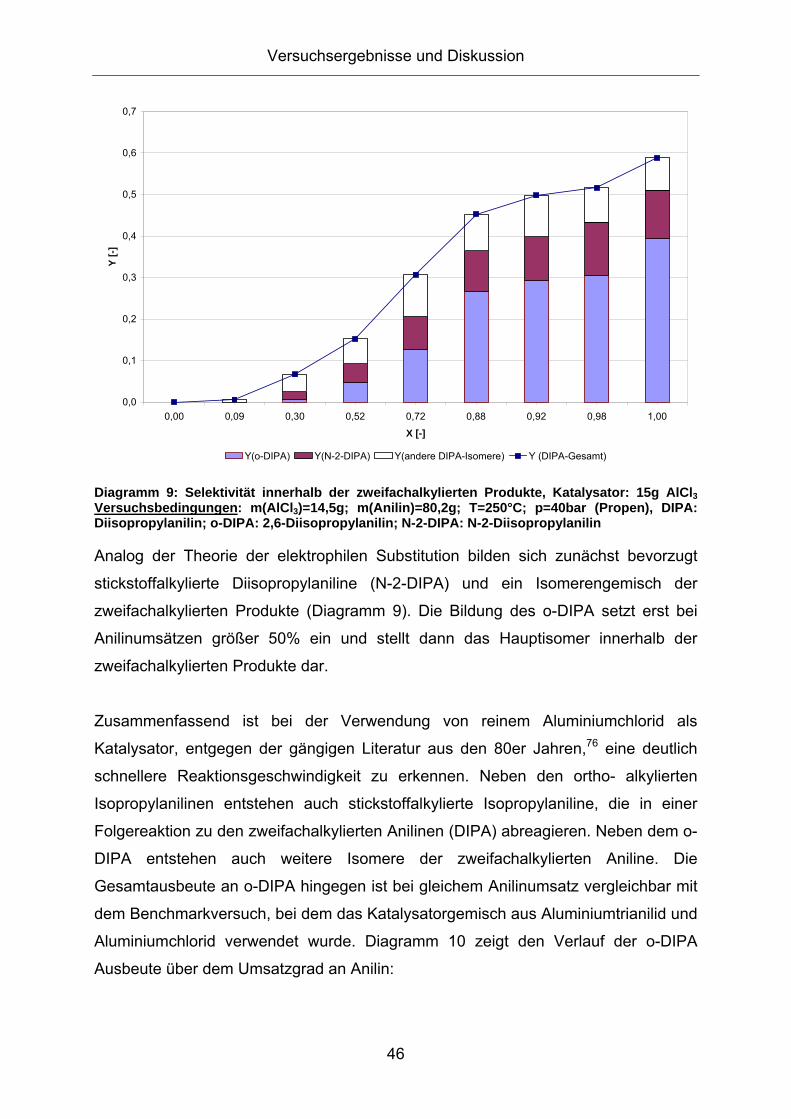
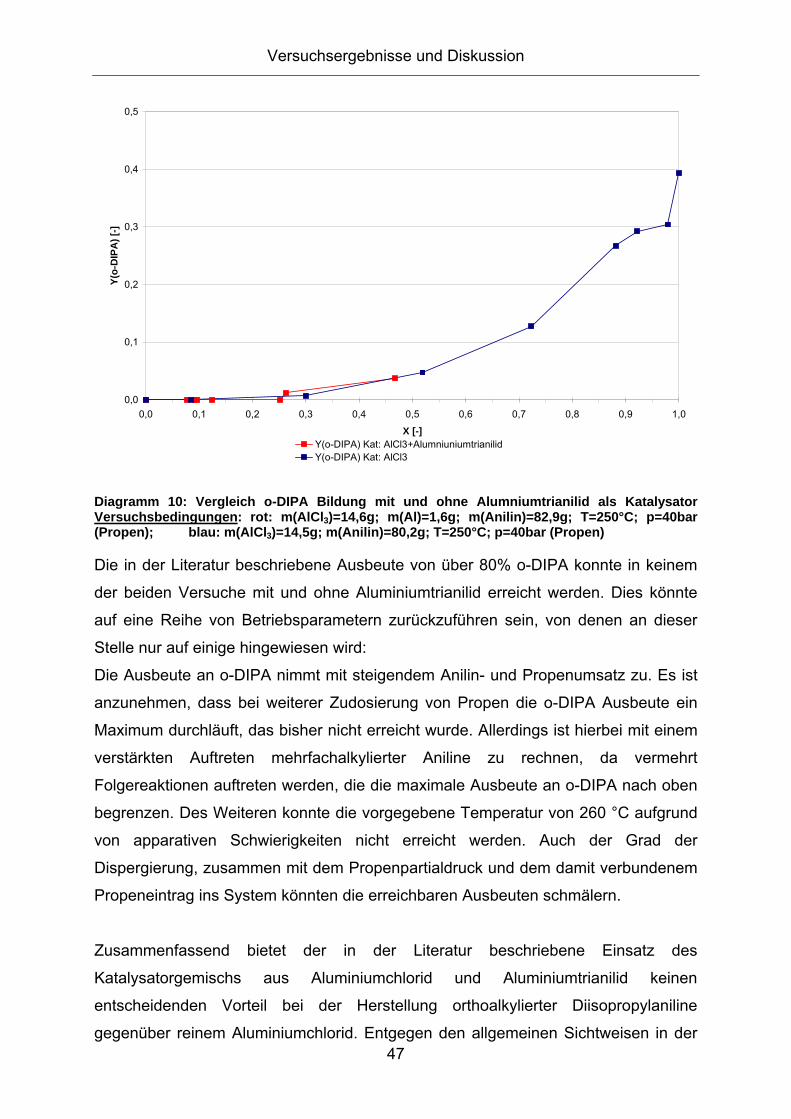
Friedel-Crafts Alkylierung von Anilin - Mechanistische Betrachtungen und Untersuchungen zur Katalysatorrezyklierung für eine nachhaltige Reaktionsführung Technische Fakultät der Universität Erlangen-Nürnberg Zur Erlangung des Grades Doktor Ingenieur vorgelegt von Diplom Ingenieur Viktor Ladnak Erlangen 2010
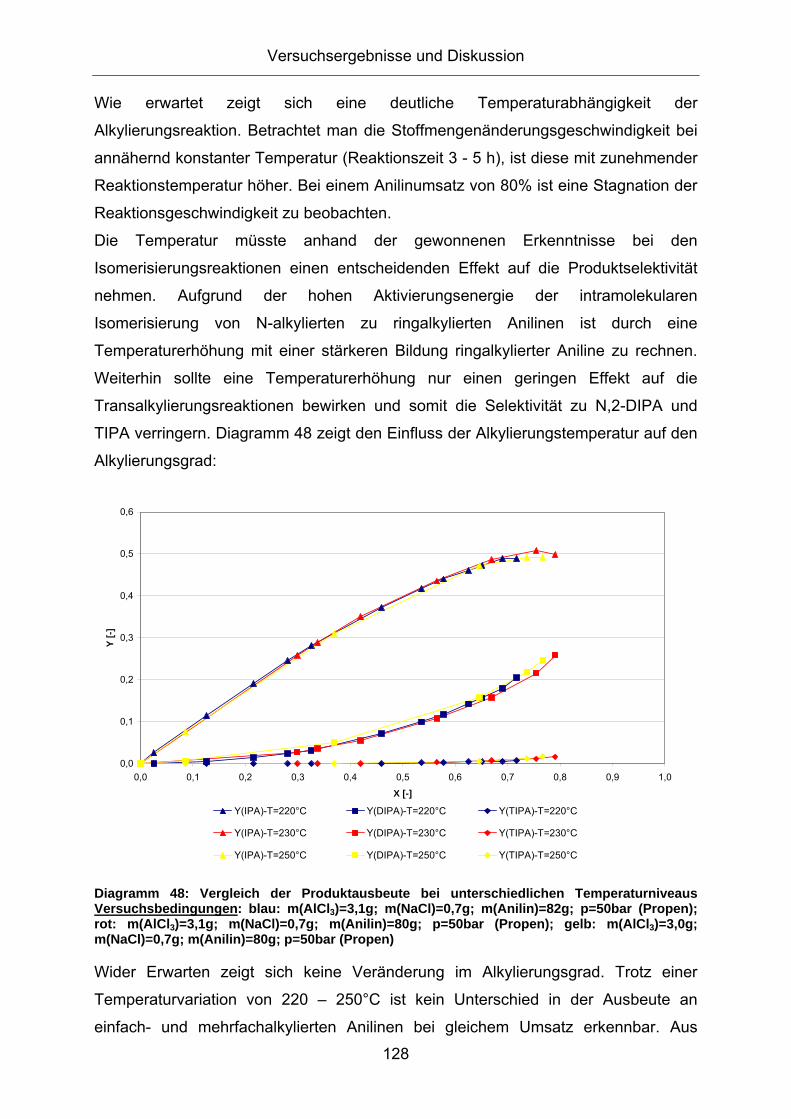
Transcript
Friedel-Crafts Alkylierung von Anilin - Mechanistische Betrachtungen und Untersuchungen zur
Katalysatorrezyklierung für eine nachhaltige Reaktionsführung
Technische Fakultät der Universität Erlangen-Nürnberg
Zur Erlangung des Grades
Doktor Ingenieur
vorgelegt von
Diplom Ingenieur
Viktor Ladnak
Erlangen 2010
Als Dissertation genehmigt von
der Technischen Fakultät der
Universität Erlangen-Nürnberg
Tag der Einreichung: 05.05.2010
Tag der Promotion: 15.10.2010
Dekan: Prof. Dr.-Ing. habil. R. German
Berichterstatter: Prof. Dr. P. Wasserscheid
Prof. Dr.-Ing. habil. K.-E. Wirth
Die vorliegende Arbeit entstand in der Zeit vom Januar 2006 bis Februar 2009 am
Lehrstuhl für Chemische Reaktionstechnik (CRT) der Friedrich-Alexander-Universität
Erlangen-Nürnberg.
Herrn Professor Dr. rer. nat. Peter Wasserscheid danke ich herzlich für die
interessante Themenstellung, die hervorragende Betreuung und die stets
motivierenden Diskussionen.
Herrn Professor Dr. Wirth danke ich für die freundliche Übernahme des
Zweitgutachtens.
Der Firma Lonza, insbesondere deren Mitarbeitern Dr. Andreas Heyl, Dr. Lothar Ott
und Dr. Paul Hanselmann, danke ich für die Bereitstellung der finanziellen Mittel und
für die sehr kooperative und überaus nette Zusammenarbeit.
Michael Schmacks, Achim Mannke, danke ich für die anlagentechnische
Unterstützung und die endlose Geduld beim ständigen Austausch korrodierter
Anlagenteile. Gerhard Dommer, Karl-Heinz Ksoll und Walter Fischer danke ich für die
technische Unterstützung in elektrotechnischen und elektronischen Angelegenheiten.
Regine Müller, Katja Kreuz und Norbert Hofmann, danke ich für die zahllosen ICP-
Messungen.
Bedanken möchte ich mich auch bei meinen HiWi´s, Studien- und Diplomarbeitern
Johannes Mang, Florian Strobl und Stefan Schlenk für die engagierte Mitarbeit.
Ferner gilt mein Dank allen Mitarbeitern im Arbeitskreis, insbesondere meinen
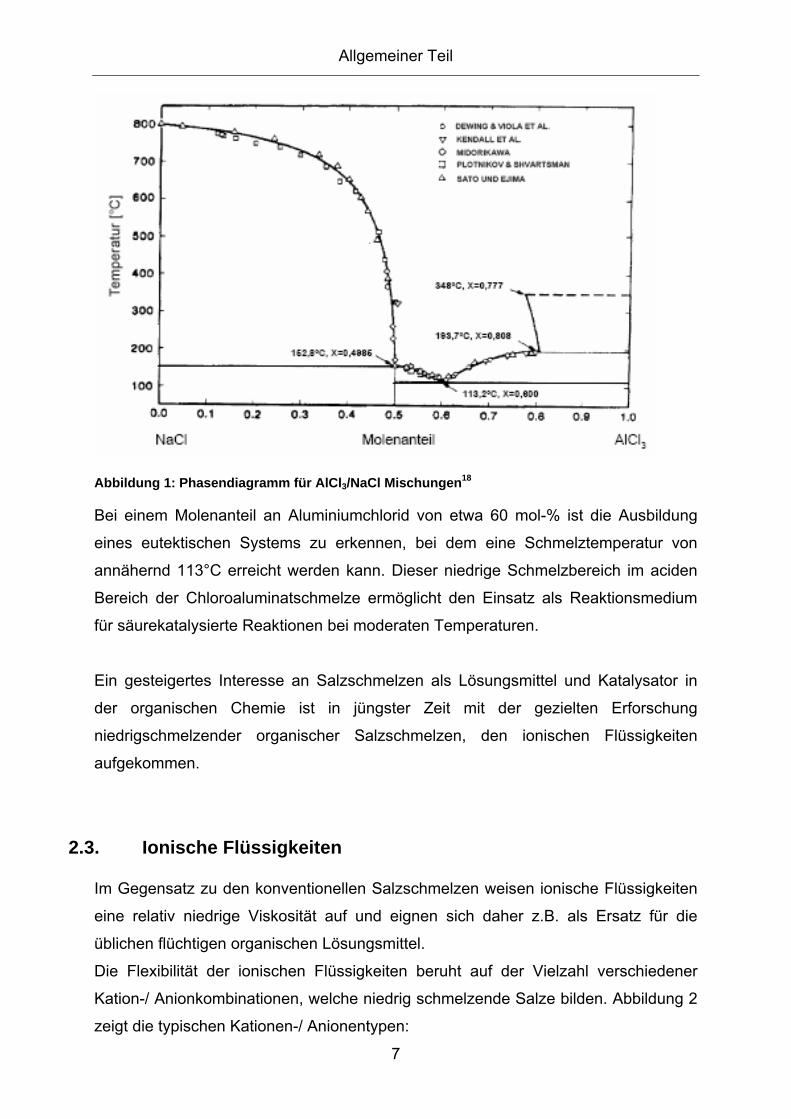
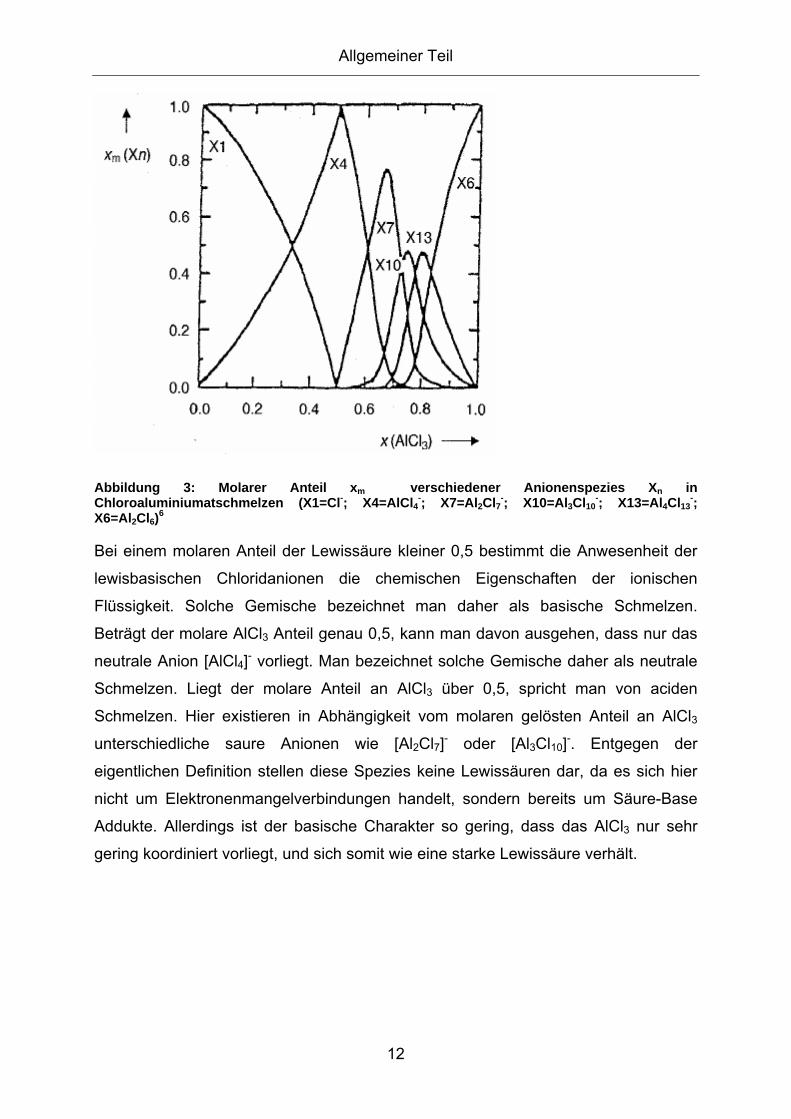
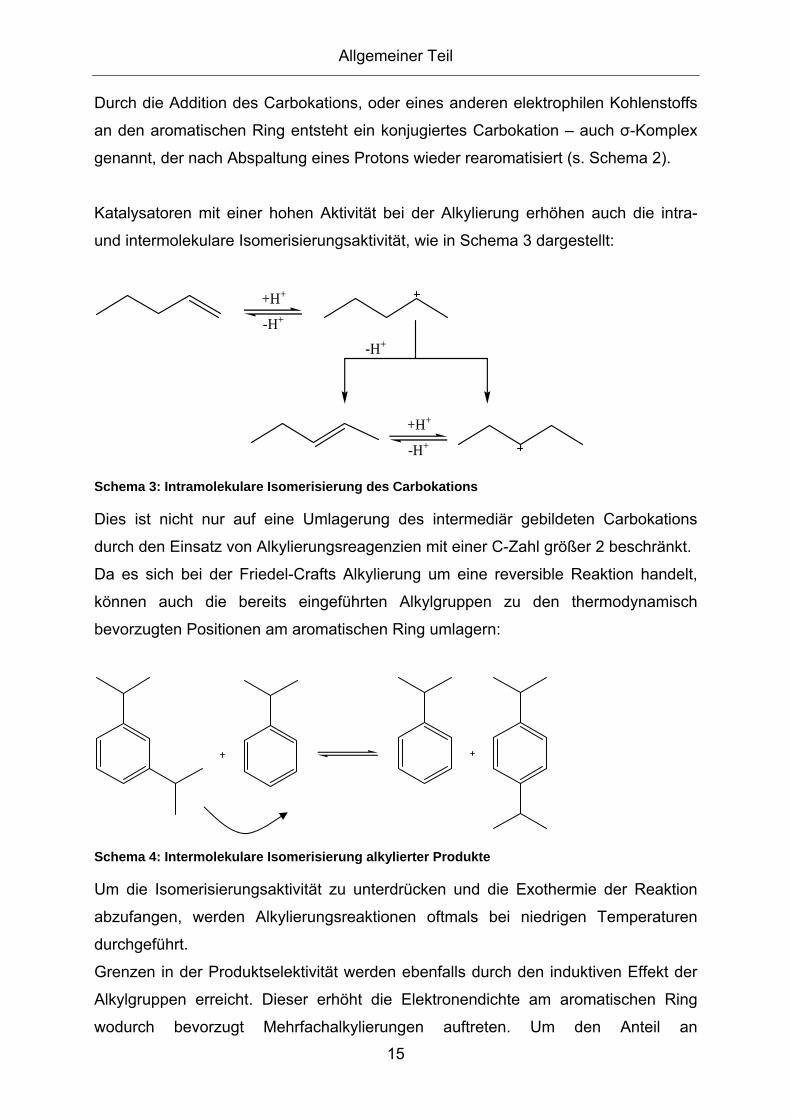
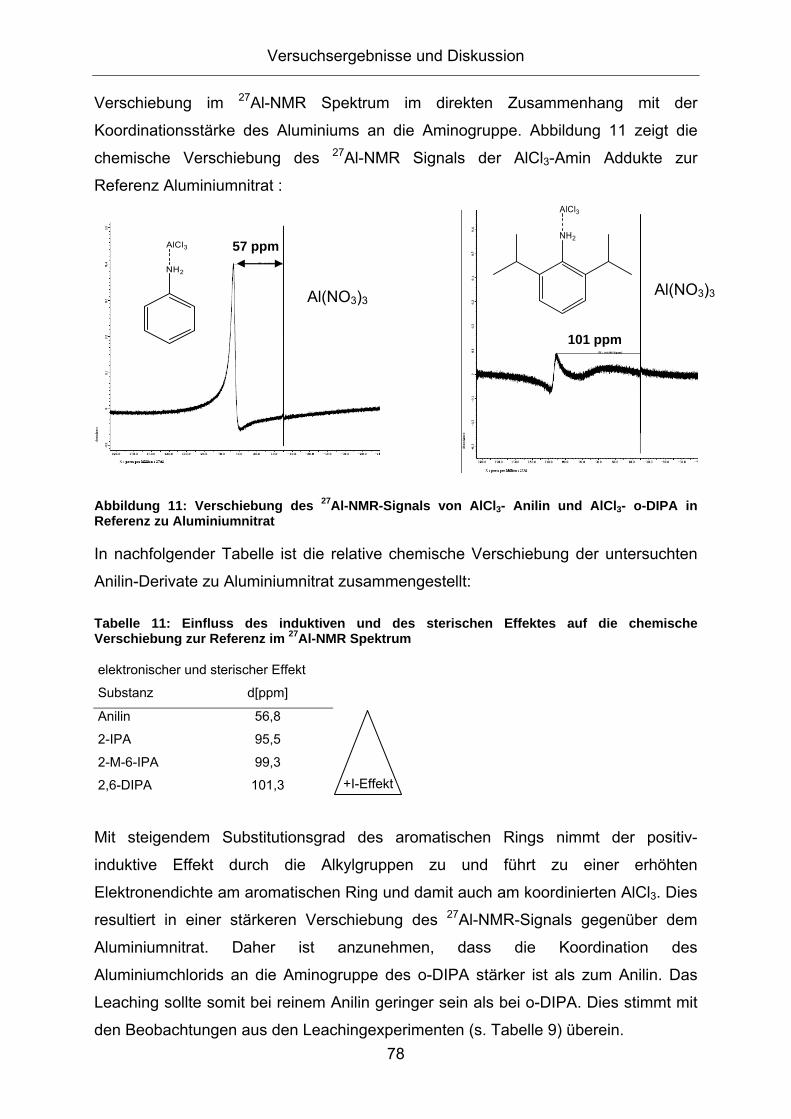
2.3.1. Eigenschaften und Anwendungen ionischer Flüssigkeiten ...................... 8 2.3.2. Acidität von ionischen Flüssigkeiten ...................................................... 11
2.4. Friedel-Crafts Alkylierung............................................................13 2.4.1. Technische Friedel-Crafts Alkylierung und deren Entwicklung .............. 17 2.4.2. Technische Friedel-Crafts Alkylierung von Anilin................................... 21 2.4.3. Friedel-Crafts Alkylierung in Salzschmelzen.......................................... 26 2.4.4. Alkylierung von Anilin in Salzschmelzen ................................................ 28
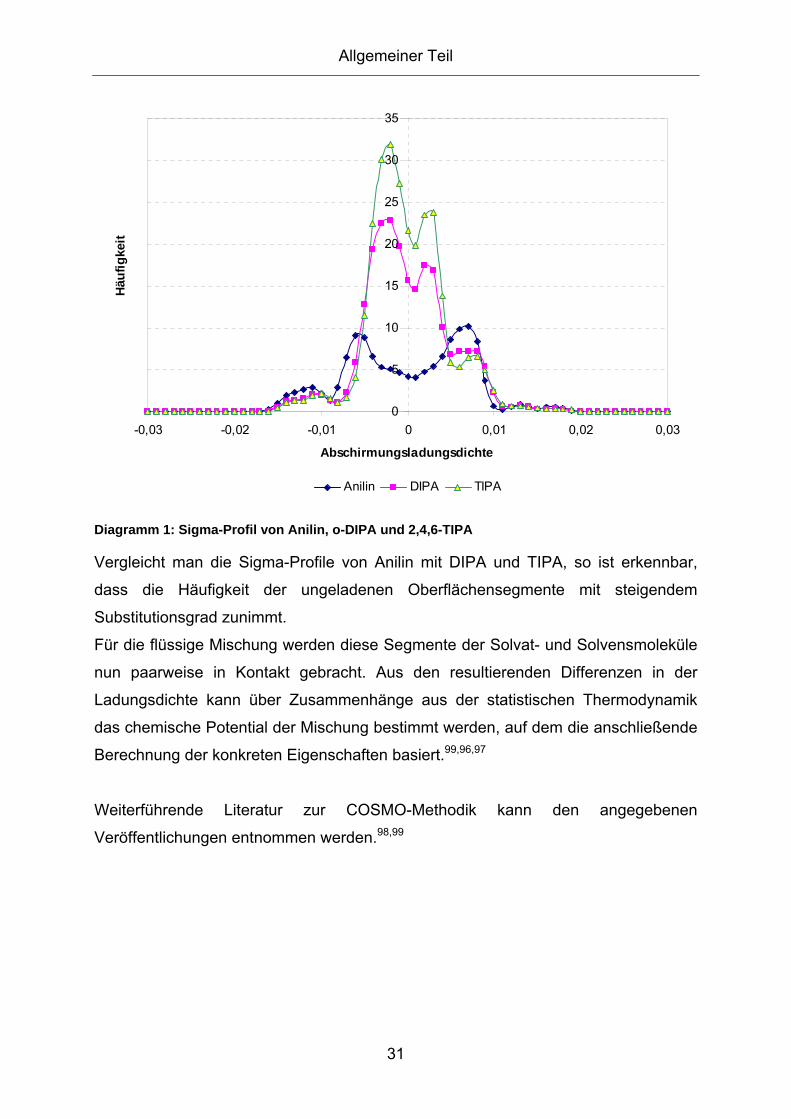
2.5. COSMOtherm zur Abschätzung von Löslichkeitseigenschaften.30 3. Versuchsergebnisse und Diskussion ...............................................35
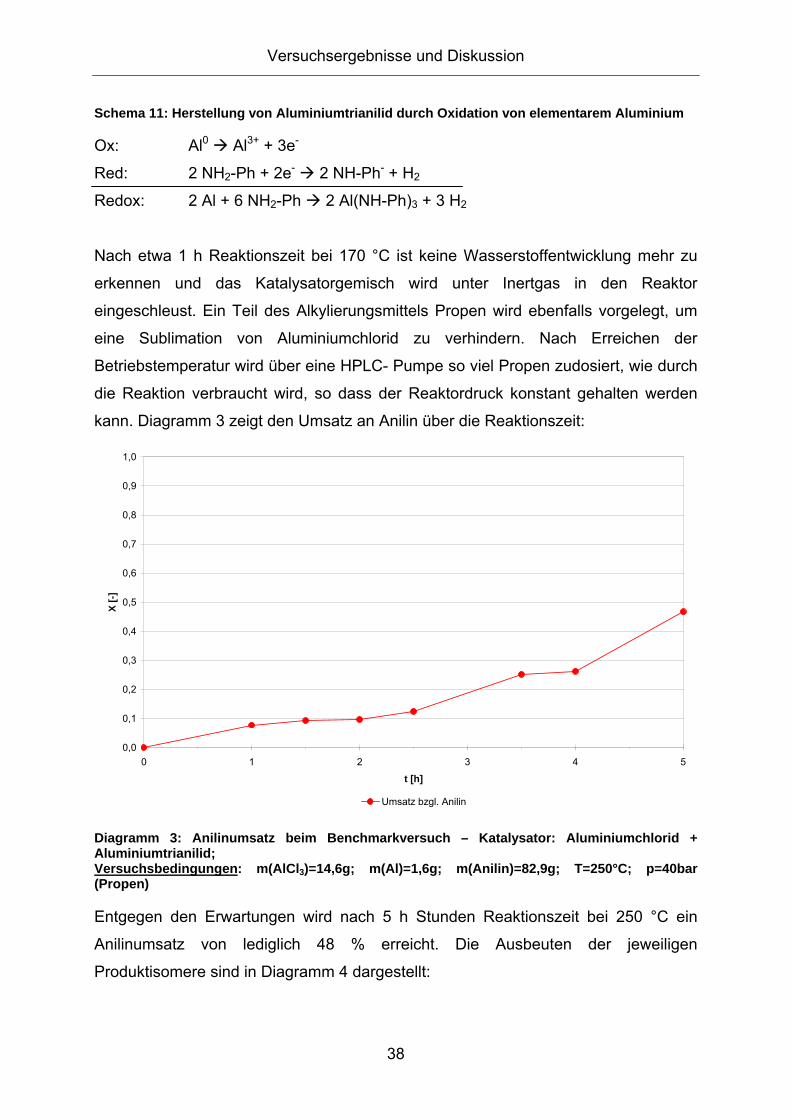
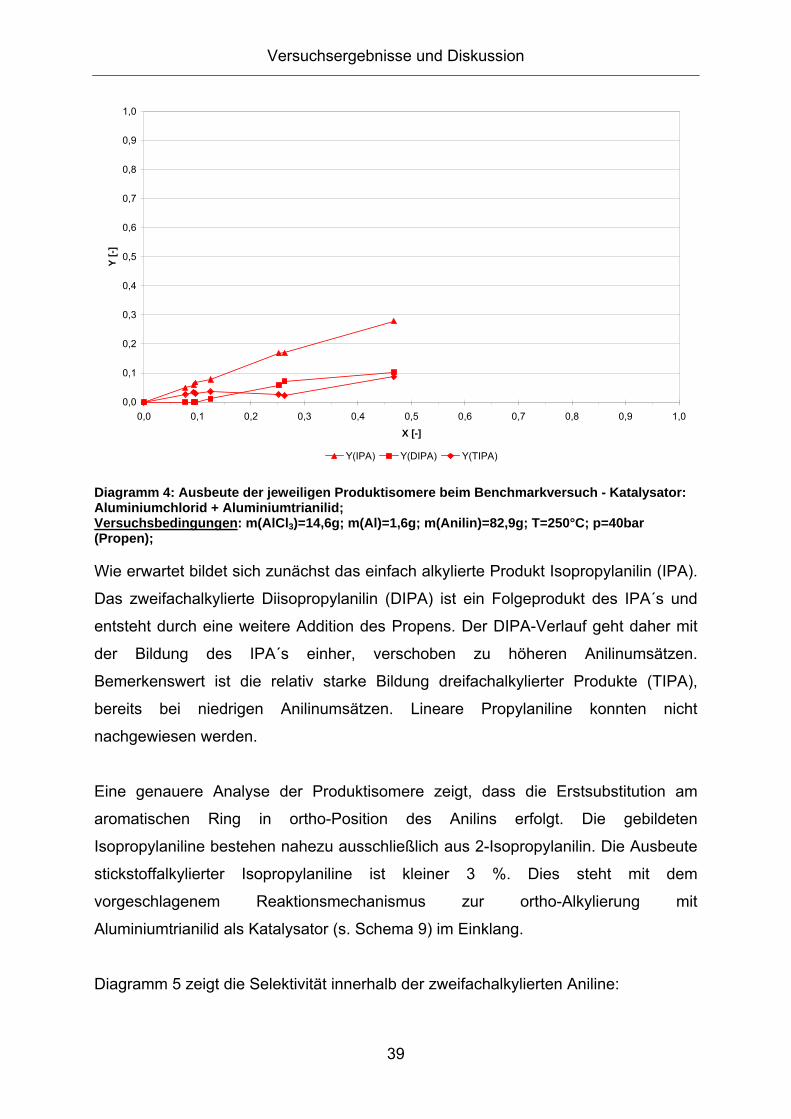
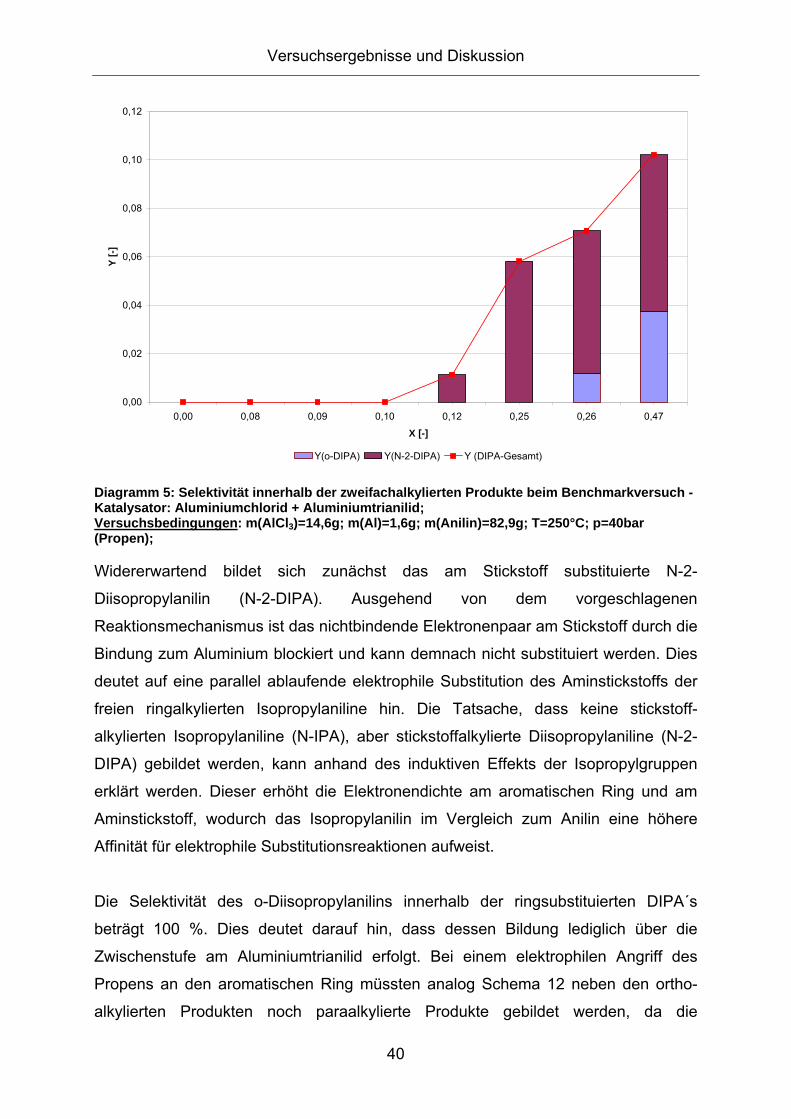
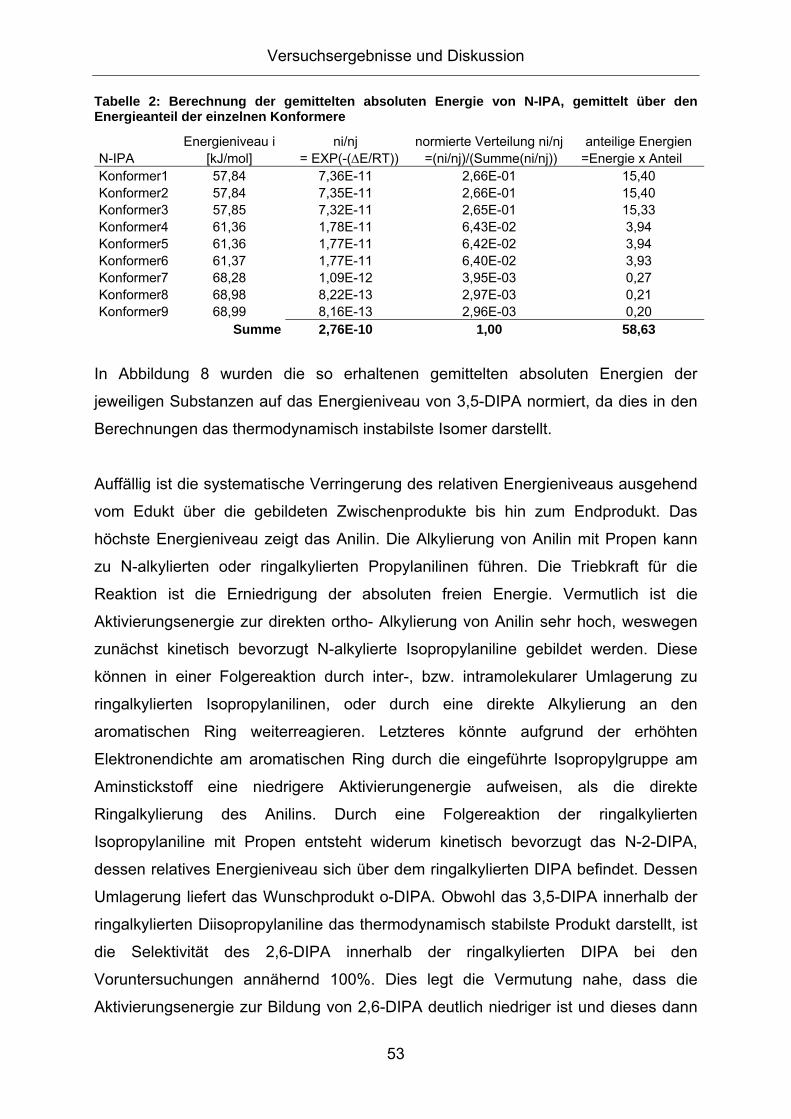
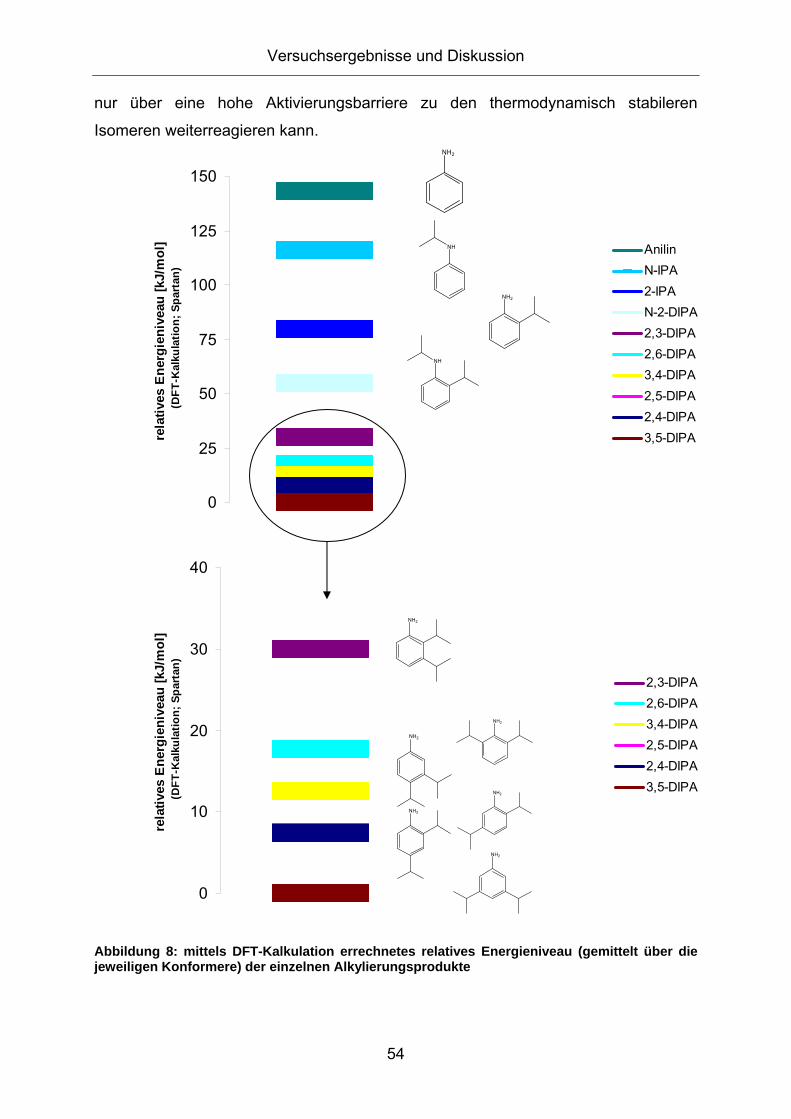
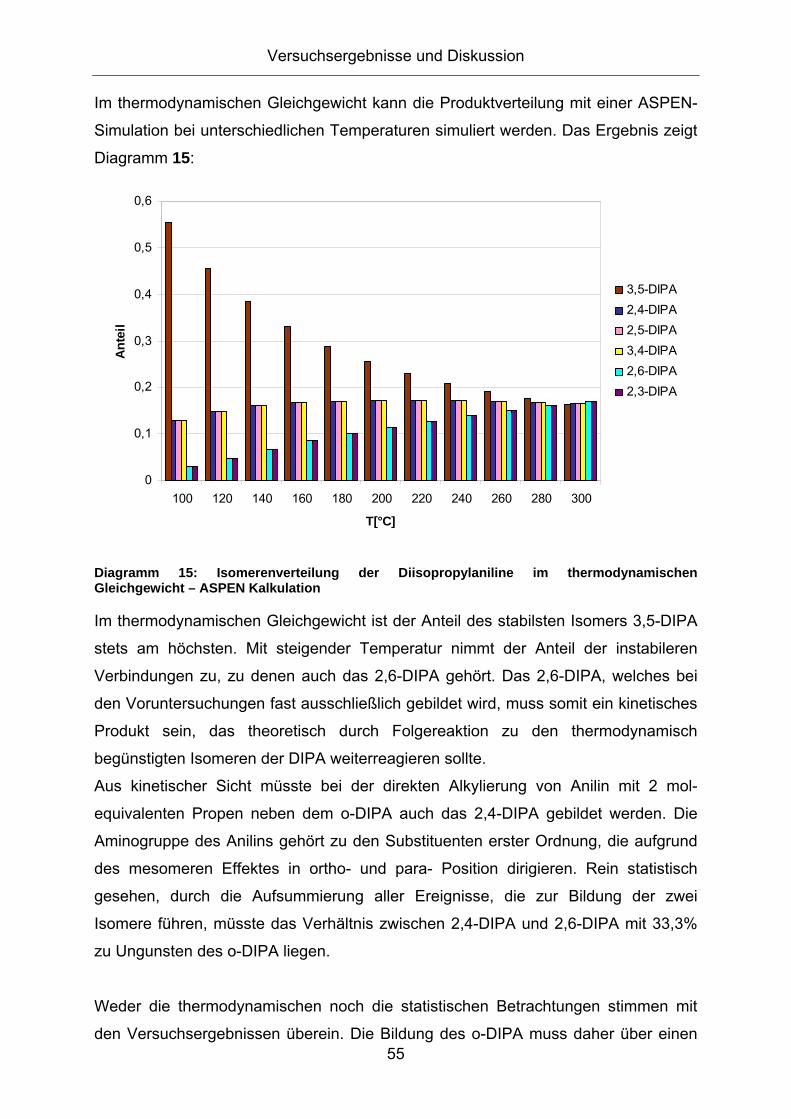
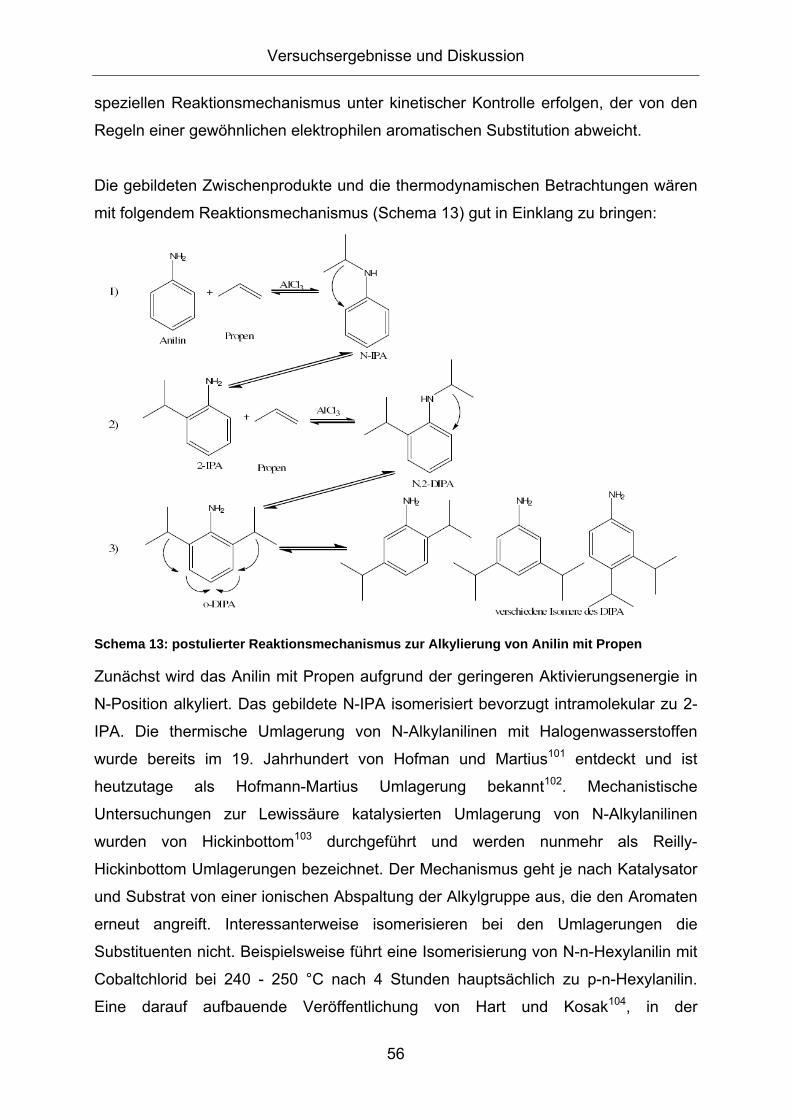
3.1. Konzeption der Arbeit .................................................................35 3.2. Vorversuche zur Auswahl geeigneter Versuchsparameter.........37
3.2.1. Aluminiumtrianilid als Katalysator .......................................................... 37 3.2.2. Aluminiumtrichlorid als Katalysator ........................................................ 43 3.2.3. Mechanistische Betrachtungen.............................................................. 52
3.3. Verfahrensentwicklung zur Katalysatorrückführung....................59 3.3.1. Einsatz ionischer Flüssigkeiten.............................................................. 59 3.3.2. Einsatz anorganischer Salzschmelzen .................................................. 74 3.3.3. Einsatz von überkritischen Gasen als Extraktionsmittel......................... 91 3.3.4. Katalysatorabtrennung durch Kristallisation........................................... 95
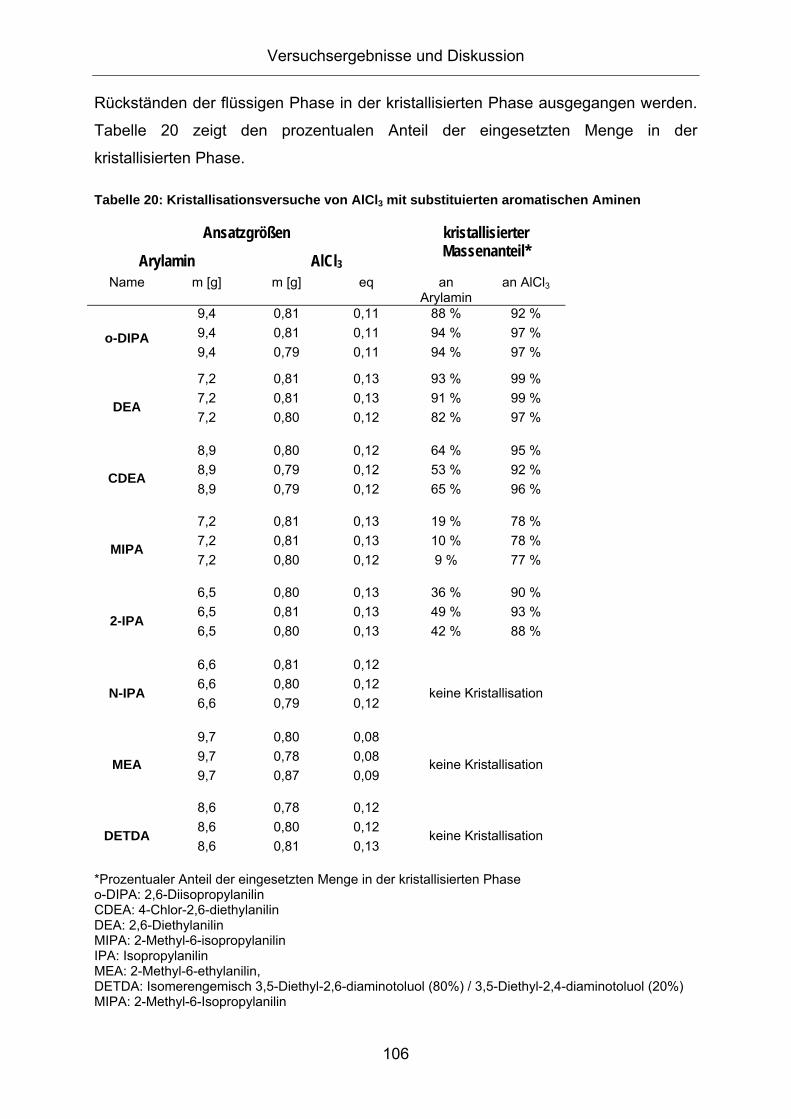
3.4. Verfahrenserweiterung auf Anilinderivate .................................105 3.4.1. Kristallisationsversuche von substituierten Anilinen..............................105 3.4.2. [AlCl3(MIPA)2] als Katalysator für die Friedel-Crafts-Alkylierung...........110
3.5. Untersuchungen zur Reaktionsoptimierung..............................112 3.5.1. Kinetische Betrachtungen .....................................................................112 3.5.2. Screening von Reaktionsparametern bei der Alkylierung .....................127
6.2.1. Arbeiten unter Schutzgas......................................................................149 6.2.2. Bereitstellung von Lösungsmitteln und Reagenzien .............................150
6.3. Alkylierungsversuche................................................................150 6.3.1. Aluminiumtrianilid als Katalysator .........................................................150 6.3.2. Versuche mit ionischer Flüssigkeit........................................................151 6.3.3. Versuche mit anorganischen Salzschmelzen .......................................151 6.3.4. Durchführung der Katalyseversuche.....................................................151 6.3.5. Rezyklierungsversuche.........................................................................153 6.3.6. Probenaufarbeitung ..............................................................................153
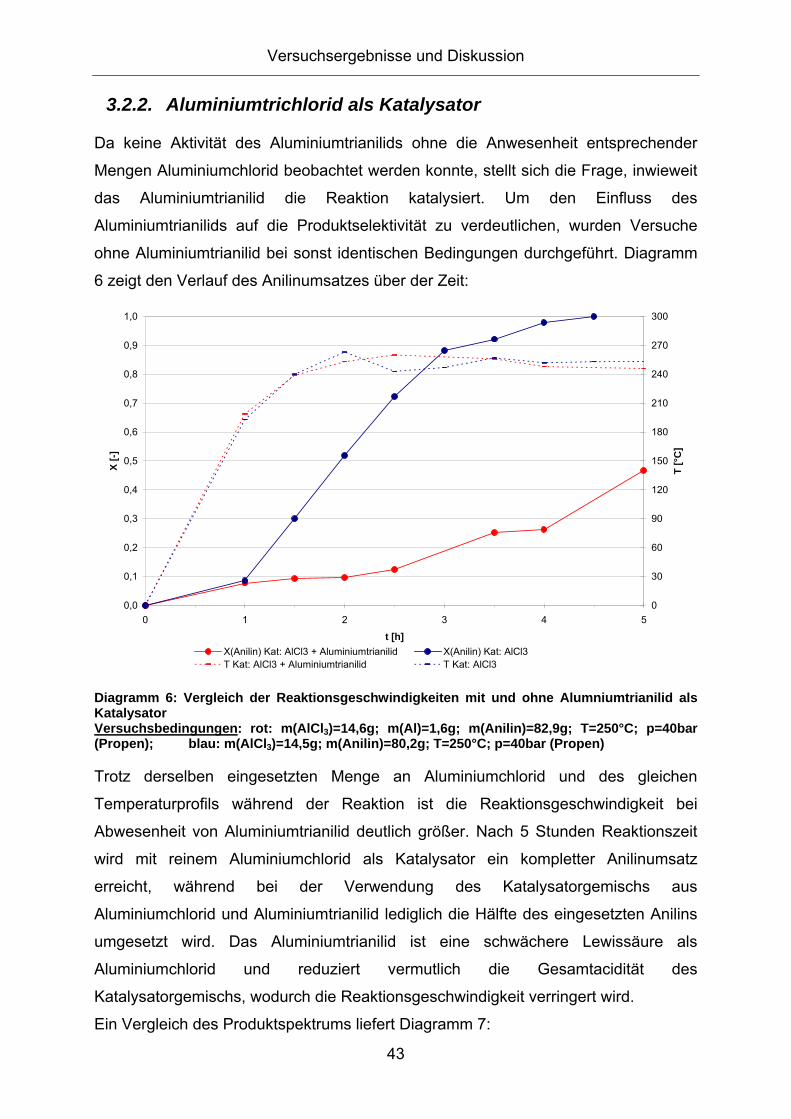
Diagramm 6: Vergleich der Reaktionsgeschwindigkeiten mit und ohne Alumniumtrianilid als Katalysator Versuchsbedingungen: rot: m(AlCl3)=14,6g; m(Al)=1,6g; m(Anilin)=82,9g; T=250°C; p=40bar (Propen); blau: m(AlCl3)=14,5g; m(Anilin)=80,2g; T=250°C; p=40bar (Propen)
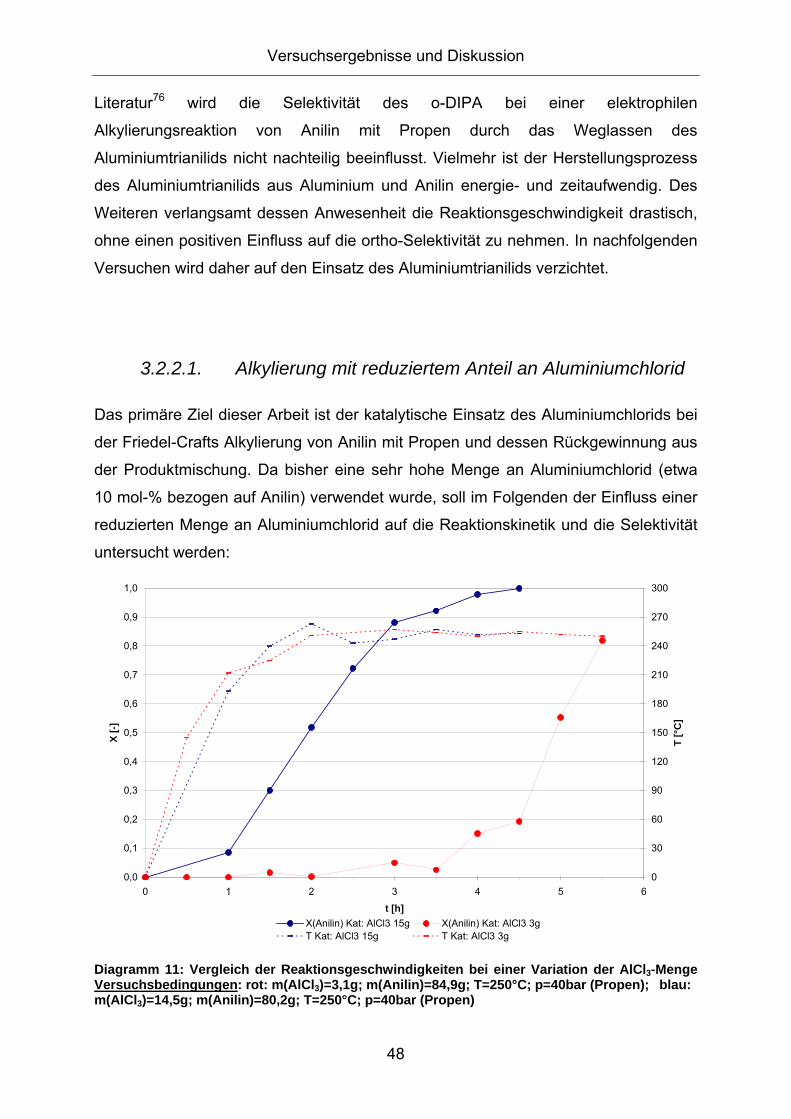
Trotz derselben eingesetzten Menge an Aluminiumchlorid und des gleichen
Temperaturprofils während der Reaktion ist die Reaktionsgeschwindigkeit bei
Abwesenheit von Aluminiumtrianilid deutlich größer. Nach 5 Stunden Reaktionszeit
wird mit reinem Aluminiumchlorid als Katalysator ein kompletter Anilinumsatz
erreicht, während bei der Verwendung des Katalysatorgemischs aus
Aluminiumchlorid und Aluminiumtrianilid lediglich die Hälfte des eingesetzten Anilins
umgesetzt wird. Das Aluminiumtrianilid ist eine schwächere Lewissäure als
Aluminiumchlorid und reduziert vermutlich die Gesamtacidität des
Katalysatorgemischs, wodurch die Reaktionsgeschwindigkeit verringert wird.
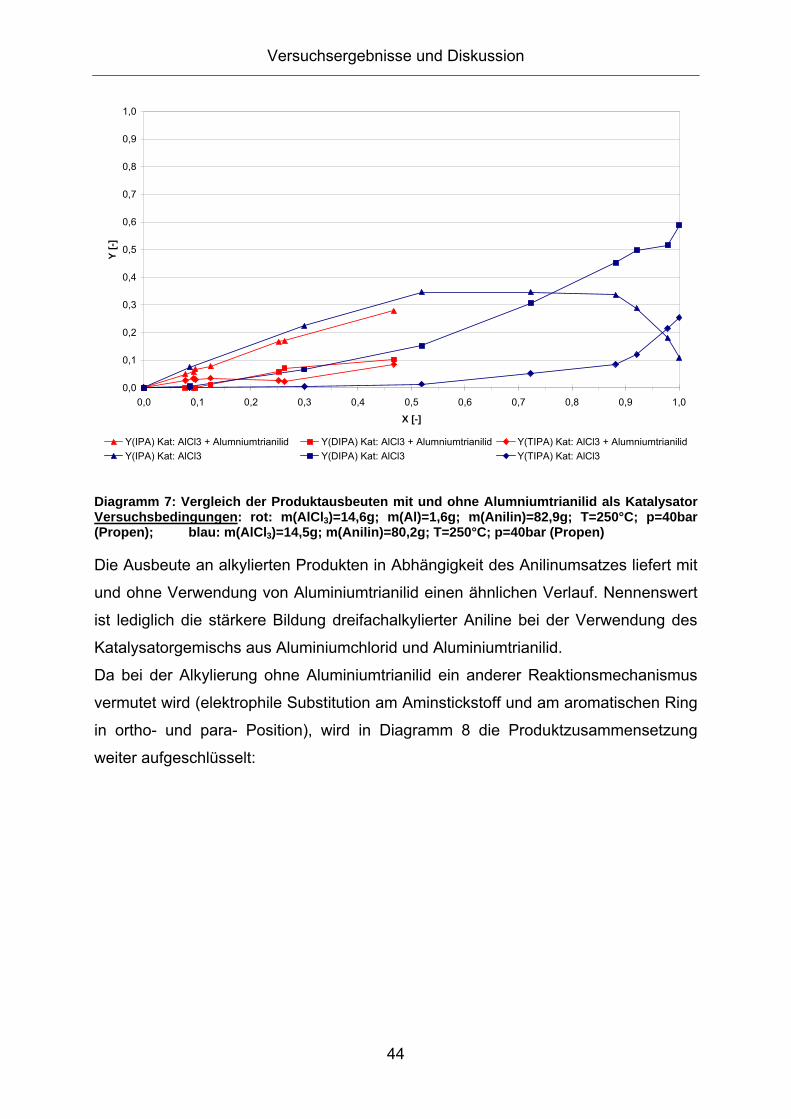
Ein Vergleich des Produktspektrums liefert Diagramm 7:
Diagramm 7: Vergleich der Produktausbeuten mit und ohne Alumniumtrianilid als Katalysator Versuchsbedingungen: rot: m(AlCl3)=14,6g; m(Al)=1,6g; m(Anilin)=82,9g; T=250°C; p=40bar (Propen); blau: m(AlCl3)=14,5g; m(Anilin)=80,2g; T=250°C; p=40bar (Propen)
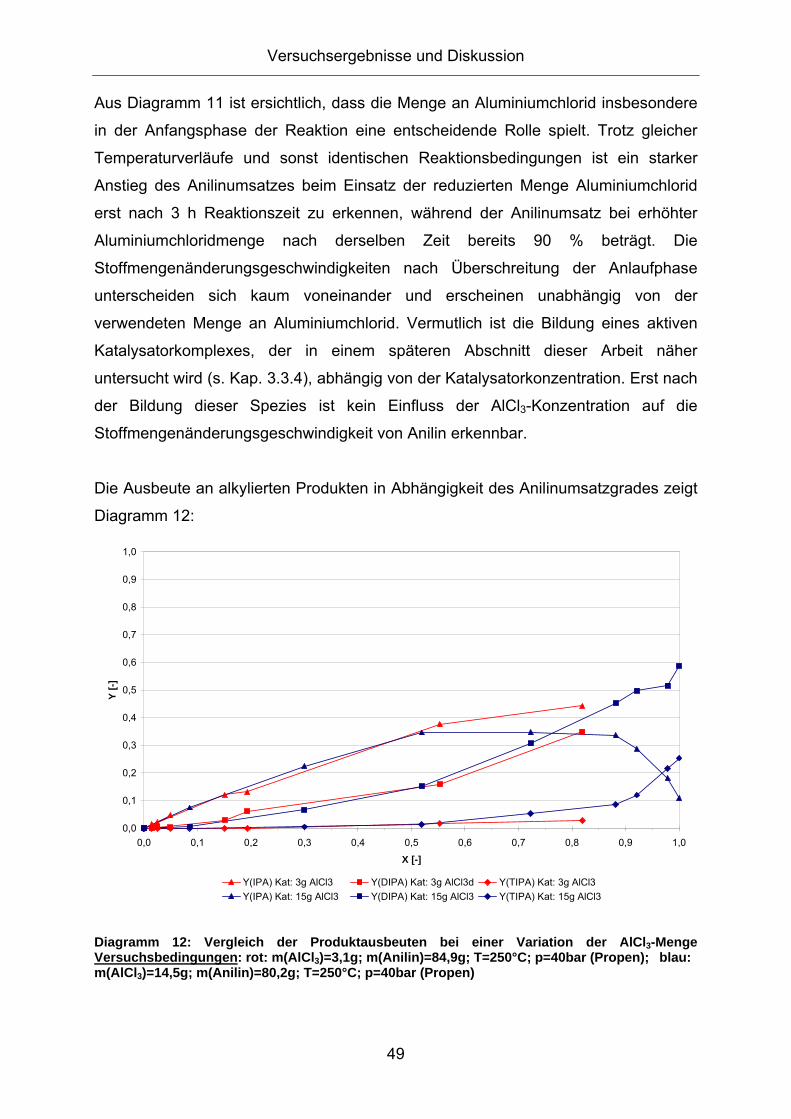
Die Ausbeute an alkylierten Produkten in Abhängigkeit des Anilinumsatzes liefert mit
und ohne Verwendung von Aluminiumtrianilid einen ähnlichen Verlauf. Nennenswert
ist lediglich die stärkere Bildung dreifachalkylierter Aniline bei der Verwendung des
Katalysatorgemischs aus Aluminiumchlorid und Aluminiumtrianilid.
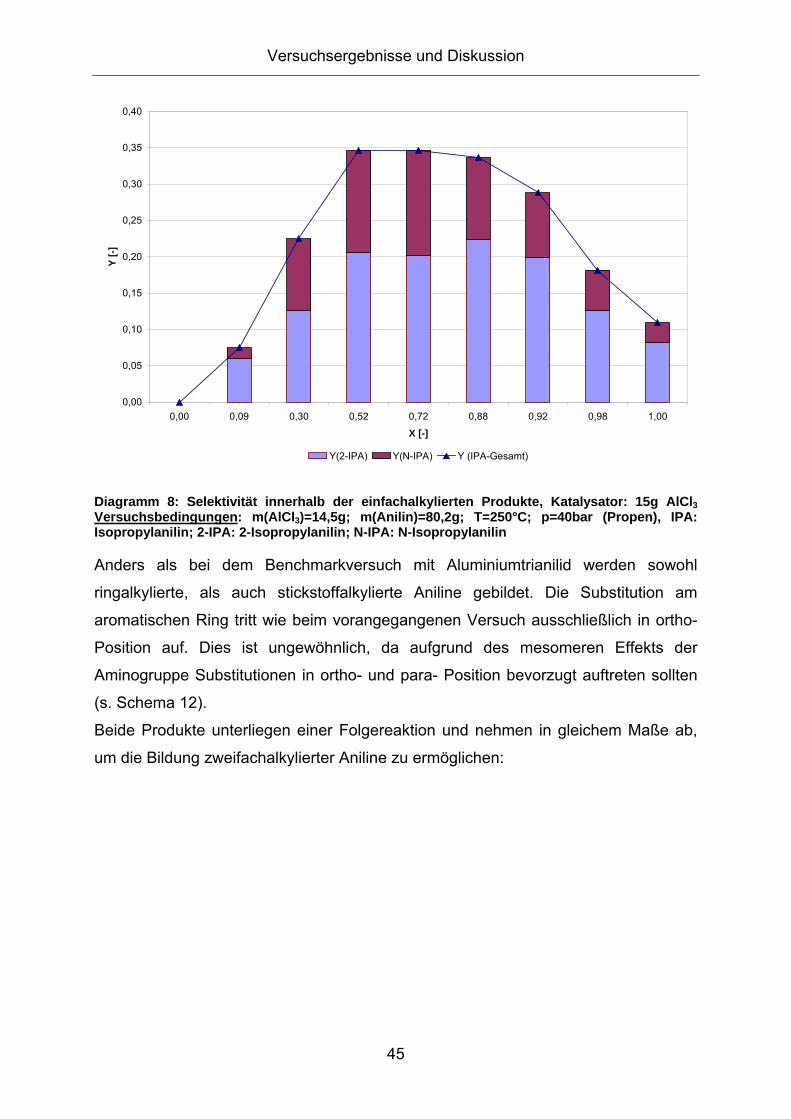
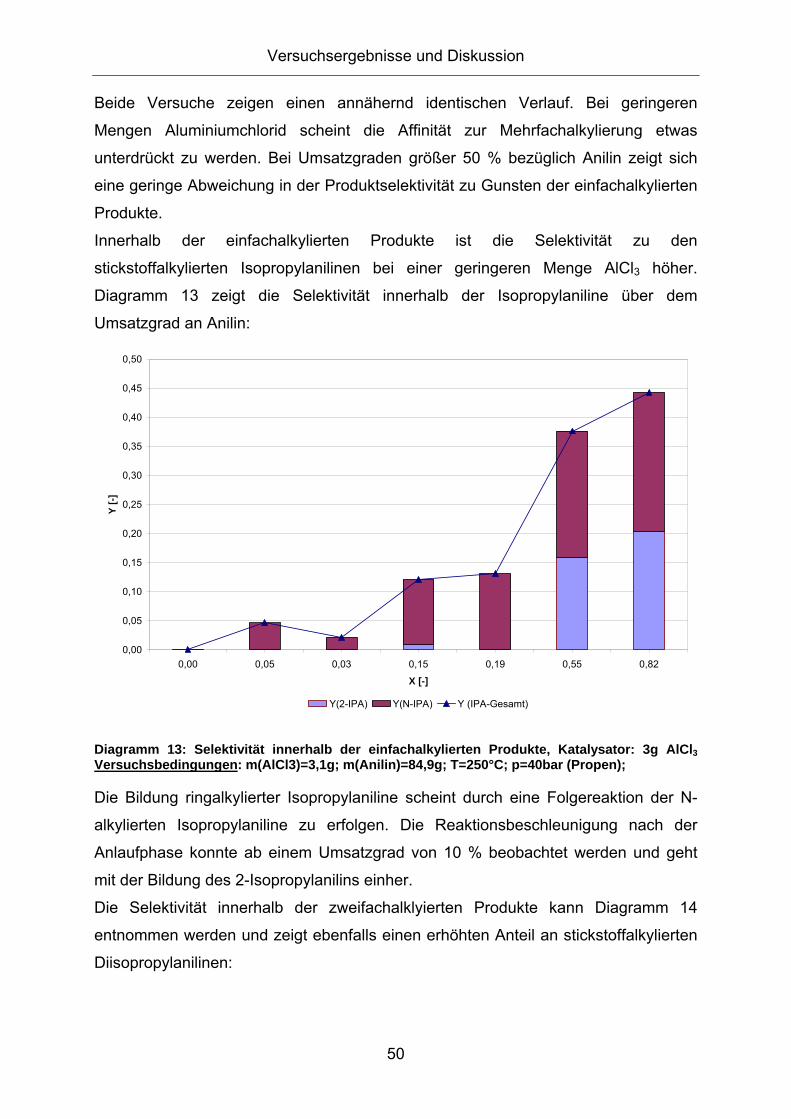
Da bei der Alkylierung ohne Aluminiumtrianilid ein anderer Reaktionsmechanismus
vermutet wird (elektrophile Substitution am Aminstickstoff und am aromatischen Ring
in ortho- und para- Position), wird in Diagramm 8 die Produktzusammensetzung
*Unter der Annahme, dass Dekalin 100% unlöslich in der IL
Versuchsergebnisse und Diskussion
65
Das Anilin zeigt nur eine geringe Löslichkeit in der organischen Extraktphase. Über
97 % des eingesetzten Anilins befinden sich in der ionischen Katalysatorphase.
Umgekehrt verhält es sich mit o-DIPA. Dieses zeigt eine hohe Affinität zur
organischen Extraktphase. Die Zugabe von o-DIPA in das System bewirkt zudem
eine Erhöhung des Anilinanteils in der organischen Extraktphase.
Der Einsatz des Systems IL/Extraktionsmittel in der Alkylierung bietet neben der
Möglichkeit zur Katalysatorrezyklierung den Vorteil, dass aufgrund der hohen
Löslichkeit des Anilins in der ionischen Katalysatorphase eine hohe
Eduktkonzentration vorliegt, was zu einer schnellen Reaktionsgeschwindigkeit führen
kann. Da die gebildeten Produkte wegen ihrer geringeren Polarität und der hohen
Affinität zum Extraktionsmittel Dekalin aus der ionischen Katalysatorphase extrahiert
werden, ist deren Konzentration in der Reaktionsphase gering. Hierdurch können
unerwünschte Folgereaktionen unterdrückt werden. Das Konzept für eine homogene
flüssig-flüssig Zweiphasenalkylierung von Anilin mit Propen scheint somit durch die
Verwendung des Systems IL/Dekalin durchführbar.
3.3.1.2. Vorversuche - Einfluss des gelösten Aluminiumchlorids
auf die Thermostabilität ionischer Flüssigkeiten
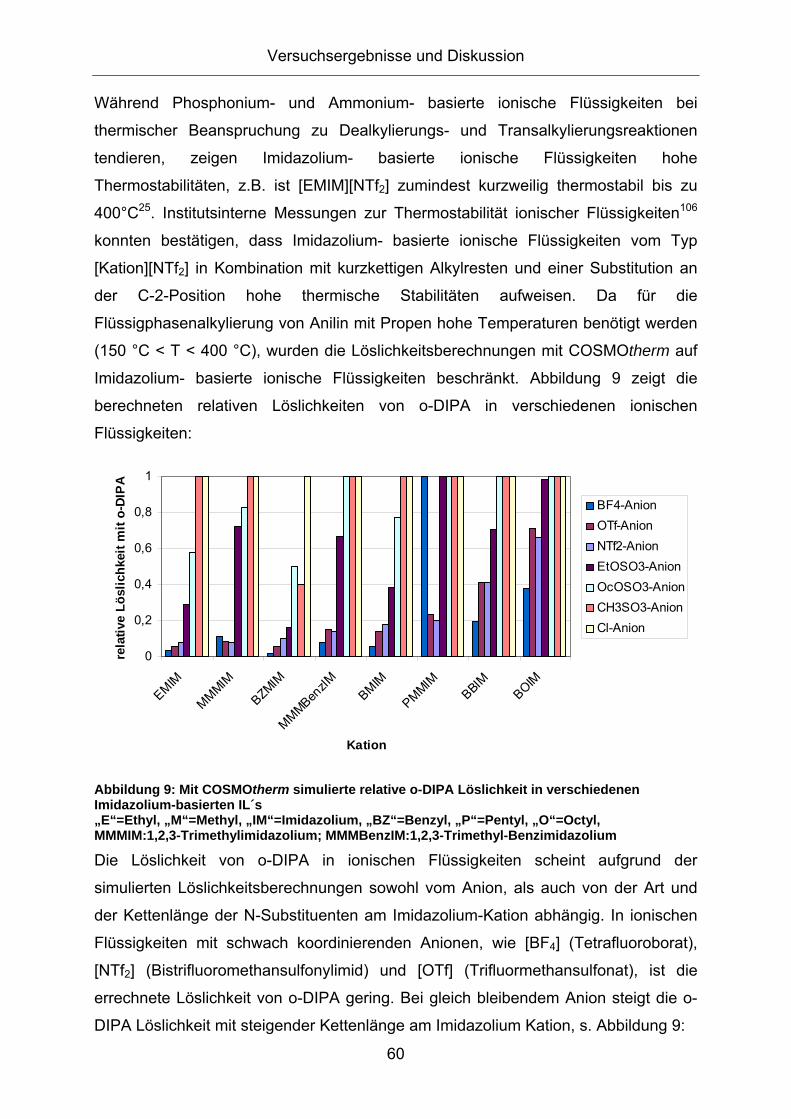
Während Phosphonium- und Ammonium- basierte ionische Flüssigkeiten bei
thermischer Beanspruchung zu Dealkylierungs- und Transalkylierungsreaktionen
tendieren, zeigen Imidazolium- basierte ionische Flüssigkeiten hohe
Thermostabilitäten, z.B. ist [EMIM][NTf2] zumindest kurzweilig thermostabil bis zu
400°C25. Allerdings können kaum Aussagen zur Langzeitstabilität während eines
kontinuierlichen Einsatzes der IL getroffen werden. Institutsinterne Messungen zur
Thermostabilität ionischer Flüssigkeiten108 konnten jedoch bestätigen, dass
Imidazolium- basierte ionische Flüssigkeiten vom Typ [Kation][NTf2] in Kombination
mit kurzkettigen Alkylresten und einer Substitution an der C-2-Position hohe
thermische Stabilitäten aufweisen. So zeigt die ionische Flüssigkeit
[MMMBenzIm][NTf2], die aufgrund der interessanten Löslichkeitseigenschaften mit o-
DIPA für nachfolgende TG-Messung ausgewählt wurde, eine Thermostabilität von
über 300 °C.
Versuchsergebnisse und Diskussion
66
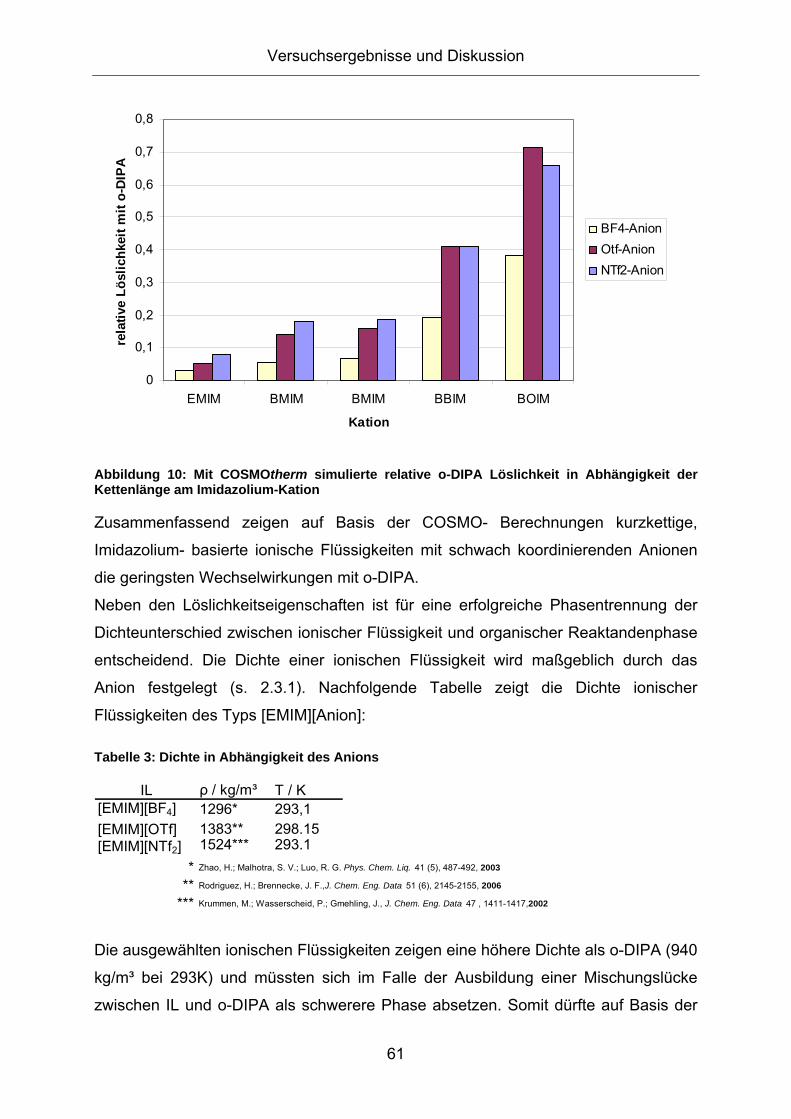
Da der Wassergehalt und der Reinheitsgrad der IL´s einen entscheidenden Einfluss
auf die Thermostabilität der ionischen Flüssigkeiten nimmt, ist davon auszugehen,
dass in einem technischen und kontinuierlichem Betrieb die im Folgenden
gemessene Thermostabilität eher die Obergrenze darstellt. Zur besseren
Vergleichbarkeit wurden in Diagramm 16 die TG-Messungen differentiell
aufgetragen:
-10
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
0 50 100 150 200 250 300 350 400 450 500
T [°C]
DTG
[m-%
/min
]
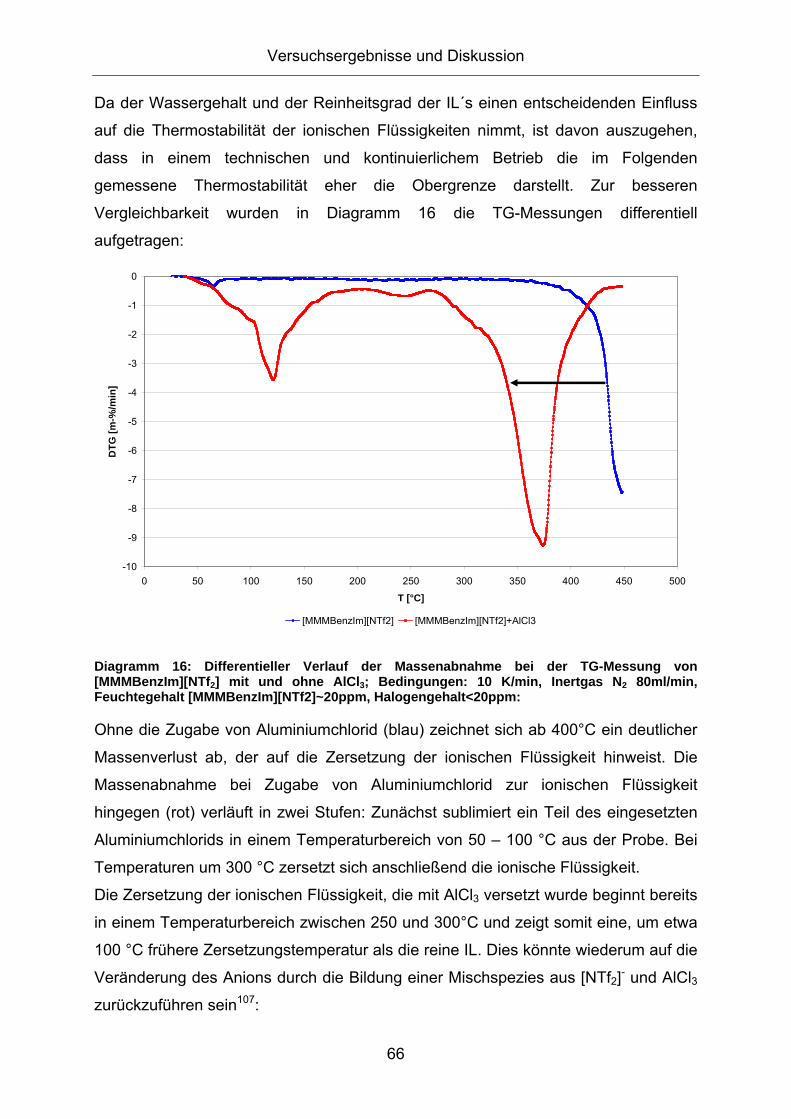
[MMMBenzIm][NTf2] [MMMBenzIm][NTf2]+AlCl3
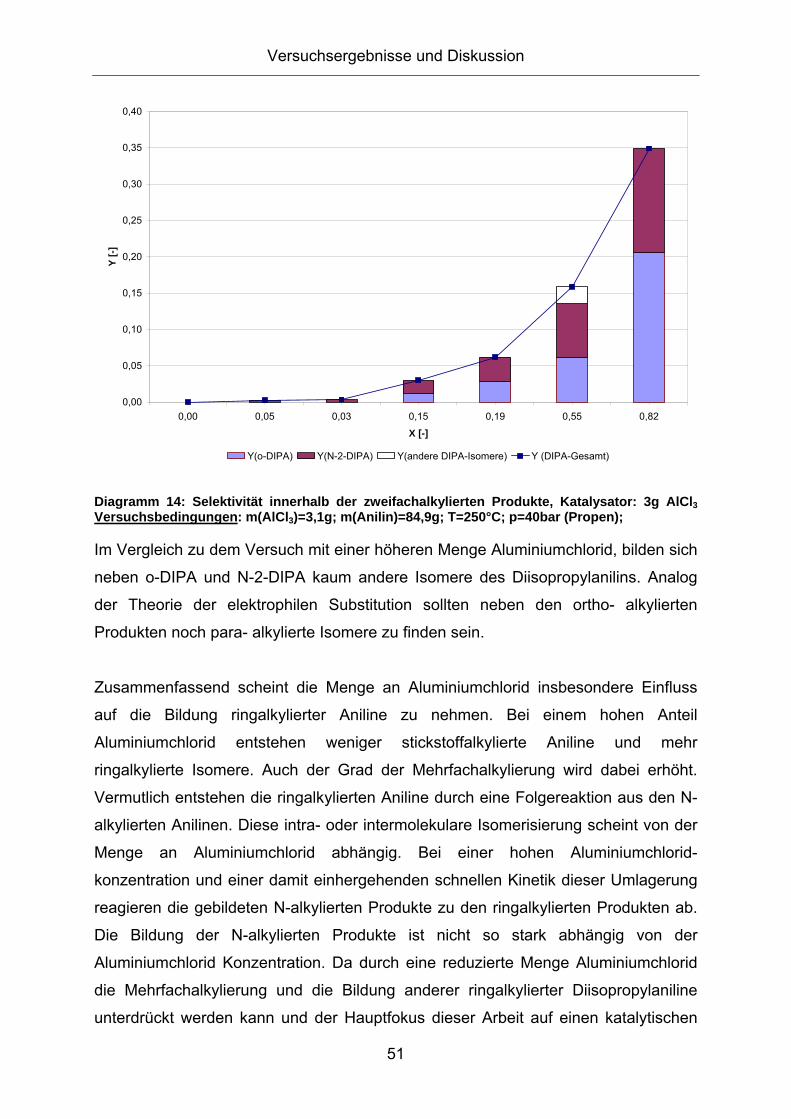
Diagramm 16: Differentieller Verlauf der Massenabnahme bei der TG-Messung von [MMMBenzIm][NTf2] mit und ohne AlCl3; Bedingungen: 10 K/min, Inertgas N2 80ml/min, Feuchtegehalt [MMMBenzIm][NTf2]~20ppm, Halogengehalt<20ppm:
Ohne die Zugabe von Aluminiumchlorid (blau) zeichnet sich ab 400°C ein deutlicher
Massenverlust ab, der auf die Zersetzung der ionischen Flüssigkeit hinweist. Die
Massenabnahme bei Zugabe von Aluminiumchlorid zur ionischen Flüssigkeit
hingegen (rot) verläuft in zwei Stufen: Zunächst sublimiert ein Teil des eingesetzten
Aluminiumchlorids in einem Temperaturbereich von 50 – 100 °C aus der Probe. Bei
Temperaturen um 300 °C zersetzt sich anschließend die ionische Flüssigkeit.
Die Zersetzung der ionischen Flüssigkeit, die mit AlCl3 versetzt wurde beginnt bereits
in einem Temperaturbereich zwischen 250 und 300°C und zeigt somit eine, um etwa
100 °C frühere Zersetzungstemperatur als die reine IL. Dies könnte wiederum auf die
Veränderung des Anions durch die Bildung einer Mischspezies aus [NTf2]- und AlCl3
zurückzuführen sein107:
Versuchsergebnisse und Diskussion
67
[Kation][NTf2] + AlCl3 [Kation][AlxNTf2yClz] Überschüssiges Aluminiumchlorid sublimiert aus der ionischen Flüssigkeit heraus,
bis ein stabiles Verhältnis zwischen Al/NTf2/Cl gebildet wird. Denkbar wäre ebenfalls,
dass die Sublimation von Al(NTf2)3 zu der Massenabnahme zwischen 50 und 150 °C
führt. Die letztlich nach der Sublimation der flüchtigen Bestandteile resultierende
ionische Flüssigkeit hat aufgrund der Strukturveränderungen am Anion eine
geringere Thermostabilität als die ursprüngliche IL ohne Zugabe von AlCl3. Die
ermittelte Thermostabilität bewegt sich dabei an der Grenze zum geforderten
Temperaturbereich für eine erfolgreiche Alkylierung.
3.3.1.3. Alkylierung mit ionischen Flüssigkeiten
In vielen Anwendungen kann durch den Einsatz ionischer Flüssigkeiten mit schwach
koordinierenden Anionen die Reaktivität und die Selektivität des gelösten
Katalysators verbessert werden, da der Katalysator in einem aktivierten und polaren
Zustand gelöst und somit besser für die Reaktanden verfügbar ist.6
Es besteht daher die Möglichkeit, dass durch die Verwendung von ionischen
Flüssigkeiten die Temperaturgrenze für eine erfolgreiche Alkylierung herabgesetzt
werden könnte. Auf diese Weise ließe sich das Problem der thermischen Stabilität
der aciden ionischen Flüssigkeit umgehen.
Diagramm 17 zeigt einen Alkylierungsversuch mit ansteigendem Temperaturprofil.
Als Katalysatorsystem wurde eine Mischung aus AlCl3 und Aluminiumtrianilid
verwendet, das in [EMIM][NTf2] gelöst wurde. Zur Phasentrennung zwischen
ionischer Katalysatorphase und organischer Produktphase wurde Decalin als
Extraktionsmittel eingesetzt:
Versuchsergebnisse und Diskussion
68
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 2 4 6 8 10 12 14 16 18 20
t [h]
Y [-]
190
200
210
220
230
240
250
260
270
280
290
T [°
C]
Y(IPA) Y(DIPA) Y(TIPA) Temperatur
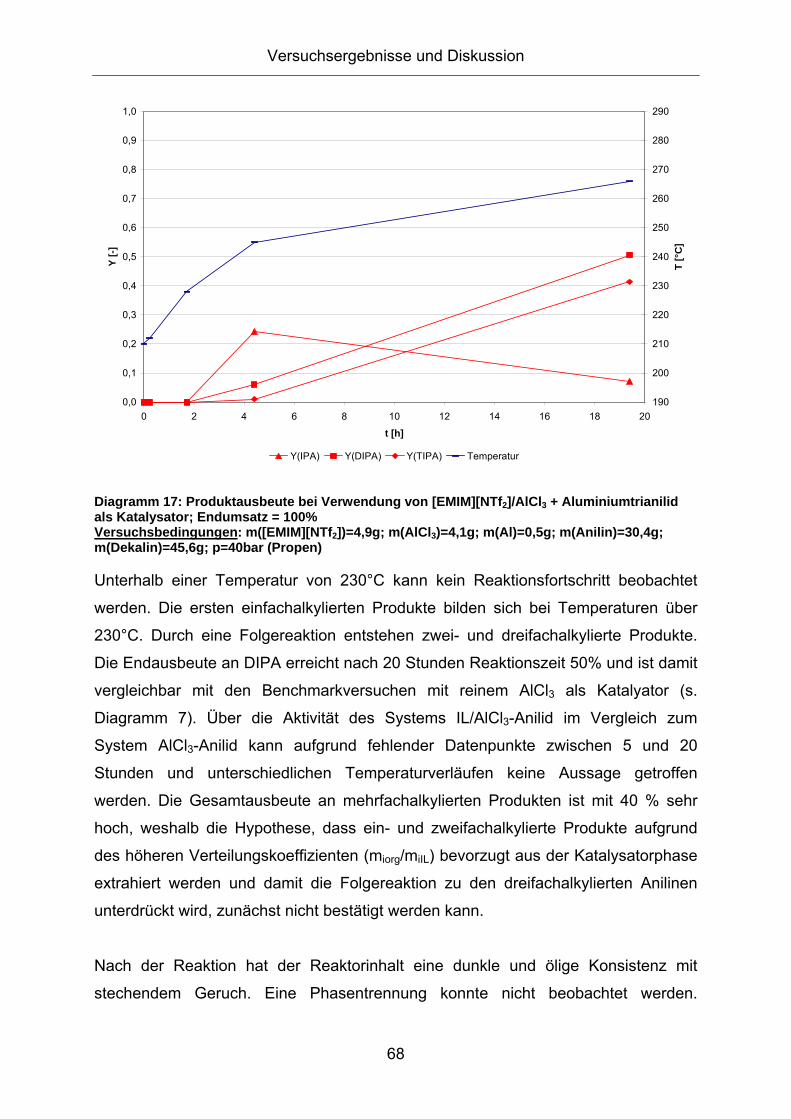
Diagramm 17: Produktausbeute bei Verwendung von [EMIM][NTf2]/AlCl3 + Aluminiumtrianilid als Katalysator; Endumsatz = 100% Versuchsbedingungen: m([EMIM][NTf2])=4,9g; m(AlCl3)=4,1g; m(Al)=0,5g; m(Anilin)=30,4g; m(Dekalin)=45,6g; p=40bar (Propen)
Unterhalb einer Temperatur von 230°C kann kein Reaktionsfortschritt beobachtet
werden. Die ersten einfachalkylierten Produkte bilden sich bei Temperaturen über
230°C. Durch eine Folgereaktion entstehen zwei- und dreifachalkylierte Produkte.
Die Endausbeute an DIPA erreicht nach 20 Stunden Reaktionszeit 50% und ist damit
vergleichbar mit den Benchmarkversuchen mit reinem AlCl3 als Katalyator (s.
Diagramm 7). Über die Aktivität des Systems IL/AlCl3-Anilid im Vergleich zum
System AlCl3-Anilid kann aufgrund fehlender Datenpunkte zwischen 5 und 20
Stunden und unterschiedlichen Temperaturverläufen keine Aussage getroffen
werden. Die Gesamtausbeute an mehrfachalkylierten Produkten ist mit 40 % sehr
hoch, weshalb die Hypothese, dass ein- und zweifachalkylierte Produkte aufgrund
des höheren Verteilungskoeffizienten (miorg/miIL) bevorzugt aus der Katalysatorphase
extrahiert werden und damit die Folgereaktion zu den dreifachalkylierten Anilinen
unterdrückt wird, zunächst nicht bestätigt werden kann.
Nach der Reaktion hat der Reaktorinhalt eine dunkle und ölige Konsistenz mit
stechendem Geruch. Eine Phasentrennung konnte nicht beobachtet werden.
Versuchsergebnisse und Diskussion
69
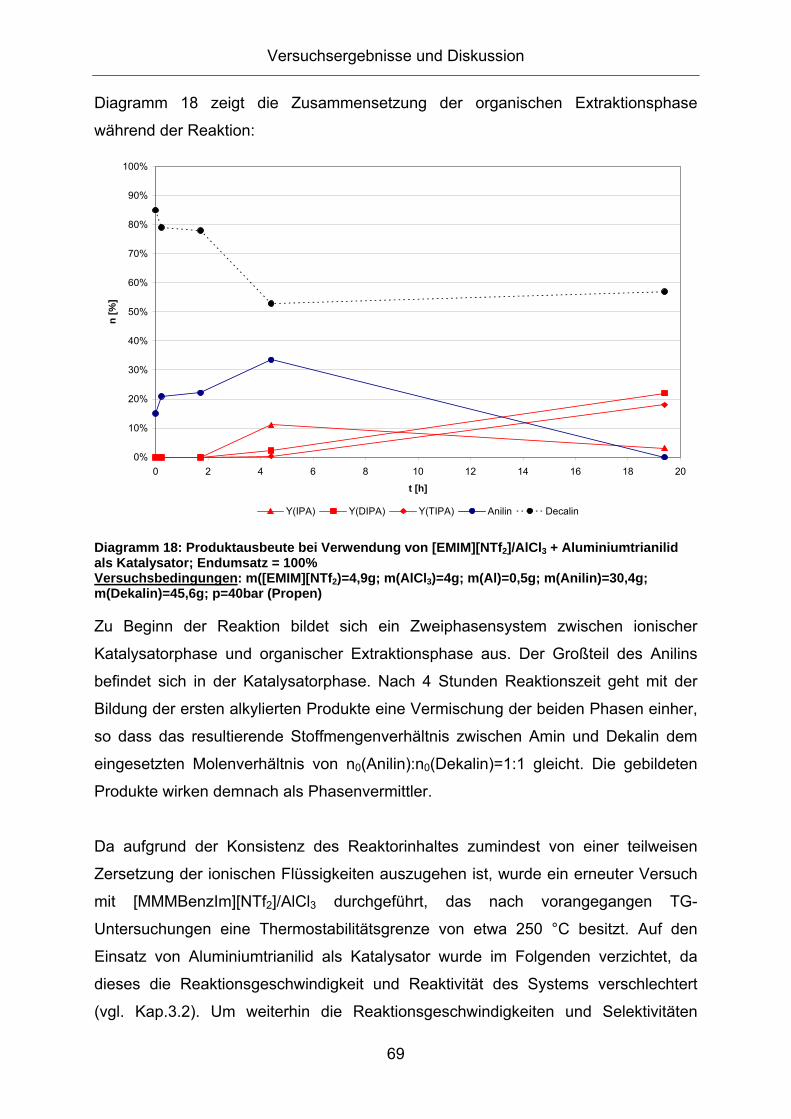
Diagramm 18 zeigt die Zusammensetzung der organischen Extraktionsphase
während der Reaktion:
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 2 4 6 8 10 12 14 16 18 20
t [h]
n [%
]
Y(IPA) Y(DIPA) Y(TIPA) Anilin Decalin
Diagramm 18: Produktausbeute bei Verwendung von [EMIM][NTf2]/AlCl3 + Aluminiumtrianilid als Katalysator; Endumsatz = 100% Versuchsbedingungen: m([EMIM][NTf2)=4,9g; m(AlCl3)=4g; m(Al)=0,5g; m(Anilin)=30,4g; m(Dekalin)=45,6g; p=40bar (Propen)
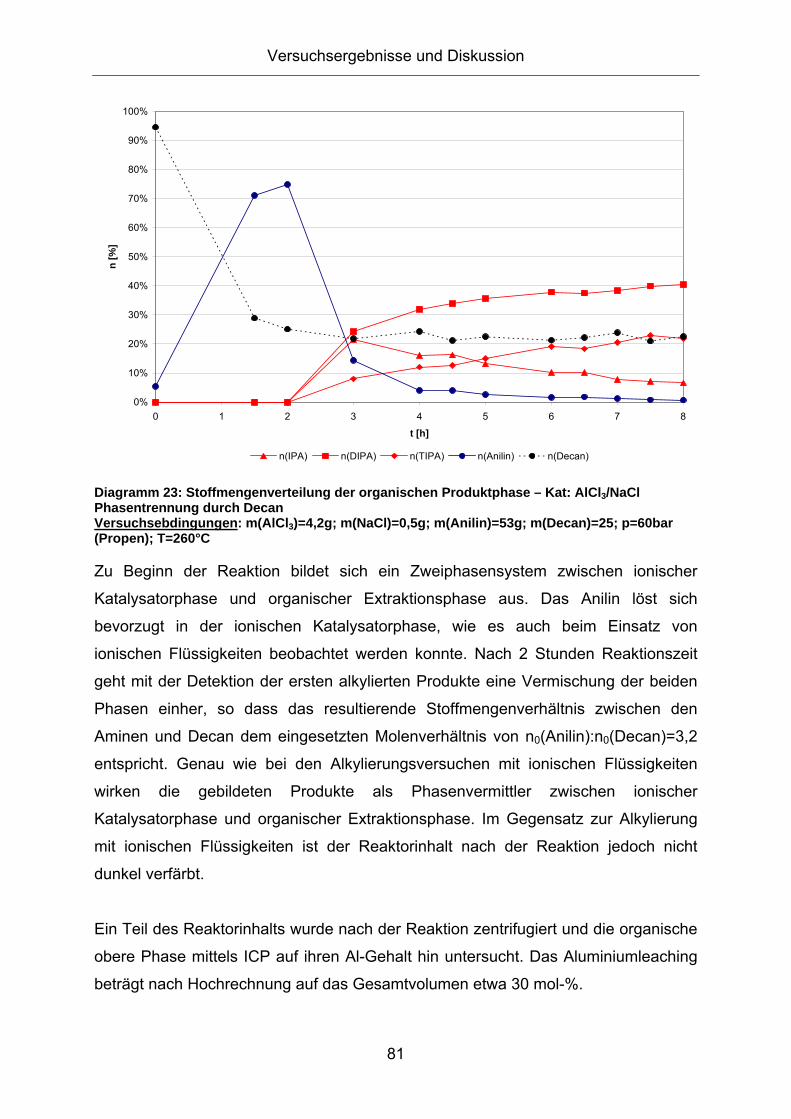
Zu Beginn der Reaktion bildet sich ein Zweiphasensystem zwischen ionischer
Katalysatorphase und organischer Extraktionsphase aus. Der Großteil des Anilins
befindet sich in der Katalysatorphase. Nach 4 Stunden Reaktionszeit geht mit der
Bildung der ersten alkylierten Produkte eine Vermischung der beiden Phasen einher,
so dass das resultierende Stoffmengenverhältnis zwischen Amin und Dekalin dem
eingesetzten Molenverhältnis von n0(Anilin):n0(Dekalin)=1:1 gleicht. Die gebildeten
Produkte wirken demnach als Phasenvermittler.
Da aufgrund der Konsistenz des Reaktorinhaltes zumindest von einer teilweisen
Zersetzung der ionischen Flüssigkeiten auszugehen ist, wurde ein erneuter Versuch
mit [MMMBenzIm][NTf2]/AlCl3 durchgeführt, das nach vorangegangen TG-
Untersuchungen eine Thermostabilitätsgrenze von etwa 250 °C besitzt. Auf den
Einsatz von Aluminiumtrianilid als Katalysator wurde im Folgenden verzichtet, da
dieses die Reaktionsgeschwindigkeit und Reaktivität des Systems verschlechtert
(vgl. Kap.3.2). Um weiterhin die Reaktionsgeschwindigkeiten und Selektivitäten
Versuchsergebnisse und Diskussion
70
besser vergleichen zu können, wurde zunächst auf den Einsatz von Dekalin als
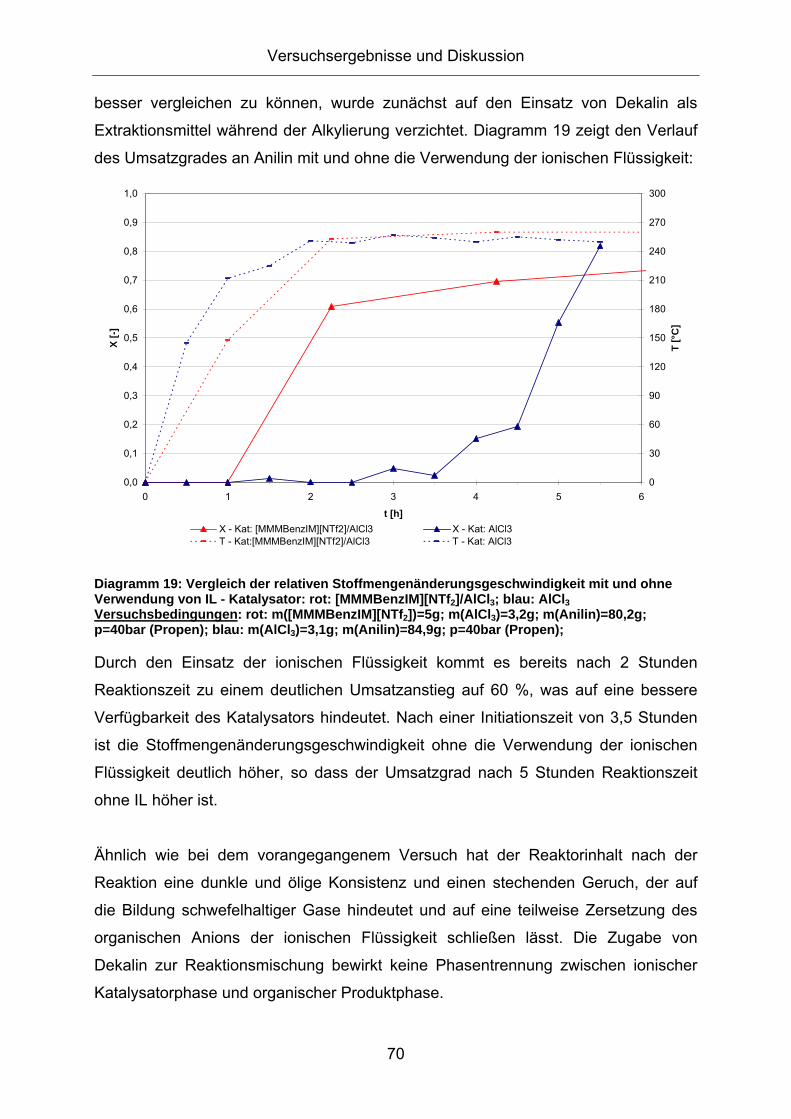
Extraktionsmittel während der Alkylierung verzichtet. Diagramm 19 zeigt den Verlauf
des Umsatzgrades an Anilin mit und ohne die Verwendung der ionischen Flüssigkeit:
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 1 2 3 4 5 6
t [h]
X [-]
0
30
60
90
120
150
180
210
240
270
300
T [°
C]
X - Kat: [MMMBenzIM][NTf2]/AlCl3 X - Kat: AlCl3T - Kat:[MMMBenzIM][NTf2]/AlCl3 T - Kat: AlCl3
Diagramm 19: Vergleich der relativen Stoffmengenänderungsgeschwindigkeit mit und ohne Verwendung von IL - Katalysator: rot: [MMMBenzIM][NTf2]/AlCl3; blau: AlCl3 Versuchsbedingungen: rot: m([MMMBenzIM][NTf2])=5g; m(AlCl3)=3,2g; m(Anilin)=80,2g; p=40bar (Propen); blau: m(AlCl3)=3,1g; m(Anilin)=84,9g; p=40bar (Propen);
Durch den Einsatz der ionischen Flüssigkeit kommt es bereits nach 2 Stunden
Reaktionszeit zu einem deutlichen Umsatzanstieg auf 60 %, was auf eine bessere
Verfügbarkeit des Katalysators hindeutet. Nach einer Initiationszeit von 3,5 Stunden
ist die Stoffmengenänderungsgeschwindigkeit ohne die Verwendung der ionischen
Flüssigkeit deutlich höher, so dass der Umsatzgrad nach 5 Stunden Reaktionszeit
ohne IL höher ist.
Ähnlich wie bei dem vorangegangenem Versuch hat der Reaktorinhalt nach der
Reaktion eine dunkle und ölige Konsistenz und einen stechenden Geruch, der auf
die Bildung schwefelhaltiger Gase hindeutet und auf eine teilweise Zersetzung des
organischen Anions der ionischen Flüssigkeit schließen lässt. Die Zugabe von
Dekalin zur Reaktionsmischung bewirkt keine Phasentrennung zwischen ionischer
Katalysatorphase und organischer Produktphase.
Versuchsergebnisse und Diskussion
71
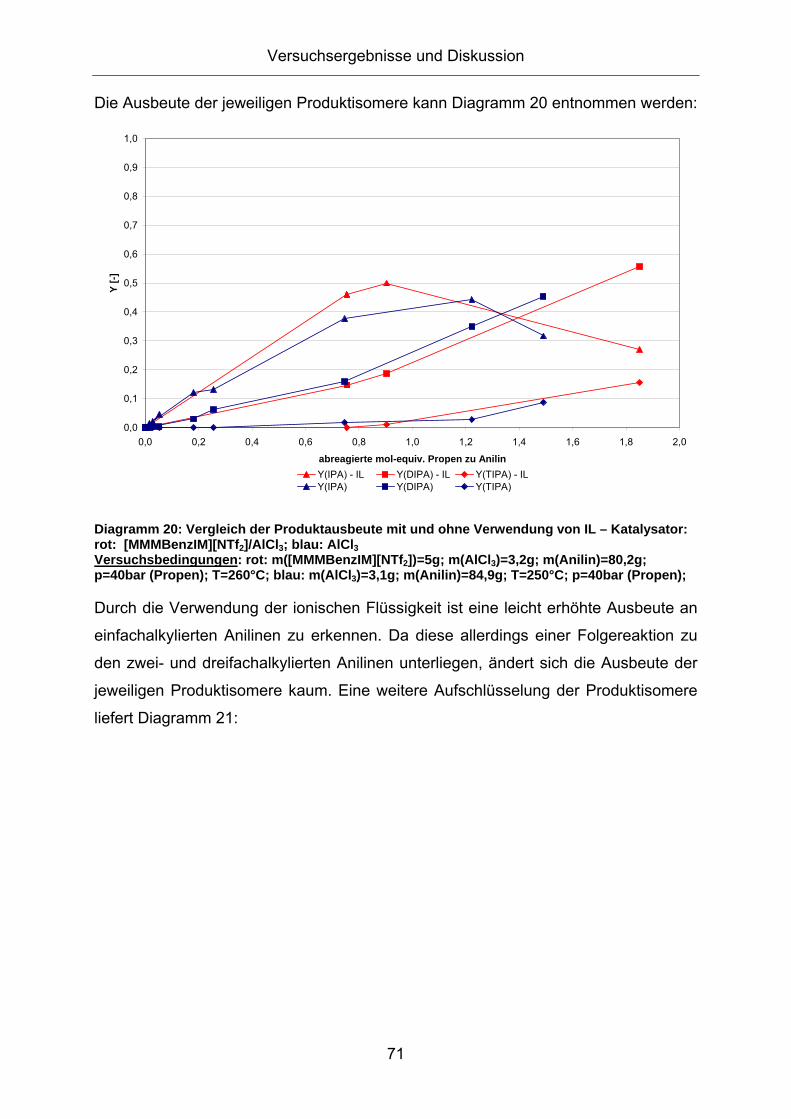
Die Ausbeute der jeweiligen Produktisomere kann Diagramm 20 entnommen werden:
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0
abreagierte mol-equiv. Propen zu Anilin
Y [-]
Y(IPA) - IL Y(DIPA) - IL Y(TIPA) - ILY(IPA) Y(DIPA) Y(TIPA)
Diagramm 20: Vergleich der Produktausbeute mit und ohne Verwendung von IL – Katalysator: rot: [MMMBenzIM][NTf2]/AlCl3; blau: AlCl3 Versuchsbedingungen: rot: m([MMMBenzIM][NTf2])=5g; m(AlCl3)=3,2g; m(Anilin)=80,2g; p=40bar (Propen); T=260°C; blau: m(AlCl3)=3,1g; m(Anilin)=84,9g; T=250°C; p=40bar (Propen);
Durch die Verwendung der ionischen Flüssigkeit ist eine leicht erhöhte Ausbeute an
einfachalkylierten Anilinen zu erkennen. Da diese allerdings einer Folgereaktion zu
den zwei- und dreifachalkylierten Anilinen unterliegen, ändert sich die Ausbeute der
jeweiligen Produktisomere kaum. Eine weitere Aufschlüsselung der Produktisomere
liefert Diagramm 21:
Versuchsergebnisse und Diskussion
72
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
X (Anilin) [-]
Y [-]
Y(IPA-Gesamt) Y(2-IPA) Y(N-IPA)Y(IPA-Gesamt) - IL Y(2-IPA) - IL Y(N-IPA) - IL
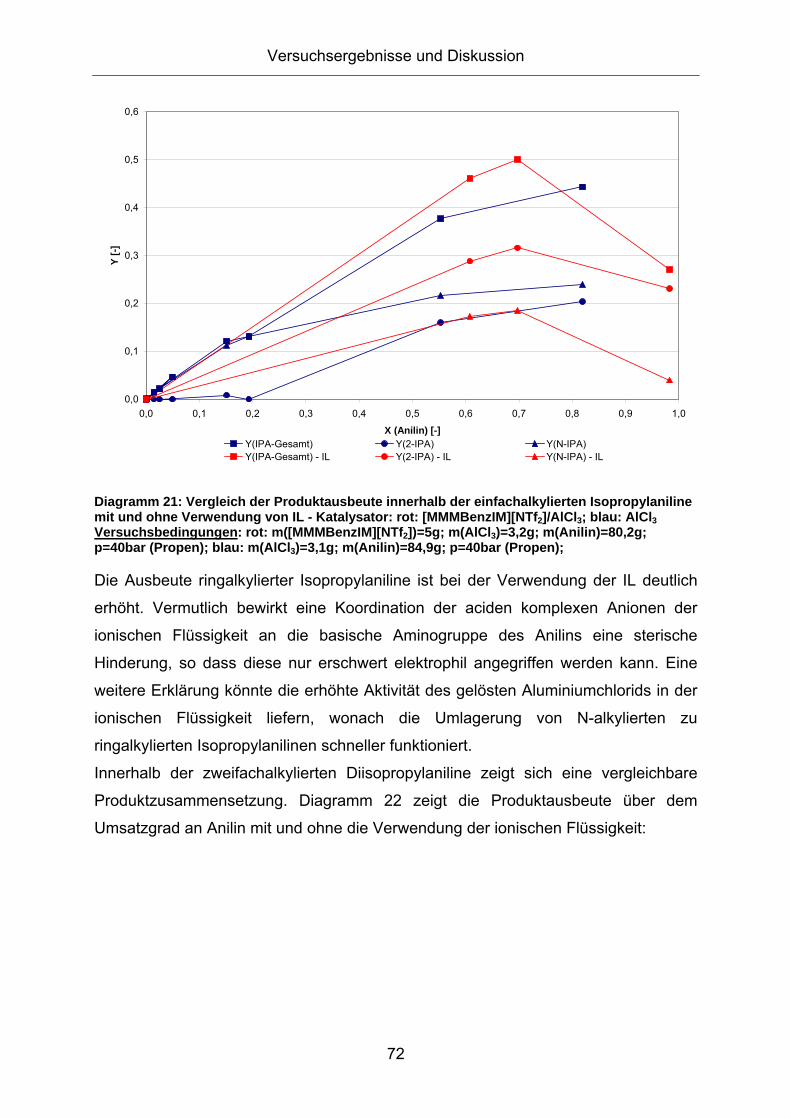
Diagramm 21: Vergleich der Produktausbeute innerhalb der einfachalkylierten Isopropylaniline mit und ohne Verwendung von IL - Katalysator: rot: [MMMBenzIM][NTf2]/AlCl3; blau: AlCl3 Versuchsbedingungen: rot: m([MMMBenzIM][NTf2])=5g; m(AlCl3)=3,2g; m(Anilin)=80,2g; p=40bar (Propen); blau: m(AlCl3)=3,1g; m(Anilin)=84,9g; p=40bar (Propen);
Die Ausbeute ringalkylierter Isopropylaniline ist bei der Verwendung der IL deutlich
erhöht. Vermutlich bewirkt eine Koordination der aciden komplexen Anionen der
ionischen Flüssigkeit an die basische Aminogruppe des Anilins eine sterische
Hinderung, so dass diese nur erschwert elektrophil angegriffen werden kann. Eine
weitere Erklärung könnte die erhöhte Aktivität des gelösten Aluminiumchlorids in der
ionischen Flüssigkeit liefern, wonach die Umlagerung von N-alkylierten zu
Innerhalb der zweifachalkylierten Diisopropylaniline zeigt sich eine vergleichbare
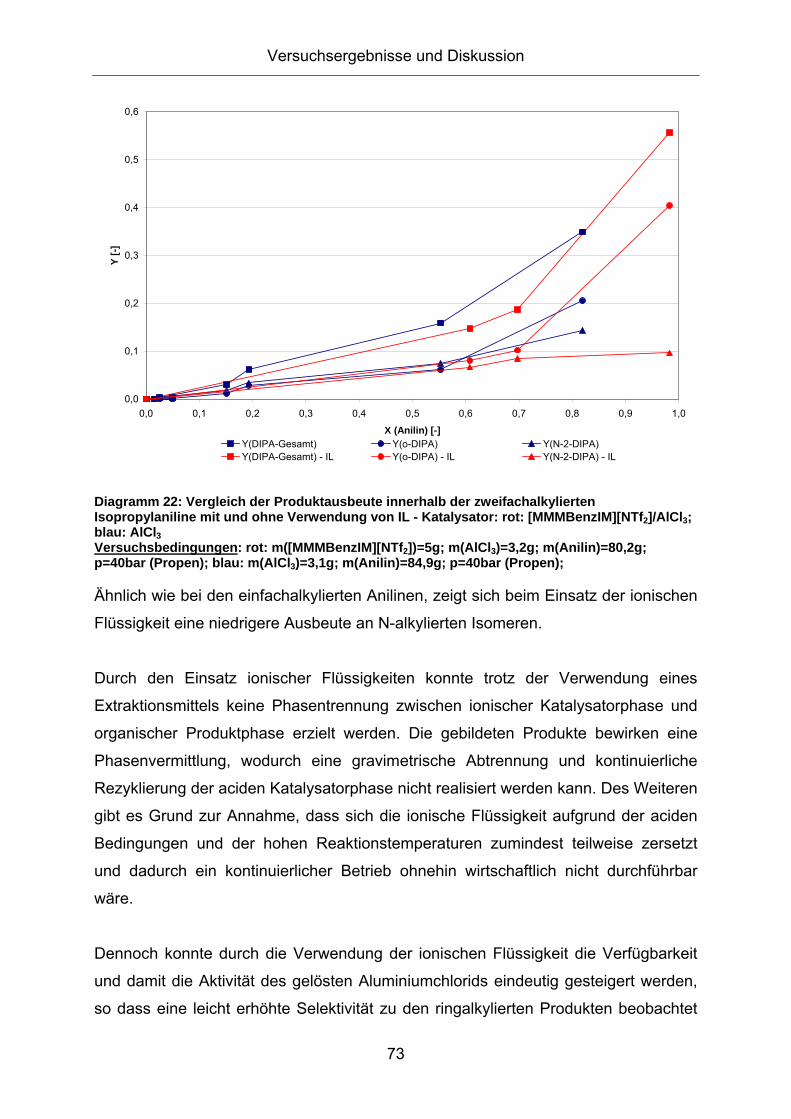
Produktzusammensetzung. Diagramm 22 zeigt die Produktausbeute über dem
Umsatzgrad an Anilin mit und ohne die Verwendung der ionischen Flüssigkeit:
Versuchsergebnisse und Diskussion
73
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
X (Anilin) [-]
Y [-]
Y(DIPA-Gesamt) Y(o-DIPA) Y(N-2-DIPA)Y(DIPA-Gesamt) - IL Y(o-DIPA) - IL Y(N-2-DIPA) - IL
Diagramm 22: Vergleich der Produktausbeute innerhalb der zweifachalkylierten Isopropylaniline mit und ohne Verwendung von IL - Katalysator: rot: [MMMBenzIM][NTf2]/AlCl3; blau: AlCl3 Versuchsbedingungen: rot: m([MMMBenzIM][NTf2])=5g; m(AlCl3)=3,2g; m(Anilin)=80,2g; p=40bar (Propen); blau: m(AlCl3)=3,1g; m(Anilin)=84,9g; p=40bar (Propen);
Ähnlich wie bei den einfachalkylierten Anilinen, zeigt sich beim Einsatz der ionischen
Flüssigkeit eine niedrigere Ausbeute an N-alkylierten Isomeren.
Durch den Einsatz ionischer Flüssigkeiten konnte trotz der Verwendung eines
Extraktionsmittels keine Phasentrennung zwischen ionischer Katalysatorphase und
organischer Produktphase erzielt werden. Die gebildeten Produkte bewirken eine
Phasenvermittlung, wodurch eine gravimetrische Abtrennung und kontinuierliche
Rezyklierung der aciden Katalysatorphase nicht realisiert werden kann. Des Weiteren
gibt es Grund zur Annahme, dass sich die ionische Flüssigkeit aufgrund der aciden
Bedingungen und der hohen Reaktionstemperaturen zumindest teilweise zersetzt
und dadurch ein kontinuierlicher Betrieb ohnehin wirtschaftlich nicht durchführbar
wäre.
Dennoch konnte durch die Verwendung der ionischen Flüssigkeit die Verfügbarkeit
und damit die Aktivität des gelösten Aluminiumchlorids eindeutig gesteigert werden,
so dass eine leicht erhöhte Selektivität zu den ringalkylierten Produkten beobachtet
Versuchsergebnisse und Diskussion
74
wird. Um diese Vorteile zu nutzen, soll im Folgenden der Einsatz anorganischer
Salzschmelzen untersucht werden, die im Vergleich zu den ionischen Flüssigkeiten
eine wesentlich höhere Thermostabilität aufweisen und zudem preisgünstiger sind.
3.3.2. Einsatz anorganischer Salzschmelzen
AlCl3 bildet mit unterschiedlichen Chloridsalzen niedrigschmelzende Salze, die
aufgrund der Ausbildung eines Eutektikums einen niedrigeren Schmelzbereich
aufweisen als die Reinsubstanzen:
eutektische Mischung (molar) Schmelzbereich
AlCl3/NaCl81 ~0,6 ~113°C
AlCl3/LiCl109 ~0,58 ~108°C
AlCl3/KCl109 ~0,65 ~123°C
Die anorganischen aciden Salzschmelzen haben den Vorteil der sehr hohen
thermischen Stabilität und sind wesentlich günstiger als organische ionische
Flüssigkeiten. Untersuchungen zum Phasenverhalten zwischen Salzschmelze und
den organischen Produkten sollen zeigen, ob die zuvor ermittelten Ergebnisse mit
ionischen Flüssigkeiten auch auf anorganische Salzschmelzen anwendbar sind.
3.3.2.1. Vorversuche - Untersuchungen zum
Mischungsverhalten mit 2,6-Diisopropylanilin
Für die Untersuchungen zum Mischungsverhalten zwischen der aciden
Salzschmelze und o-DIPA wurden die Salzkomponenten (AlCl3/NaCl) unter einer
Decanschutzschicht geschmolzen, da hierdurch die Sublimation des AlCl3 bei
Temperaturen über 100°C verringert werden kann. Anschließend wurden die
organischen Aniline unter Inertgas zugegeben und mit der aciden Salzschmelze 2
Stunden bei 170 °C dispergiert.
Die Phasentrennung zwischen der aciden Salzschmelze AlCl3/NaCl und o-DIPA
gelingt durch einfaches Überschichten der beiden Flüssigkeiten aufgrund des
vorherrschenden Dichteunterschieds. Sobald die beiden Phasen ineinander
Versuchsergebnisse und Diskussion
75
dispergiert werden, lässt sich die acide Salzschmelze nicht mehr flüssig vom o-DIPA
abtrennen. Untersucht wurden hierzu folgende Einflussparameter:
Temperaturvariation zwischen 60 und 220°C: Bei einer raschen Abkühlung
der Reaktionsmischung erstarrt die gesamte Flüssigkeit im Reaktionskolben,
ohne dass eine Trennung zwischen acider Salzschmelze und organischer
Produktphase beobachtet werden kann.
Zentrifugation der Probe bis zu dem 3000 – fachen der Erdbeschleunigung
Veränderung der Zusammensetzung der Probe von 0,6 g o-DIPA pro g
Salzschmelze bis 20 g o-DIPA pro g Salzschmelze
Anlegen eines elektrischen Feldes: elektrostatische Emulsionsspalter werden
seit vielen Jahren im großtechnischen Maßstab zur Entsalzung von Rohöl
eingesetzt.110 Untersucht wurden folgende Frequenz- und
Spannungsbereiche: f = 50 - 107 Hz; U = 0,5 - 60 V; I = 0 A.
Da sich mit keiner Variation eine flüssige Phasentrennung zwischen acider
Salzschmelze und o-DIPA einstellt, muss für eine erfolgreiche flüssig-flüssig
Zweiphasenalkylierung ein Extraktionsmittel eingesetzt werden. Ähnlich wie bei den
ionischen Flüssigkeiten weist die acide Salzschmelze eine eindeutige
Mischungslücke zu unpolaren organischen Lösungsmitteln wie Cyclohexan, Dekalin,
Decan, usw. auf.
Wie bereits in den Voruntersuchungen zum Mischungsverhalten mit ionischen
Flüssigkeiten gezeigt, besitzt das o-DIPA eine sehr gute Löslichkeit in Dekalin.
Hierdurch besteht eine Möglichkeit zur Extraktion der Produkte aus der aciden
Salzschmelze in die organische Extraktionsphase. Nachfolgende Untersuchungen
wurden mit Decan als Extraktionsmittel durchgeführt, da Dekalin als
Isomerengemisch zwischen cis- und trans vorliegt und somit die GC-Auswertung
erschwert. Des Weiteren besitzt das Decan eine niedrigere Dichte, was für eine
schnellere Phasentrennung zur aciden Salzschmelze von Vorteil sein kann.
Die Salzkomponenten (AlCl3/NaCl) wurden unter einer Decanschutzschicht
geschmolzen und die organischen Aniline anschließend unter Inertgas zugegeben.
Da nach einer 2-stündigen Dispergierung bei 170 °C keine klare Phasentrennung
beobachtet werden konnte, wurde die Probe zentrifugiert. Die Zentrifuge bewirkt eine
Aufteilung in eine hochviskose dunkle und eine hellere niederviskose Phase, deren
Versuchsergebnisse und Diskussion
76
Aluminiumgehalt mittels ICP untersucht werden kann. Phasenuntersuchungen mittels
GC bestätigen eine Phasentrennung in eine Anilin-arme, o-DIPA-reiche Decanphase
und in eine Anilin-reiche, o-DIPA-arme AlCl3-NaCl Phase. Tabelle 8 zeigt, dass durch
den Einsatz von Decan (20 mol-% bzgl. Anilin/o-DIPA) als Extraktionsmittel, der
Aluminiumaustrag in die organische Produktphase von 90 % auf 57 % reduziert wird.
Tabelle 8:Einfluss des NaCl auf die Immobilisierung des AlCl3
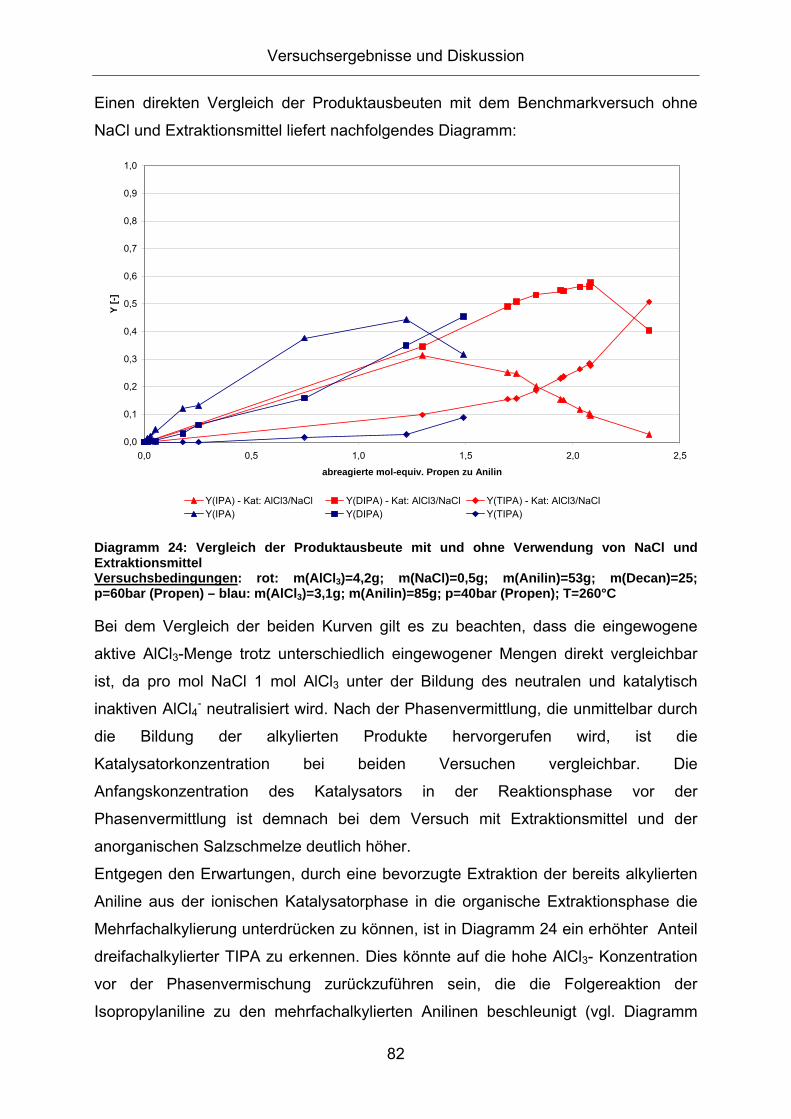
Diagramm 24: Vergleich der Produktausbeute mit und ohne Verwendung von NaCl und Extraktionsmittel Versuchsbedingungen: rot: m(AlCl3)=4,2g; m(NaCl)=0,5g; m(Anilin)=53g; m(Decan)=25; p=60bar (Propen) – blau: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen); T=260°C
Bei dem Vergleich der beiden Kurven gilt es zu beachten, dass die eingewogene
aktive AlCl3-Menge trotz unterschiedlich eingewogener Mengen direkt vergleichbar
ist, da pro mol NaCl 1 mol AlCl3 unter der Bildung des neutralen und katalytisch
inaktiven AlCl4- neutralisiert wird. Nach der Phasenvermittlung, die unmittelbar durch
die Bildung der alkylierten Produkte hervorgerufen wird, ist die
Katalysatorkonzentration bei beiden Versuchen vergleichbar. Die
Anfangskonzentration des Katalysators in der Reaktionsphase vor der
Phasenvermittlung ist demnach bei dem Versuch mit Extraktionsmittel und der
anorganischen Salzschmelze deutlich höher.
Entgegen den Erwartungen, durch eine bevorzugte Extraktion der bereits alkylierten
Aniline aus der ionischen Katalysatorphase in die organische Extraktionsphase die
Mehrfachalkylierung unterdrücken zu können, ist in Diagramm 24 ein erhöhter Anteil
dreifachalkylierter TIPA zu erkennen. Dies könnte auf die hohe AlCl3- Konzentration
vor der Phasenvermischung zurückzuführen sein, die die Folgereaktion der
Isopropylaniline zu den mehrfachalkylierten Anilinen beschleunigt (vgl. Diagramm
Versuchsergebnisse und Diskussion
83
12). Die einsetzende Phasenvermischung verhindert dabei die selektive Extraktion
der gebildeten Produkte. Allerdings erscheint es aufgrund der schnell einsetzenden
Phasenvermittlung wahrscheinlicher, dass die bevorzugte Bildung
mehrfachalkylierter Aniline auf die höhere Acidität des Aluminiumchlorids in der
Salzschmelze bzw. dessen Homogenisierung zurückzuführen ist.
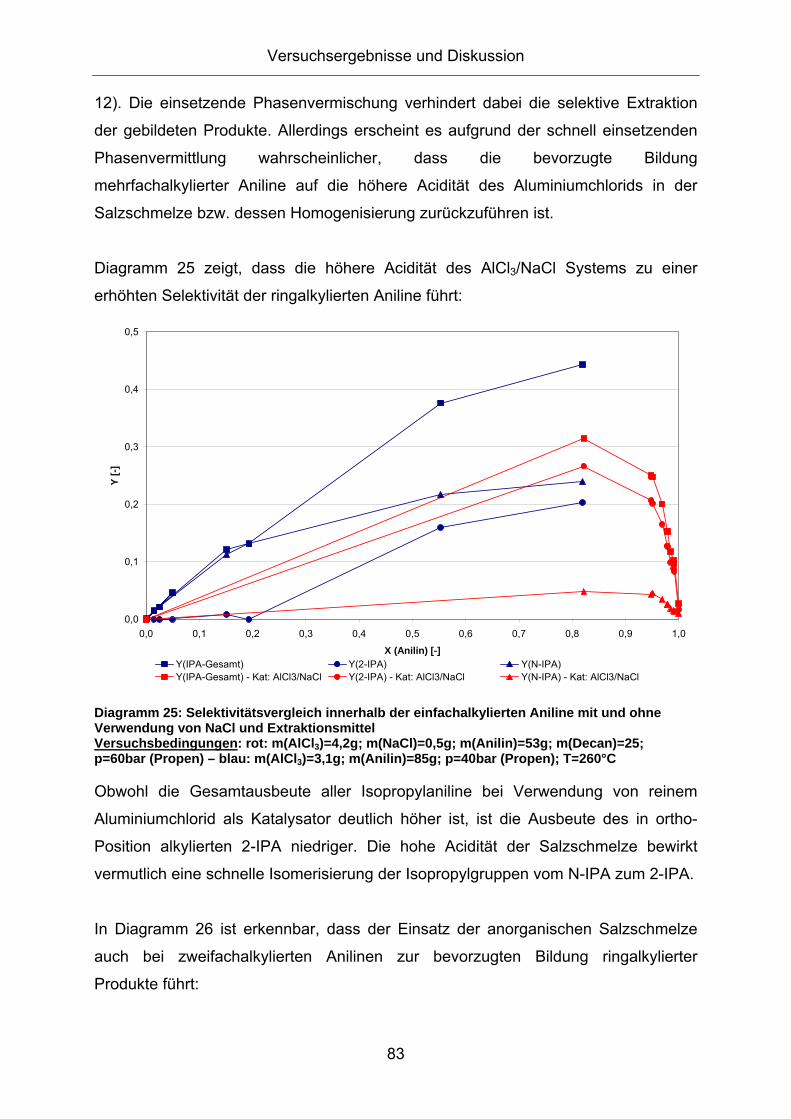
Diagramm 25 zeigt, dass die höhere Acidität des AlCl3/NaCl Systems zu einer
erhöhten Selektivität der ringalkylierten Aniline führt:
Diagramm 25: Selektivitätsvergleich innerhalb der einfachalkylierten Aniline mit und ohne Verwendung von NaCl und Extraktionsmittel Versuchsbedingungen: rot: m(AlCl3)=4,2g; m(NaCl)=0,5g; m(Anilin)=53g; m(Decan)=25; p=60bar (Propen) – blau: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen); T=260°C
Obwohl die Gesamtausbeute aller Isopropylaniline bei Verwendung von reinem
Aluminiumchlorid als Katalysator deutlich höher ist, ist die Ausbeute des in ortho-
Position alkylierten 2-IPA niedriger. Die hohe Acidität der Salzschmelze bewirkt
vermutlich eine schnelle Isomerisierung der Isopropylgruppen vom N-IPA zum 2-IPA.
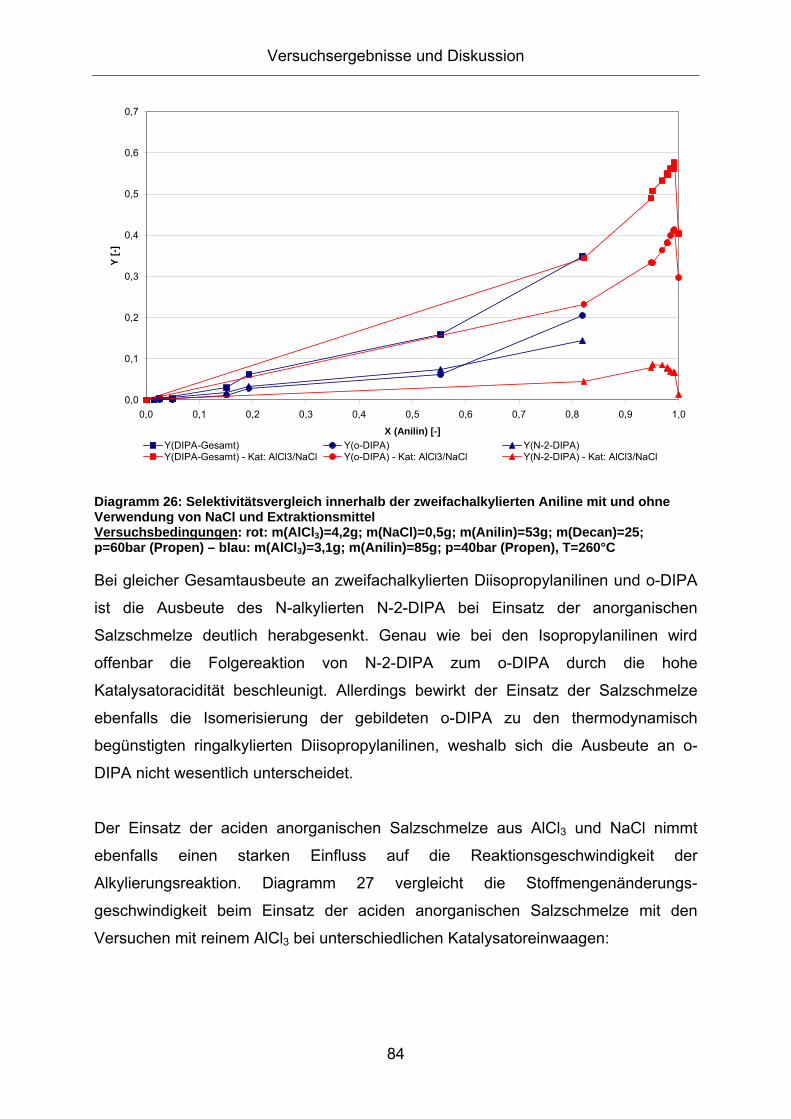
In Diagramm 26 ist erkennbar, dass der Einsatz der anorganischen Salzschmelze
auch bei zweifachalkylierten Anilinen zur bevorzugten Bildung ringalkylierter
Diagramm 26: Selektivitätsvergleich innerhalb der zweifachalkylierten Aniline mit und ohne Verwendung von NaCl und Extraktionsmittel Versuchsbedingungen: rot: m(AlCl3)=4,2g; m(NaCl)=0,5g; m(Anilin)=53g; m(Decan)=25; p=60bar (Propen) – blau: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen), T=260°C
Bei gleicher Gesamtausbeute an zweifachalkylierten Diisopropylanilinen und o-DIPA
ist die Ausbeute des N-alkylierten N-2-DIPA bei Einsatz der anorganischen
Salzschmelze deutlich herabgesenkt. Genau wie bei den Isopropylanilinen wird
offenbar die Folgereaktion von N-2-DIPA zum o-DIPA durch die hohe
Katalysatoracidität beschleunigt. Allerdings bewirkt der Einsatz der Salzschmelze
ebenfalls die Isomerisierung der gebildeten o-DIPA zu den thermodynamisch
begünstigten ringalkylierten Diisopropylanilinen, weshalb sich die Ausbeute an o-
DIPA nicht wesentlich unterscheidet.
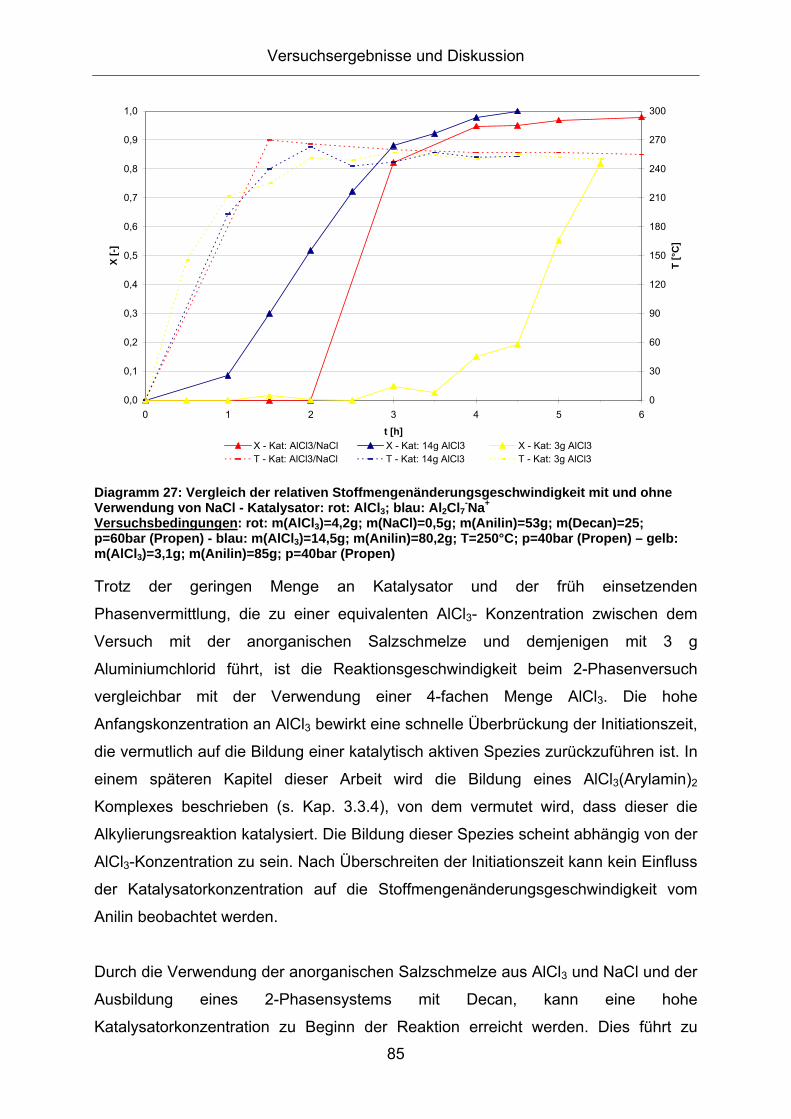
Der Einsatz der aciden anorganischen Salzschmelze aus AlCl3 und NaCl nimmt
ebenfalls einen starken Einfluss auf die Reaktionsgeschwindigkeit der
Alkylierungsreaktion. Diagramm 27 vergleicht die Stoffmengenänderungs-
geschwindigkeit beim Einsatz der aciden anorganischen Salzschmelze mit den
Versuchen mit reinem AlCl3 bei unterschiedlichen Katalysatoreinwaagen:
Versuchsergebnisse und Diskussion
85
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 1 2 3 4 5 6
t [h]
X [-]
0
30
60
90
120
150
180
210
240
270
300
T [°
C]
X - Kat: AlCl3/NaCl X - Kat: 14g AlCl3 X - Kat: 3g AlCl3T - Kat: AlCl3/NaCl T - Kat: 14g AlCl3 T - Kat: 3g AlCl3
Diagramm 27: Vergleich der relativen Stoffmengenänderungsgeschwindigkeit mit und ohne Verwendung von NaCl - Katalysator: rot: AlCl3; blau: Al2Cl7-Na+ Versuchsbedingungen: rot: m(AlCl3)=4,2g; m(NaCl)=0,5g; m(Anilin)=53g; m(Decan)=25; p=60bar (Propen) - blau: m(AlCl3)=14,5g; m(Anilin)=80,2g; T=250°C; p=40bar (Propen) – gelb: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen)
Trotz der geringen Menge an Katalysator und der früh einsetzenden
Phasenvermittlung, die zu einer equivalenten AlCl3- Konzentration zwischen dem
Versuch mit der anorganischen Salzschmelze und demjenigen mit 3 g
Aluminiumchlorid führt, ist die Reaktionsgeschwindigkeit beim 2-Phasenversuch
vergleichbar mit der Verwendung einer 4-fachen Menge AlCl3. Die hohe
Anfangskonzentration an AlCl3 bewirkt eine schnelle Überbrückung der Initiationszeit,
die vermutlich auf die Bildung einer katalytisch aktiven Spezies zurückzuführen ist. In
einem späteren Kapitel dieser Arbeit wird die Bildung eines AlCl3(Arylamin)2
Komplexes beschrieben (s. Kap. 3.3.4), von dem vermutet wird, dass dieser die
Alkylierungsreaktion katalysiert. Die Bildung dieser Spezies scheint abhängig von der
AlCl3-Konzentration zu sein. Nach Überschreiten der Initiationszeit kann kein Einfluss
der Katalysatorkonzentration auf die Stoffmengenänderungsgeschwindigkeit vom
Anilin beobachtet werden.
Durch die Verwendung der anorganischen Salzschmelze aus AlCl3 und NaCl und der
Ausbildung eines 2-Phasensystems mit Decan, kann eine hohe
Katalysatorkonzentration zu Beginn der Reaktion erreicht werden. Dies führt zu
Versuchsergebnisse und Diskussion
86
einem schnellen Anstieg des Anilinumsatzes, vermutlich durch die schnelle Bildung
einer katalytisch aktiven AlCl3(Arylamin)2- Spezies. Weiterhin ist eine verstärkte
Bildung ringalkylierter Produkte zu beobachten, was auf einen weiteren Effekt des
zugesetzten NaCl hindeutet. Vermutlich verändert sich der gebildete
Katalysatorkomplex bei Zugabe geringer Mengen NaCl durch die Bildung einer
ionischen Na+ [AlxClyArylaminz]- - Spezies, dessen veränderte Acidität die Ausbeute
beeinflusst.
Die Phasenvermischung, die mit der Bildung von alkylierten Produkten einhergeht,
bewirkt eine Erniedrigung der Katalysatorkonzentration, jedoch wird die
Folgereaktion zu unerwünschten Nebenprodukten dadurch nicht unterdrückt. Eine
flüssige Phasentrennung kann beim Einsatz der anorganischen Salzschmelze nicht
beobachtet werden.
Um eine bessere Vergleichbarkeit der Versuchsergebnisse mit und ohne den Zusatz
von NaCl zu bekommen, wurde nachfolgend auf den Einsatz von Decan als
Hilfslösungsmittel verzichtet. Somit kommt es zu keiner Veränderung der
Katalysatorkonzentration durch auftretende Phasenvermischungen im Laufe der
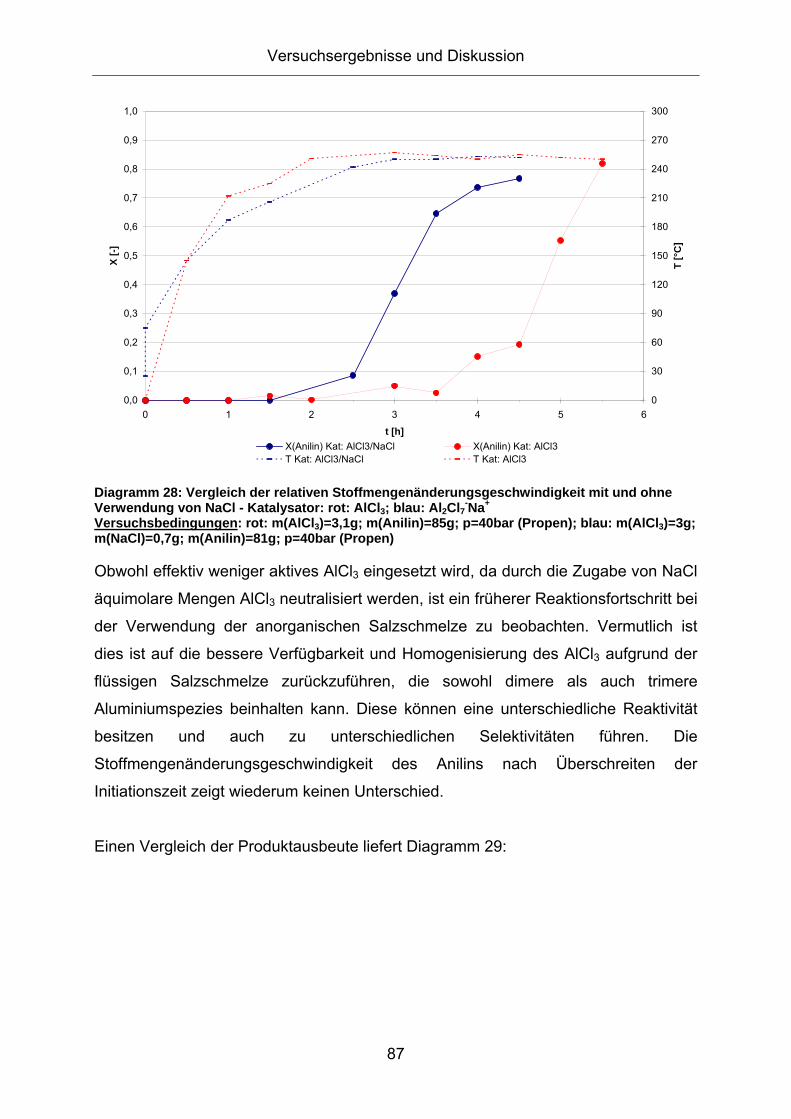
Reaktion. Diagramm 28 zeigt, dass auch bei der gleichen eingewogenen Menge
AlCl3 die Zugabe von NaCl zu einer schnelleren Überbrückung der Initiationszeit
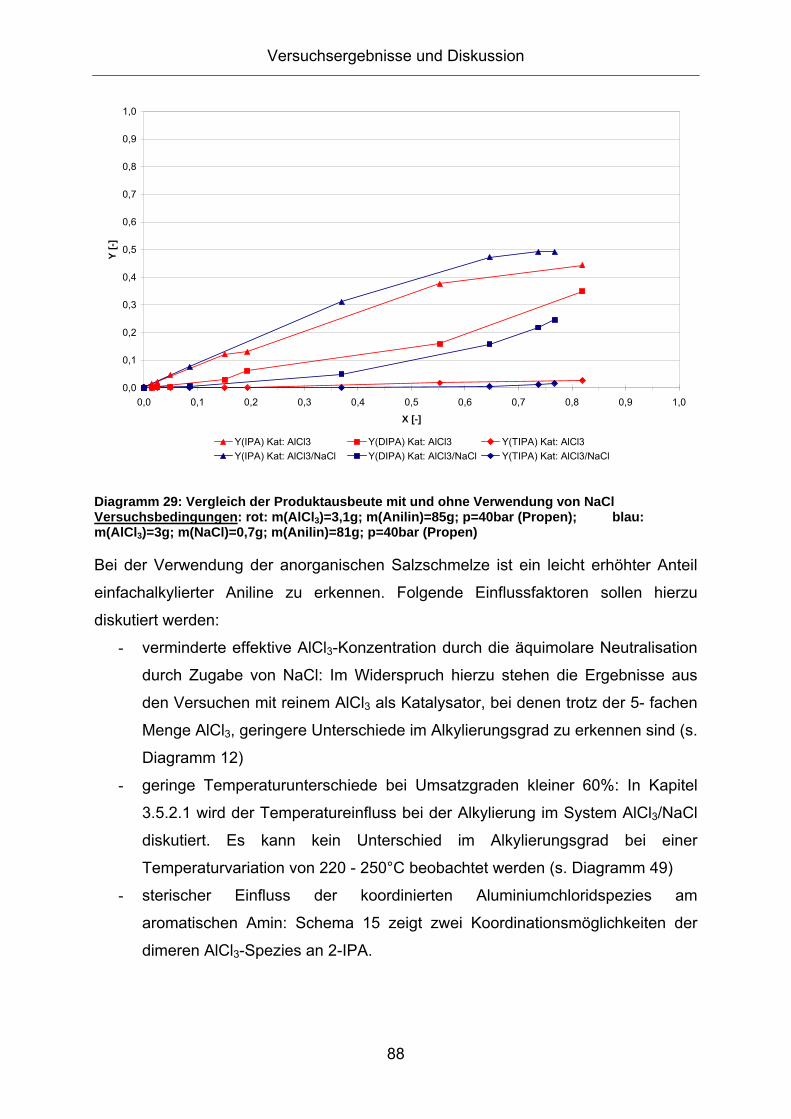
Diagramm 29: Vergleich der Produktausbeute mit und ohne Verwendung von NaCl Versuchsbedingungen: rot: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen); blau: m(AlCl3)=3g; m(NaCl)=0,7g; m(Anilin)=81g; p=40bar (Propen)
Bei der Verwendung der anorganischen Salzschmelze ist ein leicht erhöhter Anteil
einfachalkylierter Aniline zu erkennen. Folgende Einflussfaktoren sollen hierzu
diskutiert werden:
- verminderte effektive AlCl3-Konzentration durch die äquimolare Neutralisation
durch Zugabe von NaCl: Im Widerspruch hierzu stehen die Ergebnisse aus
den Versuchen mit reinem AlCl3 als Katalysator, bei denen trotz der 5- fachen
Menge AlCl3, geringere Unterschiede im Alkylierungsgrad zu erkennen sind (s.
Diagramm 12)
- geringe Temperaturunterschiede bei Umsatzgraden kleiner 60%: In Kapitel
3.5.2.1 wird der Temperatureinfluss bei der Alkylierung im System AlCl3/NaCl
diskutiert. Es kann kein Unterschied im Alkylierungsgrad bei einer
Temperaturvariation von 220 - 250°C beobachtet werden (s. Diagramm 49)
- sterischer Einfluss der koordinierten Aluminiumchloridspezies am
aromatischen Amin: Schema 15 zeigt zwei Koordinationsmöglichkeiten der
dimeren AlCl3-Spezies an 2-IPA.
Versuchsergebnisse und Diskussion
89
AlCl
Cl
Cl
ClAl
Cl
Cl
Cl
NH2Na
AlCl
ClAl
Cl
Cl
Cl
NaNH
ClCl
AlCl
Cl
Cl
ClAl
Cl
Cl
Cl
Na
NH2
Schema 15: Mögliche Koordination der dimeren AlCl3-Spezies an ein aromatisches Amin in einer aciden AlCl3/NaCl Schmelze
Bereits im Jahre 1964 konnten indische Wissenschaftler die Koordination von AlCl3
mit verschiedenen aromatischen Aminen nachweisen.113 Sie kristallisierten aus einer
benzolischen Lösung eine AlCl3(Arylamin)2- Spezies, die sie mittels atomspezifischer
Analysemethoden nachweisen konnten. Das Aluminium kann dabei eine maximale
Koordinationszahl von 6 erhalten, wenn ein dimeres Aluminiumchlorid mit 4
aromatischen Aminen komplexiert. In einer aciden Salzschmelze liegen dimere
Al2Cl7-- Spezies vor, die nach Schema 15 mit dem aromatischen Amin koordinieren
könnten. In diesem Falle könnte der höhere sterische Anspruch die
Mehrfachsubstitution unterdrücken.
In jedem Falle ist davon auszugehen, dass die Zugabe von NaCl den Katalysator
derart verändert, dass die Folgereaktion der bereits alkylierten Aniline zu den
mehrfachalkylierten Anilinen etwas unterdrückt wird.
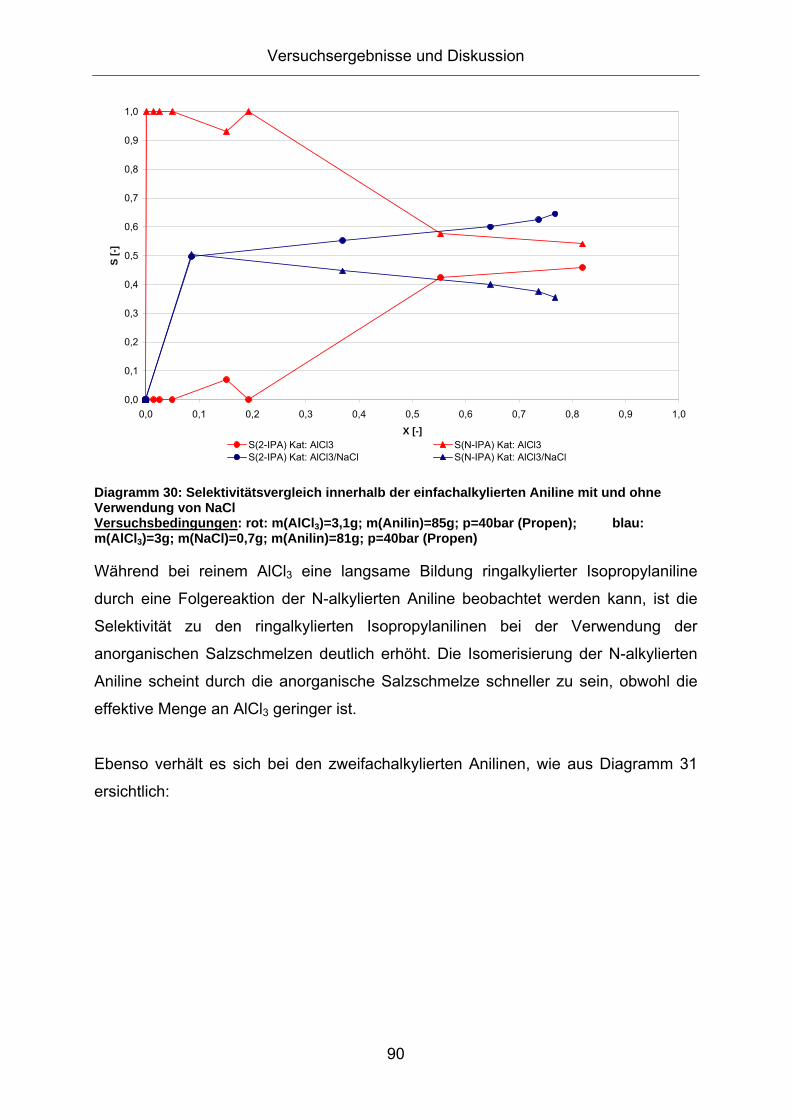
Diagramm 30 zeigt eine weitere Aufschlüsselung der Produkte und verdeutlicht die
Unterschiede bei der Alkylierung mit und ohne Zugabe von NaCl:
Diagramm 30: Selektivitätsvergleich innerhalb der einfachalkylierten Aniline mit und ohne Verwendung von NaCl Versuchsbedingungen: rot: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen); blau: m(AlCl3)=3g; m(NaCl)=0,7g; m(Anilin)=81g; p=40bar (Propen)
Während bei reinem AlCl3 eine langsame Bildung ringalkylierter Isopropylaniline
durch eine Folgereaktion der N-alkylierten Aniline beobachtet werden kann, ist die
Selektivität zu den ringalkylierten Isopropylanilinen bei der Verwendung der
anorganischen Salzschmelzen deutlich erhöht. Die Isomerisierung der N-alkylierten
Aniline scheint durch die anorganische Salzschmelze schneller zu sein, obwohl die
effektive Menge an AlCl3 geringer ist.
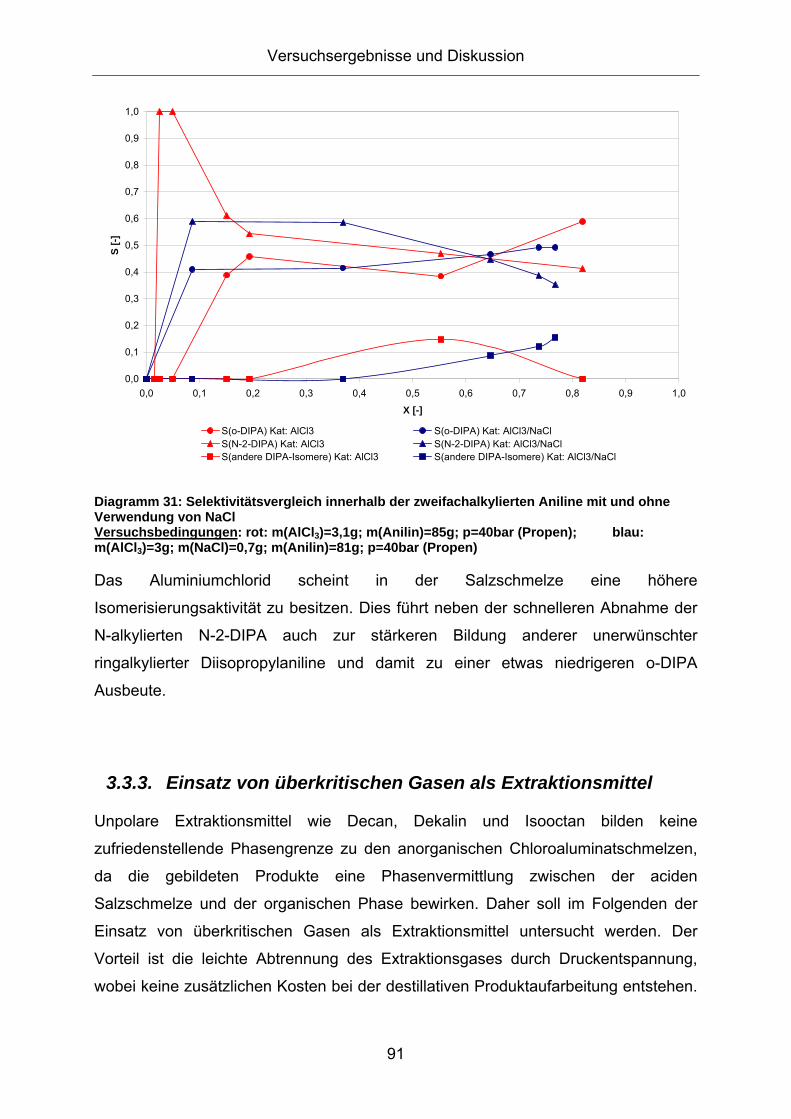
Ebenso verhält es sich bei den zweifachalkylierten Anilinen, wie aus Diagramm 31
Diagramm 31: Selektivitätsvergleich innerhalb der zweifachalkylierten Aniline mit und ohne Verwendung von NaCl Versuchsbedingungen: rot: m(AlCl3)=3,1g; m(Anilin)=85g; p=40bar (Propen); blau: m(AlCl3)=3g; m(NaCl)=0,7g; m(Anilin)=81g; p=40bar (Propen)
Das Aluminiumchlorid scheint in der Salzschmelze eine höhere
Isomerisierungsaktivität zu besitzen. Dies führt neben der schnelleren Abnahme der
N-alkylierten N-2-DIPA auch zur stärkeren Bildung anderer unerwünschter
ringalkylierter Diisopropylaniline und damit zu einer etwas niedrigeren o-DIPA
Ausbeute.
3.3.3. Einsatz von überkritischen Gasen als Extraktionsmittel
Unpolare Extraktionsmittel wie Decan, Dekalin und Isooctan bilden keine
zufriedenstellende Phasengrenze zu den anorganischen Chloroaluminatschmelzen,
da die gebildeten Produkte eine Phasenvermittlung zwischen der aciden
Salzschmelze und der organischen Phase bewirken. Daher soll im Folgenden der
Einsatz von überkritischen Gasen als Extraktionsmittel untersucht werden. Der
Vorteil ist die leichte Abtrennung des Extraktionsgases durch Druckentspannung,
wobei keine zusätzlichen Kosten bei der destillativen Produktaufarbeitung entstehen.
Versuchsergebnisse und Diskussion
92
Da bei der Reaktion bereits hohe Temperaturen und Drücke benötigt werden, ist der
zusätzliche Energieaufwand durch die Extraktion minimal.
Der Einsatz überkritischer Gase in Verbindung mit ionischen Flüssigkeiten ermöglicht
die kontinuierliche Rezyklierung homogener Katalysatoren, trotz starker
Wechselwirkungen zwischen Katalysator und Produkt.111 Aufgrund der
Unbrennbarkeit, der toxikologischen Unbedenklichkeit und der milden überkritischen
Bedingungen ((pc= 74 bar, Tc= 31,2 °C), wird in erster Linie CO2 als überkritisches
Extraktionsmittel verwendet.
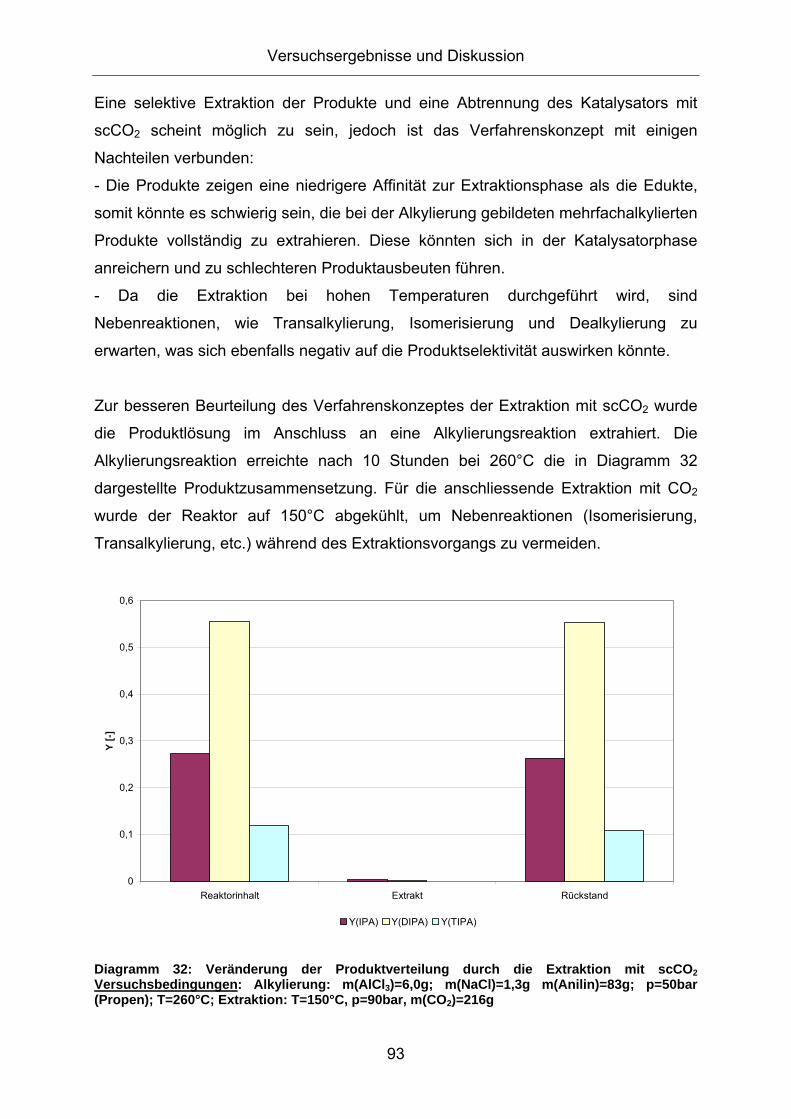
Extraktion mit überkritischem CO2 (scCO2) Als Alternative zu den bisherigen Methoden der Katalysatorabtrennung bietet sich
eine Extraktion der Produktphase mit überkritischem CO2 an. Zunächst wurde eine
Modelllösung aus Anilin, o-DIPA und AlCl3/NaCl mit überkritischem CO2 extrahiert.
Hierzu wurde dem Reaktor nach einer Dispergierung der eingesetzten Modelllösung
bei 210°C flüssiges CO2 (mittels pumpenkopfgefühlter HPLC-Pumpe) bis über den
kritischen Dampfdruck (pc= 74 bar, Tc= 31,2 °C) zudosiert. Über ein Nadelventil,
dessen Ende nicht in die flüssige Reaktandenphase eintaucht, wurde entspannt.
Während das CO2 hinter dem Ventil ausgast, konnte die austretende Extraktlösung
mittels Schlenkrohr aufgefangen werden. Bei einer anschließenden ICP-Analyse der
extrahierten Lösung konnte kein Aluminium nachgewiesen werden (Nachweisgrenze
<0,003mg/l). Tabelle 14 zeigt, dass sich die Zusammensetzung der Extraktlösung
von der eingesetzten Modelllösung unterscheidet:
Tabelle 14: Veränderung der Zusammensetzung nach einer Extraktion der Modelllösung mit scCO2 bei T=210°C und p=100bar
Tabelle 18 zeigt, dass es selbst ohne die Verwendung von NaCl möglich ist, den
Großteil an AlCl3 durch die Kristallisation einer Aluminiumspezies abzutrennen.
Neben dem Aluminium wird allerdings auch ein großer Teil der eingesetzten
organischen Phase abgetrennt, was vermutlich auf Einschlüsse durch unsaubere
Kristallisationsbedingungen zurückzuführen ist. Die entstandenen Kristalle konnten
Versuchsergebnisse und Diskussion
97
mittels DSC (differential scanning calorimetry) und Röntgenspektroskopie (XRD)
näher analysiert werden.
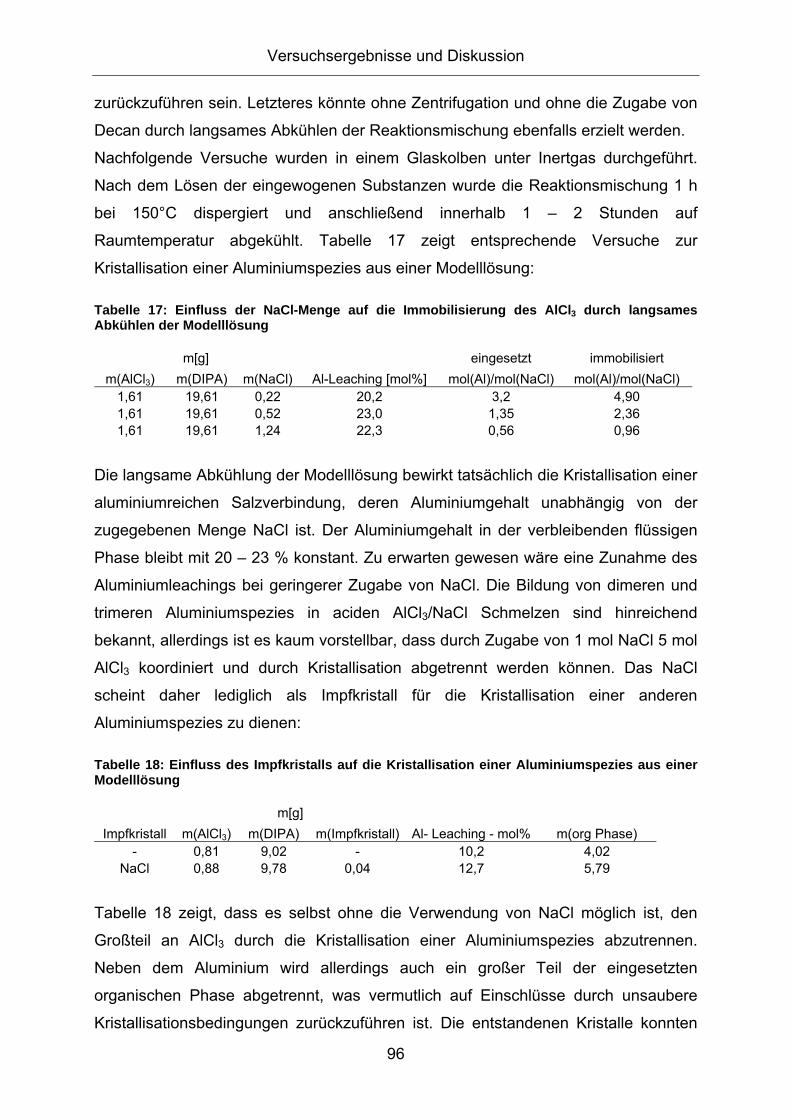
Abbildung 12: DSC-Messung der erhaltenen Kristalle bei der Kristallisation einer Aluminiumspezies aus einer Modelllösung aus AlCl3 und o-DIPA
Abbildung 12 zeigt das Ergebnis der DSC Messung. Der maximale Wärmefluss bei
der Kristallisation ist bei 30,5 °C, beim Schmelzvorgang bei 76,8 °C zu erkennen. Der
Energiebedarf für den Schmelzvorgang beträgt 92 J/g. Durch die Zugabe von
Impfkristallen, kann die Kristallisation bei einer geringen Übersättigung der
Reaktionslösung im Temperaturbereich zwischen 30 und 50°C durchgeführt werden.
Die Kristallstruktur der erhaltenen Kristalle ist in Abbildung 13 dargestellt:
Versuchsergebnisse und Diskussion
98
Abbildung 13: Kristallstruktur (XRD) der erhaltenen Kristalle bei der Kristallisation einer Aluminiumspezies aus einer Modelllösung aus AlCl3 und o-DIPA
Das o-DIPA ist ein fester Bestandteil der erhaltenen Kristalle. Zwei o-DIPA Moleküle
sind über den Aminstickstoff direkt am Aluminium des AlCl3 koordiniert. Bereits im
Jahre 1964 entdeckten indische Wissenschaftler einige AlCl3-(Arylamin)2 Spezies113,
die sie aus einer benzolischen Lösung auskristallisieren konnten. Die Kristalle
werden als leicht hygroskopisch beschrieben, die sich durch Wasser oder
Temperatureinwirkung zersetzen. Über die in Abbildung 13 beschriebene Verbindung
ist derzeit noch keine Veröffentlichung bekannt.
Durch die Kristallisation dieser Aluminiumspezies könnte es möglich sein, den
Katalysator nach einer Alkylierungsreaktion zumindest teilweise aus der
Produktlösung abzutrennen und zu rezyklieren, ohne den Reaktorinhalt durch
Zugabe von Extraktionsmitteln oder Impfkristallen zu verunreinigen. Aufgrund der
hohen Reaktionstemperatur sollte sich bei der Rezyklierung der gebildete Komplex
unter Freisetzung des eigentlichen Katalysators AlCl3 wieder zersetzen.
Versuchsergebnisse und Diskussion
99
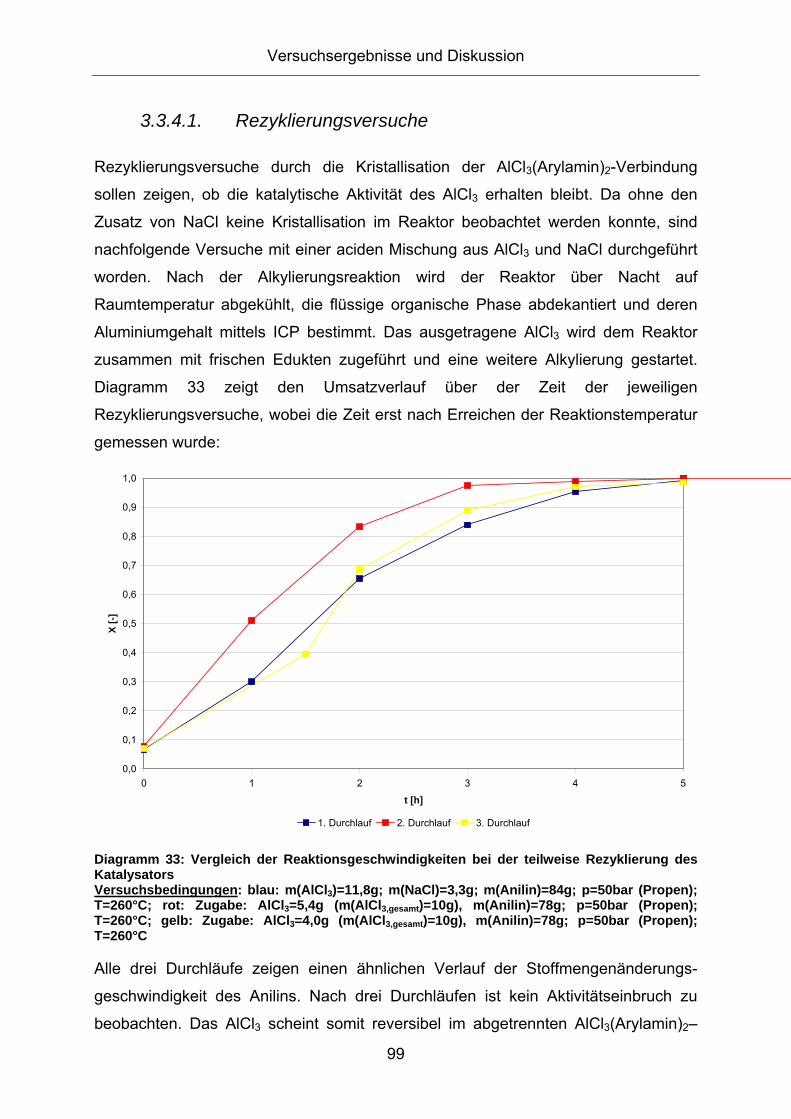
3.3.4.1. Rezyklierungsversuche
Rezyklierungsversuche durch die Kristallisation der AlCl3(Arylamin)2-Verbindung
sollen zeigen, ob die katalytische Aktivität des AlCl3 erhalten bleibt. Da ohne den
Zusatz von NaCl keine Kristallisation im Reaktor beobachtet werden konnte, sind
nachfolgende Versuche mit einer aciden Mischung aus AlCl3 und NaCl durchgeführt
worden. Nach der Alkylierungsreaktion wird der Reaktor über Nacht auf
Raumtemperatur abgekühlt, die flüssige organische Phase abdekantiert und deren
Aluminiumgehalt mittels ICP bestimmt. Das ausgetragene AlCl3 wird dem Reaktor
zusammen mit frischen Edukten zugeführt und eine weitere Alkylierung gestartet.
Diagramm 33 zeigt den Umsatzverlauf über der Zeit der jeweiligen
Rezyklierungsversuche, wobei die Zeit erst nach Erreichen der Reaktionstemperatur
gemessen wurde:
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 1 2 3 4 5
t [h]
X [-]
1. Durchlauf 2. Durchlauf 3. Durchlauf
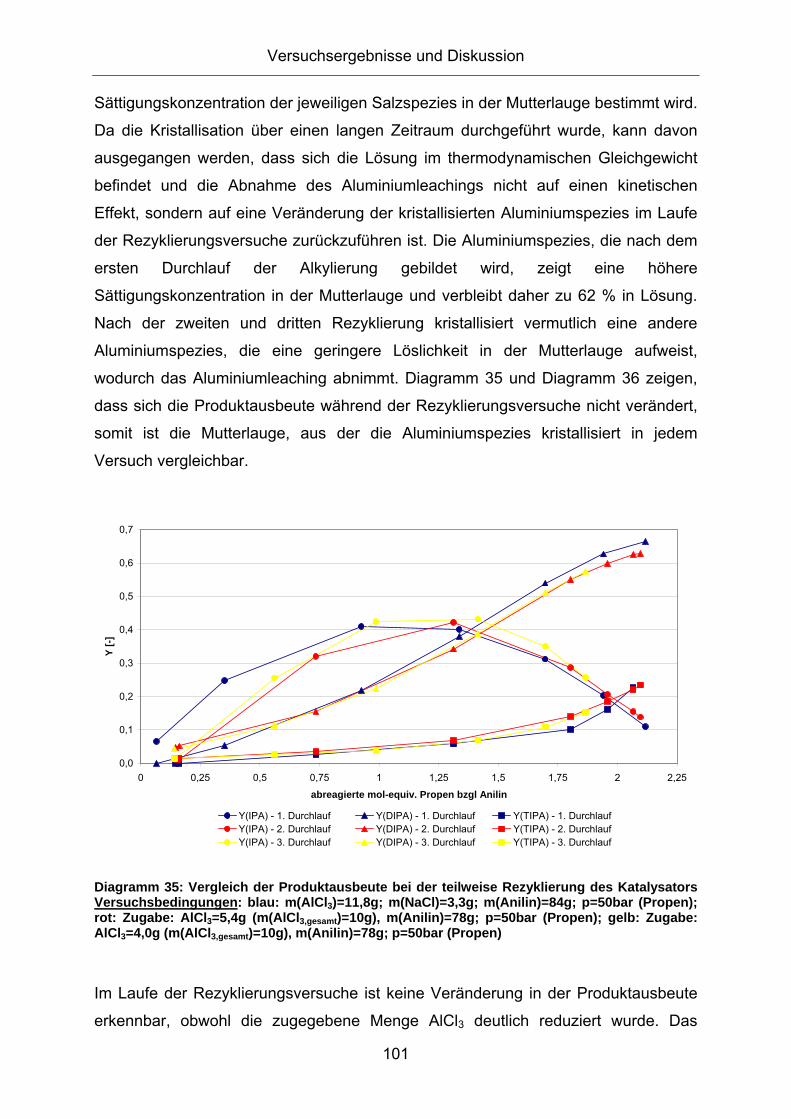
Diagramm 33: Vergleich der Reaktionsgeschwindigkeiten bei der teilweise Rezyklierung des Katalysators Versuchsbedingungen: blau: m(AlCl3)=11,8g; m(NaCl)=3,3g; m(Anilin)=84g; p=50bar (Propen); T=260°C; rot: Zugabe: AlCl3=5,4g (m(AlCl3,gesamt)=10g), m(Anilin)=78g; p=50bar (Propen); T=260°C; gelb: Zugabe: AlCl3=4,0g (m(AlCl3,gesamt)=10g), m(Anilin)=78g; p=50bar (Propen); T=260°C
Alle drei Durchläufe zeigen einen ähnlichen Verlauf der Stoffmengenänderungs-
geschwindigkeit des Anilins. Nach drei Durchläufen ist kein Aktivitätseinbruch zu
beobachten. Das AlCl3 scheint somit reversibel im abgetrennten AlCl3(Arylamin)2–
Versuchsergebnisse und Diskussion
100
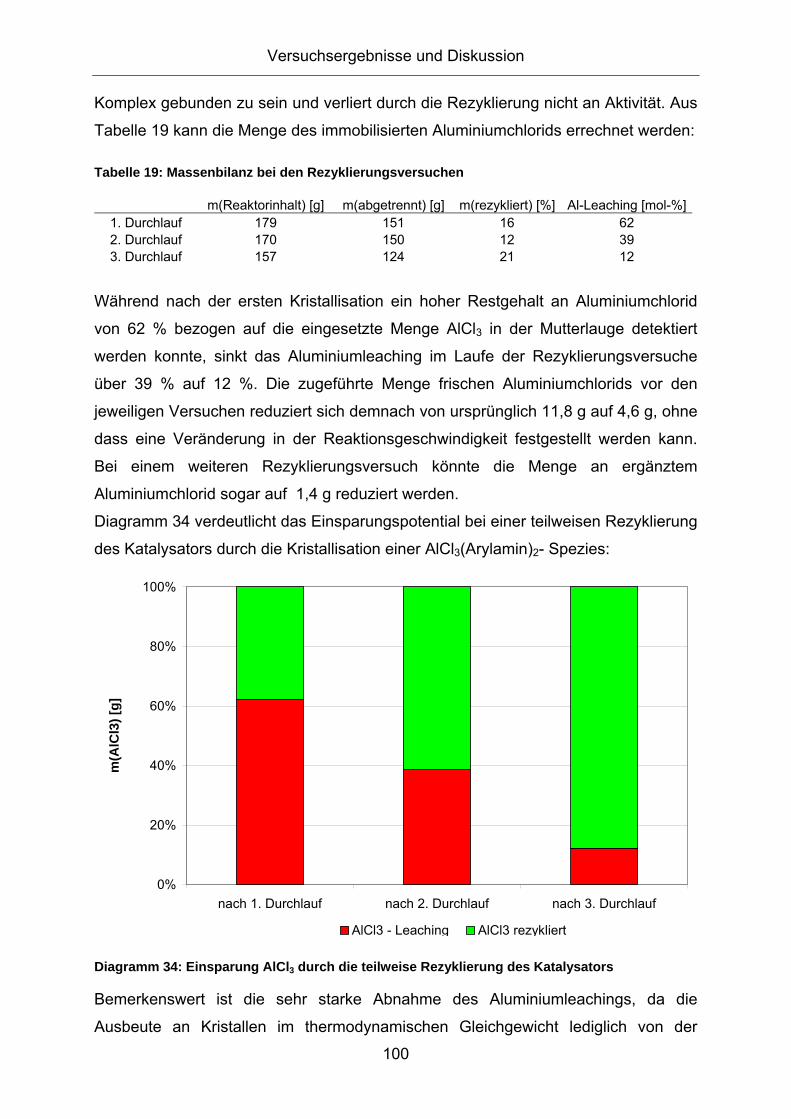
Komplex gebunden zu sein und verliert durch die Rezyklierung nicht an Aktivität. Aus
Tabelle 19 kann die Menge des immobilisierten Aluminiumchlorids errechnet werden:
Tabelle 19: Massenbilanz bei den Rezyklierungsversuchen
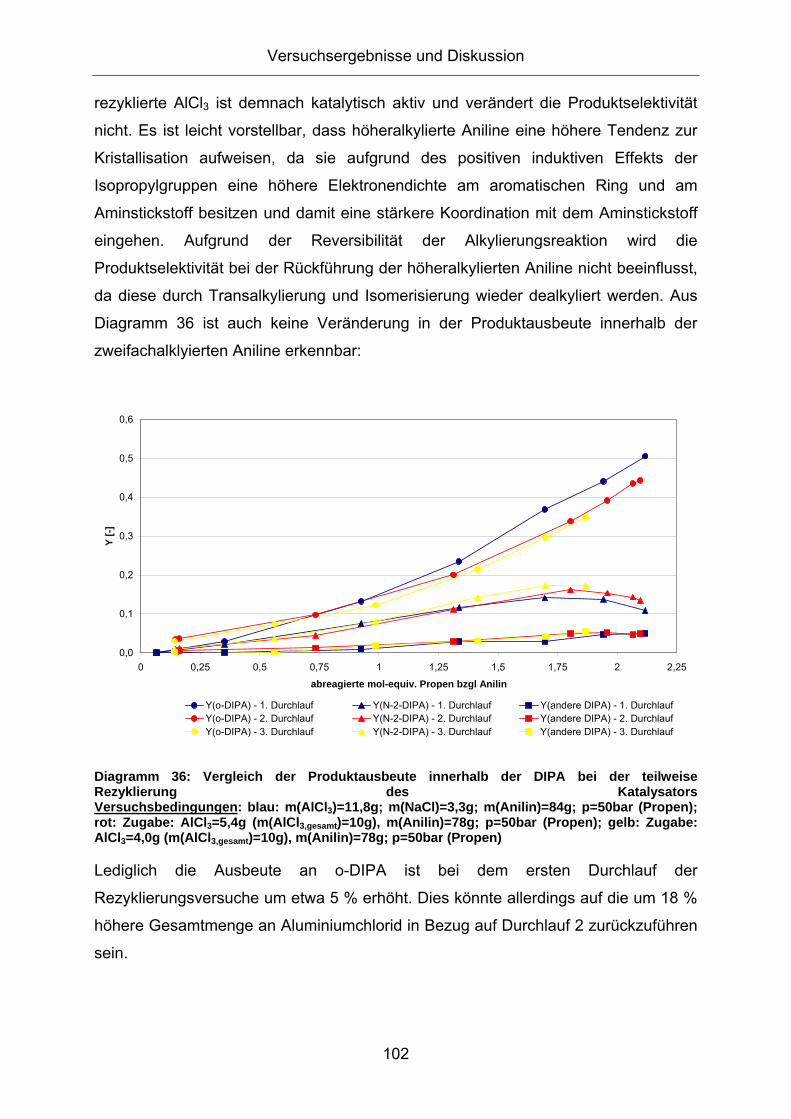
Diagramm 36: Vergleich der Produktausbeute innerhalb der DIPA bei der teilweise Rezyklierung des Katalysators Versuchsbedingungen: blau: m(AlCl3)=11,8g; m(NaCl)=3,3g; m(Anilin)=84g; p=50bar (Propen); rot: Zugabe: AlCl3=5,4g (m(AlCl3,gesamt)=10g), m(Anilin)=78g; p=50bar (Propen); gelb: Zugabe: AlCl3=4,0g (m(AlCl3,gesamt)=10g), m(Anilin)=78g; p=50bar (Propen)
Lediglich die Ausbeute an o-DIPA ist bei dem ersten Durchlauf der
Rezyklierungsversuche um etwa 5 % erhöht. Dies könnte allerdings auf die um 18 %
höhere Gesamtmenge an Aluminiumchlorid in Bezug auf Durchlauf 2 zurückzuführen
sein.
Versuchsergebnisse und Diskussion
103
3.3.4.2. Reproduktionsversuche
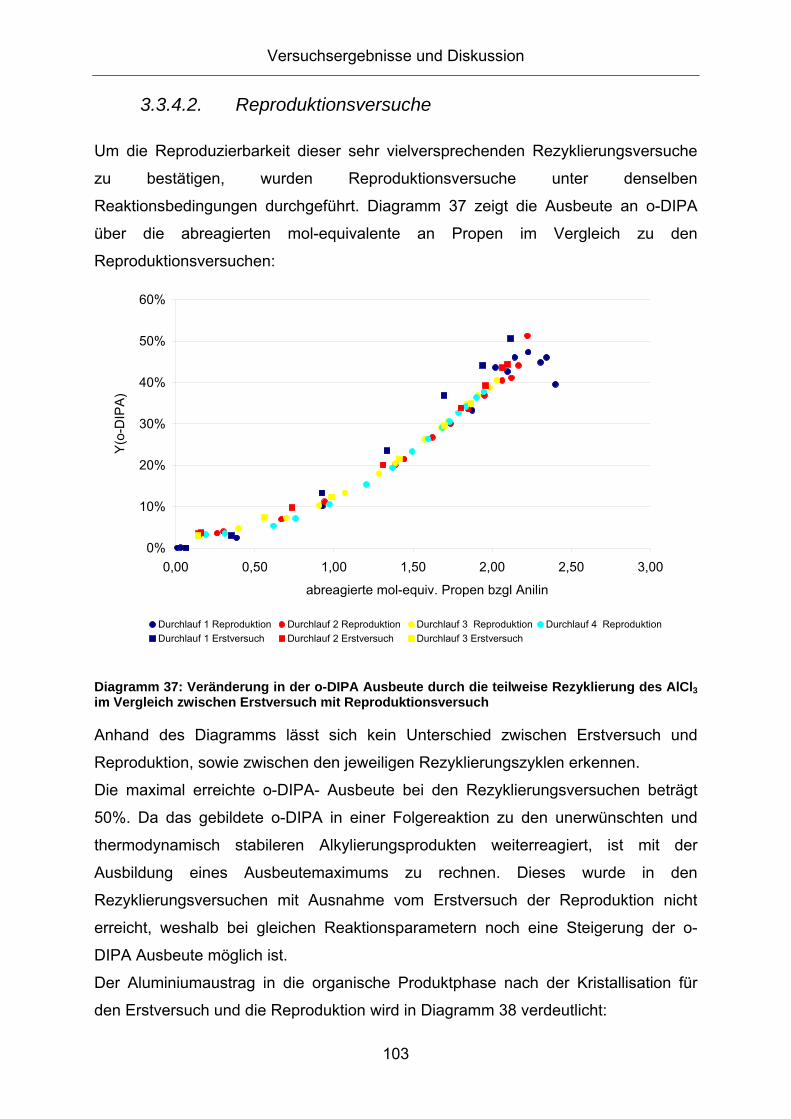
Um die Reproduzierbarkeit dieser sehr vielversprechenden Rezyklierungsversuche
zu bestätigen, wurden Reproduktionsversuche unter denselben
Reaktionsbedingungen durchgeführt. Diagramm 37 zeigt die Ausbeute an o-DIPA
über die abreagierten mol-equivalente an Propen im Vergleich zu den
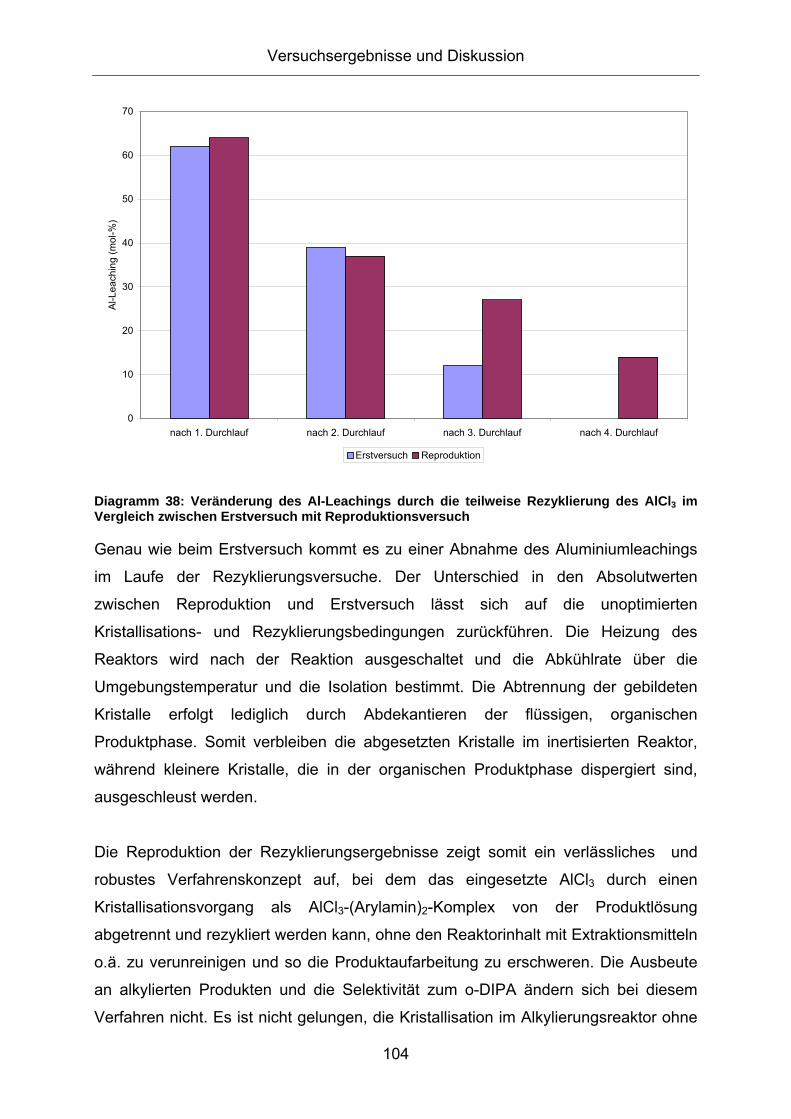
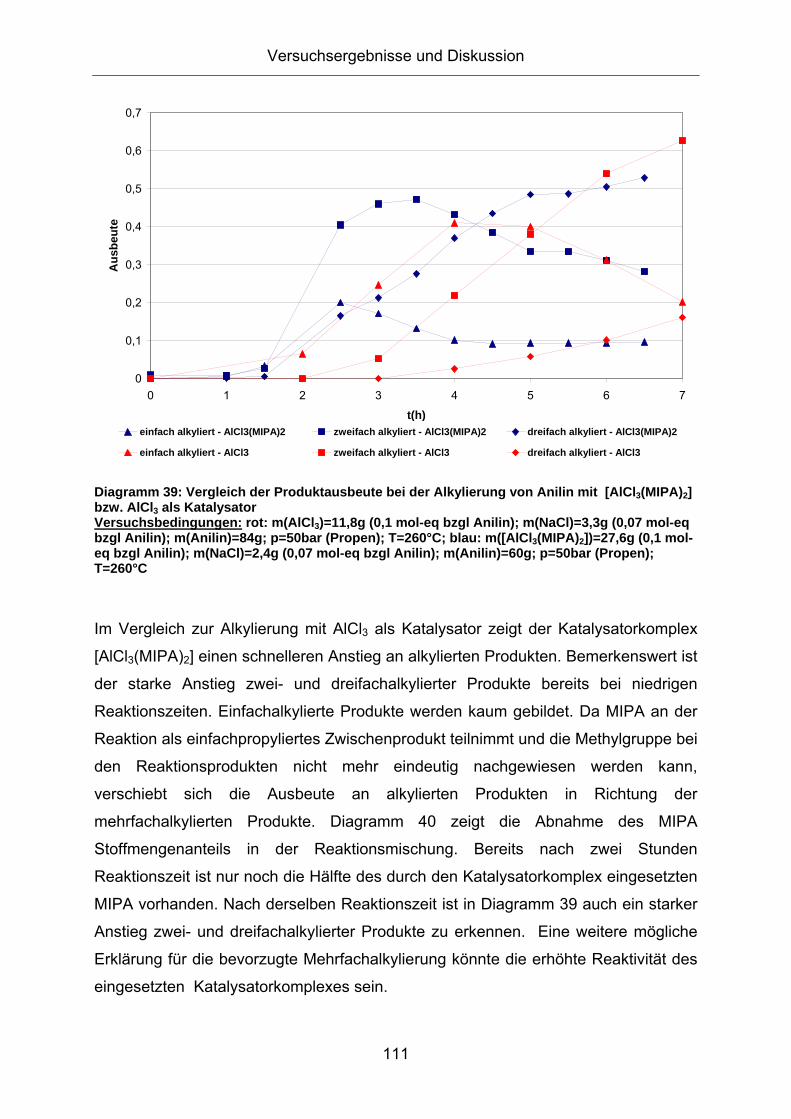
Diagramm 39: Vergleich der Produktausbeute bei der Alkylierung von Anilin mit [AlCl3(MIPA)2] bzw. AlCl3 als Katalysator Versuchsbedingungen: rot: m(AlCl3)=11,8g (0,1 mol-eq bzgl Anilin); m(NaCl)=3,3g (0,07 mol-eq bzgl Anilin); m(Anilin)=84g; p=50bar (Propen); T=260°C; blau: m([AlCl3(MIPA)2])=27,6g (0,1 mol-eq bzgl Anilin); m(NaCl)=2,4g (0,07 mol-eq bzgl Anilin); m(Anilin)=60g; p=50bar (Propen); T=260°C
Im Vergleich zur Alkylierung mit AlCl3 als Katalysator zeigt der Katalysatorkomplex
[AlCl3(MIPA)2] einen schnelleren Anstieg an alkylierten Produkten. Bemerkenswert ist
der starke Anstieg zwei- und dreifachalkylierter Produkte bereits bei niedrigen
Reaktionszeiten. Einfachalkylierte Produkte werden kaum gebildet. Da MIPA an der
Reaktion als einfachpropyliertes Zwischenprodukt teilnimmt und die Methylgruppe bei
den Reaktionsprodukten nicht mehr eindeutig nachgewiesen werden kann,
verschiebt sich die Ausbeute an alkylierten Produkten in Richtung der
mehrfachalkylierten Produkte. Diagramm 40 zeigt die Abnahme des MIPA
Stoffmengenanteils in der Reaktionsmischung. Bereits nach zwei Stunden
Reaktionszeit ist nur noch die Hälfte des durch den Katalysatorkomplex eingesetzten
MIPA vorhanden. Nach derselben Reaktionszeit ist in Diagramm 39 auch ein starker
Anstieg zwei- und dreifachalkylierter Produkte zu erkennen. Eine weitere mögliche
Erklärung für die bevorzugte Mehrfachalkylierung könnte die erhöhte Reaktivität des
eingesetzten Katalysatorkomplexes sein.
Versuchsergebnisse und Diskussion
112
0%
5%
10%
15%
20%
25%
0 1 2 3 4 5 6 7
t(h)
Stof
fmen
gena
ntei
l MIP
A
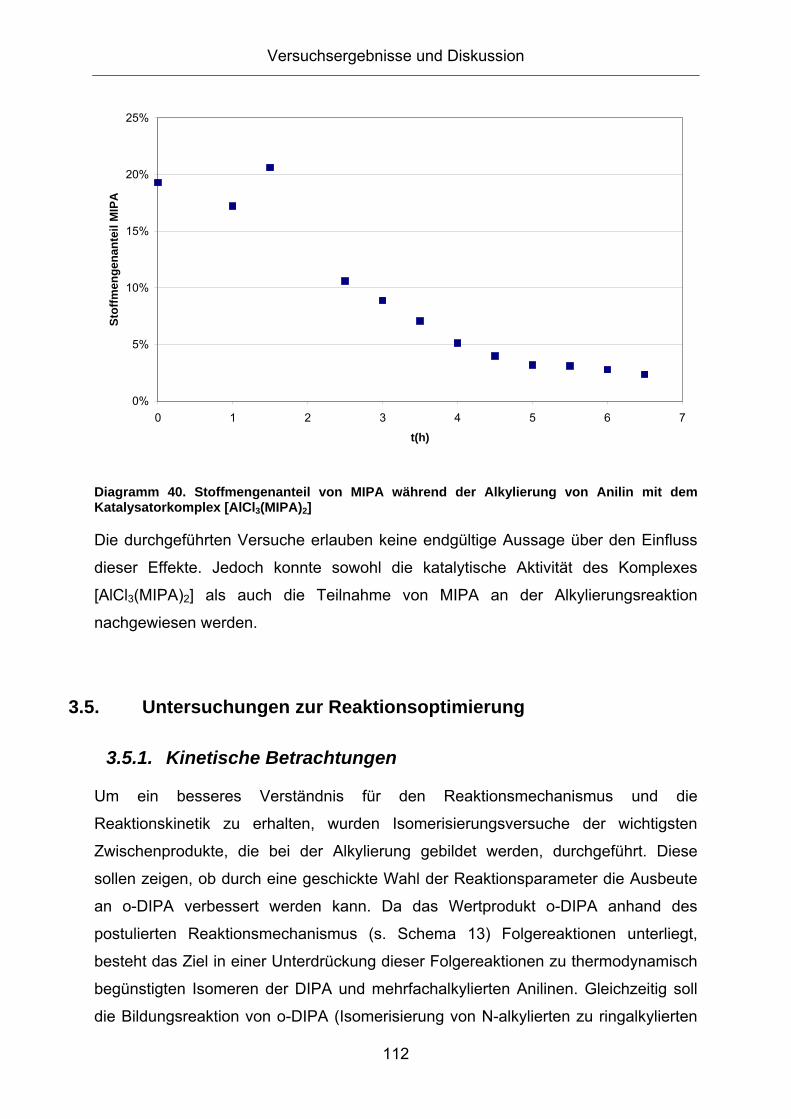
Diagramm 40. Stoffmengenanteil von MIPA während der Alkylierung von Anilin mit dem Katalysatorkomplex [AlCl3(MIPA)2]
Die durchgeführten Versuche erlauben keine endgültige Aussage über den Einfluss
dieser Effekte. Jedoch konnte sowohl die katalytische Aktivität des Komplexes
[AlCl3(MIPA)2] als auch die Teilnahme von MIPA an der Alkylierungsreaktion
nachgewiesen werden.
3.5. Untersuchungen zur Reaktionsoptimierung
3.5.1. Kinetische Betrachtungen
Um ein besseres Verständnis für den Reaktionsmechanismus und die
Reaktionskinetik zu erhalten, wurden Isomerisierungsversuche der wichtigsten
Zwischenprodukte, die bei der Alkylierung gebildet werden, durchgeführt. Diese
sollen zeigen, ob durch eine geschickte Wahl der Reaktionsparameter die Ausbeute
an o-DIPA verbessert werden kann. Da das Wertprodukt o-DIPA anhand des
postulierten Reaktionsmechanismus (s. Schema 13) Folgereaktionen unterliegt,
besteht das Ziel in einer Unterdrückung dieser Folgereaktionen zu thermodynamisch
begünstigten Isomeren der DIPA und mehrfachalkylierten Anilinen. Gleichzeitig soll
die Bildungsreaktion von o-DIPA (Isomerisierung von N-alkylierten zu ringalkylierten
Versuchsergebnisse und Diskussion
113
Anilinen) beschleunigt werden. Für den Fall, dass Bildungs- und Folgereaktion
ausreichend unterschiedliche Aktivierungsenergien besitzen, bietet sich
beispielsweise eine kinetische Kontrolle der Gesamtreaktion durch eine geschickte
Temperaturführung an.
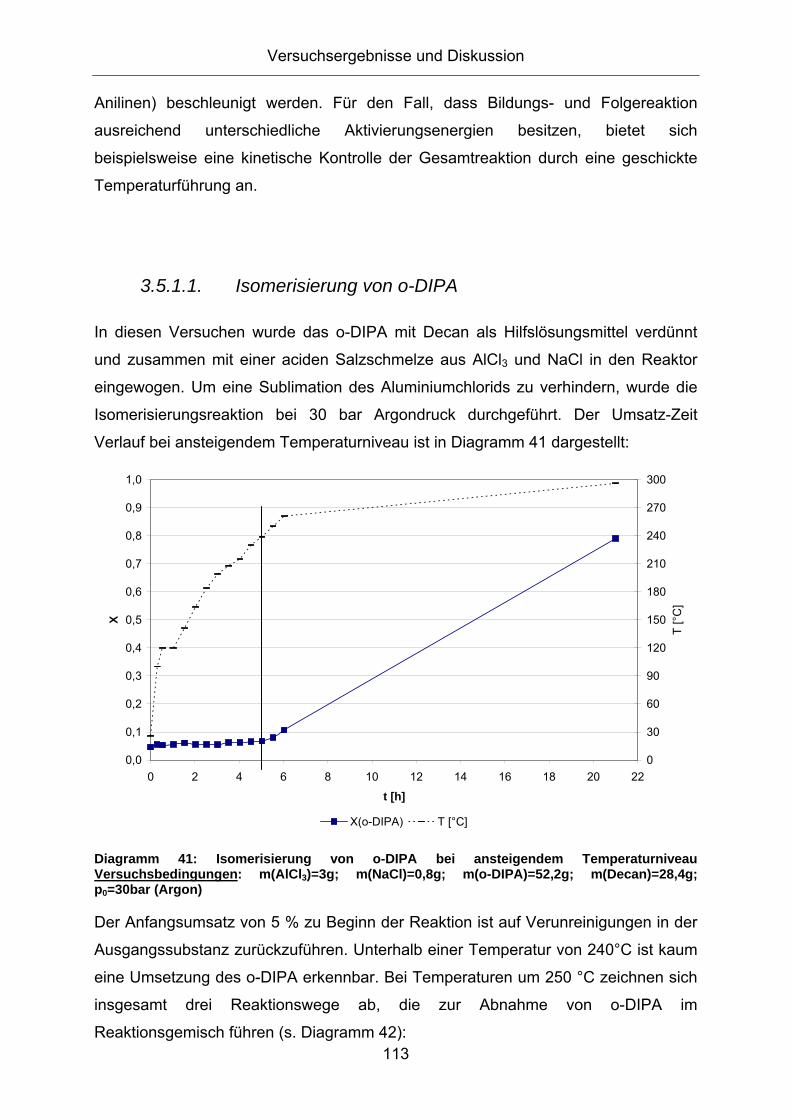
3.5.1.1. Isomerisierung von o-DIPA
In diesen Versuchen wurde das o-DIPA mit Decan als Hilfslösungsmittel verdünnt
und zusammen mit einer aciden Salzschmelze aus AlCl3 und NaCl in den Reaktor
eingewogen. Um eine Sublimation des Aluminiumchlorids zu verhindern, wurde die
Isomerisierungsreaktion bei 30 bar Argondruck durchgeführt. Der Umsatz-Zeit
Verlauf bei ansteigendem Temperaturniveau ist in Diagramm 41 dargestellt:
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 2 4 6 8 10 12 14 16 18 20 22
t [h]
X
0
30
60
90
120
150
180
210
240
270
300
T [°
C]
X(o-DIPA) T [°C]
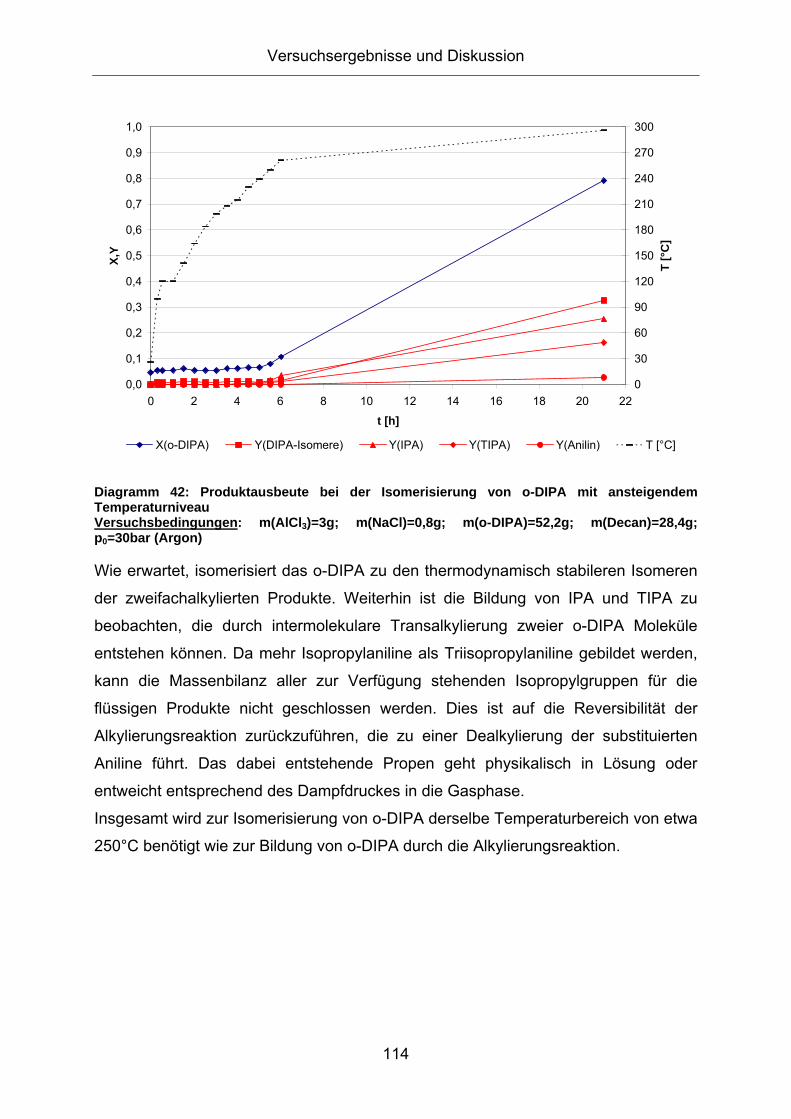
Diagramm 41: Isomerisierung von o-DIPA bei ansteigendem Temperaturniveau Versuchsbedingungen: m(AlCl3)=3g; m(NaCl)=0,8g; m(o-DIPA)=52,2g; m(Decan)=28,4g; p0=30bar (Argon)
Der Anfangsumsatz von 5 % zu Beginn der Reaktion ist auf Verunreinigungen in der
Ausgangssubstanz zurückzuführen. Unterhalb einer Temperatur von 240°C ist kaum
eine Umsetzung des o-DIPA erkennbar. Bei Temperaturen um 250 °C zeichnen sich
insgesamt drei Reaktionswege ab, die zur Abnahme von o-DIPA im
Reaktionsgemisch führen (s. Diagramm 42):
Versuchsergebnisse und Diskussion
114
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 2 4 6 8 10 12 14 16 18 20 22
t [h]
X,Y
0
30
60
90
120
150
180
210
240
270
300
T [°
C]
X(o-DIPA) Y(DIPA-Isomere) Y(IPA) Y(TIPA) Y(Anilin) T [°C]
Diagramm 42: Produktausbeute bei der Isomerisierung von o-DIPA mit ansteigendem Temperaturniveau Versuchsbedingungen: m(AlCl3)=3g; m(NaCl)=0,8g; m(o-DIPA)=52,2g; m(Decan)=28,4g; p0=30bar (Argon)
Wie erwartet, isomerisiert das o-DIPA zu den thermodynamisch stabileren Isomeren
der zweifachalkylierten Produkte. Weiterhin ist die Bildung von IPA und TIPA zu
beobachten, die durch intermolekulare Transalkylierung zweier o-DIPA Moleküle
entstehen können. Da mehr Isopropylaniline als Triisopropylaniline gebildet werden,
kann die Massenbilanz aller zur Verfügung stehenden Isopropylgruppen für die
flüssigen Produkte nicht geschlossen werden. Dies ist auf die Reversibilität der
Alkylierungsreaktion zurückzuführen, die zu einer Dealkylierung der substituierten
Aniline führt. Das dabei entstehende Propen geht physikalisch in Lösung oder
entweicht entsprechend des Dampfdruckes in die Gasphase.
Insgesamt wird zur Isomerisierung von o-DIPA derselbe Temperaturbereich von etwa
250°C benötigt wie zur Bildung von o-DIPA durch die Alkylierungsreaktion.
Versuchsergebnisse und Diskussion
115
3.5.1.2. Ermittlung kinetischer Parameter zur Isomerisierung
von o-DIPA
Mit Hilfe des formalkinetischen Potenzansatzes wurde versucht, die kinetischen
Parameter der Isomerisierungsreaktion von o-DIPA zu ermitteln.
Unter der Annahme, dass sich das Gesamtvolumen bei der Isomerisierung von o-
DIPA nicht ändert, ergibt sich die Reaktionsgeschwindigkeit durch die zeitliche
Ableitung der o-DIPA - Konzentration:
dtdcr DIPAo−=
ν1 ,
mit ν =stöchiometrischer Koeffizient, co-DIPA=o-DIPA-Konzentration
Unter Anwendung des Potenzansatzes, gilt für die Stoffmengenänderungs-
geschwindigkeit von o-DIPA:
bAlCl
aDIPAo
DIPAo cckdt
dc3•••= −
− ν ,
mit k=Geschwindigkeitskonstante, a,b=Reaktionsordnungen bzgl. der jeweiligen Substanzen Die Isomerisierungsversuche wurden bei unterschiedlichen Temperaturen und
Aciditäten (n(AlCl3)/n(NaCl)) durchgeführt. Die Acidität wird in dieser Arbeit als
Verhältnis zwischen den Stoffmengen von Lewis-Säure und Lewis-Base definiert. Sie
gibt den Überschuss an Aluminiumchlorid zu Natriumchlorid an:
NaCl
AlCl
BaseLewis
SäureLewis
nn
nn
Ac 3==−
−
Die Gesamtmasse an AlCl3 hingegen wurde nicht verändert, da das Hauptziel dieser
Arbeit in einem katalytischen Einsatz von AlCl3 besteht. Diagramm 43 zeigt die
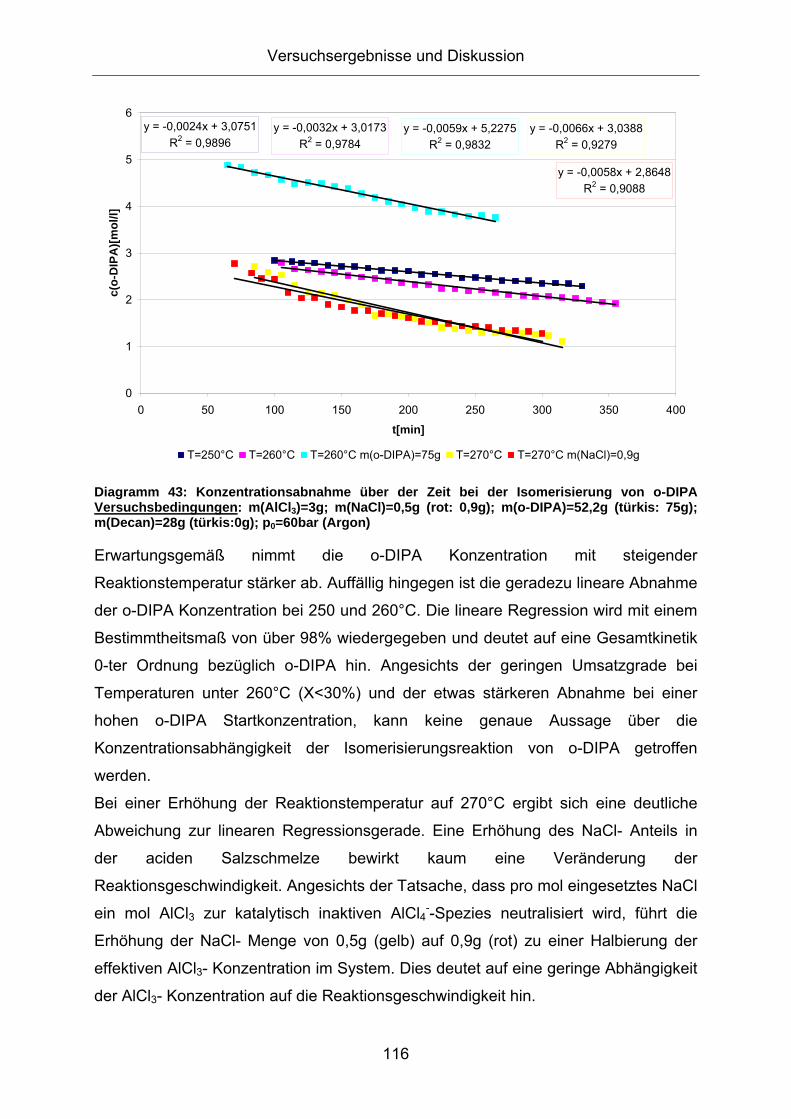
Diagramm 43: Konzentrationsabnahme über der Zeit bei der Isomerisierung von o-DIPA Versuchsbedingungen: m(AlCl3)=3g; m(NaCl)=0,5g (rot: 0,9g); m(o-DIPA)=52,2g (türkis: 75g); m(Decan)=28g (türkis:0g); p0=60bar (Argon)
Erwartungsgemäß nimmt die o-DIPA Konzentration mit steigender
Reaktionstemperatur stärker ab. Auffällig hingegen ist die geradezu lineare Abnahme
der o-DIPA Konzentration bei 250 und 260°C. Die lineare Regression wird mit einem
Bestimmtheitsmaß von über 98% wiedergegeben und deutet auf eine Gesamtkinetik
0-ter Ordnung bezüglich o-DIPA hin. Angesichts der geringen Umsatzgrade bei
Temperaturen unter 260°C (X<30%) und der etwas stärkeren Abnahme bei einer
hohen o-DIPA Startkonzentration, kann keine genaue Aussage über die
Konzentrationsabhängigkeit der Isomerisierungsreaktion von o-DIPA getroffen
werden.
Bei einer Erhöhung der Reaktionstemperatur auf 270°C ergibt sich eine deutliche
Abweichung zur linearen Regressionsgerade. Eine Erhöhung des NaCl- Anteils in
der aciden Salzschmelze bewirkt kaum eine Veränderung der
Reaktionsgeschwindigkeit. Angesichts der Tatsache, dass pro mol eingesetztes NaCl
ein mol AlCl3 zur katalytisch inaktiven AlCl4--Spezies neutralisiert wird, führt die
Erhöhung der NaCl- Menge von 0,5g (gelb) auf 0,9g (rot) zu einer Halbierung der
effektiven AlCl3- Konzentration im System. Dies deutet auf eine geringe Abhängigkeit
der AlCl3- Konzentration auf die Reaktionsgeschwindigkeit hin.
Versuchsergebnisse und Diskussion
117
Da von einer Überlagerung mehrerer Reaktionsmechanismen auszugehen ist, wurde
die Kinetik anhand postulierter parallel ablaufender Abbaureaktionen von o-DIPA
aufgestellt:
Folgende Abbaureaktionen des o-DIPA sind aufgrund der identifizierten
Nebenprodukte bei den Voruntersuchungen wahrscheinlich:
Schema 17: postulierte Folgereaktionen des o-DIPA
Reaktion 1: Intramolekulare Isomerisierung:
Reaktion 2: Dealkylierung:
Reaktion 3: Transalkylierung:
Die gebildeten Isomerisierungsprodukte sind keineswegs stabil unter den gewählten
Reaktionsbedingungen. Sie unterliegen ihrerseits weiteren Folgereaktionen, so dass
es zu einer Ausbildung eines komplexen Reaktionsnetzwerks kommt, in dem die
einzelnen Teilreaktionen voneinander abhängig sind. Um diese Folgereaktionen der
aus o-DIPA gebildeten Isomerisierungsprodukte zu unterdrücken, muss bei niedrigen
Produktkonzentrationen, respektive bei geringen o-DIPA Umsatzgraden gearbeitet
werden. Dies macht eine sinnvolle Bestimmung der Reaktionsordnungen zu den
jeweiligen Teilreaktionen nahezu unmöglich. Durch den Einsatz eines
Hilfslösungsmittels könnten bei gleichem Reaktorfüllgrad verschiedene o-DIPA-
Anfangskonzentrationen realisiert und bei geringen Umsatzgraden die
Konzentrationsabhängigkeit ermittelt werden. Idealerweise müsste hierzu ein inertes
NH2 NH2
NH2 NH2
NH2 NH2 NH2
2
Versuchsergebnisse und Diskussion
118
Lösungsmittel gefunden werden, welches die Stoffeigenschaften (Viskosität,
Löslichkeiten, etc.) bei unterschiedlichen o-DIPA- Konzentrationen nicht verändert.
Die Konzentrationsänderung von o-DIPA nach einer bestimmten Zeit ergibt sich bei
konstantem Volumen durch die Summe aller Konzentrationsänderungen der
jeweiligen Teilreaktionen:
Gleichung 2: Stoffmengenänderungsgeschwindigkeit von o-DIPA
dtdc
dtdc
dtdc
dtdc RDIPAoRDIPAoRDIPAoDIPAo 3,2,1, −−−− ++=
Die Konzentrationsänderung des o-DIPA, die durch die jeweilige Teilreaktion
entsteht, kann anhand der Bildung der teilreaktionsspezifischen Produkte ermittelt
werden. Streng genommen ist dies nur bei geringen Umsatzgraden zulässig, da die
gebildeten Zwischenprodukte weiteren Folgereaktionen unterliegen. Nur bei einem
geringen Umsatzgrad, und damit bei geringen Konzentrationen der gebildeten
Zwischenprodukte, können die Folgereaktionen vernachlässigt werden:
Reaktion 1: Da die Bildung anderer ringalkylierter Isomere des DIPA (candere-DIPA)
anhand der postulierten Teilreaktionen nur auf intramolekulare Isomerisierung
zurückzuführen ist, kann die Konzentrationsänderung des o-DIPA durch Reaktion 1
direkt aus der Bildung anderer ringalkylierter Isomere berechnet werden:
c0,o-DIPA-co-DIPA,1(t)= candere-DIPA(t)
Reaktion 2: Die Bildung von IPA kann durch Teilreaktion 2 und 3 erfolgen. Dadurch
ergibt sich für die Konzentrationsänderung an o-DIPA durch Teilreaktion 2:
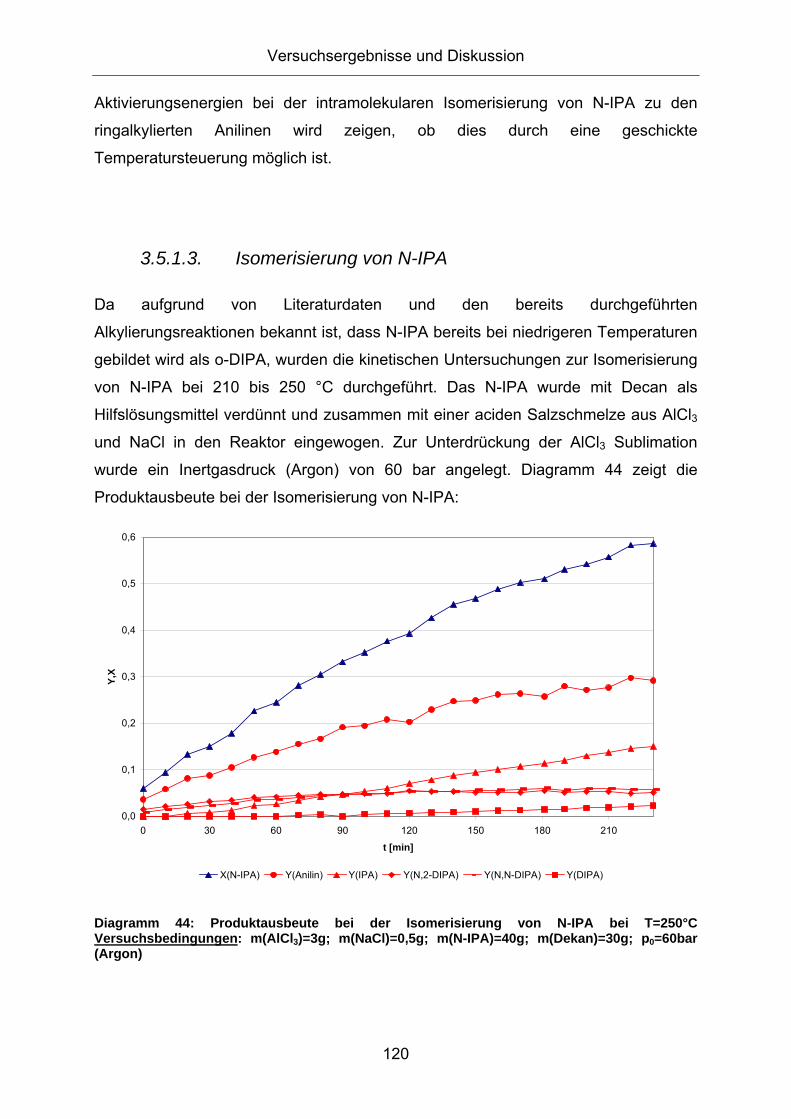
Diagramm 44: Produktausbeute bei der Isomerisierung von N-IPA bei T=250°C Versuchsbedingungen: m(AlCl3)=3g; m(NaCl)=0,5g; m(N-IPA)=40g; m(Dekan)=30g; p0=60bar (Argon)
Versuchsergebnisse und Diskussion
121
Bereits nach 3 Stunden beträgt der Umsatzgrad bezüglich N-IPA 50%. Ein Großteil
des N-IPA dealkyliert unter Bildung von Anilin und Propen. Letzteres geht
physikalisch in Lösung oder entweicht entsprechend des Dampfdruckes in die
Gasphase. Analog des vorgeschlagenen Reaktionsmechanismus entsteht durch
intramolekulare Isomerisierung das ringalkylierte 2-IPA. Des Weiteren bilden sich in
etwa gleichem Maße N-2-DIPA und N-N-DIPA, vermutlich durch eine intermolekulare
Transalkylierung, oder durch eine Addition des bei der Dealkylierung entstandenen
Propens an den Aminstickstoff. Eine geringe Menge an DIPA kann ebenfalls
detektiert werden.
Diagramm 45 zeigt die Konzentrationsabnahme über der Zeit bei unterschiedlichen
Reaktionstemperaturen:
y = -0,0073x + 3,4522R2 = 0,9456
y = -0,0078x + 3,7114R2 = 0,981
y = -0,003x + 3,5595R2 = 0,9823
y = -0,001x + 3,631R2 = 0,9581
0
1
2
3
4
5
0 30 60 90 120 150 180 210 240 270 300 330 360
t[min]
c(N
-IPA
)[mol
/l]
T=210°C T=230°C T=250°C T=250°C Ac=1,5
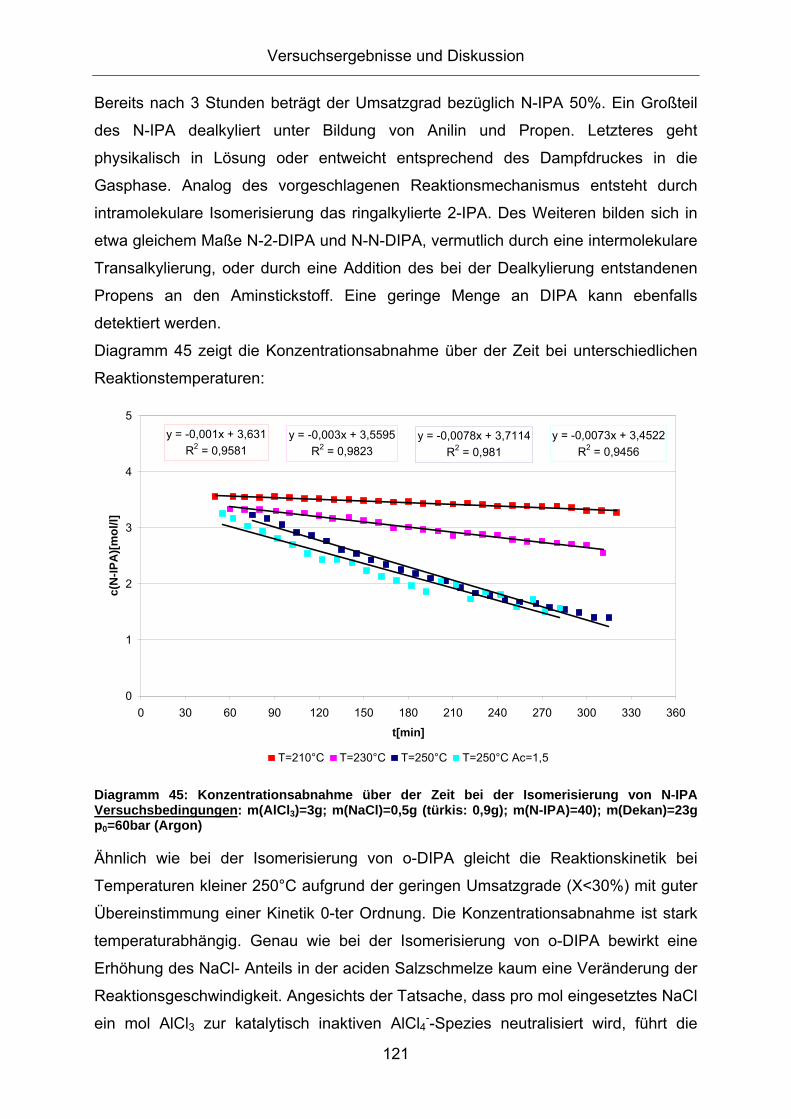
Diagramm 45: Konzentrationsabnahme über der Zeit bei der Isomerisierung von N-IPA Versuchsbedingungen: m(AlCl3)=3g; m(NaCl)=0,5g (türkis: 0,9g); m(N-IPA)=40); m(Dekan)=23g p0=60bar (Argon)
Ähnlich wie bei der Isomerisierung von o-DIPA gleicht die Reaktionskinetik bei
Temperaturen kleiner 250°C aufgrund der geringen Umsatzgrade (X<30%) mit guter
Übereinstimmung einer Kinetik 0-ter Ordnung. Die Konzentrationsabnahme ist stark
temperaturabhängig. Genau wie bei der Isomerisierung von o-DIPA bewirkt eine
Erhöhung des NaCl- Anteils in der aciden Salzschmelze kaum eine Veränderung der
Reaktionsgeschwindigkeit. Angesichts der Tatsache, dass pro mol eingesetztes NaCl
ein mol AlCl3 zur katalytisch inaktiven AlCl4--Spezies neutralisiert wird, führt die
Versuchsergebnisse und Diskussion
122
Erhöhung der NaCl- Menge von 0,5g (blau) auf 0,9g (türkis) zu einer Halbierung der
effektiven AlCl3- Konzentration im System. Dies deutet auf eine geringe Abhängigkeit
der AlCl3- Konzentration auf die Reaktionsgeschwindigkeit hin.
Dies stimmt mit den Beobachtungen aus den Alkylierungsversuchen nur zum Teil
überein. Bei einer Erhöhung der AlCl3 Konzentration in den Alkylierungsversuchen
konnte erst nach Überschreitung einer Initiationszeit kein Einfluss auf die
Reaktionsgeschwindigkeit festgestellt werden (s. Diagramm 11) .
Da von einer Überlagerung mehrerer Reaktionsmechanismen auszugehen ist, wurde
die Kinetik anhand postulierter parallel ablaufender Abbaureaktionen (s. Schema 18)
von N-IPA aufgestellt:
Schema 18: postulierte Folgereaktionen des N-IPA
Reaktion 1: Intramolekulare Isomerisierung
NH NH2
Reaktion 2: Dealkylierung
NH NH2
Reaktion 3: Transalkylierung 1
NH NH2 NH
2
Versuchsergebnisse und Diskussion
123
Reaktion 4: Transalkylierung 2
NH NH2 N
2
Die Vorgehensweise bei der Aufstellung kinetischer Gleichungen wurde bereits in
3.5.1.1 erläutert. Es soll im Folgenden auf die Berechnung der
Konzentrationsänderungen des N-IPA durch die jeweilige Teilreaktion anhand der
Bildung der teilreaktionsspezifischen Produkte eingegangen werden. Weiterhin sei
darauf verwiesen, dass dies trotz der Verwendung eines Lösungsmittels zur
Konzentrationserniedrigung nur bei geringen Umsatzgraden zulässig ist, da die
gebildeten Zwischenprodukte weiteren Folge- und Rückreaktionen unterliegen. Nur
bei einem geringen Umsatzgrad, und damit bei geringen Konzentrationen der
gebildeten Zwischenprodukte, können die Folgereaktionen vernachlässigt werden:
Reaktion 1: Da die Bildung ringalkylierter IPA anhand der postulierten Teilreaktionen
nur auf intramolekulare Isomerisierung zurückzuführen ist, kann die
Konzentrationsänderung des N-IPA durch Reaktion 1 direkt aus der Bildung von IPA
berechnet werden:
c0,N-IPA-cN-IPA,1(t)= cIPA(t)
Reaktion 2: Die Bildung von Anilin kann durch Teilreaktion 2, 3 und 4 erfolgen.
Dadurch ergibt sich für die Konzentrationsänderung an N-IPA durch Teilreaktion 2:
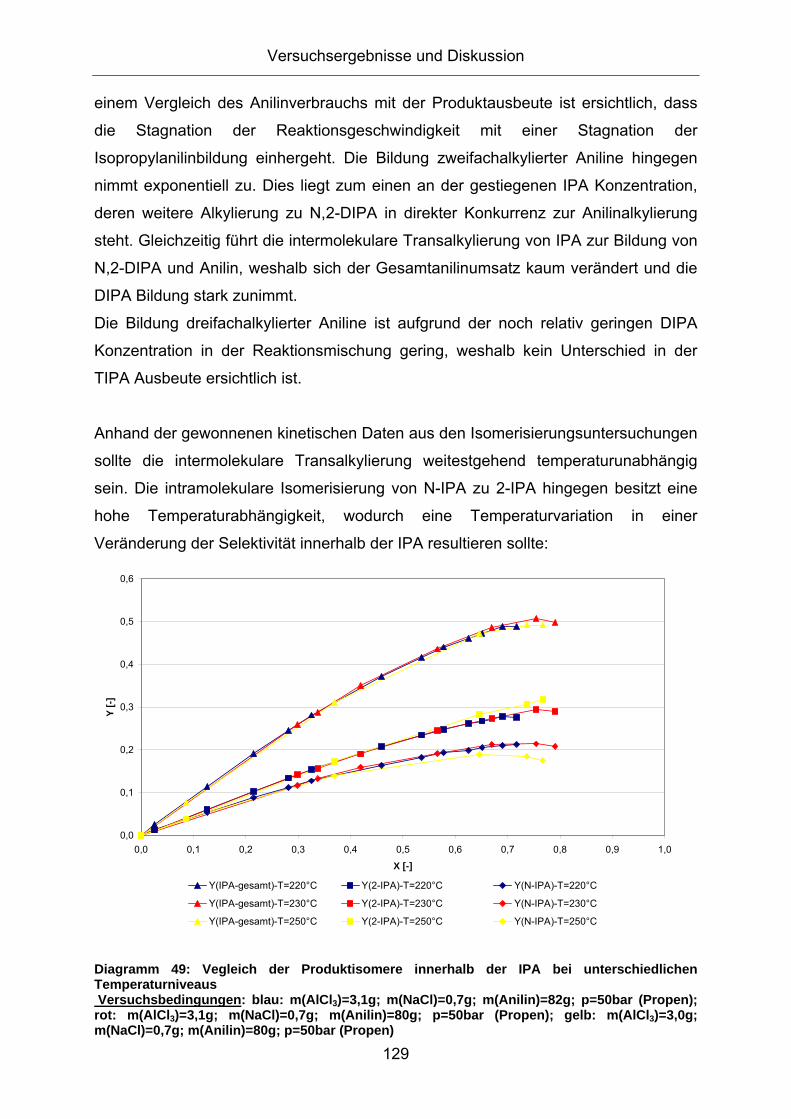
Die Aktivierungsenergien bei der intramolekularen Isomerisierung liegen für beide
Reaktanden in einem ähnlichen Bereich um 60 kJ/mol. Hier erscheint eine
Temperatursteuerung zur bevorzugten Bildung ortho- alkylierter Aniline nicht möglich,
da eine Erhöhung der Temperatur zwar zu einer schnelleren Bildung von o-DIPA aus
den N-alkylierten Anilinen, allerdings auch zu dessen schnelleren Isomerisierung zu
den thermodynamisch stabileren ringalkylierten Produktisomeren führt. Die
Dealkylierungsreaktionen von N-IPA und o-DIPA besitzen eine geringe
Aktivierungsenergie und unterscheiden sich kaum voneinander. Eine gezielte
kinetische Steuerung erscheint aufgrund der unterschiedlichen Aktivierungsenergien
lediglich bei den Transalkylierungsreaktionen möglich. Bei niedrigen
Alkylierungstemperaturen führt die Transalkylierung des N-IPA überwiegend zu N-
substituierten Diisopropylanilinen, die aufgrund des postulierten
Reaktionsmechanismus eine Vorstufe zum o-DIPA darstellen. Die Transalkylierung
von o-DIPA zu TIPA kann durch eine Temperaturerniedrigung relativ zu den anderen
Teilreaktionen verlangsamt werden und die Selektivität zu den mehrfachalkylierten
Anilinen verringert werden.
Einen direkten Vergleich der Reaktionskinetik bei der Isomerisierung von N-IPA und
o-DIPA zeigt Diagramm 46:
Versuchsergebnisse und Diskussion
126
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0 30 60 90 120 150 180 210 240
t[min]
X
X(N-IPA) T=250°C X(o-DIPA) T=250°C
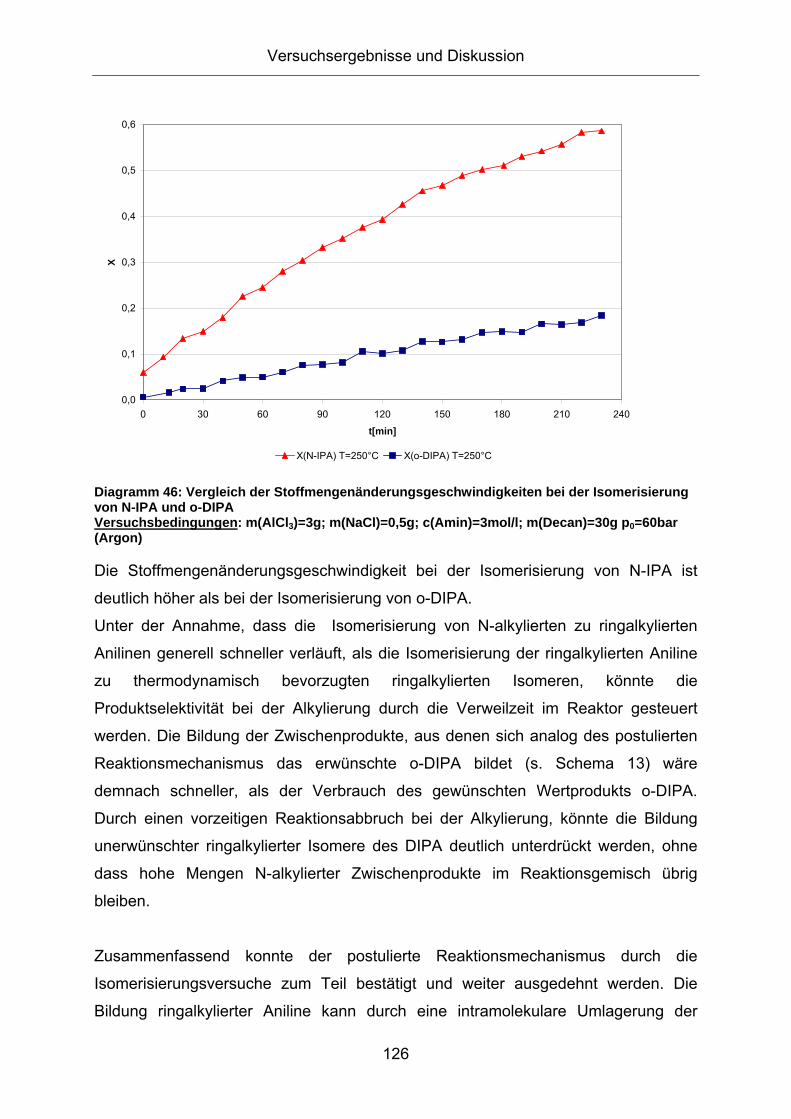
Diagramm 46: Vergleich der Stoffmengenänderungsgeschwindigkeiten bei der Isomerisierung von N-IPA und o-DIPA Versuchsbedingungen: m(AlCl3)=3g; m(NaCl)=0,5g; c(Amin)=3mol/l; m(Decan)=30g p0=60bar (Argon)
Die Stoffmengenänderungsgeschwindigkeit bei der Isomerisierung von N-IPA ist
deutlich höher als bei der Isomerisierung von o-DIPA.
Unter der Annahme, dass die Isomerisierung von N-alkylierten zu ringalkylierten
Anilinen generell schneller verläuft, als die Isomerisierung der ringalkylierten Aniline
zu thermodynamisch bevorzugten ringalkylierten Isomeren, könnte die
Produktselektivität bei der Alkylierung durch die Verweilzeit im Reaktor gesteuert
werden. Die Bildung der Zwischenprodukte, aus denen sich analog des postulierten
Reaktionsmechanismus das erwünschte o-DIPA bildet (s. Schema 13) wäre
demnach schneller, als der Verbrauch des gewünschten Wertprodukts o-DIPA.
Durch einen vorzeitigen Reaktionsabbruch bei der Alkylierung, könnte die Bildung
unerwünschter ringalkylierter Isomere des DIPA deutlich unterdrückt werden, ohne
dass hohe Mengen N-alkylierter Zwischenprodukte im Reaktionsgemisch übrig
bleiben.
Zusammenfassend konnte der postulierte Reaktionsmechanismus durch die
Isomerisierungsversuche zum Teil bestätigt und weiter ausgedehnt werden. Die
Bildung ringalkylierter Aniline kann durch eine intramolekulare Umlagerung der
Versuchsergebnisse und Diskussion
127
Isopropylgruppe vom Amin-Stickstoff an den aromatischen Ring erfolgen. Die Bildung
dreifachalkylierter Aniline könnte auf eine Transalkylierung zweier DIPA
zurückzuführen zu sein und zeigt eine höhere Temperaturabhängigkeit als die
Transalkylierungsreaktionen von N-IPA. Durch eine Erhöhung der
Reaktionstemperatur könnte die Selektivität zu den dreifachalkylierten TIPA
verringert werden, während die Ausbeute an ringalkylierten Produkten steigt.
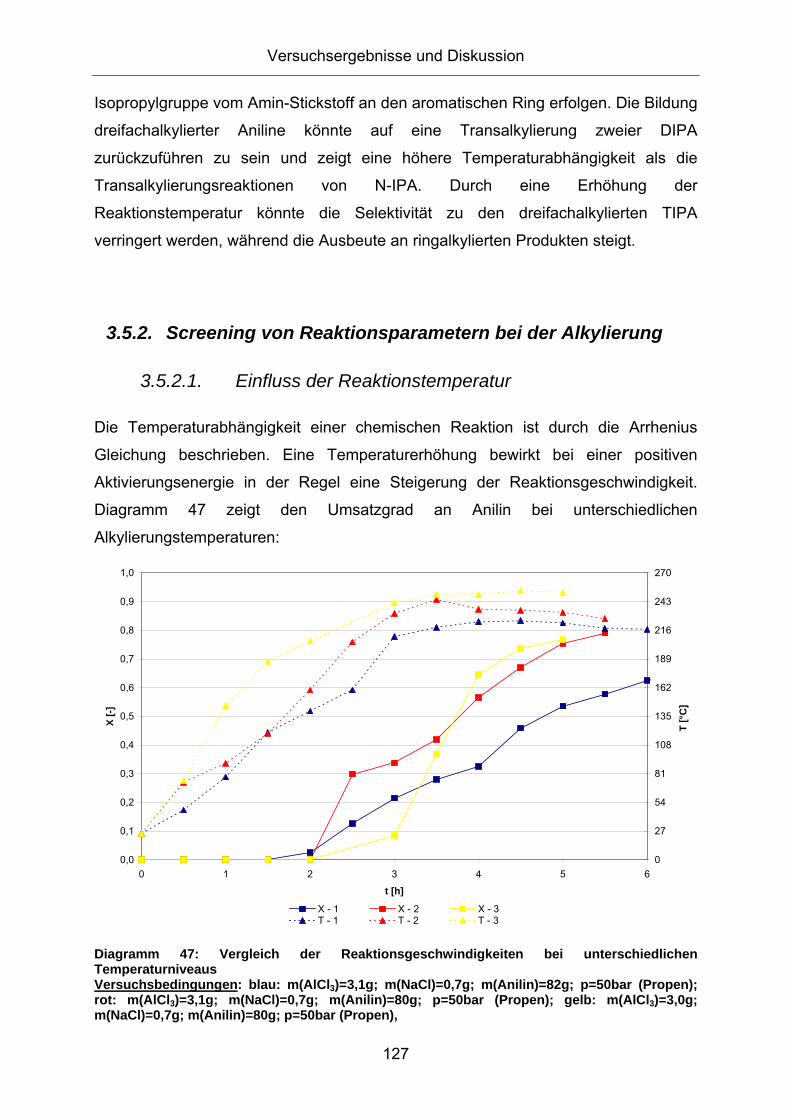
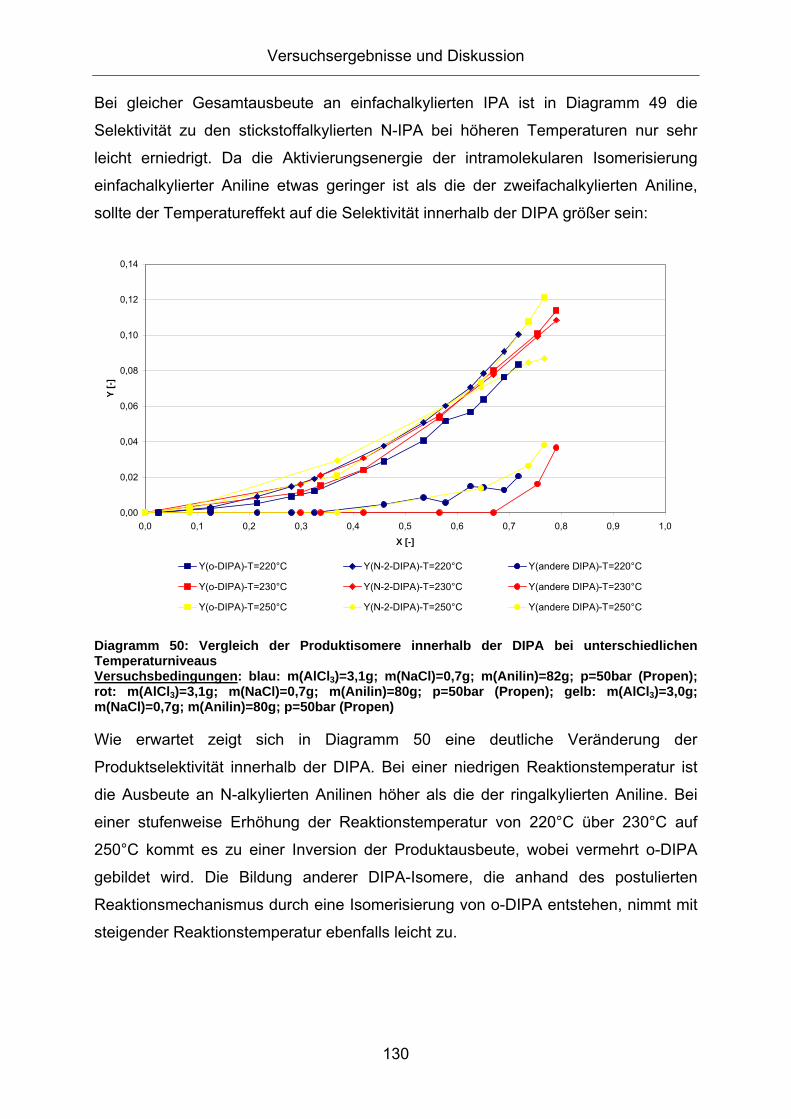
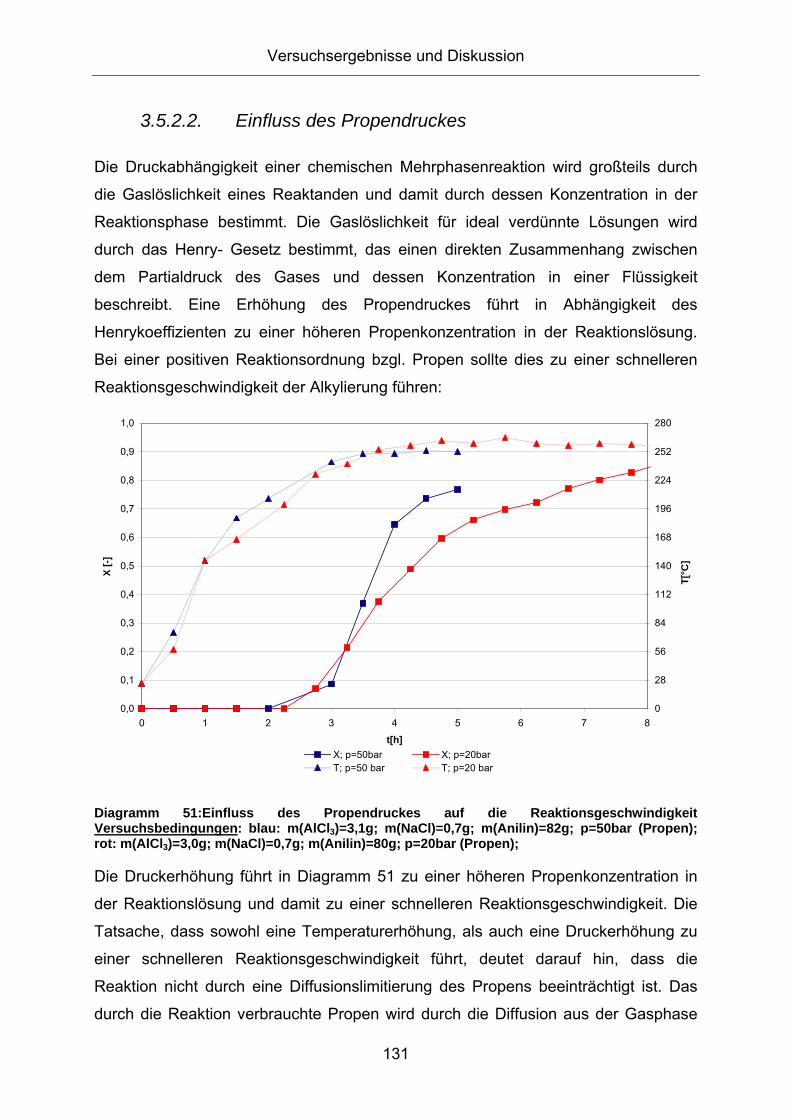
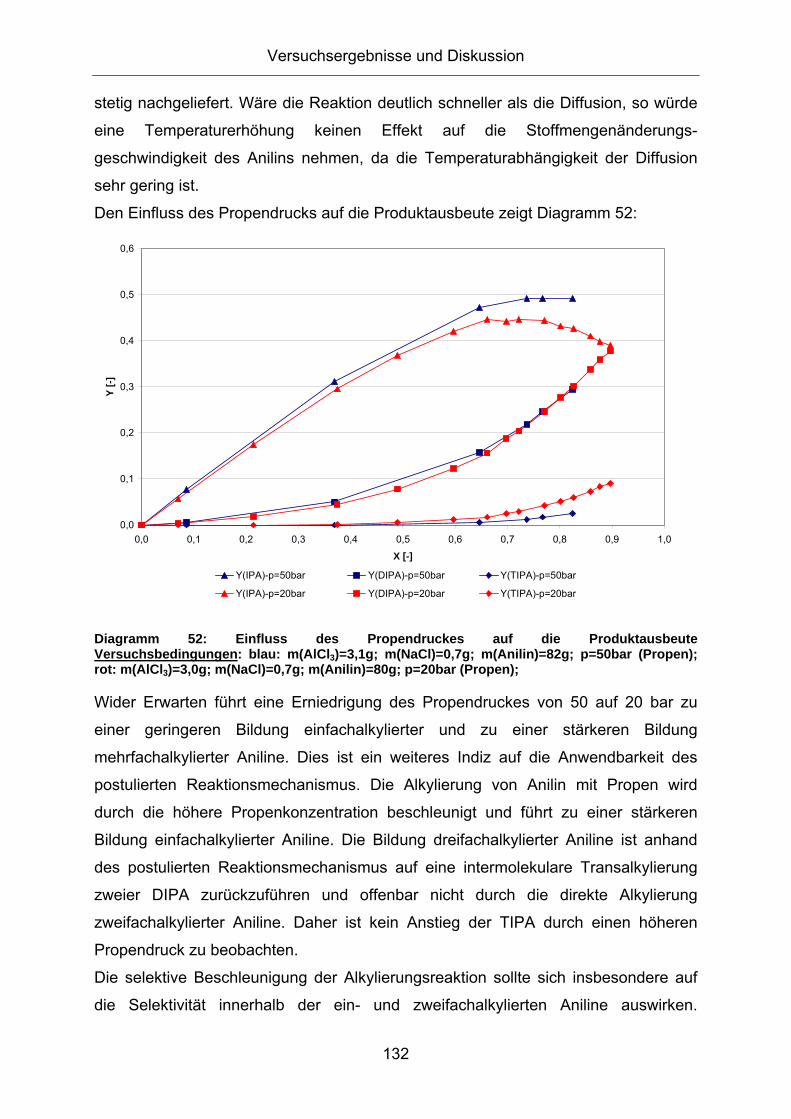
3.5.2. Screening von Reaktionsparametern bei der Alkylierung
3.5.2.1. Einfluss der Reaktionstemperatur
Die Temperaturabhängigkeit einer chemischen Reaktion ist durch die Arrhenius
Gleichung beschrieben. Eine Temperaturerhöhung bewirkt bei einer positiven
Aktivierungsenergie in der Regel eine Steigerung der Reaktionsgeschwindigkeit.
Diagramm 47 zeigt den Umsatzgrad an Anilin bei unterschiedlichen
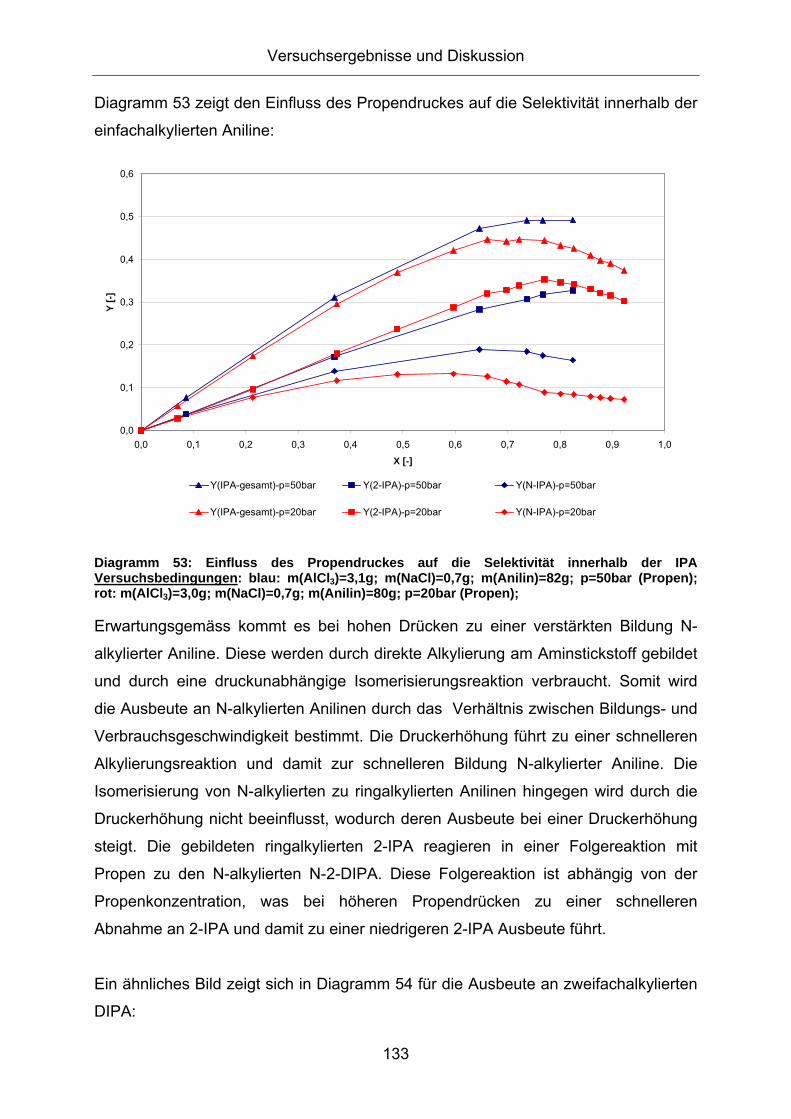
Diagramm 53: Einfluss des Propendruckes auf die Selektivität innerhalb der IPA Versuchsbedingungen: blau: m(AlCl3)=3,1g; m(NaCl)=0,7g; m(Anilin)=82g; p=50bar (Propen); rot: m(AlCl3)=3,0g; m(NaCl)=0,7g; m(Anilin)=80g; p=20bar (Propen);
Erwartungsgemäss kommt es bei hohen Drücken zu einer verstärkten Bildung N-
alkylierter Aniline. Diese werden durch direkte Alkylierung am Aminstickstoff gebildet
und durch eine druckunabhängige Isomerisierungsreaktion verbraucht. Somit wird
die Ausbeute an N-alkylierten Anilinen durch das Verhältnis zwischen Bildungs- und
Verbrauchsgeschwindigkeit bestimmt. Die Druckerhöhung führt zu einer schnelleren
Alkylierungsreaktion und damit zur schnelleren Bildung N-alkylierter Aniline. Die
Isomerisierung von N-alkylierten zu ringalkylierten Anilinen hingegen wird durch die
Druckerhöhung nicht beeinflusst, wodurch deren Ausbeute bei einer Druckerhöhung
steigt. Die gebildeten ringalkylierten 2-IPA reagieren in einer Folgereaktion mit
Propen zu den N-alkylierten N-2-DIPA. Diese Folgereaktion ist abhängig von der
Propenkonzentration, was bei höheren Propendrücken zu einer schnelleren
Abnahme an 2-IPA und damit zu einer niedrigeren 2-IPA Ausbeute führt.
Ein ähnliches Bild zeigt sich in Diagramm 54 für die Ausbeute an zweifachalkylierten
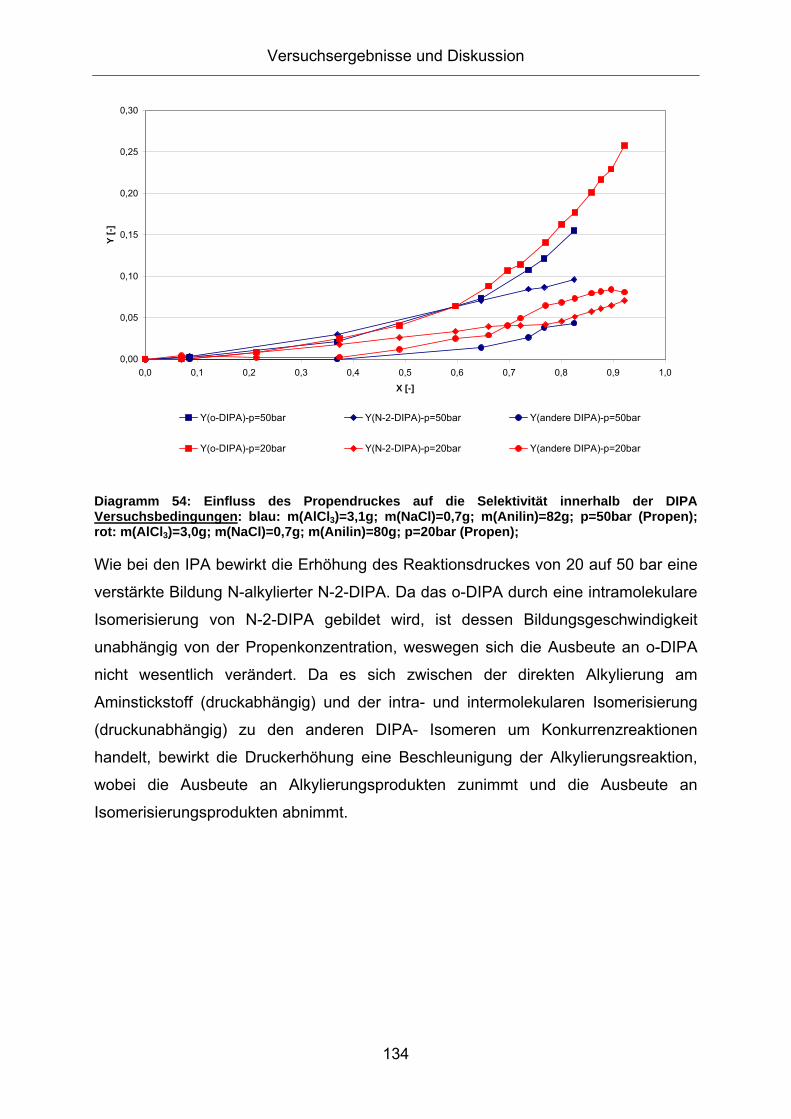
Diagramm 54: Einfluss des Propendruckes auf die Selektivität innerhalb der DIPA Versuchsbedingungen: blau: m(AlCl3)=3,1g; m(NaCl)=0,7g; m(Anilin)=82g; p=50bar (Propen); rot: m(AlCl3)=3,0g; m(NaCl)=0,7g; m(Anilin)=80g; p=20bar (Propen);
Wie bei den IPA bewirkt die Erhöhung des Reaktionsdruckes von 20 auf 50 bar eine
verstärkte Bildung N-alkylierter N-2-DIPA. Da das o-DIPA durch eine intramolekulare
Isomerisierung von N-2-DIPA gebildet wird, ist dessen Bildungsgeschwindigkeit
unabhängig von der Propenkonzentration, weswegen sich die Ausbeute an o-DIPA
nicht wesentlich verändert. Da es sich zwischen der direkten Alkylierung am
Aminstickstoff (druckabhängig) und der intra- und intermolekularen Isomerisierung
(druckunabhängig) zu den anderen DIPA- Isomeren um Konkurrenzreaktionen
handelt, bewirkt die Druckerhöhung eine Beschleunigung der Alkylierungsreaktion,
wobei die Ausbeute an Alkylierungsprodukten zunimmt und die Ausbeute an
Isomerisierungsprodukten abnimmt.
135
Kapitel 4 Zusammenfassung
136
Zusammenfassung
137
4. Zusammenfassung
Das Ziel dieser Arbeit war die Entwicklung eines nachhaltigen Verfahrenskonzepts
zur Friedel-Crafts Alkylierung von Anilin mit Propen unter katalytischem Einsatz von
Aluminiumchlorid. Um die ökologischen und ökonomischen Probleme bei den bereits
bestehenden homogenen Verfahren zu umgehen, wurden folgende
Verfahrenskonzepte zur Immobilisierung und Rezyklierung des Katalysators
eingehend untersucht:
- Einsatz ionischer Flüssigkeiten
- Einsatz anorganischer Salzschmelzen
- Katalysatorrückführung durch Kristallisation
Zum besseren Verständnis der betrachteten Alkylierungsreaktion wurden weiterhin
mechanistische und kinetische Untersuchungen durchgeführt, sowie Einflussfaktoren
auf die Selektivität und Aktivität ermittelt.
Einsatz ionischer Flüssigkeiten Auf Basis einer DFT Kalkulation mit COSMO-RS konnte eine Vorauswahl aus der
Vielzahl an möglichen Anion- und Kationkombinationen für die ionische Flüssigkeit
getroffen werden, die eine möglichst vollständige Mischunglücke mit den gebildeten
Alkylierungsprodukten aufweist. Des Weiteren war aufgrund der geforderten hohen
Temperaturen für eine erfolgreiche Alkylierung eine hohe Thermostabilität gefordert,
so dass die Wahl auf Imidazolium-basierte ionische Flüssigkeiten der Art
[Imidazolium][NTf2] fiel. Das AlCl3 löst sich in der ionischen Flüssigkeit und verändert
die Struktur des Anions unter der Bildung einer anionischen
Chloroaluminatspezies115. TG Untersuchungen zeigten allerdings, dass die
resultierende acide ionische Flüssigkeit eine verringerte Thermostabilität aufweist
und damit den hohen thermischen Belastungen bei der Alkylierung nur kurz
standhalten kann. Weiterhin zeigt die acide ionische Flüssigkeit gegenüber der
reinen IL ein verändertes Phasenverhalten mit den organischen Produkten, so dass
eine Phasentrennung nur durch den Einsatz organischer Extraktionsmittel erzielt
werden konnte. Während sich das polare Anilin überwiegend in der ionischen
Katalysatorphase löst, werden durch den Einsatz von unpolaren Extraktionsmitteln
die gebildeten alkylierten Produkte in die unpolare Extraktphase extrahiert. Die
Zusammenfassung
138
Bildung unterschiedlicher Alkylierungsprodukte bewirkt eine Phasenvermittlung
zwischen ionischer Katalysatorphase und organischer Extraktionsphase, was eine
rein gravimetrische Abtrennung der alkylierten Produkte von der ionischen
Katalysatorphase verhindert.
Einsatz anorganischer Salzschmelzen Aufgrund der begrenzten Thermostabilität organischer ionischer Flüssigkeiten wurde
weiterhin der Einsatz anorganischer Salzschmelzen mit aciden Mischungen aus AlCl3
und NaCl als Reaktionsmedium und Katalysatorphase getestet. Genau wie bei den
ionischen Flüssigkeiten kommt es zu einer Phasenvermittlung zwischen ionischer
Katalysatorphase und organischer Extraktphase. Selbst durch den Einsatz von
überkritischen Gasen, wie scCO2 und scPropan, konnte aufgrund der starken
Wechselwirkungen zwischen den Aminen und dem Aluminiumchlorid keine
zufriedenstellende Katalysator-Produkttrennung erzielt werden. Während beim
Einsatz von überkritischem CO2 nach einer Alkylierungsreaktion nur eine geringe
Extraktausbeute (<1%) erzielt werden konnte, führte die Extraktion mit
überkritischem Propan zwar zu einer hohen Ausbeute an extrahierten Produkten,
allerdings auch zu einem hohen Aluminiumleaching.
Die starken Wechselwirkungen zwischen dem AlCl3 und den jeweiligen
unterschiedlich substituierten Isomeren des Anilins konnten mittels 27Al-NMR
Messungen qualitativ miteinander verglichen werden. Aufgrund der höheren
Elektronendichte durch die eingeführten Alkylgruppen koordinieren die alkylierten
Produkte stärker an das AlCl3 als die Edukte. Dies resultiert im 27Al-NMR in einer
stärkeren Tieffeldverschiebung des Aluminiumsignals relativ zu der eingesetzten
Referenz. Die Untersuchungen haben ergeben, dass sterische Effekte bei der
Koordination an das AlCl3 eine untergeordnete Rolle spielen. Eine Abtrennung und
Rezyklierung des Katalysators von den alkylierten Produkten durch eine
Verdrängungsreaktion mit frischen Edukten ist daher kaum möglich.
Katalysatorrückführung durch Kristallisation Gerade die starken Säure-Base Wechselwirkungen zwischen den Produkten und
dem AlCl3 ermöglichen eine Katalysatorabtrennung durch Kristallisation einer
AlCl3(Arylamin)2-Spezies. Diese Lewis-Säure-Base-Addukte wurden mit mehreren
kommerziell erhältlichen Arylaminen hinsichtlich ihrer Tendenz zur Kristallisation mit
Zusammenfassung
139
Aluminiumchlorid untersucht. Bei einigen gelang die Strukturaufklärung mittels
Röntgenstrukturanalyse. Ein Versuch zur Anilinalkylierung mit [AlCl3(MIPA)2] als
Katalysator zeigt die hohe Aktivität dieser Aluminiumchlorid-Arylamin-Komplexe als
Friedel-Crafts-Katalysatoren in der Anilinalkylierung.
Die Rezyklierung einer AlCl3 Spezies nach einer Alkylierungsreaktion von Anilin mit
Propen konnte dreimal ohne Aktivitäts- und Selektivitätsverlust reproduzierbar
durchgeführt werden, wobei das Aluminiumleaching in die Produktphase im Laufe
der Rezyklierungsversuche drastisch abnimmt. Dies ist vermutlich auf die
Anreicherung einer Aminspezies zurückzuführen, die bevorzugt mit AlCl3 kristallisiert.
Es wird vermutet, dass ein bestimmtes Substitutionsmuster am Anilin eine besonders
effiziente Kristallisation durch ein optimiertes Verhältnis aus sterischer Hinderung und
Lewis-Basizität ermöglicht. Wenngleich das AlCl3 nicht komplett aus der
Reaktionsmischung kristallisiert werden konnte, ergeben sich durch das hier erstmals
vorgeschlagene Rezyklierungsverfahren erhebliche Einsparungen an Waschwasser
bei der Produktaufarbeitung und an AlCl3 durch dessen Rezyklierung. Die
Kristallisation dieser AlCl3(Arylamin)2-Spezies konnte im Reaktor nur durch Zugabe
von NaCl erzielt werden.
Mechanistische und kinetische Betrachtungen Im Vergleich zum konventionellen Prozess ohne Katalysatorrückführung mit einer
Katalysatormischung aus Aluminiumchlorid und Aluminiumtrianilid zeigt der Einsatz
von reinem Aluminiumchlorid und Mischungen aus Aluminiumchlorid und
Natriumchlorid eine wesentlich höhere Aktivität, selbst bei Verwendung einer
geringeren Menge AlCl3. Aufgrund des niedrigen Schmelzbereichs acider
Chloroaluminatschmelzen und der damit verbundenen besseren Verfügbarkeit der
aciden Spezies kann die Reaktionsgeschwindigkeit drastisch erhöht werden.
Entgegen der vorherrschenden Meinung in der Literatur bewirken das Weglassen
des Aluminiumtrianilidkatalysators und der Einsatz acider Salzschmelzen keine
Verschlechterung der Produktausbeute. Die Selektivität zu o-DIPA ist trotz der
thermodynamischen Instabilität im Vergleich zu den anderen Diisopropylanilinen am
höchsten. Dies ist umso bemerkenswerter, als bei einer statistisch kontrollierten
elektrophilen Substitution bevorzugt das 2,4-DIPA gebildet werden sollte. Um diese
Tatsache zu erklären, wurde aufgrund von thermodynamischen und kinetischen
Zusammenfassung
140
Überlegungen ein Reaktionsmechanismus postuliert, der alle experimentellen
Beobachtungen dieser Arbeit widerspiegelt (Schema 19):
NH2
NH
NH2
NH2
HN
NH2NH2
NH2
NH2
AlCl3schnell
schnell
AlCl3
langsamhohe T
begünstigt durch hohe c(AlCl3)
AlCl3
langsamhohe T
langsamhohe T
begünstigt durch hohe c(AlCl3)
AlCl3
...begünstigt durch hohe c(AlCl3)
Schema 19: postulierter Reaktionsmechanismus zur Alkylierung von Anilin mit Propen
Die Alkylierung des Anilins mit Propen unter katalytischer Wirkung von
Aluminiumchlorid scheint bei den untersuchten Reaktionsbedingungen zunächst
bevorzugt am Aminstickstoff zu erfolgen. Erst durch intramolekulare Isomerisierung
entsteht das ringalkylierte Isopropylanilin. Dieser Schritt erfordert hohe Temperaturen
und wird durch eine hohe Katalysatorkonzentration begünstigt. Das 2-IPA reagiert
durch eine Folgealkylierung am Aminstickstoff weiter zum N,2-DIPA. Das
großtechnisch wertvolle o-DIPA entsteht wiederum durch einen intramolekularen
Isomerisierungsschritt, der wiederum durch hohe Temperaturen und hohe
Katalysatorkonzentrationen beschleunigt werden kann. Als Nebenreaktionen treten
neben Dealkylierungsreaktionen auch intermolekulare Transalkylierungsreaktionen
auf, die bevorzugt zur Bildung dreifachalkylierter Aniline führen.
Kinetische Isomerisierungsuntersuchungen konnten zeigen, dass insbesondere die
intramolekularen Umlagerungen zur Bildung ringalkylierter Produkte eine starke
Temperaturabhängigkeit aufweisen. Dies steht im Einklang mit den Literaturdaten,
Zusammenfassung
141
bei denen die Bildung N-alkylierter Produkte insbesondere bei niedrigen
Temperaturen beobachtet werden kann.
Insgesamt konnte die Friedel-Crafts Alkylierung von Anilin mit Propen unter
katalytischer Wirkung von Aluminiumchlorid im Vergleich zum kommerziellen
homogenen Flüssigphasenprozess im Hinblick auf Aktivität und Nachhaltigkeit
entscheidend optimiert werden, ohne dabei einen negativen Einfluss auf die
Produktselektivität zu nehmen. Bereits bestehende Alkylierungsverfahren können
hierdurch profitieren, da keine wesentlichen Veränderungen im Stoffsystem
durchgeführt werden müssen. Während ein zusätzlicher Operationsschritt
(Kristallisation) zur Katalysatorrezyklierung hinzugefügt wird, kann ein
Operationsschritt zur Herstellung des Aluminiumtrianilidkatalysators weggelassen
werden.
Das Einsparungspotential durch die teilweise Rückführung des Katalysators ergibt
sich durch die Verringerung der insgesamt benötigten AlCl3-Menge und der damit
verbundenen Menge an Natronlauge, die für die Hydrolyse des Aluminiumchlorids
bei der Produktaufarbeitung benötigt wird. Neben den ökonomischen Vorteilen
dieses Verfahrenskonzepts kann ein Großteil des Waschwassers zur
Produktaufreinigung eingespart werden, in dem gelöste Reaktionsrückstände die
Umwelt belasten.
142
143
Kapitel 5 Ausblick
144
Ausblick
145
5. Ausblick
Die Katalysatorrückführung konnte in dieser Arbeit durch die Kristallisation einer
AlCl3-(Arylamin)2 Spezies realisiert werden. Diese wurde in separaten
Kristallisationsversuchen mit Reinsubstanzen mittels Röntgendiffraktometrie
identifiziert. Allerdings war es bisher nicht eindeutig feststellbar, welche Aminspezies
nach der Alkylierungsreaktion bevorzugt mit Aluminiumchlorid kristallisiert und zu
einer drastischen Abnahme des Aluminiumleachings führt. Durch eine genaue
Identifizierung der kristallisierten Aluminiumspezies könnte diese gezielt synthetisiert
und von vornherein für die Alkylierung eingesetzt werden. Dadurch könnten
optimierte Kristallisationsbedingungen ermittelt werden und der Aluminiumaustrag in
die Produktphase minimiert werden. Diese Friedel-Crafts aktive Aluminiumspezies
könnte in einer Vielzahl von elektrophilen Substitutionsreaktionen Verwendung
finden, bei denen eine Abtrennung des Katalysators nur schwer möglich ist.
146
147
Kapitel 6 Experimenteller Teil
148
Experimenteller Teil
149
6. Experimenteller Teil
6.1. COSMO-RS Berechnungen
Alle in dieser Arbeit verwendeten Moleküle wurden mit der DFT-Methode (Kapitel
2.3.2) unter den COSMO Randbedingungen geometrieoptimiert. Mit der
Molekülmodellierungssoftware HyperChem Release 7.51 der Firma Hypercube Inc.
wurden die Molekülgeometrien erzeugt und aus Standardvorgaben für
Bindungslängen, Bindungswinkel und Drehwinkel eine dreidimensionale
Molekülstruktur erstellt. Mit dieser angenäherten Struktur wurde eine
Konformeranalyse durchgeführt.
Da COSMO-RS auf Basis der Molekülgeometrie arbeitet, besitzen unterschiedliche
Konformere verschiedene σ –Profile und beeinflussen folglich den
Aktivitatskoeffizienten und das chemische Potential. Die Konformeranalyse wurde
ebenfalls mit dem Programm HyperChem durchgeführt. Bei der
Geometrieoptimierung innerhalb der Konformersuche wurde die Kraftfeldmethode
MM+ gewählt116. Die genaue Vorgehensweise der Konformeranalyse wird bei
Spuhl117 eingehend beschrieben.
Die ermittelten Molekülstrukturen der Konformere wurden mittels DFT durch das
Programm Turbomole118 geometrieoptimiert. Dabei wurde die RI-DFT Variante
verwendet, welche Coulomb-Wechselwirkungen einbezieht. Als Basis-Set, worin
Parameter zur Beschreibung der Basisfunktionen enthalten sind, wurde das TZVP-
(triple zeta valence polarization) Basis-Set119 verwendet. Die
Ladungsdichteverteilungen wurden im „COSMO-File“ für die COSMO-RS Rechnung
zur Verfügung gestellt und mit Hilfe von COSMOtherm die Aktivitätskoeffizienten,
chemischen Potentiale und Löslichkeiten berechnet.
6.2. Allgemeine Arbeitstechniken
6.2.1. Arbeiten unter Schutzgas
Aufgrund der Luft- und Feuchtigkeitsempfindlichkeit der eingesetzten Substanzen,
wurden die beschriebenen Versuche in einer Inertgasatmosphäre durchgeführt. Das
verwendete Argon hat die Qualität 4.6.
Experimenteller Teil
150
Die verwendeten Schlenkgefäße und der Autoklav wurden vor Gebrauch je dreimal
im Vakuum ausgeheizt und mit Argon beschickt. Während der Arbeiten an offenen
Gefäßen, wurde ein permanenter Argonstrom aufrechterhalten. Flüssige Substanzen
und Lösungsmittel wurden im Argongegenstrom mit Kunststoffspritzen, deren
Kanülen zuvor mehrmals mit Argon gespült wurden, zugegeben.
6.2.2. Bereitstellung von Lösungsmitteln und Reagenzien
Alle verwendeten aromatischen Amine wurden als Handelsprodukte von der Firma
Lonza bezogen und über einem Molsieb in einer Inertgasatmosphäre gelagert, um
Feuchtigkeitsspuren so gering wie möglich halten zu können.
Cyclohexan wurde über eine Lösungsmittelreinigungsanlage der Firma Glass
Contour getrocknet.
Die verwendeten ionischen Flüssigkeiten wurden anhand gängiger
Synthesevorschriften hergestellt und mittels NMR auf ihre Reinheit untersucht. Die
Lagerung erfolgte in einer Inertgasatmosphäre.
Alle übrigen Chemikalien wurden als Handelsprodukte bezogen und ohne weitere
Aufarbeitung verwendet.
6.3. Alkylierungsversuche
6.3.1. Aluminiumtrianilid als Katalysator
Die Katalysatormischung zwischen Aluminiumtrianilid und Aluminiumchlorid wird vor
der Reaktion in einem 100 ml Glaskolben unter Inertgas hergestellt. Dabei werden
0,6 mol-equivalente elementares Aluminium (Aluminiumflitter) und 1 mol-equivalent
Aluminiumchlorid mit 9 mol-equivalenten Anilin bei 170 °C in Reaktion gebracht. Der
dabei entstehende Wasserstoff entweicht über einen Rückflusskühler und einen
Silikonölbubbler aus der mit Inertgas gefluteten Apparatur. Dabei wird ein stetiger
Argonstrom aufrechterhalten. Nach etwa 1 h Reaktionszeit entweicht kein
Wasserstoff mehr und das Aluminium ist komplett abreagiert. Die Katalysator-
Eduktmischung wird unter Inertgas in den Reaktor eingewogen.
Experimenteller Teil
151
6.3.2. Versuche mit ionischer Flüssigkeit
Die acide ionische Flüssigkeit wird in einem 50 ml Schlenkrohr hergestellt. Dabei
werden 1,5 mol-equivalente Aluminiumchlorid unter Inertgas mit 0,75 - 1 mol-
equivalenten ionischer Flüssigkeit bei 80°C in Lösung gebracht. Die so erhaltene
acide ionische Flüssigkeit wird unter Inertgas in den Reaktor eingewogen.
6.3.3. Versuche mit anorganischen Salzschmelzen
Aufgrund der hohen Schmelztemperatur über 100 °C und der hohen
Sublimationsneigung von Aluminiumchlorid wurde die acide anorganische
Salzschmelze in-situ bei 30 bar Propendruck im Alkylierungsreaktor hergestellt.
Dabei wurden 1,5 mol-equivalente Aluminiumchlorid mit 0,75 – 1 mol-equivalenten
Natriumchlorid zusammen mit Anilin in den Reaktor eingewogen und mittels
pumpenkopfgekühlter HPLC-Pumpe Propen bis zu einem Druck von 30 bar
zudosiert. Es ist davon auszugehen, dass sich die anorganische Salzschmelze bei
Temperaturen zwischen 100 und 150 °C im Reaktor bildet.
6.3.4. Durchführung der Katalyseversuche
Alle Alkylierungsversuche wurden in einem 300 ml Hastelloy C Autoklaven der Firma
Haage mit Glasliner durchgeführt. Die Temperaturregelung erfolgt über ein
Thermoelement, das in die Reaktionsmischung eintaucht. Zur Sicherheit ist ein
weiteres Thermoelement in den Reaktormantel angebracht, das bei Überschreitung
der zulässigen Reaktorwandtemperatur von 300 °C die Begleitheizung von Horst
abschaltet. Ein über Kopf gesteuerter Begasungsrührer sorgt für eine gute
Dispergierung des Gases in der Reaktionsmischung. Nachfolgende Abbildung zeigt
den verwendeten Haage Druckautoklaven:
Experimenteller Teil
152
Abbildung 17: Alkylierungsreaktor mit kontinuierlicher Propenzudosierung
Nachdem die Katalysatormischung zusammen mit Anilin in den Reaktor eingewogen
wurde, wird der Reaktor je dreimal mit Argon und einmal mit Propen gespült, um
Feuchtigkeit aus dem System zu entfernen. Anschließend wird mittels
pumpenkopfgekühlter HPLC-Pumpe Propen bis zu einem Druck von 30 bar
zudosiert. Die Dispergierung wird gestartet und mit dem Aufheizen des Reaktors
begonnen. Dies geschieht in drei Stufen. Nachdem die Solltemperatur von 150 °C
erreicht wurde wird in 50 °C Schritten weiter bis auf die gewünschte Temperatur
aufgeheizt. Dies verhindert ein zu starkes Überschwingen der Reaktortemperatur, da
die Temperaturregelung über ein im Reaktor angebrachtes Thermoelement gesteuert
wird. Nach Erreichen der Betriebstemperatur wird über eine HPLC- Pumpe so viel
Propen hineingefördert, wie durch die Reaktion verbraucht wird, so dass der
Reaktordruck konstant gehalten werden kann. Während der Alkylierung können mit
dem angebrachten Nadelventil, das in die Reaktionslösung eintaucht, Proben
gezogen werden. Das Tauchrohr muss dazu zunächst gespült werden, so dass die
erste Probe verworfen wird, um eine repräsentative Probe aus dem Reaktorinhalt zu
bekommen.
Nach der Reaktion wird die Begleitheizung und der Rührer ausgeschaltet und nach
Abkühlung auf Raumtemperatur über den Silikonölbubbler der Druck abgelassen.
Experimenteller Teil
153
6.3.5. Rezyklierungsversuche
Um eine möglichst vollständige Kristallisation der AlCl3-(Arylamin)2 Spezies zu
bekommen wurde der Reaktor nach der Reaktion über Nacht, ohne Dispergierung,
mit ausgeschalteter, aber angebrachter Begleitheizung zur Isolation, langsam auf
Raumtemperatur abgekühlt. Nach der Druckentspannung und dem Öffnen des
Reaktors, wurde die überstehende flüssige Phase unter Inertgas abdekantiert. Die
hochviskose Phase, in der sich der kristallisierte Katalysator befindet, wurde wieder
in den Reaktor gegeben und unter Inertgas aufbewahrt, bis die nächste Reaktion
gestartet werden konnte. Hierzu musste zunächst die ausgetragene Menge an
Aluminiumchlorid in der flüssigen Phase mittels ICP detektiert werden. Die verlorene
Menge an Katalysator wurde anschließend zusammen mit neuen Edukten zu der
abgetrennten Katalysatorphase in den Reaktor eingewogen und nach erneuter
Inertisierung eine weitere Alkylierung gestartet.
6.3.6. Probenaufarbeitung
Die aus der Reaktion gezogenen Proben wurden über Magnesiumsulfat und
Natriumcarbonat gepuffert und mit Ethanol verdünnt. Gelöstes AlCl3 fällt als
Aluminiumhydroxid aus, so dass Schäden an der Analytik vermieden werden können.
Nach einer fest-flüssig Trennung, zum Teil mittels Spritzenfilter oder durch
Dekantieren erfolgte die quantitative Analyse der Produktlösung mittels GC-FID.
Zur Bestimmung des Aluminiumleachings wurden 500 µl der Reaktionslösung mit 50
ml konzentrierter Essigsäure versetzt und mittels ICP untersucht.
6.4. Kristallisationsversuche
Versuche zur Kristallisation von Lewis-Säure-Base-Addukten zwischen
Aluminiumchlorid und verschiedenen Anilinderivaten wurden mit und ohne Zusatz
von Natriumchlorid in einem Überschuss des jeweiligen Arylamins ohne weiteres
Lösungsmittel durchgeführt.
Alle Kristallisationsversuche wurden im Schlenkkolben unter Argonatmosphäre
durchgeführt. Aluminiumchlorid, evtl. Natriumchlorid und ein Überschuss des
Arylamines wurden eingewogen und anschließend bei 80 °C im Ölbad bis zur
Experimenteller Teil
154
vollständigen Auflösung des Bodensatzes gerührt. Die Kristallisation erfolgte nach
Abschaltung von Heizung und Rührer über Nacht im abkühlenden Ölbad.
Die gebildeten Kristalle wurden abgefrittet, Masse und Aluminiumgehalt der
überstehenden Flüssigkeit ermittelt und daraus quantitativ Menge und
Zusammensetzung der Kristalle bestimmt.
6.5. Analytik
6.5.1. Gaschromatographie (GC)
Die quantitative Analyse des Reaktionsgemisches wurde mittels GC-FID, mit
folgenden technischen Daten bestimmt:
GC-Gerät: Varian CP3900
GC-Säule: WCOT Fused Silica 50m x 0,21 mm Coating CP SILPONA
CB DF=0,5 µm
Temperaturprogramm: 50°C 1 min Iso, 210°C 5K/min, 250°C 10K/min 10 min iso
Trägergas: Helium
Volumenstrom: 1 ml/min
Zur Identifizierung der Peaks wurden Reinsubstanzen verglichen und anhand der
Retentionszeiten identifiziert. Zur Überprüfung wurden begleitend GC-MS
Messungen durchgeführt:
GC-Gerät: Varian Saturn 2100
GC-Säule: Varian Factor Four, VF-5ms, 30 m x 25 m, ID = 0,25 mm,
DF = 0,25 μm
Temperaturprogramm: 50 °C - 250 °C 10 K/min; 15 min iso; 275°C 15 K/min; 5
3.4.3. Als Lösungsmittel kamen d6-DMSO und CD2Cl2 zum Einsatz.
6.5.4. Röntgendiffraktometrie (XRD)
Die Röntgenstrukturanalysen wurden an einem Bruker Nonius Kappa CCD
Diffraktometer durchgeführt. Die Lösung der Kristallstrukturen und deren
Verfeinerung erfolgte mit SHELX-97.120
6.5.5. Thermogravimetrie (TG)
Die thermogravimetrischen Untersuchungen wurden an einem Netzsch TG 209 F in
Aluminiumtiegeln durchgeführt. Bei einer Aufheizrate von 10 K/min von 30°C bis auf
450°C wurde ein konstanter Stickstoffstrom von 80 ml/min aufrechterhalten.
157
Anhang
158
Anhang
159
7. Anhang
7.1. Kristallographische Daten von AlCl3-(o-DIPA)2
Empirical formula C36 H57 Al Cl3 N3 Formula weight 665.18 Temperature 150(2) K Wavelength 0.71073 A Crystal system, space group Monoclinic, P2(1)/n Unit cell dimensions a = 13.6950(13) A alpha = 90 deg. b = 19.3890(4) A beta =90.503(7) deg. c = 14.0867(9) A gamma = 90 deg. Volume 3740.3(4) A^3 Z, Calculated density 4, 1.181 Mg/m^3 Absorption coefficient 0.296 mm^-1 F(000) 1432 Crystal size 0.24 x 0.18 x 0.13 mm Theta range for data collection 2.96 to 28.50 deg. Limiting indices -18<=h<=18, -26<=k<=25, -18<=l<=18 Reflections collected / unique 92103 / 9458 [R(int) = 0.0701] Completeness to theta = 28.50 99.8 % Absorption correction Semi-empirical from equivalents Max. and min. transmission 0.960 and 0.860 Refinement method Full-matrix least-squares on F^2 Data / restraints / parameters 9458 / 0 / 559 Goodness-of-fit on F^2 1.047 Final R indices [I>2sigma(I)] R1 = 0.0371, wR2 = 0.0704 R indices (all data) R1 = 0.0673, wR2 = 0.0801 Largest diff. peak and hole 0.329 and -0.277 e.A^-3
Tabelle 24: Atomic coordinates ( x 10^4) and equivalent isotropic displacement parameters (A^2 x 10^3) for VL0801. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
1 K. Weissermehl, H.-J. Arpe, Industrial Organic Chemistry, 2003, 376. 2 SRI Consulting, S. Bizzari, A. Kishi, 2007. 3 R. Stroh, J. Ebersberger, H. Haberland, W. Hahn, Angew. Chem., 1957 (69), 124. 4 P.F. Vogt, J.J. Gerulis, Aromatic Amines, Wiley-VCH Verlag GmbH & KgaA, 2002. 5 T. Lane, A. Plagens, Namen und Schlagwortreaktionen der org. Chemie,
Teubner Studienbücher, 1998, 132-136. 6 P. Wasserscheid, W. Keim, Angewandte Chemie, International Edition, 2000,
39(21), 3772. 7 K.R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68 (4), 351. 8 J. Dupont, R.F. de Souza, P.A.Z. Suarez, Chem. Rev., 2002, 102 (10), 3667. 9 P. Wasserscheid, T. Welton (Eds.), Ionic Liquids in Synthesis, 2002, 1. Auflage,
Wiley- VCH, Weinheim. 10 V. Ladnak, N. Hofmann, N. Brausch, P. Wasserscheid, Adv. Syn. Catal., 2007,
349(4+5), 719-726. 11 U. Baudis, M. Kreutz, Technologie der Salzschmelzen, Verlag Moderne Industrie,
2001. 12 J. Richter, Equilibrium and transport properties of Low-Melting Salts, Angewandte
Chemie, international edition, 1974. 13 K.R. Seddon, Molten Salt Chemistry, Ridl Pub.Co, 1987, 365. 14 G.I. Borodkin, V.G. Shubin, Russian Journal of Organic Chemistry, 2006, 42(12),
1745-1770. 15 W. Sundemeyer, Fused salts and their use as reaction media, Angewandte
Chemie, international Edition, 1965. 16 G.J. Janz, Molten Salts Handbook, Academic Press Inc., 1967. 17 Y. Zhao, T. J. VanderNoot, Electrochimica Acta, 1997, 42, 3-13. 18 A.A. Fannin, J. Chem. Eng., 1982, 114-119. 19 K.R. Seddon, J. Chem. Technol. Biotechnol.,1997, 68 (4), 351. 20 C. Jork, M. Seiler, Y.-A. Beste, W. Arlt, Journal of Chemical and Engineering
Data, 2004, 49, 852. 21 Y. A. Beste, M. Eggersmann, H. Schoenmakers, Chemie Ingenieur Technik
2004, 76, 1407. 22 J. Gmehling, M. Krummen, (Carl V. Ossietzky Universitaet Oldenburg,
Literaturverzeichnis
172
Vertreten Durch Den Kanzler, Germany). Application: DE, 2003, p. 14 pp. 23 M.H. Valkenberg, C. DeCastro, W.F. Hölderich, Spec. Pup. – R.Soc. Chem., 2001,
266, 242. 24 J.M. Crosthwait, M.J. Mulden, J.K. Dixon, J. Landerson, J.F. Brennecke, J. Chem.
Therm., 2005, 37, 559. 25 P. Bonhote, A.P. Dias, N. Papageorgiou, K. Kalyanasundaram, M. Grätzel, Inorg.
Chem., 1996, 35(5), 1168. 26 a) N. Papageorgiou, Y. Athanassov, M. Armand, P. Bonhote, H. Pettersson, A.
b) Azam, M. Grätzel, J. Electrochem. Soc., 1996, 143, 3099;
c) H. Matsumoto, T. Matsuda, T. Tsuda, R. Hagiwara, Y. Ito, Y. Miyazaki, Chem.
Let., 2001, 30, 26;
d) W. Kubo, T. Kitamura, K. Hanabusa, S. Yanagida, Chem. Commun., 2002, 4,
374;
e) S. Murai, S. Mikoshiuba, H. Somino, T. Kato, S. Hayase, Chem. Commun.,2003,
13, 1534;
f) H. Paulsson, A. Hagfeldt,L. Kloo, J. Phys. Chem., B, 2003, 107, 13665;
g) P. Wang, S.M. Zakeeruddin, I. Exnar, M. Grätzel, J. Am. Chem. Soc., 2003, 125,
1166. 27 P. Bonhote, A.P. Dias, N. Papageorgiou, K. Kalyanasundaram, M. Grätzel, Inorg.
Chem., 1996, 35(5), 1168. 28 R.E. DelSesto, C. Corley, A. Robertson, J.S. Wilkes, J. Orgme. Chem., 2005, 690,
R.M. Pagni, C.M. Gordon, Photochemistry in ionic liquids, Handbook of organic
photochemistry and photobiology (2nd Edition), 2004. 34 D. Bansal, F. Cassel, F. Croce, M. Hendrickson, E. Plichta, M. Salomon, J. Phys.
Chem., 2005, 109 (10), 4492.
Literaturverzeichnis
173
35 P. Wang, S.M. Zakeeruddin, J.E. Moser, M. Graezel, J. Phys. Chem. B, 2003, 107
(48), 3280.
P. Wang, S.M. Zakeeruddin, J.E. Moser, R. Humphry-Baker, M. Graezel, J. Am.
Chem. Soc., 2004, 126(23), 7164. 36 S. Park, R.J. Kazlauskas, Current Opinion in Biotech., 2003, 14(4), 432;
H. Pfruender, M. Amidjojo, D. Weuster-Botz, Chemie-Ingenieur-Technik, 2004, 79
(9). 37 A. Riisager, R. Fehrmann, M. Haumann, P. Wasserscheid, European Journal
of Inorganic Chemistry 2006, 695. 38 P. Wasserscheid, Transition Metals for Organic Synthesis (2nd Edition) 2004,
2, 559. 39 T. Welton, P. J. Smith, Advances in Organometallic Chemistry 2004, 51, 251. 40 C. Jork, M. Seiler, Y.-A. Beste, W. Arlt, Journal of Chemical and Engineering
Data 2004, 49, 852. 41 M. Seiler, C. Jork, W. Arlt, Chemie Ingenieur Technik 2004, 76, 735. 42 T. Brinz, U. Simon, (Robert Bosch G.m.b.H., Germany). Application: DE, 2004,
p. 8 pp 43 C. Hilgers, M. Uerdingen, M. Wagner, P. Wasserscheid, E. Schluecker,
(Solvent Innovation GmbH, Germany). Application: WO, 2006, p. 28pp. 44 A. Jess, J. Esser, ACS Symosium Series Ionic Liquids IIIB:Fundamentals,
Progress, Challenges, and Oppertunities, 2005, 83. 45 H. Olivier-Bourbigou, L. Magna, J. Mol. Cat. A:Chem., 2002, 182, 419. 46 T. Welton, Coord. Chem. Rev.; 2004, 248, 2459. 47 M. Piquet, D. Poinsot, S. Stutzmann, I. Tkatchenko, I. Tommasi, P. Wasserscheid,
J. Zimmermann, Topics in catalysis, 2004, 29, 139. 48 M. Maase, K. Masssonne, ACS Symposium Series, Ionic LiquidsIIIB:
Fundamentals, Progress, Challenges and Oppertunities, 2005, 126. 49 H. Olivier, D. Commereuc, A. Forestiere, F. Hugues, (Institut Francais du
Petrole, Fr.). Application: EP, 1998, p. 10 pp. 50 B. Weyershausen, K. Lehmann, Green Chem. 2005, 7, 15. 51 B. Weyershausen, K. Hell, U. Hesse, Green Chem., 2005, 7(5), 283. 52 D.J. Tempel, Air Products & Chemicals, 1st International Congress on Ionic
T. Welton, Chem. Rev., 1999, 99(8), 2071. 54 H. Olivier-Bourbigou, L. Magna, J. Mol. Cat. A:Chem., 2002, 182, 419. 55G.A. Olah, Friedel-Crafts related reactions, I-IV, Interscience, Wiley, New York,
1964. 56 a) C. Friedel, J. M. Crafts, C. R. Hebd. Seances Acad. Sci., 1877, 84, 1392;
b) C. Friedel, J. M. Crafts, C. R. Hebd. Seances Acad. Sci., 1877, 84, 1450. 57 C.D. Nenitzescu, I.P. Cantuniari, Chem. Ber. 66B, 1933, 1097. 58 G. A. Olah, S. Kobayashi, M. Tashiro, J. Am. Chem. Soc. 94, 1972, 7448. 59 G. A. Olah, G. K. S. Prakash, J. Sommer, Superacids, John Wiley & Sons, New
York, 1985. 60 Beyer, Walter, Lehrbuch der organischen Chemie, S. Hirzel Verlag Stuttgart
c) A.A.K. Abdul-Sada, K.R. Seddon, N.J. Stewart, BP Chemicals Ltd., 1995,
WO9521872. 87 C.E. Song, E.J. Roh, W.H. Shim, et al., Chem. Com. (Cambridge), 2000. 88 M.H. Valkenberg, C. DeCastro, W.F.Hölderich,Spec. Publ. – R. Soc. Chem.,2001,
266, 242. 89 M.H. Valkenberg, C. DeCastro, W.F. Hölderich, Top. Catal., 2001, 14, 139. 90 X. Ying, M. Sanjay, Alkylation/Acylation reactions in pyridinium-based
ionic liquids, Am. Chem.Soc., AN 2004:658980, 2004. 91 Chiappe, Cinzia; Piccioli, Paolo; Pieraccini, Daniela., Green Chemistry, 2006,
8(3), 277-281. 92 Hobbs, Steven J.; Madabusi, Venkatramanan K.; Wang, Jin-Yun; Stieber, Joseph
F. Ring alkylation of aniline or an aniline derivative with an olefin using ionic -
liquid alkylation catalysts. U.S. Pat. Appl. Publ., 2007. 93 J. Tomasi, M. Persico, Chemical Reviews (Washington, DC, United States), 94,
(7), 1994, 2027-2094. 94 A. Klamt, G. Schürmann, Journal of the Chemical Society, Perkin
Transactions 2: Physical Organic Chemistry (1972-1999) 1993, 799. 95 P. Atkins, J. de Paula, ATKINS' Physical Chemistry, Oxford University
Press, Oxford, 2002. 96 A. Klamt, Journal of Physical Chemistry, 99, (7), 1995, 2224-2235. 97 A. Klamt, COSMO and COSMO-RS, John Wiley and Sons Ltd., New York,
1998. 98 A. Klamt, F. Eckert, Fluid Phase Equilibria, 172, 2000, 43-72. 99 F. Eckert, A. Klamt, AlChE Journal 48, 2002, 369-385. 100 Wavefunction, Inc., Irvine CA. Spartan ’04 Quantum chemistry program, 2004. 101 a) A.W. Hofmann, C.A. Martius, Ber.,4,742, 1871.
b) A.W. Hofmann, C.A. Martius, Ber.,5,704,720, 1872.
Literaturverzeichnis
177
102 J.E. Gowan, T.S. Wheeler, Name index of organic reactions, Interscience, New
York, 1960,126. 103 a) J.Reilly, W.J. Hickinbottom, J.Chem.Soc.,117,103, 1920.
i) W.J. Hickinbottom, J.Chem.Soc.,404, 1119, 1937. 104 H.Hart, J.R. Kosak, J.Org.Chem.,27(1), 1962, 116-121. 105 F. Eckert, A. Klamt, COSMOtherm manual, Version C2.1, Release 01.05;
COSMOlogic GmbH & Co. KG, Leverkusen, Germany, 2005. 106 M. Medved, Messungen zur Thermostabilität Ionischer Flüssigkeiten, nicht
veröffentlicht, 2006. 107 N. Brausch, Zur Funktionalisierung von Aromaten mit aciden Ionischen
Flüssigkeiten, Dissertation, Universität Erlangen-Nürnberg, 2006. 108 M. Medved, Messungen zur Thermostabilität Ionischer Flüssigkeiten, nicht
veröffentlicht, 2006. 109 http://www.crct.polymtl.ca/FACT/documentation/FTsalt/FTsalt_Figs.htm, 2007-05-11. 110 a) J. Draxler, R. Marr, Chem. Ing. Tech., 62, (7), 1990, 525-530.
b) T. Ichikawa, K. Itoh, et al., Colloids and Surfaces A: Physicochem. Eng.
Aspects, 242, 2004, 21–26. 111 a) D. J. Cole-Hamilton, Adv. Synth. Catal., 2006, 348, 1341.
b) W. Leitner, Multiphase Homogeneous Catalysis, ed., B. Cornils, W. A.
Herrmann, I. T. Horvath, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt,
Wiley-VCH, Weinheim, 2005, 605–750. 112 R.N. Salvatore, S. Shin, et al., J.Org.Chem.,66 (3), 2001, 1035-1037. 113 S. Prasad, L.P. Pandey, Proc. Inst Chem., (36), 1964, 293-295. 114 G. E. Ham, Journal of Polymerization Science, 61, 1962, 293-301. 115 P. Wasserscheid, A. Metlen, N. Brausch, Chem.Commun., 2004, 13, 1552.
Literaturverzeichnis
178
116 HyperChem Release 7, User Manual, Hypercube Inc., 2002. 117 O. Spuhl: Anwendung quantenchemischer Methoden in der Thermodynamik der
Stofftrennung. Dissertation. Universitat Erlangen-Nürnberg, 2006. 118 Turbomole Version 5.7. Quantum chemistry Group, University of Karlsruhe, 2004. 119 A. Schäfer, C. Huber, R. Ahlrichs, Journal of chemical Physics, 100, 1994, 5829-
5835. 120 G. M. Sheldrick, SHELX-97 - A program for automatic solution of crystal