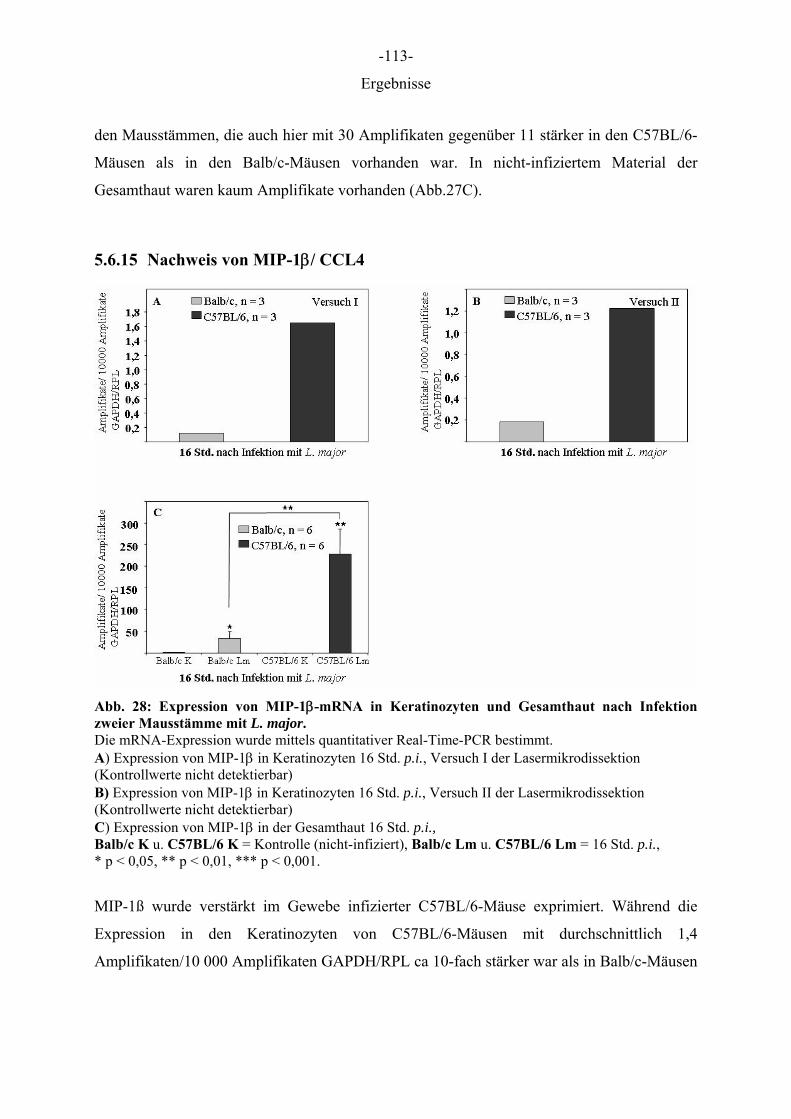
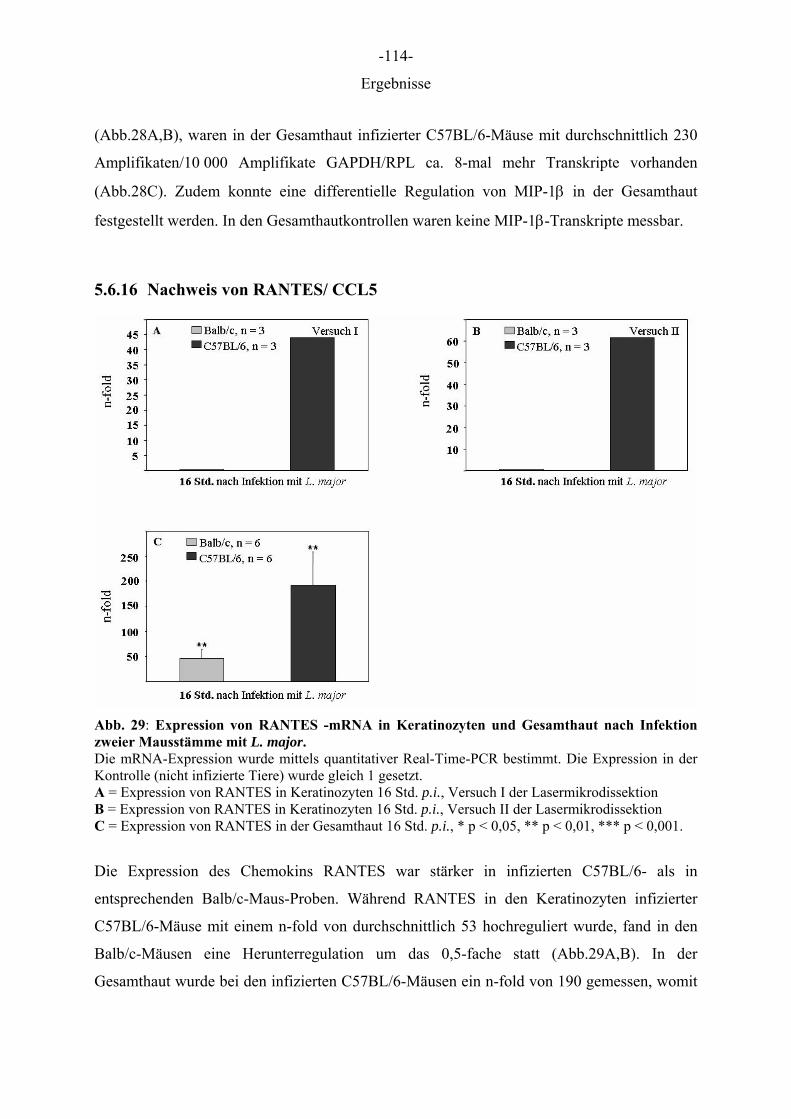
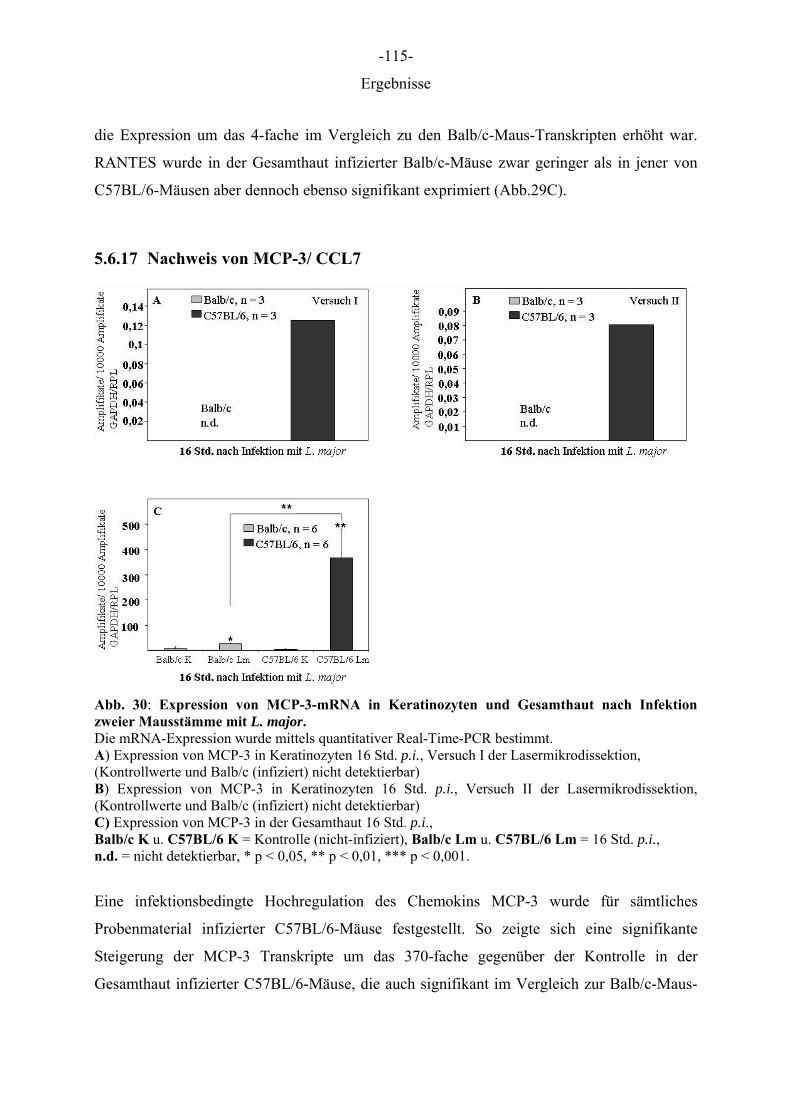
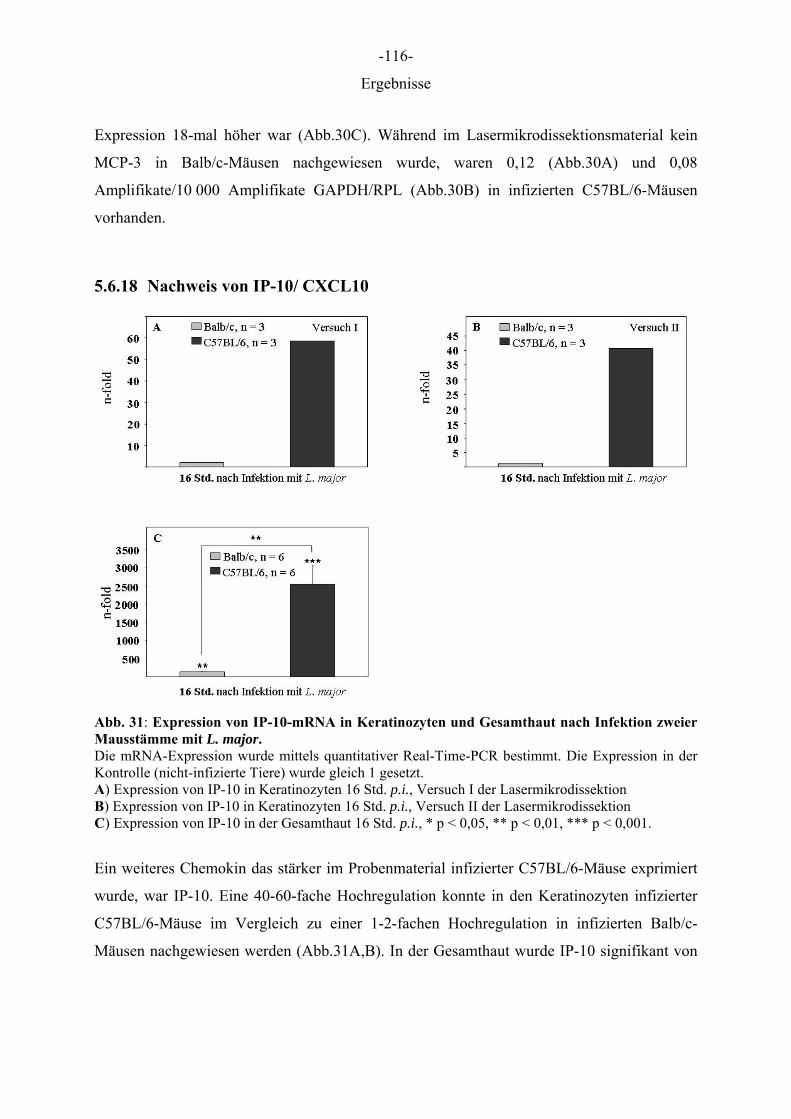
Aus dem Institut für Immunologie der Tierärztlichen Hochschule Hannover und dem Institut für Experimentelle Dermatologie der Westfälischen Wilhelms-Universität Münster Die Rolle des Epithels der Haut in der Frühphase der Leishmania-major-Infektion INAUGURAL–DISSERTATION Zur Erlangung des Grades einer Doktorin der Veterinärmedizin (Dr. med. vet.) durch die Tierärztliche Hochschule Hannover Vorgelegt von Kirsten Roebrock aus Bocholt Hannover 2006
Transcript
Aus dem Institut für Immunologie
der Tierärztlichen Hochschule Hannover
und dem Institut für Experimentelle Dermatologie
der Westfälischen Wilhelms-Universität Münster
Die Rolle des Epithels der Haut in der Frühphase der
Leishmania-major-Infektion
INAUGURAL–DISSERTATION Zur Erlangung des Grades einer Doktorin der Veterinärmedizin
(Dr. med. vet.) durch die Tierärztliche Hochschule Hannover
Vorgelegt von Kirsten Roebrock
aus Bocholt
Hannover 2006
Wissenschaftliche Betreuung: Prof. Dr. Hans-Joachim Schuberth
Prof. Dr. Johannes Roth
1. Gutachter: Prof. Dr. Hans-Joachim Schuberth
2. Gutachterin: Apl.-Prof. Dr. Astrid M. Tenter
Tag der mündlichen Prüfung: 24.11.2006
Meinen Eltern
Verzeichnis der verwendeten Abkürzungen ................................................................ 10
Abb. Abbildung A. bidest. Aqua bidestillata AD atopische Dermatitis APC antigen presenting cell (Antigen-präsentierende Zelle) BSA Bovines Serum Albumin bspw. beispielsweise ca. circa CCR Rezeptoren der C-C Chemokin Unterfamilie CD Cluster of Differentiation (System zur Bezeichnung von
Differenzierungsantigenen) CD25+ Zellen, die das Differenzierungsantigen CD25 an der Oberfläche
exprimieren CD4+ Zellen, die das Differenzierungsantigen CD4 an der Oberfläche
exprimieren CD8+ Zellen, die das Differenzierungsantigen CD8 an der Oberfläche exprimieren cDNA komplementäre DNA (Desoxyribonukleinsäure) CL cutaneous Leishmaniasis (kutane Leishmaniose) CO2 Kohlenstoffdioxid CR Complement Receptor (Komplementrezeptor) CTACK cutaneous T-cell attracting chemokine DC dendritic cells (dendritische Zellen) DEPC Diethylpyrokarbonat DMSO Dimethylsulfoxid EDTA Ethylendiamintetraessigsäure Eotaxin eosinophil chemotactic protein F forward (vorwärts) Fc Fragment cristalline (kristallisierbarer Antikörperteil, carboxy-
terminales Ende des Immunglobulins FCS fetal calf serum (foetales Kälberserum) Fcε−RI hochaffiner IgE-Rezeptor FITC Fluoreszeinisothiocyanat FU Fluorescence Units (Fluoreszenzeinheiten) IDEC inflammatorische dendritische epidermale Zellen I-309 inflammatory cytokine I-309 IFN-γ Interferon-gamma IgE/ G Immunglobulin E/ G iIFL indirekte Immunfluoreszenz IL Interleukin iNOS inducible nitric oxide synthase (induzierbare NO Synthase) IP-9 IFN-γ-inducible protein-9 IP-10 interferon (gamma)-induced protein of 10kDa ISH in situ-Hybridisierung I-TAC interferon (gamma)-induced T-cell alpha chemoattractant IU international units (internationale Einheiten)
Kb Kilobyte KC Cytokine-induced neutrophil chemoattractant L Liter LB Luria Bertani LC Langerhans cells (Langerhans-Zellen) L. major Leishmania major Lm Leishmania major Lsg. Lösung m milli (x 10-3) mA milliampere M Molar (Mol/L) MCL mucocutaneous Leishmaniasis (mukokutane Leishmaniose) MCP-1 bis 5 macrophage chemotactic proteins 1 and 3 MDC macrophage-derived chemokine MG Molekulargewicht MHC Major Histocompatibility Complex
(Haupthistokompatibilitätskomplex) MIG monokine induced by IFN-γ Min. Minute(n) MIP-1α und 1β macrophage inhibitory proteins 1α und 1β mRNA messenger-RNA (Ribonukleinsäure) MW arithmetischer Mittelwert n nano (x 10-9) (n) eine Variable, deren Werte auf natürliche Zahlen beschränkt
sind NaCl Natriumchlorid NC/Nga-Mäuse Mausinzuchtstamm der einen der AD ähnlichen Phänotyp entwickelt n.d. nicht detektierbar n-fold Regulationsfaktor NK-Zellen natürliche Killerzellen NLDC145+ DC-Zell-Marker NO Stickstoffmonoxid Nr. Nummer nt Nukleotid OCT optimal cutting temperature p Wahrscheinlichkeit mit der die Hypothese nicht zutrifft pg picogramm (x 10-12) PBS Phosphate Buffered Saline (Phosphat-gepufferte Salzlösung) PCR polymerase chain reaction (Polymerase-Kettenreaktion) p.i. post infectionem, nach Infektion PMN polymorphonuclear leukocytes (hier Granulozyten inklusive
Basophile und Eosinophile) PV parasitophore Vakuole R reverse (rückwärts) RANTES Bezeichnung eines Chemokins: “regulated upon activation,
Rt Raumtemperatur RT Reverse Transkription RT-PCR Reverse Transkriptase-Polymerase-Kettenreaktion s. siehe S. Seite s.c. subkutan SCID Severe Combined Immunodeficiency Disease Scl1 Susceptibility to cutaneous leishmaniasis 1 SD Standard Deviation (Standardabweichung) SDM Medium Schneider`s Drosophila Medium Sek. Sekunde(n) SEM Standard error of mean (Standardfehler) Std. Stunde(n) Tab. Tabelle TARC Th2-type CC chemokine thymus and activation-regulated chemokine Taq Thermophilus aquaticus TBE-Puffer Tris-Borat-EDTA-Puffer T-bet T-box expressed transcription factor (ein Th1-spezifischer Transkriptionsfaktor, der die Differenzierung vonTh2- in Th1- Lymphozyten initiieren kann). TCR T-cell-receptor (T-Zell-Rezeptor) TGF-ß Transforming growth factor ß Th1 T-Helfer Zelle vom Typ 1 Th2 T-Helfer Zelle vom Typ 2 Thy1.2+ T-Zell-Marker T-memory-Zellen T-Gedächtnis-Zellen TNF-α Tumornekrosefaktor α U Units (Einheiten) u.a. unter anderem Units Einheiten v/v Volumen pro Volumen VL visceral Leishmaniasis (viszerale Leishmaniose) well Vertiefung einer Mikrotiterplatte z.B. zum Beispiel μ mikro (x 10-6)
-13-
Einleitung
1 EINLEITUNG
Entzündliche Hautkrankheiten, die mit einem Ungleichgewicht der Immunantwort,
insbesondere mit einer gestörten Entwicklung von sogenannten Th1- und Th2-Lymphozyten,
einhergehen wie bspw. die Volkskrankheit Psoriasis und die atopische Dermatitis (AD) haben
bei Mensch und Tier im Laufe der Zeit zunehmend an Bedeutung gewonnen. So liegt die
Prävalenz der AD unter deutschen Schulkindern derzeit bei 15-20 %. Zu den betroffenen
Tieren gehören insbesondere Pferde, Katzen und Hunde, bei denen die Häufigkeit bei 3-15 %
liegt, jedoch werden bis zu 30 % in Überweisungspraxen diagnostiziert. Der genaue
Pathomechanismus dieser Erkrankung ist derzeit nicht bekannt, und zudem gibt es nur wenige
Studien, die sich mit der Bedeutung der Haut als lokal immunologisch wirksames Organ
hinsichtlich der Auswirkungen auf die T-Zell-Differenzierung in Th1- und Th2-Zellen
befassen. Zusätzlich ist trotz einiger Fortschritte auf dem Gebiet der Behandlung dieser
Erkrankung eine kausale und effiziente Therapie derzeit nicht verfügbar.
Für die Erforschung der natürlichen Infektresistenz und der zellulären Immunantwort von
Th1- und Th2-Lymphozyten ist die Infektion von Mausinzuchtstämmen mit Leishmania
major ein geeignetes Model. Bei diesem Modell führt die Infektion mit dem Erreger der
kutanen Leishmaniose in resistenten Mausstämmen zu einer Th1-Antwort, die gekennzeichnet
ist durch die Ausbildung einer lokal begrenzten, selbstheilenden kutanen Läsion, die
lebenslange Immunität hinterlässt. Bei anfälligen Mausstämmen kommt es hingegen zur
Entwicklung einer Th2-Antwort, die in einer letalen, generalisierten Infektion des gesamten
retikulo-endothelialen Systems (RES) endet.
Aufgrund der Vielzahl entzündlicher Hauterkrankungen, die mit einer veränderten T-Zell-
Differenzierung einhergehen, wird im Folgenden die AD als Beispiel für die involvierten
Mechanismen verwendet.
-14-
Literaturübersicht
2 LITERATURÜBERSICHT
2.1 Das Immunsystem der Haut
Vor nicht allzu langer Zeit war man noch der Meinung, dass die Schutzmechanismen der
Haut und das Immunsystem unabhängig voneinander arbeiten. Die Haut schien mechanisch
das Eindringen fremder Antigene in den Körper zu blockieren; das Immunsystem bekämpfte
diejenigen Antigene, die dennoch in den Körper hineingelangten. Heute ist bekannt, dass die
Haut ein immunologisch aktives Organ darstellt, welches an der immunologischen
Überwachung und Reaktivität beteiligt ist. Diese Erkenntnis führte dazu, dass die Haut heute
als Teil des Immunsystems verstanden wird; man spricht von dem Hautimmunsystem (BOS
1997).
Das Immunsystem wird in drei separate Bereiche unterteilt, nämlich in das humorale, das
zelluläre und in das unspezifische System. Die vier Typen der Hypersensibilitätsreaktionen,
wie sie von COOMBS und GELL (1963) definiert wurden, beruhen auf dieser Unterteilung
(HALLIWELL u. GORMAN 1989, TIZARD 1996, ROITT et al. 1998) und sind ein gutes
Beispiel für die Bedeutung des Hautimmunsystems im Rahmen von Hautallergien oder
chronischen Ekzemen.
Die Entzündung ist ein komplexer Vorgang, an dem Elemente unspezifischer, zellulärer und
humoraler Immunität, sowie eine Kaskade untereinander reagierender, löslicher Mediatoren-,
der Zytokine und Chemokine beteiligt sind. Antigene (Proteine/Glykoproteine) die in die
Hautschichten gelangen, werden zunächst in den meisten Fällen phagozytiert, metabolisiert
und in kleinere Untereinheiten zerlegt, die an den Haupthistokompatibilitätskomplex-(MHC)
Klasse-II binden. Dieser Peptid-MHC-Komplex wandert zur Oberfläche der Antigen
präsentierenden Zelle (APC), wo er mit T-Zell-Rezeptoren (TCR) für dieses spezifische
Antigen interagiert. Als APC dienen mononukleäre Phagozyten (Monozyten, Makrophagen),
dendritische Zellen und B-Lymphozyten (BOS 1989, NICKOLOFF 1993, TIZARD 1996).
Die dendritische Zellpopulation der Haut setzt sich aus Langerhans-Zellen (LC) und dermalen
Dendrozyten zusammen. LC, die von Knochenmarkszellen abstammen repräsentieren 3-4 %
Prozent der Zellpopulation der Epidermis. Sie exprimieren sowohl MHC-Klasse-II-Moleküle
-15-
Literaturübersicht
als auch die Rezeptoren für bestimmte Komplementfaktoren (C3b) und den Fc-Bereich von
IgG und IgE. Ihre Hauptfunktion besteht in der Verarbeitung und Präsentation von Antigenen.
Die Wechselwirkung von APC und T-Zelle über den trimolekularen TCR-Peptid-MHC-
Komplex löst eine spezifische Immunreaktion gegen das betreffende Antigen aus.
Keratinozyten können ebenfalls MHC-Klasse-II-Moleküle exprimieren, ihre Bedeutung bei
der Antigenpräsentation ist allerdings noch nicht schlüssig bewiesen (REEDY et al. 2002).
2.1.1 Immunmodulatoren: Zyto- und Chemokine
Zytokine sind lösliche Proteine mit niedrigem Molekulargewicht die als chemische
Botenstoffe das angeborene (unspezifische) und erworbene (spezifische) Immunsystems
regulieren. Sie werden von praktisch allen Zellen gebildet, die mit dem Immunsystem in
Beziehung stehen. Bei Aktivierung der Zytokin-produzierenden Zellen synthetisieren und
sezernieren diese ihre Botenstoffe, welche an spezifische Zytokinrezeptoren auf anderen
Zellen des Immunsystems binden und so deren Aktivität beeinflussen. Zytokine sind
pleiotrop, da sie auf eine Vielzahl unterschiedlicher Zellen wirken und redundant, da
unterschiedliche Zytokine die gleiche Funktion ausüben können. Zudem sind sie
multifunktional, da ein Zytokin mehrere unterschiedliche Funktionen regulieren kann. Einige
Zytokine wirken antagonistisch indem ein Zytokin eine bestimmte Abwehrfunktion stimuliert
die ein anderes Zytokin hemmt. Andere Zytokine sind synergistisch in ihrer Wirkung indem
die Kombination verschiedener Zytokine einen größeren Effekt hat als die einzelnen Proteine
(PAUL 1989, LEONARD 1994, JANEWAY et al. 2002).
Zu den Zytokinen, die in den allerersten Phasen einer Infektion in dem betroffenen Gewebe
freigesetzt werden, gehören zudem Vertreter einer Familie von chemotaktisch aktiven
Zytokinen, die man als Chemokine bezeichnet. Chemokine werden entsprechend der
Anordnung von zwei Cystein Gruppen unterteilt in die C-X-C Unterfamile (α Chemokine)
wozu das als erstes charakterisierte Interleukin-8 (IL-8) gehört sowie die C-C Unterfamile (β
Chemokine), wozu bspw. die „monocyte chemoattractant proteins“ (MCP) und das
„macrophage inflammatory protein α“ (MIP-1α) gehören. Die α Chemokine locken
hauptsächlich neutrophile Granulozyten zu Infektionsherden während die β Chemokine
-16-
Literaturübersicht
größtenteils auf Monozyten, T-Lymphozyten, und Natürliche Killerzellen (NK) wirken
(SCHALL u. BACON 1994).
2.1.2 Keratinozyten als immunologisch aktive Zellen
Da die Keratinozyten sowohl eine Vielzahl von Zytokinen und Chemokinen produzieren als
auch Adhäsionsmoleküle exprimieren, spielen sie zweifelsohne eine Schlüsselrolle bei den
entzündlichen Reaktionen (BOS et al. 1989, SUTER et al. 1996). Stimulierte Keratinozyten
sezernieren proinflammatorische Zytokine um epidermale und dermale Zellen zu aktivieren
und bilden Chemokine für die Rekrutierung von Leukozyten ins entzündliche Gewebe.
Desweiteren sind sie durch Regulierung der Proliferation und Differenzierung epidermaler
Zellen und Unterstützung der Funktion und Migration von LC wesentlich an der Homeostase
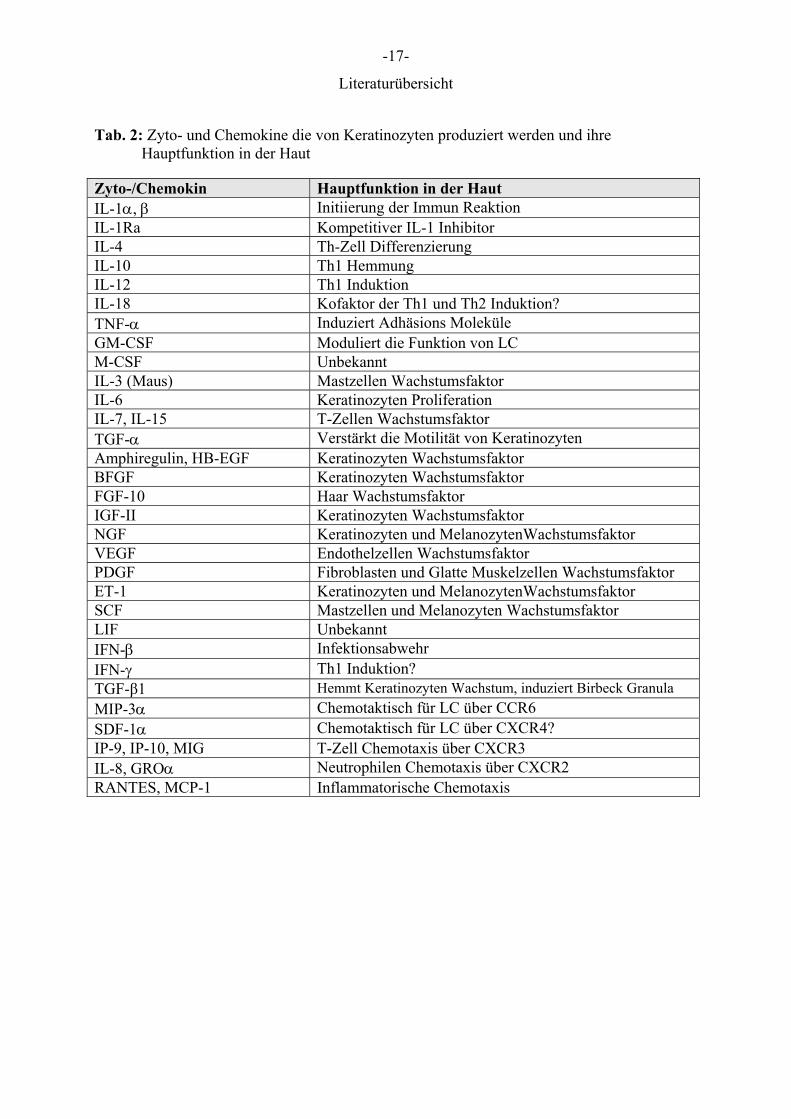
der Haut beteiligt. Die Tabellen 1 und 2 geben eine Übersicht der Rezeptoren und Zyto-
/Chemokine deren Expression oder Sekretion durch Keratinozyten bisher nachgewiesen
werden konnte (WANG et al 1999, UCHI et al. 2000).
Tab. 1: Zyto- und Chemokin-Rezeptoren auf Keratinozyten
Tab. 2: Zyto- und Chemokine die von Keratinozyten produziert werden und ihre Hauptfunktion in der Haut
Zyto-/Chemokin Hauptfunktion in der Haut IL-1α, β Initiierung der Immun Reaktion IL-1Ra Kompetitiver IL-1 Inhibitor IL-4 Th-Zell Differenzierung IL-10 Th1 Hemmung IL-12 Th1 Induktion IL-18 Kofaktor der Th1 und Th2 Induktion? TNF-α Induziert Adhäsions Moleküle GM-CSF Moduliert die Funktion von LC M-CSF Unbekannt IL-3 (Maus) Mastzellen Wachstumsfaktor IL-6 Keratinozyten Proliferation IL-7, IL-15 T-Zellen Wachstumsfaktor TGF-α Verstärkt die Motilität von Keratinozyten Amphiregulin, HB-EGF Keratinozyten Wachstumsfaktor BFGF Keratinozyten Wachstumsfaktor FGF-10 Haar Wachstumsfaktor IGF-II Keratinozyten Wachstumsfaktor NGF Keratinozyten und MelanozytenWachstumsfaktor VEGF Endothelzellen Wachstumsfaktor PDGF Fibroblasten und Glatte Muskelzellen Wachstumsfaktor ET-1 Keratinozyten und MelanozytenWachstumsfaktor SCF Mastzellen und Melanozyten Wachstumsfaktor LIF Unbekannt IFN-β Infektionsabwehr IFN-γ Th1 Induktion? TGF-β1 Hemmt Keratinozyten Wachstum, induziert Birbeck Granula MIP-3α Chemotaktisch für LC über CCR6 SDF-1α Chemotaktisch für LC über CXCR4? IP-9, IP-10, MIG T-Zell Chemotaxis über CXCR3 IL-8, GROα Neutrophilen Chemotaxis über CXCR2 RANTES, MCP-1 Inflammatorische Chemotaxis
-18-
Literaturübersicht
2.2 Atopische Erkrankungen
Der Begriff „Atopie“ (griechisch Atopia = Ungewöhnlichkeit, Seltsamkeit) wurde erstmalig
von COCA und COOKE (1923) als klinisches Bild einer Überempfindlichkeitsreaktion oder
einer allergischen Reaktion vom Typ I beschrieben. Zu den atopischen Erkrankungen gehören
das allergische Asthma, die allergische Rhinitis und Konjunktivitis und das atopische Ekzem.
Heute versteht man unter dem Begriff Atopie, eine genetisch bedingte Disposition zur
spontanen Entwicklung einer Typ-I-Überempfindlichkeitsreaktion gegenüber Inhalations-
oder perkutan aufgenommenen Allergenen, die normalerweise harmlose Substanzen
darstellen (REEDY et al. 2002). Bei der Überempfindlichkeitsreaktion vom Typ I nach
COOMBS und GELL (1963) werden B-Zellen zur Bildung von spezifischem IgE durch
T-Zell-Hilfe angeregt. Dieses antigenspezifische IgE bindet über Fc-Rezeptoren an
Mastzellen, wodurch diese sensibilisiert werden. Beim nächsten Antigenkontakt mit der
sensibilisierten Mastzelle wird das gebundene IgE kreuzvernetzt, und die Zelle degranuliert
unter Freisetzung von Mediatoren, welche die bekannten Symptome wie Asthma bronchiale,
allergische Rhinokonjunktivitis oder atopisches Ekzem hervorrufen. Bei der durch IgE
vermittelten allergischen Reaktion lassen sich eine frühe und eine späte Phase unterscheiden.
Die Sofortreaktion beruht auf der Wirkung von Histamin und anderen, von den Mastzellen
ausgeschütteten, schnell metabolisierten Effektoren. Die Entzündungseffekte von Histamin
erklären sich aus seinen pharmakologischen Eigenschaften, die über H1-Rezeptoren auf
Blutgefäße, glatte Muskulatur und die Oberfläche von Schleimhäuten einwirken. Die
Spätreaktion wird durch eingewanderte inflammatorische Leukozyten hervorgerufen, die
durch Chemokine und andere von den Mastzellen während oder nach der Sofortreaktion
freigesetzte Mediatoren angelockt werden. Immunologisch wird die verstärkte IgE-Bildung
bei der Atopie durch ein Überwiegen der Th2-Immunantwort gesteuert, bei partieller
Unterdrückung der Th1-Immunantwort (ROITT et al. 1991).
Bei Menschen und Haustieren ist die AD eine mit starkem Juckreiz einhergehende
entzündliche Hauterkrankung mit akut-entzündlichem, chronischen oder chronisch-
rezidivierenden Verläufen. Die Erkrankung ist weltweit verbreitet und tritt aufgrund der
genetischen Prädisposition bei bestimmten Rassen oder Familien gehäuft auf. Die am
-19-
Literaturübersicht
häufigsten betroffenen Stellen sind Schnauze (Gesicht), Pfoten (Handrücken), Flexor-und
Extensorflächen (REEDY et al. 2002).
Die Atopie der Hunde weist viele Gemeinsamkeiten mit ihrem Gegenstück beim Menschen
auf:
• Familiäre Disposition,
• Auftreten im Jugendalter,
• Chronischer Pruritus,
• Typische Hautveränderungen an den Beuge- oder Streckseiten der Extremitäten,
• Sofortreaktion im Hauttest (HELTON RHODES et al. 1987, RHODES et al. 1987)
2.2.1 Pathomechanismen der AD
Der der AD zugrundeliegende Pathomechanismus ist noch nicht vollständig geklärt; es liegt
wahrscheinlich eine Kombination von defekter zellulärer Immunantwort, IgE-mediierter
Immunreaktion und äußeren Faktoren die zu einer Verschlimmerung der Symptomatik führen
(z.B. durch Staphylokokkenprodukte, Malassezia Hefen) vor. Sowohl beim Menschen (VAN
DER HEYDEN et al. 1991, ZACHARY et al. 1995) als auch beim Hund (NIMMO WILKIE
et al. 1991, 1992) wurden bei atopischen Patienten Veränderungen der zellvermittelten
Immunität beobachtet. Außerdem weisen sowohl erkrankte Hunde (WILLEMSE et al. 1985,
KLEINBECK et al. 1989) als auch der Großteil betroffener Menschen (PARISH 1981,
UEHARA 1986) erhöhte Serumkonzentrationen von allergenspezifischem IgE und einigen
Untergruppen von IgE auf. Aus pathogenetischer Sicht ist die AD durch eine inadäquate T-
Zell-Antwort definiert. Die T-Zell-Reaktion hat bei Mensch und Hund (SINKE et al. 2002)
eine Prägung vom Th2-Typ, der gekennzeichnet ist durch die Bildung von Interleukin (IL)-4,
IL-5, IL-6 und IL-13. Daraus resultiert eine unangemessene und erhöhte IgE-Produktion. Die
Ekzemreaktion basiert auf erhöhtem spezifischem IgE, das über Fcε-Rezeptoren (Fcε-R)
(HAAS et al. 1992) an Langerhans-Zellen und inflammatorische, dendritische, epidermale
Zellen (IDEC) bindet. Bei Allergenexposition wird das Antigen durch diese Zellen prozessiert
und den T-Zellen präsentiert, dies induziert eine gezielte T-zelluläre Antwort. Während IDEC
bereits früh in der Entwicklung von AD betroffenen Hautbereichen in die Epidermis
einwandern, findet die Expression des Fcε-RI auf ihrer Oberfläche erst später in der
-20-
Literaturübersicht
chronischen Phase statt (KERSCHENLOHR et al 2003). Fcε-RI-aktivierte IDEC instruieren
naive T-Zellen zu IFN-γ produzierenden T-Zellen und der Ausschüttung von IL-12 und IL-18,
wodurch möglicherweise die Umwandlung von einer Th2- zu einer Th1-Antwort bedingt ist.
In der Tat werden in chronisch veränderter Haut höhere Mengen dieser Zytokine gefunden,
weshalb man auch von einem biphasischen Verlauf der AD spricht, gekennzeichnet durch
eine akute Th2-dominierte und chronische Th1-dominierte Phase (HAMID et al. 1994).
Die European Academy of Allergology and Clinical Dermatology (EAACI) hat eine neue
Nomenklatur der AD vorgeschlagen, da es eine allergische IgE-assoziierte Form der AD, das
„allergic Atopic eczema/dermatitis syndrome“ (AAEDS) welches 70–80 % der Patienten
betrifft und eine nicht-allergische Form, das „nonallergic Atopic eczema/dermatitis
syndrome“ (NAAEDS) gibt (JOHANSSON et al 2001), das bei 20–30 % der Patienten
beobachtet wird (NOVAK et al. 2003). Patienten mit NAAEDS zeigen keine respiratorischen
Symptome, wie bronchiales Asthma oder allergische Rhinitis, haben normale IgE Level im
Serum, kein spezifisches IgE und negative Haut-Prick Test Ergebnisse gegenüber Aero- oder
Nahrungsallergenen. Immunologische Unterschiede bestehen zudem zwischen diesen beiden
Formen hinsichtlich des Zytokinmusters im peripheren Blut und der betroffenen Hautbereiche
sowie in der phänotypischen Charakterisierung epidermaler dendritischer Zellen
(WUTHRICH et al. 2003). Beide Formen gehen mit einer Eosinophilie einher. Bei dem
AAEDS produzieren die T-Gedächtnis-Zellen erhöhte Mengen an Th2-Zytokinen, wie IL-4
und IL-13, die die Synthese von IgE induzieren, sowie IL-5 welches maßgeblich an der
Entwicklung von eosinophilen Granulozyten beteiligt ist (SICHERER u. LEUNG 2004).
Diese T-Zellen produzieren außerdem abnorm geringe Mengen von IFN-γ, welches als
Th1-Zytokin die Th2-Zell Funktion inhibiert. Das NAAEDS ist assoziiert mit einer
vergleichsweise geringeren Produktion von IL-4 und IL-13. In beiden Formen wird eine
IL-10 vermittelte Th2-Zell-Differenzierung vermutet (LAOUINI et al. 2003, HOWELL et al.
2005).
Beim Menschen mit AD enthält die Haut ein Infiltrat, das sich vorwiegend aus
CD4+-T-Zellen und einer kleinen Anzahl von CD8+-T-Zellen zusammensetzt, die vermutlich
über eine zytotoxische und suppressive Wirkung verfügen (LEUNG et al. 1981, LEVER et al.
1987). SINKE et al. (1997) berichteten, dass besonders in veränderter atopischer Epidermis
-21-
Literaturübersicht
des Hundes ein Anstieg von CD4+-T-Zellen zu beobachten war. In unveränderter atopischer
Haut dagegen infiltrieren sowohl CD4+- als auch CD8+-T-Zellen die Epidermis ohne
erkennbare Bevorzugung der CD4+-T-Zellen. In der Dermis atopischer Hunde war lediglich
die Anzahl der CD8+-T-Zellen erhöht. Daraus wurde geschlossen, dass CD8+-T-Zellen
wahrscheinlich wichtige Regulatoren bei der AD von Hunden sind (SINKE et al. 1997,
OLIVRY et al. 1997). Untersuchungen zeigten dass sich das Hautinfiltrat atopischer Hunde
hauptsächlich aus Mastzellen, dendritischen APC (die wahrscheinlich von LC abstammen)
und T-memory-Zellen zusammensetzt. Eosinophile und degranulierte Eosinophile wurden nur
in der Epidermis veränderter atopischer Haut nachgewiesen. Ebenso wurden T-Lymphozyten,
die den γδ-T-Zellrezeptor exprimieren in der Epidermis oder Dermis aller atopischen Hunde,
jedoch nur selten bei gesunden Tieren beobachtet (OLIVRY et al. 1997). Diese
Beobachtungen stehen im Gegensatz zu Ergebnissen beim Menschen, wo eine reduzierte
Anzahl dieser T-Zellen im peripheren Blut nachgewiesen wurde. Da die γδ-T-Zellen IFN-γ
sezernieren, könnte ihre Reduzierung die IgE–Produktion potenzieren.
Auch Chemokine prägen das Infiltrat akuter und chronischer Läsionen (BLAUVELT et al.
2003, ONO et al. 2003, LEUNG et al. 2004). So scheinen das „Th2-type CC chemokine
thymus and activation-regulated chemokine“ (TARC, CCL17), das „macrophage-derived
chemokine“ (MDC, CCL22) und das „inflammatory cytokine I-309“ (I-309, CCL1) an einer
Amplifikation der ursprünglich allergischen Entzündungsreaktion teilzuhaben. Das Chemokin
mit der Bezeichnung „regulated upon activation, normally T-expressed, and presumably
secreted“ (RANTES, CCL5), sowie das „monocyte chemoattractant protein 4“ (MCP-4,
CCL13) und das „eosinophil chemotactic protein“ (Eotaxin, CCL11) sollen darüber hinaus an
einer Rekrutierung von Eosinophilen und Th2-Zellen in die Region der Läsionen
verantwortlich sein. Auch das „cutaneous T-cell attracting chemokine“ (CTACK, CCL27),
ein hautassoziiertes Chemokin, soll am sog. „T-cell-homing“ beteiligt seint. Weiterhin ist
CTACK, bei AD-Patienten deutlich hochreguliert. Ebenfalls hochreguliert sind MDC und
TARC, die selektiv CCR4-exprimierende Th2-Zellen anlocken, wobei der Schweregrad der
AD mit den TARC-Spiegeln positiv korreliert (HIJNEN et al. 2004). Th1-Zell-Migration auf
der anderen Seite scheint durch erhöhte Level des „interferon (gamma)-induced protein of
10kDa“ (IP-10, CXCL10), sowie des „monokine induced by IFN-γ“ (MIG, CXCL9) und des
-22-
Literaturübersicht
„interferon (gamma) induced T-cell alpha chemoattractant“ (I-TAC, CXCL11) in
Keratinozyten ausgelöst zu werden (KLUNKER et al. 2003).
2.3 Leishmaniose
Die Leishmaniose ist eine Infektion mit parasitisch lebenden Protozoen der Klasse
Kinetoplastea, Ordnung Trypanosomatida, von denen bisher 30 Arten der Gattung
Leishmania bekannt sind (ASHFORD 2000). Die Leishmanien wurden von CUNNINGHAM
(1885) als erster entdeckt und nach dem britischen Tropenarzt Sir William Boog Leishman
benannt (HANDMAN 1999). Weltweit sind 12 Millionen Menschen in 88 Ländern von der
Infektionskrankheit betroffen, die sich laut WHO mit einer jährlichen Inzidenz von rund 2
Millionen Fällen abhängig von der Leishmanienspezies in einer kutanen- (CL), mukokutanen-
(MCL), viszeralen- (VL) oder diffusen-kutanen- (DCL) Leishmaniose äußert.
2.3.1 Entwicklungszyklus der Leishmanien
Im natürlichen System dienen Nager und andere Kleinsäugetiere als Reservoire für den
Erreger der CL, während der Erreger der VL vornehmlich von Hunden, aber auch Füchsen
und Schakalen beherbergt wird. Durch den Biß eines Insektenvektors, der weiblichen
Schmetterlingsmücke (Phlebotomus oder Lutzomyia) werden die Leishmanien wiederum auf
Wirbeltiere oder den Menschen übertragen. Die mit der Blutmahlzeit aufgenommenen
Leishmanien befinden sich in einem amastigoten, unbeweglichen Stadium und transformieren
im Mitteldarm des Insekten-Vektors innerhalb von 4-25 Tagen zu promastigoten,
teilungsfähigen Leishmanien, die durch Ausbildung eines Flagellums beweglich sind
(SACKS 1989). Über den Speichel und durch Regurgitation des Mageninhaltes der
Schmetterlingsmücke gelangen die nun metazyklisch promastigoten Leishmanien während
erneuter Blutmahlzeit in die Stichwunde des Wirbeltieres. Leishmanien sind in ihrer
amastigoten Form obligat intrazellulär lebende Parasiten, die vermittelt über den
Komplementrezeptor 3 (CR3) auf der Oberfläche von Phagozyten in membranständige
Phagosomen aufgenommen werden (MOSSER u. EDELSON 1985, SCHONLAU et al.
2000). Diese fusionieren mit sekundären Lysosomen zu Phagolysosomen, der so genannten
parasitophoren Vakuole (PV) (CHANG 1983). Innerhalb von 2-5 Tagen erfolgt in den
-23-
Literaturübersicht
Phagolysosomen eine Ausdifferenzierung zur replizierenden amastigoten Form. Die
Replikation in den Phagozyten erfolgt solange, bis die Zelle ruptiert und die frei werdenden
Leishmanien von neuen Makrophagen phagozytiert werden. Kann dieser Kreislauf nicht
durch körpereigene Abwehrmechanismen gestoppt werden, wie bspw. nach Infektion mit
Leishmania donovani, Leishmania infantum oder Leishmania chagasi, kommt es zur
Viszeralisierung; einer Ausbreitung des Parasiten in die lymphatischen Organe, die
unbehandelt letal endet. Eine Infektion mit Leishmania major führt zur Entwicklung lokal
begrenzter Haut-Läsionen die spontan abheilen und eine lebenslange Immunität hinterlassen.
Abb. 1: Schematische Darstellung des Entwicklungszyklus der Leishmanien (Modifiziert nach einer Abbildung der WHO, Life-cycle of Leishmania) [http://www.who.int/tdr/diseases/leish/lifecycle.htm], 2006
2.3.2 Experimentelle Leishmaniose
Die Infektion von Mausinzuchtstämmen mit dem Protozoon L. major, die experimentelle
Leishmaniose, ist ein ausgezeichnetes Modell für die Erforschung der natürlichen
Infektresistenz und der Entwicklung einer T-Helfer (Th)1/Th2-Zell-vermittelten Immun-
-24-
Literaturübersicht
antwort (ETGES u. MÜLLER 1998, BEEBE et al. 1999, LAUNOIS et al. 1999, SACKS u.
NOBEN-TRAUTH 2002).
Bei dem konventionellen L.-major-Maus-Modell wird eine hohe Dosis (2 x 105 – 2 x 107) in
vitro kultivierter promastigoter Leishmanien der metazyklischen Entwicklungsphase subkutan
in den Rücken oder die Hinterfußsohle injiziert. Eine Anzahl weiterer Varianten dieses
Modells ist beschrieben worden, wodurch Unterschiede im Verlauf der Infektion beobachtet
wurden. Dazu gehörten die Verwendung von Leishmanien unterschiedlicher
Entwicklungszustände (SACKS et al. 1985), alternative Inokulationsrouten wie bspw.
intravenös, intradermal und intranasal (NABORS et al. 1995, BELKAID et al. 2000,
CONSTANT et al. 2000), sowie eine natürliche Infektion durch den Schmetterlingsmücken-
Vektor (KAMHAWI et al. 2000). Unterschiede wurden auch durch die Verwendung
unterschiedlicher Leishmanienstämme beobachtet (NOBEN-TRAUTH et al. 1999). Zu den
meist verwendeten Stämmen gehören Friedlin (Jordan Valley, WHOM/IL/80/FN), IR173
(Iran, WHOM/IR/-173), LV39/Neal (Southern Russia, MRHO/SU/59/P), NIH/Seidman (West
Africa, MHOM/SN/74/S), und CC-1 (Iran, MHOM/IR/83/LT252). (SACKS u. NOBEN-
TRAUTH 2002). Die genannten Orte (bspw. Jordan Valley) beziehen sich hierbei auf den Ort
an dem der jeweilige Stamm isoliert wurde und die nachgestellte Bezeichnung (bspw.
WHOM/IL/80/FN) bezieht sich auf einen von der WHO vergebenen Code. Zusätzlich
bestimmen die Anzahl injizierter Parasiten (DOHERTY et al. 1996), sowie das Alter der
verwendeten Versuchstiere (EHRCHEN et al 2004) das Geschehen.
Abb. 2: Zeitliche Entwicklung des L.-major-Maus-Modells (modifiziert nach SACKS u. NOBEN-TRAUTH 2002)
-25-
Literaturübersicht
2.3.3 Resistenz und Suszeptibilität gegenüber einer Infektion mit L. major
Alle bisher untersuchten Mausstämme konnten mit Leishmanien infiziert werden, jedoch
spricht man von resistenten Mäusen bei denen die Infektion in Form einer auf die Haut und
Lymphknoten beschränkten, spontan ausheilenden Erkrankung abläuft (z.B. C56BL/6- oder
C3H-Mäuse). Als suszeptibel werden jene Stämme bezeichnet, bei denen die Infektion zu
einer nicht-abheilenden und abhängig von der Leishmanien Spezies und Dosis tödlichen
Ausbreitung des Parasiten führt (z.B. Balb/c-, C57BL/10ScCr-, DBA/2- oder SWR- Mäuse)
(BEHIN et al.1979, HANDMAN et al. 1979, NABORS u. FARRELL 1994).
Der Hintergrund der Resistenz und Suszeptibilität wird seit langem diskutiert. Die Resistenz
gegenüber L. major ist gekoppelt an die Differenzierung von CD4+-T-Lymphozyten zu
Th1-Zellen (BOGDAN u. ROLLINGHOFF 1998, JANKOVIC et al. 2001, SACKS u.
NOBEN-TRAUTH 2002, GUMY et al. 2004) und bei initial niedriger Parasitenzahl auch an
zytotoxische T-Zellen (CTL oder auch CD8+-Zellen genannt) (BELKAID et al. 2002). Diese
aktivieren vermittelt durch IL-12 die Makrophagen zur Freisetzung von Interferon-γ (IFN-γ),
zur Produktion der „inducible nitric oxide synthase“ (iNOS) und dem leishmaniziden
Stichstoffmonoxid (NO). Die Produktion von NO und reaktiven Sauerstoffverbindungen führt
letztendlich zur Abtötung der intrazellulären Parasiten (LIEW u. O´DONELL 1993, WEI et
al. 1995). In suszeptiblen Mäusen kommt es hingegegen zu einer Vermehrung L.-major-
spezifischer Th2-Zellen die unter anderem IL-4, IL-10 und IL-13 sezernieren (SACKS u.
NOBEN-TRAUTH 2002) und dadurch eine Th1-Antwort hemmen sowie die Makrophagen
deaktivieren.
Es bestehen verschiedene, genetische Unterschiede zwischen den Mausstämmen, die für die
unterschiedliche Resistenz verantwortlich gemacht werden. Die ursächlichen Resistenz-
faktoren sind aber noch nicht identifiziert worden. Zum einen wird das Gen für den
Transkriptionsfaktor des Interferon-regulatory factor 1 als Resistenzfaktor beschrieben
(LOHOFF et al. 1997) und zum anderen die Loci mit der Bezeichnung „susceptibility to
cutaneous leishmaniasis 1“ (Scl1 oder humanes 17q) auf Chromosom 11 und Scl2 auf
Chromosom 4 (GORHAM et al. 1996) sowie Gene auf sechs weiteren Loci (DEMANT et al.
1996, BEEBE et al. 1997). Der Scl1 Locus beinhaltet Gene wie Nos2 das für iNOS codiert
und eine Reihe von Zytokinen die von aktivierten Makrophagen gebildet werden, wie z.B.
-26-
Literaturübersicht
MCP-1 und MCP-2, MIP-1α und 1β sowie RANTES (ETGES u. MÜLLER 1998).
ROBERTS et al. (1997) postulierten, dass die Resistenz durch das Heterochromatin
(H-Region) auf Chromosom 17 und 9 kontrolliert wird (ROBERTS et al. 1997). Neueste
Untersuchungen erweiterten die Anzahl beteiligter Gen-Loci auf 17, von denen jeder einzelne
unterschiedliche immunologische und pathologische Faktoren kontrolliert (HAVELKOVA et
al. 2006).
Eine lebenslange Resistenz erfordert nicht nur eine Th1-Antwort, sondern eine
kontinuierliche IL-12-Produktion (SCOTT 2003). Gewiss ist auch, dass eine geringe Anzahl
an Leishmanien unbegrenzt in den DC der den Infektionsort drainierenden Lymphknoten
verbleiben können (MOLL et al. 1995). Diese DC sind vermutlich für die Antigenpräsentation
an T-memory-Zellen zuständig, wodurch die Th1-Antwort über IL-12-Produktion
aufrechterhalten wird. Die Persistenz vitaler Organismen in Wirten, die eine schützende
Immunantwort entwickelt haben, ist IL-10-abhängig (BELKAID et al. 2001). IL-10-defiziente
Mäuse werden zwar Erreger-frei, können aber keine lebenslange Immunität aufrechterhalten.
Ebenso sind CD4+CD25+-T-Zellen für die Kontrolle der schützenden Th1-Antwort sowie das
Überleben einzelner Parasiten und Aufrechterhaltung der T-memory-Antwort in resistenten
Mäusen notwendig (BELKAID et al. 2002). Trotz intensiver Forschung ist die Ursache für
die inadequate Th2-Zell-Differenzierung in den Balb/c-Mäusen noch nicht geklärt. Die
entscheidenden Mechanismen wirken sehr schnell nach Beginn der Infektion, eine
Beeinflussung des Infektionsverlaufs mit den meisten immunmodulierenden Substanzen ist
nur innerhalb der ersten Woche möglich, wobei vermutlich die ersten 2 Tage nach der
Infektion am bedeutsamsten sind (CHATELAIN et al. 1992, HEINZEL et al. 1993, LEIBY et
al. 1993, SYPEK et al. 1993, LAUNOIS et al. 1997, BIEDERMANN et al. 2001). In den
ersten 2 Tagen der Infektion findet eine Infiltration mit Zellen des angeborenen
Immunsystems, vor allem Makrophagen und Granulozyten statt, die durch die Synthese
immunmodulierender Stoffe einen entscheidenden Beitrag zum Infektionsverlauf leisten.
2.3.4 Erste Mechanismen am Infektionsort
Die obligat intrazellulären Protozoon werden neben residenten Makrophagen in ihrem
promastigoten Zustand als erstes von polymorphkernigen neutrophilen Granulozyten (PMN)
-27-
Literaturübersicht
phagozytiert, da diese die ersten Phagozyten darstellen, die den Infektionsort infiltrieren
(LAUFS et al. 2002). Sie werden durch chemotaktische Moleküle, die von den Leishmanien
produziert werden, innerhalb von 24 Std. angelockt (SORENSEN et al 1989, VAN
ZANDBERGEN 2002). Trotz Phagozytose überleben die Leishmanien in den PMN, deren
normalerweise nach 6-12 Std. eintretende Apoptose erst nach 42 Std. verzögert stattfindet.
Zudem bewirkt die Aufnahme der Parasiten eine Ausschüttung chemotaktischer Faktoren wie
bspw. MIP-1β. In resistenten Mäusen wandern im Vergleich mit suszeptiblen Mäusen nach
ca. 2 Tagen eine größere Anzahl an Makrophagen ein (BEIL et al. 1992, SUNDERKÖTTER
et al.1993, TACCHINI-COTTIER et al. 2000) die den transforming growth factor beta
(TGF-β) produzieren und die apoptotischen PMN phagozytieren, die nur promastigote nicht
replizierende Leishmanien enthalten. Deshalb wurde vermutet, dass Leishmanien PMN als
Trojanisches Pferd für das Eindringen in den Makrophagen verwenden (LASKAY et al. 2003,
VAN ZANDBERGEN et al. 2004). Die funktionelle Relevanz dieser Beobachtung wurde
dadurch demonstriert, dass eine Behandlung von Balb/c-Mäusen zum Zeitpunkt der Infektion
mit einem zytotoxischen Antikörper gegen neutrophile Granulozyten zu einer Hemmung der
Th2-Zell-Differenzierung und zu einer partiellen Resistenz gegenüber L. major führte
(TACCHINI-COTTIER et al. 2000). Die Differenzierung zu replizierenden amastigoten
Leishmanien findet unter Ausnutzung des sauren Milieus innerhalb der Phagolysosomen der
Makrophagen statt (ZILBERSTEIN u. DWYER 1985, GLASER u. MUKKADA 1992).
Chemokine sind von entscheidender Bedeutung für die Rekrutierung von Leukozyten in
entzündliches Gewebe. Für die Leishmaniose konnte bereits gezeigt werden, dass die frühe
Einwanderung von Granulozyten und NK-Zellen mit der Expression der Chemokine
„cytokine-induced neutrophil chemoattractant“ (KC) und IP-10 in der infizierten Haut
korreliert (MÜLLER et al. 2001). Die differentielle Expression von Chemokinen könnte für
den bei den Mausstämmen beobachteten unterschiedlichen Influx oder die unterschiedliche
Aktivierung von frühen Effektorzellen (z.B. Granulozyten, NK-Zellen, monozytären Zellen)
in das Gewebe verantwortlich sein. Unterschiedliche Zytokin- und Chemokinfreisetzung der
infiltrierenden Makrophagenpopulation könnten letztendlich zu einem lokal distinktivem
Zytokinmilieu mit Folgen für die T-Zell-Differenzierung führen. Tatsächlich konnte gezeigt
werden, dass es in der Haut von C57BL/6- verglichen mit Balb/c-Mäusen 24 Std. nach der
Infektion zu einer stärkeren Expression des NK-Zell-aktivierenden IP-10 kommt. Eine
-28-
Literaturübersicht
Behandlung von Balb/c-Mäusen mit IP-10 führte zu einer Zunahme der Aktivität der
NK-Zellen (VESTER et al. 1999, MÜLLER et al. 2001). Die ersten Tage nach der Infektion
sind besonders kritisch, da naive T-Zellen in dieser Zeit zu Effektorzellen differenzieren, und
somit der Typ der T-Helfer-Zell-Entwicklung festgelegt wird (SEDER et al. 1994, ABBAS et
al. 1996). Ausdifferenzierte T-Zellen sind erst ab dem vierten/fünften Tag nachzuweisen.
2.3.5 Funktion dendritischer Zellen und der T-Zell-Aktivierung
Intensive Studien des Verhaltens von DC in resistenten und suszeptiblen Mäusen konnten
keine spezifischen Unterschiede in ihrer Rekrutierung, Antigenprozessierung
und-präsentation sowie Migration nach Infektion mit L. major feststellen (VON STEBUT et
al. 2000, FILIPPI et al. 2003). In der Antigenpräsentation und der damit verbundenen
Migration sind die DC und LC den Makrophagen überlegen (DEMOTZ et al. 1990,
HARDING et al. 1990), während der Makrophage die Zelle darstellt, in der die Leishmanien
zu proliferieren vermögen und längere Zeit beherbergt werden (BOGDAN u.
ROLLINGHOFF 1998, CUNNINGHAM 2002).
Nur die amastigote Leishmanienform wird auch von DC, insbesondere LC (BLANK et al.
1993) aufgenommen. Aus diesem Grund geschieht die Infektion der DC wahrscheinlich erst
später im Verlauf der Infektion, wenn amastigote Leishmanien aus den Makrophagen ins
Gewebe ausströmen. Dieser Vorgang könnte eine Erklärung für die verzögerte Zeitdifferenz
zwischen Inokulation des Parasiten und zellulärer Immunantwort sein. Die Infektion von DC
führt zu deren Aktivierung und Reifung die verbunden ist mit einer Herunterregulation von E-
Cadherin, ein Molekül das die Adherenz von LC an Keratinozyten in der Epidermis
ermöglicht (TANG et al. 1993, VON STEBUT et al. 1998). Deshalb können aktivierte LC die
Epidermis verlassen und in die Lymphknoten auswandern.
Die infektionsbedingte Aktivierung der DC bewirkt eine Hochregulation von MHC-Klasse-I-
und -II-Molekülen (FLOHE et al. 1998, JAKOB et al. 1999, VON STEBUT et al. 2000) die
konstitutiv auf ihrer Oberfläche exprimiert sind. Auf diesen MHC-Molekülen präsentieren sie
im Lymphknoten den T-Zellen Leishmanienantigene, wodurch ruhende T-Zellen aktiviert
werden. Die T-Zell-Aktivierung verläuft über mindestens zwei Signalwege. Das erste Signal
-29-
Literaturübersicht
ist antigenspezifisch und wird über die Interaktion des MHC/Antigen-Komplexes mit dem
TCR übertragen. Das zweite Signal wird auch kostimulatorisches Signal genannt und ist
entscheidend für eine vollständige zelluläre Aktivierung. Eines der bekanntesten
kostimulatorischen Moleküle ist das Adhäsionsmolekül B7 (CD80/CD86), das mit dem
CD28-Rezeptor auf der T-Zell-Oberfläche interagiert (BRETSCHER u. COHN 1970,
LENSCHOW et al. 1996, CHAMBERS u. ALLISON 1997). Wird das Antigen auf MHC-
Klasse-I-Molekülen präsentiert, wird es von naiven CD8-T-Zellen erkannt, die sich dann in
zytotoxische Effektorzellen umwandeln. Diese sorgen für die Abtötung aller Zellen, die
solche Antigene über MHC-I präsentieren, sowie für die Sezernierung von Zytokinen, die
Makrophagen anlocken und aktivieren. Die Antigenpräsentation in Verbindung mit
MHC-Klasse-II-Molekülen bewirkt eine Erkennung von CD4-T-Zellen die dann zu
inflammatorischen und T-Helfer-Zellen werden. Es wird davon ausgegangen, dass das Maß
an Expression kostimulatorischer Moleküle, sowie die Stärke und Affinität der Interaktion
zwischen diesen Molekülen und ihren Korezeptoren auf den T-Zellen, die Antigen-spezifische
primäre Aktivierung der T-Zellen beeinflussen (THOMPSON 1995, CONSTANT u.
BOTTOMLY 1997). Die gewichtige Rolle kostimulatorischer Moleküle wie bspw.
CD80/CD86 und CD40 sind für den Verlauf der Infektion mit L. major erwiesen (CORRY et
al. 1994, CAMPBELL et al. 1996, KAMANAKA et al. 1996, ELLOSO u. SCOTT 1999).
Auch Zellen am Infektionsort beteiligen sich an der Signalübertragung durch
Herrunterregulation von CD86 auf Thy1.2+-epidermalen Keratinozyten resistenter C3H- aber
nicht auf den entsprechenden Zellen von Balb/c-Mäusen. Auf NLDC145+-Langerhans-Zellen
von Balb/c-Mäusen konnte zudem eine Herrunterregulation von CD80 und CD86
dokumentiert werden. Diese Ergebnisse legten die Vermutung nahe, dass Keratinozyten als
sogenannte „bystander“ in der Antigenpräsentation mitwirken (MBOW et al. 2001).
2.3.6 Die Rolle von IL-4 in der T-Zell-Differenzierung
Die sehr frühe Produktion von IL-4 scheint von entscheidender Bedeutung für die Th2-Zell-
Differenzierung zu sein. Es konnte gezeigt werden, dass es bereits einen Tag nach der
Infektion zur Produktion von IL-4 im lokalen Lymphknoten kommt (LAUNOIS et al. 1995,
LAUNOIS et al. 1999). Dabei scheint u. a. eine Population von CD4+-T-Zellen, die über
-30-
Literaturübersicht
einen Vβ4α8 TCR, der das L.-major-Antigen „Leishmania Homolog für Rezeptoren der
aktivierten C Kinase“ (LACK) erkennt, die Quelle des IL-4 darzustellen (LAUNOIS et al.
1997, LAUNOIS et al. 1999). Transgene Balb/c-Mäuse, denen die Vβ4 Kette des TCR fehlt,
zeigen eine Th1-dominierte Immunantwort und können die Infektion kontrollieren
(HIMMELRICH et al. 2000). Deshalb wird vermutet, dass LACK-spezifische Vβ4α8CD4+-
T-Zellen die IL-4 produzieren eine einzigartige Zelllinie in Balb/c-Mäusen repräsentieren
(MALHERBE et al. 2000). Überraschenderweise zeigte sich jedoch eine ähnliche Nutzung
des Vβ4α8-TCR in resistenten wie suszeptiblen Mausstämmen (REINER et al. 1993) und
LACK-spezifische Zellen produzierten zudem explosionsartig IL-4 in resistenten B10.D2
Mäusen (JULIA u. GLAICHENHAUS 1999) ohne eine Veränderung des Phänotyps zu
provozieren.
Studien mit IL-4-Reporter-Mäusen (Mäuse denen zur Identifizierung von IL-4 das green
fluorescence protein in den IL-4 Locus eingebaut wurde) zeigten eine ähnliche Frequenz und
Kinetik IL-4-produzierender Zellen in resistenten wie suszeptiblen Tieren (STETSON et al.
2002). Dennoch resultierte die Neutralisation von IL-4 in einer Resistenz suszeptibler Mäuse
(SADICK et al. 1990, HEINZEL et al. 1991, CHATELAIN et al. 1992), während umgekehrt
ein Überschuss an IL-4 in IL-4-transgenen C57BL/6-Mäusen zu einem suszeptiblen Phänotyp
führte (LEAL et al. 1993). Diese Ergebnisse führten zu der vorherrschenden Auffassung, dass
IL-4 und IL-4-produzierende-Th2-Zellen entscheidende Antagonisten der Th1-induzierten
Immunantwort sind (RACKE 1994, REINER et al. 1995, ABBAS et al. 1996, MOSMANN u.
SAD 1996, ROCKEN et al. 1996, KING et al. 2001). Paradoxerweise wurde in Balb/c-
Mäusen mit einer Ausschaltung des IL-4-Gens einerseits Suszeptibilität und andererseits
relative Resistenz beobachtet. So blieben Balb/c-Mäuse mit einem IL-4-Defekt suszeptibel
gegenüber einer Infektion. Sie zeigten keine Anzeichen einer Abheilung der Hautläsionen
oder Eleminierung des Parasiten und zeigten auch keinen Th1-Phänotyp (NOBEN-TRAUTH
et al. 1996). Andererseits zeigten in einer anderen Untersuchung heterozygote IL-4-knockout-
Balb/c-Mäuse im Vergleich zum Wildtyp geringere Läsionen und keine Unterschiede in der
Expression inflammatorischer Zytokine wie bspw. IL-1β, IL-1α, TNF-α, IL-12 (KOPF et al.
1996). Eine weitere Studie mit homozygoten IL-4 und IL-4-Rezeptor-knockout-Balb/c-
Mäusen demonstrierte eine reduzierte Fußschwellung und Parasitenzahl sowie eine
-31-
Literaturübersicht
Th1-assoziierte Antikörperproduktion während der ersten drei Monate nach Infektion. Danach
verschlimmerte sich das Krankheitsbild der Rezeptor-knockout-Mäuse und endete letal nach 6
Monaten, während die IL-4-knockout-Mäuse eine vergleichsweise moderate Infektions-
symptomatik zeigten, jedoch ohne den Erreger eliminieren zu können (MOHRS et al. 1999).
Ähnliche Beobachtungen konnten durch die Gruppe um BIEDERMANN (2001) gemacht
werden. Die Behandlung von Balb/c-Mäusen mit IL-4 in den ersten 8 Std. nach Infektion
führte zu einer Th1-Zell-Differenzierung und Resistenz. Dieser Effekt ging mit einer höheren
IL-12-Produktion dendritischer Zellen einher. Eine Applikation von IL-4 zu späteren
Zeitpunkten resultierte in einer Th2-Zell-Differenzierung und Suszeptibilität. Eine IL-4-
vermittelte Induktion von IL-12 durch DC wurde in weiteren Experimenten bestätigt
(HOCHREIN et al. 2000, KALINSKI et al.2000, EBNER et al. 2001, STAGER et al. 2003a
und b). Des Weiteren zeigte sich eine nur transiente Th2-Antwort in C3H-Mäusen denen IL-4
appliziert wurde. Diese Mäuse waren jedoch nicht dazu fähig, den normalerweise resistenten
Phänotyp umzukehren (CHATELAIN et al. 1992). Zusätzlich konnte die Schlüsselrolle von
Il-13 als Suszeptibilitäts-Faktor in IL-13-überexprimierenden C57BL/6-Mäusen demonstriert
werden (MATTHEWS et al. 2000).
Die Rolle von IL-4 als Hauptfaktor für die Verschlimmerung der Erkrankung ist durch
aktuellere Untersuchungen vor allem durch eine relativ häufig bestätigte Expression von IL-4
in resistenten Mausstämmen (MORRIS et al. 1992, REINER et al. 1994, HEINZEL et al.
1995, SCOTT et al. 1996, BELKAID et al. 2000,) infragegestellt worden und die Beteiligung
weiterer Mediatoren als einflussreiche Faktoren wird vermutet (SCOTT et al. 1998, SACKS
et al 2004). Es besteht die Vermutung, dass die erhöhte IL-4-Sensivität infizierter LC von
Balb/c-Mäusen auf eine erhöhte IL-4-Rezeptor-Expression zurückzuführen ist (MOLL et al.
2002). Trotz weiter Akzeptanz des Modells der IL-4-vermittelten Th2-Antwort und
IL-12-vermittelten, IFN-γ dominierenden Th1-Antwort, stellt sich heute die Frage ob dieses
Konzept nicht eine Simplifizierung der weit mehr komplex ablaufenden Mechanismen
darstellt (KELSO 1995, KELSO et al. 1995, SACKS u. NOBEN-TRAUTH 2002). Der
kritische Unterschied zwischen resistenten und suszeptiblen Mäusen scheint darin zu liegen,
ob sie dazu fähig sind, die frühe IL-4-Expression zu einer Th1-Zell-Differenzierung umleiten
zu können. Als gesichert gilt, dass eine schützende Immunantwort durch Ausbildung einer
Typ1-Immunität und somit der Differenzierung von CD4+ Lymphozyten zu T-Helfer-Zellen
-32-
Literaturübersicht
vom Typ1 entsteht, die entzündlich und cytotoxisch wirkende Mechanismen für die
Abtötungder intrazellulären Pathogene vermitteln (SACKS et al. 2002, GUMY et al. 2004,
ALEXANDER et al. 2005).
2.3.7 Die Rolle von IL-12 in der T-Zell-Differenzierung
Allgemeiner Konsens ist, dass IL-12, produziert von Antigen-präsentierenden Zellen (APC),
darunter DC, in der Expression verstärkt durch Zytokine wie bspw. IL-1α (VON STEBUT et
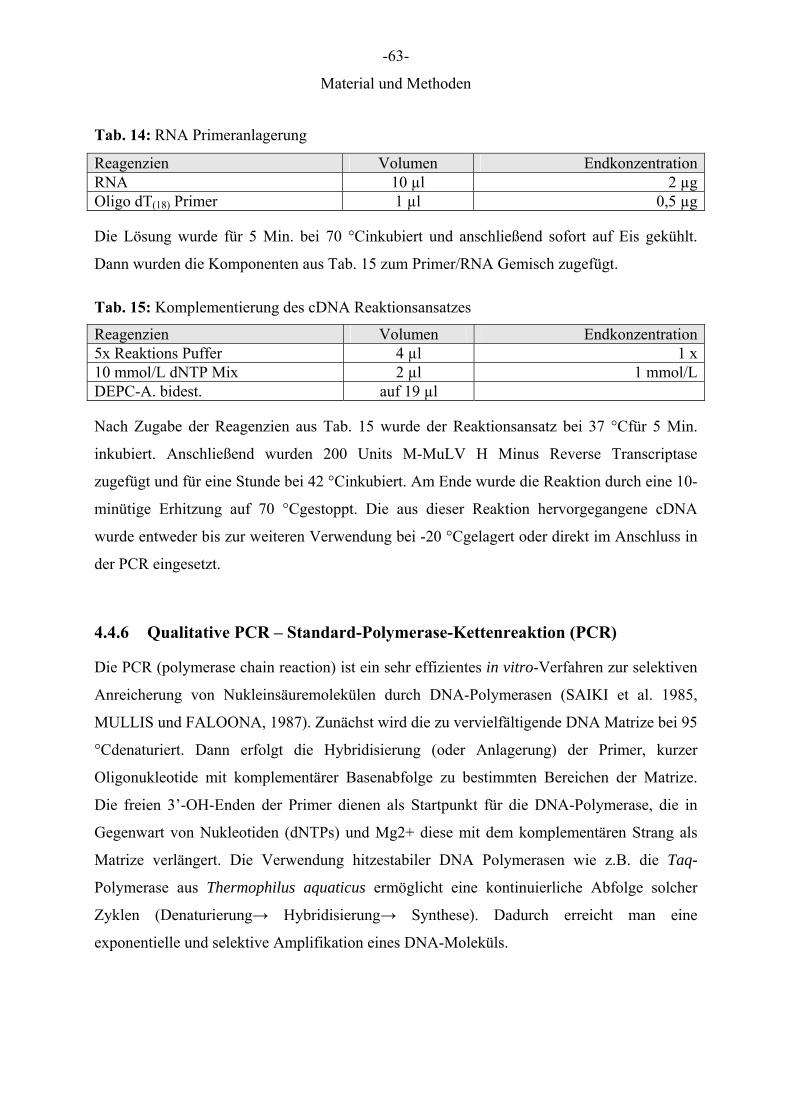
Dies wurde zusammen bei 37 °Cgelöst und unmittelbar vor Gebrauch 15 μl RNase, DNase
frei (RNase A) und 10 μl RNase T1 zugegeben.
RNA-Probenpuffer
1 ml Formamid, 350 μl Formaldehyd 37 %ig, 100 μl 20 x MOPS
-47-
Material und Methoden
Salmonsperm-DNA (ssDNA)-Lösung, 10mg/ml
Das Lyophilisat (250 mg) wurde mit 25 ml Puffer 4, pH 8,0 gelöst, so dass eine
Endkonzentration von 10mg/ml erreicht wurde. Diese Lösung wurde bei 4 °Cgelagert.
20 x SSC (“standard saline citrate“), pH 7,0 (Stammlösung)
175,3 g NaCl, 88,2 g Na-Citrat (Tri-Natrium-Di-Hydrat), ad 800 ml A. bidest.
Der pH-Wert wurde mit 1 N HCl auf 7,0 eingestellt.
2 x SSC + 5 mM EDTA-Na2
50 ml 20 x SSC, 5 ml 0,5 mol/L EDTA-Na2, 500 ml A. bidest.-DEPC
TAE-Puffer
40mM Tris pH 8,3, 2mM EDTA
1 M Tris-HCl, pH 8,0
12,11 g Tris(hydroxymethyl)aminomethan (MG 121,14), 100 ml A. bidest.
Der pH-Wert wurde mit konzentrierter HCl auf 8,0 eingestellt.
-48-
Material und Methoden
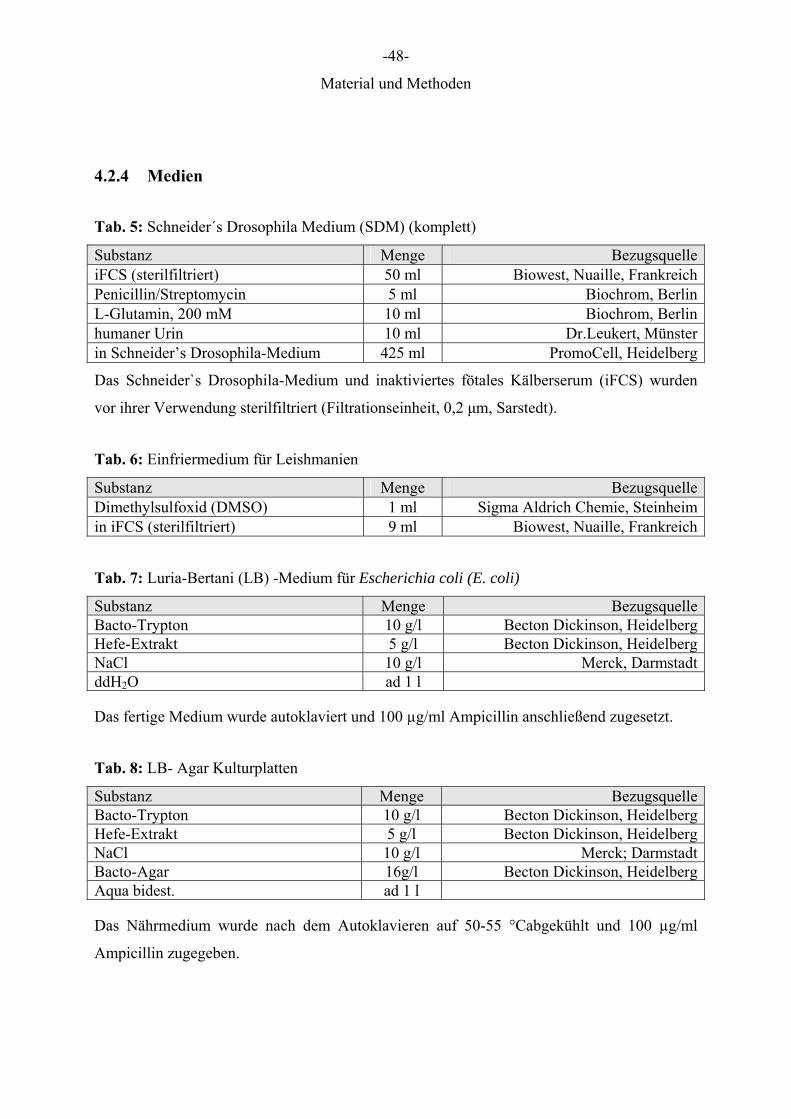
4.2.4 Medien
Tab. 5: Schneider´s Drosophila Medium (SDM) (komplett)
Substanz Menge BezugsquelleiFCS (sterilfiltriert) 50 ml Biowest, Nuaille, FrankreichPenicillin/Streptomycin 5 ml Biochrom, BerlinL-Glutamin, 200 mM 10 ml Biochrom, Berlinhumaner Urin 10 ml Dr.Leukert, Münsterin Schneider’s Drosophila-Medium 425 ml PromoCell, Heidelberg
Das Schneider`s Drosophila-Medium und inaktiviertes fötales Kälberserum (iFCS) wurden
vor ihrer Verwendung sterilfiltriert (Filtrationseinheit, 0,2 μm, Sarstedt).
Tab. 6: Einfriermedium für Leishmanien
Substanz Menge BezugsquelleDimethylsulfoxid (DMSO) 1 ml Sigma Aldrich Chemie, Steinheimin iFCS (sterilfiltriert) 9 ml Biowest, Nuaille, Frankreich
Tab. 7: Luria-Bertani (LB) -Medium für Escherichia coli (E. coli)
Substanz Menge BezugsquelleBacto-Trypton 10 g/l Becton Dickinson, HeidelbergHefe-Extrakt 5 g/l Becton Dickinson, HeidelbergNaCl 10 g/l Merck, DarmstadtddH2O ad 1 l
Das fertige Medium wurde autoklaviert und 100 µg/ml Ampicillin anschließend zugesetzt.
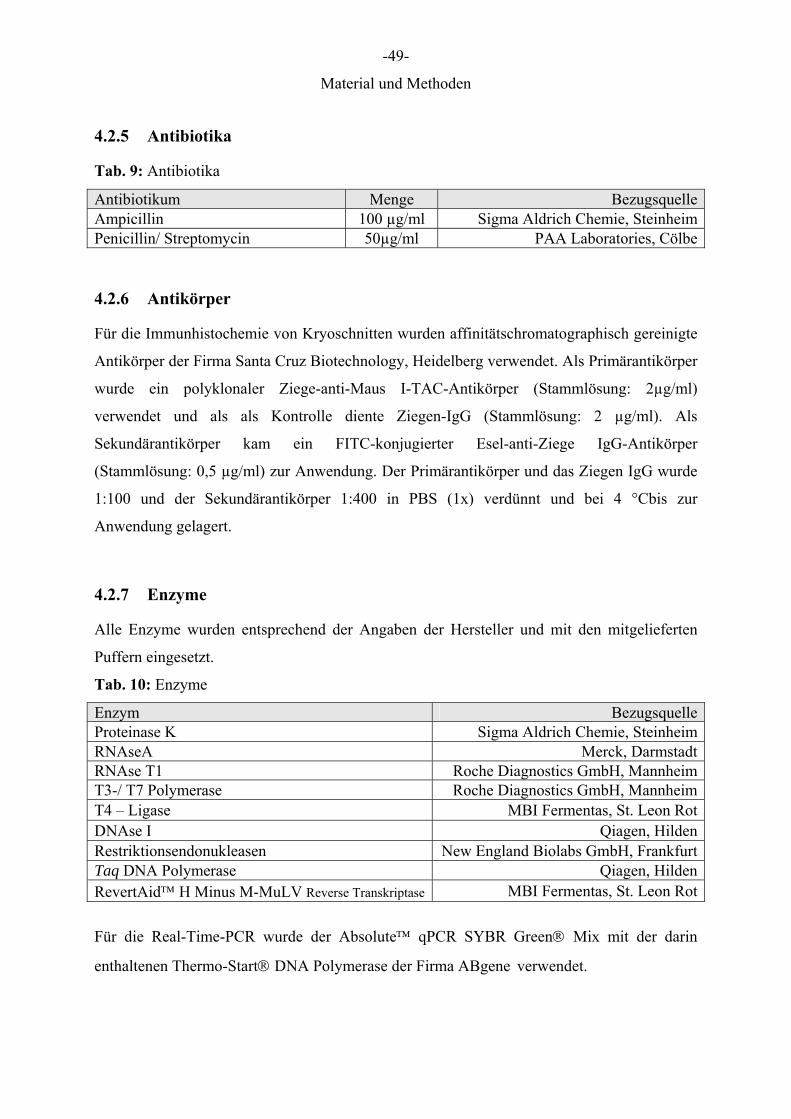
Für die Immunhistochemie von Kryoschnitten wurden affinitätschromatographisch gereinigte
Antikörper der Firma Santa Cruz Biotechnology, Heidelberg verwendet. Als Primärantikörper
wurde ein polyklonaler Ziege-anti-Maus I-TAC-Antikörper (Stammlösung: 2µg/ml)
verwendet und als als Kontrolle diente Ziegen-IgG (Stammlösung: 2 µg/ml). Als
Sekundärantikörper kam ein FITC-konjugierter Esel-anti-Ziege IgG-Antikörper
(Stammlösung: 0,5 µg/ml) zur Anwendung. Der Primärantikörper und das Ziegen IgG wurde
1:100 und der Sekundärantikörper 1:400 in PBS (1x) verdünnt und bei 4 °Cbis zur
Anwendung gelagert.
4.2.7 Enzyme
Alle Enzyme wurden entsprechend der Angaben der Hersteller und mit den mitgelieferten
Puffern eingesetzt.
Tab. 10: Enzyme
Enzym BezugsquelleProteinase K Sigma Aldrich Chemie, SteinheimRNAseA Merck, DarmstadtRNAse T1 Roche Diagnostics GmbH, MannheimT3-/ T7 Polymerase Roche Diagnostics GmbH, MannheimT4 – Ligase MBI Fermentas, St. Leon RotDNAse I Qiagen, HildenRestriktionsendonukleasen New England Biolabs GmbH, FrankfurtTaq DNA Polymerase Qiagen, HildenRevertAid™ H Minus M-MuLV Reverse Transkriptase MBI Fermentas, St. Leon Rot
Für die Real-Time-PCR wurde der Absolute™ qPCR SYBR Green® Mix mit der darin
enthaltenen Thermo-Start® DNA Polymerase der Firma ABgene verwendet.
-50-
Material und Methoden
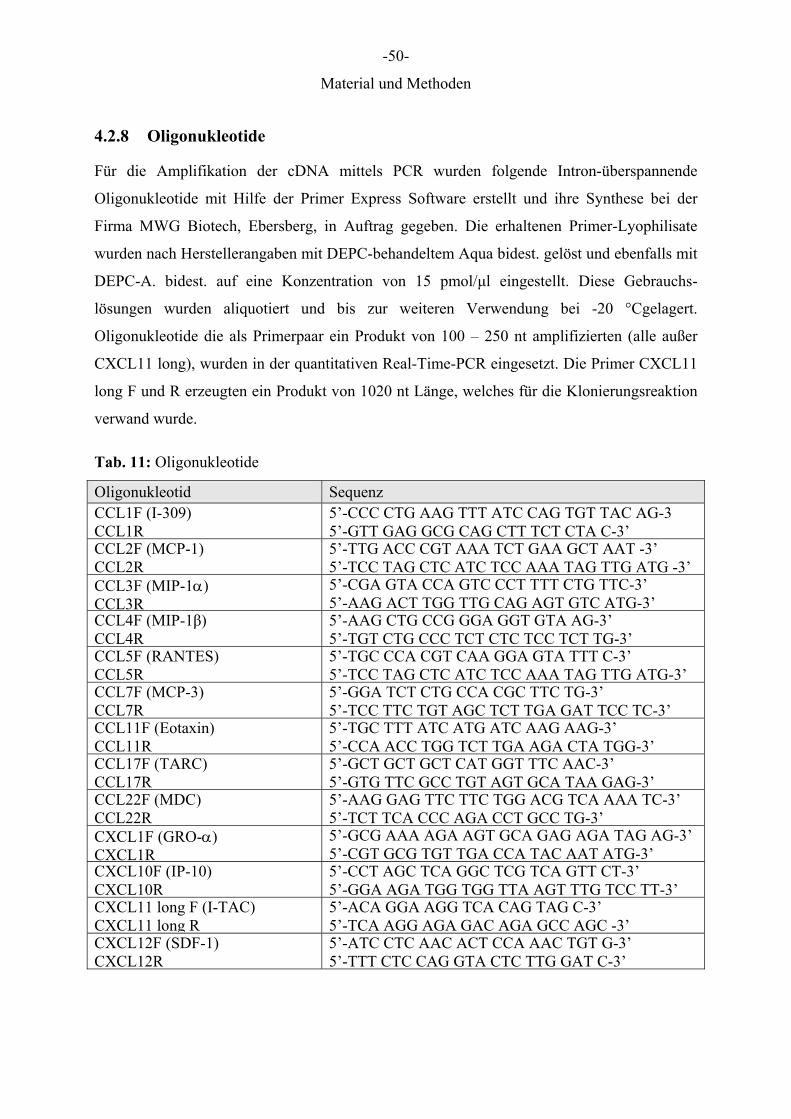
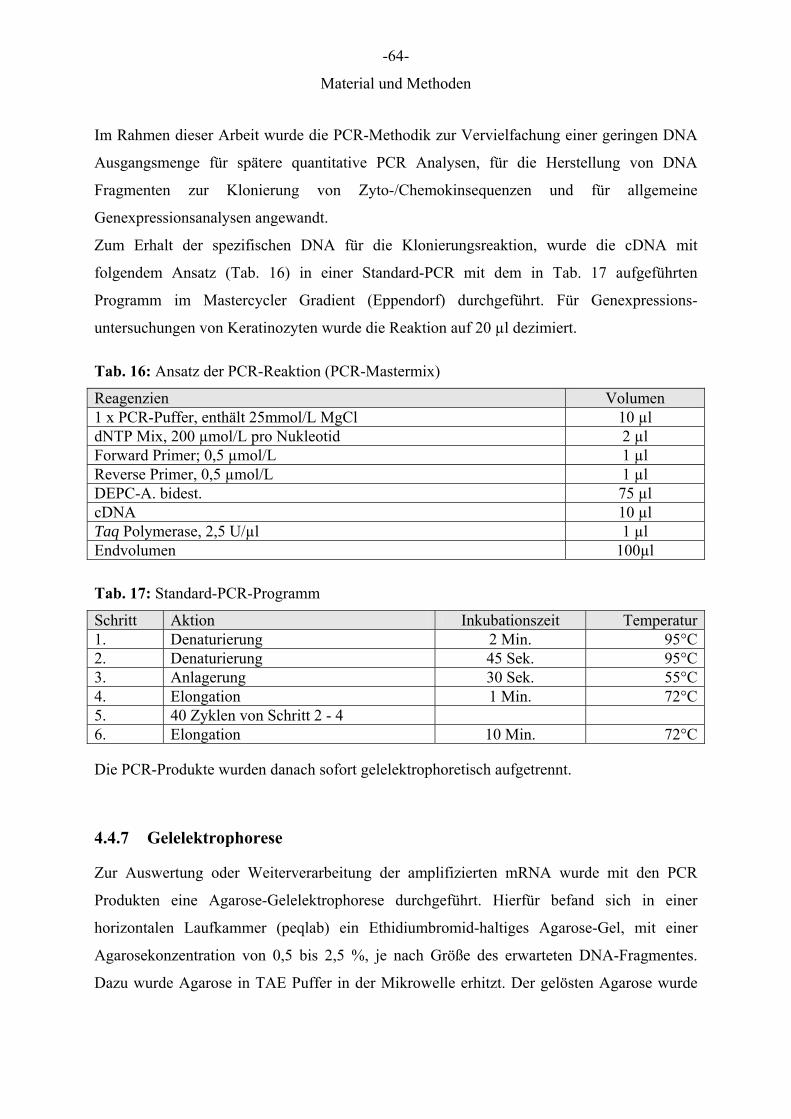
4.2.8 Oligonukleotide
Für die Amplifikation der cDNA mittels PCR wurden folgende Intron-überspannende
Oligonukleotide mit Hilfe der Primer Express Software erstellt und ihre Synthese bei der
Firma MWG Biotech, Ebersberg, in Auftrag gegeben. Die erhaltenen Primer-Lyophilisate
wurden nach Herstellerangaben mit DEPC-behandeltem Aqua bidest. gelöst und ebenfalls mit
DEPC-A. bidest. auf eine Konzentration von 15 pmol/μl eingestellt. Diese Gebrauchs-
lösungen wurden aliquotiert und bis zur weiteren Verwendung bei -20 °Cgelagert.
Oligonukleotide die als Primerpaar ein Produkt von 100 – 250 nt amplifizierten (alle außer
CXCL11 long), wurden in der quantitativen Real-Time-PCR eingesetzt. Die Primer CXCL11
long F und R erzeugten ein Produkt von 1020 nt Länge, welches für die Klonierungsreaktion
O'GeneRuler™ 100bp DNA Ladder; alle Fermentas) zum Ablesen der Länge der PCR-
Produkte aufgetragen. An die Elektrophoresekammer wurde für 24 Min. eine Spannung von 6
V/cm bei einer maximalen Stromstärke von 500 mA angelegt. Danach erfolgte die
Visualisierung des Gels mittels Durchlicht bei 312 nm auf einem UV Transilluminator. Die
photographische Dokumentation der Ergebnisse erfolgte durch ein MultiImage Light Cabinet
(Alpha Innotech).
4.4.8 Aufreinigung von DNA aus einem Agarose-Gel
Für den Erhalt des spezifischen DNA Produktes aus der PCR für die Klonierungsreaktion
wurde das sichtbare DNA-Fragment aus dem Agarose-Gel mit dem Genomed JETQUICK
Gel Extraction Spin Kit® extrahiert und gereinigt. Dazu wurde ein ausgeschnittenes Stück
Agarose-Gel in einem Reaktionsgefäß gewogen. Jeweils auf 100 mg Gel wurden 300 μl
Puffer L1 hinzugegeben. Durch 15 Min. Inkubation bei 50 °Cwurde das Gel gelöst. Danach
wurde die Lösung auf ein JETQUICK Spin Säulchen gegeben. Die Säule wurde 1 Min. bei
12000 x g zentrifugiert. Die aufgefangene Flüssigkeit wurde verworfen und die Säule mit 500
μl Waschpuffer L2 gewaschen. Der folgende Zentrifugationsschritt wurde zweimal
wiederholt, um verbleibenden Waschpuffer sicher zu entfernen. Dann wurde die DNA mit
30 μl Elutionspuffer TE in einem frischem 1,5 ml Reaktionsgefäß aufgefangen und für die
Ligation in ein Plasmid verwendet oder bei -20 °Cgelagert.
4.4.9 „Vor“-Amplifikation von lasermikrodisseziertem Material
Die reverse Transkription und Vervielfachung der Ausgangs-mRNA-Menge von
Probenmaterial nach Lasermikrodissektion wurde durch die Anwendung des Microarray
Target Amplification Kit® (Roche, Mannheim) erzielt. Die mRNA wird bei diesem
-66-
Material und Methoden
Verfahren zuerst in Einzelstrang cDNA (sscDNA) dann in Doppelstrang cDNA (dscDNA)
umgeschrieben, und diese anschließend in einer PCR vervielfältigt (s. Abb. 3) (KLUR et al.
2004).
PCR
Real Time Real-Time-PCR
Abb. 3: Ablauf der dscDNA-Synthese und „Vor“-Amplifikation. (Modifiziert nach dem „product overview“ aus dem Handbuch für das Microarray Target Amplification Kit, ROCHE APPLIED SCIENCE 2004)
-67-
Material und Methoden
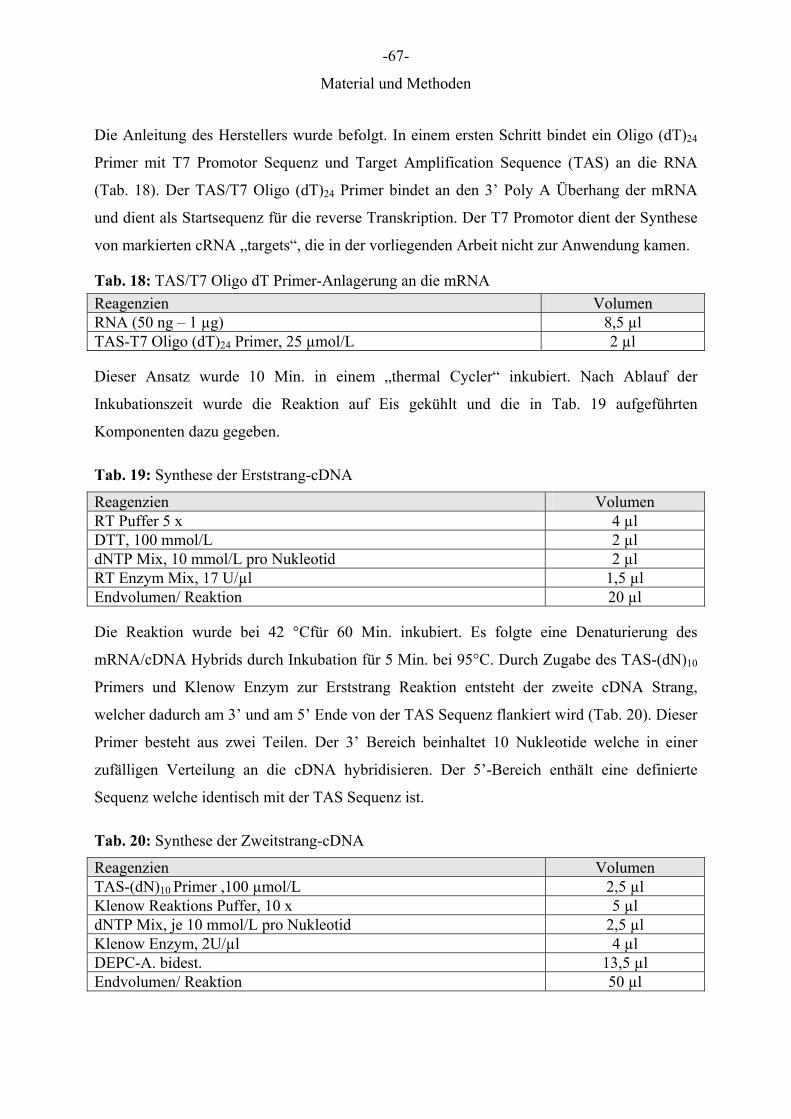
Die Anleitung des Herstellers wurde befolgt. In einem ersten Schritt bindet ein Oligo (dT)24
Primer mit T7 Promotor Sequenz und Target Amplification Sequence (TAS) an die RNA
(Tab. 18). Der TAS/T7 Oligo (dT)24 Primer bindet an den 3’ Poly A Überhang der mRNA
und dient als Startsequenz für die reverse Transkription. Der T7 Promotor dient der Synthese
von markierten cRNA „targets“, die in der vorliegenden Arbeit nicht zur Anwendung kamen.
Tab. 18: TAS/T7 Oligo dT Primer-Anlagerung an die mRNA
Dieser Ansatz wurde 10 Min. in einem „thermal Cycler“ inkubiert. Nach Ablauf der
Inkubationszeit wurde die Reaktion auf Eis gekühlt und die in Tab. 19 aufgeführten
Schritt Aktion Inkubationszeit Temperatur1. Denaturierung 2 Min. 95°C2. Denaturierung 30 Sek. 95°C3. Anlagerung 30 Sek. 55°C4. Elongation 3 Min. 72°C5. 39 Zyklen von Schritt 2-4
Während der PCR wurde ab dem 21 Zyklus und nach jedem weiteren dritten Zyklus eine
Probe entnommen und diese auf einem 1,2 %igem Agarose /EtBr- Gel analysiert. In der
exponentiellen Phase der PCR Reaktion führt jeder Zyklus zu einer Verdoppelung der
-69-
Material und Methoden
amplifizierten cDNA. Die Größe der cDNA sollte zwischen 700 – 2200 bp liegen. Es wurde
eine Zyklen-Zahl am Beginn der exponentiellen Phase (gleichmäßig sichtbare Schlieren auf
dem Gel) gewählt. Für das in Abb. 4 gezeigte Beispiel befindet sich die PCR bis zu Zyklus 24
in der exponentiellen Phase. In den Zyklen 27 – 30 ist bereits das Plateau der PCR erreicht
und es besteht die Gefahr einer negativen Auswirkung auf die Repräsentation der Transkripte.
Zyklenzahl
Abb. 4: Beispiel für die PCR-Optimierung: 10 µl Aliquots wurden nach Zyklus 18/21/24/27/30 auf einem 1,2%igem Agarose/Etbr Gel analysiert. 50 ng Kontroll-RNA ergaben nach 24 Zyklen gleichmäßig sichtbare Schlieren in der Größe zwischen 700-2200 bp. Somit war nach 24 Zyklen die PCR in der exponentiellen Phase. (Abb.: “Figure 1, First PCR” aus dem Handbuch für das Microarray Target AmplificationKit, ROCHE APPLIED SCIENCE 2004).
Diese durch die erste PCR determinierte Zyklenzahl wurde in einer zweiten PCR angewandt
um die Gesamtheit der mRNA zu amplifizieren. Der gleiche Reaktionsansatz wie in Tab. 21
wurde mit dem Programm in Tab. 22 amplifiziert. Nach Abschluss der zweiten PCR wurde
die Reaktion mit dem Microarray Target Purification Kit® wie beschrieben gereinigt. Die
amplifizierte cDNA wurde mit DEPC-A. bidest. verdünnt und für quantitative PCR Versuche
verwendet oder bei -20C gelagert.
4.4.10 Quantitative PCR – Real-Time-PCR
Bei der Real-Time-PCR wird nicht das Endprodukt untersucht, sondern das Entstehen dieses
Produktes im Laufe der PCR (FREEMAN et al. 1999). Das Prinzip dieser Methode ist, dass
mit jedem Amplifikationsschritt durch den Einsatz fluoreszierender Farbstoffe ein vom Gerät
-70-
Material und Methoden
detektierbares Signal generiert wird, welches mit der jeweils vorhandenen Menge an PCR-
Produkten korreliert. Der Anstieg der Fluoreszenz wird während eines jeden Zyklus
gemessen, mit dem Ergebnis, dass der Ablauf und nicht das Endergebnis der Reaktion
nachvollzogen wird. In der vorliegenden Arbeit wurde der interkalierende Farbstoff SYBR-
Green (absolute™ qPCR SYBR Green® MIX, ABgene) verwendet. Dieser fluoreszierende
Farbstoff bindet an doppelsträngige DNA (MORRISSON et al. 1998). Der Farbstoff wird
durch einen Laser zur Fluoreszenz, messbar bei einer Wellenlänge von 530 nm angeregt, die
wiederum von einer Fotozelle detektiert wird. Die Spezifität wird anhand einer
Dissoziationskurve überprüft (RIRIE et al. 1997) indem das PCR-Produkt nach dem letzten
Zyklus durch langsame Erhitzung bis zum Schmelzpunkt denaturiert wird. Durch den
Schmelzpunkt können die Längen der im Reaktionsgemisch vorhandenen PCR-Produkte und
damit die Spezifität der Reaktion bestimmt werden. Für die Amplifikation der sogenannten
„housekeeping“-Gene GAPDH und RPL wurde die cDNA 1:100 und für die Zielgene 1:10
mit DEPC-A. bidest. verdünnt, und die Reaktionsansätze (s. Tab. 23) im ABI prism 7900 HT-
RealTime-PCR (Applied Biosystems, Weiterstadt) als Doppelbestimmungen für jedes Gen
durchgeführt. Hierbei kam das sogenannte „two Step“- (zwei Stufen) Real-Time-PCR
Verfahren zur Anwendung. Die erste Stufe umfasst die Denaturierungsphase und die zweite
Stufe beinhaltet die Anlagerung- und Elongationsphase (s. Tab. 24). Das Pipettieren der 384
Well PCR Platten (ABgene, Hamburg) erfolgte durch den Genesis 150 Workstation
Pipettierroboter (Tecan, Crailsheim).
Tab. 23: Reaktionsansatz
Reagenzien Volumen cDNA 2,5 µl Forward Primer, 50 pmol/µl 0,2 µl Reverse Primer, 50 pmol/µl 0,2 µl SYBR-Green Mastermix, enthält SYBR® Green Dye I, Thermo -Start® DNA Polymerase, dNTPs und Puffer 5 µl
DEPC-A. bidest. 2,3 µl Endvolumen 10 µl
-71-
Material und Methoden
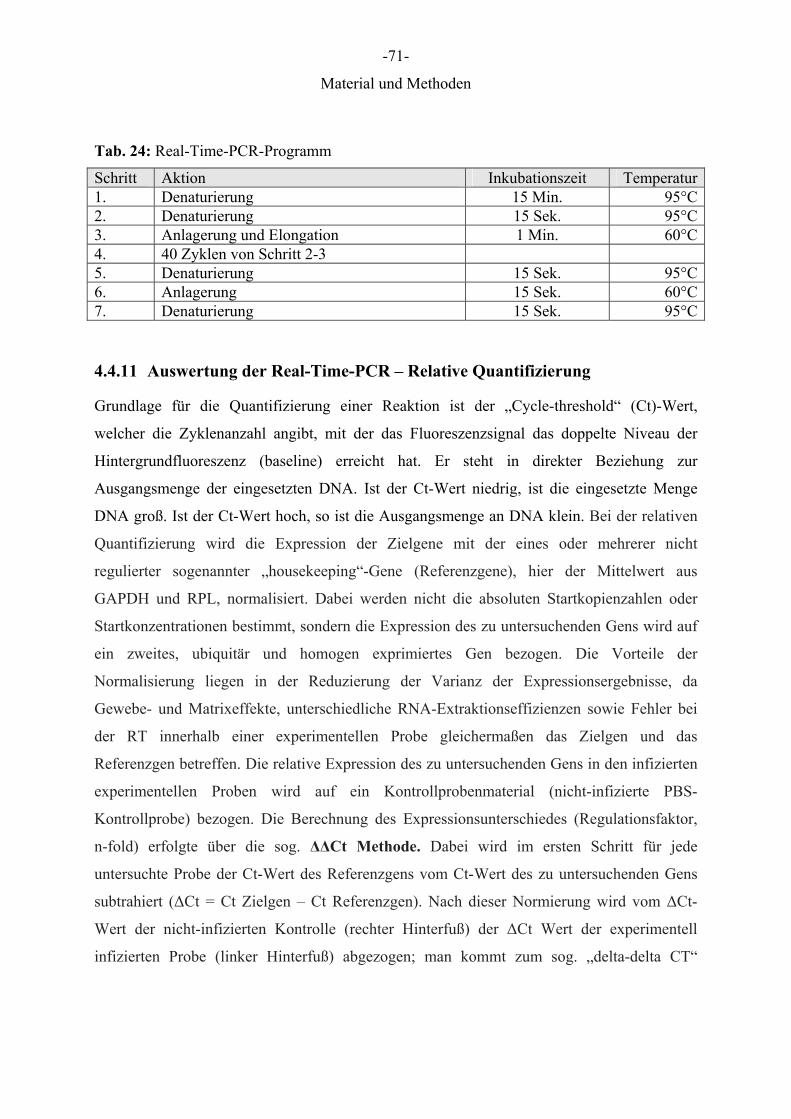
Tab. 24: Real-Time-PCR-Programm
Schritt Aktion Inkubationszeit Temperatur1. Denaturierung 15 Min. 95°C2. Denaturierung 15 Sek. 95°C3. Anlagerung und Elongation 1 Min. 60°C4. 40 Zyklen von Schritt 2-3 5. Denaturierung 15 Sek. 95°C6. Anlagerung 15 Sek. 60°C7. Denaturierung 15 Sek. 95°C
4.4.11 Auswertung der Real-Time-PCR – Relative Quantifizierung
Grundlage für die Quantifizierung einer Reaktion ist der „Cycle-threshold“ (Ct)-Wert,
welcher die Zyklenanzahl angibt, mit der das Fluoreszenzsignal das doppelte Niveau der
Hintergrundfluoreszenz (baseline) erreicht hat. Er steht in direkter Beziehung zur
Ausgangsmenge der eingesetzten DNA. Ist der Ct-Wert niedrig, ist die eingesetzte Menge
DNA groß. Ist der Ct-Wert hoch, so ist die Ausgangsmenge an DNA klein. Bei der relativen
Quantifizierung wird die Expression der Zielgene mit der eines oder mehrerer nicht
regulierter sogenannter „housekeeping“-Gene (Referenzgene), hier der Mittelwert aus
GAPDH und RPL, normalisiert. Dabei werden nicht die absoluten Startkopienzahlen oder
Startkonzentrationen bestimmt, sondern die Expression des zu untersuchenden Gens wird auf
ein zweites, ubiquitär und homogen exprimiertes Gen bezogen. Die Vorteile der
Normalisierung liegen in der Reduzierung der Varianz der Expressionsergebnisse, da
Gewebe- und Matrixeffekte, unterschiedliche RNA-Extraktionseffizienzen sowie Fehler bei
der RT innerhalb einer experimentellen Probe gleichermaßen das Zielgen und das
Referenzgen betreffen. Die relative Expression des zu untersuchenden Gens in den infizierten
experimentellen Proben wird auf ein Kontrollprobenmaterial (nicht-infizierte PBS-
Kontrollprobe) bezogen. Die Berechnung des Expressionsunterschiedes (Regulationsfaktor,
n-fold) erfolgte über die sog. ΔΔCt Methode. Dabei wird im ersten Schritt für jede
untersuchte Probe der Ct-Wert des Referenzgens vom Ct-Wert des zu untersuchenden Gens
subtrahiert (ΔCt = Ct Zielgen – Ct Referenzgen). Nach dieser Normierung wird vom ΔCt-
Wert der nicht-infizierten Kontrolle (rechter Hinterfuß) der ΔCt Wert der experimentell
infizierten Probe (linker Hinterfuß) abgezogen; man kommt zum sog. „delta-delta CT“
-72-
Material und Methoden
Berechnungsmodell ΔΔCt = (ΔCt nicht-infiziert – ΔCt infiziert). Der relative
Expressionsunterschied einer Probe zwischen der Infektion und der Kontrolle, normalisiert
zum Referenzgen und bezogen auf eine Standardprobe, ergibt sich aus der arithmetischen
Formel:
Regulationsfaktor (n-fold) = 2ΔΔCt (LIVAK u. SCHMITTGEN 2001)
Dieses Berechnungsschema setzt eine Verdoppelung der DNA Menge in jedem Zyklus
voraus. In einigen Fällen würde die Berechnung des n-folds jedoch zu falsch-positiven
Ergebnissen führen. Dies ist z.B. gegeben, wenn die Expression von Zielgenen in der nicht-
infizierten Probe sehr gering ist. In diesen Proben ist der Ct-Wert dementsprechend hoch, so
dass nach dem „delta-delta CT“ Berechnungsmodell eine stärkere Expression gemessen
würde als tatsächlich vorhanden ist. In diesem Falle, wie auch für den Fall wenn Ct–Werte
des Refernzgens außerhalb des messbaren Bereichs lagen, wurde die Amplifikate der
infizierten Probe in Bezug auf GAPDH/RPL berechnet. Dies erfolgte durch Anwendung der
Formel: 2ΔCt*-1/ Verdünnungsfaktor
Der Verdünnungsfaktor war 10, da die cDNA für die Untersuchung der Zielgene um das 10-
fache weniger verdünnt war als jene für GAPDH/RPL Untersuchungen.
In der vorliegenden Arbeit wurden die Daten von zwei unabhängig voneinander
durchgeführten Lasermikrodissektions-Versuchen und von drei Versuchen zur Genexpression
in Gesamthaut verwendet. Aus diesem Grunde wurde nur für die Ergebnisse zur Expression in
der Gesamthaut der Standardfehler und die Signifikanz, berechnet durch einen Student’s t-
Test, angegeben.
4.4.12 Klonierungsreaktion
Nach der Aufreinigung der PCR-Produkte wurden sie in die Klonierungsreaktionen
eingesetzt. Die in der PCR verwendete Taq-Polymerase fügt matrizenunabhängig einzelne
Desoxyadenosine an die 3’-terminalen-Hydroxygruppen ihrer Syntheseprodukte an (HU
1993). Dieses Phänomen bildet die Grundlage der sogenannten T/A-Klonierung, bei der
linearisierte Vektoren eingesetzt werden, die 3’-terminal ein dazu komplementäres
-73-
Material und Methoden
Desoxythymidin tragen. Im Rahmen dieser Arbeit wurde der komerziell erhältiche Vektor
pBluescript-II-SK® (Stratagene, Amsterdam) zur T/A-Klonierung nach einer Vorschrift von
HADJEB u. BERKOWITZ (1996) modifiziert. Der Vektor wurde mit EcoRV linearisiert, 2
Std. bei 72 °Cmit Taq-Polymerase in Gegenwart von 2 mM dTTP inkubiert, durch
phenolische Extraktion und ethanolische Präzipitation gereinigt und anschließend ligiert. Da
nicht vollständig mit T-Überhängen versehende Vektoren rezirkularisieren, konnten diese
dann gelelektrophoretisch abgetrennt werden.
4.4.13 Ligation
Standardmäßig wurden 7 µl PCR-Produkt in einem Klonierungsansatz (Ligation) nach
Herstellerangaben durchgeführt (s. Tab. 25) und diese in kompetente E.coli Zellen
transformiert. Das Enzym T4-DNA-Ligase (MBI Fermentas, Karlsruhe) katalysiert ATP-
abhängig die Knüpfung einer Phosphoesterbindung zwischen der 5´-Phosphatgruppe des
einen DNA-Endes und der 3´-Hydroxylgruppe des anderen Endes (ENGLER u.
Ziel der angewandten Laser Microbeam Microdissection (LMM) und der Laser Pressure
Catapulting (LPC) Methode, zusammen auch kurz LMPC oder Lasermikrodissektion genannt,
-82-
Material und Methoden
war es, Keratinozyten aus der Epidermis zu isolieren um die Genexpression von Zyto- und
Chemokinen in der Frühphase der experimentellen Leishmaniose durch Real-Time-PCR zu
untersuchen.
Die LMPC Technik musste zunächst einmal vollständig für dieses Modell etabliert werden.
Es musste geprüft werden wie viele Zellen nötig sind, um Genexpressionsanalysen mit Hilfe
der Real-Time-PCR durchführen zu können. Aus diesem Grund wurde zunächst ein Gewebe
gewählt aus dem in kürzester Zeit eine große Anzahl von Zellen isoliert werden konnte.
Hierfür wurden Gefrierschnitte der Leber aus der Maus wie beschrieben angefertigt und
nachfolgend Leberzellen selektiert. Die RNA wurde extrahiert und ihre Qualität durch
Kapillar Elektrophorese des RNA 6000 Nano Assays in Kombination mit Agilents 2100
Bioanalyzer analysiert. Nachfolgend wurde das Testverfahren auf Gefrierschnitte von
Hinterfüßen der Maus angewandt.
Sämtliche Manipulationen am Lasermikrodissektionsgerät wurden durch die PALM Robo
Software gesteuert. Der interessierende Bereich wurde im Gewebeschnitt auf der
Mikroskopierbühne (Robot Stage) per Mausklick über einen computergesteuerten
Hybridstufenmotor lokalisiert. Ein gezieltes Arbeiten im Mikrometerbereich war durch eine
Schrittgeschwindigkeit von 1 μm bis zu mehreren Millimetern pro Sekunde möglich. Im
Aufhängungsapparat (Robot Manipulator) wurde wenige Millimeter oberhalb des
Objektträgers, ein mit Lysispuffer befüllter Deckel eines Reaktionsgefäßes im Fokus des
Mikroskops in Position gebracht. Die Einstellung des Laserfokus und der Laserenergie
erfolgte über eine externe Steuereinheit. Mit Hilfe verschiedener Objektive unterschiedlicher
Vergrößerungsfaktoren (10-fach, 40-fach und 100-fach) konnte der Laserstrahl zu einem
Durchmesser von weniger als 1 μm fokussiert werden. Je kleiner der Fokusdurchmesser
gewählt wurde, desto höher war die Energiedichte im Fokus. Zum Schneiden wurde mit
einem Fokus in Folienebene gearbeitet. Die hohe Energiedichte führte für Bruchteile einer
Sekunde zur Entstehung äußerst hoher Temperaturen und ermöglichte die gezielte Zerstörung
und damit das Schneiden von Gewebe. Nach Defokussieren des Lasers wurde die
herausgelöste Gewebeprobe mit maximaler Laserenergie durch eine Photonen-Wolke in das
Reaktionsgefäß katapultiert. Über eine im Gerät installierte Kamera wurde der mikroskopierte
Ausschnitt dokumentiert und auf der Bildschirmoberfläche des Computers parallel zu den
Arbeiten abgebildet. Eine Darstellung des Systems zeigt die Abb. 5.
-83-
Material und Methoden
Robot Stage
Computer
Laser
Mikroskop
Robot Stage
Computer
Laser
Mikroskop
Abb. 5: PALM Robot Microbeam System. (Modifiziert nach einer Abb. der Firma PALM) [http://www.palm-microlaser.com/dasat/index.php?cid=100140undconid=0undsid=dasat], 2006
Es wurden verschiedene Möglichkeiten der Gewinnung von epidermalen Zellen getestet. Zum
einen wurden Zellverbände von 10–100 Zellen isoliert und zum anderen einzelne
Keratinozyten aus der Epidermis geschnitten und anschließend in einen mit Lysis Puffer
benetzten Deckel eines Reaktionsgefäßes katapultiert. PEN-beschichtete Objektträger
(PALM, Bernried) erleichtern das Katapultieren, indem Gewebe und Folie eine Einheit
bilden, die gemeinsam geschnitten und katapultiert wird. Verschiedene LMPC Funktionen
wurden angewendet (s. Tab. 29).
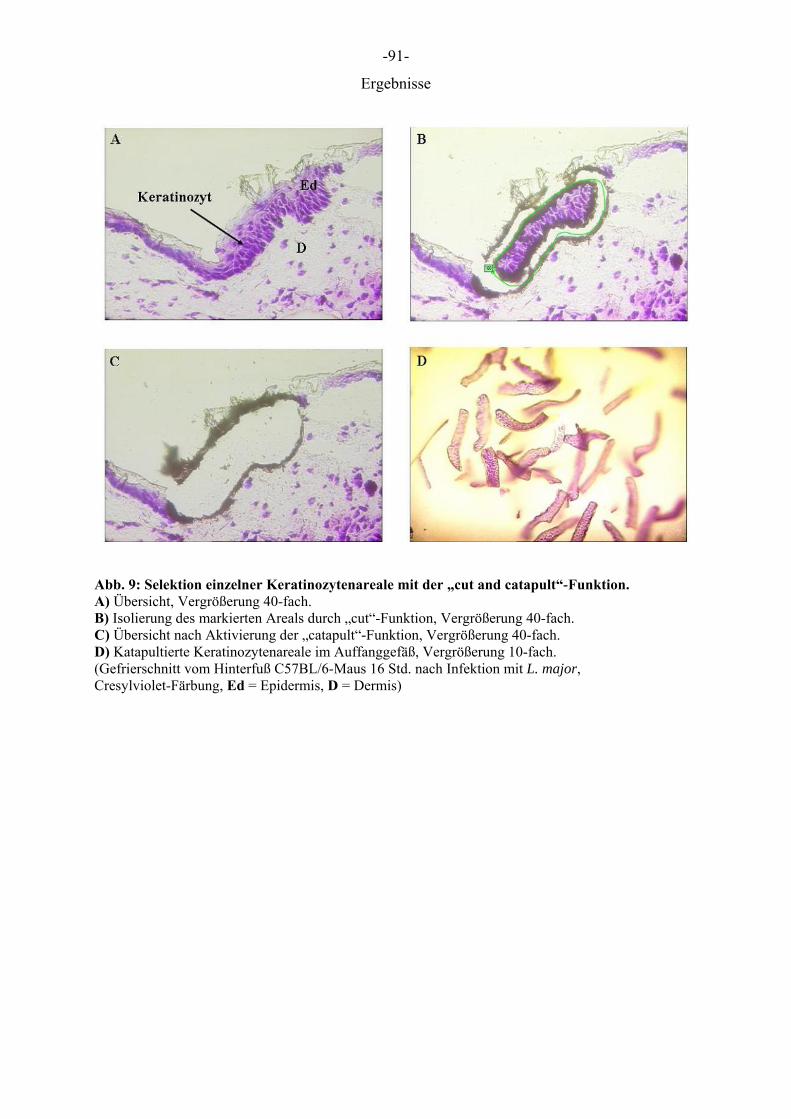
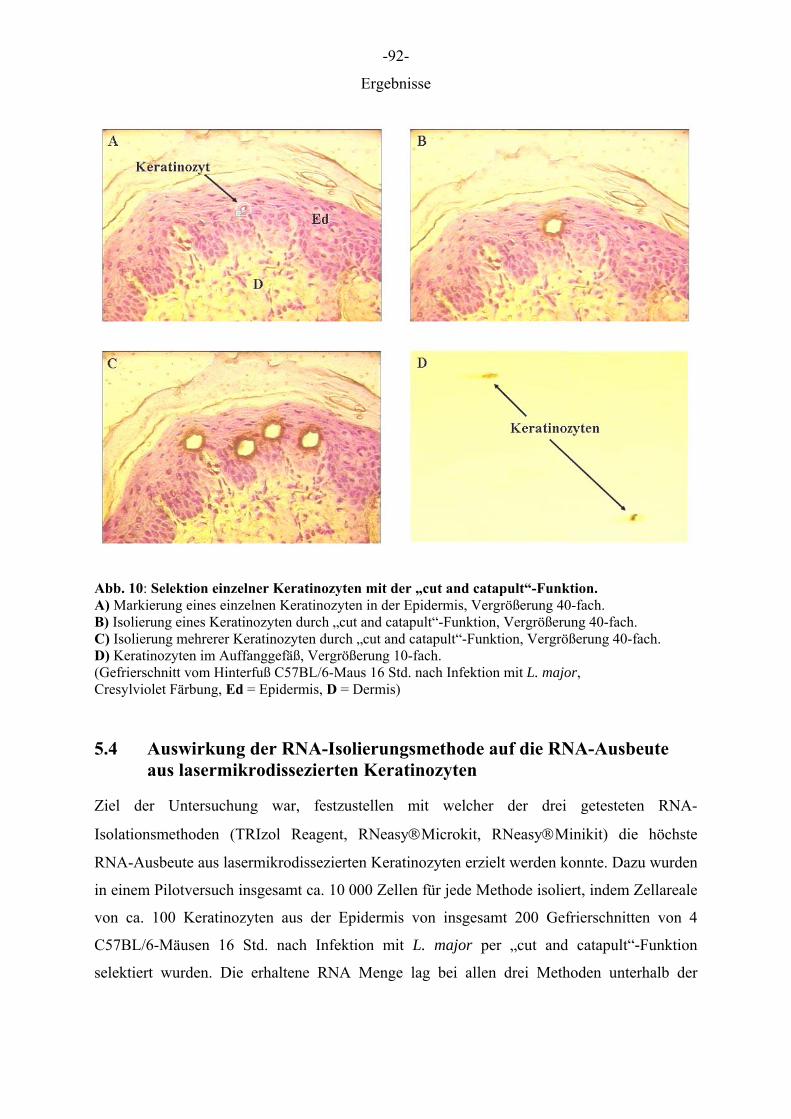
Tab. 29: LMPC Funktionen
Schnitte auf PEN beschichteten Objektträgern Beschreibung der Funktion Schneiden entlang einer vordefinierten Linie; trennt den ausgewählten Bereich vom umliegenden Material. Manuelles setzen eines Katapultpunktes am Rand der Linie
Cut and Catapult
Schneiden und Katapultieren in einem Schritt RoboLPC
Schnitte auf unbeschichteten Objektträgern Beschreibung der Funktion
AutoLPC Automatisches Katapultieren großer Areale durch Anlegen eines shot grid (shots/µm2).
-84-
Material und Methoden
4.6 Cytogenetische Methoden
4.6.1 In situ-Hybridisierung (ISH)
Die ISH ist ein Verfahren um die mRNA in einem Gewebe sichtbar zu machen. Es werden
bei einer solchen Färbung nur diejenigen Zellen angefärbt (hybridisiert), in denen das zu
untersuchende Gen aktiv ist und in denen daher die mRNA im Cytoplasma vorliegt. Für diese
Sichtbarmachung der mRNA wurden die unter 4.4.20 synthetisierten DIG-markierten RNA-
Sonden verwendet. Das Digoxigenin kann mit Hilfe eines Antikörpers, der mit einem Enzym
gekoppelt ist, erkannt werden. Das Enzym, meistens Alkalische Phosphatase oder Peroxidase,
kann dann durch Zusatz von Reagenzien einen Farbstoff umsetzen, der kovalent im Gewebe
gebunden bleibt und sich daher nicht durch Diffusion verteilt. Sämtliche genannten Puffer
und Lösungen sind unter 4.2.3 im Material Teil aufgeführt und die verwendeten cDNA
Fragmente unter 4.2.9 Plasmide, genannt.
Fixierung und Prähybridisierung
Die Gefrierschnitte wurden 30 Min. luftgetrocknet und anschließend in 4 %iger Para-
formaldehyd/PBS Lösung in einer Kuvette für 1 Std. unter Rühren bei Rt fixiert. Durch
dreimaliges Eintauchen für je 10 Min. in 1 x PBS wurden die Schnitte gewaschen. Eine
Behandlung mit Proteinase K (1 µg/ml) Lösung bei Rt für 10 Min. bewirkte eine Proteolyse
sowie den Aufschluss der Zellmembran. Nach erneutem Waschen in A. bidest. wurde für 10
Min. mit 4 %igem Paraformaldehyd nachfixiert. Die Acetylierung fand nach Waschen in
Aqua bidest. und PBS durch 10-minütige Inkubation in 0,1 mol/L TEA und 0,25 %igem
Essigsäureanhydrid statt. Nach weiteren A. bidest und PBS Waschgängen wurden die
Schnitte durch sukzessives Eintauchen in 50 %iger und 70 %iger Ethanollösung entwässert.
Die Schnitte wurden mindestens 30 Min. an der Luft getrocknet und anschließend direkt zur
Hybridisierung verwendet.
Hybridisierung
Die prähybridisierten Schnitte wurden mit den in der Hybridisierungslösung verdünnten DIG-
markierten RNA-Sonden überschichtet, als Verdunstungsschutz mit einem Deckgläschen
bedeckt und in einer feuchten Kammer über Nacht bei 56 °Cinkubiert. Die Konzentration der
eingesetzten Sonde lag durchschnittlich bei 100–200 ng/ml Hybridisierungsmix.
-85-
Material und Methoden
Posthybridisierung
Die Deckgläschen wurden durch Eintauchen in 2 x SSC entfernt, die Objektträger wurden in
2 x SSC und 1 x SSC gewaschen und anschließend 30 Min. bei 37 °Cin der RNAse Lösung
inkubiert. Es schlossen sich Waschschritte mit sinkender Ionenstärke an (bis 0,2 x SSC) und
ein Waschschritt bei hoher Temperatur (60 °Cin 0,2 x SSC). Danach wurden die Objektträger
einmal in 0,2 x SSC und anschließend in Puffer 1 (s. Lösungen und Puffer 4.2.3) gewaschen.
Es erfolgte eine Inkubation in Puffer 2 (s. Lösungen und Puffer 4.2.3) Blockierungsreagenz
zur Absättigung unspezifischer Proteinbindungsstellen. Die Proben wurden vorsichtig mit
100 µl Anti-Dig Antikörperlösung überschichtet und bei 4 °Cin einer feuchten Kammer über
Nacht inkubiert. Es schloss sich eine 2-stündige Inkubation bei 37 °Cund anschließendes
zweimaliges Waschen in Puffer 1 an. Die alkalische Phosphatase wurde durch kurzes
Waschen in Puffer 3 (s. Lösungen und Puffer 4.2.3) aktiviert und die Objektträger 3–6 Std. in
der Farblösung, im Dunkeln bei Rt inkubiert. Die Farbreaktion wurde durch Waschen in
Puffer 4 (s. Puffer und Lösungen 4.2.3) im Dunkeln bei Rt gestoppt. Danach wurden sie für
10 Sek. in Hämalaun Lösung getaucht und anschließend 3-mal für 5 Min. in H2O gewaschen.
Zum Schluss wurden die Schnitte mit Glycergel® (Dako, Hamburg) eingedeckelt. Das
Ergebnis wurde unter dem Lichtmikroskop kontrolliert und mit einer Kamera dokumentiert.
-86-
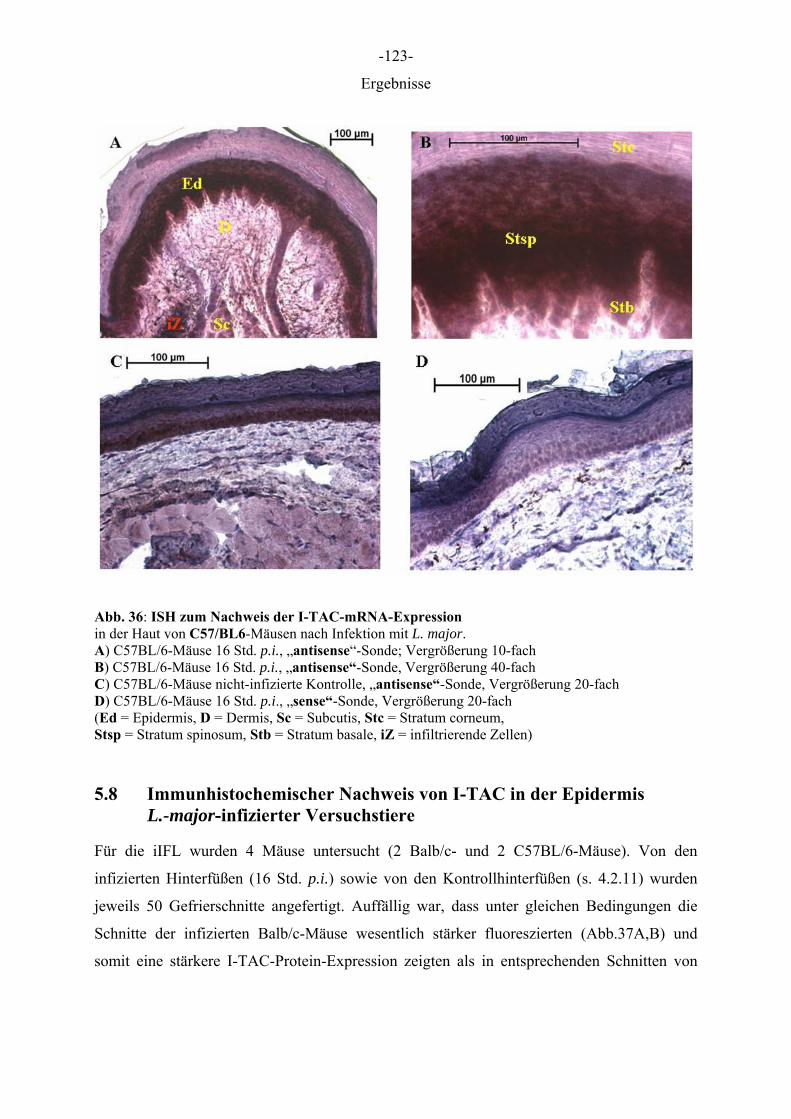
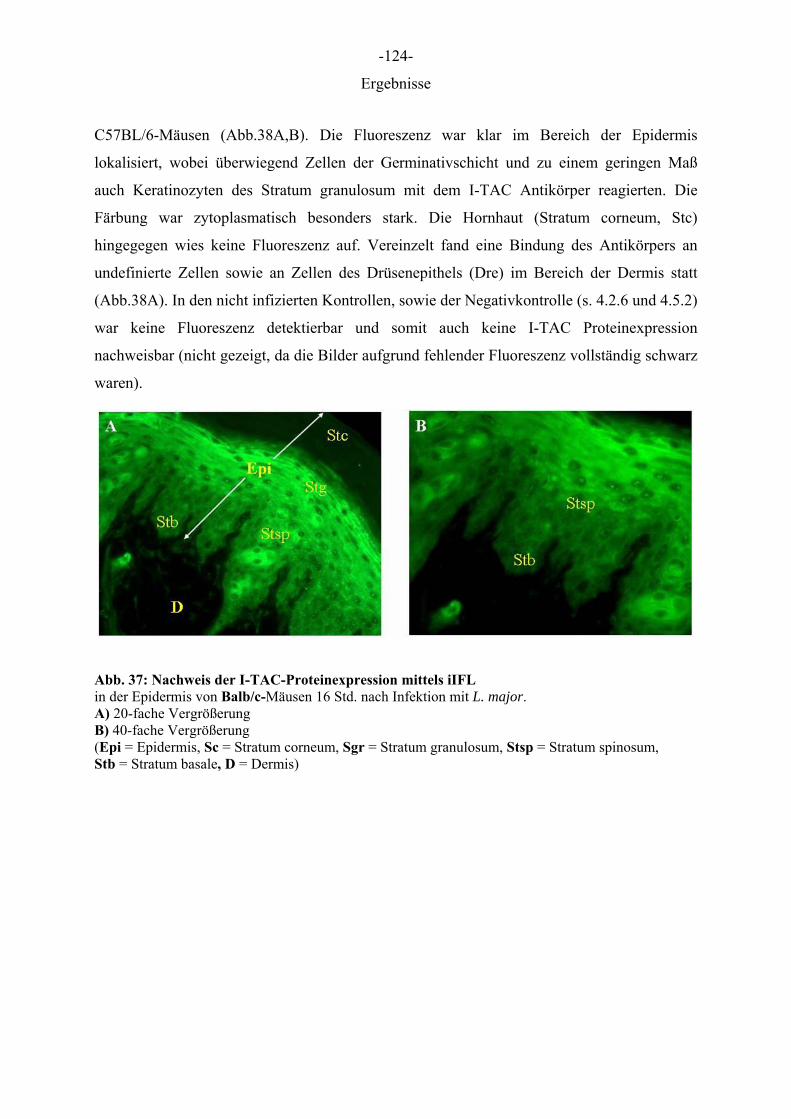
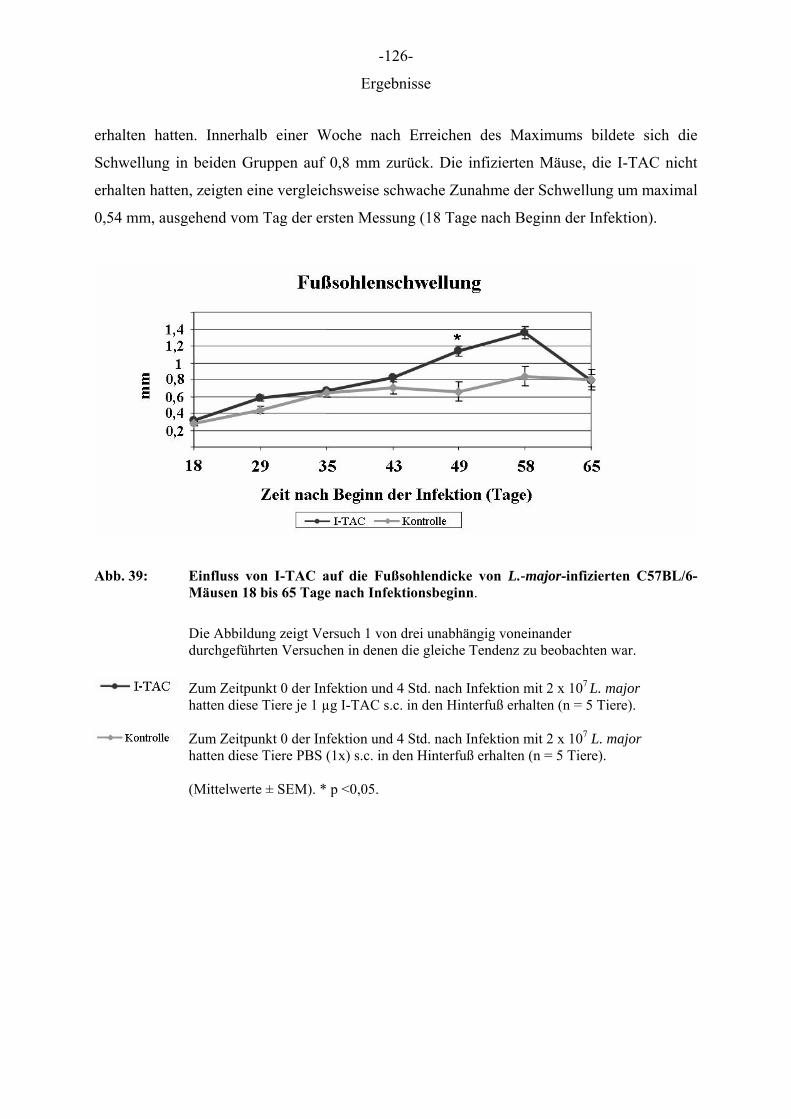
Ergebnisse
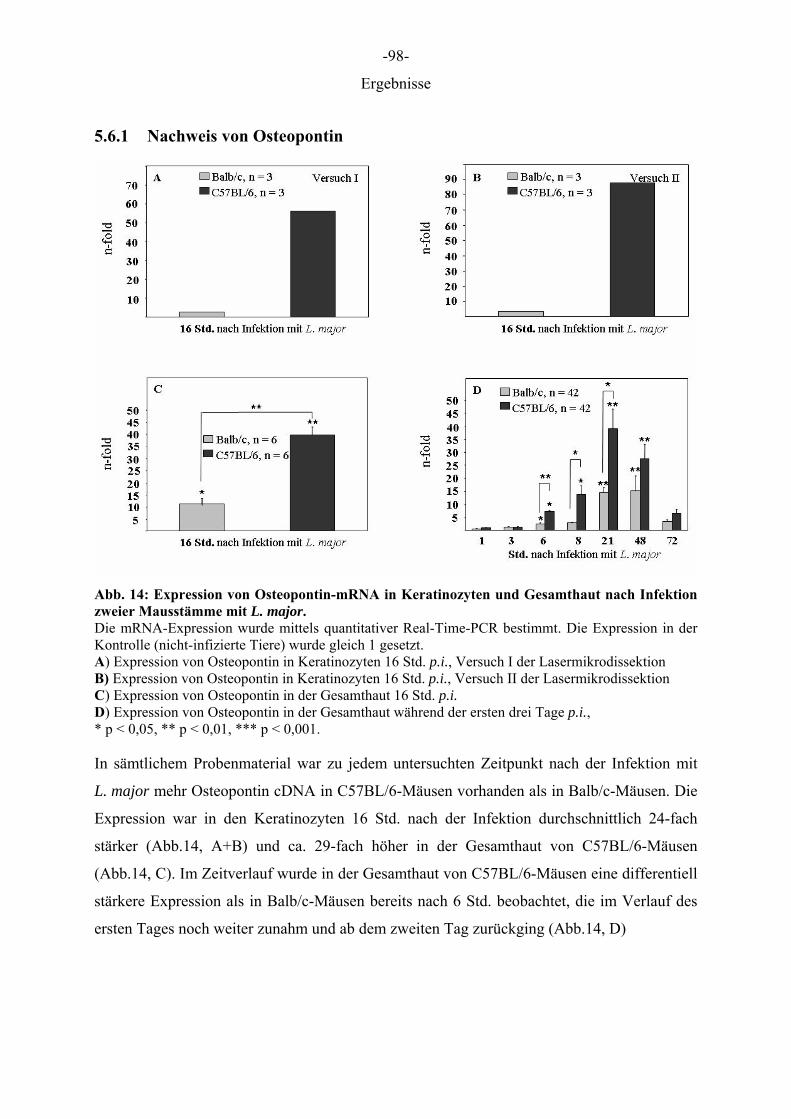
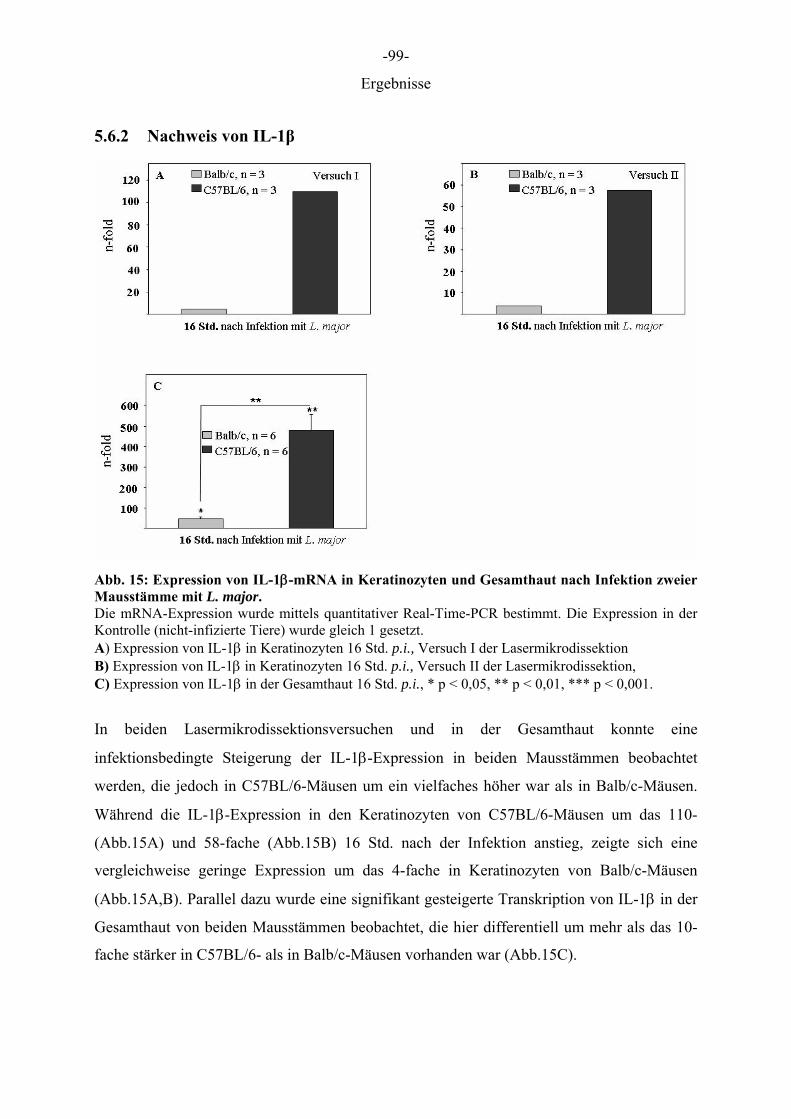
5 ERGEBNISSE
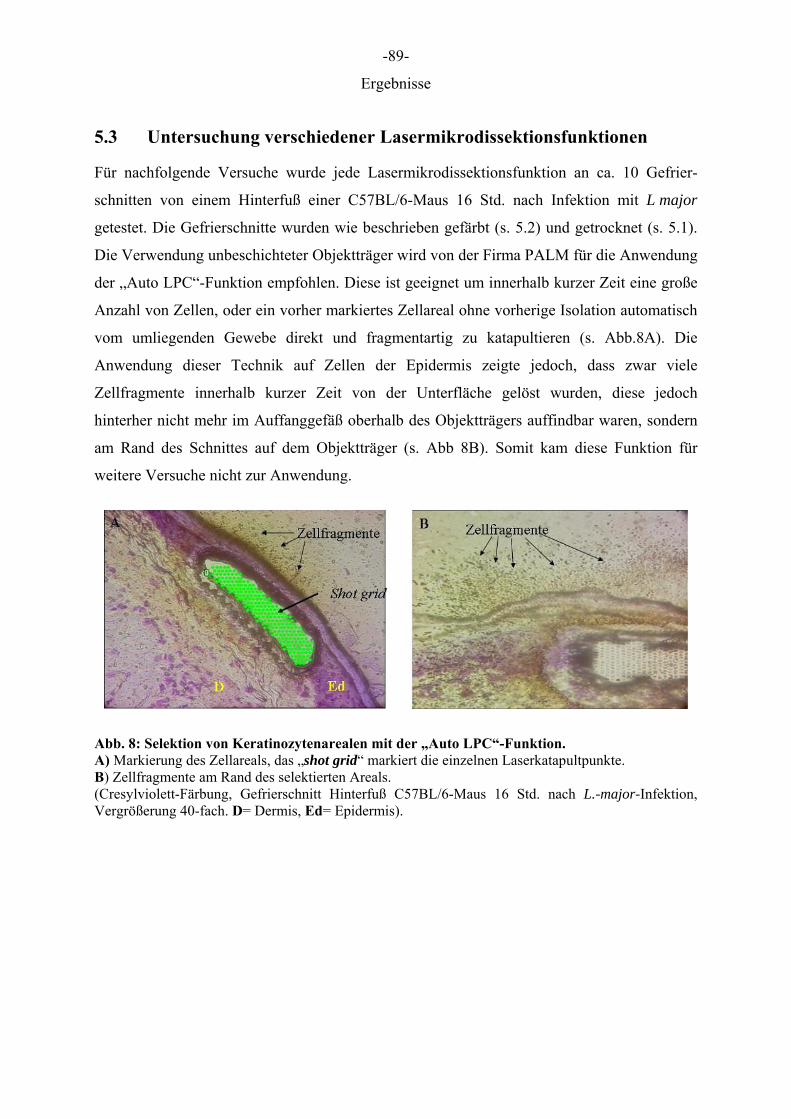
5.1 Optimierung der Präparation von Gefrierschnitten für die Lasermikrodissektion
Die Behandlung der Gefrierschnitte wurde so gewählt, dass zum einen die Epidermis vom
umliegenden Gewebe differenziert werden konnte und zweitens ein zügiges Arbeiten mit dem
Robot Laser Microbeam für eine routinemäßige Anwendung ermöglicht wurde. Da parallel
zur Schichtdicke die erforderliche Energie zur Isolation von Zellen anstieg, aber dennoch
möglichst viel Zellmaterial gewonnen werden sollte, wurde eine Schichtdicke von 12 µm
gewählt. Je höher die aufgewendete Energie desto breiter wurde der Schnittweg des Lasers,
wodurch bei Schnitten oberhalb von 12 µm ein beträchtlicher Anteil des Gewebes zerstört
wurde. Schnitte die mit Farbstoffen auf Ethanol Basis gefärbt wurden und solche, die
anschließend durch Ethanolreihen gewaschen wurden, waren sehr blass gefärbt, wodurch die
Zellen der Epidermis nicht mehr eindeutig identifiziert und differenziert werden konnten. Die
drei Farbstoffe (Toluidinblau 0,1 %, Cresyl Violet 1 %, HE 1 %) erzielten gute
morphologische Ergebnisse, wenn sie in DEPC-A. bidest. gelöst, für wenige Sekunden auf die
Schnitte gegeben und anschließend kurz mit DEPC-A. bidest. gewaschen wurden (Daten
nicht gezeigt). Entscheidend für den Prozess der Lasermikrodissektion war, dass die Schnitte
trocken waren, da jegliche Restfeuchtigkeit den Vorgang behinderte. Waren die Schnitte noch
feucht, wie im Falle einer Lagerung in Ethanol oder einer nicht ausreichenden Trocknung,
kam es zu einer großflächigen Destruktion/Koagulation des Gewebes. Das Schneiden des
Gewebes feuchter Schnitte war nicht möglich und das Katapultieren aufgrund der Haftung am
Objektträger nicht durchführbar. Aus diesen Gründen wurden die Schnitte nach der Färbung
eine halbe Stunde im Trockenschrank bei 37 °Cgetrocknet.
5.2 RNA-Qualität und -Quantität am Beispiel von lasermikrodissezierten Leberparenchymzellen
Da bisher keine Erfahrungswerte über das Verhältnis von RNA-Menge und -Qualität zur
Anzahl lasermikrodissezierter Zellen vorlagen, wurde zunächst ein Gewebe gewählt aus dem
in kurzer Zeit eine große Anzahl von Zellen isoliert werden konnte.
-87-
Ergebnisse
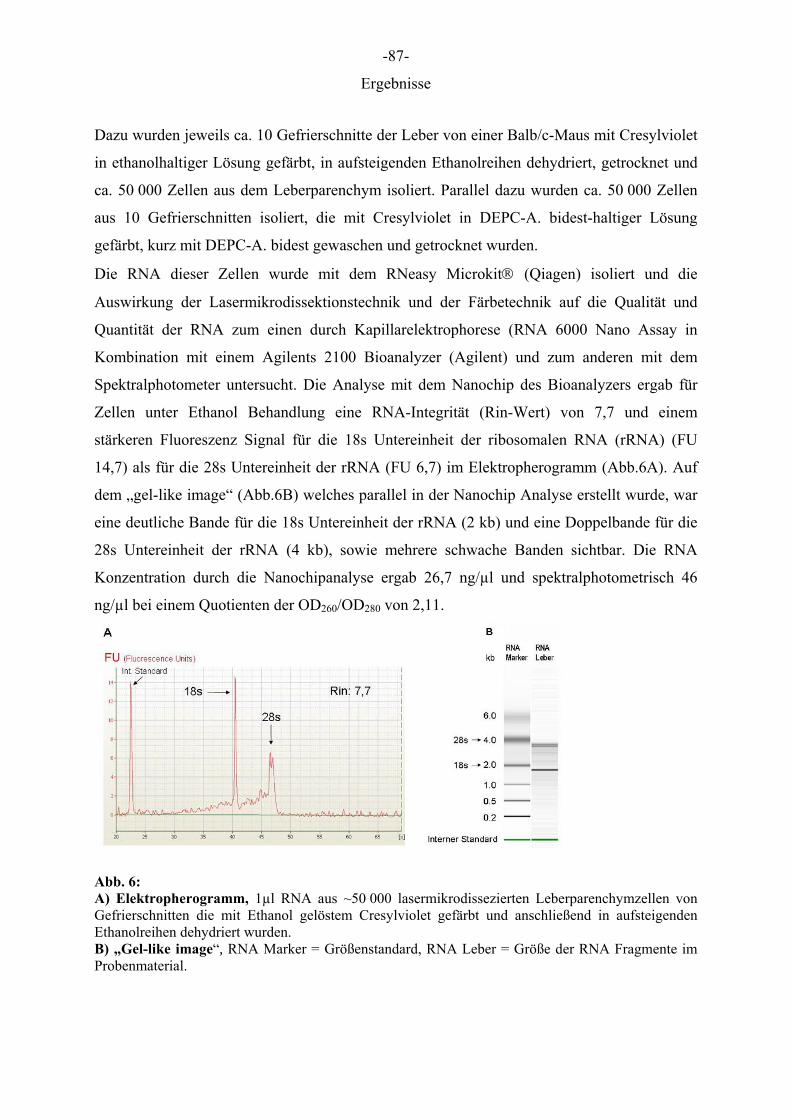
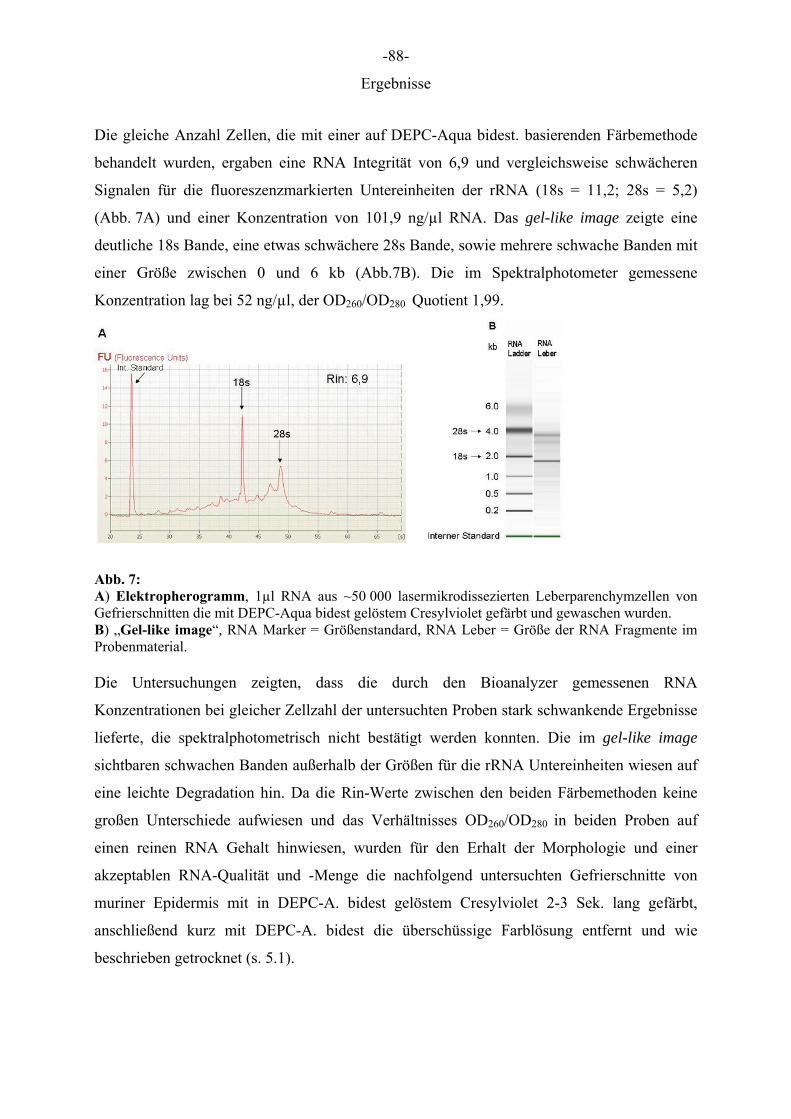
Dazu wurden jeweils ca. 10 Gefrierschnitte der Leber von einer Balb/c-Maus mit Cresylviolet
in ethanolhaltiger Lösung gefärbt, in aufsteigenden Ethanolreihen dehydriert, getrocknet und
ca. 50 000 Zellen aus dem Leberparenchym isoliert. Parallel dazu wurden ca. 50 000 Zellen
aus 10 Gefrierschnitten isoliert, die mit Cresylviolet in DEPC-A. bidest-haltiger Lösung
gefärbt, kurz mit DEPC-A. bidest gewaschen und getrocknet wurden.
Die RNA dieser Zellen wurde mit dem RNeasy Microkit® (Qiagen) isoliert und die
Auswirkung der Lasermikrodissektionstechnik und der Färbetechnik auf die Qualität und
Quantität der RNA zum einen durch Kapillarelektrophorese (RNA 6000 Nano Assay in
Kombination mit einem Agilents 2100 Bioanalyzer (Agilent) und zum anderen mit dem
Spektralphotometer untersucht. Die Analyse mit dem Nanochip des Bioanalyzers ergab für
Zellen unter Ethanol Behandlung eine RNA-Integrität (Rin-Wert) von 7,7 und einem
stärkeren Fluoreszenz Signal für die 18s Untereinheit der ribosomalen RNA (rRNA) (FU
14,7) als für die 28s Untereinheit der rRNA (FU 6,7) im Elektropherogramm (Abb.6A). Auf
dem „gel-like image“ (Abb.6B) welches parallel in der Nanochip Analyse erstellt wurde, war
eine deutliche Bande für die 18s Untereinheit der rRNA (2 kb) und eine Doppelbande für die
28s Untereinheit der rRNA (4 kb), sowie mehrere schwache Banden sichtbar. Die RNA
Konzentration durch die Nanochipanalyse ergab 26,7 ng/µl und spektralphotometrisch 46
ng/µl bei einem Quotienten der OD260/OD280 von 2,11.
Abb. 6: A) Elektropherogramm, 1µl RNA aus ~50 000 lasermikrodissezierten Leberparenchymzellen von Gefrierschnitten die mit Ethanol gelöstem Cresylviolet gefärbt und anschließend in aufsteigenden Ethanolreihen dehydriert wurden. B) „Gel-like image“, RNA Marker = Größenstandard, RNA Leber = Größe der RNA Fragmente im Probenmaterial.
-88-
Ergebnisse
Die gleiche Anzahl Zellen, die mit einer auf DEPC-Aqua bidest. basierenden Färbemethode
behandelt wurden, ergaben eine RNA Integrität von 6,9 und vergleichsweise schwächeren
Signalen für die fluoreszenzmarkierten Untereinheiten der rRNA (18s = 11,2; 28s = 5,2)
(Abb. 7A) und einer Konzentration von 101,9 ng/µl RNA. Das gel-like image zeigte eine
deutliche 18s Bande, eine etwas schwächere 28s Bande, sowie mehrere schwache Banden mit
einer Größe zwischen 0 und 6 kb (Abb.7B). Die im Spektralphotometer gemessene
Konzentration lag bei 52 ng/µl, der OD260/OD280 Quotient 1,99.
Abb. 7: A) Elektropherogramm, 1µl RNA aus ~50 000 lasermikrodissezierten Leberparenchymzellen von Gefrierschnitten die mit DEPC-Aqua bidest gelöstem Cresylviolet gefärbt und gewaschen wurden. B) „Gel-like image“, RNA Marker = Größenstandard, RNA Leber = Größe der RNA Fragmente im Probenmaterial. Die Untersuchungen zeigten, dass die durch den Bioanalyzer gemessenen RNA
Konzentrationen bei gleicher Zellzahl der untersuchten Proben stark schwankende Ergebnisse
lieferte, die spektralphotometrisch nicht bestätigt werden konnten. Die im gel-like image
sichtbaren schwachen Banden außerhalb der Größen für die rRNA Untereinheiten wiesen auf
eine leichte Degradation hin. Da die Rin-Werte zwischen den beiden Färbemethoden keine
großen Unterschiede aufwiesen und das Verhältnisses OD260/OD280 in beiden Proben auf
einen reinen RNA Gehalt hinwiesen, wurden für den Erhalt der Morphologie und einer
akzeptablen RNA-Qualität und -Menge die nachfolgend untersuchten Gefrierschnitte von
muriner Epidermis mit in DEPC-A. bidest gelöstem Cresylviolet 2-3 Sek. lang gefärbt,
anschließend kurz mit DEPC-A. bidest die überschüssige Farblösung entfernt und wie
Für nachfolgende Versuche wurde jede Lasermikrodissektionsfunktion an ca. 10 Gefrier-
schnitten von einem Hinterfuß einer C57BL/6-Maus 16 Std. nach Infektion mit L major
getestet. Die Gefrierschnitte wurden wie beschrieben gefärbt (s. 5.2) und getrocknet (s. 5.1).
Die Verwendung unbeschichteter Objektträger wird von der Firma PALM für die Anwendung
der „Auto LPC“-Funktion empfohlen. Diese ist geeignet um innerhalb kurzer Zeit eine große
Anzahl von Zellen, oder ein vorher markiertes Zellareal ohne vorherige Isolation automatisch
vom umliegenden Gewebe direkt und fragmentartig zu katapultieren (s. Abb.8A). Die
Anwendung dieser Technik auf Zellen der Epidermis zeigte jedoch, dass zwar viele
Zellfragmente innerhalb kurzer Zeit von der Unterfläche gelöst wurden, diese jedoch
hinterher nicht mehr im Auffanggefäß oberhalb des Objektträgers auffindbar waren, sondern
am Rand des Schnittes auf dem Objektträger (s. Abb 8B). Somit kam diese Funktion für
weitere Versuche nicht zur Anwendung.
Abb. 8: Selektion von Keratinozytenarealen mit der „Auto LPC“-Funktion. A) Markierung des Zellareals, das „shot grid“ markiert die einzelnen Laserkatapultpunkte. B) Zellfragmente am Rand des selektierten Areals. (Cresylviolett-Färbung, Gefrierschnitt Hinterfuß C57BL/6-Maus 16 Std. nach L.-major-Infektion, Vergrößerung 40-fach. D= Dermis, Ed= Epidermis).
-90-
Ergebnisse
Die Polyethylennaphtalat (PEN) Beschichtung von Objektträgern der Firma PALM bietet den
Vorteil, dass durch sie das Lasermikrodissektionsmaterial wie auf einem Tablett in das
Auffanggefäß katapultiert wird, wodurch Zellen und Areale von einem bis zu 50 µm
Durchmesser selektiert werden können. An diesen Objektträgern wurden die Funktionen „cut
and catapult“ sowie „Robo LPC“ angewandt. Beide Funktionen arbeiten nach dem gleichen
Prinzip, indem zuerst der interessierende Bereich markiert wird, danach isoliert- und
anschließend katapultiert wird. Der Unterschied liegt in der Automatik der einzelnen Schritte
bei Anwendung der „Robo LPC“-Funktion. Dadurch können die zu interessierenden Zellen
vorher markiert werden und somit nach und nach automatisch isoliert und katapultiert
werden. Dies bietet in der Theorie eine erhebliche Zeit- und Arbeitsersparnis. In der Praxis
war die Funktion allerdings mit erheblichen Schwierigkeiten verbunden. Die vorher gewählte
Energie reichte entweder nicht dafür aus, alle Zellen gleichmäßig zu isolieren, oder aber sie
war für einzelne Bereiche zu hoch, so dass Gewebe entweder nicht durchtrennt oder zerstört
wurde. Da das System nicht eigenmächtig die erforderliche Energie regulieren kann, ist somit
eine manuelle Korrektur nötig und keine Arbeits- und Zeitersparnis gegeben.
Funktion der Wahl war die „cut and catapult“ Funktion mit manueller Aktivierung des
Energietransfers unter Regulation der Energie- und Fokuseinstellung. Mit dieser Funktion
wurden epidermale Zellareale (Abb. 9B,C) sowie einzelne Keratinozyten (Abb. 10A,B,C)
zuerst markiert, dann aus der Epidermis isoliert, katapultiert, im Auffanggefäß erfasst (Abb.
9D und Abb. 10D) und schließlich weiteren Untersuchungen zugänglich gemacht.
-91-
Ergebnisse
Abb. 9: Selektion einzelner Keratinozytenareale mit der „cut and catapult“-Funktion. A) Übersicht, Vergrößerung 40-fach. B) Isolierung des markierten Areals durch „cut“-Funktion, Vergrößerung 40-fach. C) Übersicht nach Aktivierung der „catapult“-Funktion, Vergrößerung 40-fach. D) Katapultierte Keratinozytenareale im Auffanggefäß, Vergrößerung 10-fach. (Gefrierschnitt vom Hinterfuß C57BL/6-Maus 16 Std. nach Infektion mit L. major, Cresylviolet-Färbung, Ed = Epidermis, D = Dermis)
-92-
Ergebnisse
Abb. 10: Selektion einzelner Keratinozyten mit der „cut and catapult“-Funktion. A) Markierung eines einzelnen Keratinozyten in der Epidermis, Vergrößerung 40-fach. B) Isolierung eines Keratinozyten durch „cut and catapult“-Funktion, Vergrößerung 40-fach. C) Isolierung mehrerer Keratinozyten durch „cut and catapult“-Funktion, Vergrößerung 40-fach. D) Keratinozyten im Auffanggefäß, Vergrößerung 10-fach. (Gefrierschnitt vom Hinterfuß C57BL/6-Maus 16 Std. nach Infektion mit L. major, Cresylviolet Färbung, Ed = Epidermis, D = Dermis)
5.4 Auswirkung der RNA-Isolierungsmethode auf die RNA-Ausbeute aus lasermikrodissezierten Keratinozyten
Ziel der Untersuchung war, festzustellen mit welcher der drei getesteten RNA-
Isolationsmethoden (TRIzol Reagent, RNeasy®Microkit, RNeasy®Minikit) die höchste
RNA-Ausbeute aus lasermikrodissezierten Keratinozyten erzielt werden konnte. Dazu wurden
in einem Pilotversuch insgesamt ca. 10 000 Zellen für jede Methode isoliert, indem Zellareale
von ca. 100 Keratinozyten aus der Epidermis von insgesamt 200 Gefrierschnitten von 4
C57BL/6-Mäusen 16 Std. nach Infektion mit L. major per „cut and catapult“-Funktion
selektiert wurden. Die erhaltene RNA Menge lag bei allen drei Methoden unterhalb der
-93-
Ergebnisse
Nachweisgrenze des Spektrophotometers. Ebenso konnten in dem 1 µl der für die Analyse mit
dem RNA 6000 Nano Assay im Bioanalyzer benötigt wird, keine RNA quantifiziert und
qualitativ untersucht werden. Als nächstes wurde versucht, Aufschluss über das
Vorhandensein von RNA innerhalb der Proben zu erhalten, indem die RNA aus den
Isolationsmethoden zunächst in eine Reverse Transkriptionsreaktion eingesetzt wurde und
anschließend in einer PCR-Reaktion mit Intron-spannenden Primern für das „housekeeping“-
Gen Ribosomales Protein L (RPL) und das Zytokin Osteopontin verwendet wurde.
Amplifikate wurden von Proben erhalten, deren RNA mit der TRIzol Methode (Abb. 11,
Ziffer 2 und 5) sowie dem RNeasy®Microkit (Abb. 11, Ziffer 1 und 4) aufgereingt wurden.
Proben aus der RNeasy®Minikit-Isolation (Abb. 11, Ziffer 3 und 6) ergaben keine Produkte
in der PCR, somit war wahrscheinlich auch keine RNA vorhanden, die in cDNA
umgeschrieben werden konnte. Die Bande für RPL und Osteopontin der Probe deren RNA
mit der TRIzol Methode isoliert wurde war wesentlich stärker sichtbar auf dem
Agarose/Etbr-Gel als jene die mit dem RNeasy® Microkit extrahiert wurde.
Abb. 11: Amplifikation von RPL und Osteopontin. Aus je 10 000 Keratinozyten von C57BL/6-Mäusen infiziert mit L. major (16 Std.) wurde die RNA mit verschiedenen Techniken extrahiert, in cDNA umgeschrieben und 5 µl in eine Standard PCR mit Intron-spannenden Primern für RPL und Osteopontin eingesetzt. 10 µl Aliquots wurden auf einem 2% Agarose/Etbr Gel aufgetragen. 1 = RPL RNeasy®Microkit® 4 = Osteopontin RNeasy®Microkit® 2 = RPL TRIzol Reagent® 5 = Osteopontin TRIzol Reagent® 3 = RPL RNeasy®Minikit® 6 = Osteopontin RNeasy®Minikit® Φ = pBR322 DNA/AluI Marker
Bei einem Einsatz von 5 µl cDNA und einer vorhandenen Gesamtmenge von 20 µl cDNA,
entwickelt aus 10 000 Zellen, ist die Zahl der Gene, deren Expression durch PCR analysiert
werden kann, begrenzt. Außerdem ist eine routinemäßige Anwendung bei der Gewinnung
einer derart hohen Zellzahl nicht möglich, vor allem wenn mehrere Proben untersucht werden
-94-
Ergebnisse
sollen. Ziel dieser Arbeit war jedoch, eine umfassende Genanalyse von Zellen der Haut aus
L.-major-infizierten und nicht-infizierten Balb/c-Mäusen mit jenen von C57BL/6-Mäusen
durchzuführen und zu vergleichen. Deshalb wurde zur Vergrößerung der cDNA
Ausgangsmenge bei gleichzeitiger Dezimierung der untersuchten Zellzahl die RNA für alle
weiteren Versuche mit der TRIzol Methode extrahiert und mit dem Microarray Target
Amplifcation Kit (Roche), das im folgenden Kapitel näher erklärt wird, voramplifiziert.
5.5 Quantifizierung der für die „Vor“-Amplifikation benötigten Keratinozyten
In einem weiteren Pilotversuch wurde aus insgesamt 12 Cresylviolet-gefärbten Hinterfuß-
Gefrierschnitten von einer C57BL/6-Maus 16 Std. nach Infektion mit L. major jeweils 1000,
100 und 10 Keratinozyten einzeln lasermikrodisseziert, separat in TRIzol gesammelt, die
RNA gemäß Protokoll isoliert und anschließend voramplifiziert. Diese auf PCR basierende
Methode der „random“ cDNA-Amplifikation ist speziell für die Transkription geringer RNA-
Mengen unter Erhalt der relativen Transkript-Expressions-Profile entwickelt worden. Nach
persönlicher Mitteilung durch Dr. Harutyun Melkonyan, Institut für Experimentelle
Dermatologie in Münster, kann durch die Anwendung dieser Methode eine ca. 100 000 fache
Amplifikation der Ausgangs-mRNA erzielt werden. Dem Protokoll zur Synthese der
Doppelstrang-cDNA folgend wurde anschließend die PCR optimiert. Dazu wurde die Menge
und Größe der Produkte analysiert, indem ab dem 21. Zyklus und nach jedem darauf
folgenden 3. PCR-Zyklus ein Aliquot der Reaktion auf einem 1,2 %igen Agarose/Etbr- Gel
aufgetragen wurde. Die Amplifikation der dscDNA von 10 Zellen ergab selbst nach 36
Zyklen kein Produkt auf dem Agarose-Gel. Erst nach 39 Zyklen zeigten sich Schlieren mit
einer deutlichen Bande bei über 1 kb und mehreren kleineren und größeren, schwächeren
Banden (Abb. 12A). Ein ähnliches Bild ergab sich für die dscDNA aus 100 Keratinozyten.
Hier war nach 39 Zyklen eine starke Bande von über 1 kb und leichte Schlieren unterhalb der
Bande erkennbar (Abb. 12B). Die Amplifikation der TAS-markierten Zweitstrang-cDNA von
1000 Zellen mit dem TAS-Primer ergab gleichmäßig leichte Schlieren bereits nach 21 Zyklen
bei einer Größe von 0,4 – 1,9 kb. Nach 24 Zyklen war dieser bei gleicher Größe etwas stärker
sichtbar und nahm in den darauf folgenden PCR Zyklen an Stärke zu, jedoch wurden kleinere
Produkte überrepräsentiert (Abb. 12C). Die PCR befand sich somit im Einklang mit den
-95-
Ergebnisse
Herstellerangaben des Amplifikationskits, nach 24 Zyklen in der Phase der exponentiellen
Amplifikation. Diese Zyklenzahl fand in der darauf folgenden, zweiten „random“ PCR der
Doppelstrang-cDNA aus ca. 1000 Keratinozyten Anwendung um das Ausgangsmaterial für
nachfolgende Real-Time-PCR Analysen herzustellen.
Φ
908 bp
100 bp
3472 bp 2690 bp
421 bp
1882 bp
A
21 24 27 30 33 36 39 21 24 27 30 33 36 39
B
λ 21 24 27 30 33 36
C
Abb.12: Optimierung der PCR, TAS Produkte von 10, 100 und 1000 Keratinozyten. 10 µl Reaktionsprodukt der Zyklen 21/24/27/30/33/36/39 wurden auf einem 1,2% Agarose/Etbr Gel aufgetragen. A = 10 Keratinozyten Φ = pBR322 DNA/AluI Marker B = 100 Keratinozyten λ = Lambda DNA/Eco130I (StyI) Marker C = 1000 Keratinozyten
Um den Erfolg der „Vor“-Amplifikation noch einmal zu überprüfen, wurde das Produkt aus
der zweiten „random“ PCR 1:100 mit DEPC H2O verdünnt und in einer PCR mit GAPDH-
und Osteopontin-Primern eingesetzt. Mit beiden Primern konnte ein Produkt amplifiziert
werden, wobei jenes für GAPDH (Abb. 13, Ziffer 1) mehr als 5-mal so stark vorhanden war
als das Produkt für Osteopontin (Abb. 13, Ziffer 2). Die Ergebnisse bestätigen, dass die
Methode der „Vor“-Amplifikation zur Erhöhung geringer RNA Ausgangsmengen unter
Wahrung der Transkript-Expressions-Profile geeignet ist.
-96-
Ergebnisse
10 µl Aliquots wurden auf ein 2% Agarose/Etbr Gel aufgetragen.
(Abb.14-32 A, Versuch I und B, Versuch II). Für jeden Lasermikrodissektionsversuch wurden
3 C57BL/6-Mäuse und 3 Balb/c-Mäuse verwendet (insgesamt 12 Mäuse für 2 durchgeführte
Versuche). Pro Versuch wurden ca. 50 Gefrierschnitte von den 3 infizierten Hinterfüßen
(linke Hinterfüße der Mäuse, s. 4.2.11), sowie ca. 50 Gefrierschnitte von den 3 Kontroll-
Hinterfüßen (rechte Hinterfüße der Mäuse, s. 4.2.11) beider Mausstämme verwendet. Von den
infizierten Balb/c-und C57BL/6-Maus-Hinterfußgefrierschnitten sowie von den Balb/c- und
C57BL/6-Maus-Kontroll-Hinterfußgefrierschnitten wurden jeweils 1000 Keratinozyten durch
die „cut and catapult“-Funktion isoliert und wie beschrieben weiter verarbeitet (s. 4.4.2, 4.4.9
und 5.1-5.4).
Parallel dazu werden die Ergebnisse der Real-Time-PCR von mindestens drei Versuchen zur
Immunregulation in der Gesamthaut 16 Std. nach Infektion mit L. major dargestellt. Für
ausgewählte Gene (I-TAC, Osteopontin, IL-4 und IFN-γ), wurde zusätzlich die Expression
aus mindestens drei unabhängigen Versuchen im Verlauf der Infektion über einen Zeitraum
von einer Stunde bis zu drei Tagen dokumentiert. Für jeden untersuchten Zeitwert (1 Std., 3
Std., 6 Std., 8 Std., 16 Std., 21 Std., 48 Std. und 72 Std. nach Infektion mit L. major) wurden
Proben von der Fußsohlenhaut von 2 C57BL/6-Mäusen und 2 Balb/c-Mäusen verwendet.
Dabei wurden auch hier die linken Hinterfüße infiziert und die rechten dienten als Kontrolle.
Dies sind insgesamt 12 Mäuse für jeden der 8 Zeitwerte und für 3 unabhängig voneinander
durchgeführte Versuche, insgesamt also 96 Mäuse. Die Ergebnisse der Real-Time-PCR aus
Gesamthautproben beziehen sich somit auf den Mittelwert der 3 unabhängig voneinander
durchgeführten Versuche. Die Angaben des p-values markiert durch ein bis drei Asterixe
direkt oberhalb der Fehlerbalken (Standardabweichung) beziehen sich dabei auf die
Signifikanz der Expression des Gens innerhalb eines Mausstammes (Kontrolle/infiziert) und
die Asterixe oberhalb der Linienverbindungen beziehen sich auf die Signifikanz der
differentiellen Expression zwischen den beiden Mausstämmen.
-98-
Ergebnisse
5.6.1 Nachweis von Osteopontin
Abb. 14: Expression von Osteopontin-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von Osteopontin in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von Osteopontin in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von Osteopontin in der Gesamthaut 16 Std. p.i. D) Expression von Osteopontin in der Gesamthaut während der ersten drei Tage p.i., * p < 0,05, ** p < 0,01, *** p < 0,001. In sämtlichem Probenmaterial war zu jedem untersuchten Zeitpunkt nach der Infektion mit
L. major mehr Osteopontin cDNA in C57BL/6-Mäusen vorhanden als in Balb/c-Mäusen. Die
Expression war in den Keratinozyten 16 Std. nach der Infektion durchschnittlich 24-fach
stärker (Abb.14, A+B) und ca. 29-fach höher in der Gesamthaut von C57BL/6-Mäusen
(Abb.14, C). Im Zeitverlauf wurde in der Gesamthaut von C57BL/6-Mäusen eine differentiell
stärkere Expression als in Balb/c-Mäusen bereits nach 6 Std. beobachtet, die im Verlauf des
ersten Tages noch weiter zunahm und ab dem zweiten Tag zurückging (Abb.14, D)
-99-
Ergebnisse
5.6.2 Nachweis von IL-1β
Abb. 15: Expression von IL-1β-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von IL-1β in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von IL-1β in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, C) Expression von IL-1β in der Gesamthaut 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
In beiden Lasermikrodissektionsversuchen und in der Gesamthaut konnte eine
infektionsbedingte Steigerung der IL-1β-Expression in beiden Mausstämmen beobachtet
werden, die jedoch in C57BL/6-Mäusen um ein vielfaches höher war als in Balb/c-Mäusen.
Während die IL-1β-Expression in den Keratinozyten von C57BL/6-Mäusen um das 110-
(Abb.15A) und 58-fache (Abb.15B) 16 Std. nach der Infektion anstieg, zeigte sich eine
vergleichweise geringe Expression um das 4-fache in Keratinozyten von Balb/c-Mäusen
(Abb.15A,B). Parallel dazu wurde eine signifikant gesteigerte Transkription von IL-1β in der
Gesamthaut von beiden Mausstämmen beobachtet, die hier differentiell um mehr als das 10-
fache stärker in C57BL/6- als in Balb/c-Mäusen vorhanden war (Abb.15C).
-100-
Ergebnisse
5.6.3 Nachweis von IL-4
Abb. 16: Expression von IL-4-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von IL-4 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von IL-4 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von IL-4 in der Gesamthaut 16 Std. p.i. D) Expression von IL-4 in der Gesamthaut während der ersten drei Tage p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
16 Std. nach der Infektion wurde in der Gesamthaut eine differentielle Expression von IL-4
(Abb.16C) und höhere IL-4-Transkriptspiegel im Lasermikrodissektionsmaterial
(Abb.16A,B) von C57BL/6-Mäusen nachgewiesen als in entsprechendem Material von
Balb/c-Mäusen. Während des Infektionsverlaufs (Abb.16D) stieg die Expression in den ersten
6 Std. der Infektion in C57BL/6-Mäusen im Vergleich zu Balb/c-Mäusen stark an und ging
danach langsam zurück, allerdings zeigte sich auch nach 21 Std. noch ein signifikanter
Unterschied mit einer stärkeren IL-4-Expression in der Gesamthaut von C57BL/6-Mäusen.
-101-
Ergebnisse
Erst am dritten Tag der Infektion fand eine Umkehr dieses Verhältnis statt, erkennbar an einer
signifikanten IL-4-Expression in den Balb/c-Mäusen.
5.6.4 Nachweis von IL-10
Abb. 17: Expression von IL-10-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von IL-10 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte nicht messbar) B) Expression von IL-10 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte nicht messbar) C) Expression von IL-10 in der Gesamthaut 16 Std. p.i. , Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Insgesamt war die Menge an IL-10-cDNA in den Keratinozyten infizierter Versuchstiere im
Vergleich zu anderen untersuchten Genen, sehr gering. Sie war in Zellen von Balb/c-Mäusen
mit 1,7 (Abb.17A) und 2,5 Amplifikaten (Abb.17B) pro 10 000 Amplifikate GAPDH/RPL
etwas höher als in C57BL/6-Mäusen (1,4 bzw. 0,7 Amplifikate pro 10 000 Amplifikate
-102-
Ergebnisse
GAPDH/RPL). In der Gesamthaut (Abb.17C) stellte sich eine ca. 14–fach stärkere
differentielle Expression von IL-10 in infizierten Balb/c-Mäusen im Vergleich zu C57BL/6-
Mäusen dar. In beiden Kontroll-Gesamthautproben lag die Zahl der Amplifikate unter 0,07
Amplifikate pro 10 000 Amplifikaten GAPDH/RPL.
5.6.5 Nachweis von IL-12
Abb. 18: Expression von IL-12-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A = Expression von IL-12 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B = Expression von IL-12 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C = Expression von IL-12 in der Gesamthaut 16 Std. p.i. * p < 0,05, ** p < 0,01, *** p < 0,001. Die Ergebnisse zeigen eine starke Expression von IL-12 in Keratinozyten aus infiziertem
Probenmaterial von C57BL/6-Mäusen. Während in den Keratinozyten von Balb/c-Mäusen 16
Std. nach Infektion im ersten Versuch (Abb.18A) eine Verminderung der IL-12-cDNA um
den Faktor 0,8 und im zweiten Versuch (Abb.18B) kein Unterschied zum nicht-infizierten
Kontrollmaterial festgestellt wurde, schien in der Gesamthaut jedoch eine Regulation
-103-
Ergebnisse
stattzufinden (Abb.18C). Hier lag die Expression mit n-fold = 18 deutlich über dem Kontroll-
wert, jedoch bestand mit einem n-fold von 90 eine stärkere differentielle Expression in der
Gesamthaut von C57BL/6-Mäusen. Ähnlich war die Transkription in den Keratinozyten
infizierter C57BL/6-Mäuse durchschnittlich 73-fach höher als in Balb/c-Mäusen (Abb.18C).
5.6.6 Nachweis von IL-15
Abb. 19: Expression von IL-15-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von IL-15 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) B) Expression von IL-15 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) C) Expression von IL-15 in der Gesamthaut 16 Std. p.i., Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001. Im Vergleich zur Gesamthaut wurde in Keratinozyten mehr IL-15-cDNA in C57BL/6-
Mäusen detektiert als in Balb/c-Mäusen, allerdings waren in beiden Versuchen mit 0,04 bis
0,6 Amplifikaten (Balb/c-Mäuse) und 0,2 bis einem Amplifikat (C57BL/6-Mäuse) pro 10 000
-104-
Ergebnisse
Amplifikate GAPDH/RPL nur wenig Transkription nachweisbar (Abb.19A,B). Während
weniger IL-15-cDNA in Keratinozyten von Balb/c-Mäusen nachgewiesen wurde, schien in
der infizierten Gesamthaut kein signifikanter Unterschied zwischen den beiden Mausstämmen
zu bestehen, jedoch war die IL-15-Expression innerhalb der Mausstämme signifikant
verstärkt gegenüber der nicht-infizierten Gesamthautkontrolle (Abb.19C).
5.6.7 Nachweis von IL-18
Abb. 20: Expression von IL-18-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von IL-18 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte und Balb/c (infiziert) nicht detektierbar)) B) Expression von IL-18 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte und Balb/c (infiziert) nicht detektierbar)) C) Expression von IL-18 in der Gesamthaut 16 Std. p.i. , Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., n.d. = nicht detektierbar, * p < 0,05, ** p < 0,01, *** p < 0,001. IL-18-cDNA war in den Keratinozyten infizierter Balb/c-Mäuse nicht nachweisbar, in
C57BL/6-Mäusen lag die Expression im Bereich der Nachweisgrenze (durchschnittlich 0,08
-105-
Ergebnisse
Amplifikate pro 10 000 Amplifikate GAPDH/RPL) (Abb.20A,B). Im Gesamthautmaterial
(Abb.20C) bestand kein deutlicher, differentieller Unterschied zwischen den beiden
Mausstämmen und auch nicht zwischen den infizierten und nicht-infizierten Versuchtieren.
Allerdings fiel auf, dass in der Gesamthaut infizierter C57BL/6-Mäuse eine große Varianz in
der Anzahl der Amplifikate zwischen den drei Versuche bestand.
5.6.8 Nachweis von IFN-γ
Abb. 21: Expression von IFN-γ-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von IFN-γ in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von IFN-γ in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von IFN-γ in der Gesamthaut 16 Std. p.i. D) Expression von IFN-γ in der Gesamthaut während der ersten drei Tage p.i., Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
-106-
Ergebnisse
In den Keratinozyten infizierter Balb/c-Mäuse wurden 3-mal (Abb.21A) bzw. 2-mal
(Abb.21B) höhere IFN-γ-Transkriptspiegel gemessen als in den Keratinozyten von infizierten
C57BL/6-Mäusen. Allerdings waren die Anzahl der Amplifikate im Vergleich zu der Menge,
die später im Verlauf der Infektion gemessen wurde, in Keratinozyten und Gesamthaut 16
Std. nach der Infektion sehr gering. Die nicht infizierten Kontrollen zeigten keine IFN-γ-
Expression. In der Gesamthaut (Abb.21C) waren zudem 16 Std. nach der Infektion keine
deutlichen Unterschiede in der Anzahl der IFN-γ-Amplifikate zwischen den beiden
Mausstämmen zu erkennen, jedoch zeigte sich eine deutliche Hochregulation im Vergleich zu
den entsprechenden Kontrollwerten. Eine deutliche Erhöhung der Amplifikate fand erst nach
dem ersten Tag statt (Abb.21,D). In der Gesamthaut infizierter Balb/c-Mäuse nahm diese im
Vergleich zu C57BL/6- Mäusen ab dem zweiten Tag um das 3-fache zu und stieg am dritten
Tag um das 6-fache der IFN-γ-Transkription in Proben infizierter C57BL/6-Mäuse.
-107-
Ergebnisse
5.6.9 Nachweis von TNF-α
Abb. 22: Expression von TNF-α-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von TNF-α in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von TNF-α in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von TNF-α in der Gesamthaut 16 Std. p.i. ,* p < 0,05, ** p < 0,01, *** p < 0,001.
Während in der Gesamthaut von C57BL/6-Mäusen eine signifikante ca. 90-fache Steigerung
der TNF-α-Transkription gemessen wurde (Abb.22C), lag diese in den Keratinozyten um das
18- (Abb.22A) und 12- fache (Abb.22B) höher als in Balb/c-Mäusen. Im Gegensatz zur
Gesamthaut infizierter Balb/c-Mäuse wo eine 17-fache Hochregulation der Transkription
stattfand, wurde TNF-α in den Keratinozyten aus Balb/c-Mäusen im ersten Versuch nicht
stärker exprimiert als in der Kontrolle (n-fold = 1,3; Versuch I) und im zweiten Versuch
herunterreguliert (n-fold = 0,5; Versuch II).
-108-
Ergebnisse
5.6.10 Nachweis von TGF-β
Abb. 23: Expression von TGF-β-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von TGF-β in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) B) Expression von TGF-β in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) C) Expression von TGF-β in der Gesamthaut 16 Std. p.i., Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Das Zytokin TGF-β konnte zwar in Keratinozyten nachgewiesen werden, jedoch war die Zahl
der Amplifikate, wenn auch um etwa das drei- (Abb.23A) und 2-fache (Abb.23B) erhöht in
infizierten C57BL/6-Mäusen im Vergleich zu infizierten Balb/c-Mäusen, sehr gering. So
wurden 0,4 und 1,7 Amplifikate pro 10 000 Amplifikate GAPDH/RPL in den Keratinozyten
infizierter C57BL/6-Mäuse und 49 Amplifikate in entsprechenden Gesamthautproben
-109-
Ergebnisse
gemessen. In der Gesamthaut hingegen waren die Stammesunterschiede in der Expression
von TGF-β nicht so stark wie in den Keratinozyten (Abb.23C).
5.6.11 Nachweis von S100A8/MRP-8
Abb. 24: Expression von MRP-8-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von MRP-8 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von MRP-8 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von MRP-8 in der Gesamthaut 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Die cDNA des proinflammatorischen Moleküls myeloid-related-protein-8 (MRP-8, S100
calcium binding Protein A8, S100A8) wurde in allen untersuchten Proben in größerem
Umfang in infizierten C57BL/6-Mäusen vorgefunden. So lag die durchschnittliche Zunahme
in den Keratinozyten (Abb.24A,B) und in der Gesamthaut (Abb.24C) infizierter C57BL/6-
Mäuse um das 90 fache gegenüber der Kontrolle zu und war somit um ein vielfaches höher
als die vergleichsweise niedrige Expression in entsprechenden Proben infizierter Balb/c-
Abb. 25: Expression von MRP-14-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von MRP-14 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von MRP-14 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von MRP-14 in der Gesamthaut 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Im Vergleich mit MRP-8 wurde eine ähnliche Verteilung der MRP-14-cDNA festgestellt. In
der Gesamthaut wurde MRP-14 zudem differentiell in den beiden Mausstämmen exprimiert
(Abb.25C). Zusätzlich zeigte sich eine stärkere Hochregulation in den Keratinozyten
infizierter Balb/c-Mäuse besonders im Versuch I als von MRP-8 in entsprechenden Proben
(Abb.25A).
-111-
Ergebnisse
5.6.13 Nachweis von MCP-1/ CCL1
Abb. 26: Expression von MCP-1-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von MCP-1 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) B) Expression von MCP-1 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) C) Expression von MCP-1 in der Gesamthaut 16 Std. p.i., Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001. In den Keratinozyten infizierter C57BL/6-Mäuse wurden zum einen mit 3,5
Amplifikaten/10 000 Amplifikaten GAPDH/RPL (Abb.26A) 40-mal mehr Amplifikate als in
entsprechenden Proben von Balb/c-Mäusen gemessen und zum anderen mit 2 Amplifikaten
(Abb.26B) ca 3-mal so viele Amplifikate gemessen wie in den Keratinozyten infizierter
Balb/c-Mäuse. In der Gesamthaut wurde MCP-1 mit durchschnittlich 350 Amplifikaten pro
10 000 Amplifikate GAPDH/RPL in infizierten C57BL/6-Mäusen differentiell höher
exprimiert. In den Keratinozyten der Kontrollprobe waren keine MCP-1-Transkripte zu
-112-
Ergebnisse
detektieren und in der Gesamthautkontrolle war im Vergleich zu den infizierten Proben
deutlich weniger Amplifikate (ca. 4) nachweisbar (Abb.26C).
5.6.14 Nachweis von MIP-1α/ CCL3
Abb. 27: Expression von MIP-1α-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von MIP-1α in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) B) Expression von MIP-1α in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte nicht detektierbar) C) Expression von MIP-1α in der Gesamthaut 16 Std. p.i. , Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Ähnlich wie MCP-1 wurde auch MIP-1α mit 3 (Abb.27A) und 18 (Abb.27B)
Amplifikaten/10 000 Amplifikaten GAPDH/RPL stärker in den Keratinozyten von C57BL/6-
Mäusen exprimiert als in Balb/c-Mäusen. Eine infektionsbedingte signifikante Steigerung der
MIP-1α Transkription zeigte sich zudem auch in der Gesamthaut innerhalb sowie zwischen
-113-
Ergebnisse
den Mausstämmen, die auch hier mit 30 Amplifikaten gegenüber 11 stärker in den C57BL/6-
Mäusen als in den Balb/c-Mäusen vorhanden war. In nicht-infiziertem Material der
Gesamthaut waren kaum Amplifikate vorhanden (Abb.27C).
5.6.15 Nachweis von MIP-1β/ CCL4
Abb. 28: Expression von MIP-1β-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von MIP-1β in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion (Kontrollwerte nicht detektierbar) B) Expression von MIP-1β in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion (Kontrollwerte nicht detektierbar) C) Expression von MIP-1β in der Gesamthaut 16 Std. p.i., Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
MIP-1ß wurde verstärkt im Gewebe infizierter C57BL/6-Mäuse exprimiert. Während die
Expression in den Keratinozyten von C57BL/6-Mäusen mit durchschnittlich 1,4
Amplifikaten/10 000 Amplifikaten GAPDH/RPL ca 10-fach stärker war als in Balb/c-Mäusen
-114-
Ergebnisse
(Abb.28A,B), waren in der Gesamthaut infizierter C57BL/6-Mäuse mit durchschnittlich 230
Amplifikaten/10 000 Amplifikate GAPDH/RPL ca. 8-mal mehr Transkripte vorhanden
(Abb.28C). Zudem konnte eine differentielle Regulation von MIP-1β in der Gesamthaut
festgestellt werden. In den Gesamthautkontrollen waren keine MIP-1β-Transkripte messbar.
5.6.16 Nachweis von RANTES/ CCL5
Abb. 29: Expression von RANTES -mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht infizierte Tiere) wurde gleich 1 gesetzt. A = Expression von RANTES in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B = Expression von RANTES in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C = Expression von RANTES in der Gesamthaut 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Die Expression des Chemokins RANTES war stärker in infizierten C57BL/6- als in
entsprechenden Balb/c-Maus-Proben. Während RANTES in den Keratinozyten infizierter
C57BL/6-Mäuse mit einem n-fold von durchschnittlich 53 hochreguliert wurde, fand in den
Balb/c-Mäusen eine Herunterregulation um das 0,5-fache statt (Abb.29A,B). In der
Gesamthaut wurde bei den infizierten C57BL/6-Mäusen ein n-fold von 190 gemessen, womit
-115-
Ergebnisse
die Expression um das 4-fache im Vergleich zu den Balb/c-Maus-Transkripten erhöht war.
RANTES wurde in der Gesamthaut infizierter Balb/c-Mäuse zwar geringer als in jener von
C57BL/6-Mäusen aber dennoch ebenso signifikant exprimiert (Abb.29C).
5.6.17 Nachweis von MCP-3/ CCL7
Abb. 30: Expression von MCP-3-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. A) Expression von MCP-3 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion, (Kontrollwerte und Balb/c (infiziert) nicht detektierbar) B) Expression von MCP-3 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion, (Kontrollwerte und Balb/c (infiziert) nicht detektierbar) C) Expression von MCP-3 in der Gesamthaut 16 Std. p.i., Balb/c K u. C57BL/6 K = Kontrolle (nicht-infiziert), Balb/c Lm u. C57BL/6 Lm = 16 Std. p.i., n.d. = nicht detektierbar, * p < 0,05, ** p < 0,01, *** p < 0,001.
Eine infektionsbedingte Hochregulation des Chemokins MCP-3 wurde für sämtliches
Probenmaterial infizierter C57BL/6-Mäuse festgestellt. So zeigte sich eine signifikante
Steigerung der MCP-3 Transkripte um das 370-fache gegenüber der Kontrolle in der
Gesamthaut infizierter C57BL/6-Mäuse, die auch signifikant im Vergleich zur Balb/c-Maus-
-116-
Ergebnisse
Expression 18-mal höher war (Abb.30C). Während im Lasermikrodissektionsmaterial kein
MCP-3 in Balb/c-Mäusen nachgewiesen wurde, waren 0,12 (Abb.30A) und 0,08
Amplifikate/10 000 Amplifikate GAPDH/RPL (Abb.30B) in infizierten C57BL/6-Mäusen
vorhanden.
5.6.18 Nachweis von IP-10/ CXCL10
Abb. 31: Expression von IP-10-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von IP-10 in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von IP-10 in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von IP-10 in der Gesamthaut 16 Std. p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Ein weiteres Chemokin das stärker im Probenmaterial infizierter C57BL/6-Mäuse exprimiert
wurde, war IP-10. Eine 40-60-fache Hochregulation konnte in den Keratinozyten infizierter
C57BL/6-Mäuse im Vergleich zu einer 1-2-fachen Hochregulation in infizierten Balb/c-
Mäusen nachgewiesen werden (Abb.31A,B). In der Gesamthaut wurde IP-10 signifikant von
-117-
Ergebnisse
beiden Mausstämmen exprimiert, jedoch zeigte sich eine differentiell stärkere Expression in
C57BL/6-Mäusen als in Balb/c-Mäusen (Abb.31C).
5.6.19 Nachweis von I-TAC/ CXCL11
Abb. 32: Expression von I-TAC-mRNA in Keratinozyten und Gesamthaut nach Infektion zweier Mausstämme mit L. major. Die mRNA-Expression wurde mittels quantitativer Real-Time-PCR bestimmt. Die Expression in der Kontrolle (nicht-infizierte Tiere) wurde gleich 1 gesetzt. A) Expression von I-TAC in Keratinozyten 16 Std. p.i., Versuch I der Lasermikrodissektion B) Expression von I-TAC in Keratinozyten 16 Std. p.i., Versuch II der Lasermikrodissektion C) Expression von I-TAC in der Gesamthaut 16 Std. p.i. D) Expression von I-TAC in der Gesamthaut während der ersten drei Tage p.i., * p < 0,05, ** p < 0,01, *** p < 0,001.
Das Chemokin I-TAC wurde in lasermikrodissezierten Keratinozyten sowie in der
Gesamthaut infizierter Balb/c-Mäuse stärker exprimiert als in entsprechendem Probenmaterial
von C57BL/6-Mäusen. I-TAC wurde in den Keratinozyten infizierter Balb/c-Mäuse um das
10 bis 20-fache stärker exprimiert als in entsprechenden C57BL/6-Maus-Proben
(Abb.32A,B). 16 Std. nach der Infektion wurde eine signifikante Expression in der
-118-
Ergebnisse
Gesamthaut von Balb/c-Mäusen nachgewiesen, die mit einem n-fold von 90 neun-mal höher
lag als in der Gesamthaut infizierter C57BL/6-Mäuse (Abb.32C). Bereits nach 6 Std. war in
der Gesamthaut infizierter Balb/c-Mäuse ein signifikanter Anstieg der Expression mit einem
n-fold von 10 feststellbar. Im Verlauf des ersten Tages nahm diese um das 25-fache weiter zu
und nahm in den darauf folgenden zwei Tagen nur geringfügig ab. In der infizierten
Gesamthaut von C57BL/6-Mäusen stieg die Expression von I-TAC nie über einen n-fold von
7 hinaus. Eine signifikante Hochregulation des I-TAC-Transkriptspiegels auf einen n-fold von
6,8 konnte in C57BL/6-Mäusen nur am zweiten Tag der Infektion beobachtet werden
(Abb.32D).
Da bisher keine Studien vorliegen, die sich mit I-TAC im Rahmen einer
Leishmanieninfektion befassen und die Ergebnisse meiner Untersuchungen besonders
auffielen, bezogen sich weitere Untersuchungen insbesondere auf dieses Gen
5.7 Bestätigung der mRNA-Expression von Osteopontin und I–TAC in der Epidermis durch ISH
Es wurden insgesamt für beide untersuchten Gene 8 Mäuse untersucht (2 Balb/c- und 2
C57BL/6-Mäuse pro Gen). Von den infizierten Hinterfüßen sowie von den
Kontrollhinterfüßen (s. 4.2.11) wurden jeweils 50 Gefrierschnitte angefertigt. Für die
Untersuchung der mRNA-Expression auf zellulärer Ebene in situ wurden Osteopontin- und
I-TAC-RNA-Sonden hergestellt und mit Hinterfußgefrierschnitten von Balb/c- und C57BL/6-
Mäusen, die 8-16 Std. nach der Infektion mit L. major gewonnen wurden, hybridisiert.
Gleichzeitig wurden entsprechende Schnitte von nicht infizierten Kontrolltieren in situ-
hybridisiert. Das positive Hybridisierungssignal des Nitrobluetetrazolium/5-Brom-4-Chlor-3-
Indolyl- Phosphat (NBT/BCIP bzw. X-Phosphat) –Detektionssystems in der ISH war hell- bis
dunkelbraun. In Abhängigkeit von der verwendeten Sonde wurde eine unterschiedliche
Intensität und Verteilung des Signals in den mit L.-major-infizierten Hinterfüßen von Balb/c-
und C57BL/6-Mäusen gefunden. Als Negativkontrollen dienten neben den spezifischen
„sense“-Sonden, die nicht infizierten Kontrolltieren. Diese erbrachten, ebenfalls in
Abhängigkeit von der verwendeten Sonde, entweder kein oder im Vergleich zur „antisense“-
Sonde ein deutlich reduziertes Signal.
-119-
Ergebnisse
5.7.1 Nachweis der Osteopontin-mRNA
Die Proben aller infizierten Mäuse zeigten bereits 8 Std. nach der Infektion mit L. major bei
Hybridisierung mit der DIG-markierten „antisense“-Sonde deutliche zellassoziierte,
zytoplasmatische Reaktionen in der Epidermis (Ed) (Abb.33A,B und Abb.34A,B). Das
bräunliche Signal der „antisense“-Sonde zum Nachweis der Osteopontin–mRNA war
besonders stark in den Zellen des Stratum spinosum (Stsp), welches zusammen mit dem
Stratum basale (Stb) die epidermale Regenerationsschicht bildet (Abb.33B, Abb.34B). Die
Signale beschränkten sich bei den Balb/c-Mäusen auf diese Region (Abb.33B), während die
infizierten C57BL/6-Mäuse Osteopontin zusätzlich in den oberen Schichten der Epidermis
exprimierten (Abb.34B). Die Signale waren außerdem in letzterem Mausstamm stärker in
ihrer Farbintensität. Während die nicht infizierten Balb/c-Kontrollen eine ähnliche Anzahl
positiver Zellen in der Regenerationsschicht aufwiesen (Abb.33C), war die Signalgebung in
den C57BL/6-Maus-Kontrollschnitten wesentlich schwächer ausgeprägt (Abb.34C). Mit der
„sense“-Sonde waren keine Hybridisierungssignale nachweisbar (Abb.33D, Abb.34D). Die
entsprechenden Abbildungen befinden sich auf der folgenden Seite.
-120-
Ergebnisse
Abb. 33: ISH zum Nachweis der Osteopontin-mRNA-Expression in der Haut von Balb/c-Mäusen nach Infektion mit L. major. A) Balb/c-Mäuse 8 Std.p.i., „antisense“-Sonde; Vergrößerung 10-fach B) Balb/c-Mäuse 8 Std. p.i., „antisense“-Sonde, Vergrößerung 40-fach C) Balb/c-Mäuse nicht-infizierte Kontrolle, „antisense“-Sonde, Vergrößerung 40-fach D) Balb/c-Mäuse 8 Std. p.i., „sense“-Sonde, Vergrößerung 20-fach (Ed = Epidermis, D = Dermis, Sc = Subcutis, Stc = Stratum corneum, Stsp = Stratum spinosum, Stb = Stratum basale, iZ = infiltrierende Zellen)
-121-
Ergebnisse
Abb. 34: ISH zum Nachweis der Osteopontin-mRNA-Expression in der Haut von C57/BL6-Mäusen nach Infektion mit L. major. A) C57BL/6-Mäuse 8 Std. p.i., „antisense“-Sonde; Vergrößerung 10-fach B) C57BL/6-Mäuse 8 Std. p.i., „antisense“-Sonde, Vergrößerung 40-fach C) C57BL/6-Mäuse nicht-infizierte Kontrolle, „antisense“-Sonde, Vergrößerung 20-fach D) C57BL/6-Mäuse 8 Std. p.i., „sense“-Sonde, Vergrößerung 20-fach (Ed = Epidermis, D = Dermis, Sc = Subcutis, Stc = Stratum corneum, Stsp = Stratum spinosum, Stb = Stratum basale, iZ = infiltrierende Zellen)
5.7.2 Nachweis der I-TAC-mRNA
Bei Verwendung der „antisense“-Sonde zum Nachweis der I-TAC-mRNA wurde in beiden
Mausstämmen ein sehr deutliches dunkelbraunes Signal festgestellt, welches 16 Std. nach
Infektion zytoplasmatisch und hauptsächlich in der Epidermis lokalisiert war (Abb.35A,B und
Abb.36A,B). Besonders die Zellen im Stsp waren stärker gefärbt als die Zellen in den übrigen
Schichten der Epidermis (Abb.35B, Abb.36B). Die infiltrierenden Zellen waren schwarz
gefärbt (Abb.35A, Abb.36A). In den Kontrollen war eine schwächere Färbung zu beobachten,
-122-
Ergebnisse
welches von der Lokalisation des Signals der infizierten Probe entsprach (Abb. 35C, Abb.
36C). Eine Hybridisierung mit der „sense“-Sonde ergab in keinem der untersuchten Schnitte
ein Signal (Abb. 35D, Abb. 36D).
Abb. 35: ISH zum Nachweis der I-TAC-mRNA-Expression in der Haut von Balb/c-Mäusen nach Infektion mit L. major. A) Balb/c-Mäuse 16 Std. p.i., „antisense“-Sonde; Vergrößerung 20-fach B) Balb/c-Mäuse 16 Std. p.i., „antisense“-Sonde, Vergrößerung 40-fach C) Balb/c-Mäuse nicht-infizierte Kontrolle, „antisense“-Sonde, Vergrößerung 20-fach D) Balb/c-Mäuse 16 Std. p.i., „sense“-Sonde, Vergrößerung 20-fach (Ed = Epidermis, D = Dermis, Sc = Subcutis, Stc = Stratum corneum, Stsp = Stratum spinosum, Stb = Stratum basale, iZ = infiltrierende Zellen)
-123-
Ergebnisse
Abb. 36: ISH zum Nachweis der I-TAC-mRNA-Expression in der Haut von C57/BL6-Mäusen nach Infektion mit L. major. A) C57BL/6-Mäuse 16 Std. p.i., „antisense“-Sonde; Vergrößerung 10-fach B) C57BL/6-Mäuse 16 Std. p.i., „antisense“-Sonde, Vergrößerung 40-fach C) C57BL/6-Mäuse nicht-infizierte Kontrolle, „antisense“-Sonde, Vergrößerung 20-fach D) C57BL/6-Mäuse 16 Std. p.i., „sense“-Sonde, Vergrößerung 20-fach (Ed = Epidermis, D = Dermis, Sc = Subcutis, Stc = Stratum corneum, Stsp = Stratum spinosum, Stb = Stratum basale, iZ = infiltrierende Zellen)
5.8 Immunhistochemischer Nachweis von I-TAC in der Epidermis L.-major-infizierter Versuchstiere
Für die iIFL wurden 4 Mäuse untersucht (2 Balb/c- und 2 C57BL/6-Mäuse). Von den
infizierten Hinterfüßen (16 Std. p.i.) sowie von den Kontrollhinterfüßen (s. 4.2.11) wurden
jeweils 50 Gefrierschnitte angefertigt. Auffällig war, dass unter gleichen Bedingungen die
Schnitte der infizierten Balb/c-Mäuse wesentlich stärker fluoreszierten (Abb.37A,B) und
somit eine stärkere I-TAC-Protein-Expression zeigten als in entsprechenden Schnitten von
-124-
Ergebnisse
C57BL/6-Mäusen (Abb.38A,B). Die Fluoreszenz war klar im Bereich der Epidermis
lokalisiert, wobei überwiegend Zellen der Germinativschicht und zu einem geringen Maß
auch Keratinozyten des Stratum granulosum mit dem I-TAC Antikörper reagierten. Die
Färbung war zytoplasmatisch besonders stark. Die Hornhaut (Stratum corneum, Stc)
hingegegen wies keine Fluoreszenz auf. Vereinzelt fand eine Bindung des Antikörpers an
undefinierte Zellen sowie an Zellen des Drüsenepithels (Dre) im Bereich der Dermis statt
(Abb.38A). In den nicht infizierten Kontrollen, sowie der Negativkontrolle (s. 4.2.6 und 4.5.2)
war keine Fluoreszenz detektierbar und somit auch keine I-TAC Proteinexpression
nachweisbar (nicht gezeigt, da die Bilder aufgrund fehlender Fluoreszenz vollständig schwarz
waren).
Abb. 37: Nachweis der I-TAC-Proteinexpression mittels iIFL in der Epidermis von Balb/c-Mäusen 16 Std. nach Infektion mit L. major. A) 20-fache Vergrößerung B) 40-fache Vergrößerung (Epi = Epidermis, Sc = Stratum corneum, Sgr = Stratum granulosum, Stsp = Stratum spinosum, Stb = Stratum basale, D = Dermis)
-125-
Ergebnisse
Abb. 38: Nachweis der I-TAC Proteinexpression mittels iIFL in der Epidermis von C57BL/6-Mäusen 16 Std. nach Infektion mit L. major. A) 20-fache Vergrößerung B) 40-fache Vergrößerung. (Epi = Epidermis, Sc = Stratum corneum, Sgr = Stratum granulosum, Stsp = Stratum spinosum, Stb = Stratum basale, D = Dermis, Dre = Drüsenepithel, Sc = Subcutis).
5.9 Lokaler Effekt am Infektionsort durch die Applikation von I-TAC im Verlauf einer Infektion mit L. major in resistenten C57BL/6-Mäusen
Aufgrund der stärkeren Expression von I-TAC in L.-major-infizierten Balb/c-Mäusen, wurde
der Einfluss von I-TAC in vivo auf den Verlauf der experimentellen Leishmaniose in
C57BL/6-Mäusen untersucht (s. 4.3.6). In allen drei unabhängig voneinander durchgeführten
Versuchen war die gleiche Tendenz zu beobachten. Diese stellte sich in Form einer 2-3
Wochen andauernden zunehmenden Schwellung bei den Tieren dar, denen I-TAC appliziert
wurde. Diese Schwellungszunahme war ab der sechsten Woche nach Beginn der Infektion
messbar. Abb. 39 zeigt Versuch 1 von den drei durchgeführten Versuchen und dokumentiert
den Verlauf der Fußschwellung für beide Versuchstiergruppen. Innerhalb der ersten 35 Tage
nach Beginn der Infektion nahmen in beiden Gruppen die Hinterfüße im Bereich der Fußsohle
um 0,6 bis 0,7 mm an Umfang zu. Danach zeigte sich bei den infizierten Tieren, denen I-TAC
appliziert wurde eine im Vergleich zu den infizierten Kontrolltieren starke Zunahme der
Fußschwellung um das 5-fache der Werte der ersten Messung 18 Tage nach Beginn des
Experiments. Am Tag 58 nach Beginn der Infektion erreichte diese mit 1,36 mm ihren
Höhepunkt und war somit um 70 % stärker ausgeprägt als bei den Tieren die kein I-TAC
-126-
Ergebnisse
erhalten hatten. Innerhalb einer Woche nach Erreichen des Maximums bildete sich die
Schwellung in beiden Gruppen auf 0,8 mm zurück. Die infizierten Mäuse, die I-TAC nicht
erhalten hatten, zeigten eine vergleichsweise schwache Zunahme der Schwellung um maximal
0,54 mm, ausgehend vom Tag der ersten Messung (18 Tage nach Beginn der Infektion).
Abb. 39: Einfluss von I-TAC auf die Fußsohlendicke von L.-major-infizierten C57BL/6- Mäusen 18 bis 65 Tage nach Infektionsbeginn.
Die Abbildung zeigt Versuch 1 von drei unabhängig voneinander durchgeführten Versuchen in denen die gleiche Tendenz zu beobachten war.
Zum Zeitpunkt 0 der Infektion und 4 Std. nach Infektion mit 2 x 107 L. major hatten diese Tiere je 1 µg I-TAC s.c. in den Hinterfuß erhalten (n = 5 Tiere).
Zum Zeitpunkt 0 der Infektion und 4 Std. nach Infektion mit 2 x 107 L. major hatten diese Tiere PBS (1x) s.c. in den Hinterfuß erhalten (n = 5 Tiere). (Mittelwerte ± SEM). * p <0,05.
-127-
Ergebnisse
5.10 Einfluss von I-TAC auf die Parasitendichte in L. major infizierten C57BL/6-Mäusen
Am Tag 65 nach Beginn der Infektion von C57BL/6-Mäusen mit und ohne I-TAC wurden die
Versuchstiere aus dem in 5.9 gezeigten Experiment getötet und die Parasitendichte in den
Hinterfüßen, den poplitealen Lymphknoten und der Milz mittels eines „limiting dilution
assays“ gemessen. Zu diesem Zeitpunkt waren in der Milz keiner Gruppe Parasiten
nachweisbar (Daten nicht gezeigt). In der Gesamthaut und den Lymphknoten konnten keine
signifikanten Unterschiede in der Parasitenzahl zwischen Tieren die I-TAC erhalten hatten
und solchen denen es nicht appliziert wurde (Kontrollgruppe, Kontrolle), festgestellt werden
(Abb.40A,B). Diese Ergebnisse wurden auch in den anderen zwei unabhängig voneinander
durchgeführten Versuchen beobachtet.
Abb. 40: Zahl der Leishmanien in den Hinterfüßen und den Lymphknoten von L.-major-infizierten C57BL/6-Mäusen, denen I-TAC appliziert wurde sowie der Kontrollgruppe (Kontrolle). A) Parasitenanzahl im Hinterfuß. B) Parasitenanzahl im Lymphknoten. C57BL/6 Zum Zeitpunkt 0 der Infektion und 4 Std. nach Infektion mit 2 x 107 L. major I-TAC hatten diese Tiere je 1 µg I-TAC s.c. in den Hinterfuß erhalten (n = 5 Tiere). C57BL/6 Zum Zeitpunkt 0 der Infektion und 4 Std. nach Infektion mit 2 x 107 L. major Kontrolle hatten diese Tiere PBS (1x) s.c. in den Hinterfuß erhalten (n = 5 Tiere). Geometrisches Mittel
-128-
Diskussion
6 DISKUSSION
Ziel dieser Arbeit war es einen Beitrag zur Aufklärung der Rolle des Epithels der Haut für die
Ausbildung einer Th1/Th2-Immunreaktion zu leisten. Hierzu wurde eine vergleichende Gen-
expressionsanalyse an lasermikrodissezierten Keratinozyten von Balb/c- und C57BL/6-
Mäusen in der Frühphase der L.-major-Infektion durchgeführt. Zu diesem Zweck musste
zunächst die entsprechende Methodik etabliert werden. Die in dieser Arbeit beschriebenen
Methoden erlaubten erstmals eine semi-quantitative Bestimmung der mRNA-Expression nach
„Vor“-Amplifikation für verschiedene Immunmodulatoren aus lasermikrodissezierten
Keratinozyten. Die gewonnenen Ergebnisse lassen vermuten, dass sich die residenten Zellen
des Hautepithels durch Modulation des Zyto-und Chemokinmilieus an der Entwicklung der
adaptiven Immunantwort beteiligen.
6.1 Methodische Aspekte
6.1.1 Etablierung der Lasermikrodissektion zur Gewinnung muriner Keratinozyten aus Kryoschnitten
Die lasergestützte Isolierung mit anschließender PCR zum Expressionsnachweis bietet
gegenüber etablierten Techniken wie z. B. der ISH den Vorteil, dass die zelltypspezifische
Genexpression quantitativ und für eine Vielzahl von Genen parallel aus derselben Probe
gemessen werden kann. Allerdings erfordert die Technik umfangreiche Vorversuche um
valide Ergebnisse zu generieren.
Zum einen kann die Spezifität der Isolierung durch die Beschaffenheit des zu isolierenden
Materials eingeschränkt sein. Die Beschaffenheit des Hautepithels erfordert zur Isolierung
einzelner Zellen eine höhere Energie als bspw. Gewebe innerer Organe. Dadurch kann
teilweise ein großer Bereich entstehen der mit dem Laser zerstört wird, wodurch die Anzahl
Zellen, die pro Gewebeschnitt isoliert werden können, begrenzt wird. Somit stellt gerade die
Epidermis der Haut auf Grund ihrer festen Gewebestruktur ein schwierig zu bearbeitendes
Material dar. Zudem muss die Energiestärke, mit der die Keratinozyten isoliert werden,
immer wieder neu justiert werden, was einen automatischen Betrieb des Lasers ausschließt
-129-
Diskussion
und so einen sehr hohen Aufwand erfordert. Grundsätzlich kann man bei der Arbeit am
Mikroskop nicht ausschließen, dass auch geringe Anteile anderer Zellen als Keratinozyten,
wie bspw. LC oder Melanozyten mitisoliert werden. Diesem Problem ist in der Hinsicht
entgegengewirkt worden, indem die Keratinozyten einzeln und sehr nah am Rande der
Zellmembran isoliert wurden. Zudem ist der Anteil anderer Zellen in der Epidermis im
Vergleich mit Keratinozyten sehr gering (ca. 2-4 %) (SCHMITT 1994). Aber eine geringe
Kontamination ist prinzipiell möglich, da Keratinozyten ohne spezielle Markierung nicht
eindeutig identifiziert werden können. Es kann somit bei Genen, wie IL-10, IL-15, IL-18,
IFN-γ, TGF-β, MCP-1, MCP-3, MIP-1α und MIP-1β, die in dieser Arbeit nur niedrig von
Keratinozyten exprimiert wurden, ein Einfluß auf eine Expressionsanalyse durch
kontaminierende Zellen nicht vollständig ausgeschlossen werden. Die Ergebnisse der Real-
Time-PCR wurden daher nochmals mittels ISH und iIFL beispielhaft für Osteopontin (nur
ISH) und I-TAC überprüft. Mit diesen Methoden konnte gezeigt werden, dass die Gene
tatsächlich in Keratinozyten exprimiert wurden.
Falsch negative Ergebnisse könnten prinzipiell durch RNA Schäden bei Ablation,
Mikrodissektion und Katapultieren des Untersuchungsmaterials zustande kommen. Bei der
Umwandlung von Photonenenergie in elektrische Anregungsenergie wären DNA Schäden
prinzipiell durch photochemische, also das Aufbrechen chemischer Bindungen, oder
photophysikalische Prozesse, also temperaturbedingte Vorgänge, denkbar. Allerdings liegt die
Wellenlänge des UV-Lasers mit 337 nm so weit von dem Absorptionsmaximum der DNA
(260 nm) entfernt, dass es außerhalb des Laserfokus kaum zu photochemischen Reaktionen
kommt. Da außerdem die Temperatur exponentiell vom Wirkort des Lasers nach außen hin
abnimmt, sind auch photophysikalische Prozesse als Ursache für DNA Schäden zu
vernachlässigen (GANGNUS, Firma PALM, persönliche Mitteilung). Desweiteren ist die
Kontaminationsgefahr bei allen Arbeitsschritten zu berücksichtigen. Auch wenn die
laserunterstütze Mikrodissektion als die „no-touch“ Methode mit der geringsten
Kontaminationsgefahr des Untersuchungsmaterials angesehen wird, ist dies prinzipiell durch
Gewebsentnahme, das Schneiden am Mikrotom, der Lagerung und Färbung, der Arbeit am
Lasergerät sowie der RNA-Extraktion und PCR möglich. Einer eventuellen Kontamination
während dieser Arbeitsschritte wurde so weit wie möglich vorgebeugt und die
-130-
Diskussion
Kontaminationsgefahr streng kontrolliert (s. 4.4.1. Allgemeine Maßnahmen zur
Kontaminationsvermeidung).
Schließlich ist noch zu erwähnen, dass die Lebensdauer des Stickstofflasers begrenzt und das
gesamte System sehr kostenintensiv ist. Während der Entstehung dieser Arbeit musste die
Lasercartridge zweimal erneuert werden, was einen extrem langen Ausfall des Gerätes (>8
Monate) sowie hohe Reparaturkosten bedingte. Die Firma PALM ist sich dieser Problematik
bewusst und hat dementsprechende Maßnahmen zur Verbesserung der Technik dahingehend
unternommen, dass ein robusteres Feststofflasersytem entwickelt wurde. Eine Umrüstung des
Systems wäre jedoch mit sehr hohen Kosten verbunden.
6.1.2 Qualität und Quantität der RNA aus Probenmaterial, das mittels Lasermikrodissektion gewonnen wurde
Für die Etablierung der Lasermikrodissektion musste zunächst geprüft werden, ob die
RNA-Integrität erhalten bleibt. Dazu wurde ein Probenmaterial (Leber) gewählt, aus dem in
kurzer Zeit eine große Anzahl an Zellen isoliert werden konnte.
Die Kontrolle der RNA-Integrität mit Hilfe des Agilent-2100-Bioanalyzers (BUSTIN 2002,
SCHROEDER 2006) und die photometrisch ermittelten Werte zur RNA-Reinheit zeigten das
zu erwartende 2:1 Verhältnis von ribosomaler 28s- zu 18s-RNA, welches als ein guter
Indikator für die Intaktheit der isolierten RNA zu werten ist. Die mRNA-Menge innerhalb
einer Zelle liegt schätzungsweise zwischen 0,1 und 1 pg (EBERWINE et al. 1992). Dies
ergäbe bei denen in einem Pilotversuch untersuchten 50 000 Leberparenchymzellen
5000-50 000 pg oder 5-50 ng mRNA. Spektralphotometrisch wurden zwischen 46 und 52
ng/µl RNA gemessen, was bei 40 µl 1840-2080 ng oder ca 2 µg Gesamt RNA entspricht. 2 %
der Gesamt RNA sind mRNA, dies entspricht ungefähr 40 ng mRNA. Auch wenn die
Quantifizierung solch geringer Mengen schwierig ist, zeigen diese Daten, dass es gelungen ist
eine hohe Ausbeute bei der RNA-Extraktion aus dem dissezierten Gewebe zu erreichen.
Aufbauend auf diesen positiven Ergebnissen erfolgte die weitere Aufarbeitung der RNA aus
Keratinozyten mit diesem Protokoll (s 5.2). Bei den isolierten Keratinozyten konnte die
isolierte RNA auf Grund der Sensitivitätsgrenze nur mit Hilfe der PCR kontrolliert werden.
Es konnten Produkte der zu erwartenden Größe für RPL und Osteopontin aus 10 000
-131-
Diskussion
Keratinozyten amplifiziert werden. Dabei lieferte die verwendete Trizol-Standard-Methode
die höchste RNA-Ausbeute gegenüber kommerziellen Kit-Systemen.
Bei der Lasermikrodissektion ist das Probenmaterial begrenzt und eine entsprechende
Amplifikation der RNA in vielen Fällen wünschenswert oder sogar zwingend erforderlich.
Zudem ist es für eine routinemäßige Anwendung der sehr zeitaufwendigen
Lasermikrodissektion erforderlich, die für nachfolgende Untersuchungen benötigte Zellzahl
möglichst gering zu halten. Um dennoch genügend genetisches Material zur Verfügung zu
haben, musste ein Amplifikationsschritt vor der eigentlichen semi-quantitativen
Genexpressionsanalyse eingeschaltet werden. Dieses erstmals von VAN GELDER et al.
(1990) beschriebene Prinzip der linearen Amplifikation dient in den meisten Fällen der
Synthese markierter cRNA. In der vorliegenden Arbeit wurde eine auf PCR basierende
zufällige Amplifikation nur für die Synthese von doppelsträngiger cDNA verwendet (s. 4.4.9
„Vor“-Amplifikation geringer RNA Mengen). Der Vorteil letzterer Methode besteht in einem
besseren 3`/5`Verhältnis der cDNA. Das optimale 3`/5`Verhältnis Verhältnis ist 1 und
indiziert, dass die mRNA Sequenzen in voller Länge abgeschrieben werden, ohne jegliche
Degradation oder Kürzung während der cDNA Synthese (KLUR et al. 2004). Die
doppelsträngige cDNA wurde anschließend für Real-Time-PCR-Versuche verwendet. Die
Zuverlässigkeit dieser Applikation wurde bereits erfolgreich getestet (FREY et al. 2002) und
durch Genchipexpressionsanalysen von 1000 Zellen aus verschiedenen Bereichen des
murinen Gehirns bestätigt (KLUR et al. 2004).
Zu beachten ist, dass zu hohe PCR-Zyklen die gleichmäßige Repräsentation verschiedener
Transkripte beeinflussen können. Dies kann unter anderem dadurch erklärt werden, dass mit
zunehmender Zyklenzahl die Polymerase nicht mehr so aktiv ist wie zu Beginn. Dies kann
dazu führen, dass nicht mehr der gesamte cDNA Bereich zwischen den TAS-Sequenzen
abgelesen wird und es so zur Überrepräsentation kürzerer Abschnitte kommt. Aus diesem
Grund wurden Zyklenzahlen über 25 vermieden, was nur ab einer Zellzahl von 1000
Keratinozyten möglich war.
-132-
Diskussion
In dieser Studie konnte keine negative Beeinflussung auf den relativen Expressionslevel
festgestellt werden, wenn die PCR Zyklen in einem Bereich bis maximal 25 Zyklen lagen, da
• mit einer PCR die Expression von Osteopontin und der „housekeeping“-Gene
(GAPDH und RPL) vor und nach „Vor“-Amplifikation nachgewiesen werden
konnte,
• diese „housekeeping“-Gene vor und nach „Vor“-Amplifikation wie erwartet
stärker exprimiert wurden als Osteopontin,
• die aus Voruntersuchungen bekannte differentielle Regulation von Osteopontin in
der Gesamthaut auch noch nach „Vor“-Amplifikation in den Keratinozyten
nachweisbar war.
Weiterhin spricht die Übereinstimmung der Genexpression in den lasermikrodissezierten
Keratinozyten und den konventionell präparierten Gesamthautproben dafür, dass die
Methoden der Lasermikrodissektion und der „Vor“-Amplifikation das Endergebnis nicht
negativ beeinflussten und das Gesamtmuster der Transkriptexpression erhalten blieb.
6.1.4 Real-Time-PCR
Die im Rahmen dieser Arbeit angewendete Methode der Real-Time-PCR im ABI 7900 HT
RealTime-PCR cycler mit SYBR Green Sonden stellt eine semi-quantitative Technik dar
(MORRISON et al. 1998), die in dieser Arbeit für die Analyse von Immunmodulatoren auf
mRNA-Ebene verwendet wurde. Eine der Hauptursachen für falsch-positive PCR-Ergebnisse
sind Kontaminationen der PCR. Diese Gefahr wird durch die Echtzeit-PCR im geschlossenen
System verringert, da eine weitere Bearbeitung der Proben nach dem Ende der PCR
überflüssig ist. Hierdurch wird zusätzlich ein Zeitvorteil bei der Probenanalyse erreicht. Um
die Gefahr der Kontamination mit genomischer DNA zu minimieren, wurde bei allen
extrahierten RNA-Proben ein Verdau mit DNAse durchgeführt. Ferner wurde das
methodische Vorgehen kontrolliert, indem intron-spannende Primer verwendet wurden. Daher
konnte ich zeigen, dass in den hier verwendeten Proben keine genomische DNA amplifiziert
wurde.
-133-
Diskussion
Die Wahl eines geeigneten Referenzgens hängt von seiner Grundeigenschaft ab, eine
konstante, stabile Genexpression bei den zu untersuchenden Proben zu besitzen. Die
Expressionshöhe eines so genannten „housekeeping“-Gens kann aber nicht nur in
verschiedenen Geweben sondern auch im gleichen Gewebe bei variablen Bedingungen wie
bspw. verschiedenen Versuchstieren, Schwankungen aufweisen (SCHMITTGEN u.
ZAKRAJSEK 2000). Als interner Standard wurde deshalb die Expression von zwei
unabhängigen Genen, GAPDH und RPL, gewählt. Bei den wenigen Veröffentlichungen zu
Zytokinveränderungen bei experimentell infizierten Mäusen wurde ebenfalls GAPDH
eingesetzt (GU et al. 2000, KHASKHELY et al. 2002) und zudem zeichneten sich beide Gene
in eigenen Vorversuchen (Daten nicht gezeigt) durch geringe Expressionsunterschiede in
infiziertem und nicht-infiziertem Gesamthautgewebe von Balb/c- und C57BL/6-Mäusen aus.
6.2 Expression von Zyto- und Chemokinen im Verlauf der L.-major-Infektion
In der unveränderten, gesunden Epidermis befinden sich die Keratinozyten in einem
immunmodulatorisch wenig aktivem Zustand, indem sie langsam in der Basalschicht
proliferieren und in suprabasale Epidermisschichten differenzieren. Da sie der Außenwelt
ausgesetzt sind, müssen sie darauf vorbereitet sein, schnell auf eine Verletzung reagieren zu
können. Deshalb produzieren Keratinozyten „Wächtermoleküle“ die sofort signalisieren,
wenn eine Beschädigung stattgefunden hat und das Abwehrsystem aktiviert werden muss
(WANG et al. 1999, UCHI et al. 2000, ALBANESI et al. 2005). Ihre aktive Beteiligung an
der unspezifischen Abwehr ist heutzutage generell akzeptiert. Eine wichtige Frage, die jedoch
bislang ungeklärt war, war Gegenstand der vorliegenden Arbeit: Können Keratinozyten das
spezifische Immunsystem beeinflussen, indem sie die Th-Zell-Differenzierung modulieren?
Die Klärung dieser Frage ist von großer praktischer Relevanz für die Pathomechanismen
wichtiger Hautkrankheiten die mit einer Störung der Th-Zell-Differenzierung einhergehen.
Die Bedeutung einer adäquaten Th1/Th2-Antwort wird deutlich, wenn dieses Gleichgewicht
gestört wird und es zu einer überschießenden Reaktion eines Zelltyps kommt wie bspw. bei
der Psoriasis oder der atopischen Dermatitis. Bislang konnte nicht geklärt werden, wie es zur
Entwicklung einer derartig gestörten Immunantwort kommt, dabei scheinen jedoch besonders
-134-
Diskussion
die Abläufe in der Frühphase einer Entzündungsreaktion eine wichtige Rolle zu spielen.
Unbekannt ist zudem, welcher Zelltyp für diesen frühen Einfluss verantwortlich ist.
Um einen Beitrag zur Aufklärung dieser Mechanismen zu leisten, wurde mit der
experimentellen Leishmaniose ein etabliertes Modell für die Untersuchung der
Th1/Th2-Antwort gewählt. Besondere Aufmerksamkeit wurde dabei auf die Untersuchung
der Produktion von Immunmodulatoren durch einen bisher in diesem Modellsystem kaum
untersuchten Zelltyp, den Keratinozyten, gelegt.
Für die selektive Untersuchung dieser Zellpopulation wurde mit der Lasermikrodissektion ein
Verfahren angewendet, welches es ermöglicht, einzelne Zellen selektiv und
kontaminationsarm aus biologischem Material zu gewinnen.
Da besonders die frühe Produktion von Zyto-/Chemokinen für die Th-Zell-Differenzierung
entscheidend zu sein scheint (CHATELAIN et al. 1992, HEINZEL et al. 1993, LEIBY et al.
1993, SYPEK et al. 1993, LAUNOIS et al. 1997, BIEDERMANN et al. 2001, TAVARES et
al. 2006), wurde eine umfangreiche Genexpressionsanalyse zu einem recht frühen Zeitpunkt
nach der Infektion von Labormäusen mit L. major durchgeführt. Dazu wurden 16 Std. nach
der Infektion Keratinozyten mittels Lasermikrodissektion aus der Epidermis am Ort der
Infektion mit den Parasiten isoliert und die Genexpression von Zyto- und Chemokinen wie
beschrieben bestimmt. Die Expression im weiteren Verlauf der Infektion von bis zu drei
Tagen wurde zudem für ausgewählte Gene (Osteopontin, IL-4, IFN-γ, I-TAC) in der
Gesamthaut dokumentiert. Es konnte erstmals gezeigt werden, dass es in der Frühphase der
subkutanen Infektion mit L. major auf molekularer Ebene zu einer starken Reaktion der
Epidermis kommt, die gekennzeichnet ist durch die Produktion einer Anzahl von
immunmodulatorisch wirksamen Faktoren.
Zusammenfassend konnte gezeigt werden:
• Dass es in den ersten Std. der L.-major-Infektion zu einer starken Induktion sowie
Neoexpression einer großen Anzahl von immunmodulatorisch wirksamen Molekülen
in der Epidermis kam.
-135-
Diskussion
• Dass die Expression der meisten Gene auf die ersten Tage der Infektion beschränkt
war, ein Zeitraum von dem bekannt ist, dass in ihm die entscheidenden
Weichenstellungen für die Th-Zell-Differenzierung erfolgen.
• Dass es von entscheidender Bedeutung ist, dass deutliche Stammesunterschiede in der
Expression der Moleküle bestehen. Die Expression der meisten untersuchten Zyto-
und Chemokine (Osteopontin, IL-1β, IL-4, IL-12, IL-15, IL-18, TNF-α, TGF-β,
S100A8, S100A9, MCP-1, MIP-1α, MIP-1β, MCP-3, RANTES und IP-10) war
stärker in Labormäusen mit L.-major-resistentem genetischen Hintergrund (C57BL/6)
als in den suszeptiblen Balb/c-Mäusen. Es liegt daher die Vermutung nahe, dass die
Expression der Immunmodulatoren einen Einfluss auf Resistenz oder Suszeptibilität
für die Leishmaniose haben könnte. Die stärkere Expression der genannten
Immunmodulatoren in den resistenten Mäusen lässt einen protektiven Effekt dieser
Substanzen auf den Infektionsverlauf vermuten. Allerdings konnte auch ein Chemokin
identifiziert werden, welches nahezu ausschließlich in suszeptibelen Balb/c-Mäusen
hochreguliert wird (I-TAC).
6.2.1 Infektionsbedingte Expression von Immunmediatoren in der Haut und den Keratinozyten von resistenten Mäusen
Aufgrund der differentiellen Expression verschiedener Zytokine zwischen suszeptiblen und
resistenten Mäusen in der Frühphase der L.-major-Infektion ist davon auszugehen, dass diese
Induktion der epidermalen Genexpression einen Einfluss auf die Generierung der spezifischen
Immunantwort hat. Daten zur frühen Zytokin Expression und dem damit verbundenen
Einfluss auf die frühe Th-Differenzierung gibt es lediglich für drei Interleukine (IL-1, IL-12,
IL-4) (KOSTKA et al. 2006, VON STEBUT et al. 1998, 2002, 2003, MAROVICH et al.
2000, SCHONLAU et al. 2003, BIEDERMANN et al. 2001).
Die von mehreren unabhängigen Arbeitsgruppen (VON STEBUT et al. 2003, KOSTKA et al.
2006, DELFINO et al. 1995, BERSUDSKY et al. 2000, FILIPPI et al. 2003) bereits gezeigte
Induktion einer Th1-Antwort durch IL-1 in der Frühphase der Infektion konnte mit der in
dieser Arbeit gezeigten frühen Expression von IL-1 in der Haut und den Keratinozyten
bestätigt werden. Kontrovers diskutiert wird der Mechanismus der IL-1-vermittelten Th-Zell-
-136-
Diskussion
Differenzierung. Einerseits wird eine IL-1-induzierte klonale Expansion von Th2-Zellen
durch Erhöhung der Responsivität auf IL-4 oder Wirkung als kostimulatorisches Molekül
vermutet (GREENBAUM et al. 1988, LICHTMAN et al. 1988, HUBER et al. 1998,
SHIBUYA et al. 1998). Andererseits zeigten andere Untersuchungen, dass IL-1 für eine Th-2-
Differenzierung nicht von Bedeutung ist (SEDER u. PAUL 1994). In der etablierten Infektion
ist jedoch die Wirkung von IL-1 als Th2-Zytokin allgemein anerkannt. Als zelluläre Quelle
des IL-1 konnten Makrophagen und dendritische Zellen der Haut identifiziert werden, wobei
die Produktion von IL-1α in den genannten Zellen aus den resistenten C57BL/6-Mäusen
stärker ausgeprägt ist. (DELFINO et al. 1995, BERSUDSKY et al. 2000, FILIPPI et al. 2003,
VON STEBUT et al. 2003, BERSUDSKY et al. 2005, KOSTKA et al. 2006). IL-1 kann die
Aktivität von B und T-Zellen (SIMS u. DOWER 1994, DINARELLO 1996) sowie von DC
(JAKOB u. UDEY 1998, CUMBERBATCH et al. 2002) steigern.
In dieser Arbeit konnte nun erstmals gezeigt werden, dass zusätzlich auch ein deutlicher
Stammesunterschied in der Expression von IL-1β in der Epidermis besteht. In
Übereinstimmung mit der Th1-induzierenden Wirkung von IL-1 zeigte sich eine verstärkte
Expression in den C57BL/6-Mäusen. Es liegt daher nahe zu vermuten, dass diese Expression
von physiologischer Bedeutung für die Generation der protektiven Immunantwort ist,
insbesondere da die Expression zeitlich mit der pharmakologischen Wirkung einer
IL-1-Behandlung korreliert. Ein protektiver Effekt der sich in Form einer Reduzierung und
schnelleren Heilung der Hautläsionen in Balb/c-Mäusen darstellte, konnte durch 3-malige
Injektion von 50 ng an den ersten 3 Tagen der Infektion erreicht werden (VON STEBUT et
al. 2003). Da eine Behandlung mit IL-1β auch wirksam war, wenn die Injektion am nicht
infizierten Kontrollfuß erfolgte (VON STEBUT et al. 2003) kann von einer systemischen
Wirkung auf die Th-Zell-Differenzierung im Lymphknoten ausgegangen werden. Dies ist
aufgrund der hier präsentierten Ergebnisse auch für das von der Epidermis produzierte IL-1β
denkbar, da hohe Transkriptspiegel von IL-1β-mRNA in den Zellen detektiert wurden. Damit
konnte gezeigt werden, dass ein Zytokin, von dem bereits nachgewiesen wurde, dass es sich
über eine Induktion einer Th1-Antwort protektiv auf den Verlauf der experimentellen
Leishmaniose auswirkt in vivo verstärkt in der Epidermis resistenter Mäuse exprimiert wurde.
Dieses Ergebnis steht in sehr gutem Einklang mit der dieser Arbeit zugrunde liegenden
-137-
Diskussion
Hypothese, dass die Expression von Immunmodulatoren in der Epidermis einen Einfluss auf
die Generierung der spezifischen Immunantwort haben könnte.
Die protektive Wirkung von IL-12 hinsichtlich Heilung und Kontrolle einer
L.-major-Infektion ist hinreichend belegt (HEINZEL et al. 1993, SYPEK et al 1993, SACKS
u. NOBEN-TRAUTH 2002). IL-12 wird dabei in der Frühphase einer Immunantwort als das
wichtigste Th1-induzierende Zytokin angesehen. Während publizierte Daten eine Produktion
von IL-12 durch DC und Makrophagen in der Gesamthaut von C57BL/6-Mäusen gezeigt
haben (VON STEBUT et al. 1998, 2002, 2003, MAROVICH et al. 2000, SCHONLAU et al.
2003), ist die infektionsbedingte IL-12-Synthese von Keratinozyten zu einem solch frühen
Zeitpunkt wie 16 Std. nach der Infektion bisher nicht beschrieben. Aufgrund der deutlichen
Induktion der IL-12-mRNA ist es denkbar, dass IL-12 analog zum IL-1 direkt auf die DC-Th-
Zell-Interaktion im Lymphknoten wirkt. Somit könnten die den DC zahlenmäßig
überlegenden Keratinozyten in der Frühphase der Entzündung die IL-12-Produktion der DC
unterstützen. Das lokal produzierte IL-12 könnte zusätzlich weitere Effekte haben, die für den
Infektionsverlauf von Bedeutung sind, so z.B. eine Aktivierung von NK-Zellen.
Das bereits nach 6 Std. in C57BL/6-Mäusen stark exprimierte Osteopontin wirkt zudem als
Th1-Zytokin chemotaktisch insbesondere auf Makrophagen (PATARCA et al. 1993,
GIACHELLI et al. 1998) und DC (WEISS et al. 2001), wodurch die Sekretion von IL-12
stimuliert und gleichzeitig die Sekretion des immunsupprimierenden IL-10 gehemmt wird
(ASHKAR et al. 2000, O´REGAN 2000).
Obwohl IL-4 als das wichtigste Zytokin für die Etablierung einer Th2-Zellantwort gilt, konnte
in aktuellen Arbeiten gezeigt werden, das IL-4 in der Frühphase einer Immunantwort einen
gegenteiligen Effekt hat und als Induktor einer Th1-Antwort wirkt. Dieser Effekt scheint
dabei über eine Wirkung auf DC vermittelt zu werden, während die Wirkung als Th2-Zytokin
durch den Einfluss von IL-4 auf die proliferierenden T-Zellen zu erklären ist. IL-4 reguliert
dabei sowohl die Expression kostimulatorischer Moleküle (CD80/CD86) auf DC (BAGLEY
et al. 2000, KING et al 2003) als auch die IL-12-Produktion durch DC (BAGLEY et al. 2000,
HOCHREIN et al. 2000, KALINSKI et al. 2000, EBNER et al. 2001, KING et al 2003).
-138-
Diskussion
Dieser frühe Th1-induzierende Effekt wurde für die Leishmaniose von BIEDERMANN et al.
(2001) gezeigt, die demonstrierten, dass eine Administration von IL-4 zum Zeitpunkt der
klonalen Expansion von Th2-Zellen in Balb/c-Mäusen zu einer progressiven Leishmaniose
führt, während die sehr frühe Applikation (<8 Std.) über eine Stimulation der
IL-12-Produktion in DC zu einer Th1-Antwort sowie einer Resistenz in den normalerweise
suszeptiblen Balb/c-Mäusen führte.
Ich dokumentiere hier erstmals eine stärkere IL-4-Expression in den Keratinozyten von
C57BL/6-Mäusen als in Balb/c-Mäusen 16 Std. nach der Infektion mit L. major. Hier konnte
somit erstmals gezeigt werden, dass es in Übereinstimmung mit der pharmakologischen
Wirkung von IL-4 als frühem Induktor einer Th1-Antwort auch in vivo zu einer stärkeren
Expression von IL-4 in resistenten Mäusen kommt. Im Infektionsverlauf zeigte sich eine
besonders starke Expression von IL-4 in den ersten 8 Std. nach der Infektion in C57BL/6-
Mäusen. Dies korelliert sehr gut mit der in der Literatur beschriebenen Bedeutung einer
frühen IL-4-Expression als potentieller Modulator einer Th1-Zell-Differenzierung
(SCHULER et al. 1999, BAGLEY et al. 2000, BIEDERMANN et al 2001). Es ist daher
naheliegend davon auszugehen, dass die IL-4-Expression in der Epidermis einen Einfluss auf
die Th-Zell-Differenzierung hatte. Im Gegensatz zu IL-1β waren die Transkriptspiegel von
IL-4 sehr niedrig, demzufolge kann hier eher von einer lokalen Wirkung ausgegangen
werden. Da die IL-4 Expression noch vor einer Auswanderung der DC aus der infizierten
Gesamthaut erfolgte, liegt es nahe zu vermuten, dass die Wirkung auf die Th-Zell-
Differenzierung über eine Beeinflussung der DC zu erklären ist. Dabei könnte das epidermal
produzierte IL-4 z.B. die IL-12-Sekretion oder die Expression kostimulatorischer Moleküle
beeinflussen und so eine Th1-Zell-Differenzierung induzieren.
TNF-α ist wie IFN-γ ein wichtiges Effektorzytokin einer etablierten Th1-Antwort. Eine Reihe
von Studien konnte belegen, dass IFN-γ nur in Assoziation mit TNF-α Makrophagen dazu
aktiviert, die Leishmanien zu töten (CILLARI et al. 1989, MAUEL et al. 1991, ASSREUY et
al. 1994, JUTTNER et al. 1998, GOMES et al. 2003). Es wird vermutet, dass TNF-α ein
zweites Signal an die IFN-γ-stimulierten Makrophagen gibt, wodurch erst der
Tötungsmechanismus aktiviert wird (KAMIJO et al. 1994, MARTIN et al. 1994). Diese
Annahme wird dadurch bestätigt, dass TNF-α-knockout-C57BL/6-Mäuse die Infektion und
-139-
Diskussion
Ausbreitung von L. major nicht kontrollieren können, obwohl iNOS Protein in den
Hautläsionen und den lokalen Lymphknoten detektiert werden kann (WILHELM et al. 2001).
Dies könnte eine Begründung dafür sein, dass Balb/c-Mäuse, selbst bei normaler Produktion
von IFN-γ bei geringer TNF-α-Produktion nicht dazu in der Lage sind, den Erreger zu
eliminieren.
In der vorliegenden Arbeit zeigte sich im Untersuchungsmaterial von C57BL/6-Mäusen eine
um ein vielfaches stärkere TNF-α-Produktion als in Balb/c-Mäusen, und somit eine
gegenüber IFN-γ gegenläufige Regulation. Anhand der bisher publizierten Arbeiten lässt sich
kein konkreter Anhalt für einen möglichen Wirkmechanismus von TNF-α in der Frühphase
der L.-major-Infektion eruieren.
TGF-β wirkt ebenfalls potenzierend im Rahmen einer etablierten Th1-Antwort. Im Rahmen
der experimentellen Leishmaniose zeigt sich durch TGF-β alleine kein Effekt auf die
Infektion. In der Anwesenheit von IFN-γ wurde jedoch in vitro eine deutliche Reduzierung
L.-major-infizierter Zellen beobachtet (GOMES et al. 2003). Es wurde gezeigt, dass TGF-β
die CD4+-T-Zell-Reifung beeinflusst. Allerdings sind in diesem Zusammenhang sehr
gegensätzliche Effekte auf die T-Zell-Differenzierung in einer Vielzahl von
Veröffentlichungen dokumentiert worden, die zeigen, dass TGF-β eine Th1-Zell-
Differenzierung sowohl hemmen als auch unterstützen kann (SWAIN et al. 1991, SCHMITT
et al. 1994, HOEHN et al. 1995, PAWELEC et al. 1996).
Im Rahmen dieser Arbeit zeigte sich in den Keratinozyten und der Gesamthaut infizierter
C57BL/6-Mäuse eine stärkere TGF-β-Expresssion als in den Balb/c-Mäusen. Allerdings war
der Unterschied bei der Expression in der Gesamthaut zwischen den beiden infizierten
Mausstämmen nicht so stark wie in den Keratinozyten. Aufgrund der schwer zu einem
Gesamtbild zusammenzufassenden Wirkungen auf die Th-Zellen sind Aussagen über einen
möglichen Wirkungsmechanismus auch hier nur schwer zu treffen.
Die S100-Proteine S100A8 und S100A9 werden nicht zu den klassischen Zyto- oder
Chemokinen gezählt, sie sind jedoch auch Moleküle mit niedrigem Molekulargewicht, die
eine proinflammatorische Wirkung vermitteln und für die Rekrutierung von Leukozyten ins
entzündliche Gewebe verantwortlich sind (ROTH et al. 2003). Die erste Beschreibung dieser
-140-
Diskussion
Moleküle zeigte, dass sie von Zellen myeloiden Ursprungs, nämlich Granulozyten,
Monozyten und Makrophagen gebildet werden, wobei ihnen ein Einfluss auf die Aktivität von
Phagozyten zugeschrieben wird. Während sie in der unveränderten Gesamthaut nicht gebildet
werden, ist eine Hochregulation in der Epidermis entzündlicher Dermatosen (KELLY et al.
1989, 1991), Kontaktdermatitis (ROTH et al. 1992), Lichen planus, Lupus erythematosus und
Psoriasis (KUNZ et al. 1992, BROOME et al. 2003, ZENZ et al 2005) beschrieben.
Unbekannt war bisher, dass die beiden Proteine auch im Rahmen einer Infektion mit
Leishmanien reguliert werden. Zudem wurde eine stärkere Hochregulation beider Gene im
Untersuchungsmaterial von C57BL/6-Mäusen im Vergleich zu Balb/c-Mäusen entdeckt. Dies
ist ein möglicher Hinweis auf eine proinflammatorische Wirkung der S100A8-/9-Proteine, die
über die Rekrutierung von Makrophagen an den Infektionsort einen positiven Effekt für die
Resistenzentwicklung in der experimentellen Leishmaniose haben könnten
(SUNDERKÖTTER et al. 1993, TACCHINI-COTTIER et al. 2000).
Einem wesentlichen Augenmerk galt der Produktion von Chemokinen, die die Infiltration
(Makrophagen, Granulozyten) oder die Emigration (DC) von Leukozyten beeinflussen
können. Dies wäre somit ein Effekt im Rahmen der frühen unspezifischen Immunantwort.
Eine Reihe von Publikationen haben sich mit dem Chemokinexpressionsmuster im
Lymphknoten im Verlauf der experimentellen Leishmaniose befasst (MÜLLER et al. 2001,
2003, ROUSSEAU et al. 2001, VAN ZANDBERGEN et al. 2002, JI et al. 2003). So wurde
2-4 Wochen nach der Infektion eine stärkere Expression der Chemokine MIP-1α ,MIP-1β,
RANTES und ihrer Rezeptoren CCR1, CCR2, CCR5 im Lymphknoten resistenter Mäuse
beobachtet. Dagegen wurde innerhalb des ersten Tages (VESTER 1999, ZAPH u. SCOTT
2003) im Lymphknoten von resistenten und suszeptiblen Mäusen signifikante Mengen an
IP-10, MCP-1, RANTES, MIP-1α und Lymphotaktin gefunden. Es liegen jedoch bisher kaum
Studien vor, die sich mit der Expression von Chemokinen direkt am Infektionsort Haut
befassen. Untersucht wurde die Expression von Chemokinen in DC nach Stimulation mit
L. major in vitro, in denen bspw. MCP-1 und MIP-1α in beiden Mausstämmen
herunterreguliert werden, während IP-10 nur in DC resistenter Mäuse hochreguliert wird
(STEIGERWALD u. MOLL 2005). Die Expression des MCP-Chemokin-Rezeptor CCR2 in
DC scheint für die Migration der DC in den Lymphknoten von Bedeutung zu sein und wurde
-141-
Diskussion
deshalb in Verbindung mit einer schützenden, zellvermittelten Immunantwort gebracht
(SATO et al. 2000). Eine späte stärkere Expression in den resistenten Mäusen konnte auch für
MIG (CXCL9) und IP-10 (CXCL10) im Hinterfuß bestätigt werden, während diese
Chemokine einen Tag nach der Infektion in beiden Mausstämmen in gleichem Umfang
exprimiert werden (SANTIAGO et al. 2004).
Im Gegensatz zu den genannten Daten zur Expression im Lymphknoten konnte in der
vorliegenden Arbeit eine stärkere Expression der Chemokine MCP-1 (CCL1), MCP-3
(CCL8), MIP-1α (CCL2), MIP-1β (CCL3), RANTES (CCL5) und IP-10 (CXCL10) in der
Haut und den Keratinozyten von C57BL/6-Mäusen 16 Std. nach der Infektion als in
Balb/c-Mäusen nachgewiesen werden.
Durch die verstärkte Chemokin-Synthese in der Gesamthaut von C57BL/6-Mäusen könnte die
beschriebene stärkere Infiltration von Makrophagen in der Frühphase der Infektion
(SUNDERKÖTTER et al. 1993) erklärt werden, von der angenommen wird dass sie
bedeutsam für die Resistenzentwicklung ist. Überwiegen hingegen wie bei den Balb/c-
Mäusen Granulozyten im frühen Infiltrat, so scheint dies zu einer erhöhten Suszeptibilität zu
führen (TACCHINI-COTTIER et al. 2000). Zusätzlich könnten die in der Epidermis
produzierten Zytokine die Migration von DC stimulieren. Eine effizientere Migration von DC
in die lokalen Lymphknoten, in denen die Präsentation des Antigens an die T-Zellen
stattfindet, könnte durchaus einen Effekt auf den Typ der generierten Th-Zell-Antwort haben.
Weiterhin könnte die Migration von NK-Zellen sowie von T-Zellen in das infizierte Gewebe
beeinflusst werden. Außerdem können die Keratinozyten durch die verstärkte Expression
dieser Moleküle in der Gesamthaut möglicherweise auch autokrin aktiviert werden, da sie
teilweise über die entsprechenden Rezeptoren verfügen (UCHI et al. 2000).
6.2.2 Infektionsbedingte Expression von Immunmediatoren in der Haut und den Keratinozyten von suszeptiblen Mäusen
In dieser Arbeit zeigte sich, dass lediglich zwei Zytokine (IL-10 und IFN-γ) sowie ein
Chemokin (I-TAC) stärker in der Haut und den Keratinozyten von Balb/c-Mäusen als in
entsprechendem Material von C57BL/6-Mäusen exprimiert wurde. Die geringe Expression
von Immunmodulatoren in den suszeptiblen Mäusen legt die Vermutung nahe, dass
-142-
Diskussion
bestimmte Zyto-/Chemokine für die Heilung und Kontrolle der Infektion von großer
Bedeutung sind und ihre Produktion für die Etablierung einer Th1-Antwort notwendig ist. Am
ehesten ist aufgrund der publizierten Wirkungsmechanismen der Chemokine davon
auszugehen, dass dieser Effekt indirekt über die Beeinflussung des zellulären Infiltrats, d.h.
der emigrierenden DC erfolgt. Andererseits sind auch direkte Einflüsse auf die
expandierenden T-Zellen nicht auszuschließen, da einige Chemokine sehr stark im Rahmen
der Infektion induziert werden.
Die immunsuppressive Wirkung von IL-10 in der experimentellen Leishmaniose und in der
humanen Leishmaniose ist bekannt. In Läsionen der humanen, chronischen, kutanen
Leishmaniose (MELBY et al. 1994), im Gewebe von Kala-Azar Patienten (KARP et al. 1993)
und im Plasma von Patienten mit Post-Kala-Azar dermaler Leishmaniose wurde eine
Überproduktion von IL-10 festgestellt (KARP et al. 1993, MELBY et al. 1994, GASIM et al.
1998, KANE et al. 2001). Während die Gabe von anti-IL-10 Antikörpern nur wenig Effekt
auf das Fortschreiten der Erkrankung in Balb/c-Mäusen hat (SYPEK et al. 1993, POWRIE et
al. 1994), reagierten IL-10-transgene resistente Mäuse suszeptibel auf eine Infektion
(GROUX et al. 1994).
Meine Ergebnisse bestätigen somit die immunsuppressive Wirkung von lokal am
Infektionsort produziertem IL-10 auf die zellvermittelte Immunität. Im Gegensatz zu der
bereits beschriebenen, späten (> 20 Wochen nach Beginn der Infektion), lokalen Expression
von IL-10 durch CD4+CD25+-T-Zellen (BELKAID et al. 2002), sind dies die ersten Daten,
die eine frühe IL-10-Expression in der Gesamthaut im Rahmen einer experimentellen
Leishmaniose dokumentieren.
Erstaunlicherweise zeigte sich im Rahmen dieser Arbeit einer stärkere Expression von IFN-γ
in den Keratinozyten infizierter Balb/c-Mäuse. Allerdings waren nur wenig Amplifikate
nachweisbar, so dass unklar war, ob relevante Mengen des wirksamen Proteins produziert
wurden. IFN-γ ist das Leitzytokin einer Th1-Antwort. Im Rahmen der Leishmaniose ist es für
die Aktivierung der infizierten Makrophagen von entscheidener Bedeutung. IFN-γ stimuliert
die Makrophagen, wodurch es zur Aktivierung von iNOS und zur Produktion reaktiver
Sauerstoffverbindungen kommt, wodurch letztendlich die Parasiten abgetötet werden
(REINER et al 1995, CUNNUNGHAM et al 2002). Eine frühe IFN-γ-Produktion könnte
-143-
Diskussion
jedoch einen auto- oder parakrinen Effekt darstellen, da IFN-γ die Expression des Chemokins
I-TAC in der Gesamthaut und in Keratinozyten induziert.
Da I-TAC zum einen ein relativ unerforschtes Chemokin ist, welches bisher nicht in der
experimentellen Leishmaniose untersucht wurde und zudem das einzige Chemokin war, dass
von den suszeptiblen Mäusen exprimiert wurde, bezogen sich weitere Untersuchungen
speziell auf dieses Gen.
Real-Time-PCR-Ergebnisse zeigten, dass die I-TAC-Expression bereits nach 6 Std. in der
Gesamthaut infizierter Balb/c-Mäuse signifikant anstieg, und im Verlauf des ersten Tages
noch weiter an Stärke zunahm. Die starke Expression in den Keratinozyten von Balb/c-
Mäusen 16 Std. nach der Infektion, wurde zudem mittels iIFL und spezifischer
Hybridisierungssonden auf mRNA- und auf Protein-Ebene in der Epidermis bestätigt. Die
ISH und die iIFL sind sehr spezifische Verfahren für den morphologischen Nachweis
bestimmter Proteine oder mRNA-Abschnitte, allerdings ist die Sensitivität begrenzt und eine
Quantifizierung nur bedingt möglich. Die Signalgebung speziell in der Basal- und
Suprabasalschicht (Stratum spinosum, Stratum granulosum) der Epidermis 16 Std. nach der
Infektion spricht für eine aktive Produktion des Chemokins in den lebenden, teilungsaktiven
Keratinozyten. Charakteristisch dafür war, dass kein Signal in der oberflächlichen Schicht,
der Hornhaut (Stratum corneum) detektiert wurde, da es sich hier um abgestorbene
Korneozyten ohne Zellorganellen handelte, die nicht mehr zu einer Proteinbiosynthese
befähigt waren. Immunhistochemisch wurde die in der Real-Time-PCR gemessene stärkere
Expression in der Epidermis infizierter Balb/c-Mäuse durch eine stärkere Fluoreszenz als in
entsprechenden Schnitten von C57BL/6-Mäusen bestätigt. In den entsprechenden, nicht
infizierten Kontrollproben war kein Fluoreszenzsignal detektierbar, während die DIG
markierten mRNA-Sonden ein schwaches Signal gleicher Lokalisation wie in den infizierten
Proben zeigten. Dies war möglicherweise dadurch bedingt, dass in den Kontrollen zwar
mRNA exprimiert wurde, diese aufgrund regulatorischer Mechanismen jedoch nicht
translatiert wurde oder aber es bestanden hier unterschiedliche Sensitivitäten der Methoden.
Quantitative Unterschiede in der Expression zwischen den beiden Mausstämmen ließen sich
auf Grund der methodischen Beschränkungen der ISH mit dieser Technik nicht nachweisen.
Für diese Fragestellung war jedoch die quantitative Real-Time-PCR aussagekräftiger, die in
-144-
Diskussion
Keratinozyten und Gesamthautpräparaten gleichsinnige Ergebnisse einer differentiellen
Expression in den beiden Mausstämmen zeigte.
Erstmals entdeckt in IFN-γ aktivierten primären humanen Astrozyten wurde dieses Protein
„interferon-inducible T cell alpha chemoattractant“ (I-TAC) benannt. Die Tatsache, dass
I-TAC durch IFN hochreguliert wird sowie seine selektive chemotaktische Wirkung auf
T-Zellen lässt vermuten, dass es eine Rolle bei T-Zell-vermittelten, entzündlichen
Erkrankungen spielt (COLE et al. 1998). I-TAC ist auch ein starker chemotaktischer Faktor
für NK-Zellen (CAMPBELL et al. 2001, ROBERTSON 2002) und wird in IFN-γ stimulierten
Keratinozyten (TENSEN et al. 1999, ALBANESI et al. 2000), Monozyten (COLE et al.
1998), bronchialen Epithelzellen (SAUTY et al 1999), Endothelzellen (MAZANET et al.
2000) und PMN (GASPERINI et al. 1999) hochreguliert. Ein Jahr nach der Entdeckung
wurde die Expression eines Gens in der epidermalen Basalschicht von Gesamthautbioptaten
aus allergischer Kontaktdermatitis, Lichen Planus, und Mycosis Fungoides nachgewiesen,
dass als „IFN-γ-inducible protein-9“ (IP-9) bezeichnet wurde und welches sich später als
identisch zum bereits identifizierten I-TAC herausstellte (TENSEN et al. 1999). Die
Lokalisation dieses Chemokins in der Epidermis wurde auch in weiteren entzündlichen
Hautkrankheiten wie dem chronischem discoidalen Lupus Erythematosus und Jessner's
lymphozytärer Infiltration bestätigt (FLIER et al. 2001).
Zusammen mit IP-10 und MIG interagiert es mit dem CXC-Rezeptor 3 (CXCR3), wobei
I-TAC die höchste Affinität besitzt und daher den dominanten Liganden für diesen Rezeptor
darstellt (COLE et al. 1998). CXCR3 wird vornehmlich und in hohem Maße auf Th1-Zellen
exprimiert, welche aus dem Lymphknoten an den Ort ihres korrespondierenden Chemokins
migrieren (BONECCHI et al. 1998, SALLUSTO et al. 1998, EBERT et al. 2001). CXCR3
wurde auf CD3+-T-Zellen in der Dermis, und auf Lymphozyten, die einen speziellen T-Zell-
homing-Rezeptor besitzen, in der Epidermis von Psoriasisbioptaten nachgewiesen
(ROTTMAN et al 2001). CXCR3-exprimierende-T-Zellen sind auch in anderen
Autoimmunerkrankungen wie bspw. Rheumatoider Arthritis und Multipler Sklerose
nachgewiesen worden (LOETSCHER et al. 1998, BALASHOV et al. 1999, ARIMILLI et al.
2000, PATEL et al. 2001). Bei dem Großteil der genannten Erkrankungen wird ein
Überwiegen von Th1-Zellen beobachtet.
-145-
Diskussion
Neuere Untersuchungen haben gezeigt, dass alle drei CXCR3-Agonisten dazu befähigt sind,
den Chemokinrezeptor CCR3 zu inhibieren und I-TAC darüber hinaus spezifisch CCR5
blockieren kann (LOETSCHER et al. 2001, XANTHOU et al. 2003, PETKOVIC 2004).
CCR3 ist der Rezeptor für alle MCPs und zu den Liganden für CCR5 gehören RANTES,
MIP-1α und MIP-1β. Sie werden beide von T-Zellen und Epithelzellen sowie einer Vielzahl
weiterer Zellen exprimiert (MURPHY et al. 2000).
Bisher gibt es nur eine einzige Studie, die sich mit der Bedeutung des I-TAC-Rezeptors für
den Verlauf der experimentellen Leishmaniose, jedoch nicht mit der biologischen Funktion
des Chemokins befasst (ROSAS et al. 2005). Eine Infektion von C57BL/6-Mäusen, die kein
CXCR3 besitzen (CXCR3-knockout-Mäuse) mit L. major in die Hinterfußsohle führte zu
einer erhöhten Produktion von IFN-γ und IL-12 im lokalen Lymphknoten. Allerdings konnte
die Replikation der Leishmanien am Infektionsort nicht unter Kontrolle gebracht werden und
es entwickelten sich chronische, nicht-heilende Läsionen. Parallel dazu war die Zahl CD4+-,
CD8+-T-Zellen sowie die IFN-γ-Sekretion lokal am Infektionsort signifikant niedriger als in
den Wildtyp-Mäusen. Für die hier vorgelegten Untersuchungen sind allerdings Daten, die an
knockout-Mäusen gewonnen wurden, nur begrenzt aussagekräftig, da selektiv die Wirkung
der Substanzen speziell in dem kleinen Zeitfenster kurz nach der Infektion analysiert werden
sollte.
Obwohl MIG, IP-10 und I-TAC nah verwandte Chemokine mit großer Sequenzidentität sind,
die alle durch IFN-γ induziert werden, bestätigt die vorliegende Arbeit frühere
Untersuchungen, in denen das Expressionsmuster dieser Moleküle nicht identisch war
(FLIER et al. 2001). Grund dafür könnten Unterschiede in der Promotorregion der Gene sein,
die für diese drei Chemokine kodieren, in Kombination mit zelltypspezifischer Expression
von regulierenden Proteinen, welche entweder selektiv die IFN-induzierte Genexpression
modulieren oder an regulatorische Sequenzen in der entsprechenden Promotorregion binden
(LUSTER et al. 1987, WRIGHT et al. 1991, TENSEN et al. 1999).
-146-
Diskussion
6.2.3 Der pharmakologische Einfluss von I-TAC in L.-major-infizierten, resistenten Mäusen
Da es bei den Balb/c-Mäusen zu einer massiven Vermehrung und Ausbreitung der
Leishmanien kommt, wurde vermutet, dass I-TAC auf diesen Verlauf Einfluss nimmt.
Deshalb wurde der Krankheitsverlauf unter I-TAC-Applikation in C57BL/6-Mäusen, bei
denen die Vermehrung und Ausbreitung normalerweise kontrolliert wird, mit jener
unbehandelter C57BL/6-Mäuse verglichen. Die alleinige, lokale Applikation von 1 µg I-TAC
simultan mit der Infektion mit L. major und einer weiteren Applikation 4 Std. nach der
Infektion, zeigte in C57BL/6-Mäusen einen signifikanten jedoch transienten Effekt auf die
Entzündungsreaktion, der sein Maximum erst nach 60 Tagen erreichte. Dieser verzögerte
Effekt ist ein eindeutiger Hinweis darauf, dass die frühe I-TAC-Applikation
Differenzierungswege auslösen muss, die über Wochen Einfluss auf die sich entwickelnde
Entzündungsreaktion nehmen. Dieser Effekt wurde in drei unabhängig voneinander
durchgeführten Versuchen bestätigt. Die Tatsache, dass die I-TAC-Applikation am Anfang
der Infektion nicht zu einer vollständigen Umkehr des Infektionsverlaufs führte, kann zum
einen darin begründet sein, dass der optimale Zeitpunkt für dieses Experiment nicht bekannt
ist. Zudem müssen wir aufgrund der stärkeren Expression der meisten Zyto- und Chemokine
(16 von 19 untersuchten Genen, s. S. 135) in den resistenten Mäusen davon ausgehen, dass
die Resistenz gegenüber Leishmanien multifaktoriell bedingt ist, so das nicht davon
auszugehen ist, dass bei der Zufuhr eines einzelnen potentiell aggravierenden Faktors
(I-TAC), die gegenläufige Wirkung sämtlicher übriger potentiell protektiven Faktoren
aufgehoben werden kann. Diese Tatsache verdeutlicht die Signifikanz des beobachteten
Effektes. In Bezug auf die Anzahl der lebenden Parasiten konnte kein signifikanter Effekt
beobachtet werden, welches am ehesten durch die wesentlich stärkere Varianz der
Parasitenbestimmung im Vergleich zu den Fußdickenmessungen zu erklären ist. Im Rahmen
dieser Arbeit war es nicht möglich umfangreichere zeit- und kostenintensive in vivo Studien
durchzuführen. Durch eine Optimierung des Versuchsprotokolls hinsichtlich Dosierung und
Kinetik der Applikation sollte es jedoch in weiterführenden Arbeiten möglich sein, endgültige
Aussagen bezüglich des in vivo Effektes einer I-TAC-Applikation zu treffen. Zunächst sollte
untersucht werden, ob eine I-TAC-Applikation einen Effekt auf die Th-Zell-Differenzierung
-147-
Diskussion
hat. Anhand des mit einer starken Latenz eintretenden Effektes der I-TAC-Applikation im
Rahmen dieser Arbeit, erscheint ein Einfluss auf die Th-Zell-Differenzierung wahrscheinlich,
da bis zur vollständigen Etablierung der spezifischen Immunantwort mehrere Wochen
vergehen. Eine direkte Beeinflussung der Parasitendezimierung durch I-TAC erscheint sehr
unwahrscheinlich, da lediglich am ersten Tag der Infektion I-TAC appliziert wurde und nicht
davon auszugehen ist, dass durch diese Applikation längerfristig wirksame I-TAC-Mengen
am Infektionsort vorhanden waren.
Das Ziel zukünftiger in vivo Experimente sollte somit die Entschlüsselung des möglichen
Wirkungsmechanismus der lokalen, epidermalen I-TAC-Expression sein. Denkbar wäre zum
einen eine Beeinflussung der infiltrierenden Zellen, wobei auch negative Effekte durch die
antagonistischen Wirkungen auf CCR3 und CCR5 beispielsweise in Bezug auf die geringere
Infiltration von Makrophagen in den Balb/c-Mäusen denkbar sind. Weiterhin ist aufgrund der
starken Induktion von I-TAC-mRNA und Protein in den Balb/c-Mäusen ein direkter Effekt
auf die Th-Zellen im lokalen Lymphknoten denkbar. Eine zusätzliche Möglichkeit besteht wie
für IL-4 beschrieben in einer Beeinflussung der emigrierenden DC. Im Gegensatz zu IL-4
müßte allerdings eine deaktivierende Wirkung auf die DC gefordert werden, um einen
möglichen Zusammenhang zwischen I-TAC-Expression und Generierung einer Th2-Antwort
zu erklären. Beispielsweise könnte die IL-12 Sekretion oder die Expression
kostimulatorischer Oberflächenproteine gehemmt werden. Hier sollten entsprechende in vitro
Experimente folgen.
6.2.4 Können Keratinozyten das spezifische Immunsystem beeinflussen, indem sie die Th-Zell-Differenzierung modulieren?
Im Rahmen der experimentellen Leishmaniose ist unklar, wie es zur Induktion von Zyto- und
Chemokinen in der Epidermis kommt. Eine direkte Interaktion der subkutan applizierten
Erreger und der Keratinozyten erscheint unwahrscheinlich, ist aber möglich. Eine
Phagozytose von Leishmanien durch Keratinozyten wurde bisher nie beschrieben. In der
Initialphase der Infektion kann eine Interaktion der Leishmanien mit den gewebsständigen
Makrophagen, sowie mit den sehr schnell infiltrierenden Granulozyten erfolgen. Zusätzlich
kann in begrenztem Maße eine Phagozytose der promastigoten Leishmanien durch DC
-148-
Diskussion
erfolgen. Die Phagozytose durch Makrophagen und DC führt dabei rasch zur Expression von
Immunmodulatoren in diesen Zellen (CHAUSSABEL et al. 2003). Diese sezernierten
Botenstoffe könnten dann wiederum die Keratinozyten beeinflussen und so zur Produktion
von weiteren immunologisch wirksamen Molekülen führen. Physiologisch würde solch eine
Interaktion sehr sinnvoll sein, da in der Frühphase der Infektion nur wenige Phagozyten am
Ort der Infektion vorhanden sind. Die in großer Anzahl vorhandenen Keratinozyten könnten
eine Art zellulären Verstärker darstellen, über den die Immunantwort schnell eingeleitet
werden könnte.
Eine Beeinflussung des Immunsystems ist auf vielschichtige Art und Weise möglich. Durch
Auswirkungen auf die Migration von DC kann ein indirekter Effekt auf die T-Zell-
Differenzierung und somit die spezifische Immunantwort erfolgen. Zusätzlich kann die
kostimulatotrische Aktivität sowie die Zytokinproduktion der DC beeinflusst werden. Dies
wiederum nimmt Einfluss auf die Interaktion der DC mit den Th-Zellen im lokalen
Lymphknoten und so die Th-Differenzierung bzw Polarisierung in Richtung Th1 oder Th2.
Eine weitere indirekte Beeinflussung von DC wäre auch über Makrophagen denkbar, welche
durch die von den Keratinozyten produzierten Zytokine stimuliert werden könnten. Da die
Induktion der epidermalen mRNA-Expression einiger Immunmodulatoren im Rahmen der
Infektion sehr stark ist, kann vermutet werden, dass dies auch zur Sekretion großer Mengen
der entsprechenden Proteine führt. Die Immunmodulatoren könnten somit neben lokalen
Effekten auch direkt auf die T-Zellen im lokalen Lymphknoten wirken und somit die Th-Zell-
Differenzierung direkt beeinflussen (JANEWAY et al. 2002).
Dass Keratinozyten grundsätzlich in der Lage sind, die spezifische Immunantwort zu
beeinflussen wurde im letzten Jahr durch ZENZ et al. (2005) an einem murinen Psoriasis-
Modell gezeigt. Da in Psoriasishautläsionen beim Menschen die Expression der Proteine JunB
und c-Jun herunterreguliert wird, wurde ein transgenes Maus-Modell entwickelt, in dem beide
Proteine selektiv nicht mehr epidermal exprimiert werden können. Dies führte zu einer
deutlichen Störung des epidermalen Zytokin-Metabolismus und zur Entwicklung von
psoriasiformen Hautläsionen. Unter anderem kam es zu einer sehr starken, frühen Zunahme
der S100A8- und S100A9-Expression in der Epidermis. Interessanterweise entwickelten die
transgenen Mäuse auch arthritische Läsionen. Dieser Effekt war T-Zell-abhängig. Somit
-149-
Diskussion
wurde gezeigt, dass eine Veränderung des epidermalen Zytokinmetabolismus zur Generierung
autoreaktiver T-Zellen führt und somit einen eindeutigen Effekt auf die spezifische
Immunantwort hat (ZENZ et al. 2005).
Die im Rahmen dieser Arbeit anhand der experimentellen Leishmaniose erhaltenen Resultate
können auch für das bessere Verständnis der Pathogenese nicht-infektiöser entzündlicher
Dermatose bedeutsam sein.
Bei vielen entzündlichen Dermatosen unklarer Pathogenese, wie z.B. der atopischen
Dermatitis und der Psoriasis liegen gleichzeitig sowohl Defekte der Th-Zell-Differenzierung
als auch ein gestörter Metabolismus der Epidermis vor. Generell wird momentan davon
ausgegangen, dass der Defekt der spezifischen Immunantwort dabei das primäre
pathogenetische Prinzip ist. Die jüngst von ZENZ et al. (2005) publizierten Daten sowie die
in dieser Arbeit präsentierten Ergebnisse sprechen aber dafür, dass prinzipiell auch primäre
Störungen des Keratinozytenmetabolismus die spezifische Immunantwort beeinflussen
können. Dies besitzt große Relevanz für die Entwicklung ursächlicher Therapien. Momentan
erfolgt bei der atopischen Dermatitis und der Psoriasis primär eine immunsuppressive
Therapie, wobei es nach Absetzen der Therapie meist schnell zu einem Rezidiv kommt. Die
Entwicklung von Therapiekonzepten, die ihre Wirkung über die Keratinozyten vermitteln,
könnte von entscheidenem Nutzen für eine optimierte Behandlung dieser wichtigen
Erkrankungen sein. Zunächst jedoch muss überprüft werden, ob die am Mausmodell
erhaltenen Ergebnisse auf andere Species (z.B. Hund, Katze, Mensch) und auf
nicht-infektiöse Dermatosen übertragen werden können. Insofern konnten durch diese Arbeit
vielfältige Ansätze für weitere Untersuchungen zur Pathogenese entzündlicher Dermatosen
liefern. So könnten mit Hilfe der Lasermikrodissektion spezifische Zielzellen (Makrophagen,
DC, Granulozyten, Endothelzellen etc.) gezielt isoliert und untersucht werden. Außerdem
könnte die Rolle der Keratinozyten unter Berücksichtung kinetischer Gesichtspunkte in
weiteren entzündlichen Dermatosen untersucht werden, wie bspw. bei Mäusen, in denen eine
Kontaktallergie induziert wird oder bei NC/Nga Mäusen (MATSUDA et al. 1997) oder im
Psoriasis-SCID-Maus-Modell (NICKOLOFF et al. 2000). Außerdem bietet sich mit dieser
Methodik die Untersuchung der Keratinozyten von Bioptaten aus entzündlich veränderter
Haut beim Menschen oder Tier an (usw.).
-150-
Zusammenfassung
7 ZUSAMMENFASSUNG
Kirsten Roebrock
Die Rolle des Epithels der Haut in der Frühphase der Leishmania-major-Infektion
Entzündliche Dermatosen wie die Psoriasis und die atopische Dermatitis, die mit einem
Ungleichgewicht in der T-Helfer-Zell-Differenzierung, insbesondere mit einer über-
schießenden Proliferation von Th2-Lymphozyten, einhergehen, haben bei Mensch und Tier
aufgrund zunehmender Inzidenz und unbefriedigenden Therapiemöglichkeiten vermehrt an
Bedeutung gewonnen. Die allgemeinen Prinzipien der T-Zell-Differenzierung sind gut
untersucht worden, jedoch sind die frühen Vorgänge in der Haut vor der endgültigen
T-Zell-Differenzierung im Lymphknoten weitestgehend unbekannt. Es gibt Hinweise darauf,
dass Keratinozyten in diesem Zusammenhang eine bislang unterbewertete Rolle spielen.
Gerade das frühe Stadium einer Entzündung ist aber offensichtlich richtungweisend dafür, ob
es zur Ausbildung einer Th1- oder Th2-Antwort kommt.
Um einen Beitrag zur Aufklärung der Rolle der Keratinozyten in der Frühphase einer
Entzündung zu leisten, wurde mit der experimentellen Leishmaniose ein etabliertes
Standardmodell für die Erforschung der T-Zell-Differenzierung verwendet. Die Infektion mit
L. major induziert bei den meisten Mausstämmen (z.B. C57BL/6) eine Th1-Antwort, die in
Form einer auf die Haut und die lokalen Lymphknoten beschränkten, selbst limitierenden
Erkrankung verläuft. Bei einigen Mausstämmen (z.B. Balb/c) führt die Infektion zu einer
Th2-Antwort und einer letal endenden Ausbreitung der Parasiten.
Der Schwerpunkt dieser Arbeit lag auf der selektiven Untersuchung der von residenten Zellen
der Epidermis (Keratinozyten) produzierten Immunmodulatoren in Balb/c- und C57BL/6-
Mäusen in vivo unter Verwendung der „laser microbeam microdissection und laser pressure
catapulting“ Technologie. Die Methode musste dazu vollständig für dieses Modell etabliert
werden. Mit Hilfe einer auf PCR basierenden Amplifikation wurde die cDNA von ca. 1000
lasermikrodissezierten Keratinozyten zunächst „vor“-amplifiziert, um anschließend
umfassende Genexpressionsanalysen durch quantitative Real-Time-PCR durchführen zu
können.
-151-
Zusammenfassung
Erstmals konnte so gezeigt werden, dass Keratinozyten sehr schnell nach einer Infektion mit
einer aktiven Produktion von Immunmodulatoren reagieren. Bereits innerhalb der ersten 16
Stunden der Infektion wurde eine Vielzahl von Immunmodulatoren von den Keratinozyten
produziert (Zytokine, Chemokine, S100-Proteine, Interferon-γ und Tumornekrosefaktor-α,
etc.). Einige dieser Gene wurden bereits eine Stunde nach der Infektion „angeschaltet“, und
auch bald wieder „abgeschaltet“ (Osteopontin, IL-4, I-TAC). Die Expression der meisten
Gene war deutlich stärker in den Keratinozyten von resistenten C57BL/6-Mäusen als in den
suszeptiblen Balb/c-Mäusen.
Eine Ausnahme bildete in diesem Zusammenhang das ‚Interferon (gamma) induced T-cell
alpha chemoattractant“ (I-TAC), das bereits nach 6 Stunden signifikant stärker in suszeptiblen
Balb/c-Mäusen im Vergleich zu den resistenten C57BL/6-Mäusen exprimiert wurde. Die
Expression von I-TAC wurde hier zum ersten Mal in der experimentellen Leishmaniose
dokumentiert. Aufgrund dieser Beobachtungen bezogen sich nachfolgende Untersuchungen
auf die Bestätigung der I-TAC-Expression in der Epidermis durch indirekte
Immunfluoreszenz und in situ-Hybridisierung sowie auf die Untersuchung der biologischen
Relevanz dieser differentiellen Expression von I-TAC. Diese wurde durch einen negativen
Einfluß einer I-TAC-Applikation in den ersten 4 Stunden der Entzündung auf den Verlauf der
Infektion bestätigt, die in resistenten C57BL/6-Mäusen noch 6 Wochen nach der Infektion zu
einer stärkeren Fußschwellung als in den Kontrolltieren führte, welche sich erst 2 Wochen
danach normalisierte. Die Tatsache, dass die Resistenzlage von C57BL/6-Mäusen in diesen
Experimenten nicht vollständig umgekehrt wurde, liegt zum einen darin begründet, dass
wahrscheinlich eine Vielzahl weiterer Immunmodulatoren den negativen I-TAC-Effekt
kompensieren können. Zum anderen sind weitere Dosis- und Zeitkinetiken der I-TAC-
Applikation erforderlich, um die optimalen Effekte von I-TAC auf die T-Zell-Differenzierung
und Resistenzentwicklung in vivo zu ermitteln.
Die hier gezeigte differentielle Expression von Immunmodulatoren in Keratinozyten
zwischen suszeptiblen und resistenten Mäusen ist ein Beleg dafür, dass die Induktion der
epidermalen Expression solcher Immunmodulatoren einen Einfluss auf die Generierung der
spezifischen Immunantwort und somit auf Resistenz und Suszeptibilität in der
experimentellen Leishmaniose hat. Die molekularen Mechanismen, die für die Induktion der
-152-
Zusammenfassung
Genexpression in den Keratinozyten während der Frühphase der experimentellen
Leishmaniose verantwortlich sind, sind derzeit nicht geklärt. Zum einen können die subkutan
injizierten Parasiten einen direkten Einfluss auf die Keratinozyten haben. Zum anderen
können Interaktionen der Keratinozyten mit gewebsständigen Makrophagen und früh
infiltrierenden Granulozyten der Auslöser für die Synthese immunologisch wirksamer
Substanzen in der Epidermis sein. In diesem Fall läge die physiologische Bedeutung in einer
Verstärkerwirkung oder Modulation der Frühphase der Entzündung durch Keratinozyten, da
die Keratinozyten besonders in dieser Initialphase zahlenmäßig den Phagozyten überlegen
sind. Die dann folgenden Mechanismen, mit denen die Keratinozyten die spätere T-Zell-
Differenzierung beeinflussen können, sind vielfältig. Dabei sind direkte Effekte auf T-Zellen,
eine Beeinflussung der Differenzierung und der Migrationsfähigkeit dendritischer Zellen oder
indirekte Effekte vermittelt durch Makrophagen auf die dendritischen Zellen oder
T-Lymphozyten denkbar.
Die im Rahmen dieser Arbeit anhand der experimentellen Leishmaniose erhaltenen Resultate
können auch für das bessere Verständnis der Pathogenese nicht infektiöser, entzündlicher
Dermatosen bedeutsam sein. Bei vielen relevanten Erkrankungen wird eine pathologische
Th1/Th2-Zell-Differenzierung als das primäre pathogenetische Prinzip angesehen. Meine
Daten deuten darauf hin, dass prinzipiell auch primäre Störungen der Keratinozyten-
aktivierung diese Differenzierungswege beeinflussen können. Diese Tatsache besitzt
möglicherweise große Relevanz für die Entwicklung neuer, causaler Therapien entzündlicher,
nicht-infektiöser Dermatosen. Die mehr oder weniger ungezielte immunsuppressive Therapie
stellt zurzeit die einzige Behandlungsmöglichkeit dieser Erkrankungen dar. Die Entwicklung
von Therapiekonzepten, die gezielt in diese Differenzierungswege eingreifen, möglicherweise
über die Modulation von Keratinozyten, könnte von entscheidenem Nutzen für die optimierte
Behandlung weit verbreiteter Erkrankungen wie Psoriasis und atopischer Dermatitis sein.
Insofern ergeben sich durch diese Arbeit vielfältige Ansätze, die Pathogenese entzündlicher
Dermatosen weiter aufzuklären.
-153-
Summary
8 SUMMARY
Kirsten Roebrock
The role of the cutaneous epithelium in the early stage of a Leishmania-major-infection
Inflammatory skin disorders such as psoriasis and atopic dermatitis are associated with an
imbalance of the T-helper-cell-subsets and in particular, a dominance of Th2-cells. These
disorders have gained increasing importance in humans and animals due to rising incidences
and insufficient treatment. The overall concept of T-cell differentiation is well-investigated,
but early events in the skin before final T-cell priming take place in lymphnodes, are largely
unknown. There is evidence that keratinocytes play a thus far, underestimated role in the
process of T-cell-differentiation. During the events taking place at early stages of
inflammation, crucial factors direct the response the immune system will take such as the
development of a Th2 or Th1 response.
To further clarify the role keratinocytes play at the initial stage of inflammation, an
established standard model for the investigation of T-cell-differentiation, experimental
Leishmaniose, was applied. Infection with L. major induces in the majority of inbred mice
(e.g. C57BL/6) a Th1 response, resulting in a disease which is limited to the skin and the local
lymphnodes and which heals spontaneously. Some mice strains (e.g. Balb/c) develop a Th2
response after infection, a process that is characterized by lethal propagation of the parasite.
The main focus of this work involved the specific investigation of resident cells in the
epidermis (keratinocytes) and the subsequent production of immune modulators in Balb/c and
C57BL/6 mice in vivo. The techniques employed in this study involved the use of laser
microbeam microdissection and laser pressure catapulting and required full establishment for
this model. The cDNA of approximately 1000 keratinocytes was preamplified by means of
random based PCR amplification, prior to gene expression analysis by Real-Time-PCR.
Analysis of gene expression showed for the first time, that keratinocytes are involved in the
production of immune modulators rapidly after infection. As early as 16 hours after infection,
a multitude of immune modulators were expressed (cytokines, chemokines, S100-proteins,
interferon-γ, tumor necrosis factor-α, etc.). The expression of some of these genes was
-154-
Summary
evident after as little as one hour after infection and was soon switched off again
(Osteopontin, IL-4, I-TAC). The expression level of the majority of genes was clearly higher
in keratinocytes and the skin of resistant C57BL/6 mice than in Balb/c mice.
An exception to this was the expression of interferon inducible T-cell chemoattractant
(I-TAC), which showed a significant increase in expression levels within 6 hours of infection
in Balb/c compared to C57BL/6 mice. This is the first report of the expression of I-TAC in
experimental Leishmaniose. These results were substantiated by the confirmation of I-TAC
expression in the epidermis by immunofluorescence and in situ hybridization. The biological
relevance of this differential expression was confirmed by a negative influence on the course
of infection in C57BL/6 mice after I-TAC administration in the first 4 hours of infection. In
these animals, six weeks p.i. a stronger footpad swelling which lasted for two weeks was
observed when compared to control animals. An explanation as to why resistant mice don’t
become completely susceptible by the application of a single protein that seems to be
associated with susceptibility may be, that the strong expression of a variety of other factors
in these mice are able to compensate for this negative effect. However, further I-TAC dose
response experiments will be necessary in order to determine optimal effects on T-cell-
differentiation and development of resistance.
The demonstrated differential expression of immune modulators in keratinocytes of
susceptible and resistant Leishmania infected mice, indicates that this induction of the
epidermal gene expression has an impact on the generation of the specific immune response
and hence on resistance and susceptibility in experimental Leishmaniasis. However, the
molecular mechanisms that are involved in the induction of gene expression in keratinocytes
at the early stage of experimental Leishmaniasis remain unexplained. It is possible that the
subcutaneous administration of these parasites could have a direct influence on keratinocytes.
Alternatively, the interactions between keratinocytes and resident macrophages, as well as the
fast infiltrating granulocytes, could be a catalyst for the synthesis of immunologically active
substances in the epidermis. In this case, the physiological significance is expressed by an
amplifying function and/or modulation of the initial inflammatory phase by keratinocytes,
especially since they are far more abundant at the early stage of inflammation than
phagocytes.
-155-
Summary
The consecutive mechanisms, by which keratinocytes are able to influence the later T-cell-
differentiation, are manifold. Direct effects on T-cells as well as an influence on the
differentiation and migration capacity of dendritic cells or indirect effects mediated by
macrophages on dendritic cells or T-lymphocytes, are possible.
The results gained in this work from experimental Leishmaniasis, could also contribute to a
better understanding of the pathogenesis of non-infectious inflammatory skin diseases. A
pathologic Th1/Th2 differentiation is so far considered to be the major pathogenetical
principle in many relevant diseases. The presented data suggests that a primary dysfunction of
the activation of keratinocytes could, in principle, have an influence on these pathways of
differentiation. This fact has potential relevance for the development of new and causal
therapies for inflammatory, non-infectious skin disorders. Immunosuppressive therapy, which
has significant downfalls, is currently the only possible treatment. The development of new
therapeutics by rational drug design, which target these pathways of differentiation and
possibly the modulation of keratinocytes, could be of pivotal benefit for an effective treatment
of common diseases such as psoriasis and atopic dermatitis. Therefore, from this work
presented in this study, many aspects arise on how to further clarify the pathogenesis of
inflammatory skin diseases.
-156-
Literaturverzeichnis
9 LITERATURVERZEICHNIS
ABBAS, A. K., K. M. MURPHY u. A. SHER (1996): Functional diversity of helper T lymphocytes. Nature 383, 787-793
ALBANESI, C., C. SCARPONI, S. SEBASTIANI, A. CAVANI, M. FEDERICI, P. O. DE, P. PUDDU u. G. GIROLOMONI (2000): IL-4 enhances keratinocyte expression of CXCR3 agonistic chemokines. J. Immunol. 165, 1395-1402
ALBANESI, C., C. SCARPONI, M. L. GIUSTIZIERI u. G. GIROLOMONI (2005): Keratinocytes in inflammatory skin diseases. Curr. Drug Targets. Inflamm. Allergy 4, 329-334
ALEXANDER, J. u. K. BRYSON (2005): T helper (h)1/Th2 and Leishmania: paradox rather than paradigm. Immunol. Lett. 99, 17-23
ALTSCHUL, S. F., W. GISH, W. MILLER, E. W. MYERS u. D. J. LIPMAN (1990): Basic local alignment search tool. J. Mol. Biol. 215, 403-410
ARIMILLI, S., W. FERLIN, N. SOLVASON, S. DESHPANDE, M. HOWARD u. S. MOCCI (2000): Chemokines in autoimmune diseases. Immunol. Rev. 177, 43-51
ARNOLDI, J. u. H. MOLL (1998): Langerhans cell migration in murine cutaneous leishmaniasis: regulation by tumor necrosis factor alpha, interleukin-1 beta, and macrophage inflammatory protein-1 alpha. Dev. Immunol. 6, 3-11
ARTIS, D., L. M. JOHNSON, K. JOYCE, C. SARIS, A. VILLARINO, C. A. HUNTER u. P. SCOTT (2004): Cutting edge: early IL-4 production governs the requirement for IL-27-WSX-1 signaling in the development of protective Th1 cytokine responses following Leishmania major infection. J. Immunol. 172, 4672-4675
ASHFORD, R. W. (2000): The leishmaniases as emerging and reemerging zoonoses. Int. J. Parasitol. 30, 1269-1281
-157-
Literaturverzeichnis
ASHKAR, S., G. F. WEBER, V. PANOUTSAKOPOULOU, M. E. SANCHIRICO, M. JANSSON, S. ZAWAIDEH, S. R. RITTLING, D. T. DENHARDT, M. J. GLIMCHER u. H. CANTOR (2000): Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287, 860-864
ASSREUY, J., F. Q. CUNHA, M. EPPERLEIN, A. NORONHA-DUTRA, C. A. O'DONNELL, F. Y. LIEW u. S. MONCADA (1994): Production of nitric oxide and superoxide by activated macrophages and killing of Leishmania major. Eur. J. Immunol. 24, 672-676
AUSUBEL, F. M., R. BRENT, R. E. KINGSTON, D. D. MOORE, I. G. SEIDMAN, J. A. SMITH, u. K. STRUHL (1989): Current protocols in molecular biology. Verlag Greene Publishing Associates and Wiley-Interscience, New York BAGLEY, J., T. SAWADA, Y. WU u. J. IACOMINI (2000): A critical role for interleukin 4 in activating alloreactive CD4 T cells. Nat. Immunol. 1, 257-261
BALASHOV, K. E., J. B. ROTTMAN, H. L. WEINER u. W. W. HANCOCK (1999): CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. U.S.A 96, 6873-6878
BECKER, I., K. F. BECKER, M. H. ROHRL u. H. HOFLER (1997): Laser-assisted preparation of single cells from stained histological slides for gene analysis. Histochem. Cell Biol. 108, 447-451
BEEBE, A. M., S. MAUZE, N. J. SCHORK u. R. L. COFFMAN (1997): Serial backcross mapping of multiple loci associated with resistance to Leishmania major in mice. Immunity 6, 551-557
BEEBE, A. M., D. J. CUA u. R. L. COFFMAN (1999): Genetic control of T helper subset differentiation in Leishmania major infection of mice. Microbes. Infect. 1, 89-94
BEHIN, R., J. MAUEL u. B. SORDAT (1979): Leishmania tropica: pathogenicity and in vitro macrophage function in strains of inbred mice. Exp. Parasitol. 48, 81-91
BEIL, W. J., G. MEINARDUS-HAGER, D. C. NEUGEBAUER u. C. SORG (1992): Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J. Leukoc. Biol. 52, 135-142
-158-
Literaturverzeichnis
BELKAID, Y., S. MENDEZ, R. LIRA, N. KADAMBI, G. MILON u. D. SACKS (2000): A natural model of Leishmania major infection reveals a prolonged "silent" phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165, 969-977
BELKAID, Y., K. F. HOFFMANN, S. MENDEZ, S. KAMHAWI, M. C. UDEY, T. A. WYNN u. D. L. SACKS (2001): The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194, 1497-1506
BELKAID, Y., S. E. VON, S. MENDEZ, R. LIRA, E. CALER, S. BERTHOLET, M. C. UDEY u. D. SACKS (2002): CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol. 168, 3992-4000
BELKAID, Y., C. A. PICCIRILLO, S. MENDEZ, E. M. SHEVACH u. D. L. SACKS (2002): CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502-507
BERSUDSKY, M., R. N. APTE u. J. EL-ON (2000): Interleukin 1alpha activity of peritoneal and bone marrow macrophages infected with Leishmania major and Leishmania donovani in vitro. Exp. Parasitol. 94, 150-157
BERSUDSKY, M., A. LABAN u. J. EL-ON (2005): Immunological responses and the pattern of disease in mice infected with transfected Leishmania major constitutively expressing active IL-1alpha. Vector. Borne. Zoonotic. Dis. 5, 324-329
BIANCHI, G., S. SOZZANI, A. ZLOTNIK, A. MANTOVANI u. P. ALLAVENA (1996): Migratory response of human natural killer cells to lymphotactin. Eur. J. Immunol. 26, 816-824 BIEDERMANN, T., S. ZIMMERMANN, H. HIMMELRICH, A. GUMY, O. EGETER, A. K. SAKRAUSKI, I. SEEGMULLER, H. VOIGT, P. LAUNOIS, A. D. LEVINE, H. WAGNER, K. HEEG, J. A. LOUIS u. M. ROCKEN (2001): IL-4 instructs TH1 responses and resistance to Leishmania major in susceptible Balb/c mice. Nat. Immunol. 2, 1054-1060
BIRNBOIM, H. C. u. J. DOLY (1979): A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513-1523
-159-
Literaturverzeichnis
BLANK, C., H. FUCHS, K. RAPPERSBERGER, M. ROLLINGHOFF u. H. MOLL (1993): Parasitism of epidermal Langerhans cells in experimental cutaneous leishmaniasis with Leishmania major. J. Infect. Dis. 167, 418-425
BLAUVELT, A., S. T. HWANG u. M. C. UDEY (2003): 11. Allergic and immunologic diseases of the skin. J. Allergy Clin. Immunol. 111, S560-S570
BOGDAN, C., H. MOLL, W. SOLBACH u. M. ROLLINGHOFF (1990): Tumor necrosis factor-alpha in combination with interferon-gamma, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur. J. Immunol. 20, 1131-1135
BOGDAN, C. u. M. ROLLINGHOFF (1998): The immune response to Leishmania: mechanisms of parasite control and evasion. Int. J. Parasitol. 28, 121-134
BOGDAN, C. (2000): The function of type I interferons in antimicrobial immunity. Curr. Opin. Immunol. 12, 419-424
BONECCHI, R., G. BIANCHI, P. P. BORDIGNON, D. D'AMBROSIO, R. LANG, A. BORSATTI, S. SOZZANI, P. ALLAVENA, P. A. GRAY, A. MANTOVANI u. F. SINIGAGLIA (1998): Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187, 129-134
BOS, J. D., M. B. TEUNISSEN u. M. L. KAPSENBERG (1989): Immunology of contact dermatitis. Acta Derm. Venereol. Suppl (Stockh) 151, 84-87
BOS, J. D. (1997): The skin as an organ of immunity. Clin. Exp. Immunol. 107, 3-5
BRETSCHER, P. u. M. COHN (1970): A theory of self-nonself discrimination. Science 169, 1042-1049
BROOME, A. M., D. RYAN u. R. L. ECKERT (2003): S100 protein subcellular localization during epidermal differentiation and psoriasis. J. Histochem. Cytochem. 51, 675-685
-160-
Literaturverzeichnis
BUSTIN, S. A. (2002): Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J. Mol. Endocrinol. 29, 23-39
CACERES-DITTMAR, G., F. J. TAPIA, M. A. SANCHEZ, M. YAMAMURA, K. UYEMURA, R. L. MODLIN, B. R. BLOOM u. J. CONVIT (1993): Determination of the cytokine profile in American cutaneous leishmaniasis using the polymerase chain reaction. Clin. Exp. Immunol. 91, 500-505
CAMPBELL, J. J., S. QIN, D. UNUTMAZ, D. SOLER, K. E. MURPHY, M. R. HODGE, L. WU u. E. C. BUTCHER (2001): Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J. Immunol. 166, 6477-6482
CAMPBELL, K. A., P. J. OVENDALE, M. K. KENNEDY, W. C. FANSLOW, S. G. REED u. C. R. MALISZEWSKI (1996): CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity 4, 283-289
CARRERA, L., R. T. GAZZINELLI, R. BADOLATO, S. HIENY, W. MULLER, R. KUHN u. D. L. SACKS (1996): Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J. Exp. Med. 183, 515-526
CARSON, W. E., M. E. ROSS, R. A. BAIOCCHI, M. J. MARIEN, N. BOIANI, K. GRABSTEIN u. M. A. CALIGIURI (1995): Endogenous production of interleukin 15 by activated human monocytes is critical for optimal production of interferon-gamma by natural killer cells in vitro. J. Clin. Invest 96, 2578-2582
CHAMBERS, C. A. u. J. P. ALLISON (1997): Co-stimulation in T cell responses. Curr. Opin. Immunol. 9, 396-404
CHANG, K. P. (1983): Cellular and molecular mechanisms of intracellular symbiosis in leishmaniasis. Int. Rev. Cytol. Suppl 14, 267-305
CHATELAIN, R., K. VARKILA u. R. L. COFFMAN (1992): IL-4 induces a Th2 response in Leishmania major-infected mice. J. Immunol. 148, 1182-1187
-161-
Literaturverzeichnis
CHAUSSABEL, D., R. T. SEMNANI, M. A. MCDOWELL, D. SACKS, A. SHER u. T. B. NUTMAN (2003): Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 102, 672-681
CHOMCZYNSKI, P. u. N. SACCHI (1987): Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156-159
CILLARI, E., M. DIELI, E. MALTESE, S. MILANO, A. SALERNO u. F. Y. LIEW (1989): Enhancement of macrophage IL-1 production by Leishmania major infection in vitro and its inhibition by IFN-gamma. J. Immunol. 143, 2001-2005
COCA, A. F. u. R. A. COOKE (1923): The classification of the phenomena of hypersensitiveness. J. Immunol. 8, 163-182 COLE, K. E., C. A. STRICK, T. J. PARADIS, K. T. OGBORNE, M. LOETSCHER, R. P. GLADUE, W. LIN, J. G. BOYD, B. MOSER, D. E. WOOD, B. G. SAHAGAN u. K. NEOTE (1998): Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 187, 2009-2021
CONSTANT, S. L. u. K. BOTTOMLY (1997): Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15, 297-322
CONSTANT, S. L., K. S. LEE u. K. BOTTOMLY (2000): Site of antigen delivery can influence T cell priming: pulmonary environment promotes preferential Th2-type differentiation. Eur. J. Immunol. 30, 840-847
COOMBS, R. R. A. u. P. G. H. GELL (1963): The classification of allergic reactions underlying disease. In: COOMBS, R. R. A. u. P. G. H. GELL (Hrsg.): Clinical aspects of immunology. 2. Aufl. Verlag Blackwell Scientific Publications, Oxford, London S. 317-320 CORRY, D. B., S. L. REINER, P. S. LINSLEY u. R. M. LOCKSLEY (1994): Differential effects of blockade of CD28-B7 on the development of Th1 or Th2 effector cells in experimental leishmaniasis. J. Immunol. 153, 4142-4148
-162-
Literaturverzeichnis
CUMBERBATCH, M., R. J. DEARMAN, R. W. GROVES, C. ANTONOPOULOS u. I. KIMBER (2002): Differential regulation of epidermal langerhans cell migration by interleukins (IL)-1alpha and IL-1beta during irritant- and allergen-induced cutaneous immune responses. Toxicol. Appl. Pharmacol. 182, 126-135
CUNNINGHAM, D. D. (1885): On the presence of peculiar parasitic organisms in the tissue of a specimen of Delhi boil. Sci. Mem. Med. Offic. Army India 1, 21-31
CUNNINGHAM, A. C. (2002): Parasitic adaptive mechanisms in infection by leishmania. Exp. Mol. Pathol. 72, 132-141
DELFINO, D., M. S. CHIOFALO, G. RIGGIO, M. C. ANGELICI, M. GRAMICCIA, L. GRADONI u. D. IANNELLO (1995): Induction of interleukin 1 alpha in murine macrophages infected in vitro with different species and strains of Leishmania. Microb. Pathog. 18, 73-80
DEMANT, P., M. LIPOLDOVA u. M. SVOBODOVA (1996): Resistance to Leishmania major in mice. Science 274, 1392-1393
DEMOTZ, S., H. M. GREY u. A. SETTE (1990): The minimal number of class II MHC-antigen complexes needed for T cell activation. Science 249, 1028-1030
DINARELLO, C. A. (1996): Biologic basis for interleukin-1 in disease. Blood 87, 2095-2147
DINARELLO, C. A. (2002): The IL-1 family and inflammatory diseases. Clin. Exp. Rheumatol. 20, 1-13
EBERT, L. M. u. S. R. MCCOLL (2001): Coregulation of CXC chemokine receptor and CD4 expression on T lymphocytes during allogeneic activation. J. Immunol. 166, 4870-4878
EBERWINE, J., H. YEH, K. MIYASHIRO, Y. CAO, S. NAIR, R. FINNELL, M. ZETTEL u. P. COLEMAN (1992): Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. U.S.A 89, 3010-3014
-163-
Literaturverzeichnis
EBNER, S., G. RATZINGER, B. KROSBACHER, M. SCHMUTH, A. WEISS, D. REIDER, R. A. KROCZEK, M. HEROLD, C. HEUFLER, P. FRITSCH u. N. ROMANI (2001): Production of IL-12 by human monocyte-derived dendritic cells is optimal when the stimulus is given at the onset of maturation, and is further enhanced by IL-4. J. Immunol. 166, 633-641
EHRCHEN, J., C. SORG u. C. SUNDERKÖTTER (2002): Resistance in experimental Leishmaniasis is associated with early expression of the Th1-inducing cytokine osteopontin. J. Invest. Dermatol. 119, 603 Part 2 meeting abstract.
EHRCHEN, J., A. SINDRILARU, S. GRABBE, F. SCHONLAU, C. SCHLESIGER, C. SORG, K. SCHARFFETTER-KOCHANEK u. C. SUNDERKOTTER (2004): Senescent BALB/c mice are able to develop resistance to Leishmania major infection. Infect. Immun. 72, 5106-5114
ELLOSO, M. M. u. P. SCOTT (1999): Expression and contribution of B7-1 (CD80) and B7-2 (CD86) in the early immune response to Leishmania major infection. J. Immunol. 162, 6708-6715
ENGLER, M. J. u. C. C. RICHARDSON (1983): Bacteriophage T7 DNA replication. Synthesis of lagging strands in a reconstituted system using purified proteins. J. Biol. Chem. 258, 11197-11205
ETGES, R. u. I. MULLER (1998): Progressive disease or protective immunity to Leishmania major infection: the result of a network of stimulatory and inhibitory interactions. J. Mol. Med. 76, 372-390
FEHNIGER, T. A. u. M. A. CALIGIURI (2001): Interleukin 15: biology and relevance to human disease. Blood 97, 14-32
FILIPPI, C., S. HUGUES, J. CAZARETH, V. JULIA, N. GLAICHENHAUS u. S. UGOLINI (2003): CD4+ T cell polarization in mice is modulated by strain-specific major histocompatibility complex-independent differences within dendritic cells. J. Exp. Med. 198, 201-209
FINK, L., W. SEEGER, L. ERMERT, J. HANZE, U. STAHL, F. GRIMMINGER, W. KUMMER u. R. M. BOHLE (1998): Real-time quantitative RT-PCR after laser-assisted cell picking. Nat. Med. 4, 1329-1333
-164-
Literaturverzeichnis
FINK, L., T. KINFE, W. SEEGER, L. ERMERT, W. KUMMER u. R. M. BOHLE (2000): Immunostaining for cell picking and real-time mRNA quantitation. Am. J. Pathol. 157, 1459-1466
FINK, L. u. R. M. BOHLE (2002): Internal standards for laser microdissection. Methods Enzymol. 356, 99-113
FLIER, J., D. M. BOORSMA, P. J. VAN BEEK, C. NIEBOER, T. J. STOOF, R. WILLEMZE u. C. P. TENSEN (2001): Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J. Pathol. 194, 398-405
FLOHE, S. B., C. BAUER, S. FLOHE u. H. MOLL (1998): Antigen-pulsed epidermal Langerhans cells protect susceptible mice from infection with the intracellular parasite Leishmania major. Eur. J. Immunol. 28, 3800-3811
FREEMAN, W. M., S. J. WALKER u. K. E. VRANA (1999): Quantitative RT-PCR: pitfalls and potential. Biotechniques 26, 112-115
FREY, B., U. BREHM, G. KÜBLER, S. DEPNER, R. WARTBICHLER, C. EICHNER, H. DONNER, G. SEIDL u. M. GOERDE (2002): Gene Expression Arrays: Highly Sensitive Detection of Expression Patterns with Improved Tools for Target Amplification. Biochemica 2, 27-29 GASIM, S., A. M. ELHASSAN, E. A. KHALIL, A. ISMAIL, A. M. KADARU, A. KHARAZMI u. T. G. THEANDER (1998): High levels of plasma IL-10 and expression of IL-10 by keratinocytes during visceral leishmaniasis predict subsequent development of post-kala-azar dermal leishmaniasis. Clin. Exp. Immunol. 111, 64-69
GASPERINI, S., M. MARCHI, F. CALZETTI, C. LAUDANNA, L. VICENTINI, H. OLSEN, M. MURPHY, F. LIAO, J. FARBER u. M. A. CASSATELLA (1999): Gene expression and production of the monokine induced by IFN-gamma (MIG), IFN-inducible T cell alpha chemoattractant (I-TAC), and IFN-gamma-inducible protein-10 (IP-10) chemokines by human neutrophils. J. Immunol. 162, 4928-4937
GIACHELLI, C. M., D. LOMBARDI, R. J. JOHNSON, C. E. MURRY u. M. ALMEIDA (1998): Evidence for a role of osteopontin in macrophage infiltration in response to pathological stimuli in vivo. Am. J. Pathol. 152, 353-358
-165-
Literaturverzeichnis
GLASER, T. A. u. A. J. MUKKADA (1992): Proline transport in Leishmania donovani amastigotes: dependence on pH gradients and membrane potential. Mol. Biochem. Parasitol. 51, 1-8
GOMES, I. N., A. F. CALABRICH, R. S. TAVARES, J. WIETZERBIN, L. A. DE FREITAS u. P. S. VERAS (2003): Differential properties of CBA/J mononuclear phagocytes recovered from an inflammatory site and probed with two different species of Leishmania. Microbes. Infect. 5, 251-260
GORAK, P. M., C. R. ENGWERDA u. P. M. KAYE (1998): Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur. J. Immunol. 28, 687-695
GORHAM, J. D., M. L. GULER, R. G. STEEN, A. J. MACKEY, M. J. DALY, K. FREDERICK, W. F. DIETRICH u. K. M. MURPHY (1996): Genetic mapping of a murine locus controlling development of T helper 1/T helper 2 type responses. Proc. Natl. Acad. Sci. U.S.A 93, 12467-12472
GREEN, S. J., R. M. CRAWFORD, J. T. HOCKMEYER, M. S. MELTZER u. C. A. NACY (1990): Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J. Immunol. 145, 4290-4297
GREENBAUM, L. A., J. B. HOROWITZ, A. WOODS, T. PASQUALINI, E. P. REICH u. K. BOTTOMLY (1988): Autocrine growth of CD4+T cells. Differential effects of IL-1 on helper and inflammatory T cells. J. Immunol. 140, 1555-1560
GROUX, H., F. COTTREZ, M. ROULEAU, S. MAUZE, S. ANTONENKO, S. HURST, T. MCNEIL, M. BIGLER, M. G. RONCAROLO u. R. L. COFFMAN (1999): A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J. Immunol. 162, 1723-1729
GU, L., S. TSENG, R. M. HORNER, C. TAM, M. LODA u. B. J. ROLLINS (2000): Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 404, 407-411
GUMY, A., J. A. LOUIS u. P. LAUNOIS (2004): The murine model of infection with Leishmania major and its importance for the deciphering of mechanisms underlying differences in Th cell differentiation in mice from different genetic backgrounds. Int. J. Parasitol. 34, 433-444
-166-
Literaturverzeichnis
HAAS, N., K. HAMANN, J. GRABBE, B. CREMER u. B. M. CZARNETZKI (1992): Expression of the high affinity IgE-receptor on human Langerhans' cells. Elucidating the role of epidermal IgE in atopic eczema. Acta Derm. Venereol. 72, 271-272
HADJEB, N. u. G. A. BERKOWITZ (1996): Preparation of T-over-hang vectors with high PCR product cloning efficiency. Biotechniques 20, 20-22
HALLIWELL, R. E. u. N. T. GORMAN (1989): Nonatopic allergic skin diseases. In: HALLIWELL, R. E., N. T. GORMAN u. W. B. SAUNDERS (Hrsg.): Veterinary Clinical Immunology. Verlag Saunders, Philadelphia, U.S.A., S. 261–267.
HAMID, Q., M. BOGUNIEWICZ u. D. Y. LEUNG (1994): Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Invest 94, 870-876
HANDMAN, E., R. CEREDIG u. G. F. MITCHELL (1979): Murine cutaneous leishmaniasis: disease patterns in intact and nude mice of various genotypes and examination of some differences between normal and infected macrophages. Aust. J. Exp. Biol. Med. Sci. 57, 9-29
HANDMAN, E. (1999): Cell biology of Leishmania. Adv. Parasitol. 44, 1-39
HARDING, C. V. u. E. R. UNANUE (1990): Cellular mechanisms of antigen processing and the function of class I and II major histocompatibility complex molecules. Cell Regul. 1, 499-509
HAVELKOVA, H., J. BADALOVA, M. SVOBODOVA, J. VOJTIKOVA, I. KUREY, V. VLADIMIROV, P. DEMANT u. M. LIPOLDOVA (2006): Genetics of susceptibility to leishmaniasis in mice: four novel loci and functional heterogeneity of gene effects. Genes Immun. 7, 220-233
HEINZEL, F. P., M. D. SADICK, S. S. MUTHA u. R. M. LOCKSLEY (1991): Production of interferon gamma, interleukin 2, interleukin 4, and interleukin 10 by CD4+ lymphocytes in vivo during healing and progressive murine leishmaniasis. Proc. Natl. Acad. Sci. U.S.A 88, 7011-7015
HEINZEL, F. P., D. S. SCHOENHAUT, R. M. RERKO, L. E. ROSSER u. M. K. GATELY (1993): Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 177, 1505-1509
-167-
Literaturverzeichnis
HEINZEL, F. P., R. M. RERKO, F. AHMED u. E. PEARLMAN (1995): Endogenous IL-12 is required for control of Th2 cytokine responses capable of exacerbating leishmaniasis in normally resistant mice. J. Immunol. 155, 730-739
HIJNEN, D., M. DE BRUIN-WELLER, B. OOSTING, C. LEBRE, J. E. DE, C. BRUIJNZEEL-KOOMEN u. E. KNOL (2004): Serum thymus and activation-regulated chemokine (TARC) and cutaneous T cell- attracting chemokine (CTACK) levels in allergic diseases: TARC and CTACK are disease-specific markers for atopic dermatitis. J. Allergy Clin. Immunol. 113, 334-340
HIMMELRICH, H., P. LAUNOIS, I. MAILLARD, T. BIEDERMANN, F. TACCHINI-COTTIER, R. M. LOCKSLEY, M. ROCKEN u. J. A. LOUIS (2000): In BALB/c mice, IL-4 production during the initial phase of infection with Leishmania major is necessary and sufficient to instruct Th2 cell development resulting in progressive disease. J. Immunol. 164, 4819-4825
HOCHREIN, H., M. O'KEEFFE, T. LUFT, S. VANDENABEELE, R. J. GRUMONT, E. MARASKOVSKY u. K. SHORTMAN (2000): Interleukin (IL)-4 is a major regulatory cytokine governing bioactive IL-12 production by mouse and human dendritic cells. J. Exp. Med. 192, 823-833
HOEHN, P., S. GOEDERT, T. GERMANN, S. KOELSCH, S. JIN, N. PALM, E. RUEDE u. E. SCHMITT (1995): Opposing effects of TGF-beta 2 on the Th1 cell development of naive CD4+ T cells isolated from different mouse strains. J. Immunol. 155, 3788-3793
HOLSCHER, C. (2004): The power of combinatorial immunology: IL-12 and IL-12-related dimeric cytokines in infectious diseases. Med. Microbiol. Immunol. (Berl) 193, 1-17
HONDOWICZ, B. D., A. Y. PARK, M. M. ELLOSO u. P. SCOTT (2000): Maintenance of IL-12-responsive CD4+ T cells during a Th2 response in Leishmania major-infected mice. Eur. J. Immunol. 30, 2007-2014
HOWARD, J. G, C. HALE u. F. Y. LIEW (1980): Immunological regulation of experimental cutaneous Leishmaniasis. III. Nature and significance of specific suppression of cellmediated immunity in mice highly susceptible to Leishmani tropica. J. Exp. Med. 152, 594–607
-168-
Literaturverzeichnis
HOWELL, M. D., N. NOVAK, T. BIEBER, S. PASTORE, G. GIROLOMONI, M. BOGUNIEWICZ, J. STREIB, C. WONG, R. L. GALLO u. D. Y. LEUNG (2005): Interleukin-10 downregulates anti-microbial peptide expression in atopic dermatitis. J. Invest Dermatol. 125, 738-745
HU, G. (1993): DNA polymerase-catalyzed addition of nontemplated extra nucleotides to the 3' end of a DNA fragment. DNA Cell Biol. 12, 763-770
HUBER, M., H. U. BEUSCHER, P. ROHWER, R. KURRLE, M. ROLLINGHOFF u. M. LOHOFF (1998): Costimulation via TCR and IL-1 receptor reveals a novel IL-1alpha-mediated autocrine pathway of Th2 cell proliferation. J. Immunol. 160, 4242-4247
JAKOB, T. u. M. C. UDEY (1999): Epidermal Langerhans cells: from neurons to nature's adjuvants. Adv. Dermatol. 14, 209-258
JANEWAY, C. A., P. TRAVERS, M. WALPORT u. M. SHLOMCHIK (2002): Immunologie 5. Aufl. Verlag Spektrum, Heidelberg, Berlin
JANKOVIC, D., Z. LIU u. W. C. GAUSE (2001): Th1- and Th2-cell commitment during infectious disease: asymmetry in divergent pathways. Trends Immunol. 22, 450-457
JI, J., J. SUN u. L. SOONG (2003): Impaired expression of inflammatory cytokines and chemokines at early stages of infection with Leishmania amazonensis. Infect. Immun. 71, 4278-4288
JOHNSON, P. H. u. L. I. GROSSMAN (1977): Electrophoresis of DNA in agarose gels. Optimizing separations of conformational isomers of double- and single-stranded DNAs. Biochemistry 16, 4217-4225
JULIA, V., M. RASSOULZADEGAN u. N. GLAICHENHAUS (1996): Resistance to Leishmania major induced by tolerance to a single antigen 1. Science 274, 421-423
JULIA, V. u. N. GLAICHENHAUS (1999): CD4(+) T cells which react to the Leishmania major LACK antigen rapidly secrete interleukin-4 and are detrimental to the host in resistant B10.D2 mice. Infect. Immun. 67, 3641-3644
-169-
Literaturverzeichnis
JUTTNER, S., J. BERNHAGEN, C. N. METZ, M. ROLLINGHOFF, R. BUCALA u. A. GESSNER (1998): Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. J. Immunol. 161, 2383-2390
KALINSKI, P., H. H. SMITS, J. H. SCHUITEMAKER, P. L. VIEIRA, E. M. VAN, E. C. DE JONG, E. A. WIERENGA u. M. L. KAPSENBERG (2000): IL-4 is a mediator of IL-12p70 induction by human Th2 cells: reversal of polarized Th2 phenotype by dendritic cells. J. Immunol. 165, 1877-1881
KAMANAKA, M., P. YU, T. YASUI, K. YOSHIDA, T. KAWABE, T. HORII, T. KISHIMOTO u. H. KIKUTANI (1996): Protective role of CD40 in Leishmania major infection at two distinct phases of cell-mediated immunity. Immunity. 4, 275-281
KAMHAWI, S., Y. BELKAID, G. MODI, E. ROWTON u. D. SACKS (2000): Protection against cutaneous leishmaniasis resulting from bites of uninfected sand flies. Science 290, 1351-1354
KAMIJO, R., H. HARADA, T. MATSUYAMA, M. BOSLAND, J. GERECITANO, D. SHAPIRO, J. LE, S. I. KOH, T. KIMURA, S. J. GREEN u. . (1994): Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science 263, 1612-1615
KANE, M. M. u. D. M. MOSSER (2001): The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166, 1141-1147
KARP, C. L., S. H. EL-SAFI, T. A. WYNN, M. M. SATTI, A. M. KORDOFANI, F. A. HASHIM, M. HAG-ALI, F. A. NEVA, T. B. NUTMAN u. D. L. SACKS (1993): In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma. J. Clin. Invest 91, 1644-1648
KELLINA, O. I. (1973): Differences in the sensitivity of inbred mice of different lines to Leishmania tropica major. Med. Parazitol. (Mosk.) 42, 279–285
KELLY, S. E., D. B. JONES u. S. FLEMING (1989): Calgranulin expression in inflammatory dermatoses. J. Pathol. 159, 17-21
-170-
Literaturverzeichnis
KELLY, S. E., J. A. HUNTER, D. B. JONES, B. R. CLARK u. S. FLEMING (1991): Morphological evidence for calcium-dependent association of calgranulin with the epidermal cytoskeleton in inflammatory dermatoses. Br. J. Dermatol. 124, 403-409
KELSO, A. (1995): Th1 and Th2 subsets: paradigms lost? Immunol. Today 16, 374-379
KELSO, A., P. GROVES, A. B. TROUTT u. K. FRANCIS (1995): Evidence for the stochastic acquisition of cytokine profile by CD4+ T cells activated in a T helper type 2-like response in vivo. Eur. J. Immunol. 25, 1168-1175
KERSCHENLOHR, K., S. DECARD, B. PRZYBILLA u. A. WOLLENBERG (2003): Atopy patch test reactions show a rapid influx of inflammatory dendritic epidermal cells in patients with extrinsic atopic dermatitis and patients with intrinsic atopic dermatitis. J. Allergy Clin. Immunol. 111, 869-874
KHASKHELY, N. M., M. MARUNO, H. UEZATO, A. TAKAMIYAGI, S. T. RAMZI, K. M. AL-KASEM, K. KARIYA, T. TODA, Y. HASHIGUCHI, E. A. GOMEZ LANDIRES u. S. NONAKA (2002): Low-dose UVB contributes to host resistance against Leishmania amazonensis infection in mice through induction of gamma interferon and tumor necrosis factor alpha cytokines. Clin. Diagn. Lab Immunol. 9, 677-686
KING, C., H. R. MUELLER, C. M. MALO, K. MURALI-KRISHNA, R. AHMED, E. KING u. N. SARVETNICK (2001): Interleukin-4 acts at the locus of the antigen-presenting dendritic cell to counter-regulate cytotoxic CD8+ T-cell responses. Nat. Med. 7, 206-214
KLEINBECK, M. L., M. J. HITES, J. L. LOKER, R. E. HALLIWELL u. K. W. LEE (1989): Enzyme-linked immunosorbent assay for measurement of allergen-specific IgE antibodies in canine serum. Am. J. Vet. Res. 50, 1831-1839
KLUNKER, S., A. TRAUTMANN, M. AKDIS, J. VERHAGEN, P. SCHMID-GRENDELMEIER, K. BLASER u. C. A. AKDIS (2003): A second step of chemotaxis after transendothelial migration: keratinocytes undergoing apoptosis release IFN-gamma-inducible protein 10, monokine induced by IFN-gamma, and IFN-gamma-inducible alpha-chemoattractant for T cell chemotaxis toward epidermis in atopic dermatitis. J. Immunol. 171, 1078-1084
-171-
Literaturverzeichnis
KLUR, S., K. TOY, M. P. WILLIAMS u. U. CERTA (2004): Evaluation of procedures for amplification of small-size samples for hybridization on microarrays. Genomics 83, 508-517
KOPF, M., F. BROMBACHER, G. KOHLER, G. KIENZLE, K. H. WIDMANN, K. LEFRANG, C. HUMBORG, B. LEDERMANN u. W. SOLBACH (1996): IL-4-deficient Balb/c mice resist infection with Leishmania major. J. Exp. Med. 184, 1127-1136
KOSTKA, S. L., J. KNOP, A. KONUR, M. C. UDEY u. S. E. VON (2006): Distinct Roles for IL-1 Receptor Type I Signaling in Early Versus Established Leishmania major Infections. J. Invest Dermatol. 126, 1582-1589
KUMMER, W., L. FINK, M. DVORAKOVA, R. HABERBERGER u. R. M. BOHLE (1998): Rat cardiac neurons express the non-coding R-exon (exon 1) of the cholinergic gene locus. Neuroreport 9, 2209-2212
KUNZ, M., J. ROTH, C. SORG u. G. KOLDE (1992): Epidermal expression of the calcium binding surface antigen 27E10 in inflammatory skin diseases. Arch. Dermatol. Res. 284, 386-390
LANGRISH, C. L., B. S. MCKENZIE, N. J. WILSON, M. R. DE WAAL, R. A. KASTELEIN u. D. J. CUA (2004): IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol. Rev. 202, 96-105
LAOUINI, D., H. ALENIUS, P. BRYCE, H. OETTGEN, E. TSITSIKOV u. R. S. GEHA (2003): IL-10 is critical for Th2 responses in a murine model of allergic dermatitis. J. Clin. Invest 112, 1058-1066
LASKAY, T., Z. G. VAN u. W. SOLBACH (2003): Neutrophil granulocytes--Trojan horses for Leishmania major and other intracellular microbes? Trends Microbiol. 11, 210-214
LAUFS, H., K. MULLER, J. FLEISCHER, N. REILING, N. JAHNKE, J. C. JENSENIUS, W. SOLBACH u. T. LASKAY (2002): Intracellular survival of Leishmania major in neutrophil granulocytes after uptake in the absence of heat-labile serum factors. Infect. Immun. 70, 826-835
-172-
Literaturverzeichnis
LAUNOIS, P., T. OHTEKI, K. SWIHART, H. R. MACDONALD u. J. A. LOUIS (1995): In susceptible mice, Leishmania major induce very rapid interleukin-4 production by CD4+ T cells which are NK1.1-. Eur. J. Immunol. 25, 3298-3307
LAUNOIS, P., K. G. SWIHART, G. MILON u. J. A. LOUIS (1997): Early production of IL-4 in susceptible mice infected with Leishmania major rapidly induces IL-12 unresponsiveness. J. Immunol. 158, 3317-3324
LAUNOIS, P., I. MAILLARD, S. PINGEL, K. G. SWIHART, I. XENARIOS, H. CHA-ORBEA, H. DIGGELMANN, R. M. LOCKSLEY, H. R. MACDONALD u. J. A. LOUIS (1997): IL-4 rapidly produced by V beta 4 V alpha 8 CD4+ T cells instructs Th2 development and susceptibility to Leishmania major in BALB/c mice. Immunity 6, 541-549
LAUNOIS, P., H. HIMMELRICH, F. TACCHINI-COTTIER, G. MILON u. J. A. LOUIS (1999): New insight into the mechanisms underlying Th2 cell development and susceptibility to Leishmania major in BALB/c mice. Microbes. Infect. 1, 59-64
LEAL, L. M., D. W. MOSS, R. KUHN, W. MULLER u. F. Y. LIEW (1993): Interleukin-4 transgenic mice of resistant background are susceptible to Leishmania major infection. Eur. J. Immunol. 23, 566-569
LEIBY, D. A., R. D. SCHREIBER u. C. A. NACY (1993): IFN-gamma produced in vivo during the first two days is critical for resolution of murine Leishmania major infections. Microb. Pathog. 14, 495-500
LENSCHOW, D. J., T. L. WALUNAS u. J. A. BLUESTONE (1996): CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14, 233-258
LEONARD, W. J. (1994): The defective gene in X-linked severe combined immunodeficiency encodes a shared interleukin receptor subunit: implications for cytokine pleiotropy and redundancy. Curr Opin. Immunol. 6, 631-635 LEUNG, D. Y., A. R. RHODES u. R. S. GEHA (1981): Enumeration of T cell subsets in atopic dermatitis using monoclonal antibodies. J. Allergy Clin. Immunol. 67, 450-455
-173-
Literaturverzeichnis
LEUNG, D. Y., M. BOGUNIEWICZ, M. D. HOWELL, I. NOMURA u. Q. A. HAMID (2004): New insights into atopic dermatitis. J. Clin. Invest 113, 651-657
LEVER, R., M. TURBITT, A. SANDERSON u. R. MACKIE (1987): Immunophenotyping of the cutaneous infiltrate and of the mononuclear cells in the peripheral blood in patients with atopic dermatitis. J. Invest Dermatol. 89, 4-7
LI, J., P. SCOTT u. J. P. FARRELL (1996): In vivo alterations in cytokine production following interleukin-12 (IL-12) and anti-IL-4 antibody treatment of CB6F1 mice with chronic cutaneous leishmaniasis. Infect. Immun. 64, 5248-5254
LICHTMAN, A. H., J. CHIN, J. A. SCHMIDT u. A. K. ABBAS (1988): Role of interleukin 1 in the activation of T lymphocytes. Proc. Natl. Acad. Sci. U.S.A 85, 9699-9703
LIEW, F. Y., Y. LI u. S. MILLOTT (1990): Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J. Immunol. 145, 4306-4310
LIEW, F. Y. u. C. A. O'DONNELL (1993): Immunology of leishmaniasis. Adv. Parasitol. 32, 161-259
LIVAK, K. J. u. T. D. SCHMITTGEN (2001): Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-408
LOETSCHER, M., B. GERBER, P. LOETSCHER, S. A. JONES, L. PIALI, I. CLARK-LEWIS, M. BAGGIOLINI u. B. MOSER (1996): Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J. Exp. Med. 184, 963-969
LOETSCHER, P., A. PELLEGRINO, J. H. GONG, I. MATTIOLI, M. LOETSCHER, G. BARDI, M. BAGGIOLINI u. I. CLARK-LEWIS (2001): The ligands of CXC chemokine receptor 3, I-TAC, Mig, and IP10, are natural antagonists for CCR3. J. Biol. Chem. 276, 2986-2991
-174-
Literaturverzeichnis
LOHOFF, M., D. FERRICK, H. W. MITTRUCKER, G. S. DUNCAN, S. BISCHOF, M. ROLLINGHOFF u. T. W. MAK (1997): Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. Immunity 6, 681-689
LUSTER, A. D. u. J. V. RAVETCH (1987): Genomic characterization of a gamma-interferon-inducible gene (IP-10) and identification of an interferon-inducible hypersensitive site. Mol. Cell Biol. 7, 3723-3731
MAGHAZACHI, A. A., A. AL-AOUKATY u. T. J. SCHALL (1994): C-C chemokines induce the chemotaxis of NK and IL-2-activated NK cells. Role for G proteins. J. Immunol. 153, 4969-4977
MAGHAZACHI, A. A., B. S. SKALHEGG, B. ROLSTAD u. A. AL-AOUKATY (1997): Interferon-inducible protein-10 and lymphotactin induce the chemotaxis and mobilization of intracellular calcium in natural killer cells through pertussis toxin-sensitive and -insensitive heterotrimeric G-proteins. FASEB J. 11, 765-774
MALHERBE, L., C. FILIPPI, V. JULIA, G. FOUCRAS, M. MORO, H. APPEL, K. WUCHERPFENNIG, J. C. GUERY u. N. GLAICHENHAUS (2000): Selective activation and expansion of high-affinity CD4+ T cells in resistant mice upon infection with Leishmania major. Immunity. 13, 771-782
MARTIN, E., C. NATHAN u. Q. W. XIE (1994): Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J. Exp. Med. 180, 977-984
MATSUDA, H., N. WATANABE, G. P. GEBA3, J. SPERL, M. TSUDZUKI, J. HIROI, M. MATSUMOTO, H. USHIO, S. SAITO, P. W. ASKENASE u C. RA (1997): Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int. Immunol. 9, 461–466 MATTE, C. u. M. OLIVIER (2002): Leishmania-induced cellular recruitment during the early inflammatory response: modulation of proinflammatory mediators. J. Infect. Dis. 185, 673-681
MATTHEWS, D. J., C. L. EMSON, G. J. MCKENZIE, H. E. JOLIN, J. M. BLACKWELL u. A. N. MCKENZIE (2000): IL-13 is a susceptibility factor for Leishmania major infection. J. Immunol. 164, 1458-1462
-175-
Literaturverzeichnis
MATTNER, F., J. MAGRAM, J. FERRANTE, P. LAUNOIS, P. K. DI, R. BEHIN, M. K. GATELY, J. A. LOUIS u. G. ALBER (1996): Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur. J. Immunol. 26, 1553-1559
MAUEL, J., S. B. CORRADIN u. R. Y. BUCHMULLER (1991): Nitrogen and oxygen metabolites and the killing of Leishmania by activated murine macrophages. Res. Immunol. 142, 577-580
MAZANET, M. M., K. NEOTE u. C. C. HUGHES (2000): Expression of IFN-inducible T cell alpha chemoattractant by human endothelial cells is cyclosporin A-resistant and promotes T cell adhesion: implications for cyclosporin A-resistant immune inflammation. J. Immunol. 164, 5383-5388
MBOW, M. L., G. K. DEKREY u. R. G. TITUS (2001): Leishmania major induces differential expression of costimulatory molecules on mouse epidermal cells. Eur. J. Immunol. 31, 1400-1409
MELBY, P. C., F. J. NDRADE-NARVAEZ, B. J. DARNELL, G. VALENCIA-PACHECO, V. V. TRYON u. A. PALOMO-CETINA (1994): Increased expression of proinflammatory cytokines in chronic lesions of human cutaneous leishmaniasis. Infect. Immun. 62, 837-842
MOHRS, M., B. LEDERMANN, G. KOHLER, A. DORFMULLER, A. GESSNER u. F. BROMBACHER (1999): Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J. Immunol. 162, 7302-7308
MOLL, H., S. FLOHE u. M. ROLLINGHOFF (1995): Dendritic cells in Leishmania major-immune mice harbor persistent parasites and mediate an antigen-specific T cell immune response. Eur. J. Immunol. 25, 693-699
MOLL, H. (1997): The role of chemokines and accessory cells in the immunoregulation of cutaneous leishmaniasis. Behring Inst. Mitt. 99, 73-78
MOLL, H., A. SCHARNER u. E. KAMPGEN (2002): Increased interleukin 4 (IL-4) receptor expression and IL-4-induced decrease in IL-12 production by Langerhans cells infected with Leishmania major. Infect. Immun. 70, 1627-1630
-176-
Literaturverzeichnis
MONTEFORTE, G. M., K. TAKEDA, M. RODRIGUEZ-SOSA, S. AKIRA, J. R. DAVID u. A. R. SATOSKAR (2000): Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. J. Immunol. 164, 5890-5893
MORRIS, L., A. B. TROUTT, E. HANDMAN u. A. KELSO (1992): Changes in the precursor frequencies of IL-4 and IFN-gamma secreting CD4+ cells correlate with resolution of lesions in murine cutaneous leishmaniasis. J. Immunol. 149, 2715-2721
MORRISON, T. B., J. J. WEIS u. C. T. WITTWER (1998): Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. Biotechniques 24, 954-8, 960, 962
MOSMANN, T. R., H. CHERWINSKI, W. BOND, M. A. GIEDLIN u. R. L. COFFMAN (1986): Two types of murine helper T-cell clones. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348-2357 MOSMANN, T. R. u. S. SAD (1996): The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 17, 138-146
MOSSER, D. M., J. F. WEDGWOOD u. P. J. EDELSON (1985): Leishmania amastigotes: resistance to complement-mediated lysis is not due to a failure to fix C3. J. Immunol. 134, 4128-4131
MULLER, K., Z. G. VAN, B. HANSEN, H. LAUFS, N. JAHNKE, W. SOLBACH u. T. LASKAY (2001): Chemokines, natural killer cells and granulocytes in the early course of Leishmania major infection in mice. Med. Microbiol. Immunol. (Berl) 190, 73-76
MULLER, K., S. BISCHOF, F. SOMMER, M. LOHOFF, W. SOLBACH u. T. LASKAY (2003): Differential production of macrophage inflammatory protein 1gamma (MIP-1gamma), lymphotactin, and MIP-2 by CD4(+) Th subsets polarized in vitro and in vivo. Infect. Immun. 71, 6178-6183
MULLIS, K. B. u. F. A. FALOONA (1987): Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155, 335-350
-177-
Literaturverzeichnis
MURPHY, P. M., M. BAGGIOLINI, I. F. CHARO, C. A. HEBERT, R. HORUK, K. MATSUSHIMA, L. H. MILLER, J. J. OPPENHEIM u. C. A. POWER (2000): International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 52, 145-176
NABORS, G. S. u. J. P. FARRELL (1994): Site-specific immunity to Leishmania major in SWR mice: the site of infection influences susceptibility and expression of the antileishmanial immune response. Infect. Immun. 62, 3655-3662
NABORS, G. S., T. NOLAN, W. CROOP, J. LI u. J. P. FARRELL (1995): The influence of the site of parasite inoculation on the development of Th1 and Th2 type immune responses in (BALB/c x C57BL/6) F1 mice infected with Leishmania major. Parasite Immunol. 17, 569-579
NASSERI, M. u. F. Z. MODABBER (1979): Generalized infection and lack of delayed hypersensitivity in BALB/c mice infected with Leishmania tropica major. Infect. Immun. 26, 611-614
NATHAN, C. (2006): Role of iNOS in human host defense. Science 312, 1874-1875
NICKOLOFF, B. J. (1993): Dermal Immune System. Verlag CRC Press, Boca Raton, U.S.A.
NICKOLOFF B. J., B. BONISH, B. B. HUANG u. S. A. PORCELLI (2000): Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J. Dermatol. Sci. 24, 212–225
NIMMO WILKIE, J. S., J. A. YAGER, B. N. WILKIE u. W. M. PARKER (1991): Abnormal cutaneous response to mitogens and a contact allergen in dogs with atopic dermatitis. Vet. Immunol. Immunopathol. 28, 97-106
NIMMO WILKIE, J. S., J. A. YAGER, B. N. WILKIE u. P. J. PASCOE (1992): Changes in cell-mediated immune responses after experimentally-induced anaphylaxis in dogs. Vet. Immunol. Immunopathol. 32, 325-338
NISHIKOMORI, R., S. GURUNATHAN, K. NISHIKOMORI u. W. STROBER (2001): BALB/c mice bearing a transgenic IL-12 receptor beta 2 gene exhibit a nonhealing phenotype to Leishmania major infection despite intact IL-12 signaling. J. Immunol. 166, 6776-6783
-178-
Literaturverzeichnis
NOBEN-TRAUTH, N., P. KROPF u. I. MULLER (1996): Susceptibility to Leishmania major infection in interleukin-4-deficient mice. Science 271, 987-990
NOBEN-TRAUTH, N., W. E. PAUL u. D. L. SACKS (1999): IL-4- and IL-4 receptor-deficient BALB/c mice reveal differences in susceptibility to Leishmania major parasite substrains. J. Immunol. 162, 6132-6140
NOVAK, N., T. BIEBER u. D. Y. LEUNG (2003): Immune mechanisms leading to atopic dermatitis. J. Allergy Clin. Immunol. 112, S128-S139
O'REGAN, A. W., G. J. NAU, G. L. CHUPP u. J. S. BERMAN (2000): Osteopontin (Eta-1) in cell-mediated immunity: teaching an old dog new tricks. Immunol. Today 21, 475-478
OHKUSU, K., T. YOSHIMOTO, K. TAKEDA, T. OGURA, S. KASHIWAMURA, Y. IWAKURA, S. AKIRA, H. OKAMURA u. K. NAKANISHI (2000): Potentiality of interleukin-18 as a useful reagent for treatment and prevention of Leishmania major infection. Infect. Immun. 68, 2449-2456
OLIVRY, T., D. K. NAYDAN u. P. F. MOORE (1997): Characterization of the cutaneous inflammatory infiltrate in canine atopic dermatitis. Am. J. Dermatopathol. 19, 477-486
ONG, P. Y., Q. A. HAMID, J. B. TRAVERS, I. STRICKLAND, K. M. AL, M. BOGUNIEWICZ u. D. Y. LEUNG (2002): Decreased IL-15 may contribute to elevated IgE and acute inflammation in atopic dermatitis. J. Immunol. 168, 505-510
ONO, S. J., T. NAKAMURA, D. MIYAZAKI, M. OHBAYASHI, M. DAWSON u. M. TODA (2003): Chemokines: roles in leukocyte development, trafficking, and effector function. J. Allergy Clin. Immunol. 111, 1185-1199
PARISH, W. E. (1981): The clinical relevance of heat-stable, short-term sensitizing anaphylactic IgG antibodies (IgG S-TS) and of related activities of IgG4 and IgG2. Br. J. Dermatol. 105, 223-231
PATARCA, R., R. A. SAAVEDRA u. H. CANTOR (1993): Molecular and cellular basis of genetic resistance to bacterial infection: the role of the early T-lymphocyte activation-1/osteopontin gene. Crit Rev. Immunol. 13, 225-246
-179-
Literaturverzeichnis
PATEL, D. D., J. P. ZACHARIAH u. L. P. WHICHARD (2001): CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin. Immunol. 98, 39-45
PAUL, W. E. (1989): Pleiotropy and redundancy: T cell-derived lymphokines in the immune response. Cell 57, 521-524
PAWELEC, G., A. REHBEIN, E. SCHLOTZ, H. FRICCIUS u. H. POHLA (1996): Cytokine modulation of TH1/TH2 phenotype differentiation in directly alloresponsive CD4+ human T cells. Transplantation 62, 1095-1101
PETKOVIC, V., C. MOGHINI, S. PAOLETTI, M. UGUCCIONI u. B. GERBER (2004): I-TAC/CXCL11 is a natural antagonist for CCR5. J. Leukoc. Biol. 76, 701-708
PRESTON, P. M. u. D. C. DUMONDE (1976): Experimental cutaneous Leishmaniasis. Protective immunity in subclinical and self-healing infection in the mouse. Clin. Exp. Immunol. 23, 126-138
RACKE, M. K., A. BONOMO, D. E. SCOTT, B. CANNELLA, A. LEVINE, C. S. RAINE, E. M. SHEVACH u. M. ROCKEN (1994): Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J. Exp. Med. 180, 1961-1966
REEDY, L. M, W. H. MILLER u. T. WILLEMSE (2002): Allergische Hauterkrankungen bei Hund und Katze. Verlag Schlütersche, Hannover, S. 14-49
REINER, S. L., Z. E. WANG, F. HATAM, P. SCOTT u. R. M. LOCKSLEY (1993): TH1 and TH2 cell antigen receptors in experimental leishmaniasis. Science 259, 1457-1460
REINER, S. L., S. ZHENG, Z. E. WANG, L. STOWRING u. R. M. LOCKSLEY (1994): Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. J. Exp. Med. 179, 447-456
REINER, S. L. u. R. M. LOCKSLEY (1995): The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13, 151-177
RHODES, K. H., F. KERDEL, N. A. SOTER u. R. CHINNICI (1987): Investigation into the immunopathogenesis of canine atopy. Semin. Vet. Med. Surg. (Small Anim) 2, 199-201
-180-
Literaturverzeichnis
RHODES, K. H., F. KERDEL u. N. A. SOTER (1987): Comparative aspects of canine and human atopic dermatitis. Semin. Vet. Med. Surg. (Small Anim) 2, 166-172
RIRIE, K. M., R. P. RASMUSSEN u. C. T. WITTWER (1997): Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal. Biochem. 245, 154-160
RITTER, U., H. MOLL, T. LASKAY, E. BROCKER, O. VELAZCO, I. BECKER u. R. GILLITZER (1996): Differential expression of chemokines in patients with localized and diffuse cutaneous American leishmaniasis. J. Infect. Dis. 173, 699-709
RITTER, U. u. H. MOLL (2000): Monocyte chemotactic protein-1 stimulates the killing of leishmania major by human monocytes, acts synergistically with IFN-gamma and is antagonized by IL-4. Eur. J. Immunol. 30, 3111-3120
RITTER, U. u. H. KORNER (2002): Divergent expression of inflammatory dermal chemokines in cutaneous leishmaniasis. Parasite Immunol. 24, 295-301
ROBERTS, L. J., T. M. BALDWIN, J. M. CURTIS, E. HANDMAN u. S. J. FOOTE (1997): Resistance to Leishmania major is linked to the H2 region on chromosome 17 and to chromosome 9. J. Exp. Med. 185, 1705-1710
ROBERTSON, M. J. (2002): Role of chemokines in the biology of natural killer cells. J. Leukoc. Biol. 71, 173-183
ROBINSON, D. S. u. A. O'GARRA (2002): Further checkpoints in Th1 development. Immunity. 16, 755-758
ROCKEN, M., M. RACKE u. E. M. SHEVACH (1996): IL-4-induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol. Today 17, 225-231
ROITT, I., J. BROSTOFF u. D. MALE (1991): Kurzes Lehrbuch der Immunologie 2. Verlag Thieme, Stuttgart, S. 253-273
ROSAS, L. E., J. BARBI, B. LU, Y. FUJIWARA, C. GERARD, V. M. SANDERS u. A. R. SATOSKAR (2005): CXCR3-/- mice mount an efficient Th1 response but fail to control Leishmania major infection. Eur. J. Immunol. 35, 515-523
ROTH, J., C. SUNDERKOTTER, M. GOEBELER, J. GUTWALD u. C. SORG (1992): Expression of the calcium-binding proteins MRP8 and MRP14 by early infiltrating cells in experimental contact dermatitis. Int. Arch. Allergy Immunol. 98, 140-145
ROTH, J., T. VOGL, C. SORG u. C. SUNDERKOTTER (2003): Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 24, 155-158
ROTTMAN, J. B., T. L. SMITH, K. G. GANLEY, T. KIKUCHI u. J. G. KRUEGER (2001): Potential role of the chemokine receptors CXCR3, CCR4, and the integrin alphaEbeta7 in the pathogenesis of psoriasis vulgaris. Lab Invest 81, 335-347
ROUSSEAU, D., S. DEMARTINO, F. ANJUERE, B. FERRUA, K. FRAGAKI, F. Y. LE u. J. KUBAR (2001): Sustained parasite burden in the spleen of Leishmania infantum-infected BALB/c mice is accompanied by expression of MCP-1 transcripts and lack of protection against challenge. Eur. Cytokine Netw. 12, 340-347
SACKS, D. u. N. NOBEN-TRAUTH (2002): The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2, 845-858
SACKS, D. u. C. ANDERSON (2004): Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunol. Rev. 201, 225-238
SACKS, D. L., S. HIENY u. A. SHER (1985): Identification of cell surface carbohydrate and antigenic changes between noninfective and infective developmental stages of Leishmania major promastigotes. J. Immunol. 135, 564-569
-182-
Literaturverzeichnis
SACKS, D. L. (1989): Metacyclogenesis in Leishmania promastigotes. Exp. Parasitol. 69, 100-103
SADICK, M. D., F. P. HEINZEL, B. J. HOLADAY, R. T. PU, R. S. DAWKINS u. R. M. LOCKSLEY (1990): Cure of murine leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell-dependent, interferon gamma-independent mechanism. J. Exp. Med. 171, 115-127
SAIKI, R. K., S. SCHARF, F. FALOONA, K. B. MULLIS, G. T. HORN, H. A. ERLICH u. N. ARNHEIM (1985): Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230, 1350-1354
SALLUSTO, F., D. LENIG, C. R. MACKAY u. A. LANZAVECCHIA (1998): Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 187, 875-883.
SAMBROOK, J. u. D. W. RUSSEL (2001): Molecular Cloning: A Laboratory Manual. 3. Aufl. Verlag Cold Spring Harbor, New York, U.S.A.
SANTIAGO, H. C., C. F. OLIVEIRA, L. SANTIAGO, F. O. FERRAZ, D. G. DE SOUZA, L. A. DE-FREITAS, L. C. AFONSO, M. M. TEIXEIRA, R. T. GAZZINELLI u. L. Q. VIEIRA (2004): Involvement of the chemokine RANTES (CCL5) in resistance to experimental infection with Leishmania major. Infect. Immun. 72, 4918-4923
SATO, N., S. K. AHUJA, M. QUINONES, V. KOSTECKI, R. L. REDDICK, P. C. MELBY, W. A. KUZIEL u. S. S. AHUJA (2000): CC chemokine receptor (CCR)2 is required for langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J. Exp. Med. 192, 205-218
SATOSKAR, A. R., M. OKANO, S. CONNAUGHTON, A. RAISANEN-SOKOLWSKI, J. R. DAVID u. M. LABOW (1998): Enhanced Th2-like responses in IL-1 type 1 receptor-deficient mice. Eur. J. Immunol. 28, 2066-2074
-183-
Literaturverzeichnis
SAUTY, A., M. DZIEJMAN, R. A. TAHA, A. S. IAROSSI, K. NEOTE, E. A. GARCIA-ZEPEDA, Q. HAMID u. A. D. LUSTER (1999): The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J. Immunol. 162, 3549-3558
SCHALL, T. J. u. K. B. BACON (1994): Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 6, 865-873
SCHMITT, D. (1994): Cutaneous immune system. C. R. Seances Soc. Biol. Fil. 188, 207-21
SCHMITT, E., P. HOEHN, C. HUELS, S. GOEDERT, N. PALM, E. RUDE u. T. GERMANN (1994): T helper type 1 development of naive CD4+ T cells requires the coordinate action of interleukin-12 and interferon-gamma and is inhibited by transforming growth factor-beta. Eur. J. Immunol. 24, 793-798
SCHMITTGEN, T. D. u. B. A. ZAKRAJSEK (2000): Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J. Biochem. Biophys. Methods 46, 69-81
SCHONLAU, F., K. SCHARFFETTER-KOCHANEK, S. GRABBE, B. PIETZ, C. SORG u. C. SUNDERKOTTER (2000): In experimental leishmaniasis deficiency of CD18 results in parasite dissemination associated with altered macrophage functions and incomplete Th1 cell response. Eur. J. Immunol. 30, 2729-2740
SCHROEDER, A., O. MUELLER, S. STOCKER, R. SALOWSKY, M. LEIBER, M. GASSMANN, S. LIGHTFOOT, W. MENZEL, M. GRANZOW u. T. RAGG (2006): The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC. Mol. Biol. 7, 3
SCHULER, T., Z. QIN, S. IBE, N. NOBEN-TRAUTH u. T. BLANKENSTEIN (1999): T helper cell type 1-associated and cytotoxic T lymphocyte-mediated tumor immunity is impaired in interleukin 4-deficient mice. J. Exp. Med. 189, 803-810
SCHUTZE, K. u. G. LAHR (1998): Identification of expressed genes by laser-mediated manipulation of single cells. Nat. Biotechnol. 16, 737-742
SCOTT, P., A. EATON, W. C. GAUSE, Z. DI, X u. B. HONDOWICZ (1996): Early IL-4 production does not predict susceptibility to Leishmania major. Exp. Parasitol. 84, 178-187
-184-
Literaturverzeichnis
SCOTT, P., P. NATOVITZ, R. L. COFFMAN, E. PEARCE u. A. SHER (1988): Immunoregulation of cutaneous Leishmaniasis. T-cell lines that transfer protective immunity or exacerbation belong to different T-helper subsets and respond to distinct parasite antigens. J. Exp. Med. 168, 1675–1684
SCOTT, P. u. J. P. FARRELL (1998): Experimental cutaneous leishmaniasis: induction and regulation of T cells following infection of mice with Leishmania major. Chem. Immunol. 70, 60-80
SCOTT, P. (2003): Development and regulation of cell-mediated immunity in experimental leishmaniasis. Immunol. Res. 27, 489-498
SEDER, R. A. u. W. E. PAUL (1994): Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12, 635-673
SEDER, R. A. (1996): High-dose IL-2 and IL-15 enhance the in vitro priming of naive CD4+ T cells for IFN-gamma but have differential effects on priming for IL-4. J. Immunol. 156, 2413-2422
SHIBUYA, K., D. ROBINSON, F. ZONIN, S. B. HARTLEY, S. E. MACATONIA, C. SOMOZA, C. A. HUNTER, K. M. MURPHY u. A. O'GARRA (1998): IL-1 alpha and TNF-alpha are required for IL-12-induced development of Th1 cells producing high levels of IFN-gamma in BALB/c but not C57BL/6 mice. J. Immunol. 160, 1708-1716
SICHERER, S. H. u. D. Y. LEUNG (2004): Advances in allergic skin disease, anaphylaxis, and hypersensitivity reactions to foods, drugs, and insect stings. J. Allergy Clin. Immunol. 114, 118-124
SIMS, J. E. u. S. K. DOWER (1994): Interleukin-1 receptors. Eur. Cytokine Netw. 5, 539-546
SINKE, J. D., T. THEPEN, I. C. BIHARI, V. P. RUTTEN u. T. WILLEMSE (1997): Immunophenotyping of skin-infiltrating T-cell subsets in dogs with atopic dermatitis. Vet. Immunol. Immunopathol. 57, 13-23
SINKE, J. D., V. P. RUTTEN u. T. WILLEMSE (2002): Immune dysregulation in atopic dermatitis. Vet. Immunol. Immunopathol. 87, 351-356
-185-
Literaturverzeichnis
SORENSEN, A. L., A. KHARAZMI u. H. NIELSEN (1989): Leishmania interaction with human monocytes and neutrophils: modulation of the chemotactic response. APMIS 97, 754-760
SOUTHERN, E. (1979): Gel electrophoresis of restriction fragments. Methods Enzymol. 68, 152-176
STAGER, S., J. ALEXANDER, K. C. CARTER, F. BROMBACHER u. P. M. KAYE (2003a): Both interleukin-4 (IL-4) and IL-4 receptor alpha signaling contribute to the development of hepatic granulomas with optimal antileishmanial activity. Infect. Immun. 71, 4804-4807
STAGER, S., J. ALEXANDER, A. C. KIRBY, M. BOTTO, N. V. ROOIJEN, D. F. SMITH, F. BROMBACHER u. P. M. KAYE (2003b): Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat. Med. 9, 1287-1292
STEIGERWALD, M. u. H. MOLL (2005): Leishmania major modulates chemokine and chemokine receptor expression by dendritic cells and affects their migratory capacity. Infect. Immun. 73, 2564-2567
STENGER, S., H. THURING, M. ROLLINGHOFF u. C. BOGDAN (1994): Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. J. Exp. Med. 180, 783-793
STETSON, D. B., M. MOHRS, V. MALLET-DESIGNE, L. TEYTON u. R. M. LOCKSLEY (2002): Rapid expansion and IL-4 expression by Leishmania-specific naive helper T cells in vivo. Immunity. 17, 191-200
STUEHR, D. J. u. O. W. GRIFFITH (1992): Mammalian nitric oxide synthases. Adv. Enzymol. Relat Areas Mol. Biol. 65, 287-346
SUNDERKOTTER, C., M. KUNZ, K. STEINBRINK, G. MEINARDUS-HAGER, M. GOEBELER, H. BILDAU u. C. SORG (1993): Resistance of mice to experimental leishmaniasis is associated with more rapid appearance of mature macrophages in vitro and in vivo. J. Immunol. 151, 4891-4901
-186-
Literaturverzeichnis
SUTER, M., F. M. CRAMBERI, T. OLIVRY, E. MUELLER, C. TSCHARNER u. P. JENSEN (1996): Keratinocyte biology and pathology. Vet. Dermatol. 8, 67-100
SWAIN, S. L., G. HUSTON, S. TONKONOGY u. A. WEINBERG (1991): Transforming growth factor-beta and IL-4 cause helper T cell precursors to develop into distinct effector helper cells that differ in lymphokine secretion pattern and cell surface phenotype 1. J. Immunol. 147, 2991-3000
SYPEK, J. P., C. L. CHUNG, S. E. MAYOR, J. M. SUBRAMANYAM, S. J. GOLDMAN, D. S. SIEBURTH, S. F. WOLF u. R. G. SCHAUB (1993): Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 177, 1797-1802
SZABO, S. J., B. M. SULLIVAN, C. STEMMANN, A. R. SATOSKAR, B. P. SLECKMAN u. L. H. GLIMCHER (2002): Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295, 338-342
SZABO, S. J., B. M. SULLIVAN, S. L. PENG u. L. H. GLIMCHER (2003): Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 21, 713-758
TACCHINI-COTTIER, F., C. ZWEIFEL, Y. BELKAID, C. MUKANKUNDIYE, M. VASEI, P. LAUNOIS, G. MILON u. J. A. LOUIS (2000): An immunomodulatory function for neutrophils during the induction of a CD4+ Th2 response in BALB/c mice infected with Leishmania major. J. Immunol. 165, 2628-2636
TANG, A., M. AMAGAI, L. G. GRANGER, J. R. STANLEY u. M. C. UDEY (1993): Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature 361, 82-85
TAUB, D. D., T. J. SAYERS, C. R. CARTER u. J. R. ORTALDO (1995): Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J. Immunol. 155, 3877-3888
TAVARES, D., R. R. DA CONCEICAO u. S. A. CARLOS DA (2006): Inflammatory lesion and parasite load are inversely associated in Leishmania amazonensis infected mice genetically selected according to oral tolerance susceptibility. Microbes. Infect. 8, 957-964
-187-
Literaturverzeichnis
TENSEN, C. P., J. FLIER, VAN DER RAAIJ-HELMER EM, S. SAMPAT-SARDJOEPERSAD, R. C. VAN DER SCHORS, R. LEURS, R. J. SCHEPER, D. M. BOORSMA u. R. WILLEMZE (1999): Human IP-9: A keratinocyte-derived high affinity CXC-chemokine ligand for the IP-10/Mig receptor (CXCR3). J. Invest Dermatol. 112, 716-722
THOMPSON, C. B. (1995): Distinct roles for the costimulatory ligands B7-1 and B7-2 in T helper cell differentiation? Cell 81, 979-982
TITUS, R. G., M. MARCHAND, T. BOON u. J. A. LOUIS (1985): A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol. 7, 545-555
TIZARD, I. (1996): Veterinary Immunology, an Introduction. 6. Aufl. Verlag WB Saunders, Philadelphia, U.S.A.
UCHI, H., H. TERAO, T. KOGA u. M. FURUE (2000): Cytokines and chemokines in the epidermis. J. Dermatol. Sci. 24, S29-S38
UEHARA, M. (1986): Heterogeneity of serum IgE levels in atopic dermatitis. Acta Derm. Venereol. 66, 404-408
VAN DER HEIJDEN, F. L., E. A. WIERENGA, J. D. BOS u. M. L. KAPSENBERG (1991): High frequency of IL-4-producing CD4+ allergen-specific T lymphocytes in atopic dermatitis lesional skin. J. Invest Dermatol. 97, 389-394
VAN GELDER, R. N., M. E. VON ZASTROW, A. YOOL, W. C. DEMENT, J. D. BARCHAS u. J. H. EBERWINE (1990): Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. U.S.A 87, 1663-1667
VAN ZANDBERGEN, G., N. HERMANN, H. LAUFS, W. SOLBACH u. T. LASKAY (2002): Leishmania promastigotes release a granulocyte chemotactic factor and induce interleukin-8 release but inhibit gamma interferon-inducible protein 10 production by neutrophil granulocytes. Infect. Immun. 70, 4177-4184
-188-
Literaturverzeichnis
VAN ZANDBERGEN, G., M. KLINGER, A. MUELLER, S. DANNENBERG, A. GEBERT, W. SOLBACH u. T. LASKAY (2004): Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J. Immunol. 173, 6521-6525
VESTER, B., K. MULLER, W. SOLBACH u. T. LASKAY (1999): Early gene expression of NK cell-activating chemokines in mice resistant to Leishmania major. Infect. Immun. 67, 3155-3159
VON STEBUT, E., Y. BELKAID, T. JAKOB, D. L. SACKS u. M. C. UDEY (1998): Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J. Exp. Med. 188, 1547-1552
VON STEBUT, E., Y. BELKAID, B. V. NGUYEN, M. CUSHING, D. L. SACKS u. M. C. UDEY (2000): Leishmania major-infected murine langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous Leishmaniasis. Eur. J. Immunol. 30, 3498-3506
VON STEBUT, E., Y. BELKAID, B. NGUYEN, M. WILSON, D. L. SACKS u. M. C. UDEY (2002): Skin-derived macrophages from Leishmania major-susceptible mice exhibit interleukin-12- and interferon-gamma-independent nitric oxide production and parasite killing after treatment with immunostimulatory DNA. J. Invest Dermatol. 119, 621-628
VON STEBUT, E., J. M. EHRCHEN, Y. BELKAID, S. L. KOSTKA, K. MOLLE, J. KNOP, C. SUNDERKOTTER u. M. C. UDEY (2003): Interleukin 1alpha promotes Th1 differentiation and inhibits disease progression in Leishmania major-susceptible BALB/c mice. J. Exp. Med. 198, 191-199
WANG, B., P. AMERIO u. D. N. SAUDER (1999): Role of cytokines in epidermal Langerhans cell migration. J. Leukoc. Biol. 66, 33-39
WEI, X. Q., I. G. CHARLES, A. SMITH, J. URE, G. J. FENG, F. P. HUANG, D. XU, W. MULLER, S. MONCADA u. F. Y. LIEW (1995): Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 375, 408-411
-189-
Literaturverzeichnis
WEI, X. Q., B. P. LEUNG, W. NIEDBALA, D. PIEDRAFITA, G. J. FENG, M. SWEET, L. DOBBIE, A. J. SMITH u. F. Y. LIEW (1999): Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. J. Immunol. 163, 2821-2828
WEINHEBER, N., M. WOLFRAM, D. HARBECKE u. T. AEBISCHER (1998): Phagocytosis of Leishmania mexicana amastigotes by macrophages leads to a sustained suppression of IL-12 production. Eur. J. Immunol. 28, 2467-2477.
WEISS, J. M., A. C. RENKL, C. S. MAIER, M. KIMMIG, L. LIAW, T. AHRENS, S. KON, M. MAEDA, H. HOTTA, T. UEDE u. J. C. SIMON (2001): Osteopontin is involved in the initiation of cutaneous contact hypersensitivity by inducing Langerhans and dendritic cell migration to lymph nodes. J. Exp. Med. 194, 1219-1229
WILHELM, P., U. RITTER, S. LABBOW, N. DONHAUSER, M. ROLLINGHOFF, C. BOGDAN u. H. KORNER (2001): Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 166, 4012-4019
WILLEMSE, A., A. NOORDZIJ, V. P. RUTTEN u. W. E. BERNADINA (1985): Induction of non-IgE anaphylactic antibodies in dogs. Clin. Exp. Immunol. 59, 351-358
WRIGHT, T. M. u. J. M. FARBER (1991): 5' regulatory region of a novel cytokine gene mediates selective activation by interferon gamma. J. Exp. Med. 173, 417-422
WUTHRICH, B. u. P. SCHMID-GRENDELMEIER (2003): The atopic eczema/dermatitis syndrome. Epidemiology, natural course, and immunology of the IgE-associated ("extrinsic") and the nonallergic ("intrinsic") AEDS. J. Investig. Allergol. Clin. Immunol. 13, 1-5
XANTHOU, G., C. E. DUCHESNES, T. J. WILLIAMS u. J. E. PEASE (2003): CCR3 functional responses are regulated by both CXCR3 and its ligands CXCL9, CXCL10 and CXCL11. Eur. J. Immunol. 33, 2241-2250
ZACHARY, C. B., M. H. ALLEN u. D. M. MACDONALD (1985): In situ quantification of T-lymphocyte subsets and Langerhans cells in the inflammatory infiltrate of atopic eczema. Br. J. Dermatol. 112, 149-156
-190-
Literaturverzeichnis
ZAPH, C. u. P. SCOTT (2003): Interleukin-12 regulates chemokine gene expression during the early immune response to Leishmania major. Infect. Immun. 71, 1587-1589
ZENZ, R., R. EFERL, L. KENNER, L. FLORIN, L. HUMMERICH, D. MEHIC, H. SCHEUCH, P. ANGEL, E. TSCHACHLER u. E. F. WAGNER (2005): Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 437, 369-375
ZILBERSTEIN, D. u. D. M. DWYER (1985): Protonmotive force-driven active transport of D-glucose and L-proline in the protozoan parasite Leishmania donovani. Proc. Natl. Acad. Sci. U.S.A 82, 1716-1720
-191-
Danksagung
10 DANKSAGUNG
Mein besonderer Dank gilt Herrn Prof. Dr. Johannes Roth für die Überlassung des
interessanten Dissertationsthemas und die jederzeit gewährte freundliche Unterstützung bei
der wissenschaftlichen Betreuung und Förderung dieser Arbeit. Bei Herrn Prof. Dr. Hans-Joachim Schuberth möchte ich mich ganz herzlich für die sehr
engagierte und freundliche Betreuung dieser externen Doktorarbeit bedanken. Danke auch für
die aufmunternden Worte in der heißen Phase der Abfassung. Dr. Jan Ehrchen danke ich für viele sinnvolle Diskussionen und Ideen, sowie der
Unterstützung bei der Anfertigung der vorliegenden Arbeit. Ohne meine lustigen Kollegen PD Dr. Thomas Vogl und insbesondere Dr. Lars Steinmüller-
Magin, wäre diese Arbeit so wohl nicht zustande gekommen. Ob Statistik, Formatierung,
Korrekturlesen, anregende Diskussion oder schlaue Sprüche, für all dies hattet ihr Zeit und
wurdet nie müde mir mit Rat und Tat zur Seite zu stehen. Ich danke euch vom ganzen Herzen. Sehr bedanken möchte ich mich bei all den lieben Kollegen, die zum Gelingen dieser Arbeit
beigetragen haben: Allen voran denk ich da an Eva, durch deren hervorragende Erfahrung im
Umgang mit Leishmanien und Labormäusen ich sehr profitiert habe und an Ulla, die mich
Real-Time-PCR-technisch so prima begleitet hat. Außerdem danke an Harut und Nadja für
die molekularbiologischen Tricks und Kniffe und Claudia für ihre freundliche Hilfe bei der
Anfertigung des Literaturverzeichnisses.
Nicht vergessen zu danken möchte ich Prof. Dr. Dorothee Nashan, die so freundlich war die
Kooperation herzustellen und die Förderung dieses Projektes zu initialisieren. Meiner Mutter möchte ich für ihren ungebremsten Einsatz an der Zuhör-und Nachfrag-Front
und ihre stete liebevolle Unterstützung in allen Lebenslagen danken, und meinem Vater für
das rege Interesse an meiner Arbeit sowie der jahrelangen finanziellen Unterstützung.
Last but not least, cheers to Harvey, Paul and especially Mazzy for your perennial friendship