S. Langer1 · S. Rudnik-Schöneborn2 · K. Zerres2 · T. Grimm1
1 Abteilung für Medizinische Genetik im Institut für Humangenetik, Universität Würzburg, Biozentrum, Am Hubland, Würzburg
2 Institut für Humangenetik, RWTH, Aachen
Genetisches Modell der autosomal-rezessiv erblichen proximalen spinalen Muskelatrophie
Hintergrund
Grundlage der familiären Risikoberech-nung ist ein genetisches Modell für die betreffende Krankheit. Die Basis des ge-netischen Modells sind u. a. die Mendel-Regeln, das Hardy-Weinberg-Gleichge-wicht, Genfrequenz, Selektion und Muta-tion sowie die Penetranz. Wichtig ist, dass alle Parameter des genetischen Modells sich gegenseitig beeinflussen. Daher kann ein genetisches Modell auch als mathema-tische Gleichung erstellt werden und die Schätzung von unbekannten Parametern ist möglich.
Spinale Muskelatrophie
Bei der spinalen Muskelatrophie (SMA) handelt es sich um eine klinisch und ge-netisch heterogene Gruppe von Erkran-kungen, die durch einen progredienten Untergang der motorischen Vorderhorn-zellen und teilweise auch der motorischen Hirnnervenkerne des Hirnstamms ge-kennzeichnet ist. Diese Arbeit befasst sich ausschließlich mit der häufigsten Form, der proximalen infantilen und juvenilen SMA, die eine der häufigsten autosomal-rezessiv erblichen Erkrankungen darstellt. Man teilt die proximalen spinalen Mus-kelatrophien in 3 Formen ein, die sich v. a. nach den erreichten Meilensteinen und damit dem Krankheitsbeginn und Todesalter unterscheiden. Bei der häufigs-ten der 3 Formen, der akuten infantilen Form Werdnig-Hoffmann (SMA Typ I), wird Sitzen nicht erlernt. Patienten mit der chronischen infantilen intermediären
Form (SMA Typ II), können sitzen, aber nicht gehen, während Erkrankte der chro-nischen juvenilen Form vom Typ Kugel-berg-Welander (SMA Typ III) auch Ge-hen erlernen.
Für das Krankheitsbild ist primär das Survival-motor-neuron(SMN)-Gen ver-antwortlich. Die Region um das SMN-Gen hat sich im Laufe der Evolution beim Menschen dupliziert und liegt spiegelbild-lich vor, sodass mehrere Gene als Pseudo-gene vorliegen. Das SMN-Gen kommt da-her in 2 Kopien vor: telSMN (entspricht SMN1) und cenSMN (entspricht SMN2, . Abb. 1). Beide sind zwar von der DNA-Sequenz annähernd identisch, jedoch ist das SMN1-Gen das für die Krankheitsent-stehung bestimmende Gen, da das SMN2-Gen im Vergleich zum SMN1-Gen nur einen sehr kleinen Anteil an vollständi-gem SMN-Protein kodiert.
In der Normalbevölkerung findet man häufig pro Allel jeweils eine SMN1-Kopie, in einigen Fällen kann das Normalallel auch als Duplikation (meist 2 SMN1-Ko-pien) vorliegen. Bei den Erkrankten ist die häufigste Mutation ein Verlust der Exons 7 und 8 im SMN1-Gen. Selten liegen auch Punktmutationen im SMN1-Gen vor, oder große Deletionen, die über das SMN1-Gen hinausgehen.
Die Anzahl der SMN2-Kopien kann den Krankheitsverlauf der SMA beein-flussen [23, 45]. Darüber hinaus sind we-nige SMA-Familien beobachtet worden, bei denen eine homozygote SMN1-De-letion nicht zu einem kranken Phänotyp führte [12, 27].
Literaturdaten – Normalallele
In Anlehnung an vorhandene Litera-tur wurden für die Allelfrequenzen a, b, c und d die Bezeichnungen beibehalten und weitere Parameter neu benannt bzw. hinzugefügt (. Tab. 1), wie im Folgenden aufgeführt. Die beiden Normalallele sind a (1 SMN1-Kopie) und b (2 SMN1-Kopien; . Abb. 2).
Verschiedene Studien, in denen die Gesamtkopienzahl in der Normalbevölke-rung untersucht wurde, sind in . Tab. 2 aufgelistet. Es wurde darauf geachtet, dass hier nur Arbeiten angeführt werden, die von echten Kontrollpersonen ohne Fami-liengeschichte mit SMA berichten. Bei al-len untersuchten Personen lag mindestens eine SMN1-Kopie vor.
Da signifikante Unterschiede zwischen verschiedenen ethnischen Zugehörigkei-ten in der Heterozygotenfrequenz erkannt wurden, werden nur Daten der europäi-schen bzw. kaukasischen Bevölkerung be-rücksichtigt. Bei der amerikanischen Stu-die von Sugarman et al. [36] wird nur der Anteil der Kaukasier und bei der südafri-kanischen Studie von Labrum et al. [19] nur der Anteil der weißen Bevölkerung verwendet. Da es sich bei den Arbeiten der Autoren Hendrickson et al. [15] so-wie Sugarman et al. [36] mit hoher Wahr-scheinlichkeit um ähnliches bzw. gleiches Probandengut handelt, wird nur die zu-letzt genannte größere Studie dargestellt.
Bei fast 98% der Normalbevölkerung (25.496 von 26.040) liegen 2 oder mehr SMN1-Kopien vor, während bei den übri-gen etwa 2% (544 von 26.040) der Kont-
337Medizinische Genetik 3 · 2013 |
Schwerpunktthema: Ausgewählte Aspekte von Motoneuronerkrankungen
rollpersonen nur eine SMN1-Kopie gefun-den werden konnte. Letzteres entspricht einer Rate von etwa 1:48. Bei einem Kon-fidenzniveau von 95% bedeutet das für die Grenzen des Vertrauensbereichs 1:52–1:44. Dieser Prozentsatz wird häufig als Heterozygotenfrequenz der SMA bezeich-net. Die Berechnung ist so jedoch nicht ganz korrekt, da es auch möglich ist, dass Personen mit einer SMN1-Kopienzahl von 2 oder gar 3 heterozygot sein können (. Tab. 7).
Literaturdaten – Defektalallele
Bei den mutierten Allelen wird zwischen den Anteilen der einfachen Deletion A und der großen Deletion G sowie der Punktmutation D unterschieden. Die ent-sprechenden Allelfrequenzen werden im Folgenden a, g und d genannt (. Abb. 3).
SMA-Patienten mit einer homozygo-ten Deletion (aa) haben 0 SMN1-Kopien. Dagegen haben Patienten mit einer De-letion und einer Punktmutation (com-pound Heterozygote, z. B. ad) eine SMN1-Kopie und lassen sich daher mithilfe von MLPA („multiplex ligation-dependent probe amplification“) nicht von Heterozy-goten (ab) unterscheiden. . Tab. 3 zeigt verschiedene Studien, die Patienten hin-sichtlich ihrer SMN1-Kopienzahl unter-sucht haben.
Bei der Arbeit von Jedrzejowska et al. [18] wurden bei weiteren 4 von insgesamt 269 Patienten je 2 SMN1-Kopien gefun-den. Da diese jedoch nicht näher unter-
sucht wurden und somit eine SMN-un-abhängige Erkrankung nicht auszuschlie-ßen ist, werden sie hier in der Gesamtzahl nicht berücksichtigt. Ebenso wurden wei-tere 23 der insgesamt 525 Patienten mit mindestens 2 SMN1-Kopien von Wirth et al. [43] nicht mit angeführt, bei denen auch nach genauerer Untersuchung keine Mutationen im SMN-Gen gefunden wer-den konnten. Aus der Studie von Labrum et al. [19] werden hier aus oben genannten Gründen nur Patienten der weißen Bevöl-kerung berücksichtigt.
Es erwiesen sich 1780 von insgesamt 1864 Patienten als homozygot deletiert oder compound heterozygot für die klas-sische Deletion und eine große Deletion, was etwa 95,5% (94,5–96,4%) der Patien-ten entspricht.
Bei der Überprüfung der Gesamtko-pienzahl kann allein noch nicht zwischen einfacher und großer Deletion unter-schieden werden. Ein homozygotes Auf-treten einer solchen „large scale“ Deletion (gg) wird in der Literatur nicht beschrie-ben und ist sehr wahrscheinlich nicht mit dem Leben vereinbar. Auch über hetero-zygote große Deletionen bei SMA-Patien-ten gibt es nur sehr wenige Arbeiten, die in . Tab. 4 zusammengefasst sind. Von den 355 Patienten sind demnach 25 com-pound heterozygot für eine große Dele-tion. Dies entspricht einer Rate von etwa 7,0% (4,6–10,2%).
Genotypen
Unter der Annahme von 2 Normalallelen (b und c; . Abb. 2) und 3 Defektallelen (a, d und g; . Abb. 3) können 15 verschie-dene Genotypen vorliegen (. Tab. 5). Pa-tienten ohne SMN1-Kopie können als Ge-notypen sowohl aa als auch ag haben. Bei einer Person aus der Normalbevöl-kerung mit einer SMN1-Kopienzahl von 2 handelt es sich am häufigsten um den Genotyp bb. Es könnte sich jedoch auch um einen Überträger mit dem Genotyp ac, bd oder cg handeln. Aufgrund der un-vollständigen Penetranz ist darüber hin-aus, wenn auch äußerst selten, dd trotz ge-sunden Phänotyps denkbar. SMA-Patien-ten können neben der häufigsten Form mit 0 SMN1-Kopien (aa und ag) auch eine SMN1-Kopie (ad oder gd) oder sogar 2 SMN1-Kopien (dd) haben.
. Tab. 6 soll einen Überblick über die verschiedenen Genotypen und ihrer mög-lichen Phänotypen verschaffen, unter der Voraussetzung, dass eine unvollständi-ge Penetranz vorliegt, die für alle in Frage kommenden Genotypen gleichermaßen ausgeprägt ist.
Bestimmung der Parameter im genetischen Modell
Schätzung der AllelfequenzenDie homozygot und compound hete-
rozygot Gesunden (p2)sind:
(1)
Die heterozygoten Personen (2pq) setzen sich zusammen aus:
(2)Die homozygoten und compound hetero-zygoten SMA-Patienten (q2) sind:
S. Langer · S. Rudnik-Schöneborn · K. Zerres · T. GrimmGenetisches Modell der autosomal-rezessiv erblichen proximalen spinalen Muskelatrophie
ZusammenfassungDie proximale infantile und juvenile spina-le Muskelatrophie (SMA) ist eine der häu-figsten autosomal-rezessive Erbkrankheiten. Man unterteilt die Patienten in 3 Gruppen, SMA Typ I-III, abhängig von der Schwere der Erkrankung (den erreichten Meilensteinen). Das hauptsächlich verantwortliche Gen, das Survival-motor-neuron(SMN1)-Gen, ist auf Chromosom 5 lokalisiert. Während das Nor-malallel meist mit einer oder 2 SMN1-Kopien vorliegt, sind die Defektallele bei den meisten Patienten von einer Deletion betroffen; bei einigen liegen Punktmutationen vor. Bei den Deletionen wiederum unterscheidet man zwischen einfacher und großer Deletion, die über das SMN1-Gen hinausgeht. Ein homo-zygotes Auftreten letzterer führt zu pränata-ler Letalität.
Für die vorliegende Arbeit wurden zahl-reiche in der Literatur verfügbare Daten zur
SMA Typ I-III zusammengetragen und in ihrer Abhängigkeit in einem genetischen Modell zusammengefasst. So war es möglich, fehlen-de Parameter zu schätzen, um genauere Aus-sagen über Genotypen machen zu können. Die einzelnen Allelfrequenzen konnten wie folgt geschätzt werden:
Normalallel b (1 SMN1-Kopie): ≈0,9527; Normalallel c (2 SMN1-Kopien): ≈0,0362; ein-fache Deletion a (0 SMN1-Kopien): ≈0,0104; Punktmutation d (1 SMN1-Kopie): ≈0,0003; große Deletion g (0 SMN1-Kopien): ≈0,0004. Die Genhäufigkeit beträgt etwa 1:90 mit einer Heterozygtenfrequenz von 1:46.
Genetic model of autosomal recessive proximal spinal muscular atrophy
AbstractProximal spinal muscular atrophy (SMA) is one of the most common autosomal reces-sive diseases. According to the achieved milestones, SMA is divided into 3 groups: SMA types I–III. SMA is caused by mutations in the survival motor neuron 1 (SMN1) gene, which is located on chromosome 5. Wild type alleles usually have one or two SMN1 gene copies, disease alleles may show deletions, large scale deletions, or point mutations.
The proposed genetic model is based on published data on SMA types I–III. The com-plex genetic model of SMA allows all param-eters—even those which have not been as-sessed so far—to be calculated. The SMN1 al-
lele frequencies included the following: nor-mal allele b (1 copy of SMN1): ≈0.9527; nor-mal allele c (2 copies of SMN1): ≈0.0362; de-letion a (0 copies of SMN1): ≈0.0104; point mutation d (1 copy of SMN1): ≈0.0003; large scale deletion g (0 copies of SMN1): ≈0.0004. The result is a gene frequency of approxi-mately 1:90 and a carrier frequency of about 1:46.
Gleichung (23) mit Gleichung (15) gleich-gesetzt ergibt:
(24)
Die Gleichung (24) nach A aufgelöst er-gibt folgende quadratische Gleichung:
339Medizinische Genetik 3 · 2013 |
(25)
(26)
(27)
(28)
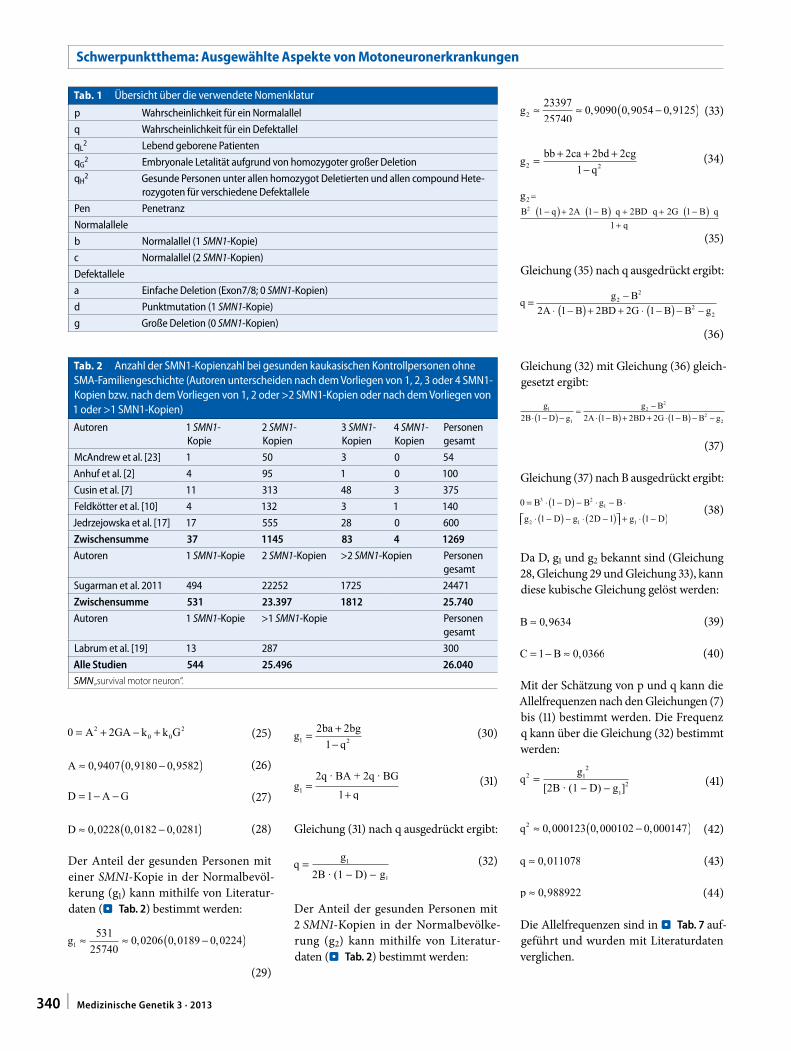
Der Anteil der gesunden Personen mit einer SMN1-Kopie in der Normalbevöl-kerung (g1) kann mithilfe von Literatur-daten (. Tab. 2) bestimmt werden:
(29)
Tab. 2 Anzahl der SMN1-Kopienzahl bei gesunden kaukasischen Kontrollpersonen ohne SMA-Familiengeschichte (Autoren unterscheiden nach dem Vorliegen von 1, 2, 3 oder 4 SMN1-Kopien bzw. nach dem Vorliegen von 1, 2 oder >2 SMN1-Kopien oder nach dem Vorliegen von 1 oder >1 SMN1-Kopien)
Alle Studien 544 25.496 26.040SMN „survival motor neuron“.
Tab. 1 Übersicht über die verwendete Nomenklatur
p Wahrscheinlichkeit für ein Normalallel
q Wahrscheinlichkeit für ein Defektallel
qL2 Lebend geborene Patienten
qG2 Embryonale Letalität aufgrund von homozygoter großer Deletion
qH2 Gesunde Personen unter allen homozygot Deletierten und allen compound Hete-
rozygoten für verschiedene Defektallele
Pen Penetranz
Normalallele
b Normalallel (1 SMN1-Kopie)
c Normalallel (2 SMN1-Kopien)
Defektallele
a Einfache Deletion (Exon7/8; 0 SMN1-Kopien)
d Punktmutation (1 SMN1-Kopie)
g Große Deletion (0 SMN1-Kopien)
(30)
(31)
Gleichung (31) nach q ausgedrückt ergibt:
(32)
Der Anteil der gesunden Personen mit 2 SMN1-Kopien in der Normalbevölke-rung (g2) kann mithilfe von Literatur-daten (. Tab. 2) bestimmt werden:
(33)
(34)
(35)
Gleichung (35) nach q ausgedrückt ergibt:
(36)
Gleichung (32) mit Gleichung (36) gleich-gesetzt ergibt:
(37)
Gleichung (37) nach B ausgedrückt ergibt:
(38)
Da D, g1 und g2 bekannt sind (Gleichung 28, Gleichung 29 und Gleichung 33), kann diese kubische Gleichung gelöst werden:
(39)
(40)
Mit der Schätzung von p und q kann die Allelfrequenzen nach den Gleichungen (7) bis (11) bestimmt werden. Die Frequenz q kann über die Gleichung (32) bestimmt werden:
(41)
(42)
(43)
(44)
Die Allelfrequenzen sind in . Tab. 7 auf-geführt und wurden mit Literaturdaten verglichen.
340 | Medizinische Genetik 3 · 2013
Schwerpunktthema: Ausgewählte Aspekte von Motoneuronerkrankungen
Inzidenz
In der Literatur findet man Werte für die Inzidenz der SMA Typ I von 1:400 in Is-rael [11] und 1:1263 auf Reunion [29] bis hin zu 1:25.708 in Nordengland [30]. Die sehr hohe Anzahl an SMA-Patienten in den ersten beiden Studien erklärt sich durch ethnische Isolate mit hoher Bluts-verwandtschaft in Israel sowie dem Grün-dereffekt auf Reunion und ist nicht reprä-sentativ. Auch für SMA Typ II und III va-riiert die Anzahl der Neuerkrankungen sehr. Bei der SMA Typ III muss bei der Auswertung der Studien v. a. darauf ge-achtet werden, ob alle Altersklassen be-rücksichtigt wurden, da die Erkrankung unter Umständen erst spät diagnostiziert wird. Da darüber hinaus auch große re-gionale Unterschiede bei den Inzidenzra-ten bestehen, werden hier ausschließlich
europäische Daten verwendet. Geeigne-te Studien, die die Anzahl der SMA-Pati-enten unter allen Neugeborenen in einem bestimmten Zeitraum beobachtet haben, zeigt . Tab. 8.
Es ergeben sich hieraus für die SMA Typ I insgesamt 413 Patienten auf 6.520.316 Lebendgeborene und damit eine Inzidenz von etwa 1:15.788 (1:17428–1:14336). Für die SMA Typ II/III findet man 197 Patienten auf 4.946.700 Lebend-geborene, was einer Inzidenz von etwa 1:25.110 (1:29.021–1:21.838) entspricht. Das bedeutet eine Gesamthäufigkeit für alle 3 Typen (qL
2) von etwa:
(45)
(46)
Embryonale Letalität
Um von der anhand von Patienten be-rechneten Inzidenz qL
2 auf die theoreti-sche Inzidenz q2 zu schließen, muss be-achtet werden, dass in der Literatur nur von Personen berichtet wird, die auch le-bend geboren wurden. Hierbei wird die embryonale Letalität aufgrund von homo-zygoten großen Deletionen nicht berück-sichtigt. Da zur embryonalen Letalität (qG
2) keine direkten Literaturdaten vor-liegen, wird diese für das genetische Mo-
Tab. 3 Anzahl der SMN1-Kopien bei SMA-Patienten
Autoren Patienten Patien-ten ge-samt
Mit 0 SMN1-Ko-pien
Mit 1 SMN1-Kopie
Davon gesicherte Punktmutationen
Bussaglia et al. [4] 50 4 4 54
Chang et al. [5] 48 0 0 48
Cobben et al. [6] 96 7 0 103
Hasanzad et al. [14] 91 5 0 96
Jedrzejowska et al. [18]
260 5 5 265
Labrum et al. [19] 23 1 0 24
Lefebvre et al. [20] 226 3 3 229
Liang et al. [21] 62 0 0 62
Parsons et al. [28] 206 23 11 229
Rodrigues et al. [32] 175 12 0 187
Velasco et al. [39] 60 5 0 65
Wirth et al. [43] 483 19 18 502
Alle Studien 1780 84 41 1864SMN „survival motor neuron“, SMA spinale Muskelatrophie.
Tab. 4 Anteil von Patienten mit hetero-zygoter großer Deletion
Autoren Patienten mit heterozygoter großer Deletion
Patienten gesamt
Lefebvre et al. [20]
9 201
Wirth et al. [42]
16 154
Gesamt 25 355
Tab. 5 Übersicht der verschiedenen Genotypen bei unterschiedlicher SMN1-Kopienzahl
SMN1-Kopien-zahl
Mögliche Genotypen
Homozygot oder bzw.(Normalallele, p2)
Heterozy-got (Über-träger, pq)
Homozygot oder compound heterozygot(Defektallele, q2)
0 aa, ag,gg (nicht lebensfähig)
1 ab, bg ad, gd
2 bb ac, bd, cg dd
3 bc cd
4 cc SMN „survival motor neuron“.
Tab. 6 Genotyp-Phänotyp-Beziehung
Geno-typ
SMN1-Kopien
Möglicher Phänotyp
bb 2 Gesund
2 bc 3 Gesund
cc 4 Gesund
p2 Alle Homozygoten (Normalalle)
2 ba 1 Gesund
2 ca 2 Gesund
2 bd 2 Gesund
2 cd 3 Gesund
2 bg 1 Gesund
2 cg 2 Gesund
2pq Alle Heterozygoten
aa 0 Er-krankt*
Ge-sund
2ad 1 Er-krankt*
Ge-sund
2ag 0 Er-krankt*
Ge-sund
dd 2 Er-krankt*
Ge-sund
2dg 1 Er-krankt*
Ge-sund
gg 0 Letal
q2 qL2* qG
2 qH2
Alle Homozygoten und compound Heterozygoten (Defektallele)
*entsprechen in der Literatur den SMA-Patienten, SMN „survival motor neuron“.
341Medizinische Genetik 3 · 2013 |
dell über die Allelfrequenz g der großen Deletionen berechnet (qG
2 = g2).
(47)
(48)
Unvollständige Penetranz
Im Rahmen von SMA-Studien wurden auch Angehörige der Patienten hinsicht-lich ihres Heterozygotenstatus untersucht. Dabei konnten einige vermeintliche An-lageträger entdeckt werden, die trotz ge-sunden Phänotyps keine SMN1-Kopien aufwiesen. . Tab. 9 zeigt eine Zusam-menstellung der besagten Personen, wo-bei nur Elternteile von SMA-Patienten be-rücksichtigt wurden, da bei Geschwistern je nach Alter weiterhin noch ein Erkran-kungsrisiko vorliegen kann und es sich bei ihnen nicht sicher um Überträger handelt. Eine Ausnahme hierbei stellt die Arbeit von Prior et al. [31] dar, in der sowohl von einem Vater als auch von einem Bruder eines SMA-Patienten berichtet wird, wo-bei letzterer schon 40 Jahre alt und ge-sund ist.
Von den 2326 (vermeintlichen) Über-trägern erwiesen sich 8 als homozygot de-letiert für das SMN1-Gen bzw. compound heterozygot für eine einfache und eine große Deletion, obgleich sie nicht an SMA erkrankt sind.
Es muss jedoch beachtet werden, dass bisher vergleichsweise wenige Eltern und Verwandte genauer hinsichtlich ihrer
SMN1-Kopienzahl untersucht wurden. Darüber hinaus bekommt ein gesundes Paar, bestehend aus einem heterozygo-ten Partner und einem Partner mit 2 De-fektallelen, mit einer annähernd doppelt so hohen Wahrscheinlichkeit ein kran-kes Kind wie 2 heterozygote Partner. Des-halb werden diese Paare als Eltern häufi-ger erfasst.
Bisher ist ungeklärt, welche Faktoren zusammenspielen müssen, dass eine Per-son trotz homozygoter oder compound heterozygoter Mutationen nicht erkrankt. Um diese Faktoren in den Berechnun-gen mit erfassen zu können, werden sie in einem Parameter zusammengefasst: der (unvollständigen) Penetranz. Diese scheint jedoch nicht alle Personen glei-chermaßen zu betreffen, sondern in ein-zelnen Familien bevorzugt vorzuliegen. Das bedeutet, dass es wahrscheinlich Fa-milien mit SMA gibt, die eine vollständi-ge Penetranz zeigen und Familien mit un-vollständiger Penetranz. Um den Parame-ter berechnen und in das genetische Mo-dell einfügen zu können, wird er aller-dings im Folgenden für die Gesamtbevöl-kerung gleichermaßen angenommen. Der Anteil der aufgrund einer unvollständigen Penetranz gesunden SMA-Patienten wird als qH
2 bezeichnet.
(49)
(50)
(51)
(52)
(53)
(54)
Segregation
In dem Modell der SMA muss, wie bereits erwähnt, beachtet werden, dass in einigen Familien aufgrund der unvollständigen Penetranz ein Teil der Personen mit 2 De-fektallelen nicht erkrankt und ein weite-rer kleiner Teil aufgrund einer homozy-goten großen Deletion pränatal verstirbt. Die Segregationsziffer für kranke Nach-kommen muss daher unter den erwarte-ten 25% liegen, wie es bereits Zerres [44] zeigte.
In 2 Studien wurden die Ergebnisse von Pränataldiagnostik in SMA-Famili-en erfasst und es wurde ebenfalls festge-stellt, dass die Segregation zu Ungunsten
Defektallele
a Del.Ex.7/8
d
g große Deletion
q = a + d + g
SMN1-Kopien
1
0
0
Punktmutation
Abb. 3 8 Defektallele im Survival-motor-neuron-1(SMN1)-Gen
SMA Ratsuchender
Abb. 4 8 Stammbaum der Beispielfamilie mit spinaler Muskelatrophie
Tab. 7 Geschätzte Allelfrequenzen im Vergleich mit den Daten von Ogino et al. [26]
Allele Frequenzen
Vorliegende Arbeit
Ogino et al. [26]
Normalallele
b 0,952679 0,946976
c 0,036243 0,037745
p 0,988922 0,984721
Defektallele
a 0,010421 0,013167
d 0,000252 0,000241
g 0,000404 –
q 0,011078 0,013408
342 | Medizinische Genetik 3 · 2013
Schwerpunktthema: Ausgewählte Aspekte von Motoneuronerkrankungen
der Defektallele verschoben ist [1, 3]. Eine vollständige Erklärung kann bisher nicht gegeben werden.
Beispiel für eine Risikoberechnung
Bei der molekulargenetischen Diagnostik von SMA wird i. d. R. „multiplex ligation dependent probe amplification“ (MLPA) eingesetzt. Mithilfe von MLPA kann je-doch nur die Anzahl der SMN1-Kopien gemessen werden. Daher muss berück-sichtigt werden, dass in einigen Fällen der Befund zweideutig sein kann (. Tab. 10).
Beispielbefunde
Der Bruder einer SMA-Patientin fragt nach seinem Heterozygotenrisiko. Es lie-gen die nachfolgenden Befunde vor.F Stammbaum (. Abb. 4)F Molekulargenetik:1 Indexpatientin hat SMA und ist
verstorben, es liegen keine moleku-largenetischen Untersuchungen vor, Ratsuchender hat 2 SMN1-Kopien
Tab. 10 Mögliche Genotypen bei bestimmten SMN1-Kopien
Phänotyp und Genotyp Prozentualer Anteil
0 SMN1-Kopien
Homozygot Defektallel (Deletion:aa) 100
1 SMN1-Kopie
Heterozygot, gesund (2 ba) 96,1
Heterozygot, gesund (2 bg) 3,8
Compound heterozygot, krank (2 ad) 0,03
Compound heterozygot, krank (2 gd) 0,001
2 SMN1-Kopien
Homozygot normal, gesund (bb) 99,7
Heterozygot, gesund (2 ca) 0,14
Heterozygot, gesund (2 bd) 0,12
Heterozygot, gesund (2 cg) 0,01
3 SMN1-Kopien
Compound heterozygot normal, gesund (2 bc) 0,999
Heterozygot, gesund (2 cd) 0,001
4 SMN1-Kopien
Homozygot normal, gesund (cc) 100SMN „survival motor neuron“.
Tab. 8 Inzidenz der spinalen Muskelatrophie (SMA) Typ I-III
Autoren Land SMA Typ I SMA Typ II/III
Anzahl der Patienten
Anzahl der Lebendge-burten
Anzahl der Patienten
Anzahl der Lebendge-burten
Czeizel und Ha-mula [8]
Ungarn 91 1.376.928 – –
Darin und Tulinius [9]
Schweden 13 320.902 3 296.601
Jedrzejowska et al. [17]
Polen 209 2.963.783 95 2.963.783
Ludvigsson et al. [22]
Island 4 65.584 5 65.584
Merlini et al. [24] Italien 8 150.978 9 150.978
Mostacciuolo et al. [25]
Italien 35 859.891 32 859.891
Pearn [30] Großbritan-nien
9 231.370 – –
Spiegler et al. [35] Polen 11 214.217 11 214.217
Thieme et al. [37] Deutschland 33 336.663 – –
Thieme et al. [38] Deutschland – – 42 395.646
Alle Studien 413 6.520.316 197 4.946.700
Tab. 9 Anzahl der gesunden Personen mit 0 SMN1-Kopien unter allen Übertägern
Autoren Anzahl (n) der gesunden Personen mit 0 SMN1-Kopien
Anzahl (n) der unter-suchten Eltern/Überträger insgesamt
Bussaglia et al. [4]
0 111
Hahnen et al. [12]
1 348
Harada et al. [13]
0 24
Jedrzejow-ska et al. [16]
2 386
Lefebvre et al. [20]
0 127
Prior et al. [31]
2 409
Rodrigues et al. [31]
0 373
Simard et al. [33]
0 109
Somerville et al. [34]
1 225
Wang et al. [41]
2 214
Alle Studien 8 2326
343Medizinische Genetik 3 · 2013 |
Tab. 11 Risikoberechnung anhand des Beispiels mithilfe eines Bayes-Tableaus
Schwerpunktthema: Ausgewählte Aspekte von Motoneuronerkrankungen
(MLPA), Eltern sind nicht unter-sucht
F Vereinfachtes genetisches Model:1 autosomal-rezessiver Erbgang im
Hardy-Weinberg-Gleichgewicht: keine Neumutation, keine Phäno-kopie.
Risikoberechnung
Im Bayes-Tableau (. Tab. 11) wird be-rücksichtigt, dass bei der verstorbenen Indexpatientin unterschiedliche moleku-largenetische Befunde vorliegen könnten (Deletion, Punktmutation oder große De-letion im homozygoten oder compound heterozygoten Zustand), sodass der Rat-suchende auch mit 2 SMN1-Kopien hete-rozygot für SMA sein kann. Sein Hetero-zygotenrisiko (Het) beträgt in dieser Situ-ation etwa 11% (NHet =0,89). Bei dieser Berechnung wurde nicht berücksichtigt, dass der Ratsuchende auch gesund ist, obwohl er 2 Defektallele besitzen könnte (unvollständige Penetranz).
Dieses Beispiel zeigt, dass zur Hete-rozygotendiagnostik bei der SMA oft die molekulargenetische Diagnostik alleine nicht ausreicht, sondern zusätzlich Risi-koabschätzungen erforderlich sind.
Diskussion
In der vorliegenden Arbeit sind die wich-tigsten für die SMA bedeutenden Parame-ter anhand von Literaturdaten geschätzt worden, indem ein genetisches SMA-Mo-dell erstellt wurde. Darüber hinaus war es mithilfe dieses Modells möglich, weitere Parameter zu bestimmen, über die bislang noch keine ausreichenden Literaturdaten vorliegen. So konnte z. B. die Wahrschein-lichkeit für embryonale Letalität aufgrund einer homozygoten großen Deletion so-wie für das Vorliegen einer homozygo-ten Punktmutation berechnet werden. Es wird in der Literatur bisher über keinen Fall mit homozygoter Punktmutation be-richtet, was nicht nur an deren Seltenheit (0,05% aller Patienten) liegen mag, son-dern auch daran, dass in der Diagnostik beim Vorliegen von 2 SMN1-Kopien eine Erkrankung an SMA i. d. R. ausgeschlos-sen wird [40]. Ebenfalls nicht ausreichend untersucht wurde bisher die Penetranz, auch wenn in vielen Studien von gesun-
den Personen mit homozygoter Deletion berichtet wird.
Mithilfe des hier angeführten geneti-schen Modells und dem Bayes-Rechen-tableau können molekulare Befunde be-rücksichtigt werden, um in der geneti-schen Beratung gerade im Hinblick auf die Familienplanung ein möglichst ge-naues und individuelles Risiko berechnen zu können.
Fazit für die Praxis
F Die molekulargenetische Diagnostik von Erbkrankheiten erfordert neben dem molekulargenetischen Laborwis-sen auch grundlegende Kenntnisse in der formalen Humangenetik.
F Die Heterozygotendiagnostik bei SMA bedarf häufig neben der Mole-kulargenetik auch des Einsatzes von Risikoberechnungen.
Korrespondenzadresse
Prof. Dr. T. GrimmAbteilung für Medizinische Genetik im Institut für Humangenetik, Universität Würzburg, Biozentrum, Am Hubland97074 Wü[email protected]
Einhaltung ethischer Richtlinien
Interessenkonflikt. S. Langer, S. Rudnik-Schöneborn, K. Zerres und T. Grimm geben an, dass kein Interessen-konflikt besteht. Dieser Beitrag beinhaltet keine Stu-dien an Menschen oder Tieren.
Literatur
1. Alias L, Barceló MJ, Gich I et al (2007) Evidence of a segregation ratio distortion of SMN1 alleles in spi-nal muscular atrophy. Eur J Hum Genet 5:1090–1093
2. Anhuf D, Eggermann T, Rudnik-Schöneborn S, Zer-res K (2003) Determination of SMN1 and SMN2 co-py number using TaqMan TM technology. Hum Mutat 22:74–78
3. Botta A, Tacconelli A, Bagni I et al (2005) Transmis-sion ratio distortion in the spinal muscular atro-phy locus:data from 314 prenatal tests. Neurology 65:1631–1635
4. Bussaglia E, Clermont O, Tizzano E et al (1995) A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat Genet 11:335–337
5. Chang JG, Jong YJ, Huang JM et al (1995) Molecu-lar basis of spinal muscular atrophy in Chinese. Am J Hum Genet 57:1503–1505 (Letters to the editor)
6. Cobben JM, Steege G van der, Grootscholten P et al (1995) Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am J Hum Genet 57:805–808
7. Cusin V, Clermont O, Gérard B et al (2003) Preva-lence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet 40:e39
8. Czeizel A, Hamula J (1989) A Hungarian study on Werdnig-Hoffmann disease. J Med Genet 26:761–763
9. Darin N, Tulinius M (2000) Neuromuscular disor-ders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord 10:1–9
10. Feldkötter M, Schwarzer V, Wirth R et al (2002) Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly relia-ble carrier testing and prediction of severity of spi-nal muscular atrophy. Am J Hum Genet 70:358–368
11. Fried K, Mundel G (1977) High incidence of spinal muscular atrophy type I (Werdnig-Hoffmann di-sease) in the Karaite community of Israel. Clin Ge-net 12:250–251
12. Hahnen E, Forkert R, Marke C et al (1995) Mole-cular analysis of candidate genes on chromoso-me 5q13 in autosomal recessive spinal muscular atrophy:evidence of homozygous deletions of the SMN gene in uneffected individuals. Hum Mol Ge-net 4:1927–1933
13. Harada Y, Sutomo R, Sadewa AH et al (2002) Cor-relation between SMN2 copy number and clinical phenotype of spinal muscular atrophy:three SMN2 copies fail to rescue some patients from the disea-se severity. J Neurol 249:1211–1219
14. Hasanzad M, Golkar Z, Kariminejad R et al (2009) Deletions in the survival motor neuron gene in Ira-nian patients with spinal muscular atrophy. Ann Acad Med Singapore 38:139–141
15. Hendrickson BC, Donohoe C, Akmaev VR et al (2009) Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet 46:641–644
16. Jedrzejowska M, Borkowska J, Zimowski J et al (2008) Uneffected patients with a homozygous abscence of the SMN1 gene. Eur J Hum Genet 16:930–934
17. Jedrzejowska M, Milewski M, Zimowski J et al (2010) Incidence of spinal muscular atrophy in Po-land – more frequent than predicted? Neuroepi-demiology 34(3):152–157
18. Jedrzejowska M, Wiszniewski W, Zimowski J et al (2005) Application of a rapid non-invasive techni-que in the molecular diagnosis of spinal muscular atrophy (SMA). Neurol Neurochir Pol 39:89–94
19. Labrum R, Rodda J, Krause A (2007) The mole-cular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromuscul Disord 17:684–692
20. Lefebvre S, Bürglen L, Reboullet S et al (1995) Iden-tification and characterization of a spinal muscular atrophy-determing gene. Cell 80:155–165
21. Liang YH, Chen XL, Yu ZS et al (2009) Deletion ana-lysis of SMN1 and NAIP genes in Southern Chinese children with spinal muscular atrophy. J Zhejiang Univ Sci B 10:29–34
22. Ludvigsson P, Olafsson E, Hauser WA (1999) Spinal muscular atrophy. Incidence in Iceland. Neuroepi-demiology 18:265–269
345Medizinische Genetik 3 · 2013 |
23. McAndrew PE, Parsons DW, Simard LR et al (1997) Identification of proximal spinal muscular atro-phy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 60:1411–1422
24. Merlini L, Stagni SB, Marri E, Granata C (1992) Epi-demiology of neuromuscular disorders in the un-der-20 population in Bologna Province, Italy. Neu-romuscul Disord 2:197–200
25. Mostacciuolo ML, Danieli GA, Trevisan C et al (1992) Epidemiology of spinal muscular atrophies in a sample of the Italian population. Neuroepide-miology 11:34–38
26. Ogino S, Wilson RB, Gold B (2004) New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and ha-plotype frequency calculations. Eur J Hum Genet 12:1015–1023
27. Oprea GE, Kröber S, McWhorter ML et al (2008) Plastin 3 is a protective modifier of autosomal re-cessive spinal muscular atrophy. Science 320:524–527
28. Parsons DW, McAndrew PE, Iannaccone ST et al (1998) Intragenetic telSMN mutations:frequency, distribution, evidence of a founder effect, and mo-dification of the spinal muscular atrophy pheno-type by cenSMN copy number. Am J Hum Genet 63:1712–1723
29. Pascalet-Guidon MJ, Bois E, Feingold J et al (1984) Cluster of acute infantile spinal muscular atrophy (Werdnig-Hoffmann disease) in a limited area of Reunion Island. Clin Genet 26:39–42
30. Pearn JH (1973) The gene frequency of acute Werdnig-Hoffmann disease (SMA type I). A total population survey in North-East England. J Med Genet 10:260–265
31. Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ (2004) Homozygous SMN1 deletions in unaffected family members and modification of the phenoty-pe by SMN2. Am J Med Genet 130A:307–310
32. Rodrigues NR, Owen N, Talbot K et al (1996) Gene deletions in spinal muscular atrophy. J Med Genet 33:93–96
33. Simard LR, Rochette C, Semionov A et al (1997) SMNT and NAIP mutations in Canadian families with spinal muscular atrophy (SMA): genotype/phenotype correlations with disease severity. Am J Med Genet 72:51–58
34. Somerville MJ, Hunter AGW, Aubry HL et al (1997) Clinical application of the molecular diagnosis of spinal muscular atrophy: deletions of neuro-nal apoptosis inhibitor protein and survival motor neuron genes. Am J Med Genet 69:159–165
35. Spiegler AW, Hausmanowa-Pertrusewicz I, Bor-kowska J, Klopocka A (1990) Population data on acute infantile and chronic childhood spinal mu-scular atrophy in Warsaw. Hum Genet 85:211–214
36. Sugarman EA, Nagan N, Zhu H et al (2012) Pan-et-hic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analy-sis of >72400 specimens. Eur J Hum Genet 20:27–32
37. Thieme A, Mitulla B, Schulze F, Spiegler AW (1993) Epidemiological data on Werdnig-Hoffmann di-sease in Germany (West-Thüringen). Hum Genet 91:295–297
38. Thieme A, Mitulla B, Schulze F, Spiegler AW (1994) Chronic childhood spinal muscular atrophy in Ger-many (West-Thüringen)- an epidemiological study. Hum Genet 93:344–346
39. Velasco E, Valero C, Valero A et al (1996) Molecu-lar analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and corre-lation between number of copies of cBCD541 and SMA phenotype. Hum Mol Genet 5:257–263
40. Wang CH, Finkel RS, Bertini ES et al (2007) Consen-sus statement for standard of care in spinal muscu-lar atrophy. J Child Neurol 22:1027–1049
41. Wang CH, Xu J, Carter TA et al (1996) Characteriza-tion of survival motor neuron (SMNT) gene dele-tions in asymptomatic carriers of spinal muscular atrophy. Hum Mol Genet 5:359–365
42. Wirth B, Hahnen E, Morgan K et al (1995) Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: association of marker genotype with disease severity and candi-date cDNAs. Hum Mol Genet 4:1273–1284
43. Wirth B, Herz M, Wetter A et al (1999) Quantitati-ve analysis of survival motor neuron copies: identi-fication of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counse-ling. Am J Hum Genet 64:1340–1356
44. Zerres K (1989) Klassifikation und Genetik spinaler Muskelatrophien. Thieme-Copythek, Stuttgart
45. Zerres K, Grimm T, Rudnik-Schöneborn S (2005) Modifikation des Phänotyps der proximalen spina-len Muskelatrophie (SMA) durch die SMN2-Genko-pie. Medgen 17:161–165
Bisher unbekannter B-Lymphozyten-Defekt entschlüsselt