

Steffen BöttcherAbteilung II
Dienstag, 02.12.2008
32-jährige Patientin mit Splenomegalieund Thrombozytopenie
-ein Fallbericht der Station 65.

Anamnese
• Vorstellung KH Nürtingen aufgrund zunehmender Schmerzen im Mittelbauch, Nachweis einer deutlichen Splenomegalie sowie einer ausgeprägten Thrombozytopenie (PLT < 10.000/µl)
• keine Blutungs-, Infektzeichen, kein Fieber, keine B-Symptome
• HWI sowie „Nierenentzündung“, antibiotisch behandelt mit Cefaclor und Norfloxacin
• keine Medikamenteneinnahme, keine Allergien, kein Nikotin / Alkohol
• Z.n. Appendektomie in der Kindheit
Mitte September 2008

Körperliche Untersuchung
• guter AZ, EZ (164cm, 60kg)
• keine Petechien, keine Hämatome
• keine Lymphadenopathie
• unauffälliger kardiopulmonaler Untersuchungsbefund
• Milz ca. 3cm unter Rippenbogen tastbar, druckschmerzhaft
• Klopfschmerz über den Nierenlogen
• WS / Gelenke unauffällig
• keine Ödeme, keine Varicosis, Pulsstatus unauffällig
• Neurologie unauffällig

Labor bei Aufnahme
Leukos: 6630/µlHb: 11.2 g/dlPLT: 20.000/µl
Quick: 93 %aPTT: 35 sFibrinogen: 391 mg/dlAT III: 104 %
Krea: 0.7 mg/dlBili: 0.9 mg/dlGOT: 13 U/lGPT: 13 U/lLDH: 127 U/lAP: 62 U/lgGT: 6 U/l
CRP: 1.55 mg/dl

Leitsymptome
Splenomegalie
&
Thrombozytopenie

Differentialdiagnose - Splenomegalie
(Harrisons Innere Medizin, 17. Auflage, McGraw-Hill und ABW Wissenschaftsverlag, 2008)

Differentialdiagnose - Thrombozytopenie
Störungen der Thrombozytopoese-medikamentös/toxisch (Thiazide, Thiamazol, Alkohol) -Infekte (CMV, EBV, HIV, Mycoplasmen, Sepsis)-Aplastische Störungen (acquired pure megakaryocytic aplasia)-Reifungsstörungen (Myelodysplasie, Vit. B12 / Folsäuremangel-Knochenmarkinfiltrationen (Leukämien, Lymphome, solide Tumoren)-Hereditäre Störungen (TAR-Syndrome, Bernard-Soulier-Syndrom)
beschleunigter Thrombozytenumsatz-Immunthrombozytopenien (primär, sekundär)-DIC-HUS/TTP-HIT-Extrakorporale Zirkulation-Herzklappen- und Gefäßprothesen
Hypersplenismus-Portale Hypertension (Milzvenen-, Pfortaderthrombose, Leberzirrhose, Rechtsherzinsuffizienz, Budd-Chiari-Syndrom)

Pseudothrombozytopenie
(Beutler et al, Hematology, 5th ed, McGraw-Hill, New York, 1995.)
Inzidenz: ca. 1:1000 – 1:50
Auto-Antikörper gegen ein Epitop des GP IIa/IIIb – Komplexes
EDTA � Konformationsänderung � Exposition des Epitops
Thrombozyten-Bestimmung in Citrat- oder Heparin-Blut

Arzneimittelinduzierte Thrombozytopenie
�������������� ������ ����������������������
http://www.ouhsc.edu/platelets/ditp.html

Primäre ITP: erworbene Thrombozytopenie unklarer Ätiologie mit normaler/gesteigerter Megakaryopoese
Sekundäre ITP: bekannte Grunderkrankung (Lymphome, Autoimmunerkrankungen, HIV, Medikamente, nach KMT)
Immunthrombozytopenie (ITP)
� Ausschlussdiagnose
� (begrenzte) Suche nach einer Grunderkrankung
Therapie: akut: IVIG 2g/kgKG über 2-3 d + Methylprednisolon 1-2g für 3 d
Primärtherapie: Prednison 1-2 mg/kgKG/d für 2 Wochen
Rezidivtherapie: Splenektomie, Immunsuppressiva, Thrombopoetika
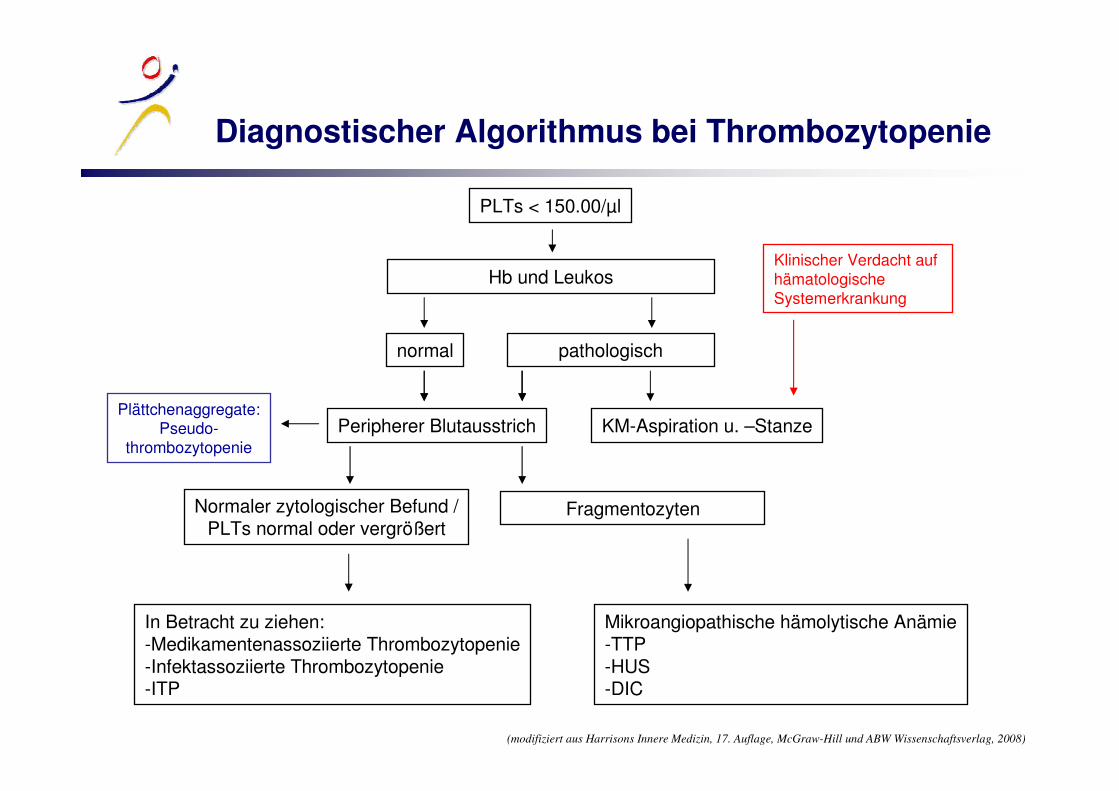
Diagnostischer Algorithmus bei Thrombozytopenie
PLTs < 150.00/µl
Hb und Leukos
normal pathologisch
KM-Aspiration u. –StanzePeripherer Blutausstrich
Normaler zytologischer Befund /PLTs normal oder vergrößert
In Betracht zu ziehen:-Medikamentenassoziierte Thrombozytopenie-Infektassoziierte Thrombozytopenie-ITP
Fragmentozyten
Mikroangiopathische hämolytische Anämie-TTP-HUS-DIC
Plättchenaggregate:Pseudo-
thrombozytopenie
(modifiziert aus Harrisons Innere Medizin, 17. Auflage, McGraw-Hill und ABW Wissenschaftsverlag, 2008)
Klinischer Verdacht aufhämatologische Systemerkrankung

Weitere Diagnostik (1)
Differenzialblutbild: normale Zellverteilung. Einzelne Riesentrombozyten. Keine Fragmentozyen. Keine Thrombozytenaggregate.
Hämolyseparameter: Haptoglobin 127 mg/dl, freies Hb i. Plasma 3.18 mg/dl, Coombs Test negativ.
Serum-El‘pho: geringe Hypergammaglobulinämie mit 22.2%
Thrombozytäre Autoantikörper: neg.
Virusserologien: CMV IgM neg., EBV VCA IgM neg., HHV-6 IgM neg., HBs-Agneg., anti-HBs neg., anti-HBc neg., anti-HCV neg., HIV Ag/Ak neg.
Autoantikörper: ANAs neg., Anti-Phospholipid-AK neg.
Laboruntersuchungen: Vit. B12 22 ng/dl, Folsäure 328 ng/dl

Weitere Diagnostik (2)
bei V.a. hämatologische Systemerkrankung ���� KMP
Sonographie-Abdomen: Leber: grenzwertig groß (15.6 cm in MCL), Oberfläche glatt, homogenes Binnenmuster, normale Gefäßstrukturen. Milz: vergrößert (19.7 cm x 12.9 cm x 7.7 cm), verplumpt, normale Parenchymstruktur.
GK-CT: Thorax: am ehesten unspezifische axilläre Lymphknoten bis 1.1 cm Größe. Abdomen: Leber homogen, leicht vergrößert. Ausgeprägt inhomogen kontrastierte Milz mit fraglich hypodensen Läsionen, vergrößert (18.5 cm).Interaortokaval Lymphknoten bis 1.4 cm Größe. Deutliche Dichteanhebung der femoralen Markräume.

Weitere Diagnostik (3)
1. KMP ���� Punctio sicca ( PLTs 6.000/µl)
Therapieversuch mit IVIG: 1g/kgKG für 2 Tage� binnen 2 Tage Anstieg der Thrombozyten auf 43.000/µl� keine Steroid-Gabe !
2. KMP ���� Punctio sicca, KM-Stanze
Zytologie: Abrollpräparat zur suffizienten Diagnostik nicht ausreichend.
Histologie: …(OA Dr. Adam)

Pathologie

ambulante Wiedervorstellung 10 Tage nach Entlassung� erneuter Thrombozytenabfall auf 12.000/µl
stationäre Wiederaufnahme� PLTs 8.000/µl� IVIG 1g/kgKG für 2 Tage, Anstieg der PLTs auf 41.000/µl
Weiterer Verlauf (1)
���� hochgradiger V.a. splenisches Lymphom mit sekundärer ITP
���� Indikation zur diagnostischen ggfs. therapeutischen Splenektomie

Verdachtsdiagnose - Splenisches Marginalzonen-Lymphom
Epidemiologie: < 5% aller NHL, Altersmedian 65 Jahre, m=w
Pathogenese: unklar, Ausgang von Post-Keimzentrums-Memory-B-Zellen
Klinik: Splenomegalie, Lymphozytose, Zytopenien (Hypersplenismus, sek. ITP)häufig geringe monoklonale Gammopathie, leichtgradiger KM-Befallselten Lymphadenopathie oder extralymphatischer Befallselten B-Symptome oder LDH-Erhöhungen
Diagnose: klinisch + Differenzial-BB + KM-Histologie oder histologisch nach Splenektomie
Therapie & Prognose: - indolenter Verlauf, medianes OS 10 Jahre- wait & see, Therapie bei Symptomen- Splenektomie (häufig sehr gutes Ansprechen mit
Langzeit-Überleben)- Radiatio- Chemotherapie

Weiterer Verlauf (2)
Durchführung der Splenektomie am 22.10.2008
� zwischenzeitlich erneuter Abfall der Thrombozyten auf 26.000/µl
� Intraoperative Gabe von 4 Pool-TKs
� Komplikationsloser postoperativer Verlauf
� Prompter Anstieg der Thrombozytenzahlen auf 105.000/µl noch am OP-Tag
� Weitere Normalisierung der Thrombozytenzahlen bis auf 156.000/µl
� Erstmalig pathologisch erhöhte LDH 297 U/l
� Erneuter Thrombozytenabfall, 88.000/µl am Entlasstag, LDH 354 U/l
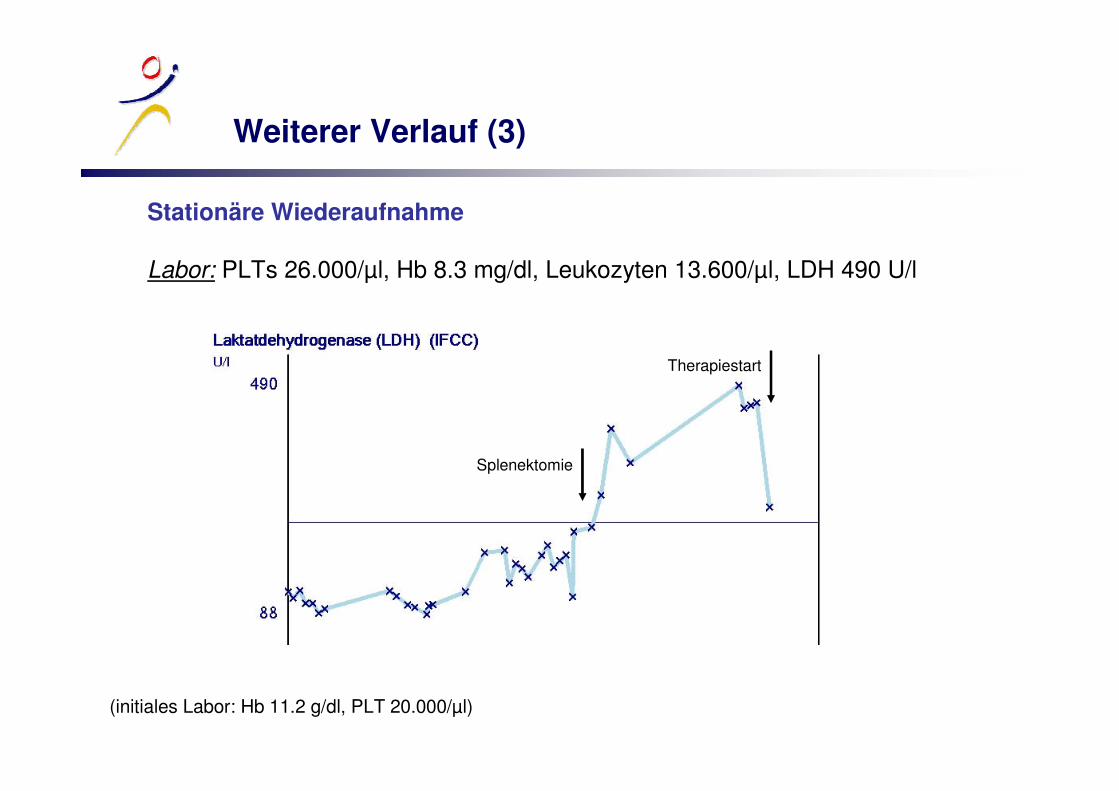
Weiterer Verlauf (3)
Stationäre Wiederaufnahme
Labor: PLTs 26.000/µl, Hb 8.3 mg/dl, Leukozyten 13.600/µl, LDH 490 U/l
(initiales Labor: Hb 11.2 g/dl, PLT 20.000/µl)
Splenektomie
Therapiestart

Weiterer Verlauf (4)
StagingGK-CT: axilläre Lymphknoten im Vgl. zur VU etwas größenprogredient mit vermehrter KM-Aufnahme und rundlicher Konfiguration und erscheinen somit suspekt. Zunehmende Hepatomegalie.
KMPZytologie: Hyperzelluläres KM, alle drei Zellreihen reifen weitgehend regelgerecht aus. Daneben finden sich am ehesten lymphatische Zellen (35%) mit schmalem Zytoplasmasaum, teils mit Nukleoli. KM-Infiltration durch ein NHL.
Histologie (Milz, KM): … (OA Dr. Adam)

Pathologie

hepatosplenisches ����� - T-Zell-Lymphom (Epidemiologie)
• extrem selten
• < 5% aller peripheren T-NHL1
• erstmals zu Beginn der 90er Jahre beschrieben2
• m>>w
• Altersmedian 34 Jahre3
1 Jaffe ES, WHO classification of tumors, 20082 Farcet JP, Blood, 19903 Belhadj K, Blood, 2003

hepatosplenisches ����� - T-Zell-Lymphom (Pathogenese)
• Immunsuppression (post transplantation lymphoproliferative disorder)1,2,3
• Autoimmunerkrankungen (SLE4, M. Behcet5)
• chronische Antigenstimulation (EBV, Malaria)4
1 Roncella S, Haematologica, 20002 Khan WA, Am J Clin Pathol, 20013 Ambramson JS, NEJM, 20084 Belhadj K, Blood, 20035 Amado A, Leuk Lymphoma, 2008

hepatosplenisches ����� - T-Zell-Lymphom (Klinik)
• Splenomegalie (97.5%, 39/40)
• Thrombozytopenie (85%, 35/41)
• Anämie (84%, 32/38)
• Hepatomegalie (80%, 32/40)
• KM-Infiltration (72%, 26/36)
• LDH-Erhöhung (62%, 16/26)
• Lymphozytose (50%, 16/32)
1 Weidman E, Leukemia, 2000
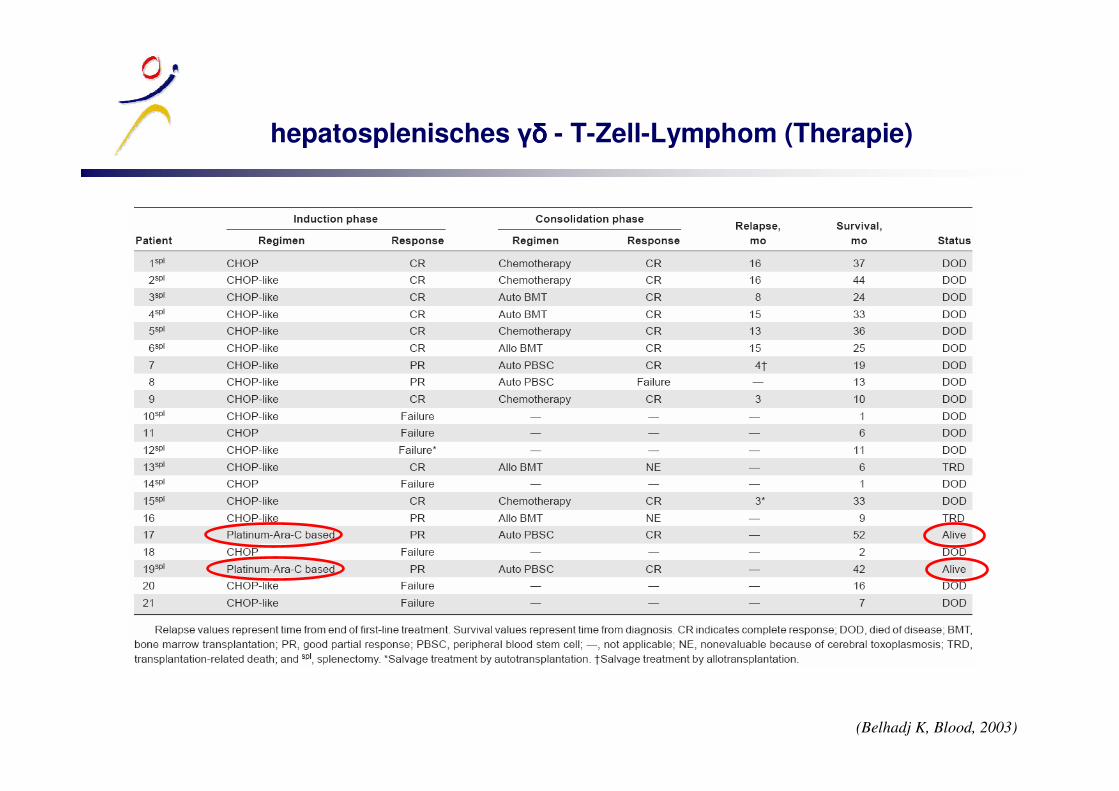
hepatosplenisches ����� - T-Zell-Lymphom (Therapie)
(Belhadj K, Blood, 2003)

Bisherige und geplante Therapie
CHOP-14 (ggfs. mehrere Zyklen)
DHAP + Leukapherese
HD-BEAM + PBSCT
ggfs. allogene Stammzelltransplantation
Eskalation

Vielen Dank für Ihre Aufmerksamkeit !







