∗ Auswertung von Chromatogrammen (qualitativ, quantitativ)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 2
Das Prinzip chromatographischer Methoden
Definition: Chromatographische Methoden sind physikalisch-chemische Trennmethoden, die alle im Prinzip darauf beruhen, dass Substanzen zwischen einer ruhenden (stationären) und einer sich an dieser vorbeibewegenden (mobilen) Phase verteilt werden.
Beispiel zur Veranschaulichung: Dünnschichtchromatographie
Start
· ·
Fließmittelfront
Substanz Fall A Fall B Fall C
Fall A, B: keine Chromatographie; Fall C: Chromatographie
Verhalten von Substanzen in einem gegebenen chromatograph. System → spezifische Substanzeigenschaft; geeignet zur Charakterisierung und Identifizierung von Substanzen (bzw. Identitätsprüfung)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 4
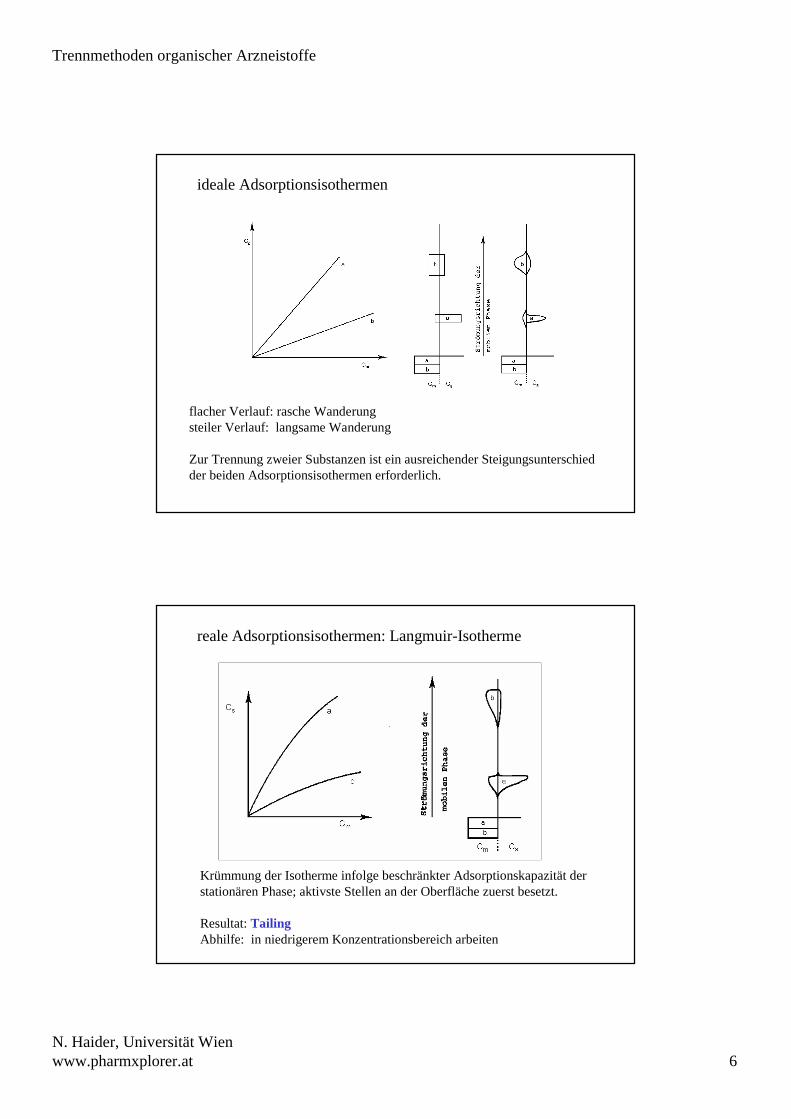
Charakterisierung des Wanderungsverhaltens (DC)
a
Glasplatte
Sorbensschicht
Fließmittelfront
RF = c
RX = RSt =ab
c
b
a
. . .
St Pr St
3 mm
20 mm
5 mm
• RF-Wert
• RX-Wert (RSt-Wert)
Messpunkte:
• Startpunkt
• Fleckenmittelpunkt bzw.
• Fließmittelfront
Grundlagen chromatographischer Trennungen
Trennprinzipien (Trennmechanismen):
• Adsorption
• Verteilung
• Molekularsiebwirkung (Ausschluss)
• Ionenaustausch
• Bioaffinität
manchmal keine 100%ige Abgrenzung möglich
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 5
Grundlagen chromatographischer Trennungen
AdsorptionDefinition: Anreicherung eines gelösten Stoffes oder eines gasförmigen Stoffes ( = ADSORBAT) an der Oberfläche eines Feststoffes ( = ADSORBENS = SORBENS);
• Oberflächenreaktion
• reversibler Prozess → Desorption (Konkurrenz mit anderenSubstanzen, v.a. mit Lösungsmittel!)
Bindung erfolgt bei der physikalischen Adsorption durch:
• Adsorptionschromatographie: apolare Substanzen wandern immer schneller als polarew Substanzen
• Verteilungschromatographie: Umkehrung des Wanderungsverhaltens möglich
o o o oStart
FM-Front
Verteilungs-DC Adsorptions-DC
"normal" mit Phasen-umkehr
aktives Sorbens,apolares Fließmittel
wenig aktives Sorbens,polares Fließmittel
A
P
P
A
A
P
A
P
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 9
Grundlagen chromatographischer Trennungen
IonenaustauschIonenaustauscher: unlösliche Festsubstanzen mit Polyelektrolyt-Charakter, die aus Elektrolytlösungen Anionen bzw. Kationen aufnehmen können und dafür äquivalente Mengen gleichsinnig geladener Ionen in die Lösung abgeben. Je nachdem, ob Anionen oder Kationen abgegeben werden →Anionen- bzw. Kationenaustauscher.
• Häufig verwendet: Co-Polymerisate aus Styrol und Divinylbenzol mit ionischen funktionellen Gruppen an den Phenylresten.
• starke IAT: bei jedem pH-Wert in ionischer Form
• starke Kationenaustauscher: z.B. R-SO3- Na+
• starke Anionenaustauscher: z.B. R-N(R‘)3+ Cl-
• schwache IAT: nur in bestimmtem pH-Bereich ionisch
• schwache Kationenaustauscher: z.B. R-COO- Na+
• schwache Anionenaustauscher: z.B. R-NH3+ Cl-
Ionenaustauschchromatographie
Trennung infolge unterschiedlichstarker Wechselwirkungskräftezwischen verschiedenen ionischen Analyten und den Festionen des IAT
N. Haider, Universität Wien www.pharmxplorer.at 10
Grundlagen chromatographischer Trennungen
Molekülsiebwirkung, Ausschluss-Chrom.verwendet in Gel-Chromatographie (Gel-Permeation), Gel-Filtration
Trenneffekt durch sterischen Ausschluss (Molekülgröße bzw. Molekular-gewicht), nur für Verbindungen ab einer best. Mindestgröße geeignet
kleinere Moleküle diffundieren weiter in die Poren hinein
→ längerer Aufenthalt in der stationären Phase
→ langsamere Wanderung
mobile Phase: geeignetes LM (meist wässrige Pufferlösungen)stationäre Phase: das selbe LM, und zwar dessen Anteil innerhalb der Poren
Grundlagen chromatographischer Trennungen
Bioaffinitäts-ChromatographieAn der Oberfläche eines inerten, polymeren Trägers sind Substanzen chemisch gebunden, die zu bestimmten Biomolekülen eine besonders hohe Affinität besitzen und diese daher hervorragend retinieren können.
z.B.:
Antikörper ↔ Antigen
Enzym ↔ Substrat, Inhibitor
Rezeptor ↔ Arzneistoff, Hormon
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 11
Einteilung chromatographischer Verfahren
• nach dem Trennprinzip
• nach der Ausführungstechnik:
• säulenchromatographische Verfahren
• flat-bed-Verfahren
• nach dem Phasenaufbau:stationäre Phase fest (S) oder flüssig (L), mobile Phase flüssig (L) oder gasförmig (G)→ Bezeichnungen wie LSC, LLC, GSC, GLC
• danach, wo die Substanzen nachgewiesen werden:
• innerhalb der Trennstrecke → inneres Chromatogramm
• nach Verlassen der Trennstrecke → äußeres Chrom.
Theorie des chromatographischen Prozesses
Mehrere Theorien zur Beschreibung der relevanten Vorgänge:
Ziel: besseres Verständnis, Möglichkeit der Vorhersage
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 12
kinetische Theorie
Verhalten einzelner Moleküle verschiedenen Typs wird betrachtet;Boots-Modell (verschiedene Typen von Booten auf einem Fluss)
Beispiel: 3 Substanzen (I, a, b)I Inertsubstanz (ohne Retention), wandert gleich schnell wie mobile Phasea mit einer bestimmten Retentionb mit Retention größer als a (wandert langsamer als a)
am Säulenkopf aufgegeben, dann LM-Gemisch zugeführt (wandert infolge Schwerkraft durch die Säule), Subst. I, a und be werden mittransportiert und verlassen nacheinander die Säule am unteren Ende;
Zeit vom Beginn der Entwicklung gemessen, Subst.-Konzentration am Ende der Trennstrecke laufend gemessen → Darstellung als zeitabhängiges Konzentrationsprofil: Elutionskurve
N. Haider, Universität Wien www.pharmxplorer.at 13
Theorie der Böden (Trennstufen-Modell)
Verteilungsgleichgewicht (Substanz ↔ mobile/stationäre Phase) stellt sich nicht unendlich schnell ein. Trennstrecke wird in gedachte Abschnitte zerlegt, die gerade lang genug sind, damit Gleichgewichtseinstellung erfolgen kann:Länge so eines Abschnittes: Höhenäquivalent eines theoretischen Bodens(Begriff aus der Destillation) bzw. height equivalen to a theoretical plate(HETP) bzw. Trennstufenhöhe (H)
Ursachen für „nicht ideale“ Chromatographie:• Viskosität der mobilen Phase• Diffusionseffekte:
• Longitudinale Diffusion: durch unterschiedlich lange Aufenthaltszeitengleichartiger Substanzmoleküle in der stationären Phase
• Streudiffusion = Eddy-Diffusion: durch unterschiedlich lange Aufenthalts-zeiten glaichartiger Substanzmoleküle in der mobilen Phase
Je kleiner die Trennstufenhöhe, umso mehr Trennstufen pro Trennstreckenlänge→ umso besser die Trennleistung des Systems
Kenngrößen eines chromatographischen Peaks
Basisbreite:• Ermittlung der Wendepunkte• Anlegen der Wendetangenten• Abstand der Schnittpunkte der
Wendetangenten mit der Basislinie
Halbwertsbreite:• keine Wendetangenten erforderlich• Schnittpunkt einer horizontalen
Linie auf halber Peakhöhe mit denbeiden Peakflanken
• bei unsymmetrischen oder überlappenden Peaks leichter zuermitteln als Basisbreite
Halbwertsbreite ist nichtdie halbe Basisbreite!!!
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 14
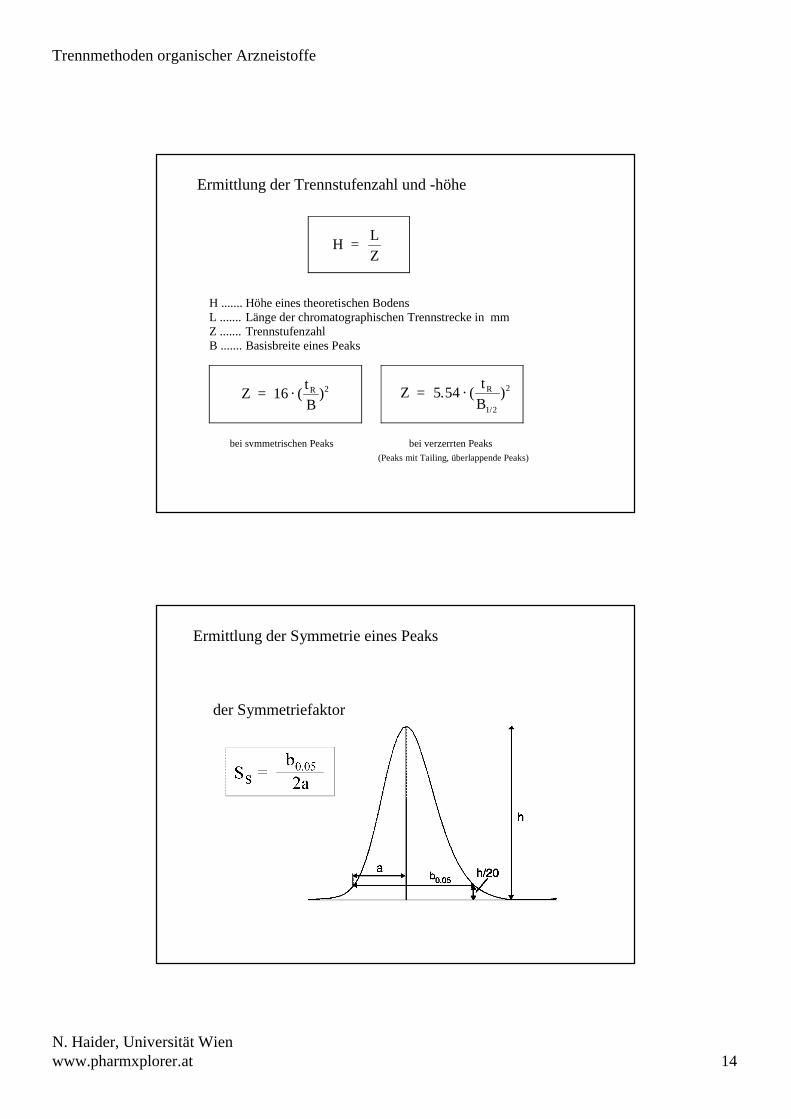
Ermittlung der Trennstufenzahl und -höhe
H ....... Höhe eines theoretischen BodensL ....... Länge der chromatographischen Trennstrecke in mmZ ....... TrennstufenzahlB ....... Basisbreite eines Peaks
bei symmetrischen Peaks bei verzerrten Peaks
H = LZ
Z = 16 · (tBR )2
Z = 5.54 · (
tB
R
1/2
)2
(Peaks mit Tailing, überlappende Peaks)
Ermittlung der Symmetrie eines Peaks
der Symmetriefaktor
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 15
dynamische Theorie
• Erweiterung der Theorie der Böden• Zusammenhang zwischen der theoretischen Trennstufenhöhe H
und dynamischen Erscheinungen (Diffusions-, Strömungseffekte und Nichtgleichgewichtseinstellung): van Deemter-Gleichung
dynamische Theorie
• van Deemter-Gleichung stellt Trennstufenhöhe H der linearen Strömungsgeschwindigkeit u gegenüber
• H umso kleiner (→ mehr Trennstufen!), wenn A, B, Cmöglichst klein sind
A: berücksichtigt Eddy-Diffusion, ist klein bei:gleichmäßiger Packungeinheitlichem Teilchendurchmesser der stationären Phasekleiner Teilchengröße der stat. Phase
B: berücksichtigt longitudinale Diffusion, ist klein bei weiten und unverzweigten Poren in der stationären Phase
C: berücksichtigt Geschwindigkeit der Gleichgewichts-einstellung, ist klein bei niedriger Viskosität der mobilen Phase
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 16
• geringer apparativer Aufwand (billig)• geringer Zeitaufwand• hohe Trennleistung• niedrige Nachweisgrenzen• hohe Selektivität des Nachweises
charakteristische Merkmale:
Trennstrecke besteht aus dünner Schicht stationärer Phase, die sich auf einer geeigneten inerten Unterlage (Glas, Alu-, Plastikfolie) befindet. Die Trennung erfolgt näherungsweise in zweiter Dimension (Unterschied zu SC).→ flat-bed-Methode.
Dünnschichtchromatographie
Bezüglich Trennmechanismen: am häufigsten Adsorption, Verteilung (ev. auch Ionen-austausch)
2) F: mit FluoreszenzindikatorP: für präparative ChromatographieR: besonders gereinigt (z.B. für quant. in situ Auswertung)Zahlenangaben: mittlerer Porendurchmesser in Å (z.B. Typ 60 ..... 60 Å)
Dünnschichtchromatographie
Adsorptionskraft des Sorbensmaterials → Aktivität
hängt ab von:
• Teilchengröße (kleine Teilchen → große spezifischeOberfläche (z.B. Kieselgel 60: 500 m2/g); in DC ca. 0.5-25 µm
• Zahl und Durchmesser der Kanäle (Poren), die bei derHerstellung entstehen
• Wassergehalt des Sorbens
Herstellung der Schichten: heute prakt. nur mehr industriell
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 18
Dünnschichtchromatographie
Auftragen der Substanzenetwa 5 - 20 µg Substanz in einem LM, das die Substanz gut löst: z.B. Aceton, Methanol, Ethanol, Dichlormethan0.1 - 5%ige Lösungen, 2 - 20 µl mit Mikropipetten, MikroliterspritzenBelastbarkeit: 10-5 g/g Sorbens
Entwicklung des Chromatogramms erst nach völligem Abdunsten des zumAuftragen verwendeten LM
• punktförmiges Auftragen: v.a. in analytischer DC
• strichförmiges (bandförmiges) Auftragen: v.a. in präparativer DC
Fleckenform und -größe beeinflussen Ergebnis der Trennung
Dünnschichtchromatographie
Thermomikro-Auftrageverfahren nach Stahl (TAS)spezielles Verfahren zum Auftragen leicht flüchtiger Substanzen aus komplexer Matrix (z.B. pflanzl. Material)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 19
Dünnschichtchromatographie
Fließmittel-Auswahl
• Nacharbeiten vorgegebener Arbeitsvorschriften: LM mit definierterReinheit und def. Wassergehalt verwenden (p.A.-Qualität)
• eigene Methodenentwicklung
• Struktur der Probenmoleküle bekannt → Abschätzung der Polarität→ Abschätzung der erforderlichen ElutionskraftReihung der Lösungsmittel nach steigender Elutionskraft in Verbindungmit einem bestimmten Sorbens, bezogen auf n-Pentan als Nullpunkt→ eluotrope Reihe
• unbekannte Eigenschaften der Analysensubstanz → Auswahl desFließmittels durch „Versuch und Irrtum“; experimentell rasch durchzu-führen mittels Mikrozirkulartechnik
N. Haider, Universität Wien www.pharmxplorer.at 20
Dünnschichtchromatographie
Fließmittel-Auswahl mittels dem Stahl‘schen Dreieck:
Dünnschichtchromatographie
Fließmittel-Auswahl mittels Mikrozirkulartechnik
Startfleck
FM zu apolar
FM zu polar
optimales FM
mehrere Versuche neben-einander auf einer einzigen DC-Platte möglich(geringer Platzbedarf)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 21
Dünnschichtchromatographie
Fließmittel-Auswahl
• Gemische von Lösungsmitteln mit sehr großem Polaritätsunterschiedvermeiden → Möglichkeit der Ausbildung einer β-Front (teilweiseEntmischung des Fließmittels auf der DC-Platte)
Dünnschichtchromatographie
Entwicklung des Chromatogrammshäufigste und einfachste Form: aufsteigende Entwicklung; 2 Möglichkeiten:• ohne Kammersättigung (Problem der nicht konstanten FM-Zusammensetzung)• mit Kammersättigung (gibt besser reproduzierbare RF-Werte)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 22
Dünnschichtchromatographie
Entwicklung des Chromatogramms
• Laufstrecken meist 5-15 cm
• Trennstufenzahl meist zwischen 500 und 3000
• Laufzeit meist 10-30 minbei aufsteigender DC unter Kammersättigung verhalten sich dieLaufzeiten wie die Quadrate der Laufstrecken
neben normalen Kammern auch Horizontal-Entwicklungskammern,Sandwich-Kammern (S-Kammern), Doppeltrog-Kammern
Dünnschichtchromatographie
Sandwich-Kammer Doppeltrog-Kammer
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 23
weitere DC-Entwicklungstechniken
Mehrfach-Entwicklung
bei ungenügender Trennung, vor allem bei präp. DC;Trocknen, neuerliche Entwicklung mit gleichem FM
→ Trennstufenzahl ist dadurch zu vergrößern.
Berechnung, dass bei der DC die Auflösung am besten bei einem RF von ca. 0.3 ist. (kleiner → zu geringe Trennstufenzahl; größer → Auflösungs-verschlechterung durch Fleckenverbreiterung)
Berechnung des RF-Wertes nach Mehrfach-Entwicklung:
nRF = 1-(1-RF)n n ... Anzahl derEntwicklungen
weitere DC-Entwicklungstechniken
Stufentechniknach Entwicklung mit FM 1 → Trocknen → Entwickeln mit FM 2; vor allem, wenn präp. Trennung von mehreren polaren und mehrerenunpolaren Verbindungen erfolgen soll: stark polares FM bis zur halbenPlattenhöhe → dann schwach polares FM bis oben.
Keilstreifentechnikvor allem bei knapp beieinander liegenden RF-Werten.Nach der Verjüngung tritt zusätzlich zur Strömung in Laufrichtung eine dazu senkrechte auf, welche die Substanz-Zonen quer zur Laufrichtung und damit auseinander zieht.
ZirkulartechnikIn der Plattenmitte wird FM mit einem Docht auf die Schicht gebracht, welche nach unten liegt; kreisförmige Ausbreitung in alle Richtungen.
F(lin)F(circ) RR =
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 24
weitere DC-Entwicklungstechniken
weitere DC-Entwicklungstechniken
zweidimensionale EntwicklungArt der Ausführung:• mit zwei verschiedenen FM zur besseren Trennung schwer trennbarer
Gemische (z.B. Arzneistoff in biolog. Proben)• mit gleichem FM:
falls während der Analyse keine Veränderung der Substanzen eintritt → Punkte nach der 2. Entwicklung auf einer Diagonalentritt Reaktion auf aktiver Oberfläche während der Chromatographie (oder beim Zwischentrocknen) ein → Punkte liegen nicht auf Diagonale
TRT-Technik (Trennung - Reaktion - Trennung)damit kann die chemische Reaktionsfähigkeit von Substanzen untersucht werden. Mögliche Reaktionstypen:• Oxidation• Hydrolyse• Photochemische Reaktionen• Halogenierungen (z.B. mittels Br2-Dampf)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 25
Dünnschichtchromatographie
Detektion der Substanzen
• Eigenfarbe
• Fluoreszenza) bei Tageslichtb) durch Anregung mit UV366 (UV-Lampe)
• chemische Farbreaktionen (→ Sprühreagentien), z.B.:I2 (Lsg. od Dämpfe) braune Flecken bei basischen N-VerbindungenKMnO4 weiße Flecken auf rosa Grund bei reduzierenden Verb.Dragendorff orange-gelbe Flecken bei AlkaloidenFeCl3 zart-violette Flecken bei Phenolen (int. grün bei 1,2-Diphenolen)Ninhydrin blauviolette Flecken bei Aminosäurenetc. etc.
Dünnschichtchromatographie
a
Glasplatte
Sorbensschicht
Fließmittelfront
RF = c
RX = RSt =ab
c
b
a
. . .
St Pr St
3 mm
20 mm
5 mm
Charakterisierung desMigrationsverhaltens
• RF-Wert
• RX-Wert (RSt-Wert)
RF-Wert immer zwischen 0 und 1
RX-Wert auch > 1 möglich
hRF-Wert = RF-Wert x 100
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 26
Dünnschichtchromatographie
Abhängigkeit des RF-Wertes
RF-Wert wäre eigentlich substanzspezifische Kenngröße, wären alle Einfluss-faktoren vollständig kontrollierbar; dies ist aber nicht der Fall, daher gewisserSchwankungsbereich. Experimentelle Faktoren, die den RF-Wert beeinflussenkönnen:
• durchschnittliche Korngröße• Aktivierungsgrad• Luftfeuchtigkeit• Schichtdicke• Art der Schichtherstellung• Sättigungsgrad der Kammeratmosphäre• Entwicklungstechnik• Länge der Laufstrecke• Substanzmenge u. a.
N. Haider, Universität Wien www.pharmxplorer.at 27
Hochleistungs-Dünnschichtchromatographie
Auftragen der Substanzen
in Lösung mit geeigneten Mikroliterspritzen; spezielle Auftrage-vorrichtung (z.B. „Linomat“)
Hochleistungs-Dünnschichtchromatographie
Entwicklung
meist horizontal in Flachkammern, sodass praktisch kein Gasraum → daher hervorragende Reproduzierbarkeit. Phasenfluss ist konstant; Zufuhr des FM mittels Kapillareffekt (hydrostatisch mit FM versorgt).
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 28
Hochleistungs-Dünnschichtchromatographie
Detektion
meist mittels Densitometer = DC-Scanner (bzw. HPTLC-Scanner)→ rel. genaue quantitative Auswertung möglich: Platte wird von Lichtstrahl abgetastet, Intensität des durchtretenden oder diffus reflektierten Lichtes wird gemessen und als Kurve aufgezeichnet
Durchführung in vertikal montierter Glasröhre (=„Säule“; Länge einige cm bis m), Verhältnis Länge/Durchmesser ca. 40 : 1
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 29
Säulenchromatographie
Säulenchromatographie
Sorbentien: alle schon bei der DC angeführten Materialien; Teilchendurchmesser meist ca. 100 µm; Sorbens-Aktivität kann hier vergleichsweise einfach festgelegt werden.
Aktivitätsstufen nach Brockmann:
Wassergehalt (%)
Stufe Al2O3 Kieselgel
I*)
IIIIIIVV
0361015
010121520
*) nach Erhitzen auf 150°C
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 30
Säulenchromatographie
Problem der gleichmäßigen Dampfraumsättigung (s. DC) entfällt bei SC; Gleichmäßigkeit der Säulenfüllung wichtig für Trenn-ergebnis → Einschlämmen des Füllmaterials; Fließmittel wandert infolge der Schwerkraft, ev. geringer Über-druck (bis ca. 5 bar, MPLC = "medium pressure liquid chromatography"), falls Strömungsgeschwindigkeit bei Verkleinerung der Teilchengröße zu stark abnimmt.
In der Adsorptions-SC gelten die bekannten Elutionsreihen.Übliche Druchflussrate 1-10 ml/min.Auffangen von Eluatfraktionen (automatisierbar).Man erhält ein äußeres Chromatogramm
Säulenchromatographie
Entwicklungstechniken
• Elutionstechnik: am meisten verwendetSubstanzgemisch in konzentrierter Lsg. aufgetragen, mit FM so lange entwickelt, bis alle Substanzen nacheinander die Säule verlassen haben
• Gradienten-Elution: Elutionskraft wird stetig erhöht
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 31
Säulenchromatographie
Gradientenelution:
• Verkürzung der Analysenzeit• Verringerung des LM-Verbrauchs• Verbesserung des Trennergebnisses durch Zurückdrängen von „tailing“• Verbesserung der Peakform (schmäler, dafür höher), wichtig bei Peaks
• Frontaltechnik:Verdünnte Lösung des zu trennenden Gemisches im Fließmittel wird kontinuierlich auf die Säule aufgegeben. Substanz mit geringster Retention erscheint als erste im Eluat rein, weitere Fraktionen sind dann Mischfraktionen.Anwendungsbeispiele:Entfernung von Peroxiden aus LM (Ether)Entfernung von Stabilisator-EtOH aus ChloroformEntwässern von LM
• Verdrängungstechnik:mobile Phase enthält „Displacer“, um stark retinierte Substanzen von der stat. Phasezu verdrängen; Anwendung v.a. in der Ionenaustauschchromatographie
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 32
Säulenchromatographie
Detektion
• diskontinuierlichZeit- oder Volumengesteuerte Fraktionskollektoren; Untersuchungder einzelnen Fraktionen mittels Farbreaktionen, DC, GC,...
Weiterentwicklung der klassischen SC; mit abnehmender Teilchengröße steigt erforderlicher Druck exponentiell an; Schwerkraft reicht nicht mehr aus, FM muss mittels geeigneter Einrichtungen unter Druck gesetzt werden (Pumpen): 20-200 bar.
Feinkörnigeres Material der stationären Phase (ca. 5 µm) → wesentliche Steigerung der Trennstufenzahl pro Säule gegenüber klass. SC erzielbar
ca. 50000-100000 Trennstufen/m
typische Säulenlänge 12.5 cm → ca. 10000 Trennstufen / Säule
Hochleistungs-Flüssigchromatographie (HPLC)
GeräteaufbauSystem für Gradientenelution (schematisch)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 34
Hochleistungs-Flüssigchromatographie (HPLC)
GeräteaufbauSystem für Gradientenelution
Hochleistungs-Flüssigchromatographie (HPLC)
GeräteaufbauPumpen: möglichst gleichmäßige Fließgeschwindigkeit der mobilen Phase; häufigster Pumpen-Typ: Kurzhub-Kolbenpumpe → Kolben (meist aus künstl. Saphir) über Nockenwelle angetrieben; hohe Anforderungen an Dichtungen (bes. bei Verwendung aggressiver Medien: Puffer, Salzlösungen); Einlass- u. Auslassventil (Kugeln); Förderleistung meist 0-10 ml/min
Pulsationen: müssen gedämpft werden (wichtig bei best. Detektoren) → Pulsationsdämpfer oder Doppelkolbenpumpen (Gegentakt)Gradientenerzeugung:a) Niederdruck-Gradient: nur 1
N. Haider, Universität Wien www.pharmxplorer.at 35
Hochleistungs-Flüssigchromatographie (HPLC)
GeräteaufbauProbenaufgabe: druckloses Einbringen der Probe in eine "Probenschleife" (Volumen 1 µl - 2000 µl, exakt reproduzierbar!), mittels Mehrweghahn-Vorrichtung (Probenventil) in Trennsäule gespült
Hochleistungs-Flüssigchromatographie (HPLC)
GeräteaufbauProbenaufgabe mittels automat. Probenwechsler (Autosampler): Magazin mit einer größeren Anzahl an vorbereiteten Probenfläschchen beladen; Roboterarm mit Nadel, sticht durch Gummiseptum in das gewünschte Fläschchen, saugt Probelösung auf und injiziert automatisch über ein Probenventil; Gerät muss entsprechend programmiert werden; ermöglicht unbeaufsichtigte Serienanalysen (z.B. bei Qualitätskontrolle in pharm. Industrie, in diagnostischen Labors, bei Behörden etc.)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 36
Hochleistungs-Flüssigchromatographie (HPLC)
SäulenMaterial: meist Edelstahl, innen poliert, ev. glasbeschichtet; Endverschraubung mit Stahlfritte; Stahlkapillare mit Klemmring-Anschluss (z.B. "Swagelok").
Andere Variante: auswechselbare Kartuschen-Säulen aus gehärtetem Spezialglas (kostengünstiger als Edelstahlsäulen) + Kartuschenhalterung aus Stahl (einmalige Anschaffung).
Hochleistungs-Flüssigchromatographie (HPLC)
SäulenInnendurchmesser: meist ca. 4 mm; präp. HPLC: auch 20 mm und mehrLänge: 5-50 cm; meist 12.5 oder 25 cm (Vorsäulen 2-5 cm)Säulenfüllen: Slurry-Verfahren → entgaste Aufschlämmung des Füll-Materials in geeignetem LM unter hohem Druck eingepresst
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 37
Hochleistungs-Flüssigchromatographie (HPLC)
SäulenMontage: in entsprechender Halterung; vorteilhaft: thermostatisierbarer Säulenofen (Trennungen besser reproduzierbar, bes. bei Verteilungschromatographie)
Hochleistungs-Flüssigchromatographie (HPLC)
Detektorenprakt. nur kontinuierliche Detektion selektive - unselektive Detektoren; unterschiedliche Empfindlichkeiten
• UV/Vis-Durchflussphotometera) mit konstanter Wellenlängeb) mit variabler Wellenlängec) Diodenarray-Detektor
• Fluorimeter
• Differentialrefraktometer
• Polarograph
• Leitfähigkeitsdetektor
• Massenspektrometer
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 38
Hochleistungs-Flüssigchromatographie (HPLC)
UV/Vis-Detektorselektiv, nur UV/Vis-absorbierende Substanzen erfassbar; trotzdem relativ universell einsetzbar (Detektoren mit variabler Wellenlänge); Nachweisgrenze ca. 10-9 g/mlmeist 2-Strahl-Prinzip: Lichtquelle, 2 Strahlen durch Blenden/Linsen-Kombination, Küvetten aus Quarzglas, Photodiode, elektr. Verstärkung; statt (austauschbarem) Filter: Monochromator (stufenlose Selektion der Wellenlänge möglich); Bereich: 200-400 nm (mit Deuteriumlampe), 350-800 nm (mit Wolframlampe)
Fließmittel darf beigewählter Wellenlänge nicht absorbieren;
Gradientenelution möglich
Hochleistungs-Flüssigchromatographie (HPLC)
Diodenarray-DetektorWeiterwentwicklung des UV/Vis-Detektors: viele Wellenlängen gleichzeitig erfasst; zusätzliche Information über substanzspezifische Eigenschaft (UV-Spektrum); graphische Darstellung in 3-dimensionaler Matrix; Array = Reihe (hier: Reihe von Photodioden; jede Diode misst Licht einer bestimmte Wellenlänge nach Auffächerung des polychromatischen Lichtstrahls mittels Beugungsgitter)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 39
Hochleistungs-Flüssigchromatographie (HPLC)
Diodenarray-Detektorauch Aussage über Peakreinheit (= Einheitlichkeit) möglich → Vergleich des UV-Spektrums an ansteigender Peakflanke mit UV-Spektrum an abfallender Peakflanke → müssen bei einem einheitlichenSubstanzpeak übereinstimmen (X-Achse: Chromatogramm, Y-Achse: UV-Spektrum)
Hochleistungs-Flüssigchromatographie (HPLC)
Fluoreszenz-Detektor (Fluorimeter)
noch selektiver als UV/Vis
Prinzip: best. Substanzen durch UV-Licht zur Fluoreszenz angeregt (Eigenstrahlung bei höherer Wellenlänge); Messung der Intensität des emittierten Lichtes erfolgt rechtwinkelig zur Richtung der UV-Anregungsstrahlung
Nachweisgrenze ca. 10-10 g/ml; ev. Derivatisierung mit fluoreszenzfähigen Reagentien → auch Verbindungen erfassbar, die von Haus aus nicht fluoreszieren
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 40
Hochleistungs-Flüssigchromatographie (HPLC)
Brechungsindex-Detektor (Differential-Refraktometer)unselektiv, dafür universell verwendbar; rel. unempfindlich;Lichtquelle, Blende/Linse, Referenz- und Messzelle mit schräggestellter Trennwand (Winkel beeinflusst Empfindlichkeit), Spiegel, opt. Nullpunkt, Photodiode;Referenzzelle: mit reinem Eluenten gefüllt; genaue Thermostatisierung erforderlich (Brechungsindex ist temperaturabhängig!), kein Gradientenbetrieb möglich; Anwendung: z.B. für Zucker; Nachweisgrenze ca. 10-7 g/ml
Hochleistungs-Flüssigchromatographie (HPLC)
Elektrochemischer Detektor (Polarograph)Oxidierbarkeit oder Reduzierbarkeit von Substanzen genutzt; mobile Phase muss gewisse Leitfähigkeit besitzen (ev. Zusatz geeigneter Elektrolyte)Messung des Stromflusses; sehr empfindlich (leider auch auf Pulsationen); selektivNachweisgrenze ca. 10-11 g/ml
Leitfähigkeits-Detektorbei leitender mobiler Phase zur Detektion von Ionen: 2 Elektroden, Spannung, Messung des Widerstandes
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 41
Hochleistungs-Flüssigchromatographie (HPLC)
MS-Detektor (Massenspektrometer)Vorteil: hoher Informationsgehalt → Hochleistungstrennverfahren + Verfahren zur StrukturaufklärungGroßer apparativer Aufwand zur Kopplung der beiden Geräteteile (HPLC + MS) → Fließmittel muss entfernt werden (versch. Spray-Verfahren, z.B. „Electrospray“); Einschränkungen z.B. bei Wahl von Pufferlösungen
Hochleistungs-Flüssigchromatographie (HPLC)
Detektor-Output:
• analoges elektr. Signal (Ausnahme: Diodenarray, MS)
Kieselgel (Si-OH-Gruppen): schwach sauerAluminiumoxid: meist basisch - neutralapolare mobile Phase: z.B. Kohlenwasserstoffe, Ether: definierter Wassergehalt! (z.B. 50% Sättigung)langsame Gleichgewichtseinstellung; Problem: Reproduzierbarkeit
• Verteilung: meist als Reversed-Phase-Chromatographie: RP-Säulena) Flüssig-flüssig-Verteilung: apolare flüssige stationäre Phase (auf inertem porösem Trägermaterial); polare (wässrige) mobile Phase;Gefahr des „Ausblutens“ der stat. Phaseb) chemisch gebundene Phasen (heute üblich): Alkylketten kovalent mit Kieselgel verknüpft, z.B. RP-18
Cl3Si-(CH2)17-CH3- HCl
Si O-SiCl2-(CH2)17-CH3
OH2
Si O-Si(OH)2-(CH2)17-CH3
(CH3)3SiClSi O Si
O-Si(CH3)3
O-Si(CH3)3
(CH2)17-CH3
Si OH +
Hochleistungs-Flüssigchromatographie (HPLC)
Trennmechanismen• Gelchromatographie: Trennung nach Molekülgröße
soft-gels: wenig druckbeständig (z.B. Polyacrylamid), nur für SCsemi-rigid-gels: bis ca. 60 bar (vernetztes Polystyrol)rigid-gels: druckstabil (z.B. poröses Glas)
• Ionenaustausch-Chromatographie: Kationen-, Anionenaustausch; mobile Phase: Pufferlösung
• Ionenpaarchromatographie:Ionenpaar: Alternative zu Ionenaustausch und IonensuppressionSysteme für Kationen und Anionen verfügbar; Zusatz eines geeigneten Gegenions, z.B. Alkylsulfonate für Kationen, Tetraalkylammoniumverbindungen für Anionen; gemeinsame Solvathülle; chromatograph. Verhalten wie Neutralstoff; Trennung auf RP-Säulen möglich
• Bioaffinitätschromatographie: für biologische Problemstellungen; stationäre Phase: makromolekulare Verbindung mit „biologisch aktiven“ Gruppen; Wechselwirkungsprinzip: z.B. Antigen -Antikörper; Enzym - Ligand (Inhibitor)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 43
Hochleistungs-Flüssigchromatographie (HPLC)
Variationsmöglicgkeiten• stationäre Phase: Trennmechanismus (und damit Selektivität)• mobile Phase: bei Adsorption und Verteilung: Polarität; bei Puffern
(Ionenaustausch): pH, Ionenstärke• Gradientenelution: binär, ternär; auch Flussgradient
Vorteil: Reaktionsbedingungen frei wählbar; chrom. Eigenschaftender Probe u.U. verbesserbarNachteil: Gefahr von Artefakten, ev. keine quant. Umsetzung
• nach der Elution (kurz vor Detektor): post-columnVorteil: Nebenprodukte irrelevant; zusätzlich 2. Detektor möglich Nachteil: technisch aufwendig, Reaktion muss in mobiler Phase ablaufen können, Reagens selbst darf kein Signal liefern
Hochleistungs-Flüssigchromatographie (HPLC)
Anwendungen
• Analytik:universelle Methode, sehr flexibel, gut standardisierbar, auch für sehr geringe Probenkonzentrationen geeignet; quantitative Bestimmungen gut reproduzierbar; durch Kombination mit speziellen Detektoren (Diodenarray, Massenspektrometer) großer Gewinn an Information
• präparative Trennungen:HPLC sehr leistungsfähig, trotzdem weniger verbreitet infolge hoher Investitions- und Betriebskosten (Säulen,Lösungsmittel)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 44
chromatographische Trennmethoden
Gaschromatographie (GC)besondere Form der Säulenchromatographie, bei der als mobile Phase ein Gas fungiert; einsetzbar zur Trennung und qual. und quant. Bestimmung von Substanzen, die unzersetzt in die Dampfform übergeführt werden können, bzw. aus denen sich unzersetzt verdampfbare Derivate herstellen lassen. Verflüchtigungstemperatur 50-300°C.
Besonderheiten infolge niedriger Viskosität der mobilen Phase:• kurze Analysenzeiten• hohe Trennschärfe• sehr geringe Substanzmengen trennbar• Substanzen im Gaszustand → hohe Diffusionsgeschwindigkeiten,
daher rasche Einstellung der Gleichgewichte zwischen mobiler und stationärer Phase
Gaschromatographie (GC)
Zeit (min)
Trägergas
Säulenofen (thermostatisiert)
Proben- einlaß
Spritze Detektor
Schreiber
Chromatogramm
Regelventil
Säule
Prinzip des Geräteaufbaus
Probe wird mittels Injektionsspritze in den geheizten Probeneinlass eingebracht, dort verdampft sie augenblicklich und wird vom Trägergas durch die Trennsäule transportiert. Die mit dem Trägergas in den Detektor gelangenden getrennten Substanzen werden vom Schreiber/Integrator/PC als Peak erfasst.
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 45
Gaschromatographie (GC)
Geräte
Die meist sehr lange, aber dünne Trennsäule ist nicht langgestreckt montiert, sondern aus Platzgründen aufgewickelt (s. rechtes Bild).
Gaschromatographie (GC)
Trägergase:He, Ar, H2, N2 (wichtig!), CO2 (zunehmende Polarität)
H2: niedrigste Viskosität, daher auch bei längeren Säulen optimale Strömungs-geschwindigkeit gewährleistet, jedoch Explosionsgefahr;Durchflussgeschwindigkeit: 30-80 ml/min
Trennsäulen:gepackte Säulen: Edelstahl bzw. Glassäulen, Belastbarkeit bis 10 µl Lösung;
stationäre Phase ist auf Oberfläche eines inerten gekörnten Träger-materials aufgezogen; meist 2-3 mm Durchmesser, Länge: einige m
Kapillarsäulen: stationäre Phase befindet sich als dünner Film an der Innenwandeiner Quarzglaskapillare; meist 0.1-1 mm Durchmesser, Länge: 30-300 mBelastbarkeit 10-1 bis 10-3 µl Lösung (spezielle Injektoren: Split)wesentlich höhere Z erzielbar, wesentlich bessere Trennergebnisse• Dünnfilmkapillare: 2-3 µm dünner Film der stat. Phase• Dünnschichtkapillare: an Innenwand dünne Schicht von
mit stat. Phase belegtem Trägermaterial
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 46
Probenaufgabe:Mit Mikroliterspritze durch ein Silikongummiseptum; muss so erfolgen, dass die Probe augenblicklich verdampft. Gase: 0.1 - 10 ml; Flüssigkeiten:gepackte Säulen: 0.1 - 10 µl, Kapillarsäulen um Faktor 1000 weniger; Feststoffe: nur wenn ausreichende Flüchtigkeit gegeben ist, in LM gelöst, in Injektor wie Flüssigkeit eingebracht;
Für nicht unzersetzt verflüchtigbare Verbindungen → Derivatisierung zu flüchtigen Derivatena) Acetylierung: OH, NH2, NHR, z.B. mit Acetanhydrid, Trifluoracetanhydrid
(mit hochempfindlichem ECD Fluor gut erfassbar)b) Silylierung: H in OH, NH, COOH, SH durch Trimethylsilylgruppe ersetzt
(z.B. mit N,O-bis-Trimethylsilylacetamid)
Trennung: isotherm (bei konstanter Temp.) oder mit programmiertem Temperaturanstieg → Temperaturgradient; Obergrenze, wenn stationäre Phase zu verdampfen beginnt („Säulenbluten“ = "bleeding")
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 47
Detektoren für die Gaschromatographie
Wärmeleitfähigkeitsdetektor (WLD):Vergleich der Wärmeleitfähigkeit des Trägergases allein und des Trägergases, das Komponenten der Probe enthält → Differential-Detektor. Beruht auf der Änderung des Widerstandes eines Heizdrahtes je nach Ausmaß, in dem ein ihn umgebendes Gas Wärme an die Detektorwand abtransportiert. Signal ist konzentrationsabhängig. Erfasst werden alle (auch C-freie) Verbindungen, welche die WLF des Trägergases verändern → universell
Detektoren für die Gaschromatographie
Flammenionisationsdetektor (FID):Eluat wird in eine Wasserstoff-Flamme geleitet und dort verbrannt. Gemessen wird die Zunahme der Ionen in der Flamme dadurch, dass der zwischen 2 Elektroden fließende Strom registriert wird. Angezeigt werden nur kohlenstoffhältige verbrennbareVerbindungen (also nicht Wasser, Edelgase, Stickstoff, CO, CO2, O2, CCl4, NH3)Das Signal ist massenstromabhängig; auch Halogen-, N-, P-selektive FIDs verfügbar.
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 48
Detektoren für die Gaschromatographie
Elektroneneinfangdetektor,Electron Capture Detector (ECD):Im Detektor befindet sich ein Beta-Strahler (63Ni, 3H). Gasraum ist durch freie Elektronen leitend. Substanzen, die Elektronen einfangen können (vor allem halogenhältige) führen zur Abnahme des gemessenen Elektronenstroms → zeigt Substanzen mit hoher Elektronenaffinität an
Massenspektrometer (MS):hochspezifisch (Kopplung on-line, heute nur mehr mit Kapillar-GC); Trägergas muss vollständig entfernt werden → leichter bei Kapillar-GC realisierbar;Aufzeichnung des Totalionenstroms (total ion current, TIC); zu jedem GC-Peak wird das entsprechende Massenspektrum erhalten (Vergleich mit Spektrenbibliotheken möglich)Bei fixer Einstellung des MS auf ein bestimmtes m/z Verhältnis → massenspezifische Detektion, "single ion monitoring" (SIM)
Gaschromatographie (GC)
Anwendungen:
qualitative und quantitative Analyse von Gemischen flüchtiger Verbindungen; auch wenn sehr komplexe Gemische vorliegen (z.B. äther. Öle)
wenig verbreitet: präparative GC
Kopplung: Trennverfahren mit Nachweisverfahrenon-line: direkte Verbindung z.B. GC-MS, auch GC-IR (hier FT-IR)off-line (Transfer erforderlich), z.B. GC-NMR
Normierung des Retentionsverhaltens von Verbindungen möglich (Kovats-Index, McReynolds-Konstante)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 49
weitere chromatographische Trennmethoden
Supercritical Fluid Chromatography (SFC):
Zwischenstellung zwischen HPLC und GCmobile Phase: überkritisches Fluid
Kompression eines Gases → Verflüssigung, aber nur wenn Temperatur unterhalb der sog. kritischen Temperatur liegt. Andernfalls → Gas immer dichter und dichter (Fluid), aber keine Kondensation zu flüssiger Phase.
• geringe Viskosität (ähnl. GC)• hohe Beladbarkeit (ähnl. HPLC)
mobile Phase: meist überkritisches CO2, ev. mit polarem “Modifier”, z.B. MeOH (bis zu 30%); ev. + “Additiva” (geringe Mengen Säure oder Base)
stat. Phase: z.B. stat. Phasen aus HPLC verwendbar
weitere chromatographische Trennmethoden
Supercritical Fluid Chromatography (SFC):
Kapillar-SFC: Erwartungen nicht ganz erfüllt → zurück zur SFC in gepackten Säulen
Druck mittels Restriktor bis nach dem Detektor aufrechterhalten(z.B. 30°C, 200 bar)
Detektoren: modifizierte HPLC-Detektoren, ev. auch FID
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 50
kommt ohne festes Trägermaterial für die flüssige stationäre Phase aus. Frühe Versionen: Craig-Apparatur, DCCC (Droplet Counter-Current Chromatography), RLCCC (Rotation Locular Counter-Current Chromatography);
neuere Entwicklung: HSCCC (High-Speed Counter-Current Chromatography) → stationäre Phase in spiralförmig aufgewickelten Kunststoff-schläuchen, die in einem Exzenter-System um zwei Achsen gleichzeitig rotieren; mobile Phase langsam durchgepumpt; gute Trennleistung, auch für präparative Trennungen geeignet (z.B. Phytochemie)
N. Haider, Universität Wien www.pharmxplorer.at 51
chromatographische Trennmethoden
Auswertung von Chromatogrammen
• Beurteilung der Trennleistung eines chrom. Systems
• Qualitative Auswertung eines Chromatogramms
• Quantitative Auswertung eines Chromatogramms
• äußere Chromatogramme (Elutionskurven)
• innere Chromatogramme
• Validierung von Analysenmethoden
Beurteilung der Trennleistung eines chrom. Systems
die Auflösung (A)
angestrebt wird optimale Trennung (nicht unbedingt „maximale“ Trennung), nähere Definition über den Begriff der Auflösung:
Auflösung A = Maßzahl dafür, wie weit die Peaks zweier Substanzen (a, b) voneinander getrennt sind;
A ist eine rechnerische Größe, aus einem äußeren Chromatogramm ermittelbar:
A = 2 ·tR(b) - tR(a)
Bb + Ba
tR = Gesamtretentionszeit, B = Basisbreite des jeweiligen Peaks
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 52
Beurteilung der Trennleistung eines chrom. Systems
die Auflösung (A)
Peaks können mit Hilfe der Wendetangenten zu Dreiecken vereinfachtdargestellt werden:
A = 0 : keine Trennung
A < 1 : unvollständige Trennung
A = 1 : Dreiecksformen getrennt, aberinfolge der Glockenform der Peaksnoch teilweise Überlappung
A = 1.5 und darüber: gute Trennung(Basislinien-Trennung)
Beurteilung der Trennleistung eines chrom. Systems
die Auflösung (A)
Bei unvollständiger Trennung: Basisbreiten d. Peaks schwierig zu ermitteln→ Auflösung kann auch mit Hilfe der Halbwertsbreiten d. Peaks berechnetwerden.
A = 1.18 ·tR(b) - tR(a)
B0.5(b) + B0.5(a)
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 53
Beurteilung der Trennleistung eines chrom. Systems
Qualität einer Trennung
Eine Verbesserung der Trennung zweier Substanzen (= Vergrößerung der Auflösung) kann erzielt werden durch:
Erhöhung der Selektivität mehr Trennstufen(z.B.: andere mobile Phase) (z.B.: kleinere Korngröße d. stat. Phase)
Beurteilung der Trennleistung eines chrom. Systems
Zusammenhang zwischen der Auflösung (A) und der Zahl der Trennstufen (Z):
A = 41 . Z . .r - 1
r
k´b
k´b + 1
r : relative Retention
k´ : Kapazitätsfaktor
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 54
Qualitative Auswertung eines Chromatogramms
• Anzahl der Substanzen in einem Gemisch:Anzehl der Flecken auf innerem Chrom. (DC, HPTLC)Anzahl der Peaks in äußerem Chrom. (HPLC, GC, ...)
• Wanderungsverhalten der einzelnen Substanzen:RF-Wert bzw. RX-Wert bei innerem Chrom.k´-Wert bei äußerem Chrom.
• Kombination mit weiterer Information aus Detektionsmethode:spezifische Nachweisreaktionen auf DC-Plattespektroskopische Information, z.B. aus Diodenarray-Detektor (HPLC), Massenspektrometer (GC, HPLC)
Quantitative Auswertung eines Chromatogramms
äußere Chromatogramme (Elutionskurven)
Fläche unter dem Peak ist proportional der Substanzmenge, die eluiert wurde; Dagegen ist die Peakhöhe ebenso wie die Peakbreite auch abhängig von der Retentionszeit ( → Peakhöhe ist nur inbestimmten Fällen zur Quantifizierung geeignet; besser: Fläche).
Ermittlung der Peakfläche:• elektronische Integration (Integrator, PC)
• Ausschneiden der Peaks (aus Kopie des Chromatogramms) und Wägen → Gewicht ist proportional der Fläche
• Berechnung mit Hilfe der Peakabmessungen:
F = h . B0.5. 1.0632
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 55
Quantitative Auswertung eines Chromatogramms
Standardisierungsmethoden:
Interner Standard: eine weitere Substanz in bekannter Konz. wird dem Analysengemisch zugesetzt; Flächenverhältnis für Analysensubstanz-Peak zu Standardsubstanz-Peak separat ermitteln (in mehreren Konzentrationen) → Eichgerade
Externer Standard: Authentische Analysensubstanz wird in getrennten Analysenläufen in jeweils bekannter Konz. vermessen → Eichgerade; hier: kein zusätzlicher Peak im Chromatogramm, aber höhere Anforderungen an Reproduzierbarkeit der Bestimmung (Probenvorbereitung, Detektor-Empfindlichkeit, Genauigkeit des injizierten Probenvolumens, usw.)
Quantitative Auswertung eines Chromatogramms
innere Chromatogramme
Fleckengröße (visuell): wichtig für quantitative bzw. semiquant. Auswertung, jedoch kein linearer Zusammenhang mit Substanz-menge:
= prop. log M F : Fleckengröße
M : Substanzmenge
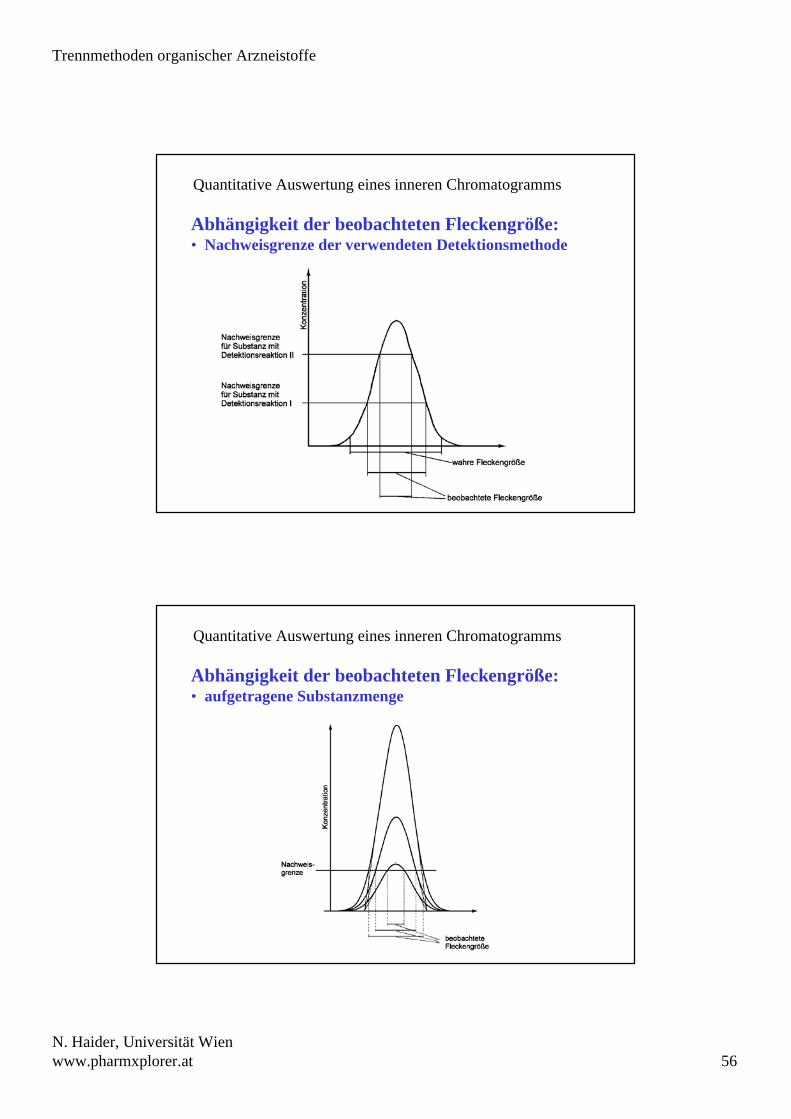
beobachtete Fleckengröße hängt von verschiedenen Faktoren ab
F
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 56
Quantitative Auswertung eines inneren Chromatogramms
Abhängigkeit der beobachteten Fleckengröße:• Nachweisgrenze der verwendeten Detektionsmethode
Quantitative Auswertung eines inneren Chromatogramms
Abhängigkeit der beobachteten Fleckengröße:• aufgetragene Substanzmenge
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 57
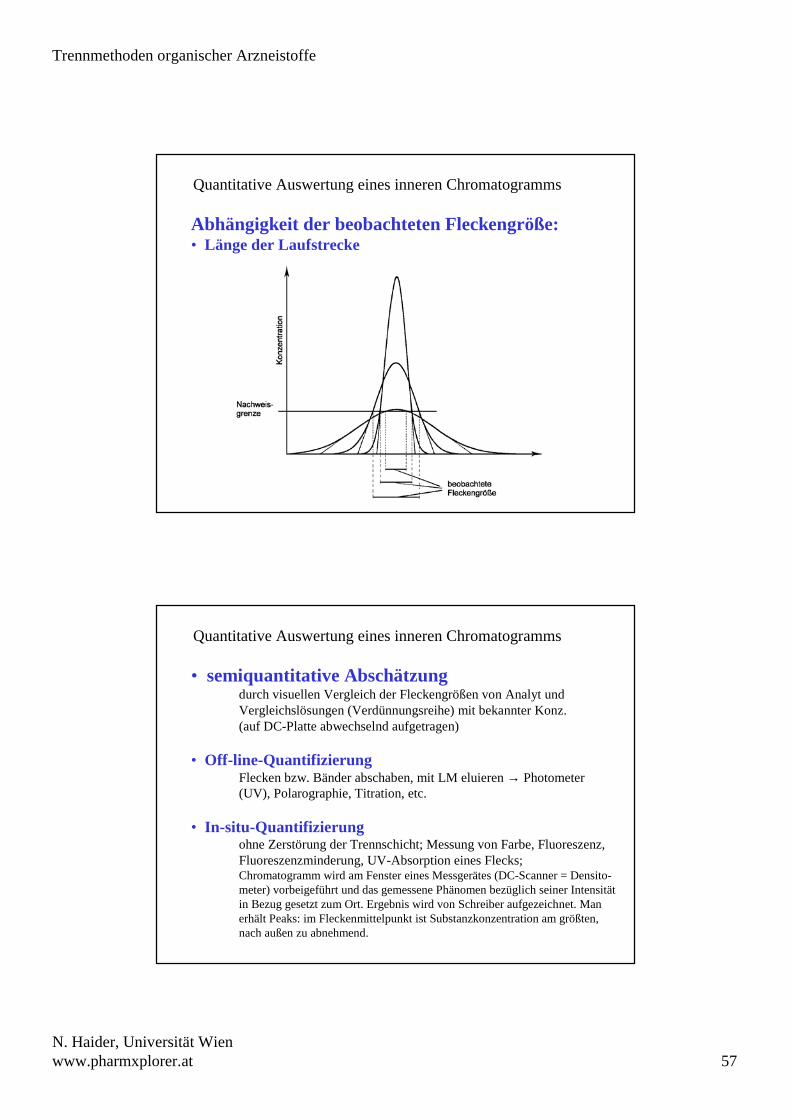
Quantitative Auswertung eines inneren Chromatogramms
Abhängigkeit der beobachteten Fleckengröße:• Länge der Laufstrecke
Quantitative Auswertung eines inneren Chromatogramms
• semiquantitative Abschätzungdurch visuellen Vergleich der Fleckengrößen von Analyt undVergleichslösungen (Verdünnungsreihe) mit bekannter Konz.(auf DC-Platte abwechselnd aufgetragen)
• Off-line-QuantifizierungFlecken bzw. Bänder abschaben, mit LM eluieren → Photometer(UV), Polarographie, Titration, etc.
• In-situ-Quantifizierungohne Zerstörung der Trennschicht; Messung von Farbe, Fluoreszenz, Fluoreszenzminderung, UV-Absorption eines Flecks;Chromatogramm wird am Fenster eines Messgerätes (DC-Scanner = Densito-meter) vorbeigeführt und das gemessene Phänomen bezüglich seiner Intensität in Bezug gesetzt zum Ort. Ergebnis wird von Schreiber aufgezeichnet. Man erhält Peaks: im Fleckenmittelpunkt ist Substanzkonzentration am größten, nach außen zu abnehmend.
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 58
Quantitative Auswertung eines inneren Chromatogramms
Messprinzipien bei der in-situ-Quantifizierung(häufig angewandt: Messung der Lichtabsorption im UV/Vis-Bereich)
• Messung in Transmission
Messung im Durchlicht; infolge von Streueffekten gilt hier das Lambert-Beer'sche Gesetz nicht. Es muss mit Eichfunktionen gearbeitet werden; nicht unter 330 nm möglich (mangelnde UV-Durchlässigkeit der Glasplatte).
• Messung in Remission
Licht wird in einem best. Winkel auf Schicht gestrahlt, wird dort diffusreflektiert. Man misst Intensität des reflektierten Lichtes und vergleicht Intensität an leerer Stelle der Schicht und an Stelle mit Substanzfleck; empfindlicher; bis hinunter zu 220 nm möglich
Quantitative Auswertung eines Chromatogramms
Validierung von chrom. AnalysenverfahrenÜberprüfung und Bewertung der Aussagekraft des jeweiligen Verfahrens anhand einer Reihe von Parametern, die experimentell bestimmt werden.
Parameter: Aussage über:Genauigkeit systematische und zufällige FehlerRichtigkeit systematische FehlerPräzision zufällige FehlerWiederholpräzision laborinterne ReproduzierbarkeitVergleichspräzision Reprod. von Labor zu LaborLinearität Zusammenhang Signal/KonzentrationWiederfindungsrate Ausbeute der ProbenvorbereitungSelektivität Störungen durch BegleitstoffeRobustheit Störanfälligkeit durch veränderte ParameterNachweisgrenze kleinste nachweisbare MengeBestimmungsgrenze kleinste quantifizierbare Menge
Dokumentation, GLP, ISO9000
Trennmethoden organischer Arzneistoffe
N. Haider, Universität Wien www.pharmxplorer.at 59
Trenn- und Analysenmethoden organischer Arzneistoffe
Literatur zum Thema „Trennmethoden“
• J. BöckerChromatographieVogel Buchverlag, Würzburg, 1997
• A. Dominik, D. SteinhilberInstrumentelle AnalytikDeutscher Apotheker Verlag, Stuttgart, 2002
• G. Rücker, M. Neugebauer, G. G. WillemsInstrumentelle Pharmazeutische AnalytikWissenschaftliche Verlagsgesellschaft, Stuttgart, 2001