Heterologe Expression und biochemische Charakterisierung von extrazellulären halotoleranten bzw. halophilen Peptidasen in Hinblick auf ihre biotechnologische Nutzung Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) Dem Fachbereich Biologie und Chemie (FB2) der Universität Bremen vorgelegt von Helge Mühl Januar 2007
Transcript
Heterologe Expression und biochemische Charakterisierung von extrazellulären halotoleranten
bzw. halophilen Peptidasen in Hinblick auf ihre biotechnologische Nutzung
Dissertationzur Erlangung des Grades eines
Doktors der Naturwissenschaften (Dr. rer. nat.)
Dem Fachbereich Biologie und Chemie (FB2)
der Universität Bremen vorgelegt von
Helge Mühl
Januar 2007
1. Gutachter: Prof. Dr. Ulrich Fischer 2. Gutachter: Prof. Dr. Michael G. Lorenz
Danksagung
DDaannkkssaagguunnggDie vorliegende Arbeit wurde in der Firma Molzym GmbH & Co. KG durchgeführt. Ich
möchte mich an dieser Stelle bei Prof. Dr. Ulrich Fischer und Prof. Dr. Michael
Lorenz für die Bereitstellung des Themas bedanken. Bei Prof. Dr. Lorenz bedanke
ich mich besonders für die Betreuung, Diskussionsbereitschaft und die hilfreichen
Hinweise beim Verfassen dieser Arbeit. Der Firma Molzym GmbH & Co. KG gilt mein
Dank für die Möglichkeit einen hervorragend ausgestatteten Arbeitsplatz zu nutzen.
Den Mitarbeitern, besonders Dr. Claudia Disqué, Katja Bergholz und Silke
Bohnacker, möchte ich für die gute Arbeitsatmosphäre und den fruchtbaren Ideen-
austausch danken. Für die zeitweise Nutzung von Arbeitsplatz, Laborausstattung
und Labormaterialien spreche ich der Arbeitsgruppe Marine Mikrobiologie der
Universität Bremen, im Besonderen Prof. Dr. U. Fischer, meinen Dank aus. Ein
herzliches Dankeschön gilt Petra Herzog und Kristina Schmanke für das Korrektur-
lesen der vorliegenden Arbeit.
Meiner Familie und meinen Freunden bin ich für die bedingungslose Unterstützung in
allen Lebenslagen sehr verbunden. Ganz besonders möchte ich mich an dieser
Stelle auch bei meiner Freundin Madeleine bedanken, die mich immer unterstützt hat
1.1 Peptidasen 11.1.1 Terminologie und Klassifizierung der Peptidasen 21.1.1.1 Serinpeptidasen 61.1.1.2 Metallopeptidasen 71.2 Halophile Bakterien, Adaption an hohe Salzkonzentrationen 81.2.1 Halophile Enzyme 91.2.1.1 Halophile Peptidasen 101.2.2 Halophile Peptidasen in der Biotechnologie 11
2 16Material und Methoden2.1 Mikroorganismen 162.1.1 Halotolerante und halophile Bakterienstämme 162.1.2 Klonier- & Expressionsstämme 162.1.3 Sonstige Stämme 172.2 Plasmide 172.3 Synthetische Oligonukleotide (Primer) 182.4 Medien, Lösungen & Puffer 202.4.1 Luria-Bertani (LB) Medium (Sambrook & Russell, 2001) 202.4.2 Salinenmedium-Lanzarote (SML) (Sinn-Meyer, 2003) 202.4.3 M9 Minimal Medium (Sambrook & Russell, 2001) 202.4.4 M9-Salzlösung (5x) (Sambrook & Russell, 2001) 212.4.5 RM-Medium (Sambrook & Russell, 2001) 212.4.6 Feste Medien für den Nachweis extrazellulärer Peptidasen 212.4.6.1 Skimmilk-Agarose für den Peptidasenachweis in PA-Gelen 212.4.7 Antibiotika und Medien-Zusätze 212.4.8 Puffer und Lösungen 222.4.9 Chemikalien 232.5 Mikrobiologische Methoden 232.5.1 Anzucht der halophilen Isolate 232.5.2 Anzucht der Klonier- und Expressionsstämme 232.6 Proteinbiochemische Methoden 232.6.1 Gewinnung von zellfreien Kulturüberständen 232.6.2 Protein-Fällung der Kulturüberstände 232.6.3 Dialyse gegen Polyethylenglykol 3500 242.6.4 Proteinbestimmung mit BCA (Bicinchoninic Acid)-Methode 242.6.5 Heterologe Proteinexpression in E. coli 242.6.6 Zellernte und Zellaufschluss 252.6.7 Säulenchromatografie 252.6.7.1 Anionenaustauschchromatografie 252.6.7.2 Hydrophobe Interaktionschromatografie 262.6.7.3 Gelfiltrationschromatografie 262.6.8 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (Laemmli, 1970,
modifiziert) 272.6.9 Detektion von Peptidase-Aktivität auf festen Medien 282.6.10 Peptidasenachweis im Polyacrylamid-Gel (Kontakt-Zymogramm) 282.6.11 Quantitative Peptidase-Aktivitätsbestimmung 292.6.11.1 Azocasein-Assay (Charney & Tomarelli, 1947, modifiziert) 292.6.11.2 EnzCheck Protease Assay Kit (Invitrogen) 292.6.11.3 Casein-Assay (Kunitz, 1947, modifiziert) 302.6.11.4 Bestimmung der pH-Abhängigkeit der Peptidase-Aktivität 312.6.11.5 Einfluss von chaotropen Salzen auf die Peptidase-Aktivität 31
Inhaltsverzeichnis
2.6.11.6 Einfluss von NaCl und Metallionen auf die Peptidase-Aktivität 312.6.11.7 Einfluss der Temperatur auf die Peptidase-Aktivität 322.7 Molekularbiologische Methoden 322.7.1 Nukleinsäure-Extraktion aus Bakterien 322.7.1.1 Extraktion chromosomaler DNA 322.7.1.2 Isolierung von RNA aus E. coli 322.7.1.3 Isolierung von Plasmid-DNA aus E. coli 322.7.1.4 Isolierung von Fosmid-DNA aus E. coli 322.7.2 Agarose-Gelelektrophorese und Dokumentation 332.7.3 Southern-Blot 332.7.4 Northern-Blot 342.7.5 Kolonie-Blot 342.7.6 Hybridisierung 352.7.7 Herstellung von DNA-Sonden 352.7.8 Herstellung von Genbanken 352.7.9 Subklonierung 362.7.10 Ligation 362.7.11 Herstellung von elektrokompetenten E. coli-Zellen 362.7.12 Transformation 372.7.13 Replikaplattierung (Tatum & Lederberg, 1947) 382.7.14 Polymerasekettenreaktion (PCR) 382.7.15 Sequenzierung und Auswertung der Sequenzdaten 39
3 40Ergebnisse3.1 Extrazelluläre Peptidasen aus halotoleranten und halophilen Bakterien 403.1.1 Vorauswahl und Charakterisierung von Peptidase produzierenden Isolaten 403.1.2 Systematische Einordnung der ausgewählten Peptidase-Produzenten
mittels 16S-rRNA Analyse 433.1.3 Physiologie der Enzymbildung 453.1.4 In vitro Peptidase-Assays - Möglichkeiten und Grenzen der verwendeten
Assays 463.1.5 Anreicherung der Peptidasen aus Kulturüberständen der halophilen
Stämme 493.1.5.1 Säulenchromatografie 513.2 Biochemische Charakterisierung extrazellulärer Peptidasen 533.2.1 Einfluss des pH-Wertes auf die Peptidase-Aktivität 543.2.2 Einfluss der Temperatur auf die Peptidase-Aktivität 553.2.3 Salzabhängigkeit der Peptidasen 563.2.4 Einfluss von Metallionen auf die Peptidase-Aktivität 583.2.5 Einfluss chaotroper Agenzien auf die Peptidasen 603.2.6 Einfluss von Inhibitoren auf die Peptidasen 613.2.7 Gewebeauflösende Aktivität 633.2.8 Analyse der Peptidasen mittels PAGE 653.3 Molekularbiologische Identifizierung von Subtilisin-ähnlichen Peptidasen 673.3.1 Entwicklung der Sonde für Subtilisin-ähnliche Peptidasen 693.3.2 Identifizierung von Klonen und Subklonen mit homologen Sequenzen zu
Subtilisin-ähnlichen Peptidasen 703.3.3 Identifizierung und Sequenzanalyse der Peptidasegene 723.3.4 Identifizierung des Signalpeptids in ProAS(#748) 743.4 Heterologe Expression der Subtilisin-ähnlichen Peptidasen in E. coli 753.4.1 Peptidase-Überexpression mit Expressionsvektoren 763.4.1.1 Modellsystem: Expression von Subtilisin E in E. coli 763.4.1.1.1 Konstruktion der Expressionsvektoren 773.4.1.1.2 Expressionsanalyse 783.4.1.2 Heterologe Expression von ProAS(#748) 813.4.1.2.1 Analyse der Expression von ProAS(#748) durch pBAD 833.4.2 Heterologe Expression einer aktiven Peptidase durch den Fosmidklon F114 84
Inhaltsverzeichnis
3.4.3 Übereinstimmung der Peptidase F114 mit Peptidase B aus B3/1-2 (#418) 863.4.3.1 Reinigung der rekombinanten Peptidase F114 873.4.3.2 Biochemische Eigenschaften der Peptidase F114 im Vergleich zur Wildtyp-
Peptidase B 883.4.3.3 Bestimmung des Molekulargewichtes von Peptidase F114 91
4 95Diskussion4.1 Zugehörigkeit der Peptidase produzierenden moderat halophilen Stämme
zur Gattung Salinivibrio 974.2 Salinivibrio bildet zwei Typen von extrazellulären Peptidasen 994.2.1 Typ A: Metallopeptidase 1004.2.2 Typ B: Serinpeptidase 1024.2.2.1 Biochemische Charakteristika 1024.2.2.2 Molekularbiologische Charakteristika 1044.3 Heterologe Expression der Serinpeptidase ProAS in E. coli 1074.4 Einsatz der Metallopeptidase und der Serinpeptidase (ProAS) aus
Salinivibrio in der Biotechnologie 1165 121Zusammenfassung6 123Literaturverzeichnis7 140Anhang Abkürzungsverzeichnis: Abkürzung Bedeutung Abkürzung Bedeutung A. dest. Destilliertes Wasser Mr Relative molekulare Größe [kb] AzU Azocaseinunit MOPS 3-Morpholinopropansulfonsäure BLAST Basic Local Alignment Search
Tool n.b. Nicht bestimmt
Bp Basenpaare OD Optische Dichte BSA Rinderserumalbumin ORF Offenes Leseraster Cfu Kolonie bildende Einheiten PA Polyacrylamid DNA Desoxyribonukleinsäure PAGE Polyacrylamid Gel Elektrophorese dNTP Desoxyribonukleotidtriphosphat Phenanthrolin 1,10-Phenanthrolin E. coli Escherichia coli PMSF Phenylmethylsulphonylfluorid EDTA Ethylendiamintetraessigsäure RNA Ribonukleinsäure ERG Eppendorfreaktionsgefäß RT Raumtemperatur (18-25°C) FlU Fluoreszenzunit SDS Natriumdodecylsulfat GuHCl Guanidinhydrochlorid SML 12/8 Salinemedium Lanzarote (12%
Tetramethylethylendiamin Kbp Kilobasenpaare Tris Tris(hydroxymethyl)-aminomethan kFlU Kilo-Fluoreszenzunit v/v Volumen pro Volumen KuU Kunitz-Unit w/v Gewicht pro Volumen mAU Milli Absorption Units WT Wildtyp
Einleitung
11 Einleitung Einleitung
1.11.1 Peptidasen Peptidasen
Peptidasen katalysieren die Hydrolyse von Peptidbindungen und sind daher in der
Lage Proteine und Peptide zu spalten, wobei kürzere Peptidfragmente oder freie
Aminosäuren als Spaltprodukte entstehen können. Peptidasen sind für alle Pro- und
Eukaryoten essentiell und beeinflussen die Funktionalität sowohl einzelner Zellen als
auch ganzer Organe bzw. vielzelliger Organismen. Übereinstimmend mit ihrer hohen
biologischen Relevanz kodieren etwa 2% aller Gene für Peptidasen (Rawlings et al.,
2006).
Abhängig von der Sequenzspezifität der Peptidasen erfolgt eine schematische
Unterteilung in eine limitierte und eine unlimitierte Proteolyse (Hooper, 2002). Bei der
unlimitierten Proteolyse werden Proteine durch unspezifische Peptidasen zumeist im
Rahmen des Protein-Turnover in der Zelle oder digestorischer Vorgänge außerhalb
der Zelle zu Oligopeptiden oder Aminosäuren abgebaut. Katalysieren Peptidasen
dagegen eine spezifische Spaltung, spricht man von einer limitierten Proteolyse,
wobei es zumeist zur Spaltung einzelner Peptidbindungen kommt. Diese selektive
Modifikation wird von vielen Proteinen zur Entwicklung ihrer biologisch aktiven Form
benötigt. So sind Viren auf die proteolytische Segmentierung ihrer Polyproteine
angewiesen, während Peptidasen bei Pro- und Eukaryoten zur Abtrennung der
Signalpeptide sekretierter Proteine notwendig sind. Fernerhin katalysieren sie die
Prozessierung von inaktiven Enzymvorstufen (Proenzyme oder Zymogene) zu
aktiven Enzymen durch Abspaltung der Propeptide. Beispielsweise werden selbst
viele Peptidasen als Proenzym synthetisiert und bedürfen einer proteolytischen
Aktivierung (Khan & James, 1998). Ort und Zeitpunkt dieser Aktivierung stellen
hierbei einen wichtigen Mechanismus der biologischen Regulation dar. Dement-
sprechend besitzen Peptidasen eine entscheidende Funktion in Regulations-
prozessen, u.a. in komplexen Abläufen wie Sporulation, Zellvermehrung, Zell-
differenzierung, Blutgerinnung, Blutdruck- und Immunregulation (Mann & Lorand,
1993; Rao et al., 1998). Ebenso können Peptidasen als integrale Membranproteine
auch Rezeptorfunktionen übernehmen und eine interzelluläre Kommunikation sowohl
über ihre Substrate als auch über den direkten Zell-Zell-Kontakt gewährleisten
(Albiston et al., 2001). Gleichbedeutend mit der Aktivierung ist für die Regulation aber
1
Einleitung
ebenso die Inaktivierung der Enzyme, welche wiederum durch eine proteolytische
Spaltung erfolgen kann (Barrett, 2000).
Eine Vielzahl von Peptidasen aus Bakterien wurde bereits beschrieben und näher
charakterisiert. Neben den ubiquitären intrazellulären Peptidasen synthetisieren viele
Bakterien proteolytische Enzyme zur Sekretion in das umgebende Medium (Rao et
al., 1998). Einige dieser extrazellulären Peptidasen sind Toxine oder virulenz-
vermittelnde Faktoren (Matsumoto, 2004). Ausgehend von ihrer medizinischen
Bedeutung bei bakteriellen Infektionen sind sie Gegenstand intensiver Forschung.
Ein Beispiel für extrazelluläre Peptidasen, die eine Invasion von bakteriellen Krank-
heitserregern in ihre Wirte ermöglichen, sind die IgA-Peptidasen. Meningitis-Erreger,
wie Streptococcus pneumoniae, Neisseria meningitidis und Haemophilus influenzae,
können durch spezifische Proteolyse der IgA-Antikörper des Wirtes die Immun-
barriere der Mukosa überwinden (Poulsen et al., 1996). Weit verbreitet ist die
Fähigkeit der Bakterien durch sekretierte Peptidasen proteinhaltige Substrate, die
außerhalb der Zelle lokalisiert sind, für ihren zentralen Metabolismus zu nutzen. Die
unlimitierte Hydrolyse extrazellulärer Proteine durch unspezifisch wirkende
Peptidasen stellt den Bakterien dabei leicht aufnehmbare Oligopeptide und Amino-
säuren zu Verfügung (Kalisz, 1988). Im Hinblick auf ihr breites Substratspektrum sind
diese extrazellulären Peptidasen aus Bakterien von herausragendem Interesse für
den Proteinabbau in diversen industriellen Prozessen. In Zusammenhang mit ihrer
hohen Toleranz gegenüber extremen Bedingungen haben sich dabei besonders
Enzyme von extremophilen Mikroorganismen in biotechnologischen Anwendungen
durchgesetzt (Hough & Danson, 1999).
1.1.11.1.1 Terminologie und Klassifizierung der Peptidasen Terminologie und Klassifizierung der Peptidasen
Die Bezeichnung für die Peptidbindungshydrolasen ist in der Literatur nicht
einheitlich. Es werden Bezeichnungen wie Proteasen, Proteinasen oder Peptidasen
häufig nach Belieben eingesetzt. Obwohl schon Bergmann und Ross (1936) eine
klare Definition erstellten und diese auch seit 1992 von der IUBMB (International
Union of Biochemistry and Molecular Biology - Nomenclature Committee, 1992)
vertreten wird, ist von einer Umsetzung in der aktuellen Literatur nur wenig zu
erkennen (vergleiche hierzu z.B.: Arnorsdottir et al., 2005; Hiraga et al., 2005; Lama
et al., 2005; Venugopal & Saramma, 2006). Nach der geltenden Definition der
IUBMB werden alle proteolytisch aktiven Enzyme (= Peptidbindung spaltende
2
Einleitung
Hydrolasen) als Peptidasen bezeichnet. Protease ist nunmehr ein Synonym für
Peptidase. Der Definition folgend können die Peptidasen aufgrund ihrer katalytischen
Eigenschaften in Exopeptidasen (Synonyme: Exoprotease) und Endopeptidasen
(Synonyme: Proteinase, Endoprotease) unterteilt werden (siehe Abb. 1). Ist die
Aktivität der Exopeptidasen an die Nähe der N- und C-Termini des Substrates
gebunden, sind die Endopeptidasen in der Regel entfernt von diesen Termini aktiv.
Diese Gruppierung ist gleichzeitig die Grundlage zur Klassifizierung der Peptidasen
nach dem System der Enzyme Commission (EC) der IUBMB.
Abb. 1: Terminologie und Klassifizierung der proteolytischen Enzyme nach der IUBMB (verändert nach: Barrett, 2001 und http://www.chem.qmul.ac.uk/iubmb/enzyme/). Die weitere Untergliederung (siehe Abb. 1) der Peptidasen im EC-System erfolgt
nach der Reaktionsart des Substratumsatzes (in Amino-, Dipeptidyl-, Tripeptidyl-,
Peptidyldi-, Carboxy- und Omegapeptidasen) und anhand der Merkmale der
katalytischen Domänen (in Serin-, Aspartat-, Cystein-, Threonin- und Metallo-
peptidasen).
Die Klassifizierung der Peptidasen nach dem EC-System weist allerdings für den
wissenschaftlichen Gebrauch Schwächen auf. So werden genealogisch
verschiedenartige Peptidasen in der gleichen Gruppe zusammengefasst und die
überaus zahlreichen Endopeptidasen durch nur fünf Untergruppen repräsentiert.
Zudem finden im EC-System strukturelle und evolutionäre Gemeinsamkeiten
zwischen den einzelnen Enzymen keine Beachtung (Barrett, 2001). Infolgedessen
wurde von Rawlings und Barrett (1993) das MEROPS-System aufgestellt, welches
aus den grundlegenden Enzymstrukturen und den evolutionären Verwandtschafts-
3
Einleitung
verhältnissen der Peptidasen auf Basis ihrer Aminosäuresequenzen hervorgeht. In
diesem hierarchischen System kommt es zur Einteilung der Peptidasen in Clans und
Familien. Ein Clan fasst dabei verschiedene Familien zusammen, deren Vertreter
sich jeweils von einem gemeinsamen „Vorfahren“ ableiten lassen. Die Bezeichnung
der zurzeit 49 verschiedenen Clans wird durch zwei Großbuchstaben vorgenommen,
wobei der jeweils erste Buchstabe auf den Aminosäurerest der katalytischen Gruppe
Unterscheiden sich diese Aminosäurereste innerhalb der Familien eines Clans, wird
dieser zur Gemischt-Gruppe (P) eingeordnet. Der zweite Buchstabe der Clan-
bezeichnung ist dagegen frei wählbar. Proteolytische Enzyme, die sich bisher nicht in
die bestehenden Gruppen einordnen lassen, werden hingegen in einer Unbekannt-
Gruppe (U) zusammengefasst. Die einzelnen Familien sind ebenfalls durch den
Großbuchstaben der katalytischen Gruppe (A, C, G, M, S, T und U) und eine
fortlaufende Nummer gekennzeichnet. Jede Familie ist dabei um ein Typenenzym
herum aufgestellt. Weitere Vertreter dieser Familie müssen signifikante Überein-
stimmungen mit der Primärstruktur des Typenenzyms oder der schon in der Familie
vorhandenen Peptidasen aufweisen. Dabei ist im Gegensatz zu dem EC-System zu
beachten, dass sowohl Exopeptidasen als auch Endopeptidasen in der gleichen
Familie vertreten sein können.
Eine treffende Benennung der jeweiligen Clans und Familien hat sich aufgrund der
dynamischen Entwicklung ihrer Klassifizierung erst in wenigen Fällen durchgesetzt.
Eine zahlenmäßige Übersicht der bislang (Stand: November 2006) bekannten Clans,
Familien und Peptidasen nach MEROPS ist in Tab. 1 zusammengefasst. Auf eine
komplette Darstellung aller Clans wurde aus Gründen der Übersichtlichkeit
verzichtet. Eine ständig aktualisierte Übersicht der momentan 49 Clans und 185
Familien, in denen die über 48.000 bekannten Peptidasen zusammengefasst
werden, ist unter http://merops.sanger.ac.uk/ einzusehen.
4
Einleitung
Tab. 1: Übersicht über die Anzahl der Clans, Familien und Enzyme der bislang bekannten Peptidasen nach MEROPS (Rawlings et al., 2006). Zu den jeweiligen katalytischen Gruppen wurden wenige Beispiele zur Verdeutlichung der MEROPS-Klassifizierung eingestellt.
Katalytische Gruppe (Kürzel)
Clan
Beispiele
Familie Enzyme
Typenenzym (Herkunft) 7 14 2.915
A1 Pepsin A (Homo sapiens) A2 Retropesin (HIV 1) AA
Abb. 3: Einige halotolerante und halophile Bakterien, ihre Klassifizierung und mögliche Biotope (aus Mühl, 2002). *Prozentbereich in Klammern hinter den Organismen gibt ihre Salztoleranz in % NaCl (w/v) an.
8
Einleitung
Halotolerante und halophile Bakterien finden sich in diversen Ordnungen der
Bacteria und Archaea, wobei halotolerante und moderat halophile Vertreter vermehrt
unter den Bacteria und extrem halophile häufig unter den Archaea zu finden sind.
Diese Verteilung basiert vermutlich auf den unterschiedlichen resistenzvermittelnden
Strategien der Organismen. Extrem halophile Archaea besitzen ein Cytoplasma,
welches isoton mit dem umgebenden Medium ist. Durch Akkumulation von Kalium-
ionen und gleichzeitiger Ausschleusung von Natriumionen wird ein osmotisches
Gleichgewicht zwischen Cytoplasma und umgebendem Medium hergestellt. Da
Kaliumionen weniger Wasser in ihrer Hydrathülle binden als Natriumionen, steht der
Zelle damit mehr freies Wasser zur Verfügung. Im Gegensatz dazu halten die
Vertreter der halotoleranten und halophilen Bacteria mit Ionen-Pumpen die cyto-
plasmatische Salzkonzentration verhältnismäßig niedrig. Mit der Anreicherung so
genannter kompatibler Solute in der Zelle werden die Wasseraktivitätsunterschiede
zum Außenmedium ausgeglichen. Diese kompatiblen Solute, wie beispielsweise
Trehalose, Ectoin und Betain, funktionieren dabei als organische Osmotika, ohne die
biochemischen Prozesse der Zelle zu hemmen (Galinski, 1993). Diese
verschiedenen Strategien führen dazu, dass bei halophilen Bacteria nur Enzyme mit
Kontakt zum Außenmedium (extrazelluläre und membranassoziierte Enzyme) einen
halophilen Charakter aufweisen. Im Gegensatz dazu besteht bei den extrem
halophilen Archaea die Notwendigkeit sowohl bei intra- als auch bei extrazellulären
Enzymen speziell an hohe Salzkonzentrationen angepasst zu sein (Kushner, 1988).
1.2.11.2.1 Halophile Enzyme Halophile Enzyme
Aufklärung über die Adaption an höhere Salzkonzentrationen bei halophilen
Enzymen konnten Untersuchungen an einigen extrem halophilen Enzymen aus
Archaeen geben (Madern et al., 2000). Diese Enzyme sind übereinstimmend durch
eine erhöhte Anzahl von sauren Aminosäureresten auf ihrer Oberfläche und somit
durch ein negatives Oberflächenpotential charakterisiert (Bieger et al., 2003; Madern
et al., 2000; Mevarech et al., 2000). Die daraus abzuleitende Halophilie ist am besten
über das Solvatisierungs-Stabilisations-Modell zu erklären (Premkumar et al., 2005).
Hydratisierte Ionen binden an der negativen Enzymoberfläche und bilden eine so
genannte Solvatisierungs-Hülle aus. Diese Hülle verhindert eine Anreicherung von
Wasser oder Salzen auf der Proteinoberfläche, was einen Zusammenbruch der
Enzymstruktur oder ein Aussalzen durch Aggregation der Enzyme verhindert
9
Einleitung
(Costenaro et al., 2002; Ebel et al., 2002). Bei salzarmer bzw. salzfreier Umgebung
kommt es hingegen zur gegenseitigen Abstoßung der negativen Ladungen auf der
Enzymoberfläche und damit zur Entfaltung des Enzyms. Einhergehend mit der
Entfaltung ist die zumeist irreversible Inaktivierung der extrem halophilen Enzyme
(Adams & Kelly, 1995).
Demgegenüber müssen die strukturellen Mechanismen von halotoleranten und
moderat halophilen Enzymen, die es ermöglichen sowohl bei niedrigen als auch bei
höheren Salzkonzentrationen stabil und katalytisch aktiv zu sein, noch aufgeklärt
werden (vergl. hierzu: Deutch, 2002; Polosina et al., 2002; Wejse et al., 2003;
Bestandteile RM-Medium (1l): A. dest. 776 ml 5x M9 Salzlsg. 200 ml 20% Casamino
Acids 100 ml
20% Glucose-Lsg. 10 ml 1 M MgCl2 1 ml
2.4.62.4.6 Feste Medien für den Nachweis extrazellulärer Peptidasen Feste Medien für den Nachweis extrazellulärer Peptidasen
Als Grundlage für die Peptidase-Plattentests wurden je nach verwendetem Stamm
und Versuch die festen Medien aus 2.4.1; 2.4.2 und 2.4.3 verwendet und die
Substrate aus Tab. 7 hinzugefügt.
Tab. 7: Peptidase-Plattentests, ihre Substrate und Zusammensetzung.
Plattentest Substrat Menge [g/l] modifiziert nach Bemerkungen
Skimmilk-Agar
Magermilch-pulver (Sucofin)
20 (DeShazer et al., 1999)
Dem Medium wurde nach Autoklavieren 2% Magermilch aus einer 20% Mager-milch-Stammlösung unter Rühren hin-zugegeben und die Platten sofort gegossen. Schnelles Abkühlen verhindert die Ausflockung der Magermilch.
Gelatine-Hydrolyse
Gelatine (Merck)
10 (Klemme & Pfleiderer, 1977)
Die Gelatine wurde vor dem Autoklavieren zugegeben.
Casein-Agar
Casein (Carl Roth)
10 (Cowan & Daniel, 1982)
Das Casein wurde vor dem Auto-klavieren dem Medium zugegeben.
2.4.6.11 Skimmilk-Agarose für den Peptidasenachweis in PA-Gelen elen2.4.6. Skimmilk-Agarose für den Peptidasenachweis in PA-G
Zum Nachweis von Peptidase-Aktivität in Polyacrylamidgelen wurden dünne
Skimmilk-Agaroseplatten in Petrischalen gegossen. Agarose (2%) wurde in
Aktivitätspuffer (30 mM Tris-HCl, pH 9, 50 mM NaCl) durch Kochen gelöst und beim
Abkühlen unter Rühren mit 0,5-1% Magermilchlösung versetzt. Um eine Ausflockung
der Magermilch zu vermeiden, wurden die Platten sofort nach dem Gießen gekühlt.
2.4.72.4.7 Antibiotika und Medien-Zusätze Antibiotika und Medien-Zusätze
Zur Kultivierung von Trägern resistenzvermittelnder Plasmide wurden entsprechende
Antibiotika dem Medium, wie in Tab. 8 aufgeführt, zugegeben. Die sterilfiltrierten
21
Material und Methoden
Stammlösungen wurden dem Medium nach dem Autoklavieren bei etwa 55°C hinzu-
gefügt. Ebenso wurde mit den Medienzusätzen zur Induktion von plasmidlokalisierten
IPTG 1 M 1 mMAnhydrotetracyclin* 400 μg/ml 0,05-0,2 μg/ml
Vitamine: Thiamin 100 mg/ml 0,1 mg/ml
*Anhydrotetracyclin wurde in dieser Arbeit als Induktor für den Expressionsvektor pASK-IBA44 verwendet und besitzt eine wesentlich schwächere antibiotische Wirkung als Tetracyclin (Degenkolb et al., 1991).
2.4.82.4.8 Puffer und Lösungen Puffer und Lösungen
- Denaturierungslösung (Kolonieblot): 0,5 M NaOH, 1,5 M NaCl
- FA-Elektrophoresepuffer (1x): 20 mM MOPS (free acid); 5 mM Na-Acetat;
1 mM EDTA; 0,74% Formaldehyd; pH 7,0
- Hybridisierungslösung: 1μl DIG-markierte Sonde pro 1 ml
Prähybridisierungslösung
- Magermilch (20%): 20% (w/v) Magermilchpulver (Sucofin) wurde in A. dest.
vollständig gelöst und kurz autoklaviert (max. 7 min, 121°C). Lagerung: 4°C.
- Neutralisierungslösung (Kolonieblot): 1 M Tris-HCl pH7,5; 1,5 M NaCl
- Osmotic Shock-Lösung 1 (OS1): 20 mM Tris-HCl, pH 8; 2,5 mM EDTA;
20% Saccharose
- Osmotic Shock-Lösung 2 (OS2): 20 mM Tris-HCl, pH 8; 2,5 mM EDTA
- PA-Gel-Waschpuffer: 0,1% Tween 20, 30 mM Tris-HCl pH 9
- Peptidase-Reaktionspuffer (1x): 30 mM Tris-HCl, pH 9; 5mM NaCl
Zur Analyse der extrazellulären Proteine und Peptidasepräparationen wurden diese
mittels 1,5 mm dicken 10% oder 12% Tris-Glycin-SDS-Polyacrylamidgelen getrennt.
Die Herstellung der PA-Gele erfolgte mit den in Tab. 9 und Tab. 10 angegeben
Bestandteilen gemäß Anleitung (Sambrook & Russell, 2001) in Minigel-Kassetten mit
10-Taschen Kamm (Invitrogen). Die Auftrennung erfolgte in einer Minigelkammer
(XCell SureLock, Invitrogen) für vertikale Gele mit 125 V in Tris-Glycin-Puffer. Die
Proben wurden, wenn nicht anders angegeben, vor dem Auftragen mit SDS-Proben-
puffer versetzt und für 5 min bei 85°C denaturiert. Als Größenmarker dienten
entweder PageRuler-Prestained-Protein-Ladder (Fermentas) oder RotiMark Standard
(Carl Roth).
Tab. 9: Zusammensetzung von 25 ml Lösung zur Herstellung von ca. 3 Trenngelen für die Tris-Glycin SDS-Polyacrylamid Gel Elektrophorese (Sambrook & Russell, 2001). Substanz 10% 12% A. dest. 9,9 ml 8,2 ml30% Acrylamid-Mix (Sigma) 8,3 ml 10 ml1,5 M Tris-HCl (pH 8,8) 6,3 ml 6,3 ml10% SDS 0,25 ml 0,25 ml10% Ammoniumpersulfat 0,25 ml 0,25 mlTEMED 0,01 ml 0,01 ml Tab. 10: Zusammensetzung von 8 ml Lösung zur Herstellung von ca. 3 Sammelgelen (5%) für die Tris-Glycin SDS-Polyacrylamid Gel Elektrophorese (Sambrook & Russell, 2001). Bestandteile A. dest. 5,5 ml30% Acrylamid-Mix (Sigma) 1,3 ml1,0 M Tris-HCl (pH 6,8) 1,0 ml10% SDS 0,08 ml10% Ammoniumpersulfat 0,08 mlTEMED 0,008 ml Die Proteinbanden wurden nach der Auftrennung mittels Silberfärbung (Roti-Black P,
Carl Roth) sichtbar gemacht.
Für die Herstellung von Zymogramm-Gelen wurden 1% (w/v) Gelatine oder Casein
dem Trenngel vor der Polymerisation hinzugegeben (Teo et al., 2003).
27
Material und Methoden
2.6.92.6.9 Detektion von Peptidase-Aktivität auf festen Medien Detektion von Peptidase-Aktivität auf festen Medien
Die extrazelluläre Peptidase-Aktivität der Isolate und Expressionsklone wurde mittels
verschiedener Plattenassays (2.4.6) detektiert (Tab. 11). Im Gegensatz zum
Gelatine-Hydrolyse-Assay konnte die Peptidase-Aktivität beim Skimmilk- und Casein-
Agar während des Wachstums der Stämme beobachtet werden. Als empfindlichste
Detektionsmethode erwies sich der Skimmilk-Agar, der hauptsächlich in dieser
Studie eingesetzt wurde.
Tab. 11: Peptidase-Plattenassays Plattentest Auswertung Skimmilk-Agar Klarzonen im milchigtrüben Skimmilk-Agar zeigen Peptidase-Aktivität an. Gelatine-Hydrolyse Nach Überschichten der Agarplatte mit gesättigter Pikrinsäure in Ethanol
(50%) sind hydrolysierte Bereiche als durchscheinende Klarzonen von dem gelblichtrüben Gelatineagar zu unterscheiden.
Casein-Agar Klarzonen im trüben Casein-Agar zeigen Peptidase-Aktivität an.
2.6.102.6.10 Peptidasenachweis im Polyacrylamid-Gel (Kontakt-Zymogramm) Peptidasenachweis im Polyacrylamid-Gel (Kontakt-Zymogramm)
In Vorversuchen erwies sich die Verwendung von SDS-PA-Gelen mit copoly-
merisierten Proteinen (Casein- oder Gelatine-Zymogramm-Gele) mit anschließender
Aktivitätsfärbung für die Zuordnung von Peptidase-Aktivität zu einzelnen Protein-
banden als ungeeignet. Große ungefärbte proteolytische Aktivitätsbereiche im hoch-
bis mittelmolekularen Bereich des Gels ließen auf Peptidase-Aktivität während der
Elektrophorese schließen.
Die Auftrennung der Proben musste demzufolge getrennt vom anschließend einge-
setzten Substrat für den Peptidasenachweis erfolgen.
So erfolgte die Auftrennung der Proben durch Tris-Glycin-PAGE (siehe Abschnitt
2.6.8). Allerdings wurden die Proben vor dem Auftragen nicht erhitzt und die Kammer
während des Gel-Laufes mit Eis gekühlt.
Nach dem Lauf wurde das im Gel enthaltene SDS mit 2 x 30 min Bädern in 0,1%
Tween 20 entfernt. Das Gel wurde nach der Wäsche mittig geteilt und die eine Hälfte
zur Detektion der Proteinbanden silbergefärbt (siehe 2.6.8), während mit der anderen
Hälfte der Aktivitätsnachweis erfolgte. Das Gelstück wurde auf Skimmilk-Agarose
(siehe 2.4.6.1) in einer Petrischale platziert und die Lage des Gels, der Taschen und
des vorgefärbten Proteinstandards markiert. Während der anschließenden Inkubation
bei 37°C erfolgte eine ständige Kontrolle der Peptidase-Aktivität. Enzymatische
Aktivität war durch das Aufklären des milchigen Skimmilk-Agars zu erkennen. Zur
Verdeutlichung der Klarzonen erfolgte eine abschließende Gegenfärbung der
28
Material und Methoden
unverdauten Proteine mittels Coomassie-Färbung (PageBlue, Fermentas). Ein
Vergleich mit der silbergefärbten Hälfte des Gels ermöglichte die Zuordnung der
Standardmäßig wurde Taq-DNA-Polymerase (Moltaq und Moltaq-MastermixRed,
beide MOLZYM) für die PCR-Reaktionen mit einer Magnesium-Endkonzentration von
1,5 mM in 25 μl Volumen eingesetzt. Die Amplifikation der PCR-Produkte erfolgte in
einem Mastercycler gradient (Eppendorf) nach dem Schema in Tab. 12. Für die
Amplifikation von Abschnitten des 16S rRNA-Gens wurde DNA-freie Taq-DNA-
Polymerase (Moltaq 16S, MOLZYM) verwendet. Als Template diente 0,2-2 μl DNA-
Probe.
Tab. 12: Zyklenschema für die Standard-Amplifikation von PCR-Produkten im Mastercycler gradient (Eppendorf).
PCR-Schritt Temperatur Dauer Zyklen Anfangsdenaturierung 94°C 4 min 1 Denaturierung 94°C 0,5 minAnnealing Primerabhängig, siehe Tab. 6 0,5 minElongation 72°C 1 min / 1 kb
35
Endelongation 72°C 10 min 1 PCR-Amplifikate für die Klonierung in Expressionsvektoren wurden mit Pfu-DNA-
Polymerase (Thermorase, MOLZYM) in 50 μl Volumen nach Zyklenschema (Tab. 13)
im Mastercycler gradient (Eppendorf) durchgeführt. Die hier verwendete Magnesium-
sulfat-Konzentration variierte je nach Reaktionsbedingung zwischen 2 und 6 mM.
Tab. 13: Zyklenschema für die Amplifikation von PCR-Produkten mit Pfu-DNA-Polymerase im Mastercycler gradient (Eppendorf).
PCR-Schritt Temperatur Dauer Zyklen Anfangsdenaturierung 94°C 4 min 1 Denaturierung 94°C 0,5 min Annealing Primerabhängig, siehe Tab. 6 0,5 min Elongation 72°C 1min / 0,5 kb
35
Endelongation 72°C 10 min 1
38
Material und Methoden
Wenn nötig erfolgte die Entfernung der Polymerase für folgende Anwendungen
mittels Ethanolfällung der Amplifikate oder mit dem QuickClean EnzymRemoval
Resin (Clontech).
2.7.152.7.15 Sequenzierung und Auswertung der Sequenzdaten Sequenzierung und Auswertung der Sequenzdaten
Die Sequenzierung von Plasmid-DNA und PCR-Produkten wurde von der Firma
GATC (Konstanz) durchgeführt. PCR-Produkte wurden zuvor mit dem QIAquick
PCR-Purification-Kit (QIAGEN, Hilden) von dNTPs und Primern gereinigt. Als Primer
der Sequenzierreaktion dienten die gekennzeichneten Oligonukleotide aus Tab. 6.
Die gelieferten Sequenzdaten wurden mittels Lasergene (DNASTAR 5.00)
ausgewertet und die Alignments mit öffentlichen Sequenzdatenbanken (NCBI) mittels
BLASTN (Version 2.2.14 Altschul et al., 1997) durchgeführt. Eine Analyse der
abgeleiteten Aminosäuresequenzen auf Signalpeptide wurde mit Hilfe des
Programms Signal P 3.0 Server (Bendtsen et al., 2004) durchgeführt.
39
Ergebnisse
33 Ergebnisse Ergebnisse
3.13.1 Extrazelluläre Peptidasen aus halotoleranten und halophilen Bakterien Extrazelluläre Peptidasen aus halotoleranten und halophilen Bakterien
In der vorliegenden Studie wurde nach extrazellulären Peptidasen aus halotoleranten
bzw. halophilen Bakterien gesucht, deren proteolytische Aktivität eine hohe Toleranz
gegenüber hochchaotropen Salzen aufweist. Dies geschah im Hinblick auf einen
möglichen Einsatz der neuen Enzyme zur Zelllyse in Puffern mit hoher Guanidin-
isothiocyanat- oder Guanidinhydrochlorid-Konzentration für ein Nukleinsäure-
Extraktionsverfahren.
Eine biochemische Charakterisierung sollte nähere Informationen über die noch
wenig bekannten Peptidasen aus halotoleranten und moderat halophilen Bakterien
erbringen. Bisher wurden nur wenige Peptidasen aus moderat halophilen Bacteria
(Hiraga et al., 2005; Kamekura & Onishi, 1974; Karbalaei-Heidari et al., 2006; Lama
et al., 2005; Sánchez-Porro et al., 2003b; Setyorini et al., 2006) isoliert und bio-
chemisch charakterisiert. Untersuchungen zur Toleranz gegenüber chaotropen
Reagenzien dieser Peptidasen fehlten bis zum jetzigen Zeitpunkt.
3.1.13.1.1 Vorauswahl und Charakterisierung von Peptidase produzierenden Vorauswahl und Charakterisierung von Peptidase produzierendenIsolaten Isolaten
Für das Auffinden von Peptidasen, deren Aktivität eine hohe Toleranz gegenüber
hochchaotropen Salzen aufweist, standen 355 halotolerante bzw. halophile Isolate
zur Verfügung, die auf Plattentests Peptidase-Aktivität gezeigten hatten (Sinn-Meyer,
2003). Weitere Daten über die Peptidasebildung oder deren biochemische
Eigenschaften lagen nicht vor. Eine Auswahl sollte daher die Anzahl der Stämme für
eine Vorcharakterisierung reduzieren und dabei möglichst verschiedene Peptidase-
Produzenten erfassen. Die Kriterien hierfür wurden in Bezug auf die Zielsetzung und
eine erleichterte Probengewinnung wie folgt getroffen:
- Wachstum: Für eine zügige Gewinnung von Probenmaterial wurden halophile
Stämme mit einem relativ schnellen Wachstum (mind. ~108 Zellen nach max.
5 Tagen) auf 12 % NaCl ausgewählt.
- Extremophilie: Die Isolate sollten möglichst unter haloalkaliphilen
Bedingungen isoliert worden sein. Extrazelluläre Enzyme von Bakterien, die
auf eine haloalkaliphile Umgebung angewiesen sind, besitzen möglicherweise
40
Ergebnisse
Aktivitäten mit hoher Toleranz gegenüber extremen Bedingungen, wie hohe
Salzkonzentrationen und alkalische pH-Werte.
- Peptidasebildung: Zur Vereinfachung der Probengewinnung sollten Isolate
bevorzugt werden, die in Voruntersuchungen auf Plattentests starke
Peptidase-Aktivität zeigten. Dabei kann es sich um Stämme handeln, die
große Mengen an Enzymen oder Enzyme mit hoher spezifischer Aktivität
ausschütten. Beide Eigenschaften waren in Hinblick auf den Einsatz des
Enzyms (s.o.) wünschenswert.
- Diversität: Um die Verschiedenartigkeit der untersuchten Peptidasen zu
gewährleisten und damit die Wahrscheinlichkeit zu erhöhen ein hinsichtlich
der Fragestellung interessantes Enzym zu finden, sollten die ausgewählten
Stämme eine möglichst hohe Diversität aufweisen.
Insgesamt wurden so 27 Stämme ausgewählt (Tab. 35, im Anhang). Darunter
befanden sich 17 Stämme, die auf Medium mit 12% NaCl und pH 8 isoliert wurden,
7 Stämme, die bei einem pH-Wert von 9,5 und 3 Stämme, die bei einem NaCl-Gehalt
von 20% wuchsen. Mit 17 dieser Stämme waren systematische Untersuchungen
(ARDRA-Gruppen1)) durchgeführt worden (Vogt, 2000): Sechs Stämme gehörten zur
ARDRA-Gruppe 2, drei zur Gruppe 1 und jeweils ein Stamm zu den Gruppen 3, 6-9,
13 und 16.
Die Peptidasen der in Tab. 35 aufgeführten Stämme wurden nun hinsichtlich ihrer
Einsatzmöglichkeit in einem Verfahren zur RNA-Isolierung getestet. Da das
Verfahren zur RNA-Isolierung eine sehr große Enzym-Menge benötigte und sehr
hohen Arbeitsaufwand beanspruchte, wurde die Eignung der Peptidasen in der
Vorauswahl stellvertretend in einem vereinfachten Enzymassay getestet. Dieser
bestand aus der Messung der Peptidase-Aktivität in einem Puffer, der zur Zelllyse
zwecks anschließender RNA-Isolierung geeignet erschien. Der Lysepuffer bestand
aus 1 M GuSCN als essentieller Bestandteil zur RNA-Stabilisierung, 0,1% SDS,
einem Detergenz zur Unterstützung der Zelllyse und 5 mM CaCl2, einem Stabilisator
vieler bekannter Peptidasen. Der pH-Wert der Lösung wurde in Anbetracht der
alkalischen Umwelt der untersuchten Peptidase-Produzenten auf pH 8 (50 mM Tris-
HCl, pH 8) eingestellt. 1)Taxonomische Untersuchungen an etwa ¼ der Stämme aus der hier zugrunde liegenden Stamm-sammlung durch Vogt (2000). 232 Stämme wurden anhand von amplified ribosomal DNA restriction analysis (ARDRA), Fluoreszenz-in-situ-Hybridisierung (FISH) und Gram-Färbung in 26 ARDRA-Gruppen eingeteilt.
41
Ergebnisse
Da eine Sekretion der Peptidasen zu verschiedenen Wachstumsphasen (Gupta et
al., 2002b; Rao et al., 1998) bekannt war, wurde vor der Probengewinnung der Zeit-
punkt der höchsten Peptidase-Aktivität im Wachstumsverlauf der Stämme
(Inkubationsbedingungen: SML12/8 Medium, 200 rpm, 30°C) im Azocasein-Assay
bestimmt (Tab. 35, im Anhang). Dabei zeigte sich, dass 6 Stämme, im Gegensatz zu
der Kultivierung auf Agarplatten, keine und in 2 Fällen kaum messbare proteolytische
Aktivität in Flüssigkultur ausbildeten. Möglicherweise besteht hier ein Zusammen-
hang zwischen Kulturmethode (fest/flüssig) und der Enzymproduktion. Im Gegensatz
zur Flüssigkultur wachsen die Bakterien auf der Agarplatte zumeist auf der Ober-
fläche und sind dabei, nebst geringerer Luftfeuchtigkeit, heterogenen Umweltein-
flüssen ausgesetzt. Aufgrund der fehlenden Durchmischung auf festen Medien
kommt es lokal zu Substratmangel und zur Anreicherung von Stoffwechselprodukten.
Scherkräfte, wie in der Schüttelinkubation der Flüssigkulturen, fehlen als möglicher
Stressfaktor in der Plattenkultur. So wurden die differierenden Enzymproduktionen in
Fest- und Flüssigkultur bei diversen Organismen auf die Unterschiede zwischen den
Kulturmethoden zurückgeführt (Sato & Sudo, 1999). Die Zugabe von Protein (hier:
5% Gelatine) als möglicher Induktor für die Peptidaseproduktion, wie bei Lama et al.
(2005) beschrieben, führte bei den 8 oben genannten Stämmen zu keiner Erhöhung
der Peptidaseproduktion in Flüssigkultur.
Die übrigen 19 Stämme zeigten mit Wachstumsraten von 0,8 bis 1,3 ein für die
Kriterien entsprechendes Wachstum und im Azocasein-Assay gemessene extra-
zelluläre Peptidase-Aktivität von 68 bis 236 AzU/ml (siehe Tab. 35, im Anhang).
Im RNA-Lysepuffer zeigten 17 der 19 Kulturüberstände proteolytische Aktivität (Tab.
35), wovon 10 noch über 1/3 Restaktivität besaßen, verglichen zu Puffern ohne SDS
und GuSCN-Zusatz. Zwei Stämme (B3/1-2 (#418) und SJ5Ü/12 (#140)) wiesen
sogar über 70% (72 bzw. 76%) Peptidase-Aktivität im Lysepuffer auf. Letztlich
verblieben 9 Stämme mit einer Restaktivität im Lysepuffer von mehr als 33%, die für
die weiterführenden Untersuchungen verwendet wurden (Tab. 35 im Anhang, türkis
markiert).
42
Ergebnisse
3.1.23.1.2 Systematische Einordnung der ausgewählten Peptidase-Produzenten Systematische Einordnung der ausgewählten Peptidase-Produzentenmittels 16S-rRNA Analyse mittels 16S-rRNA Analyse
Phylogenetische Untersuchungen der 9 ausgewählten Stämme wurden anhand von
Sequenzanalysen von Teilen des 16S-rRNA-Gens vorgenommen. Bei 7 Stämmen
wurden über 600 bp der 16S-rDNA Sequenz ermittelt, bei 2 Stämmen (B3/1-2 (#418)
und B12/10-1 (#748)) lag mit über 1400 bp die Sequenz des beinahe gesamten
Gens vor. Die Sequenzierung des Gens der Stämme B3/14 (#430) und B12/16
(#795) brach wiederholt nach 130 bp ab; die vorliegenden Daten wurden dennoch für
die Analyse verwendet.
Ein Vergleich der Sequenzen mit 16S-rRNA-Genen aus öffentlichen Datenbanken
(BLAST) zeigte hohe Sequenzhomologien aller Stämme untereinander. Die nächsten
Verwandten waren ein unidentifiziertes Bakterium mit der Bezeichnung K2-30-15 (96-
99% Sequenzidentität) und Vertreter der Gattung Salinivibrio (95-100% Sequenz-
identität; siehe Tab. 14).
Tab. 14: Nächstverwandte Arten der halophilen Stämme (NCBI-BLAST Datenbankanalyse der 16S rRNA-Teilsequenzbestimmungen).
Beim Alignment des ersten Drittels (590 bp) der 16S-rRNA-Sequenzen (bzw. 130 bp
der Stämme B3/14 (#430) und B12/16 (#795)) stellte sich heraus, dass eine hohe
Übereinstimmung von min. 96,1% der untersuchten Sequenzabschnitte unter-
einander und zu den beiden nächst verwandten Organismen Salinivibrio costicola
alcaliphilus (Romano et al., 2005) und Salinivibrio aegyptiaca sharmensis bestand
(siehe Distanzmatrix Tab. 15). Die Sequenz (590 bp) von Stamm SJ2/3-1 (#073),
SJ5Ü/12 (#140) und SJ6S/14 (#148) war identisch, ebenso die Sequenz (590 bp)
von B8/26-2 (#504) und vom Referenzorganismus Salinivibrio costicola alcaliphilus
(siehe Tab. 15).
Tab. 15: Ähnlichkeits- bzw. Distanztabelle der 9 untersuchten Stämme und den beiden nächst verwandten Vertretern der Gattung Salinivibrio, basierend auf dem Alignment von je 590 bp der 16S-rDNA-Teilsequenzen (Ausnahme 5 & 9: hier nur 130 bp). Alignment-Methode: CLUSTALV (weighted).
Divergenz [%] Zuordnung der Nummerierung: 1: SJ2/3-1 (#073); 2: SJ5Ü/8 (#098); 3: SJ5Ü/12 (#140); 4: SJ6S/14 (#148); 5: B3/1-2 (#418); 6: B3/14 (#430); 7: B8/26-2 (#504); 8: B12/10-1 (#748); 9: B12/16 (#795); 10: Salinivibrio costicola alcaliphilus; 11: Salinivibrio aegyptiaca sharmensis Ausgehend von der Analyse der 16S-rRNA-Teilsequenzabschnitte ist zu vermuten,
dass die untersuchten 9 Stämme zur Gattung Salinivibrio gehören. Anhand des
phylogenetischen Stammbaums (Abb. 4) und ausgehend von 97% Sequenzidentität
als allgemein angesehenem Grenzwert (Ludwig et al., 1998) für die Einordnung zu
einer Spezies, lässt sich des Weiteren die Artzugehörigkeit einiger Stämme festlegen
(siehe Tab. 15 und Abb. 4). Mit �98,5% Sequenzidentität können 4 Stämme (SJ2/3-1
(#073), SJ5Ü/8 (#098), SJ5Ü/12 (#140) und SJ6S/14 (#148)) der Art Salinivibrio
aegyptiaca zugeordnet werden. Drei weitere Stämme (B8/26-2 (#504), B12/10-1
(#748) und B12/16 (#795)) gehören der Spezies Salinivibrio costicola an (98,1-100%
Sequenzidentität). Mit einer Sequenzidentität von über 97% zu zwei verschiedenen
44
Ergebnisse
Arten (S. costicola: 97,1% bzw. S. aegyptiaca: 97,6%) bildet der Stamm B3/1-2
(#418) im Phylogramm einen eigenen Zweig aus, ohne einer der beschriebenen
Arten eindeutig zuzugehören. Ob er einer der beiden Salinivibrio-Spezies zugeordnet
werden kann oder eine eigene neue Art darstellt, müssten weitere systematische
Untersuchungen zeigen. Ebenso konnte keine eindeutige Zuordnung des Stammes
B3/14 (#430) erfolgen, vermutlich aufgrund des zur Verfügung stehenden kurzen
Sequenzabschnittes (130 bp). Eine Zuordnung dieses Stammes und weitere
Aussagen über die Verwandtschaft der hier untersuchten Bakterien könnten nur
Bestimmungen der kompletten 16S rRNA-Sequenzen und weitere systematische
Untersuchungen zeigen. Diese Fragestellung lag allerdings außerhalb des Fokus
dieser Arbeit.
Abb. 4: Phylogenetischer Stammbaum der untersuchten Stämme* und der beiden nächst verwandten Vertreter der Gattung Salinivibrio, basierend auf 16S-rDNA-Teilsequenzen (590 bp). Die Verwandtschaftsverhältnisse wurden mit CLUSTALW untersucht und der Stammbaum mit der Bootstrap-Methode auf seine Konsistenz und Robustheit überprüft (Felsenstein, 1988). Die Länge der Verzweigungen repräsentiert die Divergenz zwischen zwei Sequenzen, die Zahlen bezeichnen die Anzahl der Substitutionsereignisse. * Aufgrund der abweichenden Sequenzlänge von 130 bp konnten die Stämme B3/14 (#430) und B12/16 (#795) nicht in die Bootstrap-Analyse einbezogen werden.
3.1.33.1.3 Physiologie der Enzymbildung Physiologie der Enzymbildung
Die enzymatische Aktivität, gemessen als spezifische Aktivität im Azocasein-Assay
(Units pro μg extrazellulärem Protein), in Abhängigkeit des Zellwachstums wurde
bestimmt. Abb. 5 zeigt ein typisches Beispiel anhand des Stammes B12/10-1 (#748).
Proteolytische Aktivität war bereits in der frühen logarithmischen Wachstumsphase
nachweisbar und stieg im weiteren Wachstumsverlauf stetig an. Die höchste
spezifische Aktivität trat am Ende der stationären Phase auf. Dies lässt eine
konstitutive Enzymexpression vermuten. Die anderen Stämme zeigten ähnliche
Enzymbildung (Daten nicht gezeigt). Damit ist der beste Erntezeitpunkt für
Peptidasen aus diesen Stämmen die späte stationäre Phase. Die maximalen
45
Ergebnisse
Enzymaktivitäten im Azocasein-Assay (AzU/ml) und der Zeitpunkt ihres Auftretens
Abb. 5: Bildung extrazellulärer Peptidase während des Wachstums des halophilen Stammes B12/10-1 (#748) (SML-Medium 12/8; bei 30°C, 200 rpm). Spezifische Aktivität: Aktivität im Azocasein-Assay pro μg extrazellulären Proteins.
Es wurde anhand von Stamm B12/10-1 (#748) geprüft, ob eine Steigerung der
Enzymausbeute durch Veränderung der Kulturbedingungen möglich war. Die
Senkung des NaCl-Gehaltes im SML-Medium von 12% auf 10%, welche in einer
Erhöhung der Wachstumsrate resultierte, führte zu keiner Änderung der
proteolytischen Aktivität und ließ bei weiterer Senkung der NaCl-Konzentration auf
6% sogar die Gesamtaktivität auf 45% absinken. Bei 2% NaCl im Medium erfolgte
zwar ein gutes Wachstum des Stammes, aber es war keine proteolytische Aktivität in
der zellfreien Kultur nachzuweisen. Für die maximale Peptidaseproduktion bedarf
dieser Stamm demnach eines erhöhten Salzgehaltes von 10-12% im Medium. Ein
Vergleich der Peptidaseproduktion der restlichen Stämme bei 6% und 12% NaCl im
Medium bestätigte das Produktionsoptimum bei 12% NaCl.
Zur Gewinnung der peptidasehaltigen Kulturüberstände der 9 Stämme erfolgte die
optimierte Produktion in SML 12/8 Medium bis zur späten stationären Phase.
3.1.43.1.4 In vitro Peptidase-Assays - Möglichkeiten und Grenzen der verwendeten Peptidase-Assays - Möglichkeiten und Grenzen der verwendeteIn vitro nAssays Assays
Zur Bestimmung der proteolytischen Aktivität standen eine Reihe von in vitro
Peptidase-Assays zu Verfügung (Appel, 1981; Sarath et al., 2001). In dieser Studie
46
Ergebnisse
wurden zwei der gebräuchlichsten spektrophotometrischen Assays verwendet, der
Azocasein-Assay (2.6.11.1) und der Casein-Assay nach Kunitz (1947) (2.6.11.3).
Hinzu kam noch ein gewerblich vertriebener Fluoreszenzassay, das EnzCheck
Protease Assay Kit (Invitrogen) (2.6.11.2).
Der anfänglich ausschließlich benutzte Azocasein-Assay wurde nach der Vorauswahl
der Stämme (3.1.1) im Vergleich zu weiteren Assays (Casein-Assay und EnzCheck
Protease Assay Kit) getestet. Die drei Assays basierten auf verschiedenen Mess-
verfahren, die teilweise unterschiedliche Substratumsätze detektierten. Im Azo-
casein- und Casein-Assay erfolgte eine Endpunktbestimmung durch eine
Säurefällung des Ansatzes. Erfasst wurden bei der anschließenden spektrophoto-
metrischen Messung nur säurelösliche aromatische Peptide und säurelösliche Oligo-
peptide, die aromatische Pepitdreste enthielten. Während im Casein-Assay die
aromatischen Aminosäuren direkt über eine Absorptionsmessung bei 280 nm
ermittelt wurden, erfolgte im Azocasein-Assay eine Detektion des an die aroma-
tischen Aminosäuren gekoppelten Azofarbstoffes bei 340 nm. Beide Assays konnten
aufgrund des Verfahrens keine Aktivität erfassen, wenn mögliche Abbauprodukte aus
fällbaren Oligopeptiden oder aus nicht-aromatischen Aminosäuren bestanden. Beim
EnzCheck Protease Assay erfolgte die Bestimmung der Peptidase-Aktivität über eine
Fluoreszenzmessung, ohne zuvor die enzymatische Reaktion zu stoppen. Durch eine
starke Markierung des Caseins mit dem Fluoreszenzfarbstoffes BODYPY FL wurde
die Fluoreszenz im intakten Konjugat durch einen Quench-Effekt unterdrückt. Sowohl
die Abspaltung von einzelnen markierten Peptiden als auch die Abspaltung größerer
Oligopeptide erzeugte eine Fluoreszenzsteigerung im Assay (Welder et al., 2002).
Die Fluoreszenzentwicklung im EnzCheck Protease Assay Kit ist daher relativ unab-
hängig von der Spezifität der Peptidasen (Molecular Probes, 2003).
Bei späteren Untersuchungen zur GuSCN-Abhängigkeit der Peptidase-Aktivität
zeigten sich große Diskrepanzen der mit dem Fluoreszenz- und Casein-Assay auf
der einen Seite erhaltenen Daten und denen aus dem Azocasein-Assay auf der
anderen Seite. Im direkten Vergleich (Abb. 6) der drei Assays zeigte der Azocasein-
Assay bei Zugabe von GuSCN in steigender Konzentration eine abweichende
Tendenz zu den anderen beiden Testverfahren. So schien die Peptidase-Aktivität im
Azocasein-Assay eine Aktivierung mit 0,2 und 0,5 M GuSCN (115% bzw. 132%
relative Aktivität) zu erfahren, während die beiden anderen Testverfahren hier eine
Inhibition (50-78% relative Aktivität) anzeigten. Tatsächlich handelte es sich hier um
47
Ergebnisse
ein Artefakt: Wurde eine hydrolysierte Probe Azocasein mit 0,5 M bzw. 1 M GuSCN
versetzt, erhöhte sich die Absorption um das 1,3-1,8-fache, während Kontrollen mit
unverdautem Azocasein keine Änderung zeigten. Messungen mit dem Azocasein-
Assay über den Einfluss von chaotropen Salzen auf die Peptidase-Aktivität lieferten
somit verfälschte und in der Tendenz irreführende Ergebnisse (Abb. 6). Dies konnte
mit peptidasehaltigen Überständen von drei weiteren Stämmen (SJ2/3-1 (#073),
B3/1-2 (#418) und SJ6S/14 (#148)) bestätigt werden. Der Azocasein-Assay wurde
daraufhin für die weiterführenden Untersuchungen nicht mehr eingesetzt.
Abb. 6: Vergleich der relativen Peptidase-Aktivität* in den drei zur Verfügung stehenden Assays (Azocasein-, Casein-, Fluoreszenz-Assay) unter Einfluss von Guanidinisothiocyanat (GuSCN). Als Probe wurden 5μl der zellfreien Kultur von Stamm B12/10-1 (#748) verwendet. *Die Peptidase-Aktivität im Ansatz ohne GuSCN wurde für jeden Assay gleich 100% gesetzt.
Des Weiteren führte der Einsatz von GuSCN und GuHCl im Fluoreszenzassay
regelmäßig zu Ausfällen der Aktivitätsmessungen, oft in Zusammenhang mit höheren
Salzkonzentrationen. Der Casein-Assay zeigte sich hierbei viel verlässlicher, daher
wurden die Tests zur Toleranz gegenüber chaotropen Salzen mit diesem durch-
geführt. Ebenso ungeeignet erwies sich der Fluoreszenzassay bei Verwendung von
SDS. Hier konnten in der Kontrolle nicht reproduzierbare starke Anstiege der
Fluoreszenz über die Inkubationszeit ohne Zusatz von Peptidasen festgestellt
werden. Daher kam zur Messung der SDS-Toleranz der Peptidasen auch hier der
Casein-Assay zum Einsatz. Allerdings wiesen die Messergebnisse, trotz mehrfacher
Wiederholungen der Tests, sehr hohe Schwankungsbreiten auf.
Bei Untersuchungen der proteolytischen Aktivität von zwei über hydrophobe Inter-
aktionschromatografie aufgereinigten enzymhaltigen Fraktionen A und B (siehe
48
Ergebnisse
3.1.5.1) zeigten sich sehr deutliche Unterschiede zwischen dem Casein-Assay und
dem Fluoreszenzassay (siehe Tab. 16 als Beispiel).
Tab. 16: Vergleich der Peptidase-Aktivität der aufkonzentrierten enzymhaltigen Phenylsepharose-Fraktionen A und B im Fluoreszenz- und Casein-Assay der Stämme SJ5Ü/8 (#098) und SJ2/3-1 (#073) unter Standardbedingungen.
Peptidase-Aktivität Stamm Fraktion FlU/µl KuU/µlSJ5Ü/8 (#098) A 3,1 1,10SJ5Ü/8 (#098) B 25,5 0,19Faktor A/B 0,12 5,79SJ2/3-1 (#073) A 2,97 1,74SJ2/3-1 (#073) B 18,03 0,28Faktor A/B 0,16 6,21 Während im Fluoreszenzassay die Aktivität der Fraktion A etwa 1/6 bis 1/8 der
Aktivität von Fraktion B entsprach, verhielt es sich im Casein-Assay genau
umgekehrt, die Aktivität der Fraktion A entsprach etwa dem 5-8-fachen der sehr
schwachen Aktivität der Fraktion B. Es ist anzunehmen, dass mögliche
unterschiedliche Substratspezifitäten der Peptidasefraktionen aufgrund der
verschiedenen Funktionsweisen der Assays (s.o.) für diese Unterschiede ursächlich
waren. Dies sollte beim direkten Vergleich der beiden Assays beachtet werden.
Aus Gründen der Handhabbarkeit einer großen Anzahl von Proben, einer hohen
Sensitivität und der relativen Unabhängigkeit gegenüber der Spezifität der
Peptidasen wurden die biochemischen Charakterisierungen der Enzyme der 9
Stämme (siehe 3.2) mit dem Fluoreszenzassay durchgeführt (mit Ausnahme der
Messungen in 3.2.5). Ein Abgleich der Ergebnisse des Fluoreszenz-Assays mit
denen des Casein-Assays von einigen exemplarisch ausgewählten Proben zeigte
eine sehr gute Übereinstimmung in der Tendenz und überwiegend auch eine gute
Übereinstimmung der relativen Aktivitäten.
3.1.53.1.5 Anreicherung der Peptidasen aus Kulturüberständen der halophilen Anreicherung der Peptidasen aus Kulturüberständen der halophilenStämme Stämme
Um Peptidasen aus salzhaltigen Kulturüberständen anzureichern, wurden
verschiedene in der Proteinbiochemie gängige Verfahren (siehe Abschnitte 2.6.2 und
2.6.3) verwendet und auf ihre Eignung untersucht: 1. Ammoniumsulfat-Fällung,
2. Ethanolfällung und 3. Dialyse gegen Polyethylenglykol.
Für die Vorversuche wurde die zellfreie Kultur des Stammes B12/10-1 (#748)
verwendet. Die Ergebnisse dieser Versuche sind in Tab. 17 zusammengefasst.
49
Ergebnisse
Früh wurde deutlich, dass die angewendeten Verfahren aufgrund des hohen Salz-
gehaltes (12% NaCl) in der zellfreien Kultur modifiziert werden mussten. Für die
Fällung mit gesättigtem Ammoniumsulfat musste zunächst die Sättigungsgrenze bei
0°C experimentell ermittelt werden, sie lag bei ca. 490 g/l Ammoniumsulfat
(Sättigungsgrenze ohne Salz im Medium: ca. 650 g/l Ammoniumsulfat). Nach der
Fällung konnten nur ca. 6% Restaktivität im gelösten Fällungspellet nachgewiesen
werden, während 54 % der Aktivität im Fällungsüberstand messbar waren. Neben
dem Verlust von 40% der Gesamtaktivität nach der Fällung konnte gleichfalls eine
Reduzierung der spezifischen Aktivität um 18 bzw. 38% in den beiden Fällungs-
fraktionen beobachtet werden (vergleiche Tab. 17). Auch die vorhergehende
Entsalzung der zellfreien Kultur mittels Dialyse und anschließender Ammoniumsulfat-
Fällung erbrachte ähnlich geringe Ausbeuten (Daten nicht gezeigt). Offensichtlich
konnte mit der Ammoniumsulfat-Fällung keine nennenswerte Präzipitation von
aktiven extrazellulären Peptidasen erfolgen.
Tab. 17: Anreicherung von Peptidase aus der zellfreien Kultur von B12/10-1 (#748)a).Anreicherungs-
Dialyse gegen PEG 3500 Dialysat 45 34 10,31 464,3 13,7
(167%) 94 a) Der Proteingehalt und die Peptidase-Aktivität (Fluoreszenzassay) einer stationären Kultur (72h bei 30°C und 200 rpm
Schüttelfrequenz) wurden bestimmt. Alle Proben wurden nach der Anreicherung gegen 30 mM Tris-HCl, pH 9, dialysiert. b) kFlU = Kilo-Fluoreszenzunits C) 0°C, gesättigt d) 0°C, 85% v/v Die Ethanolfällung (85% EtOH Endkonzentration; siehe 2.6.2) der zellfreien Kultur
(900 ml) erbrachte eine nahezu quantitative Protein-Fällung (90-94%). Die
Peptidase-Aktivität des gelösten Sedimentes wies 76% der Aktivität der zellfreien
Kultur auf (Tab. 17). Im Überstand war keine Aktivität mehr vorhanden. Das Volumen
des dialysierten und gelösten Fällungspellets war allerdings mit 170 ml sehr groß.
Das durch das Ethanol mitgefällte Salz aus dem Medium (12% NaCl) verhinderte
eine Verringerung des Resuspensionsvolumens des Protein/Salzpellets. Aufgrund
des großen Materialaufwandes und der geringen Aufkonzentration der Probe wurde
diese Methode nicht weiter verwendet.
50
Ergebnisse
Mit Hilfe der Dialyse gegen Polyethylenglykol 3500 (2.6.3) konnte eine starke
Aufkonzentrierung um den Faktor 18,7 und leichte Anreicherung (1,67-fach) der
Enzyme aus der zellfreien Kultur von Stamm B12/10-1 (#748) bei gleichzeitig hoher
Ausbeute (94%) erzielt werden. Durch Variation der Dialysezeit konnte die zellfreie
Kultur beliebig stark aufkonzentriert werden. Aufgrund der sehr geringen Verluste an
der Gesamtaktivität der Peptidasen wurde diese Methode für weitere Versuche
Extinktionsmessung bei 280 nmrelative PeptidaseaktivitätAmmoniumsulfatgradient (100% = 1 M Ammoniumsulfat)
A B
Abb. 7: Phenylsepharose-Chromatografie von zellfreier Kultur des Stammes B12/10-1 (#748). �� Fraktionsbereiche mit Haupt-Peptidase-Aktivität (A bzw. B). Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität in einer Fraktion im Vergleich zur maximalen Peptidase-Aktivität im Peak. Die Fraktionen des Bereichs A wurden vereint (Pool A), genauso wie die des
Bereichs B (Pool B). Tab. 18 zeigt die Charakteristika dieser Pools. Die Summe der
proteolytischen Aktivität beider Pools ergab mit 22,9 kFlU (1,7 kFlU plus 21,2 kFlU)
eine Ausbeute von 19,4 % der Aktivität der zellfreien Kultur.
Tab. 18: Proteingehalt und Peptidase-Aktivitäta) der zellfreien Kultur B12/10-1 (#748) (42h, 30°C, 200 rpm) und der vereinigten aktiven Fraktionen A und B aus der hydrophoben Interaktions-chromatografie.
Fraktionen Volumen[ml]
Gesamt-protein
[mg]
Gesamt-aktivität [kFlU]b)
Spezifische Aktivität
[kFlU/mg]
Ausbeute [%] an
Aktivität
Enzym-Anreicherungs-
faktor zellfreie Kultur 350 36 118,6 3,3 100 1
Fraktionspool A 24 0,4 1,7 4,25 1,5 1,3 Fraktionspool B 52 1,5 21,2 14,1 17,9 4,3
a) Aktivitätsmessung im Fluoreszenzassay. b) kFlU= Kilo-Fluoreszenz-Units Während die frühere Fraktion (A) eine schwächere Bindung zur Phenylsepharose
aufweist, ist die Bindung zur zweiten eluierten Enzymfraktion so stark, dass diese
erst mit Erreichen des Gradienten-Endes eluiert. Es wird vermutet, dass wegen der
starken Bindung der Peptidase an die Matrix die zweite enzymhaltige Fraktion nur
unvollständig eluiert wird und dies den Verlust von über 75% der Gesamtaktivität der
Peptidasen begründet. Hinweise für eine mögliche Inaktivierung von Enzymen durch
eine vorübergehende Bindung an Phenylsepharose konnten in der Literatur nicht
gefunden werden.
52
Ergebnisse
Wegen der Möglichkeit mit dieser Methode Peptidasen anzureichern und Gemische
aus Peptidasen zu trennen, wurde dieses Verfahren im Verlauf der Studie auf
zellfreie Kulturen von weiteren Stämmen angewendet.
Bei der chromatografischen Auftrennung der Kulturüberstände von 8 weiteren
Für die Untersuchungen wurden Peptidasen aus den zellfreien Kulturen durch
hydrophobe Interaktionschromatografie (siehe Abschnitt 3.1.5.1) angereichert. Die
Ergebnisse deuteten an, dass 7 der 9 Stämme mindestens 2 verschiedene
Peptidasen besitzen. So wurden bei 7 Stämmen (Tab. 36, im Anhang) jeweils zwei
aktive Fraktionspools erhalten, Fraktion A und B. Bei den Stämmen B8/26-2 (#504)
und B12/16 (#795) trat nur eine Fraktion auf (B). Um die Eigenschaften der einzelnen
Fraktionen zu bestimmen, wurden die nachfolgenden biochemischen
Untersuchungen daher an den beiden Fraktionen eines jeden Stammes
vorgenommen. Zur Vereinfachung und in Vorgriff auf weitere Ergebnisse wird im
Folgenden von Peptidase A (Fraktion A) und Peptidase B (Fraktion B) gesprochen.
Die eingesetzten Enzymmengen im Fluoreszenzassay betrugen 1-5 FlU (unter
Standardbedingungen).
53
Ergebnisse
3.2.13.2.1 Einfluss des pH-Wertes auf die Peptidase-Aktivität Einfluss des pH-Wertes auf die Peptidase-Aktivität
Bei Untersuchungen zum Einfluss des pH-Wertes auf die Peptidasen zeigten sich
Unterschiede zwischen Peptidase A und B. Interessanterweise glichen sich die
pH-Profile von Peptidase A der verschiedenen Stämme hinsichtlich des Optimums
(siehe Tab. 19).
Für Peptidase A lagen die Aktivitätsoptima aller Stämme zwischen pH 8 und 8,5.
Hohe Aktivität (90% des Optimums) besaßen die Enzyme zwischen pH 7,5 und 8,5
(Abb. 8a). Der pH-Bereich, in dem Peptidase A Aktivität besaß (mind. 10% Rest-
aktivität vom Optimum), lag zwischen pH 6 und 11 (siehe Tab. 19 und Abb. 8a, bzw.
Abb. 33 bis Abb. 36 im Anhang).
Anders verhielt es sich mit dem Aktivitätsoptimum der Peptidase B der 9
untersuchten Stämme. So wurde bei allen Proben höchste Aktivität zwischen pH 8
und pH 9,5 festgestellt (siehe Tab. 19). Bei 4 Stämmen bildete sich ein breites
Plateau mit einer relativen Peptidase-Aktivität von 91-100% aus, bei 5 Stämmen lag
das Optimum bei pH 9 bzw. 9,5 (siehe Tab. 19). Der pH-Bereich, in dem Peptidase B
noch Aktivität zeigte, lag zwischen pH 6,0 und 12,0 (siehe Tab. 19 und Abb.8b, bzw.
Abb. 33 bis Abb. 36 im Anhang).
Tab. 19: pH-Optimum und Aktivitätsbereich (mind. 10% Restaktivität vom Optimum) der Peptidasen A und B (Fraktionen der hydrophoben Interaktionschromatografie). Stamm:/Peptidase:
Bei beiden Peptidasen handelt es sich um moderat alkaliphile Peptidasen mit einem
Aktivitätsoptimum von pH 8-8,5 (Peptidase A) und pH 9-9,5 bzw. pH 8-9,5 (Peptidase
B).
Aufgrund dieser Untersuchungsergebnisse erfolgte eine Anpassung des pH-Wertes
der Assaybedingungen für die weiteren biochemischen Untersuchungen auf pH 9,0.
54
Ergebnisse
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
B12/10-1 (#748) B3/1-2 (#418)
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
B12/10-1 (#748) B3/1-2 (#418)
a.
b.
Abb. 8: Einfluss des pH-Wertes auf die proteolytische Aktivität* der Peptidase A (a.) und B (b.) der Stämme B3/1-2 (#418) und B12/10-1 (#748). *Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
3.2.22 Einfluss der Temperatur auf die Peptidase-Aktivität 3.2. Einfluss der Temperatur auf die Peptidase-Aktivität
Die Temperaturabhängigkeit der Peptidase A und B wurde an drei ausgewählten
Stämmen getestet. Als Stämme kamen SJ6S/14 (#148), B3/1-2 (#418) und B12/10-1
(#748) zum Einsatz, die zueinander aufgrund von 16S-rRNA-Sequenzvergleichen
(siehe 3.1.2) die meisten Unterschiede aufwiesen. In Abb. 9 sind die Ergebnisse der
Versuche zusammengefasst.
Die getesteten Enzyme zeigten bei gemäßigten und erhöhten Temperaturen
proteolytische Aktivität. Sie tolerierten ein breites Temperaturspektrum und besaßen
noch bei 15°C 2-15,2% und bei 70°C 8-21% relative Aktivität.
Bezüglich ihres Optimums und Temperaturverlaufs zeigte die Peptidase B der 3
Stämme ein sehr einheitliches Bild. Das Optimum der Stämme SJ6S/14 (#148) und
B3/1-2 (#418) lag bei ca. 50°C bzw. 55°C für Stamm B12/10-1 (#748).
55
Ergebnisse
Ebenso besaß die Peptidase A der Stämme B3/1-2 (#418) und B12/10-1 (#748) ihr
Aktivitäts-Optimum bei ca. 50°C. Im Gegensatz dazu zeigte die Peptidase A des
Stammes SJ6S/14 (#148) bei 50°C nur ca. 76% relative Aktivität, ihr Optimum lag bei
einer Temperatur von 40°C.
0%
20%
40%
60%
80%
100%
0 10 20 30 40 50 60 70Temperatur [°C]
rela
tive
Pept
idas
e-A
ktiv
ität
SJ6/14 (#148)B3/1-2 (#418)B12/10-1 (#748)
0%
20%
40%
60%
80%
100%
0 10 20 30 40 50 60 70 80Temperatur [°C]
rela
tive
Pept
idas
e-A
ktiv
ität
SJ6/14 (#148)B3/1-2 (#418)B12/10-1 (#748)
a.
b.
Abb. 9: Einfluss der Temperatur auf die proteolytische Aktivität* der Peptidase A (a.) und B (b.) der Stämme SJ6S/14 (#148), B3/1-2 (#418) und B12/10-1 (#748). *Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
3.2.33.2.3 Salzabhängigkeit der Peptidasen Salzabhängigkeit der Peptidasen
Bei der Bestimmung der NaCl-Toleranz zeigte sich überraschenderweise, dass
sowohl Peptidase A als auch Peptidase B salzempfindlich war. Mit steigender Salz-
konzentration ging ihre Aktivität zurück (siehe Abb. 10 mit Stamm SJ5Ü/8 (#098) als
Beispiel), und bei 2 M NaCl waren noch 2-33% der Aktivität, abhängig vom Stamm,
nachweisbar (Tab. 20). Dennoch brauchten, abhängig vom Stamm, beide
Peptidasen geringe Mengen an Salz. Zum Beispiel besaß Peptidase A von Stamm
SJ2/3-1 (#073) ohne Salz nur noch 31% ihrer Aktivität, während bei 10 mM NaCl
volle Aktivität vorhanden war. Peptidase B desselben Stammes war eher salz-
unabhängig. Demgegenüber benötigten beide Peptidasen des Stammes SJ5Ü/8
56
Ergebnisse
(#098) Salz, allerdings in unterschiedlicher Konzentration, um höchste Aktivität zu
erringen: Peptidase A 200 mM NaCl und Peptidase B 1 mM NaCl. Diese Ergebnisse
zeigen im Allgemeinen einen Trend zur Übereinstimmung des Einflusses von Salz
auf die Peptidasen der untersuchten Stämme. Allerdings bilden sich auf dieser
Ebene auch Unterschiede zwischen den Peptidasen verschiedener Stämme heraus
(Tab. 20; vergleiche Peptidase A des Stammes SJ5Ü/8 (#098) mit denen von den
Stämmen SJ2/3-1 (#073), SJ5Ü/12 (#140), SJ6S/14 (#148) und B3/1-2 (#418)). Wie
schon vorher in der pH-Abhängigkeit verdeutlichen diese Untersuchungen fernerhin,
dass es sich bei Peptidase A und B um unterschiedliche Enzyme handelt.
Tab. 20: Einfluss der NaCl-Konzentration auf die Aktivität* von Peptidase A und B im Fluoreszenzassay.
B8/26-2 (#504) B 5 89 % 7 %A 100 95 % 9 %B12/10-1 (#748) B 1 97 % 12 %
B12/16 (#795) B 5 88 % 7 %*Die Probe mit maximaler Peptidase-Aktivität wurde gleich 100% gesetzt (entspricht etwa 1-5 FlU pro Ansatz).
57
Ergebnisse
0%
20%
40%
60%
80%
100%
0 200 400 600 800 1000 1200 1400 1600 1800 2000
NaCl [mM]
Rel
ativ
e A
ktiv
ität
Kulturüberstand Peptidase A Peptidase B
0%
20%
40%
60%
80%
100%
0 5 10 15 20 25 30 35 40 45 50
NaCl [mM]
Rel
ativ
e A
ktiv
ität
Kulturüberstand Peptidase A Peptidase B
a.
b.
Abb. 10: Einfluss der NaCl-Konzentration auf die relative Aktivität* der Peptidasen aus Stamm SJ5Ü/8 (#098). a.) Gesamtansicht; b.) Ausschnittsvergrößerung im Bereich von 0-50 mM NaCl. *Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
3.2.43.2.4 Einfluss von Metallionen auf die Peptidase-Aktivität Einfluss von Metallionen auf die Peptidase-Aktivität
Der Einfluss monovalenter und divalenter Kationen (KCl, MnCl2, MgCl2, FeCl2,
CaCl2, CuSO4) auf die Aktivität der Peptidasen wurde untersucht. Die Ergebnisse
sind in Tab. 21 zusammengefasst. Es konnte keine augenfällige Aktivierung (>1,5-
fache Aktivitäts-Steigerung) der Peptidasen durch die untersuchten Kationen
festgestellt werden. Der Einsatz von KCl (2 mM) im Assay beeinflusste die Aktivität
der untersuchten Peptidasen nur gering (88-123% relative Aktivität). Dabei zeigte die
KCl-Zugabe auf die Aktivität der Peptidase A aller Stämme (Ausnahme: SJ2/3-1
(#073): 96% rel. Aktivität) einen positiven Effekt, wahrscheinlich aufgrund der
Steigerung der Salinität (vergl. 3.2.3). Der Zusatz von MnCl2 zeigte auf die Aktivität
der untersuchten Peptidasen eine hemmende Wirkung (44-83% relative Aktivität).
Die Peptidase B der Stämme SJ2/3-1 (#073), SJ5Ü/8 (#098) und SJ6S/14 (#148)
zeigten allerdings mit MnCl2 eine schwach erhöhte Aktivität (mit 117-128% relativer
58
Ergebnisse
Aktivität). Keine oder eine schwach aktivierende Wirkung auf die Peptidase A-
Aktivität (99-147%) war auch bei Zusatz von MgCl2 und CaCl2 messbar. Die
Peptidase B der Stämme SJ2/3-1 (#073), SJ5Ü/8 (#098), SJ6S/14 (#148), B3/14
(#430) und B12/10-1 (#748) zeigten allerdings bei MgCl2-Zugabe gehemmte Aktivität
(mit 74-90% rel. Aktivität), ebenso die Peptidase B der Stämme SJ5Ü/8 (#098), B3/1-
2 (#418), B8/26-2 (#504) und B12/10-1 (#748) und Peptidase A von Stamm B3/14
FeCl2 die
s-
ngen wurde daher auf den
insatz der untersuchten Kationen im Assay verzichtet.
w Peptid
(#430) bei Zusatz von CaCl2.
Eine hemmende Wirkung von Kupfersulfat konnte bei allen untersuchten Peptidasen
nachgewiesen werden. Der Einsatz von 2 mM FeCl2 führte sowohl im Fluoreszenz-
test als auch im spektrophotometrischen Test zu einem totalen Ausfall der Aktivität
bei allen Proben, es konnte nicht geklärt werden, ob das verwendete
Assays inhibierte oder eine komplette Inhibition der Enzymaktivität vorlag.
Eine stichprobenartige Wiederholung der Versuche ergab eine hohe Schwankung
breite der Ergebnisse, jedoch mit gleich bleibender Tendenz (Daten nicht gezeigt).
Da die untersuchten Peptidasen keine eindeutige Aktivierung durch die untersuchten
Kationen zeigten, konnte eine Abhängigkeit oder mögliche Co-Faktoren der Enzyme
nicht identifiziert werden. Bei den weiteren Untersuchu
E
Tab. 21: Abhängi tä hgkeit der re
und Blativen Pe der
ptidase-Aktiviwählten S
t* von versce unter
iedenen Metallionen. Es urden die ase A ausge tämm sucht.
B12/16 (#795) B 55% 40% 24% 8% 88% 13% 10%* Die Peptidase-Aktivität einer Probe ohne den Zusatz von chaotropen Salzen wurde gleich 100% gesetzt. n.b.= nicht bestimmt. Ein Großteil der Proben zeigte unter Einfluss von 3M GuHCl oder 0,8 M GuSCN
noch Peptidase-Aktivität. So zeigte die Peptidase A der Stämme B3/1-2 (#418) und
B12/10-1 (#748) bei der höchsten getesteten GuHCl-Konzentration von 3 M noch
eine Restaktivität von 21% bzw. 37%. Bei 0,8 M GuSCN zeigten die Peptidase A der
Stämme SJ5Ü/8 (#098), SJ6S/14 (#148) und B3/1-2 (#418) Restaktivitäten von 22%
bis 26%, bzw. die Peptidase B von Stamm SJ5Ü/8 (#098) und B3/1-2 (#418) je 31%.
60
Ergebnisse
Auffällig war die höhere GuSCN und GuHCl-Resistenz der Peptidase A gegenüber
der Peptidase B, mit Ausnahme von Stamm B3/1-2 (#418) und B12/10-1 (#748).
Die Toleranz der Peptidase A und B gegenüber SDS wurde exemplarisch an drei
Stämmen gezeigt (Abb. 11). Die Stämme SJ2/3-1 (#073), B3/1-2 (#418) und
B12/10-1 (#748) stellten eine repräsentative Auswahl aus jeweils einer der 3
Verwandtschaftsgruppen (vergl. 3.1.2) der 9 bearbeiteten Stämme dar. Waren die
einzelnen Messergebnisse einer hohen Schwankungsbreite unterworfen (siehe
3.1.4), wiesen die Mittelwerte jedoch eine Tendenz in der SDS-Toleranz der
Peptidasen auf (Abb. 11). So zeigte eine Konzentration von 0,1% SDS nur wenig
Einfluss auf die Aktivität der Peptidase A der hier untersuchten Stämme, wobei bei
steigender SDS-Konzentration (0,5% und 1% SDS) ein Absinken der relativen
Aktivität z.T. bis auf 15% zu beobachten war. Die Peptidase B hingegen zeigte mit
0,1% SDS eine Steigerung der Aktivität von 143%-187%, wohingegen sie bei den
öheren Konzentrationen einen Rückgang der Aktivität bis auf 27% (bei 1% SDS)
aufwies. Beide Peptidasen (A und B) besaßen also eine Toleranz gegenüber SDS,
was auch durch proteolytische Aktivität der Proben während der SDS-PAGE
demonstriert wurde (siehe 2.6.8).
h
0%
50%
100%
rela
ti
150%
200%
250%
300%
- B12/10-1(#748) -Peptidase A
B12/10-1(#748) -Peptidase B
ve P
eptid
ase-
Akt
ivitä
t
0,1% SDS0,5% SDS1% SDS
SJ2/3-1(#073) -Peptidase A
SJ2/3-1(#073) -Peptidase B
B3/1-2(#418) -Peptidase A
B3/1-2(#418)Peptidase B
Abb. 11: Relative Peptidase-Aktivität* der Peptidase A und B in Abhängigkeit zur SDS-Konzentration. * Die Peptidase-Aktivität einer Probe ohne den Zusatz von SDS entspricht 100%.
Der Einsatz von spezifischen Peptidase-Inhibitoren sollte die Zuordnung der
untersuchten Peptidasen zu bekannten Gruppen von Peptidasen ermöglichen.
61
Ergebnisse
Insbesondere wurde nach Serinpeptidasen und Metallopeptidasen gesucht (siehe
Einleitung, Abschnitt 1.2.1.1). Als Inhibitor für Serinpeptidasen (und einigen wenigen
nanthrolin und EDTA. Der
erinpeptidase-Inhibitor PMSF besaß nur geringe hemmende Wirkung (16-39%
r Peptidase A der
genannten Stämme um eine Metallopeptidase handelt.
s von Inhib r ie Aktivi r Peptidase A und B.
Cysteinpeptidasen) wurde PMSF benutzt, Phenanthrolin bzw. EDTA als Inhibitoren
für Metallo- und Metallionen-aktivierte Peptidasen (Salvesen & Nagase, 2001). Die
Ergebnisse dieser Untersuchungen sind in Tab. 23 zusammengefasst.
Die Peptidase A der Stämme SJ2/3-1 (#073), SJ5Ü/12 (#140), B3/14 (#430) und
B12/10-1 (#748) zeigte eine komplette Inhibition bzw. starke Inhibition (58-68%
Inhibition) durch die Metallopeptidase-Inhibitoren Phe
S
Inhibition). Dies legt die Vermutung n s sich bei deahe, dass e
Tab. 23: Einflus ito en auf d tät de
Inhibi n [%] tioStamm Peptidase PMSF
(10 mM)Phe nna throlin
(10EDTA
M) m (5 mM)A 16 �5 >9 9a) 68SJ2/3-1 (#073) B >9 9a) -b) 11A 54 �18 >99a)
58SJ5Ü/8 (#098)
B >9 a)9 -b) 54A 36 �8 >9 n9a) .b.SJ5Ü/12 (#140) B >9 n9a) -b) .b.A 56 �9 >99a)
63SJ6S/14 (#148)
B >9 a)9 -b) 71A 69 �22 >99a)
29B3/1-2 (#418) B >99a) 18 40 � 62
A 61 �16 >9 9a) 68B3/14 (#430) B >99 - 50a)
b)
B8/26-2 (#504) b)B 91 �1 - 51A 39 �21 >99a)
58B12/10-1 (#748) a)
b)
B >99 - 91B12/16 (#795) B >99a)
-b) 35a)Peptidase-Aktivität unter der Nachweisgrenze; b)Aktivitätssteigerung bis zu 168% Restaktivität durch Phenanthrolin. n.b.: nicht bestimmt Ebenfalls starke (58-68% Inhibition) bis komplette Inhibition konnte durch die
Metallopeptidase-Inhibitoren bei der Peptidase A der Stämme SJ5Ü/8 (#098),
SJ6S/14 (#148) und B12/10-1 (#748) beobachtet werden. Schwierigkeiten für die
sichere Zuordnung zu den Metallopeptidasen bereitete aber die starke inhibitorische
Wirkung (54-61% Inhibition) von PMSF auf die Aktivität. Gleiches gilt für Peptidase A
des Stammes B3/1-2 (#418) mit 69% Inhibition durch PMSF und geringer Wirkung
von EDTA mit 29% Inhibition, aber kompletter Inhibition durch Phenanthrolin.
62
Ergebnisse
Die Peptidase B aller untersuchten Stämme wurde stark (91% Inhibition bei Stamm
B8/26-2 (#504)) bis komplett durch 10 mM PMSF inhibiert. Dagegen zeigte
SJ2/3-1 (#073) und B12/16 (#795) zu den Serin-
B aller Stämme sehr deutlich heraus, dass es
sich hierbei um zwei verschiedene Enzyme handelt. Insofern stehen diese
en des Einflusses des pH-Wertes, des NaCl-
Phenanthrolin nur bei Stamm B3/1-2 (#418) eine Inhibition der Peptidase B-Aktivität
von 40%, bei allen anderen Stämmen konnte keine Inhibition der Peptidase B
festgestellt werden.
Mit der geringen Inhibition von 11% bzw. 35% durch EDTA konnte eine Zuordnung
der Peptidase B der Stämme
peptidasen erfolgen. Die Peptidase B der restlichen Stämme zeigte eine Inhibition
von 50-71% durch 5 mM EDTA. Eine eindeutige Zuordnung dieser Peptidasen
konnte deshalb nicht erfolgen.
Zusammenfassend stellt die unterschiedliche Wirkung von PMSF und Phenanthrolin
Auf Plattentests konnte nachgewiesen werden, dass die Peptidasen der 9
untersuchten Stämme sowohl Magermilch und reines Casein, als auch Gelatine als
Substrate verwerten konnten (Daten nicht gezeigt). Die Tauglichkeit der Peptidasen
für den Einsatz in Nukleinsäure-Aufreinigungskits wurde hier exemplarisch anhand
y) unter Standard-
des Lyse-Verhaltens gegenüber tierischem Gewebe untersucht. Für die Versuche
standen Mäuseschwanzstückchen zur Verfügung. Als Kontrolle diente die
kommerziell verwendete Proteinase K (10 μg/μl, MOLZYM).
Dieser Versuch wurde mit jeweils 450 μl Konzentrat aus 8 Kulturüberständen (siehe
Tab. 24) durchgeführt. Das Lyse-Verhalten der Peptidasen an tierischem Gewebe
wurde zunächst unter den zuvor gewonnenen Optimalbedingungen untersucht. Die
Gewebestückchen wurden in Puffer (30mM Tris-HCl, pH 9, 5 mM NaCl) mit
zellfreiem Kultur-Konzentrat bei 50°C inkubiert, wobei die enthaltene Peptidase-
Gesamtaktivität zwischen 0,4-1,08 kFl-Units (Fluoreszenzassa
bedingungen lag. Die nach Betriebsanweisung (PrestoSpin D Tissue-Kit, MOLZYM)
eingesetzte Gesamtaktivität der Proteinase K betrug dagegen 75 kFlU. Die Proben
wurden alle 10 min kräftig gemischt und ihr Zustand protokolliert.
63
Ergebnisse
Die Lyse der Gewebeproben erfolgte, abhängig vom Stamm, mit unterschiedlicher
Geschwindigkeit (siehe Tab. 24). Anfängliche Lyseerscheinungen, wie Trübung des
Puffers, traten bei den Stämmen SJ6S/14 (#148), B3/14 (#430) und B12/16 (#795)
schon nach 20 min Inkubation auf. Eine vollständige Zersetzung des Muskelgewebes
konnte bei diesen 3 Stämmen nach 120 min Inkubation beobachtet werden (siehe
Abb. 12). Eine Zersetzung der Mäuseschwanzhaut konnte nur in geringem Maße
festgestellt werden; sie wurde am Versuchsende zur Überprüfung der Muskel-
gewebelyse entfernt. Das Konzentrat von Stamm SJ5Ü/8 (#098) zeigte unter diesen
Bedingungen kein Anzeichen von gewebelytischem Verhalten, während die
Proteinase K aufgrund nicht optimaler Reaktionsbedingungen nur eine verlangsamte
Lyse zeigte.
a. b.
Abb. 12: Wirkung des zellfreien Kultur-Konzentrates von Stamm B12/16 (#795) auf ein Mäuseschwanzstückchen in Reaktionspuffer (30 mM Tris-HCl pH 9, 5 mM NaCl) und Inkubation bei 50°C. a. 0 min; b. 120 min: Haut wurde manuell entfernt, das Muskelgewebe hat sich von den Schwanzwirbeln (Pfeil) gelöst und wurde zersetzt. Beachte sung in b. aufgrund der Zersetzung des Muskelgewebes. die Trübung der Lö
kung der ko trie Ku rübers de auf ein tückchen (ca la ) nach 0, 60, 90 und 120 min
Reaktionspuffe mM ris-H H 9, M NaC bei
Tab. 24: Wir nzen rten ltu tänMäuseschwanzs . 1 cm ng 3Inkubation in r (30 T Cl p 5 m l)50°C.
Konzentrat Gesamtaktivität 30 min 60 min 90 min 120 min [kFlU]1)
Zur Abschätzung des Molekulargewichts der Peptidasen und Analyse der
kontaminierenden Proteine wurden zellfreie Kulturen und Präparate aus der Phenyl-
sepharose-Chromatografie mit Hilfe der Polyacrylamid-Gelelektrophorese analysiert.
Die Bestimmung der nativen Größe der Peptidasen sollte dabei über einen direkten
Aktivitätsnachweis im Gel erfolgen. Die Analyse erfolgte anhand einer
repräsentativen Auswahl, als Stämme kamen SJ2/3-1 (#073), B3/1-2 (#418) und
B12/10-1 (#748) zum Einsatz, die zueinander aufgrund von 16S-rRNA-Sequenz-
vergleichen (siehe 3.1.2) die meisten Unterschiede aufwiesen.
65
Ergebnisse
Für den Aktivitätsnachweis wurden die Proben unter denaturierenden Bedingungen
(2% SDS) im Gel aufgetragen, ohne sie vorher zu erhitzen. Eine Hitzedenaturierung
der Proben (5 min bei 85°C) im SDS-Probenpuffer deaktivierte jegliche proteolytische
Aktivität. Der Peptidasenachweis erfolgte nach der Auftrennung im Kontakt-
en großen
s Stammes B3/1-2 (#418) eine ~55 kDa-Bande mit schwacher
Aktivität und eine Doppelbande unterhalb von ~30 kDa mit ebenfalls schwacher
Aktivität. Es wird vermutet, dass durch Autoproteolyse eine Zersetzung der Peptidase
B in kleinere aktive Enzymteile stattgefunden hat. Dies ist auch von anderen
Serinpeptidasen bekannt (Arnorsdottir et al., 2002). Eine Verifizierung wurde nicht
vorgenommen.
Zymogramm (siehe 2.6.10). Eine direkte Zymografie, bei der die Auftrennung und der
Aktivitätsnachweis in PA-Gelen mit copolymerisiertem Substrat erfolgten, war nicht
erfolgreich. Die Peptidasen konnten offensichtlich aufgrund ihrer hohen Affinität zu
den copolymerisierten Proteinen im Gel nicht aufgetrennt werden und bildeten einen
hochmolekularen Schmier im aktivgefärbten Gel.
In allen drei genannten Stämmen konnten bei der Analyse mittels SDS-PAGE und
anschließender Kontakt-Zymografie in den Kulturüberständen zwei Aktivitätszonen
festgestellt werden (siehe bspw. Abb. 13). Das Molekulargewicht ließ sich mit ca. 55
kDa und ca. 35 kDa nur ungefähr ermitteln, da die Aktivitätszonen ein
Wirkungsbereich aufwiesen. Diese Aktivitäts-Banden konnten auf die beiden aktiven
Fraktionspools durch Phenylsepharose-Chromatografie zurückgeführt werden. Somit
lässt sich eine direkte Zuordnung der Aktivitätsbande, die bei ca. 35 kDa auftritt, zu
der Peptidase A bei den drei untersuchten Stämmen schlussfolgern. Dasselbe gilt für
die Peptidase B, die der Aktivität bei 55 kDa zugeordnet werden konnte.
Eine längere Lagerung der Proben in 30 mM Tris-HCl >4 Monate reduzierte nicht nur
die Aktivität, sondern es konnten auch Veränderungen hinsichtlich des Musters der
Aktivitätsbanden beobachtet werden. So zeigte die angereicherte Probe von
Peptidase B de
66
Ergebnisse
Abb. 13: SDS-PAGE der zellfreien Kultur (ZK) und den Phenylsepharosefraktionspools A (PA) und B (PB) des Stammes SJ2/3-1 (#073) und B12/10-1 (#748). a: Silbergefärbtes Polyacrylamidgel; b: Kontakt-Zymogramm mit Peptidase-Aktivitätszonen, durch grafische Aufhellung hervorgehoben. Die Position einiger Banden der Proteinleiter wurde mit weißen Strichen markiert. Marker = PageRuler Prestained Protein ladder.
3.33.3 Molekularbiologische Identifizierung von Subtilisin-ähnlichen Peptidasen Molekularbiologische Identifizierung von Subtilisin-ähnlichen Peptidasen
Für die weitere Charakterisierung der untersuchten Peptidasen bedurfte es einer
Identifizierung der unbekannten Peptidasegene. Das Screening nach
Peptidasegenen in den Genbanken wurde auf zwei Weisen durchgeführt: Zum einen
durch Identifizierung von Peptidase produzierenden Klonen auf Plattenassays (siehe
2.6.9), und zum anderen durch Kolonie-Hybridisierung mit Sonden.
Zum Auffinden von Klonen mit proteolytischer Aktivität wurden Genbanken der 9
Stämme im Vektor pNOFMCS auf Skimmilk-Agar untersucht, ebenso wie die
Genbanken von 4 Stämmen im Vektor pUC13 (Daten nicht gezeigt). Zur
Identifizierung kamen neben dem Skimmilk-Agar zwei weitere sensitive
Plattenassays (siehe 2.6.9) zum Einsatz, die auf zwei verschiedenen Substraten
(Casein und Gelatine) basierten. Es wurden u.a. Verfahren angewendet, um eine
Sekretion der rekombinanten Proteine zu erleichtern (z.B. Kälteschock oder
Inkubation der Klone bei Raumtemperatur). Ebenfalls erfolgte eine Exposition der
Klone mit Salz (Überschichtung der Kolonien mit 2% und 6% NaCl-Lsg.), um bei
möglicher Salzabhängigkeit der Peptidasen die proteolytische Aktivität auf den
ZK (#
073)
ZK (#
748)
Mar
ker
PB (#
073)
PA (#
073)
PB (#
748)
PA (#
748)
~100
kDa
~70 ~55
~40
~35
~25
~100
kDa
~70
~55
~40
a
b
ZK (#
073)
ZK (#
748)
Mar
ker
PB (#
073)
PA (#
073)
PB (#
748)
PA (#
748)
67
Ergebnisse
ansonsten salzarmen (1% NaCl) Agarplatten zu detektieren. Es konnte jedoch im
Screening kein Klon gefunden werden, der proteolytische Aktivität zeigte.
Das Screening nach Peptidasegenen in den Genbanken erfolgte daher im Kolonie-
Blot-Verfahren mit Sonden, deren Sequenz homolog zu bekannten Peptidasegenen
war. Die biochemische Charakterisierung ergab Hinweise auf die Zugehörigkeit der
untersuchten Enzyme zu zwei großen Peptidasegruppen: den Metallopeptidasen und
den Serinpeptidasen (siehe Abschnitt 3.2.6). Da über Metallopeptidasen
vergleichsweise wenige Sequenzinformationen vorlagen, wurde die Suche auf
Serinpeptidasen beschränkt. Innerhalb der Serinpeptidasen bilden die Subtilisin-
ähnlichen Peptidasen eine der größten Gruppen, mit Proteinase K und Subtilisin E
als bekannteste Vertreter. Sie besitzen ein breites Substratspektrum und viele
biotechnologische Anwendungen (Siezen, 1996). Sehr viele Enzyme dieser Familie
sind gut charakterisiert und einige schon rekombinant hergestellt worden. Anhand
von Sequenzinformationen der bekannten Peptidasen wurde eine Sonde für
Subtilisin-ähnliche Peptidasen konstruiert.
Die Genbanken für den Kolonie-Blot wurden mit dem CopyControl Fosmid Library
Production-Kit (Epicentre) in E. coli Epi300 erstellt. Fosmide bieten aufgrund der
großen Insertgröße und des Vorliegens als Einzelkopie im Wirt folgende Vorteile
gegenüber konventionellen Vektoren (z.B. pUC13, pNOFMCS):
1.) Die verringerte Anzahl der zu untersuchenden Klone durch die große Insertgröße
erbringt eine erhebliche Arbeitserleichterung. Bei einer durchschnittlichen Insertgröße
von ca. 40 kbp und Annahme einer Genomgröße von ca. 4,7 Mbp und 99%
Wahrscheinlichkeit zur Klonierung des gesamten Genoms beträgt die Kolonieanzahl
nunmehr <500 pro Genbank (Clarke & Carbon, 1976).
2.) Das Vorliegen des Fosmids als Einzelkopie ermöglicht aufgrund eines geringen
Gen-Dosis-Effektes auch die Klonierung von Genen für toxische Genprodukte
(Epicentre). Rekombinante Peptidasen können aufgrund ihrer unregulierten
Hydrolyse von essentiellen Proteinen und Destabilisierung der Bakterienzelle toxisch
auf die Wirtszelle wirken (Arnorsdottir et al., 2002; Bott & Betzel, 1996; Goldberg,
1992). So wurde eine Stabilisierung von Klonen mit funktionellen Peptidasegenen
durch einen geringen Gen-Dosis-Effekt erhofft.
68
Ergebnisse
3.3.13.3.1 Entwicklung der Sonde für Subtilisin-ähnliche Peptidasen Entwicklung der Sonde für Subtilisin-ähnliche Peptidasen
Für die Sondensynthese wurden Primer gesucht, deren DNA-Sequenz homolog zu
konservierten Genabschnitten der bisher bekannten Subtilisin-ähnlichen (subtilisin-
like) Serinpeptidasen (MEROPS-Nr: S8 (Rawlings et al., 2006)) war. Beim Alignment
der Aminosäuresequenzen von 6 Subtilisin-ähnlichen Peptidasen verschiedenster
Organismen (Gram-positive, Gram-negative Bakterien und ein Vertreter der
Ascomycota) wurden drei stark konservierte Bereiche gefunden (Abb. 14A). Sie
stellten sich als die Regionen um die hochkonservierten Aminosäurereste Aspartat
(Asp, D), Histidin (His, H) und Serin (Ser, S) heraus, die charakteristisch für die
Familie der Subtilisin-ähnlichen Peptidasen eine katalytische Triade im aktiven
Zentrum ausbilden (Siezen & Leunissen, 1997).
Aus dem Alignment der DNA-Sequenzen dieser drei konservierten Regionen konnten
drei degenerierte Primer entwickelt werden (Abb. 14B): SubDfor, SubHfor und
SubSrev.
Abb. 14: Sequenzvergleich von 6 verschiedenen Subtilisin-ähnlichen Peptidasen1).A. Alignment der Aminosäuresequenzen in der Umgebung der drei Aminosäurereste(*) des
katalytischen Zentrums. Identische und homologe Aminosäuren wurden schwarz bzw. grau hinterlegt. Die grün-gestrichelten Kästen stellen den abgeleiteten Bereich der unter B angezeigten DNA-Sequenzen dar.
B. Alignment der DNA-Sequenzen der konservierten Peptidase-Regionen. Oberhalb der jeweiligen Alignments sind die Sequenzen der daraus entwickelten degenerierten Primer2) vermerkt (forward Primer (5’-3’):SubDfor, SubHfor; reverse Primer (3’-5’): SubSrev). Homologe Nukleotide sind mit einem Kasten umschlossen. Die blau gestrichelten Kästen stellen den Bereich dar, an dessen Sequenzen die Primer entwickelt wurden. 1) Enzym-Kürzel: 1.Taq: Aqualysin I, Thermus aquaticus, 2.Bsu: Subtilisin E, Bacillus subtilis 168; 3.Tal: Proteinase K,
Zusätzlich wurden zwei degenerierte Primer (hier bezeichnet als subtilisin_for, ein
forward-Primer in der H-Region und subtilisin_rev, ein reverse-Primer in der
S-Region) für Subtilisin-ähnliche Peptidasen von Jang et al. (2002) in die
Untersuchungen einbezogen.
Zur Amplifikation von Genabschnitten der Subtilisin-ähnlichen Peptidasen wurde als
Template chromosomale DNA der 9 moderat halophilen Stämme (siehe Tab. 35 im
Anhang, türkis markiert) und als Positivkontrolle DNA von Bacillus subtilis,
Geobacillus stearothermophilus und Thermus aquaticus in der PCR-Reaktion
eingesetzt. Aus den möglichen Kombinationen der 5 Primer trat das Primerpaar
SubHfor und subtilisin_rev hervor, das eine starke Amplifikation eines etwa 500 bp
großen Produktes bei allen genannten Stämmen ermöglichte. Andere mögliche
Kombinationen der 5 Primer erzielten entweder keine bis schwache Amplifikate oder
enthielten zusätzlich unspezifische Nebenamplifikate. Aufgrund der Übereinstimmung
mit der Größe des Amplifikates der Positivkontrollen und der halophilen Stämme
wurde vermutlich ein Peptidase-Sequenzabschnitt zwischen der Region H und
Region S (siehe Abb. 14 A) amplifiziert.
Das DIG-markierte PCR-Produkt (2.7.7), welches mit den Primern subHfor und
subtilisin_rev von Stamm B12/16 (#795) amplifiziert wurde, stellte die Sonde sub795
für das Screening der Genbanken nach Subtilisin-ähnlichen Peptidasen dar. Die
Auswahl des Stammes für die Sonde wurde beliebig getroffen, da über eine niedrige
Stringenz im Southern-Blot auch eine Hybridisierung von weniger homologen
Zielsequenzen erreicht werden sollte.
3.3.22 Identifizierung von Klonen und Subklonen mit homologen Sequenzen zu ogen Sequenzen z3.3. Identifizierung von Klonen und Subklonen mit homol uSubtilisin-ähnlichen Peptidasen Subtilisin-ähnlichen Peptidasen
Es wurden insgesamt Fosmid-Genbanken von 3 der halophilen Peptidase-
Produzenten mit der Sonde sub795 im Kolonie-Blot-Verfahren (2.7.5) untersucht. Die
Anzahl der untersuchten Klone (ca. 900) pro Genbank entsprach der etwa 1,8-fachen
Genomgröße. Die Fosmid-Genbanken der Stämme B3/1-2 (#418), B12/10-1 (#748)
und B12/16 (#795) erbrachten insgesamt 4 Klone mit positiven
Hybridisierungssignalen: F114 für den Stamm B3/1-2 (#418), F13 und F14 für
B12/10-1 (#748) und F9 für B12/16 (#795).
Stellvertretend wird eine Klonanalyse des Klons F13 mit anschließender
Hybridisierung gezeigt. In der Klonanalyse von F13 konnte eine zur Sonde sub795
homologe Sequenz u.a. auf zwei SalI-Fragmenten (Größe ca. 1,5 kb und 0,6 kb)
70
Ergebnisse
bzw. einem 8 kb BamHI-Fragment der restringierten Fosmid-DNA (Größe >38kb)
lokalisiert werden (Abb. 15).
Abb. 15: Klonanalyse und Southern-Blot des Fosmidklons F13 (Stamm B12/10-1 (#748)). a.) Gelelektrophorese der restringierten Fosmid-DNA mit folgenden Enzymen: 1. EcoRI, 2. SalI, 3. HindIII, 4. BamHI, 5. XhoI, 6. HaeI, 7. EcoRV, 8. XbaI, 9. SmaI. M= Marker-DNA �/HindIII-EcoRI (DIG-markiert). b.) Southern-Blot zur Klonanalyse (a.) mit der Sonde sub795.
M 1 2 3 4 5 6 7 8 9 M 1 2 3 4 5 6 7 8 9
a b
bp 21226-
5148- 4973- 4268- 3530-
2027- 1904- 1584- 1375-
947- 831- 564-
Über eine Subklonierung (Tab. 25) des 8 kb BamHI-Fragmentes vom Fosmidklon
F13 in pNOFMCS wurden zwei SalI-Fragmente wiederum in pNOFMCS subkloniert
und erbrachten die Subklone NOF13-1-7 und NOF13-1-10 für die Sequenzierung
(3.3.3).
Tab. 25: Charakteristika des Fosmidklons F13 und seiner Subklone. Größe der hybridisierungspositiven Fragmente Klon Klonierungs-
NOF13-1 pNOFMCS 0,6 kb + 1,5 kb 8 kb - NOF13-1-7 pNOFMCS 1,5 kb - - NOF13-1-10 pNOFMCS 0,6 kb - - 1)Peptidase-Aktivität auf Skimmilk-Agar. 2)Aktivität nur nach Induktion der Zellen mit L-Arabinose (0,5 mM) und Inkubation für 4 d bei RT (~23°C). Die im Southern-Blot identifizierten Klone wurden auf Peptidase-Aktivität im
Plattenassay (2.6.9) überprüft. Es konnte keine proteolytische Aktivität der Klone und
Subklone festgestellt werden. Demgegenüber konnte bei dem Fosmidklon F13 nach
Erhöhung der Kopieanzahl durch eine L-Arabinose-Induktion (0,5 mM) und
anschließender Inkubation für 4 d bei RT (~23°C) im Plattenassay geringe
Peptidase-Aktivität festgestellt werden. Gleiches galt für Fosmidklon F114. Die
Fosmidklone F9 und F14 sowie mehrere beliebig ausgewählte Fosmidklone aus den
Genbanken zeigten hingegen keine Aktivität nach Induktion.
71
Ergebnisse
3.3.33.3.3 Identifizierung und Sequenzanalyse der Peptidasegene Identifizierung und Sequenzanalyse der Peptidasegene
Die Sequenzierung der Subklone (NOF13-1-7 und NOF13-1-10) von Fosmidklon F13
erbrachten eine 2293 bp große Nukleotidsequenz (Abb. 37). Die Analyse der
abgeleiteten Aminosäuresequenzen erbrachte ein offenes Leseraster (ORF) von
1584 Nukleotiden, das vom vermuteten Startcodon (ATG) bei Position 261 bp bis
zum vermuteten Stoppcodon (TAG) bei 1842 bp reichte. Eine vermutliche ribosomale
Bindungsstelle (AGGA) wurde 8 bp stromaufwärts des Leserasters identifiziert. Die
Übersetzung der Nukleotide des ORF in eine Aminosäuresequenz ergab ein
mögliches Protein von 528 AS Länge (Abb. 16). Eine BLAST-Analyse der AS-
Sequenz ergab Übereinstimmungen mit hinterlegten Sequenzen von Subtilisin-
ähnlichen Peptidasen (Tab. 26). Die höchste Übereinstimmung mit 65% (348 von
532 Übereinstimmungen) ergab sich mit der alkalischen Serinpeptidase A (Alkaline
Serine Exoprotease Precursor A, ProA) von Vibrio alginolyticus. Aufgrund der
niedrigen Homologie der abgeleiteten Aminosäuresequenz von <66% zu bekannten
Proteinen handelt es sich bei der hier identifizierten DNA-Sequenz vermutlich um
eine neue Subtilisin-ähnliche Peptidase. Die Benennung der neuen Peptidase erfolgt
in Anlehnung zur nächst verwandten Peptidase ProA aus Vibrio alginolyticus und
dem Ursprung aus Salinivibrio spec. Stamm B12/10-1 (#748) als ProAS(#748). Der
Anhang des Buchstaben S im Enzymnamen soll dabei auf die Herkunft aus
Salinivibrio und die Nummer auf die des Stammes hinweisen.
Die Peptidasebezeichnung ProA wurde aus Prioritätsgründen übernommen, obwohl
von Deane (1987) unglücklicherweise die missverständliche Peptidasegen-
Bezeichnung proA verwendet wurde. Die Bezeichnung proA steht bekanntlich für das
Gen der Glutamat-5-Semialdehyd-Dehydrogenase in der Prolin-Biosynthese von E.
coli (Kuo & Stocker, 1969) und vielen anderen Organismen.
Abb. 16: Abgeleitete Aminosäuresequenz des Peptidasegens proAS(#748) von Stamm B12/10-1 (#748).
Vibrio sp. PA-44 336 von 528 (63%) 409 von 528 (77%)
Alkaline serine protease (Vco157)
Vibrio cholerae 295 von 535 (55%) 397 von 535 (74%)
Alkaline serine protease Photobacterium sp. SKA34 274 von 487 (56%) 359 von 487 (73%) Anhand der Sequenz von proAS(#748) wurden Primer (subHraus301 und
subHraus61) entwickelt, die eine direkte Sequenzierung der Fosmidklone F114 und
F9 der Stämme B3/1-2 (#418) und B12/16 (#795) ermöglichten. Somit konnten ohne
Subklonierung die vollständigen Sequenzen zweier weiterer Peptidasegene
proAS(#418) aus Stamm B3/1-2 (#418) und proAS(#795) aus Stamm B12/16 (#795)
identifiziert werden (siehe Abb. 38 & Abb. 39).
Des Weiteren wurden 400 bp große Peptidasegen-Teilsequenzen der übrigen 7
Stämme durch Sequenzierung von PCR-Produkten mit dem Primer subHfor
bestimmt.
Eine BLAST-Analyse der 8 abgeleiteten Nukleotidsequenzen und -teilsequenzen
erbrachte eine ähnlich hohe Übereinstimmung von 64 bzw. 65% mit alkalischen
Serinpeptidasen aus Vibrio, wie zuvor schon die Peptidase ProAS(#748). Dies lässt
die Schlussfolgerung zu, dass wahrscheinlich alle untersuchten halophilen Stämme
Subtilisin-ähnliche Peptidase besitzen. Sie wurden der oben angewandten
Bezeichnung folgend benannt.
Ausgehend von dem Alignment der proAS-Teilsequenzen (Abb. 17) lassen sich die
untersuchten 9 Peptidasegene in drei Gruppen unterteilen. Die Gruppen bestehen
jeweils aus sehr homologen oder identischen proAS-Sequenzen (99,3-100%
Identität), wobei Gruppe I die Stämme SJ2/3-1 (#073), SJ5Ü/8 (#098), SJ5Ü/12
(#140), SJ6S/14 (#148) und B3/14 (#430), Gruppe II den Stamm B3/1-2 (#418) und
Gruppe III die Stämme B8/26-2 (#504), B12/10-1 (#748) und B12/16 (#795)
einschließen. Zwischen diesen 3 proAS-Gruppen bestehen deutliche Unterschiede,
so weist die Gruppe I zu den Gruppen II und III ca. 14% bzw. 19% und Gruppe II zu
III 21% Divergenz auf.
73
Ergebnisse
Nukleotid-Austausch (x100)024681012141618
proAS(#098)proAS(#430)proAS(#140)proAS(#148)
78.9%67.5%
proAS(#073)38.0%
proAS(#418)
97.9%
proAS(#795)proAS(#748)
99.9%
proAS(#504)100.0%
69.9%
proA (V.alginolyticus)
Gruppe I
Gruppe II
Gruppe III
Abb. 17: Phylogenetischer Stammbaum der untersuchten Peptidasegene proAS der 9 untersuchten halophilen Stämme und des nächst verwandten Peptidasegens proA aus Vibrio alginolyticus, basierend auf DNA-Teilsequenzen (400 bp). Die Verwandtschaftsverhältnisse wurden mit CLUSTALW untersucht und der Stammbaum mit der Bootstrap-Methode (trials: 1000; seed: 111) auf seine Konsistenz und Robustheit überprüft (Felsenstein, 1988). Die Länge der Verzweigungen repräsentiert die Divergenz zwischen zwei Sequenzen, die Zahlen bezeichnen die Anzahl der Substitutionsereignisse. Die Prozentzahlen geben den Anteil der im Bootstrap-Verfahren bestätigten Verzweigungen wieder. Eine Gruppierung der nächst verwandten Sequenzen wurde in 3 Gruppen vorgenommen (Gruppe I, II und III). Ein Vergleich der ProAS-Sequenzabschnitte auf Aminosäureebene führt ebenfalls
zur oben genannten Gruppierung, jedoch mit erheblich geringeren Sequenz-
Unterschieden. Es besteht im Vergleich der Gruppe I zu den Gruppen II und III ca.
2% bzw. 5% Sequenzunterschied und beim Vergleich der Gruppe II zu III ca. 6%
Divergenz. Dies lässt auf eine enge Verwandtschaft der untersuchten ProAS-
Peptidasen schließen.
3.3.43.3.4 Identifizierung des Signalpeptids in ProAS(#748) Identifizierung des Signalpeptids in ProAS(#748)
Proteine, die über die bakterielle Cytoplasma-Membran transportiert werden müssen,
werden in den meisten Fällen als größere Prä-Proteine mit N-terminalem
Signalpeptid synthetisiert (Fekkes & Driessen, 1999; Müller et al., 2001).
Untersuchungen an der N-terminalen Seite von ProAS(#748), ProAS(#418) und
ProAS(#795) sollten Aufschluss über das Vorhandensein eines Signalpeptids
erbringen.
Die Analyse der Aminosäuresequenzen der drei Peptidasen und die der
nächstverwandten ProA aus Vibrio alginolyticus mit dem Programm SignalP 3.0
Server ergab eine wahrscheinliche Zugehörigkeit der ersten 23 N-terminalen AS zu
einem Signalpeptid (Abb. 18). Die zugewiesene Schnittstelle für ProA zwischen der
23. und 24. Aminosäure differierte allerdings um 2 AS von der von Deane et al.
(1989) theoretisch postulierten Stelle. Möglicherweise wurde ein anderes
Rechenmodell für die Analyse zu Grunde gelegt.
74
Ergebnisse
Bemerkenswert ist die Homologie der Signalpeptide der drei ProAS-Sequenzen mit
der Sequenz von ProA: 11 von 23 Aminosäuren sind identisch. Das Vorhandensein
von Signalpeptiden lässt die Schlussfolgerung zu, dass ein Signalpeptid-vermittelter
Transport der drei untersuchten ProAS Peptidasen im Wildtyp über die
Cytoplasmamembran erfolgt.
Abb. 18: Alignment der N-terminalen Sequenzen und vermutete Signalpeptide von Peptidasen dreier halophiler Stämme.
Zeichen und farbige Markierungen: �= Wahrscheinliche Spaltungsstelle des Signalpeptids nach Signal P 3.0 Server. �=Wahrscheinliche Spaltungsstelle des Signalpeptids von ProA nach Deane et al. (1989). Gelb hervorgehoben: Signalpeptid nach Signal P 3.0 Server. Rot: Identische Aminosäuren im Alignment. Orange: Homologe Aminosäuren im Alignment.
3.43.4 Heterologe Expression der Subtilisin-ähnlichen Peptidasen in E. coliHeterologe Expression der Subtilisin-ähnlichen Peptidasen in E. coli
Für die heterologe Expression der ProAS-Peptidasen wurden zwei verschiedene
Ansätze verfolgt.
1. (siehe 3.4.1) Eine Überexpression der Subtilisin-ähnlichen Peptidasen in E. coli
sollte durch den Einsatz von speziellen Expressionsvektoren die Herstellung von
größeren Enzymmengen erleichtern. Die Schwierigkeit bestand darin, dass keine
Daten für die Überexpression von Peptidasen aus moderat halophilen Bakterien in E.
coli vorlagen. Zudem wurde mehrfach von Problemen, vermutlich aufgrund der
Toxizität des Expressionsproduktes auf den Wirt, bei der Expression von Peptidasen
aus mesophilen Bakterien in E. coli berichtet (Arnorsdottir et al., 2002; Bott & Betzel,
1996; Sakamoto et al., 1996). Eine Modellstudie sollte daher zuvor die
Expressionssysteme mit einer bekannten Peptidase evaluieren.
2. (siehe 3.4.2) Die heterologe Expression der ProAS-Peptidase sollte ebenfalls
anhand der Fosmidklone F114 und F13 untersucht werden. Ohne Verwendung von
speziellen Expressionvektoren konnte bei diesen Klonen durch Erhöhung der
Fosmidkopiezahl pro Zelle eine Expression von aktiver Peptidase erlangt werden.
Eine Optimierung der Expressionsbedingungen sollte gewährleisten, dass genügend
rekombinantes Enzym für die folgende biochemische Untersuchung bereitstand.
75
Ergebnisse
3.4.13.4.1 Peptidase-Überexpression mit Expressionsvektoren Peptidase-Überexpression mit Expressionsvektoren
Es standen 3 verschiedene Expressionsvektoren zur Verfügung. Der Vektor
pJF118EH zeichnet sich durch einen starken tac-Promotor aus, der durch IPTG
induziert werden konnte. Er wurde bereits erfolgreich zur Expression von Enzymen
mit toxischer Wirkung (z.B. Endonukleasen, Disqué, pers. Mitteilung) eingesetzt.
Die Expressionsvektoren pBAD/gIII und pASKIBA44 wurden für die Sekretion der
rekombinanten Proteine ins Periplasma entwickelt. Die Sekretion wird über die
Fusion der rekombinanten Proteine mit E. coli-spezifischen Signalpeptiden erreicht.
Eine Abspaltung des Signalpeptides vom rekombinanten Protein erfolgt beim
Transport über die Cytoplasmamembran. Das Anhängen eines C-terminalen
Oligohistidinrestes sollte eine spätere Aufreinigung des rekombinanten Proteins
durch eine Nickel-Affinitätschromatografie erleichtern (Ni-NTA Spin Handbook,
QIAGEN, 2003). Zudem wird die heterologe Expression bei beiden
Expressionsvektoren über streng regulierte Promotoren gesteuert. So steht die
Transkription des klonierten Gens bei pASK-IBA44 unter Kontrolle des tet-Promotors.
Darüber hinaus befindet sich auf dem Plasmid ein Gen des tet-Repressors, der über
die entsprechende Promotor-/Operatorregion die Transkription des unter seiner
Kontrolle stehenden Gens reprimiert (Skerra, 1994). Die Induktion des tet-Promotors
erfolgt mit geringsten Mengen Anhydrotetracyclin, welche sich unterhalb der
antibiotischen Wirksamkeit befinden. Eine Induktion löst den Repressor von der
Promotor-/Operatorregion und erlaubt so die Expression des klonierten Gens.
Im Vektor pBAD wird die Expression über den araBAD-Promotor gesteuert. Dieser
Promotor wird positiv und negativ durch das plasmidkodierte Protein AraC reguliert.
In Abwesenheit von L-Arabinose reprimiert ein AraC-Dimer die
Transkriptionsinitiation im Promotor/Operator-Bereich. Nach der Bindung von L-
Arabinose an das AraC-Dimer wird diese Repression aufgehoben. Ein weiterer
Vorteil dieses Systems liegt in der dosisabhängigen Aktivierung des Promotors.
Somit ist es möglich, das Expressionsniveau durch die Menge an zugegebenem
Induktor zu regulieren.
3.4.1.13.4.1.1 Modellsystem: Expression von Subtilisin E in E. coliModellsystem: Expression von Subtilisin E in E. coli
Eine Evaluation und Optimierung der vorhandenen Expressionssysteme für die
extrazelluläre Expression in E. coli erfolgte mit einer bekannten Peptidase. Die Wahl
für diese Studien fiel auf eine mesophile Peptidase, da die heterologe Expression
76
Ergebnisse
einer Peptidase aus moderat halophilen Bakterien in E. coli bisher nicht publiziert
wurde. Als die bestuntersuchte Subtilisin-ähnliche Peptidase wurde Subtilisin E aus
Bacillus subtilis als Modellenzym ausgewählt. Ihre Expression in E. coli ist sehr gut
Grüne Kästen repräsentieren relevante Elemente der Expressionsvektoren. Rote Kästen repräsentieren die kodierende Region des Prä-Pro-Subtilisin E. Blaue Kästen repräsentieren die verwendeten Restriktionsenzyme und ihre Schnittstellen für die Klonierung. In dieser Studie wurde mit dem kompletten Peptidasegen gearbeitet, welches für ein
Prä-Pro-Subtilisin E kodierte. Die korrekte Insertion der Gene in den jeweiligen
Vektor wurde durch Sequenzierung der Klonierungsübergänge überprüft.
Die Vektoren wurden für eine erste Überprüfung ihrer Expressionsleistung in E. coli
Top10 transformiert. Expressionsstudien am Plasmid pJF-subE konnten nicht
durchgeführt werden, da äußerst wenig Transformanten auftraten (1-2
cfu/Transformationsansatz) und eine hohe Plasmidinstabilität beobachtet wurde.
Klonanalysen zeigten durch Fehlen von charakteristischen Restriktionsschnittstellen
zusätzlich eine Veränderung des klonierten Inserts.
Mit den beiden Vektoren pASK-subE und pBAD-subE hingegen gelang die
heterologe Expression von aktivem Subtilisin E. Die beiden hierfür benutzten
Expressionsvektoren zeichnen sich durch eine besonders starke Regulation der
Promotoren aus (siehe 3.4.1).
Zur Analyse der Expressionsleistungen der zwei Vektoren erfolgte die heterologe
Expression unter verschiedenen Inkubationstemperaturen (23°, 30° und 37°C) und
Induktionsbedingungen. Eine Analyse der Zellfraktionen auf Peptidase-Aktivität, wie
unter 2.6.5 und 2.6.6 beschrieben, sollte Aufschluss über die Lokalisation des
Subtilisin E geben.
Die mit pASK-subE transformierten E. coli Top10-Zellen reagierten auf die Induktor-
Zugabe von 0,1 und 0,2 μg/ml Anhydrotetracyclin mit sofortigem Wachstumsstillstand
bei allen überprüften Inkubationstemperaturen (s.o.). Die ebenfalls mit
Anhydrotetracyclin induzierte Kontrolle (pASK-IBA44 in E. coli Top10) zeigte
dagegen keinerlei Wachstumshemmung. Die wachstumshemmende Wirkung auf
E. coli interpretierten Bott und Betzel (1996) mit der toxischen Wirkung des
exprimierten Subtilisin E. Geringe Mengen an aktivem Subtilisin E konnten nach
Induktion ausschließlich im Periplasma gefunden werden. Die höchste Peptidase-
Aktivität von 0,6 FlU/ml im Fluoreszenztest wurde nach Induktion mit 0,2 μg/ml
Anhydrotetracyclin und 4 h Inkubation bei 30°C erreicht.
Mit pBAD-subE transformierte E. coli Top10-Zellen reagierten bei Induktor-Zugabe
von � 1,3 mM L-Arabinose mit einer Verlangsamung des Wachstums. Nach 5 h
Inkubation wurde 1/3 bis ½ der Zelldichte (OD600 nm) von uninduzierten Kulturen
erreicht. Die Expression bei 37°C führte zu keiner Sekretion von aktivem Subtilisin E
(Tab. 28). Erst nach Temperatursenkung auf 30°C bzw. 23°C konnte Peptidase-
Aktivität nachgewiesen werden. Die höchste extrazelluläre Peptidase-Aktivität von
159,8 FlU/ml im Fluoreszenzassay wurde 5 h nach Induktion mit 12 mM L-Arabinose
bei 23°C erreicht (Tab. 28).
78
Ergebnisse
Tab. 28: Expression von Subtilisin E in E. coli Top10 mit pBAD-subE bei verschiedenen Temperaturen und Induktorkonzentrationen. Inkubation: 5 h bei 23°C und 200 rpm.
*nach Induktion Aufgrund des unterschiedlichen genetischen Hintergrundes der E. coli-Stämme kann
sich die Expressionsleistung der einzelnen Stämme je nach Genprodukt und
Expressionssystem deutlich unterscheiden (Sørensen & Mortensen, 2005). Eine
Evaluation von verschiedenen E. coli-Stämmen sollte zeigen, welcher Stamm sich
am besten für die heterologe Expression von extrazellulär lokalisiertem Subtilisin E
mit pBAD-subE eignete. Dabei zeigte der Stamm BL21(DE3) mit 255,8 FlU/ml 5 h
nach Induktion mit 12 mM L-Arabinose die höchste Peptidase-Aktivität in der
zellfreien Kultur (Abb. 20). Peptidase-Aktivität, wenn auch wesentlich geringer,
konnte ebenfalls sowohl im Periplasma als auch im Cytoplasma nach Induktion bei
den verschiedenen Stämmen in unterschiedlicher Stärke nachgewiesen werden.
Eine Analyse der periplasmatischen und extrazellulären Proteine im SDS-PAGE
(Abb. 19) zeigte eine stark überexprimierte Proteinbande im Bereich von ~37 kDa der
induzierten Kultur. Aufgrund der Größe handelt es sich hier wahrscheinlich um Pro-
Subtilisin E. Ohne Induktion konnte diese Bande im Gel nicht gefunden werden. Die
Evaluation zeigt, dass eine größere Expression an aktivem Subtilisin E nur durch den
Expressionsvektor pBAD-subE erfolgte. Eine Kombination von Expressionsstamm
E. coli BL21(DE3), geringer Induktorkonzentration und Inkubation bei
Raumtemperatur führte zur optimierten Produktion von aktiver Subtilisin E Peptidase.
79
Ergebnisse
E- E+ M P- P+
~100 kDa
~70
~55
~40
~35
~25
Abb. 19: SDS-PAGE (12% PA, silbergefärbt) der Expression von pBAD-SubE in BL21(DE3). E= zellfreie Kultur; P= Periplasmatische Zellfraktion; M= PageRuler Prestained Protein ladder; += Kultur induziert; -= Kultur uninduziert. Der Pfeil markiert die überexprimierte Proteinbande. Proben wurden vor dem Auftrag hitzedenaturiert.
0 50 100 150 200 250 300 350
FlU/ml
extrazellulärperiplasmatischcytoplasmatisch
Top10 1,2 mM O17
C600 BL21(DE3)
Top10 - (Kontrolle) O17
C600 BL21(DE3)
Top10 12 mM O17
C600 BL21(DE3)
Induktion Stamm (L-Arabinose) (E. coli)
Peptidase-Aktivität [FlU/ml]
Abb. 20: Vergleich der Peptidaseaktivität (Fluoreszenzassay) der Zellfraktionen von pBAD-subE transformiert in verschiedenen E. coli-Stämmen. Die Kulturen wurden nach Induktion mit drei verschiedenen L-Arabinose-Konzentrationen bei 23°C für 5 h inkubiert.
80
Ergebnisse
3.4.1.23.4.1.2 Heterologe Expression von ProAS(#748) Heterologe Expression von ProAS(#748)
Bei der heterologen Expression der untersuchten ProAS-Peptidasen mit den 3
vorhandenen Expressionssystemen sollten die Erkenntnisse der Modelluntersuchung
Anwendung finden (3.4.1.1). Die Konstruktion der Expressionsvektoren beschränkte
sich auf ProAS(#748). Bei dieser Peptidase handelte es sich um die bestuntersuchte
der drei vollständig sequenzierten ProAS-Peptidasen, bei der auch Kontrollelemente
außerhalb des Gens sequenziert worden waren (Abb. 37 im Anhang). Die
Konstruktion der Expressionsvektoren für ProAS(#748) erfolgte mit der gleichen
Strategie wie für die Subtilisin E-Expressionsvektoren in 3.4.1.1 beschrieben wurde.
Neben der Klonierung der vollständigen Sequenz des proAS(#748) wurde auch das
Gen ohne den kodierenden Bereich für das Wildtyp-Signalpeptid (3.3.4) kloniert.
Diese Variante wurde konstruiert, um zu vermeiden, dass eine fehlende Erkennung
und anschließende Prozessierung des fremden Signalpeptids in E. coli stattfindet
und es so zur Akkumulation von inaktivem rekombinantem Enzym (Baneyx, 1999)
kommen könnte. Ein Austausch des fremden gegen ein E. coli-spezifisches
Signalpeptid konnte in einigen Fällen zur Expression von aktivem rekombinanten
Enzym führen (Baneyx, 1999; Sørensen & Mortensen, 2005). Der Aufbau der
insgesamt 5 konstruierten Vektoren ist schematisch in Tab. 29 wiedergegeben.
Tab. 29: Schematische Darstellung der Konstruktion der Expressionsvektoren für ProAS(#748).
Grüne Kästen repräsentieren relevante Elemente der Expressionsvektoren. Prä-ProAS(#748) repräsentiert die kodierende Region der ProAS(#748)-Peptidase inklusive hypothetisches Signalpeptid. ProAS(#748) repräsentiert die kodierende Region ProAS(#748)-Peptidase ohne hypothetisches Signalpeptid. Blaue Kästen repräsentieren die verwendeten Restriktionsenzyme und ihre Schnittstellen für die Klonierung. In Anlehnung an den Modellversuch in Abschnitt 3.4.1.1 wurde die Expression der
rekombinanten ProAS(#748)-Peptidase durch die in E. coli Top10 transformierten
81
Ergebnisse
Vektoren (Tab. 29) untersucht. Die Parameter Inkubationstemperatur,
Induktorkonzentration und Expressionsstamm wurden wie zuvor variiert. Die
Messung der Peptidase-Aktivität erfolgte mittels Fluoreszenzassay in der
cytoplasmatischen und periplasmatischen Zellfraktion sowie in der zellfreien Kultur.
Es konnte bei der Expressionsanalyse kein aktives ProAS(#748) nachgewiesen
werden. Auch die Anwendung der im Modellversuch optimierten
Expressionsparameter für pBAD-subE erbrachten mit den Vektoren pBAD-
proAS(#748) und pBAD-proAS-ol(#748) in E. coli BL21(DE3) keine messbare
Peptidase-Aktivität.
Der Einsatz weiterer E. coli-Stämme für die Expression, Änderung der
Medienzusammensetzung, Induktion von Chaperonen durch EtOH-Zugabe und
Rückfaltung der möglicherweise inkorrekt gefalteten Proteine ergab ebenfalls kein
aktives Enzym. Eine Zusammenfassung der wichtigsten Expressions-Parameter,
Beobachtungen und angewendeten Methoden gibt Tab. 30 wieder.
Tab. 30: Expressionsversuche mit ProAS(#748) in E. coli. Stichwortartige Zusammenfassung der Versuche und ihrer Ergebnisse. Die Messung der Peptidase-Aktivität erfolgte jeweils in den cytoplasmatischen und periplasmatischen Zellfraktionen und in der zellfreien Kultur.
�Wachstumsstillstand nach Induktion, keine Peptidase-Aktivität
�Rückfaltung von Gesamt-Proteinextrakten nach Parry (2002) �Expressionsanalyse in E. coli-Stämmen: BL21(DE3); C600; DH5�, Epi300; HB101; LMG194; O17; SF8; Top10 und XL1 �Chaperon-Induktion durch 6% EtOH Zusatz ins Medium nach Thomas et al. (1997) �Variation der Medien: LB, M9-Medium, RM und Pepton-Wasser. 5 mM Calcium-Zugabe.
- - - -
Um Hinweise auf das Fehlen einer aktiven Peptidase bei den Expressionsansätzen
zu finden, wurde die Expression mit den Vektoren pBAD-proAS(#748) und pBAD
proAS-ol(#748) in E. coli BL21(DE3) näher untersucht. Die Induktions- und
Inkubationsparameter entsprachen dabei der optimierten Expression von Subtilisin E
durch pBAD-subE (Induktion mit 12 mM L-Arabinose, 23°C).
82
Ergebnisse
3.4.1.2.13.4.1.2.1 Analyse der Expression von ProAS(#748) durch pBAD Analyse der Expression von ProAS(#748) durch pBAD
Eine vollständige Sequenzierung der Klonierungsübergänge und des Inserts von
pBAD-proAS(#748) und pBAD-proAS-ol(#748) erwiesen den korrekten Sitz des
Inserts in Frame zu den Kontrollelementen des Expressionsvektors. Ebenso stimmte
die Sequenz von proAS(#748) mit der Insert-Sequenz des Vektors überein, so dass
eine Mutation im Gen ausgeschlossen werden konnte.
Zur Aufklärung des Fehlens von aktiver Peptidase bei den Expressionsansätzen
wurde die mRNA-Bildung im Northern-Blot überprüft. Eine fehlende Transkription
oder Instabilität der mRNA der Expressionsklone könnte ein Ausbleiben der
entsprechenden Proteine begründen. Der Nachweis der Transkription des proAS-
Gens in mRNA erfolgte mit der Sonde sub748, einem markierten 500 bp Fragment
des Peptidasegens proAS(#748) (siehe 2.7.7). Die Bildung von proAS(#748)-
spezifischer mRNA konnte nach Induktion der Expressionsklone (pBAD-proAS(#748)
und pBAD-proAS-ol(#748)) nachgewiesen werden (Abb. 21). Dabei wies die mRNA
70 min nach Induktion starke Degradationserscheinungen auf, die 30 min nach
Induktion noch nicht so deutlich hervortraten.
Kein Nachweis erfolgte dagegen in uninduzierten Kulturen der genannten Stämme,
ebenso wie in den Kontrollen (pBAD-Calmodulin und pBAD-subE). Vermutlich
aufgrund stringenter Hybridisierungs-Bedingungen und der Sequenzunterschiede
von der mRNA des Subtilisin E zur eingesetzten Sonde konnte bei pBAD-subE kein
Abb. 21: RNA-Präparation und Northern-Blot-Analyse für die proAS(#748)-Expression in E. coliBL21(DE3). A. Denaturierende Gelelektrophorese der RNA-Präparation. B. Northern-Blot mit Sonde sub795. Erntezeitpunkt für die Zellen war 70 min nach Induktion (1-4.), bzw. 30 min (5.). Proben: - = uninduzierte Kulturen; + = induzierte Kulturen (12 mM L-Arabinose); 1. Kontrolle (pBad-Calmodulin); 2. Kontrolle (pBad-subE); 3. pBad-proAS-ol(#748); 4. & 5. pBad- proAS(#748). Die Analyse der extrazellulär und in das Periplasma sekretierten Proteine über SDS-
PAGE zeigte deutliche Unterschiede im Proteinmuster der induzierten zur
uninduzierten Kultur. Auffällig war eine deutliche Proteinbande bei ca. 60 kDa in den
83
Ergebnisse
induzierten Proben. Die ungefähre Größe des Proteins entsprach dem 60,1 kDa
großen unprozessierten rekombinanten Expressionsprodukt des Vektors pBAD-
proAS(#748).
E- E+ P- P+ M
~100
kDa
~130
~70
~55
~40
~35
Abb. 22: SDS-PAGE (12% PA, Coomassie gefärbt) der Expression von pBAD-proAS(#748) in BL21(DE3). Die Proben wurden vor dem Auftragen hitzedenaturiert. E= zellfreie Kultur; P= Periplasmatische Zellfraktion; M= PageRuler Prestained Protein ladder; += Kultur induziert; -= Kultur uninduziert. Der � markiert die überexprimierte Proteinbande. Über eine Affinitätschromatografie (Ni-NTA Spin Kit, QIAGEN) mittels des am
rekombinanten Protein fusionierten Oligohistidins sollte eine Aufreinigung der
rekombinanten Peptidase erfolgen. Es konnte jedoch kein Protein von der Säule
eluiert werden. Möglicherweise verhinderte eine inkorrekte Faltung des Proteins oder
die gewählten Pufferbedingungen eine Bindung der His-tags.
Abschließend lässt sich sagen, dass es klare Hinweise auf eine induzierbare
Transkription des rekombinanten Gens und vage Hinweise auf die Expression von
rekombinantem Protein gibt. Es konnte nicht geklärt werden, warum keine aktive
Peptidase exprimiert wurde.
3.4.23.4.2 Heterologe Expression einer aktiven Peptidase durch den Fosmidklon Heterologe Expression einer aktiven Peptidase durch den FosmidklonF114 F114
Um das Problem der fehlenden Expression aktiver Peptidasen bei den Versuchen mit
Plasmidvektoren zu untersuchen, wurde die heterologe Expression mit den
Fosmidklonen F13 und F114 analysiert. Die Klone hatten unter bestimmten
Induktions- und Inkubations-Bedingungen (Induktion mit 0,5 mM L-Arabinose;
Inkubation bei RT: ~23°C für >4 d) Aktivität im Plattenassay gezeigt (3.3.2). Dabei ist
84
Ergebnisse
zu bedenken, dass es sich bei den Fosmiden nicht um Expressionsvektoren handelt.
Die Fosmide besitzen keine Promotoren und andere Kontrollelemente für eine
Überexpression der Gene auf dem Insert. Die Induktion mit L-Arabinose erhöht die
Kopiezahl des bis zu 40 kb großen Fosmids auf bis zu 50, wobei die Kopiezahl von
der Menge des Induktors abhängt.
Eine Überprüfung, ob die Peptidase-Aktivität von Peptidasegenen resultierte, die auf
dem Fosmid kodieren, erfolgte durch Retransformation. Die isolierten Fosmide
wurden durch Elektroporation in E. coli Epi300 transformiert. Da die
Retransformanten unter den speziellen Induktionsbedingungen (s.o.) eine Peptidase-
Aktivität aufwiesen, kann davon ausgegangen werden, dass die Peptidase-Aktivität
von Genprodukten der Fosmidsequenz der Klone F13 und F114 resultierte. Die
weiteren Untersuchungen wurden am Fosmidklon F114 durchgeführt, da sich der
Klon F13 als instabil erwies.
Die optimale Induktionsmenge und Inkubationstemperatur wurde anhand der täglich
ermittelten Peptidase-Aktivitäten in 20 ml Kulturen bestimmt. Die Expression von
aktiver extrazellulärer Peptidase in Klon F114 zeigte eine Abhängigkeit zur
Inkubationstemperatur und Induktorkonzentration (Tab. 31). Eine Hemmung des
Wachstums konnte schon durch die kleinste zugesetzte Induktormenge ab einer
L-Arabinose-Konzentration von 0,05 mM beobachtet werden. Die höchste
extrazelluläre Peptidase-Aktivität (43,7 FlU/ml) konnte nach Induktion mit 0,5 mM
L-Arabinose und anschließender Inkubation für 5 Tage bei 23°C beobachtet werden.
Bei höheren Temperaturen oder Induktorkonzentrationen konnte keine Peptidase-
Aktivität mehr nachgewiesen werden. Aktive Peptidase konnte nicht im Cytoplasma
und Periplasma des induzierten Klons F114 im Fluoreszenzassay nachgewiesen
werden. Wie es schien, wurde das Enzym in der aktiven Form sekretiert.
Tab. 31: Peptidase-Aktivität (Fluoreszenzassay) in der zellfreien Kultur des Fosmidklons F114 nach 5 d Inkubation bei 23° und 37°C und verschiedenen Induktorkonzentrationen.
*Inkubationstemperatur nach Induktion; -= Peptidase-Aktivität unter der Nachweisgrenze (0,00001 FlU/ml)
85
Ergebnisse
0
0,5
1
1,5
2
2,5
3
3,5
4
0 24 48 72 96 120 144 168 192
Inkubationszeit [h]
OD
600n
m
0
10
20
30
40
50
60
70
80
90
Pept
idas
eakt
ivitä
t [Fl
U/m
l]
Wachstum Kontrolle (0mM ara) Wachstum (0,5 mM ara)Peptidaseaktvität Kontrolle Peptidaseaktvität (0,5 mM ara)
Abb. 23: Bildung extrazellulärer Peptidase1) während des Wachstums des Fosmidklons F114. Vergleich einer uninduzierten (Kontrolle) mit einer induzierten2) Kultur (LB-Medium; bei 23°C, 200 rpm).1)Messung der Peptidase-Aktivität im Fluoreszenzassay in FlU/ml2)Induktion der Kultur bei OD= 0,5 mit 0,5 mM L-Arabinose (ara)
Die Produktion von aktiver Peptidase des Fosmidklons F114 in Abhängigkeit zur
Inkubationszeit wurde anhand einer 300 ml Kultur ermittelt. Die Induktionsperiode
dauerte lang: Klon F114 zeigte erst 110 h nach Induktion mit 0,5 mM L-Arabinose
geringe Peptidase-Aktivität in der zellfreien Kultur (Abb. 23), die dann stetig zunahm
mit einer maximalen Aktivität nach 180 h (81,9 FlU/ml).
Zusammenfassend ist zu sagen, dass mit dem Fosmidklon F114 eine heterologe
Expression einer aktiven Peptidase in E. coli zu beobachten war. Geringe Mengen
an aktivem Enzym konnten nur nach Erhöhung der Fosmidkopiezahl und Inkubation
bei RT nach über 4 Tagen detektiert werden. Im Folgenden wird das rekombinante
Enzym aus Stamm B3/1-2 (#418) Peptidase F114 genannt.
3.4.33.4.3 Übereinstimmung der Peptidase F114 mit Peptidase B aus B3/1-2 (#418) Übereinstimmung der Peptidase F114 mit Peptidase B aus B3/1-2 (#418)
Als Nachweis für den Ursprung der rekombinanten Peptidase F114 aus dem Wildtyp-
Stamm B3/1-2 (#418) sollte ein Vergleich der biochemischen Merkmale und des
Molekulargewichtes des rekombinanten Enzyms mit denen des Wildtyp-Enzyms
durchgeführt werden. Von den beiden Peptidasen A und B aus dem Wildtyp B3/1-2
(#418) wurde vermutet, dass Peptidase B identisch mit F114 ist. Gefolgert wurde
dies aufgrund des übereinstimmenden Molekulargewichtes von etwa 55 kDa der
Peptidase B und der abgeleiteten Aminosäuresequenz von proAS(#418). Des
86
Ergebnisse
Weiteren stimmte die Zuordnung der Peptidase B zu den Serinpeptidasen mit der der
BLAST-Analyse von ProAS(#418) überein. Bei Peptidase A handelt es sich hingegen
um eine ~35 kDa große vermutliche Metallopeptidase.
3.4.3.13.4.3.1 Reinigung der rekombinanten Peptidase F114 Reinigung der rekombinanten Peptidase F114
Für die biochemische Charakterisierung erfolgte zuvor eine Anreicherung und
Reinigung der rekombinanten Peptidase F114 (siehe Tab. 32). Als Methoden zur
Aufreinigung standen die hydrophobe Interaktionschromatografie und die
anschließende Gelfiltration zur Verfügung (siehe 2.6.7.2 und 2.6.7.3). Die zellfreie
Kultur wurde im ersten Schritt über eine Phenylsepharosesäule gereinigt. Ähnlich zu
der Peptidase B des Wildtyps (siehe 3.1.5.1) eluierte die Aktivität von Peptidase
F114 nach Ende des Ammoniumsulfatgradienten (Abb. 24). Ein 30 ml Fraktionspool
aus 15 Fraktionen mit insgesamt 7500 FlU wurde gebildet. 200μl eines 10-fach
Konzentrates (mittels Dialyse gegen PEG3500, siehe 2.6.3) des Fraktionspools
wurden über eine Gelfiltrationschromatografie (Hiload Superdex200, V0=8 ml)
aufgereinigt. Es konnte Peptidase-Aktivität in 5 Fraktionen á 1 ml nachgewiesen
werden. Die Fraktion mit der höchsten Aktivität (206 FlU/ml) wurde als Probe für die
Extinktionsmessung bei 280 nmrelative PeptidaseaktivitätAmmoniumsulfatgradient (100% = 1 M Ammoniumsulfat)
Abb. 24: Phenylsepharose-Chromatografie von zellfreier Kultur des Fosmidklons F114. Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität in einer Fraktion im Vergleich zur maximalen Peptidase-Aktivität im Peak.
87
Ergebnisse
Tab. 32: Anreicherung von Peptidase F114 aus der zellfreien Kultur F1141).Anreicherungs-
methode Probe Volumen[ml]
Gesamt-protein
[mg]
Gesamt-aktivität [kFlU]
Spezifische Aktivität
[kFlU/mg]
Ausbeute [%] an
Aktivität
Enzym-Anreicherungs-
faktor keine zellfreie
Kultur 230 9,27 14,4 1,55 100 1
hydrophobe Interaktions-chromatografie
Fraktions-pool B 30 4,1 7,5 1,82 52,1 1,2
Gelfiltration2)
Fraktion höchster Peptidase-Aktivität
1 0,025 0,206 8,24 1,42) 5,3
1)Inkubationsbedingungen: 160 h; 200 rpm; 23°C nach Induktion 0,5 mM L-Arabinose. 2)Eingesetzte Probenmenge: 0,2 ml eines 10x Konzentrates des Fraktionspools B.
3.4.3.23.4.3.2 Biochemische Eigenschaften der Peptidase F114 im Vergleich zur Biochemische Eigenschaften der Peptidase F114 im Vergleich zurWildtyp-Peptidase B Wildtyp-Peptidase B
Die biochemischen Eigenschaften der Peptidase F114 wurden ermittelt. Hierzu
wurden im Vergleich die Eigenschaften der Peptidase B aus dem Wildtyp B3/1-2
(#418) dargestellt. Als Proben wurden die über Gelfiltration gereinigten Enzyme
(3.4.3.1) mit jeweils 0,5-1 FlU je Ansatz eingesetzt.
Bei den Untersuchungen über den Einfluss der Temperatur auf die Peptidase-
Aktivität wiesen sowohl die rekombinante Peptidase F114 als auch das
Wildtypenzym ein Aktivitätsoptimum bei 50-55°C auf (Abb. 25). Auch im weiteren
Aktivitätsverlauf zeigten die beiden Enzyme eine große Ähnlichkeit. So war eine
Inhibition mit ca. 50% Restaktivität bei 60°C festzustellen, bei 80°C lag die
Abb. 25: Einfluss der Temperatur auf die relative Peptidase-Aktivität* der rekombinanten Peptidase aus Klon F114 und Peptidase B aus Stamm B3/1-2 (#418). *Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
88
Ergebnisse
Untersuchungen zur Abhängigkeit der Enzymaktivität vom pH-Wert ergaben sowohl
beim rekombinant hergestellten als auch beim Wildtypenzym ein Aktivitätsoptimum
mit über 90% Restaktivität zwischen pH 8 und 9,5 in Tris-HCl-Puffer (Abb. 26). Über
10% Aktivität war im Bereich von pH 6-12 messbar.
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13 14pH
rela
tive
Pept
idas
e-A
ktiv
ität
Peptidase aus F114
Peptidase B - B3/1-2 (#418)
Abb. 26: Einfluss des pH-Wertes auf die relative Peptidase-Aktivität* der rekombinanten Peptidase aus Klon F114 und Peptidase B aus Stamm B3/1-2 (#418). *Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
Bei Untersuchungen zur NaCl-Abhängigkeit der Peptidase-Aktivität zeigten die
beiden untersuchten Enzyme ein Optimum bei 1-3 mM NaCl (Abb. 27). Mit
steigender Salzkonzentration wurde die Peptidase-Aktivität beider Enzyme
zunehmend gehemmt.
0%
20%
40%
60%
80%
100%
0 100 200 300 400 500 600NaCl (mM)
rela
tive
Pept
idas
e-Ak
tivitä
t
Peptidase B - B3/1-2 (#418)Peptidase aus F114
0%
20%
40%
60%
80%
100%
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
b.
a. Abb. 27: Einfluss der NaCl-Konzentration auf die relative Peptidase-Aktivität* der rekombinanten Peptidase aus Klon F114 und Peptidase B aus Stamm B3/1-2 (#418). a. Gesamtansicht; b. Teilausschnitt im Bereich 0-30 mM NaCl.*Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
89
Ergebnisse
Eine Steigerung der Aktivität der beiden untersuchten Peptidasen durch Zusatz von
monovalenten und divalenten Kationen konnte nur in geringem Maße (103-120%)
durch Zusatz von 2 mM KCl hervorgerufen werden (Tab. 33). Die Zugabe
verschiedener Metallionen führte zu einer Verringerung der relativen Aktivitäten. Bei
Zusatz von 2 mM MgCl2, CaCl2 oder BaCl2 zeigten die Peptidasen 28-74% relative
Aktivität. Stärkerer Aktivitätsverlust mit 14 bzw. 17% Restaktivität wurde durch 2 mM
CuSO4 erreicht, wobei nach Zugabe von 2 mM MnCl2 oder ZnSO4 keine Aktivität im
Fluoreszenz-Assay nachweisbar war. Mehrfache Wiederholungen bestätigten diese
Ergebnisse.
Der Zusatz von 0,1% SDS resultierte in einer Aktivitätssteigerung mit 188% bzw.
139% relativer Aktivität. Eine Steigerung der SDS-Konzentration auf 1% ließ die
Aktivität auf ca. 20% sinken.
Tab. 33: Abhängigkeit der relativen Peptidase-Aktivität von Peptidase F114 und Wildtyp Peptidase B auf verschiedene Metallionen, SDS und Inhibitoren.
Zusatz Konzentration Relative Aktivität
Peptidase F114
Relative Aktivität Peptidase B –B3/1-2 (#418)
- - 100 % 100 %KCl 2 mM 120% 103%MnCl2 2 mM 0% 0%MgCl2 2 mM 46% 74%CaCl2 2 mM 28% 50%BaCl2 2 mM 59% 77%CuSO4 2 mM 14% 17%ZnSO4 2 mM 0% 0%SDS 0,1 % 188% 139%SDS 1% 21% 22%EDTA 2 mM 0% 27%PMSF 10mM 0% 0%Phenanthrolin 10mM 73% 76% Die beiden untersuchten Peptidasen zeigten in Gegenwart von Peptidase-Inhibitoren
(Tab. 33) eine hohe Toleranz gegenüber Phenanthrolin (73-76% relative Aktivität),
während PMSF und EDTA ihre Aktivität stark bis vollständig hemmte (0% bzw. 27%
relative Aktivität).
Peptidase F114 zeigte eine hohe Empfindlichkeit gegen die chaotropen Salze
GuSCN und GuHCl (siehe Tab. 34). Waren bei 0,1 M GuHCl noch 12% Restaktivität
vorhanden, konnte bei >0,5 M GuHCl und bei allen getesteten GuSCN-
Konzentrationen (0,1; 0,2; 0,6; 0,8 M) keine Aktivität nachgewiesen werden. Das
Wildtypenzym zeigte im Vergleich dazu mit Restaktivitäten von 52% bei 1 M GuHCl
90
Ergebnisse
und 33% bei 0,6 M GuSCN Toleranz gegenüber diesen Substanzen. Allerdings
konnte bei Versuchen mit Konzentrationen � 0,8 M GuSCN und � 2 M GuHCl keine
Enzymaktivität nachgewiesen werden.
Tab. 34: Einfluss von chaotropen Salzen auf die Peptidase-Aktivität der Peptidase F114 und Wildtyp Peptidase B. Die Peptidase-Aktivität einer Probe ohne den Zusatz von chaotropen Salzen wurde gleich 100% gesetzt.
Zusatz Relative Aktivität
Peptidase F114
Relative Aktivität Peptidase B –B3/1-2 (#418)
0,1 M GuHCl 12 % 101 %0,5 M GuHCl 4 % 75 %1 M GuHCl 0 % 52 %2 M GuHCl 0 % 0 %0,2 M GuSCN 0 % 76 %0,6 M GuSCN 0 % 33 %0,8 M GuSCN 0 % 0 %
3.4.3.33.4.3.3 Bestimmung des Molekulargewichtes von Peptidase F114 Bestimmung des Molekulargewichtes von Peptidase F114
Eine Bestimmung des Molekulargewichtes der rekombinanten Peptidase F114 und
der Peptidase B aus dem Wildtyp erfolgte sowohl über PAGE-Analysen als auch
über eine Gelfiltration.
Eine Auftrennung der Peptidase F114 und Peptidase B in einer SDS-PAGE zeigte
einen übereinstimmenden Aktivitätsbereich der nicht hitzebehandelten Proben
(Phenylsepharosefraktionspool) in der Größe von etwa 55 kDa (Abb. 28). Eine
Zuordnung der Aktivitätsbande zu den zahlreichen Proteinbanden im silbergefärbten
Gel konnte leider nicht erfolgen.
Eine Auftrennung der Aufreinigungen der rekombinanten Peptidase F114 und der
Wildtyp Peptidase B aus B3/1-2 (#418) mittels nativer PAGE (Abb. 29) zeigte eine
Vielzahl von Proteinbanden in dem Phenylsepharosefraktionspool des Klons F114,
wobei der Fraktionspool B des Wildtyps nur wenige Banden aufwies. Jeweils eine
sehr schwache Bande in der Gelfiltrationsfraktion verwies auf den hohen
Reinigungsgrad des Enzyms. Der Aktivitätsnachweis zeigte Peptidase-Aktivität bei
allen Proben in der Ebene der schwachen Bande aus der Gelfiltration. Eine
Bestimmung des Molekulargewichtes konnte nicht erfolgen, da der Proteinstandard
trotz Eiskühlung des Gels durch proteolytische Aktivität zerstört wurde.
91
Ergebnisse
M 1 2 3 4
Abb. 28: SDS-PAGE (10% PA), Konzentrate der B-Fraktionspools aus der Phenylsepharose-Chromatografie a.) silbergefärbt; b.) Aktivitätsnachweis mit Skimmilk-Kontakt-Zymogramm. 1. + 2.: F114; 3 +4.: B3/1-2 (#418). Proben 1+3 wurden vor dem Auftragen hitzedenaturiert (85°C, 5 min). Die proteolytischen Aktivitätsbereiche aus dem Zymogramm (B.) wurden als graue Umrisse in A. eingezeichnet.
Ebenfalls Aktivitätszonen auf gleicher Höhe ergab das Kontakt-Zymogramm einer
Isoelektrischen Fokussierung (IEF-Gel, pH 3-10 in X-Cell SureLock Kammer,
Invitrogen) der rekombinanten und der Wildtyp-Peptidase (Daten nicht gezeigt).
Eine Abschätzung des Molekulargewichts von Peptidase F114 durch PAGE-Analyse
erbrachte einen Wert von ~55 kDa. Weitere Hinweise über das Molekulargewicht der
~100
~70
~55
~40
~35
~130
1 2 3 4 M
a. b.
Abb. 29: Native PAGE, (10% Tris-Glycin) der Aufreinigungsstufen der Peptidase aus F114 (1. Phenylsepharosekonzentrat; 2. Gelfiltration) und Peptidase B von Stamm B3/1-2 (#418) (3. Phenylsepharosekonzentrat; 4. Gelfiltration). a.) Silberfärbung; b.) Aktivfärbung im Kontakt-Zymogramm. Schwarze Kästen zeigen die Lage der Ausgangspunkte der sich entwickelten Peptidase-Aktivität (helle Bereiche); Pfeile verdeutlichen die Lage der Peptidase-Bande im Gel.
1 2 3 4 1 2 3 4
a. b.
92
Ergebnisse
Peptidase erfolgten über eine Gelchromatografie mit einer Hiload Superdex200-
Säule. Die Elutionsfraktionen wurden mittels Fluoreszenzassay auf ihre Peptidase-
Aktivität geprüft. Eine Kalibrationskurve (Abb. 30) mit fünf Proteinstandards sollte die
Ermittlung des Molekulargewichtes der Peptidase ermöglichen.
y = 979,45e-5,9916x
R2 = 0,99151
10
100
1000
0,2 0,3 0,4 0,5 0,6 0,7Kav
log
Mol
ekul
arge
wic
ht [k
Da]
Catalase (232 kDa) Aldolase (158 kDa)
Albumin (67 kDa)
Chymotripsinogen (25 kDa)
RNase A (13,7 kDa)
Abb. 30: Kalibrationskurve für Hiload Superdex200-Säule (1,6x60cm). Formel der Kalibrationskurve: MW = 979,45e-5,9916Kav (R2 = 0,9915). Kav=(Ve-Vo)/(Vt-Vo); Ve= Elutionsvolumen des jeweiligen Proteins; Vo= Totvolumen der Säule = Elutionsvolumen von Blue-Dextran (MW 2.000 kDa), Vt= Geometrisches Volumen der Säule. Ein 10-fach Konzentrat (200μl; mittels Dialyse gegen PEG3500, 500 FlU
Gesamtaktivität) des aktiven Phenylsepharosefraktionspools (siehe 3.4.3.1,
Gesamtaktivität 500 FlU) wurde auf die Säule aufgetragen. In 27 Elutionsfraktionen
konnten 63% der aufgetragenen Peptidase-Aktivität nachgewiesen werden (Abb. 31).
Die maximale Aktivität (>90% relative Aktivität) verteilte sich dabei breit über 8
Fraktionen. Dieser maximale Aktivitätsbereich korrelierte in etwa mit einem Peak der
Extinktionsmessung bei 280 nm (Abb. 31).
Entsprechende Elutionsprofile konnten bei zweifacher Wiederholung der Gelfiltration
Retentionsvolumen [ml] Abb. 31: Ausschnitt der Gelchromatografie (Hiload Superdex 200) der rekombinanten Peptidase aus Klon F114. Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität in einer Fraktion im Vergleich zur maximalen Peptidase-Aktivität im Peak. Für die Bestimmung des Kav-Wertes wurde das Aktivitäts-Maximum auf die Mitte des
maximalen Aktivitätsbereiches (>90% rel. Aktivität) festgelegt und ergab einen Kav-
Wert von 0,503 (Stabw: 0,07). Dies entspricht im Vergleich mit der Kalibrationskurve
(Abb. 30) einem ungefähren Molekulargewicht der rekombinanten Peptidase
ProAS(#418) von 48 kDa (nativ). Die Ermittlung des Molekulargewichtes des Wildtyp-
Enzyms (Peptidase B, Stamm B3/1-2 (#418)) durch Gelfiltration ergab mit ~50 kDa
(Kav= 0,495) eine vergleichbare molekulare Größe.
Das Molekulargewicht der Peptidase konnte mit den zur Verfügung stehenden
Methoden (SDS-PAGE und Gelfiltration) somit nur ungefähr ermittelt werden und
liegt bei ~50 kDa.
Zusammenfassend lässt die weitgehende Übereinstimmung der biochemischen
Eigenschaften und die identische Größe, ermittelt in verschiedenen PAGE-
Ausführungen und der Gelfiltration, den Schluss zu, dass die rekombinante
Peptidase F114 zu der Peptidase B aus Stamm B3/1-2 (#418) identisch ist. Weiterhin
liegt auch eine Übereinstimmung des Molekulargewichtes und der Zugehörigkeit zu
den Serinpeptidasen mit den Sequenzdaten von proAS(#418) vor. Dies lässt darüber
hinaus den Schluss zu, dass Peptidase B identisch mit ProAS(#418) ist.
94
Diskussion
44 Diskussion DiskussionExtrazelluläre Peptidasen aus halotoleranten und halophilen Bakterien sind im
Gegensatz zu den Peptidasen aus anderen extremophilen Bakterien, wie Alkaliphilen
und Thermophilen, erst wenig untersucht. Aufgrund ihrer Eigenschaft unter extremen
Umweltbedingungen noch Aktivität zu zeigen, sind halophile Enzyme von hervor-
ragendem biotechnologischen Interesse (Gomes & Steiner, 2004; van den Burg,
2003; Ventosa et al., 1998). So konnte gezeigt werden, dass extrem halophile
Enzyme nicht nur im hypersalinen Milieu, sondern auch unter Einfluss von
organischen Lösungsmitteln katalytisch aktiv sind (Marhuenda-Egea & Bonete,
2002). Beschränkte sich das Wissen über halophile Peptidasen bisher nahezu auf
wenige extrem halophile Peptidasen aus Archaeen und einige wenige biotech-
nologische Anwendungen im hypersalinen Milieu (Izotova et al., 1983; Ryu et al.,
1994), ist ein Anstieg des Interesses in den letzten 2-3 Jahren für Peptidasen aus
halophilen Bacteria zu verzeichnen (siehe Tab. 2 in der Einleitung). Die Berichte über
die Isolierung und biochemische Charakterisierung von extrazellulären Peptidasen
stehen zum Teil im Zusammenhang mit möglichen biotechnologischen
Anwendungen unter harschen Prozessbedingungen (Hiraga et al., 2005; Karbalaei-
Heidari et al., 2006; Namwong et al., 2006; Sánchez-Porro et al., 2003b; Setyorini et
al., 2006). So ist beispielsweise der Einsatz von halophilen Peptidasen aus einem
Filobacillus- und einem Halobacillus-Stamm für die beschleunigte Fermentation von
stark salzhaltigen asiatischen Fischsoßen sehr viel versprechend (Hiraga et al.,
2005; Namwong et al., 2006). Zudem sind einige halotolerante und moderat
halophile Peptidasen durch ihre breite Halotoleranz möglicherweise für die Protein-
hydrolyse in marinen bis stark salzhaltigen Flüssigkeiten oder Abwässern geeignet
(Karbalaei-Heidari et al., 2006; Lama et al., 2005; Sánchez-Porro et al., 2003b;
Setyorini et al., 2006).
Im Hinblick auf ihre außergewöhnlichen Eigenschaften standen halophile Enzyme im
Fokus dieser Arbeit. So sollten Peptidasen gefunden und charakterisiert werden, die
eine hohe Toleranz gegenüber chaotropen Bedingungen zeigen. Diese Eigenschaft
wurde hinsichtlich des Einsatzes der Enzyme in Kits für die Aufreinigung von
Nukleinsäuren gewählt. Die Peptidasen sollen hierbei die Zellen und das Gewebe
enzymatisch aufschließen und damit die darin enthaltenen Nukleinsäuren für das
weitere Extraktionsverfahren freisetzen. Die hierfür eingesetzten Lysepuffer besitzen
methodenbedingt einen alkalischen pH-Wert und hohe Konzentrationen an hoch-
95
Diskussion
chaotropen Guanidin-Salzen und Detergenzien, wie SDS. Untersuchungen, inwiefern
halophile Peptidasen diese Bedingungen im Lysepuffer tolerieren, lagen zum
Zeitpunkt der Untersuchung noch nicht vor.
Als Grundlage für das Screening nach neuen Peptidasen diente eine Stamm-
sammlung mit halotoleranten und halophilen Bakterien aus Salinen. Die Isolate
stammten aus Wasser- und Sedimentproben zweier Salinen und eines küstennahen
Sees auf Lanzarote (Kanarische Inseln, Spanien) sowie einer Saline bei La Baule
(Pays de la Loire, Frankreich) (Sinn-Meyer, 2003). Anhand von Plattenassays
konnten 355 Isolate mit extrazellulärer Peptidase-Aktivität unter halophilen
Bedingungen (12-20% NaCl) identifiziert werden. Eine taxonomische Einordnung der
Peptidase-Produzenten erfolgte bisher an 126 Stämmen aus Lanzarote, auf Basis
einer amplified ribosomal DNA-restiction analysis (ARDRA) Untersuchung (Sinn-
Meyer, 2003; Vogt, 2000). Diese zeigte, dass jeweils über 1/3 der Stämme entweder
den Pseudomonaceae (37%) oder Vibrionaceae (36%) angehören, während
geringere Anteile auf die Bacillaceae (10%), Halomonadaceae (9%), Archaea (6%)
und Alteromonadaceae (2%) entfielen.
Die Anwendung von Standardtechniken zur Gewinnung und Charakterisierung von
halophilen Enzymen kann sich als problematisch erweisen. So stellt die Proben-
konzentration und Aufreinigung von halophilen Enzymen wegen ihrer strukturellen
Besonderheiten und ihrer Salzabhängigkeit eine Herausforderung dar (Gupta et al.,
2005; Ventosa et al., 1998). Methoden zur Enzymfällung durch Aussalzen sind
zumeist aufgrund der Löslichkeit von halophilen Enzymen in hochkonzentrierten
Salzlösungen, wie gesättigtem Ammoniumsulfat, (Kirst, 1993) nicht anwendbar.
Darüber hinaus ist die Verwendung vieler chromatografischer Verfahren wegen der
Salzabhängigkeit der Enzyme ausgeschlossen (Madern et al., 2000). Durch
kristallografische Untersuchungen konnte gezeigt werden, dass ein Überschuss an
negativer Ladung auf der Enzymoberfläche typisch für extrem halophile Enzyme ist
(Frolow et al., 1996; Jolley et al., 1997). Die negative Ladung ermöglicht dabei die
Ausbildung einer sog. Solvatisierungs-Hülle durch Bindung hydratisierter Ionen.
Diese Hülle reduziert die Hydrophobizität der Enzymoberfläche und verhindert damit
die Aggregation der Enzyme bei hohen Salzkonzentrationen (Costenaro et al., 2002;
Ebel et al., 2002). Bei Entfernung des Salzes kommt es hingegen zu einer Entfaltung
der extrem halophilen Enzyme durch die gegenseitige Abstoßung der negativen
Ladungen auf der Enzymoberfläche und zum Teil zu einer irreversiblen Inaktivierung
96
Diskussion
(Eisenberg, 1992). Welche Mechanismen zur Halotoleranz bei moderat halophilen
und halotoleranten Enzymen auftreten und inwiefern diese Einfluss auf die
Anwendung von Standardtechniken nehmen, wurde bislang noch nicht untersucht.
Ähnlichkeiten zu den oben genannten Schwierigkeiten zeigten sich in der
vorliegenden Studie bei der Anwendungen von Standardtechniken (siehe 3.1.5). Eine
Ammoniumsulfat-Fällung der hier untersuchten Peptidasen sowie ihre Aufreinigung
über eine Anionenaustauschchromatografie erbrachten sehr geringe Ausbeuten. Nur
die Entwicklung von modifizierten Verfahren zur Aufkonzentrierung sowie Trennung
und Anreicherung der Peptidasen aus den zellfreien Kulturen erlaubte eine
Durchführung dieser Studie (siehe 3.1.5). So eignete sich besonders eine Dialyse
gegen PEG3500 für eine Aufkonzentrierung der Proben, sowie eine hydrophobe
Interaktionschromatografie mit Phenylsepharose über einen Ammoniumsulfat-
gradienten für die Trennung und partielle Anreicherung. Ebenso erforderten die
verwendeten Aktivitätsassays eine Evaluation und Anpassung zur Verwendung unter
den angestrebten Bedingungen der Zielsetzung (siehe Abschnitt 3.1.4).
4.11 Zugehörigkeit der Peptidase produzierenden moderat halophilen Zugehörigkeit der Peptidase produzierenden moderat halophile4. nStämme zur Gattung SalinivibrioStämme zur Gattung Salinivibrio
Zu den für diese Studie zur Verfügung stehenden 355 Peptidase-Produzenten lagen
weder Daten über die Sekretionscharakteristika der Stämme noch über die bio-
chemischen Eigenschaften der extrazellulären Peptidasen vor. Eine Vorauswahl und
anschließende Vorcharakterisierung sollte die Anzahl der Isolate auf besonders
geeignete Peptidase-Produzenten reduzieren. Die Auswahlkriterien erfolgten dabei in
Bezug auf die Zielsetzung und eine erleichterte Probengewinnung: Ein schnelles
Wachstum (mit ~108Zellen/<5d) und starke Peptidase-Aktivität sollten eine zügige
und leichte Probengewinnung ermöglichen. Zudem wurde besondere Aufmerk-
samkeit auf Bakterien gelegt, die im haloalkaliphilen Bereich (NaCl: 10-20%; pH-
Wert: 8-9,5) isoliert wurden. Extrazelluläre Enzyme aus diesen Organismen besitzen
möglicherweise eine hohe Toleranz gegenüber denaturierenden Bedingungen, wie
einer hohen Salzkonzentration und alkalischem pH-Wert. Gleichfalls sollte die
Zugehörigkeit der ausgewählten Stämme zu verschiedenen taxonomischen Gruppen
(auf Basis einer ARDRA-Studie) eine hohe Diversität innerhalb der Auswahl
gewährleisten.
Eine Auswahl von 27 Stämmen entsprach diesen Kriterien und wurde bezüglich ihrer
Sekretionscharakteristika von Peptidasen in Flüssigkultur untersucht. Somit konnte
97
Diskussion
für die spätere Probennahme der Zeitpunkt der höchsten extrazellulären Peptidase-
Aktivität bestimmt werden. Im Gegensatz zur Kultur auf festen Medien erleichtert die
Flüssigkultur die Probennahme und eine exakte Quantifizierung des Zellwachstums
und der extrazellulären Peptidaseproduktion. Diese Kultivierungsmethode wurde
auch im Hinblick auf eine spätere Gewinnung der Enzyme im Produktionsmaßstab
gewählt. So stellt die Flüssigkultur in Fermentern und anschließende Aufreinigung
der extrazellulären Enzyme aus der zellfreien Kultur eine kostengünstige und weit
verbreitete Produktionsmethode dar (Demain & Davies, 1999; Gupta et al., 2002a).
Diese gewählten Selektionsbedingungen führten zu einer weiteren Einengung der
Auswahl, da ~30% der Stämme in Flüssigkultur nur sehr geringe oder keine
messbare Peptidasesekretion zeigten. Die Vorcharakterisierung der verbliebenen 19
Stämme erfolgte an den zellfreien Kulturen mit einem in vitro Test der Peptidasen in
einem Lysepuffer (1 M GuSCN; 0,1% SDS; 5 mM CaCl2; 50 mM Tris-HCl, pH 8), der
für eine RNA-Isolierung geeignet erschien.
9 Stämme mit extrazellulären proteolytischen Enzymen zeigten letztendlich
geeignete Eigenschaften bezüglich der Zielsetzung. Aufgrund eines 16S rRNA-Teil-
sequenzvergleichs konnten alle 9 Stämme in die Gattung Salinivibrio (Familie:
Vibrionaceae) eingeordnet werden. Des Weiteren lässt sich anhand der Sequenz-
vergleiche die mögliche Zugehörigkeit der 9 untersuchten Stämme zu min. 2-3
verschiedenen Salinivibrio-Spezies festlegen (vergleiche Abschnitt 3.1.2). So können
3 Stämme (B8/26-2 (#504), B12/10-1 (#748) und B12/16 (#795)) der Spezies
Salinivibrio costicola zugeordnet werden (mit 98,1-100% Sequenzidentität zur Sub-
spezies S. costicola alcaliphilus). Alle Isolate dieser Zuordnung stammen überein-
stimmend aus La Baule (Frankreich). S. costicola wurde zuerst von gesalzenen
Lebensmitteln (Australischer Schinken) isoliert (Garcia et al., 1987), weitere Fundorte
sind Salinen auf dem Spanischen Festland und den Kanarischen Inseln (Ventosa et
al., 1998) sowie ein hypersaliner Teich in Death Valley (Kalifornien, USA) (Huang et
al., 2000). Die hier nächst verwandte Subspezies S. costicola alcaliphilus wurde aus
einer Salzquelle in der Campania-Region (Italien) isoliert (Romano et al., 2005).
Soweit bekannt, handelt es sich um moderat halophile Bakterien mit einem
Wachstumsoptimum bei 10-12% NaCl. Es sind sowohl Stämme von S. costicola mit
als auch ohne proteolytische Aktivität bekannt (Lama et al., 2005). 4 weitere Stämme
(SJ2/3-1 (#073), SJ5Ü/8 (#098), SJ5Ü/12 (#140) und SJ6S/14 (#148)), die alle den
Salinas de Janubio (Kanarische Inseln, Spanien) entstammen, gehören vermutlich
98
Diskussion
der Art Salinivibrio aegyptiaca (Romano et al., 2006) an (mit �98,5% Sequenz-
identität zur Subspezies S. aegyptiaca sharmensis). Physiologische Daten über diese
Spezies fehlen, da sie bislang nur anhand ihrer 16S rRNA-Sequenz klassifiziert
wurde (NCBI-TTaxonomy ID: 390883). Keine Aussagen konnte aufgrund des nur
unvollständig sequenzierten Genabschnittes über die Artzugehörigkeit von Stamm
B3/14 (#430) gemacht werden. Dagegen ist es möglich, dass der Stamm B3/1-2
(#418) einer neuen Salinivibrio-Art angehört, mit S. costicola (97,1% Sequenz-
identität) und S. aegyptiaca (97,6% Sequenzidentität) als nächst verwandte Arten.
Hinsichtlich der untersuchten physiologischen Eigenschaften waren die hier
behandelten Stämme sich und bekannten Salinivibrio-Stämmen sehr ähnlich. Mit
einem Wachstum bei 2-20% NaCl (Optimum bei 6-10% NaCl) und pH 8 gehören sie
ebenfalls zu den moderat halophilen Bakterien. Im Gegensatz zur Peptidase-
Produktion von Salinivibrio sp. AF-2004, bei dem extrazelluläre Peptidase erst in der
stationären Phase nachzuweisen war (Karbalaei-Heidari et al., 2006), sekretierten
die hier untersuchten Salinivibrio-Stämme schon in den frühen Wachstumsphasen
Peptidasen, was auf eine konstitutive Peptidase-Expression schließen lässt.
4.24.2 Salinivibrio bildet zwei Typen von extrazellulären Peptidasen bildet zwei Typen von extrazellulären PeptidasenSalinivibrio
Salinivibrio ist als Produzent extrazellulärer Peptidasen bekannt (Sánchez-Porro et
al., 2003a). Bislang wurden zwei Salinivibrio-Stämme untersucht, bei denen jeweils
nur eine Peptidase identifiziert und biochemisch näher charakterisiert wurde
(Amoozegar et al., 2006; Karbalaei-Heidari et al., 2006; Lama et al., 2005). Bei vielen
mesophilen und einigen extremophilen Bakterienarten wurde die Sekretion von
mehreren Peptidasen beschrieben, wobei pro Bakterium 2 bis 12 verschiedene
Peptidasen nachgewiesen werden konnten (u.a. Deane et al., 1986; Nieto & Ellis,
1991; Saeki et al., 2000; Schmidt et al., 1995). So identifizierten beispielsweise
Schmidt et al. (1995) bei Bacillus sp. LG12 vier Subtilisin-ähnliche Peptidasen,
während Deane et al. (1986) bei Vibrio alginolyticus sieben extrazelluläre Peptidasen
verschiedener Familienzugehörigkeit nachweisen konnten. Dass verschiedene
Stämme einer Spezies eine unterschiedliche Anzahl von Peptidasen sekretieren
können, zeigten Nieto & Ellis (1991) für Aeromonas hydrophila. So konnten sie
abhängig von den untersuchten Aeromonas-Stämmen zwischen 1 und 12 extra-
zelluläre Peptidasen nachweisen. Allerdings liegen bislang keine Berichte über
mehrere sekretierte Peptidasen aus Salinivibrio vor. Neu ist insofern der Befund in
99
Diskussion
dieser Arbeit, dass bei 7 der 9 untersuchten Stämme zwei extrazelluläre Peptidasen
auftraten. Mittels Trennung in der hydrophoben Interaktionschromatografie konnten
eine Peptidase A und eine Peptidase B in der zellfreien Kultur nachgewiesen werden
(Abschnitt 3.1.5.1, Abb. 7; Tab. 36 im Anhang). Bei den verbleibenden zwei
Stämmen (B8/26-2 (#504) und B12/16 (#795)) wurde nur eine Peptidase gefunden,
und zwar vom B-Typ. Die chromatografische Unterscheidung der hier untersuchten
beiden Peptidasen in zwei verschiedene Typen konnte anhand ihrer biochemischen
Eigenschaften bestätigt werden (siehe Abschnitt 3.2).
4.2.14.2.1 Typ A: Metallopeptidase Typ A: Metallopeptidase
Das Verhalten gegenüber verschiedenen Peptidase-Inhibitoren ermöglichte eine
Zuordnung der Peptidase A aller 7 Salinivibrio-Stämme, die dieses Enzym
sekretierten, zu den Metallopeptidasen. Diese Zuordnung basiert vor allem auf der
kompletten Inhibition der Peptidase A durch Phenanthrolin (siehe Abschnitt 3.2.6).
Dieser spezifische Inhibitor inaktiviert Metallopeptidasen, indem durch eine
Komplexierungsreaktion das katalytisch wirksame Zinkion aus dem aktiven Zentrum
entfernt wird (Salvesen & Nagase, 2001). Weiterhin spricht die inhibierende Wirkung
von EDTA dafür, dass es sich bei Peptidase A um eine Metallopeptidase handelt.
EDTA ist ein unspezifischer Chelator für divalente Kationen, der die Inhibition von
Metallopeptidasen bewirkt (Auld, 1988). Konträr dazu steht allerdings eine Inhibition
(bis zu 69%) der Peptidase A durch den spezifischen Inhibitor für Serinpeptidasen
PMSF. Dieser Inhibitor bewirkt die irreversible Hemmung von Serinpeptidasen durch
eine kovalente Derivatisierung des Serinrestes im aktiven Zentrum. Eine
Verunreinigung der Enzymproben durch eine Serinpeptidase scheint daher
wahrscheinlich. Zumal es sich bei der zweiten von den Stämmen sekretierten
Peptidase um eine Serinpeptidase handelt (Peptidase B, siehe Abschnitt 4.2.2). Die
unscharfe Trennung der Peptidase A und B durch die hydrophobe Interaktions-
chromatografie (siehe Abb. 7) könnte daher zu einer Verschleppung der Peptidase B
in den verwendeten Fraktionspool der Peptidase A geführt haben. Gegen diese
Hypothese spricht allerdings, dass diese Verunreinigung weder im Kontakt-
Zymogramm der Peptidase A als Aktivitätsbande detektierbar ist (siehe Abb. 13)
noch die komplette Inhibition der Peptidase A durch Phenanthrolin stört. Die
Inhibition einer Metallopeptidase durch PMSF wurde bislang nur am gereinigten
Enzym aus Pseudoalteromonas ruthenica beschrieben (83% Inhibition bei 10 mM
100
Diskussion
PMSF; (Sánchez-Porro et al., 2003b)). Der Inhibitions-Mechanismus hierfür ist
unbekannt, da keine reaktiven Serinreste in der Aminosäuresequenz der Metallo-
peptidase gefunden wurden (Sánchez-Porro Álvarez, 2005). Für eine zukünftige
Aufklärung, ob es sich bei der hier festgestellten Inhibition der Peptidase A durch
PMSF um ein Artefakt oder einen bislang unbekannten Hemmmechanismus handelt,
wird eine strukturelle Aufklärung der Peptidase vorgeschlagen.
Mit der über SDS-PAGE ermittelten ungefähren Größe von 35 kDa entspricht die
Peptidase A der Größe von anderen Metallopeptidasen, u.a. charakterisiert in
Salinivibrio sp. AF2004 (Mr: 38-43 kDa; Karbalaei-Heidari et al., 2006) und
Pseudoalteromonas ruthenica (Mr: 38 kDa; Sánchez-Porro et al., 2003b).
Gemeinsamkeiten der Peptidase A zu den Metallopeptidasen von Salinivibrio und
Pseudoalteromonas bestehen ebenfalls bei vielen biochemischen Eigenschaften
(vergl. hierzu: Amoozegar et al., 2006; Karbalaei-Heidari et al., 2006; Sánchez-Porro
et al., 2003b). So handelt es sich bei diesen Enzymen um moderat alkaliphile
Peptidasen (pH-Optimum bei 8,5) mit moderat thermoaktivem Charakter. Das
Aktivitätsoptimum der hier untersuchten Peptidase A aus den Salinivibrio-Stämmen
lag mit 40-50°C unterhalb des Temperaturoptimums der Metallopeptidase aus einem
anderen Salinivibrio-Stamm (55°C; (Karbalaei-Heidari et al., 2006)) und aus
Pseudoalteromonas ruthenica (65°C; (Sánchez-Porro et al., 2003b)). Eine
Abhängigkeit von divalenten Kationen konnte bei der Peptidase A nicht festgestellt
werden. Lediglich CaCl2 und MgCl2 zeigten eine schwach aktivierende Wirkung auf
die Peptidase A bei einer Mehrzahl der untersuchten Salinivibrio-Stämme (siehe Tab.
21). Eine ähnlich schwache Aktivierung durch 5 mM CaCl2 (109% relative Aktivität)
und 5 mM MgCl2 (124% relative Aktivität) konnte für die Metallopeptidase aus
Salinivibrio sp. AF-2004 festgestellt werden (Karbalaei-Heidari et al., 2006),
wohingegen verschiedene divalente Kationen auf die Metallopeptidase aus
Pseudoalteromonas ruthenica einen inhibitorischen Effekt hatten (Sánchez-Porro et
al., 2003a).
Hinsichtlich der NaCl-Toleranz erwies sich die Peptidase A als salzempfindlich mit
einem Aktivitätsoptimum bei 10-200 mM NaCl, abhängig vom getesteten Stamm und
einer Restaktivität von < 20% bei 2 M NaCl. Erstaunlich ist hierbei der Ursprung der
Peptidase aus moderat halophilen Stämmen (Wachstum bei 2-20% (0,34-3,4 M)
NaCl), die erst bei erhöhtem Salzgehalt im Medium (�6% (1,02 M) NaCl, Optimum
bei 10-12% (1,71-2,05 M) NaCl) extrazelluläre Peptidasen produzierten. Ähnliche
101
Diskussion
Beobachtungen liegen auch für eine extrazelluläre Peptidase aus einem halo-
alkaliphilen Bacillus-Stamm vor (Gupta et al., 2005). Möglicherweise sind diese
Charakteristika ein Hinweis auf die Anpassung an die stark schwankenden
Salinitäten der Habitate (Salinen). Die Metallopeptidase aus Salinivibrio sp. AF2004
wies ebenfalls eine gewisse Salzempfindlichkeit auf, jedoch mit einer höheren
Toleranz gegenüber hohen NaCl-Konzentrationen (Karbalaei-Heidari et al., 2006).
Das Aktivitätsoptimum dieses gereinigten Enzyms lag im Bereich von 0-0,5 M NaCl
mit einem anschließenden Aktivitäts-Abfall der Aktivität auf unter 20% bei Steigerung
der Salinität auf 4 M. Ob neben der Übereinstimmung der Größe und einiger
biochemischer Eigenschaften der Peptidase A aus den hier untersuchten Salinivibrio-
Stämmen mit der Metallopeptidase aus Salinivibrio sp. AF-2004 auch eine Sequenz-
homologie vorliegt, könnte eine zukünftige Identifizierung ihrer DNA- oder Amino-
säuresequenzen erbringen.
Die taxonomische Einordnung der Stämme (B8/26-2 (#504) und B12/16 (#795)), bei
denen keine Sekretion von Peptidase A festgestellt werden konnte, legt die
Vermutung nahe, dass die Abwesenheit der Peptidase A stamm- aber nicht
artspezifisch ist. So wurde beim Stamm B12/10-1 (#748), der gemeinsam mit den
oben genannten Stämmen einer Art (S. costicola) zugeordnet werden konnte, die
Sekretion der Peptidase A nachgewiesen.
4.2.24.2.2 Typ B: Serinpeptidase Typ B: Serinpeptidase
Abb. 32: Gegenüberstellung der phylogenetischen Stammbäume auf Basis der 16S-rDNA- und proAS-Teilsequenzen. Die Länge der Verzweigungen repräsentiert die Divergenz zwischen zwei Sequenzen, die Zahlen bezeichnen die Anzahl der Substitutionsereignisse.
Folgt man einer weiteren Untergliederung der Subtilisin-ähnlichen Peptidasen von
Siezen & Leunissen (1997), kann die ProAS-Peptidase aufgrund homologer
Sequenzabschnitte der katalytischen Domänen der Proteinase K-Familie zugeordnet
werden (Abb. 40 im Anhang, Bereiche im Alignment gelb markiert). Diese Familie
beinhaltet, neben vielen Subtilisin-ähnlichen Peptidasen aus Pilzen, einige
Peptidasen aus gram-negativen Bakterien, so auch das Aqualysin I aus Thermus
aquaticus und ProA aus Vibrio alginolyticus. Von einigen Mitgliedern dieser Familie
ist bekannt, dass sie als Präproenzyme synthetisiert werden, wobei beim Aqualysin I
neben dem N-terminalen auch ein C-terminales Propeptid vorhanden ist (Larsen et
al., 2006). Während das Präpeptid als Signalpeptid nach dem Transport ins
Periplasma abgespalten wird, fungiert das N-terminale Propeptid möglicherweise als
Chaperon für die Faltung der Peptidase zum aktiven Enzym (Lee et al., 1992). Es
gibt viele Hinweise, dass die Abspaltung des Propeptids und Faltung zum aktiven
Enzym autoproteolytisch erfolgt. Das C-terminale Propeptid und seine Abspaltung
wird dagegen mit dem Transport des Aqualysins über die äußere Membran von
Thermus aquaticus in Zusammenhang gebracht (Lee et al., 1994; Lee et al., 1996).
Allerdings ist auch zumindest eine Subtilisin-ähnliche Peptidase charakterisiert
106
Diskussion
worden, bei der keine Abspaltung von Prosequenzen erfolgt (Kannan et al., 2001).
Im Alignment mit der Proteinase K und Aqualysin I konnten für die ProAS-Peptidase
keine Hinweise auf homologe Propeptid-Sequenzen gefunden werden (siehe Abb. 40
im Anhang). Während die vermutliche katalytische Peptidase-Domäne von ProAS ca.
60% Homologie zu Aqualysin I und Proteinase K aufweist, besteht im Bereich der
Prosequenzen nur etwa 20% Übereinstimmung. Eine Abspaltung größerer
Propeptide von 10-20 kDa, wie es von den zuvor genannten Peptidasen bekannt ist,
ist anhand des übereinstimmenden Molekulargewichts der sekretierten Peptidase B
(~50 kDa) im Vergleich mit ihrer von der Sequenz errechneten Größe (55 kDa,
abzüglich 2 kDa des hypothetischen Präpeptids) nicht ersichtlich. Das Vorhanden-
sein und die Größe von Prosequenzen bei der ProAS-Peptidase könnten zukünftige
Sequenzierungen der terminalen Enden des sekretierten Enzyms aufklären.
4.34.3 Heterologe Expression der Serinpeptidase ProAS in E. coliHeterologe Expression der Serinpeptidase ProAS in E. coli
Die heterologe Expression einer Vielzahl von Serinpeptidasen aus mesophilen und
extremophilen (insbesondere thermophilen und alkaliphilen) Bakterien und Pilzen in
E. coli wurde bereits beschrieben (u.a.: Arnorsdottir et al., 2002; Deane et al., 1987;
Gödde et al., 2005; Gunkel & Gassen, 1989; Gupta et al., 2002a; Ikemura et al.,
1987; Jang et al., 2002; Kwon et al., 1995; Maciver et al., 1994). Dahingegen fehlen
bislang Berichte über eine erfolgreiche heterologe Expression von halophilen
Peptidasen in ihrer aktiven Form in E. coli. So führte die Expression einer Peptidase
aus dem moderat halophilen Bakterium Pseudoalteromonas ruthenica in E. coli zur
Akkumulation von inaktivem Enzym in Inclusion Bodies (Sánchez-Porro Álvarez,
2005). Ebenfalls liegen keine Berichte über eine heterologe Expression von
Peptidasen in halophilen Vertretern der Bacteria vor. Die heterologe Expression von
halophilen Peptidasen aus Archaeen in halophilen Archaeen-Wirtsstämmen wurde
hingegen schon mehrfach beschrieben (Kamekura et al., 1992; Kamekura et al.,
1996; Shi et al., 2006). Ein Beispiel hierfür ist die Expression von aktivem Halolysin,
eine halophile Serinpeptidase von Haloferax mediterranei, in Haloferax volcanii
(Kamekura et al., 1992; Kamekura et al., 1996).
In dieser Studie gelang die Klonierung und heterologe Expression einer Subtilisin-
ähnlichen Peptidase aus dem moderat halophilen Salinivibrio sp. Stamm B3/1-2
(#418) in E. coli. Mit dem Fosmid F114 transformierte E. coli-Zellen zeigten eine
Sekretion von aktiver Peptidase. Diese erwies sich in der Analyse der biochemischen
107
Diskussion
Merkmale und des Molekulargewichtes als übereinstimmend mit der Wildtyp-
Peptidase ProAS(#418), die der chromatografisch definierten Peptidase B entspricht.
Das Fosmid F114 besaß ein ~40 kb großes DNA-Fragment von Stamm B3/1-2
(#418) als Insert, auf dem das Peptidasegen proAS(#418) identifiziert wurde. Dem
verwendeten Fosmidvektor fehlen eigene Promotoren und andere Kontrollelemente
für eine Expression der auf dem Insert lokalisierten Gene. Deshalb liegt der Schluss
nahe, dass die Expression von ProAS(#418) mit den wildtypeigenen Kontroll-
Stoppcodon) in E. coli gelang. Aktives Enzym konnte nur unter sehr spezifischen
Bedingungen nachgewiesen werden. So trat Aktivität nur nach einer geringen
Erhöhung der Fosmidkopiezahl pro Zelle (durch Induktion mit L-Arabinose) und
Absenkung der Temperatur auf 23°C in der zellfreien Kultur auf. Der Einsatz von
Fosmiden und die hier ausgearbeiteten Bedingungen zur heterologen Expression
einer Peptidase wurde bisher noch nicht beschrieben. Er könnte ein allgemein
gangbarer Weg zu optimierten Expressionssystemen für Klonierungen von letalen
Genen aus halotoleranten/halophilen Bakterien sein.
Für die heterologe Überexpression der Subtilisin-ähnlichen Peptidase ProAS wurden
in dieser Arbeit drei verschiedene üblich verwendete Expressionsvektoren
(pJF118EH; PBAD/gIII; pASK-IBA44) benutzt. In einer vorbereitenden Modellstudie
zur Auswahl eines bestgeeigneten Vektors und Optimierung der Expression in E. coli
wurde Subtilisin E aus Bacillus subtilis verwendet. Demnach kam es mit den
Vektoren pBAD/gIII und pASK-IBA44 zur erfolgreichen Expression von aktivem
Subtilisin E. Der Vektor pBAD/gIII schien nach Optimierung der Expressions-
Bedingungen als am besten geeignet.
Eine heterologe Expression von aktiver Peptidase ProAS(#748) aus dem moderat
halophilen Stamm B12/10-1 (#748) gelang jedoch mit keinem der zur Verfügung
stehenden Expressionsvektoren. Es konnten jedoch eindeutige Hinweise auf eine
induzierbare Transkription des rekombinanten Gens bei zwei Expressionsklonen
(pBAD-proAS(#748) und pBAD-proAS-ol(#748)) durch das Auftreten von
proAS(#418)-spezifischer mRNA nachgewiesen werden (siehe Abschnitt 3.4.1.2.1).
Vage Hinweise auf die Expression von rekombinantem Protein gab der Nachweis
eines Peptids mit ähnlicher Größe zum unprozessierten rekombinanten Expressions-
produkt in der PAGE-Analyse des Klons pBAD-proAS(#748) (siehe Abb. 22 /
108
Diskussion
Abschnitt 3.4.1.2.1). Der Grund für das Fehlen von aktiver rekombinanter Peptidase
ist unklar.
Für das Fehlschlagen der heterologen Expression von aktiver extrazellulärer ProAS-
Peptidase in E. coli mittels Expressionsvektoren werden hier folgende Hypothesen
aufgestellt:
1.) Toxizität der rekombinanten ProAS auf den Wirtsstamm:
Ein generelles Problem bei der heterologen Expression von aktiven Peptidasen stellt
die zumeist toxische Wirkung der gebildeten Enzyme auf den Wirtsstamm dar
(Wladyka et al., 2005). Je nach Lokalisation und Aktivität der rekombinanten
Peptidasen in der Wirtszelle kommt es zur Wachstumshemmung bzw. zum Zelltod
(Arnorsdottir et al., 2002; Bott & Betzel, 1996; Goldberg, 1992). Die toxische Wirkung
resultiert dabei aus der unregulierten Hydrolyse von essentiellen wirtseigenen
Proteinen. Diese führt im Cytoplasma zur Störung bzw. Zerstörung des zellulären
Metabolismus, während periplasmatisch oder extrazellulär sekretierte Peptidasen
eine Destabilisierung der Bakterienzellen durch Degradation von Membran- und
Zellwandbestandteilen bewirken können. Die Instabilität einiger Expressionsklone
und die beobachtete Wachstumshemmung aller hier untersuchten Expressionsklone,
schon ab einer Induktion auf niedrigstem Niveau, legt die Vermutung nahe, dass ein
toxischer Effekt vom Expressionsprodukt ausgeht. Gleiches scheint auch für die
Etablierung von Peptidase produzierenden Klonen in Genbanken mittels
konventioneller Vektoren zu gelten. Ein vollständiges Fehlen solcher Klone in
Screenings von Genbanken konnte auch an anderer Stelle beobachtet werden (u.a.
Herzog, 2002; Lämmle, 2004). Bei bakteriellen Peptidase-Produzenten wurden zwei
Mechanismen beschrieben, die dem Wildtyp eine Kontrolle über die proteolytische
Aktivität der gebildeten Enzyme ermöglichen (Wladyka et al., 2005). So werden
sekretierte Peptidasen zumeist als inaktive Zymogene ausgeschieden, die eine
Aktivierung durch Abspaltung der Proregionen außerhalb der Zelle erfahren (Khan &
James, 1998). Es wird vermutet, dass die Proregion als Chaperon für die korrekte
Faltung zum aktiven Enzym fungiert und z.B. beim Subtilisin E autokatalytisch
abgespalten wird (Yabuta et al., 2001). Des Weiteren ist die Produktion von
spezifischen Peptidase-Inhibitoren bekannt, die die Aktivität der zelltoxischen
Peptidase in der Zelle regulieren (Taguchi et al., 1997). Der letzt genannte
Mechanismus fand bei Wladyka et al. (2005) erstmalig Anwendung, eine heterologe
109
Diskussion
Co-Expression der Peptidase Staphopain A mit ihrem Inhibitor Staphostatin A wurde
in E. coli mit Erfolg durchgeführt. Die heterologe Expression von extrazellulären
Peptidasen als Zymogene findet dagegen allgemeine Anwendung und ist zumeist
notwendig zum Erhalt des aktiven rekombinanten Enzyms (Anderson et al., 1999;
Eder et al., 1993; Yabuta et al., 2003). Allerdings zeigen viele Studien, dass auch bei
der heterologen Expression der Peptidasen als Zymogene ein toxischer Effekt auf
den Wirt die Expression hemmt oder komplett inhibiert (Arnorsdottir et al., 2002; Bott
& Betzel, 1996; Goldberg, 1992). Ob dieser Effekt vom Zymogen selber oder einer
Aktivierung desselbigen zum aktiven Enzym durch Autoproteolyse resultiert, ist
bislang noch nicht untersucht worden. Ein Vorhandensein eines Propeptids in der
hier untersuchten Peptidase konnte nicht abschließend geklärt werden (siehe
4.2.2.2).
2.) Fehlende Stringenz in der Regulation der heterologen Expression
Bedingt durch den vermutlich toxischen Charakter von ProAS sollte die heterologe
Expression streng reguliert sein. So kann bei weniger dichten (leaky) Promotoren
und auch im Zusammenhang mit einer hohen Kopiezahl des Vektors eine geringe
basale Expression (background expression) im uninduzierten Zustand bereits
toxische Konzentrationen von ProAS entstehen lassen. Dies kann zum Absterben
der Zelle und damit zur Instabilität eines Klons führen (Arnorsdottir et al., 2002;
Sørensen & Mortensen, 2005). Der tac-Promotor des in dieser Arbeit verwendeten
Vektors pJF118EH besaß offensichtlich ein zu hohes basales Niveau der Expression:
Die Klonierung von Subtilisin E und der Peptidase ProAS(#748) in diesem Vektor
führte zu instabilen Klonen. Es bedarf somit stark regulierter Promotoren auf den
verwendeten Expressionsvektoren, die mit pASKIBA44 und pBAD/gIII in dieser Arbeit
zur Verfügung standen.
Neben der Dichtigkeit stellt die Stärke von Promotoren ein allgemeines Problem bei
der heterologen Expression von Proteinen dar. Hohe Expressionsraten führen dabei
häufig zu einer Proteinaggregation im Cytoplasma (Villaverde & Carrio, 2003), den so
genannten Inclusion Bodies, welche die Sekretion aktiver Enzyme verhindert. Abhilfe
kann eine Senkung der Expressionsrate durch Verringerung der Induktormenge
schaffen. Die Ausbildung von Inclusion Bodies konnte bei Verwendung des Vektors
pASK-IBA44 in dieser Studie beobachtet werden. In mit 0,2 μg/ml Anhydrotetracyclin
induzierten E. coli-Zellen konnten bei der proAS-Expression Inclusion Bodies
110
Diskussion
mikroskopisch nachgewiesen werden. Eine Verringerung der Induktormenge führte
zum Verschwinden der unlöslichen Proteinaggregate, allerdings nicht zur Sekretion
von aktivem Enzym. Möglicherweise wurde die Peptidase als inaktives Enzym
sekretiert, wie ein Protein ähnlicher Größe zur Wildtyp-Peptidase in der PAGE-
Analyse der zellfreien Kultur vermuten ließ.
In einigen Expressionsstrategien ist die Bildung von Inclusion Bodies Teil des
Verfahrens (Baneyx, 1999). Ein Vorteil ergibt sich hierbei durch Verringerung des
zelltoxischen Effektes durch die Inaktivität der aggregierten rekombinanten
Peptidasen. Eine Aktivierung des gereinigten Enzyms wird in einigen Fällen über
eine Denaturierung und anschließende Renaturierung unter geeigneten Puffer-
bedingungen erreicht (Parry, 2002; Villaverde & Carrio, 2003). Thermophile
Peptidasen können dagegen einfach über Erhitzen der Enzymaufreinigung aktiviert
werden (Fu et al., 2003; Gödde et al., 2005; Morikawa et al., 1994; Wu et al., 2004).
Die Aktivierung der rekombinanten Peptidase erfolgt dabei in vielen Fällen über eine
Prozessierung durch Autoproteolyse und Faltung zum reifen und aktiven Enzym
(Gödde et al., 2005; Villaverde & Carrio, 2003). Eine Anwendung der
Renaturierungs-Verfahren für Serinpeptidasen nach Parry (2002) führte allerdings in
dieser Studie zu keiner aktiven Peptidase in der Aufreinigung der Inclusion Bodies.
Womöglich konnten die Bedingungen für eine Renaturierung zum aktiven Enzym
nicht eingestellt werden. Möglicherweise waren die Enzyme auch irreversibel
denaturiert.
3.) Abweichende Codon-Usage durch den Wirtsstamm
Ebenfalls ein allgemeines Problem bei der heterologen Expression von Proteinen ist
die unterschiedliche Codon-Usage von Wirtsstamm und Wildtyp. Eine Verminderung
der Expressionsrate kann durch eine hohe Anzahl von Codons in der heterologen
DNA verursacht werden, die in E. coli selten genutzt werden (Makrides, 1996). Eine
computergestützte Analyse mit dem Programm codon usage analyser 2.0 (Fuhrmann
et al., 2004) deckte für das proAS-Gen aus 3 Salinivibrio-Stämmen 5 (bei
proAS(#418) und proAS(#795)) bzw. 8 (bei proAS(#748)) Codons auf, die von E. coli
K12-Stämmen wenig genutzt werden (<10% Anpassungsfähigkeit). Möglicherweise
resultierte daraus eine verminderte Syntheserate des heterologen Proteins. Auf das
vollständige Ausbleiben von aktiver Peptidase bei den Expressionsklonen hatte die
abweichende Codon-Usage jedoch vermutlich keinen ausschlaggebenden Einfluss.
111
Diskussion
Zum einen konnte die Sekretion einer aktiven ProAS-Peptidase durch den
Fosmidklon F114 in E. coli gezeigt werden. Zum anderen legte ein nach der
Induktion gebildetes Peptid in der PAGE-Analyse der Expressionsklone (pASK-
proAS(#748) und pBAD-proAS(#748)), welches eine ähnliche Größe zum erwarteten
Genprodukt besaß, die Vermutung nahe, dass eine Translation stattgefunden hatte.
Für eine spätere Produktionsoptimierung wird die Verwendung von speziellen E. coli-
Stämmen, die zusätzliche Genkopien für seltene tRNA-Codons besitzen,
vorgeschlagen. Ebenso wäre eine Mutagenese der betreffenden Codons in der
proAS-Sequenz vorstellbar, um diese durch für E. coli häufig genutzten Codons zu
ersetzen.
4. Transport, Prozessierung und Faltung der rekombinanten Peptidase
Für die extrazelluläre Lokalisation von Proteinen muss in gramnegativen Bakterien
ein Durchqueren von zwei Membranen (Cytoplasma- und äußere Membran)
gewährleistet sein. Mehrere Transportsysteme für die Membran-Translokation in
Prokaryoten wurden beschrieben (Pugsley et al., 2004). Die Sekretion kann dabei
schematisch in einen Sec-abhängigen und einen Sec-unabhängigen Weg
unterschieden werden (Coutte et al., 2001). Sekretierte Proteine des Sec-
unabhängigen Wegs beinhalten dabei kein abspaltbares N-terminales Signalpeptid
und werden in einem Schritt durch beide Membranen transportiert (Fath & Kolter,
1993; Plano et al., 2001). Beim Sec-abhängigen Weg erfolgt der Transport
zweistufig. Der Transport ins Periplasma wird über ein N-terminales Signalpeptid
vermittelt, welches während des Transports proteolytisch abgespalten wird. Im
zweiten Schritt kann das Enzym über verschiedene Transportwege durch die äußere
Membran exportiert werden (Coutte et al., 2001). Hierfür sind einige Transport-
systeme bekannt: der AT-Weg (autotransporter pathway) (Desvaux et al., 2004;
Henderson et al., 1998), TPS-Weg (two-partner secretion pathway)(Jacob-Dubuisson
et al., 2004), der Typ II Sekretionsweg (general secretory pathway) (Sandkvist, 2001)
und Typ IV-Sekretionssysteme (Backert & Meyer, 2006).
Bei der Analyse der proAS-Gene konnte ein hypothetisches N-terminales Signal-
peptid identifiziert werden. Dies lässt auf den Signalpeptid-vermittelten posttrans-
lationalen Transport der Proteine ins Periplasma schließen. Bisher wurden zwei
dieser Transportwege in Bakterien beschrieben: Der gut untersuchte und weit
verbreitete Sec-Transportweg (Sec: secretory) und der Tat-Transportweg (Tat: twin-
112
Diskussion
arginine-translocation). Die markante Consensus-Sequenz (R-R-X-F-L-K*) im
Signalpeptid des Tat-Wegs (Blaudeck et al., 2003) lag in den 3 untersuchten
Signalpeptiden der Salinivibrio-Stämme dieser Studie nicht vor.
Die ProAS-Sequenz der hier untersuchten moderat halophilen Bakterien besaß
dagegen die typischen Eigenschaften (von Heijne, 1983) für eine Translokation über
den Sec-Weg. Dieser wird hier als Transportweg der Peptidase ProAS ins
Periplasma angenommen. Die Abspaltung des Signalpeptides vom zumeist inaktiven
Vorläufer (Winnacker, 1985) nach dem Transport über den Sec-Weg ist
entscheidend für die Expression der rekombinanten Proteine mit authentischem N-
terminalen Ende (Sørensen & Mortensen, 2005). Der Sec-Weg ist ebenfalls im
Wirtsstamm E. coli vorhanden. Einige heterologe Signalpeptide erwiesen sich dabei
als voll funktional in E. coli (Sørensen & Mortensen, 2005). Die Sekretion von ProAS
Peptidase durch den Fosmidklon lässt vermuten, dass das Wildtyp-Signalpeptid von
E. coli erkannt wird. Möglicherweise wurde die Sekretion aber auch durch Proteine
ermöglicht, die ebenfalls auf dem Fosmid kodieren und eine transportvermittelnde
bzw. sekretionserleichternde Funktion besitzen, so wie es bei der Sekretion einer
Pektat-Lyase aus Erwinia chrysanthemi in E. coli beschrieben wurde (He et al.,
1991a; He et al., 1991b). Die Sequenzierung eines Fosmidabschnittes (~1000 bp)
stromaufwärts vom proAS(#418) ergab jedoch keinen Hinweis auf Sequenzen, die
homolog zu bekannten Transportergenen waren.
Um die Sekretion von rekombinanten Proteinen ins Periplasma bei E. coli zu
gewährleisten, wird für die Überexpression das Wildtyp-Signalpeptid häufig gegen
ein E. coli-spezifisches ersetzt. Die hier verwendeten Expressionsvektoren pASK-
IBA44 und pBAD/gIII beinhalteten dementsprechend die kodierende Sequenz des
ompA- bzw. gIII-Signalpeptides für E. coli. Dass sogar die Fusion eines Wildtyp-
signalpeptides mit dem gIII-Signalpeptid des Vektors in E. coli zur heterologen
Expression von aktivem periplasmatischen bzw. extrazellulären Subtilisin E führt,
konnte hier im Modellversuch und bei Sroga & Dordick (2002) gezeigt werden. Das
Vorhandensein eines Proteins ähnlicher Größe zum Expressionsprodukt im
Periplasma des Klons pBAD-proAS(#748) legt die Vermutung nahe, dass in diesem
Klon ebenfalls ein Transport der rekombinanten ProAS(#748) ins Periplasma
stattfand. Im Vektor pBAD-proAS(#748) liegt die kodierende Sequenz des vektor-
eigenen gIII-Signalpeptides fusioniert mit dem kompletten proAS(#748)-Gen vor,
*X steht hier für eine hochvariable Stelle in der Aminosäuresequenz.
113
Diskussion
welches am 5’-Ende für ein hypothetisches Signalpeptid des Wildtyps kodiert. Ob
und welches der beiden Signalpeptide des rekombinanten Proteins im Periplasma
abgespalten wurde, konnte nicht aufgeklärt werden.
Ebenfalls wichtig für die Expression eines aktiven Enzyms ist die Ausbildung von
möglichen Disulfidbrücken im Periplasma. Ein Alignment mit der kristallografisch gut
untersuchten Peptidase Aqualysin I (Takagi et al., 1990) ergab für die ProAS-
Peptidase eine hohe Homologie von zwei Bereichen, die im Aqualysin I an der
Ausbildung von Disulfidbrücken beteiligt sind (Kwon et al., 1988). So besitzt die
Peptidase ProAS vermutlich zwei Disulfidbrücken (208. + 240. AS und 309. + 340.
AS*, siehe Abb. 40 im Anhang), die auch in der nächst verwandten Peptidase ProA
vermutet werden (Siezen & Leunissen, 1997). Somit scheint die Sekretion ins
Periplasma eine Voraussetzung zur Ausbildung von korrekt gefalteter ProAS-
Peptidase zu sein.
Der weitere Transport aus dem Periplasma über die äußere Membran ist weniger gut
untersucht. Die bislang bekannten Systeme, die diesen Transport gewährleisten,
wurden schon weiter oben genannt. E. coli K12 fehlen offensichtlich solche
Transportwege. Eine extrazelluläre Lokalisation der rekombinanten Proteine wird
zumeist mit einem passiven Transport aufgrund einer Destabilisierung der äußeren
Membran erklärt (Sørensen & Mortensen, 2005). Dieser Mechanismus wird auch für
die extrazelluläre Lokalisation von aktivem Subtilisin E im Modellsystem vermutet.
Möglicherweise spielte daher ein aktiver Transport durch die äußere Membran für die
heterologe Expression von kleineren Mengen der ProAS-Peptidasen in dieser Studie
keine Rolle.
Folglich ist nicht nur die extrazelluläre Lokalisation der heterologen Peptidasen an
sich bedeutend, sondern ebenfalls die darin eingeschlossenen Transport-,
Prozessierungs- und Faltungsvorgänge zum Erhalt eines aktiven Enzyms.
Die Notwendigkeit von höheren Salzkonzentrationen bei der heterologen Expression
zur Sekretion von aktiven Enzymen wird für einige halophile Peptidasen vermutet. So
kennzeichnet eine sehr geringe Enzym-Stabilität unter salzarmen Bedingungen
einige halophile Peptidasen (Izotova et al., 1983; Kamekura et al., 1992). Die relative
Salzempfindlichkeit der hier untersuchten Peptidasen und die gelungene Expression
von aktiver ProAS-Peptidase in E. coli (bei ~170 mM NaCl im Medium) lässt eine
*Die Nummern geben die Position der Cysteinreste im Alignment der Aminosäuresequenzen an.
114
Diskussion
ausgeprägte Salzabhängigkeit bei der Expression der ProAS-Peptidasen zum
jetzigen Forschungsstand allerdings nicht erkennen.
Bei der zukünftigen Klonierung von stark salzabhängigen Peptidasen würde eine
heterologe Expression in halophilen Bakterien sinnvoll erscheinen. Zumal wenn
diese über notwendige Transporter für die extrazelluläre Sekretion verfügen. Seit
kurzem stehen Vektoren für die heterologe Expression von Proteinen in den moderat
halophilen Bakterien aus der Gattung Halomonas und Chromohalobacter zur
Verfügung (Afendra et al., 2004; Vargas & Nieto, 2004).
Entscheidend für die heterologe Expression von aktiver Peptidase in E. coli
(Fosmidklon) zeigte sich letztendlich die Kombination von niedriger Syntheserate und
niedriger Inkubationstemperatur (~23°C). Wie schon an anderer Stelle vermutetet
(Ikemura et al., 1987; Sroga & Dordick, 2002) sind diese Bedingungen
wahrscheinlich bei einigen rekombinanten Peptidasen Voraussetzung für eine
korrekte Prozessierung und Faltung zum aktiven Enzym. Möglicherweise liegt unter
diesen spezifischen Bedingungen ein Gleichgewicht vor, so dass es zur Expression,
zum Transport und zur Prozessierung der aktiven Enzyme kommen kann, allerdings
ohne die dafür zugehörigen Systeme im Wirtsstamm zu inaktivieren. Die Ergebnisse
der Modellstudie mit Subtilisin E und die Expression von aktiver ProAS-Peptidase
durch den Fosmidklon F114 begründen jedenfalls diese Annahme, auch wenn im
Fall des Fosmidklons ein anderes System verwendet wurde. Die schwache
Anhebung der Fosmidkopiezahl entspricht hier durch Vervielfältigung der proAS-
Gene und ihrer Kontrollelemente ebenfalls einer leichten Steigerung der Synthese-
rate. Dies und die Inkubation bei 23°C führten letztendlich nach einer erstaunlich
langen Induktionsphase von 110h zur aktiven ProAS-Peptidase in der zellfreien
Kultur. Auch bei Deane et al. (1987) konnte bei der heterologen Expression der
nächstverwandten ProA in E. coli eine Verzögerung nach der Induktion von 18 h bis
zum Nachweis von aktiver Peptidase festgestellt werden. Warum es zu dieser extrem
langen Induktionsphase kam, kann zum jetzigen Zeitpunkt nicht beantwortet werden.
Möglicherweise trat eine eher spontane, nicht völlig korrekte Faltung zum aktiven
Enzym in der zellfreien Kultur auf. Hinweise auf eine eventuelle labile Struktur gab
der Vergleich der biochemischen Eigenschaften. So zeigte die rekombinante
Peptidase im Gegensatz zum Wildtypenzym eine hohe Empfindlichkeit gegenüber
stark chaotropen Salzen.
115
Diskussion
Letztendlich scheint für die heterologe Expression von größeren Mengen an aktiver
ProAS-Peptidase kein gängiges Expressionssystem geeignet. Die toxische Wirkung
der rekombinanten Peptidase auf den Wirt oder auf essentielle Proteine bei zellfreien
Expressionssystemen scheint bislang unvereinbar mit einer ökonomischen
Produktion. Die heterologe Expression als inaktives Enzym und spätere
Prozessierung zur aktiven Peptidase bietet sich hierbei als mögliche Lösung an. Eine
Möglichkeit ist hierfür die Überexpression als inaktives Fusionsprotein (Makrides,
1996). Um allerdings die anschließenden Verfahren und Bedingungen zur korrekten
Prozessierung und Faltung des Enzyms herauszufinden, bedarf es in Zukunft weiter-
gehender Analysen der Sequenz und Struktur der reifen Peptidasen aus den
Wildtypen.
4.44.4 Einsatz der Metallopeptidase und der Serinpeptidase (ProAS) aus Einsatz der Metallopeptidase und der Serinpeptidase (ProAS) ausSalinivibrio in der Biotechnologie in der BiotechnologieSalinivibrio
Extrazelluläre Peptidasen gehören zu der bedeutendsten Enzym-Gruppe aus
extremophilen Bakterien, die eine weite Anwendung zur Modifikation oder
Degradation von Proteinen in diversen industriellen Prozessen finden. Ihre Einsatz-
gebiete reichen dabei von der Reinigungs- und Waschmittelindustrie, der Abwasser-
und Abfallbehandlung, Lederverarbeitung, Nahrungs- und Futtermittelindustrie zur
Pharmaindustrie (Gupta et al., 2002b; Rao et al., 1998).
Ein Einsatzgebiet für Peptidasen in der Molekularbiologie ist u.a. der enzymatische
Aufschluss von Zellen und Geweben sowie die Hydrolyse kontaminierender
Zellproteine. Die Verwendung von Peptidasen in Kits zur Nukleinsäureaufreinigung
setzt dabei eine Toleranz gegenüber alkalischem Milieu, hochchaotropen Salzen
(Guanidin) und Detergenzien wie SDS voraus. In diesen Verfahren kommt es zum
Einsatz von stark chaotropen Salzen zum Schutz der zu gewinnenden Nukleinsäuren
vor nukleolytischen Enzymen. Werden zelluläre Desoxyribonukleasen in der Regel
schon durch GuHCl-Konzentrationen von 3-6 M oder GuSCN-Konzentrationen von
0,5 M im Zellaufschluss inaktiviert (Bowtell, 1987; Jeanpierre, 1987; Pramanick et al.,
1976; Sambrook & Russell, 2001; Verma, 1988), besitzen Ribonukleasen im
allgemeinen eine höhere Toleranz gegenüber chaotropen Salzen (Arnold & Ulbrich-
Hofmann, 2000; Sambrook & Russell, 2001). Der Einsatz des stärker chaotropen
GuSCN in Konzentrationen oberhalb von 1-1,5 M hat sich dagegen für eine effektive
Inaktivierung der Ribonukleasen aus den aufgeschlossenen Zellen bewährt
116
Diskussion
(AMBION, 2006; Boom et al., 1990; Chirgwin et al., 1979; Chomczynski & Sacchi,
1987). Zudem werden verschiedene Detergenzien, vor allem das sehr chaotrope
SDS, den Lysepuffern dieser Kits für den Zellaufschluss und die Protein-
denaturierung zugesetzt. Überwiegend wird zur enzymatischen Zelllyse unter diesen
harschen Bedingungen Proteinase K eingesetzt (Hilz et al., 1975; Hofstetter et al.,
1997; Sambrook & Russell, 2001; Sweeney & Walker, 1993). Nur wenige Verfahren
zur Verwendung anderer Peptidasen bei der Nukleinsäureaufreinigung sind bekannt
(z.B. Firma QIAGEN), wurden aber nicht publiziert. Ebenso fehlen eingehende
Untersuchungen zur Toleranz der Aktivität von Peptidasen gegenüber chaotropen
Salzen, mit Ausnahme von Proteinase K (Arnold & Ulbrich-Hofmann, 2000; Yang &
Tsou, 1995).
Die Proteinase K, eine Serinpeptidase aus dem saprophytischen Pilz Tritirachium
album, zeigt unter Einfluss von chaotropen Salzen und Detergenzien eine Steigerung
ihrer enzymatischen Aktivität. So liegt ihr Aktivitätsoptimum bei 0,2-1% SDS (Hilz et
al., 1975) und sie zeigt unter Einfluss von 3 M GuHCl oder 0,8 M GuSCN noch
gesteigerte Aktivität gegen Hämoglobin (Produktdatenblatt Proteinase K, QIAGEN,
1997). Des Weiteren ist sie thermoaktiv mit einem Temperaturoptimum bei 65°C
(Ebeling et al., 1974). In dieser Arbeit sollte untersucht werden, ob sich weitere
Peptidasen finden lassen, die sich für eine Nukleinsäureaufreinigung eignen.
Beweggründe dafür waren zum einen technische Erwägungen, zum anderen
ökonomische Betrachtungen. Zu dem technischen Fokus gehörte das Ziel, möglichst
günstige Eigenschaften über diejenigen der Proteinase K hinaus reichenden zu
erlangen. Konkret wurde angestrebt, Peptidasen bei erhöhten chaotropen
Verhältnissen einsetzen zu können. Denn gerade bei der RNA-Reinigung ist der
Einsatz der Proteinase K sehr limitiert, da RNasen ähnlich resistente Eigenschaften
gegenüber GuSCN zeigen. Für diesen Einsatzbereich gilt es, neue Peptidasen zu
suchen, die erhöhte chaotrope Verhältnisse tolerieren. Ökonomisch ist es für eine
Firma attraktiv, sich von der Abhängigkeit gegenüber Zulieferern zu lösen, vor allem,
wenn deren Zahl sehr beschränkt ist. Hier geht es um Qualitätssicherung im
Produktionsprozess ebenso wie um eine Kostenreduzierung und damit erhöhte
Wertschöpfung. Ansatzpunkt für die Suche nach neuen Peptidasen mit extremen
Eigenschaften waren deshalb halotolerante, halophile und alkaliphile Bakterien, da
im salzhaltigen Milieu Enzyme stark denaturierenden Bedingungen ausgesetzt sind.
117
Diskussion
Im Laufe der Evolution sollten Organismus und externe Enzyme optimal an diese
Bedingungen angepasst sein.
Erwartungsgemäß zeichneten sich die beiden in dieser Arbeit charakterisierten
Enzyme Peptidase A und Peptidase B durch eine Toleranz gegenüber GuHCl und
GuSCN aus. So besaßen die Peptidasen der untersuchten Stämme im Casein-Assay
größtenteils noch über 25% Aktivität in 2 M GuHCl und über 40% in 0,6 M GuSCN.
Weiterhin waren die moderat alkaliphilen (Optimum bei pH 8-9,5) und thermoaktiven
(Optimum bei 40-55°C) Eigenschaften beider Peptidasen, sowie die Toleranz
gegenüber SDS ideal für den Einsatz bei der Nukleinsäureaufreinigung. Eine direkte
Quantifizierung der Enzymaktivität im Lysepuffer eines Kits (MOLZYM) erwies sich
wegen der Bestandteile als nicht durchführbar. Die Kombination von SDS und hohen
Konzentrationen chaotroper Salze führte zu Präzipitaten in den Assays, die
vergleichbare Aktivitätsmessungen verhinderten. Die Eignung der Enzyme wurde
deshalb anwendungsbezogen anhand eines Aufschlusses von Mäuseschwanz-
gewebe getestet. Zellfreie Konzentrate von 3 der 8 untersuchten Stämme zeigten
unter Optimalbedingungen (30mM Tris-HCl, pH 9, 5 mM NaCl) die Fähigkeit Mäuse-
schwänze innerhalb von 2 h zu lysieren. Sie wiesen sogar eine bessere muskel-
gewebeauflösende Wirkung auf, als die mit über 75-fach höherer Aktivität
eingesetzte Proteinase K. Das Konzentrat eines Stammes (B12/16 (#795)) hatte
auch in einem für DNA-Extraktionen üblichen Lysepuffer mit stark chaotropen
Bedingungen (0,5 % Tween, 0,5 % SDS, 0,5 M GuSCN) die Eigenschaft Muskel-
zellen zu lysieren. Allerdings ergab sich aufgrund der inhibierenden Substanzen eine
verlangsamte Lyse (über 18 h). Dieser Befund entsprach den Ergebnissen mit der
Proteinase K: mit gleicher Enzymaktivität (~ 1 kFlU) wurde eine komplette Lyse der
Mäuseschwanzstücke in vergleichbarem Zeitraum erreicht (ca. 12 h). Zukünftige
Studien müssten zeigen, ob bei Einsatz größerer Peptidasemengen aus den hier
untersuchten Stämmen eine wie bei Proteinase K beobachtete schnelle Lyse (inner-
halb von 3 h) auch im chaotropen Milieu zu erreichen ist. Zudem wäre es interessant
zu untersuchen, ob beide in den Konzentraten enthaltenen Peptidasen zelllytisches
Verhalten zeigen. Die hervorragenden Lyseeigenschaften des Kulturkonzentrats von
Stamm B12/16 (#795), welches in der chromatografischen Auftrennung keine
Peptidase A zeigte, weist daraufhin, dass für die Gewebeauflösung die Peptidase B
verantwortlich war, also wie Proteinase K eine Serinpeptidase. Zusammenfassend
kann aus dieser Studie gefolgert werden, dass die hier charakterisierten Peptidasen
118
Diskussion
Eigenschaften aufweisen, die ihren Einsatz in der DNA-Extraktion ermöglichen. Ihre
Verwendung unter stärker chaotropen Bedingungen, wie sie für eine RNA-Extraktion
erforderlich sind, scheint dagegen eher nicht angezeigt zu sein.
Auch wenn sich die hier untersuchten Peptidasen wegen ihrer Eigenschaften noch
nicht für einen breiten Einsatz in der Nukleinsäurereinigung als geeignet zeigten,
weist diese Studie doch auf das Vorhandensein eines hohen Potentials von
bakteriellen Peptidasen unter den halotoleranten/halophilen Bakterien hin. Für
zukünftige Studien würde es sich deswegen lohnen weitere Screenings an
Peptidasen aus halotoleranten und halophilen Bakterien vorzunehmen. Im Hinblick
auf das Auffinden von Peptidasen mit einer noch höheren Toleranz gegenüber
chaotropen Bedingungen als die hier beschriebenen Enzyme scheint die Auswahl
von extrem halophilen/alkaliphilen Bakterien sinnvoll. Wie zuvor schon beschrieben
ist anzunehmen, dass die sekretierten Enzyme dieser Organismen neben ihrer sehr
hohen Halotoleranz auch hervorragende Toleranzen gegenüber anderen
denaturierenden Bedingungen besitzen. Im Hinblick auf das mitunter langsame
Wachstum extrem halophiler Bakterien, einhergehend mit einer geringen Peptidase-
produktion, kann mit einer erschwerten Probengewinnung gerechnet werden. Die in
dieser Arbeit entwickelten Verfahren zur Aufkonzentrierung und chromatografischen
Anreicherung/Trennung von halophilen Peptidasen stellen möglicherweise hilfreiche
Werkzeuge für die Gewinnung geeigneten Probenmaterials dar. Weiterhin ist zu
überlegen, ob die Probengewinnung für die Enzym-Charakterisierungen auf Platten-
kulturen ausgeweitet werden sollte. Die Gewinnung von Probenmaterial erfolgte in
dieser Arbeit gemäß den Auswahlkriterien (siehe Abschnitt 3.1.1) ausschließlich aus
Flüssigkulturen. Dabei zeigte sich, dass einige Peptidase-Produzenten nicht erfasst
werden konnten, da sie, im Gegensatz zur Agarplattenkultur, in der Flüssigkultur
keine oder nur sehr geringe Mengen an Peptidasen sekretierten (siehe Abschnitt
3.1.3). Somit könnte bei einer veränderten Probengewinnung eine erweiterte
Auswahl an Peptidasen für die Charakterisierung zur Verfügung stehen.
Darüber hinaus zeigten sowohl Peptidase A als auch Peptidase B aufgrund ihrer bio-
chemischen Eigenschaften ein Potential für weitere Einsatzfelder der Biotechnologie:
Ihr breiter Aktivitätsbereich zwischen pH 6-11, ihr moderat thermoaktiver Charakter
(Optimum zwischen 40°C und 55°C), ihr breites Substratspektrum und ihre Toleranz
gegenüber höheren SDS-Konzentrationen legen den Einsatz bei der Entfernung von
proteinhaltigen Verschmutzungen in der Abfall- und Abwasserbehandlung sowie
119
Diskussion
möglicherweise als Zusatz in Wasch- und Reinigungsmitteln nahe. Ihre Fähigkeiten
sowohl ohne Salz als auch bis zu 2 M NaCl proteolytisch aktiv zu sein, unterscheidet
sie deutlich von Peptidasen aus extrem halophilen Bakterien. Diese erfahren bei
NaCl-Konzentrationen unterhalb ihres Aktivitätsoptimums einen sehr starken Abfall
und eine irreversible Inaktivierung (Adams & Kelly, 1995), was den Einsatz der
extrem halophilen Peptidasen im Gegensatz zu den hier untersuchten Peptidasen
aus moderat halophilen Bakterien stark einschränkt. Somit könnten die hier
untersuchten Peptidasen ebenfalls zur Proteinhydrolyse in schwach bis moderat
salzhaltigen Flüssigkeiten mit stark schwankendem Chemismus Anwendung finden,
wie es beispielsweise für stark proteinverschmutzte Abwässer der leder-
verarbeitenden Industrie beschrieben wird (Park et al., 2004).
120
Zusammenfassung
55 Zusammenfassung ZusammenfassungDas Ziel der vorliegenden Arbeit war das Auffinden von extrazellulären Peptidasen
aus halotoleranten bzw. halophilen Bakterien, deren proteolytische Aktivität eine
hohe Toleranz gegenüber chaotropen Bedingungen aufweist. Dies geschah im
Hinblick auf den möglichen Einsatz der neuen Enzyme zur Zelllyse in Puffern mit
hohen Konzentrationen von hochchaotropen Guanidin-Salzen (Guanidinisothio-
cyanat oder Guanidinhydrochlorid) und Detergenzien, wie Natriumdodecylsulfat
(SDS), für ein Nukleinsäure-Extraktionsverfahren.
Eine Auswahl an 27 Stämmen mit verschiedenen physiologischen Eigenschaften aus
355 Peptidase-Produzenten einer Stammsammlung von halophilen und
halotoleranten Isolaten aus Salinen wurde auf ihre Sekretionscharakteristika für
Peptidasen untersucht. Eine weiterführende biochemische Charakterisierung wurde
an den sekretierten Peptidasen von 9 moderat halophilen Stämmen vorgenommen,
deren zellfreie Kulturen in einem in vitro Test bezüglich der Zielsetzung besonders
geeignete Peptidase-Merkmale aufwiesen.
Alle 9 Isolate gehörten der Gattung Salinivibrio an, wobei eine Unterscheidung der
Stämme auf Basis einer 16S-rRNA-Teilsequenzanalyse in mindestens 2-3
verschiedene Arten vorgenommen werden konnte. So konnten 8 der 9 Stämme den
Arten Salinivibrio costicola, S. aegyptiaca und möglicherweise einer neuen
Salinivibrio-Spezies zugeordnet werden.
Speziell angepasste Verfahren zur Aufkonzentrierung und Aufreinigung der
Peptidasen aus den salzhaltigen zellfreien Kulturen wurden in dieser Arbeit
entwickelt. Bei 7 der 9 hier untersuchten Stämme konnten mittels Trennung in der
hydrophoben Interaktionschromatografie zwei verschiedene Typen von extra-
zellulären Peptidasen (Peptidase A und B) nachgewiesen werden, während bei den
übrigen zwei Stämmen nur ein Peptidasetyp (Peptidase B) gefunden wurde. Die
chromatografische Unterscheidung der beiden Peptidasen in zwei verschiedene
Typen konnte anhand ihrer biochemischen Eigenschaften bestätigt werden. Die
inhibitorische Wirkung von 1,10-Phenanthrolin und EDTA auf die Aktivität der
Peptidase A legte nahe, dass es sich hierbei um eine Metallopeptidase handelt. Das
etwa 35 kDa große Enzym (SDS-PAGE-Analyse) wies einen moderat alkaliphilen
und thermoaktiven Charakter (Aktivitätsoptimum: pH 8,5; Temp.: 40-50°C) mit einem
niedrigen Salzoptimum (10-200 mM NaCl) und abnehmender Aktivität bei steigender
Salzkonzentration (<20% Restaktivität bei 2 M NaCl) auf. Die komplette Inhibition der
121
Zusammenfassung
zweiten Peptidase (Peptidase B) durch Phenylmethylsulphonylfluorid (PMSF) wies
auf eine Zugehörigkeit des etwa 50 kDa großen Enzyms zu den Serinpeptidasen hin.
Ein Aktivitätsoptimum bei 50-55°C und pH 9-9,5 zeichnete es ebenfalls als moderat
thermoaktives und alkaliphiles Enzym aus, welches ein Aktivitätsoptimum bei 0-
100 mM NaCl aufwies. Eine Klonierung und Identifizierung des Peptidase B-Gens,
hier als proAS bezeichnet, erfolgte mit Hilfe von Sonden, ausgehend von
Peptidasen. Die Bestimmung der Nukleotidsequenz des proAS-Gens zeigte, dass es
für eine etwa 55 kDa große Subtilisin-ähnliche Peptidase kodiert, die 65%
Übereinstimmung zur nächst verwandten Peptidase ProA von Vibrio alginolyticus
aufweist. Peptidase ProAS besitzt ein hypothetisches Signalpeptid von 23
Aminosäuren Länge. Eine hohe Homologie (>94% Aminosäuresequenzidentität)
bestand zwischen den ProAS-Teilsequenzen der 9 Stämme, wobei die
zwischenstammlichen Unterschiede der Peptidasesequenzen mit der taxonomischen
Einordnung der Stämme (16S rRNA-Analyse) korrelierten.
Über eine Fosmid-Klonierung gelang die heterologe Expression der Subtilisin-
ähnlichen Peptidase ProAS(#418) aus dem moderat halophilen Salinivibrio sp.
Stamm B3/1-2 (#418) in E. coli. Aktives Enzym zeigte sich dabei nur unter sehr
spezifischen Bedingungen (Induktion: 0,5 mM L-Arabinose; Inkubation für >110h bei
23°C, 200 rpm). Die Expression der ProAS-Peptidase mittels induzierbarer
Promotoren von Expressionsvektoren gelang auf Transkriptionsebene, führte aber zu
keinem aktiven Enzym. Die Ursachen hierfür wurden diskutiert.
Beide Peptidasen erwiesen sich durch ihre Eigenschaften, u.a. eine breite Substrat-
spezifität, Toleranz gegenüber hochchaotropen Guanidin-Salzen mit größtenteils
noch über 25% Aktivität in 2 M GuHCl und über 40% in 0,6 M GuSCN, sowie
Toleranz gegenüber höheren SDS-Konzentrationen, als geeignet für den Einsatz bei
der DNA-Isolierung. Dies zeigten auch erste anwendungsbezogene Studien am
gewebelytischen Verhalten in einem angepassten Lysepuffer, wobei sich die
Peptidase B aus Stamm B12/16 (#795) als besonders geeignet erwies.
122
Literaturverzeichnis
66 Literaturverzeichnis Literaturverzeichnis Adams, M. W., Perler, F. B. & Kelly, R. M. (1995). Extremozymes: expanding the limits of biocatalysis. Biotechnology (N Y) 13, 662-668. Adams, M. W. W. & Kelly, R. M. (1995). Enzymes from microorganisms in extreme environments. Chem Eng News 73, 32-42. Afendra, A. S., Vargas, C., Nieto, J. J. & Drainas, C. (2004). Gene transfer and expression of recombinant proteins in moderately halophilic bacteria. Methods Mol Biol 267, 209-223. Albiston, A. L., McDowall, S. G., Matsacos, D. & other authors (2001). Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem 276, 48623-48626. Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389-3402. AMBION (2006). Guanidine Thiocyanate. Produktdatenblatt der Firma AMBION. Amoozegar, M. A., Fatemi, A. Z., Karbalaei-Heidari, H. R. & Razavi, M. R. (2006). Production of an extracellular alkaline metalloprotease from a newly isolated, moderately halophile, Salinivibrio sp. Strain AF-2004. Microbiol Res in Press. Anderson, D. E., Peters, R. J., Wilk, B. & Agard, D. A. (1999). Alpha-lytic protease precursor: characterization of a structured folding intermediate. Biochemistry 38, 4728-4735. Appel, W. (1981). Methods of Enzyme Analysis, 2nd edn. Deerfield Beach, FL: Verlag Chemie. Arnold, U. & Ulbrich-Hofmann, R. (2000). Differences in the denaturation behavior of ribonuclease A induced by temperature and guanidine hydrochloride. J Protein Chem 19, 345-352. Arnorsdottir, J., Smaradottir, R. B., Magnusson, O. T., Thorbjarnardottir, S. H., Eggertsson, G. & Kristjansson, M. M. (2002). Characterization of a cloned subtilisin-like serine proteinase from a psychrotrophic Vibrio species. Eur J Biochem 269, 5536-5546. Arnorsdottir, J., Kristjansson, M. M. & Ficner, R. (2005). Crystal structure of a subtilisin-like serine proteinase from a psychrotrophic Vibrio species reveals structural aspects of cold adaptation. Febs J 272, 832-845. Auld, D. S. (1988). Use of chelating agents to inhibit enzymes. Methods Enzymol 158, 110-114.
123
Literaturverzeichnis
Auld, D. S. (1995). Removal and replacement of metal ions in metallopeptidases. Methods Enzymol 248, 228-242. Bachmann, B. J. (1972). Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev 36, 525-557. Backert, S. & Meyer, T. F. (2006). Type IV secretion systems and their effectors in bacterial pathogenesis. Curr Opin Microbiol 9, 207-217. Bajorath, J., Hinrichs, W. & Saenger, W. (1988). The enzymatic activity of proteinase K is controlled by calcium. Eur J Biochem 176, 441-447. Baneyx, F. (1999). Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10, 411-421. Barrett, A. J. (2000). Proteases. In Current protocols in protein science. Edited by G. Taylor: John Wiley & Sons, Inc. Barrett, A. J. (2001). Proteolytic enzymes: nomenclature and classification. In Proteolytic Enzymes: a practical approach. Edited by R. Beynon & J. S. Bond. Oxford: Oxford University Press. Bendlin, R. (2006). Ein Bündnis für die Weiße Biotechnologie im Norden. Pressemitteilung, Technische Universität Hamburg-Harburg, 29052006. Bendtsen, J. D., Nielsen, H., von Heijne, G. & Brunak, S. (2004). Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340, 783-795. Bergmann, M. & Ross, W. F. (1936). On proteolytic enzymes. J Biol Chem 114, 717. Bieger, B., Essen, L. O. & Oesterhelt, D. (2003). Crystal structure of halophilic dodecin: a novel, dodecameric flavin binding protein from Halobacterium salinarum. Structure 11, 375-385. Blaudeck, N., Kreutzenbeck, P., Freudl, R. & Sprenger, G. A. (2003). Genetic analysis of pathway specificity during posttranslational protein translocation across the Escherichia coli plasma membrane. J Bacteriol 185, 2811-2819. Boom, R., Sol, C. J., Salimans, M. M., Jansen, C. L., Wertheim-van Dillen, P. M. & van der Noordaa, J. (1990). Rapid and simple method for purification of nucleic acids. J Clin Microbiol 28, 495-503. Bott, R. & Betzel, C. (1996). Subtilisin enzymes: Practical protein engineering. New York: Plenum Press. Bowtell, D. D. (1987). Rapid isolation of eukaryotic DNA. Anal Biochem 162, 463-465. Boyer, H. W. & Roulland-Dussoix, D. (1969). A complementation analysis of the restriction and modification of DNA in Escherichia coli. J Mol Biol 41, 459-472.
124
Literaturverzeichnis
Brock, T. D. & Freeze, H. (1969). Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J Bacteriol 98, 289-297. Burkholder, P. R. & Giles, N. H. (1947). Induced biochemical mutations in Bacillus subtilis. Am J Bot 34, 345-348. Camacho, M., Rodriguez-Arnedo, A. & Bonete, M. J. (2002). NADP-dependent isocitrate dehydrogenase from the halophilic archaeon Haloferax volcanii: cloning, sequence determination and overexpression in Escherichia coli. FEMS Microbiol Lett 209, 155-160. Charney, J. & Tomarelli, R. M. (1947). A colorimetric method for the determination of proteolytic activity of duodenal juice. J Biol Chem 171, 501-505. Chirgwin, J. M., Przybyla, A. E., MacDonald, R. J. & Rutter, W. J. (1979). Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18, 5294-5299. Chomczynski, P. & Sacchi, N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162, 156-159. Clarke, L. & Carbon, J. (1976). A colony bank containing synthetic Col El hybrid plasmids representative of the entire E. coli genome. Cell 9, 91-99. Coligan, J. E., Dunn, B. M., Ploegh, H. L., Speicher, D. W. & Wingfield, P. T. (1998). Current protocols in protein science: John Wiley & Sons, Inc. Costenaro, L., Zaccai, G. & Ebel, C. (2002). Link between protein-solvent and weak protein-protein interactions gives insight into halophilic adaptation. Biochemistry 41, 13245-13252. Coutte, L., Antoine, R., Drobecq, H., Locht, C. & Jacob-Dubuisson, F. (2001). Subtilisin-like autotransporter serves as maturation protease in a bacterial secretion pathway. Embo J 20, 5040-5048. Cowan, D. A. & Daniel, R. M. (1982). A modification for increasing the sensitivity of the casein-agar plate assay: a simple semiquantitative assay for thermophilic and mesophilic proteases. J Biochem Biophys Methods 6, 31-37. DasSarma, S. & Arora, P. (2001). Halophiles. In Encyclopedia of life sciences, pp. 1-9. London: Nature Publishing Group. De Castro, R. E., Maupin-Furlow, J. A., Gimenez, M. I., Herrera Seitz, M. K. & Sanchez, J. J. (2006). Haloarchaeal proteases and proteolytic systems. FEMS Microbiol Rev 30, 17-35. de Kreij, A., Venema, G. & van den Burg, B. (2000). Substrate specificity in the highly heterogeneous M4 peptidase family is determined by a small subset of amino acids. J Biol Chem 275, 31115-31120.
125
Literaturverzeichnis
Deane, S. M., Robb, F. T. & Woods, D. R. (1986). Isolation and characterization of Vibrio alginolyticus mutant that overproduces extracellular proteases. J Gen Microbiol 132, 893-898. Deane, S. M., Maharaj, R., Robb, F. T. & Woods, D. R. (1987). Cloning, expression and release of a Vibrio alginolyticus SDS-resistant Ca2+-dependent exoprotease in Escherichia coli. J Gen Microbiol 133, 2295-2302. Deane, S. M., Robb, F. T., Robb, S. M. & Woods, D. R. (1989). Nucleotide sequence of the Vibrio alginolyticus calcium-dependent, detergent-resistant alkaline serine exoprotease A. Gene 76, 281-288. Deane, S. R., FT; Woods, DR (1987). Production and activation of an SDS-resistant alkaline serine exoprotease of Vibrio alginolyticus. J Gen Microbiol 133, 391-398. Degenkolb, J., Takahashi, M., Ellestad, G. A. & Hillen, W. (1991). Structural requirements of tetracycline-Tet repressor interaction: determination of equilibrium binding constants for tetracycline analogs with the Tet repressor. Antimicrob Agents Chemother 35, 1591-1595. Demain, A. L. & Davies, J. E. (1999). Manual of industrial microbiology and biotechnology, second edition edn. Washington, D.C.: ASM Press. DeShazer, D., Brett, P. J., Burtnick, M. N. & Woods, D. E. (1999). Molecular characterization of genetic loci required for secretion of exoproducts in Burkholderia pseudomallei. J Bacteriol 181, 4661-4664. Desvaux, M., Parham, N. J. & Henderson, I. R. (2004). The autotransporter secretion system. Res Microbiol 155, 53-60. Deutch, C. E. (2002). Characterization of a salt-tolerant extracellular a-amylase from Bacillus dipsosauri. Lett Appl Microbiol 35, 78-84. Diaz, S., Perez-Pomares, F., Pire, C., Ferrer, J. & Bonete, M. J. (2006). Gene cloning, heterologous overexpression and optimized refolding of the NAD-glutamate dehydrogenase from Haloferax mediterranei. Extremophiles 10, 105-115. Duong Van Qua, Simidu, U. & Taga, N. (1981). Purification and some properties of halophilic protease produced by a moderately halophilic marine Pseudomonas sp. Can J Microbiol 27, 505-510. Ebel, C., Costenaro, L., Pascu, M., Faou, P., Kernel, B., Proust-De Martin, F. & Zaccai, G. (2002). Solvent interactions of halophilic malate dehydrogenase. Biochemistry 41, 13234-13244. Ebeling, W., Hennrich, N., Klockow, M., Metz, H., Orth, H. D. & Lang, H. (1974). Proteinase K from Tritirachium album Limber. Eur J Biochem 47, 91-97. Eder, J., Rheinnecker, M. & Fersht, A. R. (1993). Folding of subtilisin BPN': role of the pro-sequence. J Mol Biol 233, 293-304.
126
Literaturverzeichnis
Eichler, J. (2001). Biotechnological uses of archaeal extremozymes. Biotechnol Adv 19, 261-278. Eisenberg, H. (1992). Halophilic malate dehydrogenase - a case history of biophysical investigations: ultracentrifugation, light-, X-ray- and neutron scattering. Biochem Soc Symp 58, 113-125. Fath, M. J. & Kolter, R. (1993). ABC transporters: bacterial exporters. Microbiol Rev 57, 995-1017. Fekkes, P. & Driessen, A. J. (1999). Protein targeting to the bacterial cytoplasmic membrane. Microbiol Mol Biol Rev 63, 161-173. Felsenstein, J. (1988). Phylogenies from molecular sequences: inference and reliability. Annu Rev Genet 22, 521-565. Frolow, F., Harel, M., Sussman, J. L., Mevarech, M. & Shoham, M. (1996). Insights into protein adaptation to a saturated salt environment from the crystal structure of a halophilic 2Fe-2S ferredoxin. Nat Struct Biol 3, 452-458. Fu, Z., Hamid, S. B., Razak, C. N., Basri, M., Salleh, A. B. & Rahman, R. N. (2003). Secretory expression in Escherichia coli and single-step purification of a heat-stable alkaline protease. Protein Expr Purif 28, 63-68. Fuhrmann, M., Hausherr, A., Ferbitz, L., Schodl, T., Heitzer, M. & Hegemann, P. (2004). Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Mol Biol 55, 869-881. Fukushima, T., Mizuki, T., Echigo, A., Inoue, A. & Usami, R. (2005). Organic solvent tolerance of halophilic alpha-amylase from a Haloarchaeon, Haloarcula sp. strain S-1. Extremophiles 9, 85-89. Fürste, J. P., Pansegrau, W., Frank, R., Blöcker, H., Scholz, P., Bagdasarian, M. & Lanka, E. (1986). Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48, 119-131. Galinski, E. A. (1993). Halophile und halotolerante Eubakterien. In Extremophile: Mikroorganismen in ausgefallenen Lebensräumen. Edited by K. Hausmann & B. P. Kremer. Weinheim: VCH. Garcia, M. T., Ventosa, A., Ruiz-Berraquero, F. & Kocur, M. (1987). Taxonomic study and amended description of Vibrio costicola. Int J Syst Bacteriol 37, 251–256. Gimenez, M. I., Studdert, C. A., Sanchez, J. J. & De Castro, R. E. (2000). Extracellular protease of Natrialba magadii: purification and biochemical characterization. Extremophiles 4, 181-188. Gödde, C., Sahm, K., Brouns, S. J., Kluskens, L. D., van der Oost, J., de Vos, W. M. & Antranikian, G. (2005). Cloning and expression of islandisin, a new thermostable subtilisin from Fervidobacterium islandicum, in Escherichia coli. Appl Environ Microbiol 71, 3951-3958.
127
Literaturverzeichnis
Goldberg, A. L. (1992). The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur J Biochem 203, 9-23. Gomes, J. & Steiner, W. (2004). The biocatalytic potential of extremophiles and extremozymes. Food Technol Biotechnol 42, 223-235. Greisen, K., Loeffelholz, M., Purohit, A. & Leong, D. (1994). PCR primers and probes for the 16S rRNA gene of most species of pathogenic bacteria, including bacteria found in cerebrospinal fluid. J Clin Microbiol 32, 335-351. Gunkel, F. A. & Gassen, H. G. (1989). Proteinase K from Tritirachium album Limber. Characterization of the chromosomal gene and expression of the cDNA in Escherichia coli. Eur J Biochem 179, 185-194. Gupta, A., Roy, I., Patel, R. K., Singh, S. P., Khare, S. K. & Gupta, M. N. (2005). One-step purification and characterization of an alkaline protease from haloalkaliphilic Bacillus sp. J Chromatogr A 1075, 103-108. Gupta, R., Beg, Q. K., Khan, S. & Chauhan, B. (2002a). An overview on fermentation, downstream processing and properties of microbial alkaline proteases. Appl Microbiol Biotechnol 60, 381-395. Gupta, R., Beg, Q. K. & Lorenz, P. (2002b). Bacterial alkaline proteases: molecular approaches and industrial application. Appl Microbiol Biotechnol 59, 15-32. Guzman, L. M., Belin, D., Carson, M. J. & Beckwith, J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177, 4121-4130. Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166, 557-580. Hanahan, D. (1985). In DNA cloning: a practical approach. Edited by D. M. Glover. McLean, Virginia: IRL Press. Harrington, D. J. (1996). Bacterial collagenases and collagen-degrading enzymes and their potential role in human disease. Infect Immun 64, 1885-1891. Häse, C. C. & Finkelstein, R. A. (1993). Bacterial extracellular zinc-containing metalloproteases. Microbiol Rev 57, 823-837. He, S. Y., Lindeberg, M., Chatterjee, A. K. & Collmer, A. (1991a). Cloned Erwinia chrysanthemi out genes enable Escherichia coli to selectively secrete a diverse family of heterologous proteins to its milieu. Proc Natl Acad Sci U S A 88, 1079-1083. He, S. Y., Schoedel, C., Chatterjee, A. K. & Collmer, A. (1991b). Extracellular secretion of pectate lyase by the Erwinia chrysanthemi out pathway is dependent upon Sec-mediated export across the inner membrane. J Bacteriol 173, 4310-4317.
128
Literaturverzeichnis
Henderson, I. R., Navarro-Garcia, F. & Nataro, J. P. (1998). The great escape: structure and function of the autotransporter proteins. Trends Microbiol 6, 370-378. Herzog, P. (2002). Klonierung von Genen für extrazelluläre Enzyme aus halophilen Bakterien. Diplomarbeit, Universität Bremen; FB2-Biologie/Chemie. Hilz, H., Wiegers, U. & Adamietz, P. (1975). Stimulation of proteinase K action by denaturing agents: application to the isolation of nucleic acids and the degradation of 'masked' proteins. Eur J Biochem 56, 103-108. Hiraga, K., Nishikata, Y., Namwong, S., Tanasupawat, S., Takada, K. & Oda, K. (2005). Purification and characterization of serine proteinase from a halophilic bacterium, Filobacillus sp. RF2-5. Biosci Biotechnol Biochem 69, 38-44. Hofstetter, J. R., Zhang, A., Mayeda, A. R., Guscar, T., Nurnberger, J. I., Jr. & Lahiri, D. K. (1997). Genomic DNA from mice: a comparison of recovery methods and tissue sources. Biochem Mol Med 62, 197-202. Hooper, N. M. (1994). Families of zinc metalloproteases. FEBS Lett 354, 1-6. Hooper, N. M. (2002). Proteases in Biology and Medicine. London: Portland Press. Hough, D. W. & Danson, M. J. (1999). Extremozymes. Curr Opin Chem Biol 3, 39-46. Huang, C. Y., Garcia, J. L., Patel, B. K., Cayol, J. L., Baresi, L. & Mah, R. A. (2000). Salinivibrio costicola subsp. vallismortis subsp. nov., a halotolerant facultative anaerobe from Death Valley, and emended description of Salinivibrio costicola. Int J Syst Evol Microbiol 50 Pt 2, 615-622. Ikemura, H., Takagi, H. & Inouye, M. (1987). Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J Biol Chem 262, 7859-7864. International Union of Biochemistry and Molecular Biology. Nomenclature Committee. & Webb, E. C. (1992). Enzyme nomenclature 1992 : recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes. San Diego: Published for the International Union of Biochemistry and Molecular Biology by Academic Press. Izotova, L. S., Strongin, A. Y., Chekulaeva, L. N., Sterkin, V. E., Ostoslavskaya, V. I., Lyublinskaya, L. A., Timokhina, E. A. & Stepanov, V. M. (1983). Purification and properties of serine protease from Halobacterium halobium. J Bacteriol 155, 826-830. Jacob-Dubuisson, F., Fernandez, R. & Coutte, L. (2004). Protein secretion through autotransporter and two-partner pathways. Biochim Biophys Acta 1694, 235-257. Jang, H. J., Kim, B. C., Pyun, Y. R. & Kim, Y. S. (2002). A novel subtilisin-like serine protease from Thermoanaerobacter yonseiensis KB-1: its cloning, expression, and biochemical properties. Extremophiles 6, 233-243.
129
Literaturverzeichnis
Jeanpierre, M. (1987). A rapid method for the purification of DNA from blood. Nucleic Acids Res 15, 9611. Jolley, K. A., Russell, R. J., Hough, D. W. & Danson, M. J. (1997). Site-directed mutagenesis and halophilicity of dihydrolipoamide dehydrogenase from the halophilic archaeon, Haloferax volcanii. Eur J Biochem 248, 362-368. Kalisz, H. M. (1988). Microbial proteinases. Adv Biochem Eng Biotechnol 36, 1-65. Kamekura, M. & Onishi, H. (1974). Protease formation by a moderately halophilic Bacillus strain. Appl Microbiol 27, 809-810. Kamekura, M. & Seno, Y. (1990). A halophilic extracellular protease from a halophilic archaebacterium strain 172 P1. Biochem Cell Biol 68, 352-359. Kamekura, M., Seno, Y., Holmes, M. L. & Dyall-Smith, M. L. (1992). Molecular cloning and sequencing of the gene for a halophilic alkaline serine protease (halolysin) from an unidentified halophilic archaea strain (172P1) and expression of the gene in Haloferax volcanii. J Bacteriol 174, 736-742. Kamekura, M. & Seno, Y. (1993). Partial sequence of the gene for a serine protease from a halophilic archaeum Haloferax mediterranei R4, and nucleotide sequences of 16S rRNA encoding genes from several halophilic archaea. Experientia 49, 503-513. Kamekura, M., Seno, Y. & Dyall-Smith, M. (1996). Halolysin R4, a serine proteinase from the halophilic archaeon Haloferax mediterranei; gene cloning, expression and structural studies. Biochim Biophys Acta 1294, 159-167. Kannan, Y., Koga, Y., Inoue, Y., Haruki, M., Takagi, M., Imanaka, T., Morikawa, M. & Kanaya, S. (2001). Active subtilisin-like protease from a hyperthermophilic archaeon in a form with a putative prosequence. Appl Environ Microbiol 67, 2445-2452. Karbalaei-Heidari, H. R., Ziaee, A.-A., Schaller, J. & Amoozegar, M. A. (2006). Purification and characterization of an extracellular haloalkaline protease produced by the moderately halophilic bacterium, Salinivibrio sp. strain AF-2004. Enzyme and Microbial Technology in Press. Kauffmann, F. (1944). Zur Serologie der Coli-Gruppe. Acta Pathol Microbiol Scand 21, 20-45. Khan, A. R. & James, M. N. (1998). Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci 7, 815-836. Kirst, G. O. (1993). Halophile Mikroalgen. In Extremophile: Mikroorganismen in ausgefallenen Lebensräumen. Edited by K. Hausmann & B. P. Kremer. Weinheim: VCH. Klemme, J. H. & Pfleiderer, C. (1977). Production of extracellular proteolytic enzymes by phototrophic bacteria. FEMS Microbiology Letters 1, 297-299.
130
Literaturverzeichnis
Kunitz, M. (1947). Crystalline soybean Trypsin Inhibitor, II. General properties. Journal of General Physiology 30, 291-310. Kuo, T. T. & Stocker, B. A. (1969). Suppression of proline requirement of proA and proAB deletion mutants in Salmonella typhimurium by mutation to arginine requirement. J Bacteriol 98, 593-598. Kushner, D. J. (1988). Halophilic bacteria: their life in and out salt. In Microbiology of extreme environments and its potential for biotechnology, pp. 280-309. Edited by M. S. Da Costa, J. C. Duarte & R. A. D. Williams. London: Elsevier Applied Science. Kwon, S. T., Matsuzawa, H. & Ohta, T. (1988). Determination of the positions of the disulfide bonds in aqualysin I (a thermophilic alkaline serine protease) of Thermus aquaticus YT-1. J Biochem (Tokyo) 104, 557-559. Kwon, Y. T., Kim, J. O., Moon, S. Y., Yoo, Y. D. & Rho, H. M. (1995). Cloning and characterization of the gene encoding an extracellular alkaline serine protease from Vibrio metschnikovii strain RH530. Gene 152, 59-63. Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. Lama, L., Romano, I., Calandrelli, V., Nicolaus, B. & Gambacorta, A. (2005). Purification and characterization of a protease produced by an aerobic haloalkaliphilic species belonging to the Salinivibrio genus. Res Microbiol 156, 478-484. Lämmle, K. (2004). Neue Enzyme für industrielle Anwendungen aus Boden-Genbanken. Dissertation, Universität Stuttgart; Institut für Grenzflächenverfahrenstechnik der Universität Stuttgart. Larsen, A. N., Moe, E., Helland, R., Gjellesvik, D. R. & Willassen, N. P. (2006). Characterization of a recombinantly expressed proteinase K-like enzyme from a psychrotrophic Serratia sp. Febs J 273, 47-60. Lee, M. H. & Murphy, G. (2004). Matrix metalloproteinases at a glance. J Cell Sci 117, 4015-4016. Lee, Y. C., Ohta, T. & Matsuzawa, H. (1992). A non-covalent NH2-terminal pro-region aids the production of active aqualysin I (a thermophilic protease) without the COOH-terminal pro-sequence in Escherichia coli. FEMS Microbiol Lett 71, 73-77. Lee, Y. C., Koike, H., Taguchi, H., Ohta, T. & Matsuzawa, H. (1994). Requirement of a COOH-terminal pro-sequence for the extracellular secretion of aqualysin I (a thermophilic subtilisin-type protease) in Thermus thermophilus. FEMS Microbiol Lett 120, 69-74. Lee, Y. C., Terada, I., Ohta, T. & Matsuzawa, H. (1996). Roles of the N- and C- terminal pro-sequences of aqualysin I precursor in the processing and extracellular secretion of the enzyme. Adv Exp Med Biol 379, 155-157.
131
Literaturverzeichnis
Lesk, A. M. & Fordham, W. D. (1996). Conservation and variability in the structures of serine proteinases of the chymotrypsin family. J Mol Biol 258, 501-537. Ludwig, W., Strunk, O., Klugbauer, S., Klugbauer, N., Weizenegger, M., Neumaier, J., Bachleitner, M. & Schleifer, K. H. (1998). Bacterial phylogeny based on comparative sequence analysis. Electrophoresis 19, 554-568. Maciver, B., McHale, R. H., Saul, D. J. & Bergquist, P. L. (1994). Cloning and sequencing of a serine proteinase gene from a thermophilic Bacillus species and its expression in Escherichia coli. Appl Environ Microbiol 60, 3981-3988. Madern, D., Ebel, C. & Zaccai, G. (2000). Halophilic adaptation of enzymes. Extremophiles 4, 91-98. Makowski, K., Bialkowska, A., Szczesna-Antczak, M., Kalinowska, H., Kur, J., Cieslinski, H. & Turkiewicz, M. (2006). Immobilized preparation of cold-adapted and halotolerant Antarctic beta-galactosidase as a highly stable catalyst in lactose hydrolysis. FEMS Microbiol Ecol. Makrides, S. C. (1996). Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 60, 512-538. Mann, K. G. & Lorand, L. (1993). Introduction: blood coagulation. Methods Enzymol 222, 1-10. Margesin, R. & Schinner, F. (2001). Potential of halotolerant and halophilic microorganisms for biotechnology. Extremophiles 5, 73-83. Marhuenda-Egea, F. C. & Bonete, M. J. (2002). Extreme halophilic enzymes in organic solvents. Curr Opin Biotechnol 13, 385-389. Matsumoto, K. (2004). Role of bacterial proteases in pseudomonal and serratial keratitis. Biol Chem 385, 1007-1016. Maurer, K. H. (2004). Detergent proteases. Curr Opin Biotechnol 15, 330-334. Mevarech, M., Frolow, F. & Gloss, L. M. (2000). Halophilic enzymes: proteins with a grain of salt. Biophys Chem 86, 155-164. Mijts, B. N. & Patel, B. K. (2002). Cloning, sequencing and expression of an alpha-amylase gene, amyA, from the thermophilic halophile Halothermothrix orenii and purification and biochemical characterization of the recombinant enzyme. Microbiology 148, 2343-2349. Molecular Probes (2003). Product Information: EnzChek Protease Assay Kit. Morikawa, M., Izawa, Y., Rashid, N., Hoaki, T. & Imanaka, T. (1994). Purification and characterization of a thermostable thiol protease from a newly isolated hyperthermophilic Pyrococcus sp. Appl Environ Microbiol 60, 4559-4566.
132
Literaturverzeichnis
Mühl, H. (2002). Screening von halotoleranten Bakterien nach extrazellulären Enzymen mit biotechnischer Bedeutung (Peroxidasen, Laccasen, Cellulasen, Amylasen) und Isolierung der Gene. Diplomarbeit, Universität Bremen; FB2-Biologie/Chemie. Müller, M., Koch, H. G., Beck, K. & Schäfer, U. (2001). Protein traffic in bacteria: multiple routes from the ribosome to and across the membrane. Prog Nucleic Acid Res Mol Biol 66, 107-157. Namwong, S., Hiraga, K., Takada, K., Tsunemi, M., Tanasupawat, S. & Oda, K. (2006). A halophilic serine proteinase from Halobacillus sp. SR5-3 isolated from fish sauce: purification and characterization. Biosci Biotechnol Biochem 70, 1395-1401. Nazina, T. N., Tourova, T. P., Poltaraus, A. B. & other authors (2001). Taxonomic study of aerobic thermophilic bacilli:descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermoglucosidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int J Syst Evol Microbiol 51, 433-446. Neumann, L. (2005). Phylogenetische Untersuchungen von halophilen Bakterien. Diplomarbeit, Universität Bremen; FB2-Biologie/Chemie. Nieto, T. P. & Ellis, A. E. (1991). Heterogeneity of extracellular proteases produced by different isolates of Aeromonas hydrophila and A. sobria pathogenic for fish. Journal of Fish Diseases 14, 229-235. Norberg, P. & von Hofsten, B. (1969). Proteolytic enzymes from extremely halophilic bacteria. J Gen Microbiol 55, 251-256. Ollis, D. L., Cheah, E., Cygler, M. & other authors (1992). The alpha/beta hydrolase fold. Protein Eng 5, 197-211. Ordas, M. C., Novoa, B., Faisal, M., McLaughlin, S. & Figueras, A. (2001). Proteolytic activity of cultured Pseudoperkinsus tapetis extracellular products. Comp Biochem Physiol 130, 199-206. Park, H.-Y., Son, K.-H., Kwon, Y.-K., Shin, D.-H. & Min, S.-G. (2004). Method for preparing leather using protease and method for treating wastes derived from leather processing. In freepatentsonline #20040214309. Parry, M. A. A. (2002). Over-expression and purification of active serine proteases and their variants from Escherichia coli inclusion bodies. In Current protocols in protein science. Edited by G. Taylor: John Wiley & Sons, Inc. Plano, G. V., Day, J. B. & Ferracci, F. (2001). Type III export: new uses for an old pathway. Mol Microbiol 40, 284-293.
133
Literaturverzeichnis
Polosina, Y. Y., Zamyatkin, D. F., Kostyukova, A. S., Filimonov, V. V. & Fedorov, O. V. (2002). Stability of Natrialba magadii NDP kinase: comparisons with other halophilic proteins. Extremophiles 6, 135-142. Poulsen, K., Reinholdt, J. & Kilian, M. (1996). Characterization of the Streptococcus pneumoniae immunoglobulin A1 protease gene (iga) and its translation product. Infect Immun 64, 3957-3966. Pramanick, D., Forstova, J. & Pivec, L. (1976). 4 M guanidine hydrochloride applied to the isolation of DNA from different sources. FEBS Lett 62, 81-84. Premkumar, L., Greenblatt, H. M., Bageshwar, U. K., Savchenko, T., Gokhman, I., Sussman, J. L. & Zamir, A. (2005). Three-dimensional structure of a halotolerant algal carbonic anhydrase predicts halotolerance of a mammalian homolog. Proc Natl Acad Sci U S A 102, 7493-7498. Pugsley, A. P., Francetic, O., Driessen, A. J. & de Lorenzo, V. (2004). Getting out: protein traffic in prokaryotes. Mol Microbiol 52, 3-11. QIAGEN (1997). Produktdatenblatt Proteinase K: Introducing QIAGEN Proteinase K - the robust proteinase. QIAGEN News. QIAGEN (2003). Ni-NTA Spin Handbook. QIAGEN, Hilden. Ramos-Cormenzana, A. (1988). Ecological distribution and biotechnological potential of halophilic microorganisms. In Microbiology of extreme environments and its potential for biotechnology, pp. 289-309. Edited by M. S. Da Costa, J. C. Duarte & R. A. D. Williams. London: Elsevier Apllied Science. Rao, M. B., Tanksale, A. M., Ghatge, M. S. & Deshpande, V. V. (1998). Molecular and biotechnological aspects of microbial proteases. Microbiol Mol Biol Rev 62, 597-635. Rawlings, N. D. & Barrett, A. J. (1993). Evolutionary families of peptidases. Biochem J 290 ( Pt 1), 205-218. Rawlings, N. D. & Barrett, A. J. (1994). Families of serine peptidases. Methods Enzymol 244, 19-61. Rawlings, N. D., Morton, F. R. & Barrett, A. J. (2006). MEROPS: the peptidase database. Nucleic Acids Res 34, D270-272. Redecke, L., Brehm, M. A. & Bredehorst, R. (2006). Cloning and characterization of dihydrofolate reductase from a facultative alkaliphilic and halotolerant bacillus strain. Extremophiles. Rodriguez-Valera, F. (1992). Biotechnological potential of halobacteria. Biochem Soc Symp 58, 135-147.
134
Literaturverzeichnis
Romano, I., Gambacorta, A., Lama, L., Nicolaus, B. & Giordano, A. (2005). Salinivibrio costicola subsp. alcaliphilus subsp. nov., a haloalkaliphilic aerobe from Campania Region (Italy). Syst Appl Microbiol 28, 34-42. Romano, I., Giordano, A., Lama, L. & Gambacorta, A. (2006).Salinivibrio species distribution in Sinai peninsula.: unpublished. Ruiz, D. M. & De Castro, R. E. (2006). Effect of organic solvents on the activity and stability of an extracellular protease secreted by the haloalkaliphilic archaeon Natrialba magadii. J Ind Microbiol Biotechnol. Ryu, K., Kim, J. & Dordick, J. S. (1994). Catalytic properties and potential of an extracellular protease from an extreme halophile. Enzyme Microb Technol 16, 266-275. Saeki, K., Okuda, M., Hatada, Y., Kobayashi, T., Ito, S., Takami, H. & Horikoshi, K. (2000). Novel oxidatively stable subtilisin-like serine proteases from alkaliphilic Bacillus spp.: enzymatic properties, sequences, and evolutionary relationships. Biochem Biophys Res Commun 279, 313-319. Sakamoto, S., Terada, I., Lee, Y. C., Uehara, K., Matsuzawa, H. & Iijima, M. (1996). Efficient production of Thermus protease aqualysin I in Escherichia coli: effects of cloned gene structure and two-stage culture. Appl Microbiol Biotechnol 45, 94-101. Salvesen, G. S. & Nagase, H. (2001). Inhibition of proteolytic enzymes. In Proteolytic enzymes: a practical approach. Edited by R. Beynon & J. S. Bond. Oxford: Oxford University Press. Sambrook, J. & Russell, D. W. (2001). Molecular cloning: A laboratory manual, 3rd edition. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. Sánchez-Porro Álvarez, C. (2005). Caracterización bioquimíca y molecula de la haloproteasa CP1 producida por Pseudoalteromonas ruthenica. Dissertation, Universidad de Sevilla, Departmento de microbiología y parasitología, Facultad de farmacia. Sánchez-Porro, C., Martin, S., Mellado, E. & Ventosa, A. (2003a). Diversity of moderately halophilic bacteria producing extracellular hydrolytic enzymes. J Appl Microbiol 94, 295-300. Sánchez-Porro, C., Mellado, E., Bertoldo, C., Antranikian, G. & Ventosa, A. (2003b). Screening and characterization of the protease CP1 produced by the moderately halophilic bacterium Pseudoalteromonas sp. strain CP76. Extremophiles 7, 221-228. Sandkvist, M. (2001). Biology of type II secretion. Mol Microbiol 40, 271-283. Sarath, G., Zeece, M. G. & Penheiter, A. R. (2001). Protease assay methods. In Proteolytic enzymes: a practical approach. Edited by R. Beynon & J. S. Bond. Oxford: Oxford University Press.
135
Literaturverzeichnis
Sato, K. & Sudo, S. (1999). Small-scale solid-state fermentations. In Manual of industrial microbiology and biotechnology, pp. 61-79. Edited by A. L. Demain & J. E. Davies. Washington, D.C.: ASM Press. Schmidt, B. F., Woodhouse, L., Adams, R. M., Ward, T., Mainzer, S. E. & Lad, P. J. (1995). Alkalophilic Bacillus sp. strain LG12 has a series of serine protease genes. Appl Environ Microbiol 61, 4490-4493. Setyorini, E., Takenaka, S., Murakami, S. & Aoki, K. (2006). Purification and characterization of two novel halotolerant extracellular proteases from Bacillus subtilis strain FP-133. Biosci Biotechnol Biochem 70, 433-440. Shi, W., Tang, X. F., Huang, Y., Gan, F., Tang, B. & Shen, P. (2006). An extracellular halophilic protease SptA from a halophilic archaeon Natrinema sp. J7: gene cloning, expression and characterization. Extremophiles. Siezen, R. J., de Vos, W. M., Leunissen, J. A. & Dijkstra, B. W. (1991). Homology modelling and protein engineering strategy of subtilases, the family of subtilisin-like serine proteinases. Protein Eng 4, 719-737. Siezen, R. J. (1996). Subtilases: subtilisin-like serine proteases. Adv Exp Med Biol 379, 75-93. Siezen, R. J. & Leunissen, J. A. (1997). Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci 6, 501-523. Sinn-Meyer, B. (2003). Physiologische und molekularbiologische Untersuchungen an hydrolytischen extrazellulären Enzymen aus extremophilen marinen Mikroorganismen unter besonderer Berücksichtigung von Nucleasen. Dissertation, Universität Bremen; FB2-Biologie/Chemie. Skerra, A. (1994). Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 151, 131-135. Smith, C. A., Toogood, H. S., Baker, H. M., Daniel, R. M. & Baker, E. N. (1999). Calcium-mediated thermostability in the subtilisin superfamily: the crystal structure of Bacillus Ak.1 protease at 1.8 A resolution. J Mol Biol 294, 1027-1040. Sørensen, H. P. & Mortensen, K. K. (2005). Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol 115, 113-128. Sroga, G. E. & Dordick, J. S. (2002). A strategy for in vivo screening of subtilisin E reaction specificity in E. coli periplasm. Biotechnol Bioeng 78, 761-769. Stepanov, V. M., Rudenskaya, G. N., Revina, L. P., Gryaznova, Y. B., Lysogorskaya, E. N., Filippova, I. & Ivanova, II (1992). A serine proteinase of an archaebacterium, Halobacterium mediterranei. A homologue of eubacterial subtilisins. Biochem J 285 ( Pt 1), 281-286.
136
Literaturverzeichnis
Studdert, C. A., Herrera Seitz, M. K., Plasencia Gil, M. I., Sanchez, J. J. & de Castro, R. E. (2001). Purification and biochemical characterization of the haloalkaliphilic archaeon Natronococcus occultus extracellular serine protease. J Basic Microbiol 41, 375-383. Studier, F. W. & Moffatt, B. A. (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189, 113-130. Sweeney, P. & Walker, J. (1993). Proteinase K (EC 3.4.21.14). In Enzymes of molecular biology, pp. 305-312. Edited by M. M. Burell. Totowa, NJ: Humana Press. Taguchi, S., Kojima, S., Terabe, M., Kumazawa, Y., Kohriyama, H., Suzuki, M., Miura, K. & Momose, H. (1997). Molecular phylogenetic characterization of Streptomyces protease inhibitor family. J Mol Evol 44, 542-551. Takagi, H., Takahashi, T., Momose, H., Inouye, M., Maeda, Y., Matsuzawa, H. & Ohta, T. (1990). Enhancement of the thermostability of subtilisin E by introduction of a disulfide bond engineered on the basis of structural comparison with a thermophilic serine protease. J Biol Chem 265, 6874-6878. Tatum, E. L. & Lederberg, J. (1947). Gene Recombination in the Bacterium Escherichia coli. J Bacteriol 53, 673-684. Teo, J. W., Zhang, L. H. & Poh, C. L. (2003). Cloning and characterization of a novel lipase from Vibrio harveyi strain AP6. Gene 312, 181-188. Terada, I., Kwon, S. T., Miyata, Y., Matsuzawa, H. & Ohta, T. (1990). Unique precursor structure of an extracellular protease, aqualysin I, with NH2- and COOH-terminal pro-sequences and its processing in Escherichia coli. J Biol Chem 265, 6576-6581. Thomas, J. G., Ayling, A. & Baneyx, F. (1997). Molecular chaperones, folding catalysts, and the recovery of active recombinant proteins from E. coli. To fold or to refold. Appl Biochem Biotechnol 66, 197-238. Tichy, H.-V. & Simon, R. (1994). Effiziente Analyse von Microorganismen mit PCR-Fingerprintverfahren. BIOforum, 499-505. Turner, A. J. (2003). Exploring the structure and function of zinc metallopeptidases: old enzymes and new discoveries. Biochem Soc Trans 31, 723-727. Usami, R., Fukushima, T., Mizuki, T., Yoshida, Y., Inoue, A. & Horikoshi, K. (2005). Organic solvent tolerance of halophilic archaea, Haloarcula strains: effects of NaCl concentration on the tolerance and polar lipid composition. J Biosci Bioeng 99, 169-174. van den Burg, B. (2003). Extremophiles as a source for novel enzymes. Curr Opin Microbiol 6, 213-218. Vargas, C. & Nieto, J. J. (2004). Genetic tools for the manipulation of moderately halophilic bacteria of the family Halomonadaceae. Methods Mol Biol 267, 183-208.
137
Literaturverzeichnis
Ventosa, A., Nieto, J. J. & Oren, A. (1998). Biology of moderately halophilic aerobic bacteria. Microbiol Mol Biol Rev 62, 504-544. Venugopal, M. & Saramma, A. V. (2006). Characterization of alkaline protease from Vibrio fluvialis strain VM10 isolated from a mangrove sediment sample and its application as a laundry detergent additive. Process Biochemistry 41, 1239-1243. Verma, M. (1988). High molecular weight DNA isolation by guanidine hydrochloride or guanidinium isothiocyanate treatment. Biotechniques 6, 848, 850, 853. Vidyasagar, M., Prakash, S., Litchfield, C. & Sreeramulu, K. (2006a). Purification and characterization of a thermostable, haloalkaliphilic extracellular serine protease from the extreme halophilic archaeon Halogeometricum borinquense strain TSS101. Archaea 2, 51-57. Vidyasagar, M., Prakash, S. B. & Sreeramulu, K. (2006b). Optimization of culture conditions for the production of haloalkaliphilic thermostable protease from an extremely halophilic archaeon Halogeometricum sp. TSS101. Lett Appl Microbiol 43, 385-391. Vieira, J. & Messing, J. (1987). Production of single-stranded plasmid DNA. Methods Enzymol 153, 3-11. Villaverde, A. & Carrio, M. M. (2003). Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol Lett 25, 1385-1395. Vogt, M. (2000). Taxonomische Untersuchungen von halophilen Bakterien aus marinen Standorten mit erhöhter Salzkonzentration. Diplomarbeit, Universität Bremen, FB2-Biologie/Chemie von Heijne, G. (1983). Patterns of amino acids near signal-sequence cleavage sites. Eur J Biochem 133, 17-21. Wang, J., Hartling, J. A. & Flanagan, J. M. (1997). The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 91, 447-456. Wejse, P. L., Ingvorsen, K. & Mortensen, K. K. (2003). Purification and characterisation of two extremely halotolerant xylanases from a novel halophilic bacterium. Extremophiles 7, 423-431. Welder, F., McCorquodale, E. M. & Colyer, C. L. (2002). Proteinase assay by capillary electrophoresis employing fluorescence-quenched protein-dye conjugates. Electrophoresis 23, 1585-1590. Wilk, S. (2001). Purification of proteolytic enzymes. In Proteolytic enzymes: a practical approach. Edited by R. Beynon & J. S. Bond. New York: Oxford University Press.
138
Literaturverzeichnis
Winnacker, E.-L. (1985). Gene und Klone - Eine Einführung in die Gentechnologie. Weinheim: VCH Verlagsgesellschaft. Wladyka, B., Puzia, K. & Dubin, A. (2005). Efficient co-expression of a recombinant staphopain A and its inhibitor staphostatin A in Escherichia coli. Biochem J 385, 181-187. Wu, J., Bian, Y., Tang, B., Chen, X., Shen, P. & Peng, Z. (2004). Cloning and analysis of WF146 protease, a novel thermophilic subtilisin-like protease with four inserted surface loops. FEMS Microbiol Lett 230, 251-258. Yabuta, Y., Takagi, H., Inouye, M. & Shinde, U. (2001). Folding pathway mediated by an intramolecular chaperone: propeptide release modulates activation precision of pro-subtilisin. J Biol Chem 276, 44427-44434. Yabuta, Y., Subbian, E., Oiry, C. & Shinde, U. (2003). Folding pathway mediated by an intramolecular chaperone. A functional peptide chaperone designed using sequence databases. J Biol Chem 278, 15246-15251. Yang, H. J. & Tsou, C. L. (1995). Inactivation during denaturation of ribonuclease A by guanidinium chloride is accompanied by unfolding at the active site. Biochem J 305 ( Pt 2), 379-384. Yoshimune, K., Yamashita, R., Masuo, N., Wakayama, M. & Moriguchi, M. (2004). Digestion by serine proteases enhances salt tolerance of glutaminase in the marine bacterium Micrococcus luteus K-3. Extremophiles 8, 441-446.
139
Anhang
77 Anhang AnhangTab. 35: Für die Voruntersuchungen ausgewählte Stämme und einige ihrer Eigenschaften. Türkis unterlegt sind 9 Stämme, die aufgrund ihrer Eigenschaften für weitere Untersuchungen ausgewählt wurden.Stamm Isolations-
bedingungen (% NaCl/ pH)1)
ARDRA-Gruppe2)
Wachstums-rate(12% NaCl, pH 8) 3)
Peptidase-produktion aufAgarplatte3)
Submerse Peptidase-produktion bei 12% NaCl; Maximale Aktivität (Inkubationszeit) 3)
Restaktivität bei 1 M GuSCN, 0,1% SDS, 5 mM CaCl2, 50 mM Tris-HCl pH 8 im Azocaseinassay3)
Quellen: 1)(Sinn-Meyer, 2003); 2)(Vogt, 2000); 3)Ergebnisse dieser Arbeit. 4)Aufgrund des gleichen Ursprungs der Isolate B12/10-1 (#748) und B12/10-2 (#749) aus Ansatz B12/10 wurde letzterer nicht in die Auswahl übernommen. Zeichenerklärung: AzU: Azocaseinunits; n.b.: nicht bestimmt. Emerse Peptidase-Aktivität: -= keine Aktivität; += kleiner Aktivitätsbereich ~1 mm; ++=mittlerer Aktivitätsbereich ~2-3 mm ; +++= großer Aktivitätsbereich >3 mm
140
Anhang
Tab. 36: Fraktionspools mit proteolytischer Aktivität (Fluoreszenzassay) in der hydrophoben Interaktionschromatografie.
Stamm Kulturvolumen [ml]
Gradientenlänge [min] Fraktionspool Vereinigte
Fraktionen Aktivität [FlU/ml]
A 120-134 ml 11,9SJ2/3-1 (#073) 125 120 B 140-174 ml 104,3
A 120-134 ml 7,6SJ5Ü/8 (#098) 125 120 B 144-178 ml 38,8
A 120-134 ml 19,1SJ5Ü/12 (#140) 150 120 B 144-176 ml 157,3
A 120-134 ml 20,5SJ6S/14 (#148) 150 120 B 140-174 ml 163,7
A 112-126 ml 30,2B3/1-2 (#418) 270 120 B 136-176 ml 198,7
A 122-136 ml 17,8B3/14 (#430) 120 120 B 144-176 ml 165,4
A - -B8/26-2 (#504) 150 120 B 144-176 ml 178,5
A 128-152 ml 70,8B12/10-1 (#748) 350 180 B 190-242 ml 407,7
A - -B12/16 (#795) 150 120 B 144-176 ml 119,5
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
SJ2/3-1 (#073) SJ5Ü/8 (#098)
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
SJ2/3-1 (#073) SJ5Ü/8 (#098) Abb. 33: Einfluss des pH-Wertes auf die proteolytische Aktivität der Peptidase A (a) und B (b) der Stämme SJ2/3-1 (#073) und SJ5Ü/8 (#098). Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
SJ5Ü/12 (#140) SJ6S/14 (#148)
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
SJ5Ü/12 (#140) SJ6S/14 (#148)
b
b
a
a
Abb. 34: Einfluss des pH-Wertes auf die proteolytische Aktivität der Peptidase A (a) und B (b) der Stämme SJ5Ü/12 (#140) und SJ6S/14 (#148). Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
141
Anhang
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
B3/14 (#430)
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
B3/14 (#430) B8/26-2 (B8/26-2 (#504))
a b
Abb. 35: Einfluss des pH-Wertes auf die proteolytische Aktivität der Peptidase A (a) des Stammes B3/14 (#430) und B (b) der Stämme B3/14 (#430) und B8/26-2 (#504). Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
0%
20%
40%
60%
80%
100%
3 4 5 6 7 8 9 10 11 12 13pH
Rel
ativ
e Pe
ptid
ase-
Akt
ivitä
t
B12/16 (#795) Abb. 36: Einfluss des pH-Wertes auf die proteolytische Aktivität der Peptidase B des Stammes B12/16 (#795). Die relative Peptidase-Aktivität entspricht der prozentualen Aktivität des Optimums.
142
Anhang
Abb. 37: Nukleotidsequenz eines Teilbereiches des Inserts von Klon F13 aus Stamm B12/10-1 (#748). Erklärung zu den farbigen Nukleotiden: rot: Start- und Stoppcodon; türkis: mögliche Ribosomenbindungsstelle; gelb hervorgehoben: kodierende Sequenz des hypothetischen Signalpeptids der Peptidase ProAS(#748).
Abb. 38: Nukleotidsequenz eines Teilbereiches des Inserts von Klon F114 aus Stamm B3/1-2 (#418). Erklärung zu den farbigen Nukleotiden: rot: Start- und Stoppcodon; gelb hervorgehoben: kodierende Sequenz des hypothetischen Signalpeptids der Peptidase ProAS(#418).
Abb. 39: Nukleotidsequenz eines Teilbereiches des Inserts von Klon F9 aus Stamm B12/16 (#795). Erklärung zu den farbigen Nukleotiden: rot: Start- und Stoppcodon; gelb hervorgehoben: kodierende Sequenz des hypothetischen Signalpeptids der Peptidase ProAS(#795).
Abb. 40: Alignment der Aminosäuresequenz von ProAS(#748), Aqualysin I und Proteinase K. Homologe Bereiche sind mit einem schwarzen Kasten umschlossen. Die Sequenzen der in Aqualysin I und Proteinase K nachgewiesenen N-terminalen Propeptide (Siezen & Leunissen, 1997) und des in Aqualysin I nachgewiesenen C-terminalen Propeptids (Terada et al., 1990) wurden rot markiert. Die Cysteinreste für eine hypothetische (in ProAS(#748)) bzw. nachgewiesene (in Aqualysin I) SH-Bindungsstelle wurden blau hervorgehoben. Grün markiert sind die Aminosäurereste der katalytischen Triade. Die konservierten Regionen, die der Einteilung der Subtilisin-ähnlichen Peptidasen in 6 Familien nach Siezen & Leunissen (1997) zugrunde liegen, wurden gelb hervorgehoben.
Hiermit versichere ich, dass ich die vorliegende Dissertation mit dem Titel Heterologe Expression und biochemische Charakterisierung von extrazellulären Peptidasen aus halotoleranten bzw. halophilen Bakterien in Hinblick auf ihre biotechnologische Nutzung selbstständig verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt habe. 28355 Bremen, im Januar 2007





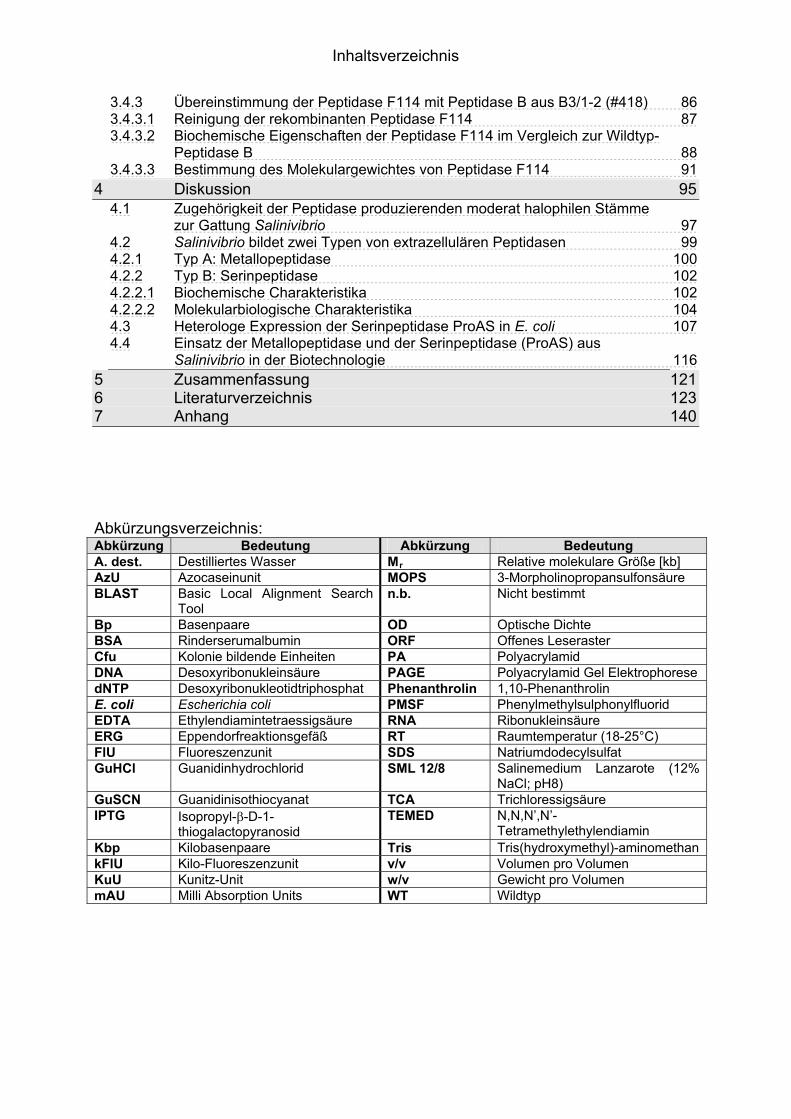











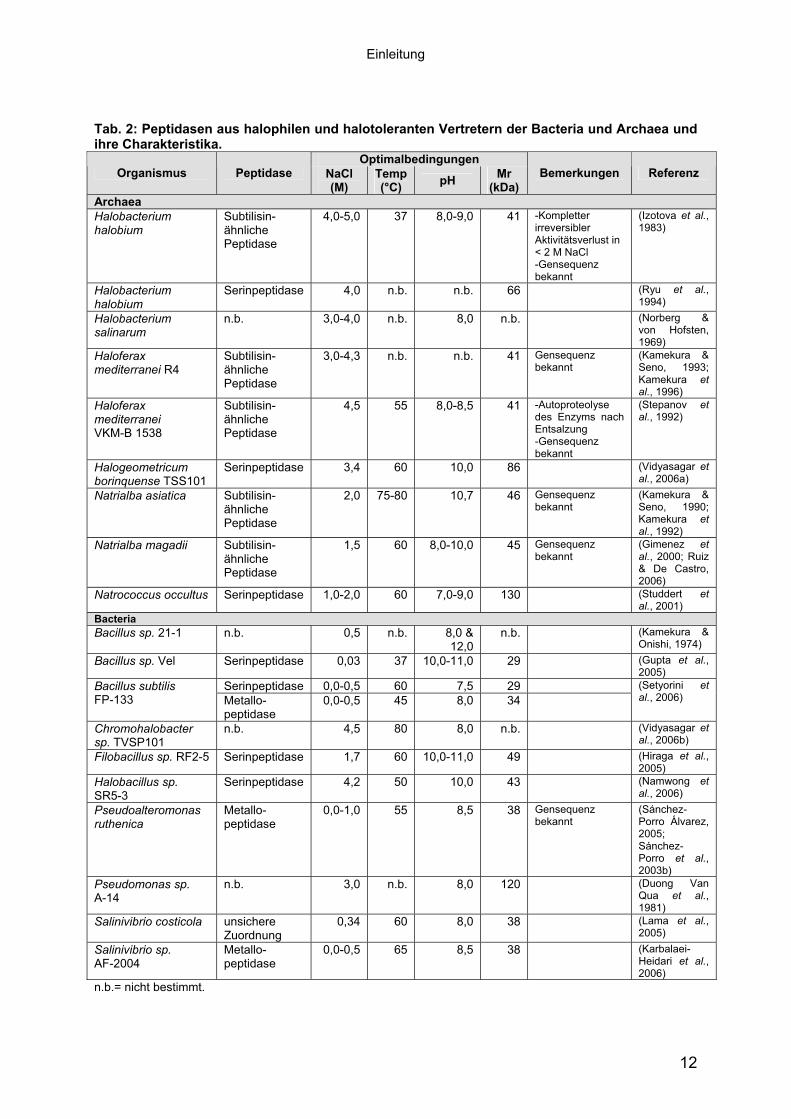












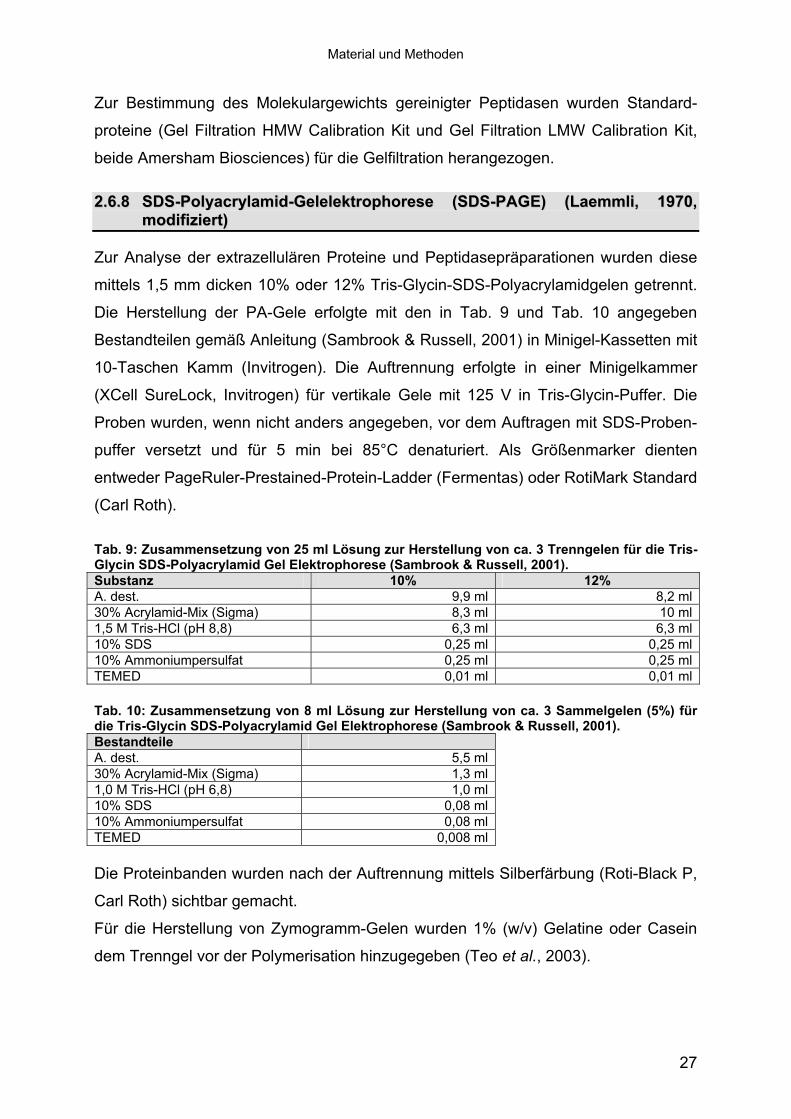










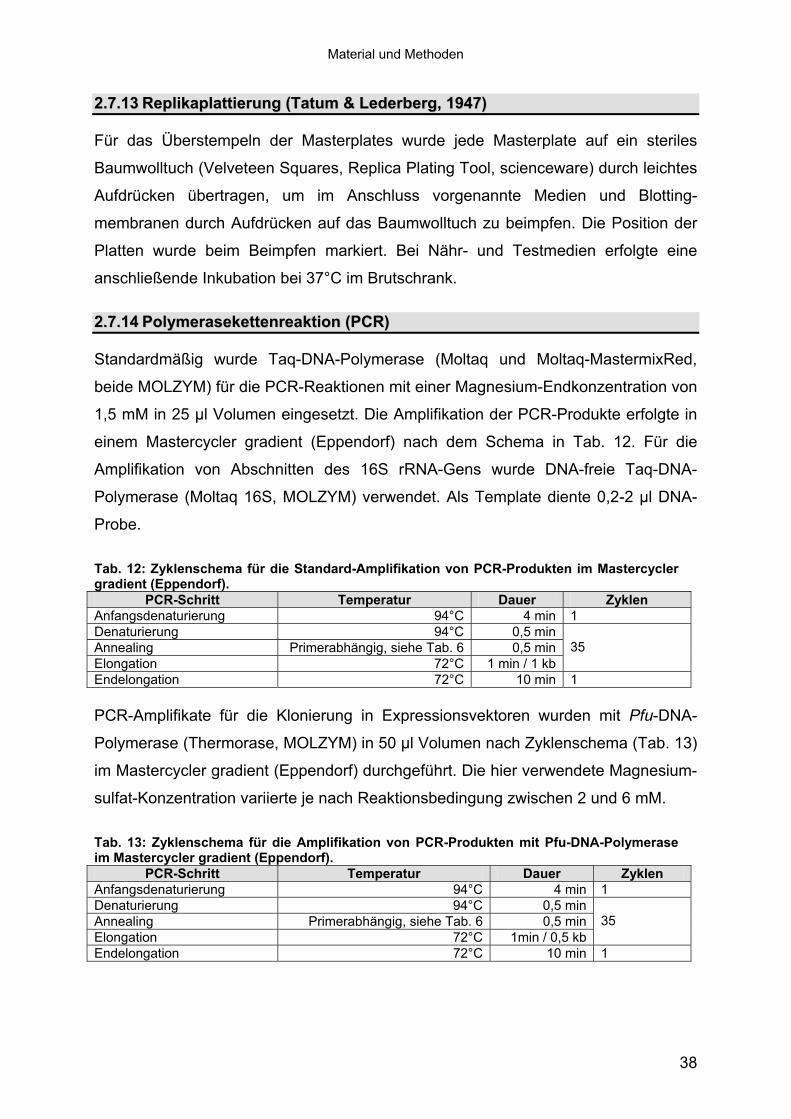




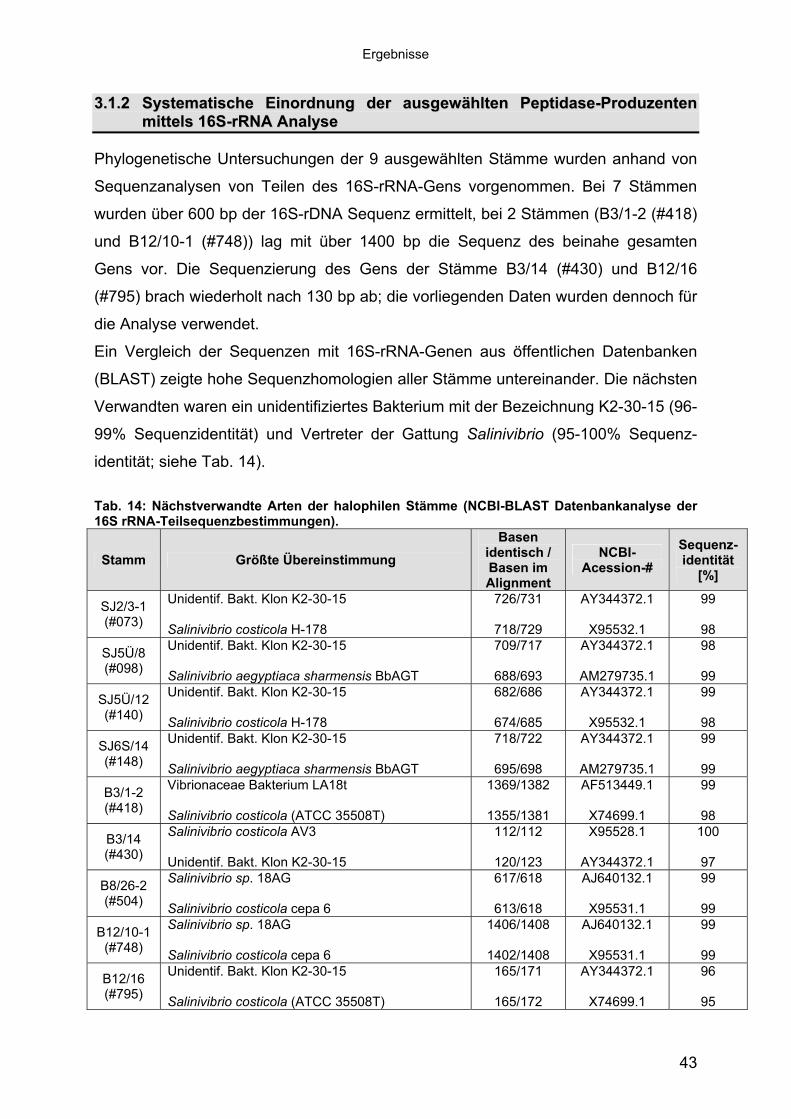
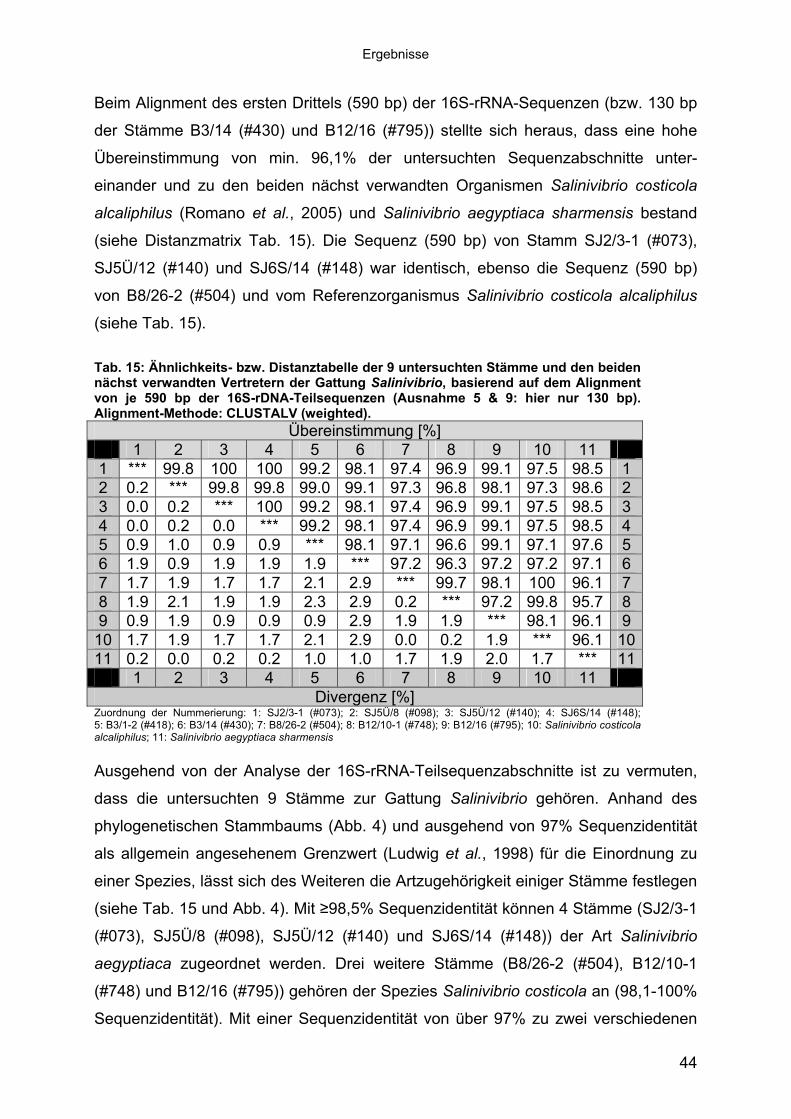












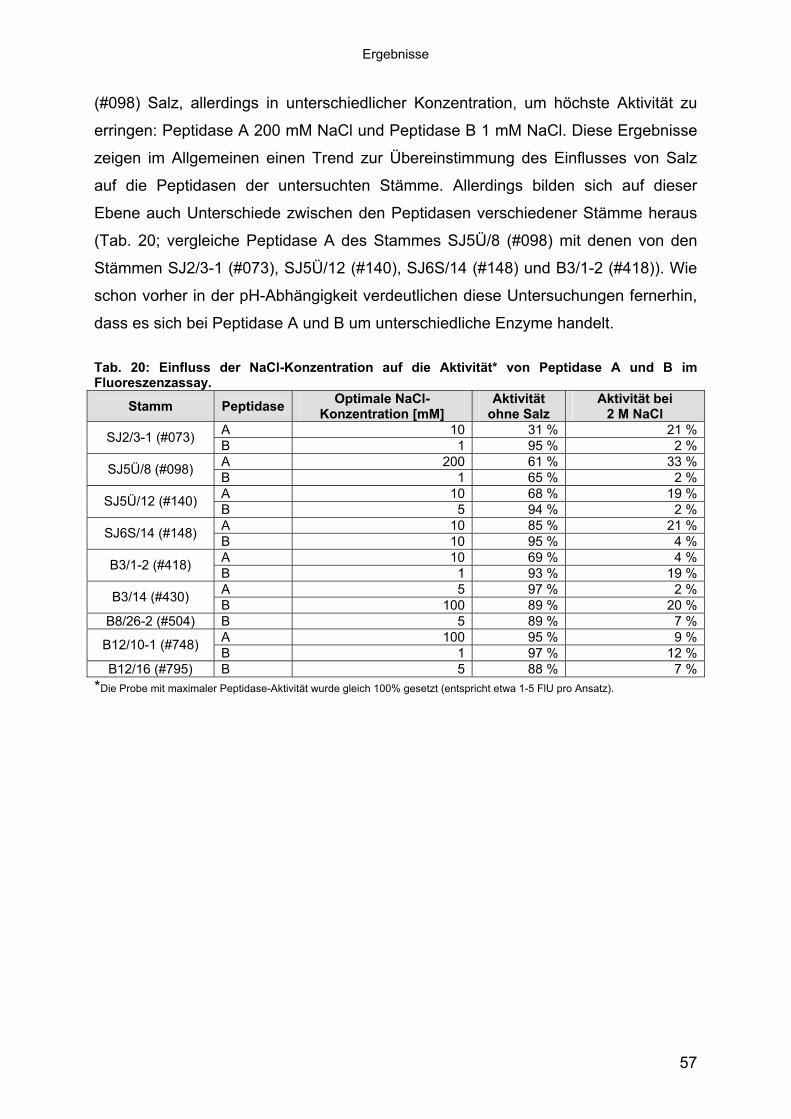

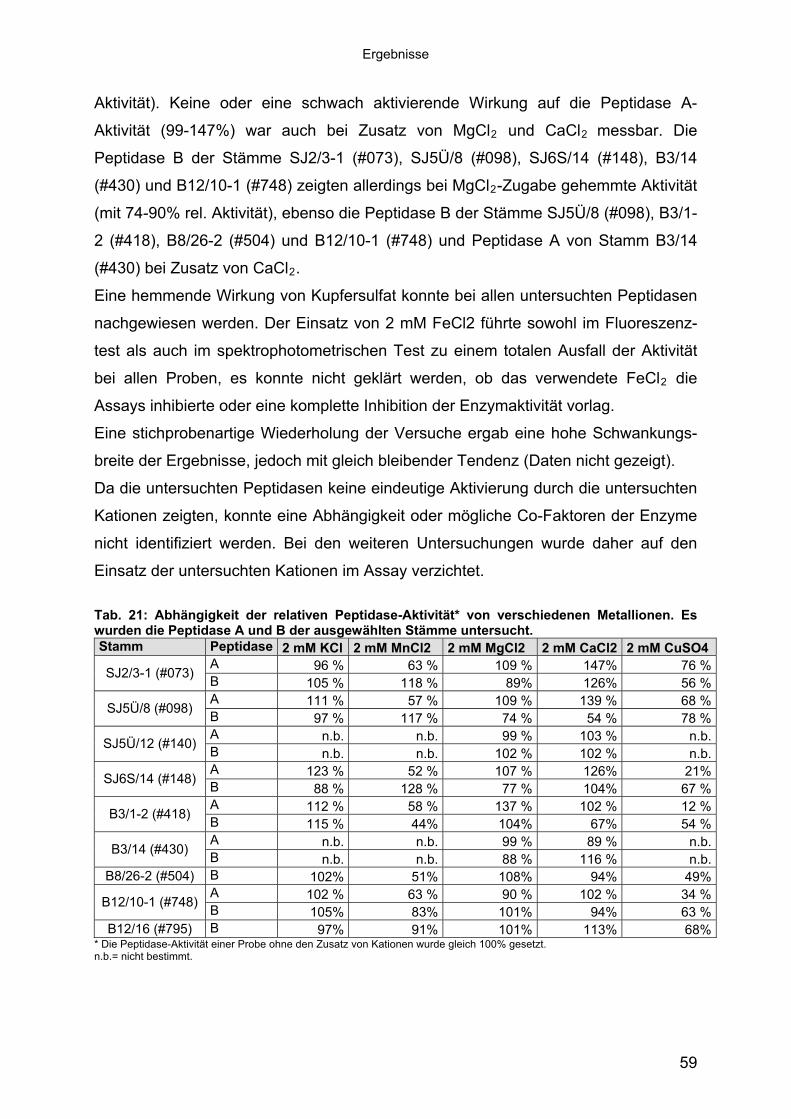

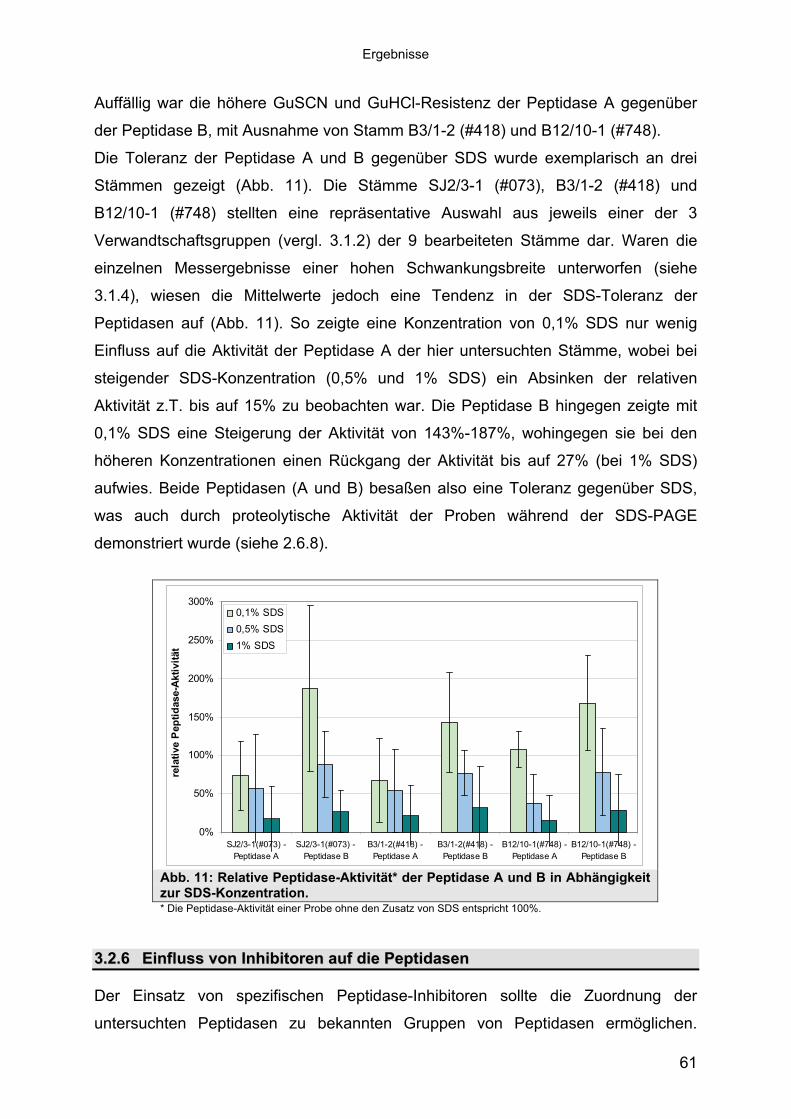
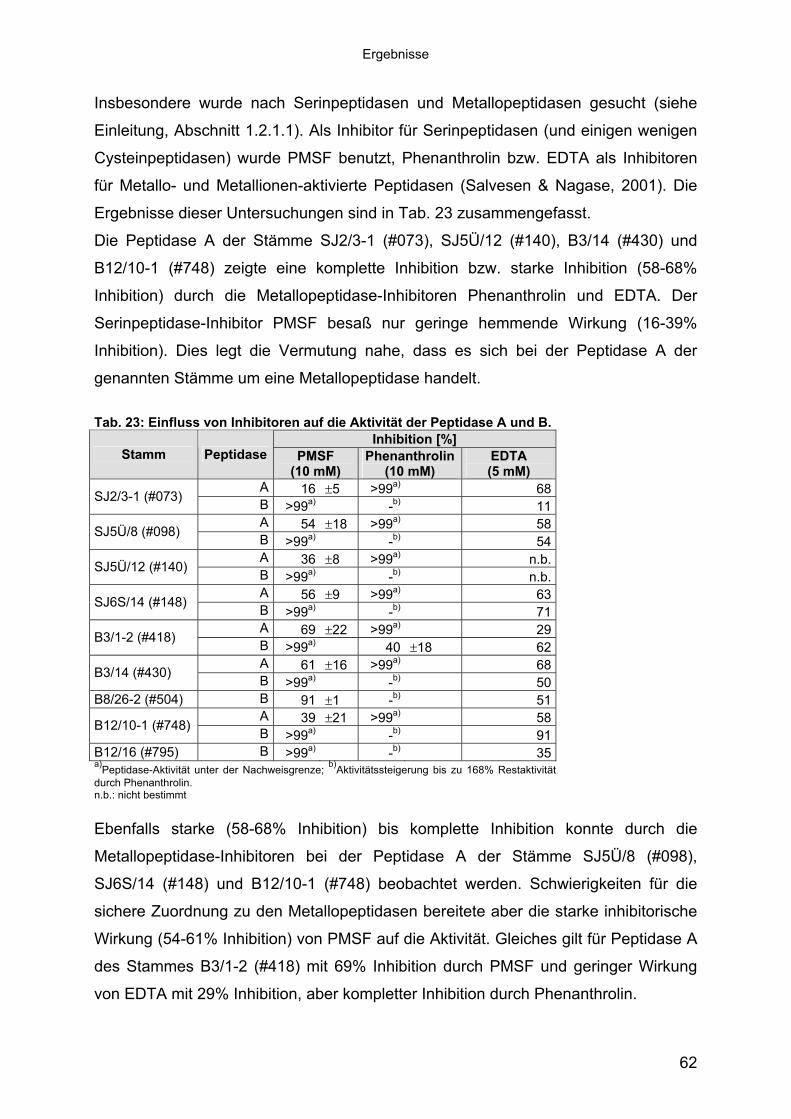

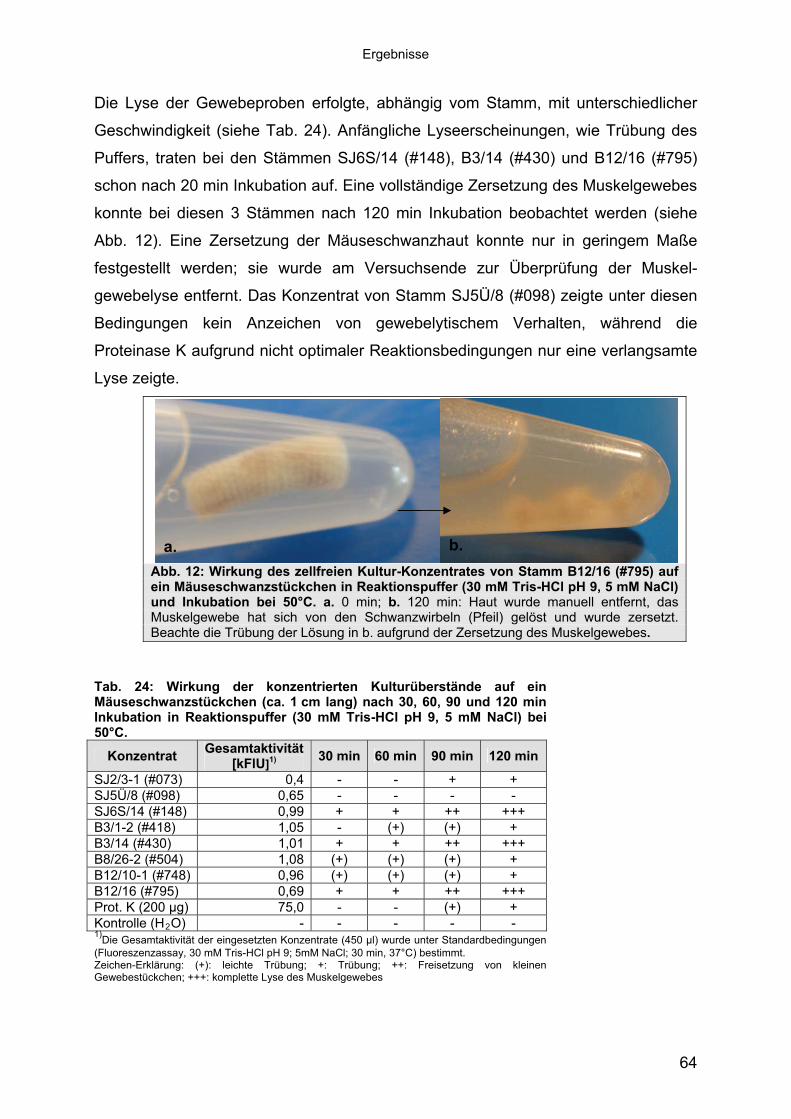


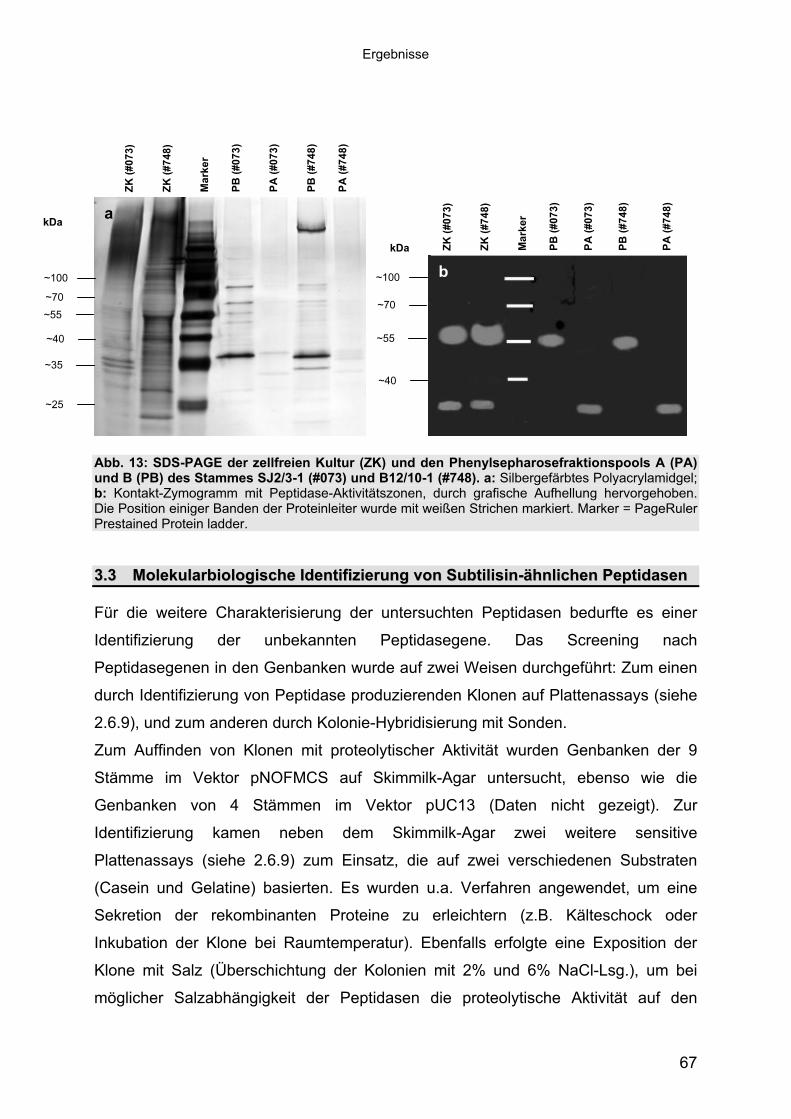












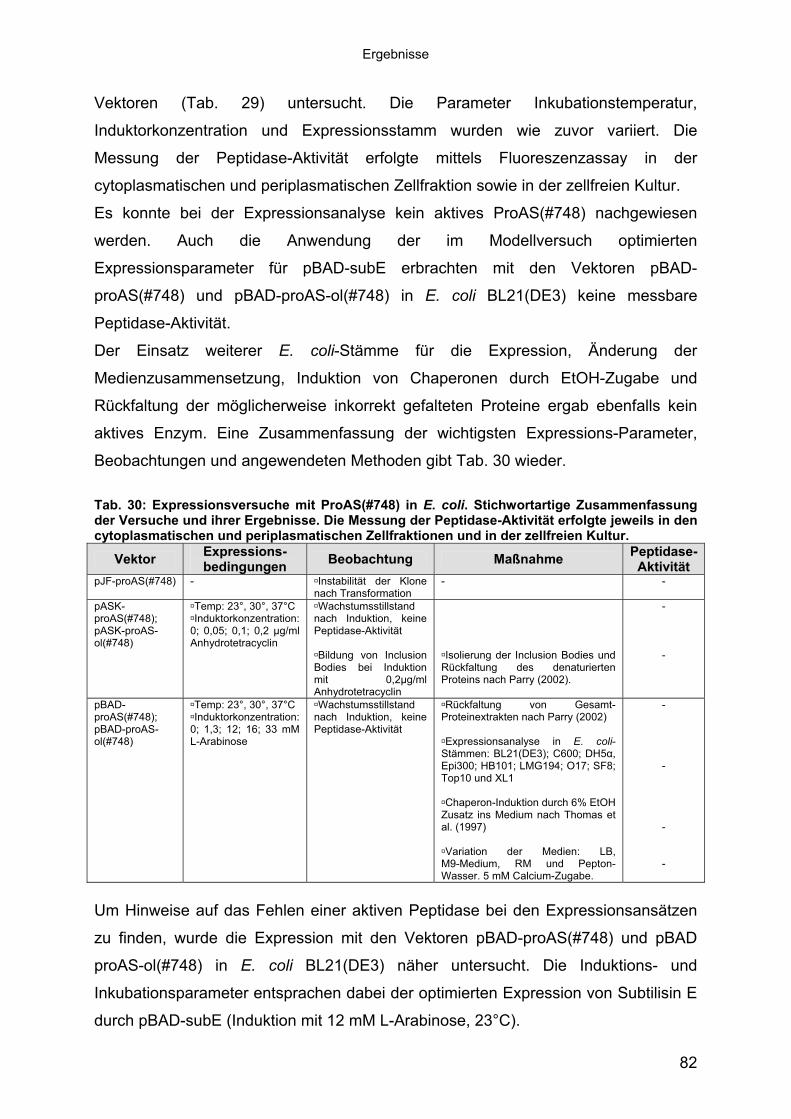



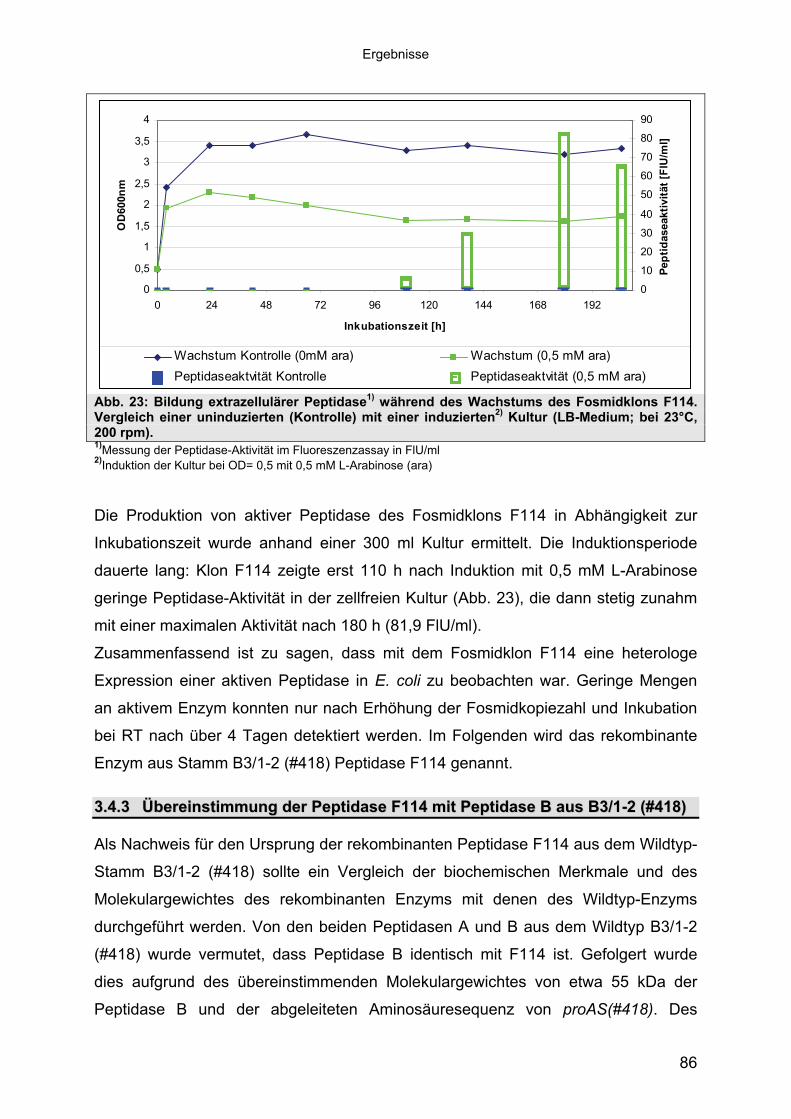


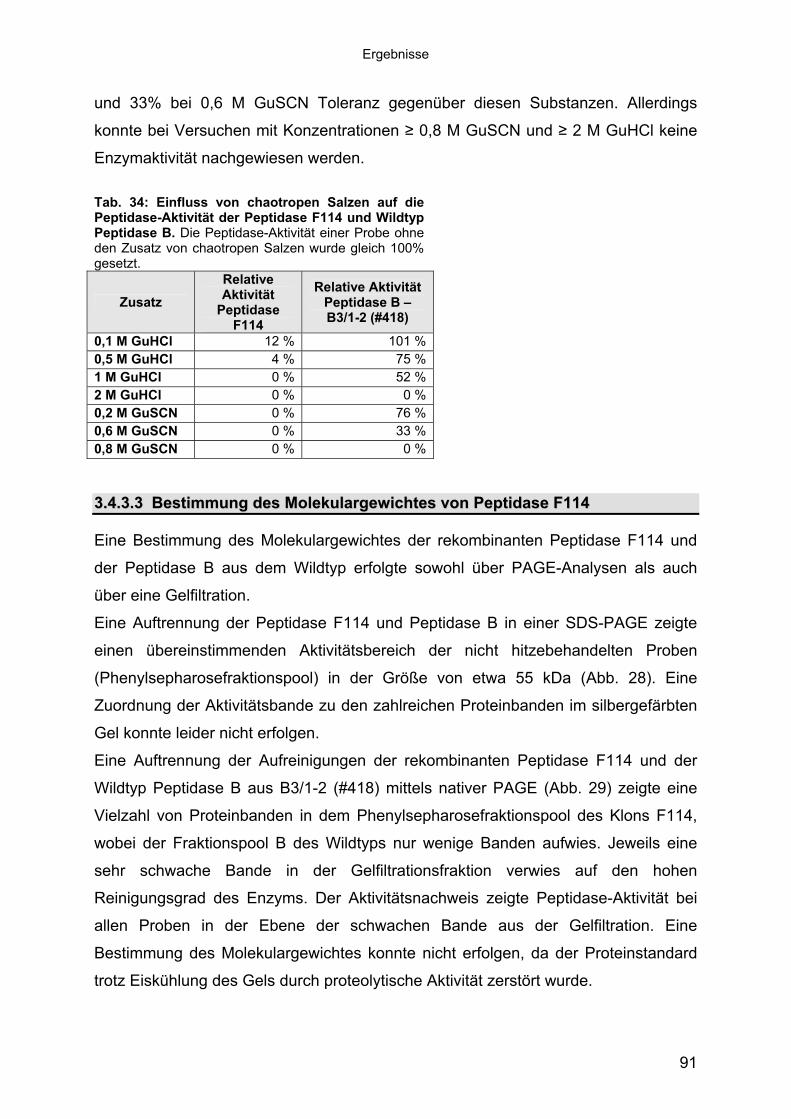
















































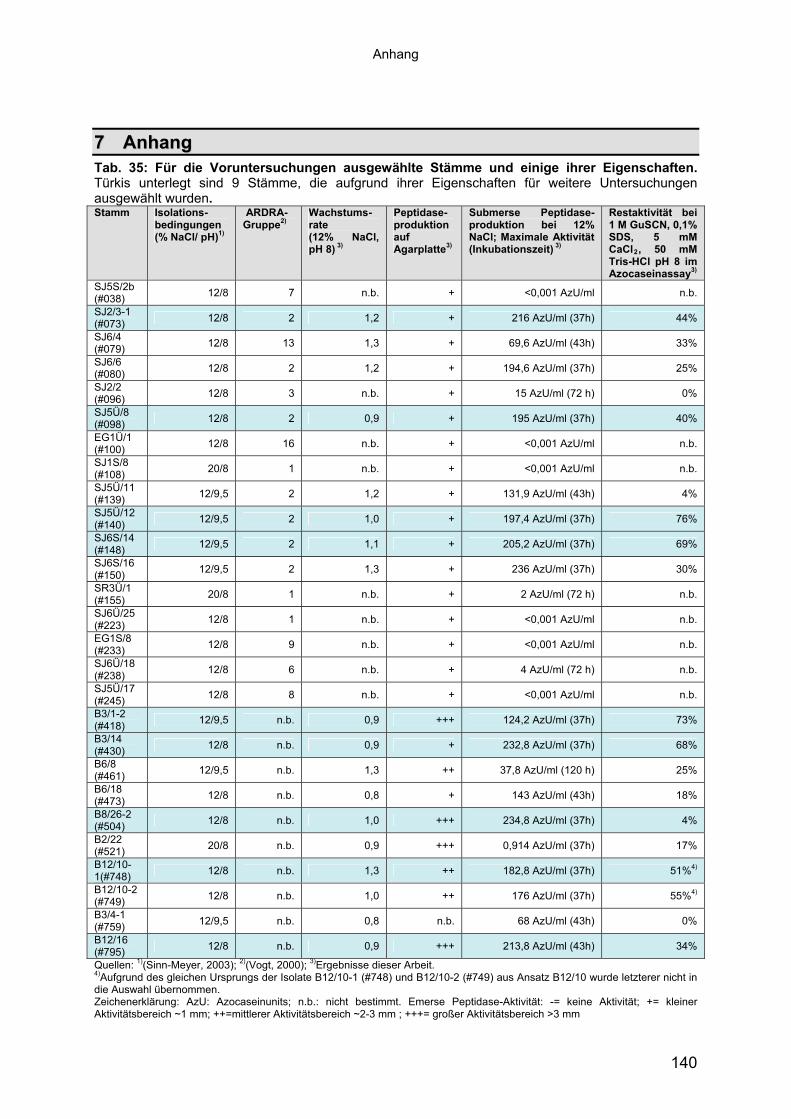

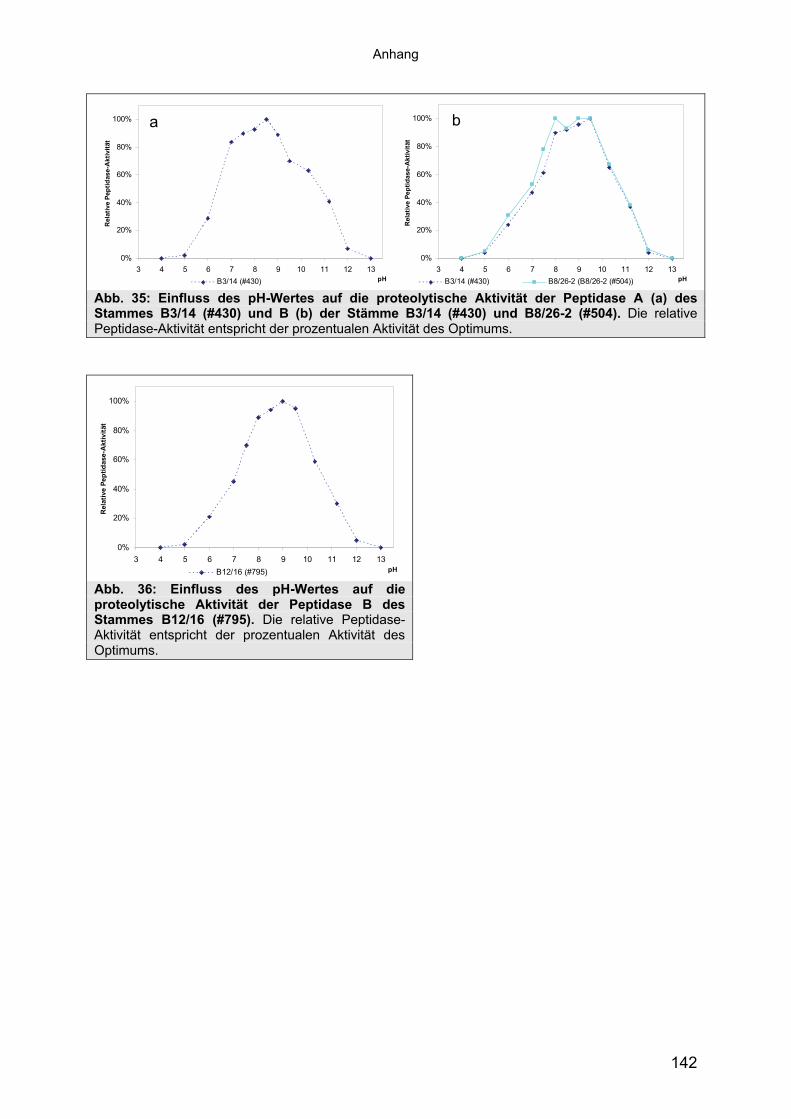


















![Modul AC III - ac2-weber.uni-bayreuth.de · phen Phenanthrolin dabco 1,4-Diazabicyclo[2.2.2]octan salen 2− Bis(salicyliden)ethylendiamin cyclam 1,4,8,12-Tetraazacyclotetradecan](https://static.unterlagen.site/doc/80x56/5d56891088c993c6438b70d7/modul-ac-iii-ac2-weberuni-phen-phenanthrolin-dabco-14-diazabicyclo222octan.jpg)



