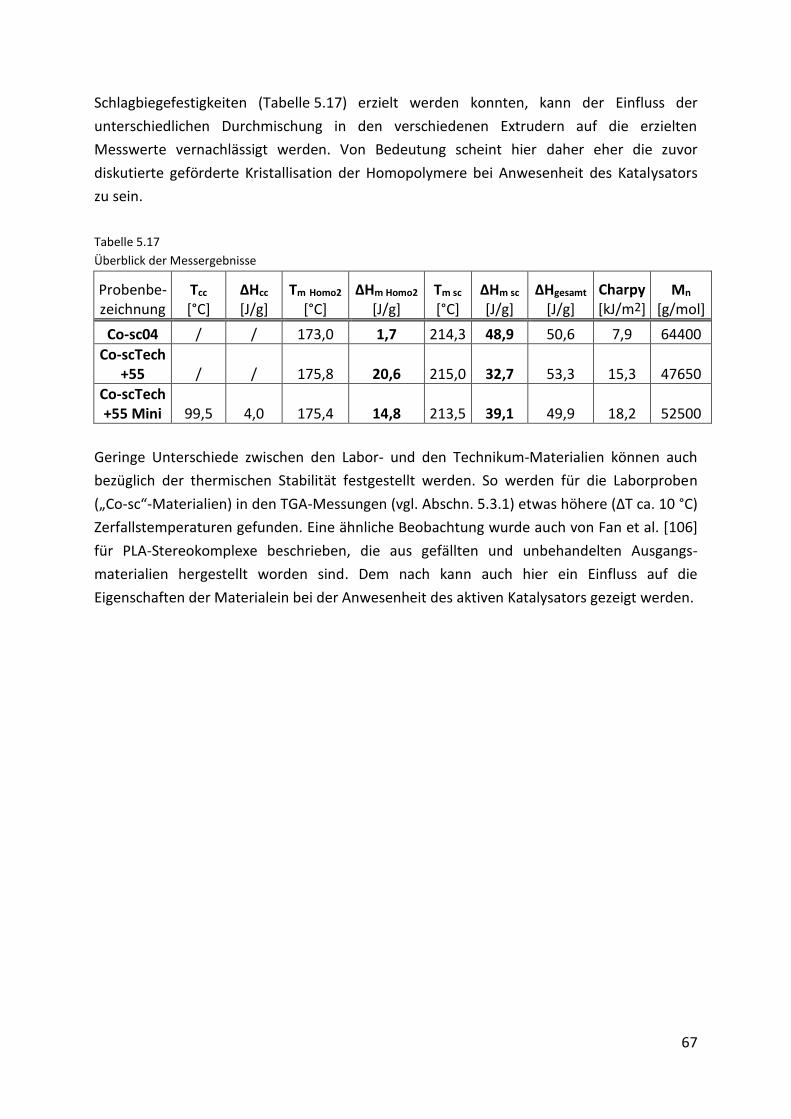
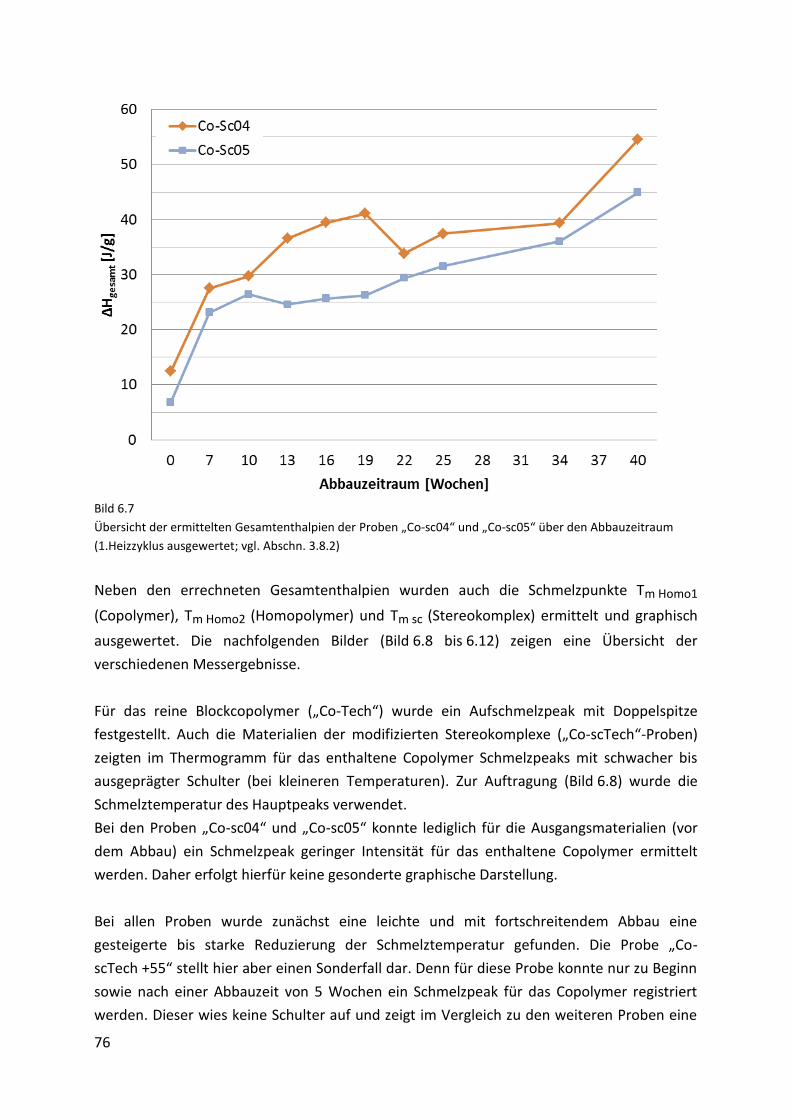
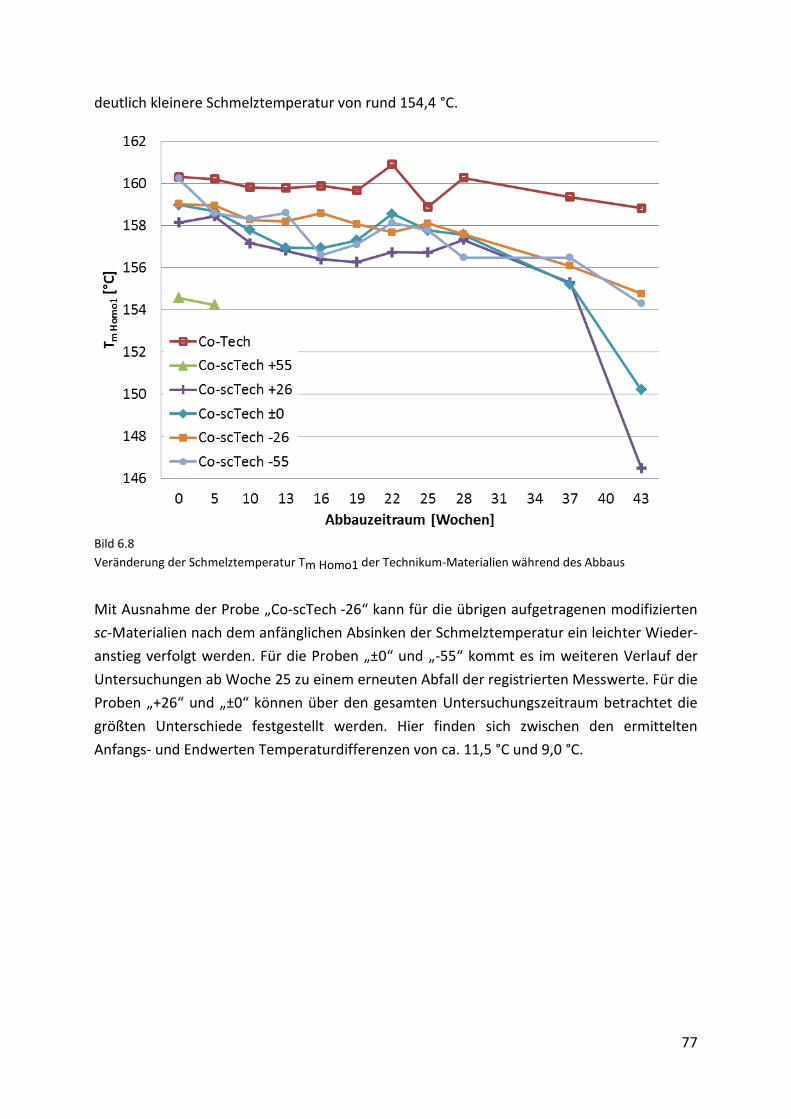
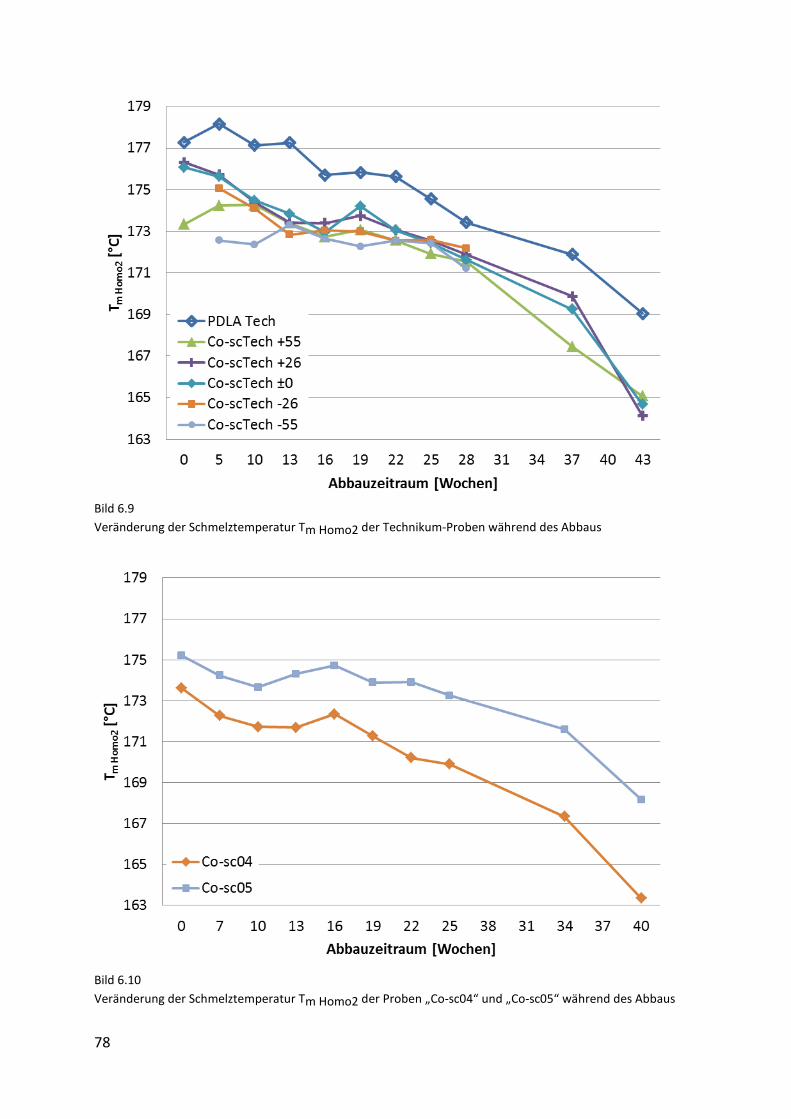
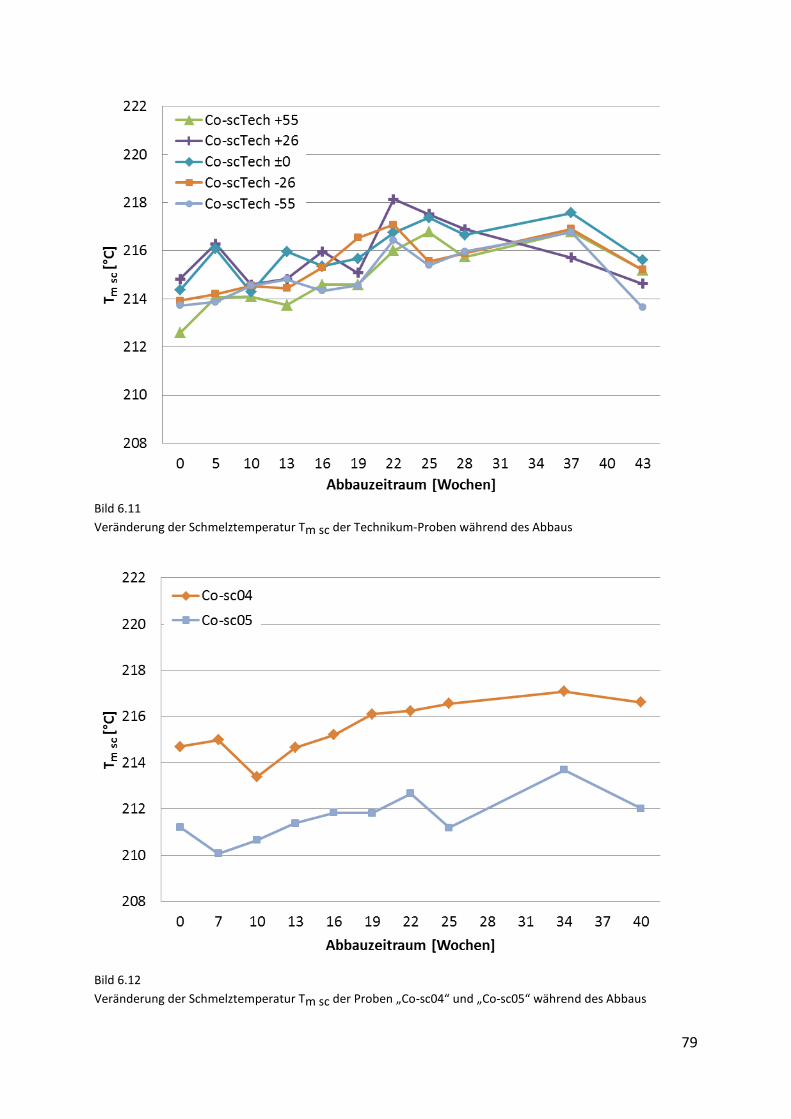
Page 1
Entwicklung eines thermisch stabilen und flexiblen
Polymers auf Basis von PLA für medizinische
Anwendungen
vorgelegt von
Dipl.-Chem. Jeanett Köhn
geb. in Kyritz
von der Fakultät III - Prozesswissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktorin der Naturwissenschaften
- Dr. rer. nat. -
genehmigte Dissertation
Prüfungsvorsitzende: Prof. Dr.-Ing. Claudia Fleck
Gutachter: Prof. Dr.-Ing. Manfred H. Wagner
Prof. Dr. Hans-Peter Fink
Tag der wissenschaftlichen Aussprache: 03. Juni 2015
Berlin 2015
Page 3
III
Inhaltsverzeichnis
1. Einleitung 1
2. Aufgabenstellung und Lösungskonzept 3
3. Grundlagen und Methoden 5
3.1. Polylactid (PLA) ............................................................................................................ 5
3.2. Stereokomplexe ........................................................................................................... 6
3.3. Weichmacher ............................................................................................................... 8
3.4. PLA-Synthese ................................................................................................................ 8
3.5. PLA-Abbau .................................................................................................................. 11
3.6. Partikelbildung ........................................................................................................... 13
3.7. Selektives Laserschmelzen ......................................................................................... 14
3.8. Analytische Mess- und Nachweismethoden .............................................................. 15
3.8.1. GPC – Gel Permeations Chromatographie ................................................................. 15
3.8.2. DSC – Differential Scanning Calorimetry .................................................................... 16
3.8.3. TGA – Thermogravimetrische Analyse ....................................................................... 17
3.8.4. Titration (Säurezahl) ................................................................................................... 17
3.8.5. Schlagbiegefestigkeit nach Charpy ............................................................................ 18
3.8.6. Shore Härte ................................................................................................................ 18
3.8.7. HDT – Wärmeformbeständigkeitstemperatur ........................................................... 19
3.8.8. Röntgenbeugungsexperimente.................................................................................. 19
3.8.9. DMA – Dynamisch Mechanische Analyse .................................................................. 20
Page 4
IV
4. Polymersynthese 21
4.1. Synthesestrategien..................................................................................................... 21
4.1.1. Festphasen-Polykondensation ................................................................................... 21
4.1.2. 3-Stufen-Synthese ...................................................................................................... 23
4.1.3. 2-Stufen-Synthese ...................................................................................................... 24
4.2. Arbeiten im Labormaßstab ........................................................................................ 25
4.2.1. Homopolymer-Synthese ............................................................................................ 25
4.2.2. Blockcopolymer-Synthese .......................................................................................... 26
4.2.3. Probencharakterisierung ........................................................................................... 27
4.2.3.1. Homopolymere .......................................................................................................... 27
4.2.3.2. Blockcopolymere ........................................................................................................ 28
4.2.4. Probenübersicht ......................................................................................................... 32
4.3. Arbeiten im Technikum-Maßstab .............................................................................. 32
4.3.1. Homopolymer-Synthese ............................................................................................ 33
4.3.2. Blockcopolymer-Synthese .......................................................................................... 34
4.3.3. Entmonomerisierung von Homo- und Blockcopolymer ............................................ 35
4.3.4. Probencharakterisierung ........................................................................................... 36
4.3.4.1. Homopolymer „PDLA Tech“ ....................................................................................... 36
4.3.4.2. Blockcopolymer „Co-Tech“ ........................................................................................ 37
4.3.5. Probenübersicht ......................................................................................................... 37
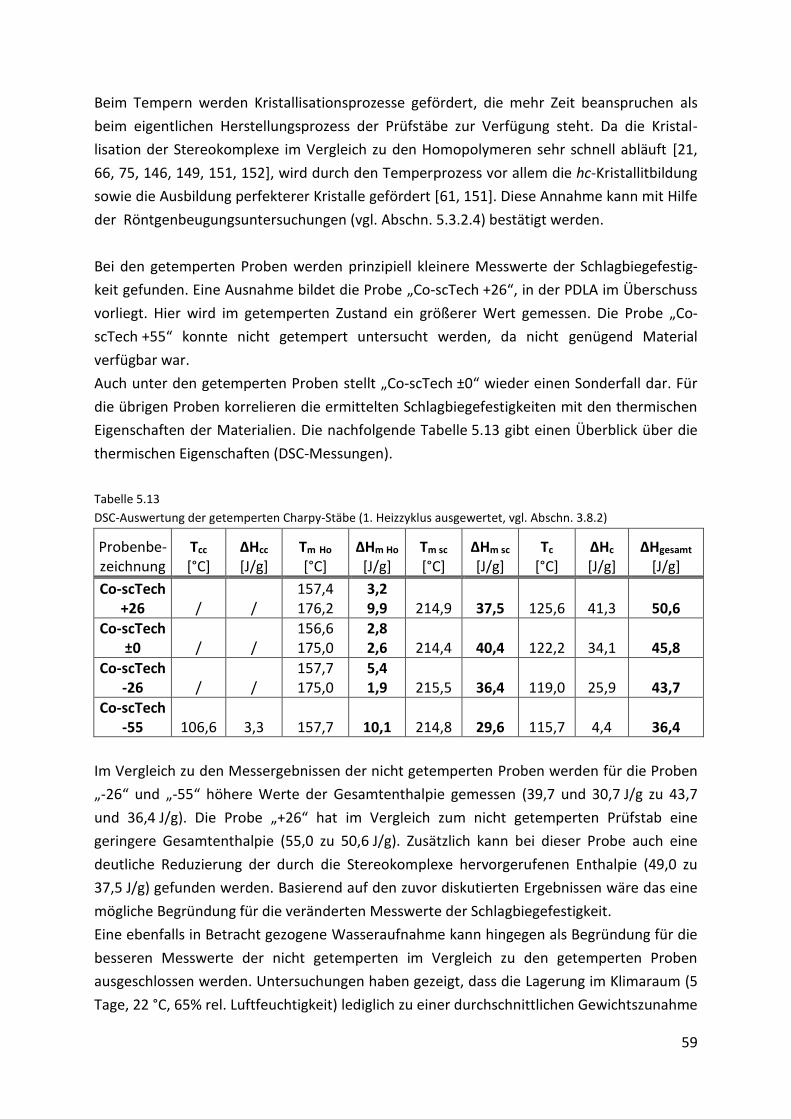
5. Stereokomplexe 39
5.1. Extruder „HAAKE MiniLab“ ........................................................................................ 39
5.1.1. Probenvorbereitung ................................................................................................... 39
5.1.2. Durchführung ............................................................................................................. 40
5.1.3. Probenübersicht ......................................................................................................... 40
5.1.4. Reproduzierbarkeitsuntersuchungen ........................................................................ 41
5.2. Mini-Compounder „Brabender KETSE 12/36“ ........................................................... 42
5.2.1. Durchführung ............................................................................................................. 42
5.2.2. Probenübersicht ......................................................................................................... 43
5.3. Probencharakterisierung ........................................................................................... 44
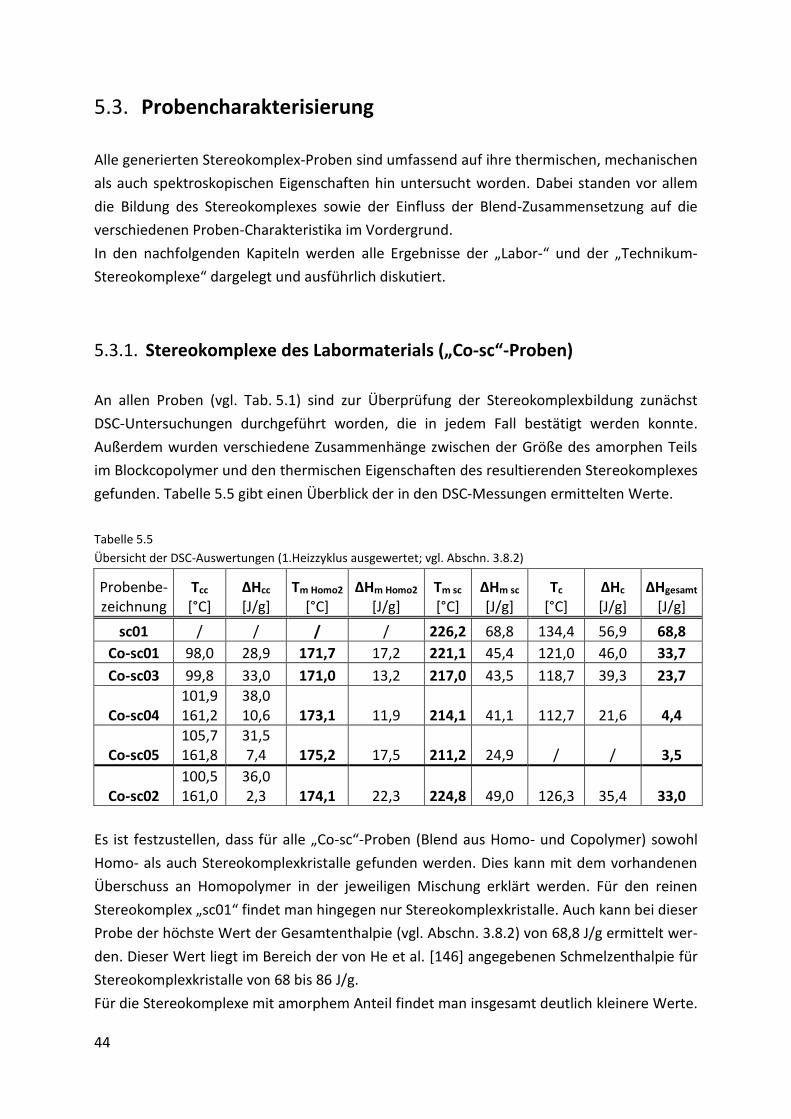
5.3.1. Stereokomplexe des Labormaterials („Co-sc“-Proben) ............................................. 44
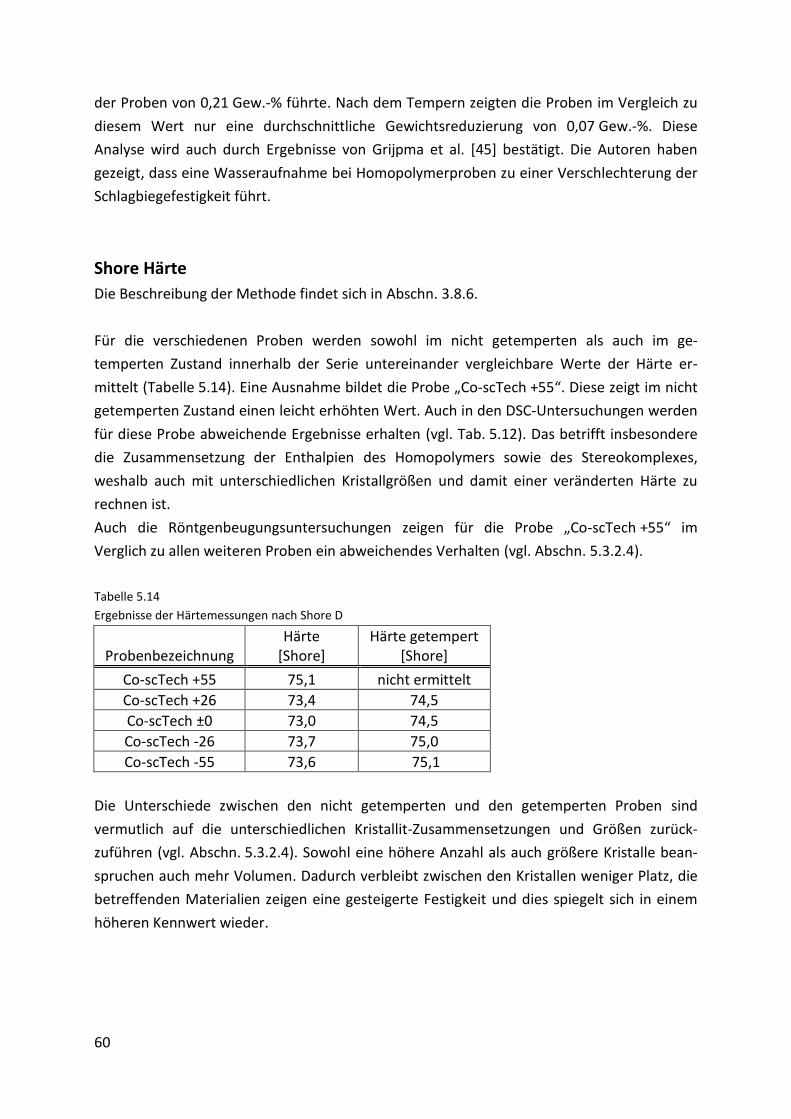
Page 5
V
5.3.1.1. Prüfstabherstellung .................................................................................................... 46
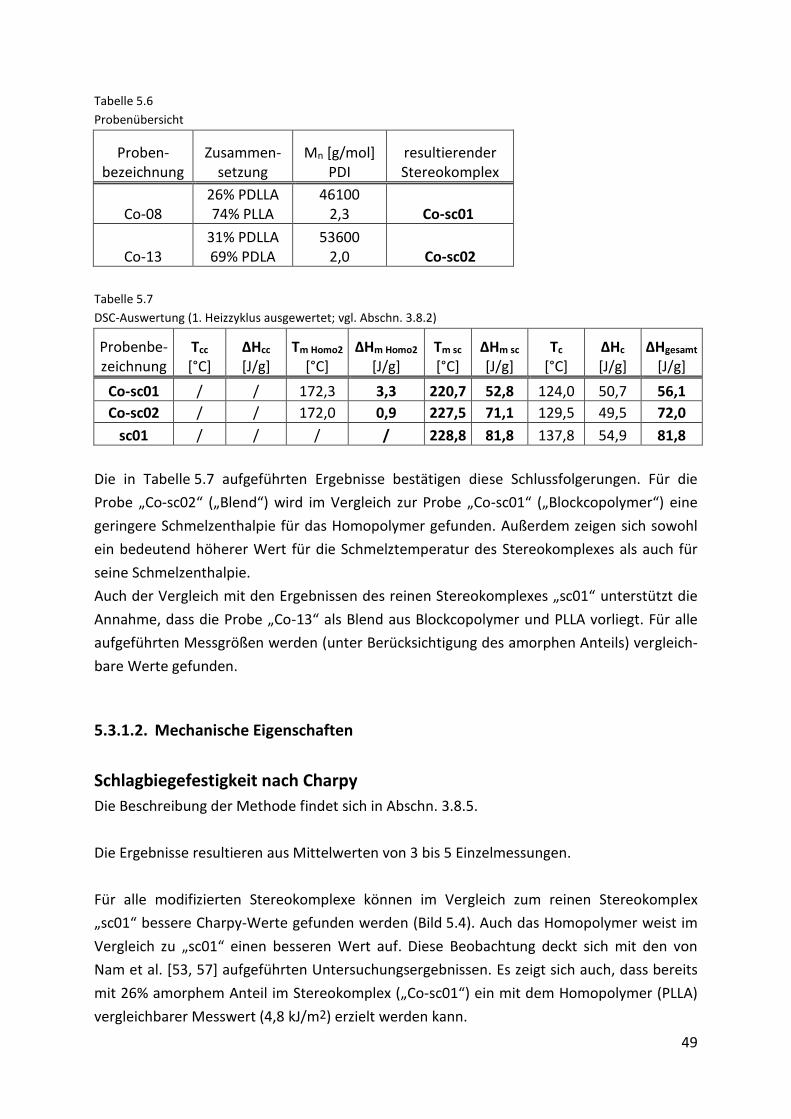
5.3.1.2. Mechanische Eigenschaften....................................................................................... 49
5.3.1.3. Röntgenbeugungsexperimente.................................................................................. 52
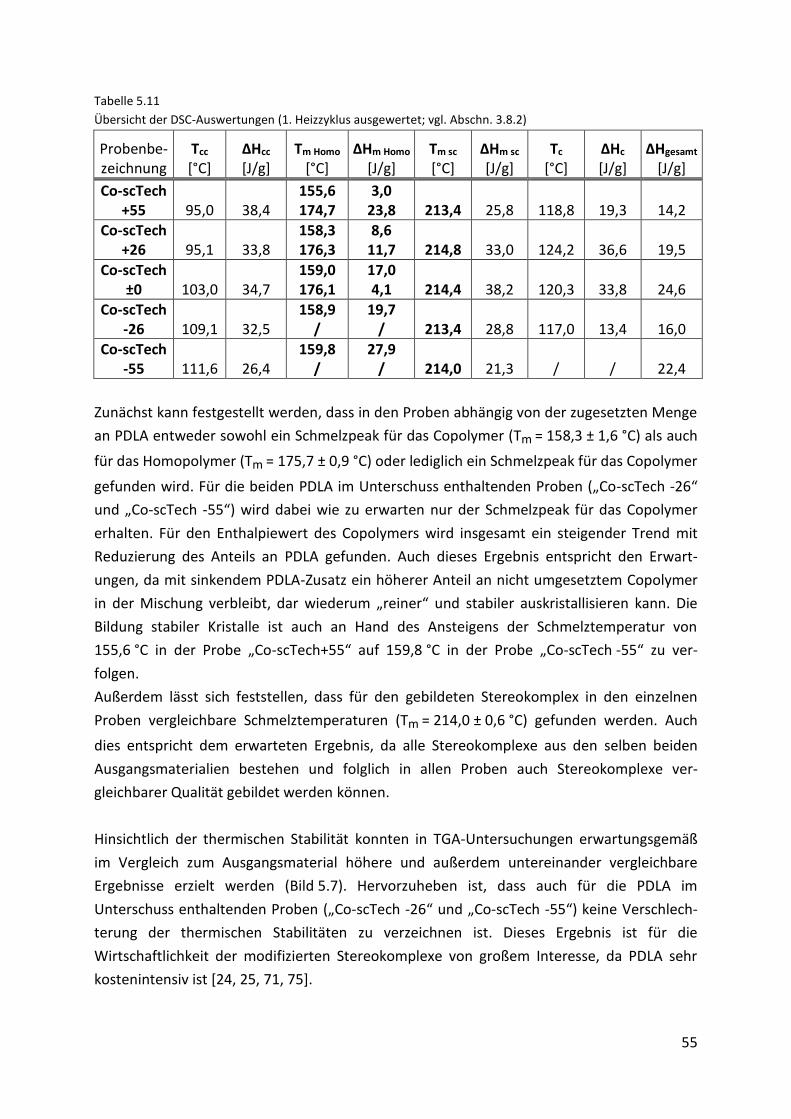
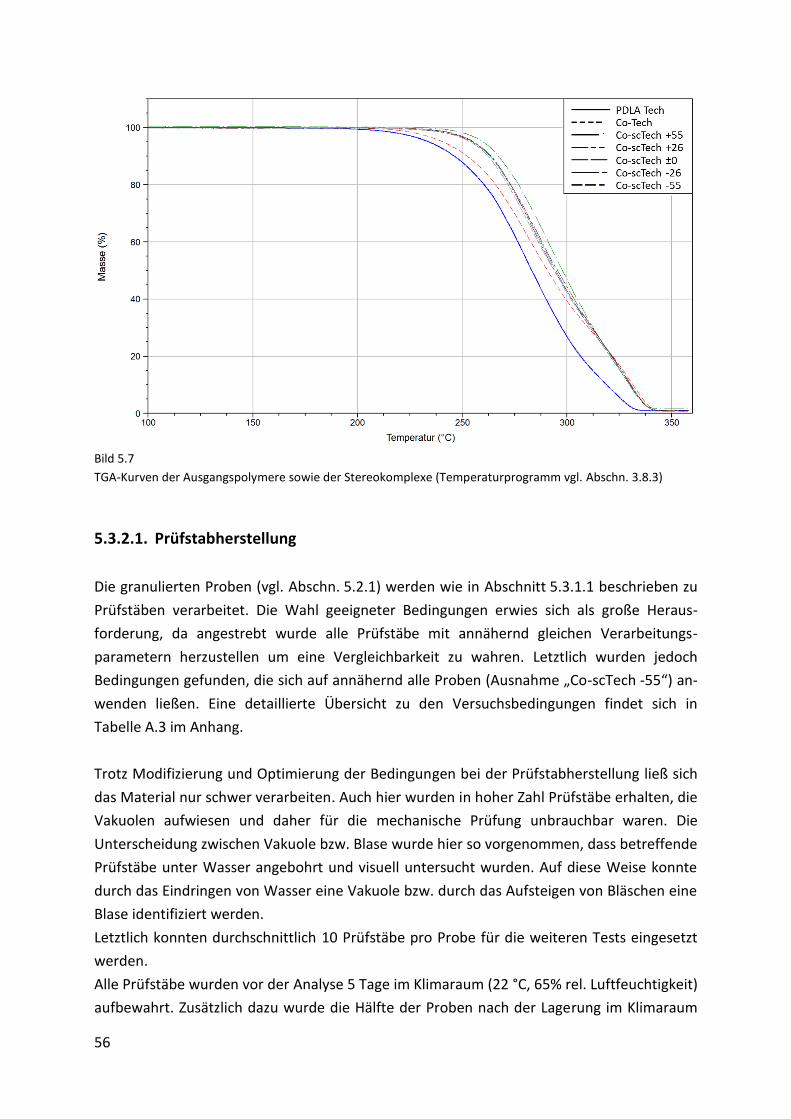
5.3.2. Stereokomplexe des Technikum-Materials (“Co-scTech“-Proben) ........................... 54
5.3.2.1. Prüfstabherstellung .................................................................................................... 56
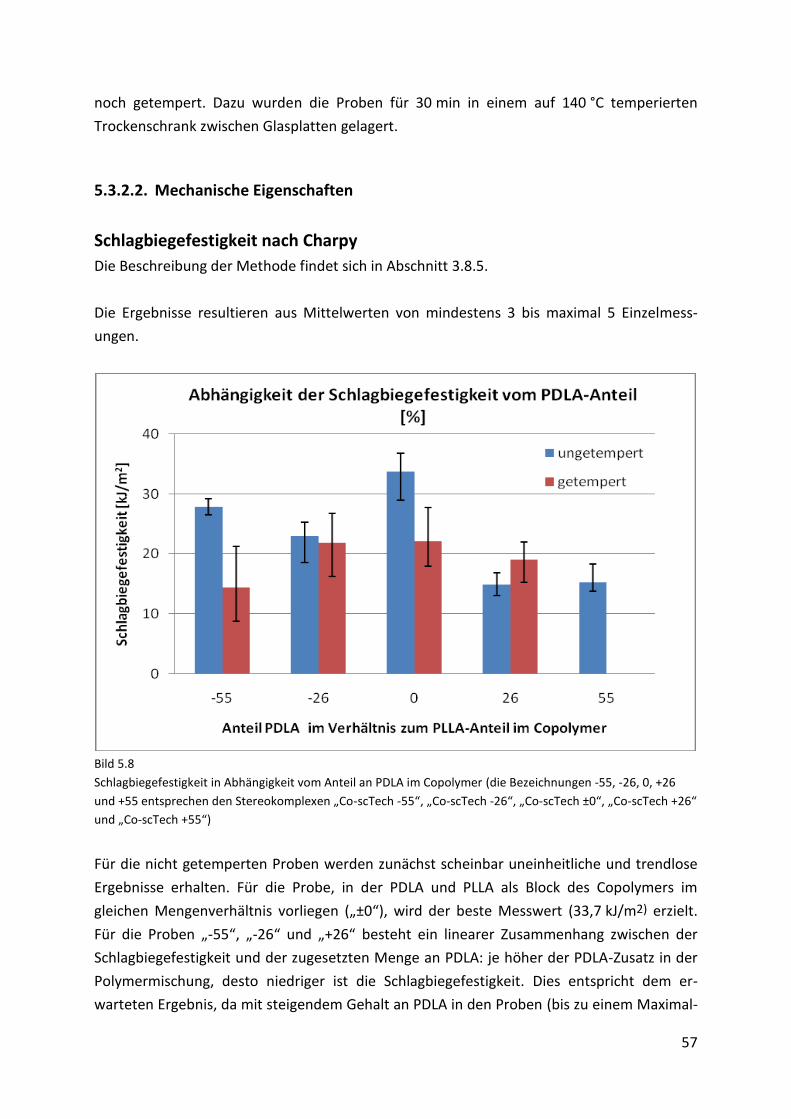
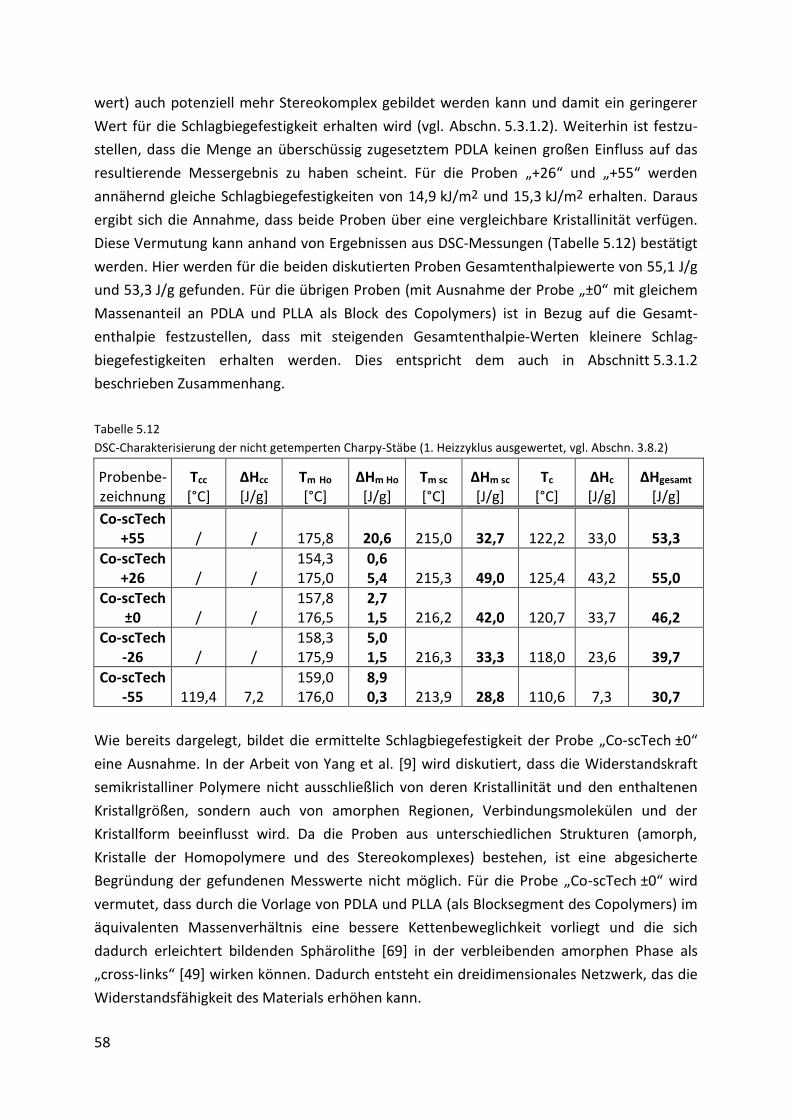
5.3.2.2. Mechanische Eigenschaften....................................................................................... 57
5.3.2.3. Thermomechanische Eigenschaften .......................................................................... 61
5.3.2.4. Röntgenbeugungsexperimente.................................................................................. 63
5.4. Gegenüberstellung des Labor- und Technikum-Materials ........................................ 65
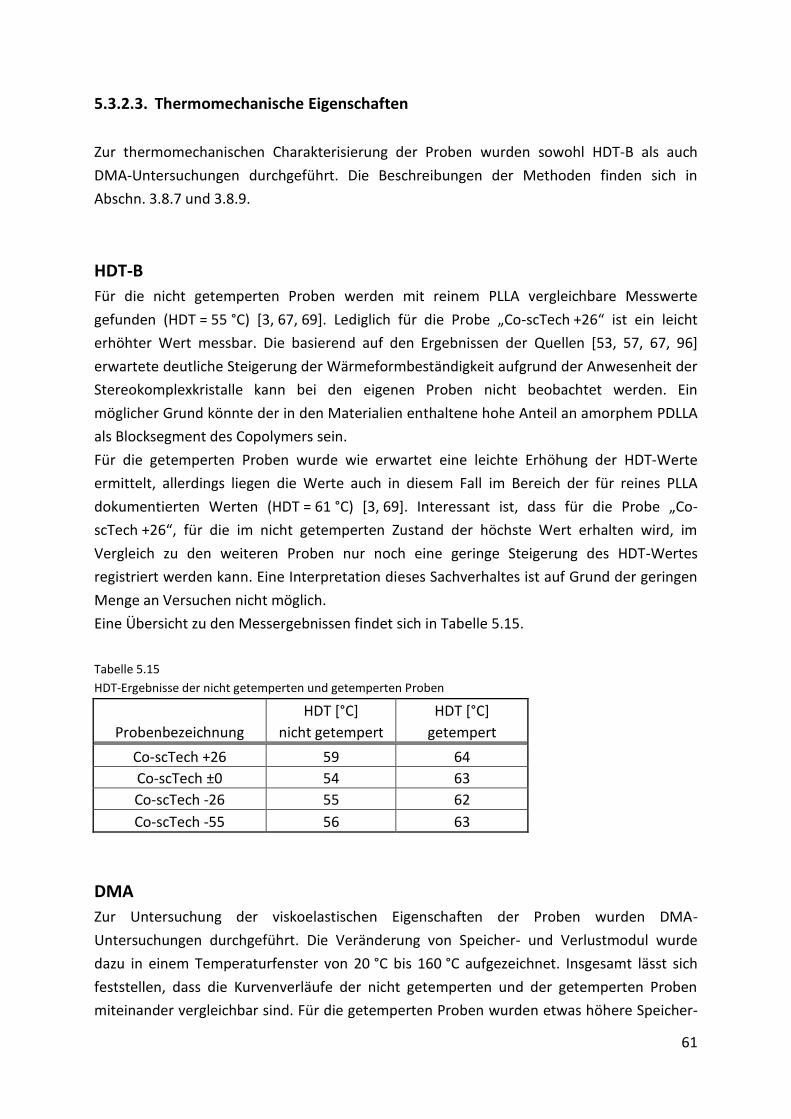
6. Abbauuntersuchungen 68
6.1. Probenvorbereitung und Pufferherstellung .............................................................. 68
6.2. Probenentnahme ....................................................................................................... 69
6.3. Charakterisierung der Abbauprodukte ...................................................................... 69
6.3.1. Visuelle Kontrolle ....................................................................................................... 70
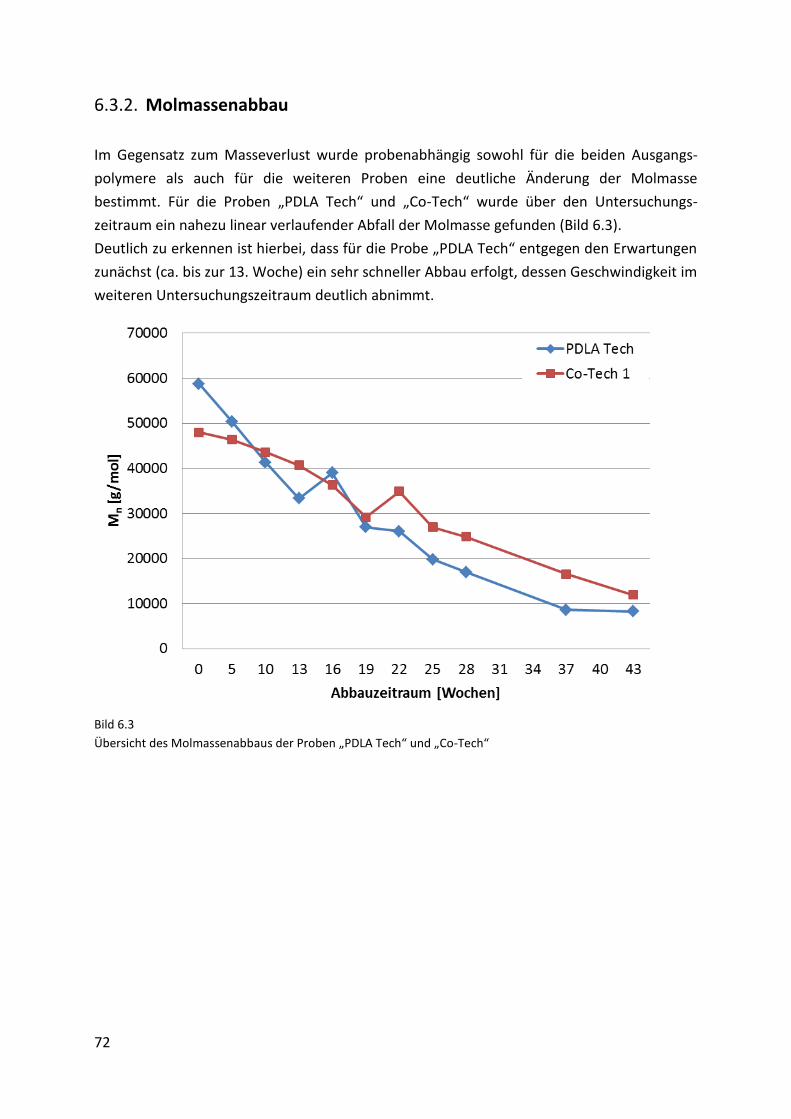
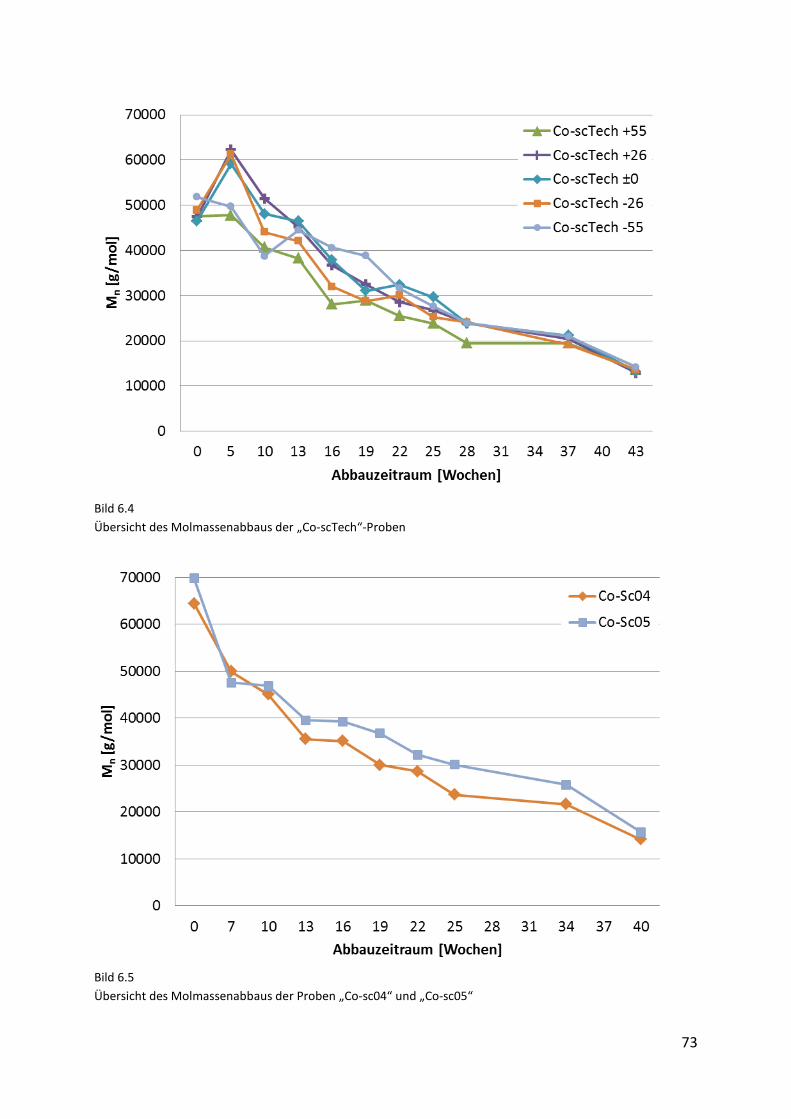
6.3.2. Molmassenabbau ....................................................................................................... 72
6.3.3. Thermische Eigenschaften ......................................................................................... 74
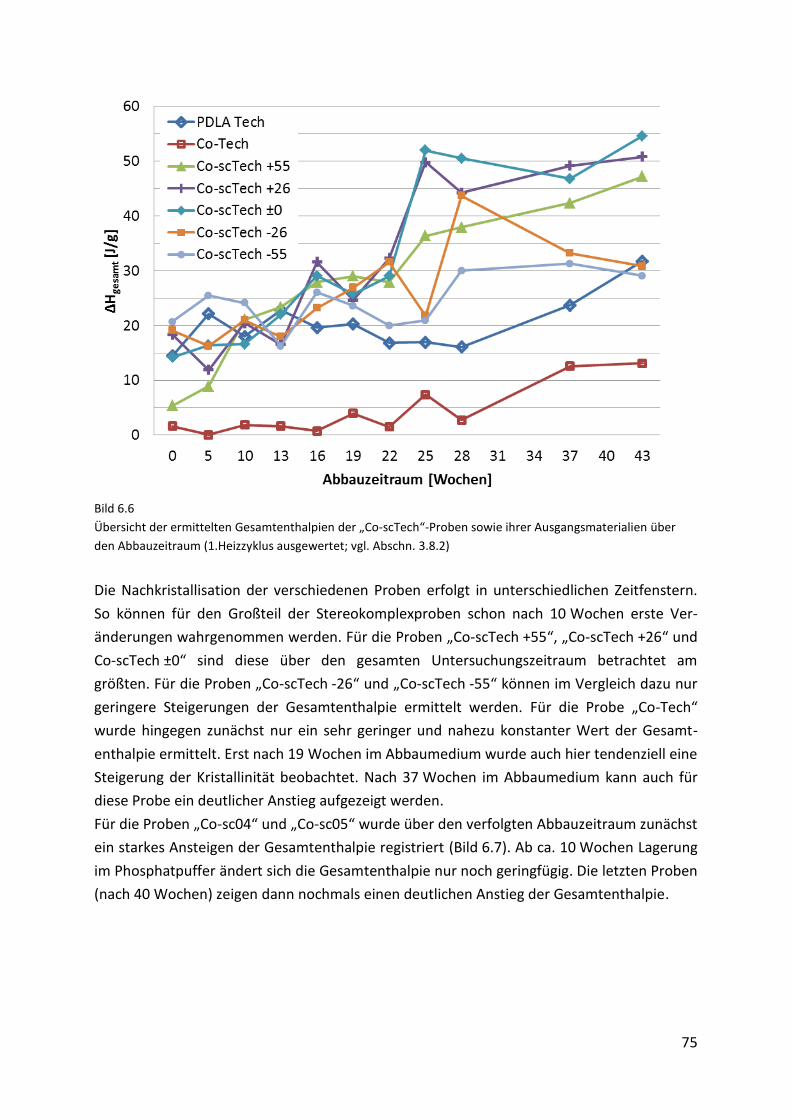
6.4. Diskussion der Ergebnisse .......................................................................................... 80
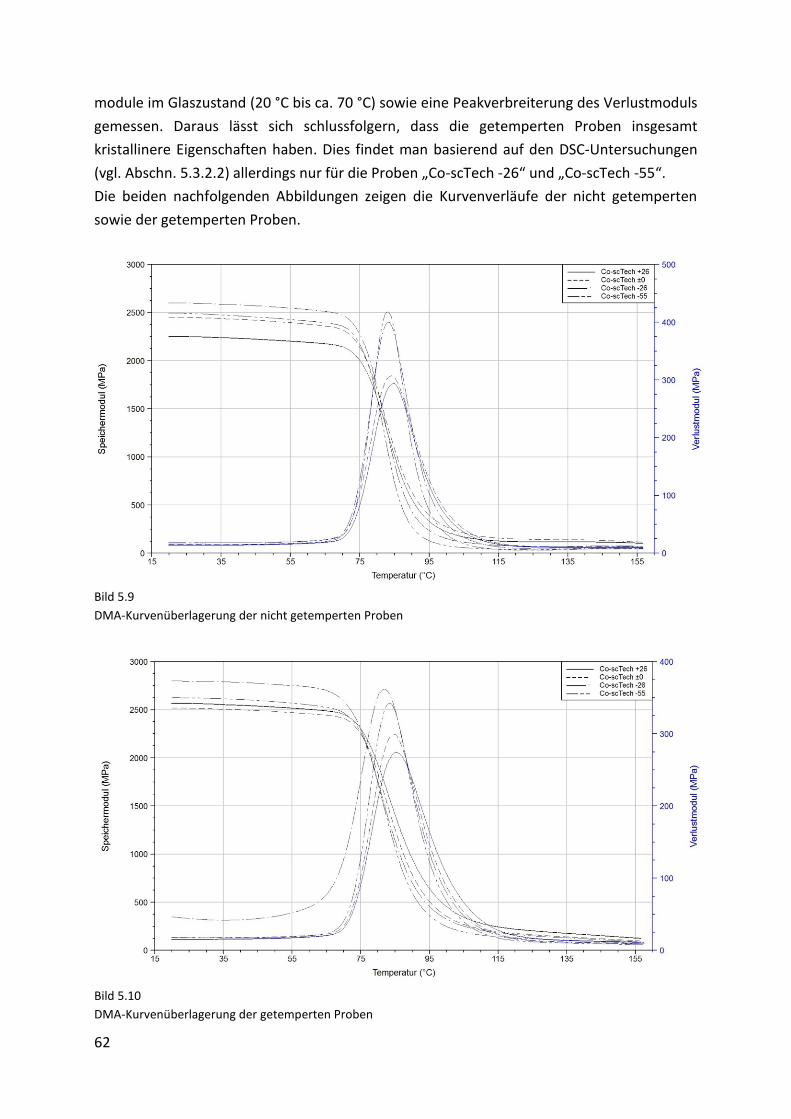
7. Partikelherstellung und -verarbeitung 82
7.1. Verfahrensoptimierung des Schmelze-Verfahrens .................................................... 82
7.2. Partikelherstellung auf Basis der optimierten Vorschrift .......................................... 84
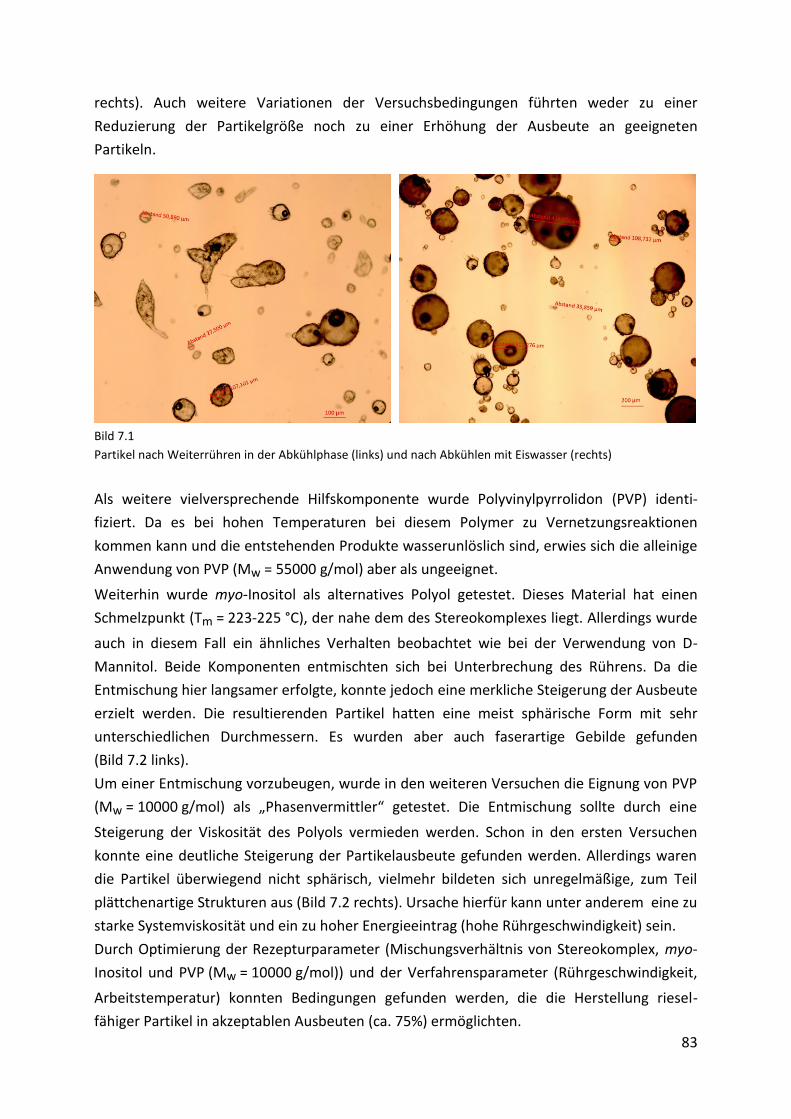
7.3. Charakterisierung der Mikropartikel ......................................................................... 85
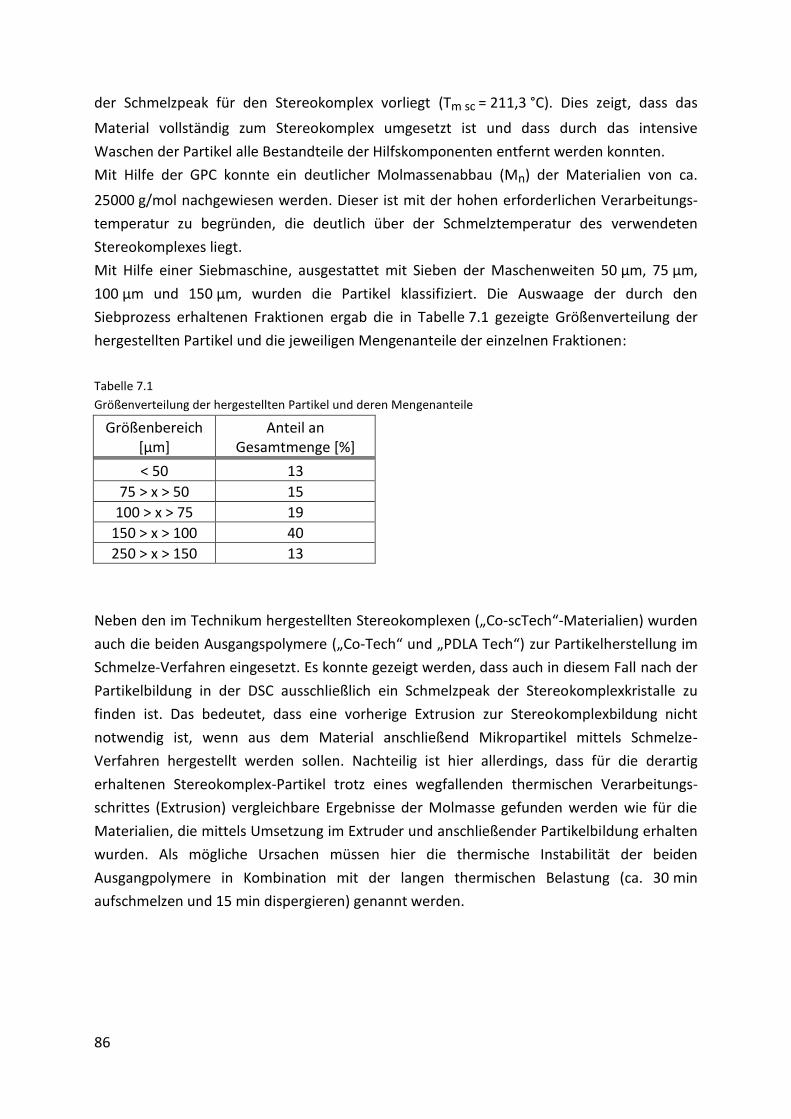
7.4. Selektives Laserschmelzen ......................................................................................... 87
7.4.1. Prototypen ................................................................................................................. 87
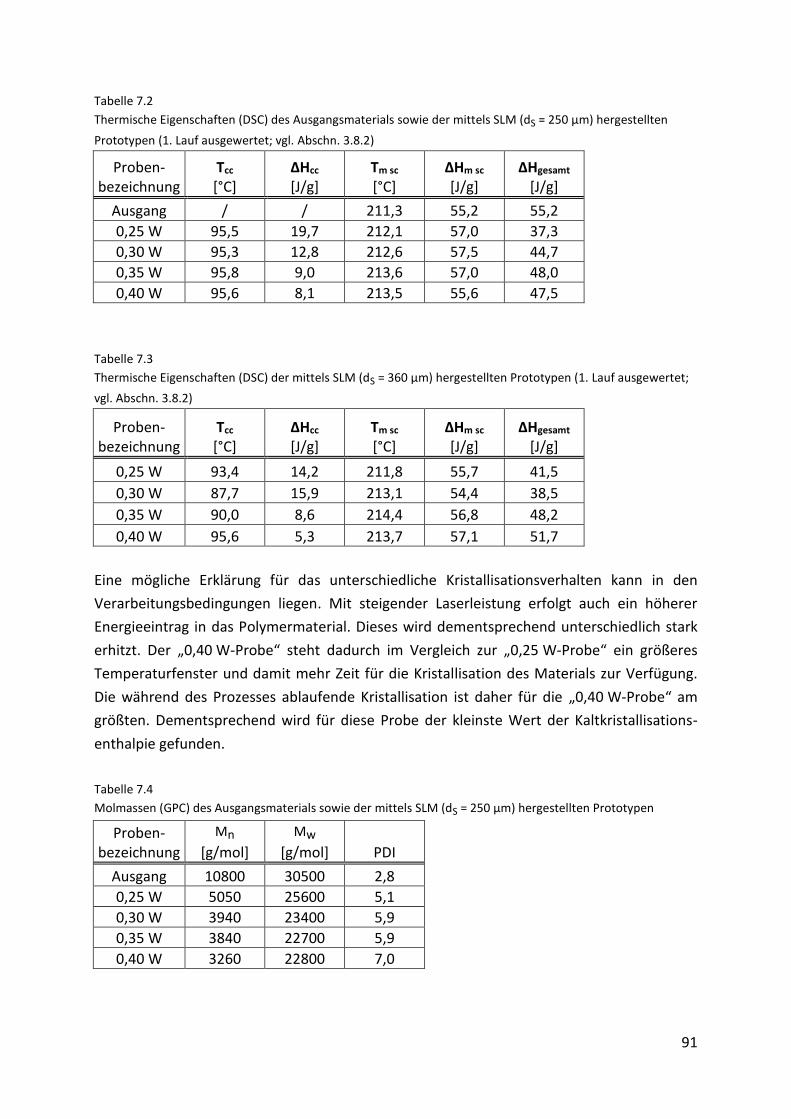
7.4.2. Charakterisierung der Prototypen ............................................................................. 90
8. Zusammenfassung 93
Literaturverzeichnis X
A. Anhang XXIV
Danksagung XXX
Page 6
VI
Abkürzungsverzeichnis
Abschn. Abschnitt
Bsp. Beispiel
bzw. beziehungsweise
ca. circa
CH2Cl2 Dichlormethan
CHCl3 Chloroform
CO2 Kohlenstoffdioxid
CT Computertomographie
d Tag
DSC Dynamische Differenzkalorimetrie
Fa. Firma
ff. folgenden
g Gramm
Gew.-% Gewichtsprozent
GG Gleichgewicht
ggf. gegebenenfalls
Gl. Gleichung
GPC Größenausschlusschromatographie
H Enthalpie
h Stunden
H2O Wasser
hc homochiral
HFIP Hexafluoroisopropanol
Hz Hertz
J Joule
Kat. Katalysator
kg Kilogramm
Page 7
VII
KOH Kaliumhydroxid
kV Kilovolt
L Liter
mA Miliampere
mbar Millibar
MeOH Methanol
μequ Mikroäquivalente
min Minuten
mL Milliliter
mm Milimeter
mol Mol
MRT Magnetresonanzthomographie
Ncm Newtonzentimeter
N2 Stickstoff
nm Nanometer
MS Milchsäure
PEG Polyethylenglykol
PDI Polydispersitätsindex
PLA Polylactid
P2O5 Phosphorpentoxid
PVA Polyvinylalkohol
PVP Polyvinylpyrrolidone
rel. relativ
ROP Ringöffnungspolymerisation
U/min Umdrehungen pro Minute
rh relative Luftfeuchtigkeit
RT Raumtemperatur
s Sekunde
sb Stereoblock
sc stereochiral
Sn(Oct)2 Zinn(II)bis-2-ethylhexanoat
Tc Kristallisationstemperatur
Tcc Kalt-Kristallisationstemperatur
Tg Glasübergangstemperatur
TGA Thermogravimetrische Analyse
Tm Schmelztemperatur
vgl. vergleiche
Vol% Volumenprozent
z.B. zum Beispiel
Page 9
1. Einleitung
Der Einsatz bioresorbierbarer Polymere wie Polylactid, Polycaprolacton und verschiedenen
Blends sowie Copolymeren daraus ist auf Grund ihres vielseitigen Anwendungsspektrums
z.B. als Schrauben, Platten, Naht- und Stentmaterial sowie ihrer Abbaubarkeit im Körper zu
biokompatiblen Produkten aus der modernen Medizin nicht mehr wegzudenken. Im Gegen-
satz zu herkömmlichem chirurgischem Befestigungsmaterial oder Implantaten entfällt
hierbei eine weitere Operation, die lediglich der Entfernung dieser dienen würde. Dem
Patienten bleiben dadurch zusätzliche eingriffbedingte Strapazen erspart und eine Unter-
brechung des Heilungsprozesses wird vermieden. Ein weiterer Vorteil liegt in der Möglichkeit
das Polymer mit unterschiedlichen Medikamenten zu beladen. So können unter anderem
z.B. Schmerzmittel oder Entzündungshemmer direkt am Einsatzort den Heilungsprozess
unterstützen und die Heilung vorantreiben.
Herkömmliche Stents werden aus Metall hergestellt. Eine Schwachstelle dieser ist die „In-
Stent Restenose“ (Wiederverschluss innerhalb des Stents). Durch Einsatz von mit Medi-
kamenten beschichteten Stents konnte dieses Risiko deutlich reduziert werden, aber andere
Probleme wie das Auftreten von Spätthrombosen wurden beobachtet. Ein weiterer ent-
scheidender Nachteil von Metallstents besteht darin, dass betroffene Patienten nicht mehr
mittels der medizinischen Standardmethoden Computertomographie (CT) und Magnet-
resonanzthomographie (MRT) untersucht werden können. Es wird über die Bildung von
Artefakten in den Aufnahmen berichtet und im Fall des MRTs kann es durch die
verwendeten Magnetfelder zu einer starken gesundheitlichen Gefährdung des Patienten
kommen. Abhilfe können hier vollständig resorbierbare Stents schaffen. Diese haben neben
der Metallfreiheit die Vorteile, dass chronische Entzündungen, Spätthrombosen und
Wiederverschlüsse vermieden werden können. Auch stabilisiert sich das betroffene Blut-
gefäß in einem notwendigen Stützzeitraum von etwa 6 bis 9 Monaten soweit, dass ein
Gerüst unnötig wird. Durch die Abbaubarkeit des Polymermaterials kann dem behandelten
Gefäß nach Vollendung der Stützfunktion seine freie physiologische Beweglichkeit wieder
zurückgegeben werden. Darüber hinaus ist es möglich das Stentmaterial über den gesamten
Querschnitt mit Medikamenten zu beladen, die dann während des Abbaus nach und nach
Page 10
2
freigesetzt werden. Ein zusätzlicher nicht zu vernachlässigender psychischer Vorteil des
abbaubaren Materials besteht darin, dass die bioresorbierbaren Stents von den Patienten
besser akzeptiert werden.
Für den medizinischen Einsatz muss das Polymermaterial einige Voraussetzungen erfüllen.
Zum einen müssen die Materialien selbst als auch die Abbauprodukte biokompatibel sein.
Auch müssen die Stabilität und die Abbaubarkeit in einem festgelegten Zeitfenster
gewährleistet sein. Zum anderen müssen Entzündungsreaktionen möglichst vermieden
werden. Für die Verwendung als Stentmaterial müssen darüber hinaus noch weitere An-
forderungen erfüllt werden. Das eingesetzte Material muss zum Beispiel einerseits stabil
sein, andererseits aber eine gewisse Flexibilität aufweisen um dem Gefäß sowohl den
notwendigen Halt geben zu können, als auch unter Belastungen nicht seine Form zu ver-
lieren. Auch muss ein Abbau innerhalb von maximal 12 Monaten gewährleistet sein.
Mit der Technologie des selektiven Laserschmelzens (SLM) steht der Polymerverarbeitung in
der jüngsten Zeit eine neue Verarbeitungstechnik zur Verfügung. Mit dieser ist es möglich
dreidimensionale Formen ohne geometrische Einschränkungen schichtweise und unter
sparsamem Einsatz des Rohmaterials herzustellen. An die zu verarbeitenden Polymere wird
dabei allerdings auch eine Reihe von spezifischen Anforderungen gestellt. So müssen diese in
einem rieselfähigen, bevorzugt sphärischen Zustand vorliegen um einen gleichmäßigen
Schichtauftrag und damit einen reibungslosen Verarbeitungsprozess sowie qualitativ
hochwertige Bauteile ohne Fehlstellen und mit guter Oberflächenbeschaffenheit garantieren
zu können. Außerdem sollten die Polymerpartikel in einem Größenbereich von etwa 25 bis
75 µm liegen um ein vollständiges Aufschmelzen des Materials mit dem scharf fokussierten
Laserstrahl in jeder Bauschicht zu gewährleisten. Neben der partikulären Struktur ist auch
eine hohe thermische Stabilität erforderlich um den Schmelzprozess ohne Verlust der
Polymereigenschaften zu durchlaufen.
Zusammenfassend betrachtet steht der Medizin mit den bioresorbierbaren Polymeren eine
gute und erprobte Materialgrundlage zur Verfügung. Unter Verwendung des SLM-Prozesses
wird es möglich z.B. Stents und andere Implantatgeometrien ohne räumliche Restriktionen
passgenau herzustellen. Limitierend für diesen Prozess sind lediglich die Eigenschaften der
eingesetzten Polymermaterialen. Die fortwährende Neu- und Weiterentwicklung sowie die
intensive Eigenschaftscharakterisierung resorbierbaren Polymermaterialien sind aus diesem
Grund aktuell und auch zukünftig von großem Interesse.
Page 11
3
2. Aufgabenstellung und Lösungskonzept
Das Ziel der vorliegenden Arbeit besteht darin, ein thermisch stabiles und flexibles
Polymermaterial auf Polylactidbasis für medizinische Anwendungen zu entwickeln und in der
Verarbeitung zu erproben. Die Materialentwicklung wird dazu zunächst im Labormaßstab
durchgeführt und anschließend die Übertragbarkeit der Syntheseprozesse in den Tech-
nikum-Maßstab untersucht. Geeignete Polymere werden darüber hinaus mit einem inner-
halb der Arbeit weiterentwickelten lösungsmittelfreien Verfahren zu Mikropartikeln ver-
arbeitet, um anschließend mittels Selektivem Laserschmelzen zu dreidimensionale Struk-
turen aufgebaut zu werden. Die Verarbeitung mittels Laserschmelzprozess wird am
Fraunhofer Institut für Lasertechnik (Aachen) durchgeführt.
Darüber hinaus werden alle hergestellten Materialien sowie die mittels SLM-Prozess
generierten Modelle umfassend charakterisiert sowie ausgewählte Proben hinsichtlich ihres
Abbauverhaltens analysiert.
Die Verbesserung der thermischen Stabilität wird durch die Bildung von Stereokomplexen
(PLLA/PDLA) angestrebt. Da PLA-Stereokomplexe aber durch eine sehr hohe Sprödigkeit
charakterisiert sind [53, 57], liegt der Forschungsansatz darin, zur Erhöhung der Flexibilität
des Materials eines der beiden Homopolymere (PLLA) durch ein Blockcopolymer aus PDLLA
und PLLA zu ersetzen und so einen internen Weichmacher in das System einzubringen.
Hierbei soll sowohl die Länge des amorphen Blocks als auch das Mischungsverhältnis von
Homo- und Blockcopolymer variiert werden, um den Einfluss dieser Größen auf die
resultierenden Materialeigenschaften zu bestimmen.
PDLLA wird als Flexibilität gebende Komponente in die Konzeption aufgenommen, da so ein
vollständig PLA-basiertes Material generiert wird und bei der Weiterverarbeitung zum
Stereokomplex eine gute Mischbarkeit vorausgesetzt werden kann.
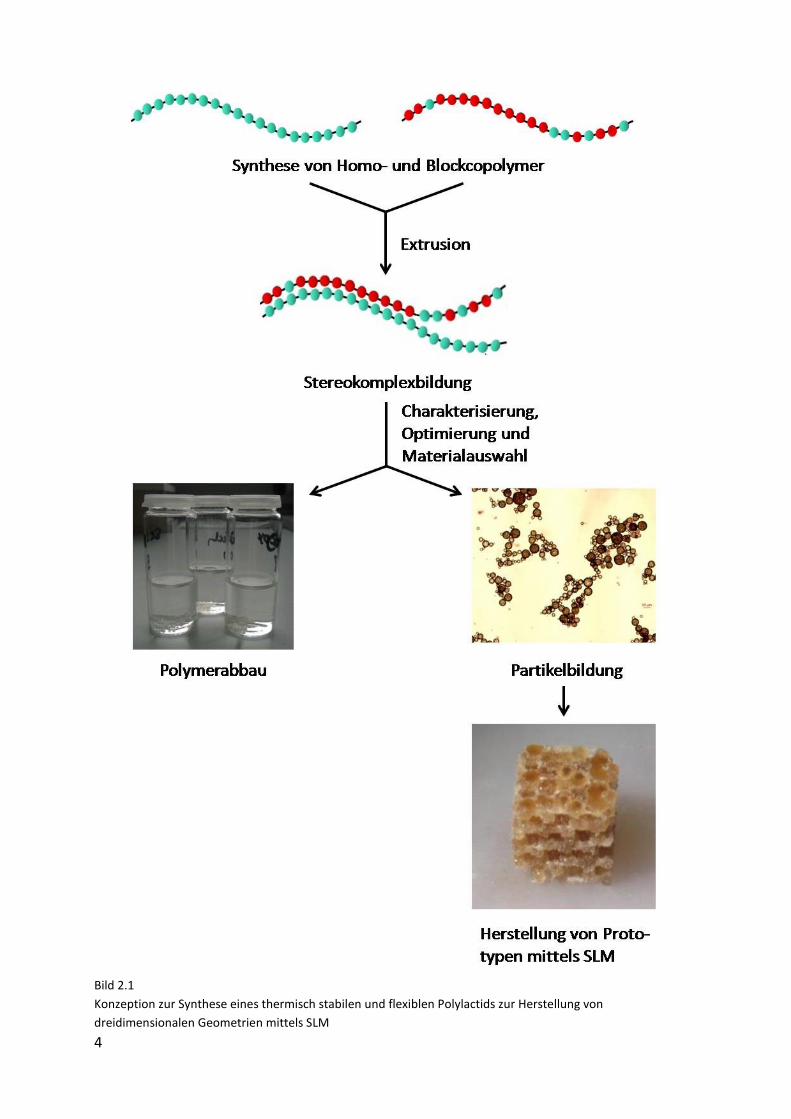
Das Lösungskonzept sowie die Vorgehensweise sind in Bild 2.1 nochmals schematisch
dargestellt.
Page 12
4
Bild 2.1
Konzeption zur Synthese eines thermisch stabilen und flexiblen Polylactids zur Herstellung von
dreidimensionalen Geometrien mittels SLM
Page 13
5
3. Grundlagen und Methoden
3.1. Polylactid (PLA)
Polylactid (PLA) ist ein bioabbaubares und biokompatibles Polymer, das vollständig aus
jährlich nachwachsenden Rohstoffen (z. B. Korn, Zuckerrohr, Zuckerrüben, Mais, Maniok)
hergestellt werden kann [1-28]. Es ist kein natürlich vorkommendes Polymer, sondern wird
synthetisch aus Monomerbausteinen, die aus nachwachsenden Rohstoffen überwiegend
biotechnologisch erzeugt werden, hergestellt [29, 30].
Auf Grund seiner Abbaubarkeit und Biokompatibilität (Abbau zu nicht toxischen Produkten)
sowie seiner mit weiteren synthetischen Polymeren ähnlichen Eigenschaften (Tranzparenz,
thermische/ mechanische Eigenschaften) besitzt PLA ein breites Anwendungsspektrum. So
wird es zum einen im medizinischen Bereich beispielsweise als Naht- [1, 6, 11, 28, 30-36] und
Stentmaterial [37-43], für Schrauben/Nägel, Implantate [11, 13, 18, 32, 34, 44-47] oder für
Drug Delivery Systeme [1, 3, 6, 11-13, 18, 28, 30, 32-34, 37, 46-52] verwendet, zum anderen
kommt es im industriellen Bereich als Verpackungsmaterial [1, 4, 7, 10, 13, 19, 20, 22, 46, 51,
53-55] wie z. B. Becher, Flaschen, Folien [3], Tüten, als Papierbeschichtung [1, 4], in der
Autoindustrie [7, 53, 56, 57] oder in Form von Fasern zur Textilherstellung [1, 3, 7, 13, 18, 19]
zum Einsatz. Für die Verarbeitung von PLA kann eine Vielzahl an unterschiedlichen
Methoden genutzt werden. Dazu zählen das Spritzgießen, die Folienextrusion, die Film-
bildung, das Blasformen, das Thermoformen und das Faserspinnen [9, 15, 33, 58]. In den
letzten Jahren wird auch das Selektive Laserschmelzen (SLM) als Verarbeitungsmethode
genutzt [41].
Um den Anforderungen der verschiedenen Einsatzgebiete und Verarbeitungsmethoden an
das Polymer gerecht zu werden, wird zur Einflussnahme der Eigenschaften des Endprodukts
zum einen auf die Copolymerisation und zum anderen auf das Blenden unterschiedlicher
Polymere zurückgegriffen. So ist es möglich Materialien zu erhalten, die für ihre Anwendung
und Verarbeitungsform maßgeschneidert sind.
Page 14
6
Durch das breite Anwendungs- und Verarbeitungsspektrum wird PLA als vielversprechende
Alternative zu erdölbasierten Polymeren gesehen. Die noch verbleibenden Fossilen-Res-
sourcen könnten so geschont und die Abhängigkeit von diesen verringert werden. Außerdem
würde die Umwelt durch die Reduzierung alternativer, erdölbasierter Plastikmaterialien und
der CO2-Emissionen deutlich entlastet werden [5-8, 15, 20-22, 25, 26, 37, 50, 56, 59-67].
Wesentliche Nachteile von PLA sind eine geringe thermische Stabilität, eine lange Kristal-
lisationszeit [2, 9, 13, 17, 19, 25, 27, 29, 57, 58, 68-71] sowie eine hohe Sprödigkeit [6, 9, 10,
28, 62, 53, 72]. Daraus resultieren nicht nur Beschränkungen in der Anwendung, sondern
auch in der Verarbeitung. Dies zeigt sich z.B. deutlich bei der Verarbeitung mittels Spritz-
gießen. Hier kann es nicht nur zum thermisch bedingten Molmassenabbau während des
Prozesses kommen, auch werden auf Grund der geringen Kristallisationsgeschwindigkeit
lange Zykluszeiten benötigt. Dadurch wird die Wirtschaftlichkeit dieser Herstellungsroute
stark negativ beeinflusst. Eine Möglichkeit, sowohl die thermische Stabilität zu verbessern
als auch die Kristallisationszeit zu verkürzen liegt in der Verwendung von Stereokomplexen
[8, 14, 24, 25, 62, 65, 70, 73-75]. Diese weisen zum einen eine höhere Schmelztemperatur im
Vergleich zu den Homopolymeren auf (vgl. Abschn. 3.2) und zum anderen fungieren sie als
Nukleierungsmittel für die Homopolymere (vgl. Abschn. 5.3.1). Zur Erhöhung der häufig
unzureichenden Materialflexibilität können z.B. Weichmacher (vgl. Abschn. 3.3) eingesetzt
werden [6, 9, 10].
3.2. Stereokomplexe
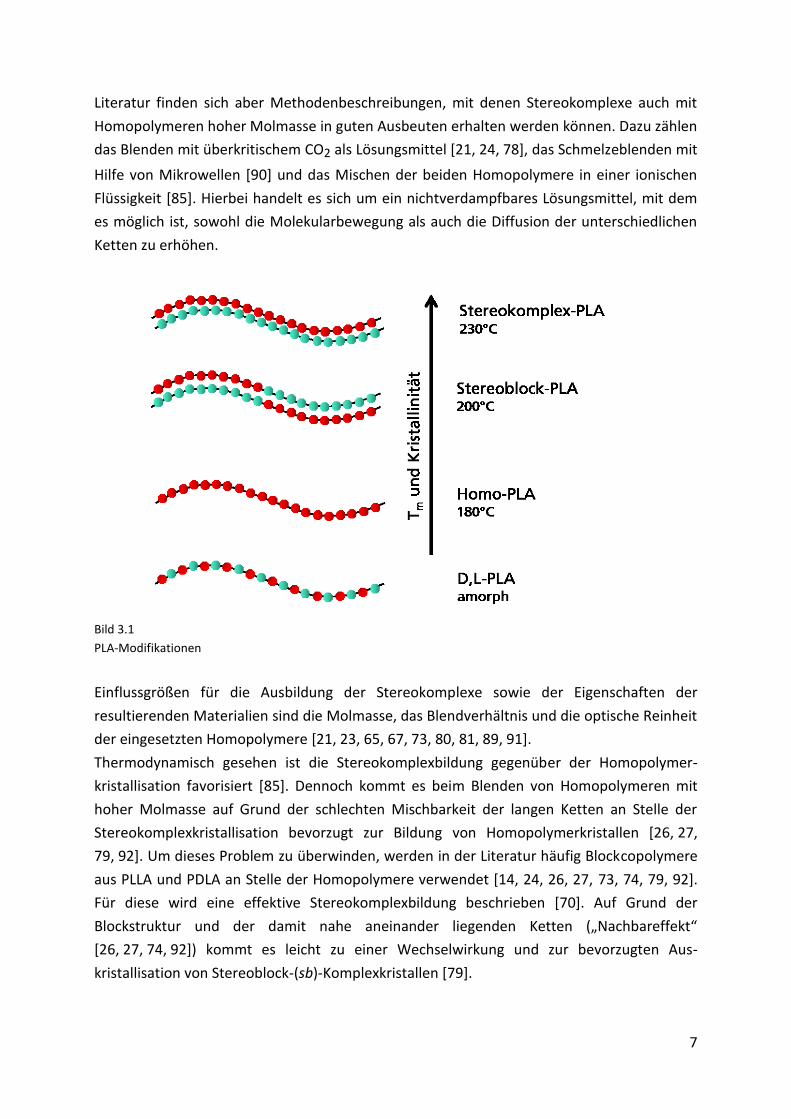
Erstmals berichtete Ikada et al. [76] im Jahre 1987 über die Bildung von PLA-Stereokom-
plexen. Zur Stereokomplexbildung werden PLLA und PDLA geblendet. Die PLA-Komplexe
besitzen einen Schmelzpunkt, der 50 °C (Tm sc = 230 °C) über dem der Homopolymere liegt
[3, 14, 20, 21, 23, 27, 28, 47, 51, 53, 67, 72, 73, 77-84]. Außerdem wird auf Grund der inne-
ren Struktur eine höhere Kristallinität und damit eine gesteigerte thermische als auch
Hydrolysestabilität beschrieben.
Die Ursache hierfür ist, dass sich die enantiomere Form der PLLA- und PDLA-Ketten
besonders dicht aneinander lagern können [70, 72, 81, 85-87] (Bild 3.1 oben). Dadurch
kommt es im Vergleich zu den Homopolymeren zur Ausbildung einer höheren Anzahl an
Wasserstoffbrückenbindungen, die außerdem einen kürzeren Abstand besitzen [51]. Zusätz-
lich dazu wirken ausgeprägte Dipol-Dipol-Wechselwirkungen in Form von stereoselektiven
van der Waals-Kräften [21, 28, 51, 57, 80, 81, 88, 89].
Das Blenden der beiden Ausgangsstoffe kann über unterschiedliche Wege erfolgen. Dazu
zählen zum Beispiel das Blenden in der Schmelze oder das Mischen in Lösung mit
anschließender Verdampfung des Lösungsmittels. Mit diesen Methoden können allerdings
nur für Homopolymere geringer Molmasse gute Ergebnisse erzielt werden. In der jüngeren
Page 15
7
Literatur finden sich aber Methodenbeschreibungen, mit denen Stereokomplexe auch mit
Homopolymeren hoher Molmasse in guten Ausbeuten erhalten werden können. Dazu zählen
das Blenden mit überkritischem CO2 als Lösungsmittel [21, 24, 78], das Schmelzeblenden mit
Hilfe von Mikrowellen [90] und das Mischen der beiden Homopolymere in einer ionischen
Flüssigkeit [85]. Hierbei handelt es sich um ein nichtverdampfbares Lösungsmittel, mit dem
es möglich ist, sowohl die Molekularbewegung als auch die Diffusion der unterschiedlichen
Ketten zu erhöhen.
Bild 3.1
PLA-Modifikationen
Einflussgrößen für die Ausbildung der Stereokomplexe sowie der Eigenschaften der
resultierenden Materialien sind die Molmasse, das Blendverhältnis und die optische Reinheit
der eingesetzten Homopolymere [21, 23, 65, 67, 73, 80, 81, 89, 91].
Thermodynamisch gesehen ist die Stereokomplexbildung gegenüber der Homopolymer-
kristallisation favorisiert [85]. Dennoch kommt es beim Blenden von Homopolymeren mit
hoher Molmasse auf Grund der schlechten Mischbarkeit der langen Ketten an Stelle der
Stereokomplexkristallisation bevorzugt zur Bildung von Homopolymerkristallen [26, 27,
79, 92]. Um dieses Problem zu überwinden, werden in der Literatur häufig Blockcopolymere
aus PLLA und PDLA an Stelle der Homopolymere verwendet [14, 24, 26, 27, 73, 74, 79, 92].
Für diese wird eine effektive Stereokomplexbildung beschrieben [70]. Auf Grund der
Blockstruktur und der damit nahe aneinander liegenden Ketten („Nachbareffekt“
[26, 27, 74, 92]) kommt es leicht zu einer Wechselwirkung und zur bevorzugten Aus-
kristallisation von Stereoblock-(sb)-Komplexkristallen [79].
Page 16
8
3.3. Weichmacher
Bei Weichmachern handelt es sich verallgemeinert um Zusatzstoffe, die Polymere flexibler
und elastischer machen und die Sprödigkeit reduzieren sollen. Nach dem Wirkprinzip wird in
innere und äußere Weichmacher unterschieden. Bei der inneren Weichmachung sind die
eingesetzten Strukturen Teil der Polymermatrix. Über kovalente Bindungen sind sie in Form
von Co- oder Graftpolymeren mit dem Orginalpolymer verbunden. Bei der äußeren Weich-
machung werden Weichmacher und Polymermatrix miteinander geblendet. Es liegt daher
keine kovalente Bindung vor. Die Eigenschaftsänderung erfolgt hier auf Grund von physi-
kalischen Wechselwirkungen zwischen Weichmacher und Polymermatrix [93, 94].
Unabhängig vom Wirkprinzip wird durch den zur Polymermatrix zugesetzten Weichmacher
der Molekülabstand vergrößert. Dadurch kommt es zu einer gesteigerten Flexibilität der
Polymerketten, die eine Deformation des Materials erlaubt, ohne dieses zu zerstören. Neben
den veränderten mechanischen Eigenschaften kann auch die Absenkung der Glas-, Kristal-
lisations- und gegebenenfalls der Schmelztemperatur beobachtet werden [6, 93-95].
Erprobte Weichmacher für PLA sind Polyethylenglykole (PEG), Glucose-Monoester, Citrat-
ester oder Oligomere der Michsäure [6, 94].
3.4. PLA-Synthese
Ausgangspunkt aller entwickelten und in dieser Arbeit vorgestellten sowie diskutierten Syn-
thesestrategien (vgl. Kapitel 4) ist die Ringöffnungspolymerisation (ROP) von Lactid. Diese
Syntheseroute ist die in der Industrie nahezu ausschließlich angewandte [59] und mit
Zinn(II)bis-2-ethylhexanoat (Zinnoktoat, Sn(Oct)2, Bild 3.2) als Katalysator die am intensiv-
sten studierte [64].
O
O
-O
O
Sn2+
Bild 3.2
Initiator Zinnoktoat
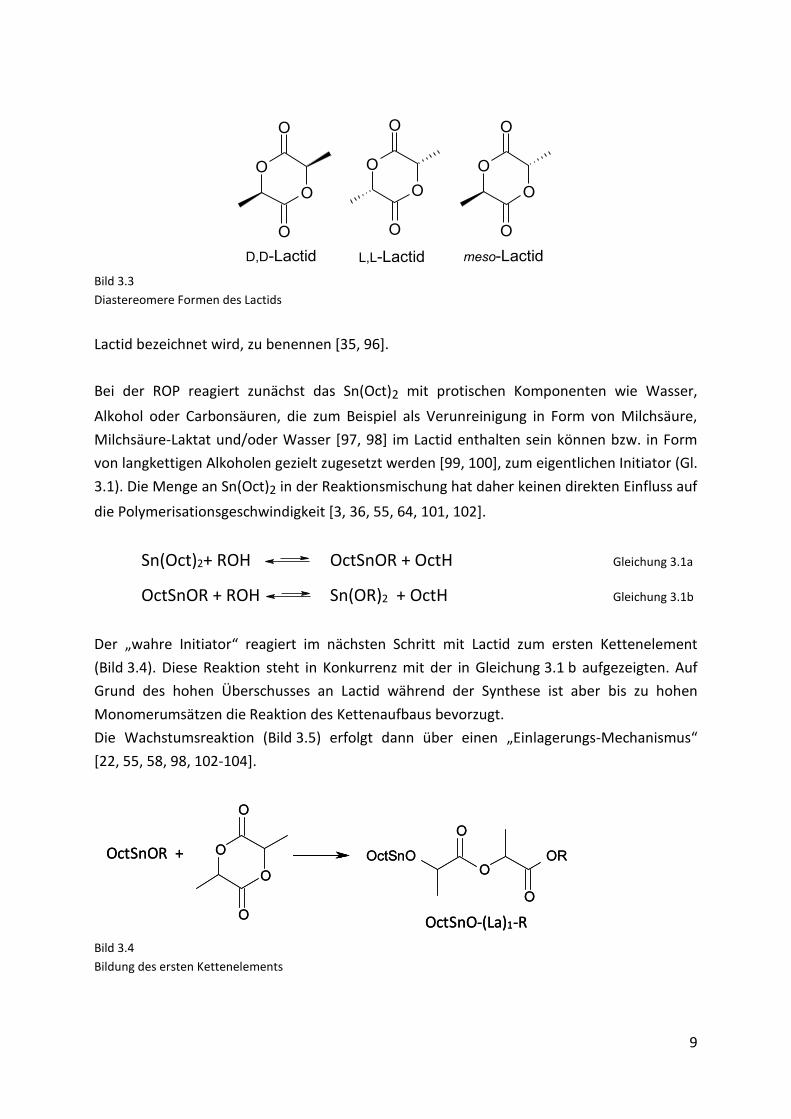
Bei der ROP wird Lactid, das zyklische Dimer der Milchsäure, als monomere Einheit
verwendet. Dieses kann auf Grund seiner zwei asymmetrischen Kohlenstoffe in drei unter-
schiedlichen diastereomeren Formen vorliegen: D,D-Lactid, L,L-Lactid und meso-Lactid
(Bild 3.3). Weiterhin ist noch die racemische Mischung aus L,L- und D,D-Lactid, die als D,L-
Page 17
9
O
O
O
O
O
O
O
O
O
O
O
O
D,D-Lactid L,L-Lactid meso-Lactid Bild 3.3
Diastereomere Formen des Lactids
Lactid bezeichnet wird, zu benennen [35, 96].
Bei der ROP reagiert zunächst das Sn(Oct)2 mit protischen Komponenten wie Wasser,
Alkohol oder Carbonsäuren, die zum Beispiel als Verunreinigung in Form von Milchsäure,
Milchsäure-Laktat und/oder Wasser [97, 98] im Lactid enthalten sein können bzw. in Form
von langkettigen Alkoholen gezielt zugesetzt werden [99, 100], zum eigentlichen Initiator (Gl.
3.1). Die Menge an Sn(Oct)2 in der Reaktionsmischung hat daher keinen direkten Einfluss auf
die Polymerisationsgeschwindigkeit [3, 36, 55, 64, 101, 102].
Sn(Oct)2+ ROH OctSnOR + OctH Gleichung 3.1a
OctSnOR + ROH Sn(OR)2 + OctH Gleichung 3.1b
Der „wahre Initiator“ reagiert im nächsten Schritt mit Lactid zum ersten Kettenelement
(Bild 3.4). Diese Reaktion steht in Konkurrenz mit der in Gleichung 3.1 b aufgezeigten. Auf
Grund des hohen Überschusses an Lactid während der Synthese ist aber bis zu hohen
Monomerumsätzen die Reaktion des Kettenaufbaus bevorzugt.
Die Wachstumsreaktion (Bild 3.5) erfolgt dann über einen „Einlagerungs-Mechanismus“
[22, 55, 58, 98, 102-104].
OctSnOR +
OctSnO-(La)1-R
O
O
O
OctSnO ORO
O
O
O
OctSnOR +
OctSnO-(La)1-R
O
O
O
OctSnO ORO
O
O
O
Bild 3.4
Bildung des ersten Kettenelements
Page 18
10
OctSnO-(La)1-R + n-1 OctSnO-(La)n-RO
O
O
O
OctSnO-(La)1-R + n-1 OctSnO-(La)n-RO
O
O
O
Bild 3.5
Wachstumsreaktion
Die in der Reaktionsmischung enthaltenen protischen Komponenten wirken aber nicht
ausschließlich als Co-Initiatoren. Sie agieren in der Synthese zusätzlich als Überträgerstoffe
(„transfer agent“) und rufen damit sowohl Konvertierungs- (wachsende in ruhende Ketten;
Gl. 3.2) als auch Terminierungsreaktionen hervor (Gl. 3.3) [101].
OctSnO-(La)n-R + ROH OctSnOR+ HO-(La)n-R Gleichung 3.2
HO-(La)n-R + OctH Oct-(La)n-R + H2O Gleichung 3.3
Um hohe Molmassen zu erzielen bzw. überhaupt gezielt Einfluss auf die Molmasse nehmen
zu können, ist es daher notwendig, das Lactid vor der Synthese mittels Umkristallisation
(Labormaßstab) bzw. Rektifikation und anschließender Schmelzekristallisation (Industrie-
maßstab) aufzureinigen. Durch definierte Zugabe von protischen Komponenten in Form von
langkettigen Alkoholen (z.B. Dodecanol) bei der Synthese können dann Polymere mit maßge-
schneiderten Molmassen erzielt werden.
Da es sich bei der Wachstumsreaktion (Bild 3.5) um eine Gleichgewichtsreaktion handelt,
verbleibt immer ein geringer Teil Monomer im Polymer. Auf Grund der gesteigerten Visko-
sität der langen Polymerketten findet man für die diffusionskontrollierte Reaktion bei
Arbeitstemperaturen zwischen 180 °C und 200 °C Werte um 5% bis 7% an nicht umge-
setztem Monomer. Bei geringeren Temperaturen (unterhalb der Schmelztemperatur des
Polymers) beträgt der Gehalt noch 0,5% bis 1%. Ursache für diesen Unterschied ist das
Auskristallisieren langer Ketten. Durch die daraus resultierende Reduzierung der Viskosität
kann es wieder leichter zu einer Reaktion von Monomer und aktiver Kette kommen [97].
Diese Systemeigenschaft wird in übertragener Form bei der Festphasen-Polykondensation
genutzt (vgl. Abschn. 4.1.1).
Im Polymer verbleibende Monomerreste beeinflussen sowohl die Eigenschaften dieses als
auch die Verarbeitung negativ [58, 64, 68, 94, 95, 105-107]. Daher ist es notwendig den Rest-
monomergehalt so weit wie möglich zu senken. In der vorliegenden Arbeit wurden zwei
unterschiedliche Ansätze zur Entmonomerisierung realisiert. Zum einen wurde der Gehalt an
Restmonomer mittels Lösen (CHCl3) und Fällen (MeOH) reduziert (Labormaßstab), zum
anderen mittels Vakuum-Destillation (Technikum-Maßstab).
Page 19
11
3.5. PLA-Abbau
Die Degradation von PLA kann über unterschiedliche Wege wie z.B. thermischen Abbau,
Photooxidation, Verwitterung in der Natur, Thermooxidation bei hohen Temperaturen oder
mittels Hydrolyse erfolgen [31, 106, 108]. Der Einfluss von Enzymen auf den PLA-Abbau wird
in der Literatur kontrovers diskutiert. Einige Autoren schreiben den Enzymen einen Einfluss
zu, andere stellen diesen als zu vernachlässigen dar [4, 31, 109, 110]. Auf Grund der unein-
heitlichen Meinung der Autoren sowie der Tatsache, dass PDLA im Körper nicht meta-
bolisiert werden kann [111], soll nachfolgend nur der Mechanismus der hydrolytischen
Spaltung als Abbaumechanismus näher betrachtet werden.
Unter dem hydrolytischen Abbau von PLA versteht man das Aufspalten der Esterbindungen
durch den Angriff von Wasser [1, 109, 110, 112, 113]. Hierbei entstehen kurzkettige Abbau-
produkte, die ihrerseits wieder der Hydrolyse unterliegen, bis sie final zu wasserlöslichen
Teilstücken (z. B. Lactoylmilchsäure und Milchsäure) abgebaut werden. Der Abbau kann
dabei über zwei verschiedene Mechanismen ablaufen: der Bulk- und der Oberflächenerosion
[89, 112-115]. Die beiden Abbaumechanismen unterscheiden sich insbesondere durch die
Geschwindigkeiten der Prozesse „Wasserdiffusion in die Polymermatrix“ und „Degradations-
prozess im Polymer“ [112, 115].
Bulkerosion
Hierbei erfolgt die Diffusion des Wassers in die Polymermatrix mit einer höheren
Geschwindigkeit als der Abbau des Polymers. Dadurch kommt es zu einem Polymerabbau
innerhalb der gesamten Matrix. Charakteristisch für diesen Abbaumechanismus ist, dass das
Material über einen weiten Zeitraum hinweg sowohl seine Größe als auch seine Form behält.
Außerdem kann lange vor Eintritt eines Masseverlustes ein ausgeprägter Molmassenverlust
verfolgt werden. Nach dem Überschreiten einer kritischen Stufe kommt es letztlich zum
vollständigen Geometrieverlust des Materials [112, 115].
Oberflächenerosion
Hierbei schreitet der Abbau der Polymermatrix schneller voran als die Diffusion des Wassers
in diese erfolgen kann. Das Wasser verursacht ausschließlich an der Oberfläche Hydrolyse
und kann nicht weiter in die Polymermatrix eindringen. Bei diesem Mechanismus können die
Abbauprodukte abtransportiert werden. Dadurch verliert das Material seine Form und wird
mit fortschreitender Degradation immer kleiner. Der Kern des Materials bleibt davon
zunächst unberührt und es findet kein Molmassenabbau statt. Für den Masseverlust wird bei
diesem Mechanismus ein linearer Zusammenhang beschrieben [112, 115].
Page 20
12
Bild 3.6
Schematische Darstellung der Oberflächen- und der Bulkerosion (in Anlehnung an [112])
Weiteren Einfluss auf den Abbau haben sowohl die äußeren Bedingungen (Temperatur [1, 4,
54, 116, 117]; pH-Wert [30, 116]; ruhendes/bewegtes Medium [112]) und die Probendimen-
sion, als auch die innere Struktur (amorph/kristallin; hc/sc) und die Architektur (linear/
verzweigt [70]; Mono-/Multiblock [118]) der untersuchten Materialen (Bild 3.7). So kann bei
höheren Temperaturen des Umgebungsmediums auch ein beschleunigter Abbau verfolgt
werden. Weiterhin wird in der Arbeit von Tartakowska [112] gezeigt, dass der Abbau in
einem ruhenden Medium schneller abläuft als in einem bewegten. Der Grund hierfür liegt in
autokatalytischen Prozessen, die im Inneren des Materials ablaufen. Im Bulk ist der
Abtransport der entstehenden sauren Abbauprodukte behindert, wodurch es zu deren
Anreicherung kommt. Hieraus resultiert ein pH-Wert-Gradient, der den weiteren Abbau
katalysiert [30, 54, 109, 110, 115, 119]. Im bewegten Medium kann das Abführen der kataly-
sierend wirkenden Abbauprodukte gefördert und die Abbaugeschwindigkeit so im Vergleich
zum ruhenden Medium vermindert werden.
Bild 3.7
Einflussfaktoren des PLA-Abbaus
Page 21
13
Autokatalytische Effekte werden entscheidend von den Probendimensionen beeinflusst. In
den Arbeiten von Li et al. [30, 34, 119, 120] konnte gezeigt werden, dass bei größeren
Probenkörpern der Abbau im Kern deutlich schneller abläuft als an der Oberfläche.
Der Einfluss der Kristallinität auf den Abbau wird in einer Vielzahl von Quellen beschrieben
[4, 34, 50, 51, 78, 89, 100, 109, 112-114, 116-122, 123, 124]. Es wird eindeutig aufgezeigt,
dass der Abbau in den amorphen Teilen der Probe schneller abläuft als in den kristallinen [4,
34, 51, 78, 100, 109, 110, 112, 117, 119-122]. Weiterhin wurde der Einfluss von Stereo-
komplexkristallen auf die Abbaugeschwindigkeit des Materials untersucht. Es konnte eine
weitere Verzögerung des Abbaus im Vergleich zu Homopolymeren festgestellt werden [50,
51, 89, 116, 117, 123, 124]. Der Grund hierfür liegt im hohen Grad an Kristallinität, den
starken Wechselwirkungen zwischen den PLLA- und PDLA-Ketten (vgl. Abschn. 3.2), sowie
dem Vorliegen eines 3D-Mikronetzwerks [50, 114, 116, 123]. Die Diffusionsrate der Wasser-
moleküle in die Polymermatrix ist hierdurch gesenkt [50, 78, 116, 123] und der Abtransport
der Hydrolyseprodukte auf Grund des Mikronetzwerks gleichzeitig erleichtert. Hieraus
resultiert eine deutliche Einschränkung des autokatalytischen Effekts in der Probenmatrix
[116, 123].
Während des Abbaus kann sowohl bei den Homo- als auch bei den Sterokomplexmaterialien
eine Steigerung der Kristallinität verfolgt werden [34, 112, 113, 116-118, 120, 121, 123]. Auf
Grund der sich reduzierenden Kettenlänge und der damit einhergehenden besseren
Beweglichkeit der resultierenden Ketten kann es zu einer Neuordnung dieser und damit zu
einer erhöhten Kristallisation kommen [116, 121]. Dieser Effekt beeinflusst die Abbau-
geschwindigkeit zu einem fortgeschrittenen Zeitpunkt des Abbaus. Auf Grund der erhöhten
Kristallisation und der dadurch reduzierten Diffusionsrate der Wassermoleküle wird zum
Abbau der verbliebenen Polymerketten mehr Zeit beansprucht.
3.6. Partikelbildung
Zur Herstellung von Partikeln bzw. Mikropartikeln (Ø bis 1000 µm [115, 125]) stehen
verschiedene Methoden zur Auswahl. Dazu gehören unter anderem die Koazervation, die
Sprühtrocknung und die Lösungsmittelverdampfung [115, 125, 126]. Der große Nachteil
dieser Methoden liegt im Fall von PLA in der Notwendigkeit der Verwendung (halogenierter)
organischer Lösungsmittel zur Herstellung der Polymerlösungen. Bei Materialien für die
körpernahe Anwendung möchte man jedoch auf den Gebrauch dieser verzichten. Daher
wurde in der vorliegenden Arbeit das lösungsmittelfreie Schmelze-Verfahren zur Bildung der
Mikropartikel eingesetzt und an das Material angepasst.
Beim Schmelze-Verfahren kommen nur das Polymer selbst sowie eine wasserlösliche Hilfs-
Page 22
14
komponente zum Einsatz. Hierfür werden nach dem Stand der Technik z.B. Polyethylen-
glykole, Polyvinylalkohole oder auch Oligosaccharide verwendet. Das Polymer und die
Hilfskomponente werden gemeinsam aufgeschmolzen und in der Schmelze miteinander
dispergiert. Beim Abkühlen werden die entstandenen Polymertröpfchen durch die
umgebende Matrix der Hilfskomponente stabilisiert und können nach dem vollständigen
Erkalten der Dispersion durch das Lösen des Polyols gewonnen werden. Die Polymerpartikel
werden abschließend mittels Filtration von der Lösung abgetrennt, gründlich mit Wasser
gewaschen und getrocknet [126, 127].
3.7. Selektives Laserschmelzen
Das selektive Laserschmelzen (SLM) zählt zu den Rapid-Manufacturing-Methoden. Eingesetzt
wurde es zunächst für die Verarbeitung von Metallpulvern [128]. In der neueren Zeit ist aber
auch die Verarbeitung von Polymeren von zunehmendem Interesse. Für den Prozess werden
rieselfähige Materialien mit bevorzugten Korngrößen von ca. 25 bis 50 µm benötigt. Aus
diesen können mit Hilfe des schichtweisen Aufbau-Prozesses komplexe dreidimensionale
Objekte hergestellt werden.
Bild 3.8
Aufbringen der Partikel-Schicht auf einen Träger (links) und selektives Aufschmelzen mittels Computerpro-
gramm berechneter Punkte (rechts) [162]
Bei dem SLM wird zunächst eine gleichmäßige, etwa 50 µm starke Schicht der Material-
Partikel auf einen Träger aufgetragen (Bild 3.8 links). Im nächsten Schritt werden die mit
Hilfe eines Computers errechneten Punkte/Formen mittels eines Lasers selektiv aufge-
schmolzen (Bild 3.8 rechts). Im Anschluss wird die Arbeitsfläche um den Betrag der
gewünschten Schichtdicke abgesenkt und eine neue Pulverschicht aufgetragen. Durch Auf-
schmelzen der für die nächste Schicht berechneten Punkte verbinden sich die beiden
Bauschichten und das Bauteil gewinnt an Höhe. Die Prozessschritte „Absenken der Bauplatt-
Page 23
15
form“, „Aufbringen einer neuen Pulverschicht“ und „Aufschmelzen der berechneten Punkte“
werden so lange wiederholt, bis der gewünschte Körper aufgebaut ist. Durch diese
Arbeitsweise ist es möglich einen dreidimensionalen Körper Schicht für Schicht und ohne
geometrische Einschränkungen aufzubauen.
Bild 3.9
Mögliche Einflussgrößen während der Verarbeitung mittels SLM [162]
Im Prozess können eine Vielzahl an Parametern variiert werden. Dadurch ist es möglich
einen Schmelzprozess zu realisieren, der für das gewünschte Verarbeitungsmaterial
optimiert ist. Zu den Einflussgrößen zählen die Laserleistung (PL), der Strahldurchmesser (dS)
sowie die Scangeschwindigkeit (vscan). Außerdem können auch der Spurabstand (ΔyS) und
wie schon benannt die Schichtdicke (DS) variiert werden (Bild 3.9).
3.8. Analytische Mess- und Nachweismethoden
Die zur Charakterisierung der Zwischen- und Endprodukte eingesetzten Methoden werden
nachfolgend vorgestellt.
3.8.1. GPC – Gel Permeations Chromatographie
Die GPC zählt zu den chromatographischen Methoden und wird zur Bestimmung der Mol-
Page 24
16
massenverteilung und der Polydispersität von Polymeren eingesetzt. Die Auftrennung erfolgt
aufgrund der Größenunterschiede der in der Probe enthaltenen Polymerketten. Kleinere
Moleküle werden dabei durch eine diffusionskontrollierte Reaktion stärker retardiert als
große. Bei der GPC handelt es sich um eine Relativmethode. Zur Auswertung der mittels RI-
Detektor erhaltenen Messergebnisse wird eine Universalkalibrierung mit Polystyrol als
Standard verwendet.
In der vorliegenden Arbeit wurden GPC-Systeme der Firma WGE Dr. Bures mit Dichlor-
methan (CH2Cl2) und Hexafluoroisopropanol (HFIP) als Laufmittel verwendet.
3.8.2. DSC – Differential Scanning Calorimetry
Mit Hilfe der DSC ist es möglich die thermischen Eigenschaften der Proben zu ermitteln.
Dazu zählen sowohl die Glasübergangstemperatur Tg, die Kristallisationstemperaturen Tcc
und Tc sowie die Schmelztemperaturen Tm als auch die Enthalpiewerte ΔH der jeweiligen
thermischen Ereignisse.
Bei der DSC wird die Probe in einen Aluminium-Tiegel eingewogen, gemeinsam mit einem
leeren Referenztiegel in einen Ofen gegeben und einem Temperaturprogramm unterworfen.
Dieses Temperaturprogramm beinhaltet das Erwärmen und Abkühlen der Probe mit einer
konstanten oder dynamischen Heizrate. Während des gesamten Prozesses wird die
Temperatur der beiden auf einer wärmeleitenden Metallscheibe befindlichen Tiegel
kontinuierlich gemessen. Dadurch kann die durch Kristallisations- oder Schmelzprozesse
begründete unterschiedliche Wärmeaufnahme und -abgabe von Probe und Referenz
aufgezeigt und im Thermogramm grafisch dargestellt werden.
Zum Vermessen der Proben wurden im Rahmen dieser Arbeit 2 identische Messzyklen
durchlaufen. Je nachdem, welcher Parameter im Fokus stand wurde der 1. oder der 2. Mess-
zyklus zur Bewertung der Proben herangezogen. Der 1. Messzyklus wird dabei dazu genutzt
eine Aussage über den durch die Verarbeitungsprozesse hervorgerufenen Istzustand der
Probe zu treffen. Während der anschließenden geregelten Abkühlung wird eine „neue“
Probenvorgeschichte generiert, die für alle zu charakterisierenden Proben gleich ist. Im
2. Messzyklus können aus diesem Grund vor allem materialspezifische Eigenschaften ge-
messen werden [129].
Zur besseren Vergleichbarkeit der Proben untereinander werden in der vorliegenden Arbeit
häufig nicht die Enthalpien der einzelnen thermischen Vorgänge, sondern die Gesamt-
enthalpien verwendet. Zur Ermittlung dieser subtrahiert man die Enthalpiewerte der wäh-
rend der Messung ablaufenden Kristallisation von der Summe der für die Proben ermittelten
Schmelzenthalpien. Eine allgemeine Berechnungsformel findet sich in Gleichung 3.4.
Page 25
17
Gleichung 3.4
Für die Messungen wurde ein Gerät von TA Instruments (DSC Q1000) verwendet. Alle
Proben wurden mit einer Heizrate von 10 K/min und im Stickstoffstrom vermessen. Die
verschiedenen verwendeten Temperaturprogramme finden sich im Anhang.
3.8.3. TGA – Thermogravimetrische Analyse
Mit Hilfe der TGA kann die Masseänderung einer Probe bei Durchlaufen eines Temperatur-
programms verfolgt werden. Die Veränderung der Probenmasse während des Heizprozesses
wird dabei mittels einer außerhalb des Ofens liegenden Waage registriert. Masse-
änderungen können aus unterschiedlichen Prozessen resultieren. Zu diesen Prozessen
zählen zum Beispiel: Verdampfung, Zersetzung oder chemische Reaktionen. Die Ver-
änderung der Probe kann dabei ein- oder auch mehrstufig ablaufen [129, 130].
In der vorliegenden Arbeit ist mit Hilfe der TGA die Zersetzung der hergestellten Stereo-
komplexe untersucht worden. Dazu wurde ein Gerät der Firma TA Instruments (TGA Q500)
verwendet. Alle Proben wurden mit einer Heizrate von 5 K/min in einem Temperaturberiech
von 30 °C bis 360 °C und im Stickstoffstrom vermessen.
3.8.4. Titration (Säurezahl)
Bei der Titration handelt es sich um eine Maßanalyse. Hierbei wird ganz allgemein einer
Lösung des zu untersuchenden Stoffes unbekannter Konzentration (Analyt) so viel einer
Lösung bekannten Gehalts (Titrant) zugesetzt, bis beide im Gleichgewicht liegen. Der
Gleichgewichts- bzw. Äquivalenzpunkt kann je nach System entweder durch eine chemische
Reaktion oder durch die Änderung einer physikalischen Größe identifiziert werden [131].
Hier wurde die Maßanalyse zur Ermittlung der Säurezahl der zur Polymerisation eingesetzten
Monomere (L,L-, D,D- und D,L-Lactid) verwendet. Diese reflektiert die Menge an in Form von
Verunreinigungen enthaltener Milchsäure.
Für die Messung wurden die Proben in absolutem Ethanol gelöst und in einer Säure-Base-
Tiration mit methanolischer KOH-Lösung bis zum Endpunkt titriert. Der Titer der Maßlösung
wurde mit Benzoesäure ermittelt. Für die Titrationen wurde ein automatisierter Titrierstand
der Firma Metrohm verwendet.
Page 26
18
3.8.5. Schlagbiegefestigkeit nach Charpy
Die Schlagbiegefestigkeit ist eine materialzerstörende Methode aus der Werkstoffprüfung.
Hierbei wird die zur Zerschlagung des Materials benötigte Arbeit in Abhängigkeit zum
Materialquerschnitt gemessen. Für die Messung wird ein Pendel verwendet, das mit einer
bestimmten kinetischen Energie auf die Probe trifft. Beim Auftreffen auf die Probe wird die
Energie von dieser absorbiert und das Pendel dem entsprechend abgebremst. Der
resultierende Messwert kann an Hand des vom Pendel mitgetragenen Schleppzeigers an
einer Skala abgelesen werden (Bild 3.10).
Bild 3.10
Pendelschlagwerk
Bei den eigenen Arbeiten wurde ein Pendelschlagwerk der Firma Ohst verwendet. Je nach
erwartetem Messwert wurde ein 0,5 J oder 4,0 J Pendel genutzt. Die Prüfkörper hatten eine
Größe von 8,0 x 1,0 x 0,4 cm. Alle Messungen erfolgten bei einer Temperatur von 22 °C und
einer relativen Luftfeuchtigkeit von 65%.
3.8.6. Shore Härte
Die „Shore Härte“ ist eine relative Kennzahl. Die für das Probenmaterial ermittelte Kennzahl
steht in direktem Zusammenhang mit der Eindringtiefe in das Material und ist daher ein Maß
für die Werkstoffhärte. Je nach verwendeter Masse und Formgebung des Eindringkörpers
wird zwischen den Methoden Shore D, Shore A und Shore C unterschieden.
Zur Ermittlung der Shore Härte wird eine Skala verwendet, bei der 0 Shore der Eindringtiefe
von 2,5 mm und 100 Shore der Eindringtiefe von 0,0 mm entsprechen. Das bedeutet, dass
Page 27
19
Materialien mit einer kleinen Shore Härte dem Eindringen des Prüfwerkzeugs wenig Wider-
stand und Materialien mit großer Shore Härte dem Eindringen einen sehr hohen Widerstand
entgegensetzen [132].
Bei den eigenen Arbeiten wurde nach der Methode Shore D gearbeitet. Hierbei wird ein
Eindringkörper mit einer kugelförmigen Spitze (r = 0,1 mm) und einem Öffnungswinkel von
30° verwendet. Alle Messungen wurden bei einer Temperatur von 22 °C und einer relativen
Luftfeuchtigkeit von 65% durchgeführt.
3.8.7. HDT – Wärmeformbeständigkeitstemperatur
Mit Hilfe der Wärmeformbeständigkeitstemperatur ist es möglich eine Aussage über die
thermische Belastbarkeit der untersuchten Proben zu treffen. Bei der Messung wird eine
Dreipunktbiegung durchgeführt. Hierzu wird der Prüfkörper mit der flachen Seite an den
äußeren Enden auf die Halterung aufgelegt und mittig mit einer konstanten Kraft belastet.
Die Probe befindet sich während der Messung in einem Ofen, der mit einer konstanten
Temperaturrampe beheizt wird. Als Wärmeformbeständigkeitstemperatur (HDT) wird die
Temperatur bezeichnet, bei der die Probe eine Randfaserdehnung von 0,2% aufweist. Je
nach angewendeter Kraft wird zwischen den Methoden HDT-A, HDT-B und HDT-C
unterschieden.
In den eigenen Arbeiten wurde die Methode HDT-B verwendet. Hier vermisst man die
Proben mit einer Biegebelastung von 0,45 MPa. Die Messungen wurden an einer Anlage der
Firma TA Instruments mit einer Heizrate von 2 K/min durchgeführt. Die Starttemperatur lag
bei 23 °C.
3.8.8. Röntgenbeugungsexperimente
Mit Hilfe von Röntgenbeugungsexperimenten ist es möglich eine Aussage über die Kristall-
stuktur der untersuchten Proben zu treffen.
Bestrahlt man eine Probe mit Röntgenstrahlen (elektromagnetische Wellen im Bereich von
10-8 bis 10-11 m), so werden die Elektronen der Probe zu Schwingungen angeregt und
können ihrerseits elektromagnetische Wellen aussenden. Dabei kann es zu Interferenzen
kommen, wodurch ein Beugungsbild entsteht. Dieses Beugungsbild kann mit einem Fotofilm
aufgenommen und anschließend entwickelt werden. Vollständig isotrope Materialien zeigen
kreisförmige Beugungsmuster. Bei steigender Orientierung entarten die kreisförmigen
Beugungsbilder zu sichelförmigen. Für Proben hoher Kristallitordnung werden auf den er-
Page 28
20
zeugten Planfilmaufnahmen punktförmige Interferenzen gefunden.
Durch Ausmessen der registrierten Reflexe mit einem Glaslineal und anschließender
Berechnung können die für das Material charakteristischen Beugungswinkel (2θ) erhalten
werden [130, 133, 134].
Zur Erzeugung der Röntgenstrahlen wurde eine Röntgenröhre mit Kupferanode und ein
Heizstrom sowie eine Heizleistung von 40 kV und 40 mA verwendet. Geregelt wurden beide
Parameter mittels eines Bruker AXS Generators. Zur Monochromatisierung der erzeugten
Kupfer-Kα-Strahlung (λ = 0,154 nm) wurde ein Nickelfilter verwendet. Um ein Überstrahlen
des Fotofilms (BioMax, Fa. Kodak) zu vermeiden, kam ergänzend ein Primärstrahlfänger zum
Einsatz. Der Abstand zwischen Probe und Filmebene betrug 60 cm. Das Filmmaterial wurde
naßchemisch entwickelt.
3.8.9. DMA – Dynamisch Mechanische Analyse
Mit Hilfe der Dynamisch Mechanischen Analyse (DMA) können Rückschlüsse auf das mech-
anische Verhalten von viskoelastischen Probenmaterialien unter geringer dynamischer
Belastung in Abhängigkeit von der Temperatur gezogen werden. Dank der schwachen
mechanischen Beanspruchung ist es möglich das Probenmaterial zerstörungsfrei und ohne
Veränderung der Probeneigenschaften zu vermessen. Als Antwort der Probe auf die
angelegte Belastung (Spannung oder Deformation) werden der Speichermodul E´ und der
Verlustmodul E´´ als charakteristische Größen erhalten. Der Speichermodul E´ stellt dabei
den elastischen und der Verlustmodul E´´ den viskosen Anteil der Materialeigenschaften dar.
Durch den Speichermodul kann die mechanische Energie wiedergegeben werden, die
während der Messung vom Probenmaterial gespeichert und wieder zurückgewonnen
werden kann. Der Verlustmodul kennzeichnet den Teil der mechanischen Energie, der vom
Probensystem in Wärmeenergie umgewandelt wird und daher verloren geht. Mit Hilfe von
DMA-Messungen kann außerdem der Glasübergangspunkt Tg der untersuchten Materialien
ermittelt werden. Dazu wird entweder der Wendepunkt der E´- oder der Hochpunkt der E´´-
Kurve ermittelt [129, 135, 136].
Die Messungen wurden deformationsgesteuert (Grenzwert: 15 µm) an einer Anlage der
Firma TA Instruments mit einer Frequenz von 10 Hz, einer Heizrate von 5 K/min und unter
Verwendung eines single Cantilever-Probenhalters durchgeführt.
Page 29
21
4. Polymersynthese
4.1. Synthesestrategien
Zur Synthese der Blockcopolymere wurden unterschiedliche Verknüpfungsansätze unter-
sucht. Dazu zählen die Festphasen-Polykondensation, eine 3-Stufen und eine 2-Stufen-
Synthese. Das Merkmal der Festphasen-Polykondensation ist, dass unterhalb der Schmelz-
temperatur des Polymers gearbeitet wird. Die 3- und 2-Stufen-Synthese beinhalten die ROP
als wesentlichen Syntheseschritt. Der Unterschied dieser beiden Ansätze liegt in der
Aufarbeitung des Prepolymers.
4.1.1. Festphasen-Polykondensation
Wie bereits erläutert (vgl. Abschn. 3.4), ist das entscheidende Kriterium bei der Festphasen-
Polykondensation das Auskristallisieren der Polymerketten. Um das zu erreichen, wird bei
Temperaturen unterhalb der Schmelztemperatur von Polylactid (z.B. PLLA; 180 °C) ge-
arbeitet [64, 137]. Bei üblichen Synthesetemperaturen der ROP (180-220°C; [73, 79, 87])
kann es auf Grund des thermodynamischen Gleichgewichts zwischen Monomer und Polymer
(vgl. Bild 3.4) zu keinem 100%igen Umsatz kommen. Zusätzlich wird die diffusionskontrol-
lierte Reaktion infolge der hohen Schmelzviskosität behindert. Durch die Reduzierung der
Reaktionstemperatur verbleiben das Monomer, der Katalysator und die aktiven Kettenenden
in einer amorphen Phase geminderter Viskosität, während die Polymerketten auskristalli-
sieren [105, 138]. Auf Grund der Anreicherung des Monomers in der amorphen Phase
kommt es zu einer Verschiebung des Gleichgewichts und damit zu einer weiteren Umsetzung
des Monomers. Der neu entstehende Polymerstrang kristallisiert wiederum aus. Dadurch
stellt sich das Gleichgewicht ständig neu ein [64]. Die besten Ergebnisse können dabei laut
den Untersuchungen von Moon et al. [139] erzielt werden, wenn im Bereich der Kristalli-
Page 30
22
sationstemperatur (Tc) des Polymers gearbeitet wird.
Diese Erkenntnis ist von Fukushima et al. [74, 92, 138] auf die PLA-Stereo-Blockbildung, also
die Verknüpfung zweier Homopolymere unterschiedlicher Konfiguration (PLLA und PDLA),
übertragen worden. Hier wird bei einer Temperatur oberhalb des Schmelzpunktes (Tm) der
Homopolymere und unterhalb des Schmelzpunktes des PLA-Stereokomplexes (sc) gearbeit-
et. Dadurch kommt es zum Auskristallisieren des sc, wobei die Kettenenden der Homo-
polymere sowie der Katalysator in der amorphen Phase verbleiben. Da die Umsetzung in
dem benannten Temperaturfenster durchgeführt wird, können sich lediglich sc- und keine
Homo (hc)-Kristalle bilden. Aus diesem Grund ist es möglich, dass sich die reaktiven Ketten-
enden unterschiedlicher Konfiguration zu Stereoblock (sb)-PLA verbinden können.
Die von Fukushima et al. beschriebene Synthese besteht demzufolge aus drei wesentlichen
Schritten: der Homopolymersynthese von PLLA und PDLA, dem Blenden dieser beiden Kom-
ponenten in der Schmelze zur Stereokomplexbildung und der Festphasen-Polykondensation
zur Verknüpfung der Ketten zu Stereo-Blockcopolymeren.
Diese Vorgehensweise wurde in übertragener Form für die im Rahmen dieser Arbeit durch-
zuführende Blockcopolymer-Synthese genutzt. In Anlehnung an [138] wurde ein Vorversuch
zur Überprüfung der beschriebenen Verknüpfungsmöglichkeit zweier Homopolymere über
diesen Syntheseweg durchgeführt. Bekannte Schwächen der Festphasen-Polykondensation
sind die in den Arbeiten von Fukushima et al. [74, 138] und Moon et al. [139] beschriebenen
Nebenreaktionen. Zum einen kommt es angesichts der langen thermischen Belastung zu
Abbaureaktionen. Dies wird infolge des angelegten Vakuums und einer durch die Entfernung
des Monomers begründeten Verschiebung des Monomer/Polymer-Gleichgewichts (vgl. Bild
3.4) noch verstärkt [27, 92]. Zum anderen kommt es auf Grund von Umesterungsreaktionen
zu einer Vermischung von einzelnen Teilen der verschiedenen Blöcke (D,L- und L-Block) des
sich bildenden Copolymers. Dies ist ungünstig für eine enantiomerenreine Blockbildung, die
wiederum für die anschließende Stereokomplexbildung von höchster Priorität ist.
Dennoch sind modellhaft zwei identisch konfigurierte Homopolymere (PLLA) unterschied-
licher Molmasse (ca. 15000 g/mol und 47000 g/mol) zu äquivalenten Teilen nach folgender
Versuchsvorschrift miteinander umgesetzt worden:
Die beiden als Flocken vorliegenden Homopolymere werden vorgemischt, in einen 3-
Halskolben überführt und anschließend für je 2 h bei RT und bei 110 °C unter Vakuum
getrocknet. Nachfolgend wird die Mischung in N2-Atmosphäre auf 200 °C erhitzt und nach
dem Schmelzen für 20 min unter mechanischem Rühren (Magnetrührstäbchen) miteinander
geblendet. Anschließend wird die Temperatur auf 100 °C reduziert und die Reaktions-
mischung für 2 h ruhen gelassen, bevor die eigentliche Festphasen-Polykondensation durch-
geführt wird. Diese erfolgt durch die thermische Behandlung der Polymere unter Vakuum für
20 h bei 150 °C. Die Reaktionsmischung lässt man letztlich auf RT abkühlen und führt sie
dann der Analyse zu.
Eine GPC-Untersuchung der resultierenden Beispielsubstanz ergab eine Molmasse von ca.
Page 31
23
29000 g/mol. Da dies annähernd dem Mittelwert der beiden eingesetzten Polymere ent-
spricht, kann davon ausgegangen werden, dass es lediglich zu einer Verblendung und zu
keiner Verknüpfung der beiden modellhaft eingesetzten Polymere gekommen ist.
Die Festphasen-Polykondensation ist demzufolge für den Aufbau des angestrebten Co-
polymers ungeeignet.
4.1.2. 3-Stufen-Synthese
Diese Synthesevariante zum Aufbau von Blockcopolymeren basiert auf den Arbeiten von
Hirate et al. [79]. Sie besteht aus 3 Verfahrensstufen:
Zunächst wird der kürzere Polymerblock (PDLLA) des aufzubauenden Copolymers über eine
ROP (vgl. Abschn. 3.4) als Prepolymer aus D,L-Lactid synthetisiert. Dies ist notwendig, da bei
einer umgekehrten Reihenfolge (zunächst Synthese des längeren Blocks) das zur zweiten
ROP hinzugegebene Lactid auf Grund der hohen Viskosität des Prepolymers nicht gleich-
mäßig in das System eingemischt werden könnte und die Reaktion des Blockaufbaus mit
Nebenreaktionen wie Homo-Polymerisation und Umesterungen konkurrieren würde [55, 73,
79]. Das im ersten Schritt erhaltene Prepolymer wird dann mittels Lösen in CHCl3 und Fällen
in MeOH von seinem Restmonomer befreit, bevor es im dritten Schritt (einer zweiten ROP)
als Makroinitiator der L,L-Lactid-Umsetzung verwendet wird. Der Zwischenschritt der Auf-
reinigung ist wiederum notwendig, um D,D-Lactid (Restmonomer des ersten Synthese-
schritts - Monomer/Polymer-GG (vgl. Bild 3.4)) aus dem System zu entfernen, welches
ansonsten in den kristallinen PLLA-Block mit eingebaut werden würde [79, 138]. Auf Grund
der so entstehenden Fehlstellen wäre eine Kristallisation des L,L-Blocks unterdrückt [140]
und somit auch eine später angestrebte Stereokomplexbildung bei Umsetzung mit PDLA
behindert oder im ungünstigsten Fall sogar nicht mehr möglich.
Bei Berücksichtigung der beiden wesentlichen Faktoren (Entfernung Monomer; Verwendung
des kürzeren Blocks als Makroinitiator) konnten mit dieser Synthesevariante in eigenen Ver-
suchen viel versprechende, aber schlecht reproduzierbare Ergebnisse erzielt werden. So ist
es zum Beispiel gelungen ein Copolymer mit einem gewünschten Blockverhältnis von 30%
amorphem zu 70% kristallinem Anteil aufzubauen. Die Molmasse des Prepolymers betrug
hierbei rund 13800 g/mol bei einem PDI von 1,8. Nach Aufbau des kristallinen Blocks
erhöhte sich die Molmasse auf 44000 g/mol bei einem PDI von 2,0. In einem zweiten
Beispiel, in dem das gleiche Blockverhältnis angestrebt war, konnte hingegen nur ein Aufbau
der Molmasse von ca. 10500 g/mol (PDI 1,4) des Prepolymers auf ca. 24700 g/mol (PDI 1,4)
des Copolymers realisiert werden. Das entspricht einem erzielten Blockverhältnis von rund
43% amorph zu 57% kristallin. Bei höheren Molmassen des Prepolymers (ca. >40000 g/mol)
konnte aus den oben benannten Gründen (hohe Viskosität, schlechte Einmischung des
zugefügten Lactids) gar kein Aufbau festgestellt werden.
Page 32
24
Da das Prepolymer aus amorphem PDLLA besteht, gestaltete sich das Fällen und spätere
Verarbeiten als umständlich. Wie schon von Rafler und Dahlmann [103] beobachtet, erhält
man bei der Fällung von PDLLA keinen Feststoff, sondern lediglich eine Gelphase, die sich bei
einer weiteren Verarbeitung nur sehr schlecht handhaben lässt. Zudem ist für die Entmono-
merisierung des Prepolymers bei dieser Synthesevariante neben dem Lösen und Fällen auch
ein zeitintensiver Vakuum-Trocknungsschritt (ca. 20h und 4 Temperaturstufen) erforderlich.
Aus dieser Art der Entmonomerisierung resultiert außerdem, dass das Prepolymer vor der
weiteren Verarbeitung als Feststoff vorliegt und erst wieder in eine Schmelze überführt
werden muss. Dies bringt eine zusätzliche thermische Belastung mit sich und erschwert die
homogene Einmischung des L,L-Lactids zur Bildung des zweiten Blocks zusätzlich. Mit hoher
Wahrscheinlichkeit kann dieser Umstand auch als Grund der oben beschriebenen nicht
reproduzierbaren Ergebnisse benannt werden.
Aus den genannten Punkten wurde daher nach einer weiteren Synthesevariante gesucht, mit
der sich insbesondere der Prepolymer-Entmonomerisierungsschritt effektiver gestalten lässt.
4.1.3. 2-Stufen-Synthese
Die 2-Stufen-Synthese unterscheidet sich nur unwesentlich von der zuvor beschriebenen
Syntheseroute. Auch bei dieser Synthesevariante wird in einer ersten ROP zunächst ein
Prepolymer hergestellt, das bei einer zweiten ROP als Makroinitiator fungiert. Der ent-
scheidende Unterschied besteht in der Verfahrensweise zur Entmonomerisierung des Pre-
polymers. Die Entfernung des Monomers erfolgt hier mittels Vakuum. Das hat im Vergleich
zur 3-Stufen-Synthese die Vorteile, dass das synthetisierte Prepolymer nicht aus dem Reak-
tionsgefäß entfernt werden muss und durch das lösungsmittelfreie Arbeiten keine zu-
sätzliche Feuchtigkeit ins System eingebracht wird. Außerdem liegt das Prepolymer während
der gesamten Synthese in der Schmelze vor, wodurch das zur Blockbildung zudosierte L,L-
Lactid wesentlich besser eingemischt und umgesetzt werden kann.
Zur Entmonomerisierung des PDLLA-Blocks im Reaktor wird nach der ersten ROP zunächst
ein leichtes Vakuum angelegt, welches dann in kleinen Schritten immer weiter reduziert
wird. Durchschnittlich waren die synthetisierten Prepolymere bei eigenen Versuchen im
Labormaßstab 17 min einem Endvakuum von 16 mbar ausgesetzt.
Da diese Synthesevariante neben den oben genannten Vorteilen und guten Ergebnissen (vgl.
weitere Erläuterungen in Abschn. 4.2.2 und Abschn. 4.3.2) auch noch deutlich weniger
aufwendig ist, kam sie sowohl für die Synthese der Blockcopolymere im Labor- als auch im
Technikum-Maßstab zum Einsatz.
Page 33
25
4.2. Arbeiten im Labormaßstab
Alle verwendeten Lactide (D,L-, L,L- und D,D-Lactid) wurden vor der Synthese aus den bereits
diskutierten Gründen (vgl. Abschn. 3.4) umkristallisiert (Versuchsvorschrift im Anhang). Zur
Kontrolle der Reinheit wurden titrimetrische Untersuchungen (vgl. Abschn. 3.8.4) zur
Bestimmung der vorhandenen Endgruppenzahl vorgenommen. Zur Synthese von Polymeren
mit hoher Molmasse ist ein Endgruppengehalt <10 µequ/g erforderlich [141], so dass in
jedem Fall so oft umkristallisiert wurde, bis der Wert erreicht war.
Für den amorphen Teil des Copolymers war zunächst der Einsatz von meso-Lactid vor-
gesehen. Dieses ist jedoch sehr hydrolyseanfällig [35] und lässt sich unter Laborbedingungen
(keine ständige Schutzgasatmosphäre) nicht verarbeiten. Die Umkristallisation des meso-
Lactids gestaltete sich entsprechend schwierig und nur in sehr wenigen Fällen konnten
Endgruppenwerte unter 40 µequ/g erreicht werden. D,L-Lactid verhält sich hingegen
vergleichbar zu L,L- und D,D-Lactid und konnte daher ohne Probleme unter Laborbe-
dingungen zur Synthese eingesetzt werden.
4.2.1. Homopolymer-Synthese
Versuchsvorschrift
Zur Synthese im Labormaßstab kamen Kondensationsgefäße mit einem Volumen von 100 mL
zum Einsatz. Diese sind mit Stickstoff-Zu- und Ableitung sowie einem Schneckenrührer
ausgestattet und werden in einem Ölbad temperiert. Das Ölbad wird auf die Reaktions-
temperatur von 180 °C eingestellt und das System für etwa 1h darin konditioniert. Unter
moderatem N2-Fluss (Kontrolle über Blasenzähler) werden zur Entfernung der Restfeuchtig-
keit im System einige mL Toluol aus dem Kondensationsgefäß abdestilliert. Nachfolgend
werden 65 g D,D-Lactid und 253 mg 1-Dodecanol (für eine angestrebte Molmasse von
45000 g/mol) eingewogen und mittels eines Trichters in das Reaktionsgefäß überführt. Das
Einfüllen wird ggf. mit einem Heißluftgebläse unterstützt. Wenn das gesamte Lactid auf-
geschmolzen und die Systemtemperatur wieder stabil bei 180 °C ist, werden 2,108 mL 1%ige
Katalysator-Lösung in Toluol (1x10-4 mol Katalysator je 1 mol Monomer) hinzugegeben. Der
Katalysator wird in verdünnter Form verwendet, da sich zum einen die geringen Einsatz-
mengen sehr schlecht handhaben lassen und zum anderen die Löslichkeit und Verteilung so
besser gewährleistet werden kann. Laut Purnama et al. [102] ist die Löslichkeit des Kataly-
sators von entscheidender Bedeutung für die Reproduzierbarkeit der Polymerisation.
Die Reaktion wird mittels visueller Kontrolle (deutliches Ansteigen der Viskosität) nach 2h
durch die Zugabe von CHCl3 beendet. Das Reaktionsgefäß verbleibt zunächst noch im
(ausgeschalteten) Ölbad, um die Durchmischung von Polymer und Lösungsmittel zu unter-
stützen. Das Polymer wird über Nacht gelöst und die resultierende Polymerlösung am
Page 34
26
nächsten Tag in einem Überschuss an MeOH ausgefällt. Das gefällte Polymer wird zunächst
getrocknet und dann zur weiteren Reduzierung des Restmonomergehalts noch ein zweites
Mal in CHCl3 gelöst und abermals in einem Überschuss an MeOH gefällt. Die so erhaltenen
Polymerflocken werden abschließend getrocknet und bis zur weiteren Verarbeitung über
P2O5 gelagert.
4.2.2. Blockcopolymer-Synthese
Versuchsvorschrift
Zur Synthese im Labormaßstab wurden Kondensationsgefäße mit einem Volumen von
100 mL verwendet. Die Vorbereitung dieser entspricht der in Abschnitt 4.2.1 beschriebenen
Verfahrensweise. Nach der Entfernung möglicher Restfeuchtigkeit aus dem System werden
entsprechend dem angestrebten Blockverhältnis zwischen amorphem und kristallinem Anteil
und der damit verbundenen Molmasse die benötigten Mengen an D,L-Lactid und 1-Do-
decanol eingewogen und mittels eines Trichters in das Reaktionsgefäß überführt. Das
Einfüllen wird ggf. mit einem Heißluftgebläse unterstützt. Wenn das gesamte Lactid auf-
geschmolzen und die Systemtemperatur wieder stabil bei 180 °C ist, wird die benötigte
Menge an 1%iger Katalysator-Lösung in Toluol (1x10-4 mol Katalysator je 1 mol Monomer)
hinzugegeben.
Die Reaktion der ersten ROP wird mittels visueller Kontrolle (Beobachtung des Viskositäts-
anstiegs) verfolgt. Nach ca. 1 h kann keine weitere Viskositätssteigerung mehr registriert
werden und die Reaktion wird beendet. Nachfolgend wird zur Entmonomerisierung des Pre-
polymers insgesamt für ca. 30 min Vakuum gezogen. Dabei wird mit einem schwachen
Vakuum begonnen. Unter Beobachtung der Polymerschmelze (es kann zu einem starken
Hochkochen kommen) wird der Druck dann langsam reduziert. Nach erfolgter Entmono-
merisierung wird eine kleine Probe zur Bestimmung der Molmasse entnommen.
In einem weiteren Schritt wird die in Abhängigkeit von der gewünschten Blockzusammen-
setzung ermittelte Menge an L,L-Lactid zum entmonomerisierten Prepolymer gegeben. Das
Monomer wird ca. 10 min lang intensiv unter die Schmelze gerührt, bevor mit der Dosierung
von weiterer Kat.-Lösung (auf die Menge an zugegebenem Lactid berechnet) die zweite ROP
gestartet wird. Nach 1h wird die Reaktion durch Zugabe von CHCl3 beendet. Das Polymer
wird über Nacht gelöst und am Folgetag in einem Überschuss an MeOH ausgefällt. Das
gefällte Polymer wird zunächst getrocknet und dann zur weiteren Reduzierung des Rest-
monomers noch ein zweites Mal in CHCl3 gelöst und in einem Überschuss an MeOH aus-
gefällt. Die erhaltenen Polymerflocken werden abschließend getrocknet und bis zur weiteren
Verarbeitung über P2O5 gelagert.
Eine Übersicht der für die Synthese der Stereokomplexe verwendeten Blockcopolymere wird
in Abschnitt 4.2.4 gegeben.
Page 35
27
Limitierende Synthesefaktoren
Lange thermische Belastungen des Prepolymers müssen vermieden werden, da es sonst zu
deutlichen Abbaureaktionen kommt, wie sie unter anderem in den Arbeiten von Wang et al.
[29], Purnama et al. [102], Rafler et al. [103] und Moon et al. [139] beschrieben werden. Aus
diesem Grund kann auch der Vakuum-Entmonomerisierungsschritt vor der zweiten ROP nur
in einem begrenzten Zeitfenster durchgeführt werden. Das bestätigt sich in eigenen
Untersuchungen. So beträgt die Molmasse eines Prepolymers vor der Entmonomerisierung
beispielsweise ca. 62000 g/mol und bereits nach 15 min Entmonomerisierung ist eine
Reduzierung der Mn von 7000 g/mol auf ca. 55000 g/mol zu verzeichnen.
Eine weitere wichtige Kenngröße ist die Molmasse des Prepolymers. Wie schon in Abschnitt
4.1.2 beschrieben, darf diese nicht zu hoch sein, da das zur zweiten ROP zugefügte Lactid
ansonsten auf Grund der hohen Viskosität nicht ausreichend in das System eingemischt
werden kann. Die Blockbildung steht dann in Konkurrenz zu Nebenreaktionen und ein
gezielter Blockaufbau kann nicht mehr gewährleistet werden. So werden zum einen Block-
copolymere mit unerwünschten Blockverhältnissen (amorph zu kristallin) und zum anderen
geringe Umsätze während der zweiten ROP erhalten. Als Beispiel sei hier ein Prepolymer mit
einer vergleichsweise hohen Molmasse (Mn) von 57000 g/mol angeführt, das mit L,L-Lactid
zu einem 50/50-Blockcopolymer umgesetzt werden sollte. Erzielt wurde hingegen ein
Blockverhältnis von 88/12 (amorph zu kristallin). In der zweiten ROP konnte die Molmasse
nur um rund 12% auf 65000 g/mol erhöht werden. Dies zeigt deutlich, dass bei zu hohen
Molmassen des Prepolymers das Endergebnis der zweiten ROP nicht aktiv beeinflusst
werden kann.
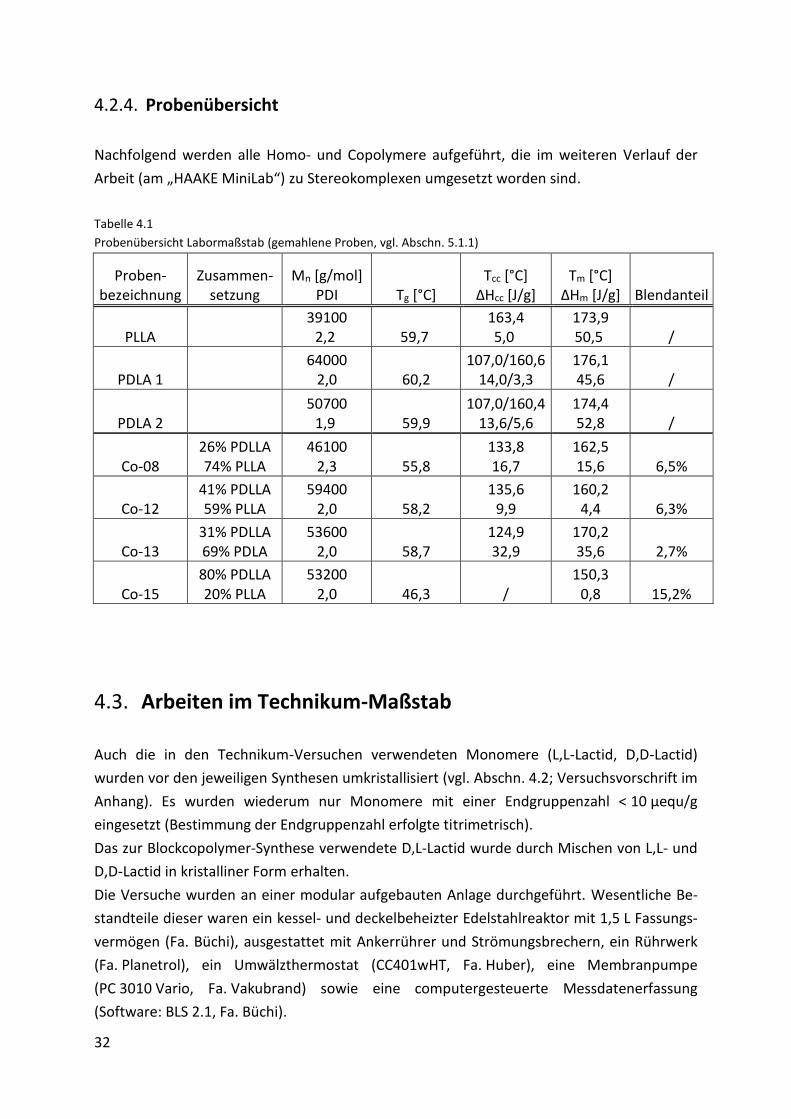
4.2.3. Probencharakterisierung
Alle Homo- und Copolymere wurden mittels GPC (Molmasse und Verteilung) und DSC
(thermische Eigenschaften) untersucht. Zur Charakterisierung der Copolymere wurden
weiterhin Lösetests durchgeführt.
4.2.3.1. Homopolymere
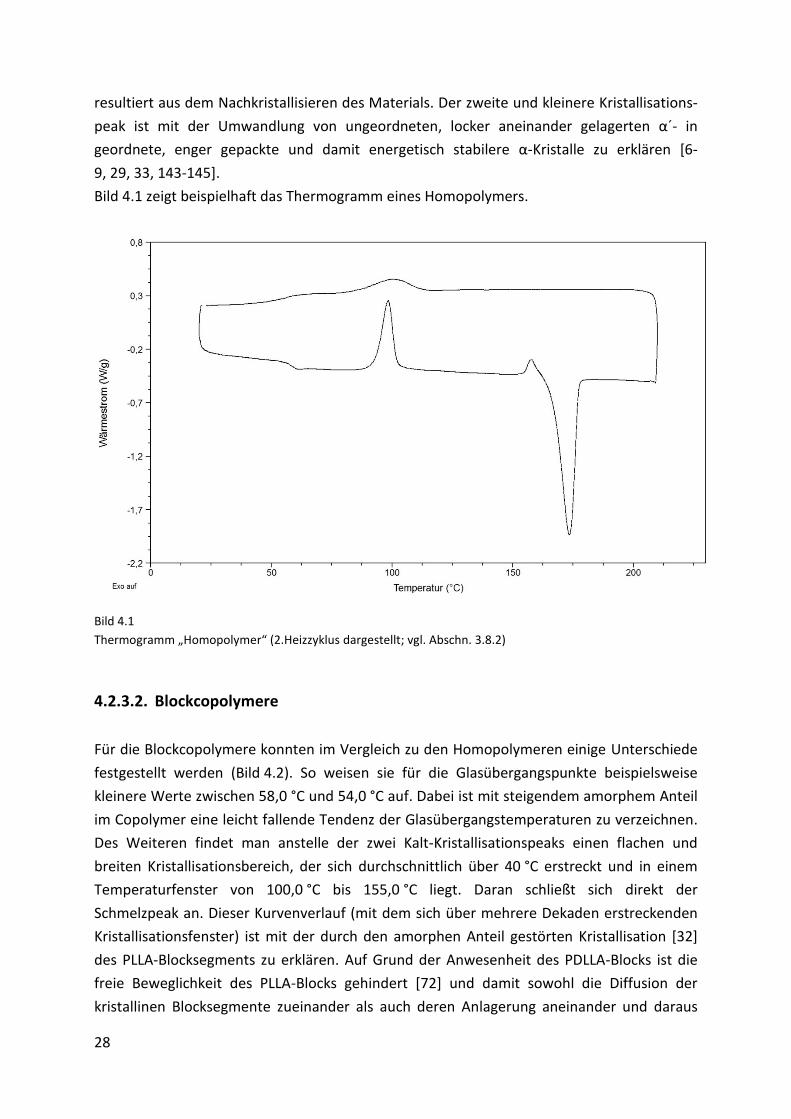
Die DSC-Kurven der Homopolymere zeigen den aus der Literatur [8, 142] bekannten Verlauf.
Für die Polymere mit durchschnittlichen Molmassen von 60000 g/mol und einem PDI von 2,1
konnten nachfolgende charakteristische Temperaturen gefunden werden: einen Glas-
übergangspunkt (Tg) bei ca. 60,0 °C, zwei Kristallisationspeaks während des Aufheizens (Tcc1;
Tcc2) bei ca. 104,0 °C und 160,1 °C, sowie einen Schmelzpeak (Tm) bei etwa 175,3 °C. Tcc1
Page 36
28
resultiert aus dem Nachkristallisieren des Materials. Der zweite und kleinere Kristallisations-
peak ist mit der Umwandlung von ungeordneten, locker aneinander gelagerten α´- in
geordnete, enger gepackte und damit energetisch stabilere α-Kristalle zu erklären [6-
9, 29, 33, 143-145].
Bild 4.1 zeigt beispielhaft das Thermogramm eines Homopolymers.
Bild 4.1
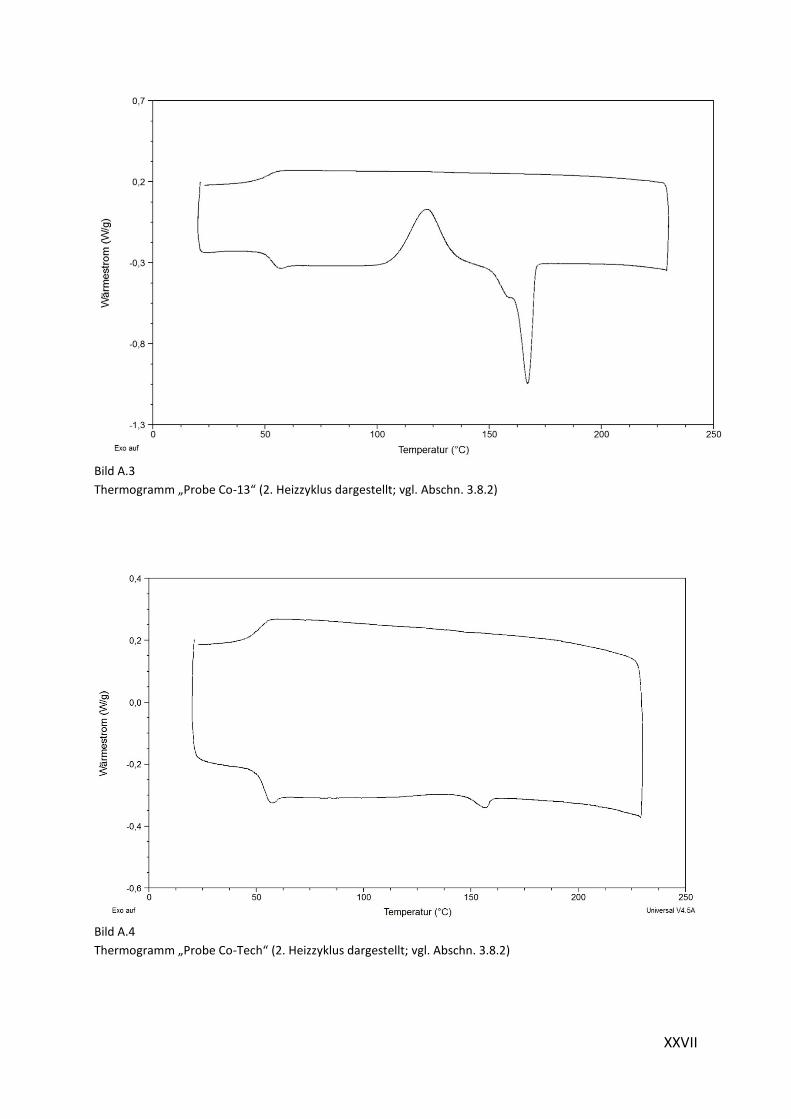
Thermogramm „Homopolymer“ (2.Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
4.2.3.2. Blockcopolymere
Für die Blockcopolymere konnten im Vergleich zu den Homopolymeren einige Unterschiede
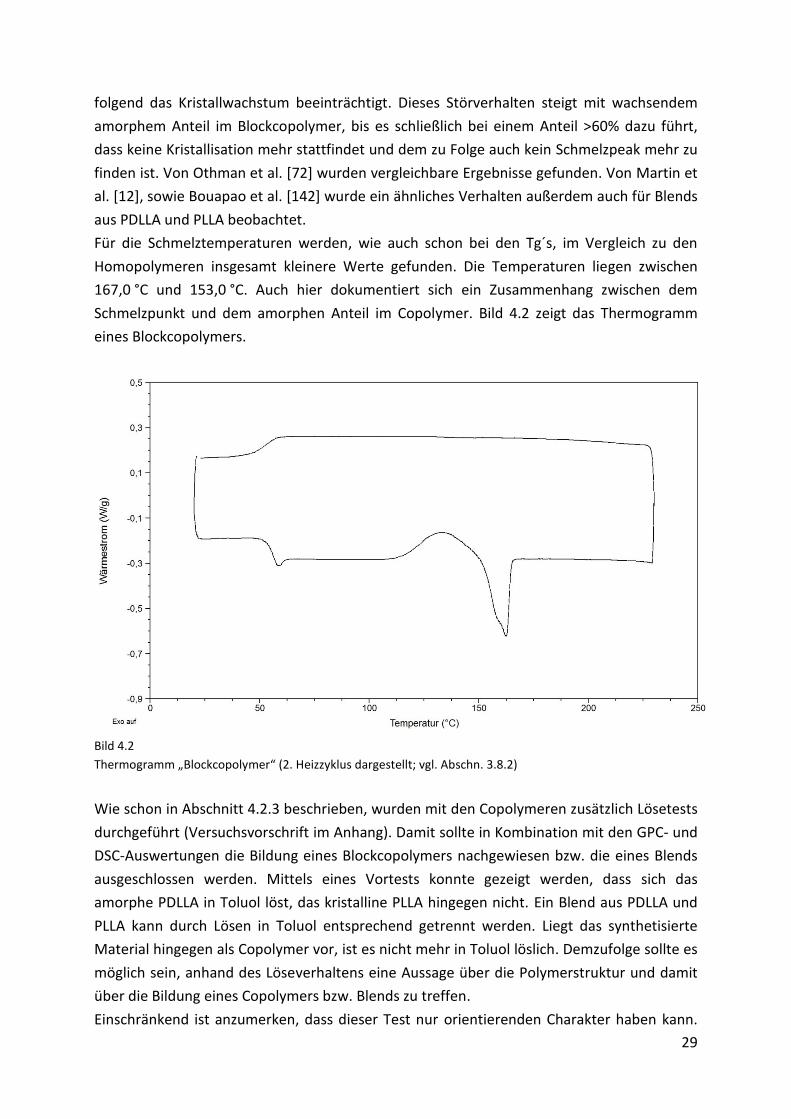
festgestellt werden (Bild 4.2). So weisen sie für die Glasübergangspunkte beispielsweise
kleinere Werte zwischen 58,0 °C und 54,0 °C auf. Dabei ist mit steigendem amorphem Anteil
im Copolymer eine leicht fallende Tendenz der Glasübergangstemperaturen zu verzeichnen.
Des Weiteren findet man anstelle der zwei Kalt-Kristallisationspeaks einen flachen und
breiten Kristallisationsbereich, der sich durchschnittlich über 40 °C erstreckt und in einem
Temperaturfenster von 100,0 °C bis 155,0 °C liegt. Daran schließt sich direkt der
Schmelzpeak an. Dieser Kurvenverlauf (mit dem sich über mehrere Dekaden erstreckenden
Kristallisationsfenster) ist mit der durch den amorphen Anteil gestörten Kristallisation [32]
des PLLA-Blocksegments zu erklären. Auf Grund der Anwesenheit des PDLLA-Blocks ist die
freie Beweglichkeit des PLLA-Blocks gehindert [72] und damit sowohl die Diffusion der
kristallinen Blocksegmente zueinander als auch deren Anlagerung aneinander und daraus
Page 37
29
folgend das Kristallwachstum beeinträchtigt. Dieses Störverhalten steigt mit wachsendem
amorphem Anteil im Blockcopolymer, bis es schließlich bei einem Anteil >60% dazu führt,
dass keine Kristallisation mehr stattfindet und dem zu Folge auch kein Schmelzpeak mehr zu
finden ist. Von Othman et al. [72] wurden vergleichbare Ergebnisse gefunden. Von Martin et
al. [12], sowie Bouapao et al. [142] wurde ein ähnliches Verhalten außerdem auch für Blends
aus PDLLA und PLLA beobachtet.
Für die Schmelztemperaturen werden, wie auch schon bei den Tg´s, im Vergleich zu den
Homopolymeren insgesamt kleinere Werte gefunden. Die Temperaturen liegen zwischen
167,0 °C und 153,0 °C. Auch hier dokumentiert sich ein Zusammenhang zwischen dem
Schmelzpunkt und dem amorphen Anteil im Copolymer. Bild 4.2 zeigt das Thermogramm
eines Blockcopolymers.
Bild 4.2
Thermogramm „Blockcopolymer“ (2. Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
Wie schon in Abschnitt 4.2.3 beschrieben, wurden mit den Copolymeren zusätzlich Lösetests
durchgeführt (Versuchsvorschrift im Anhang). Damit sollte in Kombination mit den GPC- und
DSC-Auswertungen die Bildung eines Blockcopolymers nachgewiesen bzw. die eines Blends
ausgeschlossen werden. Mittels eines Vortests konnte gezeigt werden, dass sich das
amorphe PDLLA in Toluol löst, das kristalline PLLA hingegen nicht. Ein Blend aus PDLLA und
PLLA kann durch Lösen in Toluol entsprechend getrennt werden. Liegt das synthetisierte
Material hingegen als Copolymer vor, ist es nicht mehr in Toluol löslich. Demzufolge sollte es
möglich sein, anhand des Löseverhaltens eine Aussage über die Polymerstruktur und damit
über die Bildung eines Copolymers bzw. Blends zu treffen.
Einschränkend ist anzumerken, dass dieser Test nur orientierenden Charakter haben kann.
Page 38
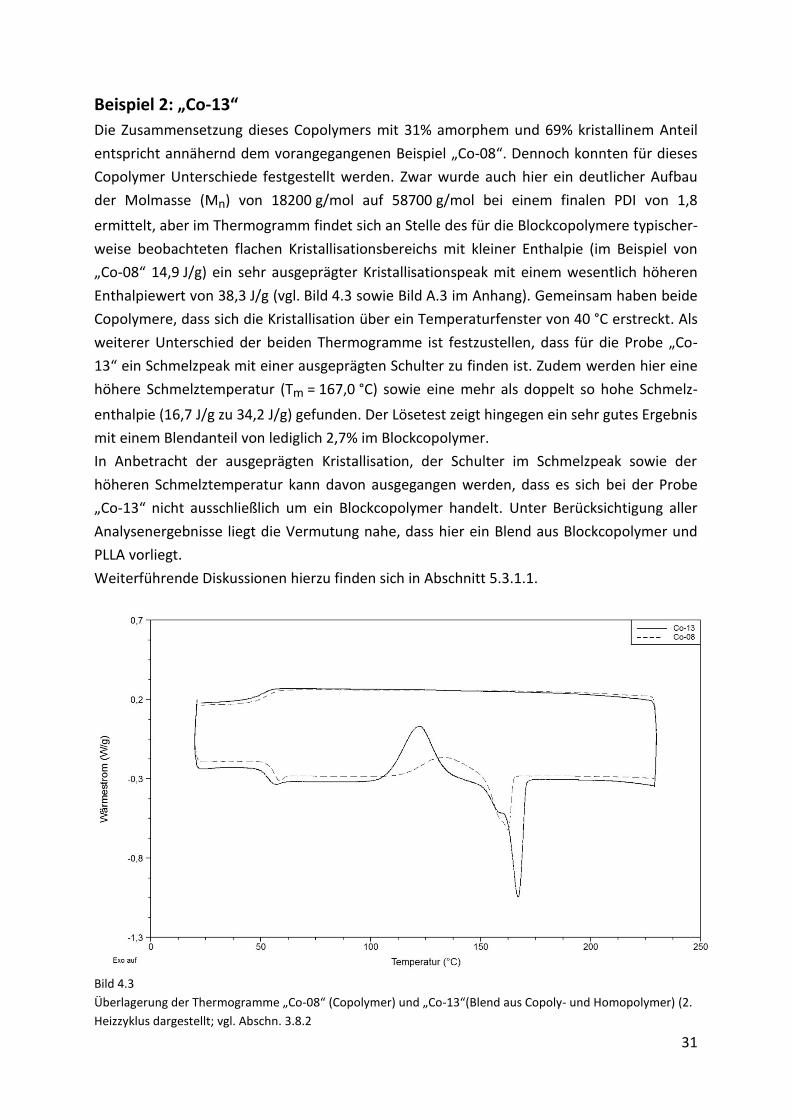
30
Bei der Probenentnahme nach der ersten ROP (vgl. Synthese in Abschnitt 4.2.2) ist es nicht
vollständig zu vermeiden, dass geringe Mengen des Prepolymers an der Glaswand des
Kondensationsgefäßes haften bleiben und aus diesem Grund nicht mehr an der zweiten ROP
teilnehmen können. Beim Lösen des finalen Blockcopolymers wird das an der Wandung ver-
bliebene PDLLA mit gelöst und dadurch mit in die Blockcopolymerlösung eingemischt. Daher
wird dann im Lösetest ein zu hoher Wert an nicht umgesetztem PDLLA ermittelt. Dies soll
nachfolgend an einem Beispiel gezeigt werden. Hier wurden das zu Beginn und das am Ende
aus dem Kondensationsgefäß gelöste Copolymer gesondert untersucht. Für das zuerst
gelöste Polymer wurde ein Blendanteil von 20,6% ermittelt. Im zum Ende gelösten Polymer
wurde lediglich ein Wert von 8,8% gefunden.
Eine weitere potenzielle Fehlerquelle ist, dass sich während der zweiten ROP unerwünscht
gebildetes PLLA nicht aus dem Polymerisationsgemisch herauslösen lässt. Liegt also ein
Blend aus Blockcopolymer und PLLA vor, kann dieser mit dem Lösetest nicht aufgetrennt und
das PLLA demzufolge nicht nachgewiesen werden.
Aus diesen Gründen ist es wichtig, nicht ausschließlich den Lösetest zur Beweisführung der
Blockbildung heranzuziehen. Bei Verknüpfung der Ergebnisse aus dem Lösetest, den GPC-
und DSC-Messungen sowie der vergleichenden Analyse eines gezielt hergestellten Blends
aus PDLLA und PLLA können aber belastbare Einschätzungen über die Polymerstruktur vor-
genommen werden.
Dies soll an zwei Beispielen („Co-08“ und „Co-13“) nachfolgend gezeigt werden:
Beispiel 1: „Co-08“
Hierbei handelt es sich um ein Copolymer mit 26% amorphem und 74% kristallinem Anteil.
Mit Hilfe der GPC kann ein deutlicher Molmassenaufbau von der ersten hin zur zweiten ROP
dokumentiert werden. So beträgt die Mn nach der ersten ROP 15100 g/mol und nach der
zweiten ROP 58200 g/mol. Der PDI liegt final bei 1,9.
In der DSC findet sich außerdem ein wie oben beschrieben für die Copolymere „typisches“
Thermogramm (vgl. Bild 4.3 sowie Bild A.1 im Anhang) mit dem charakteristischen flachen
Kristallisationsbereich in einem Temperaturfenster von rund 110 °C bis 150 °C und dem sich
direkt anschließenden Schmelzpeak Tm von 162,5 °C. Über den Lösetest konnte ein Blend-
anteil von 6,5% ermittelt werden.
Wenn man zusätzlich noch den Vergleich zu den Messergebnissen des Blends aus PDLLA und
PLLA zieht, kann mit sehr hoher Wahrscheinlichkeit davon ausgegangen werden, dass es sich
bei „Co-08“ tatsächlich um ein Blockcopolymer handelt. Das Thermogramm des Blends (Bild
A.2 im Anhang) ist bis auf eine geringfügig kleinere Temperatur des Tg´s mit dem der
Homopolymere vergleichbar. Es zeigt also nicht den für die Copolymere typischen breiten
Kristallisationsbereich.
Page 39
31
Beispiel 2: „Co-13“
Die Zusammensetzung dieses Copolymers mit 31% amorphem und 69% kristallinem Anteil
entspricht annähernd dem vorangegangenen Beispiel „Co-08“. Dennoch konnten für dieses
Copolymer Unterschiede festgestellt werden. Zwar wurde auch hier ein deutlicher Aufbau
der Molmasse (Mn) von 18200 g/mol auf 58700 g/mol bei einem finalen PDI von 1,8
ermittelt, aber im Thermogramm findet sich an Stelle des für die Blockcopolymere typischer-
weise beobachteten flachen Kristallisationsbereichs mit kleiner Enthalpie (im Beispiel von
„Co-08“ 14,9 J/g) ein sehr ausgeprägter Kristallisationspeak mit einem wesentlich höheren
Enthalpiewert von 38,3 J/g (vgl. Bild 4.3 sowie Bild A.3 im Anhang). Gemeinsam haben beide
Copolymere, dass sich die Kristallisation über ein Temperaturfenster von 40 °C erstreckt. Als
weiterer Unterschied der beiden Thermogramme ist festzustellen, dass für die Probe „Co-
13“ ein Schmelzpeak mit einer ausgeprägten Schulter zu finden ist. Zudem werden hier eine
höhere Schmelztemperatur (Tm = 167,0 °C) sowie eine mehr als doppelt so hohe Schmelz-
enthalpie (16,7 J/g zu 34,2 J/g) gefunden. Der Lösetest zeigt hingegen ein sehr gutes Ergebnis
mit einem Blendanteil von lediglich 2,7% im Blockcopolymer.
In Anbetracht der ausgeprägten Kristallisation, der Schulter im Schmelzpeak sowie der
höheren Schmelztemperatur kann davon ausgegangen werden, dass es sich bei der Probe
„Co-13“ nicht ausschließlich um ein Blockcopolymer handelt. Unter Berücksichtigung aller
Analysenergebnisse liegt die Vermutung nahe, dass hier ein Blend aus Blockcopolymer und
PLLA vorliegt.
Weiterführende Diskussionen hierzu finden sich in Abschnitt 5.3.1.1.
Bild 4.3
Überlagerung der Thermogramme „Co-08“ (Copolymer) und „Co-13“(Blend aus Copoly- und Homopolymer) (2.
Heizzyklus dargestellt; vgl. Abschn. 3.8.2
Page 40
32
4.2.4. Probenübersicht
Nachfolgend werden alle Homo- und Copolymere aufgeführt, die im weiteren Verlauf der
Arbeit (am „HAAKE MiniLab“) zu Stereokomplexen umgesetzt worden sind.
Tabelle 4.1
Probenübersicht Labormaßstab (gemahlene Proben, vgl. Abschn. 5.1.1)
Proben-bezeichnung
Zusammen-setzung
Mn [g/mol] PDI Tg [°C]
Tcc [°C] ΔHcc [J/g]
Tm [°C] ΔHm [J/g] Blendanteil
PLLA 39100
2,2 59,7 163,4 5,0
173,9 50,5 /
PDLA 1 64000
2,0 60,2 107,0/160,6
14,0/3,3 176,1 45,6 /
PDLA 2 50700
1,9 59,9 107,0/160,4
13,6/5,6 174,4 52,8 /
Co-08 26% PDLLA 74% PLLA
46100 2,3 55,8
133,8 16,7
162,5 15,6 6,5%
Co-12 41% PDLLA 59% PLLA
59400 2,0 58,2
135,6 9,9
160,2 4,4 6,3%
Co-13 31% PDLLA 69% PDLA
53600 2,0 58,7
124,9 32,9
170,2 35,6 2,7%
Co-15 80% PDLLA 20% PLLA
53200 2,0 46,3 /
150,3 0,8 15,2%
4.3. Arbeiten im Technikum-Maßstab
Auch die in den Technikum-Versuchen verwendeten Monomere (L,L-Lactid, D,D-Lactid)
wurden vor den jeweiligen Synthesen umkristallisiert (vgl. Abschn. 4.2; Versuchsvorschrift im
Anhang). Es wurden wiederum nur Monomere mit einer Endgruppenzahl < 10 µequ/g
eingesetzt (Bestimmung der Endgruppenzahl erfolgte titrimetrisch).
Das zur Blockcopolymer-Synthese verwendete D,L-Lactid wurde durch Mischen von L,L- und
D,D-Lactid in kristalliner Form erhalten.
Die Versuche wurden an einer modular aufgebauten Anlage durchgeführt. Wesentliche Be-
standteile dieser waren ein kessel- und deckelbeheizter Edelstahlreaktor mit 1,5 L Fassungs-
vermögen (Fa. Büchi), ausgestattet mit Ankerrührer und Strömungsbrechern, ein Rührwerk
(Fa. Planetrol), ein Umwälzthermostat (CC401wHT, Fa. Huber), eine Membranpumpe
(PC 3010 Vario, Fa. Vakubrand) sowie eine computergesteuerte Messdatenerfassung
(Software: BLS 2.1, Fa. Büchi).
Page 41
33
4.3.1. Homopolymer-Synthese
Versuchsvorschrift
Zur Vorbereitung des Reaktors wird dieser mit Toluol ausgekocht und dreimal sekuriert.
800 g frisch umkristallisiertes D,D-Lactid werden in ein Becherglas eingewogen und an-
schließend im N2-Gegenstrom mit Hilfe eines Trichters in den auf RT abgekühlten Reaktor
überführt. Außerdem werden noch 1,88 g 1-Dodecanol zum Einstellen der final gewünschten
Molmasse von 75000 g/mol hinzugegeben. Die Molmasse wird dabei etwas höher
angestrebt, als für die Weiterverarbeitung erforderlich, da sowohl beim Ausfahren der
Schmelze aus dem Reaktor als auch bei der anschließenden Entmonomerisierung im
Extruder (vgl. Abschn. 4.3.3) ein Abbau zu erwarten ist. In den Arbeiten von Jaszkiewicz et al.
[68] und Perego et al. [69] wird eine Reduzierung der Molmasse für jeden Prozess be-
schrieben, bei dem das Polymer in die Schmelze überführt wird.
Wenn sich beide Komponenten im Reaktor befinden, wird dieser zunächst auf eine Tem-
peratur von 150 °C (Öltemperatur) aufgeheizt. Nach ca. 15 min Heizdauer (Reaktor-
temperatur 122 °C) ist das gesamte Lactid aufgeschmolzen und es kann mit dem Rühren (ca.
25 U/min) begonnen werden. Nach ca. 20 min (Reaktortemperatur 136 °C) wird der Reaktor
nochmals kurz für die im N2-Gegenstrom erfolgende Zugabe von 1,945 mL 10%iger
Katalysator-Lösung (in Toluol) geöffnet und die eigentliche Reaktion gestartet. Die Ölbad-
temperatur wird auf 180 °C erhöht und die Rührgeschwindigkeit auf 55 U/min eingestellt.
Der Verlauf der Reaktion wird am Monitor über das Ansteigen des Drehmoments verfolgt.
Schon nach einer Reaktionszeit von 6 min muss die Rührgeschwindigkeit auf Grund des
starken Viskositätsanstiegs auf 10 U/min reduziert werden, um so ein Aufsteigen der
Schmelze am Rührer zu unterbinden. Nach 9 min wird der N2-Fluss abgestellt, um ein
partielles Austragen des entstehenden Polymers am Temperaturfühler zu vermeiden. Etwa
20 min nach der Katalysatorzugabe wird die Regelung der Temperatur vom Ölbad des
Thermostaten auf den Prozess umgestellt, um eine bessere Kontrolle der Reaktions-
temperatur zu gewährleisten. Das Drehmoment hat zu diesem Zeitpunkt bereits seinen
Endwert erreicht (600 Ncm). Die Reaktion wird noch für weitere 20 min fortgeführt und
dann mit dem Ausfahren des Polymers über eine Dreilochdüse begonnen. Um ein Einfrieren
der Schmelze beim Ausfahren zu vermeiden, wird die Düse von außen mit einem
Heißluftgebläse erwärmt. Der ausgetragene Polymerstrang wird über ein ca. 2 m langes
Förderband geführt und dabei mit einer „N2-Dusche“ auf RT abgekühlt. Etwa alle 8 min
werden die 3 Stränge abgetrennt und das bis dahin ausgetragene Polymer zu einer
Probenfraktion vereint. Insgesamt können über diesen Austragungsweg auf Grund der
hohen Polymerviskosität in einem Zeitraum von 50 min nur 374 g (Ausbeute: 46,8%) Produkt
gewonnen werden.
Um die Ausbeute zu erhöhen, wird der Reaktor nach dem Ausfahren geöffnet und so viel wie
möglich des verbliebenen Polymers mit Hilfe eines Spatels aus dem Reaktor entnommen.
Page 42
34
Das so erhaltene Polymer (129 g, Ausbeute: 16%) kann im Technikum-Maßstab nicht weiter
verarbeitet werden und wird aus diesem Grund mittels zweimaligen Lösens in CHCl3 und
Fällen in MeOH entmonomerisiert und dann unter der Probenbezeichnung „PDLA Tech
gefällt“ der Stereokomplexbildung im Labormaßstab (vgl. Abschn. 5.1 und Unterpunkte) zu-
geführt.
Alle mittels Ausfahren der Schmelze erhaltenen Probenfraktionen werden mit einem Granu-
lierer zu Mikrogranulat verarbeitet und bis zur weiteren Verarbeitung über P2O5 gelagert.
4.3.2. Blockcopolymer-Synthese
Versuchsvorschrift
Zur Vorbereitung des Reaktors wird dieser mit Toluol ausgekocht, dreimal sekuriert und
anschließend unter N2-Atmosphäre auf RT abgekühlt. Die frisch umkristallisierten Mono-
mere D,D- und L,L-Lactid werden zu gleichen Teilen miteinander vermischt um 364 g D,L-
Lactid zu erhalten. Das Monomer wird wie in Abschnitt 4.3.1 beschrieben in den Reaktor
überführt. Außerdem werden noch 2,26 g 1-Dodecanol zur Einstellung der Molmasse auf
30000 g/mol hinzugegeben. Nach dem Verschließen des Reaktors wird auf eine Temperatur
von 180 °C (Ölbadtemperatur) geheizt. Nach 15 min Heizdauer (bei ca. 140 °C) werden
11,8 mL 1%ige Katalysator-Lösung (in Toluol) zur Lactidschmelze gegeben und damit die
Reaktion (t = 0) gestartet. Die Rührgeschwindigkeit beträgt dabei 24 U/min. Der Reaktions-
verlauf wird über das Ansteigen des Drehmoments am Monitor verfolgt. Nach Erreichen
eines konstant bleibenden Drehmoments (etwa 15 min bei ca. 22 Ncm) wird eine erste
Probe entnommen. Im nachfolgenden Schritt wird der Reaktor wieder verschlossen und wie
bereits im Laborversuch (vgl. Abschn. 4.2.2) zur Entmonomerisierung des Prepolymers
Vakuum angelegt. Dabei wird zunächst mit einem moderaten Vakuum begonnen, das dann
langsam bis auf 60 mbar erhöht wird. Die Entmonomerisierung wird insgesamt über einen
Zeitraum von 40 min durchgeführt. Das Drehmoment erhöht sich dabei leicht auf 28 Ncm.
Nach Abschluss der Entmonomerisierung wird eine zweite Probe entnommen.
Parallel zu diesen Arbeiten werden die für die zweite ROP benötigten 540 g L,L-Lactid in
einer Schott-Flasche im Trockenschrank bei 160 °C aufgeschmolzen.
Das aufgeschmolzene Lactid sowie 5 mL 1%ige Katalysator-Lösung (in Toluol) werden über
einen Trichter in den Reaktor gegeben. Dieser wird anschließend wieder verschlossen und
die Rührgeschwindigkeit für wenige Minuten zur intensiven Durchmischung der Kom-
ponenten auf 80 U/min gesteigert. Der Verlauf der Reaktion wird wiederum über das An-
steigen des Drehmoments am Monitor verfolgt. Nach Erreichen eines konstanten Wertes
(nach etwa 1 h) wird mit dem Ausfahren der Polymerschmelze begonnen. Hierfür wird eine
Dreilochdüse verwendet. Diese wird mit einem Heißluftgebläse extern beheizt, um ein
Einfrieren der Schmelze am Düsenausgang zu vermeiden. Das ausgetragene Produkt wird
Page 43
35
über ein ca. 2 m langes Förderband geführt und dabei mit einer „N2-Dusche“ auf RT
abgekühlt. Etwa alle 5 min werden die 3 Probenstränge abgetrennt und die bis dahin aus-
getragene Probe zu einer Fraktion vereint. Über einen Zeitraum von 1 h werden so 536 g
(Ausbeute: 60%) Produkt erhalten.
Alle Probenfraktionen werden mit einem Granulierer zu Mikrogranulat verarbeitet und bis
zur weiteren Verarbeitung über P2O5 gelagert.
Die Anteile der Probe, die z.B. auf Grund des Verklebens der Polymerstränge nicht zu Mikro-
granulat verarbeitet werden können, werden mittels zweimaligen Lösens in CHCl3 und Fällen
in MeOH entmonomerisiert und dann unter der Probenbezeichnung „Co-Tech gefällt“ der
Stereokomplexbildung im Labormaßstab (vgl. Abschn. 5.1 und Unterpunkte) zugeführt.
4.3.3. Entmonomerisierung von Homo- und Blockcopolymer
Die Vakuum-Entmonomerisierung des im Technikum hergestellten Homo- und Blockco-
polymers wird an einem Mini-Compounder (Brabender KETSE 12/36) durchgeführt, der in
der weiteren Arbeit auch für die Stereokomplexbildung zum Einsatz kommt (vgl.
Abschn. 5.2).
Versuchsvorschrift
Für das Homopolymer wird der Extruder in allen Zonen auf 188 °C, für das Copolymer auf
180 °C aufgeheizt. Das Polymergranulat wird in eine Dosierwaage gegeben und mit einem
Durchsatz von 150 g/h dem Extruder zugeführt. Mit dieser Dosiereinstellung wird bei der
gewählten Extruderkonfiguration (Schneckenaufbau vgl. Bild A.5 im Anhang) und einer
Schneckendrehzahl von 300 U/min eine Verweilzeit von 6 min erzielt. Nach vollständiger
Befüllung des Extruders wird an zwei Entgasungspunkten zunächst ein schwaches Vakuum
angelegt, das sukzessive auf die Drücke von 820 mbar und 375 mbar (im Fall des Homo-
polymers) reduziert wird. Bei dem Copolymer werden Endwerte zwischen 850-800 mbar und
470-420 mbar eingestellt. Zum Erreichen dieser Werte werden 2 Vakuumpumpen
verwendet, die jeweils mit 2 Kühlfallen ausgestattet sind. Die Erste wird dabei mit einer
Trockeneis-Aceton-Mischung, die Zweite mit Eiswasser gekühlt.
Der aus dem Extruder geförderte Polymerstrang (kleine Düse; Ø 2 mm) wird über ein etwa
2 m langes Förderband gefördert und mit einer „N2-Dusche“ auf RT abgekühlt. Sobald die
oben genannten Endvakuumwerte stabil sind, kann mit der Probenentnahme begonnen
werden. Dazu wird der Polymerstrang etwa alle 20 min abgetrennt und die bis dahin
aufgefangene Probe zu einer Fraktion vereint.
Insgesamt können (inklusive Vorlauf) rund 90% der eingesetzten Probenmenge zurück-
gewonnen werden.
Page 44
36
4.3.4. Probencharakterisierung
Alle Probenfaktionen der Polymere „PDLA Tech“ und „Co-Tech“ wurden sowohl nach der
Synthese (vgl. Abschn. 4.3.1 und Abschn. 4.3.2) als auch nach der Entmonomerisierung (vgl.
Abschn. 4.3.3) mittels GPC und DSC untersucht. So können Veränderungen der Molmasse,
des Restmonomergehalts und der thermischen Eigenschaften während der Synthese und
nach der Entmonomerisierung dokumentiert werden.
4.3.4.1. Homopolymer „PDLA Tech“
Die einzelnen Probenfraktionen wurden unmittelbar nach der Synthese untersucht. Für das
Homopolymer „PDLA Tech“ wurde eine durchschnittliche Molmasse (Mn) von rund
77000 g/mol und ein PDI von 2,0 ermittelt. Der Restmonomergehalt (GPC) betrug zunächst
rund 4,9%. Durch den Entmonomerisierungsschritt konnte der Restmonomergehalt (GPC)
auf 0,6% reduziert werden. Allerdings resultierte aus dem Entmonomerisierungsschritt auch
eine Molmassenreduzierung auf durchschnittlich 53600 g/mol bei einem PDI von 2,6.
Die Thermogramme zeigen sowohl vor als auch nach der Entmonomerisierung den schon in
Abschnitt 4.2.3.1 beschriebenen Verlauf, charakterisiert durch einen Glasübergangspunkt,
zwei Kalt-Kristallisationspeaks und einen Schmelzpeak. Nach der Synthese findet man hier
durchschnittliche Werte von Tg = 53,5 °C, Tcc1 = 97,4 °C und Tcc2 = 154,2 °C sowie
Tm = 173,2 °C. Nach der Entmonomerisierung werden geringe Änderungen gefunden. Für
den Tg wird hier ein Wert von 50,5 °C, für Tcc1 und Tcc2 Werte von 87,9 °C und 156,6 °C und
für Tm ein Wert von 174,0 °C ermittelt. Die unterschiedlichen Werte des Glasübergangs
sowie des ersten Kristallisationspeaks sind basierend auf Erfahrungswerten auf die
voneinander abweichenden Molmassen zurückzuführen. Der geringfügig höhere
Schmelzpunkt sowie der höhere Wert von Tcc2 lassen sich mit der höheren Reinheit des
entmonomerisierten Polymers erklären. Die äußerst geringe Steigerung von Tm ist mit der
gleichzeitig gesunkenen Molmasse zu begründen.
Der höhere Reinheitsgrad und die damit gesteigerte Kristallinität des Materials nach der
Entmonomerisierung spiegeln sich auch in der Gesamtenthalpie (vgl. Abschn. 3.8.2) des Poly-
mers wieder. Hier konnte ein Wert von 51,1 J/g ermittelt werden. Vor der Entmono-
merisierung wurde lediglich ein Wert von durchschnittlich 8,2 J/g gefunden.
Für die Probe „PDLA Tech gefällt“ (vgl. Abschn. 4.3.1) beträgt die Molmasse rund
77000 g/mol bei einem PDI von 2,4. Der mit der GPC ermittelte Restmonomergehalt liegt
ebenfalls bei 0,6%. Die DSC-Analyse zeigt für das gefällte Homopolymer einen Tg von 61,0 °C
sowie die beiden Kalt-Kristallisationspeaks bei 101,0 °C und 158,6 °C. Für den Schmelzpunkt
wird ein Wert von 175,7 °C bestimmt.
Page 45
37
4.3.4.2. Blockcopolymer „Co-Tech“
Die vereinten Fraktionen des Blockcopolymers haben direkt nach der Synthese eine durch-
schnittliche Molmasse von 72400 g/mol bei einem PDI von 2,2. Der Restmonomergehalt
beträgt ebenso wie beim Homopolymer (vgl. Abschn. 4.3.4.1) 4,9%. Nach der Entmonomeri-
sierung liegt die Molmasse bei durchschnittlich 62100 g/mol mit einem PDI von 1,7, der
Restmonomergehalt bei 0,6%.
Die DSC-Untersuchungen nach der Synthese zeigen im Prinzip das für die synthetisierten
Blockcopolymere typische Bild (vgl. Abschn. 4.2.3.2). Abweichend hiervon ist allerdings, dass
die Kristallisation während des Hochheizens, die sich wie gewöhnlich über ein weites Tem-
peraturfenster von ca. 40 °C erstreckt, nicht so ausgeprägt ist. Auch der sich unmittelbar an
die Kristallisation anschließende Schmelzpeak ist im Vergleich zu den im Labor hergestellten
Copolymeren wesentlicher kleiner. Lediglich für den Glasübergangspunkt zeigt sich eine
deutlich stärker ausgeprägte Stufe (vgl. Bild A.4 im Anhang). Für den Tg wird dabei eine
durchschnittliche Temperatur von 53,7 °C ermittelt. Für den Schmelzpunkt wird ein Wert
von 155,7 °C bestimmt.
Nach der Entmonomerisierung haben sowohl der Kristallisations- als auch der Schmelzpeak
die gewohnt ausgeprägte Form bei insgesamt unveränderten Temperaturen.
Für die Probe „Co-Tech gefällt“ (vgl. Abschn. 4.3.2) wird eine Molmasse von rund
69800 g/mol und ein PDI von 2,3 ermittelt. Der Restmonomergehalt beträgt hier 1,1%. Das
erhaltene Thermogramm der Probe gleicht dem der ungefällten und noch nicht
entmonomerisierten Probe. Der Glasübergangspunkt zeigt für das gefällte Copolymer einen
Wert von 56,7 °C. Die Schmelztemperatur liegt bei 157,1 °C. Die Kristallisation ist bei der
Probe „Co-Tech gefällt“ so schwach ausgeprägt, dass sie nicht ausgewertet werden kann.
4.3.5. Probenübersicht
Die nachfolgende Tabelle 4.2 gibt einen Überblick der Charakteristika der beiden Technikum-
Polymere sowie der entsprechenden, mittels Lösen und Fällen entmonomerisierten Proben.
Page 46
38
Tabelle 4.2
Probenübersicht Technikum-Maßstab
Proben-bezeichnung
Zusammen-setzung
Mn [g/mol] PDI Tg [°C]
Tcc [°C] ΔHcc [J/g]
Tm [°C] ΔHm [J/g] Blendanteil
PDLA Tech. 53600
2,6 50,5 87,9/156,6
3,6/5,4 174,0 60,0 /
PDLA Tech. gefällt
77000 2,4 61,0
101,0/158,6 29,9/7,6
175,7 55,5 /
Co-Tech 55% PDLLA 45% PLLA
62100 1,7 53,7
121,1 19,0
155,7 17,7 9,0%
Co-Tech gefällt
55% PDLLA 45% PLLA
69800 2,3 56,7
nicht auswertbar
157,1 1,0 8,8%
Page 47
39
5. Stereokomplexe
Die Polymere aus den Labor- (vgl. Abschn. 4.2.4), als auch aus den Technikum-Versuchen
(vgl. Abschn. 4.3.4.1 und 4.3.4.2) wurden mit Hilfe zweier unterschiedlicher Extruder zu Ste-
reokomplexen umgesetzt. Ausschlaggebend für die Wahl des jeweiligen Extruders war dabei
die vorhandene Probenmenge. Die Laborproben wurden aus diesem Grund mit dem „HAAKE
MiniLab“ verarbeitet, die Technikum-Proben mit dem „Brabender Mini-Compounder“.
5.1. Extruder „HAAKE MiniLab“
Der Laborextruder „HAKKE MiniLab“ arbeitet mit zwei konischen Schnecken und besitzt
einen integrierten Rückflusskanal. Über ein Bypassventil, mit dem der Rückflusskanal zu- und
weggeschalten werden kann, ist es möglich eine definierte Verweilzeit des Probenmaterials
einzustellen. Das Fassungsvermögen des Extruders liegt bei ca. 10 g.
5.1.1. Probenvorbereitung
Die in den Laborversuchen erhaltenen Polymerproben (vgl. Abschn. 4.2.4) liegen nach dem
zweiten Fällen als „Flocken“ und teils faserartige Gebilde vor (Bild 5.1 links). Für eine
Mischung von Homo- und Copolymer sowie die anschließende Verarbeitung im Extruder,
dessen Öffnungsdurchmesser ca. 1,5 cm beträgt, ist das Material in dieser Form nicht ein-
setzbar. Aus diesem Grund wurden beide Polymertypen vor der Extrusion in einer Zentri-
fugalmühle (Retsch ZM 200) zu feinem Mahlgut (Bild 5.1 rechts) verarbeitet. Dazu wurde
eine Rotorgeschwindigkeit von 8000 U/min sowie ein Ringsieb mit Trapezlochung der Größe
1,5 mm verwendet. Während des gesamten Mahlvorgangs wurden sowohl die Polymere als
auch die Mühle mit flüssigem Stickstoff gespült, um so eine thermische Belastung des Mate-
rials während des Mahlvorgangs zu vermeiden.
Page 48
40
Bild 5.1
Polymer vor (links) und nach (rechts) dem Vermahlen
Das erhaltene Mahlgut wurde abschließend in Petrischalen über Nacht bei 40 °C mit P2O5 im
Vakuumtrockenschrank getrocknet.
Die Molmasse der Proben reduzierte sich durch diesen Aufarbeitungsschritt um ca. 9%.
5.1.2. Durchführung
Die gemahlenen und getrockneten Proben werden in einem Verhältnis von 5 g Copolymer zu
5 g Homopolymer bzw. 10 g Homopolymer (ohne Homogenisierung) als Vergleichsmuster in
einer 250 mL Schott-Flasche eingewogen und für ca. 5 min in einem Turbula-Mischer
homogenisiert. Der Extruder wird auf die Verarbeitungstemperatur von 230 °C vorgeheizt
und die Schnecken auf eine Geschwindigkeit von 200 U/min kalibriert. Nachfolgend werden
die Probenmischung /das Homopolymer über einen Zeitraum von durchschnittlich 15 min in
das vorbereitete System eingefüllt und für 4 min im Kreislauf gefahren. Abschließend
werden der resultierende Blend und das Homopolymer ausgefahren und bis zur weiteren
Verarbeitung über P2O5 gelagert.
Die gesamte Prozedur wird je nach vorhandener Menge der jeweiligen Proben mit frischem
Material zwei bis dreimal wiederholt.
5.1.3. Probenübersicht
In der nachfolgenden Tabelle 5.1 findet sich eine Auflistung der im Labormaßstab her-
gestellten Stereokomplexe sowie deren Ausgangsstoffe. Wie schon in Abschnitt 5.1.2 be-
schrieben, werden die jeweils eingesetzten Edukte in gleichen Gewichtsverhältnissen
miteinander umgesetzt. Die charakterisierenden Kenngrößen der resultierenden Stereo-
komplexe werden in Abschnitt 5.3.1 beschrieben und diskutiert.
Page 49
41
Tabelle 5.1
Mittels „Haake MiniLab“ hergestellte Stereokomplexe (Mischungsverhältnis 1:1)
Homopolymer Copolymer resultierender Stereokomplex
PLLA PDLA02 sc01
PDLA02 Co-08 Co-sc01
PDLA01 Co-12 Co-sc03
PDLA Tech gefällt Co-Tech gefällt Co-sc04
PDLA Tech gefällt Co-15 Co-sc05
PDLA01 Co-13 Co-sc02
5.1.4. Reproduzierbarkeitsuntersuchungen
Zur Untersuchung der Reproduzierbarkeit der Verblendung im Extruder wurden die Pro-
dukte der einzelnen Durchläufe einer Beispielprobe nicht vereint, sondern separat auf-
gefangen und anschließend mittels GPC und DSC untersucht. Die Ergebnisse dieser
Untersuchungen sind in den beiden nachfolgenden Tabellen 5.2 und 5.3 dargestellt.
Tabelle 5.2
Ergebnisse der GPC-Auswertungen (HFIP-GPC; vgl. Abschn. 3.8.1)
Probenbezeichnung Mn
[g/mol] Mw
[g/mol] PDI
Mini 1 48920 124200 2,54
Mini 2 46790 116900 2,55
Mini 3 51610 124600 2,42
Mini 4 62590 142300 2,27
Mittelwert 52478 127000 2,45
Standardabweichung 7024 10797 0,13
Tabelle 5.3
Ergebnisse der DSC-Auswertungen (1.Heizzyklus ausgewertet; vgl. Abschn. 3.8.2)
Probenbezeichnung Tm Homo1
[°C] ΔHm Homo1
[J/g] Tm Homo2
[°C] ΔHm Homo2
[J/g] Tm sc [°C]
ΔHm sc [J/g]
Mini 1 157,5 5,4 175,4 13,0 214,6 31,0
Mini 2 157,2 5,0 174,7 14,4 214,1 33,0
Mini 3 157,8 6,7 175,9 13,3 214,6 34,0
Mini 4 157,6 7,2 175,4 13,3 214,1 32,5
Mittelwert 157,5 6,1 175,4 13,5 214,3 32,6
Standardabweichung 0,28 1,05 0,49 0,62 0,30 1,22
Page 50
42
Wie aus den Ergebnissen zu entnehmen ist, weist die Verarbeitung am „MiniLab“-Extruder
eine gute Reproduzierbarkeit auf.
Die vorhandenen Abweichungen bei den Molmassen sind mit hoher Wahrscheinlichkeit auf
eine etwas schnellere bzw. langsamere Befüllung des Extruders zurückzuführen (vgl.
Abschn. 5.1.2). Beim Aufgeben der Probe kann es zum Verkleben des frisch eingefüllten
Polymers kommen, wodurch eine weitere Befüllung erschwert und verzögert wird.
Die Analyse der thermischen Eigenschaften zeigt für die vier Proben vergleichbare Werte.
Für die Schmelztemperaturen der beiden „Homopolymere“ (Tm Homo1, Tm Homo2; vgl.
Abschn. 5.3) sowie des Stereokomplexes wird eine durchschnittliche Standardabweichung
von 0,36 gefunden. Die dazu gehörenden Enthalpiewerte zeigen eine Standardabweichung
von durchschnittlich 0,96.
5.2. Mini-Compounder „Brabender KETSE 12/36“
Der Mini-Compounder „Brabender KETSE“ ist ein gleichläufiger Doppelschneckenextruder,
der über eine Hauptdosieröffnung sowie über zwei weiteren Dosieröffnungen entlang des
Fließkanals verfügt, die auch als Entgasungspunkte genutzt werden können (vgl.
Abschn. 4.3.3). Der Compounder verfügt über 8 Temperaturzonen. Die beiden Schnecken
lassen sich aus Förder-, Stau- und Mischelementen modular konfigurieren und können so
den jeweiligen Prozessbedingungen (z.B. lange Verweilzeit oder intensive Durchmischung)
angepasst werden. Die Dosierung kann in einer Geschwindigkeit von 50 bis 500 g/h erfolgen.
Mit den gewählten Einstellungen sollten zur besseren Vergleichbarkeit die Bedingungen am
„Haake MiniLab“ nachempfunden werden. Vorrangig diente der Extrusionsprozess aber der
Sterokomplexbildung. Im Rahmen der praktischen Arbeiten wurden keine Optimierungen
des Extrusionsprozesses (Schneckenkonfiguration, -drehzahl, Dosierung) vorgenommen.
5.2.1. Durchführung
Die entmonomerisierten und granulierten Technikum-Proben (vgl. Abschn. 4.3.3) werden in
unterschiedlichen Verhältnissen (vgl. Tab. 5.4) in eine 250 mL Schott-Flasche eingewogen
und für ca. 5 min im Turbola-Mischer homogenisiert. Eine längere Mischzeit ist nicht
sinnnvoll, da sich das Material dabei statisch auflädt und dann keine effektive Durch-
mischung mehr garantiert werden kann. Außerdem ist auch eine verlustfreie Überführung
des Materials in die Dosierwaage des Extruders nicht mehr möglich.
Der Extruder wird in allen Zonen auf die Verarbeitungstemperatur von 230 °C geheizt,
Page 51
43
außerdem wird die Dosierwaage kalibriert. In der Anlaufphase wird zur Befüllung des Ex-
truders eine Dosiergeschwindigkeit von 150 g/h bei einer Schneckendrehzahl von 200 U/min
gewählt. Nachdem die erste Probe aus dem Extruder ausgetreten ist (nach ca. 7 min), wird
die Dosierung auf 400 g/h erhöht. Die Schneckendrehzahl bleibt dabei unverändert. Für die
gewählten Einstellungen sowie die verwendete Schneckenkonfiguration (vgl. Bild A.6 im
Anhang) wurde eine Verweilzeit des Polymers im Extruder von 4 min ermittelt.
Alle Mischungsverhältnisse werden kontinuierlich, beginnend mit der Probe „Co-scTech +55“
und chronologisch der Tabelle 5.4 folgend verarbeitet ohne den Extruder zwischen den ein-
zelnen Proben zu öffnen bzw. zu säubern. Um die jeweils aus den unterschiedlichen
Mischungsverhältnissen resultierenden Proben identifizieren zu können, werden zwischen
den verschiedenen Chargen farbige PLLA-Masterbatches dosiert. Wenn keine Verfärbung
mehr sichtbar ist, wird mit dem Auffangen der jeweils neuen Probe begonnen. Die
resultierenden Polymerstränge werden mit Hilfe eines Granulierers zu Mikrogranulat ver-
arbeitet und mittels DSC und TGA analysiert (Ergebnisse vgl. Abschn. 5.3.2).
Auf Grund von gleichbleibenden bzw. (durch die thermische Belastung begründet) leicht
reduzierten Molmassen (Ergebnisse vgl. Tab. A.1 im Anhang) der Produkte kann ange-
nommen werden, dass die beiden Ausgangsstoffe während des Extrusionsprozesses nicht
miteinander reagieren, sondern wie gewünscht geblendet werden. In der Arbeit von
Fukushima et al. [138] werden im Falle einer Reaktion für die Produkte Molmassen be-
schrieben, die sich deutlich von denen der Edukte unterscheiden.
Bis zur weiteren Verarbeitung werden die Proben über P2O5 gelagert.
5.2.2. Probenübersicht
Die nachfolgende Tabelle 5.4 enthält eine Auflistung der eingestellten Mischungsverhältnisse
(in Gew.-%) zwischen Homo- und Copolymer sowie die Bezeichnungen der daraus gebildeten
Stereokomplexe. Die jeweiligen Produkteigenschaften werden in Abschnitt 5.3.2 be-
schrieben und diskutiert.
Tabelle 5.4
Mischungsverhältnisse (in Gew.-%) der Edukte und Bezeichnung der daraus mittels „Brabender Mini-
Compounder“ hergestellten Stereokomplexe
Homopolymer PDLA Tech
Copolymer Co-Tech
resultierender Stereokomplex
50 50 Co-scTech +55
38 62 Co-scTech +26
31 69 Co-scTech ±0
25 75 Co-scTech -26
18 83 Co-scTech -55
Page 52
44
5.3. Probencharakterisierung
Alle generierten Stereokomplex-Proben sind umfassend auf ihre thermischen, mechanischen
als auch spektroskopischen Eigenschaften hin untersucht worden. Dabei standen vor allem
die Bildung des Stereokomplexes sowie der Einfluss der Blend-Zusammensetzung auf die
verschiedenen Proben-Charakteristika im Vordergrund.
In den nachfolgenden Kapiteln werden alle Ergebnisse der „Labor-“ und der „Technikum-
Stereokomplexe“ dargelegt und ausführlich diskutiert.
5.3.1. Stereokomplexe des Labormaterials („Co-sc“-Proben)
An allen Proben (vgl. Tab. 5.1) sind zur Überprüfung der Stereokomplexbildung zunächst
DSC-Untersuchungen durchgeführt worden, die in jedem Fall bestätigt werden konnte.
Außerdem wurden verschiedene Zusammenhänge zwischen der Größe des amorphen Teils
im Blockcopolymer und den thermischen Eigenschaften des resultierenden Stereokomplexes
gefunden. Tabelle 5.5 gibt einen Überblick der in den DSC-Messungen ermittelten Werte.
Tabelle 5.5
Übersicht der DSC-Auswertungen (1.Heizzyklus ausgewertet; vgl. Abschn. 3.8.2)
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Homo2 [°C]
ΔHm Homo2 [J/g]
Tm sc [°C]
ΔHm sc [J/g]
Tc [°C]
ΔHc [J/g]
ΔHgesamt
[J/g]
sc01 / / / / 226,2 68,8 134,4 56,9 68,8
Co-sc01 98,0 28,9 171,7 17,2 221,1 45,4 121,0 46,0 33,7
Co-sc03 99,8 33,0 171,0 13,2 217,0 43,5 118,7 39,3 23,7
Co-sc04 101,9 161,2
38,0 10,6 173,1 11,9 214,1 41,1 112,7 21,6 4,4
Co-sc05 105,7 161,8
31,5 7,4 175,2 17,5 211,2 24,9 / / 3,5
Co-sc02 100,5 161,0
36,0 2,3 174,1 22,3 224,8 49,0 126,3 35,4 33,0
Es ist festzustellen, dass für alle „Co-sc“-Proben (Blend aus Homo- und Copolymer) sowohl
Homo- als auch Stereokomplexkristalle gefunden werden. Dies kann mit dem vorhandenen
Überschuss an Homopolymer in der jeweiligen Mischung erklärt werden. Für den reinen
Stereokomplex „sc01“ findet man hingegen nur Stereokomplexkristalle. Auch kann bei dieser
Probe der höchste Wert der Gesamtenthalpie (vgl. Abschn. 3.8.2) von 68,8 J/g ermittelt wer-
den. Dieser Wert liegt im Bereich der von He et al. [146] angegebenen Schmelzenthalpie für
Stereokomplexkristalle von 68 bis 86 J/g.
Für die Stereokomplexe mit amorphem Anteil findet man insgesamt deutlich kleinere Werte.
Page 53
45
Außerdem ist hier ein Zusammenhang zwischen der Größe des amorphen Anteils im
eingesetzten Blockcopolymer und der Gesamtenthalpie feststellbar. Zusätzlich sind die
Stereokomplexe mit steigendem amorphen Anteil durch eine sinkende Schmelztemperatur
charakterisiert. So wird für den reinen Stereokomplex eine Schmelztemperatur von 226,2 °C
ermittelt. Für den modifizierten Stereokomplex mit einem amorphen Anteil von 26% („Co-
sc01“) findet man nur noch einen Wert von Tm = 221,1 °C. Bei den Materialien mit 41% („Co-
sc03“), 55% („Co-sc04“) und 80% („Co-sc05“) amorphem Anteil im Copolymer reduzieren
sich die Schmelztemperaturen auf Tm = 217,0 °C, Tm = 214,1 °C und Tm = 211,2 °C. Das
Absinken der Schmelztemperaturen kann auf die schlechtere Kristallisationsfähigkeit der
Materialien auf Grund der Einlagerung der amorphen Kettensegmente bzw. der Ausbildung
kleinerer Kristalle zurückgeführt werden [24, 54, 87].
Von Tsuji und Tajima [24] wird eine Abhängigkeit der Tcc vom sc-Anteil in sb-PLA be-
schrieben. Die diskutierten Analysen zeigen ein Absinken der Tcc mit steigender Menge an
Stereokomplex auf. Dieser Zusammenhang wurde auch bei den eigenen Untersuchungen
gefunden (Tabelle 5.5). Grund hierfür ist, dass Stereokomplexkristalle als Nukleierungsmittel
fungieren [5, 6, 17-19, 23, 24, 26, 70, 71, 75, 80, 93, 144, 147-150] und die Kristallisation
daher bei niedrigeren Temperaturen ablaufen kann.
Bei den Schmelzpunkten der Homopolymere ist hingegen eine gegenläufige Tendenz
feststellbar. Hier steigt die Schmelztemperatur mit wachsendem amorphem Anteil im
Blockcopolymer. Diese Ergebnisse waren zu erwarten, da mit steigendem amorphem Anteil
in den Copolymeren vergleichbarer Molmassen der kristalline Anteil in diesen sinken muss.
Dies hat zur Folge, dass bei Umsetzungen von PDLA mit den verschiedenen Copolymeren
(sinkenden kristallinen Anteils) im ausgewogenen Verhältnis (50:50; vgl. Abschn. 5.1.2)
immer weniger Stereokomplex gebildet werden kann und so ein höherer Anteil an nicht zum
Stereokomplex umgesetztem Homopolymer im System verbleibt. Dieses Homopolymer kann
wiederum immer besser geordnete Kristalle ausbilden, wodurch die Schmelztemperatur
steigt [5].
Außerdem wird auch für die Temperatur der Kristallisation in der Abkühlphase (Tc) sowie für
die entsprechende Kristallisationsenthalpie (ΔHc) eine Abhängigkeit vom amorphen Anteil
gefunden. Sowohl die Kristallisationstemperatur als auch die –enthalpie sinken mit
steigendem PDLLA-Anteil im Copolymer. Dies kann mit der gehinderten Kristallisation erklärt
werden.
Zur Charakterisierung der thermischen Stabilität der verschiedenen Stereokomplexe wurden
TGA-Messungen durchgeführt. Es kann gezeigt werden (Bild 5.2), dass unabhängig vom ent-
haltenen Anteil an PDLLA annähernd gleiche Werte erzielt werden. Die thermische Stabilität
kann nach diesen Beobachtungen also auch für Materialien gewährleistet werden, die
weniger sc-Kristallinität enthalten. Dieses Ergebnis ist so nicht zu erwarten gewesen. Zu
vermuten war, dass für die modifizierten Stereokomplexe auf Grund des enthaltenen
Page 54
46
amorphen Anteils verringerte Werte gemessen werden. Aber angesichts der Startpunkte des
Masseverlustes kann für alle Materialien festgestellt werden, dass der Abbau erst oberhalb
der Schmelztemperatur der Sterokomplex-Kristalle erfolgt [106].
Bild 5.2
TGA-Kurven der unterschiedlichen Stereokomplexe (Temperaturprogamm vgl. Abschn. 3.8.3)
5.3.1.1. Prüfstabherstellung
Zur vergleichenden Analyse der mechanischen Eigenschaften wurde die Methode der
„Schlagbiegefestigkeit nach Charpy“ gewählt. Dazu war es zunächst notwendig genormte
Prüfstäbe herzustellen. Hierzu wurde die Kolbenspritzgießmaschine „HAAKE MiniJet“ ver-
wendet. Zur Formgebung der Prüfstäbe wurde ein Spritzwerkzeug nach ISO 179 eingesetzt.
Versuchsvorschrift
Die als Polymerstrang vorliegenden Proben (vgl. Abschn. 5.1.2) werden mit einem Seiten-
schneider in ca. 1 cm lange Segmente geteilt und zu je 4,7 g Portionen abgewogen. Die
Probenportionen werden nacheinander in den vorgeheizten Zylinder überführt und dort für
3 min aufgeschmolzen. Nachfolgend wird der gefüllte Zylinder in den Arbeitsraum des
„MiniJets“ überführt und die Schmelze über einen Kolben in das temperierte Werkzeug
gepresst. Nach Ablauf der Nachdruckzeit wird das Werkzeug aus der Anlage entnommen und
der fertige Prüfstab aus diesem entfernt. Die genauen Einstellungen des „MiniJets“ mussten
auf die jeweiligen Proben angepasst werden. Eine Auflistung der verwendeten Versuchs-
bedingungen findet sich in der Tabelle A.2 im Anhang.
Die Prüfstäbe wurden bis zur Analyse 4 Tage bei 22 °C und 65% rel. Luftfeuchtigkeit
Page 55
47
konditioniert.
Das Material ließ sich insgesamt nur sehr schwer zu Prüfstäben verarbeiten. Oft waren
Vakuolen vorhanden oder die Prüfstäbe teilweise eingefallen. Aus diesen Gründen konnten
nur wenige Prüfstäbe final der Analyse zugeführt werden. Bei der Probe „sc01“ konnten
zusätzlich dazu nur wenige Prüfstäbe vollständig aus dem Formteil entnommen werden. Auf
Grund der hohen Brüchigkeit des reinen Stereokomplexes kam es schon hierbei oft zur Zer-
störung der Prüfkörper.
Mit dem Material „Co-sc02“ ist es nicht gelungen messbare Prüfkörper herzustellen. Alle
Stäbe enthielten eine Vielzahl an kleinen Bläschen, die ein zu starkes Aufkochen der
Schmelze vermuten ließen. Eine Reduzierung der Zylindertemperatur um 5 °C zog aber schon
das Einfrieren der Austrittsdüse nach sich. So war es auch nach Variation der Versuchs-
bedingungen nicht möglich verwendbare Stäbe zu fertigen. Als Ursache für das im Vergleich
zu den anderen modifizierten Stereokomplexen veränderte Eigenschaftsprofil bei der
Prüfstabherstellung wird die schon in Abschnitt 4.2.3.2 im Zusammenhang mit dem Löse-
verhalten diskutierte Blendzusammensetzung des Ausgangsmaterials „Co-13“ aus Block-
copolymer und PLLA-Homopolymer vermutet. Auf Grund der verschiedenen im System ent-
haltenen Polymere und der daraus gebildeten jeweiligen Stereokomplexe (sc aus
Blockcopolymer und PDLA sowie aus PLLA-Homopolymer und PDLA) mit unterschiedlichen
thermischen Eigenschaften kommt es zu einem inhomogenen Aufschmelzverhalten der
Probe „Co-sc02“. Außerdem kühlen die einzelnen Bestandteile unterschiedlich schnell ab,
was Fehlstellen im Material induzieren kann. Auch konnte an den Prüfstäben eine leichte
opake Verfärbung beobachtet werden. Diese ist laut der Arbeit von Jacobsen et al. [10] auf
das separate Auskristallisieren verschiedener Probenbestandteile zurückzuführen. Das
unterstützt die Hypothese des Vorliegens unterschiedlicher kristalliner Komponenten.
Die Zusammensetzung der Probe „Co-13“ aus mehreren Komponenten kann in diesem
Zusammenhang auch mit den Ergebnissen weiterführender Untersuchungen gezeigt werden.
Es wurde zunächst vermutet, dass es beim Aufschmelzen der Probe „Co-sc02“ im Zylinder
des „MiniJets“ zu einem starken Abbau des amorphen Teils des enthaltenen Block-
copolymers („Co-13“) kommt und dies der Grund für die schlechte Verarbeitbarkeit des
Materials zu Prüfstäben ist. Zur Prüfung dieser Hypothese wurde das Copolymer zweimal im
Zylinder des „MiniJets“ aufgeschmolzen und anschließend mittels GPC und DSC untersucht.
In der GPC konnte nur eine geringe Veränderung festgestellt werden, was einen starken
Molmassenabbau als Begründung ausschließt. In der DSC konnte hingegen eine deutliche
Veränderung des Materials verfolgt werden. So wird aus dem in Abschnitt 4.2.3.2 beschrie-
benen Schmelzpeak mit ausgeprägter Schulter (vgl. Bild 3.7 bzw. Bild A.3 im Anhang) ein
Doppelpeak, dessen Aufspaltung mit jedem Aufschmelzvorgang steigt (Bild 5.3). Auch läuft
die Kristallisation in einem engeren Temperaturfenster ab. Beide Beobachtungen können als
Bestätigung der mehrkomponentigen Zusammensetzung der Probe „Co-13“ herangezogen
Page 56
48
Bild 5.3
Überlagerung der Thermogramme „Co-13 1x aufgeschmolzen“ und „Co-13 2x aufgeschmolzen“ (jeweils 2.
Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
werden. Einen weiteren Hinweis auf eine Blendzusammensetzung der Probe „Co-13“ gibt ein
Vergleich der Thermogramme der Proben „Co-sc02“ sowie „Co-sc01“ nach der Verarbeitung
mit dem „MiniJet“. Beide Stereokomplexe beruhen auf Blockcopolymeren annähernd ver-
gleichbarer Zusammensetzung (Tabelle 5.6). Dennoch werden hinsichtlich der thermischen
Eigenschaften (ΔHm Homo, Tm sc, ΔHm sc) sowohl für die Homopolymere, als auch die resul-
tierenden Stereokomplexe unterschiedliche Werte gefunden (Tabelle 5.7). Unter der
Annahme, dass das Blockcopolymer „Co-13“ wie vermutet als Blend aus PLLA und Co-
polymer vorliegt, sind diese Unterschiede erklärbar.
Stereokomplexe kristallisieren beim Abkühlen im Vergleich zu Homopolymeren wesentlich
schneller [21, 66, 70, 75, 146, 149-152], außerdem bilden Homopolymere mittlerer Mol-
masse (Mn < 40000 g/mol) nahezu ausschließlich Stereokomplexe [87, 149, 153]. Vor-
handenes PLLA in der Probenmischung würde mit dem im Überschuss zugesetzten PDLA also
bevorzugt als Stereokomplex anstelle des Homopolymers auskristallisieren. Da zudem auf
Grund des fehlenden PDLLA-Blocks keine sterische Hinderung vorhanden ist, sollten auch
stabilere Kristalle ohne bzw. mit wenigen Fehlstellen und damit eine höhere Schmelztem-
peratur für den Stereokomplex gefunden werden.
Page 57
49
Tabelle 5.6
Probenübersicht
Proben-bezeichnung
Zusammen-setzung
Mn [g/mol] PDI
resultierender Stereokomplex
Co-08 26% PDLLA 74% PLLA
46100 2,3 Co-sc01
Co-13 31% PDLLA 69% PDLA
53600 2,0 Co-sc02
Tabelle 5.7
DSC-Auswertung (1. Heizzyklus ausgewertet; vgl. Abschn. 3.8.2)
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Homo2 [°C]
ΔHm Homo2 [J/g]
Tm sc [°C]
ΔHm sc [J/g]
Tc [°C]
ΔHc [J/g]
ΔHgesamt
[J/g]
Co-sc01 / / 172,3 3,3 220,7 52,8 124,0 50,7 56,1
Co-sc02 / / 172,0 0,9 227,5 71,1 129,5 49,5 72,0
sc01 / / / / 228,8 81,8 137,8 54,9 81,8
Die in Tabelle 5.7 aufgeführten Ergebnisse bestätigen diese Schlussfolgerungen. Für die
Probe „Co-sc02“ („Blend“) wird im Vergleich zur Probe „Co-sc01“ („Blockcopolymer“) eine
geringere Schmelzenthalpie für das Homopolymer gefunden. Außerdem zeigen sich sowohl
ein bedeutend höherer Wert für die Schmelztemperatur des Stereokomplexes als auch für
seine Schmelzenthalpie.
Auch der Vergleich mit den Ergebnissen des reinen Stereokomplexes „sc01“ unterstützt die
Annahme, dass die Probe „Co-13“ als Blend aus Blockcopolymer und PLLA vorliegt. Für alle
aufgeführten Messgrößen werden (unter Berücksichtigung des amorphen Anteils) vergleich-
bare Werte gefunden.
5.3.1.2. Mechanische Eigenschaften
Schlagbiegefestigkeit nach Charpy
Die Beschreibung der Methode findet sich in Abschn. 3.8.5.
Die Ergebnisse resultieren aus Mittelwerten von 3 bis 5 Einzelmessungen.
Für alle modifizierten Stereokomplexe können im Vergleich zum reinen Stereokomplex
„sc01“ bessere Charpy-Werte gefunden werden (Bild 5.4). Auch das Homopolymer weist im
Vergleich zu „sc01“ einen besseren Wert auf. Diese Beobachtung deckt sich mit den von
Nam et al. [53, 57] aufgeführten Untersuchungsergebnissen. Es zeigt sich auch, dass bereits
mit 26% amorphem Anteil im Stereokomplex („Co-sc01“) ein mit dem Homopolymer (PLLA)
vergleichbarer Messwert (4,8 kJ/m2) erzielt werden kann.
Page 58
50
Der Zusammenhang zwischen dem amorphen Anteil im Copolymer und den ermittelten
Charpy-Werten ist annähernd linear.
Bild 5.4
Bildliche Darstellung der Messwerte zur Schlagbiegefestigkeit (die Bezeichnungen 0, 26, 41, 55 und 80
entsprechen den Stereokomplexen „sc01“, „Co-sc01“, „Co-sc03“, „Co-sc04“ und „Co-sc05“)
Zur genaueren Analyse der Schlagbiegefestigkeitswerte wurden ergänzend DCS-Messungen
an den Charpy-Stäben durchgeführt. Die Auswertung der resultierenden Thermogramme
findet sich in Tabelle 5.8.
Tabelle 5.8
DSC-Charakterisierung der Charpy-Stäbe (1. Heizzyklus ausgewertet, vgl. Abschn. 3.8.2)
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Ho
[°C] ΔHm Ho [J/g]
Tm sc [°C]
ΔHm sc [J/g]
Tc [°C]
ΔHc [J/g]
ΔHgesamt [J/g]
PLLA / / 171,3 58,0 / / 106,4 42,6 58,0
sc01 / / / / 228,8 81,8 137,8 54,9 81,8
Co-sc01 / / 172,3 3,3 220,7 52,8 124,0 50,7 56,1
Co-sc03 / / 170,7 2,4 220,0 51,7 122,1 42,7 54,1
Co-sc04 / / 173,0 1,7 214,3 48,9 113,5 25,6 50,6
Co-sc05 / / 171,8 11,1 211,1 29,2 / / 40,3
Co-sc02 / / 172,0 0,9 227,5 71,1 129,5 49,9 72,0
Aus verschiedenen Quellen [1, 3, 9, 22, 31, 33, 48, 49, 56, 61, 69, 93, 100, 153] ist bekannt,
dass die mechanischen Eigenschaften stark von der vorhandenen Kristallinität sowie den
Page 59
51
generierten Kristallgrößen in den Proben beeinflusst werden. Allgemein gilt, dass kleinere
Kristalle eine höhere Schlagbiegefestigkeit ermöglichen. Für reines Polylactid gilt außerdem,
dass mit steigender Kristallinität auch die Schlagbiegefestigkeit zunimmt. Für stereo-
komplexes PLA wird hingegen ein gegenläufiger Trend gefunden. Mit steigender sc-
Kristallinität werden kleinere Werte für die Schlagbiegefestigkeit angegeben [53]. Dieser
Zusammenhang kann auch bei den eigenen Untersuchungen beobachtet werden. Auf Grund
der Proportionalität zwischen Kristallinität und Schmelzenthalpie [5, 154] kann mit Hilfe der
für die Proben ermittelten Gesamtenthalpien auf deren Kristallinität geschlossen werden.
Insgesamt wird für die modifizierten Stereokomplexe mit steigenden Charpy-Werten eine
Abnahme der Gesamtenthalpie gefunden.
Außerdem können verschiedene Abhängigkeiten zwischen der Menge an PDLLA als
Blocksegment des Copolymers, welches als Weichmacher fungiert und den thermischen
Eigenschaften der Materialien beobachtet werden. So sinkt mit steigendem Gehalt an
„Weichmacher“ die Schmelztemperatur. Weiterhin kann auch eine Reduzierung der
Kristallisationstemperatur Tc mit steigendem Weichmacheranteil beobachtet werden. Die
amorphen Kettenenden verursachen sowohl wachsende Abstände zwischen als auch immer
mehr Fehlstellen in den Kristallen. Die Ausbildung weniger perfekter Kristallite und z.B.
sinkende Kristallisationstemperaturen sind die Folge.
Für die Probe „Co-sc01“ (mit Enthalpiewert des Homopolymers) wird im Vergleich zum
reinen Stereokomplex „sc01“ (ohne Enthalpiewert des Homopolymers) ein mehr als 3,5-mal
so großer Wert für die Schlagbiegefestigkeit (4,8 kJ/m2 zu 1,3 kJ/m2) gefunden. Daher kann
das Vorliegen von Homokristallinität bei der Betrachtung der Ergebnisse nicht vernachlässigt
werden.
Auf Grund ihrer kurzen Induktionsphase sowie ihrer hohen „Radius-Wachstumsrate“ [75]
kristallisieren beim Abkühlen der Probenschmelze bevorzugt Stereokomplexe (vgl.
Abschn. 5.3.1.1) aus. Da diese zusätzlich als Nukleierungsmittel der Homopolymere wirken
(vgl. Abschn. 5.3.1), findet man die dadurch hervorgerufene und auch mit der DSC nach-
gewiesene Mischung der beiden Kristallformen. Die Homopolymere bilden nach der
Induktion mit Stereokomplexkristallen an deren Oberfläche [17, 18, 75, 149] viele kleine
Sphärolithe [5, 17, 75], wodurch die gesteigerte Schlagbiegefestigkeit bei Anwesenheit der
Homopolymere bzw. der schlechte Messwert für den reinen Stereokomplex möglicherweise
erklärt werden kann.
Shore Härte
Die Beschreibung der Methode findet sich in Abschnitt 3.8.6.
Die ermittelten Härte-Werte entsprechen den basierend auf den Gesamtenthalpien der
Proben (vgl. Tab. 5.8) erwarteten Ergebnissen. Hohe Enthalpiewerte sprechen für eine hohe
Kristallinität der Probe. Das wiederum bedeutet, dass wenig freies Volumen in der Probe
Page 60
52
vorhanden ist, was eine gesteigerte Festigkeit mit sich bringt [33]. Daher wird für den reinen
Stereokomplex „sc01“ mit dem höchsten Wert der Gesamtenthalpie (81,8 J/g) auch die
größte Härte von 84,3 Shore ermittelt. Für das Homopolymer mit der zweithöchsten
Gesamtenthalpie (58,0 J/g) wird eine Härte von 82,5 Shore gemessen. Für die übrigen
Proben findet sich mit Ausnahme der Probe „Co-Sc03“ ein linearer Zusammenhang zwischen
der Größe des amorphen Teils und der gefunden Härte.
Tabelle 5.9
Ergebnisse der Härtemessungen nach Shore D
Probenbezeichnung Härte
[Shore]
PLLA 82,5
sc01 84,3
Co-sc01 81,3
Co-sc03 81,0
Co-sc04 75,5
Co-sc05 72,9
5.3.1.3. Röntgenbeugungsexperimente
Zur weiterführenden Diskussion der in den Proben enthaltenen Kristallformen wurden an
den Prüfstäben Röntgenbeugungsuntersuchungen (vgl. Abschn. 3.8.8) durchgeführt. Die ent-
sprechenden Planfilmaufnahmen des Homopolymers sowie der verschiedenen Stereo-
komplexe sind in den Bildern 5.5 und 5.6 dargestellt.
Über die Intensität der beobachteten Beugungsringe ist es möglich eine Aussage zur
Kristallinität der Proben zu machen. Je ausgeprägter und klarer (wenig Rauschen) das
jeweilige Interferenzbild, desto mehr Kristalle (hc und/oder sc) sind in der Probe vorhanden.
Bei amorphen Proben können dem entsprechend keine Beugungsringe beobachtet werden.
Zusätzlich dazu kann an Hand der Breite der Beugungsringe eine Aussage über die Kristall-
größe getroffen werden. Auf Grund des reziproken Zusammenhangs zwischen Kristallitgröße
und Ringstärke werden bei schmalen Beugungsringen große Kristalle und bei breiten
Reflexen dem entsprechend kleine Kristalle in der Probe vorliegen.
In verschiedenen Quellen [65, 123, 147, 153, 155-157] werden für das Homopolymer (α-
oder α´-Form) die Beugungswinkel bei 2θ = 15, 17 und 19° als intensivste Reflexe be-
schrieben. Zusätzlich dazu findet man die Angaben von 2θ = 16, 18,5 und 22,5° als typisch für
das Homopolymer [76, 148, 149]. Für den Stereokomplex finden sich in den Quellen ein-
heitliche Aussagen über die Lage der charakteristischen Beugungswinkel bei 2θ = 12, 21 und
24° [5, 20, 26, 28, 65-67, 75, 76, 118, 147, 148, 149, 153, 155-159].
Page 61
53
α-Phase sc-Phase
Bild 5.5
Planfilmaufnahmen der Proben „PLLA“ und „sc01“
26% 41% 55% 80% D,L-Anteil
„Co-sc01“ „Co-sc03” „Co-sc04” „Co-sc05”
Bild 5.6
Planfilmaufnahmen der Proben „Co-sc01“, „Co-sc03“, „Co-sc04“ und „Co-sc05“
Bei den selbst hergestellten Polymeren findet man für die Homopolymerprobe „PLLA“ den
intensitätsstärksten Beugungswinkel bei 2θ = 16,2°, sowie einen zweiten bei 2θ = 18,6°.
Beide Reflexe können der α-Phase zugeordnet werden. Das geringe Rauschen lässt außer-
dem darauf schließen, dass es sich insgesamt um eine kristalline Probe handelt.
Für die Probe „sc01“ wird der intensitätsstärkste Beugungswinkel für 2θ = 11,9° ermittelt.
Außerdem werden Beugungswinkel mit mittlerer und schwacher Intensität bei 2θ = 20,8 °
und 23,7° gefunden. Damit zeigt die Probe „sc01“ das typische Beugungsbild eines voll-
ständig umgesetzten Stereokomplexes. Auf Grund des klaren, wenig verrauschten Hinter-
grundes kann auch hier darauf geschlossen werden, dass es sich um ein kristallines Material
handelt.
Für die Probe „Co-sc01“ wird der intensitätsstärkste Reflex wiederum bei 2θ = 11,9° ge-
funden. Außerdem können auch die anderen für den Stereokomplex typischen Reflexe bei
2θ = 20,8° in schwacher und 23,7° in sehr schwacher Intensität beobachtet werden. Darüber
hinaus werden zusätzlich zwei typische Reflexe der α-Phase bei 2θ = 17,2° und 19,3° in
schwacher und sehr schwacher Intensität gefunden. Diese Ergebnisse decken sich mit den
kalorimetrischen Beobachtungen der DSC-Untersuchungen.
Auch für die Probe „Co-sc03” wird der intensitätsstärkste Reflex bei 2θ = 11,9° ermittelt.
Page 62
54
Außerdem lassen sich Beugungswinkel mittlerer Intensität bei 2θ = 20,8° bzw. mit sehr
schwacher Intensität bei 2θ = 23,7° feststellen. Alle gefundenen Winkel sowie die Intensi-
tätsverteilung stellen wieder das typische Beugungsbild von Stereokomplexkristallen dar. Die
aus der DSC erwartete geringe Homokristallinität kann hier mit Hilfe der Röntgenbeugung
nicht gezeigt werden.
In der Probe „Co-sc04“ findet man neben den in Probe „Co-sc03“ beobachteten Reflexen
auch einen sehr schwachen bei 2θ = 16,2°. Dieser ist hc-Kristallen zuzuordnen. Das hier
gefundene Bild ist demnach vergleichbar mit dem in der DSC ermittelten Ergebnis.
Die Probe „Co-sc05“ zeigt ähnlich wie Probe „Co-sc03“ die typischen Stereokomplex-Reflexe
bei 2θ = 11,9°, 20,8° sowie 23,7° mit vergleichbarer Intensitätsverteilung. Interessante Inter-
ferenzen, die Rückschlüsse auf die Anwesenheit von hc-Kristallen zulassen würden, werden
hier durch die amorphe Untergrundstreuung überschattet.
Die identifizierten Reflexe werden in Tabelle 5.10 zusammengefasst.
Tabelle 5.10
Zusammenstellung der ermittelten Reflexe
sc hc hc hc hc sc sc
Probenbezeichnung 11,9° 16,2° 17,2 18,6° 19,3 20,8° 23,7°
PLLA
m s
sc01 st
m s
Co-sc01 m
s
ss s ss
Co-sc03 st
m s
Co-sc04 st ss
m ss
Co-sc05 st st s
Legende: st stark; m mittel; s schwach; ss sehr schwach
Das Gesamtbild der untersuchten Proben zeigt, dass das Grundrauschen wie erwartet vom
reinen Stereokomplex „sc01“ über die Proben „Co-sc01“, „Co-sc03“ und „Co-sc04“ hin zur
Probe „Co-sc05“ stetig zunimmt und die Materialen daher als zunehmend amorpher
identifiziert werden können.
Zusätzlich dazu konnte festgestellt werden, dass bei keiner der Proben eine Orientierung der
Kristalle vorliegt. Es handelt sich also ausnahmslos um isotrope Materialien.
5.3.2. Stereokomplexe des Technikum-Materials (“Co-scTech“-Proben)
Auch in diesem Fall wurden zur Überprüfung der Stereokomplexbildung zunächst DSC-
Messungen durchgeführt, deren Ergebnisse die Komplexbildung nachgewiesen haben.
Außerdem können Zusammenhänge zwischen den eingestellten Mischungsverhältnissen und
den resultierenden Enthalpiewerten der Homopolymere aufgezeigt werden (Tabelle 5.11).
Page 63
55
Tabelle 5.11
Übersicht der DSC-Auswertungen (1. Heizzyklus ausgewertet; vgl. Abschn. 3.8.2)
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Homo [°C]
ΔHm Homo [J/g]
Tm sc [°C]
ΔHm sc [J/g]
Tc [°C]
ΔHc [J/g]
ΔHgesamt
[J/g]
Co-scTech +55 95,0 38,4
155,6 174,7
3,0 23,8 213,4 25,8 118,8 19,3 14,2
Co-scTech +26 95,1 33,8
158,3 176,3
8,6 11,7 214,8 33,0 124,2 36,6 19,5
Co-scTech ±0 103,0 34,7
159,0 176,1
17,0 4,1 214,4 38,2 120,3 33,8 24,6
Co-scTech -26 109,1 32,5
158,9 /
19,7 / 213,4 28,8 117,0 13,4 16,0
Co-scTech -55 111,6 26,4
159,8 /
27,9 / 214,0 21,3 / / 22,4
Zunächst kann festgestellt werden, dass in den Proben abhängig von der zugesetzten Menge
an PDLA entweder sowohl ein Schmelzpeak für das Copolymer (Tm = 158,3 ± 1,6 °C) als auch
für das Homopolymer (Tm = 175,7 ± 0,9 °C) oder lediglich ein Schmelzpeak für das Copolymer
gefunden wird. Für die beiden PDLA im Unterschuss enthaltenden Proben („Co-scTech -26“
und „Co-scTech -55“) wird dabei wie zu erwarten nur der Schmelzpeak für das Copolymer
erhalten. Für den Enthalpiewert des Copolymers wird insgesamt ein steigender Trend mit
Reduzierung des Anteils an PDLA gefunden. Auch dieses Ergebnis entspricht den Erwart-
ungen, da mit sinkendem PDLA-Zusatz ein höherer Anteil an nicht umgesetztem Copolymer
in der Mischung verbleibt, dar wiederum „reiner“ und stabiler auskristallisieren kann. Die
Bildung stabiler Kristalle ist auch an Hand des Ansteigens der Schmelztemperatur von
155,6 °C in der Probe „Co-scTech+55“ auf 159,8 °C in der Probe „Co-scTech -55“ zu ver-
folgen.
Außerdem lässt sich feststellen, dass für den gebildeten Stereokomplex in den einzelnen
Proben vergleichbare Schmelztemperaturen (Tm = 214,0 ± 0,6 °C) gefunden werden. Auch
dies entspricht dem erwarteten Ergebnis, da alle Stereokomplexe aus den selben beiden
Ausgangsmaterialien bestehen und folglich in allen Proben auch Stereokomplexe ver-
gleichbarer Qualität gebildet werden können.
Hinsichtlich der thermischen Stabilität konnten in TGA-Untersuchungen erwartungsgemäß
im Vergleich zum Ausgangsmaterial höhere und außerdem untereinander vergleichbare
Ergebnisse erzielt werden (Bild 5.7). Hervorzuheben ist, dass auch für die PDLA im
Unterschuss enthaltenden Proben („Co-scTech -26“ und „Co-scTech -55“) keine Verschlech-
terung der thermischen Stabilitäten zu verzeichnen ist. Dieses Ergebnis ist für die
Wirtschaftlichkeit der modifizierten Stereokomplexe von großem Interesse, da PDLA sehr
kostenintensiv ist [24, 25, 71, 75].
Page 64
56
Bild 5.7
TGA-Kurven der Ausgangspolymere sowie der Stereokomplexe (Temperaturprogramm vgl. Abschn. 3.8.3)
5.3.2.1. Prüfstabherstellung
Die granulierten Proben (vgl. Abschn. 5.2.1) werden wie in Abschnitt 5.3.1.1 beschrieben zu
Prüfstäben verarbeitet. Die Wahl geeigneter Bedingungen erwies sich als große Heraus-
forderung, da angestrebt wurde alle Prüfstäbe mit annähernd gleichen Verarbeitungs-
parametern herzustellen um eine Vergleichbarkeit zu wahren. Letztlich wurden jedoch
Bedingungen gefunden, die sich auf annähernd alle Proben (Ausnahme „Co-scTech -55“) an-
wenden ließen. Eine detaillierte Übersicht zu den Versuchsbedingungen findet sich in
Tabelle A.3 im Anhang.
Trotz Modifizierung und Optimierung der Bedingungen bei der Prüfstabherstellung ließ sich
das Material nur schwer verarbeiten. Auch hier wurden in hoher Zahl Prüfstäbe erhalten, die
Vakuolen aufwiesen und daher für die mechanische Prüfung unbrauchbar waren. Die
Unterscheidung zwischen Vakuole bzw. Blase wurde hier so vorgenommen, dass betreffende
Prüfstäbe unter Wasser angebohrt und visuell untersucht wurden. Auf diese Weise konnte
durch das Eindringen von Wasser eine Vakuole bzw. durch das Aufsteigen von Bläschen eine
Blase identifiziert werden.
Letztlich konnten durchschnittlich 10 Prüfstäbe pro Probe für die weiteren Tests eingesetzt
werden.
Alle Prüfstäbe wurden vor der Analyse 5 Tage im Klimaraum (22 °C, 65% rel. Luftfeuchtigkeit)
aufbewahrt. Zusätzlich dazu wurde die Hälfte der Proben nach der Lagerung im Klimaraum
Page 65
57
noch getempert. Dazu wurden die Proben für 30 min in einem auf 140 °C temperierten
Trockenschrank zwischen Glasplatten gelagert.
5.3.2.2. Mechanische Eigenschaften
Schlagbiegefestigkeit nach Charpy
Die Beschreibung der Methode findet sich in Abschnitt 3.8.5.
Die Ergebnisse resultieren aus Mittelwerten von mindestens 3 bis maximal 5 Einzelmess-
ungen.
Bild 5.8
Schlagbiegefestigkeit in Abhängigkeit vom Anteil an PDLA im Copolymer (die Bezeichnungen -55, -26, 0, +26
und +55 entsprechen den Stereokomplexen „Co-scTech -55“, „Co-scTech -26“, „Co-scTech ±0“, „Co-scTech +26“
und „Co-scTech +55“)
Für die nicht getemperten Proben werden zunächst scheinbar uneinheitliche und trendlose
Ergebnisse erhalten. Für die Probe, in der PDLA und PLLA als Block des Copolymers im
gleichen Mengenverhältnis vorliegen („±0“), wird der beste Messwert (33,7 kJ/m2) erzielt.
Für die Proben „-55“, „-26“ und „+26“ besteht ein linearer Zusammenhang zwischen der
Schlagbiegefestigkeit und der zugesetzten Menge an PDLA: je höher der PDLA-Zusatz in der
Polymermischung, desto niedriger ist die Schlagbiegefestigkeit. Dies entspricht dem er-
warteten Ergebnis, da mit steigendem Gehalt an PDLA in den Proben (bis zu einem Maximal-
Page 66
58
wert) auch potenziell mehr Stereokomplex gebildet werden kann und damit ein geringerer
Wert für die Schlagbiegefestigkeit erhalten wird (vgl. Abschn. 5.3.1.2). Weiterhin ist festzu-
stellen, dass die Menge an überschüssig zugesetztem PDLA keinen großen Einfluss auf das
resultierende Messergebnis zu haben scheint. Für die Proben „+26“ und „+55“ werden
annähernd gleiche Schlagbiegefestigkeiten von 14,9 kJ/m2 und 15,3 kJ/m2 erhalten. Daraus
ergibt sich die Annahme, dass beide Proben über eine vergleichbare Kristallinität verfügen.
Diese Vermutung kann anhand von Ergebnissen aus DSC-Messungen (Tabelle 5.12) bestätigt
werden. Hier werden für die beiden diskutierten Proben Gesamtenthalpiewerte von 55,1 J/g
und 53,3 J/g gefunden. Für die übrigen Proben (mit Ausnahme der Probe „±0“ mit gleichem
Massenanteil an PDLA und PLLA als Block des Copolymers) ist in Bezug auf die Gesamt-
enthalpie festzustellen, dass mit steigenden Gesamtenthalpie-Werten kleinere Schlag-
biegefestigkeiten erhalten werden. Dies entspricht dem auch in Abschnitt 5.3.1.2
beschrieben Zusammenhang.
Tabelle 5.12
DSC-Charakterisierung der nicht getemperten Charpy-Stäbe (1. Heizzyklus ausgewertet, vgl. Abschn. 3.8.2)
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Ho
[°C] ΔHm Ho [J/g]
Tm sc [°C]
ΔHm sc [J/g]
Tc [°C]
ΔHc [J/g]
ΔHgesamt [J/g]
Co-scTech +55 / / 175,8 20,6 215,0 32,7 122,2 33,0 53,3
Co-scTech +26 / /
154,3 175,0
0,6 5,4 215,3 49,0 125,4 43,2 55,0
Co-scTech ±0 / /
157,8 176,5
2,7 1,5 216,2 42,0 120,7 33,7 46,2
Co-scTech -26 / /
158,3 175,9
5,0 1,5 216,3 33,3 118,0 23,6 39,7
Co-scTech -55 119,4 7,2
159,0 176,0
8,9 0,3 213,9 28,8 110,6 7,3 30,7
Wie bereits dargelegt, bildet die ermittelte Schlagbiegefestigkeit der Probe „Co-scTech ±0“
eine Ausnahme. In der Arbeit von Yang et al. [9] wird diskutiert, dass die Widerstandskraft
semikristalliner Polymere nicht ausschließlich von deren Kristallinität und den enthaltenen
Kristallgrößen, sondern auch von amorphen Regionen, Verbindungsmolekülen und der
Kristallform beeinflusst wird. Da die Proben aus unterschiedlichen Strukturen (amorph,
Kristalle der Homopolymere und des Stereokomplexes) bestehen, ist eine abgesicherte
Begründung der gefundenen Messwerte nicht möglich. Für die Probe „Co-scTech ±0“ wird
vermutet, dass durch die Vorlage von PDLA und PLLA (als Blocksegment des Copolymers) im
äquivalenten Massenverhältnis eine bessere Kettenbeweglichkeit vorliegt und die sich
dadurch erleichtert bildenden Sphärolithe [69] in der verbleibenden amorphen Phase als
„cross-links“ [49] wirken können. Dadurch entsteht ein dreidimensionales Netzwerk, das die
Widerstandsfähigkeit des Materials erhöhen kann.
Page 67
59
Beim Tempern werden Kristallisationsprozesse gefördert, die mehr Zeit beanspruchen als
beim eigentlichen Herstellungsprozess der Prüfstäbe zur Verfügung steht. Da die Kristal-
lisation der Stereokomplexe im Vergleich zu den Homopolymeren sehr schnell abläuft [21,
66, 75, 146, 149, 151, 152], wird durch den Temperprozess vor allem die hc-Kristallitbildung
sowie die Ausbildung perfekterer Kristalle gefördert [61, 151]. Diese Annahme kann mit Hilfe
der Röntgenbeugungsuntersuchungen (vgl. Abschn. 5.3.2.4) bestätigt werden.
Bei den getemperten Proben werden prinzipiell kleinere Messwerte der Schlagbiegefestig-
keit gefunden. Eine Ausnahme bildet die Probe „Co-scTech +26“, in der PDLA im Überschuss
vorliegt. Hier wird im getemperten Zustand ein größerer Wert gemessen. Die Probe „Co-
scTech +55“ konnte nicht getempert untersucht werden, da nicht genügend Material
verfügbar war.
Auch unter den getemperten Proben stellt „Co-scTech ±0“ wieder einen Sonderfall dar. Für
die übrigen Proben korrelieren die ermittelten Schlagbiegefestigkeiten mit den thermischen
Eigenschaften der Materialien. Die nachfolgende Tabelle 5.13 gibt einen Überblick über die
thermischen Eigenschaften (DSC-Messungen).
Tabelle 5.13
DSC-Auswertung der getemperten Charpy-Stäbe (1. Heizzyklus ausgewertet, vgl. Abschn. 3.8.2)
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Ho
[°C] ΔHm Ho [J/g]
Tm sc [°C]
ΔHm sc [J/g]
Tc [°C]
ΔHc [J/g]
ΔHgesamt [J/g]
Co-scTech +26 / /
157,4 176,2
3,2 9,9 214,9 37,5 125,6 41,3 50,6
Co-scTech ±0 / /
156,6 175,0
2,8 2,6 214,4 40,4 122,2 34,1 45,8
Co-scTech -26 / /
157,7 175,0
5,4 1,9 215,5 36,4 119,0 25,9 43,7
Co-scTech -55 106,6 3,3 157,7 10,1 214,8 29,6 115,7 4,4 36,4
Im Vergleich zu den Messergebnissen der nicht getemperten Proben werden für die Proben
„-26“ und „-55“ höhere Werte der Gesamtenthalpie gemessen (39,7 und 30,7 J/g zu 43,7
und 36,4 J/g). Die Probe „+26“ hat im Vergleich zum nicht getemperten Prüfstab eine
geringere Gesamtenthalpie (55,0 zu 50,6 J/g). Zusätzlich kann bei dieser Probe auch eine
deutliche Reduzierung der durch die Stereokomplexe hervorgerufenen Enthalpie (49,0 zu
37,5 J/g) gefunden werden. Basierend auf den zuvor diskutierten Ergebnissen wäre das eine
mögliche Begründung für die veränderten Messwerte der Schlagbiegefestigkeit.
Eine ebenfalls in Betracht gezogene Wasseraufnahme kann hingegen als Begründung für die
besseren Messwerte der nicht getemperten im Vergleich zu den getemperten Proben
ausgeschlossen werden. Untersuchungen haben gezeigt, dass die Lagerung im Klimaraum (5
Tage, 22 °C, 65% rel. Luftfeuchtigkeit) lediglich zu einer durchschnittlichen Gewichtszunahme
Page 68
60
der Proben von 0,21 Gew.-% führte. Nach dem Tempern zeigten die Proben im Vergleich zu
diesem Wert nur eine durchschnittliche Gewichtsreduzierung von 0,07 Gew.-%. Diese
Analyse wird auch durch Ergebnisse von Grijpma et al. [45] bestätigt. Die Autoren haben
gezeigt, dass eine Wasseraufnahme bei Homopolymerproben zu einer Verschlechterung der
Schlagbiegefestigkeit führt.
Shore Härte
Die Beschreibung der Methode findet sich in Abschn. 3.8.6.
Für die verschiedenen Proben werden sowohl im nicht getemperten als auch im ge-
temperten Zustand innerhalb der Serie untereinander vergleichbare Werte der Härte er-
mittelt (Tabelle 5.14). Eine Ausnahme bildet die Probe „Co-scTech +55“. Diese zeigt im nicht
getemperten Zustand einen leicht erhöhten Wert. Auch in den DSC-Untersuchungen werden
für diese Probe abweichende Ergebnisse erhalten (vgl. Tab. 5.12). Das betrifft insbesondere
die Zusammensetzung der Enthalpien des Homopolymers sowie des Stereokomplexes,
weshalb auch mit unterschiedlichen Kristallgrößen und damit einer veränderten Härte zu
rechnen ist.
Auch die Röntgenbeugungsuntersuchungen zeigen für die Probe „Co-scTech +55“ im
Verglich zu allen weiteren Proben ein abweichendes Verhalten (vgl. Abschn. 5.3.2.4).
Tabelle 5.14
Ergebnisse der Härtemessungen nach Shore D
Probenbezeichnung Härte
[Shore] Härte getempert
[Shore]
Co-scTech +55 75,1 nicht ermittelt
Co-scTech +26 73,4 74,5
Co-scTech ±0 73,0 74,5
Co-scTech -26 73,7 75,0
Co-scTech -55 73,6 75,1
Die Unterschiede zwischen den nicht getemperten und den getemperten Proben sind
vermutlich auf die unterschiedlichen Kristallit-Zusammensetzungen und Größen zurück-
zuführen (vgl. Abschn. 5.3.2.4). Sowohl eine höhere Anzahl als auch größere Kristalle bean-
spruchen auch mehr Volumen. Dadurch verbleibt zwischen den Kristallen weniger Platz, die
betreffenden Materialien zeigen eine gesteigerte Festigkeit und dies spiegelt sich in einem
höheren Kennwert wieder.
Page 69
61
5.3.2.3. Thermomechanische Eigenschaften
Zur thermomechanischen Charakterisierung der Proben wurden sowohl HDT-B als auch
DMA-Untersuchungen durchgeführt. Die Beschreibungen der Methoden finden sich in
Abschn. 3.8.7 und 3.8.9.
HDT-B
Für die nicht getemperten Proben werden mit reinem PLLA vergleichbare Messwerte
gefunden (HDT = 55 °C) [3, 67, 69]. Lediglich für die Probe „Co-scTech +26“ ist ein leicht
erhöhter Wert messbar. Die basierend auf den Ergebnissen der Quellen [53, 57, 67, 96]
erwartete deutliche Steigerung der Wärmeformbeständigkeit aufgrund der Anwesenheit der
Stereokomplexkristalle kann bei den eigenen Proben nicht beobachtet werden. Ein
möglicher Grund könnte der in den Materialien enthaltene hohe Anteil an amorphem PDLLA
als Blocksegment des Copolymers sein.
Für die getemperten Proben wurde wie erwartet eine leichte Erhöhung der HDT-Werte
ermittelt, allerdings liegen die Werte auch in diesem Fall im Bereich der für reines PLLA
dokumentierten Werten (HDT = 61 °C) [3, 69]. Interessant ist, dass für die Probe „Co-
scTech +26“, für die im nicht getemperten Zustand der höchste Wert erhalten wird, im
Vergleich zu den weiteren Proben nur noch eine geringe Steigerung des HDT-Wertes
registriert werden kann. Eine Interpretation dieses Sachverhaltes ist auf Grund der geringen
Menge an Versuchen nicht möglich.
Eine Übersicht zu den Messergebnissen findet sich in Tabelle 5.15.
Tabelle 5.15
HDT-Ergebnisse der nicht getemperten und getemperten Proben
HDT [°C] HDT [°C]
Probenbezeichnung nicht getempert getempert
Co-scTech +26 59 64
Co-scTech ±0 54 63
Co-scTech -26 55 62
Co-scTech -55 56 63
DMA
Zur Untersuchung der viskoelastischen Eigenschaften der Proben wurden DMA-
Untersuchungen durchgeführt. Die Veränderung von Speicher- und Verlustmodul wurde
dazu in einem Temperaturfenster von 20 °C bis 160 °C aufgezeichnet. Insgesamt lässt sich
feststellen, dass die Kurvenverläufe der nicht getemperten und der getemperten Proben
miteinander vergleichbar sind. Für die getemperten Proben wurden etwas höhere Speicher-
Page 70
62
module im Glaszustand (20 °C bis ca. 70 °C) sowie eine Peakverbreiterung des Verlustmoduls
gemessen. Daraus lässt sich schlussfolgern, dass die getemperten Proben insgesamt
kristallinere Eigenschaften haben. Dies findet man basierend auf den DSC-Untersuchungen
(vgl. Abschn. 5.3.2.2) allerdings nur für die Proben „Co-scTech -26“ und „Co-scTech -55“.
Die beiden nachfolgenden Abbildungen zeigen die Kurvenverläufe der nicht getemperten
sowie der getemperten Proben.
Bild 5.9
DMA-Kurvenüberlagerung der nicht getemperten Proben
Bild 5.10
DMA-Kurvenüberlagerung der getemperten Proben
Page 71
63
Alle Proben zeigen bis ca. 65 °C gleichbleibende Werte für Speicher- und Verlustmodul. Die
Ursache hierfür liegt in dem festkörperartigen Verhalten, das das Material in diesem
Temperaturbereich besitzt. Die durch die Deformation zugeführte Energie kann vollständig
gespeichert und wieder abgegeben werden. Im Temperaturfenster von ca. 70 °C bis 95 °C
kann eine deutliche Veränderung der beiden Module beobachtet werden. Der Grund hierfür
ist die Glasübergangstemperatur des Materials [160].
Im Temperaturbereich von 70 °C bis 95 °C kommt es zu einer deutlichen Steigerung der
Beweglichkeit der Polymerketten und die Proben verlieren ihre elastischen Eigenschaften.
Die durch die Deformation eingebrachte mechanische Energie kann vom System nicht mehr
gespeichert werden und wird in Wärme umgewandelt. Der Verlustmodul hat sein Maximum
am Glasübergangspunkt des Materials. Hier wird zur weiteren Steigerung der Ketten-
beweglichkeit die meiste Energie benötigt. Danach fällt der Verlustmodul wieder auf seinen
Ausgangswert ab.
Die in der DSC registrierte Nachkristallisation der Probe „Co-scTech -55“ lässt sich für das
nicht getemperte Material durch einen schwachen Wiederanstieg des Speichermoduls auch
in der DMA-Untersuchung wiederfinden.
5.3.2.4. Röntgenbeugungsexperimente
Die nachfolgenden Bilder 5.11 und 5.12 zeigen die Planfilmaufnahmen der nicht ge-
temperten und der getemperten Proben aus den Technikum-Versuchen. Tabelle 5.16 gibt
eine Übersicht aller ermittelten Reflexe.
„Co-scTech +55“ „Co-scTech +26” „Co-scTech ±0” „Co-scTech -26“ „Co-scTech -55”
Bild 5.11
Planfilmaufnahmen der Proben im nicht getemperten Zustand
Page 72
64
„Co-scTech +26” „Co-scTech ±0” „Co-scTech -26“ „Co-scTech -55”
Bild 5.12
Planfilmaufnahmen der Proben im getemperten Zustand
In der Reihe der nicht getemperten Materialen hebt sich die Probe „Co-scTech +55“ durch
die hc-Reflexe starker und sehr schwacher Intensität bei 2θ = 16,2° und 18,3° vom Er-
scheinungsbild der übrigen Proben ab. Im Vergleich dazu zeigen die Proben „Co-scTech +26“
und „Co-scTech ±0“ lediglich den hc-Reflex bei 2θ = 16,2° in mittlerer Intensität und die
Proben „Co-scTech -26“ und „Co-scTech -55“ keinen hc-Reflex. Die Besonderheit der Probe
„Co-scTech +55“ zeigte sich bereits bei der Auswertung der DSC-Thermogramme (vgl.
Tab. 5.12). Hier wurde im Vergleich zu den anderen Proben ein deutlich höherer Enthalpie-
wert für das Homopolymer ermittelt.
Als intensitätsstärkster Reflex kann bei allen nicht getemperten Proben der Beugungswinkel
2θ = 11,9° identifiziert werden. Dieser stellt einen der drei typischen durch Stereokomplex-
kristalle hervorgerufenen Reflexe dar. Zusätzlich dazu kann in allen Proben auch der sc-
Reflex bei 2θ = 20,3° mit mittlerer Intensität gefunden werden. Der dritte charakteristische
sc-Reflex bei 2θ = 23,3° geht bei den meisten Proben in die Untergrundstreuung über und ist
aus diesem Grund nicht klar messbar. Lediglich für die Proben „Co-scTech ±0“ und „Co-
scTech -26“ lässt er sich in schwacher Intensität ermitteln. Auf Grund der Intensitäts-
verteilung ist klar zu erkennen, dass in den nicht getemperten Proben die Anwesenheit der
Stereokomplexkristalle überwiegt.
Tabelle 5.16 Zusammenstellung der ermittelten Reflexe
sc hc hc sc sc
Probenbezeichnung 11,9° 16,2° 18,3° 20,3° 23,3°
Co-scTech +55 st st ss m
Co-scTech +26 st m m
Co-scTech +26 getempert st st ss m
Co-scTech ±0 st m m s
Co-scTech ±0 getempert st st ss s
Page 73
65
Co-scTech -26 st m s
Co-scTech -26 getempert st st ss s
Co-scTech -55 m s
Co-scTech -55 getempert m st ss ss
Legende: st stark; m mittel; s schwach; ss sehr schwach
Nach dem Tempern ist für alle Proben eine deutliche Veränderung in den Planfilm-
aufnahmen zu verfolgen. In allen Proben lassen sich jetzt die hc-Reflexe bei 2θ = 16,2° mit
starker und bei 2θ = 18,3° mit sehr schwacher Intensität feststellen. Es lässt sich also
schlussfolgern, dass es zu einer deutlichen Zunahme an hc-Kristallen in den Proben
gekommen sein muss. Für die Probe „Co-scTech ±0“, für die der hc-Reflex bei 2θ = 16,2°
bereits im nicht getemperten Zustand identifiziert werden konnte, lässt sich außerdem eine
leichte Reduzierung der Ringstärke dokumentieren. Aus dieser Beobachtung kann ein
geringes Wachstum der diesen Reflex erzeugenden Kristalle abgeleitet werden. Rückblickend
auf die für diese Probe erzielte Schlagbiegefestigkeit (vgl. Abschn. 5.3.1.2) kann mit den
größeren Kristallen sowie der damit verbundenen Verschlechterung der mechanischen
Eigenschaften [9, 61, 93, 153] eine mögliche Erklärung für die gefundenen Messergebnisse
gegeben werden.
Für alle Proben ist in Bezug auf die Beugungsmuster weiterhin festzustellen, dass für den sc-
Reflex 2θ = 20,3° eine Intensitätsabnahme erfolgt. Demzufolge kann eine durch das Tempern
verursachte Reduzierung der betreffenden sc-Kristalle nicht ausgeschlossen werden.
Auch für die Proben aus den Technikum-Materialien lässt sich weder im nicht getemperten
noch im getemperten Zustand eine Orientierung der Kristalle ermitteln. Auch diese
Materialen liegen also ausnahmslos im isotopen Zustand vor.
5.4. Gegenüberstellung des Labor- und Technikum-Materials
Die Schlagbiegefestigkeiten der Labor- und der Technikum-Materialien weisen deutliche
Unterschiede auf. Während die ermittelten Schlagbiegefestigkeiten für die Laborproben im
Bereich von 4,8 bis 10,2 kJ/m2 liegen, werden für die im Technikum hergestellten Proben
Werte im Bereich von 14,9 bis 33,7 kJ/m2 gefunden.
Eine mögliche Ursache für die unterschiedlichen Messwerte könnte in der Art der Ent-
monomerisierung der Ausgangsmaterialien liegen. Während die Labormaterialen mittels
Lösen in Chloroform und Fällen in Methanol entmonomerisiert (vgl. Abschn. 4.2.1 und
Abschn. 4.2.2) und zeitgleich auch der Katalysator in eine unreaktive Spezies überführt
wurde, erfolgt die Entmonomerisierung der Technikum-Materialien mittels Vakuum und
unter thermischer Belastung (vgl. Abschn. 4.3.3). Der Katalysator bleibt bei dieser Art der
Entmonomerisierung unverändert. Aus der Arbeit von Grijpma und Pennings [49] ist
Page 74
66
bekannt, dass die im Polymer enthaltene Katalysatorkonzentration einen Einfluss auf die
Polymerkristallinität besitzt. Mit steigendem Katalysatorgehalt konnte hier eine höhere
Kristallinität der untersuchten Polylactidproben beobachtet werden. Auch bei den eigenen
Proben konnten Unterschiede in der Kristallinität festgestellt werden, die möglicherweise
auf die An- bzw. Abwesenheit des Katalysators zurückzuführen sind. Die Probe „Co-sc04“,
die aus den gefällten Technikum-Materialien (vgl. Tab. 5.1) hergestellt worden ist, hat
Schmelzenthalpiewerte von 1,7 J/g für das Homopolymer und 48,9 J/g für den
Stereokomplex. Die vergleichbare Technikum-Probe („Co-scTech +55“) weist einen deutlich
höheren Wert von 20,6 J/g für die Homopolymerkristalle und einen kleinerer Enthalpiewert
von 32,7 J/g für den Stereokomplex auf. Eine vergleichende Übersicht der Ergebnisse der
betreffenden Labor- und Technikum-Probe findet sich in Tabelle 5.17. Diese gefundenen
Unterschiede legen die Vermutung nahe, dass die Anwesenheit des Katalysators bevorzugt
die Kristallisationsfähigkeit des Homopolymers fördert. Zusätzlich zu den Ergebnissen aus
den DSC-Thermogrammen kann die verschiedenartige Kristallitzusammensetzung der beiden
Proben auch an Hand der Planfilmaufnahmen verfolgt werden (vgl. Abschn. 5.3.1.3 und
Abschn. 5.3.2.4).
Eine weitere mögliche Begründung für die sich voneinander unterscheidenden Messwerte
der Schlagbiegefestigkeit ist eine unterschiedliche (gute bzw. uneinheitliche) Durchmischung
der Blendschmelzen in den beiden verwendeten Extrudern. Einen Hinweis darauf geben
teilweise differierende DSC-Thermogramme, die für die Technikum-Proben erhalten wurden.
Eine schlechtere/uneinheitliche Durchmischung der Proben im Mini-Compounder
„Brabender“ lässt aufgrund der sinkenden Wahrscheinlichkeit des Aufeinandertreffens von
PDLA und PLLA (als Blocksegment des Copolymers) mit einen kleineren Schmelzenthalpie-
wert für den Stereokomplex und einen höheren Wert der Schmelzenthalpie für das
Homopolymer erwarten.
Um den Effekt der An- bzw. Abwesenheit des Katalysators bei den Betrachtungen
ausschließen zu können, wurden die Ausgangsstoffe „PDLA Tech“ und „Co-Tech“ im
Mischungsverhältnis von 1:1 (entspricht dem Mischungsverhältnis der Probe „Co-
scTech +55“) auch am „Haake MiniLab“-Extruder (vgl. Abschn. 5.1.2) zum Blend verarbeitet.
Für das resultierende Material mit der Probenbezeichnung „Co-scTech +55 Mini“ konnten
die erwarteten Enthalpieergebnisse nachgewiesen werden (Tabelle 5.17). Ergänzend dazu
kann die unterschiedliche Durchmischung in den beiden verwendeten Extrudern noch an
Hand eines weiteren Effekts gezeigt werden. So wird in der im „Haake MiniJet“ generierten
Probe eine kleine Schmelztemperatur für den Stereokomplex gefunden als in der Probe aus
dem Mini-Compounder. Hervorgerufen wird dieser Effekt durch Kristallite unterschiedlicher
Reinheit bzw. unterschiedlich perfekter Anordnung [37]. Bei einer besseren Durchmischung
werden zwar mehr, dafür aber auch Stereokomplexe minderer Qualität gebildet, da es
verstärkt zu einer Einlagerung von amorphen Blocksegmenten kommt, die die Kristallisation
hindern.
Da für die Proben „Co-scTech +55“ und „Co-scTech +55 Mini“ aber annähernd vergleichbare
Page 75
67
Schlagbiegefestigkeiten (Tabelle 5.17) erzielt werden konnten, kann der Einfluss der
unterschiedlichen Durchmischung in den verschiedenen Extrudern auf die erzielten
Messwerte vernachlässigt werden. Von Bedeutung scheint hier daher eher die zuvor
diskutierte geförderte Kristallisation der Homopolymere bei Anwesenheit des Katalysators
zu sein.
Tabelle 5.17
Überblick der Messergebnisse
Probenbe-zeichnung
Tcc [°C]
ΔHcc [J/g]
Tm Homo2
[°C] ΔHm Homo2
[J/g] Tm sc [°C]
ΔHm sc [J/g]
ΔHgesamt [J/g]
Charpy [kJ/m2]
Mn [g/mol]
Co-sc04 / / 173,0 1,7 214,3 48,9 50,6 7,9 64400
Co-scTech +55 / / 175,8 20,6 215,0 32,7 53,3 15,3 47650
Co-scTech +55 Mini 99,5 4,0 175,4 14,8 213,5 39,1 49,9 18,2 52500
Geringe Unterschiede zwischen den Labor- und den Technikum-Materialien können auch
bezüglich der thermischen Stabilität festgestellt werden. So werden für die Laborproben
(„Co-sc“-Materialien) in den TGA-Messungen (vgl. Abschn. 5.3.1) etwas höhere (ΔT ca. 10 °C)
Zerfallstemperaturen gefunden. Eine ähnliche Beobachtung wurde auch von Fan et al. [106]
für PLA-Stereokomplexe beschrieben, die aus gefällten und unbehandelten Ausgangs-
materialien hergestellt worden sind. Dem nach kann auch hier ein Einfluss auf die
Eigenschaften der Materialein bei der Anwesenheit des aktiven Katalysators gezeigt werden.
Page 76
68
6. Abbauuntersuchungen
6.1. Probenvorbereitung und Pufferherstellung
Als Proben wurden die in Abschnitt 5.2.1 beschriebenen Mikrogranulate einschließlich der
Ausgangsstoffe „PDLA Tech“ und „Co-Tech“ (Übersicht vgl. Tab. 5.4), sowie die in Ab-
schnitt 5.1.2 beschriebenen und von Hand zerkleinerten Polymerblends „Co-sc04“ und „Co-
sc05“ (vgl. Tab. 5.1) verwendet.
Als Abbaumedium wurde ein Sörensen Phosphatpuffer genutzt. Dieser wurde nach der von
Kaltofen et al. [161] beschrieben Vorschrift aus den frisch hergestellten Lösungen Dinatrium-
hydrogenphosphat und Kaliumdihyrigenphosphat gemischt und auf einen pH-Wert von 7,45
eingestellt.
Von jeder Probe wurden je 10 Probenfraktionen bereitgestellt. Dazu wurden jeweils ca. 0,2 g
Page 77
69
Probe in ein 25 mL Schnappdeckelglas eingewogen und mit 10 mL der Pufferlösung bedeckt.
Anschließend wurden die Proben für den gewünschten Untersuchungszeitraum bei 37 °C in
einem Klimaschrank (40% rh) gelagert.
6.2. Probenentnahme
Nach dem jeweils angestrebten Abbauzeitraum wurde je eine Fraktion der Proben ent-
nommen und für ca. 30 min auf RT konditioniert. Nach erfolgter pH-Wert-Messung wurden
die Polymerpartikel mittels Vakuumfiltration über ein Cellulosenitrat-Membranfilter (0,8 µm,
Ø 25 mm, Whatman) von der Pufferlösung abgetrennt und mit entionisiertem Wasser
gewaschen. Abschließend wurden die Polymerpartikel in Petrischalen überführt, einer
optischen Kontrolle unterzogen und für mind. 3 Tage bei RT über P2O5 in einem Vakuum-
trockenschrank gelagert, bevor die weiteren Analysen erfolgten.
Eine schematische Darstellung der Arbeiten findet sich in Bild 6.1.
Bild 6.1
Schematische Darstellung der Probenaufarbeitung bei den Abbauuntersuchungen
6.3. Charakterisierung der Abbauprodukte
Nach erfolgter Trocknung wurden die Proben gewogen. Im Abbauzeitraum von 43 bzw.
40 Wochen wurde unabhängig vom Probentyp kein Massenverlust registriert. Es ist also
davon auszugehen, dass bis dahin keine wasserlöslichen Abbauprodukte gebildet worden
sind [113, 114, 123] und die verbliebene Molmasse noch deutlich über 10000 g/mol liegen
muss. Nach Thelen [109] kann erst nach Unterschreiten der Molmasse (Mw) von ca. 10000
bis 20000 g/mol ein messbarer Masseverlust verfolgt werden.
Auch für den pH-Wert wurde in dem benannten Zeitraum keine Veränderung festgestellt.
Page 78
70
Hinsichtlich des Aussehens, der thermischen Eigenschaften (DSC) sowie der Molmasse (GPC)
konnten hingegen Unterschiede festgestellt werden. Diese sollen nachfolgend eingehender
diskutiert werden.
6.3.1. Visuelle Kontrolle
Für die Probe „PDLA Tech” konnte nach 22 Wochen die erste optische Veränderung fest-
gestellt werden. Bei einem geringen Teil der eingesetzten Granulate wurde im Inneren eine
leichte Verfärbung beobachtet. Nach 28 Wochen waren bereits ca. 50% der eingesetzten
Partikel weiß verfärbt. Nach einem Beobachtungszeitraum von 37 Wochen waren etwa 90%
der Partikel stark weiß verfärbt. Zum Ende der Untersuchungen (43 Wochen) zeigten nur ca.
5% der Partikel keine Verfärbung. Die Beobachtungen im getrockneten Zustand gleichen hier
denen direkt nach der Probenentnahme.
Für die Probe „Co-Tech“ konnte nach 19 Wochen bei 2 der eingesetzten Partikel an der
Oberfläche eine Verfärbung beobachtet werden. Im Gegensatz zur Probe „PDLA Tech“ nahm
die Zahl der sich farblich verändernden Partikel im weiteren Verlauf des Abbaus jedoch nicht
zu. Auch nach 37 Wochen waren nur 3 der eingesetzten Partikel verfärbt. Nach 43 Wochen
konnte mit ca. 50% weiß verfärbten Partikel erstmals eine deutliche Veränderung fest-
gestellt werden. Die optischen Änderungen nach dem Trocknen gleichen auch bei dieser
Probe denen direkt nach der Probenentnahme.
Die Probe „Co-scTech +55“ zeigt im Vergleich zu den weiteren Proben die deutlichste
optische Veränderung. Bereits nach 13 Wochen waren ca. 50% der eingesetzten Partikel
vollständig weiß-milchig verfärbt und schienen leicht aufgequollen zu sein. Nach 16 Wochen
waren ca. 90% der Partikel stark und der verbleibende Rest leicht verfärbt. Nach 19 Wochen
wurde bei allen Partikeln eine starke Verfärbung sowie eine leichte Quellung registriert.
Nach dem Trocknen konnte bei den Proben nach einer Abbauzeit von 13 Wochen für alle
eingesetzten Partikel eine stark weiß-milchige Verfärbung wahrgenommen werden.
Bei der Probe „Co-scTech +26“ konnte erstmals nach 22 Wochen für alle Partikel eine
schwach milchige Verfärbung beobachtet werden. Nach 25 Wochen wurde bei ca. der Hälfte
der Partikel eine etwas stärkere Verfärbung gefunden. Im weiteren Verlauf des Abbaus ver-
stärkte sich diese Verfärbung zunehmend. Nach dem Trocknen konnte für diese Probe
bereits nach 16 Wochen eine schwach milchig-gelbliche Verfärbung beobachtet werden.
Die Probe „Co-scTech ±0“ zeigte die erste Veränderung nach 25 Wochen. Für alle Partikel
wurde eine ganz schwache bis schwache Verfärbung festgestellt. Während des weiteren
Abbaus kam es nur zu einer geringen Zunahme des Verfärbungsgrades. Im getrockneten
Zustand lässt sich für diese Probe schon nach 16 Wochen eine schwach milchige und
gelbliche Verfärbung der Probenpartikel feststellen.
Die Probe „Co-scTech -26“ zeigte nach 25 Wochen eine erste optische Veränderung. Bei
allen Partikeln trat eine ganz schwach milchige Verfärbung auf. Im weiteren Verlauf des
Page 79
71
Abbaus ändert sich diese Verfärbung nur noch geringfügig. Zum Ende der Untersuchungen
(43 Wochen) waren alle Partikel weiß verfärbt. Im getrockneten Zustand lässt sich für die
Probe, ähnlich wie bei Probe „Co-scTech ±0“, schon nach 16 Wochen eine schwach milchige
und leicht gelbliche Verfärbung der Probenpartikel beobachten.
Bei der Probe „Co-scTech -55“ wurde nach 10 Wochen eine erste Veränderung bei wenigen
Partikeln festgestellt. Diese äußerte sich in der Verfärbung der Oberfläche. Im weiteren Ver-
lauf des Abbaus kam es zu keiner ausgeprägten Veränderung der Partikel. Nach 25 Wochen
war bei 3 Partikeln eine stark milchige und bei den übrigen eine schwach milchige Ver-
färbung zu erkennen. Nach 28 Wochen kam es bei 8 Partikeln zu einer starken und bei den
übrigen zu einer gesteigerten Verfärbung im Vergleich zu den Partikeln nach 25 Wochen.
Nach 43 Wochen zeigten alle Partikel eine weiß-milchige Verfärbung. Im getrockneten
Zustand konnte erst nach 16 Wochen eine erste Veränderung festgestellt werden.
Bild 6.2
Vergleich der Proben nach einem Abbauzeitraum von 10 (links) und 25 Wochen (rechts) – nach dem Trocknen;
bei 3 Uhr beginnend und dem Uhrzeigersinn folgend handelt es sich um die Proben: „PDLA Tech“ (1), „Co-Tech“
(2), „Co-scTech +55“ (3), „Co-scTech +26“ (4), Co-scTech ±0“ (5), „Co-scTech -26“ (6), „Co-scTech -55“ (7), „Co-
sc04“ (8) und „Co-sc05“ (9)
Für die Probe „Co-sc04“ konnte im feuchten Zustand zu keinem Zeitpunkt der Abbau-
untersuchungen eine Veränderung festgestellt werden. Lediglich im getrockneten Zustand
ließ sich nach 13 Wochen eine erste Verfärbung registrieren. Alle Probenpartikel waren zu
diesem Zeitpunkt schwach milchig und leicht gelblich verfärbt.
Im Gegensatz dazu zeigte die Probe „Co-sc05“ schon nach 10 Wochen eine erste leichte Ver-
färbung bei einem geringen Anteil der Probenpartikel. Nach 19 Wochen konnte bei der
Hälfte der Partikel eine leicht weißliche Verfärbung der Oberfläche beobachtet werden.
Nach 25 Wochen war für alle Partikel eine leicht milchig-gelbliche Verfärbung festzustellen.
Im getrockneten Zustand konnte erst nach 13 Wochen eine optische Veränderung der
Partikel wahrgenommen werden.
Page 80
72
6.3.2. Molmassenabbau
Im Gegensatz zum Masseverlust wurde probenabhängig sowohl für die beiden Ausgangs-
polymere als auch für die weiteren Proben eine deutliche Änderung der Molmasse
bestimmt. Für die Proben „PDLA Tech“ und „Co-Tech“ wurde über den Untersuchungs-
zeitraum ein nahezu linear verlaufender Abfall der Molmasse gefunden (Bild 6.3).
Deutlich zu erkennen ist hierbei, dass für die Probe „PDLA Tech“ entgegen den Erwartungen
zunächst (ca. bis zur 13. Woche) ein sehr schneller Abbau erfolgt, dessen Geschwindigkeit im
weiteren Untersuchungszeitraum deutlich abnimmt.
Bild 6.3
Übersicht des Molmassenabbaus der Proben „PDLA Tech“ und „Co-Tech“
Page 81
73
Bild 6.4
Übersicht des Molmassenabbaus der „Co-scTech“-Proben
Bild 6.5
Übersicht des Molmassenabbaus der Proben „Co-sc04“ und „Co-sc05“
Page 82
74
Auf Grund des hohen amorphen Anteils sowie der geringeren Molmasse zu Beginn der
Untersuchungen war für die Probe „Co-Tech“ ein im Vergleich zum Homopolymer deutlich
schnellerer Abbau erwartet worden. Die experimentellen Untersuchungen zeigten jedoch,
dass für das Copolymer über den gesamten Untersuchungszeitraum ein gleichbleibender
Abbau moderater Geschwindigkeit verfolgt werden kann.
Auch der Molmassenabbau für die Proben der modifizierten Stereokomplexe erfolgt nahezu
linear. In den ersten Wochen findet hier zunächst ein deutlicher Abbau statt. Nach etwa 19
(bzw. 13) Wochen kann dann, an Hand einer verminderten Geschwindigkeit, eine Änderung
im Abbauverhalten verfolgt werden. Ab diesem Zeitraum ändern sich die Molmassen bis
zum Ende der Untersuchungen nur noch wenig (Bild 6.4 und 6.5).
Der registrierte Kurvenanstieg für einen Teil der Proben in Bild 6.4 kann an Hand von
niedermolekularen Anhaftungen erklärt werden, die sich an der Oberfläche der Materialien
befinden. Diese täuschen bei der Bestimmung der Ausgangsmolmasse einen zu kleinen Wert
vor. Nach dem Aufenthalt im Abbaumedium sind die niedermolekularen Verunreinigungen
entfernt und man erhält einen höheren Messwert.
Für alle Proben kann ein insgesamt vergleichbarer Kurvenverlauf des Molmassenabbaus
beobachtet werden. Die mittels GPC erhaltenen Chromatogramme zeigen außerdem über
den gesamten Untersuchungszeitraum für alle Proben eine monomodale Verteilung.
6.3.3. Thermische Eigenschaften
Um Hinweise zur Änderung der thermischen Eigenschaften der Probenmaterialien während
des Abbaus zu erhalten, wurden DSC-Messungen durchgeführt. Da es beim Aufheizen bei
allen Proben zu einer Nachkristallisation kam, wird für die Bewertung der verschiedenen
Materialien die berechnete Gesamtenthalpie (vgl. Abschn. 3.8.2) herangezogen.
Mit Ausnahme der Probe „PDLA Tech“, die über den Untersuchungszeitraum von 28 Wochen
zunächst einen relativ konstanten Wert der Gesamtenthalpie aufweist, kann für die übrigen
Proben von Beginn an mit fortschreitendem Abbau eine leicht („Co-Tech“) bis stark aus-
geprägte Nachkristallisation registriert werden. Für die Probe „PDLA Tech“ zeigt sich erst ab
der 37. Untersuchungswoche eine deutlich steigende Tendenz (Bild 6.6).
Page 83
75
Bild 6.6
Übersicht der ermittelten Gesamtenthalpien der „Co-scTech“-Proben sowie ihrer Ausgangsmaterialien über
den Abbauzeitraum (1.Heizzyklus ausgewertet; vgl. Abschn. 3.8.2)
Die Nachkristallisation der verschiedenen Proben erfolgt in unterschiedlichen Zeitfenstern.
So können für den Großteil der Stereokomplexproben schon nach 10 Wochen erste Ver-
änderungen wahrgenommen werden. Für die Proben „Co-scTech +55“, „Co-scTech +26“ und
Co-scTech ±0“ sind diese über den gesamten Untersuchungszeitraum betrachtet am
größten. Für die Proben „Co-scTech -26“ und „Co-scTech -55“ können im Vergleich dazu nur
geringere Steigerungen der Gesamtenthalpie ermittelt werden. Für die Probe „Co-Tech“
wurde hingegen zunächst nur ein sehr geringer und nahezu konstanter Wert der Gesamt-
enthalpie ermittelt. Erst nach 19 Wochen im Abbaumedium wurde auch hier tendenziell eine
Steigerung der Kristallinität beobachtet. Nach 37 Wochen im Abbaumedium kann auch für
diese Probe ein deutlicher Anstieg aufgezeigt werden.
Für die Proben „Co-sc04“ und „Co-sc05“ wurde über den verfolgten Abbauzeitraum zunächst
ein starkes Ansteigen der Gesamtenthalpie registriert (Bild 6.7). Ab ca. 10 Wochen Lagerung
im Phosphatpuffer ändert sich die Gesamtenthalpie nur noch geringfügig. Die letzten Proben
(nach 40 Wochen) zeigen dann nochmals einen deutlichen Anstieg der Gesamtenthalpie.
Page 84
76
Bild 6.7
Übersicht der ermittelten Gesamtenthalpien der Proben „Co-sc04“ und „Co-sc05“ über den Abbauzeitraum
(1.Heizzyklus ausgewertet; vgl. Abschn. 3.8.2)
Neben den errechneten Gesamtenthalpien wurden auch die Schmelzpunkte Tm Homo1
(Copolymer), Tm Homo2 (Homopolymer) und Tm sc (Stereokomplex) ermittelt und graphisch
ausgewertet. Die nachfolgenden Bilder (Bild 6.8 bis 6.12) zeigen eine Übersicht der
verschiedenen Messergebnisse.
Für das reine Blockcopolymer („Co-Tech“) wurde ein Aufschmelzpeak mit Doppelspitze
festgestellt. Auch die Materialien der modifizierten Stereokomplexe („Co-scTech“-Proben)
zeigten im Thermogramm für das enthaltene Copolymer Schmelzpeaks mit schwacher bis
ausgeprägter Schulter (bei kleineren Temperaturen). Zur Auftragung (Bild 6.8) wurde die
Schmelztemperatur des Hauptpeaks verwendet.
Bei den Proben „Co-sc04“ und „Co-sc05“ konnte lediglich für die Ausgangsmaterialien (vor
dem Abbau) ein Schmelzpeak geringer Intensität für das enthaltene Copolymer ermittelt
werden. Daher erfolgt hierfür keine gesonderte graphische Darstellung.
Bei allen Proben wurde zunächst eine leichte und mit fortschreitendem Abbau eine
gesteigerte bis starke Reduzierung der Schmelztemperatur gefunden. Die Probe „Co-
scTech +55“ stellt hier aber einen Sonderfall dar. Denn für diese Probe konnte nur zu Beginn
sowie nach einer Abbauzeit von 5 Wochen ein Schmelzpeak für das Copolymer registriert
werden. Dieser wies keine Schulter auf und zeigt im Vergleich zu den weiteren Proben eine
Page 85
77
deutlich kleinere Schmelztemperatur von rund 154,4 °C.
Bild 6.8
Veränderung der Schmelztemperatur Tm Homo1 der Technikum-Materialien während des Abbaus
Mit Ausnahme der Probe „Co-scTech -26“ kann für die übrigen aufgetragenen modifizierten
sc-Materialien nach dem anfänglichen Absinken der Schmelztemperatur ein leichter Wieder-
anstieg verfolgt werden. Für die Proben „±0“ und „-55“ kommt es im weiteren Verlauf der
Untersuchungen ab Woche 25 zu einem erneuten Abfall der registrierten Messwerte. Für die
Proben „+26“ und „±0“ können über den gesamten Untersuchungszeitraum betrachtet die
größten Unterschiede festgestellt werden. Hier finden sich zwischen den ermittelten
Anfangs- und Endwerten Temperaturdifferenzen von ca. 11,5 °C und 9,0 °C.
Page 86
78
Bild 6.9
Veränderung der Schmelztemperatur Tm Homo2 der Technikum-Proben während des Abbaus
Bild 6.10
Veränderung der Schmelztemperatur Tm Homo2 der Proben „Co-sc04“ und „Co-sc05“ während des Abbaus
Page 87
79
Bild 6.11
Veränderung der Schmelztemperatur Tm sc der Technikum-Proben während des Abbaus
Bild 6.12
Veränderung der Schmelztemperatur Tm sc der Proben „Co-sc04“ und „Co-sc05“ während des Abbaus
Page 88
80
Die Schmelztemperatur des Homopolymers (Tm Homo2) sinkt bei allen Proben deutlich.
Außerdem kann klar aufgezeigt werden, dass es zum Ende der Untersuchungen nochmals zu
einem stärkeren Abfall der beobachteten Temperatur kommt (Bild 6.9 und 6.10). Die größte
Temperaturdifferenz von ca. 12 °C zeigt auch hier die Probe „Co-scTech +26“. Hervorzu-
heben ist, dass für die Proben „Co-scTech -26“ und „Co-scTech -55“ zu Beginn der Abbau-
untersuchungen kein Schmelzpeak des Homopolymers ermittelt werden konnte (Bild 6.9).
Im Verlauf des Abbaus wurde dann bis zu einem Abbauzeitraum von 28 Wochen für beide
Proben ein schwach ausgeprägter Peak identifiziert. Diese Beobachtung legt die Vermutung
nahe, dass es hier zu einer deutlichen Veränderung des Kristallisationsverhaltens gekommen
sein muss.
Die Schmelztemperaturen der Stereokomplexe (Bild 6.11 und 6.12) zeigen im Vergleich zu
den Schmelztemperaturen der Homopolymeren (Tm Homo1 und Tm Homo2) nahezu über den
gesamten Untersuchungszeitraum einen gegenläufigen Trend. Insbesondere für die Probe
„Co-scTech +55“ kann eine deutliche Steigerung der Schmelztemperatur beobachtet werden
(Bild 6.11). Der anschließende Abfall der untersuchten Temperatur kann als erstes für die
Probe „Co-scTech +26“ gefunden werden. Hier zeigt der Kurvenverlauf nach Durchlaufen
eines Maximums (Tm sc = 218 °C) nach 22 Wochen im Abbaumedium ein lineares Absinken
der Messwerte. Die übrigen Proben zeigen erst nach einem Untersuchungszeitraum von 43
bzw. 40 Wochen eine Reduzierung der Werte. Für keine der Proben kann dabei ein Abfallen
der Schmelztemperatur unter ihren jeweiligen Ausgangswert registriert werden.
6.4. Diskussion der Ergebnisse
Auf Grund der unveränderten Werte der Probenmasse sowie des pH-Wertes bei einer gleich-
zeitigen nahezu kontinuierlichen Reduzierung der Molmasse kann für alle Proben auf einen
Abbau mittels Bulkerosion geschlossen werden.
Die weißliche bis gelbliche Verfärbung der Proben deutet auf eine molekulare Umordnung
[116] und damit eine Kristallisation während des Abbaus hin. An Hand der durchgeführten
DSC-Untersuchungen kann aber gezeigt werden, dass bereits vor den ersten optischen
Änderungen eine Veränderung der Kristallinität bzw. der Gesamtenthalpie stattgefunden hat
(vgl. Bild 6.6 und 6.7).
Das Absinken der Schmelztemperaturen Tm Homo1 (Copolymer) und Tm Homo2 (Homopoly-
mer) ist auf den Abbau der Molmasse zurückzuführen [120]. Im Gegensatz dazu weist die
fast über den gesamten Untersuchungszeitraum beobachtete Erhöhung der Schmelz-
temperatur Tm sc auf ein Wachstum der Stereokomplexkristalle hin [118]. Durch das
Ansteigen der Schmelztemperatur bei gleichzeitigem Abbau der Molmasse wird von
Page 89
81
Rahaman und Tsuji [118] der Einfluss des Kristallwachstums im Vergleich zum Effekt des
Abbaus als bedeutender diskutiert. Zusätzlich dazu ist auch die Bildung von Stereo-
komplexen beim Abbau von D,L-PLA bekannt [89]. Eine weitere Erklärung könnte das
ausschließliche Kristallwachstum ohne Beteiligung von Stereokomplexabbau für den be-
treffenden Abbauzeitraum sein, in dem der Anstieg von Tm sc zu beobachten ist.
Die Unterschiede der Proben „Co-sc04“ und „Co-sc05“ lassen sich mit Hilfe der unter-
schiedlichen enthaltenen amorphen Anteile in den eingesetzten Copolymeren sowie dem
Mischungsverhältnis von Co- und Homopolymer erklären. Begründet durch das kleinere
amorphe Blocksegment (im Copolymer) kann für das Probenmaterial „Co-sc04“ sowohl ein
höheres Niveau der Gesamtenthalpie (vgl. Bild 6.7) als auch der Schmelztemperatur des
Stereokomplexes (vgl. Bild 6.12) gefunden werden. Für die Schmelztemperaturen des Homo-
polymers werden im Vergleich zu Probe „Co-sc05“ hingegen kleinere Werte gefunden. Dies
ist auf die gleichbleibenden Mischungsverhältnisse der Proben zurückzuführen. Beide
Proben werden in einem 1:1-Verhältnis von Co- und Homopolymer umgesetzt. Auf Grund
des höheren amorphen Anteils in Probe „Co-sc05“ verbleibt hier ein größerer Anteil an nicht
zum Stereokomplex umgesetztem PDLA im Material, das wiederum besser und „störungs-
freier“ auskristallisieren kann als in der Probe „Co-sc04“ (vgl. Abschn. 5.3.1). Durch die
hieraus resultierende Reinheit/Unreinheit der entstehenden Kristalle können die
Unterschiede der Schmelztemperatur hervorgerufen werden.
Page 90
82
7. Partikelherstellung und -verarbeitung
In der vorliegenden Arbeit wurde das Schmelze-Verfahren zur Partikelbildung verwendet.
Wie bereits in Abschnitt 3.6 beschrieben, werden hierbei das Polymer und eine wasser-
lösliche Hilfskomponente gemeinsam aufgeschmolzen und in der Schmelze miteinander
dispergiert. Während des Abkühlens werden die Polymertröpfchen durch die umschließende
Hilfskomponente stabilisiert und können nach dem vollständigen Abkühlen durch Lösen
dieser erhalten werden.
7.1. Verfahrensoptimierung des Schmelze-Verfahrens
In der Offenlegungsschrift [127] werden sowohl Polyethylenglykole und Polyvinylakohole
aber auch Oligosaccharide als mögliche Hilfskomponenten angeführt. Auf Grund der hohen
Schmelztemperatur der Stereokomplexmaterialien sind PEG und PVA als Hilfskomponente
nicht geeignet. Auch im Bereich der Oligosaccharide ist die Zahl der im angestrebten
Temperaturbereich thermisch stabilen Materialien nur gering. Ein möglicher Kandidat ist D-
Mannitol, das eine Siedetemperatur von 290-295 °C aufweist und bei RT eine Wasser-
löslichkeit von 216 g/L besitzt.
Voruntersuchungen zeigten, dass sich die Eigenschaften (Viskosität und Polarität) des Polyols
und des Stereokomplexes stark voneinander unterscheiden und es daher bei Unterbrechung
des Rührprozesses zu einer sofortigen Entmischung der beiden Komponenten kommt. Durch
Fortsetzen der Durchmischung während der Abkühlphase war es prinzipiell möglich, auch
mit diesem System im geringen Umfang Partikel herzustellen. Diese waren allerdings nicht
sphärisch und wiesen teilweise einen Schweif auf (Bild 7.1 links). Außerdem kam es zur
Bildung von Fasern und größeren Agglomeraten an der Oberfläche des Ansatzes.
Weiterhin brachte auch das rasche Abkühlen der Schmelzeemulsion mit Eiswasser nicht den
gewünschten Erfolg. Es konnten zwar mehr Partikel als zuvor erhalten werden, diese wiesen
auf Grund der sehr schnell ablaufenden Agglomeration aber große Durchmesser auf (Bild 7.1
Page 91
83
rechts). Auch weitere Variationen der Versuchsbedingungen führten weder zu einer
Reduzierung der Partikelgröße noch zu einer Erhöhung der Ausbeute an geeigneten
Partikeln.
Bild 7.1
Partikel nach Weiterrühren in der Abkühlphase (links) und nach Abkühlen mit Eiswasser (rechts)
Als weitere vielversprechende Hilfskomponente wurde Polyvinylpyrrolidon (PVP) identi-
fiziert. Da es bei hohen Temperaturen bei diesem Polymer zu Vernetzungsreaktionen
kommen kann und die entstehenden Produkte wasserunlöslich sind, erwies sich die alleinige
Anwendung von PVP (Mw = 55000 g/mol) aber als ungeeignet.
Weiterhin wurde myo-Inositol als alternatives Polyol getestet. Dieses Material hat einen
Schmelzpunkt (Tm = 223-225 °C), der nahe dem des Stereokomplexes liegt. Allerdings wurde
auch in diesem Fall ein ähnliches Verhalten beobachtet wie bei der Verwendung von D-
Mannitol. Beide Komponenten entmischten sich bei Unterbrechung des Rührens. Da die
Entmischung hier langsamer erfolgte, konnte jedoch eine merkliche Steigerung der Ausbeute
erzielt werden. Die resultierenden Partikel hatten eine meist sphärische Form mit sehr
unterschiedlichen Durchmessern. Es wurden aber auch faserartige Gebilde gefunden
(Bild 7.2 links).
Um einer Entmischung vorzubeugen, wurde in den weiteren Versuchen die Eignung von PVP
(Mw = 10000 g/mol) als „Phasenvermittler“ getestet. Die Entmischung sollte durch eine
Steigerung der Viskosität des Polyols vermieden werden. Schon in den ersten Versuchen
konnte eine deutliche Steigerung der Partikelausbeute gefunden werden. Allerdings waren
die Partikel überwiegend nicht sphärisch, vielmehr bildeten sich unregelmäßige, zum Teil
plättchenartige Strukturen aus (Bild 7.2 rechts). Ursache hierfür kann unter anderem eine zu
starke Systemviskosität und ein zu hoher Energieeintrag (hohe Rührgeschwindigkeit) sein.
Durch Optimierung der Rezepturparameter (Mischungsverhältnis von Stereokomplex, myo-
Inositol und PVP (Mw = 10000 g/mol)) und der Verfahrensparameter (Rührgeschwindigkeit,
Arbeitstemperatur) konnten Bedingungen gefunden werden, die die Herstellung riesel-
fähiger Partikel in akzeptablen Ausbeuten (ca. 75%) ermöglichten.
Page 92
84
Bild 7.2
Partikel bei Verwendung von myo-Inositol (links) und bei Verwendung eines Gemischs aus myo-Inositol und
PVP (rechts)
7.2. Partikelherstellung auf Basis der optimierten Vorschrift
Für die Partikelbildung werden Stereokomplex, Polyol (myo-Inositol) und Hilfskomponente
(PVP Mw = 10000 g/mol) in einem Mischungsverhältnis von 1 : 7,8 : 1,3 in ein zylinder-
förmiges Glasgefäß eingewogen und das Ölbad auf eine Temperatur von 250 °C erwärmt.
Nach Erreichen der gewünschten Verarbeitungstemperatur wird das Glasgefäß in das Ölbad
eingehängt und das Probenmaterial aufgeschmolzen. Nachdem etwa die Hälfte des
Materials aufgeschmolzen ist, wird ein Rührer installiert und die Schmelze-Pulver-Mischung
leicht von Hand durchmischt. Nach dem vollständigen Aufschmelzen (ca. 32 min) wird der
Rührer in Betrieb genommen. Es wird 14 min mit einer Geschwindigkeit von 430 U/min und
1 min mit einer Geschwindigkeit von 275 U/min gerührt. Diese Variation der Rührgeschwin-
digkeit wurde gewählt um zunächst möglichst keine Partikel zu erhalten, die durch die
Geschwindigkeitsreduzierung die Möglichkeit erhalten zu Polymertröpfchen mit sphärischer
Form zusammenzufließen. Dabei ist darauf zu achten, dass nicht zu lange bei verminderten
Geschwindigkeiten gerührt wird (Verhinderung der Agglomeration der Partikel, vgl.
Abschn. 7.1).
Nach Abschluss der Durchmischung wird das Gefäß aus dem Ölbad entnommen und die
Schmelze auf RT abgekühlt. Anschließend werden das Polyol und die Hilfskomponente durch
Zugabe von Wasser gelöst. Da zum Lösen ein großer Überschuss an Wasser notwendig ist,
wird der schon gelöste Anteil regelmäßig aus dem Reaktionsgefäß entfernt und die
verbliebene Schmelzemischung mit frischem Wasser aufgefüllt.
Die entstehende Suspension wird mittels Vakuumfiltration über einen Cellulosefilter
(4,0 µm, MN616, Macherey-Nagel) in Partikelfraktion und Polyol/PVP-Lösung aufgetrennt.
Die Partikel wurden anschließend mit Wasser gewaschen, in Petrischalen überführt und im
Page 93
85
Vakuumtrockenschrank bei RT über P2O5 getrocknet.
Zur Abtrennung von „Häutchen“, faserartigen Gebilden und größeren Partikel-Agglomeraten
wird das getrocknete Produkt über ein Sieb der Maschenweite 250 µm unter Verwendung
einer Siebmaschiene (AS200, Retsch) klassiert.
7.3. Charakterisierung der Mikropartikel
Zur Ermittlung von Partikelform und -größe, wurden von jedem Ansatz Lichtmikroskop-
aufnahmen angefertigt. Die nachfolgenden Bilder zeigen einige Beispiele der mittels in
Abschnitt 7.2 beschriebenen Versuchsvorschrift erhaltenen Partikel.
Bild 7.3
Aussehen der Partikel bei Verwendung der in Abschnitt 7.2 beschriebenen Versuchsvorschrift
Die durch die Partikelbildung in der Schmelze möglicherweise hervorgerufene Änderung der
thermischen Eigenschaften wurde mittels DSC untersucht. Weiterhin diente diese Methode
der Bestimmung der Reinheit der Partikel. Es ist ersichtlich, dass nach der Verarbeitung nur
Page 94
86
der Schmelzpeak für den Stereokomplex vorliegt (Tm sc = 211,3 °C). Dies zeigt, dass das
Material vollständig zum Stereokomplex umgesetzt ist und dass durch das intensive
Waschen der Partikel alle Bestandteile der Hilfskomponenten entfernt werden konnten.
Mit Hilfe der GPC konnte ein deutlicher Molmassenabbau (Mn) der Materialien von ca.
25000 g/mol nachgewiesen werden. Dieser ist mit der hohen erforderlichen Verarbeitungs-
temperatur zu begründen, die deutlich über der Schmelztemperatur des verwendeten
Stereokomplexes liegt.
Mit Hilfe einer Siebmaschine, ausgestattet mit Sieben der Maschenweiten 50 µm, 75 µm,
100 µm und 150 µm, wurden die Partikel klassifiziert. Die Auswaage der durch den
Siebprozess erhaltenen Fraktionen ergab die in Tabelle 7.1 gezeigte Größenverteilung der
hergestellten Partikel und die jeweiligen Mengenanteile der einzelnen Fraktionen:
Tabelle 7.1
Größenverteilung der hergestellten Partikel und deren Mengenanteile
Größenbereich [µm]
Anteil an Gesamtmenge [%]
< 50 13
75 > x > 50 15
100 > x > 75 19
150 > x > 100 40
250 > x > 150 13
Neben den im Technikum hergestellten Stereokomplexen („Co-scTech“-Materialien) wurden
auch die beiden Ausgangspolymere („Co-Tech“ und „PDLA Tech“) zur Partikelherstellung im
Schmelze-Verfahren eingesetzt. Es konnte gezeigt werden, dass auch in diesem Fall nach der
Partikelbildung in der DSC ausschließlich ein Schmelzpeak der Stereokomplexkristalle zu
finden ist. Das bedeutet, dass eine vorherige Extrusion zur Stereokomplexbildung nicht
notwendig ist, wenn aus dem Material anschließend Mikropartikel mittels Schmelze-
Verfahren hergestellt werden sollen. Nachteilig ist hier allerdings, dass für die derartig
erhaltenen Stereokomplex-Partikel trotz eines wegfallenden thermischen Verarbeitungs-
schrittes (Extrusion) vergleichbare Ergebnisse der Molmasse gefunden werden wie für die
Materialien, die mittels Umsetzung im Extruder und anschließender Partikelbildung erhalten
wurden. Als mögliche Ursachen müssen hier die thermische Instabilität der beiden
Ausgangpolymere in Kombination mit der langen thermischen Belastung (ca. 30 min
aufschmelzen und 15 min dispergieren) genannt werden.
Page 95
87
7.4. Selektives Laserschmelzen
7.4.1. Prototypen
Um die Verarbeitungsfähigkeit der hergestellten Stereokomplex-Partikel mittels SLM zu
untersuchen, wurden am Fraunhofer ILT (Aachen) entsprechende Versuche durchgeführt. In
einem ersten Versuch ist dabei zunächst die Laserleistung variiert worden. Die weiteren
Parameter (dS = 250 µm, vscan = 50 mm/s, ΔyS = 20 µm, DS = 50 µm; vgl. Abschn. 3.7) wurden
basierend auf Erfahrungswerten mit PLA-Materialien gewählt und konstant gehalten.
Während des laufenden Prozesses wurde die Laserleistung ständig dem aktuellen Schmelz-
bild neu angepasst. Die nachfolgenden Bilder (Bild 7.4 bis Bild 7.6) dokumentieren den
Prozessverlauf.
Bild 7.4
Probenkörper nach 10 Schichten (Bauhöhe: 0,50 mm; links) und nach 15 Schichten (Bauhöhe: 0,75 mm; rechts)
[162]
Bild 7.5
Probenkörper nach 20 Schichten (Bauhöhe: 1,00 mm; links) und nach 45 Schichten (Bauhöhe: 2,25 mm; rechts)
[162]
Page 96
88
Bild 7.6
Probenkörper nach 80 Schichten (Bauhöhe: 4,00 mm) mit (links) und ohne umgebende Pulverschicht (rechts)
[162]
An Hand des gezeigten Verlaufs dieses Versuchs kann deutlich aufgezeigt werden, dass eine
Verarbeitung des modifizierten sc-Materials mittels SLM prinzipiell möglich ist.
In einem zweiten Versuch wurde der Einfluss des Strahldurchmessers auf die Verarbeitung
des Materials untersucht. Es sollte überprüft werden, ob eine schonendere Verarbeitung für
das Polymer möglich ist. Dazu wurde der Strahldurchmesser dS von ~250 µm auf ~360 µm
vergrößert und die übrigen Parameter konstant gehalten. An Hand der nachfolgenden Bilder
(Bild 7.7 bis Bild 7.9) soll auch hier der Prozessverlauf aufgezeigt werden.
Bild 7.7
Probenkörper nach 10 Schichten (Bauhöhe: 0,50 mm; links) und nach 20 Schichten (Bauhöhe: 1,00 mm; rechts)
[163]
Page 97
89
Bild 7.8
Probenkörper nach 40 Schichten (Bauhöhe: 2,00 mm; links) und nach 80 Schichten (Bauhöhe: 4,00 mm; rechts)
[163]
Bild 7.9
Probenkörper nach 80 Schichten (Bauhöhe: 4,00 mm) ohne umgebende Pulverschicht (links) und als Draufsicht
(rechts) [163]
Wie den Abbildungen zu entnehmen ist können die Stereokomplex-Partikel auch mit einem
breiteren Strahldurchmesser des verwendeten Lasers erfolgreich verarbeitet werden.
Lediglich bei der geringsten Laserleistung von PL = 0,25 W wurde eine unregelmäßige
Oberfläche erhalten (Bild 7.9). Dieser Effekt deutet auf ein unvollständiges Aufschmelzen der
Polymerpartikel hin. Für eine gleichmäßige Verarbeitung des Materials wird daher eine
Laserleistung von mindestens PL = 0,30 W benötigt [163].
Zusätzlich zu den kompakten Geometrien wurde auch ein Würfel mit Porenstruktur
(Porendurchmesser 1,0 mm) hergestellt. Die Verarbeitung wurde in einem beschleunigten
Verfahren (Prozessparameter: dS = 360 µm, vscan = 2700 mm/s, ΔyS = 20 µm, DS = 50 µm,
PL = 4,00 W; vgl. Abschn. 3.7) durchgeführt. Das hatte zur Folge, dass letztlich keine
Optimierung der Parameter auf das Stereokomplex-Material erfolgte. Während der Ver-
Page 98
90
arbeitung kam es zu einer starken Rauchentwicklung und auf Grund einer geringen Schmelz-
viskosität zu einem starken Zerlaufen der Schmelze. Aus diesem Grund sind die Poren des
erhaltenen Bauteils teilweise verschlossen (Bild 7.10) [163].
Bild 7.10
Würfels direkt nach dem Schmelzprozess (links) und nach Entfernung der überschüssigen Partikel (rechts) [163]
Bei allen verwendeten Prozessparametern konnte die Wiederverwendbarkeit des den Form-
körper umgebendem Polymerpulvers festgestellt werden. Das im Schmelzprozess nicht zum
Aufbau der Geometrie genutzte Material muss daher nicht verworfen werden, sondern kann
für die Herstellung eines weiteren Formkörpers genutzt werden.
7.4.2. Charakterisierung der Prototypen
Die aufgebauten Geometrien wurden mittels DSC und GPC hinsichtlich ihrer Material-
eigenschaften untersucht. Es sollte geprüft werden, ob sich das thermische Verhalten des
Materials während des SLM-Prozesses ändert und ob ein Molmassenabbau stattfindet. Die
Messergebnisse sind den Tabellen 7.2 und 7.3 zu entnehmen.
Die Untersuchungen zeigen, dass auch nach dem SLM-Prozess nur der Schmelzpeak des
Stereokomplexes gefunden werden kann. Zusätzlich haben die Materialien der Prüfkörper
im Gegensatz zum Ausgangsmaterial aber einen Kaltkristallisationspeak. Die Enthalpiewerte
der Kaltkristallisation ΔHcc sinken tendenziell mit steigender Energie des Lasers.
Page 99
91
Tabelle 7.2
Thermische Eigenschaften (DSC) des Ausgangsmaterials sowie der mittels SLM (dS = 250 µm) hergestellten
Prototypen (1. Lauf ausgewertet; vgl. Abschn. 3.8.2)
Proben-bezeichnung
Tcc [°C]
ΔHcc [J/g]
Tm sc [°C]
ΔHm sc [J/g]
ΔHgesamt [J/g]
Ausgang / / 211,3 55,2 55,2
0,25 W 95,5 19,7 212,1 57,0 37,3
0,30 W 95,3 12,8 212,6 57,5 44,7
0,35 W 95,8 9,0 213,6 57,0 48,0
0,40 W 95,6 8,1 213,5 55,6 47,5
Tabelle 7.3
Thermische Eigenschaften (DSC) der mittels SLM (dS = 360 µm) hergestellten Prototypen (1. Lauf ausgewertet;
vgl. Abschn. 3.8.2)
Proben-bezeichnung
Tcc [°C]
ΔHcc [J/g]
Tm sc [°C]
ΔHm sc [J/g]
ΔHgesamt [J/g]
0,25 W 93,4 14,2 211,8 55,7 41,5
0,30 W 87,7 15,9 213,1 54,4 38,5
0,35 W 90,0 8,6 214,4 56,8 48,2
0,40 W 95,6 5,3 213,7 57,1 51,7
Eine mögliche Erklärung für das unterschiedliche Kristallisationsverhalten kann in den
Verarbeitungsbedingungen liegen. Mit steigender Laserleistung erfolgt auch ein höherer
Energieeintrag in das Polymermaterial. Dieses wird dementsprechend unterschiedlich stark
erhitzt. Der „0,40 W-Probe“ steht dadurch im Vergleich zur „0,25 W-Probe“ ein größeres
Temperaturfenster und damit mehr Zeit für die Kristallisation des Materials zur Verfügung.
Die während des Prozesses ablaufende Kristallisation ist daher für die „0,40 W-Probe“ am
größten. Dementsprechend wird für diese Probe der kleinste Wert der Kaltkristallisations-
enthalpie gefunden.
Tabelle 7.4
Molmassen (GPC) des Ausgangsmaterials sowie der mittels SLM (dS = 250 µm) hergestellten Prototypen
Proben-bezeichnung
Mn
[g/mol]
Mw
[g/mol] PDI
Ausgang 10800 30500 2,8
0,25 W 5050 25600 5,1
0,30 W 3940 23400 5,9
0,35 W 3840 22700 5,9
0,40 W 3260 22800 7,0
Page 100
92
Der Molmassenabbau der mit unterschiedlichen Verfahrensparametern verarbeiteten
Materialien wurde mittels GPC verfolgt. Für die Prototypen, die mit einem Strahlen-
durchmesser dS von ~250 µm hergestellt wurden, kann ein deutlicher Molmassenabbau
registriert werden. Tendenziell sinkt mit steigender Laserleistung die Molmasse der
Prototypen und die Werte der Polydispersitätsindices steigen (Tabelle 7.4).
Die Prototypen, die mit einem Strahlendurchmesser dS von ~360 µm generiert wurden,
zeigen ein anderes Bild. In Hinblick auf den Fehler der Methode kann man davon ausgehen,
dass sich sowohl die Molmasse als auch der PDI nicht geändert haben.
Unter Berücksichtigung der gefundenen Ergebnisse kann klar aufgezeigt werden, dass schon
geringe Variationen der möglichen SLM-Verfahrensparameter (vgl. Abschn. 3.7) deutliche
Unterschiede in den Eigenschaften der hergestellten Prototypen mit sich bringen. Nach
sorgfältiger Anpassung der SLM-Parameter sollte es daher möglich sein, die neuartigen
Stereokomplexmaterialen ohne Veränderung der stoffspezifischen Parameter zu drei-
dimensionalen Geometrien zu verarbeiten.
Page 101
93
8. Zusammenfassung
Die Nachfrage von Industrie und Gesellschaft nach Kunststoffen aus nachwachsenden
Rohstoffen steigt aus einer Reihe von Gründen in der heutigen Zeit stetig. Damit einher-
gehende neuartige Verarbeitungsmethoden und Anwendungsschwerpunkte führen zu
immer neuen Anforderungen an die Materialien. Daher ist es notwendig, die Entwicklung
verarbeitungs- und anwendungsorientierter Polymermaterialien auf der Basis nachwach-
sender Rohstoffe von der Synthese bis zur Verarbeitung weiter voranzutreiben.
In Hinblick auf die Verarbeitungstechnologie Selektives Laserschmelzen (SLM) und deren
Anwendung im medizinischen Bereich besteht die Nachfrage nach einem sowohl thermisch
stabilen, als auch im Körper abbaubaren Material. PLA bietet hier eine gute Grundlage.
Nachteilig ist, dass dieses eine geringe thermische Stabilität besitzt. Daher ist vor einer
erfolgreichen Verarbeitung mittels SLM eine Modifizierung des Polymers notwendig. Der
Forschungsansatz dieser Arbeit bestand darin, die thermische Stabilität von PLA durch den
Einsatz von Stereokomplexen zu verbessern. Die hohe Brüchigkeit dieser Stereokomplexe
sollte durch den Einsatz von Blockcopolymeren als Ausgangsmaterial reduziert werden.
Es konnte gezeigt werden, dass es möglich ist, bisher nicht in der Literatur beschriebene PLA-
Stereokomplexe aus Ausgangsmaterialien herzustellen, die amorphe Kettensegmente
enthalten. Dazu wurden zunächst Blockcopolymere bestehend aus einem kristallinen (PLLA)
und einem amorphen (PDLLA) Teil sowie kristalline Homopolymere (PDLA) synthetisiert und
diese dann in Extrudern zu Stereokomplexen umgesetzt.
Für die Synthese der Blockcopolymere musste zunächst eine geeignete Synthesevariante
gefunden werden, um Polymere mit gezielten Blockverhältnissen zu erhalten. Hierbei
wurden einschränkende Verfahrens- und Stoffparameter ermittelt. Es zeigte sich, dass
sowohl lange thermische Belastungen als auch hohe Molmassen des Prepolymers un-
geeignet sind, um gute und reproduzierbare Syntheseergebnisse zu erhalten. Unter
Beachtung dieser Randbedingungen und nach erfolgreicher Umsetzung im Labormaßstab
wurde die Synthese auch auf den Technikum-Maßstab übertragen.
Die hergestellten Polymere wurden zu Stereokomplexen umgesetzt. Dabei erfolgte eine
Page 102
94
Variation der Länge des amorphen Blocksegments des Copolymers sowie des
Mischungsverhältnisses von Blockco- und Homopolymer. Der Einfluss dieser Größen auf die
Stereokomplexbildung sowie die thermischen und mechanischen Eigenschaften der
resultierenden Materialien wurde umfassend untersucht. Auch der Einfluss des Temperns
war Bestandteil der Untersuchungen.
Es konnte mittels DSC und Röntgenbeugung gezeigt werden, dass es prinzipiell unabhängig
von der Länge des amorphen Blocksegments des Copolymers und des Mischungs-
verhältnisses von Blockco- und Homopolymer möglich ist Stereokomplexe zu bilden. Die
Ergebnisse zeigen außerdem, dass mit der Erhöhung des amorphen Teils im Copolymer auch
eine deutliche Steigerung der Schlagbiegefestigkeit des daraus hergestellten Materials erzielt
werden kann. Mit den modifizierten Stereokomplexen konnten im Vergleich zum reinen
Homopolymer bis zu 52% höhere Werte erreicht werden. Weiterhin wurde festgestellt, dass
neben dem amorphen Teil auch die in den Komplexen vorliegenden Kristallformen (sc
und/oder hc) sowie die mittels DSC für die Materialien ermittelten Enthalpiewerte von
entscheidender Bedeutung für die finalen Materialeigenschaften sind. Sowohl mit steigender
Schmelzenthalpie des Stereokomplexes als auch mit steigender Gesamtenthalpie der
hergestellten Materialien wurde eine Reduzierung der Schlagbiegefestigkeitswerte
gefunden.
Nach dem Temperprozess konnte mittels DSC über die Auswertung der Gesamtenthalpie
eine gesteigerte Kristallinität der betreffenden Proben ermittelt werden. Dadurch begründet
zeigen die Materialien einen höheren Härtekennwert sowie eine geringere Schlagbiege-
festigkeit im Vergleich zu den nicht getemperten Proben.
Die gemessenen Schlagbiegefestigkeiten der im Labor- bzw. im Technikum-Maßstab
hergestellten Materialien liegen in voneinander abweichenden Größenbereichen. Als
mögliche Ursache hierfür wurde der nach der Synthese im Labor und im Technikum noch
vorliegende unterschiedliche Gehalt an zur Polymersynthese verwendeten Katalysator
identifiziert.
Neben den Materialeigenschaften wurde auch das Abbauverhalten der verschiedenen
Stereokomplexmaterialien untersucht. Die Abbauuntersuchungen erfolgten über einem Zeit-
raum von 43 (bzw. 40) Wochen. Die Proben wurden dazu zur Nachahmung der Bedingungen
im menschlichen Körper in einem Phosphatpuffer bei 37 °C gelagert.
An Hand der Untersuchungen von pH-Wert-, Molmasse- und Masseänderung konnte ein
Abbauverhalten des Materials mittels Bulkerosion nachgewiesen werden. Diese äußerte sich
in gleichbleibenden pH- und Masse-Werten, sowie einem linearen Abbau der Molmasse.
Durch zusätzliche DSC-Messungen wurde auch die Veränderung der thermischen Eigen-
schaften während des Abbaus verfolgt. Auf Grund des Molmassenabbaus konnte hier für die
Schmelztemperaturen Tm Homo1 (Copolymer) und Tm Homo2 (Homopolymer) insgesamt eine
Reduzierung der Werte beobachtet werden. Hervorzuheben ist, dass sich die Schmelzpunkte
der Stereokomplexe (Tm sc) um bis zu 4 °C erhöhten. Begründet werden kann dies mit dem
Page 103
95
Kristallwachstum der Stereokomplexe während des Abbaus. Mit Hilfe der DSC-Messungen
konnte außerdem eine zum Teil ausgeprägte Nachkristallisation des Materials nachgewiesen
werden. Auch optisch wurde eine Veränderung der Proben registriert, die eine
Nachkristallisation sowie Chromophorenbildung der Materialien vermuten ließ. Bei allen
Proben wurde eine teilweise bis vollständige weiße bis gelbliche Verfärbung beobachtet,
wobei der Zeitpunkt dieser Verfärbung in Abhängigkeit von den Materialproben variierte.
Parallel zu den Abbauuntersuchungen wurde mittels eines lösungsmittelfreien Verfahrens
Mikropartikel aus dem im Technikum synthetisierten Material hergestellt.
Da Partikelbildungsmethoden, die auf (halogenierten) Lösungsmitteln basieren, für Mate-
rialien zur körpernahen Anwendung unerwünscht sind und zudem zur Lösung von Stereo-
komplexen nur Hexafluorisopropanol (HFIP) als mögliches Lösungsmittel zur Verfügung
steht, wurde ein Schmelze-Verfahren angewendet. Hierbei wird das Material mit einer
wasserlöslichen Hilfskomponente in der Schmelze emulgiert und nach dem Abkühlen durch
Lösen in Wasser wieder von dieser abgetrennt.
Für die Partikelbildung musste dem Material entsprechend zunächst ein geeignetes
wasserlösliches Hilfssystem und anschließend Verfahrensparameter gefunden werden, mit
denen sich Partikel im gewünschten Größenbereich herstellen ließen. Als Hilfssystem erwies
sich eine 7,8 : 1,3-Mischung aus myo-Inositol und PVP (Mw = 10000 g/mol) als praktikabel.
Die Verarbeitungstemperatur lag bei 250 °C.
Mit Hilfe der verfahrensoptimierten Methode war es möglich rieselfähige Mikropartikel aus
dem Probenmaterial herzustellen. Die anschließende Charakterisierung zeigte, dass etwa
60% der generierten Partikel im Größenbereich von 75 µm bis 150 µm lagen. Mittels DSC-
Messungen konnte gezeigt werden, dass die Verarbeitung des Materials zu Partikeln keinen
Einfluss auf die thermischen Eigenschaften hat. An Hand von GPC-Untersuchungen kann auf
Grund der hohen Arbeitstemperatur aber eine deutliche Reduzierung der Molmasse (> 50%)
verfolgt werden.
Aus den hergestellten Mikropartikeln wurden mit Hilfe der Rapid-Manufacturing-Methode
SLM Prototypen aufgebaut. Es konnte gezeigt werden, dass eine Verarbeitung des Materials
mit dieser Methode grundsätzlich möglich ist und nach Anpassung der Verarbeitungs-
bedingungen vielversprechende Ergebnisse erhalten werden können. So ist es final
gelungen, das neu entwickelte Material ohne einen durch die thermische Belastung zu
erwartenden Abbau zu Prototypen zu verarbeiten. Auch die Generierung von Poren-
strukturen in den mittels SLM aufgebauten Probenkörpern war möglich.
Schlussfolgernd aus den Ergebnissen zeigt sich, dass mit den modifizierten Stereokomplexen
ein vielversprechendes Polymermaterial zur Verfügung steht, das mittels SLM zu geometrie-
unabhängigen Objekten verarbeitet werden kann. Ein zusätzlicher und weiterführender
Ansatz wäre die Manipulation der Materialeigenschaften am fertigen Bauobjekt. So ist es
Page 104
96
denkbar eine Beschichtung auf die mittels SLM hergestellten Objekte aufzubringen, um so
zum Beispiel Einfluss auf das Abbauverhalten nehmen zu können. Auch wäre es vorstellbar
Antibiotika, Schmerzmittel oder Entzündungshemmer direkt während des Schmelzprozesses
in das Polymermaterial mit einzubauen. Im Verlauf des Abbaus würden diese Stoffe dann
gezielt freigesetzt und könnten die Heilung unterstützen.
Vorstellbare Anwendungsgebiete für das neu entwickelte Material wären auf Grund seiner
Biokompatibilität beispielsweise die Zahnmedizin, die Herstellung von Stents oder die
Nachbildung komplexer Knochenstrukturen (z.B. Becken) zur OP-Vorbereitung. Naturgetreue
Nachbildungen von Skelettteilen werden zum Beispiel dazu verwendet, um Prothesen
besser an die vorhandene Knochenstruktur anpassen zu können. In der heutigen Zeit werden
dazu Vollkörper unterschiedlicher Materialien genutzt, aus denen man das gewünschte
Objekt CNC-gesteuert herausfräst. Dieser Herstellungsprozess unterliegt jedoch geo-
metrischen Einschränkungen. Mit dem SLM steht hier ein alternatives Verfahren zur
Verfügung, mit dem es möglich ist einen passgenaueren und materialeffizienteren
Herstellungsprozess zu realisieren.
Neben der Medizin wäre aber auch die technische Anwendung denkbar. Allerdings muss
hierbei einschränkend beachtet werden, dass die Glasübergangstemperatur der modifi-
zierten Stereokomplexe auf Grund des hohen enthaltenen amorphen Anteils im Größen-
bereich der Homopolymere liegt.
Page 106
X
Literaturverzeichnis
[1] Harris, A. M., E. C. Lee; "Improving Mechanical Performance of Injection Molded PLA
by Controlling Crystallinity." Journal of Applied Polymer Science, 2008, 107, 2246-2255.
[2] Urayama, H., T. Kanamori, et al.; "Controlled crystal nucleation in the melt-
crystallization of poly(L-Lactid) and poly(L-lactid)/poly(D-lactide) stereocomplexes."
Polymer, 2003, 44, 5635-5641.
[3] Auras, R., L.-T. Lim, et al.; "Poly(lactid acid): Synthesis, Structures, Properties,
Processing and Applications." John Wiley & Sons, Inc., 2010.
[4] Auras, R., B. Harte, S. Selke; "An Overview of Polylactides as Packaging Materials."
Macromolecular Bioscience, 2004, 4, 835-864.
[5] Xu, H., S. Tang, J. Chen; "Unique Crystallization Behavior of Poly(L-lactic acid)
Nucleated by Stereocomplex with Different Fine Structure." Polymer-Plastics
Technology and Engineering, 2013, 52, 690-698.
[6] Saeidlou, S., M. A. Huneault, et al.; "Poly(lactide acid) crystallization" Progress in
Polymer Science, 2012, 37, 1657-1677.
[7] Zhang, J., K. Tashiro, et al.; "Disorder-to-Order Phase Transition and Multiple Melting
Behavior of Poly(L-lactide) Investigated by Simultaneous Measurements of WAXD and
DSC." Macromolecules, 2008, 41, 1352-1357.
[8] Gallos, A., G. Fontaine, S. Bourbigot; "Reactive Extrusion of Stereocomplexes Poly-L,D-
lactides: Processing, Characterization, and Properties." Macromolecular Materials and
Engineering, 2013, 298, 1016-1023.
[9] Yang, G., J. Su, et al.; "Toughening of Poly(L-Lactic Acid) by Annealing: The Effect of
Crystal Morphologies and Modifications." Journal of Macromolecular Science, Part B:
Physics, 2012, 51, 184-196.
Page 107
XI
[10] Jacobsen, S., H. Fritz; "Plasticizing Polylactide – The Effect of Different Plasticizers on
the Mechanical Properties." Polymer Engineering and Science, 1999, 39, 1303-1310.
[11] Wang, S., W. Cui, J. Bei; "Bulk and surface modifications of polylactide." Analytical and
Bioanalytical Chemistry, 2005, 381, 547-556.
[12] Martin, O., L. Averous; "Poly(lactic acid): plasticization and properties of biodegradable
multiphase systems." Polymer, 2001, 42, 6209-6219.
[13] Byun, Y., S. Whiteside, et al.; "The Effect of Solvent Mixture on the Properties of
Solvent Cast Polylactic Acid (PLA) Film." Journal of Applied Polymer Science, 2012, 124,
3577-3582.
[14] Masutani, K., C.-W. Lee, Y. Kimura; "Synthesis of stereo multiblock polylactides by dual
terminal couplings of poly-L-lactide and poly-D-lactide prepolymers: A new route to
high-performance polylactides." Polymer, 2012, 53, 6053-6062.
[15] Marques, D. S., M. H. Gil, C. M. S. G. Baptista; "Improving Lactic Acid Melt Poly-
condensation: The Role of Co-Catalyst." Journal of Applied Polymer Science, 2013, 128,
2145-2151.
[16] Furuhashi, Y., N. Yoshie; "Stereocomplexation of solvent-cast poly(lactic acid) by
addition of non-solvents." Polymer International, 2012, 61, 301-306.
[17] Saeidlou, S., M. A. Huneault, et al.; "Evidence of a dual network/spherulitic crystalline
morphology in PLA stereocomplexes." Polymer, 2012, 53, 5816-5824.
[18] Yamane, H., K. Sasai; "Effect of the addition of poly(D-lactic acid) on the thermal
property of poly(L-lactic acid)." Polymer, 2003, 44, 2569-2575.
[19] Saeidlou, S., M. A. Huneault, et al.; "Effect of nucleation and plasticization on the
stereocomplex formation between enantiomeric poly(lactic acid)s." Polymer, 2013, 54,
5762-5770.
[20] Bai, H., H. Liu, et al.; "Enhancing the melt stability of polylactide stereocomplexes using
a solid-state cross-linking strategy during a melt-blending process." Polymer Chemistry,
2014, 5, 5985-2993.
[21] Li, Y., X. Zhang, et al.; "Poly(L-lactide)/Poly(D-lactide)/Clay Nanocomposites: Enhanced
Dispersion, Crystallization, Mechanical Properties, and Hydrolytic Degradation."
Polymer Engineering and Science, 2014, 54, 914-924.
Page 108
XII
[22] Becker, J. M., R. J. Pounder, A. P. Dove; "Synthesis of Poly(lactide)s with Modified
Thermal and Mechanical Properties." Macromolecular Rapid Communications, 2010,
31, 1923-1937.
[23] Sun, J., J. Shao, et al.; "Thermostimulated crystallization of polylactide stereocomplex."
Materials Letters, 2012, 89, 169-171.
[24] Tsuji, H., T. Tajima; "Crystallization Behavior of Stereo Diblock Poly(Lactide)s with
Relatively Short Poly(D-Lactide) Segment from Partially Melted State." Macromolecular
Materials and Engineering, 2014, 299, 1089-1105.
[25] Quynh, T. M., H. Mitomo, et al.; "Properties of a Poly(L-lactid acid)/Poly(D-lactic acid)
Stereocomplex and the Stereocomplex Crosslinked with Triallyl Isocyanurate by
Irradiation." Journal of Applied Polymer Science, 2008, 110, 2358-2365.
[26] Fukushima, K., Y.-H. Chang, Y. Kimura; "Enhanced Stereocomplex Formation of Poly(L-
lactid acid) an Poly(D-lactide acid) in the Presence of Stereoblock Poly(lactid acid)."
Macromolecular Bioscience, 2007, 7, 829-835.
[27] Fukushima, K., M. Hirata, Y. Kimura; "Synthesis and Characterization of Stereoblock
Poly(lactic acid)s with Nonequivalent D/L sequence Ratios." Macromolecules, 2007, 7,
3049-3055.
[28] Xu, H., C. Teng, M. Yu; "Improvements of thermal property and crystallization behavior
of PLLA based multiblock copolymer by forming stereocomplex with PDLA oligomer."
Polymer, 2006, 47, 3922-3928.
[29] Wang, Y., J. F. Mano; "Influence of melting conditions on the thermal behavior of
poly(L-lactic acid)." European Polymer Journal, 2005, 41, 2335-2342.
[30] Li, S., S. McCarthy; "Further investigations on the hydrolytic degradation of poly(DL-
lactide)." Biomaterials, 1999, 20, 35-44.
[31] Ikada, Y., H. Tsuji; "Biodegradable polyesters for medical and ecological applications."
Macromolecular Rapid Communications, 2000, 21, 117-132.
[32] Kolstad, J. J.; "Crystallization Kinetics of Poly(L-lactide-co-meso-lactide)." Journal of
Applied Polymer Science, 1996, 62, 1079-1091.
[33] Sarasua, J. R., A. López Arraiza, et al.; "Crystallization and Mechanical Properties of
Optically Pure Polylactides and Their Blends." Polymer Engineering and Science, 2005,
45, 745-753.
Page 109
XIII
[34] Li, S., M. Vert; "Morphological Changes Resulting from the Hydrolytic Degradation of
Stereocopolymers Derived from L- and D,L-Lactides." Macromolecules, 1994, 27, 3107-
3110.
[35] Kohn, F. E., J. W. A. Van den Berg, et al.; "The Ring-Opening Polymerization of D,L-
Lactide in the Melt Initiated with Tetraphenyltin." Journal of Applied Polymer Science,
1984, 29, 4265-4277.
[36] Kricheldorf, H. R.; "Syntheses and application of polylactides." Chemosphere, 2001, 49-
54.
[37] Peponi, L., I. Navarro-Baena, et al.; "Effect of the molecular weight on the crystallinity
of PCL-b-PLLA di-block copolymers." Polymer, 2012, 53, 4561-4568.
[38] Ormiston, J. A., P. W. S. Serruys; "Bioabsorbable Coronary Stents." Circulation
Cardiovascular Interventions, 2009, 2, 255-260.
[39] Colombo, A., E. Karvouni; "Biodegradable Stents: Fulfilling the Mission and Stepping
Away." Circulation, 2000, 102, 371-373.
[40] Tamai, H., K. Igaki, et al.; "Initial and 6-Month Results of Biodegradable Poly-L-Lactic
Acid Coronary Stents in Humans." Circulation, 2000, 102, 399-404.
[41] Flege, Ch., F. Vogt, et al.; "Development and characterization of a coronary polylactic
acid stent prototype generated by selective laser melting." Journal of Material Science:
Materials in Medicine, 2013, 24, 241-255.
[42] Tsuji, T., H. Tamai, et al.; "Biodegradable stents as a platform to drug loading."
International Journal of Cardiovascular Interventions, 2003, 5, 13-16.
[43] Tamai, H., K. Igaki, et al.; "A Biodegradable Poly-L-lactic Acid Coronary Stent in the
Porcine Coronary Artery." Journal of International Cardiology, 1999, 12, 443-449.
[44] Grijpma, D. W., H. Altpeter, et al.; "Improvement of the mechanical properties of
poly(D,L-lactide) by orientation." Polymer International, 2002, 51, 845-851.
[45] Grijpma, D. W., A. J. Nijenhuls, et al.; "High impact strength as-polymerized PLLA."
Polymer Bulletin, 1992, 29, 571-578.
[46] Grijpma, D. W., A. J. Pennings; "Polymerization temperature effects on the properties
of L-lactide and ε-caprolactone copolymers." Polymer Bulletin, 1991, 25, 335-341.
[47] Yui, N., P. J. Dijkstra, J. Feijen; "Stereo block copolymers of L- and D-lactides." Die
Makromolekulare Chemie, 1990, 191, 481-488.
Page 110
XIV
[48] Miyata, T., T. Masuko; "Crystallization behavior of poly(L-lactide)." Polymer, 1998, 39,
5515-5521.
[49] Grijpma, D., A. J. Pennings; "(Co)polymers of L-lactide, 2 Mechanical properties."
Macromolecular Chemistry and Physics, 1994, 195, 1649-1663.
[50] Tsuji, H.; "In vitro hydrolysis of blends from enantiomeric Poly(lactide)s Part 4: well-
homo-crystallized blend and nonblended films." Biomaterials, 2003, 24, 537-547.
[51] Karst, D., Y. Yang; "Molecular modeling study of the resistance of PLA to hydrolysis
based on the blending of PLLA and PDLA." Polymer, 2006, 47, 4845-4850.
[52] Stevels, M. W., M. J. K. Ankoné, et al.; "Well defined block copolymers of ε-
caprolactone and L-lactide using Y5(µ-O)(OiPr)13 as an initiator." Macromolecular
Chemistry and Physics, 1995, 196, 1153-1161.
[53] Nam, B. U., B. S. Lee, et al.; "Thermal and Mechanical property of PLA stereocomplex
with Impact Modifiers." (www.plasticsengineering.org/node/4649 am 11.09.2014)
[54] Karjomaa, S., T. Suortti, et al.; "Microbial degradation of poly-(L-lactic acid) oligomers."
Polymer Degradation and Stability, 1998, 59, 333-336.
[55] Rafler, G., J. Lang, et al.; "Biodegradable Polymers, 9. Technologically Relevant Aspects
of Kinetics and Mechanism of Ring-Opening Polymerization of L,L-Lactid."
Macromolecular Materials and Engineering, 2001, 286, 761-768.
[56] Park, S.-D., M. Todo, K. Arakawa; "Effect of annealing on the fracture toughness of
poly(lactic acid)." Journal of Material Science, 2004, 39, 1113-1116.
[57] Nam, B. U., B. S. Lee, et al.; "Effect of a Nucleating Agent on Crystallization Behavior
and Mechanical property of PLA stereocomplex." (www.iccm-central.org/
Proceedings/ICCM18proceedings/data/3.%Poster%20Presentation/Aug22(Monday)/P
1-1~20%Application%20of%20Composites/P1-5-IK1844.pdf am 10.09.2014)
[58] Cam, D., M. Marucci; "Influence of residual monomers and metals on poly(L-lactide)
thermal stability." Polymer, 1997, 38(8), 1879-1884.
[59] Lim, L. T., R. Auras, et al.; "Processing technologies for poly(lactic acid)." Progress in
Polymer Science, 2008, 33, 820-852.
[60] Urayama, H., Y. Kimura, et al.; "Microsructure and Thermal Properties of Glasssy
Polylactides with Different optical Purity of the Lactate Units." Macromolecular
Materials and Engineering, 2001, 286, 705-713.
Page 111
XV
[61] Tang, Z., C. Zhang, et al.; "The Crystallization Behavior and Mechanical Properties of
Polylactic Acid in the Presence of a Crystal Nucleating." Journal of Applied Polymer
Science, 2012, 125, 1108-1115.
[62] Andersson, S. R., M. Hakkarainen, A.-Ch. Albertsson; "Stereocomplexation between
PLA-like substituted oligomers and the influence on the hydrolytic degradation."
Polymer, 2013, 54, 4105-4111.
[63] Mulyashov, S. A., F. S. Sirovski, S. G. Beksaev; "Depolymerisation of oligolactic acid:
Simulation and pilot plant trial." Chemical Engineering Joournal, 2011, 176-177, 225-
230.
[64] Slomkowski, S., S. Penczek, A. Duda; "Polylactides – an overview." Polymers for
Advanced Technologies, 2014, 25, 436-447.
[65] Tsuji, H., T. Wada, et al.; "Stereocomplex crystallization and spherulite growth behavior
of poly(L-lactide)-b-poly(D-lactide) stereoblock copolymers." Polymer, 2010, 51, 4937-
4947.
[66] Tsuji, H., L. Bouapao; "Stereocomplex formation between poly(L-lactic acid) and
poly(D-lactic acid) with disproportionately low and high molecular weights from the
melt." Polymer International, 2012, 61, 442-450.
[67] Liu, Y., J. Sun, et al.; "Melt stereocomplexation from poly(L-lactic acid) and poly(D-lactic
acid) with different optical purity." Polymer Degradation and Stability, 2013, 98, 844-
852.
[68] Jaszkiewicz, A., A. K. Bledzki, et al.; "Investigation of Processability of Chain-Extended
Polylactides During Melt Processing – Compounding Conditions and Polymer Molecular
Structure." Macromolecular Materials and Engineering, 2014, 229, 307-318.
[69] Perego, G., G. Domenico, C. Bastioli; "Effect of Molecular Weight and Cristallinity on
Poly(lactic acid) Mechanical Properties." Journal of Applied Polymer Science, 1996, 59,
37-43.
[70] Andersson, S. R., M. Hakkarainen, et al.; "Customizing the Hydrolytic Degradation Rate
of Stereocomplex PLA through Different PDLA Architectures." Biomacromolecules,
2012, 13, 1212-1222.
[71] Lee, H. S., E. H. Kim, J. D. Kim; "Effect of Stereocomplex Crystallite as a Nucleating
Agent on the Isothermal Crystallization Behavior of Poly(L-lactic acid)." Plastics
Engineering (digital issue), 2013, (http://read.nxtbook.com/wiley/plasticengineering/
prototype/effectof.html am 15.09.2014)
Page 112
XVI
[72] Othman, N., C. Xu, et al.; "Thermorheological and mechanical behavior of polylactide
and its enantiomeric diblock copolymers and blends." Polymer, 2012, 53, 2443-2452.
[73] Masutani, K., C.-W. Lee, Y. Kimura; "Synthesis and Thermomechanical Properties of
Stereo Triblock Polylactides with Nonequivalent Block Compositions." Macromolecular
Chemistry and Physics, 2012, 213, 695-704.
[74] Fukushima, K., Y. Kimura; "A Novel Synthetic Approach to Stereo-Block Poly(lactid
acid)." Macromolecular Symposia, 2005, 224, 133-143.
[75] Tsuji, H., H. Takai, S. K. Saha; "Isothermal and non-isothermal crystallization behavior
of poly(L-lactic acid): Effects of stereocomplex as nucleating agent." Polymer, 2006, 47,
3826-3837.
[76] Ikada, Y., K. Jamshidi, et al.; "Stereocomplex Formation between Enantiomeric
Poly(lactides)." Macromolecules, 1987, 20, 904-906.
[77] Tsuji, H.; "Crystallization from the melt of poly(lactide)s with different optical purities
and their blends." Macromolecular Chemistry and Physics, 1996, 197, 3483-3499.
[78] Kang, M. K., Y. Jung, S. H. Kim; "Biodegradable Stereocomplex Polylactide Having
Flexible ε-Caprolactone Unit." Macromolecular Research, 2013, 21, 1036-1041.
[79] Hirate, M., K. Kobayashi, Y. Kimura; "Synthesis and Properties of High-Molecular-
Weight Stereo Di-Block Polylactides with Nonequivalent D/L Ratios." Journal of
Polymer Science: Part A: Polymer Chemistry, 2010, 48, 794-801.
[80] Anderson, K. S., M. A. Hillmyer; "Melt preparation and nucleation efficiency of
polylactide stereocomplex crystallites." Polymer, 2006, 47, 2030-2035.
[81] Woo, E. M., L. Chang; "Crystallization and morphology of stereocomplexes in
nonequimolar mixtures of poly(L-lactic acid) with excess poly(D-lactic acid)." Polymer,
2011, 52, 6080-6089.
[82] Purnama, P., Y. Jung, S. H. Kim; "Stereocomplexation of Poly(L-lactide) and Random
Copolymer Poly(D-lactide-co-ε-caprolactone) to enhance Melt Stabiltity."
Macromolecules, 2012, 45, 4012-4014.
[83] Bao, R.-Y., W. Yang, et al.; "Stereocomplex formation of high-molecular-weight
polylactide: A low temperature approach." Polymer, 2012, 53, 5449-5454.
[84] Tsuji, H., Y. Ikada, et al.; "Stereocomplex Formation between Enantiomeric Poly(lactic
acid)s. VIII. Complex Fibers Spun from Mixed Solution of Poly(D-lactic acid) and Poly(L-
lactic acid)." Journal of Applied Polymer Science, 1994, 51, 337-344.
Page 113
XVII
[85] Zhu, J., B. Na, et al.; "Enhanced stereocomplex formation of high-molecular-weight
polylactides by gelation in an ionic liquid." Polymer International, 2014, 63, 1101-1104.
[86] Tsuji, H., F. Horii, et al.; "Stereocomplex Formation between Enantiomeric Poly(lactic
acid)s. 2. Stereocomplex Formation in Concentrated Solutions." Macromolecules, 1991,
24, 2719-2724.
[87] Tsuji, H., S.-H. Hyon, Y. Ikada; "Stereocomplex Formation between Enantiomeric
Poly(lactic acid)s. 3. Calorimetric Studies on Blend Films Cast from Dilute Solution."
Macromolecules, 1991, 24, 5651-5656.
[88] Brizzolara, D., H.-J. Cantow, et al.; "Mechanism of Stereocomplex Formation between
Enantiomeric Poly(lactide)s." Macromolecules, 1996, 29, 191-197.
[89] Slager, J., A. J. Domb; "Biopolymer stereocomplexes." Advanced Drug Delivery Reviews,
2003, 55, 549-583.
[90] Purnama, P., S. H. Kim; "Stereocomplex Formation of Polylactide by Microwave
Irradiation." Polymer International, 2014, 63, 741-745.
[91] Brochu, S., R. E. Purd´homme, et al.; "Stereocomplexation and Morphology of
Polylactides." Macromolecules, 1995, 28, 5230-5239.
[92] Fukushima, K., Y. Kimura; "A Efficient Solid-State Polycondensation Method for
Synthesizing Stereocomplexed Poly(Lactic Acid)s with High Molecular Weight." Journal
of Polymer Science: Part A: Polymer Chemistry, 2008, 46, 3714-3722.
[93] Siebert-Raths, A.; "Modifizierung von Polylactid (PLA) für technische Anwendungen
Verfahrenstechnische Optimierung der Verarbeitungs- und Gebrauchseigenschaften."
Dissertation, 2012, Abfall- und Stoffwirtschaft, Universität Rostock, ISBN 978 3 86009
405 1.
[94] Mekonnen, T., P. Mussone, et al.; "Progress in bio-based plastics and plasticizing
modifications." Journal of Materials Chemistry A, 2013, 1, 13379-13398.
[95] López-Rodríguez, N., J.R. Sarasua; "Plasticization of Poly-L-lactide with L-Lactide, D-
Lactid and D,L-Lactide Monomers." Polymer Engineering and Science, 2013, 53(10),
2073-2080.
[96] Ramy-Ratiarison, R., V. Lison, et al.; "Synthesis of melt-processable PLA-based
stereocomplexes through a sustainable melt-approach." Green Chemistry, 2014, 16,
1759-1763.
Page 114
XVIII
[97] Nijenhuis, A. J., D. W. Grijpma, A. J. Pennings; "Lewis Acid Catalyzed Polymerization of
L-Lactid. Kinetic and Mechanism of the Bulk Polymerization." Macromolecules, 1992,
25, 6419-6424.
[98] Zhang, X., D. A. Macdonald, et al.; "Mechanism of Lactide Polymerization in the
Presence of Stannous Octoate: The Effect of Hydroxy and Carboxylic Acid Substances."
Journal of Polymer Science: Part A: Polymer Chemistry, 1994, 32, 2965-2970.
[99] ten Breteler, M. R., J. Feijen, et al.; "Synthesis and thermal properties of hetero-
bifunctional PLA oligomers and their stereocomplexes." Reactive & Functional
Polymers, 2013, 73, 30-38.
[100] Bechthold, I.; "Technologisch relevante Aspekte der Ringöffnungspolymerisation von
L,L-Dilactid." Dissertation, 2003, Technische Universität Berlin.
[101] Kowalski, A., A. Duda, S. Penczek; "Kinetics and Mechanism of Cyclic Esters
Polymerization Initiated with Tin(II) Octoate. 3. Polymerization of L,L-Dilactide."
Macromolecules, 2000, 33, 7359-7370.
[102] Purnama, P., Y. Jung, et al.; "Synthesis of Poly(D-lactide) with Different Molecular
Weight via Melt-Polymerization." Macromolecular Research, 2012, 20, 515-519.
[103] Rafler, G., J. Dahlmann; "Biologisch abbaubare Polymere 2. Mitt.: Zur Homo- und
Copolymerisation von D,L-Lactid." Acta Polymerica, 1990, 12, 611-617.
[104] Kricheldorf, H. R., I. Krieser-Saunders; "Polylactides – Synthesis, Characterization and
Medical Application." Macromolecular Symposia, 1996, 103, 85-102.
[105] Shinno, K., M. Masatoshi, et al.; "Solid-State Postpolymerization of L-Lactide Promotes
by Crystallization of Product Polymer: An Effective Method for Reduction of Remaining
Monomer." Macromolecules, 1997, 30, 6438-6444.
[106] Fan, Y., H. Nishida, et al.; "Thermal degradation behavior of poly(lactic acid) stereo-
complex." Polymer Degradation and Stability, 2013, 98, 709-719.
[107] Hyon, S.- H., K. Jamahidi, Y. Ikada; "Effects of residual monomer on the degradation of
DL-lactide polymer." Polymer International, 1998, 46, 196-202.
[108] Rasselet, D., A. Ruellan, et al.; "Oxidative degradation of polylactide (PLA) and its
effects on physical and mechanical properties." European Polymer Journal, 2014, 50,
109-116.
Page 115
XIX
[109] Thelen, H.; "Bewertung der Biokompatibilität von Poly(L-lactid) als Wirkstoffmatrix in
Stent-Beschichtungen." Abschlussarbeit, 2005, Postgradualstudium Toxikologie,
Universität Leipzig.
[110] Wintermantel, E., S.-W. Ha; "Medizintechnik. Life Science Engineering." Springer Verlag
Berlin Heidelberg, 2009, 5. Auflage, ISBN 978-3-540-93936-8.
[111] Martinez, F. A. C., E. M. Balciunas, et al.; "Lactid acid properties, applications and
production: A review." Trends in Food Science & Technology, 2013, 30, 70-83.
[112] Tartakowska, D. J.; "Degradationskinetik von medizinisch relevanten bioabbaubaren
Copolymeren unter statischen und dynamischen Bedingungen." Dissertation, 2005,
Polymertechnik / Kunststofftechnikum, Technische Universität Berlin, ISBN 3 7983
1967 7.
[113] Tsuji, H., Y. Ikada; "Blends of Crystalline and Amorphous Poly(lactide). III. Hydrolysis of
solution-cast Blend Films." Journal of Applied Polymer Science, 1997, 63, 855-863.
[114] Tsuji, H., M. Suzuki; "In Vitro Hydrolysis of Blends from Enantiomeric Poly(lactide)s Part
2. Well-Stereocomplexed Fiber and Film." Sen´i Gakkaishi, 2001, 57, 198-202.
[115] Scheler, S.; "Polymermikropartikel. Kapitel 1.7." (www.pharmazie-lehrbuch.de/
Kapitel/1-6.pdf am 24.10.2011)
[116] Andersson, S. R., M. Hakkarainen, et al.; "Polylactide Sterocomplexation Leads to
Higher Hydrolytic Stability but More Acidic Hydrolysis Product Pattern."
Biomacromolecules, 2010, 11, 1067-1073.
[117] Tsuji, H., T. Tsuruno; "Accelerated hydrolytic degradation of Poly(L-lactide)/Poly(D-
lactide) stereocomplex up to late stage." Polymer Degradation and Stability, 2010, 95,
477-484.
[118] Rahaman, Md. H., H. Tsuji; "Hydrolytic degradation behavior of stereo multiblock and
diblock poly(lactic acid)s: Effects of block lengths." Polymer Degradation and Stability,
2013, 98, 709-719.
[119] Li, S. M., H. Garreau, M. Vert; "Structure-property relationship in the case of the
degradation of massive aliphatic poly-(α-hydroxy acids) in aqueous media, Part 1:
Poly(DL-lactic acid)." Journal of Material Science: Materilas in Medicine, 1990, 1, 123-
130.
[120] Li, S., H. Garreau, M. Vert; "Structure-property relationship in the case of the
degradation of massive aliphatic poly-(α-hydroxy acids) in aqueous media, Part 3
Page 116
XX
Influence of the morphologie of poly(L-lactic acid)." Journal of Material Science:
Materilas in Medicine, 1990, 1, 198-206.
[121] Mainil-Varlet, P., R. Curtis, S. Gogolewski; "Effect of in vivo and in vitro degradation on
molecular and mechanical properties of various low-molecular-weight polylactides."
Journal of Biomedical Materials Research, 1997, 36, 360-380.
[122] Dorff, G.; "Eigenschaftsverbesserung von Polylactid durch Copolymerisation mit
anderen Thermoplasten." Dissertation, 2012, Universität Potsdam.
[123] Tsuji, H.; "In vitro hydrolysis of blends from enantiomeric poly(lactide)s Part 1. Well-
stereo-complexed blend and non-blended films." Polymer, 2000, 41, 3621-3630.
[124] Ishii, D., T. H. Ying, et al.; "In Vivo Tissue Response and Degradation Behavior of PLLA
and Stereocomplexed PLA Nanofibers." Biomacromolecules, 2009, 10, 237-242.
[125] Spiegelberg, S.; "Herstellung von Mikropartikeln aus einem abbaubaren Multi-
blockcopolymer zur Freisetzung von protischen Wirkstoffen." Dissertation, 2004,
Rheinisch-Westfälische Technische Hochschule Aachen.
[126] Jesse, K.; "Evaluierung von Partikelbildungsverfahren zur Herstellung von
bioresorbierbaren Polymerpartikeln im µm-Bereich." Diplomarbeit, 2010, Fachbereich
Chemie, Hochschule Niederrhein.
[127] Degussa GmbH; "Polymerpulver, Verfahren zur Herstellung und Verwendung eines
solchen Pulvers und Formkörper daraus." Offenlegungsschrift, 2007, DE 10 2006 015
791 A1 2007.10.04.
[128] Wehmöller, M., P. H. Warnke, et al.; "Implant design and production – a new approach
by selective laser melting." International Congress Series, 2005, 1281, 690-695.
[129] Ehrenstein, G. W., G. Riedel, P. Trawiel; "Praxis der Thermischen Analyse von
Kunststoffen" Karl Hanser Verlag, 2003, 2. Auflage, ISBN 3-446-22340-1.
[130] Naumer, H., W. Heller, et al.; "Untersuchungsmethoden in der Chemie – Einführung in
die moderne Analytik" Georg Thieme Verlag Stuttgart, 1990, 2. Auflage, ISBN 3-13-
681402-9.
[131] Kunze, U. R., G. Schwedt; "Grundlagen der qualitativen und quantitativen Analyse"
Georg Thieme Verlag Stuttgart, 1996, 4. Auflage, ISBN 3-13-585804-9.
[132] "Shore Härte" (http://wiki.polymerservive-merseburg.de/index.php/shore-Härte am
18.06.2013)
Page 117
XXI
[133] Rihm, R. F.; "Röntgen-Strukturuntersuchungen an Celluloseregeneratfasern."
Dissertation, 2003, Technische Universität Berlin.
[134] Atkins, P. W.; "Physikalische Chemie" Wiley-VCH, 2001, 3. Auflage, ISBN 3-527-30433-
9.
[135] "Dynamisch Mechanische Analyse (DMA)" (www.mk-atomy.de/protokolle/mc/
dma.pdf am 21.05.2014)
[136] "Untersuchung der Reaktionskinetik von Photopolymeren im Dentalbereich. Kapitel 3
Funktionsprinzip von DSC, DMA und DEA" (www.fb05.h-bonn-rhein-
sieg.de/annamedia/Bilder/projekte/Dentalmassen/Funktionsprinzip_DSC_DMA_DEA.p
df am 21.05.2014)
[137] Moon, S.-I., C.-W. Lee, et al.; "Melt/solid polycondensation of L-lactic acid: an
alternative route to poly(L-lactic acid) with high molecular weight." Polymer, 2001, 42,
5059-5062.
[138] Fukushima, K., Y. Furuhashi, et al.; "Stereoblock Poly(lactid acid): Synthesis via Solid-
State Polycondensation of a Stereocomplexed Mixture of Poly(L-lactid acid) an Poly(D-
lactide acid)." Macromolecular Bioscience, 2005, 5, 21-29.
[139] Moon, S.-I., I. Taniguchi, et al.; "Synthesis and properties of high-molecular-weight
poly(L-lactic acid) by melt/solid polycondensation under different reaction conditions."
High Performance Polymers, 2001, 13, 189-196.
[140] Tsuji, H., Y. Ikada; "Stereocomplex Formation between Enantiomeric Poly(lactic acid)s.
6. Binary blends from Copolymers." Macromolecules, 1992, 25, 5719-5723.
[141] Broniecki, J.; "Bericht: Polymerisation von meso-Dilactid." Fraunhofer Institut für
Angewandte Polymerforschung, 2008.
[142] Bouapao, L., H. Tsuji, et al.; "Crystallization, spherulite growth, and structure of blends
of crystalline and amorphous poly(lactide)s." Polymer, 2009, 50, 4007-4017.
[143] Pan, P., Y. Inoue; "Polymorphism and isomorphism in biodegradable polyesters."
Progress in Polymer Science, 2009, 34, 605-640.
[144] Sarasua, J. R., A. López Arraiza; "Crystallization and thermal behavior of optically pure
polylactides and their blends." Journal of Materials Science, 2005, 40, 1855-1862.
[145] Yasuniwa, M., K. Sakamo, et al.; "Melting behavior of poly(L-lactic acid): X-Ray and DSC
analyses of the melting process." Polymer, 2008, 49, 1943-1951.
Page 118
XXII
[146] He, Y., Y. Xu, et al.; "Unique crystallization behavior of poly(L-lactide)/poly(D-lactide)
stereocomplex depending on initial melt states." Polymer, 2008, 49, 5670-5675.
[147] Tsuji, H., T. Tajima; "Relatively Short Poly(D-lactide) Segments as Intra-Crystallization-
Accelerating Moieties in Stereo Diblock Poly(lactid)s." Macromolecular Materials and
Engineering, 2014, 299, 430-435.
[148] Fukushima, K., Y. Kimura; "Review Sterocomplexed polylactides (Neo-PLA) as high-
performance bio-based polymers: their formation, properties, and application."
Polymer International, 2006, 55, 626-642.
[149] Schmidt, S. C., M. A. Hillmyer; "Polylactide Stereocomplex Crystallites as Nucleating
Agents for Isotactic Polylactide." Macromolecular Journal of Polymer Science: Part B:
Polymer Physics, 2001, 39, 300-313.
[150] Anderson, K. S.; "High Impact Polylactide Composites." Dissertation, 2004, University
of Minnesota.
[151] Tsuji, H., Y. Ikada; "Stereocomplex Formation between Enantiomeric Poly(lactic acid)s.
9. Stereocomplexation from the Melt." Macromolecules, 1993, 26, 6918-6926.
[152] Bouapao, L., H. Tsuji; "Stereocomplex Crystallization and Spherulite Growth of Low
Molecular Weight Poly(L-lactide) and Poly(D-lactide) from the Melt." Macromolecular
Chemistry and Physics, 2009, 210, 993-1002.
[153] Tsuji, H., Y. Ikada; "Stereocomplex formation between enantiomeric Poly(lactic acid)s.
XI. Mechanical properties and morphology of solution-cast films." Polymer, 1999, 40,
6699-6708.
[154] Yasuniwa, M., K. Iura, Y. Dan; "Melting behavior of poly(L-lactic acid): Effect of
crystallization temperature and time." Polymer, 2007, 48, 5398-5407.
[155] Tsuji, H.; "Poly(lactide) Stereocomplexes: Formation, Structure, Properties,
Degradation, and Applications." Macromolecular Bioscience, 2005, 5, 569-597.
[156] Tsuji, H., K. Tashiro, et al.; "Synchronous and separate homo-crystallization of
enantiomeric poly(L-lactic acid)/poly(D-lactic acis) blends." Polymer, 2012, 53, 747-754.
[157] Shao, J., J. Sun, et al.; "The formation and transition behaviors of the mesophase in
poly(D-lactide)/poly(L-lactide) blends with low molecular weights." CrystEngComm,
2013, 15, 6469-6476.
[158] Sarasua, J. R., N. López Rodríguez, et al.; "Stereoselective Crystallization and Specific
Interactions in Polylactides." Macromolecules, 2005, 38, 8362-8371.
Page 119
XXIII
[159] Chen, D., J. Li, J. Ren; "Crystal and Thermal Properties of PLLA/PDLA Blends Synthesized
by Direct Melt Polycondensation." Journal of Polymers and the Environment, 2011, 19,
574-581.
[160] Srithep, Y., P. Nealey, L.-S. Turng; "Effects of Annealing Time and Temperature on the
Crystallinity and heat resistance Behavior of Injection-Molded Poly(lactid acid)."
Polymer Engineering and Science, 2013, 53, 580-588.
[161] Kaltofen, R., R. Opitz, et al.; "Tabellenbuch Chemie" VEB Deutscher Taschenbuch Verlag
für Grundstoffindustrie, 1973, 6. Auflage.
[162] Gayer, Ch.; "1. Interner Bericht: SLM eines PLA Sterokomplexes." Fraunhofer ILT, 2014.
[163] Gayer, Ch.; "2. Interner Bericht: SLM eines PLA Sterokomplexes." Fraunhofer ILT, 2014.
Page 120
XXIV
A. Anhang
DSC-Temperatur-Zeit-Programm
1. Heizzyklus:
Ausgleichen bei 20 °C
Isotherm für 2 min
Aufheizen von 20 °C auf x °C mit einer Rate von 10 K/min
Isotherm für 5 min
Abkühlen von x °C auf 20 °C mit einer Rate von 10 K/min
2. Heizzyklus:
Isotherm für 2 min
Aufheizen von 20 °C auf x °C mit einer Rate von 10 K/min
Isotherm für 5 min
Abkühlen von x °C auf 20 °C mit einer Rate von 10 K/min
Bei den Copoly- und Homopolymeren wurde bis auf eine Endtemperatur von 230 °C geheizt
(x = 230 °C). Bei den Stereokomplexmaterialien bis auf eine Temperatur von 250 °C
(x = 250 °C).
Versuchsvorschrift Umkristallisation (Labor)
150 g Lactid sowie 150 g Toluol (Flaschenware über Molsieb (4 Å) gelagert) in einen mit
einem Rührfisch ausgestatteten 3-Halskolben einwiegen und in einem Ölbad temperieren.
Das Ölbad dazu auf 100 °C einstellen und die Lactid-Toluol-Mischung bei leichtem N2-Fluss
unter Reflux kochen. Solange der Rührfisch noch nicht beweglich ist, wird das Auflösen des
Lactids durch gelegentliches Umrühren mit einem Glasstab unterstützt. Nach dem voll-
Page 121
XXV
ständigen Auflösen und dem damit verbundenen Aufklaren der Lösung wird noch 30 min
unter Reflux weitergekocht. Die Lösung wird anschließend in eine 500 mL Schott-Flasche
überführt und das Lactid unter Rühren über Nacht auskristallisiert. Am Folgetag werden die
Kristalle zunächst mit einem Spatel aufgelockert und anschließend unter N2-Atmosphäre
über eine Nutsche abgezogen. Zur Reinigung wird zweimal mit je 100 mL n-Hexan nach-
gewaschen. Die Kristalle werden abschließend in eine Petrischale gegeben und über Nacht
bei 40 °C im Vakuumtrockenschrank getrocknet.
Die Ausbeuten liegen bei rund 95%.
Versuchsvorschrift Umkristallisation (Technikum)
1 kg Lactid und 1,3 kg Toluol werden in einen 42 L Diessel-Reaktor (Merck) gegeben. Dieser
wird anschließend verschlossen und unter N2-Atmosphäre auf 100 °C erhitzt. Sobald der
Ankerrührer beweglich ist, wird zunächst eine geringe Rührgeschwindigkeit von 5 U/min ein-
gestellt, die dann langsam auf 20 U/min gesteigert wird. Wenn das gesamte Lactid gelöst ist
(ca. 1 h), wird noch für 30 min weiter gerührt und beheizt. Danach wird die Heizung aus-
geschalten und die Rührgeschwindigkeit wieder auf 5 U/min reduziert. Man lässt die Lösung
über Nacht abkühlen und das Lactid auskristallisieren. Am Folgetag wird das Toluol vorsichtig
aus dem Reaktor abgelassen und dieser anschließend unter internem und externem N2-Fluss
geöffnet. Das Lactid wird mit Hilfe eines Becherglases aus dem Reaktor geschöpft und über
einen Feststofftrichter in einen mit N2 gefluteten Druckfilter überführt. Wenn sich das
gesamte Lactid im Druckfilter befindet, wird dieser verschlossen und durch Anlegen eines
starken N2-Flusses unter Druck gesetzt. Wenn kein Toluol mehr aus dem System fließt, wird
der N2-Fluss wieder reduziert und der Druckfilter geöffnet um das Lactid anschließend mit
300 mL Toluol zu waschen. Der Filter wird dann wieder verschlossen und mit N2 unter Druck
gesetzt. Dieser Spülschritt wird noch drei weitere Male wiederholt. Abschließend wird das
umkristallisierte und gewaschene Lactid in große Aluschalen überführt und über Nacht bei
40 °C unter Vakuum im Trockenschrank getrocknet.
Die Ausbeuten liegen hier bei rund 85%.
Versuchsvorschrift Lösetest
1g Probe in ein 200 mL Becherglas geben und mit 100 mL Toluol bedecken. Die Proben-
mischung 21 h bei RT rühren und anschließend über ein Filterpapier bei Normaldruck
abtrennen. Das Filtrat in einem 250 mL Rundkolben auffangen und den Filterkuchen zweimal
mit je 25 mL Toluol nachwaschen. Das Filtrat bis zur Trockne einrotieren und den Rückstand
auswiegen.
Page 122
XXVI
Bild A.1
Thermogramm „Probe Co-08“ (2. Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
Bild A.2
Thermogramm „Blend“ (2. Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
Page 123
XXVII
Bild A.3
Thermogramm „Probe Co-13“ (2. Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
Bild A.4
Thermogramm „Probe Co-Tech“ (2. Heizzyklus dargestellt; vgl. Abschn. 3.8.2)
Page 124
XXVIII
Bild A.5
Schneckenkonfiguration bei der Entmonomerisierung
Bild A.6
Schneckenkonfiguration bei der Stereokomplexbildung
Tabelle A.1
Übersicht der GPC-Ergebnisse der Technikumsmaterialien (HFIP-GPC; vgl. Abschn. 3.8.1)
Probenbezeichnung Mn
[g/mol]
PDLA Tech 58400
Co-Tech 53900
Co-scTech +55 47500
Co-scTech +26 47400
Co-scTech ±0 46500
Co-scTech -26 49000
Co-scTech -55 51800
Page 125
XXIX
Tabelle A.2
Bedingungen am „ HAAKE MiniJet“ (vgl. Abschn. 5.3.1.1)
Probenbe-zeichnung
T eingespritzte
Masse [°C]
T Werkzeug
[°C] Spritzdruck
[bar] Spritzdauer
[sek] Nachdruck
[bar]
Dauer Nachdruck
[sek]
PLLA 170 45 300 7 300 7
sc01 230 180 600 7 600 7
Co-sc01 230 120 180 7 400 7
Co-sc03 215 110 150 7 400 7
Co-sc04 215 125 100 2 600 7
Co-sc05 215 125 100 2 600 7
Co-sc02 230 180 600 7 600 7
Tabelle A.3
Bedingungen am „HAAKE MiniJet“ (vgl. Abschn. 5.3.2.1)
Probenbe-zeichnung
T eingespritzte
Masse [°C]
T Werkzeug
[°C] Spritzdruck
[bar] Spritzdauer
[sek] Nachdruck
[bar]
Dauer Nachdruck
[sek]
Co-scTech +55 210 135 100 2 600 7
Co-scTech +26 210 135 100 2 600 7
Co-scTech ±0 210 135 100 2 600 7
Co-scTech -26 210 135 100 2 600 7
Co-scTech -55 210 115 100 2 600 7
Page 126
XXX
Danksagung
Ich möchte mich bei Herrn Prof. Dr.-Ing. M. H. Wagner ganz herzlich für das entgegen-
gebrachte Interesse sowie die Bereitschaft zur wissenschaftlichen Betreuung meiner Arbeit
bedanken.
Herrn Prof. Dr. H.-P. Fink danke ich für die Übernahme der Gutachterfunktion sowie Frau
Prof. Dr.-Ing. C. Fleck für die Übernahme des Amts der Prüfungsvorsitzenden.
Dr. R. Rihm möchte ich für die unzähligen Diskussionen, Anregungen und Hinweise sowie
seine stete Hilfsbereitschaft bedanken. Danke, dass du immer ein offenes Ohr für mich
hattest.
Herrn Dr. M. Hahn sowie Frau Dipl.-Ing. M. Jobmann möchte ich für die interessante
Themenstellung sowie Ihre konstruktive Kritik bei der Fertigstellung der vorliegenden Arbeit
danken.
Bei meiner Kollegin Dipl.-Ing. M. Walter möchte ich mich für die nette Bürogesellschaft
sowie die unzähligen gemessenen GPC-Proben bedanken. Weiterhin gilt mein Dank
K. Hohmann, K. Callies, V. Mikulla sowie Dipl.-Ing. K. Jesse für die schönen gemeinsamen
Stunden und netten Ausflüge.
Beim gesamten Fachbereich 3 möchte ich mich für die entgegengebrachte Hilfsbereitschaft
bedanken.
Meinen Kollegen aus dem Technikum, insbesondere M. Koch, W. Fehrle† und M. Schulze,
möchte ich für die tatkräftige Unterstützung bei der Synthese und Verarbeitung sowie für
die stetige Hilfsbereitschaft und die aufmunternden Worte danken.
Weiterhin vielen Dank an Herrn Dr. A. Bohn für das Vermessen meiner Proben mittels
Röntgenbeugung sowie an Frau N. Fischer für die HDT-B als auch DMA-Messungen.
Herrn Ch. Gayer vom Fraunhofer Institut für Lasertechnik danke ich für die für die Erstellung
der Prototypen mittels SLM.
Bei Herrn Dr. U. Mühlbauer der Firma Uhde Inventa Fischer möchte ich mich für die
Bereitstellung des D,D-Lactids bedanken.
Abschließend möchte ich meiner Familie sowie meinem Partner P. Lenz für Ihre Unter-
stützung während der Bearbeitung und Fertigstellung meiner Promotion danken.