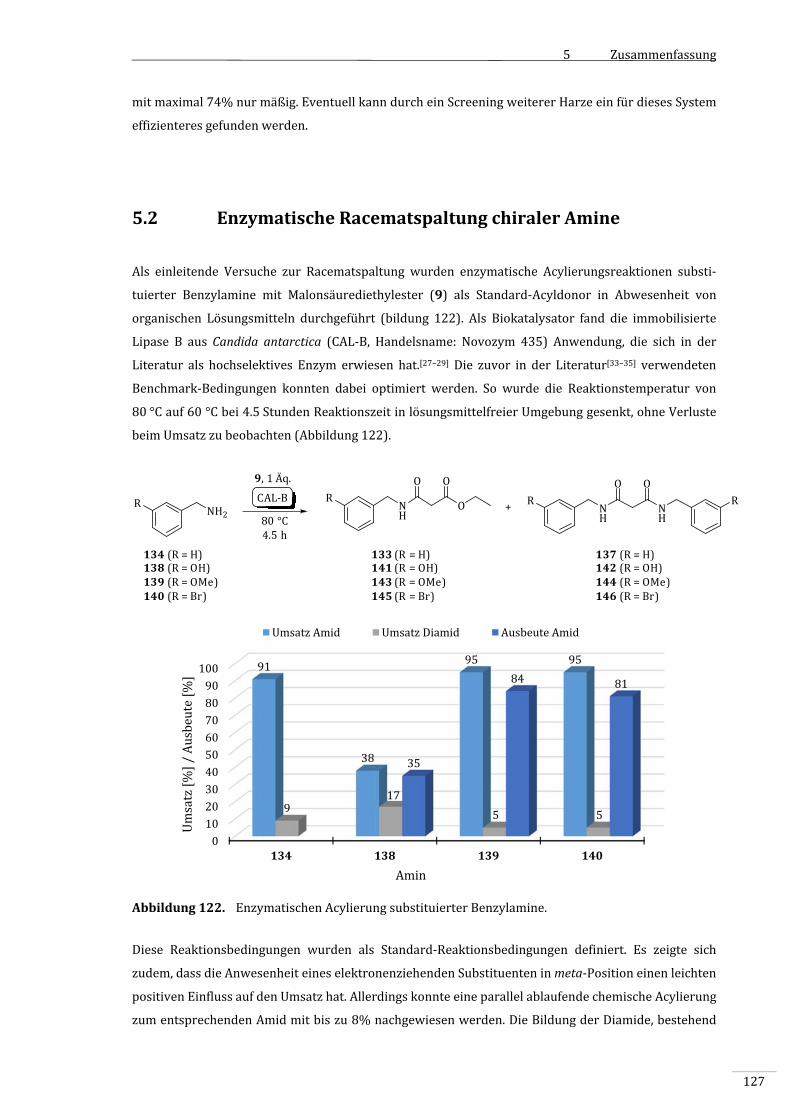
5 Zusammenfassung
127
mitmaximal74%nurmäßig.EventuellkanndurcheinScreeningweitererHarzeeinfürdiesesSystem
effizienteresgefundenwerden.
5.2 EnzymatischeRacematspaltungchiralerAmine
Als einleitende Versuche zur Racematspaltung wurden enzymatische Acylierungsreaktionen substi‐
tuierter Benzylamine mit Malonsäurediethylester (9) als Standard‐Acyldonor in Abwesenheit von
organischen Lösungsmitteln durchgeführt (bildung 122). Als Biokatalysator fand die immobilisierte
Lipase B aus Candida antarctica (CAL‐B, Handelsname: Novozym 435) Anwendung, die sich in der
Literatur als hochselektives Enzym erwiesen hat.[27–29] Die zuvor in der Literatur[33–35] verwendeten
Benchmark‐Bedingungen konnten dabei optimiert werden. So wurde die Reaktionstemperatur von
80°Cauf60°Cbei4.5StundenReaktionszeitinlösungsmittelfreierUmgebunggesenkt,ohneVerluste
beimUmsatzzubeobachten(Abbildung122).
NH2CAL‐B
NH
O
O
O
R R
134 (R=H)138 (R=OH)139 (R=OMe)140 (R=Br)
9,1Äq.
NH
O
NH
OR R+
80°C4.5h
133 (R=H)141 (R=OH)143 (R=OMe)145 (R=Br)
137 (R=H)142 (R=OH)144 (R=OMe)146 (R=Br)
Abbildung122. EnzymatischenAcylierungsubstituierterBenzylamine.
Diese Reaktionsbedingungen wurden als Standard‐Reaktionsbedingungen definiert. Es zeigte sich
zudem,dassdieAnwesenheiteineselektronenziehendenSubstituenteninmeta‐Positioneinenleichten
positivenEinflussaufdenUmsatzhat.AllerdingskonnteeineparallelablaufendechemischeAcylierung
zumentsprechendenAmidmitbiszu8%nachgewiesenwerden.DieBildungderDiamide,bestehend
0102030405060708090100
134 138 139 140
91
38
95 95
917
5 5
35
84 81
Umsatz[%
]/Ausbeute[%]
Amin
UmsatzAmid[%] UmsatzDiamid[%] AusbeuteAmid[%]

5 Zusammenfassung
128
ausMalonsäurediethylester(9)undzweiÄquivalentensubstituiertenBenzylamins,wurde innahezu
allenFällennachgewiesen.DurchEinsatzeinesleichtenÜberschusses(1.2Äquivalente)anMalonester
9konntedieBildungausgehendvonMethoxy‐Amin139nahezuunterdrücktwerden.Dabeiistaller‐
dings ein leichter Abfall des Umsatzes von 91% auf 82% zu beobachten. Bei der Verwendung des
Hydroxy‐substituiertenBenzylamins134warjedochaufgrunddeshohenSchmelzpunktesderEinsatz
vonLösungsmittelnnotwendig,umdieHomogenitätderMischungzugewährleisten.Vondenunter‐
suchtenunterschiedlichenLösungsmittelnliefertenTHF(75%Umsatzbei80°CReaktionstemperatur)
und2‐Methyl‐THF(70%Umsatzbei80°C)diebestenErgebnisse.
DieenzymatischeRacematspaltungvonracemischenAminenunter lösungsmittelfreienBedingungen
konnteanschließendanhanddesSystemsaus1‐Phenylethylamin(rac‐4)undMalonsäurediethylester
(9) zum enantiomerenreinen Amid rac‐10 untersucht werden (Abbildung 123). Dabei wurde der
EinflussvonTemperaturundReaktionsdauersowiederEnzymbeladunguntersuchtunddieReaktions‐
kinetik unter den optimierten Bedingungen dokumentiert. Das Amid rac‐10 konnte dabeimit 42%
Umsatzundmit97%Enantiomerenreinheiterhaltenwerden,waseinerexzellentenEnantioselektivität
von>100entspricht.EineVerlängerungderReaktionszeitauf24Stundenbei60°CReaktionstempe‐
ratur führte zu einemvollständigenUmsatz zumAmid (R)‐10mit 98%ee. Eine parallel ablaufende
chemischeAcylierungwurdebeirac‐4mitdemMalonester9alsAcyldonornichtbeobachtet.
Abbildung123. EnzymatischeRacematspaltung(Benchmark‐Reaktion).
EinVergleichdiesesVerfahrensmitbestehendenindustriellenProzessen[30,264]zeigte,dassdieRacemat‐
spaltung des chiralen Amins rac‐4 unter den in dieser Arbeit gewählten lösungsmittelfreien Bedin‐
gungenEnantioselektivitätendergleichenGrößenordnungliefert.WährenddasinderLiteratur[30]ver‐
wendeteMethoxyessigester11152%Umsatzund95%ee(E>100)lieferte,gelangdieRacematspal‐
tungunterdengleichen inder IndustrieverwendetenBedingungenmitMalonester9 alsDonordas
Amid (R)‐10 mit 48% Umsatz und ebenfalls 95% ee (E>100). Die Racematspaltung unter den im
Rahmen dieser Arbeit gewählten Bedingungen lieferte eine Enantioselektivität von >100. Die
verwendete größere Enzymmenge und höhere Temperatur wird folglich durch Vermeidung von
Lösungsmittel und viel kürzererReaktionszeit ausgeglichen.Der in dieserArbeit etablierte lösungs‐
mittelfreieProzessstelltsomiteinesehrguteAlternativezubestehendenVerfahrendar.
Bei den anschließend durchgeführten Recyclingversuchen der Racematspaltung von rac‐4 mit
Malonester9durchdie immobilisiertenCAL‐B (Novozym435)konnteeindeutlicherAbfalldesUm‐
satzesimVerlaufderRecyclingschrittebeobachtetwerden(Abbildung124).NachdemzweitenRecyc‐
lingschritt wurden nur noch 30% und im dritten bis fünften Durchgang nur 20‐24% Umsatz zum

5 Zusammenfassung
129
entsprechenden Amid (R)‐10 gefunden. Die ee‐Werte der einzelnen Durchgänge blieben allerdings
konstant hoch. Dieses Verhalten ist auf die Instabilität des Biokatalysators in lösungsmittelfreier
UmgebungzurückzuführenundstehtinAnalogiezurLiteratur.[36]
Abbildung124. Enzymrecycling.
In einembreitenEnzym‐Screening aus23Lipasenkonnte sowohldie CAL‐BausCandidaantarctica
(Novozym435) als auch zwei Formender CAL‐Bder Firma C‐LECTA, in immobilisierter und lyophi‐
lisierterForm,alsdieeffektivstenLipasenbeiderAcylierungderBenzylamine134und138sowieder
Racematspaltungvonrac‐4nachgewiesenwerden.BeiTitrationsuntersuchungenwiesendieLipasen
vonC‐LECTA68%bzw.1000%relativeAktivitätinBezugaufNovozym435auf.BeipräparativenVer‐
suchenjedochzeigtenurdieimmobilisierteLipasevonC‐LECTAsehrguteErgebnisse(52%Umsatz,96%
ee).Die lyophilisierteCAL‐B lieferteunteroptimiertenBedingungen(5facheEnzymmenge)nur26%
Umsatz.DieimmobilisierteCAL‐BvonC‐LECTAstelltsomiteinweiteresWerkzeugzurenzymatischen
RacematspaltungvonAminendar.
DieUntersuchungzurVerwendungalternativerAcyldonorenerfolgteinHinblickaufeineVerbesserung
derRacematspaltungmitderimmobilisiertenCAL‐B(Novozym435).DieunterStandard‐Bedingungen
enzymatisch dargestellten Amide wiesen sehr unterschiedliche Enantiomerenüberschüsse auf. So
konntenurmitdemliteraturbekannten[229]Methoxyessigester11192%eeerzieltwerden.Racemat‐
spaltungenmitweiterengetestetenAcyldonorenführtenzuee‐Wertevon10–65%,sodassdieseunter
dengewähltenStandard‐BedingungeneineeffizienteRacematspaltungnicht stattfindet.Beiden teil‐
weisesehrreaktivenAcyldonorenkonnteeineparallelablaufendechemischeAcylierungnachgewiesen
werden.SowurdemitVinylessigester125inAbwesenheitderLipasebereitsbei40°C81%Umsatzzum
entsprechendenracemischenAmiderzielt.DurchdieseHintergrundreaktionenwerdendieee‐Werte
derRacematspaltungenverringert.DasbereitsinderArbeitsgruppeetablierte[33,34]undindieserArbeit
alsStandard‐AcyldonorverwendeteMalonsäurediethylester(9)stelltesichauchinlösungsmittelfreier
UmgebungalsdereffektivsteDonordar.
ZumdirektenVergleichderAcyldonorenuntereinanderwurdenReaktionskinetikenderenzymatischen
Acylierungvonrac‐4unterlösungsmittelfreienStandard‐Bedingungenaufgenommen(Abbildung125).
0102030405060708090100
1 2 3 4 5
43
3025
20 24
92 89 87 90 94
Umsatz[%
]/ee[%]
Zyklus
Umsatz[%] ee[%]ee(R)‐10
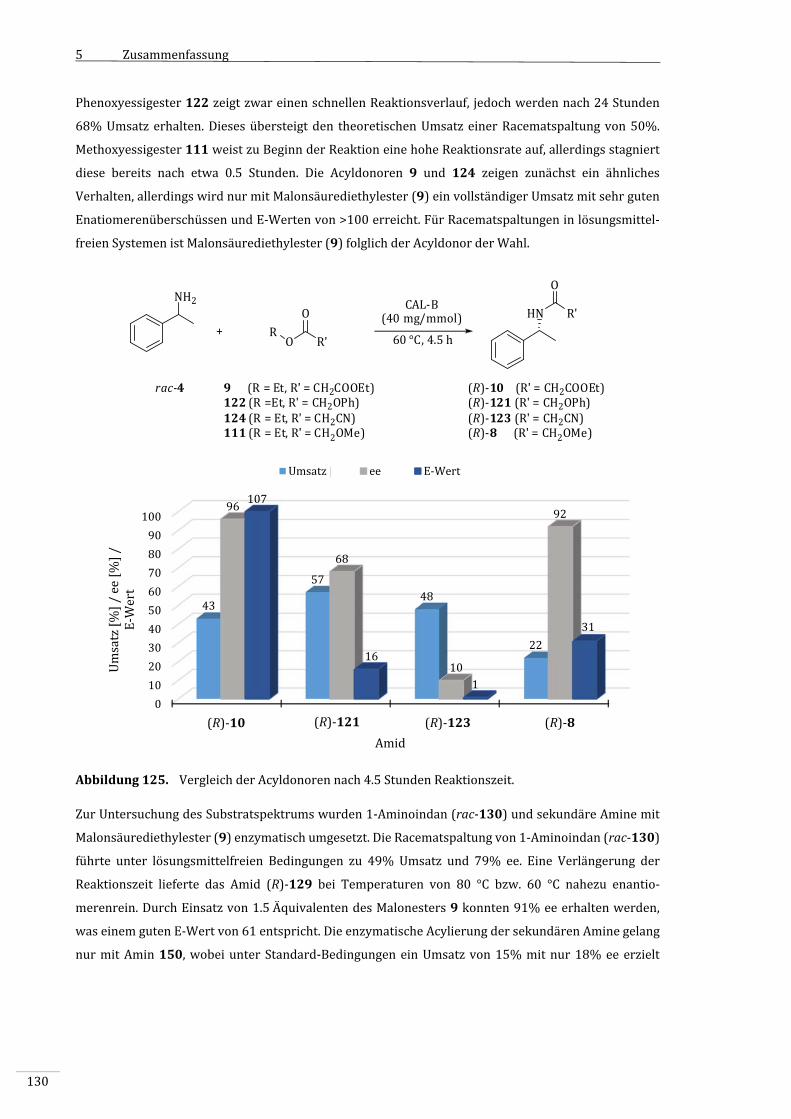
5 Zusammenfassung
130
Phenoxyessigester122zeigtzwareinenschnellenReaktionsverlauf, jedochwerdennach24Stunden
68%Umsatzerhalten.Diesesübersteigtden theoretischenUmsatzeinerRacematspaltungvon50%.
Methoxyessigester111weistzuBeginnderReaktioneinehoheReaktionsrateauf,allerdingsstagniert
diese bereits nach etwa 0.5 Stunden. Die Acyldonoren 9 und 124 zeigen zunächst ein ähnliches
Verhalten,allerdingswirdnurmitMalonsäurediethylester(9)einvollständigerUmsatzmitsehrguten
EnatiomerenüberschüssenundE‐Wertenvon>100erreicht.FürRacematspaltungeninlösungsmittel‐
freienSystemenistMalonsäurediethylester(9)folglichderAcyldonorderWahl.
RO
O R'+
NH2HN R'
O
rac‐4
CAL‐B(40 mg/mmol)
9 (R = Et, R' = CH2COOEt)122 (R =Et, R' = CH2OPh)124 (R = Et, R' = CH2CN)111 (R = Et, R' = CH2OMe)
(R)‐10 (R' = CH2COOEt)(R)‐121 (R' = CH2OPh)(R)‐123 (R' = CH2CN)(R)‐8 (R' = CH2OMe)
60 °C, 4.5 h
Abbildung125. VergleichderAcyldonorennach4.5StundenReaktionszeit.
ZurUntersuchungdesSubstratspektrumswurden1‐Aminoindan(rac‐130)undsekundäreAminemit
Malonsäurediethylester(9)enzymatischumgesetzt.DieRacematspaltungvon1‐Aminoindan(rac‐130)
führte unter lösungsmittelfreien Bedingungen zu 49% Umsatz und 79% ee. Eine Verlängerung der
Reaktionszeit lieferte das Amid (R)‐129 bei Temperaturen von 80 °C bzw. 60 °C nahezu enantio‐
merenrein.DurchEinsatzvon1.5ÄquivalentendesMalonesters9konnten91%eeerhaltenwerden,
waseinemgutenE‐Wertvon61entspricht.DieenzymatischeAcylierungdersekundärenAminegelang
nurmitAmin150,wobeiunterStandard‐BedingungeneinUmsatzvon15%mitnur18%eeerzielt
0
10
20
30
40
50
60
70
80
90
100
(R)‐10 (R)‐121 (R)‐123 (R)‐8
43
5748
22
96
68
10
92107
16
1
31
Umsatz[%
]/ee[%]/
E‐Wert
Amid
Umsatz[%] ee[%] E‐Wert
(R)‐10 (R)‐121 (R)‐123 (R)‐8

5 Zusammenfassung
131
werden konnte. Unter den gewählten Bedingungen konnte eine effiziente Racematspaltung nicht
erreichtwerden.
AlsweitererAcyldonor sollte das zuvor enzymatisch synthetisierte ɛ–Caprolacton (3) dienen.Dabei
konnte inallenFällennurgeringeUmsätzeundauchnur racemischeGemischedesentsprechenden
Amidserhaltenwerden.Somit istdasLacton3keingeeigneterAcyldonor.Allerdingskanndieenzy‐
matische Amidbildung als Startreaktion für die Polymerisation zum Polycaprolacton (PCL) genutzt
werden.
Die erhaltenen Ergebnisse zeigen, dass die lösungsmittelfreie Racematspaltung vergleichbar ist mit
Reaktionen in organischenMedien.DerUnterschied zwischen lösungsmittelfreienAcylierungen und
ReaktioneninLösungsmittelnliegeninderbenötigtenZeit,um50%Umsatzzuerreichen,sowieinder
benötigtenEnzymmenge.BesondersmitMalonsäurediethylester(9)alsAcyldonorundderLipaseBaus
Candidaantarctica(CAL‐B,Novozym435)konntenbeiderRacematspaltungchiralerAminewierac‐4
undrac‐130inAbwesenheitvonorganischenLösungsmittelnsehrguteErgebnissebei60°CReaktions‐
temperaturinnur4.5Stundenbzw.24StundenReaktionszeitimFallvonrac‐130erzieltwerden.

6 Summary
132
6 Summary
Theaimofthisworkwastoestablishefficientandsustainablesynthesisroutesofɛ‐caprolactone(3)
andchiralamines.Inthisthesistheenzymaticdoubleoxidationofcyclohexanol(1)tolactone3byusage
of CHMO from Acinetobacter calcoaceticus as well as Lk‐ADH from Lactobacillus kefir has been
successfully carried out. The synthesis was optimized in regard of its efficiency. In particular the
enzymaticresolutionofchiralaminesbyusageoflipaseBfromCandidaantarctica(CAL‐B)inasolvent‐
freesystemwasinthefocusofresearch.
6.1 Enzymaticoxidationofcyclohexanol(1)bymeansof
CHMO
Thetwo‐stepbiocatalyticoxidationofcyclohexanol(1)toɛ–caprolactone(3)performedbymeansof
ADHfromLactobacilluskefirandcyclohexanone‐monooxygenasefromAcinetobactercalcoaceticusvia
theintermediatestateoftheinsitu‐formedcyclohexanone(2)hasbeenexaminedindetail.
For a reliable determination of the conversion two methods have been developed first, the use of
urotropine(51)asanexternalstandardfor1H‐NMRspectroscopyandamethodofanalysisbyuseofa
createdstandardcurveviagaschromatography.Ithastobetakenintoaccountwhencomparingboth
methodsthattheGCmethodprovideshigherconversionsandlowerrangeoffluctuationbecausethis
method issensitiveathigherconcentrationsand themeasureof removingthesolvent iseliminated.
ThereforeanalysisviaGCisthemethodofchoice.
Afterwardstherecoveryratesoftheindividualsubstanceshadtobedeterminedwhicharedependent
ontheusedamountofenzymeaswellasthetimeofcentrifugationduringthework‐upprocedure.At
growingconcentrationofthesubstratecyclohexanol(1)anincreasedrecoveryratefrom71–80%to
90–97%isobserved. InadditionthebeforehandusedmethodofextractionviaEPPENDORFvialshas
beenimprovedandthusreductionofthelossfactorofthesubstanceswasachieved.
ThespecificactivityofthehereinusedCHMOwithregardtocyclohexanone(2)assubstratehasbeen
determinedandcomparedwithotherBVMOs,ECSMo‐03andECSMo‐05fromENZYMICALSAG.Therefore
firstlythevolumetricactivityhasbeendeterminedviaphotometertestsandsecondlyaBRADFORDtest
hasbeenperformedtoidentifytheproteincontentoftheenzymes.Inthiswaythespecificactivityof
thesemonooxygenasescouldbecalculated.ThespecificactivityoftheCHMOisrepresentedas100%.
TheothertwoBVMOs,ECSMo‐03andECSMo‐05,showed0.2U/mgand0.05U/mgspecificactivity,
whichcorrespondsto7%and2%relativeactivity,respectively.Thusbothothermonooxygenasesare
notcapableofoxidizingcyclohexanone(2)effectively.

6 Summary
133
Inadditiontheactivityoftheenzymeshavebeenassayedphotometricallywithregardtothebenchmark
substanceaswellasfurthersubstrates.TheCHMOshowedanactivityof0.082U/mLregardingcyclo‐
hexanone(2),whichisrepresentedas100%.Towardsthesubstrateisophorone(52)theCHMOshowed
anactivityofjust3%.Sonoconversioncouldbedetectedbythebiocatalyticoxidationofisophorone
(52) with the D‐glucose (48‐a)/GDH‐regeneration system. Under the terms used in this work this
activityisnotsufficientforaneconomicallyreasonableoxidation.
The activity of the Lk‐ADHhas been determinedwith regardof thebenchmark substrate 1‐phenyl‐
ethylethanol(47)andrepresentedas100%relativeactivity.Comparedtothistherelativeactivitywith
regard to cyclohexanol (1) and 3,3,5‐trimethylcyclohexanol (53) has been set at 153% and 0%,
respectively.Cyclohexanol(1)isanextremelywell‐suitedsubstrateforoxidationbyLk‐ADH.However,
theenzymaticdoubleoxidationofthethreefoldmethylatedalcohol53providesnolactone56or57
underthebenchmarkreactionconditions.
Thisresultsarefollowedbytheoptimizationofthebenchmarkreactioninviewofproductinhibitionof
theCHMObytestingdifferentorganicsolventsinatwo‐phasesystem.Itwasfoundthatthebestsolvent
isamixtureofKPi‐bufferand10%isooctane(v/v),whichledtoaconversionof87%(Scheme1).With
organicsolventswithhigherlogP‐values,ingeneral,higherconversionscanbeachieved.Besidesboth
the influenceof the amountof theLk‐ADHandof the cofactor aswell as the temperaturehasbeen
investigated.The conversionhas fallenbyone thirdby increasing theamountof cofactor from1 to
10mol‐%.ThereductionoftheamountofLk‐ADHfrom40to13Uandtheriseofthetemperaturefrom
RTto30°Chadanevenstrongernegativeimpact.
Scheme1. Enzymaticdoubleoxidationofcyclohexanol(1)with10%(v/v)isooctane.
Itwasdemonstratedthatevenwithveryhighproductconcentrationof1Mnoproductinhibitionofthe
Lk‐ADH appeared.However, by use of theTitrino titration apparatus the hydrolytic cleavage of the
lactone3tothecorrespondingacid53couldbeobserved.
Theinsitu‐productremovalviaadsorberresinsdidnotsucceedunderthechosenconditions.Alsothe
recovery rates fromthe resinsbymeansof extractive recycling ismodestwithamaximumof74%.
Eventuallyamoreefficientsystemcanbefoundbyscreeningoffurtherresins.
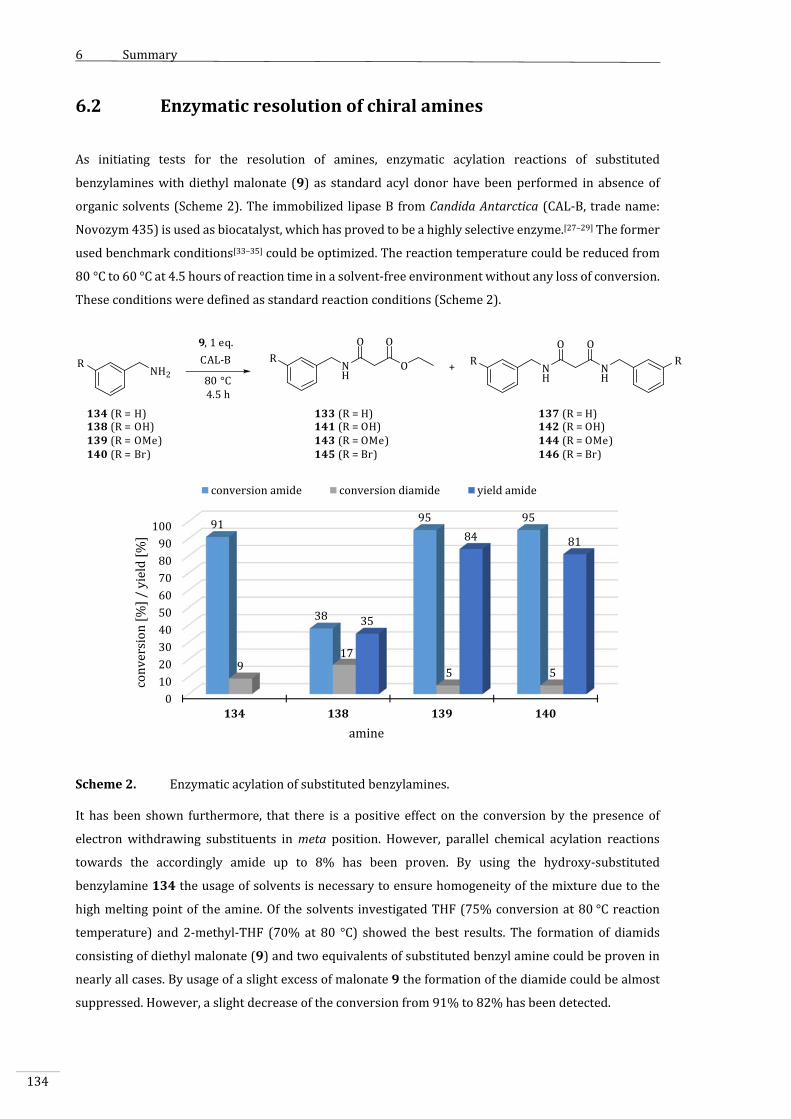
6 Summary
134
6.2 Enzymaticresolutionofchiralamines
As initiating tests for the resolution of amines, enzymatic acylation reactions of substituted
benzylamineswithdiethylmalonate (9) as standardacyl donorhavebeenperformed in absence of
organicsolvents(Scheme2).The immobilized lipaseB fromCandidaAntarctica (CAL‐B, tradename:
Novozym435)isusedasbiocatalyst,whichhasprovedtobeahighlyselectiveenzyme.[27–29]Theformer
usedbenchmarkconditions[33–35]couldbeoptimized.Thereactiontemperaturecouldbereducedfrom
80°Cto60°Cat4.5hoursofreactiontimeinasolvent‐freeenvironmentwithoutanylossofconversion.
Theseconditionsweredefinedasstandardreactionconditions(Scheme2).
NH2CAL‐B N
H
O
O
O
R R
134 (R=H)138 (R=OH)139 (R=OMe)140 (R=Br)
9,1eq.
NH
O
NH
OR R+
80°C4.5h
133 (R=H)141 (R=OH)143 (R=OMe)145 (R=Br)
137 (R=H)142 (R=OH)144 (R=OMe)146 (R=Br)
Scheme2. Enzymaticacylationofsubstitutedbenzylamines.
It has been shown furthermore, that there is a positive effect on the conversion by the presence of
electron withdrawing substituents inmeta position. However, parallel chemical acylation reactions
towards the accordingly amide up to 8% has been proven. By using the hydroxy‐substituted
benzylamine134theusageofsolventsisnecessarytoensurehomogeneityofthemixtureduetothe
highmeltingpointof theamine.Of thesolvents investigatedTHF(75%conversionat80°Creaction
temperature) and 2‐methyl‐THF (70% at 80 °C) showed the best results. The formation of diamids
consistingofdiethylmalonate(9)andtwoequivalentsofsubstitutedbenzylaminecouldbeprovenin
nearlyallcases.Byusageofaslightexcessofmalonate9theformationofthediamidecouldbealmost
suppressed.However,aslightdecreaseoftheconversionfrom91%to82%hasbeendetected.
0102030405060708090100
134 138 139 140
91
38
95 95
917
5 5
35
84 81
conversion[%
]/yield[%
]
amine
conversionamide conversiondiamide yieldamide
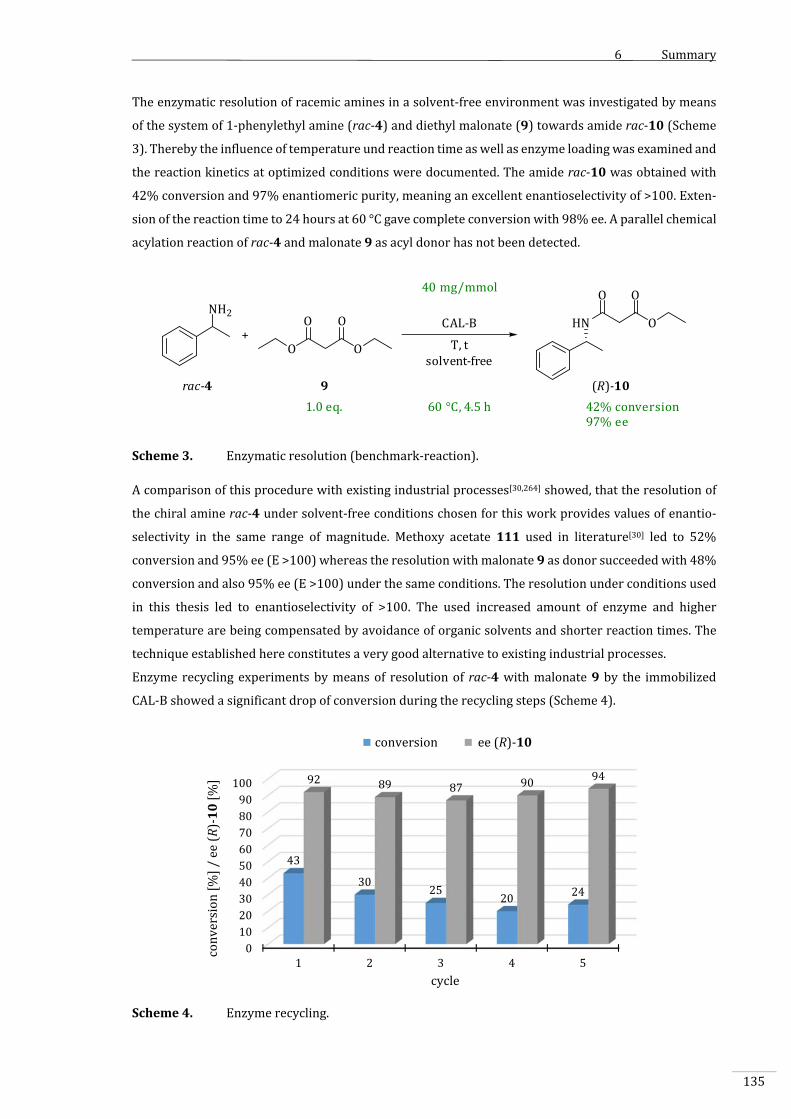
6 Summary
135
Theenzymaticresolutionofracemicaminesinasolvent‐freeenvironmentwasinvestigatedbymeans
ofthesystemof1‐phenylethylamine(rac‐4)anddiethylmalonate(9)towardsamiderac‐10(Scheme
3).Therebytheinfluenceoftemperatureundreactiontimeaswellasenzymeloadingwasexaminedand
thereactionkineticsatoptimizedconditionsweredocumented.Theamiderac‐10wasobtainedwith
42%conversionand97%enantiomericpurity,meaninganexcellentenantioselectivityof>100.Exten‐
sionofthereactiontimeto24hoursat60°Cgavecompleteconversionwith98%ee.Aparallelchemical
acylationreactionofrac‐4andmalonate9asacyldonorhasnotbeendetected.
O
O
O
O+
(R)‐10rac‐4 9
NH2HN
O
O
O
CAL‐B
T,tsolvent‐free
1.0eq.
40mg/mmol
42%conversion97%ee
60°C,4.5h
Scheme3. Enzymaticresolution(benchmark‐reaction).
Acomparisonofthisprocedurewithexistingindustrialprocesses[30,264]showed,thattheresolutionof
thechiralaminerac‐4undersolvent‐freeconditionschosenforthisworkprovidesvaluesofenantio‐
selectivity in the same range of magnitude. Methoxy acetate 111 used in literature[30] led to 52%
conversionand95%ee(E>100)whereastheresolutionwithmalonate9asdonorsucceededwith48%
conversionandalso95%ee(E>100)underthesameconditions.Theresolutionunderconditionsused
in this thesis led to enantioselectivity of >100. The used increased amount of enzyme and higher
temperaturearebeingcompensatedbyavoidanceoforganicsolventsandshorterreactiontimes.The
techniqueestablishedhereconstitutesaverygoodalternativetoexistingindustrialprocesses.
Enzymerecyclingexperimentsbymeansof resolutionofrac‐4withmalonate9 by the immobilized
CAL‐Bshowedasignificantdropofconversionduringtherecyclingsteps(Scheme4).
Scheme4. Enzymerecycling.
0102030405060708090100
1 2 3 4 5
43
3025
20 24
92 89 87 90 94
conversion[%
]/ee(R)‐10[%]
cycle
Umsatz[%] ee[%]conversion ee(R)‐10

6 Summary
136
After the second recycling step, only 30% conversion and in the following recycling steps, merely
20‐ 24% conversion towards the amide (R)‐10 could be detected. The ee‐values of the resolution
reactionsremainedconstantlyhighthough.Thisbehaviorisduetotheinstabilityofthebiocatalystina
solvent‐freeenviron‐mentandisanalogoustoliterature.[36]
Inabroadlipasescreeningfrom23lipasesCAL‐BfromCandidaantarctica(Novozym435)aswellas
two forms of CAL‐B, received from C‐LECTA in immobilized and lyophilized form, have been
demonstratedasthemosteffectivelipasesbytheacylationofbenzylamines130and132aswellasthe
resolutionofrac‐4.TitrationinvestigationsofthelipasesfromC‐LECTAshowed68%and1000%relative
activityregardingNovozym435,respectively.Inpreparativeexperimentsonlytheimmobilizedlipase
fromC‐LECTAoffered very good results (52%conversion, 96%ee).The lyophilizedCAL‐Bdelivered
merely26%conversionunderoptimizedconditions(fivefoldamountofenzyme).Thustheimmobilized
CAL‐BfromC‐LECTApresentsanimportanttoolforenzymaticresolutionofchiralamines.
Theexaminationoftheusageofalternativeacyldonorsoccurredwithregardtotheimprovementofthe
resolution by the immobilized CAL‐B. The amides, which were enzymatically synthesized under
standardconditions,presentedverydifferentenantiomericratios.Soonlywithmethoxyacetate111
knownfromliterature[229]92%eecouldbeachieved.Resolutionwiththeothertestedacyldonorsledto
ee‐valuesof10–65%.Thusthesedonorsdonotleadtoefficientresolutionundertheconditionschosen
inthisthesis.Incaseofveryreactiveacyldonors,parallelrunningchemicalacylationreactionswere
proven.Sowithvinylacetate125at40°Calready81%conversiontotherespectiveracemicamidewas
receivedinabsenceoflipase.Duetothisbackgroundreactiontheee‐valuesoftheenzymaticresolution
arebeingdecreased.Diethylmalonate(9),alreadyestablishedinthisworkinggroup[33–35]andusedas
standardacyldonor in this thesis, represented itselfas themosteffectiveacyldonor in solvent‐free
environmentalso.
Forthedirectcomparisonoftheacyldonorsamongthemselvesreactionkineticsoftheresolutionof
rac‐4undersolvent‐freeconditionsweremeasured(Scheme5).Phenoxyacetate118presentedafast
reaction process but after 24 hours 68% conversion were achieved, which surpasses the theoretic
conversion of a resolution of 50%.Methoxy acetate111 exhibited a very high reaction rate at the
beginningwhichstagnatedalreadyafter0.5hours.Theacyldonors9and124showedsimilarbehavior,
butjustdiethylmalonate(9)ledtocompleteconversionandveryhighenantiomericratio(E>100).For
resolutionreactionsinsolvent‐freesystemdiethylmalonate(9)istheacyldonorofchoice.
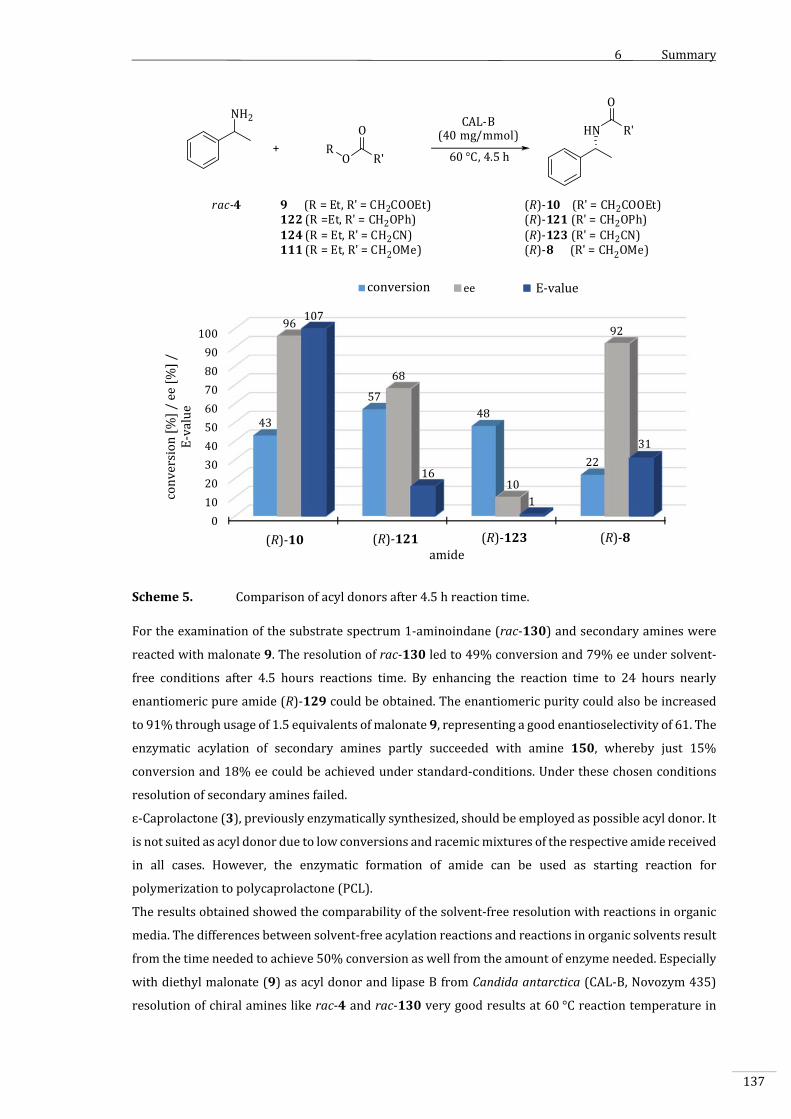
6 Summary
137
RO
O R'+
NH2HN R'
O
rac‐4
CAL‐B(40 mg/mmol)
9 (R = Et, R' = CH2COOEt)122 (R =Et, R' = CH2OPh)124 (R = Et, R' = CH2CN)111 (R = Et, R' = CH2OMe)
(R)‐10 (R' = CH2COOEt)(R)‐121 (R' = CH2OPh)(R)‐123 (R' = CH2CN)(R)‐8 (R' = CH2OMe)
60 °C, 4.5 h
Scheme5. Comparisonofacyldonorsafter4.5hreactiontime.
Fortheexaminationofthesubstratespectrum1‐aminoindane(rac‐130)andsecondaryamineswere
reactedwithmalonate9.Theresolutionofrac‐130ledto49%conversionand79%eeundersolvent‐
free conditions after 4.5 hours reactions time. By enhancing the reaction time to 24 hours nearly
enantiomericpureamide(R)‐129couldbeobtained.Theenantiomericpuritycouldalsobeincreased
to91%throughusageof1.5equivalentsofmalonate9,representingagoodenantioselectivityof61.The
enzymatic acylation of secondary amines partly succeeded with amine 150, whereby just 15%
conversionand18%eecouldbeachievedunderstandard‐conditions.Underthesechosenconditions
resolutionofsecondaryaminesfailed.
ɛ‐Caprolactone(3),previouslyenzymaticallysynthesized,shouldbeemployedaspossibleacyldonor.It
isnotsuitedasacyldonorduetolowconversionsandracemicmixturesoftherespectiveamidereceived
in all cases. However, the enzymatic formation of amide can be used as starting reaction for
polymerizationtopolycaprolactone(PCL).
Theresultsobtainedshowedthecomparabilityofthesolvent‐freeresolutionwithreactionsinorganic
media.Thedifferencesbetweensolvent‐freeacylationreactionsandreactionsinorganicsolventsresult
fromthetimeneededtoachieve50%conversionaswellfromtheamountofenzymeneeded.Especially
withdiethylmalonate(9)asacyldonorandlipaseBfromCandidaantarctica(CAL‐B,Novozym435)
resolutionofchiralamineslikerac‐4andrac‐130verygoodresultsat60°Creactiontemperaturein
0
10
20
30
40
50
60
70
80
90
100
(R)‐10 (R)‐121 (R)‐123 (R)‐8
43
5748
22
96
68
10
92107
16
1
31
conversion[%
]/ee[%]/
E‐value
amide
Umsatz[%] ee[%] E‐Wert
(R)‐10 (R)‐121 (R)‐123 (R)‐8
conversion E‐value

6 Summary
138
just4.5hoursreactiontimeand24hoursfortheaminerac‐130,respectively,havebeenachievedin
absenceoforganicsolvents.

7 ExperimentellerTeil
139
7 ExperimentellerTeil
7.1 VerwendeteChemikalienundGeräte
AutomatischesTitrationssystem:
TitrationenwurdenaufeinemTitroLinealphaPlusderFirmaDIANALYTICSdurchgeführt.AlsTitrier‐
lösungdienteverdünnteNaOH‐Lösung(0.1M).DieSpektrenwerdenmitderTitriSoft2.73Softwarevon
SCHOTTINSTRUMENTSaufgenommenundbearbeitet.
Chemikalien:
DieverwendetenkäuflichenChemikalienwurden,wennnichtandersvermerkt,vonACROSORGANICS®,
ALFAAESAR®,FLUKA®,FLUOROCHEMLTD.UNIT.®,MERCK®,ROTH®,SIGMA‐ALDRICH®bezogenundohne
weitereReinigungeingesetzt.DasLösungsmittelMTBEwurdealsHochschulspendevonEVONIKDEGUSSA
GMBHzurVerfügunggestellt.AlleweiterenverwendetenLösungsmittelwurdenvonVWRCHEMICALS®,
FISHERCHEMICAL®,ROTH®undAPPLICHEM®bezogenundwarenfürReaktionenundAnalytikvonp.a.
Qualität.DeuterierteLösungsmittelstammenvonDEUTERO.
Dünnschichtchromatographie(DC):
Dünnschichtchromatogrammewerdenmit MERCK DC‐Folien, Kieselgel 60 auf Aluminiumfolie (F245),
durchgeführt.DieDC‐FolienwerdennachdemEntwickelnmitIoddampfangefärbt.DieDetektionerfolgt
mittelsUV‐Licht.
DC‐MS‐Spektrometrie:
DC‐MS‐KopplungenwurdenamZQ2000(Normalphase;Eluens: iPrOH)miteinerESI‐Ionenquelleder
FirmaWATERS durchgeführt.DieDatenwurdenmitder SoftwareMestReNova (MESTRELABRESEARCH,
Version7.0.2‐8636)ausgewertet.
Elementaranalyse(EA):
ElementaranalysenwurdenaneinemEuroEAElementaranalysatordurchgeführt.
EnzymeundCofaktoren:
DieBAEYER‐VILLIGER‐Monooxygenasen(BVMO)ECS‐MO01sowieECS‐MO03undECS‐MO05wurden
freundlicherweise von der ENZYMICALS AG zur Verfügung gestellt. Die Alkohol‐Dehydrogenase aus
Lactobacillus kefir (Lk‐ADH, DSM 20587) wurden im Arbeitskreis von Herrn Prof. Dr. W.HUMMEL
(FORSCHUNGSZENTRUM JÜLICH) entwickelt und zur Verfügung gestellt. Die Cofaktoren NAD(P)H und
NAD(P)+wurdenvonORIENTALYEASTerworben.CandidaantarcticaLipaseB(CAL‐B)inimmobilisierter

7 ExperimentellerTeil
140
Form(Novozym435)wurdevonSIGMAALDRICH®käuflicherworben.CAL‐BinlyophilisierterFormund
immobilisiertaufeinemMethacrylat‐TrägerwurdevonC‐LECTAGMBHzurVerfügunggestellt.
ESI‐MS:
ESI‐MassenspektrenwerdenmitdemEsquire3000derFirmaBRUKERDALTONIKmitIonenfalleundESI‐
Quelleaufgenommen.DieProbenwerdendirektmiteinerautomatischenSpritzeeingeführt.Stickstoff
dient als Zerstäuber‐ und Trockengas undwird durch den Stickstoffgenerator NGM 11 von BRUKER
generiert. Zur Kühlung der Ionenfalle dient Helium. Die Spektren werden mit der BRUKER Daltonik
esquireNT4.0esquireControlSoftware(V6.04)aufgenommenundmitderDataAnalysisSoftware2.0
bearbeitet.
Gaschromatographie(GC):
UmsatzbestimmungenerfolgtenaneinemGC‐2010vonSHIMADZUmiteinemAutoinjektorAOC‐20ivon
SHIMADZUundeinerRt‐βDEXm‐SäulevonRESTEK.AlsTrägergasdienteStickstoff(100kPa).DieSäule
weisteineLängevon30mundeinenInnendurchmesservon0.25mmauf.DieSpektrenwerdenmitder
Software GCsolution Analysis (Version 2.41.00) von SHIMADZU aufgenommen und mit der Software
GCsolutionPostrun(Version2.41.00)bearbeitet.
Methode:Startbei70°C,mit7.5°C/minauf110°C,mit25°C/minauf220°C.
HPLC:
DieChromatogrammewerdenmiteinemGerät,bestehendausfolgendenBausteinenderFirmaJASCO,
aufgenommen:PumpenPU‐2080Plus,automatischerRückdruckreglerBP‐2080Plus,Säulenthermostat
CO‐2060Plus,Multiwellenlängen‐DetektorMD‐2010PlusundAutosamplerAS‐2059Plus.DieTrennung
derProbenerfolgtemitSäulenvonDAICELChiralpak®(AD‐H).DieChromatogrammewurdenmitder
GalaxyChromatographyDataSystemSoftwareaufgenommenundausgewertet.
IR‐Spektroskopie:
IR‐spektroskopischeMessungenwurdenaneinemNicolet308FT‐IR‐SpektrometerderFirmaTHERMO
ELECTRON CORPORATION durchgeführt undmit der Software Omnic (Version 2.11) aufgenommen. Die
AbsorptionwirdinWellenzahl in[cm‐1]angegeben.
NMR‐Spektroskopie:
Die 1H‐ und 13C‐NMR‐Spektrenwurden an einemBRUKER DRX‐500 oder BRUKER Advance500NMR‐
SpektrometerbeiRaumtemperaturaufgenommen.DiechemischeVerschiebungδ[ppm]der 1H‐und
13C‐Spektren werden, sofern nicht anders angegeben, in Relation zur normierten chemischen Ver‐
schiebungdesverwendetenpartiellundeuteriertenLösungsmittelsangegeben(CDCl3δ=7.26ppm).
DieSpin‐Multiplizitätenwerdenalsbr(broad),s(Singulett),d(Dublett),dd(dupliziertesDublett), t
(Triplett), td(TripletteinesDubletts),q(Quartett),qui (Quintett)undm(Multiplett)angegeben.Die
erhaltenenDatenwurdenmit der SoftwareMestReNova (MestrelabResearch, Version 8.1.0‐11315)
ausgewertet.

7 ExperimentellerTeil
141
UV/Vis‐Spektroskopie:
UV/Vis‐spektroskopischeMessungenwurden an einemV‐630 von JASCOundan einemUV‐2450der
FirmaSHIMADZUdurchgeführt.DieerhaltenenDatenwurdenmitderSoftwareSpectraManager(Version
2.008.04)ausgewertet.

7 ExperimentellerTeil
142
7.2 Synthesen,MethodenundspektroskopischeDaten
7.2.1 EnzymatischeOxidationvonCyclohexanol(1)unterEinsatzvonCHMOund
LK‐ADH
7.2.1.1 Standardreaktion:AllgemeineArbeitsvorschrift1(AAV1):Enzymatische
OxidationvonCyclohexanol(1)zuε‐Caprolacton(3)
Abbildung126. DoppeloxidationvonCyclohexanol(1)zuε‐Caprolacton(3).[26]
In einem 10 mL‐Rundkolben wird Rohextrakt der CHMO aus Acinetobacter calcoaceticus (3.82U,
bezogenaufCyclohexanon(2))inKPi‐Puffer(pH7.0,50mM,5mL)aufgenommenundgutgeschüttelt.
Cyclohexanol(1,0.2mmol,20.0mg),MgCl2(1mM),undLk‐ADH(40U,Rohextrakt/Glycerin1:1(v/v),
bezogen auf Acetophenon) werden zugegeben. Das Gemisch wird für 20 min bei Raumtemperatur
gerührt.DurchZugabevonNADP+(1mol‐%,1.57mg)wirddieReaktiongestartetunddieMischungfür
einebestimmteZeitbeiRaumtemperaturgerührt.Anschließendwerdendie5mLzuje0.7mLaufsieben
1.5mL‐EPPENDORF‐Gefäße verteilt und jeweilsmit 0.7mL DCM versetzt. Durch 30min Schütteln im
Thermomixer wird das Zweiphasensystem extrahiert. Die Phasentrennung wird durch 30 min
Zentrifugieren bei 13000 rpm erreicht.Die überstehendewässrigePhasewird abgenommenund in
siebenneueEPPENDORF‐Gefäßegegeben.DieExtraktionmitanschließenderPhasentrennungwirdnoch
zweimalwiederholt.DieorganischenPhasenwerdenineinem25mL‐Rundkolbenüberführtunddas
Lösungsmittel am Rotationsverdampfer bei 900mbar und 40 °C Badtemperatur entfernt. Der
produktbezogeneUmsatzwirdmithilfeder1H‐NMR‐Spektroskopiebestimmt.
ProduktbezogenerUmsatz:>97%.
1H‐NMR(500MHz,CDCl3):δ(ppm):1.75(m,4H,H‐4,H‐5),1.84(2H,H‐3),2.62
(t,3J=6.5Hz,2H,H‐2),4.21(t,3J=4.5Hz,2H,H‐6).
13C‐NMR(500MHz,CDCl3):23.22(C‐3),29.25(C‐5),29.57(C‐4),34.85(C‐2),
69.60(C‐6),176.52(C‐1).
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenen
Wertenüberein.[272]

7 ExperimentellerTeil
143
7.2.1.2 Analytik
7.2.1.2.1 Umsatzbestimmungmittels1H‐NMR‐Spektroskopie
FürdieUmsatzbestimmungmittels1H‐NMR‐SpektroskopiewirdUrotropin(51)alsexternerStandard
verwendet. Dazuwird der gesamte Reaktionsansatz im Anschluss an die Aufarbeitung in ein NMR‐
RöhrchengegebenundfrischangesetzteUrotropin‐Stammlösung(51,0.17MinCDCl3,0.1mL)sowie
CDCl3(0.7mL)zugegeben.DerUmsatzwirdinBezugaufdasSingulettsignaldesStandards50bestimmt.
7.2.1.2.2 StabilitätvonUrotropin(51)alsStandard
Zur Bestimmung der Stabilität des externen Standards Urotropin (51) werden Cyclohexanol (1,
0.2mmol,21µL)sowieUrotropin(51,0.1mLder0.17M‐StammlösunginCDCl3)zusammenmitCDCl3
in ein NMR‐Röhrchen gegeben und sofort vermessen. Das Verhältnis beider Komponenten wird
bestimmt.Nach24hbeiRTwirddieselbeProbeerneutvermessenunddasVerhältnisbeiderKompo‐
nentenmittels1H‐NMR‐Spektroskopiebestimmt.DieErgebnissesindinTabelle23aufgelistet.
Tabelle23. BestimmungderStabilitätvonUrotropin(51)über24h.
EintragVerhältnis51 /1
eingewogen
Verhältnis51/1
nach24
1 1:0.80 1:0.80
7.2.1.2.3 BestimmungderRückgewinnungsratemittelsNMR
Zur Bestimmung der Rückgewinnungsrate der Substanzen wird die Aufarbeitung simuliert. Dazu
werdenCyclohexanon(2,0.2mmol,20.7µL)bzw.ɛ–Caprolacton(3,0.2mmol,21.1µL)inKPi‐Puffer
(pH7,50mM,5mL)gelöstundfür24hbeiRTgerührt.DieAufarbeitungdurchExtraktionerfolgtanalog
der AAV 1. Zur Bestimmung der Substanzmenge mittels 1H‐NMR‐Analytik wird Urotropin (51) als
Standard (0.1mL der 0.17M‐Stammlösung in CDCl3) verwendet. Die Ergebnisse sind in Tabelle 24
aufgelistet.

7 ExperimentellerTeil
144
Tabelle24. RückgewinnungsratederSubstanzennachsimulierterAufarbeitung.
Eintrag Substanz Rückgewinnungsrate[%]
1 2 91
2 3 98
7.2.1.3 UmsatzbestimmungmittelsGC
7.2.1.3.1 ErstellenderEichgerade
VordemErstelleneinerEichgeradenwurdezunächsteineMethodeamGaschromatographenerstellt,
dieeineguteTrennungderSubstanzenerlaubt. ImAnschlusswurdeeineStammlösung(10g/l)von
Cyclohexanol (1),Cyclohexanon (2)undɛ–Caprolacton (3)hergestellt.Dafürwurden je1gderdrei
Substanzeninein100mL‐fassendenMaßkolbeneingewogen,mitDCMbiszumEichstrichaufgefülltund
sehrgutgeschüttelt.AusgehendvondieserStammlösungwurdeeineVerdünnungsreihedurchgeführt,
sodass Lösungen der Konzentrationen von 5 g/l, 2.5 g/l, 2.0 g/l, 1.75 g/l, 1.5 g/l, 1.25 g/l, 1.0 g/l
hergestelltwerdenkonnten.VonderStammlösungsowievondenweiterenLösungenwurdenjeweils3
Probenfläschchen abgefüllt und doppelt am Gaschromatographen vermessen. Aus den erhaltenen
WertenwurdemithilfedesProgrammsderMittelwertgebildetunddieEichgeradeerstellt.
7.2.1.3.2 BestimmungderDurchschnittsabweichungen
Zur Bestimmung der durchschnittlichen Abweichungen der zuvor erstellten Eichgerade werden
unterschiedlicheMengenderSubstanzen1,2und3ineinen100mL‐Maßkolbeneingewogenundmit
DCM aufgefüllt. Davon werden jeweils drei Proben abgefüllt und mittels Gaschromatographie ver‐
messen.DieErgebnissesindinTabelle25aufgelistet.
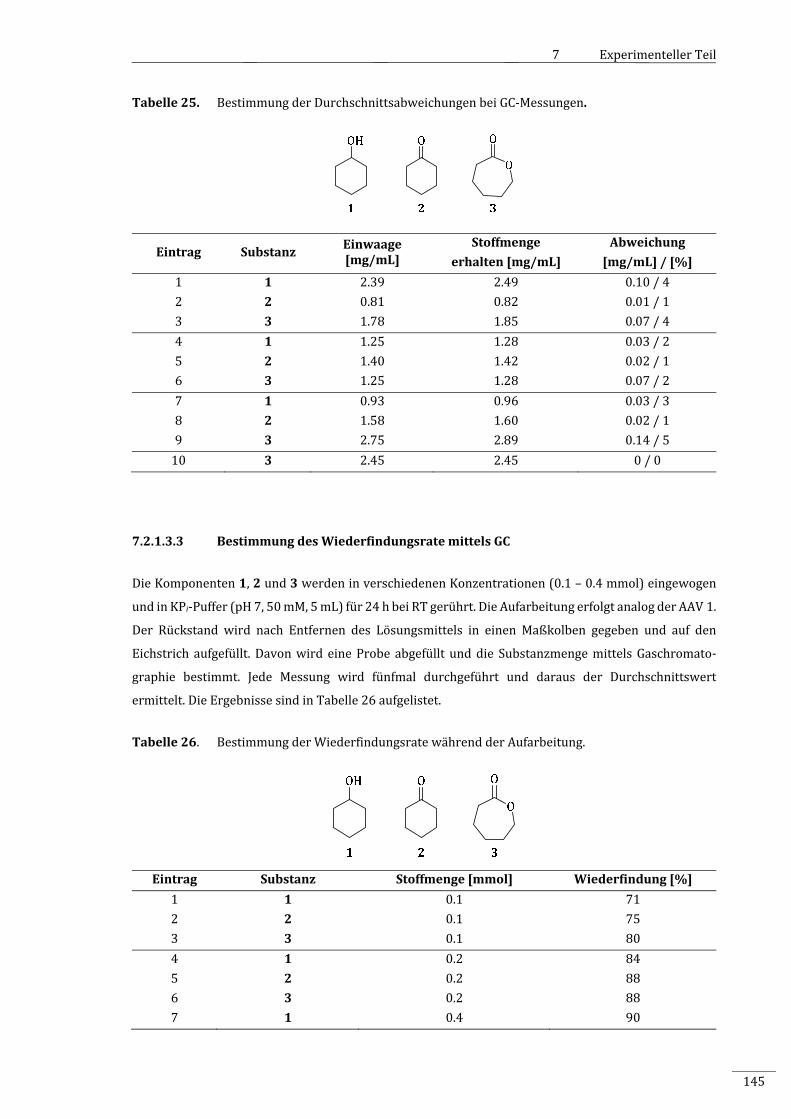
7 ExperimentellerTeil
145
Tabelle25. BestimmungderDurchschnittsabweichungenbeiGC‐Messungen.
Eintrag Substanz Einwaage[mg/mL]
Stoffmenge
erhalten[mg/mL]
Abweichung
[mg/mL]/[%]
1
2
3
1 2.39 2.49 0.10/4
2 0.81 0.82 0.01/1
3 1.78 1.85 0.07/4
4
5
6
1 1.25 1.28 0.03/2
2 1.40 1.42 0.02/1
3 1.25 1.28 0.07/2
7
8
9
1 0.93 0.96 0.03/3
2 1.58 1.60 0.02/1
3 2.75 2.89 0.14/5
10 3 2.45 2.45 0/0
7.2.1.3.3 BestimmungdesWiederfindungsratemittelsGC
DieKomponenten1,2und3werdeninverschiedenenKonzentrationen(0.1–0.4mmol)eingewogen
undinKPi‐Puffer(pH7,50mM,5mL)für24hbeiRTgerührt.DieAufarbeitungerfolgtanalogderAAV1.
Der Rückstand wird nach Entfernen des Lösungsmittels in einen Maßkolben gegeben und auf den
Eichstrich aufgefüllt.Davonwird eineProbeabgefülltunddie SubstanzmengemittelsGaschromato‐
graphie bestimmt. Jede Messung wird fünfmal durchgeführt und daraus der Durchschnittswert
ermittelt.DieErgebnissesindinTabelle26aufgelistet.
Tabelle26. BestimmungderWiederfindungsratewährendderAufarbeitung.
Eintrag Substanz Stoffmenge[mmol] Wiederfindung [%]
1 1 0.1 71
2 2 0.1 75
3 3 0.1 80
4 1 0.2 84
5 2 0.2 88
6 3 0.2 88
7 1 0.4 90

7 ExperimentellerTeil
146
Eintrag Substanz Stoffmenge[mmol] Wiederfindung[%]
8 2 0.4 95
9 3 0.4 97
7.2.1.3.4 BestimmungderWiederfindungsrateinAnwesenheitvonLk‐ADH
DieKomponenten1,2und3(0.2mmol)werdeneingewogenundinKPi‐Puffer(pH7,50mM,5mL)
zusammenmitLk‐ADH(0‐40U,0‐141µL)für1hbeiRTgerührt.DieAufarbeitungerfolgtanalogder
AAV1.DerRückstandnachEntfernendesLösungsmittelswirdineinen25mL‐Maßkolbengegebenund
davon drei Proben abgefüllt. Die Substanzmenge wird mittels Gaschromatographie bestimmt. Jede
MessungwirddreimaldurchgeführtunddarausderDurchschnittswertermittelt.DieErgebnissesindin
Tabelle27aufgelistet.
Tabelle27. BestimmungderWiederfindungsrateinAnwesenheitvonLk‐ADH.
Eintrag Substanz Lk‐ADH Wiederfindung[%]
1 1 ‐ 88
2 1 + 90
3 2 ‐ 76
4 2 + 73
5 3 ‐ 94
9 3 + 93
7.2.1.3.5 EinflussderEnzympellet‐BildungaufdieRückgewinnungsrate
ZurUntersuchungderEinflussesderPellet‐BildungaufdieRückgewinnungsratewerdenɛ‐Caprolacton
(3,0.2mmol,22.8mg)undLk‐ADH(0–500µL)inKPi‐Puffer(pH7,50mM,5mL)für10minbeiRT
gerührt.DieAufarbeitungerfolgtanalogderAAV1,mitderAbweichungderZentrifugierzeittzwischen
5–30Minuten.DieSubstanzmengevon3wirdanschließendmittelsGaschromatographiebestimmt.Die
ErgebnissesindinTabelle28aufgelistet.

7 ExperimentellerTeil
147
Tabelle28. EinflussderPellet‐BildunginAnwesenheitvonLk‐ADH.
Eintrag Lk‐ADH[µ] t[min] Rückgewinnungsrate[%]
1 0 3x30 68
2 150 3x30 61
3 500 3x30 20
4 0 3x5 93
5 150 3x5 73
6 500 3x5 69
7.2.1.3.6 VergleichderAnalytikmethoden:GCvs.NMR
DieSyntheseerfolgtanalogderAAV1.DazuwirdCHMO(3.82U,20.1mg),Cyclohexanol(1,0.2mmol),
MgCl2(1mM)sowieLk‐ADH(40U,210.5µL)undNADP+(1mol%,1.57mg)eingesetztundinKPi‐Puffer
(pH7,50mM,5mL)für24hgerührt.DieAufarbeitungerfolgtanalogderAAV1.DerRückstandwird
nach Entfernen des Lösungsmittels in einen Maßkolben gegeben und auf den Eichstrich aufgefüllt.
DavonwerdendreiProbenabgefülltundperGCvermessen.DerrestlicheInhaltderGC‐Gläschenwird
mit der restlichen Lösung zusammengegeben und das Lösungsmittel am Rotationsverdampfer bei
900mbarund40°CBadtemperaturentfernt.DergesamteReaktionsansatzwirdimAnschlussandie
AufarbeitungineinNMR‐RöhrchengegebenundfrischangesetzteUrotropin‐Stammlösung(51,0.17M
inCDCl3,0.1mL)sowieCDCl3(0.7mL)zugegeben.DerUmsatzwirdinBezugaufdasSingulettsignaldes
Standards50bestimmt.DieErgebnissesindinTabelle29aufgezeigt.
Tabelle29. VergleichderAnalytikmethoden:GCvs.NMR
Eintrag Substanz Umsatzvia GC[%] UmsatzviaNMR[%]
1 1 6 0
2 2 5 3
3 3 74 59

7 ExperimentellerTeil
148
7.2.1.4 Photometertests
7.2.1.4.1 BestimmungderEnzymaktivitätderLk‐ADHgegenüber3,3,5‐Trimethyl‐
cyclohexanol(53)
DieEnzymaktivitätderLk‐ADHwirdspektroskopischanalogeinerarbeitsgruppeninternerVorschrift
spektroskopischbeieinerWellenlängevon340nmdurchMessungdesVerbrauchsvonNADPHdurch
Oxidation zu NADP+ in Gegenwart eines Substrates bestimmt. Dafürwerden in eine 3mL fassende
Küvette 2880 µL der Substratlösung (10mMbis 2M in KPi‐Puffer (pH 7, 50mM)) und 60 µL einer
NADPH‐Lösung (12.5mM in KPi‐Puffer (pH 7, 50mM)) gegeben und gut geschüttelt. Abschließend
werden60µLderentsprechendverdünntenLk‐ADH‐Lösung(Rohextrakt)zugegeben.DieLösungwird
nocheinmalgutgeschütteltunddieMessungsofortgestartet.Eswurdeüber3MinutenjedeSekunde
einMesspunktaufgenommen.ZurBerechnungdervolumetrischenEnzymaktivitätwirddieAnfangs‐
steigungderAbsorptionskurve in folgendeGleichung4eingesetzt.DieErgebnissesind inTabelle30
aufgezeigt.
Gleichung4
U/mL: volumetrischeEnzymaktivität∆E340nm
t: Anfangssteigung/‐abnahmederAbsorptionskurve[min‐1]
Vg: GesamtvolumenderProbe[mL]
f: VerdünnungsfaktordesRohextraktes
Vp: Enzymvolumen[mL]
ɛ: molarerExtinktionskoeffizientmL
mM∙cm
d: SchichtdickederKüvette[cm]
IndieserArbeitsindd=1cmundɛ=6.3mL
mM∙cm.
Tabelle30. BestimmungderAktivitätderLk‐ADHgegenüber3,3,5‐Trimethylcyclohexanol(53)imVergleichzu1‐Phenylethanol(47)undCyclohexanol(1).
Eintrag Substrat Aktivität[U/mL] RelativeAktivität[%]
1 47 21.5 100
2 1 33.0 153
3 53 0 0

7 ExperimentellerTeil
149
7.2.1.4.2 AllgemeineArbeitsvorschrift2(AAV2):BestimmungderEnzymaktivitätder
CHMO
Die Enzymaktivität der CHMO (ECSMo‐01)wird nach einer Vorschrift der ENZYMICALSAG bei einer
Wellenlänge von 340 nm durch Messung des Verbrauchs an NADPH durch Oxidation zu NADP+ in
GegenwartvonCyclohexanon(2)alsSubstratbestimmt.DazuwerdenStammlösungendesSubstrats
Cyclohexanon(2,100mMinDMF)undeineNADPH‐Lösung(125mMindest.Wasser)hergestellt.Es
werden2670µLdesTRIS‐HCl‐Puffers(pH8.5,50mM)zusammenmit30µLderSubstratlösungund
3µLderNADPH‐Lösung ineineKüvettepipettiertunddurchSchüttelngutvermischt.Anschließend
werden300µLderjeweiligenEnzymlösung(RohextraktinPuffer)zugegebenunddieLösungnochein
weiteresMal gutdurchmischtunddieMessunggestartet. Eswirdüber3Minuten jedeSekundeein
Messpunkt aufgenommen. Zur Berechnung der volumetrischen Enzymaktivität wird die Anfangs‐
steigungderAbsorptionskurvenachGleichung5berechnet.
Gleichung5
U/mL: volumetrischeEnzymaktivität∆E340nm
t: Anfangssteigung/‐abnahmederAbsorptionskurve[min‐1]
f: VerdünnungsfaktordesRohextraktes
ɛ: molarerExtinktionskoeffizientmL
mM∙cm
d: SchichtdickederKüvette[cm]
IndieserArbeitsindd=1cmundɛ=6.3mL
mM∙cm.
Aktivität:0.082U/mg.
7.2.1.4.3 BVMO‐ScreeningundBRADFORD‐TestzurOxidationvonCyclohexanol(2)
Die Enzymaktivitäten der BVMOs ECSMo‐03 und ECSMo‐05 (ENZYMICALSAG) sollen photometrisch
analog zur AAV 2 bestimmt werden. Dazu werden Stammlösungen des Substrats Cyclohexanon (2,
100mMinDMF)undeineNADPH‐Lösung(125mMindest.Wasser)hergestellt.Eswerden2670µLdes
TRIS‐HCl‐Puffers(pH8.5,50mM)zusammenmit30µLderentsprechendenSubstratlösungund3µL
derNADPH‐LösungineineKüvettepipettiertunddurchSchüttelngutvermischt.Anschließendwerden
300µLderjeweiligenEnzymlösung(RohextraktinPuffer)zugegebenunddieLösungnocheinweiteres
MalgutdurchmischtunddieMessunggestartet.Eswirdüber3MinutenjedeSekundeeinMesspunkt
aufgenommen. Zur Berechnung der Enzymaktivitätwird die Anfangssteigung der Absorptionskurve
nachGleichung5berechnet.DieErgebnissesindinTabelle31aufgezeigt.

7 ExperimentellerTeil
150
Tabelle31. BVMO‐ScreeningzurOxidationvonCyclohexanon(2).
Eintrag BVMO Aktivität[U/mL] Aktivität[U/mg] RelativeAktivität[%]
1 ECSMo‐01 7.38 3.00 100
2 ECSMo‐03 0.50 0.20 7
3 ECSMo‐05 0.14 0.05 2
7.2.1.4.4 AktivitätsbestimmungderCHMOgegenüberIsophoron(52)
Die Enzymaktivität der CHMO soll photometrisch gegenüber Isophoron (52) als Substrat bestimmt
werden.AbweichendvonderAAV1wirddieAktivitätnacheinerVorschriftderFirmaENZYMICALSAG
dargestellt.DazuwerdenStammlösungenderSubstrateCyclohexanon(2)undIsophoron(52,100mM
in DMF) und eine NADPH‐Lösung (125 mM in dest. Wasser) hergestellt. Es werden 2670 µL des
TRIS‐HCl‐Puffers(pH8.5,50mM)zusammenmit30µLderentsprechendenSubstratlösungund3µL
derNADPH‐LösungineineKüvettepipettiertunddurchSchüttelngutvermischt.Anschließendwerden
300µLderjeweiligenEnzymlösung(RohextraktinPuffer)zugegebenunddieLösungnocheinweiteres
MalgutdurchmischtunddieMessunggestartet.Eswirdüber3MinutenjedeSekundeeinMesspunkt
aufgenommen. Zur Berechnung der Enzymaktivitätwird die Anfangssteigung der Absorptionskurve
nachGleichung5berechnet.DieErgebnissesindinTabelle32aufgezeigt.
Tabelle32. Bestimmung der Aktivität von CHMO gegenüber Isophoron (52) im Vergleich zuCyclohexanon(2).
Eintrag Substrat Aktivität[U/mg] Aktivität[%]
1 2 0.101 100
2 52 0.003 3
7.2.1.5 OptimierungderStandard‐Reaktion
7.2.1.5.1 AllgemeineArbeitsvorschrift3(AAV3):Doppeloxidationzuɛ‐Caprolacton(3)mit
optimierterAufarbeitung
Zur Optimierung der Aufarbeitung wird die Synthese analog der AAV 1 (siehe Abbildung 126)
durchgeführt.DazuwerdenCHMO(3.82U,68.2mg)inKPi‐Puffer(pH7,50mM,5mL),Cyclohexanol(1,
0.2mmol, 22.0mg),MgCl2 (1mM) sowie Lk‐ADH (40 U, 210.5 µL) undNADP+ (1mol‐%, 1.57mg)
eingesetztundfür24hbeiRTgerührt.Anschließendwerdendie5mLzuje1mLauffünf2mL‐EPPEN‐
DORF‐Gefäßeverteiltundjeweilsmit1mLDCMversetzt.Durch30minSchüttelnimThermomixerwird

7 ExperimentellerTeil
151
dasZweiphasensystemextrahiert.DiePhasentrennungwirddurch5minZentrifugierenbei13000rpm
erreicht.DieuntereorganischePhasewirdabgenommenunddiezurückbleibendewässrigePhasesowie
dasdenaturierteEnzymwerdenerneutmitje1mLDCMversetzt.DieExtraktionmitanschließender
Phasentrennung wird noch zweimal wiederholt. Die organischen Phasen werden in einem 50mL‐
MaßkolbenüberführtundderKolbenbiszurEichmarkemitDCMaufgefüllt.EswerdendreiProben‐
fläschchenabgefülltundderUmsatzmittelsGaschromatographiebestimmt.
Umsatz:74%ɛ‐Caprolacton(3),20%Cyclohexanon(2),10%Cyclohexanol(1).
7.2.1.5.2 VariationdesLösungsmittels
DieSynthesenerfolgenanalogderAAV1.DazuwerdenjeweilsCHMO(3.82U,20.1mg),Cyclohexanol
(1,0.2mmol),MgCl2(1mM)sowieLk‐ADH(40U,210.5µL)undNADP+(1mol‐%,1.57mg)eingesetzt
und für 24 h bei RT gerührt. Als Lösungsmittel dient zum einen mit organischem Co‐Solventien
versetzterKPi‐Puffer(pH7,50mM)bzw.dest.Wasser.ZumanderenwirdKPi‐Puffer(pH7,50mL)bzw.
dest.WassermitdemCo‐Solvenz(je10mL)für24hbeiRTund1200rpmgerührtumeinegesättigte
wässrigePhasezuerhalten.DieAufarbeitungerfolgtanalogderAAV1.DieUmsätzezu2und3sowie
die Stoffmenge des nicht umgesetzten Edukts 1 werden entweder mittels 1H‐NMR‐Spektroskopie
bestimmtundaufdenStandardUrotropin(51)bezogenoderviaGCermittelt.DieErgebnissesindin
Tabelle33aufgelistet.
Tabelle33. VariationdesLösungsmittelgemischesbeiderDoppeloxidationvonCyclohexanol(1).
Eintrag Lösungsmittel‐GemischStoffmenge3[mmol]
Stoffmenge2[mmol]
Stoffmenge1[mmol]
1a) KPi/MTBE8:2 1.6 1.2 18.4
2a) KPi/MTBE6:4 0.4 0.8 28.0
3a) KPi/EtOAc6:4 0 2.4 27.6
4b) KPi/Toluol9:1 23.6 3.2 8.4
5b) KPi/Methylcyclohexan9:1 25.6 2.0 2.4
6b) KPi/Isooctan9:1 34.8 2.0 2.0
7b) KPi/n‐Heptan9:1 26.0 2.0 4.4
8a) MTBE‐ges.KPi 13.2 1.2 3.6
7 ExperimentellerTeil
152
Eintrag Lösungsmittel‐GemischStoffmenge3[mmol]
Stoffmenge 2[mmol]
Stoffmenge1[mmol]
9a) EtOAc‐ges.KPi 0 2.8 24.4
10a) n‐Heptan‐ges.KPi 14.0 1.2 3.2
11a) n‐Hexan‐ges.KPi 19.6 0.8 0
12a) Methylcyclohexan‐ges.KPi 28.0 1.2 0
13a) MTBE‐ges.dest.Wasser 0.8 2.0 32.0
14a) dest.Wasser 14.0 1.2 6.8a)UmsatzbestimmtmittelsNMR‐SpektroskopiemitUrotropin(51)alsStandard;b)UmsatzbestimmtmittelsGC.
7.2.1.5.3 VariationderReaktionsbedingungen
DieSynthesenerfolgenanalogderAAV1.DazuwerdenCHMO(3.82U,20.1mg) inKPi‐Puffer(pH7,
50mM,5mL),Cyclohexanol(1,0.2mmol),MgCl2(1mM)sowieLk‐ADH(13–40U,70.2‐210.5µL)und
NADP+(2‐10mol‐%)eingesetztundfür24hbeiRT–30°Cgerührt.DieAufarbeitungerfolgtanalogder
AAV1.DieproduktbezogenenUmsätzewerdenmittels 1H‐NMR‐Spektroskopiebestimmt.DieErgeb‐
nissesindinTabelle34aufgelistet.
Tabelle34. VariationderReaktionsbedingungenderOxidationvonCyclohexanol(1).
EintragNADP+[mol‐%]
Lk‐ADH
[U]
T
[°C]
Umsatz3
[%]
Umsatz2
[%]
1 2 40 RT 97 3
2 10 40 RT 61 11
3 1 40 30 48 5
4 1 13 RT 55 3
7.2.1.6 AllgemeineArbeitsvorschrift4(AAV4):UntersuchungderStabilitätderLk‐ADH
mittelsTitrations‐Apparatur
Abbildung127. UntersuchungderStabilitätderLk‐ADHinAnwesenheitvonLacton3.
IneinerTitrino‐ApparaturwerdenAcetophenon(46,20mM,21μL),ε‐Caprolacton(3,10mM–1M)
sowieD‐Glucose(48‐a,100mM,180.1mg)in10mLdest.Wassergegeben.Anschließendwerden1mg

7 ExperimentellerTeil
153
MgCl2,40U/mmolLk‐ADHund40U/mmolGlucosedehydrogenase(GDH)„AMANO2“zugegebenunddie
ReaktionmitZugabevonNADP+(1mol%,1.57mg)alsCofaktorfür24hgestartet.
D‐Glucose(48‐a)dienthierbeialsRegenerationsmediumfürdeneingesetztenCofaktor,derdurchdie
VerwendungvonGDHzumbenötigtenNADPHreduziertwird.D‐Glucose(48‐a)selbstwirdzunächstin
Gluconolacton(49‐a)umgewandelt,welchesirreversibelzurGluconsäure(50‐a)gespaltenwird.Durch
Zugabe von Natronlauge (0.2 M) wird die gebildete Säure zum Natriumgluconat neutralisiert. Die
verbrauchteMengeanNatronlaugegibtAuskunftüberdenUmsatzderReaktion.
DurchZugabevon15mLDCMwirddieReaktionbeendet.EswirdimScheidetrichterausgeschütteltund
dieorganischePhaseabgetrennt.Anschließendwirdnochzweimalmitje15mLDCMextrahiert.Das
organische Lösungsmittel wird bei 900 mbar und 40°C Badtemperatur am Rotationsverdampfer
entfernt. DerUmsatzwird ebenfallsmittels 1H‐NMR‐Spektroskopie bestimmt und auf den Standard
Urotropin(51)bezogen.DieErgebnissesindinTabelle35aufgezeigt.
Tabelle35. UntersuchungderStabilitätvonLk‐ADHinAnwesenheitvonLacton3.
Eintrag Keton46
[M]
Lacton3
[M]
Produktkonzentration
viaTitrino[%]
ProduktkonzentrationviaNMRa)[%]
1 0.02 0 25 19
2 0.02 0.01 24 16
3 0.02 0.04 30 16
4 0.02 0.10 42 15
5b) 0.02 0.25 49 16
6b) 0.02 1.00 50 14
7b) 0 0.25 0 0a)UmsatzbestimmtmittelsUrotropin(51)alsexternerStandard;b)automatischerAbbruchderReaktionnach3.5h.
7.2.1.7 VariationdesSubstrates
7.2.1.7.1 3,5,5‐Trimethylcyclohexanol(53)alsSubstrat
Abbildung128. Doppeloxidationvon3,5,5‐Trimethylcyclohexanol(53).

7 ExperimentellerTeil
154
DieSynthesenerfolgenanalogderAAV1.DazuwerdenjeweilsCHMO(3.82U,20.1mg),3,5,5‐Trimethyl‐
cyclohexanol (53, 0.2mmol), MgCl2 (1mM) sowie Lk‐ADH (40 U, 210.5 µL) und NADP+ (1mol‐%)
eingesetztundfür24hbeiRTgerührt.DieAufarbeitungerfolgtanalogderAAV1.DieKonzentrationen
derKomponentenwird sowohlmittels 1H‐NMR‐Spektroskopie, bezogenaufdenStandardUrotropin
(51),sowiemittelsGaschromatographiebestimmt.DieErgebnissesindinTabelle36aufgelistet.
Tabelle36. 3,5,5‐Trimethylcyclohexanol(53)alsSubstrat.
Eintrag AnalytikKonzentration53
[mM]Konzntration 55
[mM]Konzentration56/57
[mM]
1 1H‐NMR 28.4 0 0
2 GC 38.8 0 1.2
7.2.1.7.2 Isophoron(52)alsSubstrat
Abbildung129. OxidationvonIsophoron(52)mitLk‐ADHalsRegenerationssystem.
Ineinem10mL‐RundkolbenwirdRohextraktderCHMO(3.82U,20.1mg)inKPi‐Puffer(pH7.0,50mM,
5mL)aufgenommenundgutgeschüttelt. Isophoron (52, 0.2mmol,27.6mg),MgCl2(1mM),Lk‐ADH
(40U,Rohextrakt/Glycerin1:1(v/v))sowiei‐PrOH(1.25ml)werdenzugegeben.DasGemischwirdfür
20minbeiRaumtemperaturgerührt.DurchZugabevonNADP+(1mol‐%,1.57mg)wirddieReaktion
gestartet und dieMischung für eine bestimmte Zeit bei Raumtemperatur gerührt. Die Aufarbeitung
erfolgtanalogderAAV1.DieKonzentrationwirdmittels1H‐NMR‐Spektroskopiebestimmtundaufden
StandardUrotropin(51)bezogen.DieErgebnissesindinTabelle37aufgelistet.
Tabelle37. Isophoron(52)alsSubstrat.
Eintrag Analytik Konzentration52[mM] Konzentration59[%]
1 1H‐NMR 23.6 0
2 GC 39.9 1

7 ExperimentellerTeil
155
7.2.1.8 VerwendungvonAdsorberharzen
7.2.1.8.1 BestimmungderRückgewinnungsrateinAnwesenheitvonXAD‐7undXAD‐1180
ZurBestimmungderRückgewinnungsratewerdenCyclohexanol(1)bzw.dasLacton3(0.2mmol)in
KPi‐Puffer (pH 7, 50mM)mit XAD‐7 bzw. XAD‐1180 (100 – 200mg) für 20 h bei RT gerührt. Die
Harzkügelchenwerdenabfiltriertundzunächstmitdest.Wasser(3x20mL)undanschließendmitDCM
bzw.MTBE(3x20mL)gewaschenunddiePhasengetrennt.DiewässrigePhasewirdmitDCMbzw.
MTBE (3 x 20 mL) extrahiert. Die organischen Phasen werden vereint und das Lösungsmittel bei
900mbarund40°Centfernt.DerUmsatzwirdmittels1H‐NMR‐Spektroskopiebestimmtundaufden
StandardUrotropin(51)bezogen.DieErgebnissesindinTabelle38aufgelistet.
Tabelle38. BestimmungderRückgewinnungsrate inAnwesenheitderAdsorberharzeXAD‐7bzw.XAD‐1180.
Eintrag Substanz Harz/[mg] Solvenz Rückgewinnung [%]
1 1 XAD‐7/100 DCM 46
2 3 XAD‐7/100 DCM 68
3 3 XAD‐7/200 DCM 74
4 3 XAD‐7/100 MTBE 30
5 1 XAD‐1180/100 DCM 89
6 1 XAD‐1180/200 DCM 41
7.2.1.8.2 DoppeloxidationvonCyclohexanol(1)inAnwesenheitvonXAD‐7
Abbildung130. DoppeloxidationvonCyclohexanol(1)inAnwesenheitvonXAD‐7.
Die Synthese erfolgt analog der AAV 1. Dazu werden CHMO (3.82 U, 20.1mg), Cyclohexanol (1,
0.2mmol),XAD‐7 (100mg),MgCl2 (1mM)sowieLk‐ADH(40U,210.5µL)undNADP+ (1mol‐%) in
KPi‐Puffer(pH7,50mM,5mL)eingesetztundfür24hbeiRTgerührt.DieAufarbeitungerfolgtanalog

7 ExperimentellerTeil
156
derAAV1.DerUmsatzwirdmittels1H‐NMR‐SpektroskopiebestimmtundaufdenStandardUrotropin
(51)bezogen.DieErgebnissesindinTabelle39aufgelistet.
Tabelle39. DoppeloxidationvonCyclohexanol(1)inAnwesenheitvonXAD‐7.
Umsatz3[%] Umsatz2[%]ZurückgewonneneStoffmenge
1+2+3[%]
24 3 50

7 ExperimentellerTeil
157
7.2.2 EnantioselektiveRacematspaltungvonAminenmittelsLipasen
7.2.2.1 AllgemeineArbeitsvorschrift5(AAV5):SynthesevonReferenzverbindungen
Zumjeweiligenrac‐AminwirdderentsprechendeAcyldonor(1Äq.)gegebenundfürdieangegebene
Reaktionszeit zum Rückfluss (Ölbad auf 180 °C) erhitzt (Abbildung 131).[253] Der produktbezogene
Umsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.DasRohproduktwirdmittelsSäulenchromato‐
graphieaufgereinigt,umdasisolierterac‐Amidzuerhalten.
Abbildung131. SynthesederReferenzverbindungen.[253]
7.2.2.1.1 SynthesedesDL‐Ethyl‐3‐oxo‐3‐(1‐phenylethylamino)propanoats(rac‐10)
Die Synthese erfolgt analog der AAV 5.[253] Zu 1‐Phenylethylamin (rac‐4, 2.7mmol, 327.3mg)wird
Malonsäurediethylester (9, 15.1mmol,2.4g)gegebenund für4hzumRückflusserhitzt.DieAufrei‐
nigungerfolgtmittelsSäulenchromatographie(Laufmittel:CH/EtOAc2:1).DasAmidrac‐10wirdals
farbloserFeststofferhalten.
Umsatz:85%;Ausbeute:71%(448.6mg,1.9mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.28(t,3J=7.1Hz,3H,H‐14),
1.51(d,3J=6.9Hz,3H,H‐9),3.32(d,3J=4.9Hz,2H,H‐11),4.19(q,
3J=7.2Hz,2H,H‐13),5.15(q,3J=7.2Hz,1H,H‐7),7.24–7.36(m,
5H,Har),7.50(m,1H,H‐8).
13C‐NMR(500MHz,CDCl3):14.16(C‐14),22.22(C‐9),41.21(C‐11),
49.04 (C‐7),61.69 (C‐13),126.19 (C‐1,C‐5),127.46 (C‐3),128.80
(C‐2,C‐4),143.16(C‐6),164.12(C‐10),169.83(C‐12).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210nm, flow:
0.8mL/min,tR=16.9min(R),26.7min(S),ee‐Wert:0%.
IR (ATR): (cm‐1) = 3276.0, 3064.4, 2977.5, 2930.4, 2359.4, 1735.8, 1642.7, 1541.0, 1448.4, 1367.9,
1152.8,1031.6,943.8,761.0,698.3,613.8,537.9.
MS(ESI):m/z(%)=258.1(100)[M+Na]+,356.3(3),437.3(5),493.2(31)[2M+Na]+.
EA(C13H17NO3):berechnet:C:61.95%,H:8.98%,N:6.57%,O:22.50%;gefunden:C:61.08%,H:8.99%,
N:6.47%,O:23.46%.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[273]
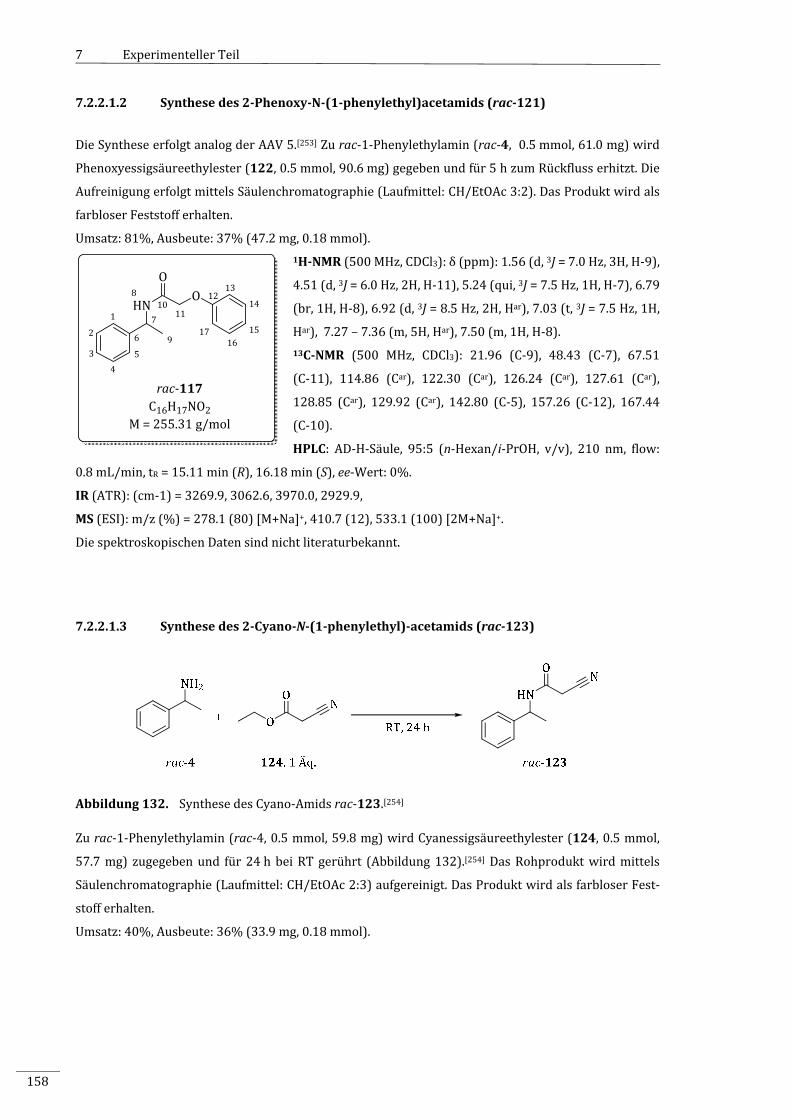
7 ExperimentellerTeil
158
7.2.2.1.2 Synthesedes2‐Phenoxy‐N‐(1‐phenylethyl)acetamids(rac‐121)
DieSyntheseerfolgtanalogderAAV5.[253]Zurac‐1‐Phenylethylamin(rac‐4,0.5mmol,61.0mg)wird
Phenoxyessigsäureethylester(122,0.5mmol,90.6mg)gegebenundfür5hzumRückflusserhitzt.Die
AufreinigungerfolgtmittelsSäulenchromatographie(Laufmittel:CH/EtOAc3:2).DasProduktwirdals
farbloserFeststofferhalten.
Umsatz:81%,Ausbeute:37%(47.2mg,0.18mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.56(d,3J=7.0Hz,3H,H‐9),
4.51(d,3J=6.0Hz,2H,H‐11),5.24(qui,3J=7.5Hz,1H,H‐7),6.79
(br,1H,H‐8),6.92(d,3J=8.5Hz,2H,Har),7.03(t,3J=7.5Hz,1H,
Har),7.27–7.36(m,5H,Har),7.50(m,1H,H‐8).
13C‐NMR (500 MHz, CDCl3): 21.96 (C‐9), 48.43 (C‐7), 67.51
(C‐11), 114.86 (Car), 122.30 (Car), 126.24 (Car), 127.61 (Car),
128.85 (Car), 129.92 (Car), 142.80 (C‐5), 157.26 (C‐12), 167.44
(C‐10).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm, flow:
0.8mL/min,tR=15.11min(R),16.18min(S),ee‐Wert:0%.
IR(ATR):(cm‐1)=3269.9,3062.6,3970.0,2929.9,
MS(ESI):m/z(%)=278.1(80)[M+Na]+,410.7(12),533.1(100)[2M+Na]+.
DiespektroskopischenDatensindnichtliteraturbekannt.
7.2.2.1.3 Synthesedes2‐Cyano‐N‐(1‐phenylethyl)‐acetamids(rac‐123)
Abbildung132. SynthesedesCyano‐Amidsrac‐123.[254]
Zurac‐1‐Phenylethylamin(rac‐4,0.5mmol,59.8mg)wirdCyanessigsäureethylester(124,0.5mmol,
57.7mg) zugegebenund für24hbeiRTgerührt (Abbildung132).[254]DasRohproduktwirdmittels
Säulenchromatographie(Laufmittel:CH/EtOAc2:3)aufgereinigt.DasProduktwirdalsfarbloserFest‐
stofferhalten.
Umsatz:40%,Ausbeute:36%(33.9mg,0.18mmol).
HN
OO
rac‐117C16H17NO2
M=255.31g/mol
1
2
3
4
5
6
7
8
9
1012
11
13
14
1516
17
7 ExperimentellerTeil
159
1H‐NMR(500MHz,CDCl3):δ(ppm):1.56(d,3J=7.0Hz,3H,H‐9),3.35
(d,3J=2.5Hz,2H,H‐11),5.12(qui,3J=7Hz,1H,H‐7),6.29(br,1H,H‐
8),7.29–7.38(m,5H,Har).
13C‐NMR (500 MHz, CDCl3): 21.60 (C‐8), 26.04 (C‐11), 50.12 (C‐7),
114.85 (C‐13), 126.27 (Car), 128.01 (Car), 129.02 (Car), 141.99 (C‐5),
160.09(C‐10).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm, flow: 0.8
mL/min,tR=17.14min(R),21.38min(S),ee‐Wert:0%.
IR (ATR): (cm‐1) = 3275.3, 3064.8, 2966.1, 2919.9, 2357.5, 2341.2, 2256.4, 1644.8, 1550.2, 1445.2,
1355.0,1241.6,1195.0,1106.6,934.9,748.5,684.3,597.8,587.6,528.2.
MS (ESI): m/z (%) = 211.0 (100) [M+Na]+, 316.2 (4), 353.3 (13), 399.1 (52) [2M+Na]+, 437.2 (6),
569.2(3).
EA (C11H12N2O):berechnetC:70.19%,H:6.43%,N:14.88%,O:8.50;gefunden:C:70.53%,H:6.66%,
N:3.93%,O:6.69%.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[254]
7.2.2.1.4 SynthesedesN‐(1‐Phenylethyl)acetamids(rac‐7)
Abbildung133. Acylierungvonrac‐4mitEssigsäurevinylester(125).
Zu1‐Phenylethylamin(rac‐4,0.5mmol,59.8mg)wirdEssigsäurevinylester(125,0.5mmol,57.7mg)
zugegebenundfür4.5Stundenbei60°Cgerührt.DasRohproduktwird indest.H2O(0.5mL)aufge‐
nommenundmitDCMextrahiert.DasProduktwirdalsgelbesÖlerhalten.
Umsatz:95%.
1H‐NMR(500MHz,CDCl3):δ(ppm):1.49(d,3J=6.9Hz,3H,H‐9),1.98(s,3H,
H‐11),5.13(qui,3J=7.2Hz,1H,H‐7),5.71(br,1H,H‐8),7.25–7.36(m,5H,
Har).
13C‐NMR (500MHz,CDCl3):21.82 (C‐9),23.56 (C‐11),48.91 (C‐7),114.85
(C‐13), 126.32 (C‐6, C‐5), 127.49 (C‐2), 128.78 (CC‐1, C‐3), 143.28 (C‐5),
169.25(C‐10).
HPLC:AD‐H‐Säule,95:5(n‐Hexan/i‐PrOH,v/v),210nm,flow:0.8mL/min,
tR=27.39min(R),37.85min(S),ee‐Wert:0%.
IR(ATR):(cm‐1)=3733.9,3278.5,2974.9,2358.8,2340.8,1646.2,1541.3,
1473.4,1447.8,1370.5,1296.7,1274.5,1209.7,1138.5,1028.3,728.9,638.2,519.8.499.2.
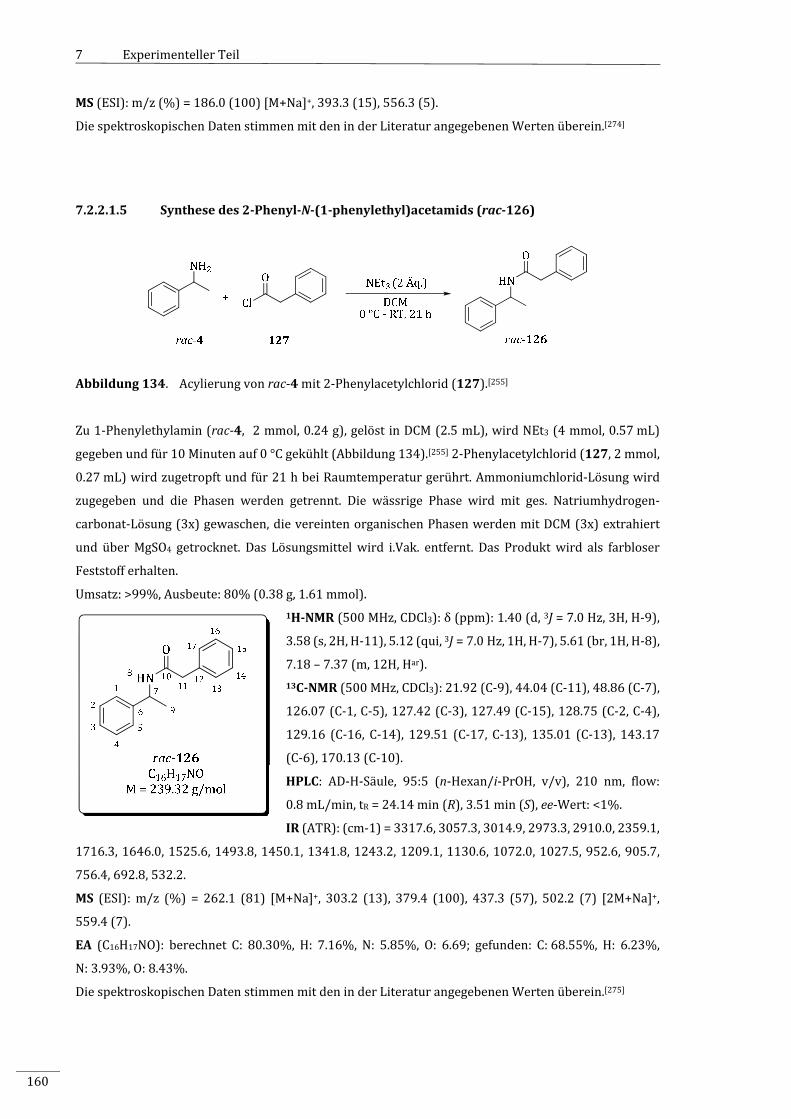
7 ExperimentellerTeil
160
MS(ESI):m/z(%)=186.0(100)[M+Na]+,393.3(15),556.3(5).
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[274]
7.2.2.1.5 Synthesedes2‐Phenyl‐N‐(1‐phenylethyl)acetamids(rac‐126)
Abbildung134. Acylierungvonrac‐4mit2‐Phenylacetylchlorid(127).[255]
Zu1‐Phenylethylamin(rac‐4, 2mmol,0.24g),gelöstinDCM(2.5mL),wirdNEt3(4mmol,0.57mL)
gegebenundfür10Minutenauf0°Cgekühlt(Abbildung134).[255]2‐Phenylacetylchlorid(127,2mmol,
0.27mL)wirdzugetropftundfür21hbeiRaumtemperaturgerührt.Ammoniumchlorid‐Lösungwird
zugegeben und die Phasen werden getrennt. Die wässrige Phase wird mit ges. Natriumhydrogen‐
carbonat‐Lösung(3x)gewaschen,dievereintenorganischenPhasenwerdenmitDCM(3x)extrahiert
und überMgSO4 getrocknet. Das Lösungsmittelwird i.Vak. entfernt. Das Produktwird als farbloser
Feststofferhalten.
Umsatz:>99%,Ausbeute:80%(0.38g,1.61mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.40(d,3J=7.0Hz,3H,H‐9),
3.58(s,2H,H‐11),5.12(qui,3J=7.0Hz,1H,H‐7),5.61(br,1H,H‐8),
7.18–7.37(m,12H,Har).
13C‐NMR(500MHz,CDCl3):21.92(C‐9),44.04(C‐11),48.86(C‐7),
126.07(C‐1,C‐5),127.42(C‐3),127.49(C‐15),128.75(C‐2,C‐4),
129.16 (C‐16, C‐14), 129.51 (C‐17, C‐13), 135.01 (C‐13), 143.17
(C‐6),170.13(C‐10).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm, flow:
0.8mL/min,tR=24.14min(R),3.51min(S),ee‐Wert:<1%.
IR(ATR):(cm‐1)=3317.6,3057.3,3014.9,2973.3,2910.0,2359.1,
1716.3,1646.0,1525.6,1493.8,1450.1,1341.8,1243.2,1209.1,1130.6,1072.0,1027.5,952.6,905.7,
756.4,692.8,532.2.
MS (ESI):m/z (%) = 262.1 (81) [M+Na]+, 303.2 (13), 379.4 (100), 437.3 (57), 502.2 (7) [2M+Na]+,
559.4(7).
EA (C16H17NO): berechnet C: 80.30%, H: 7.16%, N: 5.85%, O: 6.69; gefunden: C:68.55%, H: 6.23%,
N:3.93%,O:8.43%.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[275]

7 ExperimentellerTeil
161
7.2.2.1.6 Synthesedes2‐Methoxy‐N‐(1‐phenylethyl)acetamids(rac‐8)
Abbildung135. Acylierungvonrac‐4mit2‐Methoxyacetylchlorid(128).[256]
Zu 1‐Phenylethylamin (rac‐4, 1.25 mmol, 161.8µL) wird bei 0°C Piperidin (1.05 Äq., 103.8µL)
zugegeben(Abbildung135).[256]Eswirdmit2‐Methoxyacetylchlorid(128,1.1Äq.,131.5µL)versetzt
undfür1hunterEisbadkühlunggerührt.DaserhaltenefarbloseÖlwirdinDCMaufgenommenunddie
Phasenwerdengetrennt.DieorganischePhasewirdmitHCl(1M,2x),mitges.Natriumchlorid‐Lösung
(2x)sowiemitges.Natriumhydrogencarbonat‐Lösung(2x)gewaschenundüberMgSO4getrocknet.Das
Lösungsmittelwirdi.Vak.entfernt.DasProduktwirdalsfarbloserFeststofferhalten.
Umsatz:>99%,Ausbeute:95%(227.8mg,1.18mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.52(d,3J=7.0Hz,3H,H‐12),3.40
(s,3H,H‐12),3.90(d,3J=10.5Hz,2H,H‐11),5.18(qui,3J=7.0Hz,1H,
H‐7),6.75(br,1H,H‐8),7.25–7.28(m,1H,Har),7.31–7.37(m,4H,Har).
13C‐NMR(500MHz,CDCl3):21.97(C‐9),48.14(C‐7),59.21(C‐12),72.03
(C‐11), 126.24 (Car), 127.49 (Car), 127.78 (Car), 143.01 (C‐5), 167.71
(C‐10).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm, flow:
0.8mL/min,tR=13.09min(R),17.47min(S),ee‐Wert:<1%.
IR (ATR): (cm‐1) = 3342.7, 2971.0, 2931.3, 2828.6, 1650.5, 1521.7,
1454.9,1208.0,1112,0,1018.2,977.9,766.0,701.1,663.4,614.5,571.8,537.5,501.7.
MS(ESI):m/z(%)=216.0(100)[M+Na]+,217.0(13),409.2(8)[2M+Na]+.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[276]
7.2.2.1.7 SynthesedesEthyl‐3‐((2,3‐dihydro‐1H‐inden‐1‐yl)amino)‐3‐oxo‐propanoats
(rac‐129)
Abbildung136. SynthesedesAminoindan‐Amidsrac‐129.[257]

7 ExperimentellerTeil
162
Zu1‐Aminoindan(130,1.0mmol,132.5mg)werdenMalonsäurediethylester(9,1.0mmol,159.0mg)
undDBU (0.5mmol, 78.0mg) in DCM (0.2mL) gegeben und bei 40 °C für 3 d gerührt (Abbildung
136).[257] Der Umsatz wird mittels 1H‐NMR‐Spektroskopie bestimmt. Das Rohprodukt wird mittels
Säulenchromatographie(LaufmittelCH/EtOAc1:4)aufgereinigtumdasisolierteProduktzuerhalten.
Umsatz:43%,Ausbeute:17%(40.6mg,0.17mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.28(t, 3J=7.1Hz,3H,
H‐15),1.84(m,1H,H‐2),2.62(m,1H,H‐1),2.88(m,1H,H‐2),
3.00 (m, 1H, H‐2), 3.37 (d, 3J = 2.2 Hz, 2H, H‐12), 4.19 (q,
3J=7.2Hz,2H,H‐14),5.51(q,3J=7.8Hz,1H,H‐9),7.20–7.30
(m,4H,Har).
13C‐NMR(500MHz,CDCl3):14.19(C‐15),30.39(C‐1),34.08
(C‐2), 41.42 (C‐12), 54.89 (C‐9), 61.71 (C‐14), 124.12 (C‐4),
124.92(C‐7),126.92(C‐5),128.11(C‐6),143.00(C‐8),143.48
(C‐3),164.96(C‐11),169.58(C‐13).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm, flow: 0.8 mL/min, tR=21.89 min (R),
28.47min(S),ee‐Wert:0%.
IR (ATR): (cm‐1) = 3269.9, 3069.2, 2990.9, 2961.8, 2847.6, 2358.0, 1732.4, 1640.2, 1548.9, 1457.6,
1409.5,1363.8,1228.4,1148.2,1023.2,749.1,587.1,539.3.
MS(ESI):m/z(%)=270.1(100)[M+Na]+,315.2(3),437.2(3),517.1(5)[2M+Na]+.
EA(C14H17NO3):berechnetC:68.00%,H:6.93%,N:5.66%;gefunden:C:68.58%,H:6.96%,N:5.77%.
DiespektroskopischenDatensindnichtliteraturbekannt.
7.2.2.2 Analytik
7.2.2.2.1 ValidierungderAufarbeitungvonAAV5mittelsNMR‐Analytik
ZurValidierungderAnalytikwerdenMalonsäurediethylester(9)undrac‐10imVerhältnis1:1einge‐
wogen. Mittels 1H‐NMR‐Spektroskopie wird das Verhältnis beider Komponenten bestimmt. An‐
schließend erfolgt eine Aufarbeitung analog der AAV6, indem filtriert undmit DCM undMTBE (je
10mL)gespültwird.DasVerhältnisderbeidenKomponentenwirderneutmittels 1H‐NMR‐Spektro‐
skopieermittelt.DieErgebnissesindinTabelle40dargestellt.

7 ExperimentellerTeil
163
Tabelle40. ValidierungderAufarbeitungvonAAV6mittels1H‐NMR‐Analytik.
EintragVerhältnis9/rac‐10
vorAufarbeitung
Verhältnis9/rac‐10
nachAufarbeitung
Abweichung
[%]
1 1:0.98 1:0.99 1
2 1:1.02 1:1.06 4
3 1:1.04 1:1.05 1
7.2.2.2.2 BestimmungderEnantiomerenüberschüssedurchchemischeAcylierungdes
resultierendenAmins(S)‐4
DieSyntheseerfolgtanalogderAAV5.Zurac‐1‐Phenylethylamin(rac‐4,0.5mmol,60.1mg)werden
Malonsäurediethylester (9, 0.5mmol, 80.1mg) und CAL‐B (40mg/mmol) gegeben und für 24h bei
60°Cgerührt.DieAufarbeitungerfolgtanalogderAAV5. ImAnschlusswirddergesamteReaktions‐
ansatzzusammenmitAcetanhydrid(132,0.5mmol,47.7µL)für6hbeiRTinChloroformgerührt.Das
Lösungsmittelwirdi.Vak.entfernt.MittelsHPLCwerdendieee‐WertedesAmids(R)‐10bestimmt.Der
ee‐WertdesAmids(S)‐7wurdeebenfallsmittelsHPLCdetektiertundmitdemberechnetenWertfür
(S)‐4verglichen.DieErgebnissesindinTabelle41aufgelistet.
Tabelle41. BestimmungdesEnantiomerenüberschussesdesresultierendenAmins(S)‐4.
ee‐Wert(S)‐10detektiert ee‐Wert(S)‐4 berechnet ee‐Wert(R)‐7detektiert
97% 90% 88%
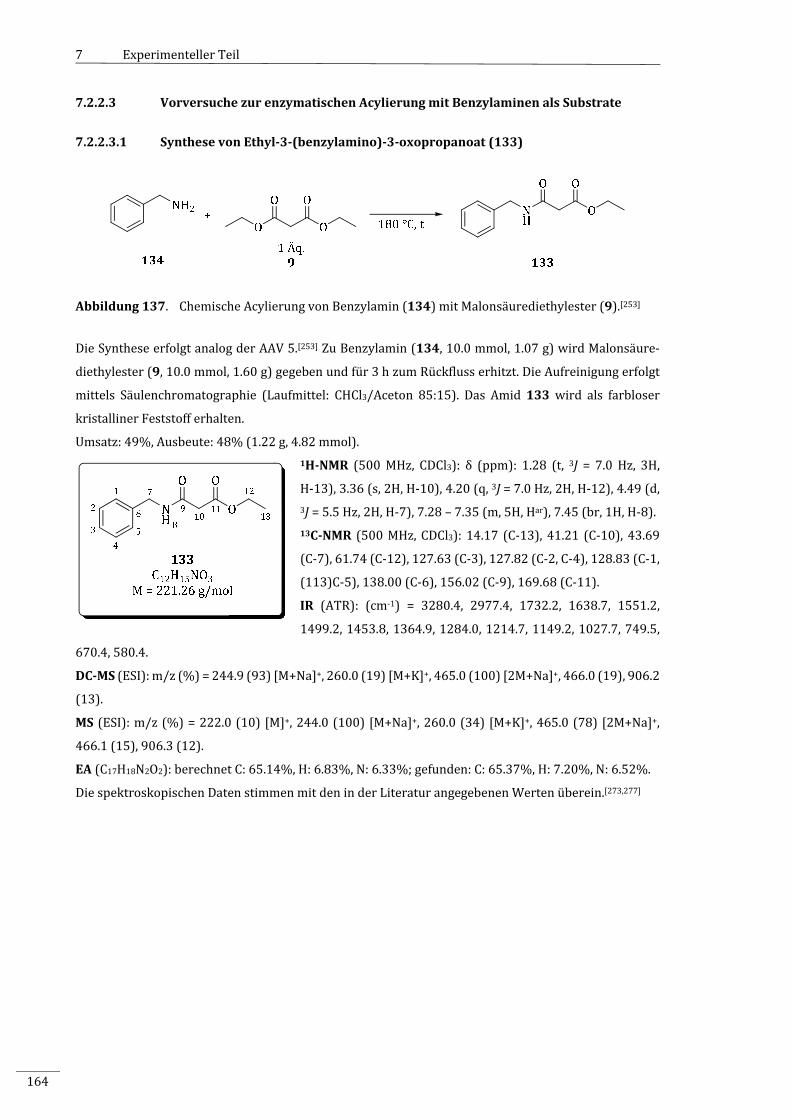
7 ExperimentellerTeil
164
7.2.2.3 VorversuchezurenzymatischenAcylierungmitBenzylaminenalsSubstrate
7.2.2.3.1 SynthesevonEthyl‐3‐(benzylamino)‐3‐oxopropanoat(133)
Abbildung137. ChemischeAcylierungvonBenzylamin(134)mitMalonsäurediethylester(9).[253]
DieSyntheseerfolgtanalogderAAV5.[253]ZuBenzylamin(134,10.0mmol,1.07g)wirdMalonsäure‐
diethylester(9,10.0mmol,1.60g)gegebenundfür3hzumRückflusserhitzt.DieAufreinigungerfolgt
mittels Säulenchromatographie (Laufmittel: CHCl3/Aceton 85:15). Das Amid133 wird als farbloser
kristallinerFeststofferhalten.
Umsatz:49%,Ausbeute:48%(1.22g,4.82mmol).
1H‐NMR (500MHz, CDCl3): δ (ppm): 1.28 (t, 3J = 7.0 Hz, 3H,
H‐13),3.36(s,2H,H‐10),4.20(q,3J=7.0Hz,2H,H‐12),4.49(d,
3J=5.5Hz,2H,H‐7),7.28–7.35(m,5H,Har),7.45(br,1H,H‐8).
13C‐NMR (500MHz,CDCl3):14.17 (C‐13),41.21 (C‐10),43.69
(C‐7),61.74(C‐12),127.63(C‐3),127.82(C‐2,C‐4),128.83(C‐1,
(113)C‐5),138.00(C‐6),156.02(C‐9),169.68(C‐11).
IR (ATR): (cm‐1) = 3280.4, 2977.4, 1732.2, 1638.7, 1551.2,
1499.2,1453.8,1364.9,1284.0,1214.7,1149.2,1027.7,749.5,
670.4,580.4.
DC‐MS(ESI):m/z(%)=244.9(93)[M+Na]+,260.0(19)[M+K]+,465.0(100)[2M+Na]+,466.0(19),906.2
(13).
MS(ESI):m/z(%)=222.0(10)[M]+,244.0(100)[M+Na]+,260.0(34)[M+K]+,465.0(78)[2M+Na]+,
466.1(15),906.3(12).
EA(C17H18N2O2):berechnetC:65.14%,H:6.83%,N:6.33%;gefunden:C:65.37%,H:7.20%,N:6.52%.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[273,277]
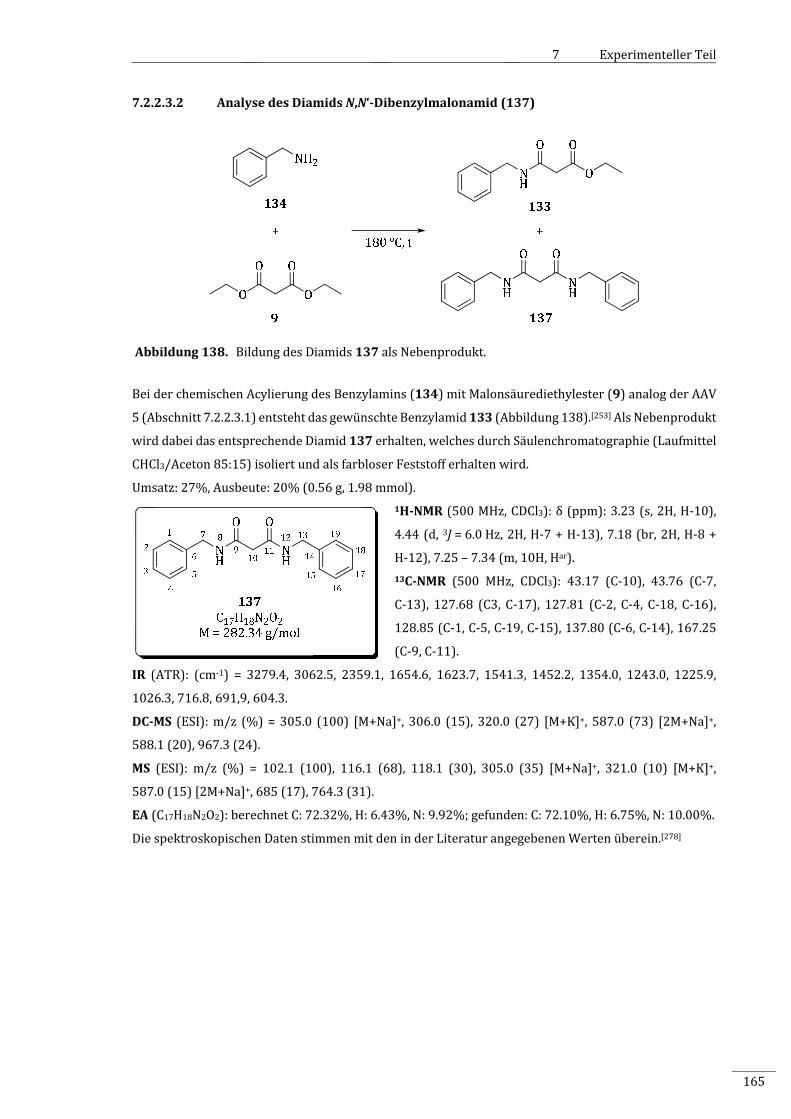
7 ExperimentellerTeil
165
7.2.2.3.2 AnalysedesDiamidsN,N‘‐Dibenzylmalonamid(137)
Abbildung138. BildungdesDiamids137alsNebenprodukt.
BeiderchemischenAcylierungdesBenzylamins(134)mitMalonsäurediethylester(9)analogderAAV
5(Abschnitt7.2.2.3.1)entstehtdasgewünschteBenzylamid133(Abbildung138).[253]AlsNebenprodukt
wirddabeidasentsprechendeDiamid137erhalten,welchesdurchSäulenchromatographie(Laufmittel
CHCl3/Aceton85:15)isoliertundalsfarbloserFeststofferhaltenwird.
Umsatz:27%,Ausbeute:20%(0.56g,1.98mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):3.23(s,2H,H‐10),
4.44(d,3J=6.0Hz,2H,H‐7+H‐13),7.18(br,2H,H‐8+
H‐12),7.25–7.34(m,10H,Har).
13C‐NMR (500 MHz, CDCl3): 43.17 (C‐10), 43.76 (C‐7,
C‐13), 127.68 (C3, C‐17), 127.81 (C‐2, C‐4, C‐18, C‐16),
128.85(C‐1,C‐5,C‐19,C‐15),137.80(C‐6,C‐14),167.25
(C‐9,C‐11).
IR (ATR): (cm‐1) = 3279.4, 3062.5, 2359.1, 1654.6, 1623.7, 1541.3, 1452.2, 1354.0, 1243.0, 1225.9,
1026.3,716.8,691,9,604.3.
DC‐MS (ESI):m/z (%)=305.0 (100) [M+Na]+, 306.0 (15), 320.0 (27) [M+K]+, 587.0 (73) [2M+Na]+,
588.1(20),967.3(24).
MS (ESI): m/z (%) = 102.1 (100), 116.1 (68), 118.1 (30), 305.0 (35) [M+Na]+, 321.0 (10) [M+K]+,
587.0(15)[2M+Na]+,685(17),764.3(31).
EA(C17H18N2O2):berechnetC:72.32%,H:6.43%,N:9.92%;gefunden:C:72.10%,H:6.75%,N:10.00%.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[278]

7 ExperimentellerTeil
166
7.2.2.3.3 AllgemeineArbeitsvorschrift6(AAV6):EnzymatischeAcylierungsubstituierter
Benzylamine
Abbildung139. EnzymatischeAcylierungsubstituierterBenzylamine.
ZusubstituiertemBenzylamin (134,138 ‐140, 0.5–2.5mmol)werdenMalonsäurediethylester (9,
1Äq.)undCAL‐B(40mg/mmol)gegebenundbei80°Cfür4.5hbei200rpmgerührtoderimThermo‐
Schüttlerbei300rpmgeschüttelt(Abbildung139).DieLipasewirdabfiltriert,mitDCMundMTBE(je
10–40mL)gewaschenunddieLösungsmitteli.Vak.entfernt.DerUmsatzwirdmithilfevon1H‐NMR‐
Spektroskopiebestimmt.DasProduktwirdmittelsSäulenchromatographieisoliert.
7.2.2.3.4 EnzymatischeSynthesevonEthyl‐3‐(benzylamino)‐3‐oxopropanoat(133)
DieSyntheseerfolgtanalogderAAV6.ZuBenzylamin(134,0.5mmol,53.6mg)werdenMalonsäure‐
diethylester(9,0.5mmol,81.3mg)undCAL‐B(40mg/mmol,20mg)gegebenundbei80°Cfür4.5h
erhitzt.DieAufarbeitungerfolgtanalogderAAV2.DasProdukt133wirdineinerMischungmitdem
Diamid137erhalten.
Umsatz:91%133+9%137.
1H‐NMR(500MHz,CDCl3):δ(ppm):1.28(t,3J=7.0Hz,3H,H‐
13),3.36(s,2H,H‐10),4.20(q, 3J=7.0Hz,2H,H‐12),4.48(d,
3J=5.5Hz,2H,H‐7),7.28–7.35(m,5H,Har),7.45(br,1H,H‐8).
Die analytischenDaten stimmenmit denVergleichsdaten aus
Abschnitt7.2.1.1undderLiteraturüberein.[277]
7.2.2.3.5 EnzymatischeSynthesevonN‐(m‐Hydroxybenzyl)‐N‐ethylmalonamid(141)
DieSyntheseerfolgtanalogderAAV6.Zu3‐Hydroxybenzylamin (138, 1.0mmol,123.8mg)werden
Malonsäurediethylester (9, 1.0mmol, 162.5mg) und CAL‐B (40mg/mmol, 40mg) gegeben und bei
80°C für 4.5h gerührt. Die Aufreinigung erfolgtmittels Säulenchromatographie (Laufmittel: CHCl3/
Aceton3:2).DasProduktwirdalsfarbloserFeststofferhalten.

7 ExperimentellerTeil
167
Umsatz:38%,Ausbeute:35%+17%(136).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.26(t,3J=7.5Hz,3H,
H‐14),3.36(s,2H,H‐11),3.80(s,3H,H‐7),4.18(q,3J=7.0
Hz,2H,H‐13),4.46(d,3J=5.5Hz,2H,H‐8),6.75–6.81(m,
3H,Har),7.18(t,3J=7.5Hz,1H,Har),7.61(br,1H,H‐7).
13C‐NMR (500 MHz, CDCl3): 14.07 (C‐14), 41.12 (C‐11),
43.66 (C‐8), 61.89 (C‐13), 114.80 (C‐3), 114.96 (C‐1),
119.31 (C‐4), 129.94 (C‐4), 139.18 (C‐6), 156.91 (C‐2),
166.03(C‐10),169.42(C‐12).
IR (ATR): (cm‐1) = 3291.6, 2980.8, 2358.9, 1720.8, 1646.4, 1588.9, 1541.5, 1455.3, 1368.9, 1330.2,
1194.0,1154.6,1020.6,946.9,866.2,780.6,731.5,694.3.
MS(ESI):m/z(%)=260.0(100)[M+Na]+,497.1(52).
EA(C13H16NO3):berechnetC:60.75%,H:6.37%,N:5.90%;gefunden;C:59.24%,H6.86%,N:5.83%.
DiespektroskopischenDatensindnichtliteraturbekannt.
7.2.2.3.6 EnzymatischeSynthesevonN‐(m‐Bromobenzyl)‐N‐ethylmalonamid(145)
Die Synthese erfolgt analog der AAV 6. Zu 3‐Brombenzylamin (140, 2.5 mmol, 468.5mg) werden
Malonsäurediethylester(9,2.5mmol,404.1mg)undCAL‐B(40mg/mmol,200mg)gegebenundbei
80°C für 4.5 h gerührt. Die Aufreinigung erfolgt mittels Säulenchromatographie (Laufmittel:
CHCl3/Aceton85/15).DasProdukt137wirdinFormvonkristallinenNadelnerhalten.
Umsatz:95%,Ausbeute:81%.
1H‐NMR(500MHz,CDCl3):δ(ppm):1.29(t,3J=7.1Hz,3H,
H‐13),3.37(s,2H,H‐10),4.20(q,3J=7.0Hz,2H,H‐12),4.46
(d,3J=6.0Hz,2H,H‐7),7.18–7.23(m,2H,Har),7.39–7.44
(m,2H,Har),7.55(br,1H,H‐8).
13C‐NMR (500 MHz, CDCl3): 14.19 (C‐13), 41.07 (C‐10),
42.97 (C‐7), 61.86 (C‐12), 122.85 (C‐2), 126.39 (C‐5),
130.37(C‐3+C‐4),130.76(C‐1),140.43(C‐6),165.17(C‐9),
169.72(C‐11).
IR (ATR): (cm‐1) = 3234.5, 3059.8, 2973.5, 2360.5, 2341.5, 1745.7, 1635.4, 1551.8, 1416.9, 1329.3,
1275.8,1234.8,1143.8,1071.7,1030.9,994.6,786.5,683.5.
MS(ESI):m/z(%)=322.0(95),324.0(100)[M+Na]+,393.3(4),437.2(6).
EA(C13H16NO3):berechnetC:48.02%,H:4.70%,N:4.67%;gefunden:C:47.71%,H4.79%,N:4.67%.
DiespektroskopischenDatensindnichtliteraturbekannt.
NH
141C12H15NO4
M = 251.28 g/mol
1
2
3
4
5
67
8
9
1011
12
13HO
O
O
O
14
NH
145C13H16BrNO3
M = 314.18 g/mol
1
2
3
4
5
6
7
8
910
11
12
13
BrO
O
O

7 ExperimentellerTeil
168
7.2.2.3.7 SynthesevonN‐(m‐Methoxybenzyl)‐N‐ethylmalonamid(143)
DieSyntheseerfolgtanalogderAAV6.Zu3‐Methoxybenzylamin(139,2.5mmol,345.5mg)werden
Malonsäurediethylester (9, 2.5mmol,403.4mg)undCAL‐B (40mg/mmol,200mg)gegebenundbei
80°C für 4.5h gerührt. Die Aufreinigung erfolgt mittels Säulenchromatographie (Laufmittel:
CHCl3/Aceton85/15).DasProdukt138wirdinFormvonkristallinenNadelnerhalten.
Umsatz:95%,Ausbeute:84%.
1H‐NMR(500MHz,CDCl3):δ(ppm):1.29(t,3J=7.0Hz,3H,
H‐14),3.36(s,2H,H‐11),3.80(s,3H,H‐7),4.20(q,3J=7.0
Hz,2H,H‐13),4.46(d,3J=5.5Hz,2H,H‐8),6.81–6.84(m,
2H,Har),6.87–6.88(d,3J=7.5Hz,1H,Har),7.25(t,3J=8.0
Hz,2H,Har),7.55(br,1H,H‐7).
13C‐NMR (500 MHz, CDCl3): 14.18 (C‐14), 41.20 (C‐11),
43.65 (C‐8), 55.37 (C‐7), 61.76 (C‐13), 113.08 (C‐1),
113.43 (C‐3), 120.03 (C‐5), 129.88 (C‐4), 139.58 (C‐6),
160.00(C‐2),165.01(C‐10),169.70(C‐12).
IR (ATR): (cm‐1) = 3229.5, 3056.8, 2978.2, 2361.0, 2341.5, 1748.1, 1635.5, 1597.8, 1553.4, 1453.1,
1325.2,1276.3,1134.6,1037.7,879.5,792.2,737.6,669.9,570.0.
MS(ESI):m/z(%)=322.0(95),324.0(100)[M+Na]+,393.3(4),437.2(6).
EA(C13H17NO4):berechnetC:62.14%,H:6.82%,N:5.57%;gefunden;C:61.96%,H7.04%,N:5.63%.
DiespektroskopischenDatensindnichtliteraturbekannt.
7.2.2.3.8 BestimmungdesVerlustsvonBenzylamin(134)undMalonsäurediethylester(9)
ZurBestimmungdesVerlustsvonBenzylamin(134)undMalonsäurediethylester(9)währendderAuf‐
arbeitungwirdeinebekannteMenge derSubstanzenübereinenbestimmtenZeitraumbei230bzw.
550mbarund40°Cgerührt.DurchAuswaagewirdderVerlustderSubstanzenbestimmt.DieErgeb‐
nissesindinTabelle42aufgelistet.
NH
143C13H17NO4
M = 251.28 g/mol
1
2
3
4
5
67
8
9
1011
12
13O
O
O
O
14

7 ExperimentellerTeil
169
Tabelle42. Bestimmung des Verlusts von Benzylamin (134) und Malonsäurediethylester (9)währendderAufarbeitung.
Eintrag SubstanzZeit
[min]
Verlust[mg] Verlust[%] Verlust[mg] Verlust[%]
230mbar 550mbar
1 134 0 0 0 0 0
2 134 15 1.1 10 0 0
3 134 30 1.8 17 0.5 3
4 134 60 2.9 28 1.2 7
5 134 120 7.2 69 2.1 12
6 9 0 0 0 0 0
7 9 15 0.4 1 0.1 0
8 9 30 0.7 2 0.1 0
9 9 60 1.3 3 0.5 1
10 9 120 4.0 9 0.9 2
7.2.2.3.9 AllgemeineArbeitsvorschrift7(AAV7):Kontrollederchemischen
ParallelreaktionderAcylierungverschiedenerBenzylamine
ZusubstituiertemBenzylamin(134,139bzw.140,0.25‐0.5mmol)wirdMalonsäurediethylester(9,
1Äq.)gegebenundbei80°Cfür4.5hgerührt.EswirdanalogderAAV6filtriert,mitDCMundMTBE(je
10–40mL)gewaschenunddieLösungsmitteli.Vak.entfernt.DerUmsatzwirdmittels1H‐NMR‐Spektro‐
skopiebestimmt.DieErgebnissesindinTabelle43aufgelistet.
Tabelle43. ParallelablaufendechemischeAcylierungsubstituierterBenzylamine.
Eintrag Amin UmsatzAmid[%] UmsatzDiamid[%]
1 134 0 0
2 140 7 0
3 139 8 2

7 ExperimentellerTeil
170
7.2.2.3.10 UntersuchungenzurDiamidbildungausgehendvon3‐Methoxybenzylamin(139)
Zu3‐Methoxybenzylamin(139,0.1mmol)werdenanalogderAAV2Malonsäurediethylester(9,0.6–
1.5mmol)undCAL‐B(40mg/mmol)zugegebenundfür4.5hbei80°Cgerührt.DieAufarbeitungerfolgt
analogderAAV6.DieUmsätzewerdenmittels1HNMR‐Spektroskopieermittelt.DieErgebnissewerden
inTabelle44aufgelistet.
Tabelle44. Untersuchungen zurBildungdesDiamids144 bei der enzymatischenAcylierung von3‐Methoxybenzylamin(139).
EintragMalonsäurediethylester(9)
[mmol]UmsatzAmid143
[%]
UmsatzDiamid144
[%]
1 0.6 53 32
2 1.0 91 9
3 1.2 84 3
4 1.5 79 1
7.2.2.3.11 LösungsmitteleffektderenzymatischenAcylierungsubstituierterBenzylamine
Die enzymatischeAcylierung substituierter Benzylamine (0.5mmol)mitMalonsäurediethylester (9,
1Äq.)undLipaseCAL‐B(40mg/mmol)wurdebeiunterschiedlichenReaktionsbedingungenuntersucht.
DieSynthesemitanschließenderAufarbeitungerfolgteanalogderAAV6.DieErgebnissesindinTabelle
45aufgelistet.

7 ExperimentellerTeil
171
Tabelle45. EinleitendeVersuchezurProzessoptimierungderenzymatischenAcylierungsubstituierterBenzylamine.
Eintrag Amin T[°C] t[h] Solvens logP Umsatz[%]
1 140 60 4.5 1,4‐Dioxan ‐1.10a)/‐0.30
75
2 140 60 4.5 n‐Heptan 4.50b) 76
3 140 60 4.5 ‐ ‐ 91
4 139 60 4.5 ‐ ‐ 93
5 138 80 4.5 MTBE 0.94b) 51
6 138 80 4.5 THF 0.46b) 75
7 138 80 4.5 2‐Me‐THF 1.00c) 70
8 138 80 4.5 MeCN ‐0.34b) 43
9 138 80 4.5 i‐PrOH 0.28b) 22
10 138 60 4.5 n‐Heptan 4.50b) 45
11 138 60 4.5 EtOAc 0.73b) 41
12 138 60 4.5 Malonester 9(100Äq.) 0.90c) 50
13 138 60 4.5 Et2O 0.89b) 30
14d) 138 RT 24 Et2O 0.89b) 30
15 138 RT 24 EtOH ‐0.24a) 5Datenentnommenaus:a)LAANEetal.[175];b)SANGSTERetal.[177];c)BestimmtdurchAdvancedChemistryDevelopment(ACD/Labs)SoftwareV11.02[263];d)Zugabevon1.5ÄquivalentenMalonsäurediethylester(9).
7.2.2.4 EnzymatischenRacematspaltungvon1‐Phenylethylamin(rac‐4)
7.2.2.4.1 Standardreaktion:Racematspaltungvon1‐Phenylethylamin(rac‐4)mit
Malonsäurediethylester(9)
Abbildung140. Racematspaltungvonrac‐4mitMalonester9.
DieSyntheseerfolgtanalogderAAV6.Zurac‐1‐Phenylethylamin(rac‐4,5.0mmol,604.1mg)werden
Malonsäurediethylester(9,5.1mmol,810.6mg)undCAL‐B(40mg/mmol)gegebenundfür4.5hbei

7 ExperimentellerTeil
172
80°C und 200 rpm gerührt (Abbildung 140). Die Aufarbeitung erfolgt analog der AAV6. Durch
AufreinigungmittelsSäulenchromatographie(Laufmittel:CH/EtOAc2:1)wirddasProduktrac‐10als
farbloserFeststofferhalten.
Umsatz:44%,Ausbeute:26%(312.0mg,1.3mmol).
1H‐NMR (500MHz, CDCl3): δ (ppm): 1.28 (t, 3J = 7.0 Hz, 3H,
H‐14),1.51(d,3J=7.0Hz,3H,H‐9),3.31(d,3J=5.0Hz,2H,H‐11),
4.19(q,3J=7.5Hz,2H,H‐13),5.15(qui,3J=7.0Hz,1H,H‐7),7.24
–7.28(m,1H,Har),7.31‐7.36(m,5H,Har),7.47(m,1H,H‐8).
HPLC:AD‐H‐Säule,95:5 (n‐Hexan/i‐PrOH,v/v),210nm, flow:
0.8mL/min,tR=17.0min(R),27.1min(S),ee‐Wert:96%.
DiespektroskopischenDatenstimmenmitdeninderLiteratur
undinAbschnitt7.2.2.1.1angegebenenWertenüberein.[279]
7.2.2.4.2 ProzessoptimierungderenzymatischenRacematspaltungvonrac‐4
Die enzymatische Racematspaltung von 1‐Phenylethylamin (rac‐4) mit Malonsäurediethylester (9,
1Äq.) und Lipase CAL‐B (40 mg/mmol) wurde in Analogie zu AAV 6 mit unterschiedlichen
Reaktionsbedingungen untersucht. Die Synthese erfolgte analog der AAV 6. Die Ergebnisse sind in
Tabelle46aufgelistet.
Tabelle46. ProzessoptimierungderenzymatischenRacematspaltungvonrac‐4.
Eintrag T[°C] t[h] Umsatz[%] eeP [%] eeS [%] a) E
1b) 80 4.5 42 92 67 48
2 80 4.5 46 96 82 >100
3 60 4.5 42 97 70 >100
4 60 8 46 89 76 39
5 60 24 50 94 94 >100
6c) 40 4.5 45 99 81 >100
7 40 6 46 85 72 27
8 20 4.5 41 90 63 36
9c) 0 4.5 24 99 n.b. >100a)berechnetmit„Enantioselectivity“[258];b)Zugabevon0.4mLMTBE;c)präperativeDurchführungderVersuchedurchT.Geisler.

7 ExperimentellerTeil
173
7.2.2.4.3 UntersuchungderParallelreaktionderAcylierungvon1‐Phenylethylamin(rac‐4)
DieSynthesenwerdenanalogderAAV7durchgeführt.Zu1‐Phenylethylamin(rac‐4,0.5mmol)wird
Malonsäurediethylester(9,1Äq.)gegebenundbeiRT–60°Cfür4.5hgerührt.Eswird,analogderAAV
6,filtriert,mitDCMundMTBE(je10mL)gewaschenunddieLösungsmitteli.Vak.entfernt.DerUmsatz
wirdmittels1H‐NMR‐Spektroskopiebestimmt.DieErgebnissesindinTabelle47aufgelistet.
Tabelle47. NegativkontrollederRacematspaltungvonrac‐4mitMalonsäurediethylester(9).
Eintrag T[°C] Umsatzrac‐10[%]
1 60 0
2 40 0
3 RT 0
7.2.2.4.4 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)beigeringerer
Enzymbeladung
Abbildung141. Enzymatische Racematspaltung von rac‐4 mit Malonsäurediethylester (9) beigeringererEnzymbeladung.[264]
Zu 1‐Phenylethylamin (rac‐4, 0.49 mmol, 59.8 mg) werden Malonsäurediethylester (9, 0.49 mmol,
79.1mg)undCAL‐B(2mg/mmol,1mg)gegebenundfür24hbeiRTund200rpmgerührt.[264]Das
EnzymwirdabfiltriertundanalogderAAV6mitDCMundMTBE(je10mL)gespült.DieLösungsmittel
werdeni.Vak.entfernt.DerUmsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.
Umsatz:25%.
HPLC:AD‐H‐Säule,95:5(CO2/i‐PrOH,v/v),210nm,flow:0.8mL/min,tR=21.38min(R),34.97min(S),
ee‐Wert:>95%.

7 ExperimentellerTeil
174
7.2.2.4.5 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)beigeringerer
EnzymbeladunginEt2O
Abbildung142. EnzymatischeRacematspaltungvonrac‐4beigeringererEnzymbeladunginEt2O.[30]
Zu1‐Phenylethylamin(rac‐4,1.25–2.5mmol)werdenderentsprechendeEster9oder111(1.5Äq.)
undCAL‐B(10mg/mmol)gegebenundinEt2O(5–10mL)für24hbeiRTgerührt(Abbildung142).[30]
Das Enzym wird abfiltriert und analog der AAV 6 mit DCM und MTBE (je 10 mL) gespült. Die
Lösungsmittelwerdeni.Vak.entfernt.DerUmsatzwirdmittels 1H‐NMR‐Spektroskopiebestimmt.Die
ErgebnissesindinTabelle48aufgelistet.
Tabelle48. EnzymatischeRacematspaltungvonrac‐4mitgeringererEnzymbeladunginEt2O.[30]
Eintrag Ester Umsatz[%] ee[%] E
1 9 48 95 >100
2 111 52 95 >100
7.2.2.5 Enzymrecycling
Das Enzymrecycling wird analog der AAV 6 durchgeführt. Dazu werden 1‐Phenylethylamin (rac‐4,
5mmol)undMalonsäurediethylester(9,5mmol)zusammenmitderCAL‐B(40mg/mmol)für4.5hbei
60°Cgerührt.DieAufarbeitungerfolgtanalogderAAV6.DiezurückerhalteneCAL‐Bwirdgetrocknet
undimnächstenZykluseingesetzt.DieErgebnissesindinTabelle49aufgelistet.

7 ExperimentellerTeil
175
Tabelle49. Enzymrecycling.
Eintrag Zyklus Umsatz[%] eeP [%] eeS[%]a) E
1 1 43 90 68 39
2 2 30 81 35 13
3 3 25 87 n.b. 19
4 4 20 90 n.b. 26
5b) 5 24 94 n.b. 45a)berechnetmit„Enantioselectivity“[258];b)Reaktionsdauer:6,5h;n.b.=nichtbestimmt.
7.2.2.6 Lipasen‐ScreeningdurchAcylierungvonAminen
7.2.2.6.1 Lipasen‐ScreeningdurchAcylierungvonBenzylamin(134)
Das Screeningwird analog derAAV6 durchgeführt.DazuwerdenBenzylamin (134, 0.5mmol) und
Malonsäurediethylester(9,0.5mmol)zusammenmitderjeweiligenLipase(20mg)inMTBE(0.3mL)
für3hbei60°CineinemThermomix‐Schüttlerbei300rpmgeschüttelt.DieAufarbeitungerfolgtanalog
derAAV6.DieErgebnissesindinTabelle50aufgelistet.
Tabelle50. Lipasen‐ScreeningmittelsAcylierungvonBenzylamin(134).
Eintrag
Lipase
Umsatz
Amid133
[%]
Umsatz
Diamid137
[%]
Gesamtumsatz
[%]
1 CAL‐B(Novozym435) 89 10 >99
2 Toyobo 11 3 14
3 Hogpancreas(FLUKA) 12 3 15
4 L„AMANO“5 17 6 20
5 Rhizopusniveus(FLUKA) 6 1 7
6 L.P.L.„AMANO“100S 6 1 7
7 GC„AMANO“4 22 10 32
8 R„AMANO“10 19 9 28
9 AY„AMANO“30 17 6 23
10 NConc(AMANO) 6 4 10

7 ExperimentellerTeil
176
Eintrag
Lipase
Umsatz
Amid133
[%]
Umsatz
Diamid137
[%]
Gesamtumsatz
[%]
11 AP6(AMANO) 6 1 7
12PSimmob.aufToyonite‐200‐P
(AMANO)15 3 18
13 PSimmob.aufDiatomite(AMANO) 4 <1 5
14 M‐AP10(AMANO) 5 <1 5
15 F‐AP15(AMANO) 6 <1 6
16 R10(AMANO) 12 1 13
17 CE„AMANO“5 18 7 25
18 D„AMANO“20 22 11 33
19 AK„AMANO“ 13 3 16
20 AYS„AMANO“ 8 1 9
21 PS„AMANO“SD 11 3 15
22 CAL‐Bimmob.(C‐LECTA) 89 11 >99
23 CAL‐Blyo.(C‐LECTA) 94 6 >99
7.2.2.6.2 Lipasen‐ScreeningdurchAcylierungvon3‐Hydroxybenzylamin(138)
Das Screening wird analog der AAV 6 durchgeführt. Dazu werden 3‐Hydroxybenzylamin (138,
0.1mmol) und Malonsäurediethylester (9, 0.1mmol) zusammen mit der jeweiligen Lipase
(40mg/mmol,4mg)inMTBE(0.3mL)für3hbei60°CineinemThermomix‐Schüttlerbei300rpm
geschüttelt.DieAufarbeitungerfolgtanalogderAAV6.DieErgebnissesindinTabelle51aufgelistet.
Tabelle51. Lipasen‐ScreeningmittelsAcylierungvon3‐Hydroxybenzylamin(138).
Eintrag Lipase
Umsatz
Amid141
[%]
Umsatz
Diamid142
[%]
Gesamtumsatz
[%]
1 CAL‐B(Novozym435) 52 12 60
2 L„AMANO“5 0 0 0
3 GC„AMANO“4 0 0 0
4 CE„AMANO“5 0 0 0
5 R„AMANO“10 0 0 0
6 D„AMANO“20 0 0 0
7 AY„AMANO“30 0 0 0
8 PSimmob.aufToyonite‐200‐P(AMANO)
47 0 47

7 ExperimentellerTeil
177
Eintrag Lipase
Umsatz
Amid141
[%]
Umsatz
Diamid142
[%]
Gesamtumsatz
[%]
9 BSA 0 0 0
10 CAL‐Bimmob.(C‐LECTA) 44 3 47
11 CAL‐Blyo.(C‐LECTA) 18 <1 18
7.2.2.6.3 Lipasen‐ScreeningdurchAcylierungvon1‐Phenylethylamin(rac‐4)
DieSynthesenwerdennachAAV6durchgeführt.Dafürwerden1‐Phenylethylamin(rac‐4,0.1mmol)
und Malonsäurediethylester (9, 0.1mmol) zusammen mit der jeweiligen Lipase (4 mg) in MTBE
(0.3mL) für3hbei60 °C ineinemThermomix‐Schüttlerbei300 rpmgeschüttelt.DieAufarbeitung
erfolgtanalogderAAV6.DieErgebnissesindinTabelle16aufgelistet.DieErgebnissesindinTabelle52
aufgelistet.
Tabelle52. Lipasen‐ScreeningmittelsAcylierungvon1‐Phenylethylamin(rac‐4).
Eintrag Lipase Umsatz[%]
1 CAL‐B(Novozym435) 46
2 L„AMANO“5 0
3 GC„AMANO“4 0
4 CE„AMANO“5 0
5 R„AMANO“10 0
6 D„AMANO“20 0
7 AY„AMANO“30 0
8 PSimmob.aufToyonite‐200‐P(AMANO) <1
9 CAL‐Bimmob.(C‐LECTA) 45
10 CAL‐Blyo.(C‐LECTA) 13

7 ExperimentellerTeil
178
7.2.2.7 EnzymatischeRacematspaltungvon1‐Phenylethylamin(rac‐4)mitalternativen
LipasenCAL‐BvonC‐LECTA
7.2.2.7.1 AllgemeineArbeitsvorschrift9(AAV9):VergleichderAktivitätvonLipasenvon
C‐LECTAmittelsTitrationmitNaOH
ZurBestimmungderEnzymaktivitätwirdPuffer(KPi,pH7,10mM)mitEssigester5gesättigt,indem
PufferundEsterfür1hbeiRTgerührtundanschließenddiePhasengetrenntwerden.Zunächstwird
alsReferenzdieAktivitätderCAL‐B(Novozym435)bestimmt.AnschließendwerdendiebeidenLipasen
vonC‐LECTA,immobilisiertaufeinemMethacrylat‐Trägersowielyophilisiert,vermessen.Dazuwerden
20mgEnzymin5mLPuffervorgelegtundgegebenenfallsaufdieentsprechendeTemperaturerwärmt.
VorBeginnderMessung istderpH‐WertdesEnzym‐enthaltenenPufferszuprüfenundmithilfeder
wässr.NaOH‐Lösung(0.05M)neueinzustellen.DurchZugabevon5mLdesEtOAc‐ges.Pufferswirddie
Titrationgestartet.Gemessenwird1MinutejedeSekunde,wobeiderVerbrauchanwässr.NaOH‐Lösung
beobachtetunddieAnfangssteigungbestimmtwird.Diesekannamgenauestenbestimmtwerden,wenn
derKurvenverlaufzuBeginnderMessungübermehrereSekundenkonstantist.Gegebenenfallsmuss
dieMengedesimmobilisiertenEnzymsreduziertbzw.dieLösungderlyophilisiertenCAL‐Bverdünnt
unddieMessungentsprechendwiederholtwerden.FürdieBerechnungderEnzymaktivitätwirddie
AnfangssteigungderTitrationmitwässr.NaOH‐Lösung(0.1M)verwendet.DieBerechnungderEnzym‐
aktivitäterfolgtnachfolgenderGleichung6:
Gleichung6
U/mg: Enzymaktivität
M: MolaritätderNaOH[µmol/mL]
: AnfangssteigungdesKurvenverlaufs[mL/sec]
f: Umrechnungsfaktor[sec/min]
m: MengederLipasein5mLPuffer[mg]
IndieserArbeitsindM=50µmol/mLundf=60sec/min.
7.2.2.7.2 BestimmungderEnzymaktivitätenderLipasenvonC‐LECTAmittels
enzymatischerSpaltungdesEssigesters5bei20°C
Die Spaltung des Essigesters 5 zu Essigsäure und Ethanol mit der immobilisierten sowie der
lyophilisiertenCAL‐BvonC‐LECTAwirdanalogderAAV7bei20°Cdurchgeführt.AlsReferenzdientdie
CAL‐BvonSIGMAALDRICH(Novozym435).DiefürNovozym435erhalteneAktivitätvon855U/gwirdals

7 ExperimentellerTeil
179
100%definiertunddiejeweilsgemessenenAktivitätenderLipasenwerdenaufdiesenWertbezogen.
DieErgebnissesindinTabelle53aufgelistet.
Tabelle53. EnzymkatalysierteSpaltungdesEssigesters5mitunterschiedlichenLipasenbei20°C.
Eintrag LipaseEnzym
[mg/mmol]
Steigung
[mL/sec]
Aktivität
[U/mg]
Rel.Aktivität
[%]
1 Novozym435 20 0.0057 0.855 100
2 C‐LECTAimmobilisiert 20 0.0039 0.585 68
3 C‐LECTAlyophilisiert 1 0.0057 17.100 2000
7.2.2.7.3 BestimmungderEnzymaktivitätenderLipasenmittelsenzymatischerSpaltung
desEssigesters5bei60°C
Die Spaltung des Essigesters 5 zu Essigsäure und Ethanol mit der immobilisierten sowie der lyo‐
philisiertenCAL‐BvonC‐LECTAwirdanalogderAAV5bei60°Cdurchgeführt.AlsReferenzdientdie
CAL‐B von SIGMA ALDRICH (Novozym435). Die für CAL‐B Novozym 435 bestimmte Aktivität von
3.180U/mgwirdals100%definiertunddiejeweilsgemessenenAktivitätenderLipasenwerdenauf
diesenWertbezogen.DieErgebnissesindinTabelle54aufgelistet.
Tabelle54. EnzymkatalysierteSpaltungdesEssigesters5mitunterschiedlichenLipasenbei60°C.
Eintrag LipaseEnzym
[mg/mmol]
Aktivität
[U/mg]
Rel.Aktivität
[%]
1 Novozym435 20 3.180 100
2 C‐LECTAimmobilisiert 20 2.190 69
3 C‐LECTAlyophilisiert 1 31.800 1000

7 ExperimentellerTeil
180
7.2.2.7.4 EnzymatischeRacematspaltungvonrac‐4mittelsimmobilisierterCAL‐Bvon
C‐LECTA
NachderAAV6werden1‐Phenylethylamin(rac‐4,0.5mmol)undMalonsäurediethylester(9,0.5mmol)
zusammenmit immobilisierterCAL‐BvonC‐LECTA(2.190U/mg,29.0mg)undMTBE(0‐0.5mL)für
4.5hbei60°Cgerührt.DieAufarbeitungerfolgtanalogderAAV1.DieErgebnissesindinTabelle55
aufgelistet.
Tabelle55. Enzymkatalysierte Racematspaltung von rac‐4 mittels immobilisierter CAL‐B vonC‐LECTA.
Eintrag MTBE[mL] Umsatz[%] eeP [%] eeS [%]a) E
1 0 52 96 96 194
2 0.5 49 84 81 28a)berechnetmit„Enantioselectivity“[258].
7.2.2.7.5 EnzymatischeRacematspaltungvonrac‐4mittelslyophilisierterCAL‐Bvon
C‐LECTA
NachderAAV6werden1‐Phenylethylamin(rac‐4,0.5mmol)undMalonsäurediethylester(9,0.5mmol)
zusammenmitlyophilisierterCAL‐BvonC‐LECTA(31.8U/mg,2‐10mg)für4.5‐24hbei20‐60°C
gerührt.GegebenenfallserfolgtnocheineZugabevonMTBE(0–0.5mL)unddest.Wasser(10µL).Es
erfolgtkeineweitereAufarbeitung.DerUmsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.Vorder
Bestimmungdesee‐WertesmittelsHPLCwirddielyophilisierteCAL‐BmittelsSpritzenfilterabfiltriert.
DieErgebnissesindinTabelle56aufgelistet.

7 ExperimentellerTeil
181
Tabelle56. EnzymkatalysierteRacematspaltungvonrac‐4mittelslyophilisierterCAL‐BvonC‐LECTA.
Eintrag CAL‐B[mg] T[°C] t[h] Umsatz [%] ee[%] E
1 2 60 4.5 9 81 10
2 10 60 4.5 26 91 29
3 2 60 24 19 74 8
4 2 20 24 6 80 10
5a) 2 60 4.5 7 87 2
6b) 2 60 4.5 6 n.b. n.b.a)Zugabevon0.5mLMTBE;cb)Zugabevon10µLdest.Wasser;n.b.=nichtbestimmt.
7.2.2.8 VariationderAcyldonoren
7.2.2.8.1 EnzymatischeRacematspaltungvonrac‐4mitalternativenAcyldonoren
Abbildung143. Vergleich von alternativenAcyldonorendurch enzymatischeRacematspaltung vonrac‐4.
Der Vergleich der alternativenAcyldonoren erfolgt analog der AAV 6. Zu 1‐Phenylethylamin (rac‐4,
0.5mmol)wirdder entsprechendeAcyldonor (1Äq.) gegeben und bei RT – 60 °C für 4.5 h gerührt
(Abbildung 143). Die Aufarbeitung erfolgt nach der AAV 6. Der Umsatz wird mittels 1H‐NMR‐
Spektroskopiebestimmt.DieErgebnissesindinTabelle57aufgelistet.
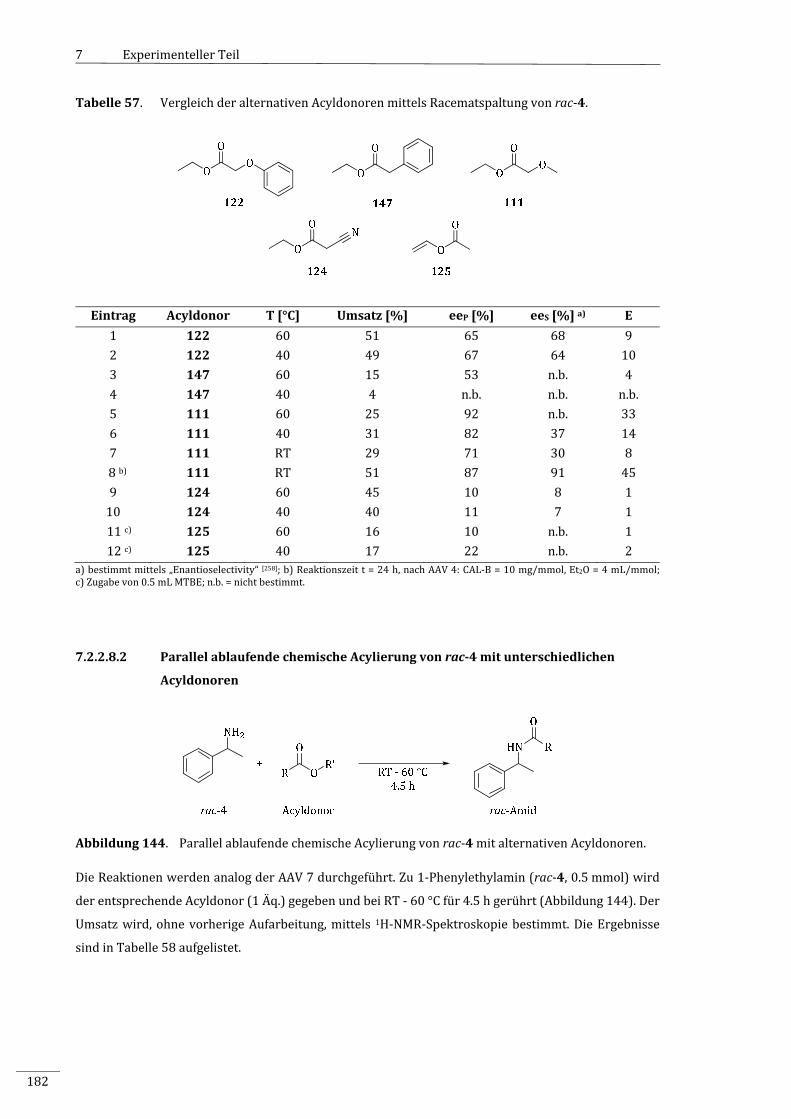
7 ExperimentellerTeil
182
Tabelle57. VergleichderalternativenAcyldonorenmittelsRacematspaltungvonrac‐4.
Eintrag Acyldonor T[°C] Umsatz[%] eeP [%] eeS [%]a) E
1 122 60 51 65 68 9
2 122 40 49 67 64 10
3 147 60 15 53 n.b. 4
4 147 40 4 n.b. n.b. n.b.
5 111 60 25 92 n.b. 33
6 111 40 31 82 37 14
7 111 RT 29 71 30 8
8b) 111 RT 51 87 91 45
9 124 60 45 10 8 1
10 124 40 40 11 7 1
11c) 125 60 16 10 n.b. 1
12c) 125 40 17 22 n.b. 2a)bestimmtmittels„Enantioselectivity“[258];b)Reaktionszeitt=24h,nachAAV4:CAL‐B=10mg/mmol,Et2O=4mL/mmol;c)Zugabevon0.5mLMTBE;n.b.=nichtbestimmt.
7.2.2.8.2 ParallelablaufendechemischeAcylierungvonrac‐4mitunterschiedlichen
Acyldonoren
Abbildung144. ParallelablaufendechemischeAcylierungvonrac‐4mitalternativenAcyldonoren.
DieReaktionenwerdenanalogderAAV7durchgeführt.Zu1‐Phenylethylamin(rac‐4,0.5mmol)wird
derentsprechendeAcyldonor(1Äq.)gegebenundbeiRT‐60°Cfür4.5hgerührt(Abbildung144).Der
Umsatzwird,ohnevorherigeAufarbeitung,mittels 1H‐NMR‐Spektroskopiebestimmt.DieErgebnisse
sindinTabelle58aufgelistet.

7 ExperimentellerTeil
183
Tabelle58. ParallelablaufendeAcylierungvonrac‐4mitalternativenAcyldonoren.
O
OO
O
ON
O
O
O
O
O
OO
122
124
147
125
111
Eintrag Acyldonor T[°C] Umsatz[%] eeP [%] eeS[%]a) E
1 122 60 0 ‐ ‐ ‐
2 122 40 0 ‐ ‐ ‐
3 147 60 0 ‐ ‐ ‐
4 147 40 0 ‐ ‐ ‐
5 111 60 2 n.b. n.b. n.b.
6 111 40 0 ‐ ‐ ‐
7 124 60 33 3 1 1
8 124 40 12 5 n.b. 1
9 125 60 93 0 13 1
10 125 40 81 0 4 1a)Bestimmtmittels„Enantioselectiviy“[258];n.b.=nichtbestimmt.
7.2.2.8.3 KinetikderRacematspaltungvonrac‐4mitMalonsäurediethylester(9)
DieSynthesenerfolgenanalogderAAV6.Zu1‐Phenylethylamin(rac‐4,0.5mmol)werdenMalonsäure‐
diethylester(9,1Äq.)undCAL‐B(40mg/mmol)gegebenundbei60°Cfür1min–24hgerührt.Die
AufarbeitungerfolgtanalogderAAV6.DieErgebnissesindinTabelle59aufgelistet.
Tabelle59. KinetikderenzymatischenRacematspaltungvonrac‐4mitMalonsäurediethylester(9).
Eintrag t[min] Umsatz[%] eeP [%] eeS[%]a) E
1 1 15 97 n.b. 78
2 5 26 95 n.b. 54
3 10 30 94 40 48
4 20 32 95 45 61
5 30 33 97 48 >100

7 ExperimentellerTeil
184
Eintrag t[min] Umsatz[%] eeP [%] eeS [%]a) E
6 45 36 97 55 >100
7 60 37 98 58 >100
8 120 41 97 67 >100
9 180 46 98 83 >100
10 270 43 96 70 >100
11 480 45 89 73 37
12 1440 49 98 94 >100
a)Bestimmtmittels„Enantioselectivity“[258];n.b.=nichtbestimmt.
7.2.2.8.4 KinetikderRacematspaltungvonrac‐4mitPhenoxyessigsäureethylester(122)
DieSynthesenerfolgenanalogderAAV6.Zu1‐Phenylethylamin(rac‐4,0.5mmol)werdenPhenoxy‐
essigsäureethylester (122,1Äq.)undCAL‐B(40mg/mmol)gegebenundbei60°C für1min–24h
gerührt.DieAufarbeitungerfolgtanalogderAAV6.DieErgebnissesindinTabelle60aufgelistet.
Tabelle60. KinetikderenzymatischenRacematspaltungvonrac‐4mitPhenoxyessigester122.
Eintrag t[min] Umsatz[%] eeP [%] eeS [%]a) E
1 1 30 79 34 12
2 5 43 74 56 12
3 10 41 73 51 11
4 20 42 71 51 10
5 30 47 71 63 11
6 45 49 64 61 8
7 60 50 62 62 8
8 120 52 64 69 9
9 180 51 65 67 9
10 270 57 68 90 16
11 480 51 69 72 12
12 1440 56 61 78 9a)Bestimmtmittels„Enantioselectiviy“[258];n.b.=nichtbestimmt.
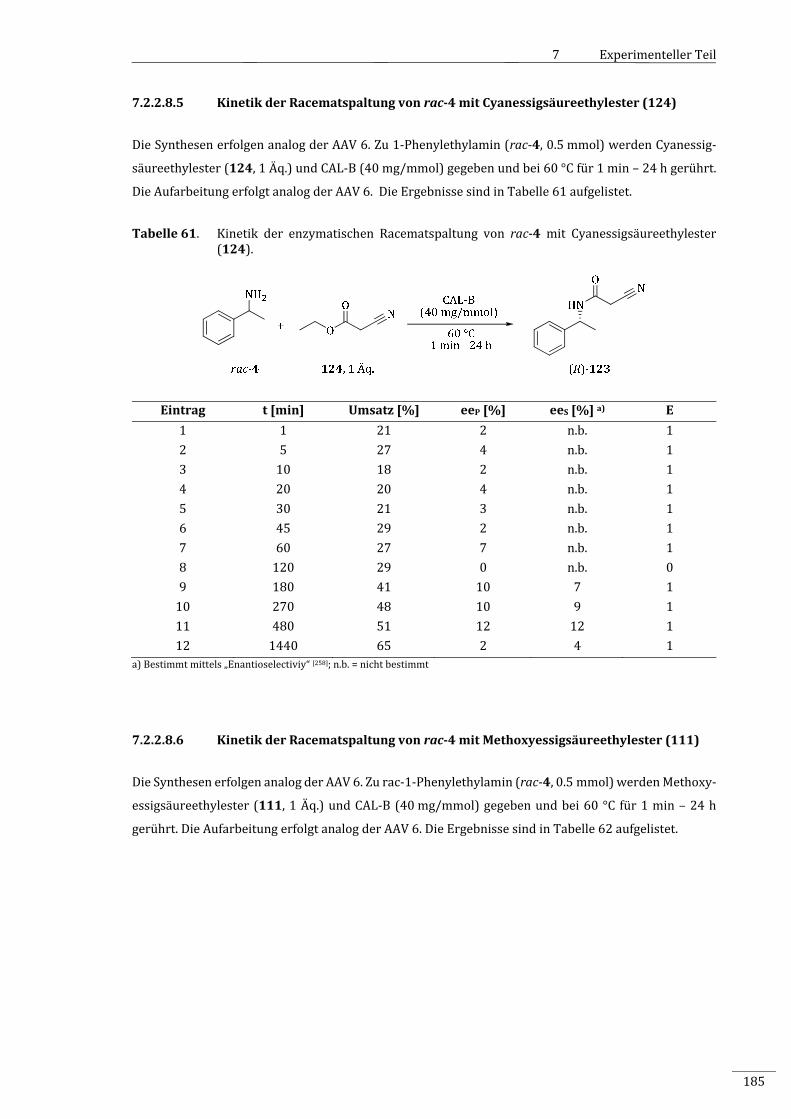
7 ExperimentellerTeil
185
7.2.2.8.5 KinetikderRacematspaltungvonrac‐4mitCyanessigsäureethylester(124)
DieSynthesenerfolgenanalogderAAV6.Zu1‐Phenylethylamin(rac‐4,0.5mmol)werdenCyanessig‐
säureethylester(124,1Äq.)undCAL‐B(40mg/mmol)gegebenundbei60°Cfür1min–24hgerührt.
DieAufarbeitungerfolgtanalogderAAV6.DieErgebnissesindinTabelle61aufgelistet.
Tabelle61. Kinetik der enzymatischen Racematspaltung von rac‐4 mit Cyanessigsäureethylester(124).
Eintrag t[min] Umsatz[%] eeP [%] eeS[%]a) E
1 1 21 2 n.b. 1
2 5 27 4 n.b. 1
3 10 18 2 n.b. 1
4 20 20 4 n.b. 1
5 30 21 3 n.b. 1
6 45 29 2 n.b. 1
7 60 27 7 n.b. 1
8 120 29 0 n.b. 0
9 180 41 10 7 1
10 270 48 10 9 1
11 480 51 12 12 1
12 1440 65 2 4 1a)Bestimmtmittels„Enantioselectiviy“[258];n.b.=nichtbestimmt
7.2.2.8.6 KinetikderRacematspaltungvonrac‐4mitMethoxyessigsäureethylester(111)
DieSynthesenerfolgenanalogderAAV6.Zurac‐1‐Phenylethylamin(rac‐4,0.5mmol)werdenMethoxy‐
essigsäureethylester(111,1Äq.)undCAL‐B(40mg/mmol)gegebenundbei60°C für1min–24h
gerührt.DieAufarbeitungerfolgtanalogderAAV6.DieErgebnissesindinTabelle62aufgelistet.

7 ExperimentellerTeil
186
Tabelle62. KinetikderenzymatischenRacematspaltungvonrac‐4mitMethoxyessigsäureethylester(111).
Eintrag t[min] Umsatz[%] ee[%] E
1 1 16 72 7
2 5 17 74 8
3 10 17 76 9
4 20 16 77 9
5 30 18 79 9
6 45 19 78 10
7 60 18 78 10
8 120 24 79 11
9 180 19 72 7
10 270 22 92 31
11 480 27 77 10
12 1440 33 83 16
7.2.2.9 1‐Aminoindan(rac‐130)alsSubstrat
7.2.2.9.1 AnalysedesDiamidsN1,N3‐bis(2,3‐dihydro‐1H‐inden‐1‐yl)malonamid(129)
Abbildung145. BildungdesDiamids131alsNebenprodukt.
Bei derReaktiondes1‐Aminoindans (rac‐130)mitMalonsäurediethylester (9, 1Äq.) alsAcyldonor
analogderAAV6entstehtdasgewünschteIndan‐Amid(rac‐129,Abbildung145).AlsNebenprodukt
wirddabeidasentsprechendeDiamid131erhalten,welchesdurchSäulenchromatographie(Laufmittel:
CH/EtOAc1:4)isoliertwerdenkann.DasDiamid131wirdalsfarbloserFeststofferhalten.

7 ExperimentellerTeil
187
Umsatz:11%,Ausbeute:9%(29.9mg,0.09mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.89(m,2H,H‐2
+H‐22),2.57(m,2H,H‐1+H‐23),2.90(m,2H,H‐2+
H‐22), 3.02 (m, 2H,H‐1+H‐23), 3.27 (s, 2H,H‐12),
5.14(q,3J=7.8Hz,2H,H‐9+H‐15),7.21–7.31(m,8H,
Har),(t,3J=7.5Hz,2H,H‐10+H‐14).
13C‐NMR(500MHz,CDCl3):30.36(C‐2+C‐22),33.82
(C‐1+C‐23),43.31(C‐12),54.90(C‐9+C‐15),124.11
(C‐4+C‐20),124.89(C‐5+C‐19),126.86(C‐6+C‐18),
128.11(C‐7+C‐17),142.81(C‐8+C‐16),143.41(C‐3+C‐21),167.24(C‐11+C‐13).
MS(ESI):m/z(%)=357.2(100)[M+Na]+,691.2(30)[2M+Na]+.
EA(C21H22N2O2):berechnetC:75.42%,H:6.63%,N:8.38%;gefunden:C:74.27%,H:6.63%,N:8.08%.
DiespektroskopischenDatensindnichtliteraturbekannt.
7.2.2.9.2 BestimmungdesVerlustsvon1‐Aminoindan(rac‐130)währendderAufarbeitung
ZurBestimmungdesVerlustsvon1‐Aminoindan(rac‐130)währendderAufarbeitungwerden49.6mg
derSubstanzeingewogenundübereinenbestimmtenZeitraumbei550mbargerührt.DurchAuswaage
wirdderVerlustvonrac‐130bestimmt.DieErgebnissesindinTabelle63aufgelistet.
Tabelle63.BestimmungdesVerlustsvon1‐Aminoindan(rac‐130)währendderAufarbeitung.
Eintrag Zeit[min] Verlustrac‐130[mg] Verlustrac‐130[%]
1 0 0 0
2 5 0.33 0.7
3 10 1.12 2.3
4 15 1.44 2.9
7.2.2.9.3 Temperatur‐undZeiteffektderenzymatischenRacematspaltungvon
1‐Aminoindan(rac‐130)
Die enzymatischeRacematspaltungvon1‐Aminoindan (rac‐130)mitMalonsäurediethylester (9,1–
5Äq.)undCAL‐B(40mg/mmol)wurdeanalogderAAV6mitunterschiedlichenReaktionsbedingungen
untersucht.DieSyntheseerfolgteanalogderAAV6.DieErgebnissesindinTabelle64dargestellt.
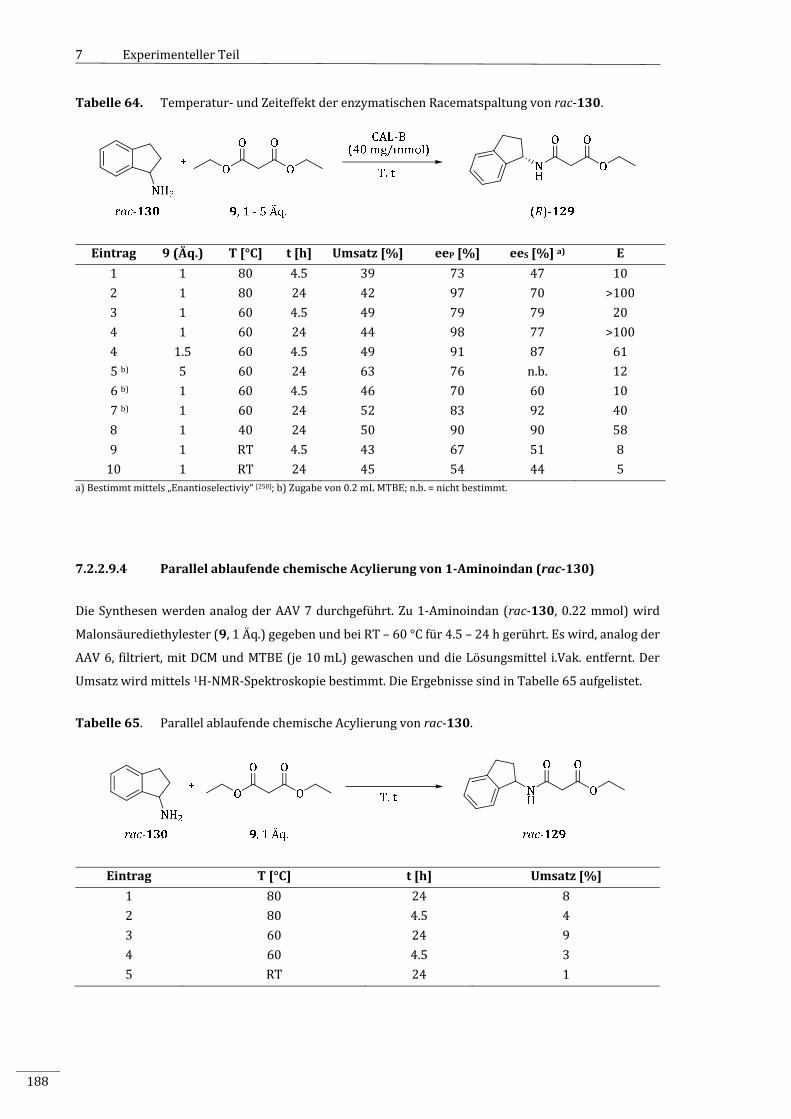
7 ExperimentellerTeil
188
Tabelle64. Temperatur‐undZeiteffektderenzymatischenRacematspaltungvonrac‐130.
Eintrag 9(Äq.) T[°C] t[h] Umsatz[%] eeP [%] eeS [%]a) E
1 1 80 4.5 39 73 47 10
2 1 80 24 42 97 70 >100
3 1 60 4.5 49 79 79 20
4 1 60 24 44 98 77 >100
4 1.5 60 4.5 49 91 87 61
5b) 5 60 24 63 76 n.b. 12
6b) 1 60 4.5 46 70 60 10
7b) 1 60 24 52 83 92 40
8 1 40 24 50 90 90 58
9 1 RT 4.5 43 67 51 8
10 1 RT 24 45 54 44 5a)Bestimmtmittels„Enantioselectiviy“[258];b)Zugabevon0.2mLMTBE;n.b.=nichtbestimmt.
7.2.2.9.4 ParallelablaufendechemischeAcylierungvon1‐Aminoindan(rac‐130)
DieSynthesenwerdenanalogderAAV7durchgeführt.Zu1‐Aminoindan(rac‐130,0.22mmol)wird
Malonsäurediethylester(9,1Äq.)gegebenundbeiRT–60°Cfür4.5–24hgerührt.Eswird,analogder
AAV6, filtriert,mitDCMundMTBE(je10mL)gewaschenunddieLösungsmittel i.Vak.entfernt.Der
Umsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.DieErgebnissesindinTabelle65aufgelistet.
Tabelle65. ParallelablaufendechemischeAcylierungvonrac‐130.
Eintrag T[°C] t[h] Umsatz[%]
1 80 24 8
2 80 4.5 4
3 60 24 9
4 60 4.5 3
5 RT 24 1

7 ExperimentellerTeil
189
7.2.2.10 SekundäreAminealsSubstrate
7.2.2.10.1 BestimmungdesVerlustsdersekundärenAmine149bzw.150
Zur Bestimmung des Verlusts von 2‐Methyl‐ bzw. 3‐Methylpiperidin (149 bzw. 150) während der
Aufarbeitung werden 43.7 bzw. 41.2 mg der Substanzen eingewogen und über einen bestimmten
Zeitraum bei 550 mbar gerührt. Durch Auswaage wird der Verlust der Substanzen bestimmt. Die
ErgebnissesindinTabelle66aufgelistet.
Tabelle66. BestimmungdesVerlustsdersekundärenAmine149und150.
Eintrag Amin Rührzeit[min] Verlust[mg] Verlust [%]
1 149 5 9.2 21
2 149 10 18.7 43
3 149 15 28.2 64
4 150 5 2.6 6
5 150 10 6.0 15
6 150 15 11.1 27
7.2.2.10.2 SynthesevonEthyl‐3‐(2‐methylpiperidin‐1‐yl)‐3‐oxopropanoat(rac‐151)
DieSyntheseerfolgtanalogderAAV5.[253]Zu2‐Methylpiperidin(149,1mmol,99.8mg)wirdDiethyl‐
Malonester (9, 1mmol, 161.2mg) gegebenund für 3 h bei 180°C erhitzt.DerUmsatzwirdmittels
1H‐NMR‐Spektroskopie bestimmt. Das Produkt rac‐151 wird mittels Säulenchromatographie (Lauf‐
mittel:EtOAc)isoliert.
Umsatz:56%,Ausbeute:41%(87.9mg,0.41mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):1.16(d,3J=7.0Hz,3H,H‐6),1.27
–1.30(t,3J=7.0Hz,3H,H‐11),1.35–1.47(m,2H,H‐3),1.52–1.69
(m,2H,H‐2),2.69(td,3J=13.5Hz,1H,H‐4),3.16(td,3J=13.5Hz,1H,
H‐4),3.43(s,2H,H‐8),3.48(m,1H,H‐5),4.19(q,3J=7.0Hz2H,H‐10),
4.49(m,1H,H‐1),4.90(m,1H,H‐1).
13C‐NMR (500MHz, CDCl3): 14.22 (C‐11), 18.66 (C‐6), 25.46 (C‐3),
26.13(C2),29.71(C‐4),36.68(C‐8),41.96(C‐1),61.41(C‐10),164.46
(C‐9),167‐96(C‐7).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm, flow: 0.8mL/min, tR=18.29 min (R),
19.68min(S),ee‐Wert:0%.

7 ExperimentellerTeil
190
IR (ATR): (cm‐1) = 2935.4, 2864.0, 2357.9, 1733.0, 1634.9, 1437.3, 1367.5, 1319.5, 1239.4, 1149.7,
1032.0,850.0,608.6.
MS(ESI):m/z(%)=236.0(100)[M+Na]+,277.1(10),449.2(23)[2M+Na]+.
EA(C14H17NO3):berechnetC:61.95%,H:8.98%,N:6.57%,O:22.50%;gefunden:C:61.08%,H:8.99%,
N:6.47%,O:23.46%.
DiespektroskopischenDatensindlautSciFinderinderLiteraturbeschriebenworden.[280]
7.2.2.10.3 SynthesevonEthyl‐3‐(3‐methylpiperidin‐1‐yl)‐3‐oxopropanoat(rac‐152)
Die Synthese erfolgt analog der AAV 5.[253] Zu 3‐Methylpiperidin (150, 1 mmol, 99.6 mg) wird
Malonsäurediethylester(9,1mmol,159.8mg)gegebenundfür3hbei180°Cerhitzt.DerUmsatzwird
mittels 1H‐NMR‐Spektroskopie bestimmt. Das Rohprodukt wird mittels Säulenchromatographie
(LaufmittelCH/EtOAc1:4)aufgereinigtumdasisolierteProduktzuerhalten.
Umsatz:63%,Ausbeute:42%(87.9mg,0.41mmol).
1H‐NMR(500MHz,CDCl3):δ(ppm):0.90(d,3J=6.5Hz,3H,H‐6),1.08
–1.18(m,1H,H‐11),1.40–1.73(m,2H,H‐3),1.80–1.84(m,1H,H‐2),
2.30(dd,3J=12.5Hz,10,5Hz,1H,H‐4),2.62(td,3J=12.5Hz,3.0Hz,
1H,H‐4),2.69(dd,3J=13.5Hz,10.5Hz,1H,H‐4),2.97–3.03(m,1H,
H‐8),3.55–3.66(m,1H,H‐5),4.38–4.45(m,1H,H‐1).
13C‐NMR (500MHz, CDCl3): 14.26 (C‐11), 18.93 (C‐6), 21.11 (C‐2),
24.74(C‐4),30.90(C‐3),32.89(C‐8),41.52(C‐1),54.00(C‐5),60.44
(C‐10),164.20(C‐7),171.19(C‐9).
HPLC:AD‐H‐Säule,95:5(n‐Hexan/i‐PrOH,v/v),210nm,flow:0.8mL/min,tR=19.11min(R),21.01min
(S),ee‐Wert:0%.
IR (ATR): (cm‐1) = 2929.0, 2852.3, 2360.1, 2341.4, 1733.0, 1603.5, 1440.1, 1366.8, 1316.8, 1266.2,
1224.5,1157.9,1139.5,1030.7,969.2,851.8,667.6,578.6.
MS(ESI):m/z(%)=214.0(5),236.1(100)[M+Na]+,277.1(3),449.2(97)[2M+Na]+.
DiespektroskopischenDatensindlautSciFinderinderLiteraturbeschriebenworden.[280]
7.2.2.10.4 EnzymatischeAcylierungvon3‐Methylpiperidin(rac‐150)
Abbildung146. EnzymatischeRacematspaltungvonrac‐150.

7 ExperimentellerTeil
191
Die Synthese erfolgt analog der AAV 6. Zu 3‐Methylpiperidin (150, 0.5 mmol, 47.4mg) werden
Malonsäurediethylester(9,0.5mmol,79.6mg)undCAL‐B(40mg/mmol,20mg)gegebenundbei60°C
für 24 h erhitzt. Die Aufarbeitung erfolgt analog zur AAV 6. Der Umsatz wird mittels 1H‐NMR‐
Spektroskopiebestimmt.
Umsatz:15%.
HPLC:AD‐H‐Säule,95:5(n‐Hexan/i‐PrOH,v/v),210nm,flow:0.8mL/min,tR=19.35min(R),21.27min
(S),ee‐Wert:10%.
DieanalytischenDatenstimmenmitdenVergleichsdatenausAbschnitt7.2.2.10.3überein.
7.2.2.10.5 ProzessoptimierungderenzymatischenRacematspaltungvon3‐Methylpiperidin
(rac‐150)
DieenzymatischeRacematspaltungvon3‐Methylpiperidin(rac‐150)mitMalonsäurediethylester(9,1
–5Äq.)undLipaseCAL‐B(40mg/mmol)wurdeanalogderAAV6mitunterschiedlichenReaktions‐
bedingungenuntersucht.DieSyntheseerfolgteanalogderAAV6.DerUmsatzwirdmittels 1H‐NMR‐
Spektroskopiebestimmt.DieErgebnissesindinTabelle67dargestellt.
Tabelle67. ProzessoptimierungderenzymatischenRacematspaltungvonrac‐150.
Eintrag MTBE[mL] T[°C] Umsatz[%] eeP [%] eeS[%]a) E
1 ‐ 80 5 10 n.b. <2
2 ‐ 60 15 10 n.b. 1
3 0.1 60 34 13 7 <2
4b) 0.1 60 75 18 54 2
5 ‐ 40 19 11 n.b. <2
6c) ‐ 40 22 10 n.b. <2a)Bestimmtmittels„Enantioselectiviy“[258];b)CAL‐B:80mg/mmol;c)Reaktionsdauer:48h,;n.b.=nichtbestimt.
7.2.2.10.6 ParallelablaufendechemischeAcylierungvonrac‐150
Die Synthesenwerden analog der AAV 6 durchgeführt. Zu 3‐Methylpiperidin (150, 0.5mmol)wird
Malonsäurediethylester(9,1Äq.)gegebenundbei40–80°Cfür24‐48hgerührt.DerUmsatzwird
ohneweitereAufarbeitungmittels1H‐NMR‐Spektroskopiebestimmt.DieErgebnissesindinTabelle68
aufgelistet.

7 ExperimentellerTeil
192
Tabelle68. ParallelablaufendechemischeAcylierungvonrac‐150.
Eintrag T[°C] t[h] Umsatz[%] ee[%] E
1 80 24 5 n.b. n.b.
2 60 24 9 <1 1
3 40 48 6 <1 1n.b.=nichtbestimmt.
7.2.2.11 Verwendungvonɛ–Caprolacton(3)alsAcyldonor
7.2.2.11.1 BestimmungderStabilitätvonɛ–Caprolacton(3)währendderReaktionund
Aufarbeitung
ɛ‐Caprolacton(3)wirdübereinenZeitraumvon24hbei60°CinAnwesenheitundohneCAL‐Bgerührt.
BeiAbwesenheitvonCAL‐BwirddirekteineNMR‐Probeentnommen.BeimAnsatzmitEnzymerfolgt
zusätzlich eine Aufarbeitung analog AAV 6. Das Verhältnis von ɛ–Caprolacton(3) zum Zersetzungs‐
produkt 6‐Hydroxyhenxansäure (54) wirdmittels 1H‐NMR‐Spektroskopie bestimmt. Die Ergebnisse
sindinTabelle69aufgelistet.
Tabelle69. BestimmungderStabilitätvonɛ–Caprolacton(3).
Eintrag Bedingungen Verhältnis3/54
1 RührenohneCAL‐B 100:0
2RührenmitCAL‐Bund
AufarbeitungnachAAV61:77
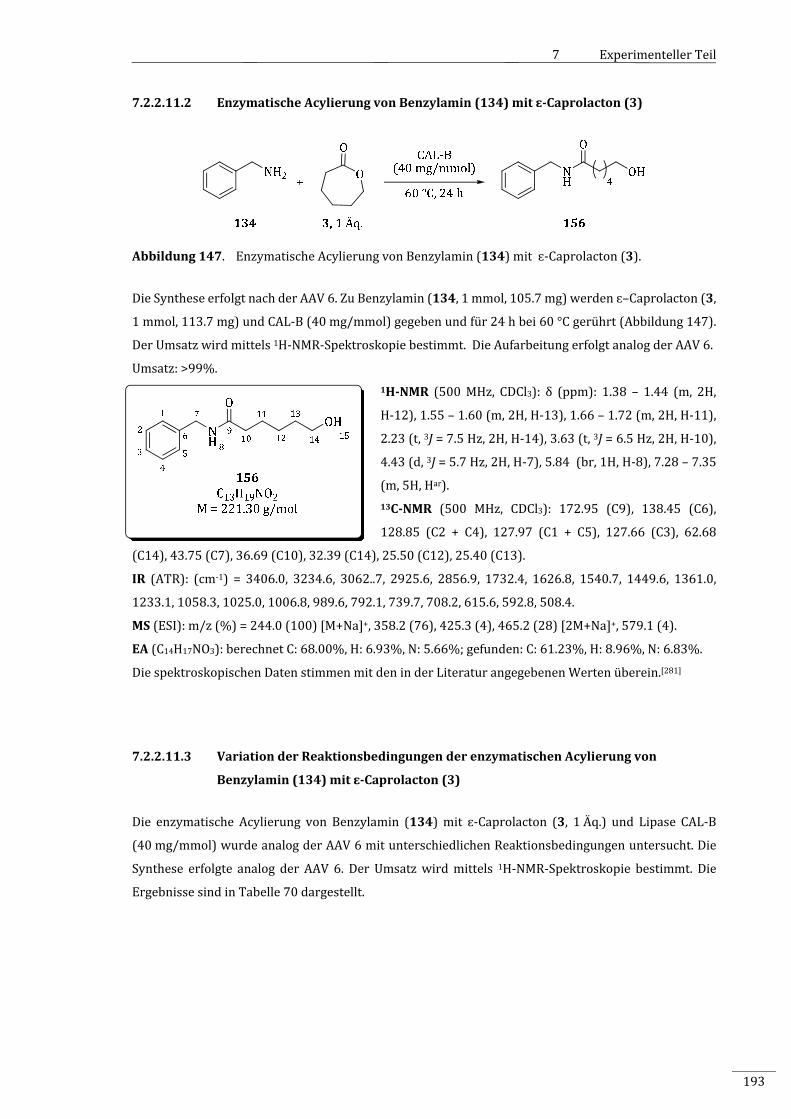
7 ExperimentellerTeil
193
7.2.2.11.2 EnzymatischeAcylierungvonBenzylamin(134)mitɛ‐Caprolacton(3)
Abbildung147. EnzymatischeAcylierungvonBenzylamin(134)mitɛ‐Caprolacton(3).
DieSyntheseerfolgtnachderAAV6.ZuBenzylamin(134,1mmol,105.7mg)werdenɛ–Caprolacton(3,
1mmol,113.7mg)undCAL‐B(40mg/mmol)gegebenundfür24hbei60°Cgerührt(Abbildung147).
DerUmsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.DieAufarbeitungerfolgtanalogderAAV6.
Umsatz:>99%.
1H‐NMR (500MHz, CDCl3): δ (ppm): 1.38 – 1.44 (m, 2H,
H‐12),1.55–1.60(m,2H,H‐13),1.66–1.72(m,2H,H‐11),
2.23(t,3J=7.5Hz,2H,H‐14),3.63(t,3J=6.5Hz,2H,H‐10),
4.43(d,3J=5.7Hz,2H,H‐7),5.84(br,1H,H‐8),7.28–7.35
(m,5H,Har).
13C‐NMR (500 MHz, CDCl3): 172.95 (C9), 138.45 (C6),
128.85 (C2 + C4), 127.97 (C1 + C5), 127.66 (C3), 62.68
(C14),43.75(C7),36.69(C10),32.39(C14),25.50(C12),25.40(C13).
IR (ATR): (cm‐1) = 3406.0, 3234.6, 3062..7, 2925.6, 2856.9, 1732.4, 1626.8, 1540.7, 1449.6, 1361.0,
1233.1,1058.3,1025.0,1006.8,989.6,792.1,739.7,708.2,615.6,592.8,508.4.
MS(ESI):m/z(%)=244.0(100)[M+Na]+,358.2(76),425.3(4),465.2(28)[2M+Na]+,579.1(4).
EA(C14H17NO3):berechnetC:68.00%,H:6.93%,N:5.66%;gefunden:C:61.23%,H:8.96%,N:6.83%.
DiespektroskopischenDatenstimmenmitdeninderLiteraturangegebenenWertenüberein.[281]
7.2.2.11.3 VariationderReaktionsbedingungenderenzymatischenAcylierungvon
Benzylamin(134)mitɛ‐Caprolacton(3)
Die enzymatische Acylierung von Benzylamin (134)mit ɛ‐Caprolacton (3,1Äq.) und Lipase CAL‐B
(40mg/mmol)wurdeanalogderAAV6mitunterschiedlichenReaktionsbedingungenuntersucht.Die
Synthese erfolgte analog der AAV 6. Der Umsatzwirdmittels 1H‐NMR‐Spektroskopie bestimmt. Die
ErgebnissesindinTabelle70dargestellt.

7 ExperimentellerTeil
194
Tabelle70. ProzessoptimierungderenzymatischenRacematspaltungvonBenzylamin (134)mitɛ‐Caprolacton(3).
Eintrag MTBE[mL] T[°C] t[h] Umsatz[%]
1 0 80 4.5 51
2 0.5 80 4.5 65
3 0 RT 4.5 57
7.2.2.11.4 ParallelablaufendechemischeAcylierungvonBenzylamin(134)mit
ɛ‐Caprolacton(3)
DieSynthesenwerdenanalogderAAV7durchgeführt.ZuBenzylamin(134,0.5mmol)wirdɛ‐Capro‐
lacton (3, 1 Äq.) gegeben und bei verschiedenen Temperaturen und Reaktionszeiten gerührt. Der
UmsatzwirdohneweitereAufarbeitungmittels1H‐NMR‐Spektroskopiebestimmt.DieErgebnissesind
inTabelle71aufgelistet.
Tabelle71. ParallelablaufendeAcylierungvonBenzylamin(134)mitɛ–Caprolacton(3).
Eintrag T[°C] t[h] Umsatz[%]
1 80 4.5 30
2 60 4.5 69
3 RT 4.5 12
4 RT 24 53
5 0 4.5 3

7 ExperimentellerTeil
195
7.2.2.11.5 Synthesevon6‐Hydroxy‐N‐(1‐phenylethyl)hexanamid(157)
Abbildung148. Synthesevon157ausgehendvonrac‐4.[253]
Die Synthese erfolgt analog der AAV 5.[253] Zu 1‐Phenylethylamin (rac‐4, 2.5mmol, 303.4mg)wird
ɛ‐Caprolacton (3,2.5mmol,288.1mg)gegebenundfür4hauf180°Cerhitzt(Abbildung148).Der
UmsatzwirdohneweitereAufarbeitungmittels1H‐NMR‐Spektroskopiebestimmt.
Umsatz:62%.
1H‐NMR (500MHz,CDCl3): δ (ppm):1.34–1.42 (m,3H,
H‐13), 1.49 (d, 3J = 6.9Hz, 2H,H‐9), 1.54 – 1.60 (m,2H,
H‐12), 1.61 – 1.71 (m, 2H,H‐14), 2.19 (t, 3J=7.4Hz, 2H,
H‐15),3.63(t,3J=6.6Hz,2H,H‐11),5.14(p,3J=7.2Hz,1H,
H‐7),5.70(br,1H,H‐8),7.25–7.35(m,5H,Har).
HPLC: AD‐H‐Säule, 95:5 (n‐Hexan/i‐PrOH, v/v), 210 nm,
flow: 0.8 mL/min, tR=32.21 min (R), 39.69 min (S),
ee‐Wert:0%.
DiespektroskopischenDatensindinderLiteraturnichtbekannt.
7.2.2.11.6 EnzymatischeAcylierungvon1‐Phenylethylamin(rac‐4)mitɛ‐Caprolacton(3)
Abbildung149. EnzymatischeAcylierungvonrac‐4mitɛ–Caprolacton(3).
Die Synthese erfolgt analog der AAV 6. Zu 1‐Phenylethylamin (rac‐4, 5.0 mmol, 0.61 g) werden
ɛ‐Caprolacton(3,4.9mmol,0.56g)undCAL‐B(40mg/mmol,200mg)gegebenundfür4.5hbei80°C
gerührt (Abbildung 149). Die Aufarbeitung erfolgt analogAAV 6. DerUmsatzwirdmittels 1H‐NMR‐
Spektroskopiebestimmt.DieAufreinigungerfolgtmittelsSäulenchromatographie(Laufmittel:CHCl3/
Aceton7:3).DasProdukt157wirdalsfarbloserFeststofferhalten.
Umsatz:51%,Ausbeute:43%(0.51g,2.15mmol).
HN
O
157C14H21NO2
M = 235.33 g/mol
1
2
3
4
5
6
7
8
9
1011
12
13
14OH
1516

7 ExperimentellerTeil
196
1H‐NMR (500MHz,CDCl3):δ (ppm):1.36–1.42 (m,3H,
H‐13),1.49(d,3J=6.9Hz,2H,H‐9),1.54–1.60
(m,2H,H‐12),1.64–1.70(m,2H,H‐14),2.19(t,3J=7.4Hz,
2H, H‐15), 3.63 (t, 3J=6.5Hz, 2H, H‐11), 5.14 (p,
3J=7.2Hz,1H,H‐7),5.73(br,1H,H‐8),7.25–7.35(m,5H,
Har).
13C‐NMR (500 MHz, CDCl3): 21.84 (C‐9), 25.36 (C‐13),
25.42(C‐12),32.33(C‐14),36.70(C‐11),48.73(C‐7),62.50
(C‐15),126.26(C‐1+C‐5),127.41(C‐3),128.73(C‐2+C‐4),143.38(C‐6),172.27(C‐10).
HPLC:AD‐H‐Säule,95:5(n‐Hexan/i‐PrOH,v/v),210nm,flow:0.8mL/min,tR=32.21min(R),39.69min
(S),ee‐Wert:0%.
IR (ATR): (cm‐1) = 3270.5, 3062.7, 2929.3, 2859.9, 2360.1, 2341.3, 1731.2, 1540.1, 1448.9, 1374.9,
1277.9,1209.4,1129.5,1051.0,1020.7,908.7,759.6,703.5,539.7.
MS(ESI):m/z(%)=258.1(100)[M+Na]+,493.2(47)[2M+Na]+.
EA(C14H21NO2):berechnetC:70.46%,H:9.00%,N:5.95%,O:16.60%;gefunden:C:70.01%,H:9.27%,
N:5.99%,O:14.73%.
DiespektroskopischenDatenstimmenmitdeneninAbschnitt7.2.2.11.5überein.
7.2.2.11.7 TemperaturabhängigkeitderenzymatischenAcylierungvon1‐Phenylethylamin
(rac‐4)mitɛ–Caprolacton(3)
DieenzymatischeAcylierungvon1‐Phenylethylamin(rac‐4,1mmol)mitɛ‐Caprolacton(3,1Äq.)und
CAL‐B(40mg/mmol)wurdeanalogderAAV6mitunterschiedlichenReaktionsbedingungenuntersucht.
DieSyntheseerfolgteanalogderAAV6.DerUmsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.Die
ErgebnissesindinTabelle72dargestellt.
Tabelle72. ProzessoptimierungderenzymatischenAcylierungvonrac‐4mitɛ‐Caprolacton(3).
Eintrag T[°C] t[h] Umsatz[%] ee[%] E
1 80 4.5 51 0 <1
2a) 80 4.5 49 0 <1
3 60 4.5 41 0 <1
4 60 6 38 0 <1
5 60 8 45 0 <1
6 40 4.5 25 3 1
7 RT 4.5 26 4 1a)Zugabevon0.5mLMTBEnach1h.
HN
O
157C14H21NO2
M = 235.33 g/mol
1
2
3
4
5
6
7
8
9
1011
12
13
14OH
1516

7 ExperimentellerTeil
197
7.2.2.11.8 ParallelablaufendechemischeAcylierungvonrac‐4mitɛ‐Caprolacton(3)
Die Synthesenwerden analog der AAV 6 durchgeführt. Zu 1‐Phenylethylamin (rac‐4, 1mmol)wird
ɛ‐Caprolacton(3,1Äq.)gegebenundbei0–80°C für4.5hgerührt.DerUmsatzwirdohneweitere
Aufarbeitungmittels1H‐NMR‐Spektroskopiebestimmt.DieErgebnissesindinTabelle73aufgelistet.
Tabelle73. ParallelablaufendechemischeAcylierungvonrac‐4mitɛ‐Caprolacton(3).
Eintrag T[°C] Umsatz[%] ee[%]
1 80 37 0
2 60 4 n.b.
3 40 2 n.b.
4 RT 1 n.b.
5 0 0 n.b.n.b.=nichtbestimmt.
7.2.2.11.9 ReaktionskinetikderAcylierungvon1‐Phenylethylamin(rac‐4)mit
ɛ‐Caprolacton(3)
DieSynthesenerfolgenanalogderAAV11.ZuPhenylethylamin(rac‐4,0.5mmol)werdenɛ‐Caprolacton
(3,1Äq.)undCAL‐B(40mg/mmol)gegebenundbei60°Cfür1min–24hgerührt.DieAufarbeitung
erfolgtanalogderAAV7.DieErgebnissesindinTabelle74aufgelistet.
Tabelle74. KinetikderenzymatischenAcylierungvonrac‐4mitɛ‐Caprolacton(3).
Eintrag t[min] Umsatz[%] ee[%] E
2 5 2 n.b. n.b.
3 10 3 n.b. n.b.
4 20 7 0 0
5 30 6 <2 1
6 45 7 0 0
7 60 12 0 0

7 ExperimentellerTeil
198
Eintrag t[min] Umsatz[%] ee[%] E
8 120 20 0 1
9 180 22 0 1
10 270 41 0 1
11 360 38 0 1
11 480 45 0 1
12 1440 65 0 1n.b.=nichtbestimmt.
7.2.2.11.10 Synthesevon6‐Hydroxy‐1‐(3‐methylpiperidin‐1‐yl)hexan‐1‐on(155)
Abbildung150. ChemischeAcylierungvon3‐Methylpiperidin(150)mitɛ‐Caprolacton(3).[253]
DieSyntheseerfolgtanalogderAAV6.[253]Zu3‐Methylpiperidin(150,1mmol,99.6mg)wirdɛ‐Capro‐
lacton(3,1mmol,115.5mg)gegebenundfür4hbei180°Cgerührt(Abbildung150).DieAufarbeitung
erfolgtanalogAAV6.DerUmsatzwirdmittels1H‐NMR‐Spektroskopiebestimmt.
Umsatz:17%.
1H‐NMR (500MHz, CDCl3): δ (ppm): 0.90 (d, 3H, 3J=6.7Hz,
H‐6),1.07–1.16(m,3H,H‐10),1.34–1.46(m,H2,H‐3),1.51–
1.71(m,6H,H‐9,H‐11,H2),2.21(t,3J=12.1Hz,2H,H‐8),2.52
– 2.59 (m, 2H, H‐5), 2.93 (t, 3J=12.5Hz, 2H, H‐4), 3.73 (d,
3J=13.6Hz,2H,H‐1),4.39–4.44(m,2H,H‐12).
13C‐NMR (500 MHz, CDCl3): 19.00 (C‐6), 22.45 (C‐2), 24.50
(C‐10), 25.49 (C‐9), 28.31 (C‐4), 31.56 (C‐11), 31.83 (C‐3),
34.03(C‐8),46.61(C‐1),52.31(C‐5),62.05(C‐12).
DC‐MS(ESI):m/z(%)=214.1(99)[M]+,236.2(40)[M+Na]+,427.2(20)[2M]+,449.2(1)[2M+Na]+.
MS(ESI):m/z(%)=236.1(9)[M+Na]+,350.3(100),464.3(59)[2M+K]+,578.3(10).
DiespektroskopischenDatensindinderLiteraturnichtbekannt.
7.2.2.11.11 EnzymatischeAcylierungensekundärerAminemitɛ‐Caprolacton(3)
DieSynthesenerfolgenanalogderAAV6.ZudemAmin(149bzw.150,0.5mmol)werdenɛ‐Caprolacton
(3,0.5mmol,57.3mg)undCAL‐B(40mg/mmol,20mg)gegebenundinMTBE(0‐0.1mL)für24hbei
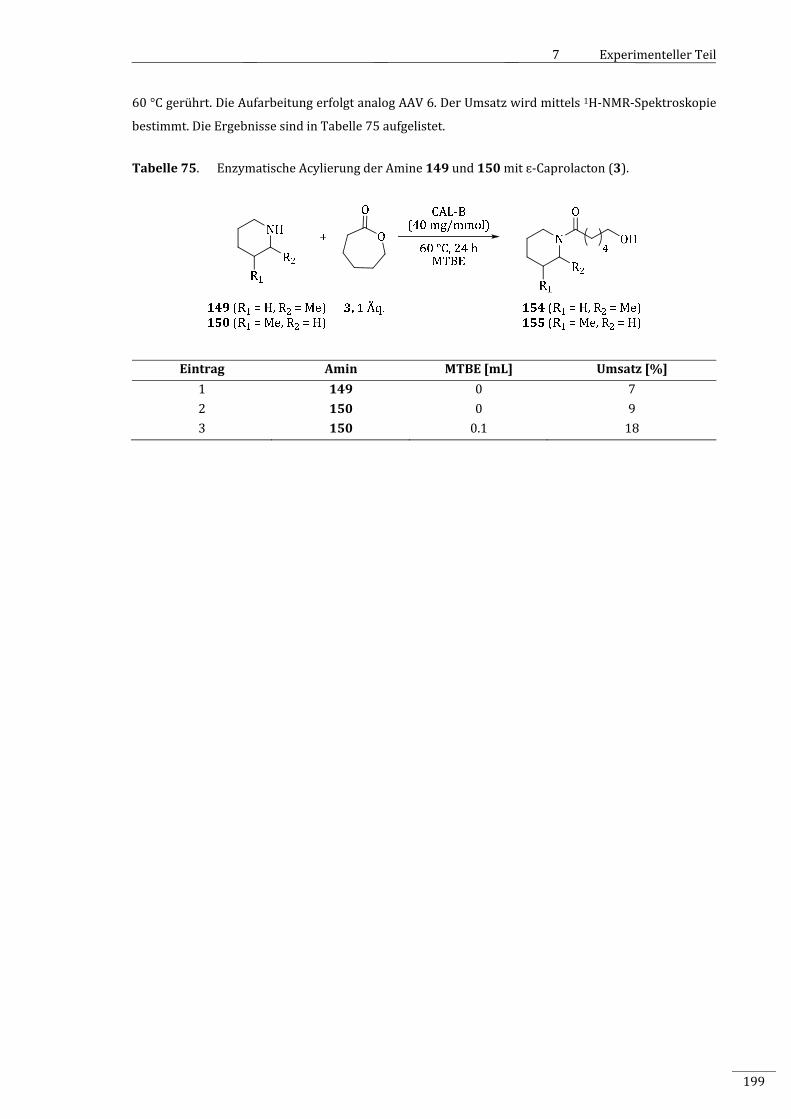
7 ExperimentellerTeil
199
60°Cgerührt.DieAufarbeitungerfolgtanalogAAV6.DerUmsatzwirdmittels1H‐NMR‐Spektroskopie
bestimmt.DieErgebnissesindinTabelle75aufgelistet.
Tabelle75. EnzymatischeAcylierungderAmine149und150mitɛ‐Caprolacton(3).
Eintrag Amin MTBE[mL] Umsatz[%]
1 149 0 7
2 150 0 9
3 150 0.1 18

200

8 Literaturverzeichnis
201
8 Literaturverzeichnis
[1] M.N.Gupta,S.Raghava,Chem.Centr.J.2007,1,17.
[2] a)BundesministeriumfürForschungundBildung(BMBF),WeißeBiotechnologie.Chancenfür
einebio‐basierteWirtschaft,Berlin,2012;b)E.Flaschel,PositionspapierderDECHEMAe.V.
WeißeBiotechnologie:ChancenfürDeutschland,FrankfurtamMain,2004.
[3] UnitedStatesDepartmentofAgriculture,2008.
[4] M.Breuer,K.Ditrich,T.Habicher,B.Hauer,M.Kesseler,R.Stürmer,T.Zelinski,Angew.Chem.Int.
Ed.2004,43,788–824.
[5] a)A.G.McDonald,K.F.Tipton,FEBSJ.2014,281,583–592;b)D.Schomburg,"BRENDA.The
ComprehensiveEnzymeInformationSystem",zufindenunterhttp://www.brenda‐
enzymes.org/.
[6] S.L.Neidleman,Biotechnol.Gen.Engin.Rev.1984,1–38.
[7] A.Illanes(Hrsg.)EnzymeBiocatalysis.PrinciplesandApplications,SpringerScience+Business
MediaB.V.,Heidelberg,2008.
[8] J.B.Jones,Tetrahedron1986,42,3351–3403.
[9] F.M.Menger,Acc.Chem.Res.1993,206–212.
[10] K.Drauz,H.Waldmann(Hrsg.)EnzymeCatalysisinOrganicSynthesis.AComprehensive
Handboook,Wiley‐VCHVerlagGmbH,Weinheim,2002.
[11] K.Faber,Biotransformationsinorganicchemistry.Atextbook,6.Aufl.,Springer‐Verlag,Berlin,
Heidelberg,2011.
[12] Z.‐G.Zhang,L.P.Parra,M.T.Reetz,Chem.Eur.J.2012,18,10160–10172.
[13] D.Brady,J.Jordaan,Biotechnol.Lett.2009,31,1639–1650.
[14] H.Gröger,F.Chamouleau,N.Orologas,C.Rollmann,K.Drauz,W.Hummel,A.Weckbecker,O.
May,Angew.Chem.Int.Ed.2006,45,5677–5681.
[15] F.Zambianchi,S.Raimondi,P.Pasta,G.Carrea,N.Gaggero,J.M.Woodley,J.Mol.Catal.B:Enzym.
2004,31,165–171.
[16] W.‐H.Lee,J.‐B.Park,K.Park,M.‐D.Kim,J.‐H.Seo,Appl.Microbiol.Biotechnol.2007,76,329–338.
[17] M.A.Woodruff,D.W.Hutmacher,Prog.Pol.Sci.2010,35,1217–1256.
[18] D.Zhu,L.Hua2009,4,1420–1431.
[19] G.A.R.Nobes,R.J.Kazlauskas,R.H.Marchessault,Macromol.1996,4829–4833.
[20] SIDSInitialAssessmentReportForSIAM19.E‐Caprolactone.UNEPPublication,Berlin,2004.
[21] a)A.R.Pohlmann,F.N.Fonseca,K.Paese,C.B.Detoni,K.Coradini,R.C.R.Beck,S.S.Guterres,
ExpertOpin.DrugDeliv.2013,10,623–638;b)T.K.Dash,V.B.Konkimalla,J.Contr.Rel.2012,
158,15–33.
[22] T.Fujiwa(DaicelChemicalIndustriesLtd.),5,116,932,1990.
[23] K.Weissermel,H.‐J.Arpe(Hrsg.)IndustrialOrganicChemistry,VCHWileyVerlagsgesellschaft
mbH,Weinheim,1997.

8 Literaturverzeichnis
202
[24] BASF,Presse‐Information.BASFerhöhtPreisefürPolyamid6Basis‐Polymere,Caprolactamund
AdipinsäureinEuropa,Ludwigshafen,2014.
[25] A.J.Willetts,C.J.Knowles,M.S.Levitt,S.M.Roberts,H.Sandey,N.F.Shipston,J.Chem.Soc.,
PerkinTrans.11991,1608–1610.
[26] S.Staudt,U.T.Bornscheuer,U.Menyes,W.Hummel,H.Gröger,EnzymeMicrob.Technol.2013,
53,288–292.
[27] I.Alfonso,V.Gotor,Chem.Soc.Rev.2004,33,201–209.
[28] V.Gotor‐Fernández,E.Busto,V.Gotor,Adv.Synth.Catal.2006,348,797–812.
[29] F.vanRantwijk,R.A.Sheldon,Tetrahedron2004,60,501–519.
[30] K.Ditrich,Synthesis2008,14,2283–2287.
[31] M.Nechab,N.Azzi,N.Vanthuyne,M.Bertrand,S.Gastaldi,G.Gil,J.Org.Chem.2007,72,6918–
6923.
[32] a)F.Balkenhohl,B.Hauer,W.Ladner,U.Pressler(BASFSE),DE4332738A1,25.09.93;b)K.
Ditrich,F.Balkenhohl,W.Ladner(BASFSE),DE19534208A1,1995.
[33] S.Simon,Dissertation,Friedrich‐Alexander‐Universität,Erlangen‐Nürnberg,2011.
[34] S.Simon,S.Oßwald,J.Roos,H.Gröger,Z.Naturforsch.2012,67,1123–1126.
[35] F.Uthoff,Bachelorarbeit,UniversitätBielefeld,Bielefeld,2012.
[36] M.Päiviö,P.Perkiö,L.T.Kanerva,Tetrahed.Asymm.2012,23,230–236.
[37] A.Ghanem,Tetrahedron2007,63,1721–1754.
[38] A.Baeyer,V.Villiger,Ber.Dtsch.Chem.Ges.1899,32,3625–3633.
[39] H.Caro,Angew.Chem.1898,36,815–846.
[40] M.Renz,B.Meunier,Eur.J.Org.Chem.1999,737–750.
[41] L.Ruzicka,M.Stoll,Helv.Chim.Acta1928,1159–1173.
[42] a)W.Dilthey,M.Inckel,H.Stephan,J.Prakt.Chem.1939,219–237;b)R.Robinson,L.HartSmith,
J.Chem.Soc.1937,371–374.
[43] G.Wittig,G.Pieper,Ber.Dtsch.Chem.Ges.1940,73,295–297.
[44] R.Criegee,JustusLiebigsAnn.Chem.1948,127–135.
[45] W.vonE.Doering,E.Dorfman,J.Am.Chem.Soc.1953,75,5595–5598.
[46] G.R.Krow,Org.React.1993,43,251–353.
[47] G.R.Krow,Tetrahedron1981,37,2697–2724.
[48] R.B.Turner,J.Am.Chem.Soc.1950,72,878–882.
[49] J.A.Berson,S.Suzuki,J.Am.Chem.Soc.1959,4088–4094.
[50] K.Mislow,J.Brenner,J.Am.Chem.Soc.1953,75,2318–2322.
[51] G.Strukul,Angew.Chem.1998,1256–1267.
[52] G.Strukul,Angew.Chem.Int.Ed.1998,1198–1209.
[53] S.E.Jacobson,R.Tang,F.Mares,J.Chem.Soc.Chem.Commun.1978,888–889.
[54] W.A.Herrmann,R.W.Fischer,J.D.G.,Correira,J.Molecul.Cat.1994,94,213–223.
[55] R.Bernini,A.Coratti,G.Fabrizi,A.Goggiamani,Tetrahed.Lett.2003,44,8991–8994.
[56] A.M.F.Philips,C.Romao,Eur.J.Org.Chem.1999,1767–1770.

8 Literaturverzeichnis
203
[57] a)G.Strukul,A.Varagnolo,F.Pinna,J.Mol.Cat.A:Chem.1997,413–423;b)M.DelTedesco
Frisone,F.Pinna,G.Strukul,Organometallics1993,148–156;c)A.Cavarzan,A.Scarso,P.
Sgarbossa,R.A.Michelin,G.Strukul,ChemCatChem2010,2,1296–1302.
[58] a)L.Syper,Synthesis1989,167–172;b)L.Yu,Y.Wu,H.Cao,X.Zhang,X.Shi,J.Luan,T.Chen,Y.
Pan,Q.Xu,GreenChem.2014,16,287–293.
[59] A.Bhaumik,P.Kumar,R.Kumar,Catal.Lett.1996,40,47–50.
[60] M.Sasidharan,S.Suresh,A.Sudalai,Tetrahed.Lett.1995,36,9071–9072.
[61] T.Yamada,T.Takai,O.Rhode,T.Mukaiyama,Tetrahed.Lett.1991,1–4.
[62] a)T.Yamada,K.Takahashi,K.Kato,T.Takai,S.Inoki,T.Mukaiyama,Chem.Lett.1991,20,641–
644;b)I.C.Chisem,J.Chisem,J.S.Rafelt,D.J.Macquarrie,J.H.Clark,K.A.Utting,J.Chem.
Technol.Biotechnol.1999,923–930.
[63] C.Bolm,G.Schlingloff,K.Weickhardt,Tetrahed.Lett.1993,34,3405–3408.
[64] Y.‐y.Yan,L.‐m.Dong,J.‐p.Guo,M.‐y.Huang,Y.‐y.Jiang,J.Macromol.Sci.,PartA1997,34,1097–
1104.
[65] S.‐I.Murahashi,Y.Oda,T.Naota,Tetrahed.Lett.1992,33,7557–7560.
[66] K.Kaneda,S.Ueno,T.Imanaka,E.Shimotsuma,Y.Nishiyama,Y.Ishii,J.Org.Chem.1994,59,
2915–2917.
[67] H.Subramanian,E.G.Nettleton,S.Budhi,R.T.Koodali,J.Mol.Cat.A:Chem.2010,330,66–72.
[68] E.‐Y.Jeong,M.B.Ansari,S.‐E.Park,ACSCatal.2011,855–863.
[69] a)A.Corma,L.T.Nemeth,M.Renz,S.Valencia,Nature2001,423–425;b)M.Renz,T.Blasco,A.
Corma,V.Fornés,R.Jensen,L.T.Nemeth,Chem.Eur.J.2002,8,4708–4717.
[70] S.P.Panchgalle,U.R.Kalkote,P.S.Niphadkar,P.N.Joshi,S.P.Chavan,G.M.Chaphekar,Green
Chem.2004,6,308.
[71] a)T.Hara,M.Hatakeyama,A.Kim,N.Ichikuni,S.Shimazu,GreenChem.2012,14,771;b)Z.Lei,
Q.Zhang,J.Luo,X.He,Tetrahed.Lett.2005,46,3505–3508;c)C.Li,Z.Lei,H.Ma,S.Wu,Q.Sun,J.
Disp.Sci.Technol.2012,33,983–989.
[72] a)K.Kaneda,S.Ueno,T.Imanaka,J.Chem.Soc.Chem.Commun.1994,787–789;b)K.Kaneda,T.
Yamashita,Tetrahed.Lett.1996,37,4555–4558;c)C.Chen,J.Peng,B.Li,L.Wang,Catal.Lett.
2009,131,618–623.
[73] M.Hamamoto,K.Nakayama,Y.Nishiyama,Y.Ishii,J.Org.Chem.1993,58.
[74] a)C.Palazzi,F.Pinna,G.Strukul,J.Mol.Cat.A:Chemical2000,245–252;b)J.Fischer,W.F.
Hölderich,Appl.Catal.A:General1999,435–443.
[75] H.‐J.Xu,F.‐F.Zhu,Y.‐Y.Shen,X.Wan,Y.‐S.Feng,Tetrahedron2012,68,4145–4151.
[76] a)W.Adam,A.Rodríguez,J.Org.Chem.1979,26,4969–4970;b)M.Suzuki,H.Takada,R.Noyori,
J.Org.Chem.1982,47,902–904.
[77] S.Baj,A.Chrobok,R.Słupska,GreenChem.2009,11,279.
[78] N.O.Mahmoodi,S.Z.D.Heirati,K.Ekhlasi‐Kazaj,J.IranChem.Soc.2012,9,521–528.
[79] R.Mello,A.Olmos,J.Parra‐Carbonell,M.E.González‐Núñez,G.Asensio,GreenChem.2009,11,
994.
[80] S.Baj,A.Chrobok,A.Siewniak,Appl.Catal.A:General2011,395,49–52.

8 Literaturverzeichnis
204
[81] X.‐H.Li,X.‐G.Meng,Y.Liu,X.Peng,GreenChem.2013,15,3332.
[82] K.Matuszek,P.Zawadzki,W.Czardybon,A.Chrobok,NewJ.Chem.2013,38,237.
[83] S.L.Jain,B.Sain,Appl.Catal.A:General2006,301,259–264.
[84] B.H.Brodsky,J.DuBois,J.Am.Chem.Soc.2005,127,15391–15393.
[85] A.Gusso,C.Baccin,F.Pinna,G.Strukul,Organometallics1994,3442–3451.
[86] a)G.Bianchini,A.Cavarzan,A.Scarso,G.Strukul,GreenChem.2009,11,1517;b)A.Cavarzan,G.
Bianchini,P.Sgarbossa,L.Lefort,S.Gladiali,A.Scarso,G.Strukul,Chem.Eur.J.2009,15,7930–
7939.
[87] T.Uchida,T.Katsuki,Tetrahed.Lett.2001,42,6911–6914.
[88] a)S.Xu,Z.Wang,X.Zhang,X.Zhang,K.Ding,Angew.Chem.Int.Ed.2008,47,2840–2843;b)S.
Xu,Z.Wang,X.Zhang,X.Zhang,K.Ding,Angew.Chem.2008,120,2882–2885;c)B.Wang,Y.‐M.
Shen,Y.Shi,J.Org.Chem.2006,71,9519–9521;d)S.‐I.Murahashi,S.Ono,Y.Imada,Angew.
Chem.2002,114,2472–2474;e)S.‐I.Murahashi,S.Ono,Y.Imada,Angew.Chem.Int.Ed.2002,
41,2366–2368.
[89] C.Bolm,K.K.Luong,G.Schlingloff,Synlett1997,1151–1152.
[90] a)A.V.Malkov,F.Friscourt,M.Bell,M.E.Swarbrick,P.Kočovský,J.Org.Chem.2008,73,3996–
4003;b)K.S.Petersen,B.M.Stoltz,Tetrahedron2011,67,4352–4357;c)K.Ito,A.Ishii,T.
Kuroda,T.Katsuki,Synlett2003,643–646.
[91] C.Paneghetti,R.Gavagnin,F.Pinna,G.Strukul,Organometallics1999,18,5057–5065.
[92] a)A.Wanatabe,T.Uchida,R.Irie,T.Katsuki,PNAS2004,101,5737–5742;b)C.Bolm,O.
Beckmann,Chirality2000,12,523–525.
[93] K.Matsumoto,A.Watanabe,T.Uchida,K.Ogi,T.Katsuki,Tetrahed.Lett.2004,45,2385–2388.
[94] C.Bolm,O.Beckmann,A.Cosp,C.Palazzi,Synlett2001,1461–1463.
[95] a)J.‐C.Frison,C.Palazzi,C.Bolm,Tetrahedron2006,62,6700–6706;b)C.Bolm,O.Beckmann,C.
Palazzi,Can.J.Chem.2001,79,1593–1597.
[96] S.C.Lemoult,P.F.Richardson,S.M.Roberts,J.Chem.Soc.PerkinTrans.11995,89–91.
[97] M.Y.Ríos,E.Salazar,H.F.Olivo,GreenChem.2007,9,459–462.
[98] O.Kirk,M.W.Christensen,T.Damhus,S.E.Godtfredsen,Biocatalysis1994,11,65–77.
[99] B.K.Pchelka,M.Gelo‐Pujic,E.Guibé‐Jampel,J.Chem.Soc.PerkinTrans.11998,2625–2628.
[100] N.A.Donoghue,D.B.Norris,P.W.Trudgill,Eur.J.Biochem.1976,175–192.
[101] S.Colonna,N.Gaggero,G.Carrea,G.Ottolina,P.Pasta,F.Zambianchi,Tetrahed.Lett.2002,43,
1797–1799.
[102] a)D.R.Light,D.J.Waxman,C.T.Walsh,Biochemistry1982,2490–2498;b)G.Carrea,B.
Redigolo,S.Riva,S.Colonna,N.Gaggero,E.Battistel,D.Bianchi,Tetrahedron:Asymmetry1992,
1063–1068;c)S.Colonna,N.Gaggero,P.Pasta,G.Ottolina,Chem.Commun.1996,2303–2307;d)
J.Beecher,P.Richardson,A.Wiletta,Biotechnol.Lett.1994,16,909–912;e)D.R.Kelly,C.J.
Knowles,J.G.Mahdi,I.N.Taylor,M.A.Wright,Tetrahedron:Asymmetry1996,7,365–368.
[103] B.P.Branchaud,C.T.Walsh,J.Am.Chem.Soc.1985,107,2153–2161.
[104] G.Ottolina,S.Bianchi,B.Belloni,G.Carrea,B.Danieli,Tetrahed.Lett.1999,40,8483–8486.
[105] C.T.Walsh,J.A.Latham,J.Chem.Soc.Chem.Commun.1986,527–528.

8 Literaturverzeichnis
205
[106] J.A.Letham,B.P.Brenchaud,Y.‐C.J.Chen,C.T.Walsh,J.Chem.Soc.Chem.Commun.1986,528–
530.
[107] M.W.Fraaije,N.M.Kamerbeek,Berkel,WillemJ.H.van,D.B.Janssen,FEBSLett.2002,43–47.
[108] N.M.Kamerbeek,D.B.Janssen,W.J.H.vanBerkel,M.W.Fraaije,Adv.Synth.Cat.2003,345,
667–678.
[109] C.T.Walsh,Y.‐C.J.Chen,Angew.Chem.1988,100,342–352.
[110] C.T.Walsh,Y.‐C.J.Chen,Angew.Chem.Int.Ed.1988,27,333–343.
[111] M.P.Beam,M.A.Bosserman,N.Noinaj,M.Wehenkel,J.Rohr,Biochemistry2009,48,4476–
4487.
[112] G.deGonzalo,M.D.Mihovilovic,M.W.Fraaije,ChemBioChem2010,11,2208–2231.
[113] G.E.Turfitt,Biochem.J.1948,376–383.
[114] J.Fried,R.W.Thoma,A.Klingsberg,J.Am.Chem.Soc.1953,5764–5765.
[115] G.E.Peterson,R.W.Thoma,D.Perlman,J.Fried,J.Bacteriol.1957,684–688.
[116] H.Leisch,K.Morley,P.C.K.Lau,Chem.Rev.2011,111,4165–4222.
[117] R.L.Prairie,P.Talaly,Biochemistry1963,203–208.
[118] M.A.Rahim,C.J.Sih,J.Biol.Chem.1996,3615–3623.
[119] a)Y.Hasegawa,Y.Nakai,T.Tokuyama,H.Iwaki,Biosci.Biotechnol.Biochem.2000,2696–2698;
b)H.H.J.Cox,B.W.Faber,W.N.M.vanHeiningen,H.Radhoe,H.J.Doddema,W.Harder,Appl.
Environ.Microbiol.1999,1471–1474.
[120] V.Champreda,N.‐Y.Zhou,D.J.Leak,FEMSMicrobiol.Lett.2004,239,309–318.
[121] C.A.Townsend,S.B.Christensen,S.G.Davis,J.Am.Chem.Soc.1982,104,6152–6153.
[122] a)L.N.Britton,A.J.Markovez,J.Biol.Chem.1977,8561–8566;b)M.Griffin,P.W.Trudgill,Eur.J.
Biochem.1976,199–209;c)N.M.Kamerbeek,M.J.H.Moonen,J.G.M.vanderVen,W.J.K.van
Berkel,M.W.Fraaije,D.B.Janssen,Eur.J.Biochem.2001,2547–2557;d)J.Rehdorf,C.L.
Zimmer,U.T.Bornscheuer,Appl.Environ.Microbiol.2009,75,3106–3114.
[123] J.D.Schumacher,R.M.Fakoussa,Appl.Microbiol.Biotechnol.1999,85–90.
[124] T.Kotani,H.Yurimoto,N.Kato,Y.Sakai,J.Bacteriol.2007,189,886–893.
[125] S.Hartmans,A.M.deBont,FEMSMicrobiol.Lett.1986,155–158.
[126] Y.C.Chen,O.P.Peoples,C.T.Walsh,J.Bacteriol.1988,781–789.
[127] a)S.Damtoft,H.Franzyk,S.RosendalJensen,Phytochemistry1995,40,773–784;b)J.Winter,B.
Schneider,S.Meyenburg,D.Strack,G.Adam,Phytochemistry1999,237–242.
[128] J.L.C.Wright,T.Hu,J.L.McLachlan,J.Needham,J.A.Walter,J.Am.Chem.Soc.1996,118,8757–
8758.
[129] A.Willetts,TrendsBiotechnol.1997,55–62.
[130] D.Sheng,D.P.Ballou,V.Massey,Biochemistry2001,40,11156–11167.
[131] D.R.Light,D.J.Waxman,C.T.Walsh,Biochemistry1982,2490–2498.
[132] G.Ottolina,G.deGonzalo,G.Carrea,J.Mol.Structure:THEOCHEM2005,757,175–181.
[133] D.E.TorresPazmiño,B.‐J.Baas,D.B.Janssen,M.W.Fraaije,Biochemistry2008,47,4082–4093.
[134] H.Iwaki,Y.Hasegawa,M.Teraoka,T.Tokuyama,H.Bergeron,P.C.K.Lau,Appl.Environ.
Microbiol.1999,5158–5162.

8 Literaturverzeichnis
206
[135] Q.Cheng,S.M.Thomas,P.Rouvière,Appl.Microbiol.Biotechnol.2002,58,704–711.
[136] M.D.Mihovilovic,B.Müller,P.Stanetty,Eur.J.Org.Chem.2002,3711–3730.
[137] E.Malito,A.Alfieri,M.W.Fraaije,A.Mattevi,PNAS2004,101,13157–13162.
[138] D.E.TorresPazmiño,H.M.Dudek,M.W.Fraaije,Curr.Op.Chem.Bio.2010,14,138–144.
[139] N.M.Kamerbeek,M.W.Fraaije,D.B.Janssen,Eur.J.Biochem.2004,271,2107–2116.
[140] I.A.Mirza,B.J.Yachnin,S.Wang,S.Grosse,H.Bergeron,A.Imura,H.Iwaki,Y.Hasegawa,P.C.K.
Lau,A.M.Berghuis,J.Am.Chem.Soc.2009,131,8848–8854.
[141] P.Muller,PureAppl.Chem.1994,1077–1184.
[142] G.Ottolina,G.deGonzalo,G.Carrea,B.Danieli,Adv.Synth.&Catal.2005,347,1035–1040.
[143] M.D.Mihovilovic,B.Müller,M.M.Kayser,J.D.Stewart,J.Fröhlich,P.Stanetty,H.Spreitzer,J.Mol.
Catal.B:Enzym.2001,349–353.
[144] K.Abokitse,W.Hummel,Appl.Microbiol.Biotechnol.2003,62,380–386.
[145] R.A.Kwiecień,F.Ayadi,Y.Nemmaoui,V.Silvestre,B.‐L.Zhang,R.J.Robins,Arch.Biochem.
Biophys.2009,482,42–51.
[146] V.Prelog,PureAppl.Chem.1964,119–130.
[147] a)R.J.Lamed,J.G.Zeikus,Biochem.J.1981,183–190;b)E.Keinan,K.K.Seth,R.J.Lamed,Ann.
NYAcad.Sci.1987,130–149.
[148] K.Niefind,J.Müller,B.Riebel,W.Hummel,D.Schomburg,J.Mol.Biol.2003,327,317–328.
[149] N.H.Schlieben,K.Niefind,J.Müller,B.Riebel,W.Hummel,D.Schomburg,J.Mol.Biol.2005,349,
801–813.
[150] C.Filling,K.D.Berndt,J.Benach,S.Knapp,T.Prozorovski,E.Nordling,R.Ladenstein,H.Jörnvall,
U.Oppermann,J.Biol.Chem.2002,277,25677–25684.
[151] W.Hummel,Appl.Microbiol.Biotechnol.1990,15–19.
[152] W.Hummel,Adv.Biochem.Engin.Biotechnol.1997,145–184.
[153] W.Hummel,TrendsBiotechnol.1999,487–492.
[154] W.Kroutil,H.Mang,K.Edegger,K.Faber,Curr.Opin.Chem.Biol.2004,8,120–126.
[155] C.‐H.Wong,D.G.Drueckhammer,H.M.Sweers,J.Am.Chem.Soc.,1985,4028–4031.
[156] C.‐H.Wong,G.M.Whitesides,J.Am.Chem.Soc.1981,103,4890–4899.
[157] R.Wichmann,C.Wandrey,A.F.Bückmann,M.‐R.Kula,Biotechnol.Bioengin.1981,2789–2802.
[158] J.M.Vrtis,A.K.White,W.W.Metcalf,vanderDonk,WilfredA.,Angew.Chem.Int.Ed.2002,41,
3257–3259.
[159] J.M.Vrtis,A.K.White,W.W.Metcalf,vanderDonk,WilfredA.,Angew.Chem.2002,114,3391–
3393.
[160] D.E.TorresPazmiño,R.Snajdrova,B.‐J.Baas,M.Ghobrial,M.D.Mihovilovic,M.W.Fraaije,
Angew.Chem.Int.Ed.2008,2275–2278.
[161] H.Gröger,F.Chamouleau,N.Orologas,C.Rollmann,K.Drauz,W.Hummel,A.Weckbecker,O.
May,Angew.Chem.2006,118,5806–5809.
[162] M.C.Hogan,J.M.Woodley,Chem.Engin.Sci.2000,55,2001–2008.
[163] A.J.Willetts,C.J.Knowles,M.S.Levitt,S.M.Roberts,H.Sandey,N.F.Shipston,J.Chem.Soc.,
PerkinTrans.11991,1608.

8 Literaturverzeichnis
207
[164] G.Grogan,S.M.Roberts,A.Willetts,Biotechnol.Lett.1992,14,1125–1130.
[165] a)"NADP+(β‐Nicotinamideadeninedinucleotidephosphatehydrate)",zufindenunter
http://www.sigmaaldrich.com/catalog/product/sigma/n5755?lang=de®ion=DE,2014;b)
"NADPH(β‐Nicotinamideadeninedinucleotidephosphate,reduced
tetra(cyclohexylammonium)salt)",zufindenunter
http://www.sigmaaldrich.com/catalog/product/sigma/n5130?lang=de®ion=DE,2014.
[166] F.Hollmann,A.Taglieber,F.Schulz,M.T.Reetz,Angew.Chem.2007,119,2961–2964.
[167] F.Hollmann,K.Hofstetter,A.Schmid,TrendsBiotechnol.2006,24,163–171.
[168] H.Mallin,H.Wulf,U.T.Bornscheuer,Enz.Microb.Technol.2013,53,283–287.
[169] J.T.Vicenzi,M.J.Zmijewski,M.R.Reinhard,B.E.Landen,W.L.Muth,P.G.Marler,Enz.Microb.
Technol.1997,494–499.
[170] a)H.D.Simpson,V.Alphand,R.Furstoss,J.Mol.Catal.B:Enzym.2001,101–108;b)I.Hilker,V.
Alphand,R.Wohlgemuth,R.Furstoss,Adv.Synth.Catal.2004,346,203–214.
[171] F.Schulz,F.Leca,F.Hollmann,M.T.Reetz,BeilsteinJ.Org.Chem.2005,1,10.
[172] A.Rioz‐Martínez,G.deGonzalo,D.E.TorresPazmiño,M.W.Fraaije,V.Gotor,Eur.J.Org.Chem.
2009,2009,2526–2532.
[173] G.deGonzalo,G.Ottolina,F.Zambianchi,M.W.Fraaije,G.Carrea,J.Mol.Catal.B:Enzym.2006,
39,91–97.
[174] A.M.Klibanov,TrendsBiotechnol.1997,15,97–101.
[175] C.Laane,S.Boeren,K.Vos,C.Veeger,Biotechnol.Bioengin.1987,81–87.
[176] S.C.Basak,G.J.Niemi,G.D.Veith,J.Math.Chem.1990,185–205.
[177] J.Sangster,J.Phys.CHem.Ref.Data1989,18,1111–1227.
[178] a)D.Sheng,D.P.Ballou,V.Massey,Biochemistry2001,40,11156–11167;b)B.J.Yachnin,T.
Sprules,M.B.McEvoy,Lau,PeterCK,A.M.Berghuis,J.Am.Chem.Soc.2012,134,7788–7795.
[179] F.Zambianchi,P.Pasta,G.Carrea,S.Colonna,N.Gaggero,J.M.Woodley,Biotechnol.Bioeng.
2002,78,489–496.
[180] MerckMillipore,"Sicherheitsdatenblatt.E‐Caproacton",zufindenunter
http://www.emdmillipore.com/DE/de/product/%CE%B5‐Caprolacton,MDA_CHEM‐
802801#anchor_MSD.
[181] J.D.Stewart,K.W.Reed,C.A.Martinez,J.Zhu,G.Chen,M.M.Kayser,J.Am.Chem.Soc.1998,
3541–3548.
[182] N.Berezina,E.Kozma,R.Furstoss,V.Alphand,Adv.Synth.Catal.2007,349,2049–2053.
[183] "Isophoron.DamitderLacknichtabist",zufindenunter
http://history.evonik.com/sites/geschichte/de/erfindungen/isophoron/Pages/default.aspx.
[184] C.Jiménez‐Sanchidrián,J.R.Ruiz,Tetrahedron2008,64,2011–2026.
[185] R.Gupta,N.Gupta,P.Rathi,Appl.Microbiol.Biotechnol.2004,64,763–781.
[186] A.Pandey,S.Benjamin,C.R.Soccol,P.Nigam,N.Krieger,V.T.Soccol,Biotechnol.Appl.Biochem.
1999,119–131.
[187] K.‐E.Jaeger,S.Ransac,B.W.Dijkstra,C.Colson,M.vanHeuvel,O.Misset,FEMSMicrobiol.Rev.
1994,29–63.

8 Literaturverzeichnis
208
[188] N.J.Turner,NatureChem.Biol.2009,5,567–573.
[189] K.‐E.Jaeger,T.Eggert,Curr.Opin.Biotechnol.2002,390–397.
[190] AmericanChemicalSociety,"SciFinder.cas.org",zufindenunterhttps://scifinder.cas.org.
[191] L.Brady,A.M.Brzozowski,Z.S.Derewenda,E.Dodson,G.Dodson,S.Tolley,J.P.Turkenburg,L.
Christiansen,B.Huge‐Jensen,L.Norskovetal.,Nature1990,767–770.
[192] F.K.Winkler,A.D'Arcy,W.Hunziker,Nature1990,771–774.
[193] K‐E.Jaeger,B.W.Dijkstra,M.T.Reetz,Annu.Rev.Microbiol.1999,315–351.
[194] J.Uppenberg,M.T.Hansen,S.Patkar,T.A.Jones,Structure1994,293–308.
[195] E.M.Anderson,K.M.Larsson,O.Kirk,Biocatcal.Biotrans.1998,181–204.
[196] F.Secundo,G.Carrea,C.Soregaroli,D.Varinelli,R.Morrone,Biotechnol.Bioeng.2001,157–163.
[197] J.Uppenberg,N.Öhrner,M.Norin,K.Hult,G.J.Kleywegt,S.Patkar,V.Waagen,T.Anthonsen,T.
A.Jones,Biochemistry1995,16838–16851.
[198] R.J.Kazlauskas,A.N.E.Weissfloch,A.T.Rappaport,L.A.Cuccia,J.Org.Chem.1991,2656–2665.
[199] V.Gotor‐Fernández,R.Brieva,V.Gotor,J.Mol.Catal.B:Enzym.2006,40,111–120.
[200] D.Xaio,X.Zhang,Angew.Chem.Int.Ed.2001,3425–3428.
[201] G.Hou,F.Gosselin,W.Li,J.C.McWilliams,Y.Sun,M.Weisel,P.D.O'Shea,C.‐y.Chen,I.W.Davies,
X.Zhang,J.Am.Chem.Soc.2009,131,9882–9883.
[202] K.‐i.Yamada,K.Tomioka,Chem.Rev.2008,108,2874–2886.
[203] J.M.Blackwell,E.R.Sonmor,T.Scoccitti,W.E.Piers,Org.Lett.2000,2,3921–3923.
[204] N.B.Johnson,M.J.Burk,G.Casy(ChirotechTechnologyLtd.),WO9918065A1I,1998.
[205] M.J.Burk,G.Casy,N.B.Johnson,J.Org.Chem.1998.
[206] C.K.Savile,J.M.Janey,E.C.Mundorff,J.C.Moore,S.Tam,W.R.Jarvis,J.C.Colbeck,A.Krebber,F.
J.Fleitz,J.Brandsetal.,Science2010,329,305–309.
[207] F.G.Mutti,J.Sattler,K.Tauber,W.Kroutil,ChemCatChem2011,3,109–111.
[208] M.Breuer,K.Ditrich,T.Habicher,B.Hauer,M.Keßeler,R.Stürmer,T.Zelinski,Angew.Chem.
2004,116,806–843.
[209] K.Tomioka,I.Inoue,M.Shindo,K.Koga,Tetrahed.Lett.1990,6681–6684.
[210] S.E.Denmark,C.M.Stiff,J.Org.Chem.2000,65,5875–5878.
[211] a)I.Sato,K.Hosoi,R.Kodaka,K.Soai,Eur.J.Org.Chem.2002,3115–3118;b)I.Sato,R.Kodaka,
K.Soai,J.Chem.Soc.,PerkinTrans.12001,2912–2914.
[212] a)M.Bao,H.Nakamura,Y.Yamamoto,Tetrahed.Lett.2000,131–134;b)R.A.Fernandes,A.
Stimac,Y.Yamamoto,J.Am.Chem.Soc.2003,125,14133–14139;c)R.A.Fernandes,Y.
Yamamoto,J.Org.Chem.2004,69,735–738;d)K.Nakamura,H.Nakamura,Y.Yamamoto,J.Org.
Chem.1999,64,2614–2615.
[213] H.‐U.Blaser,H.‐P.Buser,H.‐P.Jalett,B.Pugin,F.Spindler,Synlett1999,867–868.
[214] H.‐U.Blaser,F.Spindler,Top.Catal.1997,275–282.
[215] S.Ram,R.E.Ehrenkaufer,Synthesis1988,91–95.
[216] R.Kadyrov,T.H.Riermeier,Angew.Chem.Int.Ed.2003,42,5472–5474.
[217] X.‐M.Zhou,J.‐D.Huang,L.‐B.Luo,C.‐L.Zhang,Z.Zheng,X.‐P.Hu,Tetrahed.Asymm.2010,21,
420–424.

8 Literaturverzeichnis
209
[218] L.Pignataro,S.Carboni,M.Civera,R.Colombo,U.Piarulli,C.Gennari,Angew.Chem.2010,122,
6783–6787.
[219] X.‐P.Hu,J.‐D.Huang,Q.‐H.Zeng,Z.Zheng,Chem.Commun.2006,293–295.
[220] N.J.Turner,NatureChem.Bio.2009,5,567–573.
[221] S.Mathew,H.Yun,ACSCatal.2012,2,993–1001.
[222] D.Zhu,L.Hua,Biotechnol.J.(Biotechnologyjournal)2009,4,1420–1431.
[223] UnitedStatesEnvironmentalProtectionAward,"2010GreenerReactionConditionsAward",zu
findenunterhttp://www2.epa.gov/green‐chemistry/2010‐greener‐reaction‐conditions‐award,
2015.
[224] K.Faber,W.Kroutil,Curr.Opin.Chem.Biol.2005,9,181–187.
[225] R.Carr,M.Alexeeva,A.Enright,Eve,TomSC,M.J.Dawson,N.J.Turner,Angew.Chem.Int.Ed.
2003,42,4807–4810.
[226] G.P.Moss,PureAppl.Chem.1996,2193–2222.
[227] O.Ghisalba,M.Kittelmann,K.Laumen,P.Walser‐Volken(NovartisAG),EP0954571B1I,1997.
[228] H.Smidt,A.Fischer,P.Fischer,R.D.Schmid,U.Stelzer(BayerAG),EP0812363B1,1996.
[229] F.Balkenhohl,K.Ditrich,B.Hauer,W.Ladner,J.Prakt.Chem.1997,381–384.
[230] a)M.Cammenberg,K.Hult,S.Park,ChemBioChem2006,7,1745–1749;b)H.Ismail,R.Madeira
Lau,F.vanRantwijk,R.A.Sheldon,Adv.Synth.Catal.2008,350,1511–1516.
[231] O.Pàmies,A.H.Èll,J.S.M.Samec.,N.Hermanns,J.‐E.Bäckvall,Tetrahed.Lett.2002,4699–4702.
[232] J.Paetzold,J.E.Bäckvall,J.Am.Chem.Soc.2005,127,17620–17621.
[233] L.K.Thalén,D.Zhao,J.‐B.Sortais,J.Paetzold,C.Hoben,J.‐E.Bäckvall,Chemistry2009,15,3403–
3410.
[234] K.Ditrich,U.Block(BASFAG),WO0046177A1I,2000.
[235] A.Maestro,C.Astorga,V.Gotor,Tetrahed.Asymm.1997,3153–3159.
[236] A.Luna,I.Alfonso,V.Gotor,Org.Lett.2002,4,3627–3629.
[237] I.Alfonso,C.Astorga,F.Rebolledo,V.Gotor,Chem.Commun.1996,2471–2472.
[238] E.J.Ebbers,Ariaans,GerryJ.A.,Houbiers,JoannesP.M.,A.Bruggink,B.Zwanenburg,
Tetrahedron1997,9417–9476.
[239] M.Stirling,J.Blacker,M.I.Page,Tetrahed.Lett.2007,48,1247–1250.
[240] M.T.Reetz,K.Schimossek,Chimia1996,668–669.
[241] A.N.Parvulescu,P.A.Jacobs,D.E.deVos,Chemistry2007,13,2034–2043.
[242] M.‐J.Kim,W.‐H.Kim,K.Han,Y.K.Choi,J.Park,Org.Lett.2007,9,1157–1159.
[243] S.Gastaldi,S.Escoubet,N.Vanthuyne,G.Gil,M.P.Bertrand,Org.Lett.2007,9,837–839.
[244] a)J.Gu,M.Liu,F.Guo,W.Xie,W.Lu,L.Ye,Z.Chen,S.Yuan,H.Yu,Enz.Microb.Technol.2014,55,
121–127;b)U.Felfer,M.Goriup,M.F.Koegl,U.Wagner,B.Larissegger‐Schnell,K.Faber,W.
Kroutil,Adv.Synth.Catal.2005,347,951–961.
[245] P.Trodler,J.Pleiss,BMCStruct.Biol.2008,8,9.
[246] D.S.Clark,Phil.Trans.R.Soc.Lond.B2004,359,1299–1307.
[247] P.Tufvesson,A.Annerling,R.Hatti‐Kaul,D.Adlercreutz,Biotechnol.Bioengin.2007,97,447–
453.

8 Literaturverzeichnis
210
[248] A.B.Majumder,B.Singh,D.Dutta,S.Sadhukhan,M.N.Gupta,Bioorg.Med.Chem.Lett.2006,16,
4041–4044.
[249] a)A.Mahapatro,A.Kumar,B.Kalra,R.A.Gross,Macromol.2004,37,35–40;b)B.Guyot,M.Pina,
J.Graille,Biotechnol.Lett.1997,6,529–532;c)L.Hilterhaus,O.Thum,A.Liese,Org.ProcessRes.
Dev.2008,12,618–625;d)G.A.Hills,A.R.Macrae,R.R.Poulina,1990;e)K.Bélafi‐Bakó,F.
Kovács,L.Gubicza,J.Hancsók,Biocatal.Biotransform.2002,20,437–439;f)V.V.Kuperkar,V.G.
Lade,A.Prakash,V.K.Rathod,J.Mol.Catal.B:Enzym.2014,99,143–149;g)R.Irimescu,T.Saito,
K.Kato,J.Mol.Catal.B:Enzym.2004,27,69–73.
[250] R.Irimescu,K.Kato,Tetrahed.Lett.2004,45,523–525.
[251] N.Öhrner,C.Orrenius,A.Mattson,T.Norin,K.Hult,Enz.Microb.Technol.1996,328–331.
[252] A.K.Prasad,M.Husain,B.K.Singh,R.K.Gupta,V.K.Manchanda,C.E.Olsen,V.S.Parmar,
Tetrahed.Lett.2005,46,4511–4514.
[253] Z.Finta,Z.Hell,J.Bálint,A.Takács,L.Párkányi,L.Töke,Tetrahed.Asymm.2001,1287–1292.
[254] K.Wang,K.Nguyen,Y.Huang,A.Dömling,J.Combinat.Chem.2009,11,920–927.
[255] C.E.Rye,D.Barker,J.Org.Chem.2011,76,6636–6648.
[256] D.C.Horwell,R.A.Lewthwaite,M.C.Pritchard,G.S.Ratcliffe,J.R.Rubin,Tetrahedron1998,
4591–4606.
[257] K.E.Price,C.Larrivée‐Aboussafy,B.M.Lillie,R.W.McLaughlin,J.Mustakis,K.W.Hettenbach,J.
M.Hawkins,R.Vaidyanathan,Org.Lett.2009,11,2003–2006.
[258] K.Faber,W.Kroutil,"Enantioselectivity",zufindenunterhttp://biocatalysis.uni‐
graz.at/enantio/cgi‐bin/enantio.pl,2014.
[259] M.Fuchs,D.Koszelewski,K.Tauber,W.Kroutil,K.Faber,Chem.Commun.2010,46,5500–5502.
[260] J.Mangas‐Sánchez,M.Rodríguez‐Mata,E.Busto,V.Gotor‐Fernández,V.Gotor,J.Org.Chem.
2009,74,5304–5310.
[261] B.C.Vanwagenen,S.R.Duff,W.A.Nelson,T.E.D'Ambra,WO9602492A1I,2014.
[262] a)"Diethylmalonate",zufindenunter
http://www.sigmaaldrich.com/catalog/product/aldrich/w237507?lang=de®ion=DE,2014;
b)"Benzylamin",zufindenunter
http://www.sigmaaldrich.com/catalog/product/aldrich/185701?lang=de®ion=DE,2014.
[263] "AdvancedChemistryDevelopment(ACD/Labs)SoftwareV11.02",zufindenunter
www.scifinder.cas.org,2015.
[264] K.Ditrich(BASF),WO2006136538A1I,2006.
[265] a)c‐LEcta,"CalBlyo.immobilized,recombinant",zufindenunterhttp://www.c‐
lecta.com/?lang=de&category=products&page=lipasen,2014;b)c‐LEcta,"CalBimmo.
immobilized,recombinant",zufindenunterhttp://www.c‐
lecta.com/?lang=de&category=products&page=lipasen,2014.
[266] a)M.M.Darla,B.S.Krishna,K.UmamaheswaraRao,N.B.Reddy,M.K.Srivash,K.Adeppa,C.S.
Sundar,C.S.Reddy,K.Misra,Res.Chem.Intermed.2015,41,1115–1133;b)W.Priebe,N.
Donato,M.Talpaz,S.Szymanshi,I.Fokt,A.Levitki,WO2005058829A1,2005.

8 Literaturverzeichnis
211
[267] X.Huang,M.Ortiz‐Marciales,K.Huang,V.Stepanenko,F.G.Merced,A.M.Ayala,W.Correa,M.D.
Jesús,Org.Lett.2007,9,1793–1795.
[268] M.Binanzer,S.‐Y.Hsieh,J.W.Bode,J.Am.Chem.Soc.2011,133,19698–19701.
[269] A.Liljeblad,J.Lindborg,A.Kanerva,J.Katajisto,L.T.Kanerva,Tetrahed.Lett.2002,2471–2474.
[270] a)T.K.Houlding,K.Tchabanenko,M.T.Rahman,E.V.Rebrov,Org.Biomol.Chem.2013,11,
4171–4177;b)C.L.Allen,A.R.Chhatwal,Williams,JonathanMJ,Chem.Commun.2012,48,666–
668;c)A.Orliac,D.GomezPardo,A.Bombrun,J.Cossy,Org.Lett.2013,15,902–905.
[271] L.A.Henderson,Y.Y.Svirkin,R.A.Gross,Macromolecules1996,7759–7766.
[272] B.H.Brodsky,J.DuBois,J.Am.Chem.Soc.2005,127,15391–15393.
[273] N.Chandan,A.L.Thompson,M.G.Moloney,Org.Biomol.Chem.2012,10,7863.
[274] Y.Ksashima,A.Uzawa,K.Hashimoto,Y.Yokoyama,T.Mino,M.Sakamoto,T.Fujita,J.OleoSci.
2010,11,607–613.
[275] B.V.SubbaReddy,N.SivasankarReddy,C.Madan,J.S.Yadav,Tetrahed.Lett.2010,51,4827–
4829.
[276] M.A.J.Veld,K.Hult,A.R.A.Palmans,E.W.Meijer,Eur.J.Org.Chem.2007,2007,5416–5421.
[277] N.D.Paul,A.Chirila,H.Lu,X.P.Zhang,B.deBruin,Chem.Eur.J.2013,19,12953–12958.
[278] T.Zweifel,J.‐V.Naubron,H.Grützmacher,Angew.Chem.Int.Ed.2009,48,559–563.
[279] N.Chandan,A.L.Thompson,M.G.Moloney,Org.Biomol.Chem.2012,10,7863.
[280] N.Shindo,Bull.Fac.Agricul.1988,29.
[281] Y.Kita,O.Tamura,N.Shibata,T.Miki,Chem.Pharm.Bull.1990,1473–1478.







![Arylcalciumhalogenide: Synthesen, Eigenschaften, Strukturen und … · 2008. 7. 3. · wasserstoffen [43] erhalten, selbst die Herstellung des hochreaktiven und thermisch sehr instabilen](https://static.unterlagen.site/doc/80x56/60a83a153092781e1c1ef614/arylcalciumhalogenide-synthesen-eigenschaften-strukturen-und-2008-7-3-wasserstoffen.jpg)