48
Dr. S. Keitel Seite 1 ICH Q8, Q9 & Q 10 - welche Anforderungen werden an die pharmazeutischen Unternehmer im Zulassungsprozess gestellt? Forum Online-Seminar Dr. Susanne Keitel 2. März 2006

Dr. S. Keitel Seite 1
ICH Q8, Q9 & Q 10- welche Anforderungen werden an die
pharmazeutischen Unternehmer im Zulassungsprozess gestellt?
Forum Online-SeminarDr. Susanne Keitel
2. März 2006

Dr. S. Keitel Seite 2
Gliederung
• Stand der Entwicklung der ICH-Leitlinien • Hintergrund – ICH Q 8 „Kerndokument” und
zukünftige Entwicklungen• Konzept und Philosophie der Leitlinie• Erwartungen an die pharmazeutische
Industrie• Vorteile und Herausforderungen

Dr. S. Keitel Seite 3
"A modern harmonised approach to Pharmaceutical Quality and Manufacturing"
Chicago, Illinois, November 7-10, 2005
The International Conference on Harmonisation (ICH) Steering Committee and its expert working groups met in Chicago, Illinois on November 9th and 10th, 2005.
This meeting highlighted the progress made in the area of pharmaceutical quality and manufacturing.
A new topic, "Quality Systems" (Q10) was approved by the Steering Committee. This proposed guideline will augment existing Good Manufacturing Practices (GMPs) with modern quality systems elements.

Dr. S. Keitel Seite 4
The Steering Committee was pleased to adopt 2 related guidelines as final (Step 4 of the ICH process):
- "Pharmaceutical Development " (Q8), which describes what should be submitted to a regulatory authority in the relevant section of the Common Technical Document (ICH topic M4);
- "Quality Risk Management" (Q9), which provides principles and examples of quality risk management that can be applied to all aspects of developing a medicinal product, submitting to a regulatory authority, and for manufacturing sites inspections.
Together these guidelines form the foundation for a modern risk-based approach to pharmaceutical quality and manufacturing. Source: Press Release ICH Steering Committee

Dr. S. Keitel Seite 5
ICH Q 8: Pharmaceutical Development
“This guideline is intended to provide guidance on the contents of Section 3.2.P.2 (Pharmaceutical Development) for drug products as defined in the scope of Module 3 of the CTD. The guideline does not apply to contents of submissions for drug products during the clinical research stages of drug development. However, the principles in this guideline are important to consider during these stages, -this guideline might also be appropriate for other types of products. To determine the applicability of this guideline for a particular type of product, applicants should consult with the appropriate regulatory authorities.” (http://www.ich.org)

Dr. S. Keitel Seite 6
Aufbau von ICH Q 8
“Part 1”
Kerndokument
“Baseline expectations”
Optionale Daten
Definition “Design Space”
Regulatorische Flexibilität
“Part 2”
“Annex“ zu spezifischen Darreichungsformen
Beispiele für “baseline expectations“ vs. “optionale Daten“
Referenz zu ICH Q 9

Dr. S. Keitel Seite 7
Das Kerndokument• Gibt Empfehlungen zum Inhalt des Kapitels 3.2.P.2
(Pharm. Development) für Arzneimittel, die in den Anwendungsbereich des Moduls 3 des CTD fallen.
• Auch für andere Produktarten anwendbar zuständige Behörde konsultieren!
• Anforderungen gelten nicht für die Einreichung vom IMPDs (aber die allgemeinen Prinzipien sollten berücksichtigt werden)

Dr. S. Keitel Seite 8
Zukünftige Entwicklung• Annex (ICH Q 8 (R1)) zu spezifischen
Darreichungsformen in der Erarbeitung
• Ziel: Beispiele für die Anwendung von “Quality byDesign”-Konzepten an Hand spezifischer Darreichungsformen vermitteln, kein Lehrbuchwissen!
• Voraussichtlicher Zeitplan: Step 2 im Juni oder November 2006
• „Q8-type“ Leitlinie für Wirkstoffe??

Dr. S. Keitel Seite 9
ICH Q 9: Risk Management
“This guidelines provides principles and examples of tools on quality risk management that can be applied to all aspects of pharmaceutical quality including development, manufacturing ,distribution, and the inspection and submission/review processes throughout the lifecycle of drug substances and drug products, biological and biotechnological products, including the use of raw materials, solvents, excipients, packaging and labeling materials.” (http://www.ich.org)

Dr. S. Keitel Seite 10
Status:
• Leitlinienentwurf im März 2005 zur Kommentierung publiziert
• Im November 2005 als harmonisierte ICH-Leitlinie verabschiedet
• Veröffentlichung und Inkrafttreten in der EU?

Dr. S. Keitel Seite 11
ICH Q10 - Quality Systems
“ICH Q 10, Quality Systems is in its infancy stages with the International Conference on Harmonisation. The proposed intent of this document is to describe a Quality System that leads to a common understanding of QS terms and applications across the three regions, link together and enable the benefits of the current ICH Q8 and ICH Q9 guidelines and encourage a regulatory environment that facilitates continual improvement in pharmaceutical manufacturing…(“under construction” guideline)….” (http://www.pda.org)

Dr. S. Keitel Seite 12
Status:
• Konzeptpapier für eine ICH-Leitlinie zu Qualitätssystemen, verbunden mit dem Vorschlag, offiziell eine Expert Working Group einzurichten, wurde vom ICH-Steering Committee akzeptiert
• Erste Sitzung der EWG im November 2005 in Chicago
• Step 2 Dokument im Juni oder November 2006?

Dr. S. Keitel Seite 13
Zukünftige Entwicklung
ICH Q 8 kein “stand alone”-Dokument, sondernTeil eines Gesamtkonzepts
• ICH Q 8 Pharmaceutical Development• ICH Q 9 Quality Risk Management• ICH Q 10 Quality Systems

Dr. S. Keitel Seite 14
ICH Q 8 - Hintergrund
Unterschiedliche Erfahrungen in der ICH-Triade:
- EU- USA- Japan
Wunsch nach Entwicklung einerharmonisierten Leitlinie durch dieVerabschiedung und Implementierungdes CTD ausgelöst

Dr. S. Keitel Seite 15
Pharmazeutische Entwicklung im CTD
“The … section should contain information on the development studies conducted to establish that the dosage form, the formulation, manufacturing process, container closure system, microbiological attributes and usage instructions are appropriate for the purpose specified in the application. The studies described here are distinguished from routine control tests conducted according to specifications….”

Dr. S. Keitel Seite 16
Pharmazeutische Entwicklung im CTD
“… Additionally, this section should identify and describe the formulation and process attributes (critical parameters) that can influence batch reproducibility, product performance and drug product quality. Supportive data and results from specific studies or published literature can be included within or attached to the …. Section. Additional supportive data can be referenced to the relevant nonclinical or clinical sections of the application.”
ICH M4 Q

Dr. S. Keitel Seite 17
P2 Pharmazeutische Entwicklung im CTD
P 2.1 Components of the drug productP 2.1.1 Active substanceP 2.1.2 ExcipientsP 2.2 Drug productP2.2.1 Formulation developmentP2.2.2 OveragesP2.2.3 Physical and biological propertiesP 2.3 Manufacturing process developmentP 2.4 Container/closure systemP 2.5 Microbiological attributesP 2.6 Compatibility

Dr. S. Keitel Seite 18
Pharmazeutische Entwicklung...
• ist die Basis jeder ordentlichen Arzneimittel-entwicklung
• sollte als Risikoanalyse für die Eignung von Formulierung und Herstellungsverfahren dienen
• sollte Schwachpunkte in der Formulierung und imHerstellungsverfahren identifizieren
• sollte ausreichende Sicherheit bringen, dass dasArzneimittel reproduzierbar in der spezifiziertenQualität hergestellt werden kann
• …
Pharm. Ent. – EU-Standpunkt

Dr. S. Keitel Seite 19
Pharm. Entwicklung – die RealitätUntersuchungen zur pharm. Entwicklung ...
• werden für NCEs und innovative Arzneimittel in den meisten Fällen ordentlich durchgeführt (“baseline approach”)
• sind bei Generika manchmal darauf beschränkt, wie das Innovator-Präparat am einfachsten imitiert werden kann (Patente!)
• die im Zulassungsantrag eingereicht werden, sind meistens hilfreich, um die Herangehensweise des Antragstellers zu verstehen, aber liefern oft nicht ausreichende Informationen

Dr. S. Keitel Seite 20
ICH Q 8 – Das Konzept (I)
ICH Q 8 - Pharmaceutical Development
“The Pharmaceutical Development section providesan opportunity to present the knowledge gainedthrough the application of scientific approaches tothe development of a product and its manufacturingprocess, and quality risk management…..”

Dr. S. Keitel Seite 21
Das Konzept (II)
ICH Q 8 - Pharmaceutical Development
“… It is first produced for the original marketing application and may be updated to support new knowledge gained over the lifecycle of a product.…..”

Dr. S. Keitel Seite 22
Das Konzept (III)
ICH Q 8 - Pharmaceutical Development
“… is intended to provide a comprehensive understanding of the product and manufacturing process for reviewers and inspectors .…..”

Dr. S. Keitel Seite 23
Das Konzept (IV)
ICH Q 8 - Pharmaceutical Development
“… also indicates areas where the demonstration of greater understanding of the pharmaceutical andmanufacturing sciences can create a basis forflexible regulatory approaches. The degree of regulatory flexibility is predicated on the level of relevant scientific knowledge provided.”

Dr. S. Keitel Seite 24
Das Konzept (V)ICH Q 8 - Pharmaceutical Development
”The aim… is to design a quality product and themanufacturing process to consistently deliver the intended performance of the product. Theinformation and knowledge learned from …development studies and manufacturing experienceprovide scientific understanding necessary tosupport the establishment of the design space,specifications and manufacturing controls. …”

Dr. S. Keitel Seite 25
Das Konzept (VI)
ICH Q 8 - Pharmaceutical Development
“… Information from pharmaceutical developmentstudies can be a basis for quality risk management.It is important to recognize that quality cannot be tested into products; i.e., quality has to be built in bydesign. ”

Dr. S. Keitel Seite 26
Die Chance…....... den Behörden Informationen für eine wissenschaftliche und risikobasierte Bewertung (und Inspektion!) zu liefern
• z.B. für die Festlegung von Spezifikationen
•„regulatorische Flexibilität“ für das Life-CycleManagement, d.h. Basis für kontinuierliche Verbesserungen ohne vorherige Zustimmung
DIE BEHÖRDEN VON DER EIGENEN PHILOSOPHIE/HERANGEHENSWEISE FÜR DIE ENTWICKLUNG ZUÜBERZEUGEN!

Dr. S. Keitel Seite 27
DIE PHILOSOPHIEPharmazeutische Entwicklung….
• ist keine “Momentaufnahme”, aber ein ständigerLernprozess über den Lebenszyklus einesArzneimittels
• kann Wissen/Informationen aus missglücktenVersuchen beinhalten
• Änderungen an Formulierung und Verfahrenwährend der Entwicklung und des Lebenszyklussind zusätzliche Möglichkeiten, den “DesignSpace” zu bestätigen bzw. auszuweiten

Dr. S. Keitel Seite 28
Die Philosophie
Ein unerwartetes oder missglücktes Ergebnis kann viel mehr sein als nur eine
Enttäuschung!

Dr. S. Keitel Seite 29
ICH Q 8 – Was soll erreicht werden?• Product quality and performance achieved andassured by design of effective and efficientmanufacturing processes
• Product specifications based on mechanisticunderstanding of how formulation and process factors impact product performance
• An ability to effect Continuous Improvement and Continuous "real time" assurance of quality

Dr. S. Keitel Seite 30
Design Space
“The design space is the established range of process parameters that has been demonstrated to provide assurance of quality. In some cases design space can also be applicable to formulation attributes. Working within the design space is not generally considered as a change of the approved ranges for process parameters and formulation attributes. Movement out of the design space is considered to be a change and would normally initiate a regulatory post approval change process.”
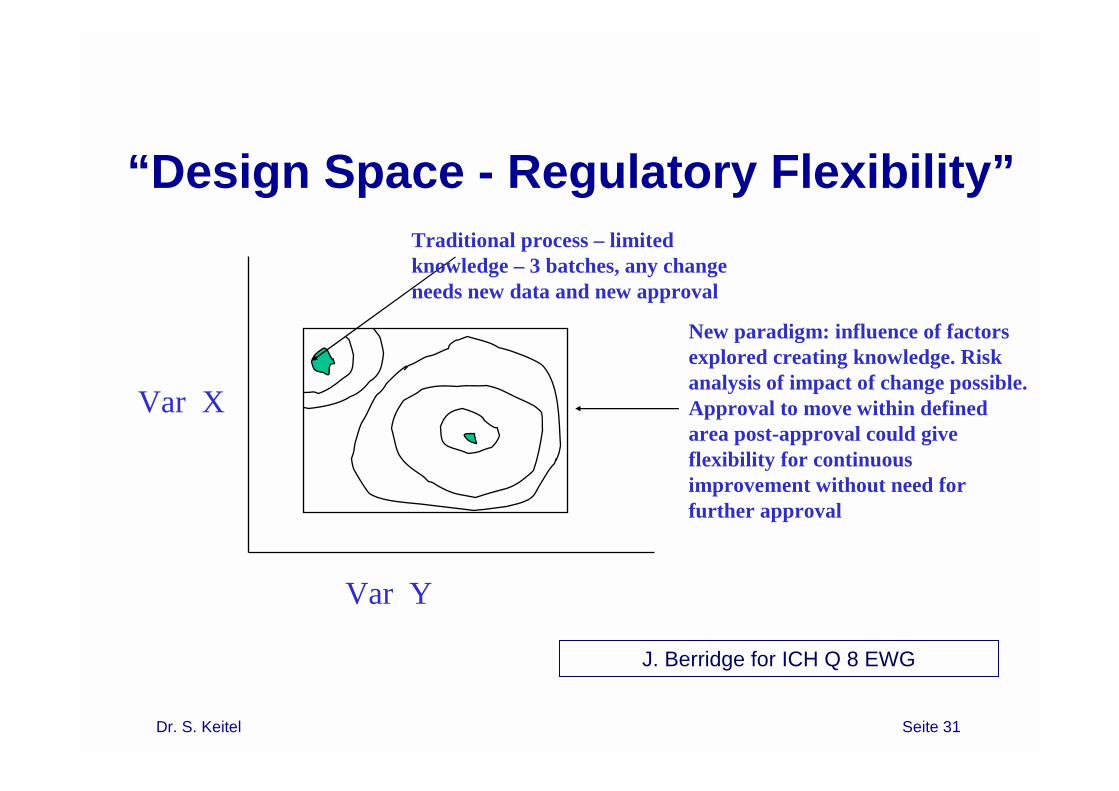
Dr. S. Keitel Seite 31
Var X
Var Y
Traditional process – limited knowledge – 3 batches, any change needs new data and new approval
New paradigm: influence of factors explored creating knowledge. Risk analysis of impact of change possible. Approval to move within defined area post-approval could give flexibility for continuous improvement without need for further approval
“Design Space - Regulatory Flexibility”
J. Berridge for ICH Q 8 EWG

Dr. S. Keitel Seite 32
“Regulatory Flexibility”
• a-b inside space• b-c inside space• c-d explores new
space – comparability protocol?
• OK to do if spec not impacted?
initial Filing
Factor space explored at initial filing
a b
cd
J. Berridge for ICH Q8 EWG

Dr. S. Keitel Seite 33
Das „Zweigleisige System”
Klare Unterscheidung zwischen Minimal-Anforderungen (“baseline expectations”) und
optionalen zusätzlichen Informationen (“opportunities”)
Ausschließlich Entscheidung des Antragstellers, wieviel Ressourcen überhaupt und zu welchem Zeitpunkt in die Entwicklung investiert werden!

Dr. S. Keitel Seite 34
Minimalanforderungen“At a minimum, those aspects of drug substances, excipients, container closure systems, and manufacturing processes that are critical to product quality should be determined and control strategies justified.
Critical formulation attributes and process parameters are generally identified through an assessment of the extent to which their variation can have impact on the quality of the drug product.”

Dr. S. Keitel Seite 35
EU: Minimalanforderungen
Anforderungen, wie derzeit in der Leitlinie
CPMP/QWP Note for Guidance on Development Pharmaceutics
definiert sind im Allgemeinen einzuhalten!

Dr. S. Keitel Seite 36
Optionale Informationen“In addition, the applicant can choose to conduct pharmaceutical development studies that can lead to an enhanced knowledge of product performance over a wider range of material attributes, processing options and process parameters. Inclusion of this additional information in this section provides an opportunity to demonstrate a higher degree of understanding of material attributes, manufacturing processes and their controls….”

Dr. S. Keitel Seite 37
Optionale Informationen“… This scientific understanding facilitates establishment of an expanded design space. In these situations, opportunities exist to develop more flexible regulatory approaches, e.g. to facilitate
• risk-based regulatory decisions
• manufacturing process improvements, within the approved design space described in thedossier, without further regulatory review

Dr. S. Keitel Seite 38
Optionale Informationen• Reduction of post-approval submission,
• Real-time quality control, leading to a reductionof end- product release testing
To realise this flexibility, the applicant should demonstrate an enhanced knowledge of product performance….. This understanding can be gained by application of, e.g., formal experimental designs, process analytical technology, and/or prior knowledge….”

Dr. S. Keitel Seite 39
EU ErwartungenAntragsteller/Zulassungsinhaber entscheidet, ob und zu welchem Zeitpunkt in Untersuchungen zu einem vertieften wissenschaftlichen Verständnis von Formulierung und Verfahren “investiert” wird, z.B.
• “Design of Experiments” vs. Informationen an Hand vonvergleichbaren Präparaten oder aus der Produktion, Literaturdaten
• Alle Aspekte von Formulierung und/oder Prozess vs.ausgewählten Aspekten (z.B. unterschiedlicheHilfsstoffquellen)

EU Erwartungen
Einreichung einer überzeugenden und plausiblen “Geschichte” im Dossier
• um Assessoren (und Inspektoren!) zuüberzeugen
• Wissen, nicht (zwingend) Daten!

Dr. S. Keitel Seite 41
Herausforderungen für die Behörden
• Zweigleisiges System: “Baseline”-Untersuchungen vs. vertieftes mechanistisches Verständnis von Formulierungs- und Prozesseigenschaften, z.B. statistische Versuchsplanung, PAT-Konzepte, Anwendung von “Risikomanagement-Werkzeugen“ (und jede Schattierung dazwischen…..)
• Modifizierung der Bewertungs-/Inspektionsstrategien

Dr. S. Keitel Seite 42
Herausforderungen für die Behörden
• Verstärkung der Zusammenarbeit zwischen Assessoren und Inspektoren zum Zeitpunkt der Ersteinreichung und während des Lebenszyklus eines Arzneimittels
• Wieviel Informationen / welcher Detaillierungsgrad muss im Dossier gefordert werden?
• Wie weit kann der Begriff “regulatorische Flexibilität“ definiert werden?
• Wie soll das Konzept der “regulatorischen Flexibilität” in den Rahmen einer bestehenden EU Variation Regulation eingefügt werden?

Dr. S. Keitel Seite 43
Mögliche Konsequenzen für den regulatorischen Prozess – “Reg. Flexibilität”☺Durch den Einsatz von z.B. statistischer
Versuchsplanung kann der pU ein besseres Verständnis für sein Produkt gewinnen
☺Größerer Sicherheitsspielraum für den Hersteller, ggf. mit einer Reduzierung von Fehlchargen verbunden
☺Ein besseres und wissenschaftsbasierteres Verständnis von Formulierung und Prozess durch Assessoren und Inspektoren kann zu wissenschafts-und risikobasierteren Bewertungs- und Inspektionsstrategien führen

Dr. S. Keitel Seite 44
Mögliche Konsequenzen für den regulatorischen Prozess – “Reg. Flexibilität”
☺Der pU kann mehr Flexibilität für z.B. Modifizierungen/Veränderungen innerhalb des im Zulassungsdossier beschriebenen Rahmens erlangen, die ohne vorherige Zustimmung durch die Behörden umgesetzte werden können

Dr. S. Keitel Seite 45
Mögliche Konsequenzen für den regulatorischen Prozess - Zusammenfassung
“You get what you pay for” – frühzeitige Investition in wissenschaftliches und mechanistisches Verständnis von Formulierung und Herstellungsverfahren während der Präparateentwicklung kann die Erstbewertung des Zulassungsdossiers beschleunigen und das Life-Cycle-Management für Industrie und Behörden erleichtern

Dr. S. Keitel Seite 46
Was wird vom pharmazeutischen Unternehmer erwartet?
EU-Standpunkt:Erfüllung der Minimalanforderungen („Baseline-Expectations“), KEINE zwingenden zusätzlichen Anforderungen für das ZulassungsdossierABER:Unternehmerische Entscheidung, ob„Investitionen“ getätigt werden

Dr. S. Keitel Seite 47
Was wird vom pharmazeutischen Unternehmer erwartet?
Prinzip “You get what you pay for” – frühzeitige Investition und Weitergabe der Kenntnisse an dieBehörden können ein höheres Maß anProzesssicherheit und regulatorische Freiräumeschaffen!

Dr. S. Keitel Seite 48
Herzlichen Dank für Ihre Aufmerksamkeit!

















