Page 1
Kapitel: Aminosäuren
1
Stoffwechsel von Aminosäuren (AS)
Repetition Die Aminosäuren besitzen im Stoffwechsel der Zellen vier Funktionen:
• Aminosäuren sind die 20 Bausteine für die Biosynthese der Proteine. Wie bei allen
Körperbausteinen mit Ausnahme der Desoxyribonucleinsäuren besteht auch bei den
Proteinen ein dynamisches Gleichgewicht zwischen Auf- und Abbau. Die
Halbwertszeit der einzelnen Proteine schwankt zwischen Minuten und Stunden bei
einigen Enzymen, Tagen bis Wochen bei Plasmaproteinen und Monaten bei
Strukturproteinen wie dem Kollagen. Die Halbwertszeit wird auch durch das Organ in
dem das betreffende Protein zu finden ist, bestimmt.
• Aus den Aminosäuren entstehen in einem Mehrschrittprozess die Proteine, die
charakteristisch für eine Zelle sind und das Proteom darstellen. Der Abbau von
Proteinen zu Aminosäuren erfolgt unter dem Einfluss von Proteinasen, die
Aminosäureseitenketten erkennen, die sich in Nachbarschaft der gespaltenen
Peptidbindung befinden. Im Cytosol sind diese Proteinasen oft Bestandteil von
Proteasomen.
• Aminosäuren wirken als Stickstoff- bzw. Aminogruppendonatoren bei der
Biosynthese anderer stickstoffhaltiger Verbindungen wie Purinen, Pyrimidinen,
Kreatinphosphat oder Stickstoffmonoxid (NO).
• Aminosäuren spielen eine grosse Rolle bei der Glucosehomöostase, da neben
Metaboliten der Glycolyse und dem aus der Lipolyse stammenden Glycerin nur die
glucogenen Aminosäuren als Substrat für die Gluconeogenese zur Verfugung stehen.
• Aminosäuren wirken im Gehirn als exzitatorische und inhibitorische Neurotransmitter
(Glutamat, GABA und Glycin).
Abbau der Proteine und Aminosäuren Alle Aminosäuren, die in Proteinen vorkommen, befinden sich auch in freier Form im Blut,
in der extrazellularen Flüssigkeit und in den Zellen. Dieser Pool von freien
Aminosäuren hat seinen Zufluss aus Nahrungsproteinen, welche im Darm in
Aminosäuren zerlegt werden, und aus dem intrazellularen Abbau von körpereigenen
Proteinen. Die Aminosäuren des Pools werden einerseits verwendet für den Aufbau
körpereigener Proteine und für die Synthese von anderen N-haltigen Verbindungen,
andrerseits werden sie bei einem Überangebot zu CO2 oxidiert und - bei Glucose-Mangel -
teilweise zu Glucose verwandelt.
Page 2
Kapitel: Aminosäuren
2
Umwelt Organismus
Aminosäurenpool
Proteine
Aminosäurenderivate:•Nucleotide•Porphyrine•Kreatin•N-haltige Lipide
C-GerüstAcetoacetat
Acetyl-CoA Pyruvat undDicarbonsäurendes Citratzyklus
Proteine
O C
NH2
NH2
NH4+
CO2 + H2O
Schema des Aminosäurenstoffwechsels
Glucose
Unter AS-Pool (70g) versteht man die Gesamtheit aller im Organismus vorhandenen
Aminosäuren. Aus dem AS-Pool gelangen die AS in die Stoffwechselwege des Anabolismus
(Protein-Synthese, etc.) oder des Katabolismus. Täglich werden im Organismus eines
erwachsenen Mannes – bei einem Gesamtbestand von etwa 10 kg Protein - ca. 300 g
Protein synthetisiert.
Ein Minimum an Aminosäuren (~30g/Tag) (obligatorischer AS-Verlust) wird auch bei
ausreichender Zufuhr von Kohlehydraten und Fett abgebaut unter ATP-Gewinn.
Der AS-Abbau wird gesteigert bei
- proteinreicher Kost und
- wenn Kohlehydrat- und Fettreserven aufgebraucht sind, d.h. im Endstadium des
Verhungerns.
Der gesunde Erwachsene befindet sich bei einer täglichen Zufuhr von 32 g hochwertigem
Protein im Stickstoffgleichgewicht, d. h. bei dieser Zufuhr halten sich Stickstoffaufnahme mit
der Nahrung und Stickstoffabgabe mit den Faeces, dem Urin und über die Haut (durch
Schweissabgabe, Epidermisabschilferung und Haarausfall) die Waage.
Page 3
Kapitel: Aminosäuren
3
Grundsätzlich bestehen 2 Möglichkeiten des Abbaus von AS
1. Zur ATP-Gewinnung (Hauptweg)
a) Entfernung von N
b) Oxydation des C-Skeletts
2. Zur Synthese von biologisch aktiven Verbindungen
c) Decarboxylierung Biogene Amine
d) Oxydation der Amine durch Amin-Oxidasen
Der Abbau der AS läuft über mehrere Reaktionen:
1. Desaminierung/Transaminierung NH2-Gruppe NH3 oder Aspartat
2. Harnstoffbildung N-Atom aus NH3 und Aspartat Harnstoff
3. Abbau des Kohlenstoffgerüstes AS und der α-Ketocarbonsäure Zwischenprodukte
Der grösste Teil des Stickstoffs der Aminosäuren wird im Urin als Harnstoff ausgeschieden.
Das eine N-Atom des Harnstoffs stammt aus NH4+, das andere aus Aspartat. In der Leber
wird der Stickstoff der meisten Aminosäuren durch Transaminierung so verschoben, dass er
schliesslich im Glutamat vorliegt. Dieses kann entweder mit Oxalacetat transaminieren zu
Aspartat oder seinen Stickstoff als Ammonium-Ion abgeben. NH4+ und die Aminogruppe von
Aspartat werden anschliessend mit HCO3- im Harnstoffzyklus zu Harnstoff kondensiert. Die
Entfernung des Stickstoffs aus den Aminosäuren hinterlässt Kohlenstoff-Skelette, welche
nun - ebenfalls in der Leber - zu Glucose und Ketonkörpern umgebaut werden können.
AS-Stickstoff wird über 3 aufeinander folgende Vorgänge eliminiert (Desaminierung):
• Intermolekulare Verschiebung der Aminogruppe (Transanimierung)
↓
• Abspaltung von NH3 vom C-Gerüst (oxidative Desanimierung)
↓
• Erzeugung einer ungiftigen Transportform (Glutamin) bzw. eines ausscheidbaren N-
haltigen Derivates (Harnstoff)
Page 4
Kapitel: Aminosäuren
4
Weg des Aminosäuren-N zum Harnstoff
COO-
CH
CH2
CH2
COO-
NH3+
COO-
C
CH2
CH2
COO-
O
COO-
CH
CH2
COO-
NH3+
N von anderen Aminosäuren
NH2
C
NH2
O
NH4+
HCO3-
Glutamat
Aspartat Harnstoff
a-Ketoglutarat
+
Transaminierung
oxidative Desaminierung
Harnstoff Bildung
Aminosäuren-Desaminierung
A. Transaminierung (Transaminasen)
B. Oxidative Desaminierung (Glutamat-Dehydrogenase, GLDH)
Die erste Reaktion beim Abbau einer Aminosäure ist fast immer die Entfernung ihrer α-
Aminogruppe mit dem Ziel, überschüssigen Stickstoff auszuscheiden und das zurückblei-
bende Kohlenstoffgerüst abzubauen.
Die Desaminierung der meisten Aminosäuren erfolgt durch Transaminierung, der
Übertragung ihrer Aminogruppe auf eine α-Ketocarbonsäure; dabei entsteht die
α-Ketocarbonsäure der ursprünglichen Aminosäure und eine neue Aminosäure. Diese
Reaktionen werden von Aminotransferasen (Transaminasen) katalysiert.
Die Transaminierung führt natürlich nicht zu einer Netto-Desaminierung. Die eigentliche
Desaminierung erfolgt vor allem über die oxidative Desaminierung von Glutamat durch
Glutamat-Dehydrogenase, wobei Ammoniak entsteht.
Page 5
Kapitel: Aminosäuren
5
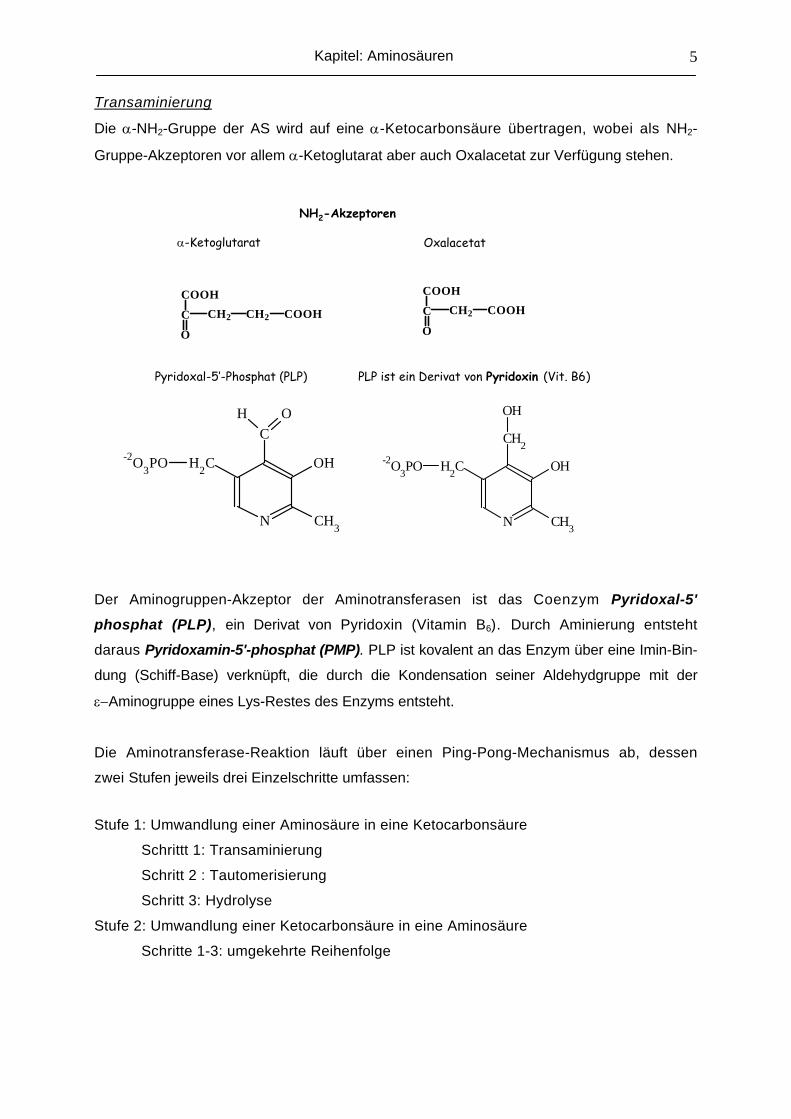
Transaminierung
Die α-NH2-Gruppe der AS wird auf eine α-Ketocarbonsäure übertragen, wobei als NH2-
Gruppe-Akzeptoren vor allem α-Ketoglutarat aber auch Oxalacetat zur Verfügung stehen.
α-Ketoglutarat Oxalacetat
NH2-Akzeptoren
C
COOH
O
CH2 CH2 COOH C
COOH
O
CH2 COOH
N
H2C
C
OH
CH3
H O
-2O3PO
N
H2C OH
CH3
-2O3PO
CH2
OH
Pyridoxal-5’-Phosphat (PLP) PLP ist ein Derivat von Pyridoxin (Vit. B6)
Der Aminogruppen-Akzeptor der Aminotransferasen ist das Coenzym Pyridoxal-5'
phosphat (PLP), ein Derivat von Pyridoxin (Vitamin B6). Durch Aminierung entsteht
daraus Pyridoxamin-5'-phosphat (PMP). PLP ist kovalent an das Enzym über eine Imin-Bin-
dung (Schiff-Base) verknüpft, die durch die Kondensation seiner Aldehydgruppe mit der
ε−Aminogruppe eines Lys-Restes des Enzyms entsteht.
Die Aminotransferase-Reaktion läuft über einen Ping-Pong-Mechanismus ab, dessen
zwei Stufen jeweils drei Einzelschritte umfassen:
Stufe 1: Umwandlung einer Aminosäure in eine Ketocarbonsäure
Schrittt 1: Transaminierung
Schritt 2 : Tautomerisierung
Schritt 3: Hydrolyse
Stufe 2: Umwandlung einer Ketocarbonsäure in eine Aminosäure
Schritte 1-3: umgekehrte Reihenfolge
Page 6
Kapitel: Aminosäuren
6
N
CH
CH2O P
N
Lys
HO
H3C
N
CHNCH
COOH
CH3
N
CH2NC
COOH
CH3
N
CH2H2N
COOH
C
CH2
CH2
COOHN
CH2 N
COOH
CH
CH2
CH2
COOHN
CH N
COOH
CH
CH3
NH2
COOH
C
CH3
O
COOH
C
CH2
CH2
COOH
O
COOH
CH
CH2
CH2
COOH
NH2
Alanin
Pyruvat
Pyridoxal-phosphat(PLP)
Pyridoxamin-phosphat (PMP)
Glutamat
α-Ketoglutarat
H2O H2O
Transaminierung
-
Stufe 1 Stufe 2
Bilanz:
Aminosäure1 + α-Ketosäure2 -----> Aminosäure2 + α-Ketosäure1
Durch Aminotransferasen, die für die jeweiligen Aminosäuren spezifisch sind, werden die
Aminogruppen auf die α-Ketosäuren α-Ketoglutarat bzw. Pyruvat unter Bildung von Glutamat
bzw. Alanin übertragen. Durch zwei Aminotransferasen, die in hohen Konzentrationen in
Leber, Myocard und Gehirn vorkommen, kann die Aminogruppe von Alanin auf
α-Ketoglutarat unter Bildung von Glutamat und Pyruvat übertragen werden. Es handelt sich
um die Aspartat-Aminotransferase oder Glutamat-Oxalacetat Transaminase (ASAT
oder GOT) bzw. die Alanin-Aminotransferase oder Glutamat-Pyruvat-Transaminase
(ALAT oder GPT).
Aspartataminotransferase (AST) GOT
Asp + α-Ketoglutarat <-----> Glu + Oxalacetat
Alaninaminotransferase (ALT) GPT
Ala + α-Ketoglutarat <-----> Glu + Pyruvat
Page 7
Kapitel: Aminosäuren
7
Oxidative Desaminierung
Glutamat wird im Mitochondrium oxidativ durch Glutamat-Dehydrogenase desaminiert;
es ist das einzige Enzym, das - zumindest in einigen Organismen - sowohl NAD+ als auch
NADP+ als Redox-Coenzym akzeptiert.
GLDH-Reaktion wird durch Koppelung mit der Transaminasereaktion zum Hauptweg für
Bildung von Ammoniak.
α-AS
a-KS
α-Ketoglutarat
Glutamat
GLDHASAT/ALAT
NADH + H+
NH4+
H2O
NAD+
3 Produkte: α-Ketoglutarat Citratzyklus, Transaminasen (Rezykl.)NADHNH4+ Harnstoff
Citratzyklus
Gleichgewicht der GLDH-Reaktion liegt stark auf der Seite von Glutamat. Reaktion läuft nur
dann in nennenswertem Umfang nach rechts ab, wenn NH4+ sofort abgefangen wird (In der
Leber geschieht das durch Synthese von Glutamin und Harnstoff).
Oxidative Desaminierung
COO-
C
CH2
CH2
COO-
NH3+H
NAD+ H2O
COO-
C
CH2
CH2
COO-
ONH4
+NADH H++ + + + +
Glutamat α-Ketoglutarat
GLDH
Page 8
Kapitel: Aminosäuren
8
Speicherung und Transport von Ammoniak als Glutamin
Da Ammoniak schon in verhältnismässig niedrigen Konzentrationen als Zellgift
(Nervensystem!) wirkt, ist eine Umwandlung in andere Verbindungen nötig. Eine wichtige
Reaktion, die in vielen Organen ablaufen kann, ist die energetisch günstige Bildung von
Glutamin aus Glutaminsäure und Ammoniak. Sie wird durch das Enzym Glutamin-
Synthetase katalysiert und erfolgt nach der Gleichung:
Glutamin dient im Stoffwechsel als NH2-Donator für die meisten Aminogruppen,
ausgenommen die α-Aminogruppen der Aminosäuren; ausserdem für die Purin-Synthese
und für die Pyrimidin-Synthese; ferner ist es eine Transportform des Ammoniaks im
Organismus. Glutamin kann nämlich leicht hydrolytisch gespalten werden zu Glutamat
und Ammoniak; das entsprechende Enzym, die Glutaminase, findet sich vor allem in der
Niere, und das Ammoniak des Harns entsteht zum erheblichen Teil durch diese
Reaktion.
Transport von Aminogruppen durch den Aspartatzyklus
ATP ADP + Pi
Glutamat Glutamin
NH4+ H2O+ +
COO-
CH
CH2
CH2
COO-
NH3+
COO-
CH
CH2
CH2
CO
NH3+
NH2
Page 9
Kapitel: Aminosäuren
9
Aspartatzyklusreaktionen. Auf eine Reihe von Ketoverbindungen kann eine Aminogruppe
dadurch übertragen werden, dass die Aminogruppe der Aminosäure Aspartat mit der
Ketoverbindung unter Energieverbrauch (ATP oder GTP) zu einem Kondensationsprodukt
zusammentritt und das Kondensationsprodukt durch eine Lyasereaktion in die aminierte
Verbindung und Fumarat gespalten wird. Aspartat wird aus Fumarat durch die Fumarase-,
Malatdehydrogenase- und Glutamat-Oxalacetat-Transaminase-Reaktion unter gleichzeitigem
Gewinn eines Reduktionsäquivalentes ( 1 mol NADH = 3 mol ATP) regeneriert und steht
für den erneuten Umlauf zur Verfügung. Alle genannten Reaktionen, auch die Teilreaktionen
des Citratzyklus (!), laufen im Cytosol ab.
Ausscheidung von Stickstoff bei Vertebraten
Lebende Organismen können überschüssigen Stickstoff, der aus dem Aminosäureabbau
stammt, auf drei Wegen ausscheiden. Viele im Wasser lebende Tiere geben einfach
Ammoniak an das umgebende Milieu ab. Wo Wasser nicht so reichlich vorhanden ist,
wurden Mechanismen entwickelt, die Ammoniak in weniger toxische Abfallprodukte
umwandeln, für deren Ausscheidung dann weniger Wasser erforderlich ist. Eines dieser
Produkte ist Harnstoff der von den meisten landlebenden Vertebraten ausgeschieden
wird; ein weiteres ist Harnsäure, die von Vögeln und landlebenden Reptilien abgesondert
wird.
AS-Stickstoff
Ammoniakausscheider ammnotelisch
Harnstoffausscheider ureotelisch
Harnsäureausscheider uricotelisch
Harnstoff-Synthese (Harnstoff-Zyklus)
Harnstoff wird in der Leber durch die Enzyme des Harnstoff-Zyklus synthetisiert. Er wird
dann in den Blutstrom abgegeben und von den Nieren mit dem Urin ausgeschieden. Die
Grundzüge des Harnstoff-Zyklus wurden 1932 von Hans Krebs und Kurt Henseleit
aufgeklärt (es war der erste bekannte Stoffwechsel-Zyklus überhaupt).
Eine 7o kg schwere Normalperson bildet in 24 h etwa 0,5 mol (30 g) Harnstoff. Bei
proteinreicher Ernährung kann die Harnstoffbildung bis auf das Dreifache ansteigen. Diese
Page 10
Kapitel: Aminosäuren
10
Steigerung ist deshalb möglich, weil die Enzyme im Überschuss vorhanden sind und weil
sich außerdem die Enzymaktivitäten bei proteinreicher Nahrung um das Zwei- bis Dreifache
erhöhen können.
HCO3- NH4
++
NH2
C O
O P
2ATP
2ADP+Pi
CH2
CH2
CH2
CH
NH3+
COO-NH3
+
CH2
CH2
CH2
CH
NH
COO-NH3
+
NH2
C O
CH2
CH2
CH2
CH
NH
COO-NH3
+
NH2+
C NH
COO-
CH
CH2
COO-
CH2
CH2
CH2
CH
NH
COO-NH3
+
NH2+
C NH2
Pi
COO-
CH NH3+
CH2
COO-
+ATP
AMP + PPi + H2O
COO-
CH
CHCOO-
H2O
O CNH2
NH2
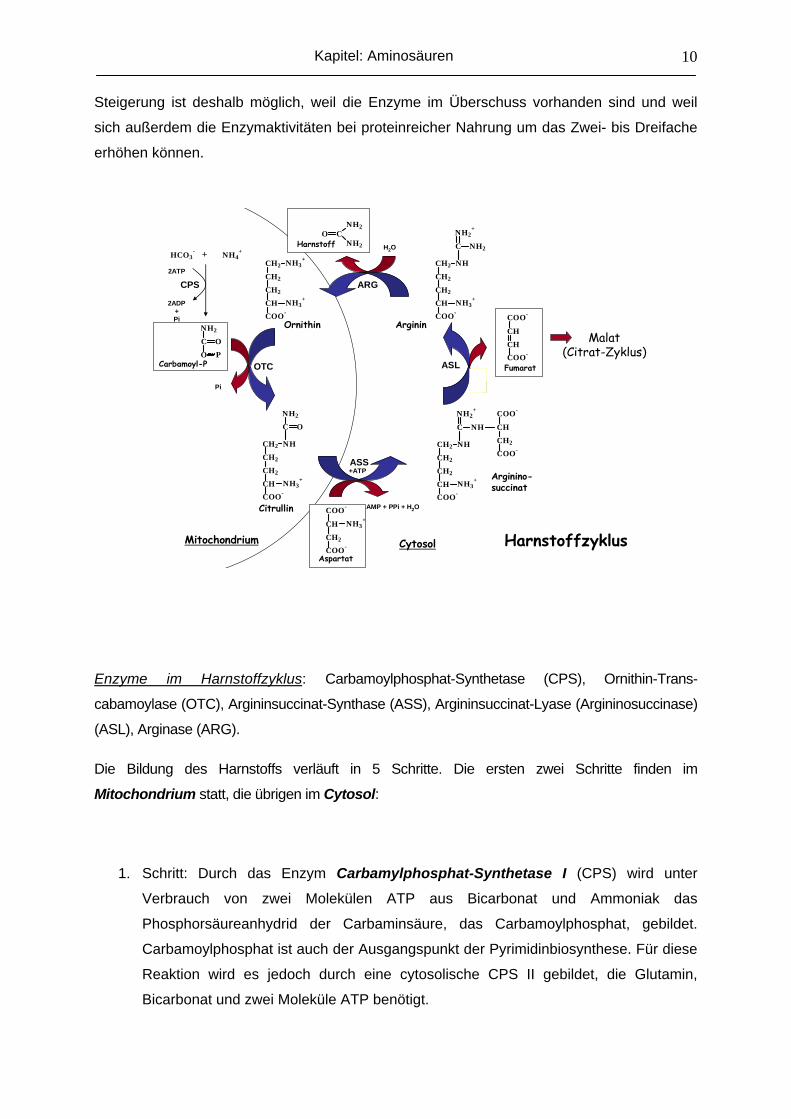
Harnstoffzyklus
Carbamoyl-P
Ornithin
Citrullin
Aspartat
Arginino-succinat
Fumarat
Arginin
Harnstoff
Mitochondrium Cytosol
ASS
ASLOTC
ARGCPS
Malat(Citrat-Zyklus)
Enzyme im Harnstoffzyklus: Carbamoylphosphat-Synthetase (CPS), Ornithin-Trans-
cabamoylase (OTC), Argininsuccinat-Synthase (ASS), Argininsuccinat-Lyase (Argininosuccinase)
(ASL), Arginase (ARG).
Die Bildung des Harnstoffs verläuft in 5 Schritte. Die ersten zwei Schritte finden im
Mitochondrium statt, die übrigen im Cytosol:
1. Schritt: Durch das Enzym Carbamylphosphat-Synthetase I (CPS) wird unter
Verbrauch von zwei Molekülen ATP aus Bicarbonat und Ammoniak das
Phosphorsäureanhydrid der Carbaminsäure, das Carbamoylphosphat, gebildet.
Carbamoylphosphat ist auch der Ausgangspunkt der Pyrimidinbiosynthese. Für diese
Reaktion wird es jedoch durch eine cytosolische CPS II gebildet, die Glutamin,
Bicarbonat und zwei Moleküle ATP benötigt.
Page 11
Kapitel: Aminosäuren
11
2. Schritt: Zunächst wird der Carbamylrest von Carbamylphosphat auf die nicht
proteinogene Aminosäure Ornithin übertragen (Ornithin-Transcarbamylase). Dabei
entstehen Citrullin, eine ebenfalls nicht proteinogene Aminosäure, und anorganisches
Phosphat. Das Reaktionsgleichgewicht liegt stark auf der Seite der Citrullinbildung.
3. Schritt: Citrullin tritt durch die Mitochondrienmembran in das Cytosol über, wo alle
weiteren Reaktionen des Kreisprozesses ablaufen. Auf den an Ornithin gebundenen
Carbamylrest (Citrullin) soll im nächsten Schritt eine weitere Aminogruppe übertragen
werden. Dabei kondensiert die Carbonylgruppe von Citrullin mit der Aminogruppe von
Aspartat unter Bildung von Argininosuccinat (Argininosuccinat-Synthetase). Auch
diese Reaktion ist ATP-abhängig, das entstehende Pyrophosphat wird durch eine
Pyrophosphatase gespalten, wobei das Reaktionsgleichgewicht in Richtung Arginino-
succinatbildung verschoben wird.
4. Schritt: Die anschließende Spaltung der C-N-Bindung (Argininosuccinat-Lyase) in
die Produkte Fumarat und die proteinogene Aminosäure Arginin ist im Gegensatz
zu den vorhergehenden Reaktionen reversibel. Durch diese beiden Reaktionen ist
die Aminogruppe von Aspartat auf Citrullin übertragen worden.
5. Schritt: Der Kreisprozess der Harnstoffbiosynthese wird durch die hydrolytische
Abspaltung der Guanidinogruppe von Arginin geschlossen. Dabei entstehen
Harnstoff und Ornithin.
Bilanz
• Energieverbrauch: 3ATP 2ADP, 2Pi, AMP, PPi(2Pi)
• Bedarf für die Regeneration: 4 ATP
• Energiekompensierung (durch Fumarat): 1 NADH = 3 ATP
Bemerkenswert an den beschriebenen Reaktionen ist, dass durch den Kreisprozess das
Trägermolekül Ornithin ständig regeneriert wird und auch der Aminogruppendonator
Aspartat durch wenige enzymatische Schritte erneuert werden kann, wobei ein Reduk-
tionsäquivalent gewonnen wird. Somit werden dem Hepatocyten bei der Biosynthese von
Harnstoff lediglich zwei Ammoniakmoleküle, Bicarbonat und Energie entzogen.
Hyperammoniämien
Homozygote bzw. gemischt heterozygote Defekte der Harnstoffzyklus-Enzyme führen zu
einem Zusammenbruch des Harnstoffzyklus mit einem Anstieg der
Ammoniakkonzentration (Hyperammoniämien). Ohne entsprechende Therapie, d.h.
Hämodialyse in der Akutphase, treten Gehirnschaden (Hirnödem mit verkleinertem Ventri-
kelsystem, Abflachung der Gyri) und der Tod der Patienten ein. Ammoniak ist extrem
Page 12
Kapitel: Aminosäuren
12
neurotoxisch in höheren Konzentrationen und eine Belastung für die GLDH (α-Ketoglutarat ↓),
Glu-Synthase (GABA ↓) und Neurotransmittersynthese (Dopamin ↓).
Die Harnstoffsynthese produziert ein Proton, welches zur Neutralisierung eines Teils des beim
AS-Abbau freigesetzten HCO3- dient. Blockierung der Harnstoffsynthese (Enzymdefekte) führt
zu Alkalose.
1. HCO3- + 2NH4
+ H‘Stoff + 2H2O + H+
2. H+ + HCO3- CO2 + H2O
Σ 2 HCO3- + 2NH4
+ H‘Stoff + 3 H2O + CO2
Enzymdefekte des Harnstoffzyklus 1. Cabamoylphosphat-Synthase (CPS)-Mangel
2. Ornithin-Transcabamoylase (OTC)-Mangel
3. Argininsuccinat-Synthase (ASS)-Mangel
4. Argininsuccinat-Lyase (Argininosuccinase) (ASL)-Mangel
5. Arginase (ARG)-Mangel
6. N-Acetyglutamat-Synthase (NAGS)-Mangel
Differentialdiagnose (Beispiel ASL-Magel)
PLASMA ASLGln ↑Ala ↑/nAsn ↑/nCit ↑↑
Arg-suc ↑↑↑Orn nLys nArg ↓/n
Page 13
Kapitel: Aminosäuren
13
Abbau des Kohlenstoffgerüstes der Aminosäuren Die Abbauprodukte von Aminosäuren sind im allgemeinen Intermediate des
Citronensäurezyklus oder deren Vorstufen, so dass sie zu CO, und H2O verarbeitet oder
in der Gluconeogenese eingesetzt werden können. Tatsachlich liefert der oxidative
Aminosäureabbau typischerweise 10 bis 15% der im Stoffwechsel erzeugten Energie eines
Tieres.
Die Standard-Aminosäuren können zu sieben verschiedenen Intermediaten des
Stoffwechsels abgebaut werden, nämlich Pyruvat, α-Ketoglutarat, Succinyl-CoA, Fumarat,
Oxalacetat, Acetyl-CoA oder Acetoacetat. Die Aminosäuren lassen sich je nach
katabolischem Weg in zwei Gruppen einteilen:
1. Glucogene (glucoplastische) AS Synthese v. Kohlehydraten
2. Ketogene (ketoplastische) AS Fettsäuren und Ketokörper
Oxalacetat
Fumarat
Citrat
a-Katoglutarat
Succinly-CoA
CO2
CO2
CO2
CO2
CO2
PyruvatPEP
IleMetThrVal
GluGlnHisProArg
TyrPheAsp
AspAsn
Ala, GlyCys, SerThr, TrpGlucose
LeuIleTrp, Thr
LeuLysPheTyr
Acetyl-CoA Acetoacetat
Lipide
Citronensäure-Zyklus
glucogene AS
ketogene AS
So ist z. B. Alanin glucogen, weil sein Transaminierungsprodukt Pyruvat über die
Gluconeogenese in Glucose umgewandelt werden kann. Leucin dagegen ist ketogen, da
sein Kohlenstoffgerüst zu Acetyl-CoA und Acetoacetat abgebaut wird.
Page 14
Kapitel: Aminosäuren
14
Ala Ser Gly Thr Cys Trp Pyruvat
Das Abbauprodukt von fünf Aminosäuren (Alanin, Cystein, Glycin, Serin und Threonin) ist
Pyruvat. Tryptophan gehört eigentlich auch zu dieser Gruppe, da eines seiner
Abbauprodukte - Alanin - durch Transaminierung zu Pyruvat konvertiert wird.
Abbau des Glycins
Glycin wird durch Serin-Hydroxymethyltransferase - ein PLP-haltiges Enzym - in Serin
umgewandelt. Dieses Enzym verwendet N5,N10-Methylen-Tetrahydrofolat (5,10-MTHF) als
Coenzym, das die für diese Umwandlung benötigte C1-Einheit liefert. Die bei der
Umwandlung von Glycin in Serin eingesetzte Methylen-Gruppe aus N5,N10-Methylen-THF
stammt aus einem zweiten Abbauweg fur Glycin; dieser wird vom glycinabbauenden
Enzymkomplex (bei der Rückreaktion als Glycin-Spaltsystem bezeichnet) katalysiert. Es
handelt sich dabei um einen Multienzymkomplex. Die Synthese des Glycins erfolgt über
eine Serin-Aldolase.
CO2+
NH4+
Gly NAD+
NADH
1
THF
5,10-MTHF
2 Ser-Aldolase
H2C OH
C NH3+H
COO-
H2C NH3+
COO- Gly
Ser Pyruvat + NH4+
Ser-Dehydratase (PLP)
1. Gly Ser ( Pyruvat) ] Ser-Hydroxymethyltransferase (PLP-Enzym)2. Gly CO2 + NH4
+ ] Gly-Spaltsystem (THF-abhängig)
Abbau des Glycins
Die menschliche Erbkrankheit nichtketotische Hyperglycinamie (NKH), die sich durch geistige
Retardierung und Akkumulation grösser Mengen von Glycin in den Körperflüssigkeiten auszeichnet,
beruht auf einem Fehlen des glycinabbauenden Enzymkomplexes.
Page 15
Kapitel: Aminosäuren
15
Ile Leu Acetoacetat Val Pyruvat
Beim Abbau von Isoleucin, Leucin und Valin, die verzweigte Ketten haben, sind die
Enzyme bei den ersten drei Reaktionen: (1) Transaminierung zur entsprechenden α-
Ketocarbonsdure, (2) oxidative Decarboxylierung zum entsprechenden Acyl-CoA und
(3) Dehydrierung durch FAD unter Bildung einer Doppelbindung. Danach verläuft der
Isoleucinabbau wie bei der Fettsäureoxidation: (4) Hydratisierung der
Doppelbindung, (5) Dehydrierung durch NAD+ und (6) thiolytische Spaltung zu Acetyl-
CoA und Propionyl-CoA, das anschliessend zu Succinyl-CoA ,verarbeitet" wird. Valin wird
analog abgebaut: Nach der Hydratisierung der Doppelbindung (7), wird (8) die CoA-
Thioester-Bindung hydrolysiert, dann erst folgt (9) die zweite Dehydrierungsreaktion. Die
Thioesterbindung wird in der letzten Reaktion (10) - einer oxidativen Decarboxylierung
anstelle einer thiolytischen Spaltung - als Propionyl-CoA regeneriert.
Abbau der Verzweigkettigten Aminosäuren und bekannte Enzymdefekte
Page 16
Kapitel: Aminosäuren
16
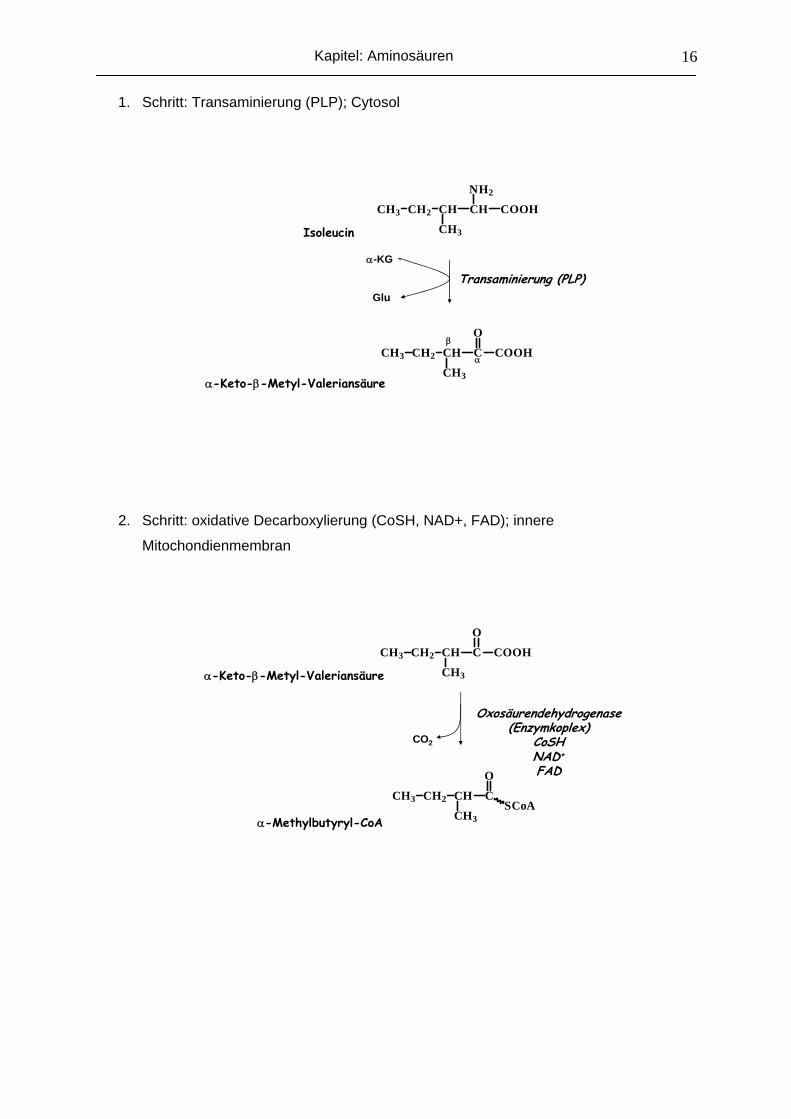
1. Schritt: Transaminierung (PLP); Cytosol
CH3 CH2 CH CH
NH2
COOH
CH3
CH3 CH2 CH C
O
COOH
CH3
Isoleucin
α-Keto-β-Metyl-Valeriansäure
Transaminierung (PLP)α-KG
Glu
α
β
2. Schritt: oxidative Decarboxylierung (CoSH, NAD+, FAD); innere
Mitochondienmembran
CH3 CH2 CH C
O
COOH
CH3
CH3 CH2 CH C
O
CH3SCoA
α-Keto-β-Metyl-Valeriansäure
α-Methylbutyryl-CoA
Oxosäurendehydrogenase(Enzymkoplex)
CoSHNAD+
FAD
CO2
Page 17
Kapitel: Aminosäuren
17
3. Schritt: Dehydrierung (FAD); Mitochondrien
CH3 CH C C
O
CH3SCoA
CH3 CH2 CH C
O
CH3SCoA
α-Methylbutyryl-CoA
Acyl-CoA-DehydrogenaseFAD
Tiglyl-CoA
Ahornsirup-Krankheit ist die Folge eines Defektes beim Abbau verzweigtkettiger Aminosäuren
α-Ketoisovalerat-Dehydrogenase, die beim Abbau von Aminosäuren mit verzweigten
Ketten Reaktion 2 katalysiert, ist ein Multienzymkomplex, der neben seinem terminal
oxidierenden Agens NAD+ die Coenzyme TPP, Lipoamid und FAD einsetzt. Ein genetischer
Defekt in diesem Enzymkomplex ist die Ursache der Ahornsirup-Krankheit (Meaplesyrup
urine disease, MSUD); die daraus folgende Akkumulation von α-Ketocarbonsäuren (α-
Keto-β-Methylvaleriansäure) mit verzweigten Ketten verleiht dem Urin den
charakteristischen Geruch von Ahornsirup (würzig-süsslich). Wird der Anteil
verzweigtkettiger Aminosäuren in der Nahrung nicht reduziert, führt die Krankheit schnell
zum Tod.
α-Ketosäuren-Dehydogenase Komplex
• 6 Genloci: 2 Proteine und 4 regulatorische Enzyme
• MSUD Häufigkeit: 1:200'000 Neugeborene (Mennoiten 1:176)
• Phenotyp: Apathie, Trinkschwäche, Krämpfe, Coma
Page 18
Kapitel: Aminosäuren
18
Phe Tyr Fumarat
Acetoacetat
Die erste Reaktion des Abbaus von Phenylalanin ist die Hydroxylierung zu Tyrosin, so
dass der weitere Abbau der beiden Aminosäuren auf einem einzigen Weg erfolgt. Die
Endprodukte des sechsstufigen Abbaus sind Fumarat, Bestandteil des Citronensäure-
Zyklus, und Acetoacetat.
Abbau der aromatischen Aminosäuren Phenylalanin und Tyrosin
CH2
CHNH2
COOH
CH2
CHNH2
COOH
OH
CH2
C
COOH
OH
O
OH
OH
CH2 COOH
COOH
O O
COOH
HOOCCOOH
O O
Phenylalanin-hydroxylase Transaminase
Tetrahydro-biopterin
+O2-H2O
α-Ketoglutarat(PLP)
Dioxygenase
Ascorbat+O2-CO2
Dioxygenase+O2
IsomeraseFumarat
Acetoacetat +H2O
Phenylalanin Tyrosin p-Hydroxy-phenylpyruvat
Homogentisinsäure
Maleylacetoacetat(cis)
Fumarylacetoacetat(trans)
Phenylketonurie (PKU) und Hyperphenylalaninämien
PKU und Hyperphenylalaninämien sind eine Gruppe von angeborenen Stoffwechsel-
Erkrankungen, die auf eine ungenügende Oxidation des Phenylalanins zurückzuführen sind.
Die gemeinsame biochemische Abnormität ist eine Anhäufung von Phenylalanin im Gewebe
und Blut. Je nach Schweregrad der Hyperphenylalaninämie werden drei Formen
unterschieden (klassische PKU, milde PKU und benigne Hyperphenylalaninämie).
Asbjörn Fölling (Norwegen) fand 1934 im Urin eines Patienten grosse Mengen
Phenylbrenztraubensäure. Drei Jahre später wurde diese Krankheit als „Phenylketonurie“
beschrieben und erst 1947 konnte mittels eines Phenylalaninbelastungstestes der
metabolische Defekt bei diesen Patienten identifiziert werden. Es wurde festgestellt, dass es
sich dabei um einen Phenylalaninhydroxylase-Mangel handelt. Bickel und Mitarbeiter
(Heidelberg) führten eine wirksame Therapie mit einer phenylalaninarmen Diät ein und
Page 19
Kapitel: Aminosäuren
19
Guthrie und Susi propagieren seit 1961 das Screening auf Hyperphenylalaninämie bei
Neugeborenen mit dem von ihnen entwickelten bakteriellen Hemmtest (Guthrietest).
CH2 CH COO-
NH3+
CH2 CH COO-
NH3+
HO
CH2 C COO-
O
Phenylketonurie (PKU): Entstehung von Metaboliten
CH2 CH COO-
OH
CH2 CO SCoACH2 COO-
Glutalat
a-Ketoglutarat
Transaminasen (PLP)
Phenylalanine Tyrosin
Phenylpyruvat
NAD+NADH +H+
CoA
CO2
NADH +H+
Dehydrogenase
PhenyllactatPhenylacetat Phenylacetyl-CoA
H2O
CoA
PAH
NAD+
Das überschüssige Phenylalanin wird auf einem sonst unbedeutenden Weg zu
Phenylpyruvat transaminiert.
Phenylalanin-Hydroxylase-Mangel erklärt auch ein anderes allgemeines PKU-Symptom:
Die Betroffenen haben hellere Haar- und Hautfarbe als ihre gesunden Geschwister. Die
Tyrosin-Hydroxylierung, die erste Reaktion bei der Bildung des schwarzen Hautpigments
Melanin, wird durch erhöhte Phenylalaninspiegel gehemmt. Phenylalanin hemmt zudem
zwei geschwindigkeitsbestimmende Enzyme in der Biosynthese von Dopamin und
Serotonin (Tyrosin- und Tryptophanhydroxylase).
Phenylketonurie
• Häufigkeit: 1:15’000 Neugeborene
• Erbgang: Autosomal-rezessiv
• Phenotyp (unbehandelt): Hypotonie der Rumpfmuskulatur, Hypertonie der
Extremitäten, Epilepsie, Ptose
• Therapie: Phenylalaninarme Diät (neu: BH4-Cofaktor Therapie)
Page 20
Kapitel: Aminosäuren
20
Neugeborenen Screeing
Ein Enzymdefekt führt zu einem Block in einer Reaktionskette des Intermediärstoffwechsels.
Die dem Block folgenden Metaboliten werden nicht oder nur in erheblich verminderter Menge
gebildet und die Vorstufen akkumulieren (Phenylalanin). Jedem Neugeborenen werden in
der ersten Lebenswoche aus der Ferse einige Tropfen Blut für den so genannten „Guthrie-
Test“ entnommen. Die Neugeborenen werden, neben der PKU, auch auf einige andere
behandelbare Stoffwechselstörungen getestet.
Bei dem „Guthrie-Test“ auf PKU werden die Bakteriensporen (B. subtilis) zusammen mit dem
Inhibitor (β-Thienylalanin; Antimetabolit) in einem phenylalanin-freien Agar auf Platten
gegossen. Auf diese Agarplatte werden Trockenblutproben (Filterpapier) aufgelegt. Durch
die Gegenwart von Phenylalanin in diesen Proben wird die Wirkung des kompetitiven
Inhibitors (β-Thienylalanin) aufgehoben und die Bakterien bilden einen Wachstumshof. Die
Größe dieses Wachstumhofes ist dem Gehalt der Blutprobe an Phenylalanin proportional.
Phenylalanin im Blut (normal) < 0.12 mmol/l
Tetrahydrobiopterin (BH4): Cofaktor der Hydroxylierung von aromatischen Aminosäuren Die Hydroxylierung von Phenylalanin durch das Fe(III)-haltige Enzym Phenylalanin-
Hydroxylase benötigt einen Cofaktor: Tetrahydrobiopterin, ein Pterin-Derivat. Pterine enthalten
ein Pteridinring, welcher auch in der Folsäure enthalten ist (siehe Kapitel: C1-
Stoffwechsel). Pterine sind wie Flavine an biologischen Oxidationen beteiligt. Die aktive
Form von Biopterin ist die vollreduzierte Form 5,6,7,8-Tetrahydrobiopterin. Sie wird
durch drei Enzyme (GTP-Cyclohydrolase, PTP-Synthase und Sepiapterin-Reduktase)
aus GTP gebildet. In der Phenylalanin-Hydroxylase-Reaktion wird 5,6,7,8-
Tetrahydrobiopterin über eine Zwischenstufe (Carbinolamin) zur chinoiden Form von 7,8-
Dihydrobiopterin oxidiert. Dieses Chinoid wird anschliessend durch das NADH-abhängige
Enzym Dihydropteridin-Reduktase reduziert und der aktive Cofaktor regeneriert.
Page 21
Kapitel: Aminosäuren
21
HN
H2N
O
NH
N
HN
OH
OH
HN
H2N
O
NH
N
N
OH
OH
O
OH
HN
H2N
O
NH
N
N
OH
OH
OHN
H2N
O
NH
N
N
OH
OH
PAH
PAH
PCD
DHPR
5,6,7,8-Tetrahydrobiopterin
Tetrahydrobiopterin-4a-carbinolamine
q-Dihydrobiopterin
+O2
-H2O
+NADH- NAD+
GTPCH2
CHNH2
COOH
CH2
CHNH2
COOH
OH
Phe
Tyr
Funktion des Tetrahydrobiopterins als Redox-Cofaktor
PCD = Pterin-4a-CarbinolamindehydrataseDHPR = Dihydropteridin-Reduktase
Enzyme, die auf diese Weise zwei miteinander gekoppelte Oxidationen katalysieren
(Oxidation von Phenylalanin zu Tyrosin und 5,6,7,8-Tetrahydrobiopterin zu q-7,8-Dihydro-
biopterin), werden als gemischt funktionelle Oxidasen (Hydroxylasen, Monooxygenasen)
bezeichnet.
Allgemeine Reaktion:
XH + CoeH2 + O2 X-OH + H2O + Coe
Tetrahydrobiopterin-Mangel
Seit 1974 wurde über einige Patienten mit atypischer Hyperphenylalaninämie berichtet, die
trotz Früherkennung und Behandlung mit phenylalaninarmer Diät progressive neurologische
Störungen und einen zunehmenden Entwicklungsrückstand zeigten und zum Teil daran
starben. Man fand bei diesen Patienten ein Defekt in der Biosynthese des BH4, das im
Gegensatz zur Folsäure, kein Vitamin darstellt, sondern im gesunden Menschen nach Bedarf
in katalytisch kleinen Mengen synthetisiert wird.
Die Bedeutung dieser Stoffwechselkrankheiten, die alle als BH4-Mangel bezeichnet werden
können, ist trotz ihrer Seltenheit (2-3 : 1’000’000) gross. Das Risiko eines Hirnschadens bei
konventioneller Behandlung eines neuentdeckten Patienten mit Hyperphenylalaninämie
muss vermieden werden.
Page 22
Kapitel: Aminosäuren
22
BH4-Mangel (Info: www.bh4.org)
• Häufigkeit: 1:1’000’000 Neugeborene
• Erbgang: Autosomal-rezessiv
• Phenotyp (unbehandelt): Hypotonie der Rumpfmuskulatur, Hypertonie der
Extremitäten, Epilepsie, Ptose,
• Therapie: BH4-Cofaktor Therapie, Neurotransmitter-Vorstufen
(L-Dopa/Carbidopa/5-hydroxytryptophan)
Abbau des Tyrosins (Abb. S. 18) Tyrosin verliert seine Aminogruppe über die Transaminierung auf a-Ketoglutarat oder
Pyruvat. Das verantwortliche Enzym, die Tyrosinaminotransferase, ist sowohl im Cytosol
als auch in den Mitochondrien nachweisbar. Das entstandene p-Hydroxyphenylpyruvat wird
durch die cytosolische p-Hydroxyphenylpyruvat-Dioxygenase, eine kupferhaltige
mischfunktionelle Oxygenase in Leber und Nieren, in Homogentisat umgewandelt. Bei
dieser Reaktion, die die Anwesenheit von Ascorbinsäure (Vitamin C) oder einem anderen
Reduktionsmittel erfordert, erfolgt gleichzeitig die Hydroxylierung des Benzolrings in
Parastellung sowie eine Wanderung der Pyruvatseitenkette, aus der durch Dehydrierung
und Decarboylierung eine Acetatseitenkette entsteht. Im nächsten Schritt wird der
Benzolring durch die Einführung von molekularem Sauerstoff gespalten. Katalysator ist die
Homogentisatdioxygenase, ein eisenabhängiges Protein. Das Reaktionsprodukt Maleyl-
acetoacetat wird durch eine Glutathion-abhängige cistrans-Isomerisierung in
Fumarylacetoacetat umgewandelt und anschliessend hydrolytisch in Fumarat und
Acetacetat gespalten. Fumarat ist ein direkter Bestandteil des Citratcyclus, Acetoacetat,
ein Ketonkörper, wird nach Aktivierung zu Acetoacetyl-CoA in 2 Moleküle Acetyl-CoA
gespalten.
Tyrosinämien Drei angeborene Stoffwechseldefekte im Tyrosinabbau sind bekannt:
• Tyrosinämie Typ 1 (Fumaryl-Acetoacetase-Mangel)
• Tyrosinämie Typ 2 (Tyrosinaminotransferase-Mangel)
• Alkaptonurie (Homogentisinsäure-Dioxigenase-Mangel) Bei der Tyrosinämie Typ 1 (Inzidenz 1:100’00) handelt sich um eine autosomal-rezessiv
vererbte Abbaustörung von Tyrosin, die zur Bildung des toxischen Metabolites
Succinylaceton (aus Fumarylacetoacetat und Maleylacetoazetat) führt. Die toxischen
Page 23
Kapitel: Aminosäuren
23
Metabolite greifen in die Hämsynthese ein und verursachen eine progrediente Hepatopathie
mit schlechter Prognose, die entweder im ersten Lebensjahr zum Leberversagen und Tode
oder - bei langsamerem, chronischen Verlauf - zur Leberzirrhose, häufig hepatozellulärem
Karzinom und Tod in der späten Kindheit führt. Während früher allein die
Lebertransplantation die einzige Möglichkeit war, das Leben zu retten, steht heute mit dem
NTBC [2-(2-Nitro-4-trifluoromethylbenzoyl)-1,3-zyklohexandion] ein potenter Inhibitor des
Abbaus des Tyrosins auf einer Stufe vor der Furmaylacetoacetase
(p-Hydroxyphenylpyruvat-Dioxygenase) zur Verfügung, der verhindert, dass die toxischen
Metaboliten anfallen.
Alkaptonurie ist die erste, zu Beginn dieses Jahrhunderts von Archibald Garrod
erkannte, angeborene Stoffwechselkrankheit, die zur Ausscheidung grosser Mengen
von Homogentisinsäure führte. Die Symptome der Alkaptonurie sind relativ milde und
treten erst im Erwachsenenalter auf. Typischerweise entwickeln Alkaptonuriker im Laufe
des Lebens eine Arthritis; ihr Urin färbt sich infolge Luftoxidation des ausgeschiedenen
Homogentisats dunkel.
Aminosäuren als Vorstufer wichtiger Biomoleküle Bestimmte Aminosäuren sind Vorstufen wichtiger Biomoleküle, einschliesslich der
Nucleotide und Nucleotid-Coenzyme, Häm, verschiedener Hormone, Neurotransmitter
und Glutathion.
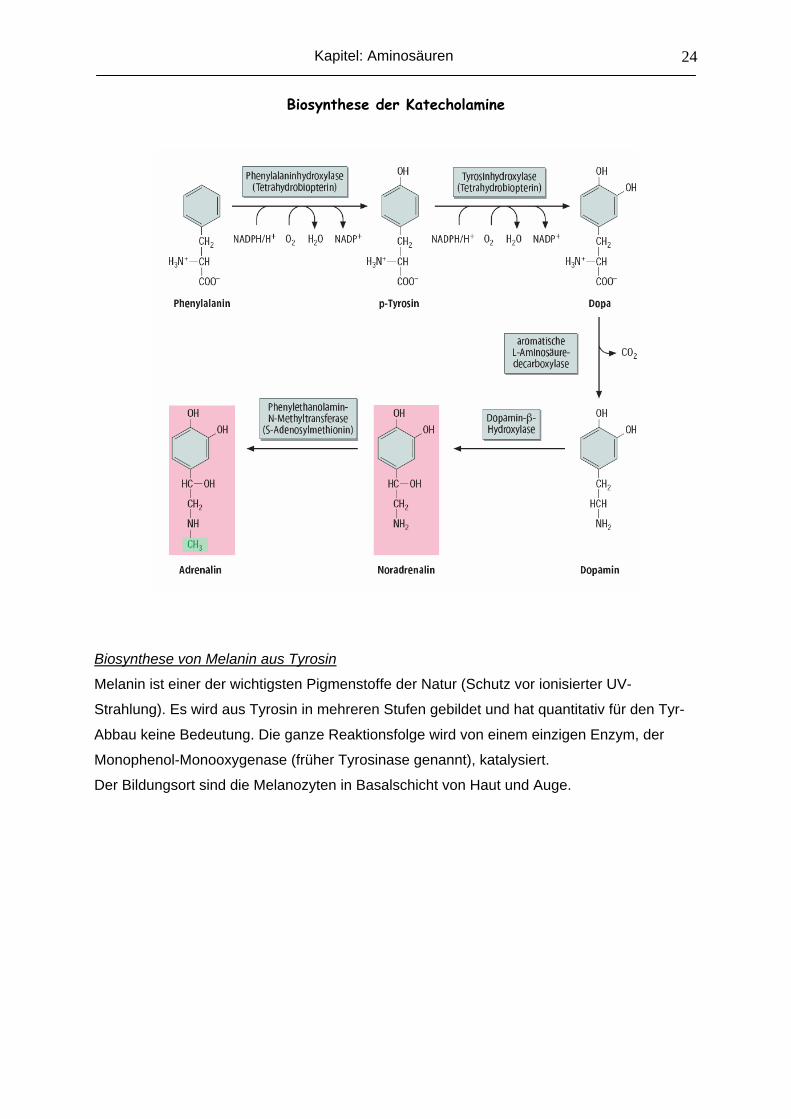
Tyr Katecholamine
Melanin Thyroxin
Biosynthese der Katecholamine aus Tyrosin
Dopamin, Noradrenalin und Adrenalin werden zusammen als Katecholamine bezeichnet,
da sie die Amin-Derivate von Katechol sind. Die Umwandlung von Tyrosin in die
verschiedenen Katecholamine läuft über eine Tyrosin-Hydroxylase (BH4-Abhängig), AA-
Decarboxylase (B6-Abhängig), Dopamin-β-Hydroxylase (Vit.C-Abhängig; Cu-Enzym) und
Phenylethanolamin-N-Methyltransferease (SAM-Abhängig; siehe Kapitel „C1-Stoffwechsel).
Page 24
Kapitel: Aminosäuren
24
Biosynthese der Katecholamine
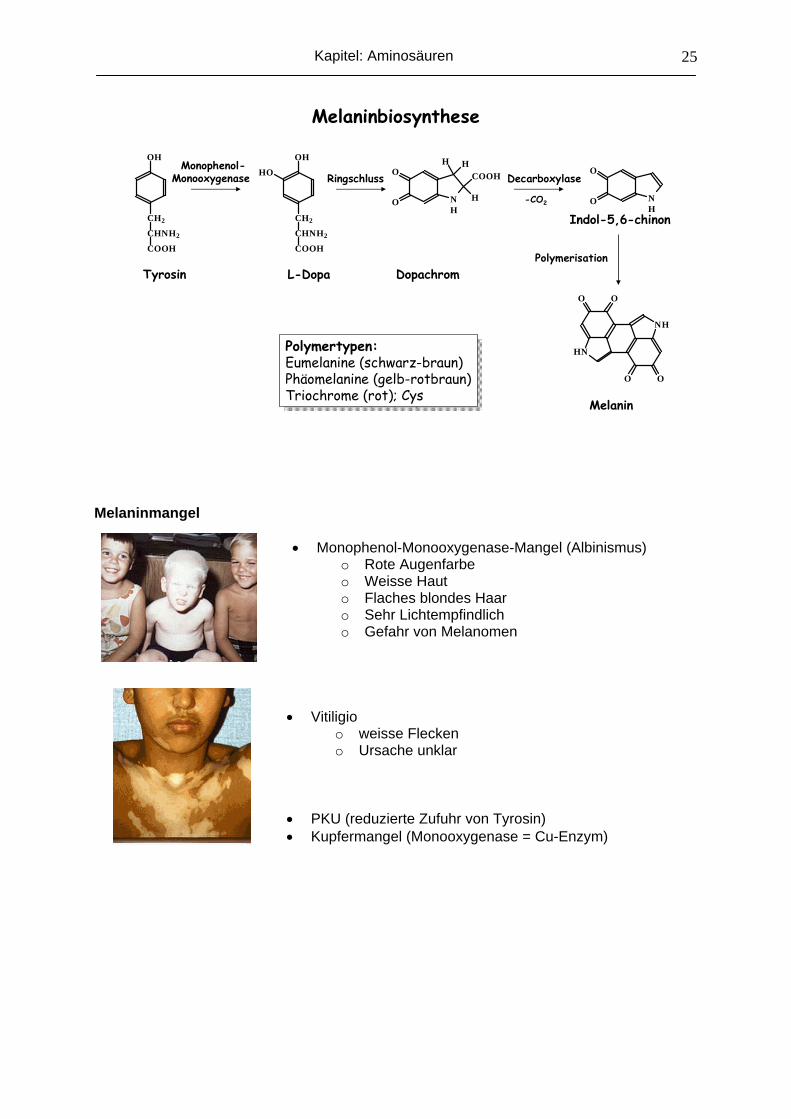
Biosynthese von Melanin aus Tyrosin
Melanin ist einer der wichtigsten Pigmenstoffe der Natur (Schutz vor ionisierter UV-
Strahlung). Es wird aus Tyrosin in mehreren Stufen gebildet und hat quantitativ für den Tyr-
Abbau keine Bedeutung. Die ganze Reaktionsfolge wird von einem einzigen Enzym, der
Monophenol-Monooxygenase (früher Tyrosinase genannt), katalysiert.
Der Bildungsort sind die Melanozyten in Basalschicht von Haut und Auge.
Page 25
Kapitel: Aminosäuren
25
CH2
CHNH2
COOH
OH
CH2
CHNH2
COOH
OH
HO O
O NH
H HCOOH
H
O
O NH
OO
HN
NH
OO
Melaninbiosynthese
Tyrosin L-Dopa Dopachrom
Indol-5,6-chinon
Melanin
Ringschluss Decarboxylase
Polymerisation
-CO2
Polymertypen:Eumelanine (schwarz-braun)Phäomelanine (gelb-rotbraun)Triochrome (rot); Cys
Polymertypen:Eumelanine (schwarz-braun)Phäomelanine (gelb-rotbraun)Triochrome (rot); Cys
Monophenol-Monooxygenase
Melaninmangel
• Monophenol-Monooxygenase-Mangel (Albinismus)
o Rote Augenfarbe o Weisse Haut o Flaches blondes Haar o Sehr Lichtempfindlich o Gefahr von Melanomen
• Vitiligio o weisse Flecken o Ursache unklar
• PKU (reduzierte Zufuhr von Tyrosin) • Kupfermangel (Monooxygenase = Cu-Enzym)
Page 26
Kapitel: Aminosäuren
26
Biosynthese von Aminosäuren Viele Aminosäuren können nur von Pflanzen und Mikroorganismen synthetisiert werden. Da
Säuger diese Aminosäuren mit der Nahrung aufnehmen müssen, bezeichnet man sie als
essentielle Aminosäuren. Aminosäuren, die von Säugern selbst gebildet werden können,
nennt man nichtessentiell. Die für Menschen essentiellen und nichtessentiellen
Aminosäuren sind unten aufgeführt. Arginin wird zwar im Harnstoff-Cyclus synthetisiert, da
aber Kinder (nicht Erwachsene) während ihrer normalen Entwicklung mehr davon
benötigen, als dieser Stoffwechselweg liefern kann, rechnet man es zu den halbessentiellen
Aminosäuren.
Essentielle Nichtessentielle
Arginin*
Histidin
Isoleucin
Leucin
Lysin
Methionin
Phenylalanin
Threonin
Tryptophan
Valin
Alanin
Asparagin
Aspartat
Cystein
Glutamat
Glutamin
Glycin
Prolin
Serin
Tyrosin
Biosynthese nichtessentieller Aminosäuren
Alle nichtessentiellen Aminosäuren, ausser Tyrosin, werden auf einfachen Wegen synthetisiert,
die von vier Grundbausteinen des Stoffwechsels ausgehen: Pyruvat, Oxalacetat,
α-Ketoglutarat und 3-Phosphoglycerat. Tyrosin entsteht in einem Schritt durch
Hydroxylierung der essentiellen Aminosäuren Phenylalanin, ist also strenggenommen
ebenfalls als essentiell anzusehen.
Biosynthese aus Pyruvat, Oxalacetat und α-Ketoglutarat durch Transaminierung
Ala: Pyruvat Ala
Asp, Asn: Ocalacetat Asp (Asn)
Glu, Gln: α-Ketoglutarat Glu (Gln)
Page 27
Kapitel: Aminosäuren
27
Biosynthese von Alanin, Aspartat, Glutamat, Asparagin und Glutamin
Pyruvat
Alanin
AS
α-KS
Oxalacetat
Aspartat
Asparagin
AS
α-KS
ATP
AMP+PPi
α-Ketoglutarat
Glutamat
Glutamin
AS
α-KS
ATP
ADP+Pi
NH3
Transaminasen Transaminasen Transaminasen
Asn-Synthase Gln-Synthase
Synthese von Prolin, Ornithin und Arginin aus Glutamat
Bei der Umwandlung von Glutamat in Prolin sind 4 Enzyme beteiligt.
Glu Glu-5-P Glu-5-semialdehyd Prolin
Bei der Umwandlung von Glutamat in Ornithin sind 5 Enzyme beteiligt.
Glu N-Acetyl-Glu N-Acetyl-Glu-P N-Acetyl-Glu-semialdehyd N-Acetyl-Orn Orn
Arginin wird im Harnstoffzyklus aus Ornithin synthetisiert.
Synthese von Serin, Cystein und Glycin aus 3-Phosphoglycerat
3-Phosphoglycerat 3-Phosphohydroxypyruvat 3-Phosphoserin Serin Serin + Homocystein Cystathionin Cystein (siehe Seite 31) Serin Glycin (siehe Seite 14)