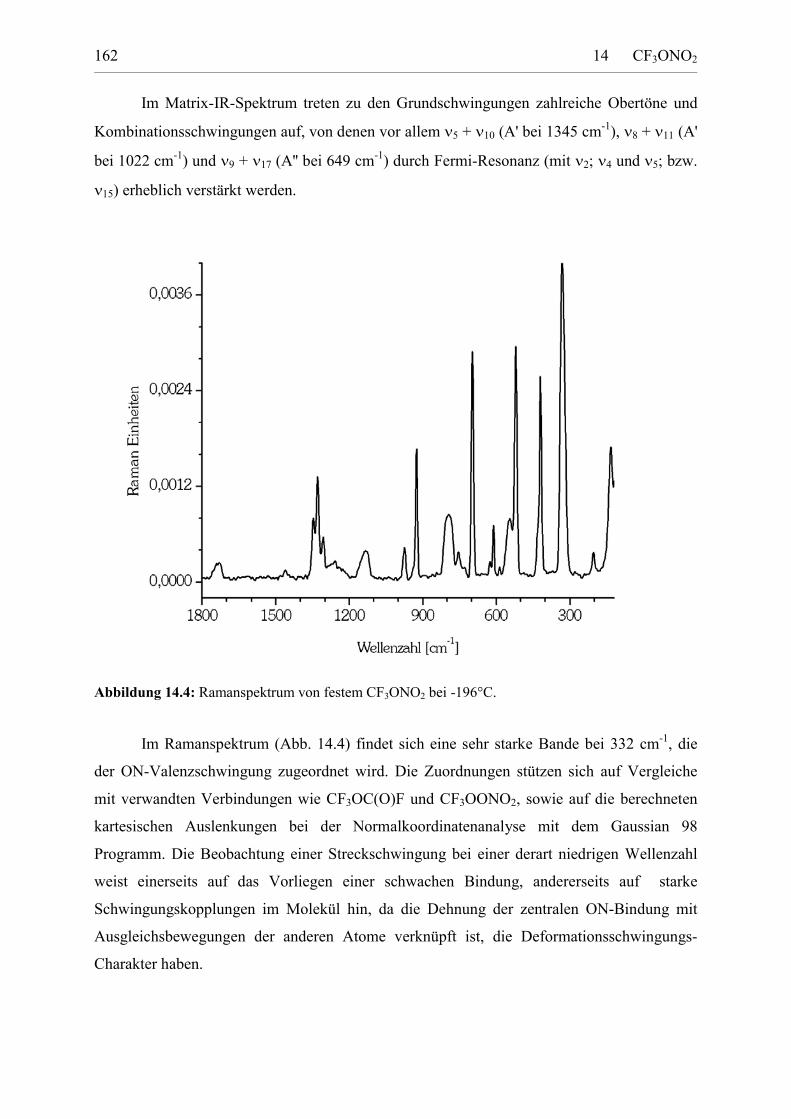
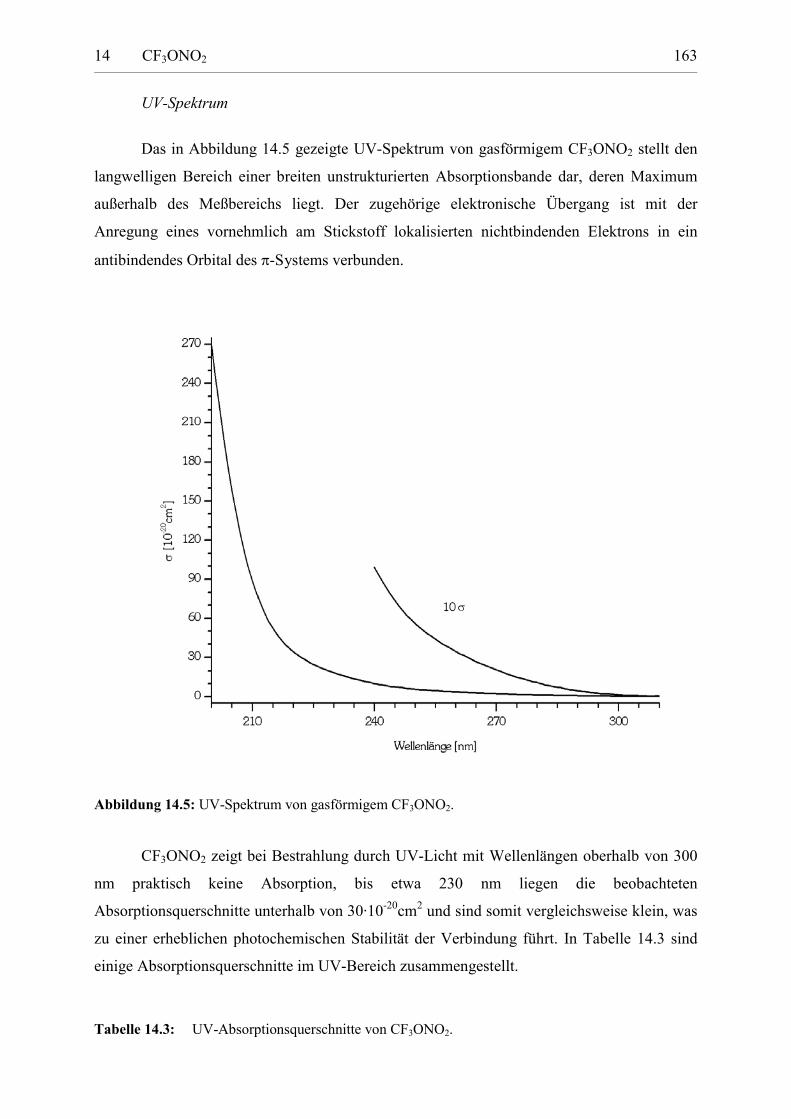
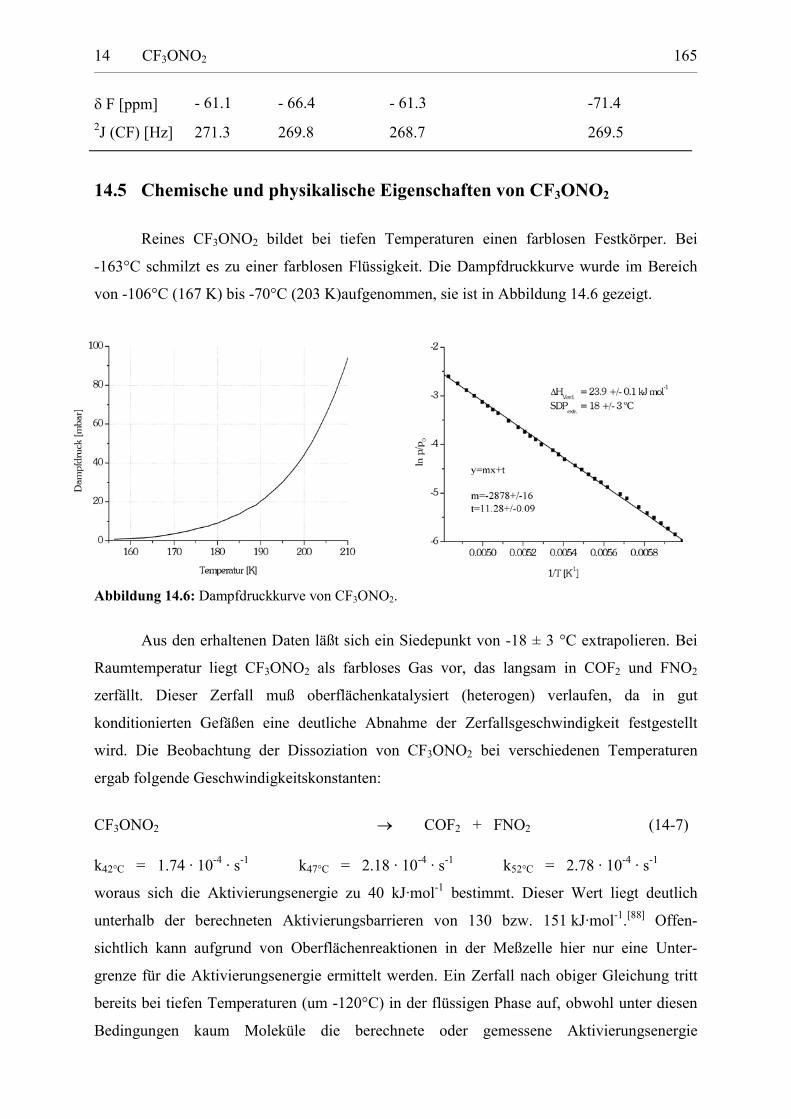
Darstellung und Charakterisierung atmosphärenrelevanter Fluorverbindungen Von der Fakultät für Naturwissenschaften der Gerhard Mercator Universität Duisburg zur Erlangung des Grades Doktor der Naturwissenschaften - Dr. rer. nat. - genehmigte Dissertation von Dipl. Chem. Stefan von Ahsen, geb. Sander geboren am 29. Dezember 1969 in Hannover * Februar 2003 Tag der mündlichen Prüfung: 4. Dezember 2002
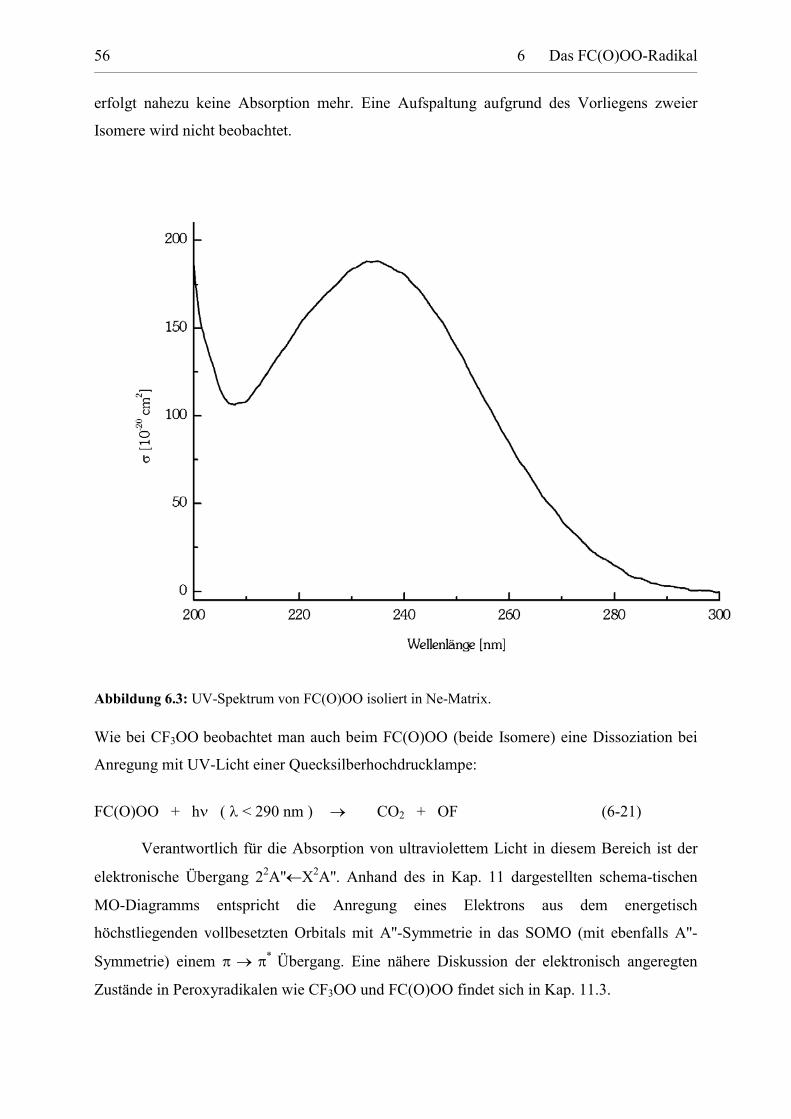
Transcript
Darstellung und Charakterisierung
atmosphärenrelevanter Fluorverbindungen
Von der Fakultät für Naturwissenschaften
der Gerhard Mercator Universität Duisburg
zur Erlangung des Grades
D o k t o r d e r N a t u r w i s s e n s c h a f t e n
- Dr. rer. nat. -
genehmigte Dissertation
von
Dipl. Chem. Stefan von Ahsen, geb. Sander
geboren am 29. Dezember 1969 in Hannover
*
Februar 2003
Tag der mündlichen Prüfung: 4. Dezember 2002
Die vorliegende Arbeit wurde im Zeitraum von Juli 1999 bis September 2002 im Fachbereich
Anorganische Chemie der Gerhard-Mercator-Universität Duisburg unter der Leitung von
Herrn Prof. Dr. H. Willner angefertigt.
Referent: Prof. Dr. H. Willner, Anorganische Chemie - Universität Duisburg
Korreferent: Prof. Dr. H.-J. Frohn, Anorganische Chemie - Universität Duisburg
Zusammenfassung
Diese Arbeit stellt einen Beitrag zur Synthese und umfassenden Charakterisierung
einfacher C, F, O-Verbindungen dar, die durch ihr Auftreten bei Abbauprozessen von FCKWs
und FCKW-Ersatzstoffen eine wichtige Rolle in der Atmosphärenchemie spielen.
Die Erzeugung der Peroxyradikale CF3OO, FC(O)OO, CF3OC(O)OO, CF3C(O)OO
und CH3C(O)OO gelingt durch Niederdruckpyrolyse von Gemischen aus geeigneten
Vorläuferverbindungen hochverdünnt mit Neon bzw. Argon und anschließender
Matrixisolation der Reaktionsprodukte. Darüber hinaus werden die Radikale CF3OCO, SF5,
und CF3OOO ebenfalls als matrixisolierte Spezies erzeugt.
Die Charakterisierung der Radikale erfolgt durch IR- und UV-Spektroskopie, und die
Eigenschaften der Fluorkohlenstoffperoxyradikale werden vergleichend diskutiert.
Weiterhin wird über die erstmalige Reindarstellung des Trifluormethylnitrats,
CF3ONO2, auf verschiedenen Synthesewegen berichtet. Neben der vollständigen
Charakterisierung der Verbindung (IR, Raman, UV, NMR, Schmelz- u. Siedepunkt,
Dampfdruckkurve, Gaselektronenbeugung) wird das grundlegende chemische Verhalten von
CF3ONO2 untersucht. Es wird gezeigt, daß Trifluormethylnitrat keine Reservoirspezies für
CF3O-Radikale und NO2 darstellt.
Mit der Synthese von Bistrifluormethyltrioxydicarbonat, CF3OC(O)OOOC(O)OCF3, ist
ein neues, unterhalb von -20°C stabiles offenkettiges Trioxid zugänglich geworden.
CF3OC(O)OOOC(O)OCF3 wird spektroskopisch (IR, Raman, UV, NMR) charakterisiert und
zur Erzeugung von CF3OC(O)OO, einem bisher noch nicht bekannten Peroxyradikal,
eingesetzt.
Zu allen untersuchten Verbindungen werden zudem quantenmechanische
Berechnungen durchgeführt, wobei die einheitliche Verwendung der Dichtefunktional-
methode B3LYP und des Basisatzes 6-311G(d,p) vergleichende Studien erlaubt. Die
Ergebnisse der Berechnungen unterstützen darüber hinaus die Bandenzuordnungen in den
Synthese von CF3OC(O)OOOC(O)OCF3 148Präparation der Matrices 149Präparation der Raman-Probe 150
14 CF3ONO2 151
14.1 Einführung 151
14.2 Die Darstellung von CF3ONO2 152
Die Reaktion von CF3OC(O)OOCF3 bzw. CF3OC(O)OOC(O)OCF3 mit NO2 152Die Reaktion von CF3OOOCF3 mit NO2 153Die Reaktion von CF3OONO2 mit O3 bzw. NO 154Die Reaktion von CF3OF mit NO2 155
14.3 Die Struktur von CF3ONO2 156
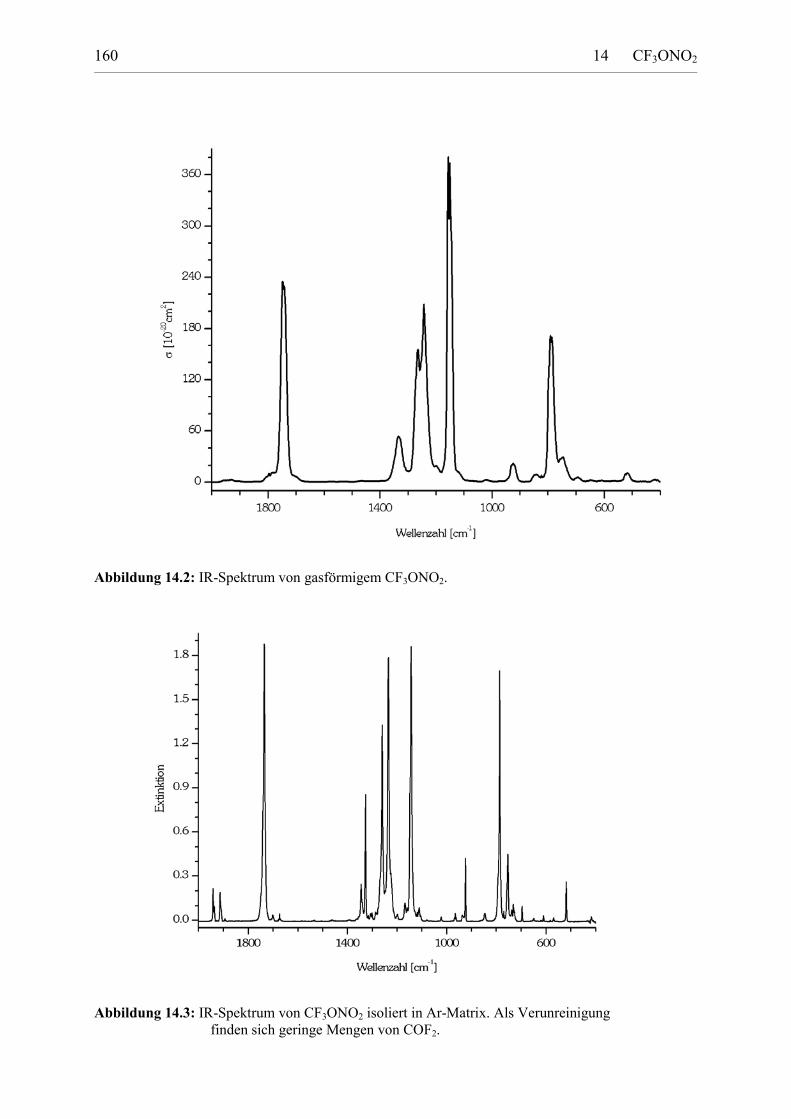
14.4 Die spektroskopische Charakterisierung von CF3ONO2 158Schwingungsspektren 158UV-Spektrum 162NMR-Spektren 163
14.5 Chemische und physikalische Eigenschaften von CF3ONO2 164
14.6 Experimentelles 167
Synthese von CF3ONO2 aus CF3OC(O)OOCF3 bzw. CF3OC(O)OOC(O)OCF3 167Synthese von CF3ONO2 aus CF3OONO2 und O3 168Synthese von CF3ONO2 aus CF3OF und NO2 168
Inhaltsverzeichnis
V
Matrixexperimente 169
15 SF5NO2 171
15.1 Einführung 171
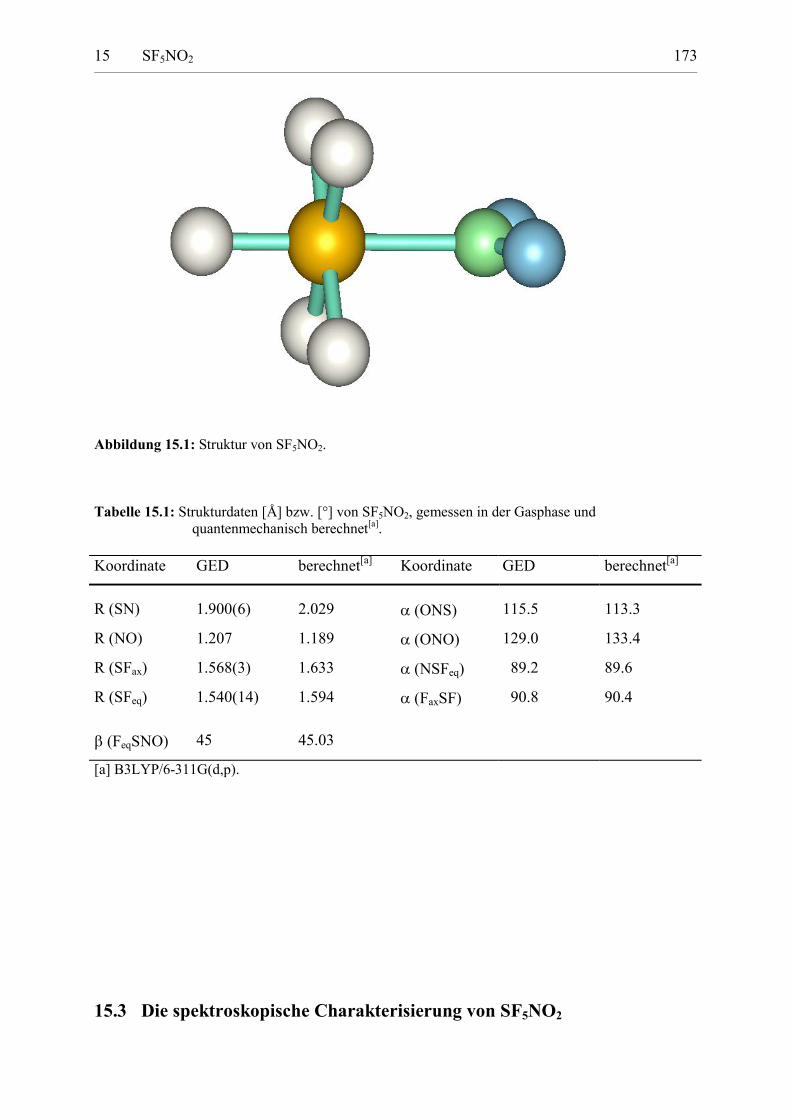
15.2 Die Struktur von SF5NO2 171
15.3 Die spektroskopische Charakterisierung von SF5NO2 173
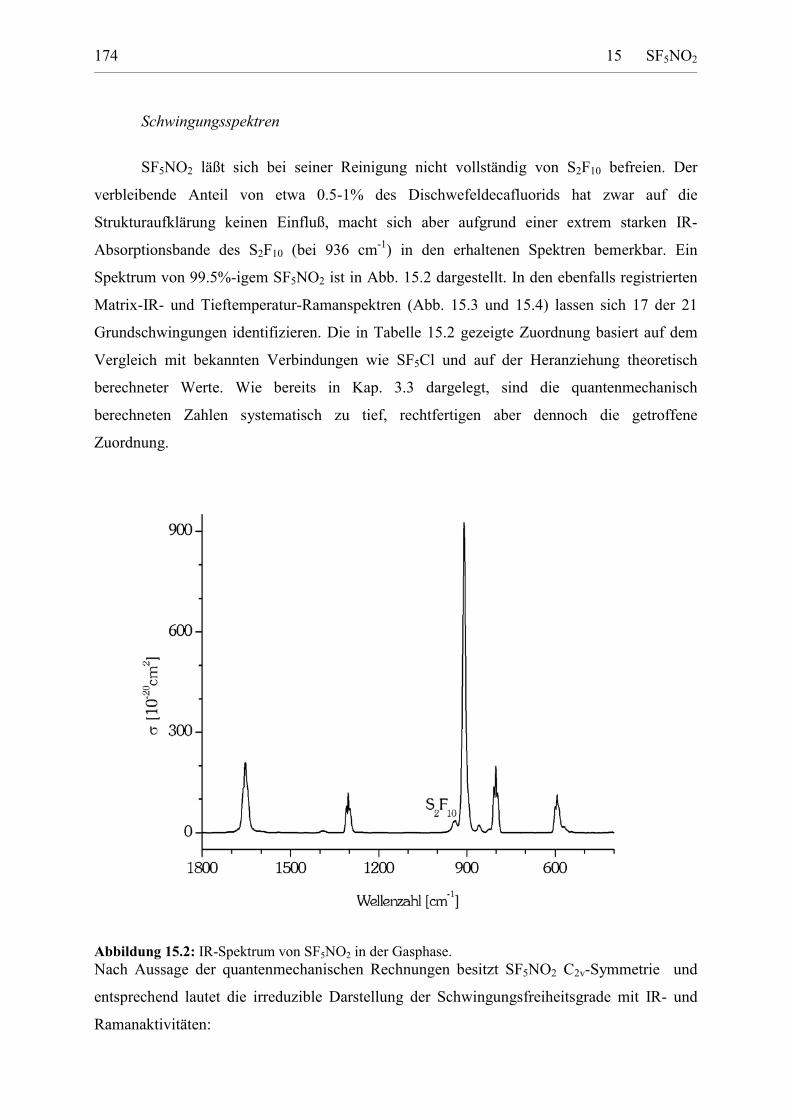
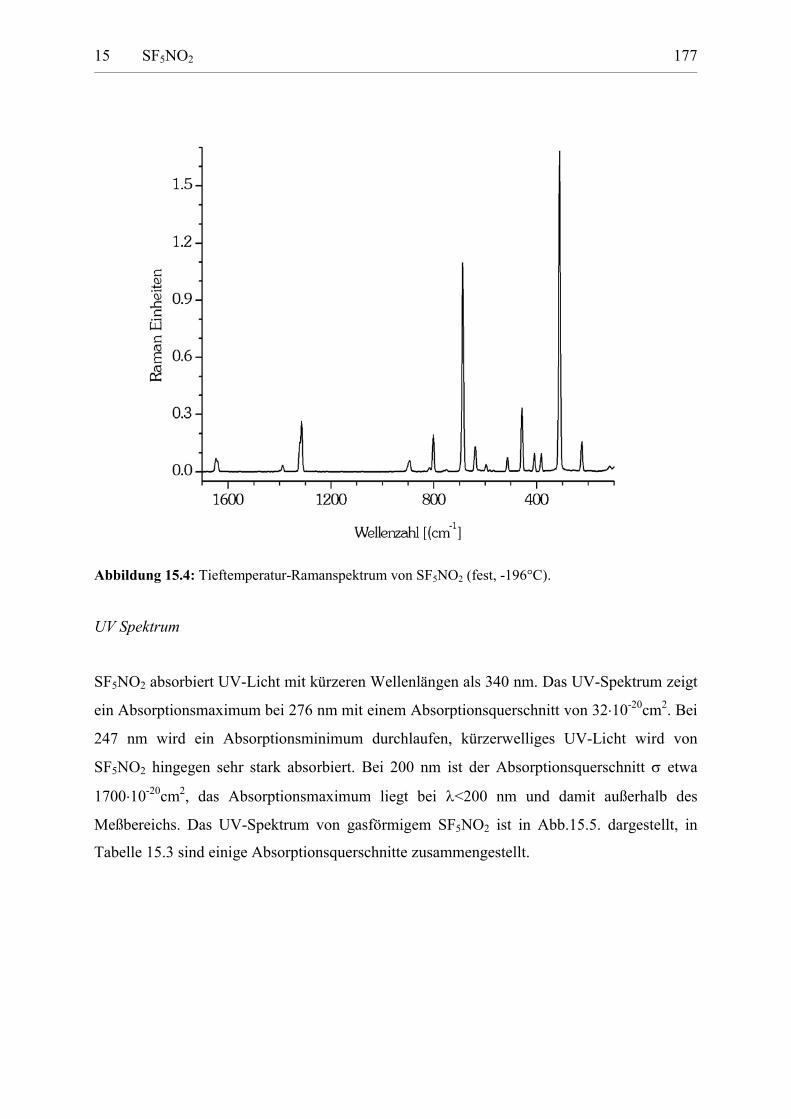
Schwingungsspektren 173UV-Spektrum 176
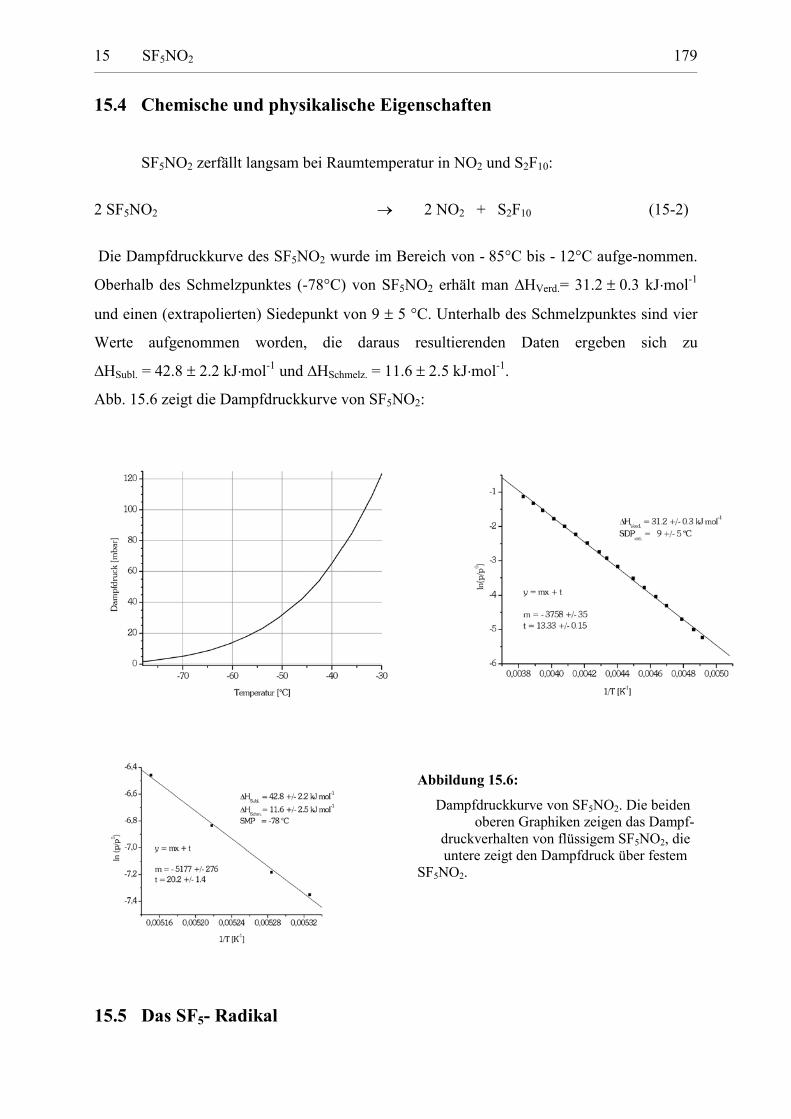
15.4 Chemische und physikalische Eigenschaften 178
15.5 Das SF5- Radikal 179
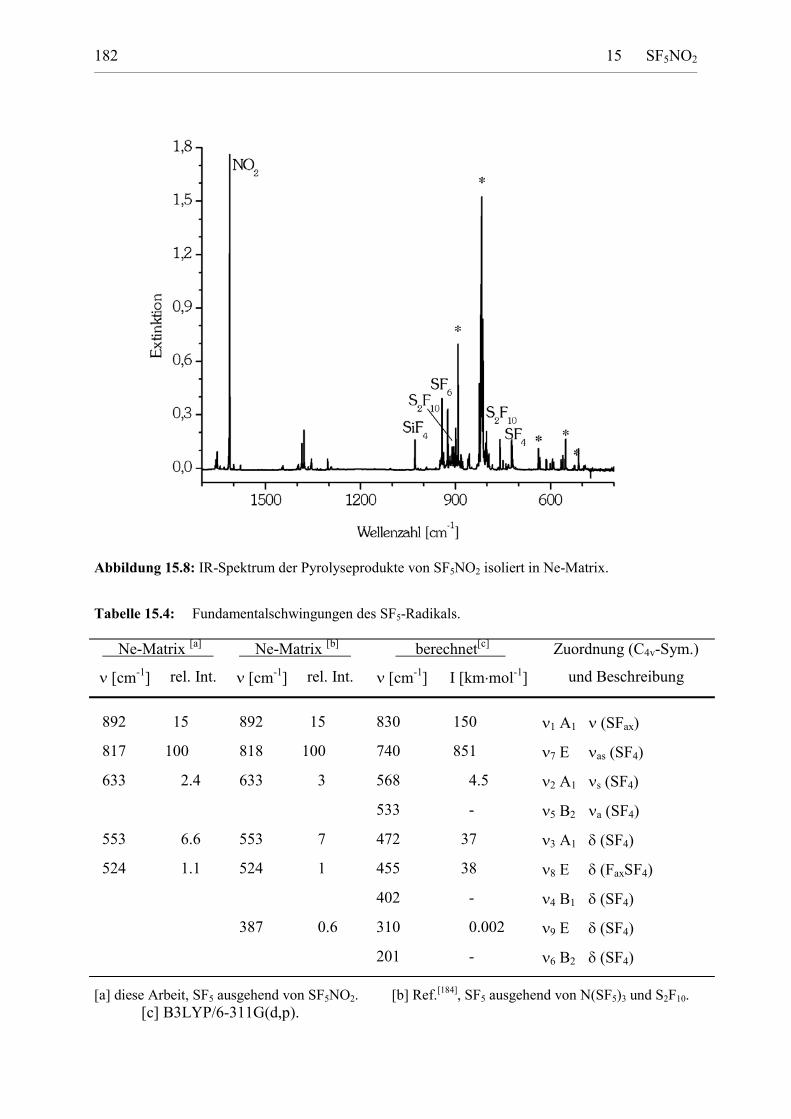
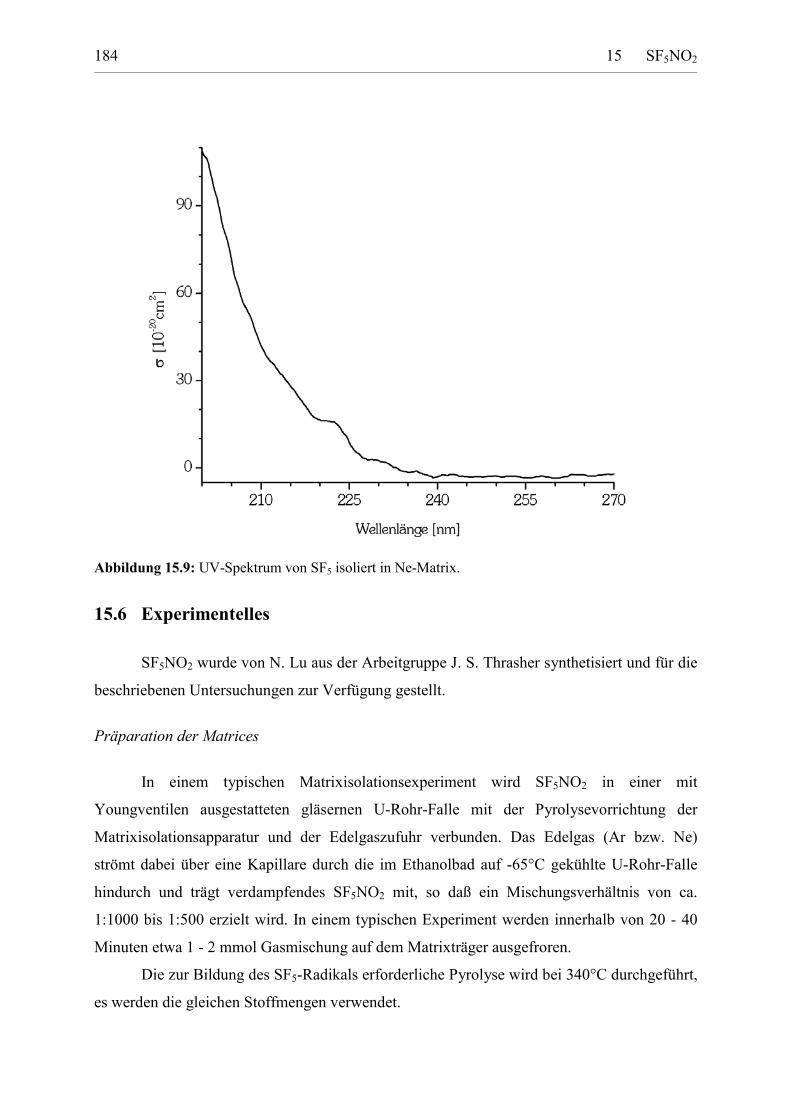
Berechnete Struktur 179Erzeugung von SF5-Radikalen aus SF5NO2 180IR-Spektrum 180UV-Spektrum 182
15.6 Experimentelles 183
16 Ausblick 185
17 Instrumentation 187
18 Literatur 189
Lebenslauf 199
Publikationsliste 200
VI
Tabellenverzeichnis VII
Tabellenverzeichnis
Tabelle 4.1: Berechnete Reaktionsenthalpien für die Teilreaktionen einesOzonabbau-Zyklus unter Beteiligung von Peroxyradikalen. 32
Tabelle 4.2: Geschwindigkeitskonstanten für Reaktionen ausgewählter Peroxy-,Oxy- und Alkylradikale mit atmosphärischen Spurengasen. 33
Tabelle 5.1: Berechnete[a] Strukturparameter von CF3OO. 37
Tabelle 5.2: Fundamentalschwingungen von CF3OO. 40
Tabelle 5.3: Zuordnung der beobachteten Absorptionsbanden des12A'�X2A'' Übergangs von CF3OO. 43
Tabelle 5.4: UV Absorptionsquerschnitte von CF3OO. 45
Tabelle 6.1: Berechnete Strukturparameter von trans- und cis-FC(O)OO. 51
Tabelle 6.2: Fundamentalschwingungen von trans-FC(O)OO. 55
Tabelle 6.3: Fundamentalschwingungen von cis-FC(O)OO. 55
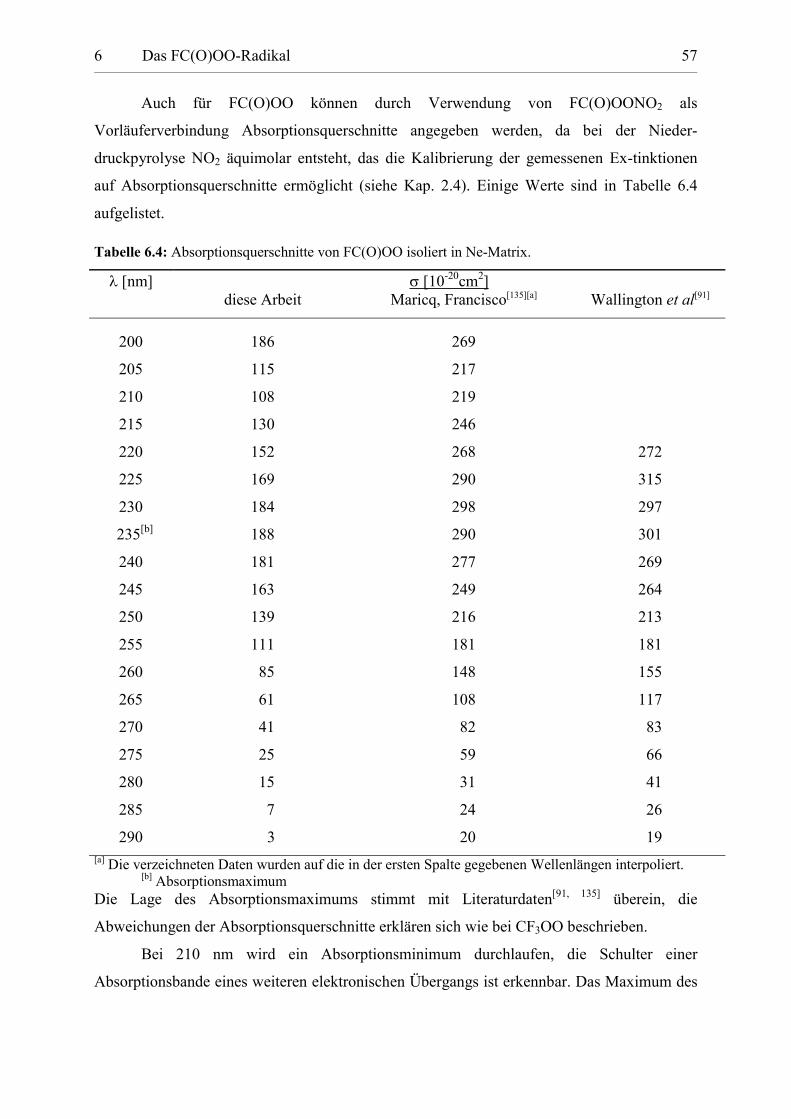
Tabelle 6.4: Absorptionsquerschnitte von FC(O)OO. 57
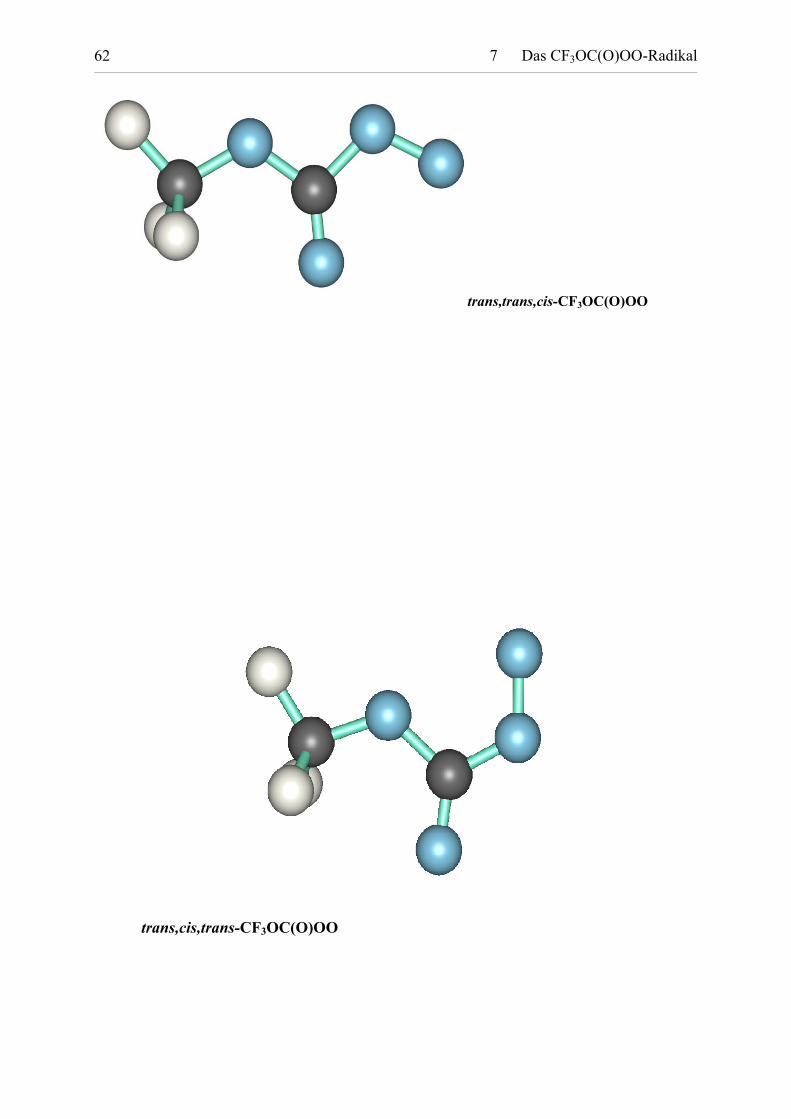
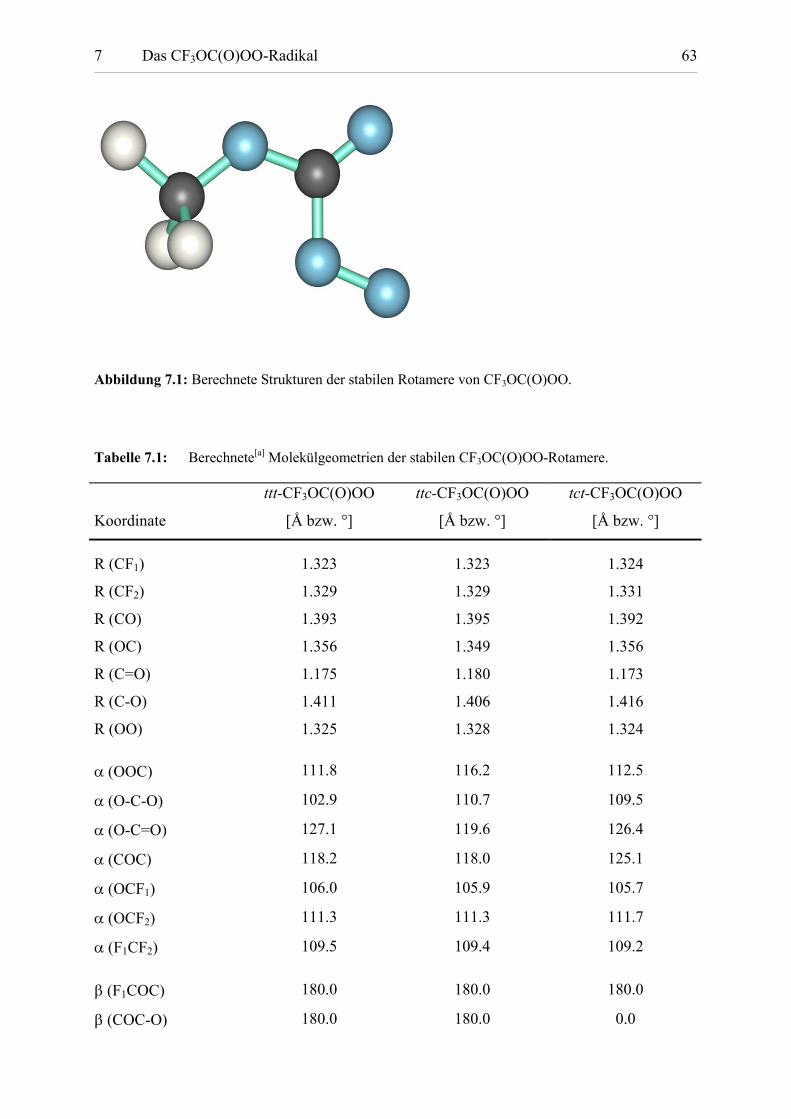
Tabelle 7.1: Berechnete Molekülgeometrien der stabilen CF3OC(O)OO-Rotamere. 63
Tabelle 7.2: Fundamentalschwingungen von t,t,t-CF3OC(O)OO. 69
Tabelle 7.3: Fundamentalschwingungen von t,t,c-CF3OC(O)OO. 70
Tabelle 7.4: UV-Extinktionen von CF3OC(O)OO. 73
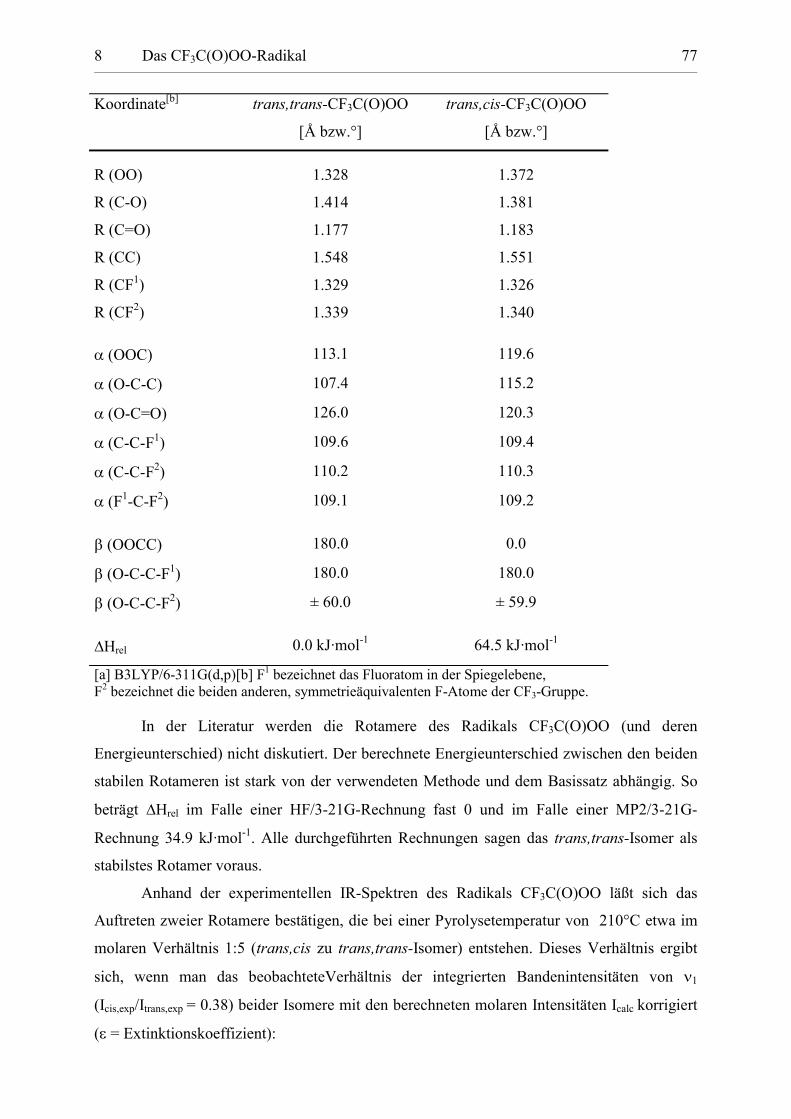
Tabelle 8.1: Berechnete Geometrien der stabilen Rotamere von CF3C(O)OO. 77
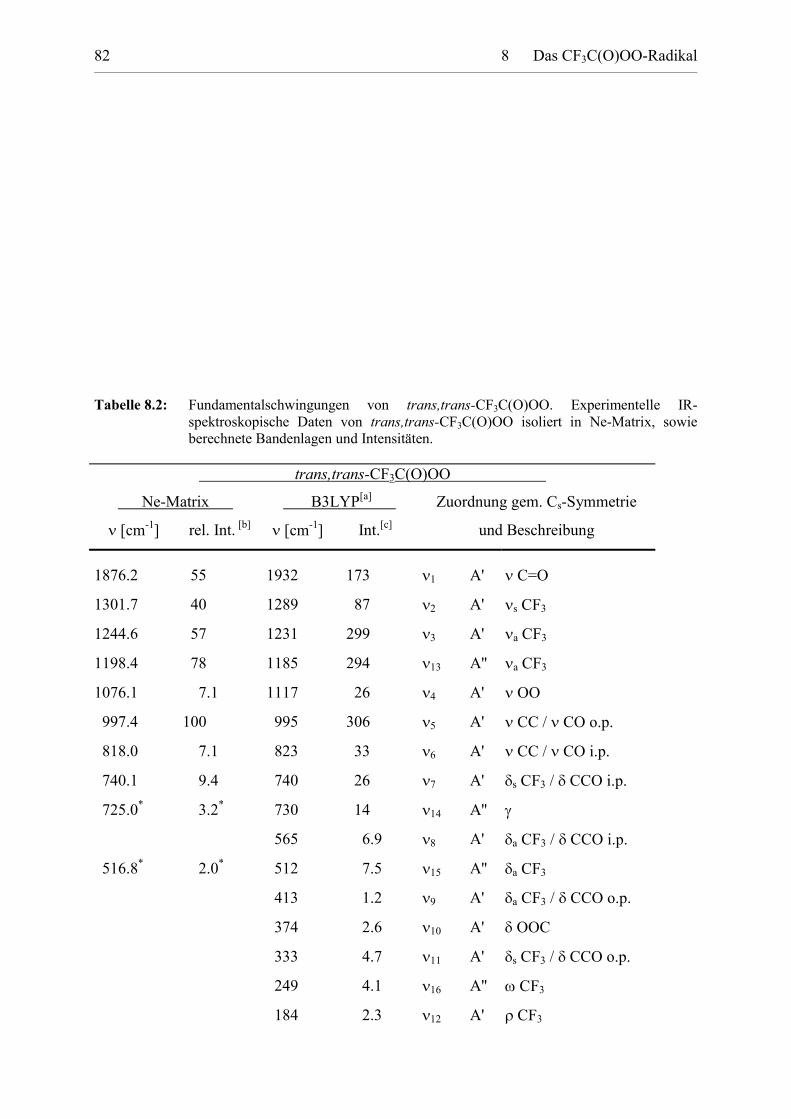
Tabelle 8.2: Fundamentalschwingungen von trans,trans-CF3C(O)OO. 82
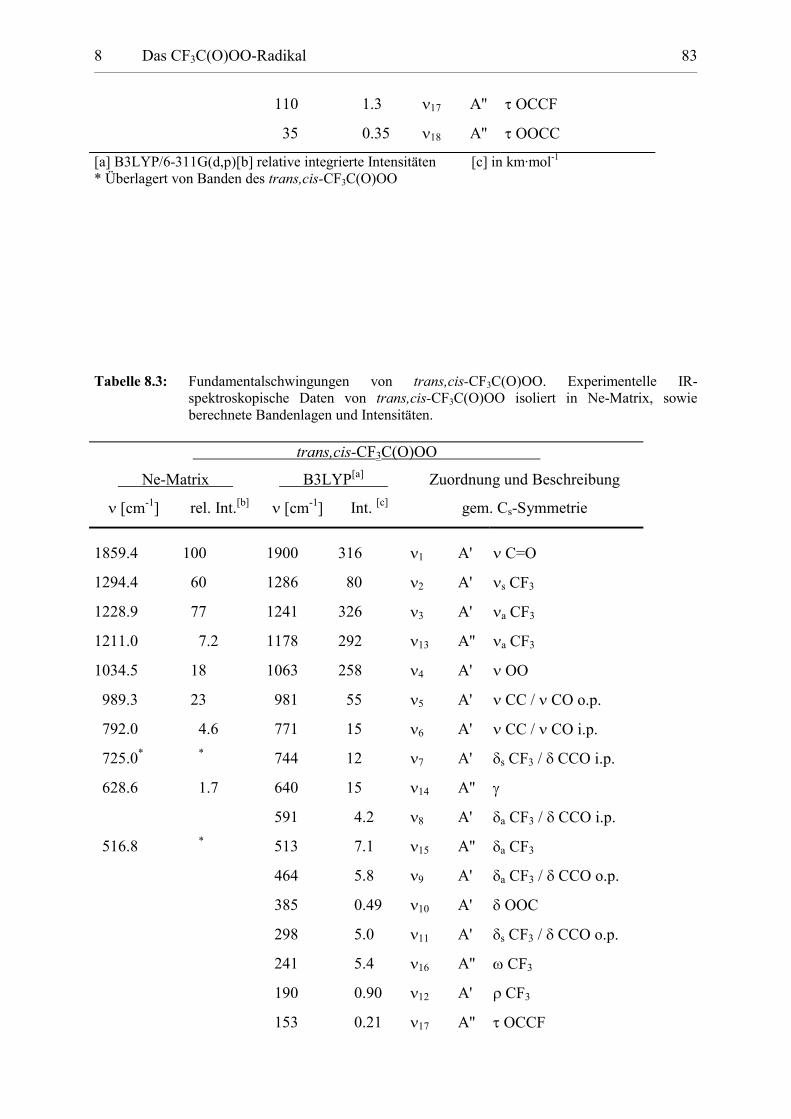
Tabelle 8.3: Fundamentalschwingungen von trans,cis-CF3C(O)OO. 83
Tabelle 8.4: UV-Extinktionen von CF3C(O)OO. 85
Tabelle 9.1: Berechnete Molekülparameter der stabilen CH3C(O)OO-Rotamere. 89
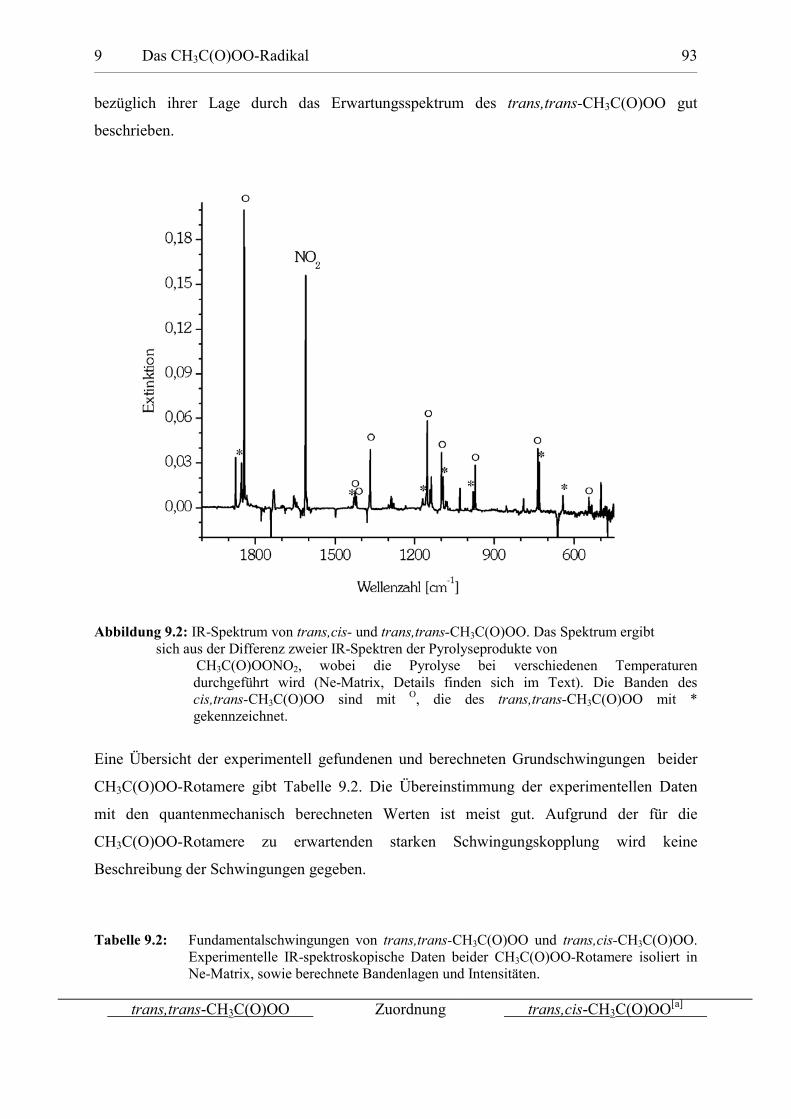
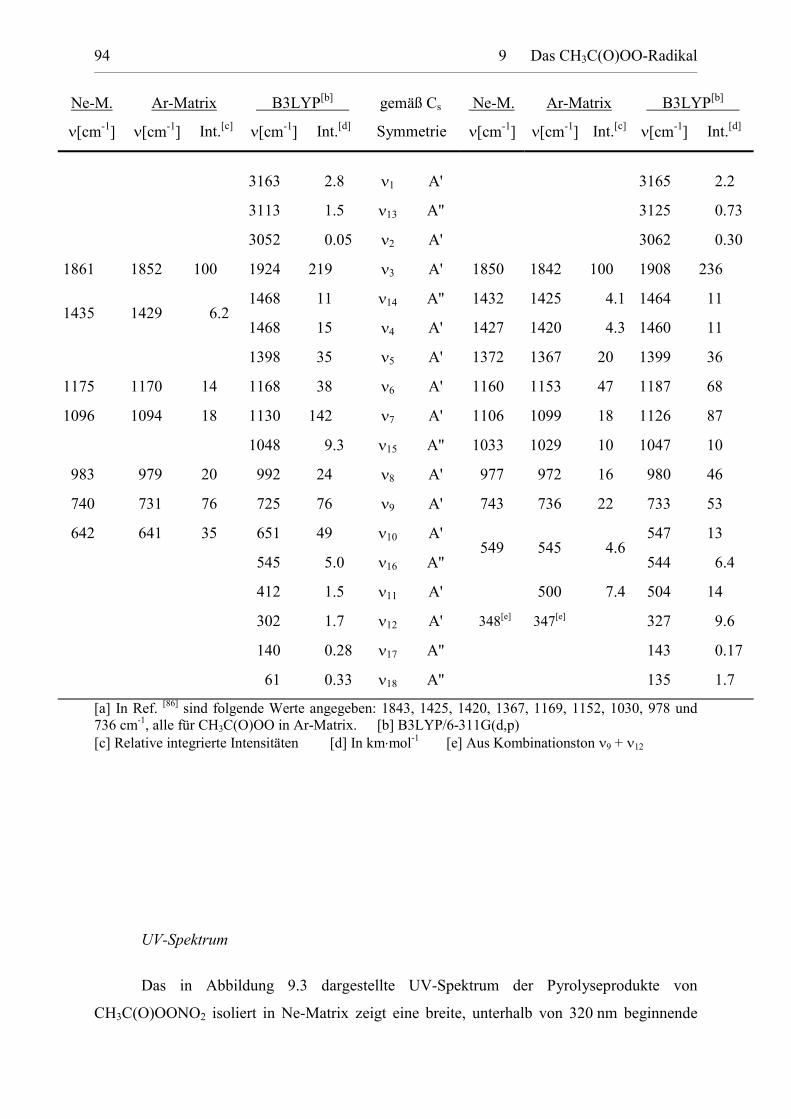
Tabelle 9.2: Fundamentalschwingungen von trans,trans-CH3C(O)OO undtrans,cis-CH3C(O)OO. 93
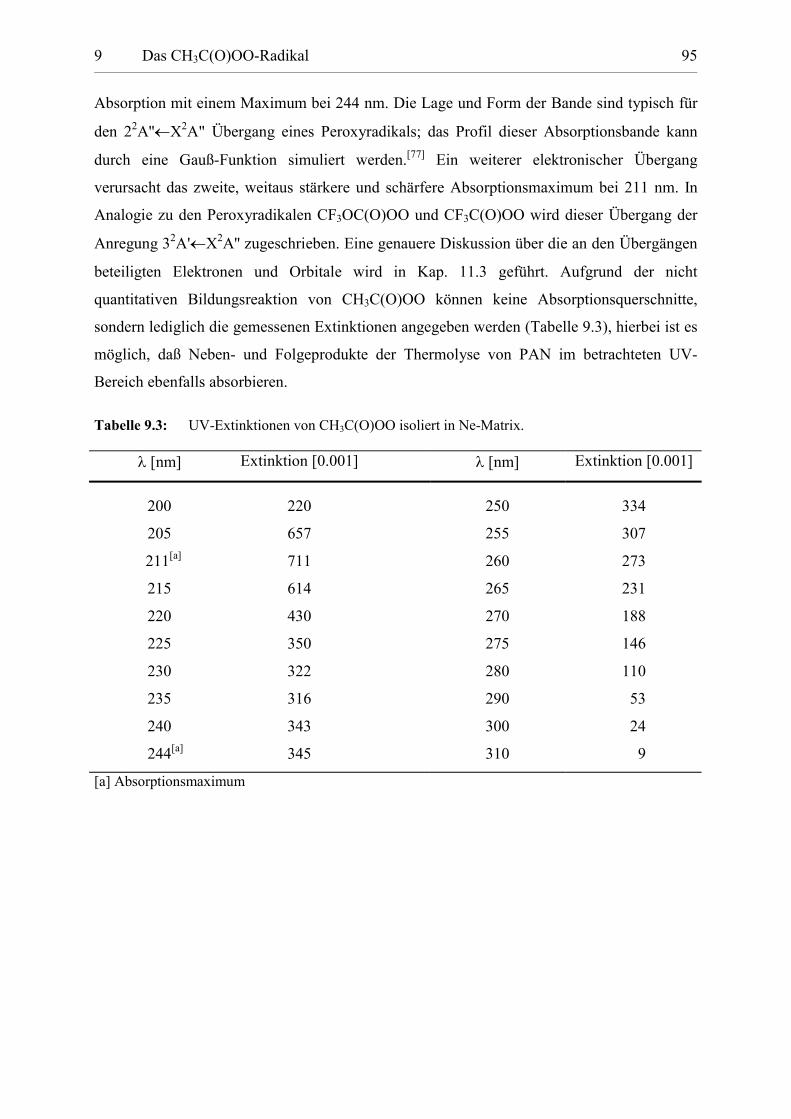
Tabelle 9.3: UV-Extinktionen von CH3C(O)OO. 94
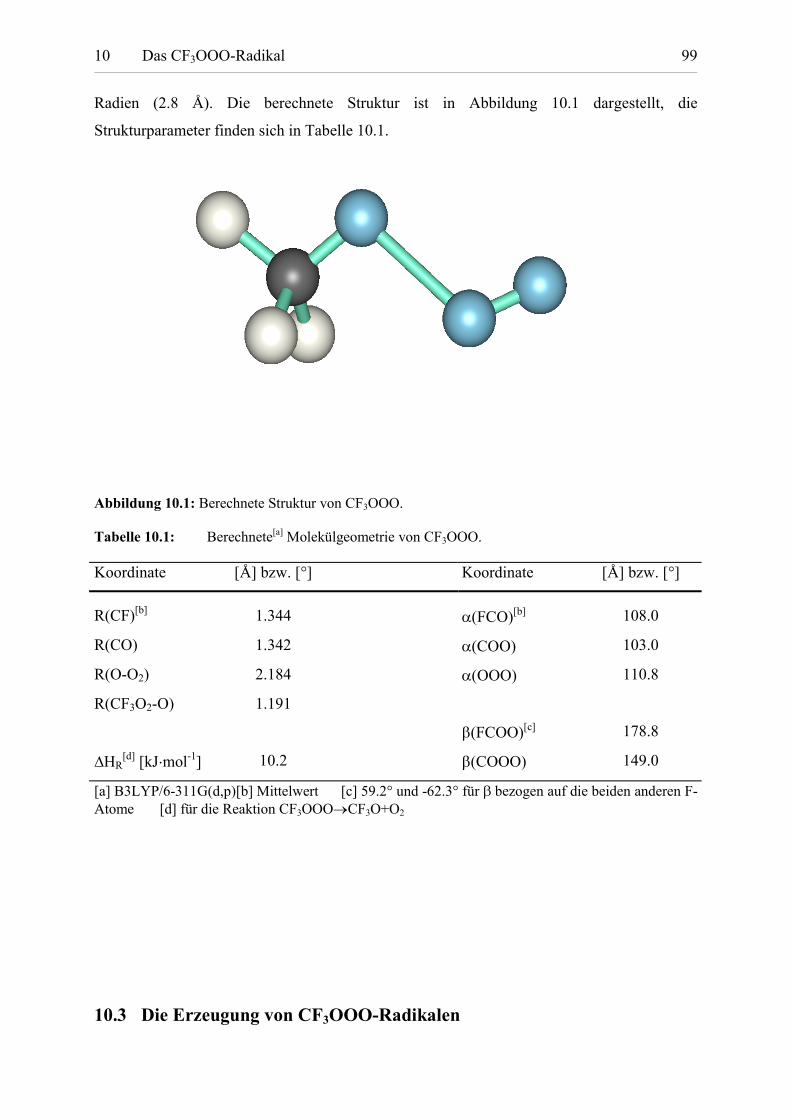
Tabelle 10.1: Berechnete Molekülgeometrie von CF3OOO. 98
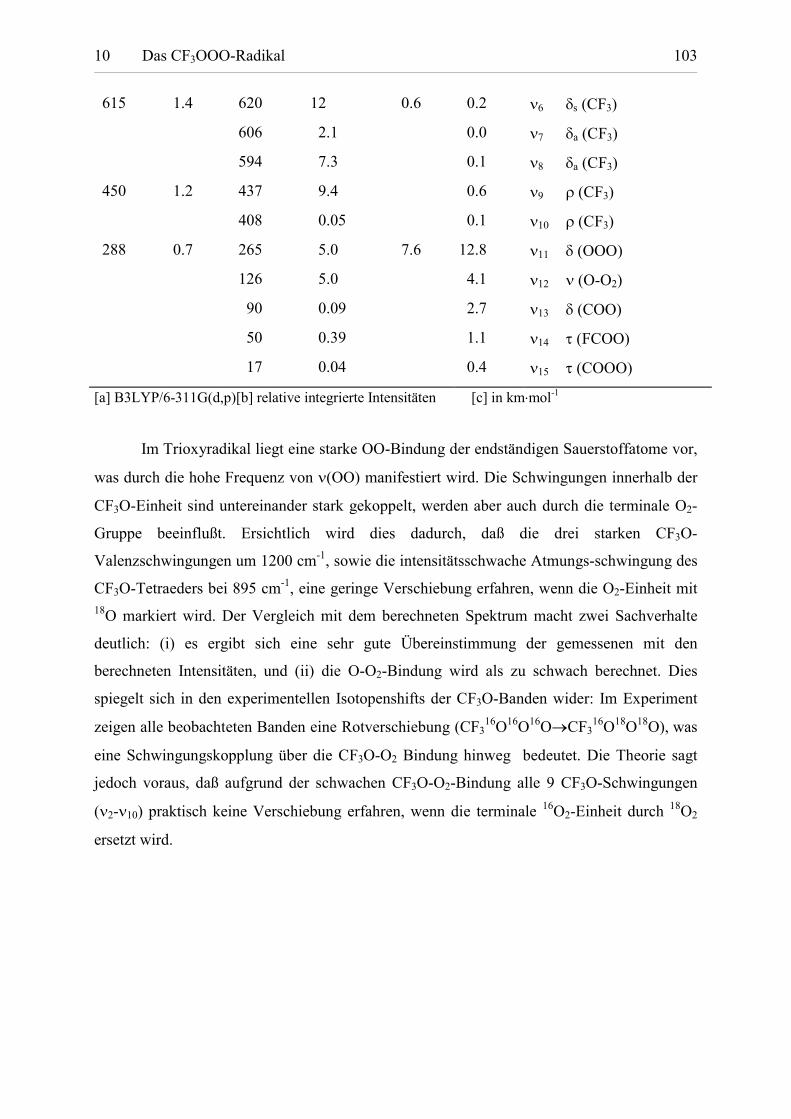
Tabelle 10.2: Fundamentalschwingungen von CF3OOO. 102
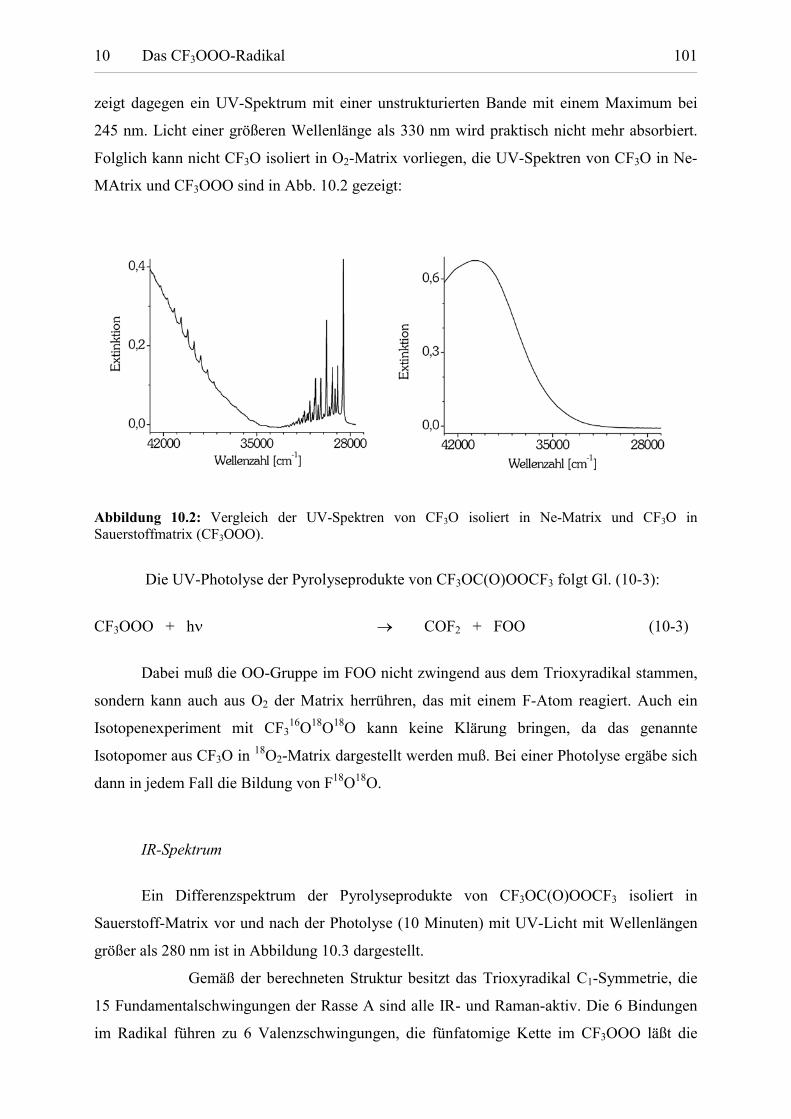
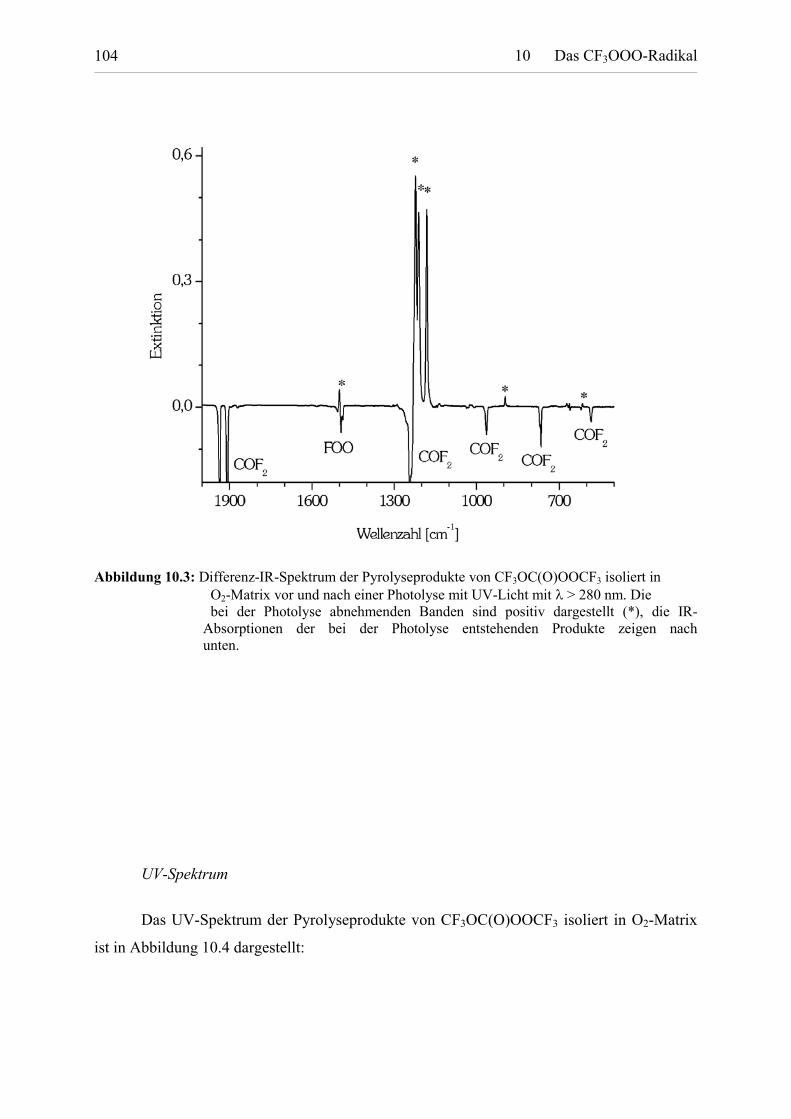
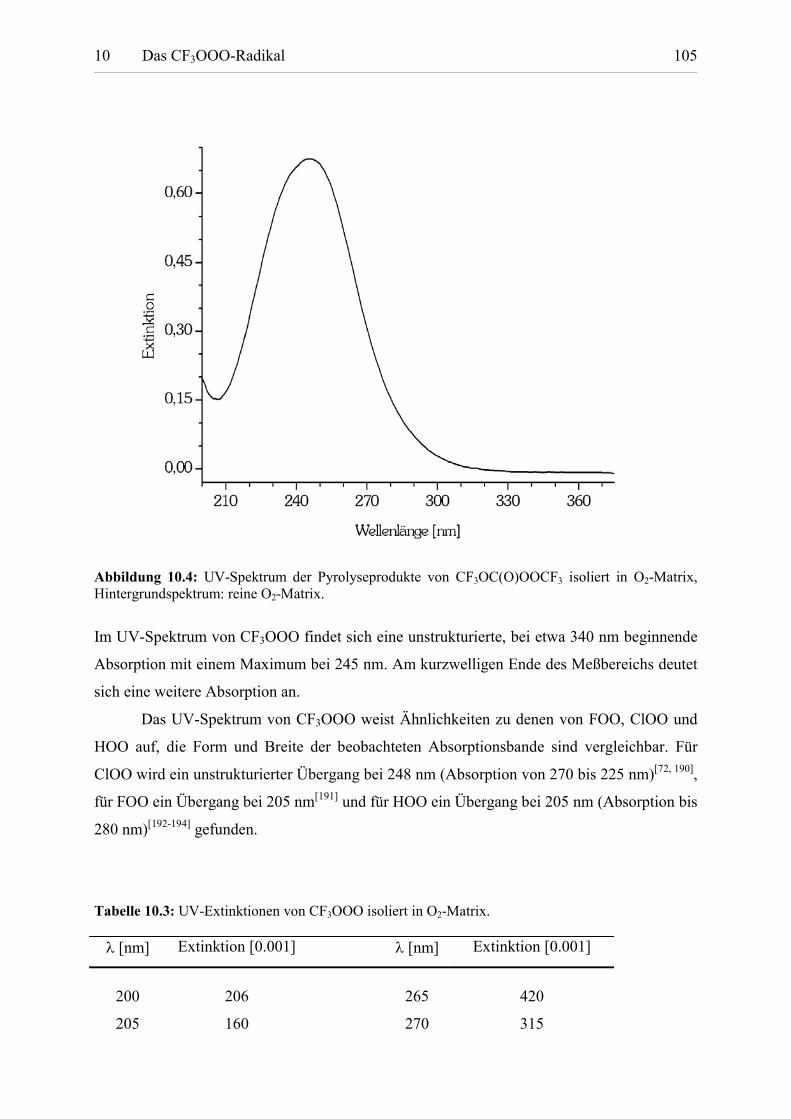
Tabelle 10.3: UV-Extinktionen von CF3OOO. 105
Tabelle 10.4: Berechnete Reaktionsenthalpien und physikalische Größenfür CF3OOO und CF3O. 106
Tabelle 11.1: Berechnete Reaktionsenthalpien und Freie Reaktionsenthalpien
Tabellenverzeichnis VIII
für die Reaktion R + O2 � ROO. 110
Tabelle 11.2: Berechnete Reaktionsenthalpien und gemessene Aktivierungs-energien für die Reaktion ROONO2 � ROO + NO2. 111
Tabelle 11.3: Berechnete Reaktionsenthalpien für die ReaktionROOOR � ROO + RO. 113
Tabelle 11.4: Berechnete ausgewählte innere Koordinaten und relativeEnergien der stabilen Rotamere einiger Peroxyradikale. 114
Tabelle 11.5: Berechnete Ladungs- und Spinverteilung in Peroxyradikalen. 115
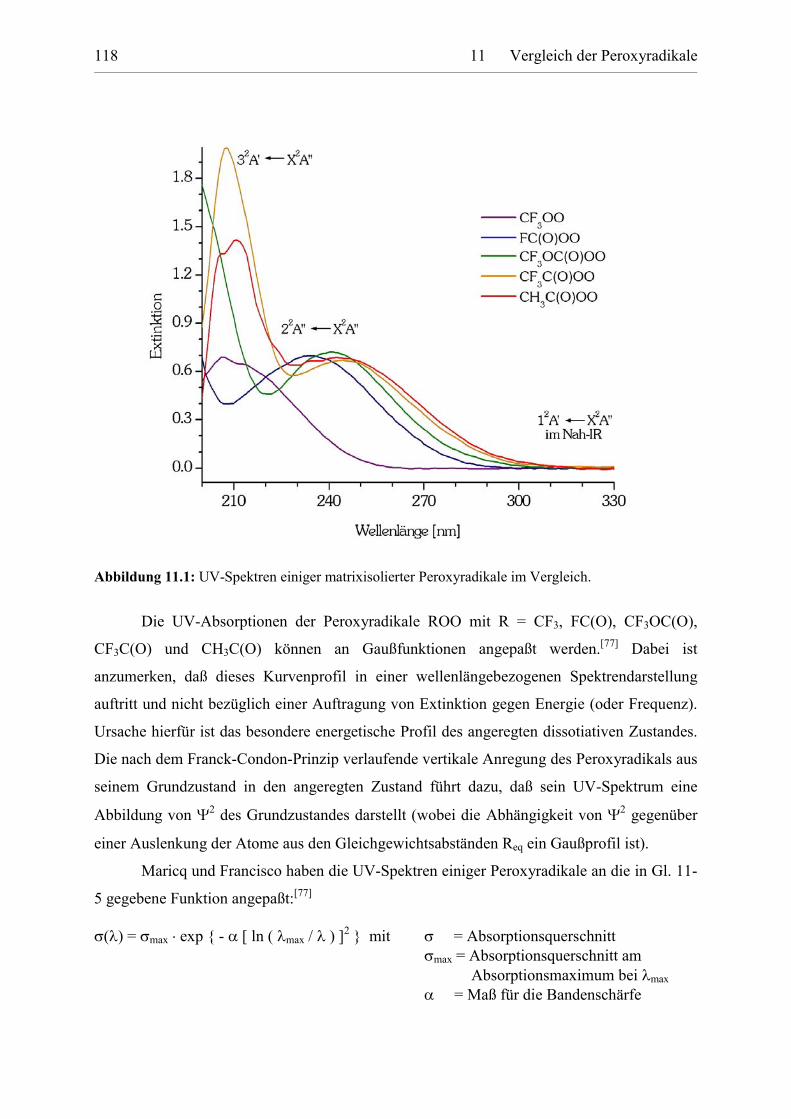
Tabelle 11.6: Parameter für die UV-Spektrenbeschreibung mit der Banden-kontourfunktion von Francisco und Maricq. 118
Tabelle 11.7: Absorptionen der 12A'�X2A'' Übergänge von Peroxyradikalen. 121
Tabelle 12.1: Berechnete Geometrie von trans- und cis-CF3OCO. 125
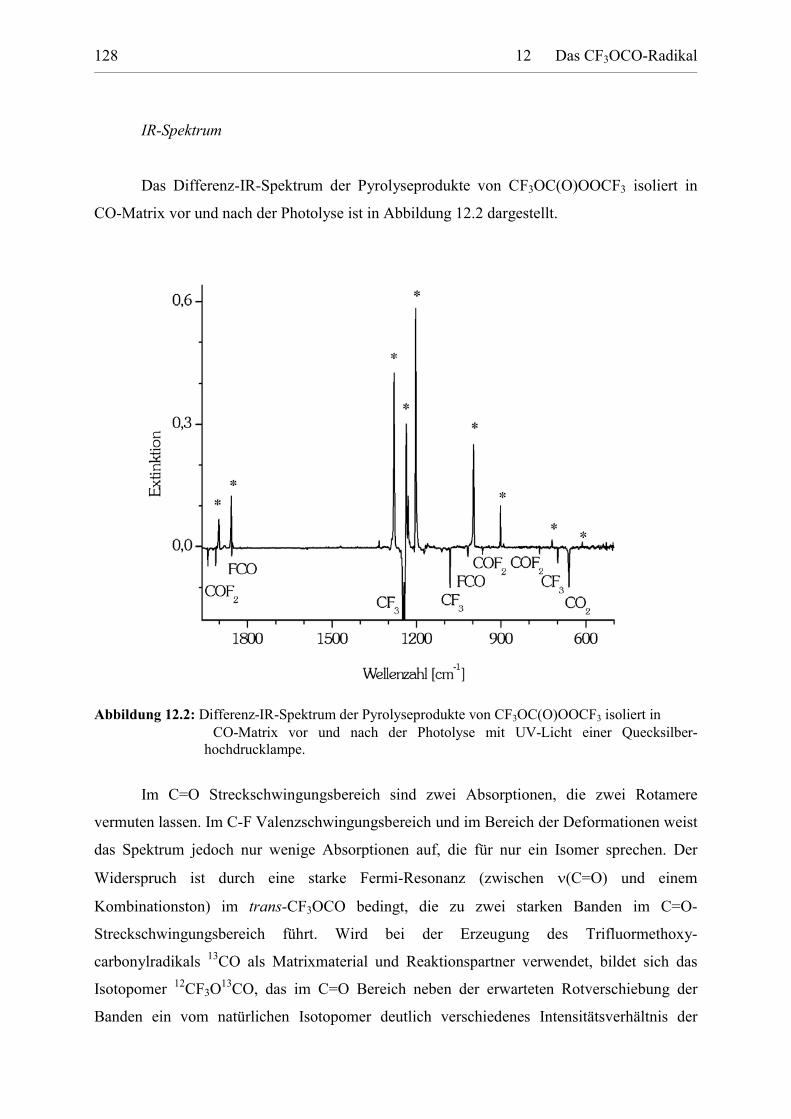
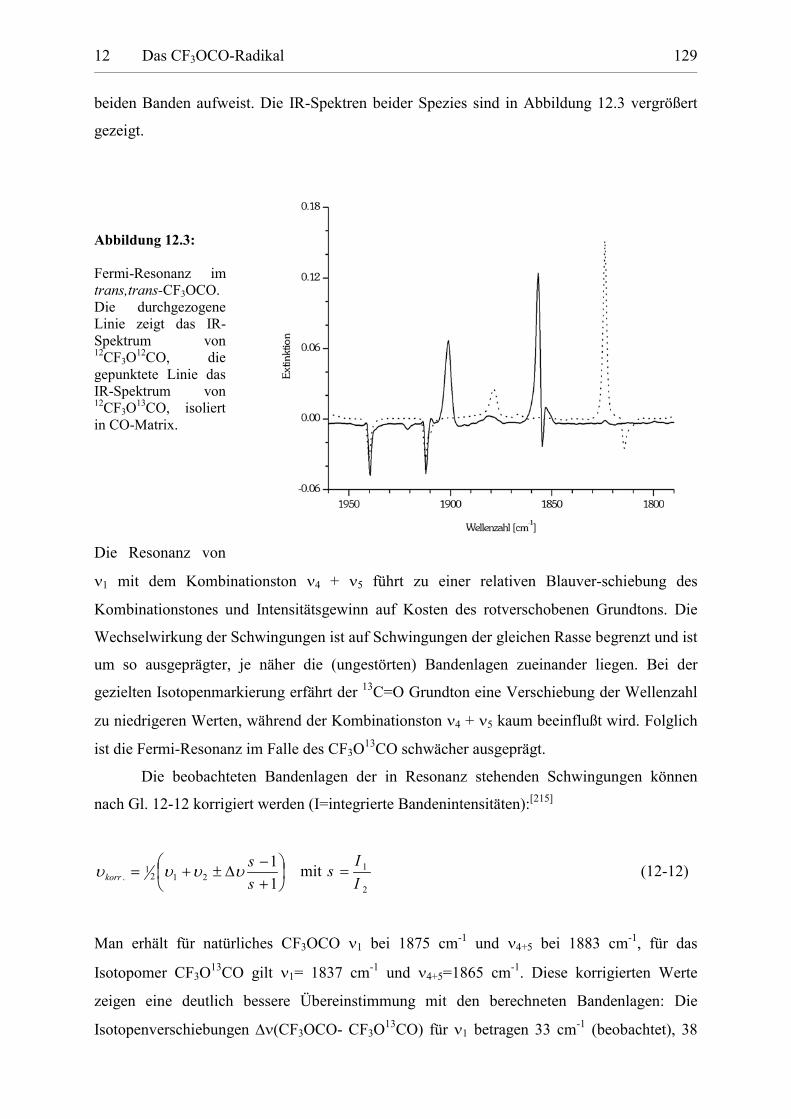
Tabelle 12.2: IR-Absorptionen des CF3OCO Radikals isoliert in CO-Matrix. 130
Tabelle 12.3: Berechnete und gemessene elektronische Übergänge der RadikaleCF3OCO, CH3OCO und HOCO. 133
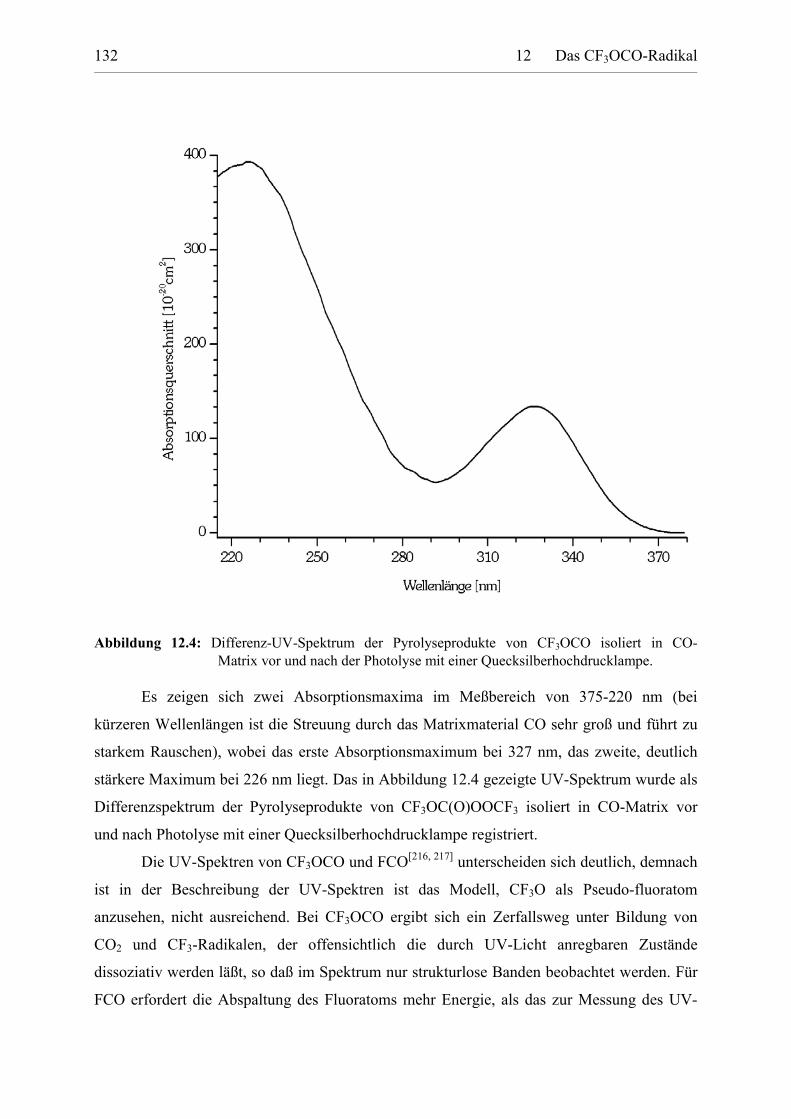
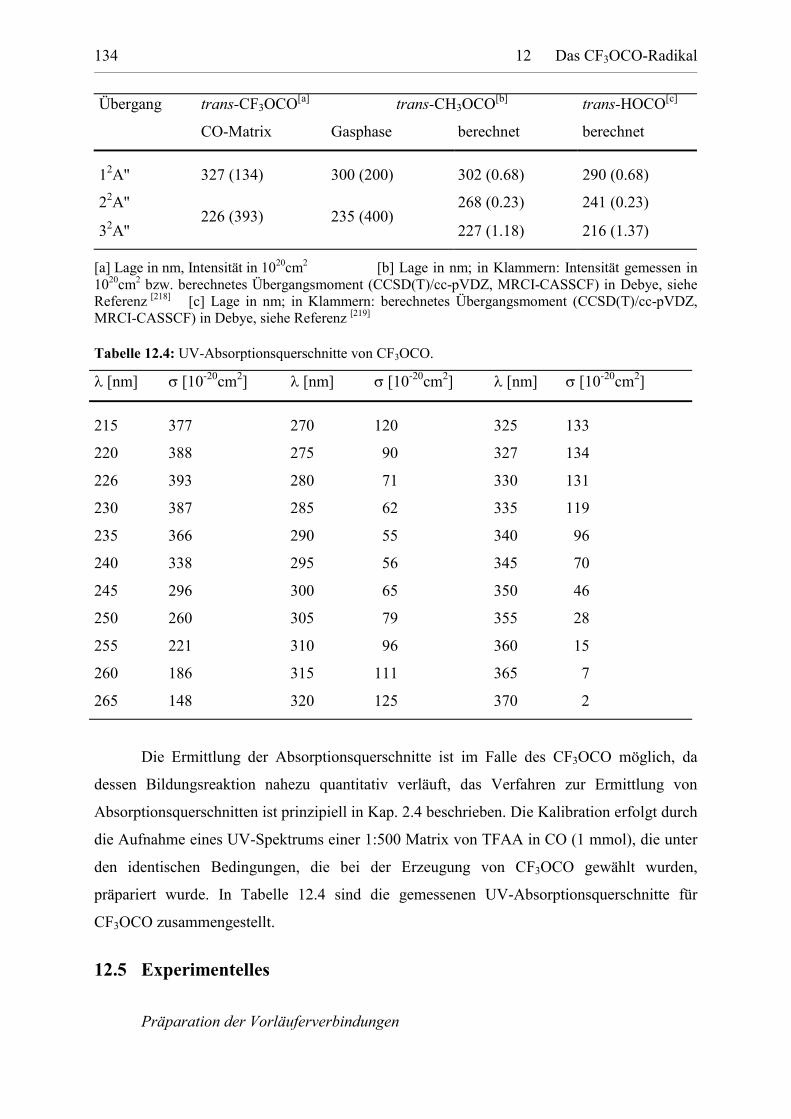
Tabelle 12.4: UV-Absorptionsquerschnitte von CF3OCO. 133
Tabelle 13.1: Berechnete Geometrien des stabilsten Rotamers vonCF3OC(O)OOOC(O)OCF3. 139
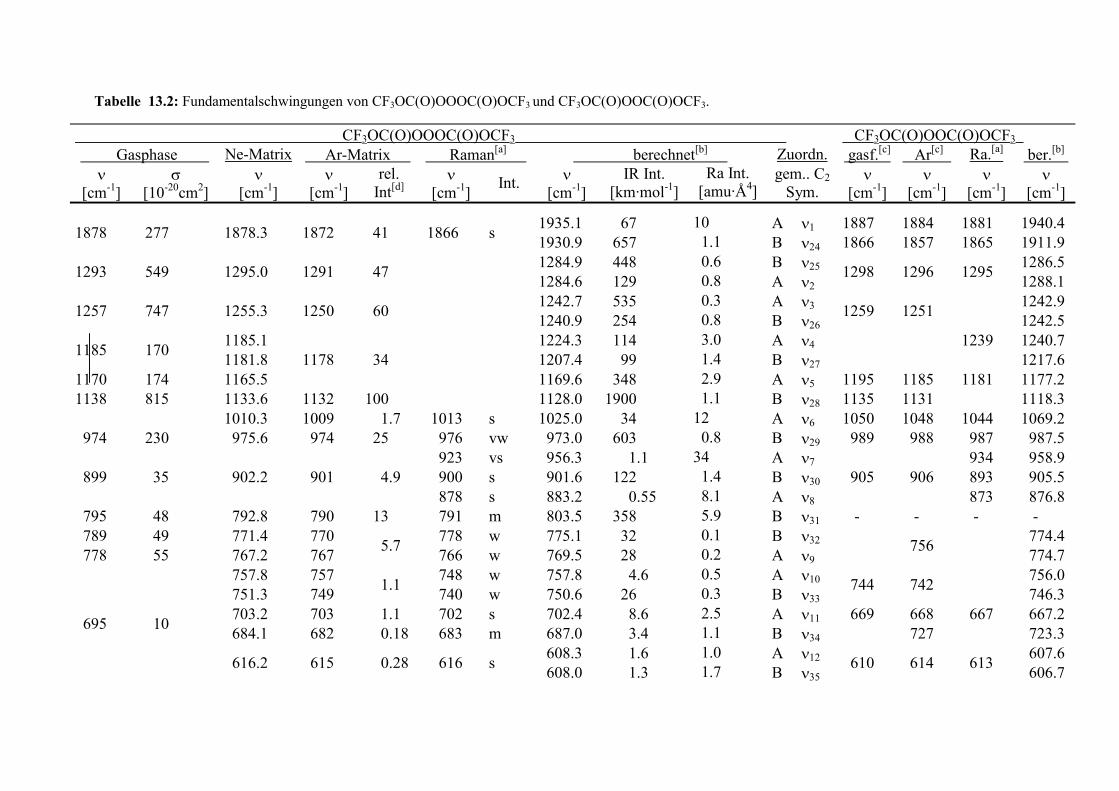
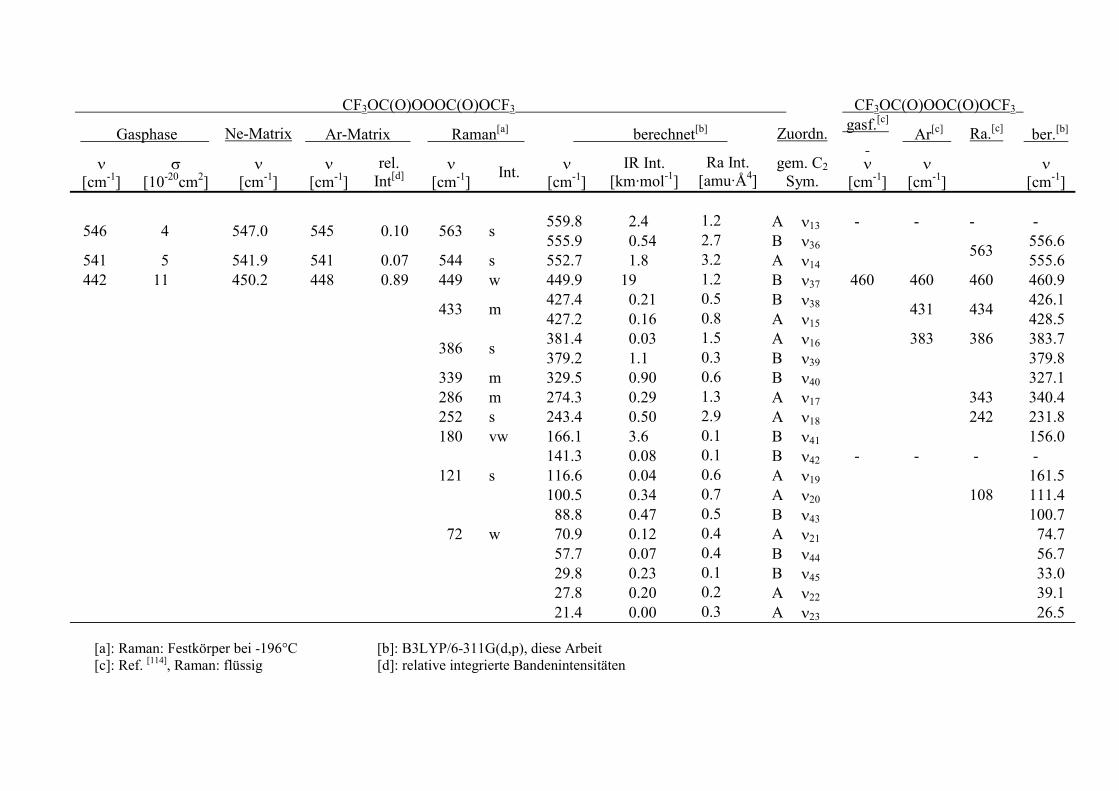
Tabelle 13.2: Fundamentalschwingungen von CF3OC(O)OOOC(O)OCF3 undCF3OC(O)OOC(O)OCF3. 144
Tabelle 13.3: NMR Daten für CF3OC(O)OOOC(O)OCF3 undCF3OC(O)OOC(O)OCF3. 147
Tabelle 14.1: Experimentelle und berechnete Strukturparameter von CF3ONO2. 157
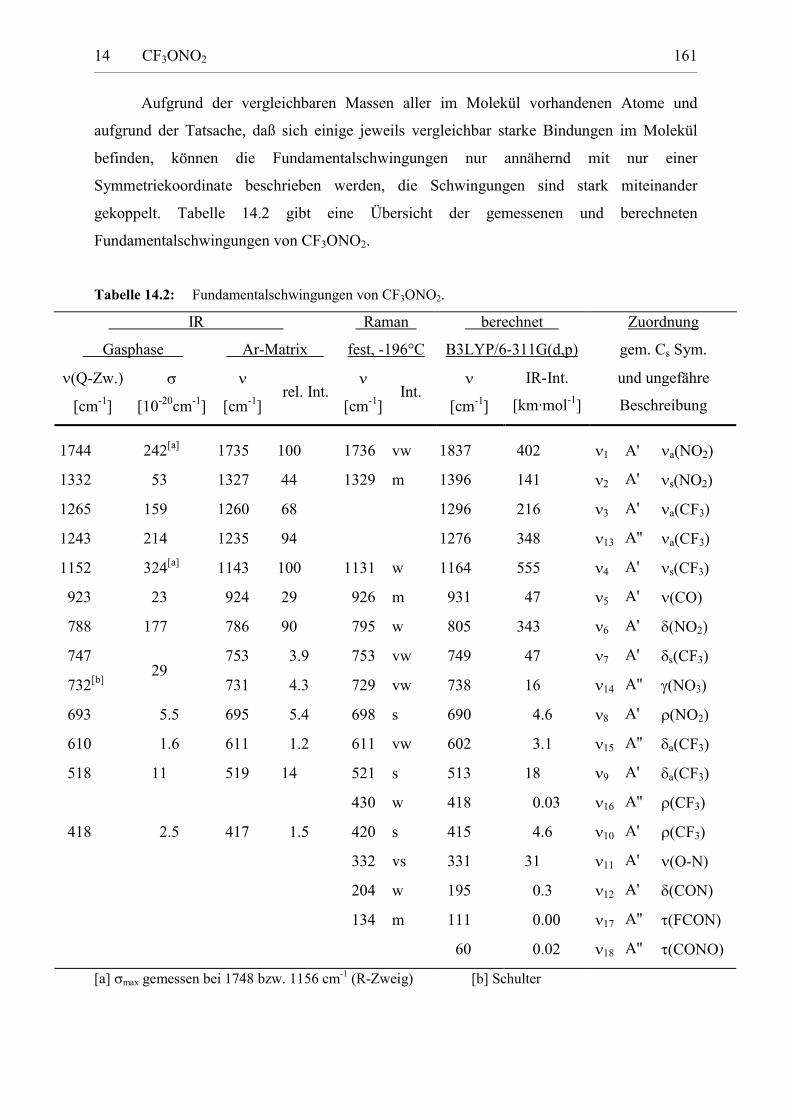
Tabelle 14.2: Fundamentalschwingungen von CF3ONO2. 160
Tabelle 14.3: UV-Absorptionsquerschnitte von CF3ONO2. 163
Tabelle 14.4: NMR-Daten von CF3ONO2 und einiger ausgewählter Spezies. 164
Tabelle 15.1: Strukturdaten von SF5NO2, gemessen in der Gasphase undquantenmechanisch berechnet. 172
Tabelle 15.2: Grundschwingungen von SF5NO2. 174
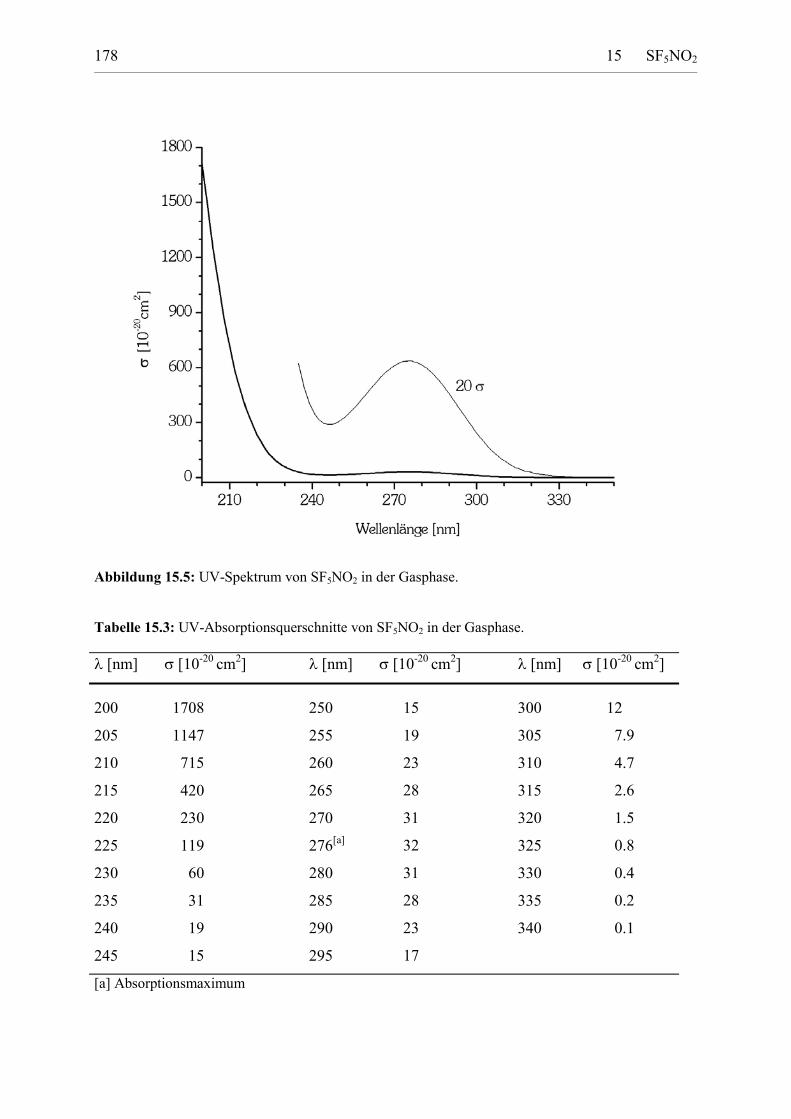
Tabelle 15.3: UV-Absorptionsquerschnitte von SF5NO2 in der Gasphase. 177
Tabelle 15.3: Berechnete Geometrien von SF5. 179
Tabelle 15.4: Fundamentalschwingungen des SF5-Radikals. 181
Tabelle 15.5: UV-Absorptionsquerschnitte von SF5 isoliert in Ne-Matrix. 182
Abbildungsverzeichnis IX
Abbildungsverzeichnis
Abbildung 1.1: Temperatur- und Druckverlauf in der Erdatmosphäre. 2
GED Elektronenbeugung an Gasen (gas electron diffraction)GWP global warming potentialI, Int. Intensitäti.p. in phasem mittelM StoßpartnerODP ozone depletion potentialo.p. off phasep polarisiertR Bindungslänge, -abstands starksh Schulter
Abkürzungsverzeichnis XII
Sym. Symmetrievs sehr starkvw sehr schwachw schwach
Atombezeichnungen in den Abbildungen
Kohlenstoff
Fluor
Sauerstoff
Stickstoff
Wasserstoff
Schwefel
1 Einleitung
1
1 Einleitung
In den 70er und 80er Jahren wurden zahlreiche dramatische Umwelt-veränderungen
festgestellt. Zu diesen gehören unter anderem das Auftreten eines massiven stratosphärischen
Ozonverlustes über der Antarktis, der Anstieg der globalen Durchschnittstemperatur und die
Schädigung von Wäldern und Gewässern. Erst zu diesem Zeitpunkt begann man durch
weltweite Forschungsprogramme die Ursachen für die genannten Phänomene umfassend zu
untersuchen und darüber hinaus vor einem industriellen Einsatz neuer Substanzen deren
Umweltverträglichkeit durch Studien wie das AFEAS-Programm[1-3] zu prüfen.
Die beobachteten Umweltveränderungen haben alle ihren Ursprung in der Emission
großer Mengen an Stoffen, die durch industrielle oder andere anthropogene Prozesse
freigesetzt werden, deren atmosphärische Abbauprozesse und Einfluß auf die Atmosphäre
entweder ignoriert wurden oder noch nicht bekannt waren.
Für den Rückgang der Ozonmenge in der Stratosphäre wurden, basierend auf
zahlreichen Studien, die Fluor-Chlor-Kohlenwasserstoffe (FCKWs) verantwortlich
gemacht.[1-5] Sie wurden über viele Jahre als Kältemittel, Treibgase, Brandschutzmittel[6] u. a.
eingesetzt, und ihre Herstellung wurde erst durch das Montreal-Protokoll[7-13] verboten. Neben
besonderen physikalischen Eigenschaften der FCKWs, welche zu einer umfangreichen
technischen Nutzung führten, zeichnet sich diese Substanzklasse durch ihre chemische
Reaktionsträgheit aus, die den Abbau in der Troposphäre verhindert und dadurch eine
Akkumulation in der Stratosphäre ermöglicht. In der Folge wurden Ersatzstoffe gefunden, im
allgemeinen als "Alternative Halocarbons" bezeichnet, deren chemische Beständigkeit durch
das Vorhandensein mindestens einer C-H Bindung stark herabgesetzt ist. Ein Abbau in
tieferen Atmosphärenschichten durch das OH Radikal ist nun möglich. Dennoch bergen die
Ersatzstoffe (wie auch die FCKWs) andere Gefahren in sich, wie zum Beispiel die
Eigenschaft, starke IR-Absorber zu sein und somit zum Treibhauseffekt beizutragen.[14, 15]
Noch zum heutigen Zeitpunkt fehlen viele elementare Informationen über wichtige
atmosphärische Spurengase, so daß ein erheblicher Forschungsbedarf besteht, um Aussagen
über die Einflußnahme von in die Atmosphäre entlassener Stoffe auf Umwelt und Klima zu
treffen.
1 Einleitung 2
Im Rahmen der vorliegenden Arbeit soll ein Beitrag zur Identifizierung und
Charakterisierung fluorierter und perfluorierter kohlenstoffhaltiger Radikale geleistet werden.
Solche Radikale sind Abbauprodukte von FCKWs oder deren Ersatzstoffe und besitzen somit
Bedeutung für die Atmosphärenchemie.
1.1 Ozonabbau in der Stratosphäre
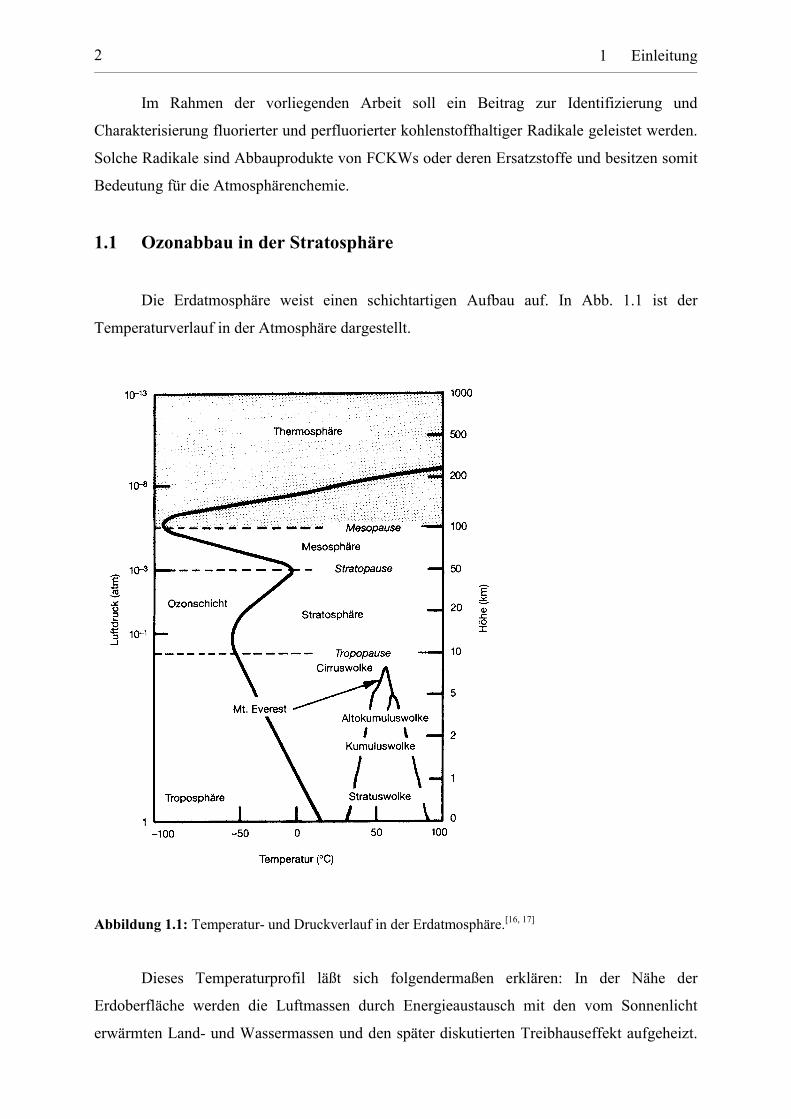
Die Erdatmosphäre weist einen schichtartigen Aufbau auf. In Abb. 1.1 ist der
Temperaturverlauf in der Atmosphäre dargestellt.
Abbildung 1.1: Temperatur- und Druckverlauf in der Erdatmosphäre.[16, 17]
Dieses Temperaturprofil läßt sich folgendermaßen erklären: In der Nähe der
Erdoberfläche werden die Luftmassen durch Energieaustausch mit den vom Sonnenlicht
erwärmten Land- und Wassermassen und den später diskutierten Treibhauseffekt aufgeheizt.
1 Einleitung
3
Die warme Luft steigt auf und kühlt sich durch adiabatische Expansion mit zunehmender
Höhe ab. Diese Konvektion bestimmt die physikalischen und chemischen Eigenschaften der
untersten Atmosphärenschicht, der Troposphäre. Die starke Durchmischung in der
Troposphäre führt zu vergleichsweise kleinen vertikalen und horizontalen
Konzentrationsgradienten von atmosphärischen Bestandteilen.
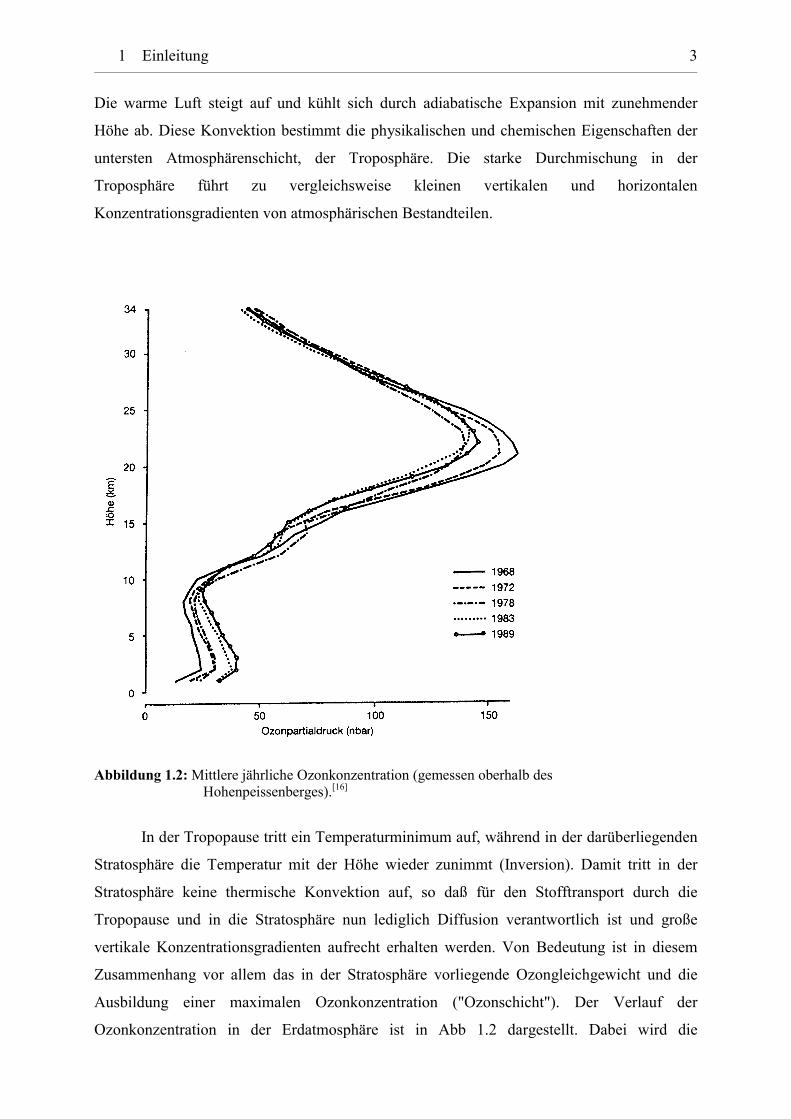
Abbildung 1.2: Mittlere jährliche Ozonkonzentration (gemessen oberhalb des Hohenpeissenberges).[16]
In der Tropopause tritt ein Temperaturminimum auf, während in der darüberliegenden
Stratosphäre die Temperatur mit der Höhe wieder zunimmt (Inversion). Damit tritt in der
Stratosphäre keine thermische Konvektion auf, so daß für den Stofftransport durch die
Tropopause und in die Stratosphäre nun lediglich Diffusion verantwortlich ist und große
vertikale Konzentrationsgradienten aufrecht erhalten werden. Von Bedeutung ist in diesem
Zusammenhang vor allem das in der Stratosphäre vorliegende Ozongleichgewicht und die
Ausbildung einer maximalen Ozonkonzentration ("Ozonschicht"). Der Verlauf der
Ozonkonzentration in der Erdatmosphäre ist in Abb 1.2 dargestellt. Dabei wird die
1 Einleitung 4
Hauptmenge an Ozon über einen Bereich von 18 bis etwa 40 km Höhe gebildet, wobei die
maximale Konzentration von ca. 15 ppmv (ca. 150 nbar bei 10 mbar Luftdruck, entsprechend
5�1013 Moleküle�cm-3) bei etwa 22 km Höhe auftritt.[16, 18]
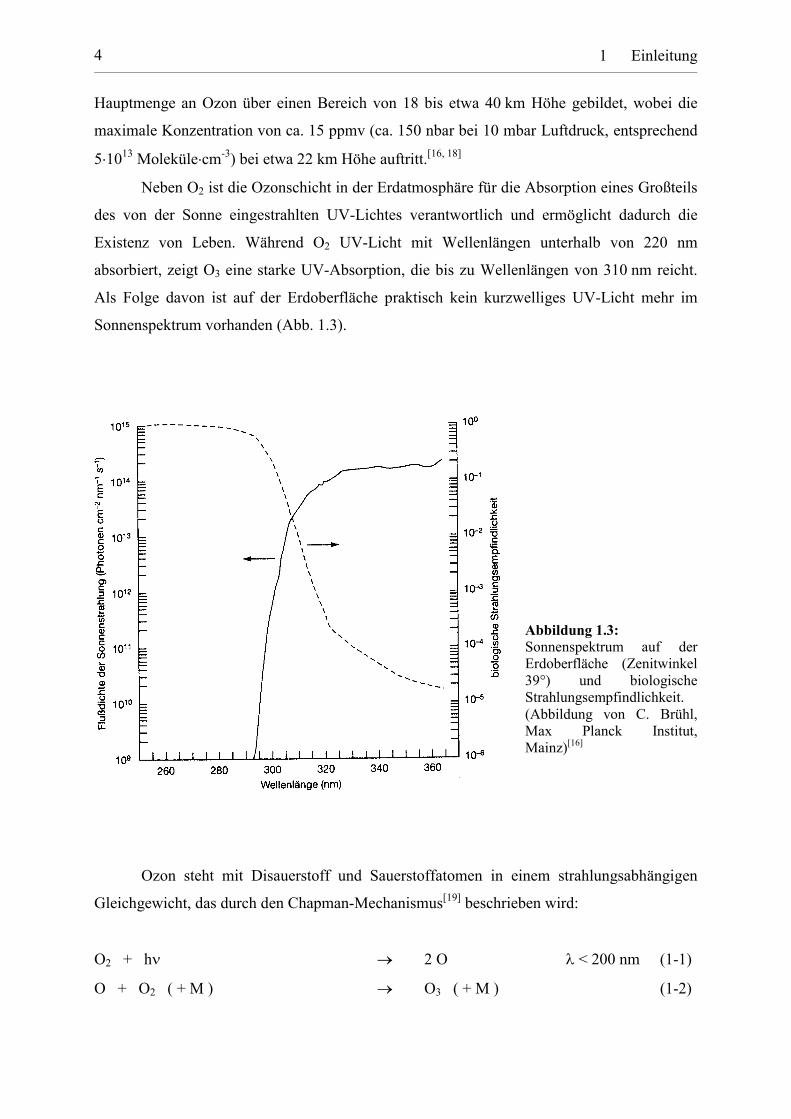
Neben O2 ist die Ozonschicht in der Erdatmosphäre für die Absorption eines Großteils
des von der Sonne eingestrahlten UV-Lichtes verantwortlich und ermöglicht dadurch die
Existenz von Leben. Während O2 UV-Licht mit Wellenlängen unterhalb von 220 nm
absorbiert, zeigt O3 eine starke UV-Absorption, die bis zu Wellenlängen von 310 nm reicht.
Als Folge davon ist auf der Erdoberfläche praktisch kein kurzwelliges UV-Licht mehr im
Sonnenspektrum vorhanden (Abb. 1.3).
Abbildung 1.3:Sonnenspektrum auf derErdoberfläche (Zenitwinkel39°) und biologischeStrahlungsempfindlichkeit.(Abbildung von C. Brühl,Max Planck Institut,Mainz)[16]
Ozon steht mit Disauerstoff und Sauerstoffatomen in einem strahlungsabhängigen
Gleichgewicht, das durch den Chapman-Mechanismus[19] beschrieben wird:
O2 + h� 2 O � < 200 nm (1-1)
O + O2 ( + M ) O3 ( + M ) (1-2)
1 Einleitung
5
O3 + h� O2 + O � < 300 nm (1-3)
O3 + O 2 O2 (1-4)
Der bestimmende Schritt für die Ozonbildung ist die Erzeugung von Sauerstoffatomen gemäß
Gl. (1-1).
Bereits 1974 wurde darauf hingewiesen,[20] daß Chloratome in dieses Gleichgewicht
Sauerstoff stellt in einigen Fällen einen Sonderfall dar, da O2 im Grundzustand als Triplett-O2
vorliegt, und Spin-Wechselwirkungen insbesondere mit matrixisolierten Radi-kalen
manchmal sogar eine Blauverschiebung bestimmter Absorptionsbanden hervor-rufen kann.
Eine theoretische Behandlung der Schwingungsspektroskopie matrixisolierter Moleküle und
der besonderen Effekte bei der Matrixisolation gibt H. E. Hallam.[36] Die Wechselwirkungen
von Gastmolekül und Umgebung führen manchmal auch dazu, daß für das freie Molekül IR-
inaktive Übergänge in Matrix IR-Absorption zeigen.
Die UV-Spektren matrixisolierter Moleküle können sich von Gasphasenspektren
unterscheiden. So wie es Verschiebungen von Absorptionsbanden bei gelösten Stoffen gibt,
bewirkt auch die Matrixumgebung eine Verschiebung, die im allgemeinen zu größeren
Wellenlängen führt. Allerdings stellen Edelgase sehr schwach koordinierende Umgebungen
dar, so daß die zu erwartende Verschiebung gering ist. Am Beispiel NO2 zeigt sich, daß die
Spektren dieser Verbindung in gasförmigem Zustand und isoliert in Neon-Matrix fast
identisch sind, eine Verschiebung der in beiden Spektren gut strukturierten
Absorptionsbanden liegt im Bereich von 1 bis 2 nm.
Nach dem Lambert-Beer'schen Gesetz gilt:
2 Matrixisolationstechnik 16
� = � � c � d mit � = Extinktion (2-1)c = Konzentration
d = optische Weglänge� = Extinktionskoeffizient
Die Extinktion ist gegeben als dekadischer Logarithmus des Quotienten der gemessenen
Lichtintensitäten mit und ohne der zu vermessenden Probe:
� = - lg ( I / I0 ) (2-2)
Die Abnahme der Lichtintensität mit fortschreitendem Durchgang durch eine absorbierende
Probe ist ein Beispiel für abklingende Phänomene wie etwa die Radioaktivität einer
zerfallenden Probe, so daß eine allgemein vergleichbare Formulierung des
Absorptionsgesetzes unter Verwendung des natürlichen Logarithmus getroffen werden kann,
wobei der Absorptionsquerschnitt � den Extinktionskoeffizienten ersetzt:
� = � � N � d � (1/ln 10) � V-1 oder I = I0 � exp [ - � � N � d � V-1 ] (2-3)
Bei Gasen ist die Teilchendichte N�V-1 nach dem idealen Gasgesetz mit p�(kT)-1
gleichzusetzen. Die Ermittlung absoluter Absorptionsquerschnitte kann bei einer
matrixisolierten Probe prinzipiell auf zwei Wegen erfolgen. Als unbekannte Größe tritt die
optische Weglänge, also die Dicke der Matrix (bei Messung in Transmission, bei Messung in
Reflexion die zweifache Dicke) auf. Falls die Dicke der Matrix bestimmt werden kann (durch
Elipsometrie: Ermittlung der Filmdicke aufgrund des veränderten Lichtreflexions-verhaltens
des Matrixträgers durch das Vorhandensein eines Films aus optisch dichterem Material - oder
durch Beobachtung von Interferenzmustern eines im spitzen Winkel zum Matrixträger
eingestrahlten Lasers während des Aufdampfens), sind die Absorptions-querschnitte direkt
zugänglich, da die Teilchendichte durch das eingestellte Mischungs-verhältnis von Probengas
zu Inertgas konstant ist. Ein zweiter Weg zu den Querschnitten ist die Eichung der
Matrixprobe mit einem Stoff bekannter Konzentration und mit bekanntem (UV-)Spektrum zu
nennen.
Im Rahmen dieser Arbeit konnten auf diese Weise UV-Absorptionsquerschnitte von
Peroxyradikalen ermittelt werden, sofern als Vorläufersubstanz ein entsprechendes
Peroxynitrat gewählt wurde. Bei den Pyrolyseexperimenten entsteht dann neben dem zu
untersuchenden Peroxyradikal zusätzlich NO2, welches zur Kalibration der UV-Spektren
genutzt wurde. Nachstehend wird diese Eichung kurz erläutert:
2 Matrixisolationstechnik 17
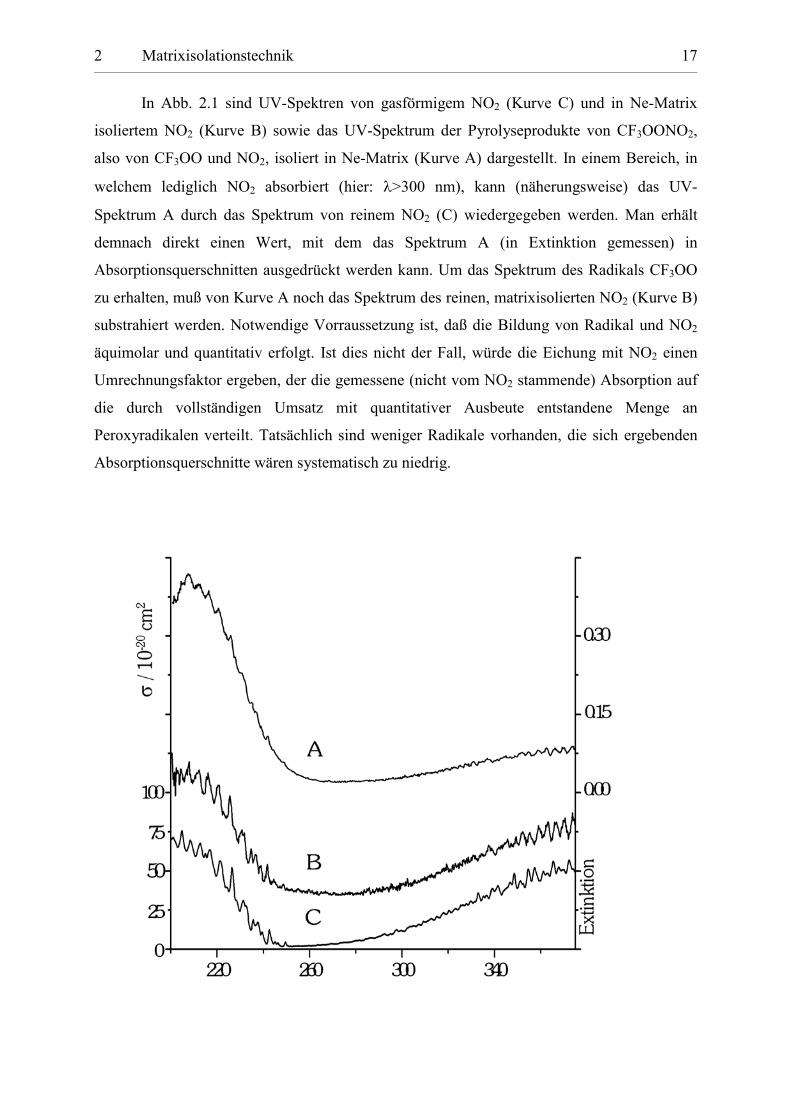
In Abb. 2.1 sind UV-Spektren von gasförmigem NO2 (Kurve C) und in Ne-Matrix
isoliertem NO2 (Kurve B) sowie das UV-Spektrum der Pyrolyseprodukte von CF3OONO2,
also von CF3OO und NO2, isoliert in Ne-Matrix (Kurve A) dargestellt. In einem Bereich, in
welchem lediglich NO2 absorbiert (hier: �>300 nm), kann (näherungsweise) das UV-
Spektrum A durch das Spektrum von reinem NO2 (C) wiedergegeben werden. Man erhält
demnach direkt einen Wert, mit dem das Spektrum A (in Extinktion gemessen) in
Absorptionsquerschnitten ausgedrückt werden kann. Um das Spektrum des Radikals CF3OO
zu erhalten, muß von Kurve A noch das Spektrum des reinen, matrixisolierten NO2 (Kurve B)
substrahiert werden. Notwendige Vorraussetzung ist, daß die Bildung von Radikal und NO2
äquimolar und quantitativ erfolgt. Ist dies nicht der Fall, würde die Eichung mit NO2 einen
Umrechnungsfaktor ergeben, der die gemessene (nicht vom NO2 stammende) Absorption auf
die durch vollständigen Umsatz mit quantitativer Ausbeute entstandene Menge an
Peroxyradikalen verteilt. Tatsächlich sind weniger Radikale vorhanden, die sich ergebenden
Absorptionsquerschnitte wären systematisch zu niedrig.
2 Matrixisolationstechnik 18
Abbildung 2.1: Ermittlung der UV-Absorptionsquerschnitte von CF3OO durch Kalibration mit NO2. Kurve A zeigt das UV-Spektrum der Pyrolyseprodukte von CF3OONO2 (CF3OO und NO2), isoliert in Ne-Matrix; Kurve B zeigt das UV-Spektrum von reinem NO2 in Ne-Matrix; C stellt ein UV-Spektrum von gasförmigem NO2 dar. Die Prozedur der Kalibration ist im Text beschrieben.
Eine Abschätzung der Absorptionsquerschnitte einer unbekannten Spezies kann
erfolgen, wenn eine bekannte Substanz in einem zusätzlichen, unter möglichst identischen
Bedingungen durchgeführten Experiment der Matrixisolation unterzogen und spektroskopisch
vermessen wird.
2.5 Matrixisolationsapparatur
An dieser Stelle wird nur auf die im Rahmen dieser Arbeit verwendete Matrix-
isolationsapparatur eingegangen. In Abb. 2.2 ist der Rezipient schematisch dargestellt, eine
Beschreibung der Apparatur darüber hinaus ist bereits in der Literatur ver-zeichnet.[37]
Die Anordnung der Matrixisolationsapparatur erlaubt es, von einer matrixisolierten Probe
nahezu den gesamten spektralen Bereich, in dem Schwingungs- und elektronische Übergänge
stattfinden, zu vermessen. Die technische Ausstattung (siehe Kap. 4) ermöglicht die
Spektroskopie von 50 cm-1 bis 50000 cm-1 (200 µm - 200 nm). Dabei erfolgt die Detektion
der Spektren in Reflexion.
2 Matrixisolationstechnik 19
Abbildung 2.2: Schematische Darstellung der verwendeten Matrixisolationsapparatur. Links ist ein vertikaler Schnitt (Seitenansicht) durch den Rezipienten gezeigt. Blau
eingefärbt erscheint der Matrixträger, ein Al-bedampfter vernickelter Cu-Block, derin Kontakt mit dem Kühlsystem steht. Die Kühlung erfolgt mit kaltem Heliumgas, dasaus einem Vorratsbehälter mit flüssigem He durch einen Wärmetauscher gepumptwird und dabei verdampft wurde. Rechts ist ein horizontaler Schnitt (Aufsicht)durch den Rezipienten gezeigt. Jeweils rechts im Bild befindet sich dieEinlaßvorrichtung mit heizbarer Zone (Pyrolysevor- r ichtung).
In Abbildung 2.2 sind die Kühlung und die Thermolyseeinrichtung schematisch
dargestellt. Durch Pumpen an der Abgasseite des Wärmetauschers strömt aus einer
Vorratskanne flüssiges Helium durch eine Kapillare in einem superisolierten Vakuum-
mantelheber und schließlich durch den Wärmetauscher. Dabei verdampft das Helium und das
kalte He-Gas kühlt den Wärmetauscher, der mit dem Matrixträger, einem Al-bedampften
vernickelten Kupferblock in Kontakt steht. Durch den He-Fluß, die durch das Kühlsystem
gepumpt wird, läßt sich jede Matrixträgertemperatur zwischen 4 und 300 K einstellen.
Die Pyrolysevorrichtung, durch die die Probe auf den Matrixträger gelangt, besteht aus
einem 6 � 1 mm Quarzrohr, das am Ende zugeschmolzen und mit einem Loch von 1 mm
Durchmesser versehen ist. Die letzten 10 mm des Quarzrohres sind durch eine Platinwicklung
beheizbar. Die Temperatur der Heizwicklung wird mit einem Pt-100-Meßwiderstand
gemessen. Durch die maximale thermische Belastbarkeit dieses Meßwiderstandes (500°C)
ergibt sich die maximale Betriebstemperatur der heizbaren Zone. Um höhere
Pyrolysetemperaturen zu ermöglichen, kann ein Thermoelement eingesetzt werden, wodurch
Betriebstemperaturen bis zu 1000°C realisierbar sind. Die Temperatur konnte weiter
gesteigert werden, indem eine neue Pyrolysevorrichtung gebaut wurde, bei dem der Heizdraht
aus Wolfram bestand und das Quarzrohr durch Sinterkorund ersetzt wurde. Experimente zur
Untersuchung von F,C,O-haltigen Radikalen erforderten meist Temperaturen unterhalb von
500°C, so daß im Rahmen dieser Arbeit stets mit der Quarzdüse und mit dem Pt-100-
Meßwiderstand gearbeitet werden konnte.
Die Zuführung der Matrixgase in die Pyrolysevorrichtung erfolgt durch eine
Stahlkapillare an einer Edelstahl-Vakuumapparatur. Instabile Substanzen werden in einer U-
Rohr-Falle aus Duranglas gehandhabt. Die Falle ist auf der einen Seite mit dem Einlaß der
Pyrolysevorrichtung der Matrixisolationsapparatur, auf der anderen Seite mit der
Stahlkapillare verbunden. Die Falle wird mittels Kältebäder so temperiert, daß sich ein
Dampfdruck der darin befindliche Probensubstanz von etwa 10-3 mbar einstellt. Über die
temperierte Probe wird das Matrixgas geleitet. Dabei stellt sich in der heizbaren Düse ein
Staudruck von etwa 1 mbar ein, bevor die Matrixgasmischung in das Hochvakuum des
Rezipienten strömt. So gelingt es, Mischungen von ca. 1:1000 auch von thermisch labilen
2 Matrixisolationstechnik 20
Stoffen zu erzeugen. Andere Mischungsverhältnisse werden durch entsprechendes
Temperieren der zu untersuchenden Probe bereitet.
Wie Abb. 2.2 zeigt, ist der Matrixträger drehbar und wird so vor die Aufdampfdüse
und die jeweilige Fenster positioniert. Folgende Fenstermaterialien wurden verwendet: KBr
(IR), PE (FIR), Quarz (NIR, VIS, UV). Das Quarzglasfenster ermöglichte außerdem
Photolyseexperimente an den matrixisolierten Spezies.
3 Quantenmechanische Berechnungen 21
3 Quantenmechanische Berechnungen
3.1 Allgemeines
Durch den rasanten Fortschritt auf dem Gebiet der Computertechnologie ist es
heutzutage möglich, Strukturen, spektroskopische Daten und physikalische Eigenschaften
kleinerer Moleküle mit bis zu etwa 20 Atomen der ersten und zweiten Langperiode des
Periodensystems innerhalb von Stunden zu berechnen. Im folgenden werden die elementaren
Grundzüge der theoretischen Berechnungen dargelegt, wobei der Schwerpunkt auf der
Anwendbarkeit für die hier untersuchten Spezies liegt.
Die statischen Eigenschaften eines Moleküls sind durch die zeitunabhängige
Schrödinger-Gleichung bestimmt, die aber bei Mehrelektronensystemen analytisch nicht
lösbar ist. Bei der numerischen Lösung der Schrödinger-Gleichung wird zunächst neben der
Born-Oppenheimer-Näherung eine weitere mathematische Vereinfachung gemacht. Die
natürlichen Wellenfunktionen, die mit einem Slater-Ansatz, also einer in der Variable x
einfachen Exponentialfunktion (N�e-ax) beschreibbar sind, werden durch numerisch einfacher
zu behandelnde Gauß-Funktionen ersetzt. Dabei erreicht man die Anpassung an das reale
Verhalten durch die Summierung über eine Reihe von Gaußfunktionen, die Anzahl und Art
der Glieder einer solchen Reihe wird mit dem Begriff Basissatz bezeichnet. Die generelle
Vorgehensweise bei der theoretischen Berechnung eines Moleküls ist durch die Methode der
Rechnung bestimmt. Neben den direkten numerischen Methoden zur Ermittlung der
Wellenfunktionen (ab initio Verfahren), ist der mathematisch einfachere Umweg über die
Berechnung von Dichtefunktionalen (DFT)[38] möglich. Details finden sich in einführenden
Lehrbüchern.[39-41]
3 Quantenmechanische Berechnungen 22
3.2 Berechnungen von F,C,O-haltigen kleinen Molekülen
Die Wahl des Verfahrens und des Basissatzes richtet sich zunächst nach dem zu
behandelnden Problem. Im Rahmen dieser Arbeit treten zunächst vier wichtige
Fragestellungen auf:
(i) Bei den matrixspektroskopischen Studien an Radikalen oder Molekülen ist es möglich,
nicht nur isotopomere Spezies, sondern auch durch das Einfrieren der Rotation bei den tiefen
Meßtemperaturen verschiedene Rotamere zu unterscheiden. Folglich ist die Information über
die Anzahl der stabilen Rotamere und deren energetische Abfolge von großer Bedeutung.
(ii) Die Bandenzuordnung von gemessenen Schwingungsspektren wird durch Vergleich
mit berechneten Spektren wesentlich vereinfacht.
(iii) Reaktions- und Aktivierungsenergien können im Rahmen dieser Arbeit mit den
verfügbaren technischen Mitteln nur abgeschätzt werden. Die Abschätzungen lassen sich
anhand der berechneten Werte überprüfen.
(iv) Nur wenige der untersuchten Spezies konnten strukturell untersucht werden. Für
Radikale sind Strukturparameter nur durch spezielle Methoden (z. B. hochauflösende IR-
Spektroskopie, Rotationsspektroskopie) ermittelbar, die aber in Duisburg nicht zur Verfügung
stehen. Die theoretische Berechnung der Gasphasenstruktur bietet somit einen alternativen
Zugang zu wichtigen Molekülparametern.
Da für die Berechnung von Energien eine hohe Genauigkeit zu fordern ist, sind auf
Variationsrechnungen bzw. Störungsrechnungen basierende ab initio Verfahren die Methoden
der Wahl. Allerdings ist der Rechenaufwand für eine MP2[42, 43]-Rechnung bereits
verhältnismäßig groß. Die Hybridmethode B3LYP[44-46] liefert in Verbindung mit einem
ausreichend großen Basissatz wie 6-311G(d,p) fast identische Werte für Reaktionsenthalpien
oder Energiedifferenzen zwischen zwei Rotameren und das bei deutlich geringerem
Rechenaufwand. Der Vergleich von gemessenem und berechnetem IR-Spektrum von
bekannten CxFyOz-Verbindungen zeigt, daß Schwingungsspektren durch B3LYP-Rechnungen
deutlich besser wiedergegeben werden als durch MP2 (bei gleichem Basissatz). Die dritte
genannte Fragestellung erfordert die Berechnung vieler Spezies mit identischem Basissatz
und identischer Methode, um vergleichbare Werte für die Berechnung von Reaktionsenergien
zu erhalten. Somit müssen die Vor- und Nach-teile der DFT- und ab initio Methoden
abgewogen werden. Zusammenfassend gilt für die hier untersuchten Moleküle, daß die DFT-
Methode neben den besten Ergebnissen bei der Spektrenberechnung gute Energiewerte liefert,
während das MP2-Verfahren zwar Energien optimiert, aber die Abweichungen zwischen
3 Quantenmechanische Berechnungen 23
experimentellen und berechneten Schwingungsspektren groß sind. Entsprechend sind alle im
Rahmen dieser Arbeit berechneten Daten mit dem B3LYP-Verfahren ermittelt worden.
Sowohl MP2- als auch B3LYP-Rechnungen für gesättigte Moleküle wie FC(O)OOC(O)F
liefern Geometrien, die mit experimentell bestimmten Gasphasenstrukturen in Einklang
stehen. Aufgrund der wenigen bekannten Strukturen F,C,O-haltiger Radikale ist eine
allgemeine Aussage über die Qualität der errechneten Molekülgeometrien in Abhängigkeit
von der Methodik an dieser Stelle nicht möglich. In einer theoretischen Studie über die
Strukturen von Sauerstofffluoriden liefert das B3LYP-Verfahren im Vergleich zu anderen
DFT- und ab initio Verfahren, darunter auch MP2, die verläßlicheren Werte.[47]
Die Wahl des optimalen Basissatzes wird durch den Vergleich von berechneten und
beobachteten schwingungsspektroskopischen Daten für das Radikal CF3OO bestimmt.
Zunächst zeigt sich die zunehmende Verbesserung der theoretischen Werte bei Vergrößerung
des Basissatzes von 3-21G bis 6-311G. Die Hinzunahme diffuser Funktionen (inklusive eines
3d-Orbitalsatzes) bewirkt eine weitere Verbesserung der Übereinstimmung. Die Verwendung
von Polarisationsfunktionen im Basissatz verschlechtert hingegen das Ergebnis, so daß für
CF3OO eine B3LYP/6-311G(d,p) Rechnung die besten Resultate liefert. Sofern keine
abweichenden Angaben gemacht werden, werden daher im Rahmen dieser Arbeit die
Ergebnisse der B3LYP/6-311G(d,p) Berechnungen diskutiert.
Die quantenmechanischen Berechnungen wurden mit dem Programm Gaussian98[48]
durchgeführt. Zu einzelnen Spezies, die im Rahmen dieser Arbeit untersucht wurden, sind
bereits theoretische Arbeiten publiziert worden,[49-60] darüber hinaus sind in der Literatur auch
umfassendere Studien an CxFyOz-Verbindungen verzeichnet.[61-66] Der Schwerpunkt dieser
Arbeiten liegt auf der Ermittlung von Standard-bildungsenthalpien bzw.
Dissoziationsenergien. Zusätzlich wurden Aussagen über Bindungsenergien und
Standardbildungsenergien von C,F,O Spezies getroffen, die auf experimentellen Daten
beruhen und Energieinkremente zur Abschätzung thermo-dynamischer Größen
heranziehen.[67-71]
3.3 Spezielle Probleme
Bei den B3LYP/6-311G(d,p) Berechnungen der Schwingungsspektren der aus F, C
und O bestehenden Radikale treten Abweichungen zum Experiment nicht gleichmäßig über
den ganzen Bereich der Fundamentalschwingungen auf. Vielmehr sind besonders die Lagen
der Valenzschwingungen, an denen Doppelbindungen beteiligt sind, systematisch bei zu
hohen Frequenzen berechnet. In den hier betrachteten Fällen wie FC(O)OO und
3 Quantenmechanische Berechnungen 24
CF3OC(O)OO zeigt sich für �(C=O) eine mittlere Abweichung von 2.7% (gleichbedeutend
mit einem Skalierungsfaktor von 0.973 für diese Schwingungs-frequenzen), für �(N=O) das
doppelte (0.946). Offensichtlich wird die Stärke der �-Bindung in der B3LYP-Rechnung
überschätzt. Andere Methoden liefern bei der Berech-nungen von �-Bindungssystemen häufig
noch größere Abweichungen zwischen experi-mentellen und theoretischen
Schwingungsfrequenzen.
Weiterhin zeigt sich, das bei Valenzschwingungen, an denen ein schwereres Atom wie
Schwefel beteiligt ist, eine Unterschätzung der Schwingungsfrequenz um etwa 10%, bei den
Deformationsschwingungen sogar mehr, vorliegt. Hier scheint in den Rechnungen ein Defizit
in der Berücksichtigung von Polarisationsfunktionen aufzutreten. Die Schwingungen, an
denen kein Schweratom beteiligt ist, werden hingegen sehr gut vorhergesagt. Daher wird, das
verwendete B3LYP-Verfahren konsequent angewandt, um die Vergleichbarkeit der
Ergebnisse sicher zu stellen. Die in dieser Arbeit angegebenen berechneten Wellenzahlen sind
nicht skaliert.
Bei schwachen Bindungen zeigt sich ein fundamentaler Nachteil der gewählten
Methodik. Sofern keine multi-reference-Verfahren eingesetzt werden, bei denen mehrere
elektronische Zustände Berücksichtigung finden, werden schwache Wechselwirkungen
unterschätzt. Dies führt zu niedrigen Bindungsenergien und Streckschwingungs-frequenzen
bezüglich der schwachen Bindung. So ist im Falle des CF3OOO Radikals ein Grenzfall
erreicht, an dem die Berechnung eher einen Komplex CF3O·O2 mit langem O-O Abstand
(etwa 2.2Å) vorhersagt, der durch eine CF3O-O2 Bindungsenergie von etwa 10 kJ·mol-1
charakterisiert ist. Die experimentellen Befunde deuten aber auf eine stärkere Bindung
(CF3OOO-Molekül) hin. Die Limitierung der theoretischen Methode zeigt sich besonders
anschaulich in der abnormalen berechneten Verteilung des Elektronenspins. Die B3LYP/6-
311G(d,p) Rechnung liefert hohe Spindichten auf allen drei O-Atomen des CF3OOO (zwei
stark positive Werte, stark negative Spindichte auf dem mittleren O). Ein Radikal CF3OOO·
sollte erwartungsgemäß den Hauptanteil an Spindichte auf dem terminalen Sauerstoff tragen,
während die anderen Atome kaum Restspindichte aufweisen, sofern es im Grundzustand X2A
vorliegt. Ein System mit marginalem Kontakt zwischen einem CF3O Radikal und Triplett-
Sauerstoff ergibt eine Spindichte auf allen Sauerstoffen. Vergleichbare Phänomene treten bei
ähnlich schwach gebundenen Spezies FOO, FOOO und ClOO auf. Für ClOO zeigt eine in der
Literatur verzeichnete theoretische Studie[72] deutlich, daß sowohl MP2 als auch DFT
Berechnungen versagen und nur die multi-reference Methode CMRCI[73] gute Ergebnisse
liefert.
3 Quantenmechanische Berechnungen 25
26
4 Peroxyradikale: Eigenschaften und Trends 27
4 Peroxyradikale: Eigenschaften und Trends
4.1 Überblick
Peroxyradikale, ROO, lassen sich gemäß der Natur des Substituenten R in
verschiedene Typen einteilen. Das direkt mit der O2-Einheit verknüpfte Atom kann (i) über
freie Elektronenpaare verfügen (wie in den Radikalen FOO und ClOO), (ii) Teil eines �-
Elektronensystems sein (z. B. FC(O)OO, CF3C(O)OO), oder (iii) durch �-Bindungen
koordinativ abgesättigt (CF3OO, CH3OO und auch HOO) sein. Es ist zu erwarten, daß die
spektroskopischen Eigenschaften von Peroxyradikalen innerhalb einer der genannten Gruppen
weniger voneinander differieren, als wenn man Peroxyradikale verschiedener Gruppen
miteinander vergleicht.
Die im Rahmen dieser Arbeit untersuchten Peroxyradikale werden in einer
Reihenfolge behandelt, die sich aus folgendem Überlegungen ergibt: Ausgehend von der
fundamentalen Spezies CF3OO (Kap. 5) können rein formal zwei Fluoratome (oder
Fluoridionen) durch einen doppelt gebundenen Sauerstoff (Oxidion) ersetzt werden, wodurch
man FC(O)OO (Kap. 6) erhält. In einem weiteren Schritt wird das am Kohlenstoff
verbleibende Fluoratom durch eine Trifluormethoxygruppe substituiert, das Resultat ist
CF3OC(O)OO (Kap. 7). Durch die Substitution von R=OCF3 durch R=CF3 wird die
Elektronegativität des Restes R in RC(O)OO deutlich verringert, und man erhält CF3C(O)OO
(Kap. 8). Schließlich folgt durch Eratz der Fluoratome durch Wasserstoff das Radikal
CH3C(O)OO (Kap. 9).
Wie nachfolgend gezeigt wird, ändern sich einige Eigenschaften der untersuchten
Peroxyradikale (wie beispielsweise die Lage von UV-Absorptionsmaxima) gerade in der
Lange Zeit wurde diskutiert, ob solche Zyklen von Peroxy/Oxyradikalen einen signifikanten
Einfluß auf den globalen Ozonhaushalt haben.[53, 68, 76, 118-124] Heute weiß man, daß fluorierte
Peroxy- und Oxyradikale praktisch kein Ozonabbaupotential (ODP) aufweisen,[24, 76] da diese
Radikale im Laufe ihrer Lebensdauer obigen Zyklus nur wenige Male durchlaufen (wie unten
näher erläutert wird), während das für den Ozonrückgang in der Stratosphäre
hauptverantwortliche Cl/ClO-System ca. 10000 Ozonmoleküle zersetzt, ehe Chlor in einer
Senke (z. B. durch Ausregnen einer langlebigen Substanz wie HCl) der Atmosphäre entzogen
wird.[20] Um ein Ozonabbaupotential zu besitzen, müssen Stoffe zwei Bedingungen erfüllen:
(i) Der katalytische Zyklus zum Ozonabbau muß aus Teilschritten bestehen, die alle
exotherm verlaufen.
(ii) Die atmosphärische Lebensdauer des Stoffes muß lang genug sein, um den Zyklus
häufig zu durchlaufen. Daraus folgt, daß Reaktionen des Stoffes oder seiner Folgeprodukte
mit anderen atmosphärischen Komponenten, die zu einer Senke führen, langsam gegenüber
der Ozonabbaureaktion sein müssen.
Die Standardbildungsenthalpien von Peroxyradikalen und den korrespon-dierenden
Oxyradikalen, sowie von Ozon und (Triplett-) O2 wurden im Rahmen dieser Arbeit berechnet.
Daraus ergeben sich für die in den Gln. (4-7) und (4-8) beschriebenen Reaktionen die in Tab.
4.1 zusammengefaßten Reaktionenthalpien unter Standardbedingungen.
Tabelle 4.1: Berechnete Reaktionsenthalpien für die Teilreaktionen eines Ozonabbau-Zyklus unterBeteiligung von Peroxyradikalen, sowie für den monomolekularen Zerfall derbeteiligten Oxyradikale.
In der linken und mittleren Tabellenspalte (Tab. 4.1) sind die Reaktionsenthalpien für
beide Schritte im katalytischen Ozonabbauzyklus zu finden, sie verlaufen alle stark exotherm.
Man erkennt, daß die Bildung der resonanzstabilisierten Carboxyradikale FCO2, CF3OCO2
und CF3CO2 den Reaktionsschritt I energetisch begünstigt. Die rechte Tabellenspalte gibt die
Wärmetönung bei einer CO2-Abspaltung aus dem Oxyradikal an. Die Radikale CF3CO2 und
CF3OCO2 sind thermodynamisch instabil gegenüber der Abspaltung von CO2. Diese
monomolekulare Reaktion ist, theoretischen Berechnungen folgend, mit einer geringen
Aktivierungsenergie[93] verbunden, so daß dieser Schritt die Lebensdauer der Carboxyl-
Radikale begrenzt. Die Zyklen mit ROx (R=CF3C(O) und CF3OC(O)) münden jedoch im
CF3O / CF3OO-Zyklus, so daß dieser Kreislauf die höchste atmosphärische Relevanz für ein
mögliches Ozonabbaupotential aufweist. Die Reaktionen von ROO bzw. RO mit
Sauerstoffatomen, die in den Bildungsweg des O3 eingreifen, sind ebenfalls alle exotherm.
Die Kinetik von Reaktionen der Peroxyradikale und ihrer korrespondierenden
Oxyradikale mit atmosphärischen Komponenten war Gegenstand zahlreicher Studien, einige
der Ergebnisse sind in Tab. 4.2 zusammengestellt.
Tabelle 4.2: Geschwindigkeitskonstanten für Reaktionen ausgewählter Peroxy-, Oxy- undAlkylradikale mit atmosphärischen Spurengasen. Alle Geschwindigkeitskonstantenbimolekularer Reaktionen sind k�-Werte und gelten für 298K.
Reaktion k [10-12·cm3·s-1][a] Ref. Reaktion k [10-12·cm3·s-1][a] Ref.
[a] B3LYP/6-311G(d,p)[b] F1 bezeichnet das Fluoratom in der Spiegelebene, F2 bezeichnet die beidenanderen, symmetrieäquivalenten F-Atome der CF3-Gruppe
Abbildung 5.1: Berechnete Struktur des stabilen Rotamers von CF3OO.
5.3 Die Erzeugung von CF3OO-Radikalen
CF3OO läßt sich in nahezu quantitativer Ausbeute aus CF3OONO2 darstellen. Die im
Rahmen der Matrixexperimente durchgeführte Niederdruckpyrolyse zeigt die beginnende
Dissoziation des Trifluormethylperoxynitrates bei Temperaturen oberhalb von 300°C. Mit
5 Das CF3OO-Radikal 38
vollständigem Umsatz und etwa 95% Ausbeute gelingt die Thermolyse bei 365°C, gemäß Gl.
(5-10):
365°C
CF3OONO2 CF3OO + NO2 (5-10)
Eine weitere Erhöhung der Reaktionstemperatur führt zu einer starken Zunahme der
Nebenprodukte COF2 und CF3OOCF3, die anhand der IR-Spektren identifiziert werden
konnten. Im wesentlichen sind die nachstehenden sekundären Reaktionsschritte anzunehmen:
[a] B3LYP/6-311G(d,p) [b] rel. integrierte Intensitäten [c] in km�mol-1
5 Das CF3OO-Radikal 41
Abbildung 5.2: Differenz-IR-Spektrum der Pyrolyseprodukte von CF3OONO2 isoliert in Ne- Matrix vor und nach der Photolyse mit einer Quecksilberniederdrucklampe. Die Absorptionsbanden des CF3OO sind positiv dargestellt und mit * markiert, die Banden der Photolyseprodukte COF2 und OF negativ (NO2-Banden bei 1613 und 750 cm-1 entstehen durch teilweisen Zerfall in NO + O).
Elektronische Übergänge
Bei CF3OO mit Cs-Symmetrie befindet sich das ungepaarte Elektron in einem Orbital,
das vornehmlich in der O2-Einheit lokalisiert ist und �*-Charakter besitzt. Theoretische
Berechnungen und experimentelle Beobachtungen[77] der Reaktivität des Radikals zeigen, daß
sich das ungepaarte Elektron im Grundzustand in einem a'' Orbital aufhält, wobei die
Aufenthaltswahrscheinlichkeit des Elektrons am äußeren Sauerstoffatom im CF3OO am
größten ist. Dieses gilt für alle Peroxyradikale mit Cs-Symmetrie, die folglich einen
X2A''Grundzustand besitzen. Eine schematische Darstellung der Molekülvalenzorbitale eines
Peroxyradikals ist Abb. 11.2 (Kapitel 11) gegeben.
Die erste elektronische Anregung wird dadurch beschrieben, daß ein Elektron aus dem
höchsten voll besetzten a' Orbital (HOMO) in das halb besetzte Orbital (SOMO, semi
occupied molecular orbital) wechselt und so den Zustand 12A' realisiert. Die Anregung des
5 Das CF3OO-Radikal 42
Elektrons aus einem nichtbindenden in ein antibindendes Orbital verringert die OO-
Bindungsordnung, führt aber noch nicht zur Dissoziation. In Ne-Matrix isoliertes CF3OO
zeigt diesen 12A'�X2A'' Übergang bei 6663 cm-1, also im Nah-Infrarot-Bereich. Darüber
hinaus können weitere Absorptionen im NIR beobachtet werden, die sich durch zusätzliche
Anregung von Schwingungszuständen des angeregten CF3OO erklären lassen. Eine
gleichzeitige elektronische und vibronische Anregung des Radikals ist für alle
Normalschwingungen des CF3OO-Radikals im 12A'-Zusatand erlaubt, die Auswahlregeln sind
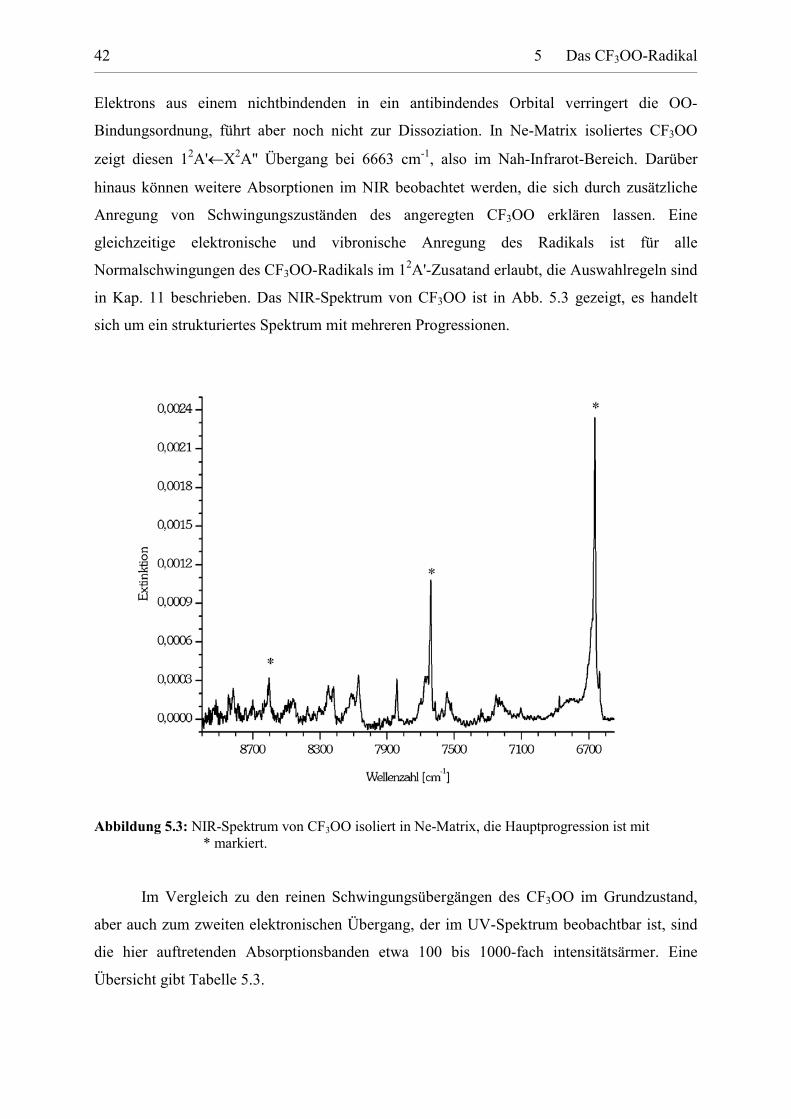
in Kap. 11 beschrieben. Das NIR-Spektrum von CF3OO ist in Abb. 5.3 gezeigt, es handelt
sich um ein strukturiertes Spektrum mit mehreren Progressionen.
Abbildung 5.3: NIR-Spektrum von CF3OO isoliert in Ne-Matrix, die Hauptprogression ist mit * markiert.
Im Vergleich zu den reinen Schwingungsübergängen des CF3OO im Grundzustand,
aber auch zum zweiten elektronischen Übergang, der im UV-Spektrum beobachtbar ist, sind
die hier auftretenden Absorptionsbanden etwa 100 bis 1000-fach intensitätsärmer. Eine
Übersicht gibt Tabelle 5.3.
5 Das CF3OO-Radikal 43
Tabelle 5.3: Zuordnung der beobachteten Absorptionsbanden des 12A'�X2A'' Übergangs vonCF3OO. Die Werte in Klammern geben die Verschiebung zu höheren Wellenzahlen inBezug zum schwingungsfreien Übergang �0 (12A'�X2A'') an.
Die Dissoziation von CF3OO und die Bildung von COF2 und OF können während der UV-
Bestrahlung von matrixisoliertem CF3OO IR-spektroskopisch verfolgt werden; dem-nach ist
der zweite angeregte Zustand dissoziativ. Das Bandenprofil ist Folge des Franck-Condon-
Prinzips.[77] Aus Gl. (5-10) folgt, das CF3OO und NO2 im Verhältnis 1:1 entstehen, sodaß
NO2 zur Ermittlung der Absorptionsquerschnitte des Peroxyradikals heran-gezogen werden
kann. Die erhaltenen Werte sind in Tab. 5.4 aufgeführt:
Tabelle 5.4: UV Absorptionsquerschnitte von CF3OO isoliert in Ne-Matrix.
Wellenlänge [nm] � (10-20cm2)diese Arbeit Maricq, Szente[148] Nielsen et al. [147]
200 160 377[a]
205 201 412
206.5[b] 207 421
210 196 427
215 189 417 340
220 170 376 297
225 141 317 267
230 110 252 206
235 79 190 162
240 51 130 127
245 30 88 101
250 16 53 66
255 6 33 41[c]
260 20 25
[a] Die Literaturdaten wurden auf die in der ersten Spalte gegebenen Wellenlängen interpoliert [b]Absorptionsmaximum [c] Wert bei � = 255 nm interpoliert
Die Lage des Absorptionsmaximums kann im Vergleich zu den Literaturdaten[147, 148]
bestätigt werden. Die ermittelten Absorptionsquerschnitte zeigen hingegen deutliche und
systematische Abweichungen. Die Ursachen hierfür sind:
5 Das CF3OO-Radikal 46
i) Mögliche Fehler bei der Übertragung der Absorptionsquerschnitte von gasförmiges
auf matrixisoliertes NO2. Vergleichsexperimente mit Aceton, gemessen in verschiedenen
organischen Lösemitteln, Wasser, in der Gasphase, sowie in Neon-Matrix zeigen aller-dings,
daß für Aceton die Unterschiede zwischen gasförmiger und matrixisolierter Probe
vernachlässigbar sind, wohingegen die Lösemitteleinflüsse stark ins Gewicht fallen.
ii) Die Absorptionsquerschnitte von CF3OO basieren auf der Annahme einer
quantitativen Ausbeute an CF3OO bezogen auf NO2. Da aber in den IR-Spektren der
Pyrolyseprodukte von CF3OONO2 stets eine kleine Menge COF2 detektiert wird, ist mit einer
Unterschätzung der hier ermittelten Absorptionsquerschnitte im Bereich von 5% bis 15% zu
rechnen.
iii) Die Hauptursache der beobachteten Abweichungen ist aber in der gänzlich
verschiedenen Methodik gegenüber den zitierten Arbeiten[147, 148] zu suchen. Dort wird das
CF3OO Radikal in der Gasphase erzeugt, indem CF3H durch Fluoratome in CF3 Radikale
überführt wird, die in Gegenwart von Sauerstoff CF3OO bilden:
F2 + h� � 2 F (5-16)
SF6 + h� � SF5 + F (5-17)
F + CF3H � CF3 + HF (5-18)
CF3 + O2 ( + M ) � CF3OO ( + M ) (5-2)
Dabei ist zu bedenken, daß neben CF3OO auch andere (Peroxy-)Radikale entstehen können,
deren UV-Spektren sich nicht immer fehlerfrei vom gemessenen UV-Spektrum der
Reaktionsmischung abziehen lassen, um die Absorptionsquerschnitte für das reine CF3OO zu
ermitteln:
F + O2 ( + M ) � FOO ( + M ) (5-19)
SF5 + O2 ( + M ) � SF5OO ( + M ) (5-20)
Weiterhin sind die Absorptionsquerschnitte für gasförmiges CF3OO[147, 148] ermittelt worden,
indem CF3H durch Ethan ersetzt wurde und so das UV-Spektrum von C2H5OO[152] als
Referenz benutzt wurde, oder indem der Anteil an gebildetem Nebenprodukt FOO zunächst
bestimmt und dann dessen Absorption zur Eichung herangezogen wurde.[147] Die publizierten
Absorptionsquerschnitte für das Ethylperoxyradikal streuen allerdings um fast 50%,[153] auch
5 Das CF3OO-Radikal 47
wenn der zur Referenzierung herangezogene Wert etwa dem Mittelwert der publizierten
Daten entspricht und somit eine gewisse statistische Zuverlässigkeit besitzt.
Letztendlich kann im Rahmen dieser Arbeit nicht abschließend geklärt werden, welche
Meßwerte richtig sind. Dennoch ist es gelungen, mit einer unabhängigen Methode
reproduzierbare Werte für die UV-Absorption der atmosphärenrelevanten Spezies CF3OO zu
erhalten.
5.5 Experimentelles
Vorläuferverbindungen
Die zur Erzeugung von CF3OO eingesetzte Ausgangsverbindung CF3OONO2 wurde
nach Literaturvorschrift[103] synthetisiert. Die Präparation einer kleinen Menge CF318O18ONO2
weicht in der Ansatzgröße von der publizierten Synthese ab und ist bereits in meiner
Diplomarbeit[146] beschrieben.
CF3OOOCF3 wurde aus Beständen des Arbeitskreises übernommen bzw. nach einer
Literaturvorschrift[154] synthetisiert.
Präparation der Matrices
In einem typischen Matrixisolationsexperiment wird reines CF3OONO2 in einer mit
Youngventilen ausgestatteten gläsernen U-Rohr-Falle mit der Pyrolysevorrichtung der
Matrixisolationsapparatur verbunden. Aufgrund des langsamen Zerfalls des Peroxynitrates bei
Raumtemperatur, kann die Substanz nicht bei 25°C mit dem Matrixmaterial, wahlweise
Argon oder Neon, in der Edelstahl-Vakuumapparatur gemischt werden. Das Edelgas strömt
über eine Kapillare durch die im Isopentanbad auf -140°C gekühlte U-Rohr-Falle hindurch
und trägt verdampfendes Peroxynitrat mit. Da der Dampfdruck des CF3OONO2 bei -140°C
etwa 10-3 mbar beträgt, ergibt sich unter Berücksichtigung des Staudruckes vor der Düse des
Pyrolysevorrichtung von etwa 1 mbar ein Mischungsverhältnis von ca. 1:1000.
Die Pyrolyse des CF3OONO2 zu CF3OO und NO2 erfolgt bei einer Düsentemperatur
von 365°C nahezu quantitativ. In einem typischen Experiment werden etwa 1 mmol Edelgas
innerhalb von 20 Minuten als Matrix auf dem Matrixträger (Metallspiegel) abgeschieden. Zur
Auffindung schwacher IR-Banden oder NIR-Absorptionen werden 5 mmol Edelgas bei
gleichbleibender Strömungsgeschwindigkeit als Matrix ausgefroren. Photolyseexperimente
werden durchgeführt, indem die matrixisolierten Pyrolyseprodukte mit einer
5 Das CF3OO-Radikal 48
Quecksilberhochdrucklampe ohne Einsatz von Kantenfiltern 15 bis 30 Minuten bestrahlt
werden.
6 Das FC(O)OO-Radikal 49
6 Das FC(O)OO-Radikal
6.1 Einführung
Die Bildung von FC(O)OO-Radikalen in atmosphärischen Prozessen verläuft über
FCO-Radikale. Diese entstehen aus CFH-Verbindungen, die eine CH2F-Einheit enthalten: [92,
155-157]
CH3F + OH � CH2F + H2O (6-1)
CH2F + O2 ( + M ) � CH2FOO ( + M ) (6-2)
CH2FOO � HC(O)F + OH (6-3)
HC(O)F + OH � FCO + H2O (6-4)
Die Reaktion des FCO-Radikals mit O2 verläuft schnell und quantitativ: [91, 97, 158]
FCO + O2 ( + M ) � FC(O)OO ( + M ) (6-5)
Zudem ist eine Beteiligung des Spurengases NO an dem in Gln. (6-1 bis 6-5) gegebenen
Reaktionsschema möglich, wobei das Peroxyradikal CH2FOO reduziert wird:[124, 159]
CH2FOO + NO � CH2FO + NO2 (6-6)
CH2FO � HC(O)F + H (6-7)
Weiterführende Studien zu atmosphärischen Zerfallswegen von CFH-Verbindungen finden
sich in der Literatur.[6, 15, 24, 25, 76, 77, 79, 92, 155-157]
In Laborexperimenten besitzt das FC(O)OO-Radikal große Bedeutung bei den
Synthesen zahlreicher CFO-Verbindungen wie z. B. FC(O)OOC(O)F. Es wurde bereits 1961
als Intermediat bei der Umsetzung von CO mit elementarem Fluor und Sauerstoff
postuliert:[160, 161]
F2 � 2 F (6-8)
F + CO ( + M ) � FCO ( + M ) (6-9)
FCO + O2 ( + M ) � FC(O)OO ( + M ) (6-5)
2 FC(O)OO � 2 FCO2 + O2 (6-10)
6 Das FC(O)OO-Radikal 50
2 FCO2 ( + M ) � FC(O)OOC(O)F (6-11)
Darüber hinaus können folgende Schritte beteiligt sein:
F + O2 ( + M ) � FOO ( + M ) (6-12)
FOO + CO � FCO + O2 (6-13)
Unter den von Arvia[161] beschriebenen Reaktionsbedingungen wurde nur das Peroxid
FC(O)OOC(O)F identifiziert. Durch Modifizierung der Reaktionsführung gelang es, ein
weiteres Produkt, das Trioxid FC(O)OOOC(O)F, das nach Gl. (6-12) entsteht, zu isolieren:[162, 163]
FC(O)OO + FCO2 ( + M ) � FC(O)OOOC(O)F ( + M ) (6-14)
Das UV-Spektrum von FC(O)OO, gemessen in der Gasphase, konnte durch Puls-Radiolyse
einer SF6/CO/O2-Mischung bzw. Blitzlichtphotolyse eines F2/CO/O2-Gemisches erhalten
werden.[91, 124, 135]
Obwohl die theoretische Berechnung von FC(O)Ox-Spezies, aufgrund der ver-
gleichsweise hohen Anzahl elektronegativer Elemente, lange Zeit eine große Heraus-
forderung darstellte, sind inzwischen für FC(O)OO quantenmechanische Berechnungen
publiziert.[51, 64, 164]
Im Rahmen meiner Diplomarbeit[146] und einer von H. Pernice vorgenommenen Studie
der Verbindungsklasse FC(O)OnC(O)F[163] wurden beide FC(O)OO-Rotamere IR-
spektroskopisch in Matrix untersucht, die Ergebnisse werden hier kurz zusammengefaßt. In
der vorliegenden Arbeit wird das UV-Spektrum von matrixisoliertem FC(O)OO mit-geteilt,
wobei zugleich ein unabhängiger Zugang zu dessen UV-Absorptionsquerschnitten vorgestellt
wird. Darüber hinaus werden quantenmechanisch berechnete Molekül-strukturen der beiden
stabilen Rotamere trans- und cis-FC(O)OO und deren Schwingungsfrequenzen diskutiert.
6.2 Die Struktur des FC(O)OO-Radikals
6 Das FC(O)OO-Radikal 51
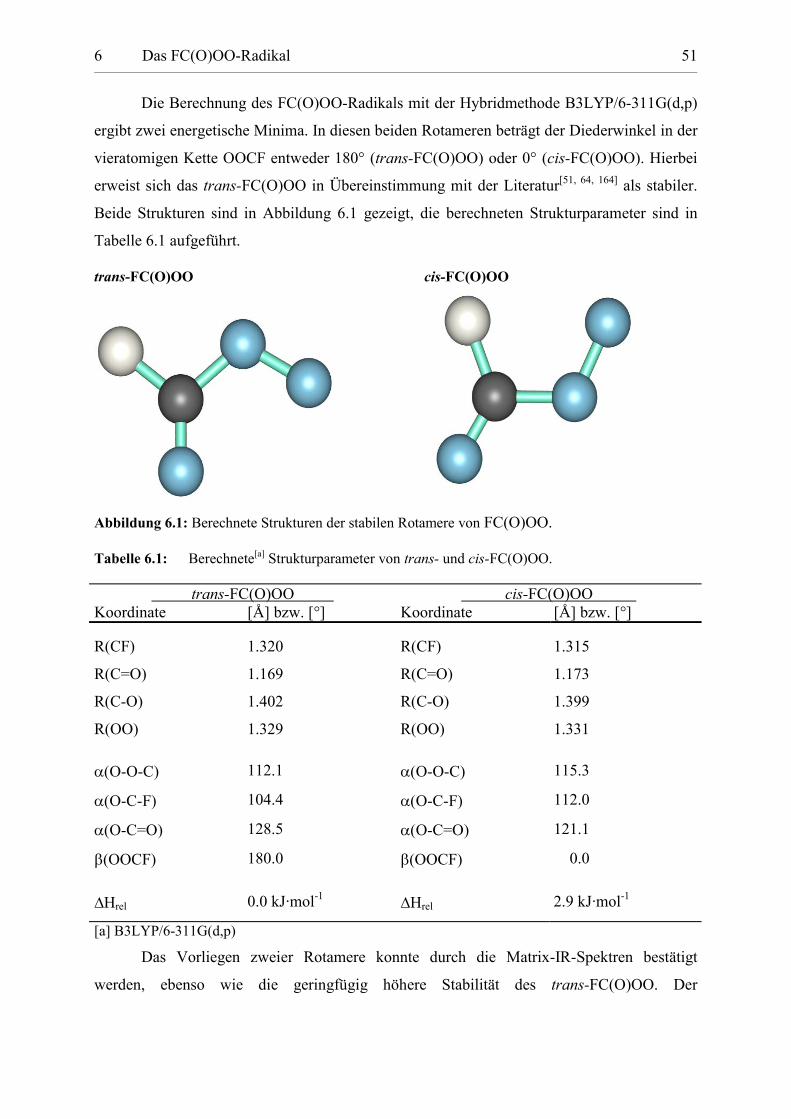
Die Berechnung des FC(O)OO-Radikals mit der Hybridmethode B3LYP/6-311G(d,p)
ergibt zwei energetische Minima. In diesen beiden Rotameren beträgt der Diederwinkel in der
vieratomigen Kette OOCF entweder 180° (trans-FC(O)OO) oder 0° (cis-FC(O)OO). Hierbei
erweist sich das trans-FC(O)OO in Übereinstimmung mit der Literatur[51, 64, 164] als stabiler.
Beide Strukturen sind in Abbildung 6.1 gezeigt, die berechneten Strukturparameter sind in
Tabelle 6.1 aufgeführt.
trans-FC(O)OO cis-FC(O)OO
Abbildung 6.1: Berechnete Strukturen der stabilen Rotamere von FC(O)OO.
Tabelle 6.1: Berechnete[a] Strukturparameter von trans- und cis-FC(O)OO.
trans-FC(O)OO cis-FC(O)OO Koordinate [Å] bzw. [°] Koordinate [Å] bzw. [°]
R(CF) 1.320 R(CF) 1.315
R(C=O) 1.169 R(C=O) 1.173
R(C-O) 1.402 R(C-O) 1.399
R(OO) 1.329 R(OO) 1.331
�(O-O-C) 112.1 �(O-O-C) 115.3
�(O-C-F) 104.4 �(O-C-F) 112.0
�(O-C=O) 128.5 �(O-C=O) 121.1
(OOCF) 180.0 (OOCF) 0.0
Hrel 0.0 kJ·mol-1Hrel 2.9 kJ·mol-1
[a] B3LYP/6-311G(d,p)
Das Vorliegen zweier Rotamere konnte durch die Matrix-IR-Spektren bestätigt
werden, ebenso wie die geringfügig höhere Stabilität des trans-FC(O)OO. Der
6 Das FC(O)OO-Radikal 52
Energieunterschied zwischen beiden Rotameren von 2.9 kJ·mol-1 stimmt mit den publizierten
Werten (0.6 kcal·mol-1 = 2.5 kJ·mol-1)[64] sehr gut überein.
6.3 Die Erzeugung von FC(O)OO-Radikalen
Analog zu CF3OO läßt sich FC(O)OO durch Niederdruckpyrolyse von FC(O)OONO2
(Fluorformylperoxynitrat[100, 165]) in einem Überschuß Inertgas (Ne, Ar) und anschließender
Matrixisolation in nahezu quantitativer Ausbeute erhalten. Bei 310°C wird eine Mischung von
trans- und cis-FC(O)OO erhalten, als Nebenprodukt entstehen nur Spuren von CO2:
310°C
FC(O)OONO2 � FC(O)OO + NO2 (6-15)
Alternativ läßt sich auch das offenkettige Trioxid FC(O)OOOC(O)F
(Bisfluorformyltrioxid)[162] unter milderen Pyrolysebedingungen von 210°C zu FC(O)OO und
FCO2 spalten. Anschließend kann hier die, durch das FCO2-Radikal tiefblau gefärbte, Matrix
durch Photolyse mit sichtbarem Licht "gebleicht" werden. Da dabei ausschließlich das FCO2
Radikal abgebaut wird, ist FC(O)OO in der bestrahlten Matrix lediglich durch CO2 und IR-
und UV-inaktive Fluoratome verunreinigt:
210°C
FC(O)OOOC(O)F � FC(O)OO + FCO2 (6-16)
FC(O)OO + FCO2 + h� (�>300nm) � FC(O)OO + CO2 + F (6-17)
Unter der Annahme, daß bei beiden Pyrolyseexperimenten (neben dem Edukt) nur die
Temperatur als Reaktionsparameter verändert wurde, läßt sich aus dem beobachteten 5%igen
Zuwachs des cis-Isomers relativ zum trans-Isomer bei Temperaturerhöhung von 210°C auf
310°C G berechnen:
Aus RTG
trans
cis eTnTn �
�
�
)()(
(6-18)
folgt bei zwei Temperaturen T1 und T2 mit T=T2-T1
6 Das FC(O)OO-Radikal 53
TTRT
G�
����ln21 mit
21
21
,,
,,
TcisTtrans
TtransTcis
nnnn
�� (6-19)
wobei für die Berechnung von � gilt, daß anstelle der (unbekannten) Stoffmengen n der
beiden Isomere auch direkt gemessene IR-Intensitäten von Absorptionsbanden der Rotamere
eingesetzt werden können (da � dimensionslos ist). Mit � = 1/1.05 und den genannten
Temperaturen ergibt sich G zu 1.2 kJ·mol-1 (höhere Stabilität von trans-FC(O)OO).
6.4 Die spektroskopische Charakterisierung von FC(O)OO
IR-Spektrum
Die IR-Spektren der Pyrolyseprodukte von FC(O)OONO2[100, 165] bzw.
FC(O)OOOC(O)F[162] isoliert in Argon- oder Neonmatrix finden sich bereits in meiner
Diplomarbeit[146] und sollen hier nur kurz zusammengefaßt werden. Nach Aussage der
quantenmechanischen Rechnungen besitzen beide FC(O)OO Rotamere Cs-Symmetrie;
demnach lautet die irreduzible Darstellung der Schwingungsfreiheitsgrade mit IR- und
Ramanaktivitäten:
�vib = 7 A' (IR, Ra p) + 2 A'' (IR, Ra dp) (6-20)
Von den jeweils 9 IR-aktiven Fundamentalschwingungen des FC(O)OO konnten für beide
Isomere 8 identifiziert und zugeordnet werden. Ein IR-Spektrum des Isomerengemisches von
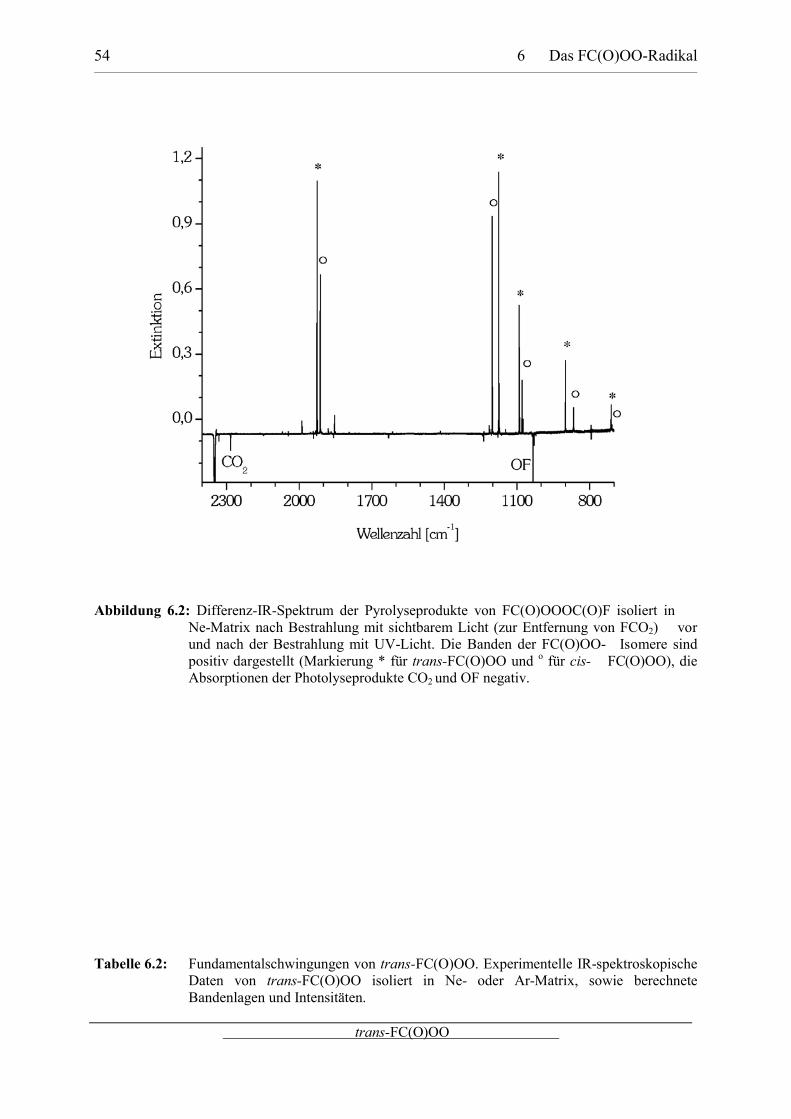
FC(O)OO isoliert in Ne-Matrix ist in Abbildung 6.2 dargestellt, eine Übersicht geben die
Tabellen 6.2 und 6.3. Die Zuordnung der beobachteten IR-Banden basiert auf dem Vergleich
mit den in dieser Arbeit theoretisch berechneten Spektren, sowie mit IR-Spektren der
verwandten Rotamere von FC(O)OF-Moleküls.[166]
6 Das FC(O)OO-Radikal 54
Abbildung 6.2: Differenz-IR-Spektrum der Pyrolyseprodukte von FC(O)OOOC(O)F isoliert in Ne-Matrix nach Bestrahlung mit sichtbarem Licht (zur Entfernung von FCO2) vorund nach der Bestrahlung mit UV-Licht. Die Banden der FC(O)OO- Isomere sindpositiv dargestellt (Markierung * für trans-FC(O)OO und o für cis- FC(O)OO), dieAbsorptionen der Photolyseprodukte CO2 und OF negativ.
Tabelle 6.2: Fundamentalschwingungen von trans-FC(O)OO. Experimentelle IR-spektroskopischeDaten von trans-FC(O)OO isoliert in Ne- oder Ar-Matrix, sowie berechneteBandenlagen und Intensitäten.
� [cm-1] rel. Int.[a]� [cm-1] � [cm-1] Int.[c] und Beschreibung
1925.9 85 1919.2 1981 358 �1 A' � (C=O)1175.0 100 1168.3 1179 260 �2 A' � (C-F)1090.1 38 1084.8 1125 189 �3 A' � (O-O) 900.8 23 895.7 892 77 �4 A' � (C-O) 712.0 9.1 707.4 721 34 �8 A'' � (FCOO2) 680.2 3.7 678.4 687 7.3 �5 A' � (O=C-O) 520.9 1.5 518.5 521 1.7 �6 A' � (F-C=O) 335.7 0.7 329 3.4 �7 A' � (C-O-O)
152 0.30 �9 A''
[a] relative integrierte Bandenintensitäten [b] B3LYP/6-311G(d,p) [c] in km�mol-1
Tabelle 6.3: Fundamentalschwingungen von cis-FC(O)OO. Experimentelle IR-spektroskopischeDaten von cis-FC(O)OO isoliert in Ne- oder Ar-Matrix, sowie berechneteBandenlagen und Intensitäten.
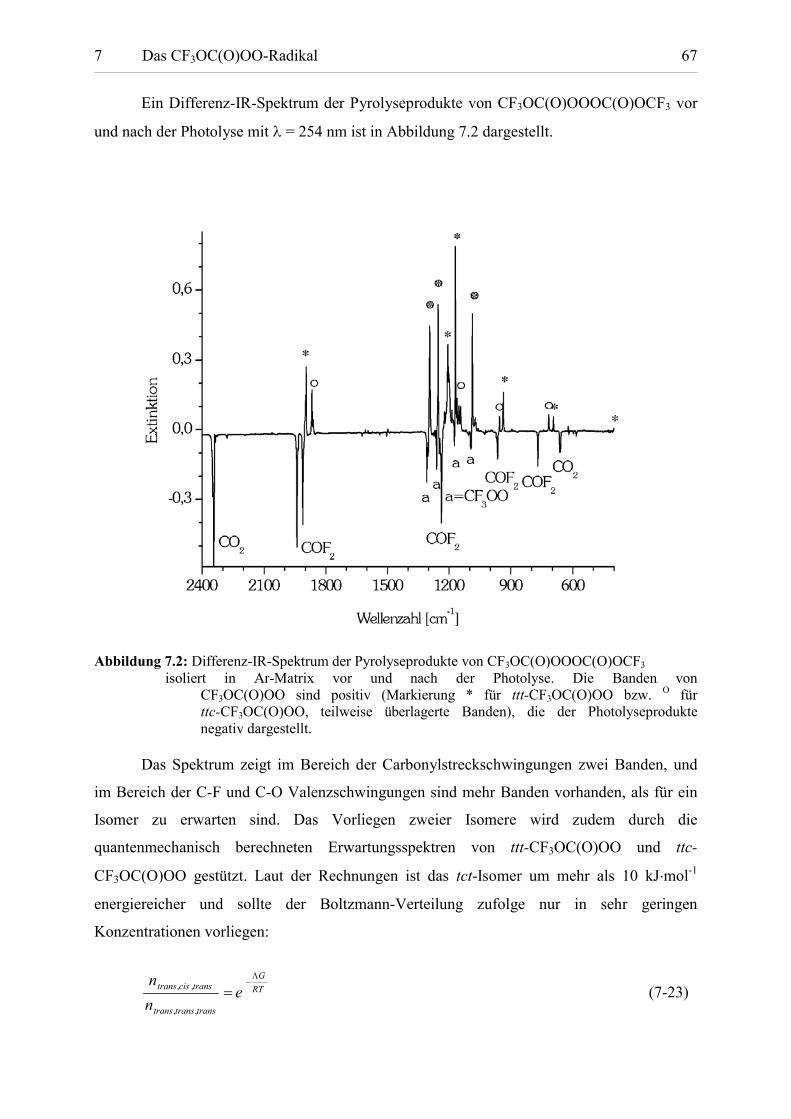
Ein Differenz-IR-Spektrum der Pyrolyseprodukte von CF3OC(O)OOOC(O)OCF3 vor
und nach der Photolyse mit � = 254 nm ist in Abbildung 7.2 dargestellt.
Abbildung 7.2: Differenz-IR-Spektrum der Pyrolyseprodukte von CF3OC(O)OOOC(O)OCF3
isoliert in Ar-Matrix vor und nach der Photolyse. Die Banden von CF3OC(O)OO sind positiv (Markierung * für ttt-CF3OC(O)OO bzw. O für ttc-CF3OC(O)OO, teilweise überlagerte Banden), die der Photolyseprodukte negativ dargestellt.
Das Spektrum zeigt im Bereich der Carbonylstreckschwingungen zwei Banden, und
im Bereich der C-F und C-O Valenzschwingungen sind mehr Banden vorhanden, als für ein
Isomer zu erwarten sind. Das Vorliegen zweier Isomere wird zudem durch die
quantenmechanisch berechneten Erwartungsspektren von ttt-CF3OC(O)OO und ttc-
CF3OC(O)OO gestützt. Laut der Rechnungen ist das tct-Isomer um mehr als 10 kJ�mol-1
energiereicher und sollte der Boltzmann-Verteilung zufolge nur in sehr geringen
Konzentrationen vorliegen:
RTG
transtranstrans
transcistrans enn �
�
�
,,
,, (7-23)
7 Das CF3OC(O)OO-Radikal 68
Mit T=433 K und G=11.8 kJ�mol-1 erhält man ein Stoffmengenverhältnis von 1:27. In den
Matrix-IR-Spektren sind keine Hinweise auf das dritte Isomer, das tct-Rotamer, vorhanden.
Die stabilen Rotamere von CF3OC(O)OO besitzen Cs-Symmetrie, demnach lautet die
irreduzible Darstellung der Schwingungsfreiheitsgrade mit IR- und Ramanaktivitäten:
�vib = 14 A’ (IR, Ra p) + 7 A’’ (IR, Ra dp) (7-24)
In beiden beobachteten Isomeren gibt es 8 Bindungen und somit 8
Valenzschwingungen. Das CF3O-Fragment enthält 6 Winkel, wobei 5 unabhängig
voneinander sind. Die CO3-Einheit wird durch zwei unabhängige Winkel charakterisiert, die
COC und COO-Kette durch je einen. Somit ergeben sich aus den inneren Freiheits-graden 9
Deformationsschwingungen für jedes CF3OC(O)OO-Rotamer. Schließlich treten noch in
beiden Isomeren eine out-of-plane-Schwingung der CO3-Gruppe sowie drei
Torsionsschwingungen auf.
Aufgrund der zumeist vergleichbaren Bindungsstärken in beiden Rotameren und der
ähnlichen Atommassen treten starke Schwingungskopplungen innerhalb der jeweiligen
Rotamere auf. Die Normalkoordinaten der Fundamentalschwingungen der CF3OC(O)OO-
Rotamere lassen sich folglich nur annähernd beschreiben. Die Zuordnung der im IR-Spektrum
detektierten Absorptionsbanden zu den Schwingungen zweier Rotamere erfolgt durch
Vergleich der Spektren mit denen von bekannten, ähnlichen Spezies wie CF3OO[35, 143-145, 164]
und FC(O)OO[164] und durch den Vergleich mit den quantenmechanisch berechneten
Spektren. Berücksichtigt werden muß hierbei das Rotamerenverhältnis von etwa 4:1 (ttt-
CF3OC(O)OO als Hauptisomer), das sich aus dem theoretisch berechneten
Energieunterschied der Isomere (G=4.7 kJ�mol-1) und aus der Temperatur bei deren Bildung
(433 K) nach der Boltzmannverteilung aus Gl. (7-23) ergibt. Eine Übersicht über die
Fundamentalschwingungen beider im Experiment beobachteter Isomere ist in den Tabellen
7.2 und 7.3 dargestellt.
Die Carbonylstreckschwingung �1 beider CF3OC(O)OO-Rotamere ist jeweils
geringfügig aufgespalten. Die Aufspaltung nimmt beim Übergang von Neon zu Argon als
Matrixmaterial zu und ist daher auf Matrixeffekte zurückführbar. Bande �7 (895 cm-1 im ttt-
und 882 cm-1 im tct-CF3OC(O)OO), die einer gemischten CF und CO-Valenzschwingung
entspricht, ist wegen der geringen IR-Intensitäten als Atmungsschwingung des CF3O-
Fragments aufzufassen. Die Bandenpositionen von �5, �15 und �16 beider Rotamere sind nach
den Berechnungen sehr ähnlich und überlagern sich somit in den beobachteten Spektren.
7 Das CF3OC(O)OO-Radikal 69
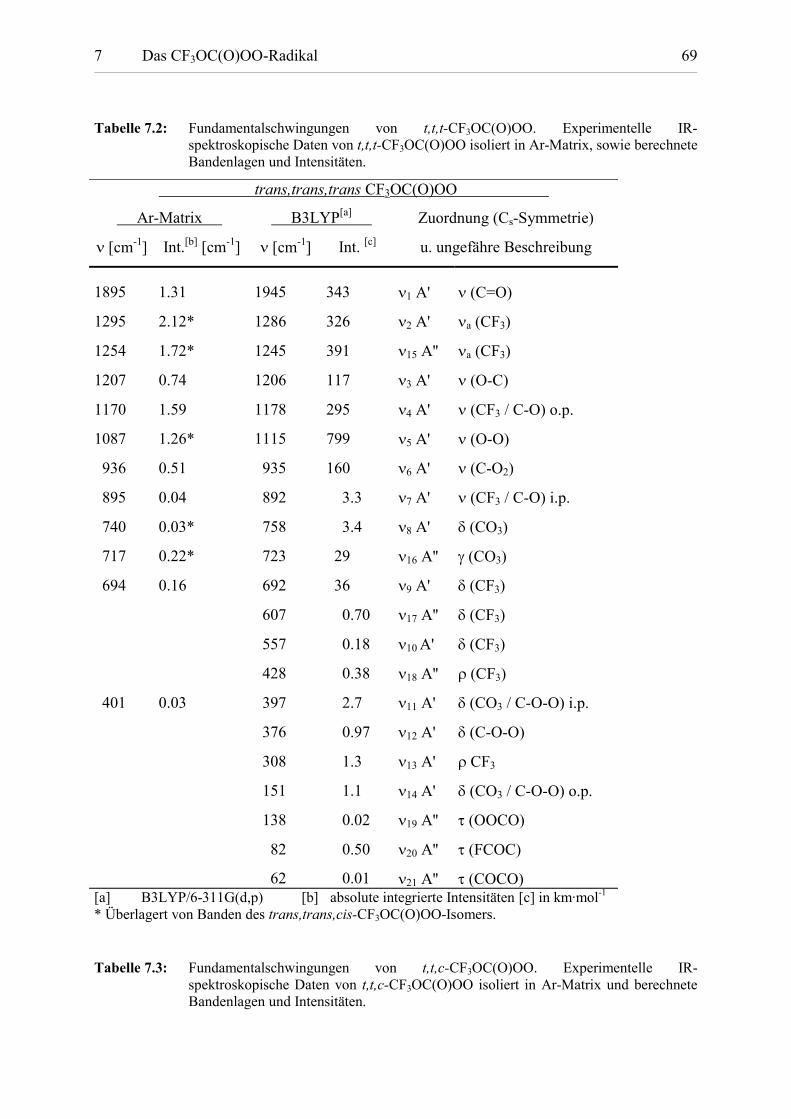
Tabelle 7.2: Fundamentalschwingungen von t,t,t-CF3OC(O)OO. Experimentelle IR-spektroskopische Daten von t,t,t-CF3OC(O)OO isoliert in Ar-Matrix, sowie berechneteBandenlagen und Intensitäten.
62 0.01 �21 A'' (COCO)[a] B3LYP/6-311G(d,p) [b] absolute integrierte Intensitäten [c] in km·mol-1 * Überlagert von Banden des trans,trans,cis-CF3OC(O)OO-Isomers.
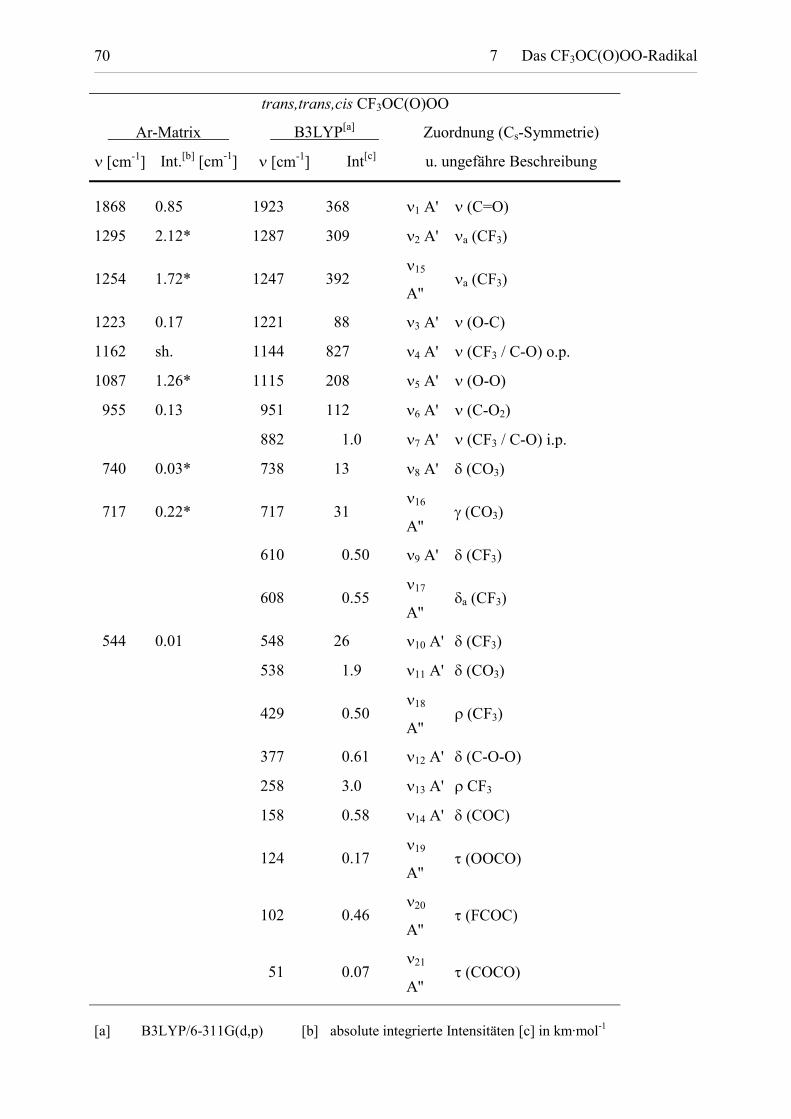
Tabelle 7.3: Fundamentalschwingungen von t,t,c-CF3OC(O)OO. Experimentelle IR-spektroskopische Daten von t,t,c-CF3OC(O)OO isoliert in Ar-Matrix und berechneteBandenlagen und Intensitäten.
� [cm-1] Int.[b] [cm-1] � [cm-1] Int[c] u. ungefähre Beschreibung
1868 0.85 1923 368 �1 A' � (C=O)
1295 2.12* 1287 309 �2 A' �a (CF3)
1254 1.72* 1247 392�15
A''�a (CF3)
1223 0.17 1221 88 �3 A' � (O-C)
1162 sh. 1144 827 �4 A' � (CF3 / C-O) o.p.
1087 1.26* 1115 208 �5 A' � (O-O)
955 0.13 951 112 �6 A' � (C-O2)
882 1.0 �7 A' � (CF3 / C-O) i.p.
740 0.03* 738 13 �8 A' � (CO3)
717 0.22* 717 31�16
A''� (CO3)
610 0.50 �9 A' � (CF3)
608 0.55�17
A''�a (CF3)
544 0.01 548 26 �10 A' � (CF3)
538 1.9 �11 A' � (CO3)
429 0.50�18
A''� (CF3)
377 0.61 �12 A' � (C-O-O)
258 3.0 �13 A' � CF3
158 0.58 �14 A' � (COC)
124 0.17�19
A'' (OOCO)
102 0.46�20
A'' (FCOC)
51 0.07�21
A'' (COCO)
[a] B3LYP/6-311G(d,p) [b] absolute integrierte Intensitäten [c] in km·mol-1
7 Das CF3OC(O)OO-Radikal 71
* Überlagert von Banden des trans,trans,trans-CF3OC(O)OO-Isomers.
UV-Spektrum
Das UV-Spektrum von CF3OC(O)OO erhält man aus der Differenz der Spektren der
Pyrolyseprodukte von CF3OC(O)OOOC(O)OCF3 vor und nach der Photolyse mit einer
Quecksilber-Niederdrucklampe (Abb. 7.3). Das in der vorangegangen Pyrolyse äquimolar zu
CF3OC(O)OO entstandene CF3O, das im Bereich um � = 350 nm eine gut strukturierte
Bande[169] besitzt, wird innerhalb der Photolysedauer von 10 Minuten praktisch nicht
abgebaut. Somit entfällt eine Korrektur des UV-Differenzspektrums bezüglich CF3O. Das
während der Photolyse nach Gl. 7-21 entstehende CF3OO absorbiert hingegen stark im UV-
Bereich, daher muß das Differenzspektrum auf Trifluor-methylperoxyradikale korrigiert
werden. Die gemessenen Differenz-IR-Intensitäten von CF3OO ergeben die während der
Photolyse entstandende Menge an Radikal, das bekannte UV-Spektrum von CF3OO wird zu
dem Differenzspektrum entsprechend addiert. Die Subtraktion des nach der Photolyse
aufgenommenen Spektrums von dem vor der Photolyse gemessenen Spektrums bedingt, daß
CF3OO-Absorptionen im Ergebnisspektrum negativ dargestellt und daher durch Addition
korrigiert werden müssen.
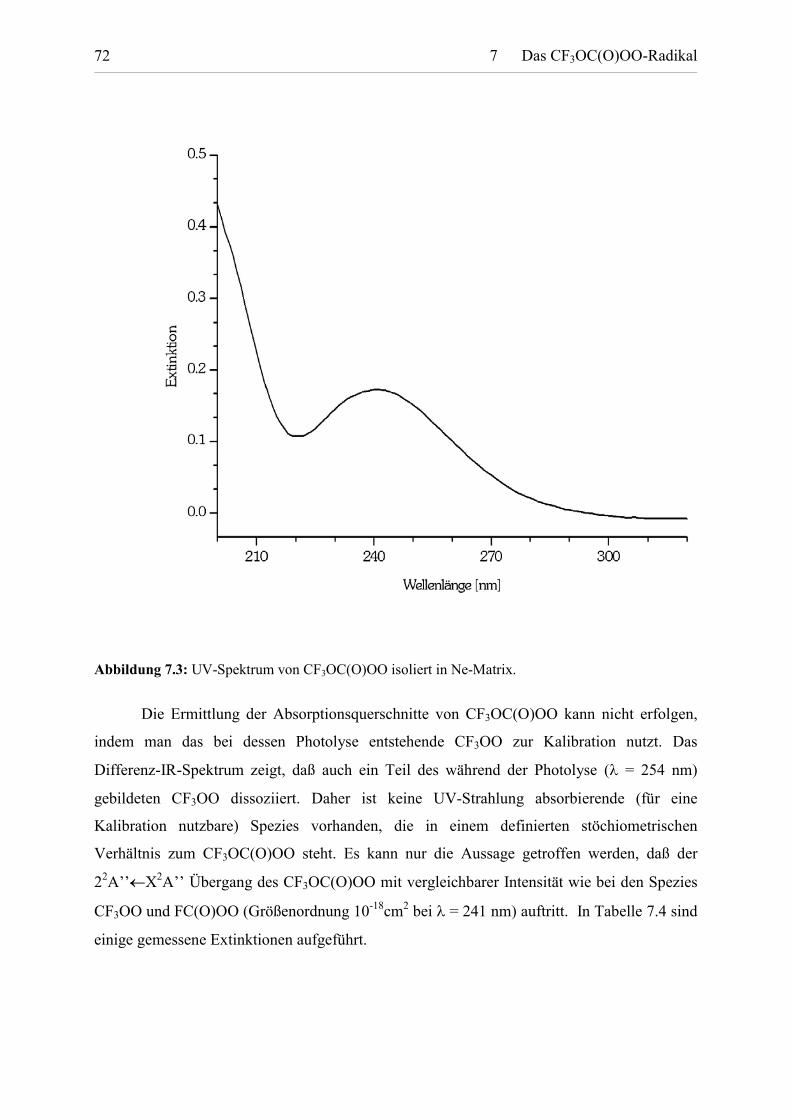
CF3OC(O)OO zeigt zwei sich überlagernde Absorptionsbanden, die beide keine
Struktur aufweisen. UV-Licht mit � > 315 nm wird praktisch nicht mehr absorbiert.
Der erste Übergang, der ein Maximum bei � = 241 nm aufweist, wird durch die
Anregung eines Elektrons aus dem höchsten besetzten a’’-Orbital in das halb besetzte a’’-
Orbital beschrieben. Dieser 22A’’�X2A’’ Übergang führt zur O-O Bindungsspaltung und
zeigt eine, für Peroxyradikale typische, �-abhängige Gaußkurvenkontur.[77] Die Photolyse des
CF3OC(O)OO, die im wesentlichen auf der Anregung des Radikals in den 22A’’ Zustand
basiert, verläuft in Matrix nach folgendem Schema, wobei die Radikale bzw. Atome in einem
Matrixkäfig isoliert bleiben:
CF3OC(O)OO + h� � [CF3OCO2 + O] (7-25)
[CF3OCO2 + O] � [CF3O + CO2 + O] (7-26)
[CF3O + CO2 + O] � CF3OO + CO2 (7-27)
7 Das CF3OC(O)OO-Radikal 72
Abbildung 7.3: UV-Spektrum von CF3OC(O)OO isoliert in Ne-Matrix.
Die Ermittlung der Absorptionsquerschnitte von CF3OC(O)OO kann nicht erfolgen,
indem man das bei dessen Photolyse entstehende CF3OO zur Kalibration nutzt. Das
Differenz-IR-Spektrum zeigt, daß auch ein Teil des während der Photolyse (� = 254 nm)
gebildeten CF3OO dissoziiert. Daher ist keine UV-Strahlung absorbierende (für eine
Kalibration nutzbare) Spezies vorhanden, die in einem definierten stöchiometrischen
Verhältnis zum CF3OC(O)OO steht. Es kann nur die Aussage getroffen werden, daß der
22A’’�X2A’’ Übergang des CF3OC(O)OO mit vergleichbarer Intensität wie bei den Spezies
CF3OO und FC(O)OO (Größenordnung 10-18cm2 bei � = 241 nm) auftritt. In Tabelle 7.4 sind
einige gemessene Extinktionen aufgeführt.
7 Das CF3OC(O)OO-Radikal 73
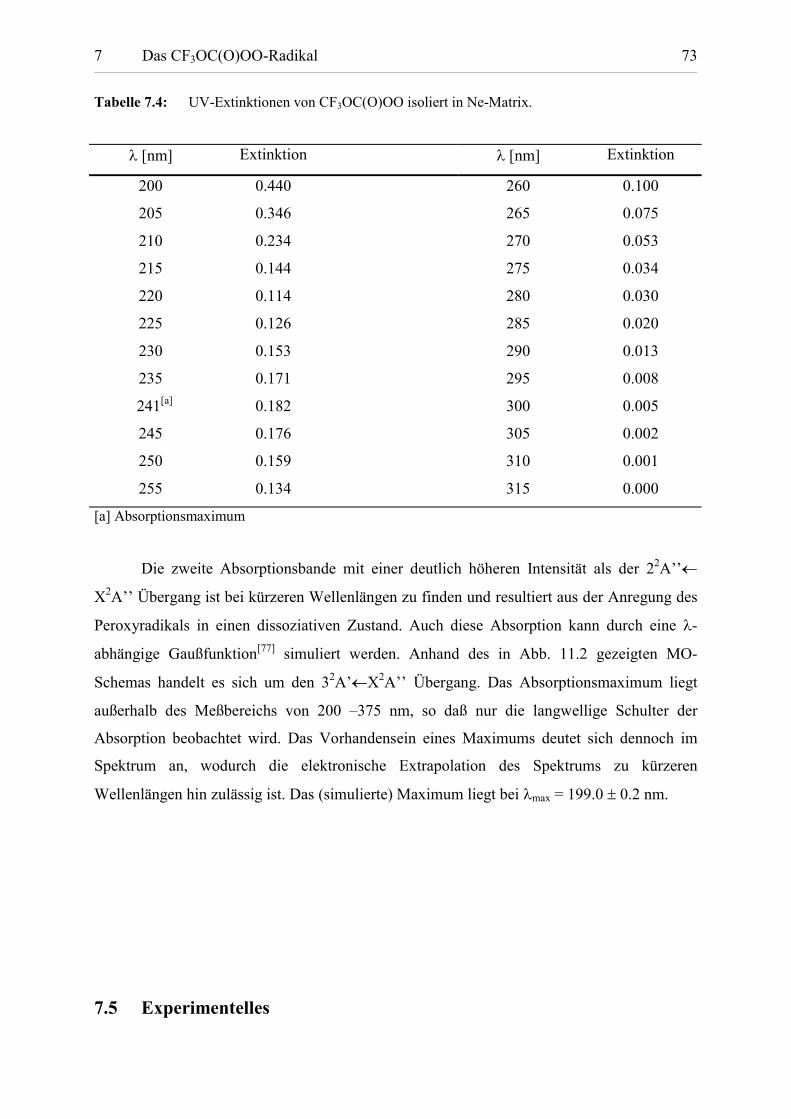
Tabelle 7.4: UV-Extinktionen von CF3OC(O)OO isoliert in Ne-Matrix.
� [nm] Extinktion � [nm] Extinktion
200 0.440 260 0.100
205 0.346 265 0.075
210 0.234 270 0.053
215 0.144 275 0.034
220 0.114 280 0.030
225 0.126 285 0.020
230 0.153 290 0.013
235 0.171 295 0.008
241[a] 0.182 300 0.005
245 0.176 305 0.002
250 0.159 310 0.001
255 0.134 315 0.000
[a] Absorptionsmaximum
Die zweite Absorptionsbande mit einer deutlich höheren Intensität als der 22A’’�
X2A’’ Übergang ist bei kürzeren Wellenlängen zu finden und resultiert aus der Anregung des
Peroxyradikals in einen dissoziativen Zustand. Auch diese Absorption kann durch eine �-
abhängige Gaußfunktion[77] simuliert werden. Anhand des in Abb. 11.2 gezeigten MO-
Schemas handelt es sich um den 32A’�X2A’’ Übergang. Das Absorptionsmaximum liegt
außerhalb des Meßbereichs von 200 –375 nm, so daß nur die langwellige Schulter der
Absorption beobachtet wird. Das Vorhandensein eines Maximums deutet sich dennoch im
Spektrum an, wodurch die elektronische Extrapolation des Spektrums zu kürzeren
Wellenlängen hin zulässig ist. Das (simulierte) Maximum liegt bei �max = 199.0 � 0.2 nm.
7.5 Experimentelles
7 Das CF3OC(O)OO-Radikal 74
Die Synthese der Vorläuferverbindung CF3OC(O)OOOC(O)OCF3 wird in Kap. 13
beschrieben.
Präparation der Matrices
In einem typischen Matrixisolationsexperiment wird reines Bistrifluormethyl-
trioxydicarbonat, CF3OC(O)OOOC(O)OCF3, in einer mit Youngventilen ausgestatteten,
gläsernen U-Rohr-Falle mit der Pyrolysevorrichtung der Matrixisolationsapparatur
verbunden. Das Edelgas strömt über eine Kapillare durch die im Ethanolbad auf -80°C
gekühlte U-Rohr-Falle hindurch und trägt verdampfendes Trioxid mit. Die
Pyrolyseexperimente werden bei 160°C durchgeführt, es wurden 1 bis 3 mmol Edelgas
innerhalb von 20 bis 60 Minuten als Matrix auf dem Matrixträger ausgefroren. Die
Produktmatrix enthält neben CF3OC(O)OO vor allem CF3O und CO2.
Bei den Photolyseexperimenten wird die Matrix der Pyrolyseprodukte von
CF3OC(O)OOOC(O)OCF3 mittels einer Quecksilberhochdrucklampe ohne Kantenfilter oder
mit einer Quecksilberniederdrucklampe (�= 254 nm), bzw. einer Quecksilberhochdrucklampe
mit 254 nm Interferenzfilter, für 10-30 Minuten bestrahlt.
Bei den nicht erfolgreichen Versuchen zur Erzeugung von CF3OC(O)OO wurden
CF3OC(O)OOCF3 bzw. CF3OC(O)OOC(O)OCF3[114] im Verhältnis 1:200:500 mit Sauer-stoff
und Kohlenmonoxid in der Edelstahl-Vakuumapparatur vermischt und jeweils 1 mmol
innerhalb von 20 Minuten durch die auf 310 bzw. 265°C geheizte Pyrolyse-vorrichtung
geleitet und als Matrix isoliert.
8 Das CF3C(O)OO-Radikal 75
8 Das CF3C(O)OO-Radikal
8.1 Einführung
In atmosphärischen Prozessen tritt das Radikal CF3C(O)OO stets als direktes
Folgeprodukt von CF3CO auf, da letzteres schnell mit molekularem Sauerstoff reagiert:
CF3CO + O2 ( + M ) � CF3C(O)OO ( + M ) (8-1)
Diese Reaktion ist, bedingt durch den großen Überschuß von O2 in der Atmosphäre, der
einzige Weg, den das CF3CO-Radikal beschreitet,[93, 139] obwohl es aufgrund seiner
Instabilität innerhalb von Mikrosekunden dissoziiert:[55, 58, 139, 170]
CF3CO � CF3 + CO (8-2)
Die Bildung von CF3CO erfolgt hauptsächlich durch Photolyse von CF3C(O)X (X=F,Cl),[113]
sowie durch Spaltung von CF3C(O)H durch OH-Radikale:
CF3C(O)X + h� � CF3CO + X (X=F,Cl) (8-3)
CF3C(O)H + OH � CF3CO + H2O (8-4)
Trifluoressigsäurechlorid wird allerdings durch Wasser in der Troposphäre schnell zu HCl
und Trifluoressigsäure umgesetzt [Gl. (8-5)], wobei für die letztgenannte Verbindung bis
heute kein biologischer Abbauweg bekannt ist.[171] Die Akkumulation von Trifluoracetaten in
der Natur kann daher zu nicht absehbaren Problemen führen.[172]
CF3C(O)Cl + H2O � CF3C(O)OH + HCl (8-5)
Die spektroskopische Charakterisierung des CF3C(O)OO-Radikals beschränkt sich in
der Literatur auf das in der Gasphase gemessene UV-Spektrum,[85, 94, 95] das jedoch von
mäßiger Qualität ist.
8 Das CF3C(O)OO-Radikal 76
Im Rahmen dieser Arbeit wurde CF3C(O)OO erstmals in Matrix isoliert und IR- und
UV-spektroskopisch charakterisiert. Darüber hinaus wurden die Molekülstruktur und die
Schwingungsfrequenzen der zwei stabilen Rotamere quantenmechanisch berechnet.
8.2 Die Struktur des CF3C(O)OO-Radikals
Das Trifluoracetylperoxyradikal CF3C(O)OO weist eine fünfatomige Kette (F-C-C-O-
O) mit zwei unabhängigen Diederwinkeln 1 = (OCCF) und 2 = (OOCC) (1 und 2 0°
bzw. 180°) auf. Die theoretischen Berechnungen zeigen, daß nur zwei der vier möglichen
Isomere energetische Minima darstellen: Die Rotamere trans,trans-CF3C(O)OO (1 = 2 =
180°) und trans,cis-CF3C(O)OO (1 = 180° und 2 = 0°), wobei das erstgenannte um 64.5
kJ·mol-1 stabiler ist. Die anderen Isomere, in denen die O2-Gruppe synplanar zu dem in der
Symmetrieebene gelegenen Fluoratom der CF3-Gruppe liegt, sind Übergangszustände mit
einer imaginären Schwingungsfrequenz. Die berechneten Strukturen der stabilen Rotamere
sind in Abb. 8.1 gezeigt.
trans,trans-CF3C(O)OO trans,cis-CF3C(O)OO
Abbildung 8.1: Berechnete Strukturen der stabilen Rotamere von CF3C(O)OO.Die berechneten Strukturparameter sind in Tabelle 8.1 zusammengestellt. Auffällig ist hierbei
der bei beiden Rotameren auftretende große CC-Abstand von etwa 1.55 Å.
Tabelle 8.1: Berechnete[a] Geometrien der stabilen Rotamere von CF3C(O)OO.
[a] B3LYP/6-311G(d,p)[b] F1 bezeichnet das Fluoratom in der Spiegelebene,F2 bezeichnet die beiden anderen, symmetrieäquivalenten F-Atome der CF3-Gruppe.
In der Literatur werden die Rotamere des Radikals CF3C(O)OO (und deren
Energieunterschied) nicht diskutiert. Der berechnete Energieunterschied zwischen den beiden
stabilen Rotameren ist stark von der verwendeten Methode und dem Basissatz abhängig. So
beträgt Hrel im Falle einer HF/3-21G-Rechnung fast 0 und im Falle einer MP2/3-21G-
Rechnung 34.9 kJ·mol-1. Alle durchgeführten Rechnungen sagen das trans,trans-Isomer als
stabilstes Rotamer voraus.
Anhand der experimentellen IR-Spektren des Radikals CF3C(O)OO läßt sich das
Auftreten zweier Rotamere bestätigen, die bei einer Pyrolysetemperatur von 210°C etwa im
molaren Verhältnis 1:5 (trans,cis zu trans,trans-Isomer) entstehen. Dieses Verhältnis ergibt
sich, wenn man das beobachteteVerhältnis der integrierten Bandenintensitäten von �1
(Icis,exp/Itrans,exp = 0.38) beider Isomere mit den berechneten molaren Intensitäten Icalc korrigiert
(� = Extinktionskoeffizient):
8 Das CF3C(O)OO-Radikal 78
trans
cisRTG
calccistrans
calctranscis
nn
eII
II��
��
,exp,
,exp, da �
In � (8-6)
Mit T=483 K resultiert ein Energieunterschied zwischen den Isomeren von 6.3 kJ·mol-1, also
zehnmal geringer als die mit B3LYP/6-311G(d,p) berechnete Energiedifferenz.
8.3 Die Erzeugung von CF3C(O)OO-Radikalen
In Analogie zu CF3OO und FC(O)OO wird CF3C(O)OO durch Niederdruckpyrolyse
von CF3C(O)OONO2[102] (FPAN) hochverdünnt in Ne oder Ar erzeugt und die
Reaktionsprodukte als Matrix isoliert. Bei Pyrolysetemperaturen oberhalb von 260°C verläuft
die Spaltung des FPAN quantitativ:
260°C
CF3C(O)OONO2 � CF3C(O)OO + NO2 (u. a. Produkte) (8-7)
Mit zunehmender Reaktionstemperatur sinkt die Ausbeute an CF3C(O)OO und es werden
CF3OO und CO gebildet:
CF3C(O)OO � CF3OO + CO (8-8)
Führt man die Pyrolyse bei tieferen Temperaturen durch, ist die Bildung von Nebenprodukten
geringer. Nachteilig ist dann jedoch das Auftreten von unzersetztem Peroxynitrat im
Produktgemisch. Insgesamt beträgt die maximale Ausbeute an CF3C(O)OO, abgeschätzt
anhand der im IR-Spektrum beobachteten Absorptionen, etwa 20% (bei 260°C
Reaktionstemperatur).
8.4 Die spektroskopische Charakterisierung von CF3C(O)OO
Das IR-Spektrum der Pyrolyseprodukte von CF3C(O)OONO2, isoliert in Ne-Matrix,
weist sehr viele Banden auf. Neben noch nicht identifizierten Banden zeigen sich
Absorptionen von CF3OO, CO2, COF2 und CO. Ein Weg zur Identifizierung des Radikals
CF3C(O)OO besteht in der Bestrahlung der Produktmatrix mit UV-Licht. Man beobachtet
eine Gruppe von Absorptionsbanden, die gleichmäßig an IR-Intensität verlieren. Die IR-
Absorptionen von CF3OO zeigen, je nach verwendeter Lichtquelle bzw. verwendetem
8 Das CF3C(O)OO-Radikal 79
Kantenfilter, ein variables Verhalten. Bei Bestrahlung mit ungefiltertem UV-Licht einer
Quecksilberhochdrucklampe nimmt die Konzentration von CF3OO ab, bei zusätzlicher
Verwendung eines 280 nm Kantenfilters nimmt sie zu. Dies steht in Einklang mit Gln. (8-9)
und (8-10):
CF3C(O)OO + h� ( � < 280 nm ) � CF3OO + CO (8-9)
CF3OO + h� ( � < 260 nm ) � COF2 + OF (8-10)
Bei Verwendung einer Quecksilberniederdrucklampe (� = 254 nm) bzw. einer Hg-
Hochdrucklampe mit 254 nm Interferenzfilter, wird die Konzentration des CF3OO annähernd
konstant gehalten, da dann etwa so viel vorhandenes Trifluormethyl-peroxyradikal zerfällt,
wie durch die Photolyse aus CF3C(O)OO neu gebildet wird.
Die bei allen Photolysen beobachtbare Zunahme der CO2-Konzentration zeigt einen
zweiten Photolyseweg an, welcher ebenfalls mit dem Vorliegen von CF3C(O)OO in Einklang
steht:
CF3C(O)OO + h� � CF3O + CO2 (8-11)
Es kann nicht unterschieden werden, ob die CO2-Abspaltung aus dem angeregten Zustand des
CF3C(O)OO erfolgt oder eine Reaktion von CF3OO mit CO in einem Matrixkäfig eintritt. Da
CF3O selbst Licht der Wellenlänge �=254 nm absorbiert, wird es bei einer Photolysedauer
von 90 Minuten in sein Folgeprodukt COF2 zerlegt:
CF3O + h� � COF2 + F (8-12)
IR-Spektrum
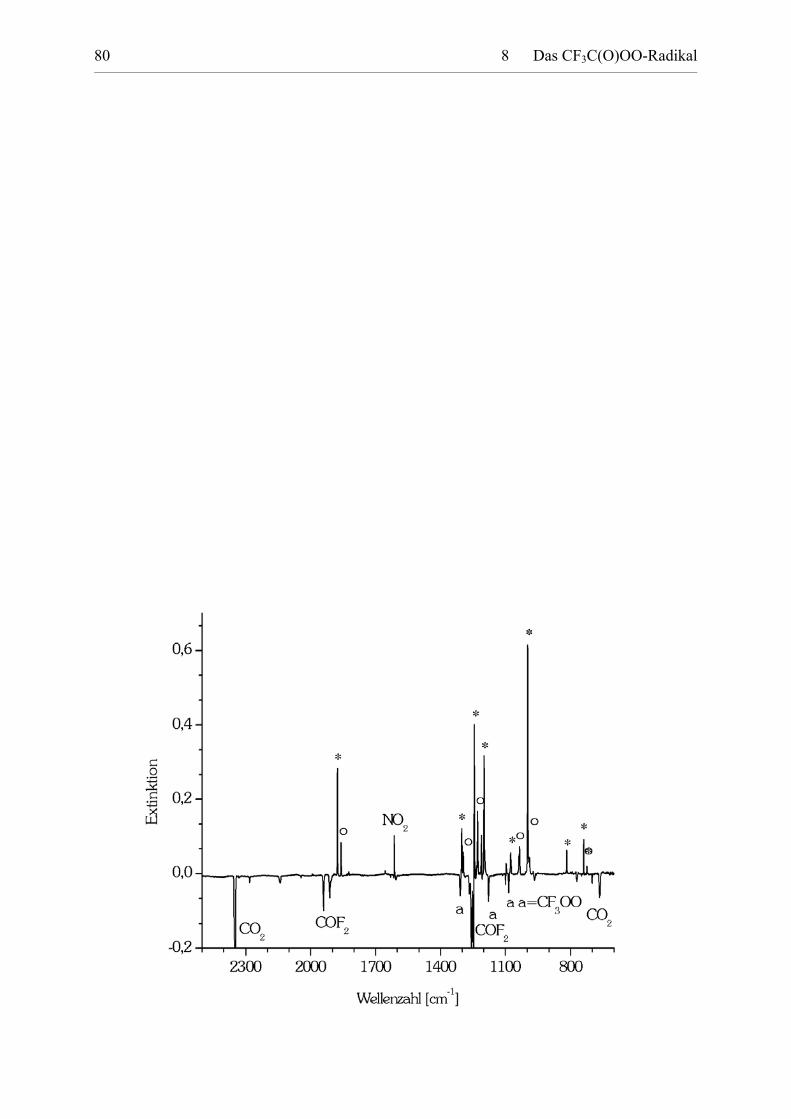
Ein Differenzspektrum der Pyrolyseprodukte von CF3C(O)OONO2 isoliert in Ne-
Matrix vor und nach der Photolyse ist in Abbildung 8.2 gezeigt.
8 Das CF3C(O)OO-Radikal 80
8 Das CF3C(O)OO-Radikal 81
Abbildung 8.2: Differenz-IR-Spektrum der Pyrolyseprodukte von CF3C(O)OONO2 vor und nachder Photolyse mit UV-Licht (� = 254 nm). Die Banden der bei der Photolysedissoziierten CF3C(O)OO-Rotamere (* für trans,trans- und o für trans,cis-CF3C(O)OO) sind positiv, die der Photolyseprodukte negativ dargestellt.
Es werden mehr Banden beobachtet, als von einem Rotamer CF3C(O)OO zu erwarten sind.
Insbesondere das Vorliegen von zwei Banden im Bereich der C=O Streckschwingungen weist
auf das Vorhandensein zweier Isomere hin.
Die Variation der Pyrolyse, die aufgrund des bereits diskutierten Verhaltens des
CF3C(O)OO Radikals nur in einem recht kleinen Temperaturfenster erfolgen kann, führt
allerdings nicht zu einer signifikanten Änderung der Intensitätsverhältnisse dieser C=O
Banden. Ein sicherer Beweis für das Vorliegen zweier Isomere (sofern eines 0 bis 10 kJ�mol-1
stabiler ist als das andere) läßt sich auf diese Weise nicht führen. Somit stützt sich die
Annahme, daß zwei Rotamere auftreten, auf die Tatsache, daß die beobachteten Banden durch
die theoretisch berechneten Spektren beider Isomere gut wiedergegeben werden. Eine
Übersicht über die den Rotameren des CF3C(O)OO Radikals zugeordneten IR-Absorptionen
findet sich in den Tabellen 8.2 und 8.3.
Nach Aussage der quantenmechanischen Rechnungen besitzen beide Isomere Cs-
Symmetrie und entsprechend lautet die irreduzible Darstellung der Schwingungsfreiheitsgrade
mit IR- und Ramanaktivitäten:
�vib = 12 A' (IR, Ra p) + 6 A'' (IR, Ra dp) (8-12)
Die Zuordnung der beobachteten Absorptionsbanden erfolgte im Vergleich mit
bekannten Spezies wie CF3OO[35, 143-146, 164] und FC(O)OO,[146, 164] und den
quantenmechanisch berechneten Spektren.
8 Das CF3C(O)OO-Radikal 82
Tabelle 8.2: Fundamentalschwingungen von trans,trans-CF3C(O)OO. Experimentelle IR-spektroskopische Daten von trans,trans-CF3C(O)OO isoliert in Ne-Matrix, sowieberechnete Bandenlagen und Intensitäten.
trans,trans-CF3C(O)OO
Ne-Matrix B3LYP[a] Zuordnung gem. Cs-Symmetrie
� [cm-1] rel. Int. [b]� [cm-1] Int.[c] und Beschreibung
1876.2 55 1932 173 �1 A' � C=O
1301.7 40 1289 87 �2 A' �s CF3
1244.6 57 1231 299 �3 A' �a CF3
1198.4 78 1185 294 �13 A'' �a CF3
1076.1 7.1 1117 26 �4 A' � OO
997.4 100 995 306 �5 A' � CC / � CO o.p.
818.0 7.1 823 33 �6 A' � CC / � CO i.p.
740.1 9.4 740 26 �7 A' �s CF3 / � CCO i.p.
725.0* 3.2* 730 14 �14 A'' �
565 6.9 �8 A' �a CF3 / � CCO i.p.
516.8* 2.0* 512 7.5 �15 A'' �a CF3
413 1.2 �9 A' �a CF3 / � CCO o.p.
374 2.6 �10 A' � OOC
333 4.7 �11 A' �s CF3 / � CCO o.p.
249 4.1 �16 A'' � CF3
184 2.3 �12 A' � CF3
8 Das CF3C(O)OO-Radikal 83
110 1.3 �17 A'' OCCF
35 0.35 �18 A'' OOCC
[a] B3LYP/6-311G(d,p)[b] relative integrierte Intensitäten [c] in km·mol-1
* Überlagert von Banden des trans,cis-CF3C(O)OO
Tabelle 8.3: Fundamentalschwingungen von trans,cis-CF3C(O)OO. Experimentelle IR-spektroskopische Daten von trans,cis-CF3C(O)OO isoliert in Ne-Matrix, sowieberechnete Bandenlagen und Intensitäten.
Das CH3C(O)OO-Radikal weist eine fünfatomige Kette (H-C-C-O-O) mit zwei
voneinander unabhängigen Diederwinkeln auf, die jeweils 180° bzw. 0° betragen können.
Laut der quantenmechanischen Berechnungen sind nur zwei der vier möglichen Rotamere
energetische Minima auf der Potentialhyperfläche, nämlich trans,cis- und trans,trans-
CH3C(O)OO, wobei das trans,cis-CH3C(O)OO, mit (HCCO) = 180° und (CCOO) = 0°,
geringfügig stabilerer ist. Eine Geometrie, bei welcher die Carbonylgruppe trans-ständig zu
einem Wasserstoffatom der CH3-Gruppe steht, erweist sich als Übergangszustand mit einer
imaginären Schwingungsfrequenz. Eine vollständig transoide Anordnung der H-C-C-O-O
Kette des Moleküls (trans,trans-CH3C(O)OO) wird um 2.2�kJ�mol-1 energiereicher berechnet
(H unter Standardbedingungen), und Grel (berechnet) ist annähernd 0. Die geringen
Energiedifferenzen bedeuten, daß im Falle des CH3C(O)OO bereits bei Raumtemperatur
nahezu eine Gleichverteilung zwischen beiden Rotameren vorliegen sollte. Die berechneten
Strukturen der stabilen Rotamere ist in Abb. 9.1 gezeigt, die geometrischen Parameter finden
sich in Tabelle 9.1.
trans,trans-CH3C(O)OO trans,cis-CH3C(O)OO
Abbildung 9.1: Berechnete Strukturen der stabilen Rotamere von CH3C(O)OO.Tabelle 9.1: Berechnete[a] Molekülparameter der stabilen Rotamere von CH3C(O)OO.
[a] B3LYP/6-311G(d,p)[b] H1 bezeichnet das Fluoratom in der Spiegelebene,H2 bezeichnet die beiden anderen, symmetrieäquivalenten H-Atome der CH3-Gruppe.[c] �Grel ergibt eine um 0.1 kJ�mol-1 höhere Stabilität des trans,trans-CH3C(O)OO, �Urel favorisiertwiederum das trans,cis-Rotamer um 1.7 kJ�mol-1, jeweils unter Standardbedingungen.
Die experimentell ermittelten IR-Spektren zeigen, daß CH3C(O)OO in zwei
Rotameren vorliegt, wobei die Banden, die anhand der quantenmechanisch berechneten
Erwartungsspektren den Schwingungen des trans,cis-CH3C(O)OO zugeordnet werden
können, deutlich höhere Intensitäten aufweisen als die des trans,trans-CH3C(O)OO. Aus den
gemessenen und berechneten IR-Intensitäten läßt sich das Rotamerenverhältnis von trans,cis
zu trans,trans-CH3C(O)OO zu etwa 4:1 ableiten, gleichbedeutend mit einem
Energieunterschied von etwa 5.6 kJ�mol-1 (trans,cis-CH3C(O)OO stabiler):
RTG
tc
tt
berechnettttc
berechnettctt enn
IIII �
�
��
,exp,
,exp, mit T= 483 K (210°C) (9-2)
9 Das CH3C(O)OO-Radikal 91
Im Vergleich mit CF3C(O)OO sagen die Berechnungen, neben der "Umkehr" der
Stabilitätsverhältnisse (CH3C(O)OO: trans,cis stabiler als trans,trans) einen weiteren
strukturellen Unterschied voraus: Die CC-Bindungslänge ist im CH3C(O)OO (beide
Rotamere) mit 1.50 Å deutlich kürzer als im CF3C(O)OO (1.55 Å).
Es ist anzumerken, daß die Nomenklatur trans,trans- und trans,cis-CH3C(O)OO, und
damit die Reihenfolge der Nennung der Diederwinkeleinstellungen (cis und trans), an die der
fluorierten Peroxyradikale angepaßt wurde, um eine einheitliche Bezeichnung der Rotamere
beizubehalten.
9.3 Die Erzeugung von CH3C(O)OO-Radikalen
CH3C(O)OO läßt sich durch Niederdruckpyrolyse des Peroxynitrates
CH3C(O)OONO2 generieren[86] und in überschüssigem Inertgas (Ar oder Ne) als
Edelgasmatrix isolieren:
210°C
CH3C(O)OONO2 � CH3C(O)OO + NO2 (9-3)
Dabei treten Folgereaktionen auf, die mit steigender Pyrolysetemperatur die ohnehin geringe
Ausbeute von etwa 10 - 20% an CH3C(O)OO weiter herabsetzen. Die Folgeprodukte konnten
nicht identifiziert werden. Die Reaktion
�
CH3C(O)OO � CH3OO + CO (9-4)
gilt durch den Nachweis von CO im IR-Spektrum als wahrscheinlich, wenngleich CH3OO
nicht zweifelsfrei IR-spektroskopisch nachgewiesen werden kann, da nicht alle zu dieser
Spezies gehörenden Absorptionen eindeutig im bandenreichen Spektrum der
Pyrolyseprodukte identifizierbar sind.
9.4 Die spektroskopische Charakterisierung von CH3C(O)OO
9 Das CH3C(O)OO-Radikal 92
IR-Spektrum
Das IR-Spektrum der Pyrolyseprodukte von CH3C(O)OONO2, isoliert als Ar- oder
Ne-Matrix, zeigt neben den Absorptionen bekannter Verbindungen wie NO2, CO und CO2
eine Reihe neuer Banden. Bei Erhöhung der Pyrolysetemperatur nimmt die Intensität einiger
dieser Banden zu, während andere an Intensität wieder abnehmen. Aus der Abhängigkeit der
Bandenintensitäten von der Reaktionstemperatur ergibt sich: Die PAN-Konzentration fällt mit
steigender Reaktionstemperatur, die NO2-Menge steigt entsprechend. Die Absorptionsbanden,
die ein temperaturabhängiges Maximum aufweisen, stammen vom CH3C(O)OO, die mit der
Temperatur stark anwachsenden Banden sind Folgeprodukten zuzuordnen. So beträgt der
relative Anteil von CH3C(O)OO 0, 100, 60 und 7% bei Reaktionstemperaturen von 140, 210,
240 und 310°C.
Das in Abb. 9.2 gezeigte IR-Spektrum von CH3C(O)OO stellt ein Differenzspektrum
dar. Hierbei wird von einem Spektrum von CH3C(O)OONO2 nach der Pyrolyse bei 210°C
eines nach Pyrolyse bei 310°C (enthält Spuren von CH3C(O)OO, aber sehr viel
Folgeprodukte und CO2) abgezogen. Dabei ist die Subtraktion so durchführbar, daß eine
Gruppe von Absorptionen einiger Folgeprodukte gerade verschwindet, da diese
Folgeprodukte bei der Temperaturerhöhung von 210°C auf 310°C gleichmäßig zunehmen. Im
Differenzspektrum verbleiben die Banden der CH3C(O)OO-Isomere, NO2, CO2 (positv
dargestellt) und einer weiteren Gruppe von Folgeprodukt-Absorptionen (negativ dargestellt).
Nach Aussage der quantenmechanischen Rechnungen besitzen beide Rotamere des
CH3C(O)OO Cs-Symmetrie und entsprechend lautet die irreduzible Darstellung der
Schwingungsfreiheitsgrade mit IR- und Ramanaktivitäten:
�VIB = 12 A' (IR, Ra p) + 6 A'' (IR, Ra dp) (9-5)
Die Zuordnung der Banden erfolgt durch Vergleich mit den berechneten Spektren der
beiden stabilen Rotamere und durch Vergleich mit Literaturdaten.[86] Die dort dem
CH3C(O)OO zugewiesenen Absorptionen konnten bestätigt und dem trans,cis-CH3C(O)OO
zugeordnet werden. Darüber hinaus wurden zusätzliche Banden gefunden, die durch trans,cis-
und trans,trans-CH3C(O)OO hervorgerufen werden. Zudem ist eine Unterscheidung zwischen
beiden Rotameren auch durch den Vergleich der Bandenformen der IR-Absorptionen im
Spektrum möglich. Ein Teil der Banden des CH3C(O)OO ist vergleichsweise scharf und
entsprechend intensiv, während der andere Teil verbreitert ist. Die breiteren Banden werden
9 Das CH3C(O)OO-Radikal 93
bezüglich ihrer Lage durch das Erwartungsspektrum des trans,trans-CH3C(O)OO gut
beschrieben.
Abbildung 9.2: IR-Spektrum von trans,cis- und trans,trans-CH3C(O)OO. Das Spektrum ergibt sich aus der Differenz zweier IR-Spektren der Pyrolyseprodukte von CH3C(O)OONO2, wobei die Pyrolyse bei verschiedenen Temperaturen
durchgeführt wird (Ne-Matrix, Details finden sich im Text). Die Banden des cis,trans-CH3C(O)OO sind mit O, die des trans,trans-CH3C(O)OO mit * gekennzeichnet.
Eine Übersicht der experimentell gefundenen und berechneten Grundschwingungen beider
CH3C(O)OO-Rotamere gibt Tabelle 9.2. Die Übereinstimmung der experimentellen Daten
mit den quantenmechanisch berechneten Werten ist meist gut. Aufgrund der für die
CH3C(O)OO-Rotamere zu erwartenden starken Schwingungskopplung wird keine
Beschreibung der Schwingungen gegeben.
Tabelle 9.2: Fundamentalschwingungen von trans,trans-CH3C(O)OO und trans,cis-CH3C(O)OO.Experimentelle IR-spektroskopische Daten beider CH3C(O)OO-Rotamere isoliert inNe-Matrix, sowie berechnete Bandenlagen und Intensitäten.
Abbildung 9.3: UV-Spektrum von CH3C(O)OO isoliert in Ne-Matrix.
9.5 Experimentelles
CH3C(O)OONO2 wurde aus Beständen des Arbeitskreises entnommen, die Synthese
ist in der Literatur[106] beschrieben.
In der an der Matrixisolationsapparatur befindlichen Edelstahl-Vakuumapparatur
werden CH3C(O)OONO2 und Argon bzw. Neon im Verhältnis 1:600 gemischt. Dabei ist zu
beachten, daß CH3C(O)OONO2 im Laufe einiger Stunden in der Edelstahlapparatur bereits
merklich zerfällt. In einem typischen Experiment werden 1 bis 3 mmol der Gasmischung
innerhalb von 20 bis 60 Minuten auf dem Matrixträger ausgefroren. Die Pyrolyse wird bei
210°C (ergibt maximale Ausbeute an CH3C(O)OO) bzw. 310°C (geringe Ausbeute an
CH3C(O)OO, hoher Anteil an Folgeprodukten) durchgeführt.
9 Das CH3C(O)OO-Radikal 97
10 Das CF3OOO-Radikal 98
10 Das CF3OOO-Radikal
10.1 Einführung
In der Chemie des Sauerstoffs sind Moleküle, die eine mehratomige Sauerstoffkette
und ein ungepaartes Elektron besitzen, fast ausnahmslos auf Peroxy-radikale begrenzt. Zu den
wenigen Ausnahmen gehören die Salze der Ionen [O2]+, [O2]2-, [O2]- und [O3]-. Darüber
hinaus sind bezüglich der nichtionischen Trioxyradikale lediglich die Spezies FO3[182] und
FS(O)2OOO[183] in der Literatur beschrieben, weiterhin gibt es Hinweise auf die Radikale
SF5OOO[184] und ClO4�O2 (entsprechend O3ClOOO).[185] Somit scheinen Oxyradikale, die
eine besonders hohe Elektronenaffinität aufweisen, Wechsel-wirkungen mit molekularem
Sauerstoff einzugehen, die über die Stärke eines Van-der-Waals Kontakts hinausgehen. Es ist
daher zu erwarten, daß auch CF3O zur Bildung des Radikals CF3OOO fähig ist. In ESR-
Experimenten mit CF3O-Spezies in Anwesenheit von Sauerstoff wurde die mögliche Existenz
von CF3OOO bereits diskutiert.[186] Darüberhinaus gibt es in der Literatur keine Hinweise auf
diese Spezies, auch nicht im Zuge UV-spektroskopischer Untersuchungen an CF3Ox
Radikalen.[147, 148, 164, 169]
Im Rahmen dieser Arbeit konnte zum ersten Mal die Existenz des CF3OOO-Radikals,
isoliert in Matrix, IR- und UV-spektroskopisch bewiesen werden. Neben der
spektroskopischen Charakterisierung werden die Ergebnisse quantenmechanischer
Berechnungen präsentiert.
10.2 Die Struktur des CF3OOO-Radikals
Die Geometrie des CF3OOO wurde unter Verwendung der B3LYP-Methode mit 6-
311G(d,p) Basissatz quantenmechanisch berechnet, was allerdings wegen der aus-
schließlichen Berücksichtigung eines elektronischen Zustands zu Fehlern führt (siehe Kap.
3.3). Multireferenz-Methoden führen sicherlich zu genaueren Resultaten. Die optimierte
Geometrie zeigt einen langen Bindungsabstand zwischen der CF3O-Gruppe und der O2-
Einheit an, der allerdings mit ca. 2.2 Å deutlich kürzer ist als die Summe der Van-der-Waals
10 Das CF3OOO-Radikal 99
Radien (2.8 Å). Die berechnete Struktur ist in Abbildung 10.1 dargestellt, die
Strukturparameter finden sich in Tabelle 10.1.
Abbildung 10.1: Berechnete Struktur von CF3OOO.
Tabelle 10.1: Berechnete[a] Molekülgeometrie von CF3OOO.
Koordinate [Å] bzw. [°] Koordinate [Å] bzw. [°]
R(CF)[b] 1.344 �(FCO)[b] 108.0
R(CO) 1.342 �(COO) 103.0
R(O-O2) 2.184 �(OOO) 110.8
R(CF3O2-O) 1.191
(FCOO)[c] 178.8
HR[d] [kJ�mol-1] 10.2 (COOO) 149.0
[a] B3LYP/6-311G(d,p)[b] Mittelwert [c] 59.2° und -62.3° für � bezogen auf die beiden anderen F-Atome [d] für die Reaktion CF3OOO�CF3O+O2
10.3 Die Erzeugung von CF3OOO-Radikalen
10 Das CF3OOO-Radikal 100
Bei der Niederdruckpyrolyse von CF3OC(O)OOCF3 in einem großen Überschuß von
O2 entsteht zunächst das Trifluormethoxyradikal:
CF3OC(O)OOCF3 � 2 CF3O + CO2 (10-1)
Nach der Pyrolyse werden die Reaktionsprodukte ins Hochvakuum expandiert und schließlich
auf einem auf 16 K gekühlten Träger als O2-Matrix ausgefroren. Man beobachtet die Bildung
des Trioxyradikals, die wahrscheinlich während des Kondensationsprozesses abläuft:
CF3O + O2 � CF3OOO (10-2)
Daneben ist auch COF2 IR-spektroskopisch nachweisbar, die Bildung des Trioxyradikals
verläuft also nicht quantitativ.
Aufgrund der durchgeführten Experimente kann keine Aussage über die Stabilität des
Radikals getroffen werden. Somit sind auch keine Rückschlüsse auf die Existenz oder die
Lebensdauer des Radikals unter stratosphärischen Bedingungen oder gar bei Raumtemperatur
möglich.
10.4 Die spektroskopische Charakterisierung von CF3OOO
Das IR-Spektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in O2-
Matrix ist verhältnismäßig bandenarm. Es finden sich neben den Absorptionen von CO2 und
COF2 nur 4 starke Banden, die keiner bekannten Verbindung zugeordnet werden können.
Eine Photolyse der Matrix mit UV-Licht einer Quecksilber-hochdrucklampe zeigt, daß diese
Banden gleichzeitig abnehmen und daher nur einer Spezies zugeordnet werden. Als
Photolyseprodukte können COF2 und FOO[187-189] anhand der bekannten IR-Spektren
identifiziert werden. Daraus läßt sich allerdings nicht das Vorliegen von CF3OOO folgern, da
auch CF3O, falls es als isoliertes Radikal in O2-Matrix vorliegt bei Bestrahlung mit UV-Licht
in COF2 und F zerfallen sollte, was schließlich in Sauerstoff-Matrix ebenfalls zu COF2 und
FOO führen muß. Der Existenzbeweis für CF3OOO gelingt aber durch die Aufnahme eines
UV-Spektrums der neuen Spezies. CF3O zeigt in Ne-Matrix vergleichsweise schwache UV-
Absorption, wobei eine Absorptionsbande im Bereich von 350 bis 310 nm eine ausgeprägte
Struktur besitzt. Ein zweiter Übergang beginnt bei 290 nm und hat ein Maximum unterhalb
von 240 nm; die Absorptionsbande ist breit und zeigt eine Progression.[169] Die neue Spezies
10 Das CF3OOO-Radikal 101
zeigt dagegen ein UV-Spektrum mit einer unstrukturierten Bande mit einem Maximum bei
245 nm. Licht einer größeren Wellenlänge als 330 nm wird praktisch nicht mehr absorbiert.
Folglich kann nicht CF3O isoliert in O2-Matrix vorliegen, die UV-Spektren von CF3O in Ne-
MAtrix und CF3OOO sind in Abb. 10.2 gezeigt:
Abbildung 10.2: Vergleich der UV-Spektren von CF3O isoliert in Ne-Matrix und CF3O inSauerstoffmatrix (CF3OOO).
Die UV-Photolyse der Pyrolyseprodukte von CF3OC(O)OOCF3 folgt Gl. (10-3):
CF3OOO + h� � COF2 + FOO (10-3)
Dabei muß die OO-Gruppe im FOO nicht zwingend aus dem Trioxyradikal stammen,
sondern kann auch aus O2 der Matrix herrühren, das mit einem F-Atom reagiert. Auch ein
Isotopenexperiment mit CF316O18O18O kann keine Klärung bringen, da das genannte
Isotopomer aus CF3O in 18O2-Matrix dargestellt werden muß. Bei einer Photolyse ergäbe sich
dann in jedem Fall die Bildung von F18O18O.
IR-Spektrum
Ein Differenzspektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in
Sauerstoff-Matrix vor und nach der Photolyse (10 Minuten) mit UV-Licht mit Wellenlängen
größer als 280 nm ist in Abbildung 10.3 dargestellt.
Gemäß der berechneten Struktur besitzt das Trioxyradikal C1-Symmetrie, die
15 Fundamentalschwingungen der Rasse A sind alle IR- und Raman-aktiv. Die 6 Bindungen
im Radikal führen zu 6 Valenzschwingungen, die fünfatomige Kette im CF3OOO läßt die
10 Das CF3OOO-Radikal 102
unabhängige Variation von zwei Diederwinkeln zu, was im Schwingungsspektrum zu zwei
Torsionsschwingungen führt. Die übrigen 7 Fundamentalschwingungen sind
Deformationsschwingungen. Die Zuordnung erfolgt in Anlehnung an CF3O und diese
Gruppierung enthaltende Moleküle, sowie durch Vergleich mit dem berechneten
Erwartungsspektrum. Zudem ist das IR-Spektrum der isotopenmarkierten Spezies
CF316O18O18O aufgenommen worden, was die Identifizierung der Schwingungen, bei denen
sich die Bewegung der Atome im Molekül auf die O3-Einheit konzentriert, erleichtert. In
Tabelle 10.2 sind die Fundamentalschwingungen von CF3OOO isoliert in Sauerstoff-Matrix
zusammengestellt.
Tabelle 10.2: Fundamentalschwingungen von CF3OOO. Experimentelle IR-spektroskopische Datenvon CF3OOO isoliert in Ne-Matrix, sowie berechnete Bandenlagen und Intensitäten.
[a] B3LYP/6-311G(d,p)[b] relative integrierte Intensitäten [c] in km�mol-1
Im Trioxyradikal liegt eine starke OO-Bindung der endständigen Sauerstoffatome vor,
was durch die hohe Frequenz von �(OO) manifestiert wird. Die Schwingungen innerhalb der
CF3O-Einheit sind untereinander stark gekoppelt, werden aber auch durch die terminale O2-
Gruppe beeinflußt. Ersichtlich wird dies dadurch, daß die drei starken CF3O-
Valenzschwingungen um 1200 cm-1, sowie die intensitätsschwache Atmungs-schwingung des
CF3O-Tetraeders bei 895 cm-1, eine geringe Verschiebung erfahren, wenn die O2-Einheit mit18O markiert wird. Der Vergleich mit dem berechneten Spektrum macht zwei Sachverhalte
deutlich: (i) es ergibt sich eine sehr gute Übereinstimmung der gemessenen mit den
berechneten Intensitäten, und (ii) die O-O2-Bindung wird als zu schwach berechnet. Dies
spiegelt sich in den experimentellen Isotopenshifts der CF3O-Banden wider: Im Experiment
zeigen alle beobachteten Banden eine Rotverschiebung (CF316O16O16O�CF3
16O18O18O), was
eine Schwingungskopplung über die CF3O-O2 Bindung hinweg bedeutet. Die Theorie sagt
jedoch voraus, daß aufgrund der schwachen CF3O-O2-Bindung alle 9 CF3O-Schwingungen
(�2-�10) praktisch keine Verschiebung erfahren, wenn die terminale 16O2-Einheit durch 18O2
ersetzt wird.
10 Das CF3OOO-Radikal 104
Abbildung 10.3: Differenz-IR-Spektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in O2-Matrix vor und nach einer Photolyse mit UV-Licht mit � > 280 nm. Die bei der Photolyse abnehmenden Banden sind positiv dargestellt (*), die IR-
Absorptionen der bei der Photolyse entstehenden Produkte zeigen nach unten.
UV-Spektrum
Das UV-Spektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in O2-Matrix
ist in Abbildung 10.4 dargestellt:
10 Das CF3OOO-Radikal 105
Abbildung 10.4: UV-Spektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in O2-Matrix,Hintergrundspektrum: reine O2-Matrix.
Im UV-Spektrum von CF3OOO findet sich eine unstrukturierte, bei etwa 340 nm beginnende
Absorption mit einem Maximum bei 245 nm. Am kurzwelligen Ende des Meßbereichs deutet
sich eine weitere Absorption an.
Das UV-Spektrum von CF3OOO weist Ähnlichkeiten zu denen von FOO, ClOO und
HOO auf, die Form und Breite der beobachteten Absorptionsbande sind vergleichbar. Für
ClOO wird ein unstrukturierter Übergang bei 248 nm (Absorption von 270 bis 225 nm)[72, 190],
für FOO ein Übergang bei 205 nm[191] und für HOO ein Übergang bei 205 nm (Absorption bis
280 nm)[192-194] gefunden.
Tabelle 10.3: UV-Extinktionen von CF3OOO isoliert in O2-Matrix.
Die Verküpfung der OONO2-Gruppe mit einem Carbonylkohlenstoff bewirkt eine
offensichtliche Stabilisierung des Peroxynitrats; unklar bleibt die abweichende Stabilitätsfolge
bei erhöhter Temperatur.
Vergleicht man die berechneten Reaktionsenthalpien für die Spaltung des
Peroxynitrats ROONO2 � ROO + NO2 mit den Enthalpien für eine mögliche Folgereaktion
11 Vergleich der Peroxyradikale 113
ROO � R + O2 (Tab. 11.1), ist der weitere thermische Abbau eines entstandenen
Peroxyradikals deutlich stärker endotherm und tritt somit erst bei höherer
Reaktionstemperatur, als für die Erzeugung von ROO notwendig ist, auf. Eine Ausnahme ist
das Radikal SF5OO, in diesem Fall sagen die Berechnungen eine weniger endotherme
Spaltung von SF5OO, als die von SF5OONO2 voraus.
Bei der thermischen Erzeugung von CF3C(O)OO und CH3C(O)OO aus ihren
korrespondierenden Peroxynitraten werden mehrere Folgereaktionen beobachtet. Neben der
möglichen Abspaltung der Peroxyeinheit, ist vor allem die Bildung von CO und CF3OO bei
der thermischen Erzeugung von CF3C(O)OO bemerkenswert. Die Berechnungen zeigen, daß
eine Reaktion
RC(O)OO � ROO + CO (11-3)
für R = CF3 nahezu thermoneutral verlaufen soll, während für R = CH3 eine endotherme
Reaktion HR(11-4) = 64 kJ�mol-1 vorhergesagt wird (zum Vergleich: für R = F, wo
experimentell kein CO gefunden wird, ist HR(11-3) = 243 kJ�mol-1). Die geringen Ausbeuten
an Peroxyradikal und der große Anteil an Folgeprodukten wird durch die Berechnungen
bestätigt.
Thermische Spaltung von ROOOR
Als weitere Quellen für die Erzeugung von Peroxy- und Oxyradikalen kommen deren
korrespondierende offenkettige Trioxide in Betracht. In diesem Fall ist ein Bindungsbruch in
der OOO-Einheit des Moleküls bevorzugt und die zugänglichen Trioxide CF3OOOCF3,[154,
203-206] FC(O)OOOC(O)F[162, 163]
und CF3OC(O)OOOC(O)CF3 zeigen, mit Ausnahme von
CF3OOOCF3, eine noch größere Neigung zur Dissoziation als die Peroxynitrate:
ROOOR � ROO + RO (11-4)
Während CF3OOOCF3 auch bei einer Pyrolysetemperatur von 500°C nicht vollständig
dissoziiert, liegen die für die Spaltung der Trioxide CF3OC(O)OOOC(O)CF3 und
FC(O)OOOC(O)F erforderlichen Temperaturen bei 160°C. Die experimentellen Befunde
werden durch quantenmechanische Berechnungen bestätigt, eine Übersicht gibt Tabelle 11.3:
Tabelle 11.3: Berechnete[a] Reaktionsenthalpien für die Reaktion ROOOR � ROO + RO.
11 Vergleich der Peroxyradikale 114
R in ROOOR H0R(11-3) [kJ · mol-1] G0
R(11-3) [kJ · mol-1]
CF3 102.4 47.0
CF3C(O) 79.5 23.1
CF3OC(O) 65.1 8.7
FC(O) 60.4 6.6
SF5 60.1 3.7
[a] B3LYP/6-311G(d,p)
Für FC(O)OO und CF3OC(O)OO stellen die korrespondierenden Trioxide die besten
(thermischen) Quellen dar.
11.2 Die Strukturen von Peroxyradikalen
Die Aufklärung der Struktur eines Radikals durch hochauflösende IR-Spektroskopie
oder Rotationsspektroskopie ist bisher nur bei kleinen und meist hochsymmetrischen oder
linearen Spezies gelungen, experimentell ermittelte Strukturen sind für HOO[207-210] und
FOO[211-213] publiziert. Um ausschließlich Schwingungsübergänge zur Strukturaufklärung
heranziehen zu können, müssen die Schwingungsspektren isotopenmarkierter Spezies bekannt
sein, damit genügend Parameter für die Berechnung von Bindungswinkeln zur Verfügung
stehen. Gerade dieser Sachverhalt bereitet bei mehrfach fluorierten Molekülen
Schwierigkeiten, da Fluor als Reinisotop vorliegt. Aufgrund des Fehlens experimenteller
Strukturdaten werden im Rahmen dieser Arbeit die berechneten Strukturen der Radikale
miteinander verglichen.
Bei der Betrachtung von Peroxyradikalen ist der OO-Abstand, sowie die Geometrie
und Anzahl der stabilen Rotamere von besonderem Interesse. In allen fluorierten
Peroxyradikalen ergeben die Berechnungen als stabilste Form eine transoide Anordnung der
längsten im Molekül vorhandenen Kette von Atomen, was experimentell durch die
Zuordnung der gemessenen Schwingungsspektren bestätigt werden kann. In den
kohlenstoffhaltigen, fluorierten Peroxyradikalen liegen laut der Berechnungen vergleichbare
OO-Bindungslängen und OOC-Bindungswinkel vor. Die Änderung der elektronischen
Eigenschaften von R in ROO mit R = CF3, FC(O), CF3C(O), CH3C(O) und CF3OC(O) haben
demnach weder einen signifikanten, noch einheitlichen Einfluß auf die Struktur des Radikals.
Tendenziell ist beim Vorliegen einer cis-Konfiguration eine COO-Bindungswinkelaufweitung
11 Vergleich der Peroxyradikale 115
zu beobachten. In Tabelle 11.4 sind einige Parameter der berechneten Molekülgeometrien für
die im Rahmen dieser Arbeit untersuchten Peroxyradikale aufgeführt:
Tabelle 11.4: Berechnete[a] ausgewählte innere Koordinaten und relative Energien der stabilen Rotamere einiger Peroxyradikale.
Radikal R (O-O)[Å]
� (OOC)[b] (OOCX)[c]
Grel, berechnet[kJ·mol-1][d]
Gexp.[kJ·mol-1][d]
CF3OO 1.327 110.7 180 - -
trans-FC(O)OO 1.329 112.1 180 0.0 0.0
cis-FC(O)OO 1.331 115.3 0 2.9 1.2
trans-CF3C(O)OO 1.328 113.1 180 0.0 0.0
cis-CF3C(O)OO 1.372 119.6 0 65.9 6.3
trans-CH3C(O)OO 1.322 113.6 180 0.0 5.6
cis-CH3C(O)OO 1.323 114.4 0 0.1 0.0
t,t,t-CF3OC(O)OO 1.325 111.8 180 0.0 0.0
t,t,c-CF3OC(O)OO 1.328 116.2 0 4.7 6.1
t,c,t-CF3OC(O)OO 1.324 112.5 180 11.3 >10
CF3OOO 1.191 110.8 149 - -
FOO 1.185 111.7 - - -
SF5OO 1.246 114.5 - - -
[a] B3LYP/6-311G(d,p)[b] � (OOO) für CF3OOO, � (FOO) für FOO, � (OOS) für SF5OO.[c] X = F für CF3OO und FC(O)OO, X = C für CF3C(O)OO, CH3C(O)OO und CF3OOO,X = O für CF3OC(O)OO. [d] �Grel = G0-G0
(stabilstes Isomer)
Deutliche Unterschiede der Bindungsparameter fallen bei den Peroxyradikalen auf, die
eine vergleichsweise schwach gebundene OO-Einheit aufweisen. Die Schwäche der R-O2
Bindung spiegelt sich in einer entsprechend starken OO Bindung mit einem sehr kurzen OO
Abstand wider.
Aus den theoretischen Berechnungen lassen sich weitere physikalische Größen
ableiten, wie z. B. die Ladungsverteilung und Spindichten (Tab. 11.5). Aus den berechneten
Ladungsverteilungen läßt sich ein Zusammenhang mit den bekannten Stabilitäten ermitteln:
Wenn sich eine deutliche negative Partialladung (mehr als -0.15 e) in der O2-Einheit befindet,
bildet sich das Peroxyradikal leicht.
Tabelle 11.5: Berechnete[a] Ladungs- und Spinverteilung in einigen Peroxyradikalen.
[a] B3LYP/6-311G(d,p) [b] Atomkennzeichnung: R-O1-O2 [c] Ladung auf demdirekt mit der OO-Einheit verknüpften Atom [d] sd=Spindichte in atomaren Einheiten
Das Trioxyradikal CF3OOO ist in dieser Arbeit bei tiefen Temperaturen in Matrix
identifiziert worden. An den ungewöhnlichen Spindichten in Peroxyradikalen mit schwacher
Bindung zur O2-Einheit (wie in CF3OOO, FOO und SF5OO) erkennt man, daß die B3LYP-
Rechnungen in diesen Fällen weniger verläßlich werden.
11.3 Die UV-Spektren von Peroxyradikalen
Die im Rahmen dieser Arbeit erhaltenen UV-Spektren der Radikale CF3OO,
FC(O)OO, CF3C(O)OO und CH3C(O)OO zeigen Abweichungen zu den bisher publizierten
Spektren. In den Fällen, in denen die Ermittlung von Absorptionsquerschnitten möglich war,
sind diese signifikant niedriger als die veröffentlichten Daten. Dieser Sachverhalt wurde
bereits in der Charakterisierung der betreffenden Radikale CF3OO und FC(O)OO diskutiert.
Für CF3C(O)OO und CH3C(O)OO sind die in der Literatur verzeichneten Spektren - bedingt
durch einen hohen Rauschpegel - von mäßiger Qualität. Insbesondere die Verläufe an den
langwelligen Flanken der jeweiligen 22A''�X2A''-Übergänge, die in den kurzwelligen
11 Vergleich der Peroxyradikale 117
Bereich des Sonnenspektrums um 300 nm reichen, sind in dieser Arbeit viel genauer
vermessen. Für die Betrachtung der photochemischen Lebensdauer eines Peroxyradikals ist
dieser Bereich entscheidend, da das unterhalb der Ozonschicht vorhandene Sonnenspektrum
Strahlung mit � > 290 nm enthält. Für CF3C(O)OO und CH3C(O)OO konnten in den
Matrixexperimenten keine Absorptionsquerschnitte ermittelt werden, die eine quantitative
Aussage ermöglichen. Dennoch kann gefolgert werden, daß die fluorierten Peroxyradikale in
Anbetracht ihrer geringen Absorptionsquerschnitte bzw. gemessenen Extinktionen keinem
nennenswerten photolytischen Abbau in der unteren Atmosphäre unterliegen. Hingegen
nimmt innerhalb der Ozonschicht und in größeren Höhen die photochemische Lebensdauer
der Radikale aufgrund des wachsenden Anteils von kurzwelligem UV-Licht im
Sonnenspektrum ab.
Die UV-Spektren der Peroxyradikale weisen einige Gemeinsamkeiten auf. So ist der
angeregte Zustand 22A'', der durch Bestrahlen des Radikals mit UV-Licht von Wellenlängen
im Bereich von etwa 200 nm bis 300 nm erreicht wird, bei allen im Rahmen dieser Arbeit
untersuchten Peroxyradikalen dissoziativ, was in den Spektren durch das Auftreten einer
breiten, unstrukturierten Absorption erkennbar ist. In Abb. 11.1 sind die UV-Spektren einiger
[a] Der Index in Klammern bezeiht sich auf den betreffenden Übergang: (3) beschreibt 32A'�X2A''und (2) 22A''�X2A''. [b] keine Absorptionsquerschnitte ermittelbar, angegeben sind relativeExtinktionen bezogen auf lg(I/I0) = 1 bei �=�max (22A'�X2A'').
Auffällig ist die Rotverschiebung des Absorptionsmaximums in der Reihe CF3OO,
FC(O)OO, CF3OC(O)OO, CF3C(O)OO, CH3C(O)OO. Zur Erklärung können die in den Abb.
11.2 und 11.3 gegebenen schematischen MO-Diagramme (für CF3OO bzw. FC(O)OO)
herangezogen werden.
Der im UV-Spektrum aller untersuchten Peroxyradikale beobachtbare Übergang
22A''�X2A'' entspricht der Anregung eines Elektrons aus dem mit �OO bezeichneten MO in
das SOMO �*OO. Beide beteiligten Orbitale besitzen A''-Symmetrie, ihr Energieinhalt und
damit die Energiedifferenz ist somit nur von anderen im Peroxyradikal vorhandenen a''-
Orbitalen abhängig (nicht in Abb. 11.2 eingezeichnet).
11 Vergleich der Peroxyradikale 120
�OO
�
��
��
�C
�OO
�CO
�OO
��CO
��OO
��OO
n
a‘
a‘‘
a‘‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘‘
a‘‘
R0
E
R R
CF3 CF3OO O2
Abbildung 11.2: Schematisches MO-Diagramm von CF3OO.[77] Dargestellt ist die Konstruktion
der MOs bei Überlappung eines -Orbitals von CF3 (linker Bildteil) mit den MOs des O2-Moleküls (rechter Bildteil) bei der Bildung von CF3OO (Mitte). Die Abbildung ist nicht maßstabsgetreu.
Im Falle des CF3OO sind diese anderen a''-Orbitale voll besetzte MOs, die vornehmlich die
"freien Elektronenpaare" der Fluoratome enthalten. Somit liegen sie auf der Energieskala
unterhalb des für die Eigenschaften der chromophoren OO-Einheit relevanten Bereichs und
beeinflussen somit die energetischen Lagen der am Übergang beteiligten Orbitale praktisch
nicht. Der Energieunterschied (bei vertikaler Anregung) zwischen �OO und �*OO im CF3OO
entspricht der Energie, die ein UV-Photon mit � = 209 nm besitzt.
Betrachtet man nun das FC(O)OO-Radikal, so müssen die Einflüsse der a''-Orbitale
berücksichtigt werden: Das �-Elektronensystem der C=O-Bindung wird durch ein bindendes
und gefülltes (hauptsächlich am Carbonylsauerstoff lokalisiertes) � a''-Orbital und ein
unbesetztes, antibindendes (hauptsächlich am Carbonylkohlenstoff lokalisiertes) �* a''-Orbital
beschrieben. Das letztgenannte Orbital zeigt eine große Wechselwirkung mit den am
22A''�X2A'' Übergang beteiligten Orbitalen, die Bildung der Molekülorbitale im FC(O)OO
aus FC(O) und O2 ist in Abb. 11.3 schematisch dargestellt.
11 Vergleich der Peroxyradikale 121
�OO
�
��
��
�C
a‘
a‘‘
a‘‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘
a‘‘
a‘‘
a‘‘��C=O
a‘‘
E
RR0
FCO FC(O)OO O2
R
Abbildung 11.3: Schematisches MO-Diagramm von FC(O)OO.[77] Dargestellt ist die Konstruktion der MOs bei Überlappung eines -Orbitals von FC(O) (linker Bildteil) mit den MOs des O2-Moleküls (rechter Bildteil) bei der Bildung von FC(O)OO (Mitte) unter Berücksichtigung des Orbitals *
C=O. Die Abbildung ist nicht maßstabsgetreu.
Als Folge dieser Wechselwirkung wird das SOMO energetisch stärker abgesenkt, was
zu einer Verkleinerung der Energiedifferenz von �OO und �*OO führt. Folglich ist der
22A'�X2A'' Übergang bei längeren Wellenlängen im UV-Spektrum beobachtbar. Die in der
Reihe FC(O)OO, CF3OC(O)OO, CF3C(O)OO und CH3C(O)OO weitere zunehmende
Rotverschiebung des 22A''�X2A'' Übergangs läßt sich durch eine zunehmende Stärke der
Beeinflussung durch das antibindende a''-Orbital der Carbonylgruppe erklären.
Auch die beobachteten 12A'�X2A'' und 32A�X2A'' Übergänge in Peroxyradikalen
lassen sich durch die oben gezeigten MO-Schemata erklären. Zwei Faktoren beeinflussen die
energetische Lage der beteiligten Orbitale und somit deren Energieunterschied:
(i) Die Absenkung des SOMO durch die Nachbarschaft der OO-Einheit zu einem �-
Elektronensystem verringert die Anregungsenergien und führt zu einer Rotverschiebung der
UV-Absorptionen. So wird der 12A'�X2A''-Übergang des CH3OO bei 7375 cm-1 beobachtet,
der des Radikals CH3C(O)OO liegt bei 5562 cm-1.[178]
11 Vergleich der Peroxyradikale 122
(ii) Die energetischen Lagen der beiden höchsten voll besetzten a'-Orbitale werden
durch die Stärke der Wechselwirkungen des Orbitals �C mit den a'-Orbitalen �OO und �*OO
beeinflußt. Eine Erniedrigung der Elektronegativität des an die OO-Einheit gebundenen
Kohlenstoffs bedeutet eine höhere Energie von �C. Folglich verstärkt sich die
Wechselwirkung mit dem �*OO-Orbital, die mit dem �OO-Orbital des O2-Moleküls nimmt ab.
Dies führt zu einer fortschreitenden energetischen Absenkung des höchsten besetzten a'-
Orbitals im ROO-Radikal und zu einer Frequenzzunahme des 12A'�X2A'' Übergangs
innerhalb der Reihe CF3OO<CH3OO<C2H5OO. Ein Überblick (Tabelle 11.7) über die
12A'�X2A'' Übergänge einiger Peroxyradikale bestätigt diese Argumentation:
Tabelle 11.7: Absorptionsbanden der 12A'�X2A'' Übergänge von Peroxyradikalen. (Die Werte in Klammern geben �(OO) im angeregten Zustand an.)
Das nächste, energetisch tiefer liegende a'-Orbital, daß am 32A'�X2A'' Übergang
beteiligt ist, wird mit abnehmender Elektronegativität des an der OO-Einheit gebundenen
Kohlenstoffs zunehmend energiereicher, der Abstand zum SOMO verringert sich und der
32A'�X2A'' Übergang zeigt sich im UV-Spektrum bei längeren Wellenlängen. Diese
Rotverschiebung zeigt sich in den UV-Spektren der Radikale CF3OC(O)OO, CF3C(O)OO und
CH3C(O)OO, in denen der 32A'�X2A'' Übergang bei 199, 208 bzw. 209 nm
(Ansorptionsmaximum) beobachtet wird.
Alle betrachteten Übergänge sind erlaubt, da die Spinmultiplizität des Radikals
erhalten bleibt. Bei gleichzeitiger elektronischer und vibronischer Anregung des 12A'�X2A''-
Übergangs (12A'�X2A''-Progressionen) verschwinden die Übergangswahr-scheinlichkeiten
weder bei der Anregung einer A'- noch einer A''-Schwingung.
11 Vergleich der Peroxyradikale 123
12 Das CF3OCO-Radikal 124
12 Das CF3OCO-Radikal
12.1 Einführung
Unter atmosphärischen Bedingungen ist die Bildung von CF3OCO auf zwei Wegen
möglich. Der Abbau des teilfluorierten Ethes CF3OCH3 wird durch OH-Radikale initiiert und
verläuft über Oxidationsprozesse am wasserstofftragenden Kohlenstoff bis zur Bildung des
Carbonylradikals CF3OCO:[25, 79]
CF3OCH3 + OH � CF3OCH2 + H2O (12-1)
CF3OCH2 + O2 ( + M ) � CF3OCH2OO ( + M ) (12-2)
CF3OCH2OO + NO � CF3OCH2O + NO2 (12-3)
CF3OCH2O + O2 � CF3OC(O)H + HOO (12-4)
CF3OC(O)H + OH � CF3OCO + H2O (12-5)
Dieser Weg ist auf Trifluormethyl-Kohlenwasserstoffether beschränkt. Sofern eine CF3-
Gruppe im Molekül vorhanden ist, vermögen jedoch auch andere CFH-Verbindungen und
FCKWs CF3O-Radikale zu bilden, die mit Kohlenmonoxid reagieren.[115-117] Wird die
Reaktionswärme durch einen Stoßpartner aufgenommen, kann sich das Radikal CF3OCO
bilden:
CF3O + CO ( + M ) � CF3OCO ( + M ) (12-6)
Über atmosphärische Reaktionen von CF3OCO ist bisher nichts bekannt. In jüngster
Zeit ist jedoch die Existenz des Peroxynitrates CF3OC(O)OONO2 nachgewiesen worden.[79]
Daher liegt die Vermutung nahe, daß die elementaren Reaktionen des CF3OCO, insbesondere
die Addition von O2 unter Bildung des korrespondierenden Peroxyradikals CF3OC(O)OO
(Kap. 7), analog zu denen der anderen fundamentalen Radikale CF3, CF3CO und FCO
verlaufen. Betrachtet man die CF3O-Gruppe als "Pseudolfluor", so ist eine chemische
Ähnlichkeit von CF3OCO zu FCO zu erwarten.
12 Das CF3OCO-Radikal 125
CF3OCO wurde in der Literatur bereits als Intermediat einiger Reaktionen unter
Laborbedingungen postuliert,[113-115, 168] unter anderem in dem in Kap. 7.1 formulierten Prozeß
der CF3O-katalysierten Oxydation von CO zu CO2.
Im Rahmen dieser Arbeit wurde das Radikal CF3OCO erstmals gezielt und nahezu
quantitativ erzeugt und umfassend spektroskopisch charakterisiert. Die experimentellen
Ergebnisse werden mit quantenchemisch berechneten Daten verglichen.
12.2 Die Struktur des CF3OCO-Radikals
Die fünfatomige Kette im Trifluormethoxycarbonylradikal erlaubt die unabhängige
Einstellung (0 bzw. 180°) zweier Diederwinkel und führt somit zu vier möglichen Rotameren.
Zunächst ergeben DFT-Berechnungen (B3LYP/6-311G(d,p)) das Vorliegen von nur zwei
energetische Minima auf der Potentialhyperfläche, die durch die trans- bzw. cis-Anordnung
der C-O-C=O Einheit bei trans-Anordnung der F-C-O-C Kette beschrieben werden.
Geometrien mit cis-Konfiguration der F-C-O-C Kette besitzen eine imaginäre
Schwingungsfrequenz und stellen somit Übergangszustände dar. Im folgenden werden
trans,trans-CF3OCO durch trans-CF3OCO und trans,cis-CF3OCO durch cis-CF3OCO
abgekürzt. Die Struktur der beiden stabilen Rotamere ist in Abbildung 12.1 dargestellt, die
Bindungslängen und -winkel beider Isomere sind in Tabelle 12.1 zusammengefaßt. Das trans-
CF3OCO ist laut der Berechnungen um 11.8 kJ·mol-1 stabiler (Hrel) als das cis-CF3OCO.
trans,trans-CF3OCO trans,cis-CF3OCO
Abbildung 12.1: Berechnete Strukturen von trans,trans-CF3OCO und trans,cis-CF3OCO.
Tabelle 12.1: Berechnete[a] Geometrie von trans- und cis-CF3OCO
12 Das CF3OCO-Radikal 126
trans-CF3OCO cis-CF3OCO
Koordinate [Å bzw. °] [Å bzw. °]
R (CF1) 1.327 1.323
R (CF2) 1.333 1.333
R (CO) 1.387 1.405
R (OC) 1.372 1.353
R (C=O) 1.171 1.176
� (O=C-O) 124.8 130.4
� (COC) 116.0 119.2
� (OCF1) 106.6 106.4
� (OCF2) 111.4 111.1
(COCO) 180.0 0.0
(F1COC) 180.0 180.0
(F2COC) ±60.7 ±60.8
Hrel 0.0 kJ·mol-1 11.8 kJ·mol-1
[a] B3LYP/6-311G(d,p)
Auffällig sind zum einen die Aufweitungen der Winkel �(O=C-O) und �(COC) beim
Übergang von trans- zu cis-CF3OCO, und zum anderen die Konstanz des C=O Abstands in
der Carbonylgruppe im Vergleich zum CF3OC(O)OO-Radikal (ca. 1.39 Å), obwohl das
ungepaarte Elektron im CF3OCO am Kohlenstoff der Carbonylgruppe, im CF3OC(O)OO am
endständigen Sauerstoff der Peroxy-Einheit lokalisiert ist.
12.3 Die Erzeugung von CF3OCO-Radikalen
Die Erzeugung des CF3OCO-Radikals erfolgt durch Reaktion von CF3O-Radikalen
mit CO. Hierbei werden die CF3O-Quellen CF3OC(O)OOC(O)OCF3 bzw. CF3OC(O)OOCF3
in einem Überschuß CO einer Niederdruckpyrolyse unterworfen und anschließend bei 16 K
als CO-Matrix isoliert. Die Bedingungen der Niederdruckpyrolyse unterdrücken bimolekulare
Reaktionen, insbesondere solche, bei denen ein Stoßpartner (M) zur Aufnahme der
Reaktionswärme benötigt wird. Die Bildung von CF3OCO erfolgt demnach nicht in der
12 Das CF3OCO-Radikal 127
Gasphase, sondern erst während des Kondensationsprozesses des CF3O/CO-Gemisches auf
dem gekühlten Matrixträger:
CF3OC(O)OOCF3 � 2 CF3O + CO2 (12-7)bzw.CF3OC(O)OOC(O)OCF3 � 2 CF3O + 2 CO2 (12-8)
[CF3O + CO]Matrix � CF3OCO (12-9)
Die beschriebene Darstellung liefert nahezu quantitative Ausbeuten an CF3OCO, lediglich
Spuren von COF2 und CF3OOCF3 sind nachzuweisen.
12.4 Die spektroskopische Charakterisierung von CF3OCO
Die Untersuchung der erhaltenen Matrices erfolgt schwingungsspektroskopisch. Das
IR-Spektrum der Pyrolyseprodukte zeigt neue Banden im Bereich der C=O und C-F
Valenzschwingungen, sowie die charakteristischen IR-Absorptionen von CO2 (und geringe
Anteile an COF2 und CF3OOCF3). Zur Identifizierung der unbekannten Spezies wird eine
Photolyse der erhaltenen Matrix mit einer Quecksilberhochdrucklampe vorgenommen. Dabei
verschwinden die unbekannten IR-Absorptionen gleichmäßig und die Absorptionen von CO2
nehmen zu. Weiterhin bildet sich äquimolar zu der Zunahme an CO2 das CF3-Radikal, das
durch sein bekanntes IR-Spektrum identifiziert wird.[214] In geringerem Umfang ist die
Bildung von COF2 und FCO zu beobachten. Aus der Stoffbilanz der photolytischen Spaltung
der neuen folgt, daß es sich um CF3OCO handeln muß:
ca. 5 % ca. 95 %
COF2 + FCO � CF3OCO + h� � CF3 + CO2 (12-10)
Bemerkenswert ist in diesem Zusammenhang, daß die bei der Photolyse entstehenden CF3-
Radikale, die noch Überschußenergie enthalten, nicht mit CO reagieren, obwohl für die
Reaktion
CF3 + CO � CF3CO (12-11)
laut Theorie ein exothermer Reaktionsverlauf erwartet wird.[65, 93] Die im Rahmen dieser
Arbeit durchgeführten theoretischen Berechnungen sagen für die Reaktion nach Gl. (12-11)
HR = - 28.6 kJ·mol-1 (also ebenfalls exotherm) voraus; GR spricht mit + 12.6 kJ·mol-1 für
eine (unter Standardbedingungen) endergonische Reaktion.
12 Das CF3OCO-Radikal 128
IR-Spektrum
Das Differenz-IR-Spektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in
CO-Matrix vor und nach der Photolyse ist in Abbildung 12.2 dargestellt.
Abbildung 12.2: Differenz-IR-Spektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in ���� ��CO-Matrix vor und nach der Photolyse mit UV-Licht einer Quecksilber-
hochdrucklampe.
Im C=O Streckschwingungsbereich sind zwei Absorptionen, die zwei Rotamere
vermuten lassen. Im C-F Valenzschwingungsbereich und im Bereich der Deformationen weist
das Spektrum jedoch nur wenige Absorptionen auf, die für nur ein Isomer sprechen. Der
Widerspruch ist durch eine starke Fermi-Resonanz (zwischen �(C=O) und einem
Kombinationston) im trans-CF3OCO bedingt, die zu zwei starken Banden im C=O-
Streckschwingungsbereich führt. Wird bei der Erzeugung des Trifluormethoxy-
carbonylradikals 13CO als Matrixmaterial und Reaktionspartner verwendet, bildet sich das
Isotopomer 12CF3O13CO, das im C=O Bereich neben der erwarteten Rotverschiebung der
Banden ein vom natürlichen Isotopomer deutlich verschiedenes Intensitätsverhältnis der
12 Das CF3OCO-Radikal 129
beiden Banden aufweist. Die IR-Spektren beider Spezies sind in Abbildung 12.3 vergrößert
gezeigt.
Abbildung 12.3:
Fermi-Resonanz imtrans,trans-CF3OCO.Die durchgezogeneLinie zeigt das IR-Spektrum von12CF3O12CO, diegepunktete Linie dasIR-Spektrum von12CF3O13CO, isoliertin CO-Matrix.
Die Resonanz von
�1 mit dem Kombinationston �4 + �5 führt zu einer relativen Blauver-schiebung des
Kombinationstones und Intensitätsgewinn auf Kosten des rotverschobenen Grundtons. Die
Wechselwirkung der Schwingungen ist auf Schwingungen der gleichen Rasse begrenzt und ist
um so ausgeprägter, je näher die (ungestörten) Bandenlagen zueinander liegen. Bei der
gezielten Isotopenmarkierung erfährt der 13C=O Grundton eine Verschiebung der Wellenzahl
zu niedrigeren Werten, während der Kombinationston �4 + �5 kaum beeinflußt wird. Folglich
ist die Fermi-Resonanz im Falle des CF3O13CO schwächer ausgeprägt.
Die beobachteten Bandenlagen der in Resonanz stehenden Schwingungen können
nach Gl. 12-12 korrigiert werden (I=integrierte Bandenintensitäten):[215]
��
���
�
�
���
11
2121
. ss
korr ���� mit 2
1
II
s � (12-12)
Man erhält für natürliches CF3OCO �1 bei 1875 cm-1 und �4+5 bei 1883 cm-1, für das
Isotopomer CF3O13CO gilt �1= 1837 cm-1 und �4+5=1865 cm-1. Diese korrigierten Werte
zeigen eine deutlich bessere Übereinstimmung mit den berechneten Bandenlagen: Die
Isotopenverschiebungen �(CF3OCO- CF3O13CO) für �1 betragen 33 cm-1 (beobachtet), 38
12 Das CF3OCO-Radikal 130
cm-1 (korrigiert) und 44 cm-1 (berechnet), sowie für den Kombinationston �4+5 12 cm-1
(beobachtet), 18 cm-1 (korrigiert) und 18 cm-1 (berechnet).
Das cis-CF3OCO ist weder bei Pyrolysetemperaturen von 310°C noch bei 460°C
nachweisbar, was durch die Bildung des CF3OCO erst während des Kondensationsprozesses
erklärt wird. Das thermische Gleichgewicht zwischen cis- und trans-CF3OCO liegt bei tiefen
Temperaturen völlig auf der Seite des stabileren trans-CF3OCO:
RTG
trans
cis enn �
�
� (12-13)
Mit G = 11.8 kJ�mol-1 (berechneter Wert) beträgt das Isomerenverhältnis etwa 1:11 (T=583
K, Entstehung des CF3OCO während der Pyrolyse) bzw. 1:2�1015 (T=40 K, Entstehung des
CF3OCO während des Kondensationsprozesses).
Den Berechnungen folgend, zeigt trans-CF3OCO Cs-Symmetrie, die irreduzible
Darstellung der Schwingungsfreiheitsgrade mit IR- und Ramanaktivitäten lautet:
�vib = 10 A' (IR, Ra p) + 5 A'' (IR, Ra dp) (12-14)
Die 15 Schwingungsfreiheitsgrade verteilen sich auf 6 Valenzschwingungen, 7
Deformationsschwingungen (ermöglicht 5 Deformationen des CF3O-Tetraeders, 2
Deformationen der C-O-C und O-C=O Winkel) und 2 Torsionsschwingungen der fünfatomige
Kette F-C-O-C=O. Aufgrund der an den Schwingungen beteiligten vergleichbar schweren
Atomen, die ebenso vergleichbar fest gebunden sind, ergibt sich eine starke Mischung der
Schwingungen, so daß die beobachteten Fundamentalschwingungen nur annähernd mit
inneren bzw. Symmetrie-Koordinaten zu beschreiben sind. Eine Übersicht über die
beobachteten IR-Absorptionen von trans-CF3OCO gibt Tabelle 12.2 .
Tabelle 12.2: IR-Absorptionen des CF3OCO Radikals isoliert in CO-Matrix. Experimentelle IR- spektroskopische Daten von CF3OCO (natürlich und des Isotopomers CF3O13CO) isoliert in Ne-Matrix, sowie berechnete Bandenlagen und Intensitäten.
1901 11 1929 1879 1911 �4 + �5 A' Fermi-Res. mit �1
12 Das CF3OCO-Radikal 131
1857 16 1927 36 1824 1883 �1 A' � C=O
1280 83 1282 84 1279 1282 �2 A' �a CF3
1236 45 1230 73 1235 1230 �11 A'' �a CF3
1203 100 1191 100 1201 1189 �3 A' �s CF3 / CO o.p.
997 52 1026 53 988 1015 �4 A' � OC
902 11 903 8.6 894 896 �5 A' �s CF3 / CO i.p.
720 1.3 718 1.9 719 717 �6 A' �s CF3 / OC=O i.p.
612 0.46 612 �12 A'' �a CF3 612 0.8
608 0.92 608
605 �7 A' �a CF3
475 0.5 472 0.56 472 469 �8 A' �s CF3 / OC=O o.p.
430 0.02 430 �13 A'' � CF3
418 2.9 415 0.56 415 413 �9 A' � CF3
191 0.40 190 �10 A' � COC
187 0.90 182 �14 A'' COCO
86 0.00 86 �15 A'' COCF3
[a] B3LYP/6-311G(d,p) [b] relative integrierte Intensitäten [c] Intensität in km·mol-1
Die Zuordnung erfolgte durch Vergleich mit bekannten Verbindungen wie CF3O[169] und
CF3ONO2 (Kap. 14), sowie auf Grundlage der berechneten Fundamentalschwingungen. Die
Betrachtung der CF3O-Gruppe als "Pseudofluor" spiegelt sich auch im Schwingungsspektrum
wider, so daß die Wellenzahlen von �(C=O) im trans-CF3OCO (1857 cm-1) und FCO (1855
cm-1), beide isoliert in CO-Matrix, nahezu identisch sind. Sogar �(O-C) bei 997 cm-1 läßt sich
mit � (CF) von FCO bei 1018 cm-1 vergleichen.
UV-Spektrum
Die CF3OCO enthaltende Matrix zeigt ein nicht strukturiertes UV-Spektrum mit
beginnender Absorption bei Wellenlängen unterhalb von 380 nm (Abb. 12.4):
12 Das CF3OCO-Radikal 132
Abbildung 12.4: Differenz-UV-Spektrum der Pyrolyseprodukte von CF3OCO isoliert in CO-��������Matrix vor und nach der Photolyse mit einer Quecksilberhochdrucklampe.
Es zeigen sich zwei Absorptionsmaxima im Meßbereich von 375-220 nm (bei
kürzeren Wellenlängen ist die Streuung durch das Matrixmaterial CO sehr groß und führt zu
starkem Rauschen), wobei das erste Absorptionsmaximum bei 327 nm, das zweite, deutlich
stärkere Maximum bei 226 nm liegt. Das in Abbildung 12.4 gezeigte UV-Spektrum wurde als
Differenzspektrum der Pyrolyseprodukte von CF3OC(O)OOCF3 isoliert in CO-Matrix vor
und nach Photolyse mit einer Quecksilberhochdrucklampe registriert.
Die UV-Spektren von CF3OCO und FCO[216, 217] unterscheiden sich deutlich, demnach
ist in der Beschreibung der UV-Spektren ist das Modell, CF3O als Pseudo-fluoratom
anzusehen, nicht ausreichend. Bei CF3OCO ergibt sich ein Zerfallsweg unter Bildung von
CO2 und CF3-Radikalen, der offensichtlich die durch UV-Licht anregbaren Zustände
dissoziativ werden läßt, so daß im Spektrum nur strukturlose Banden beobachtet werden. Für
FCO erfordert die Abspaltung des Fluoratoms mehr Energie, als das zur Messung des UV-
12 Das CF3OCO-Radikal 133
Spektrums benutzte Licht besitzt. Die beobachteten elektronischen Übergänge des FCO sind
nicht dissoziativ und zeigen ausgeprägte Schwingungsprogressionen.[216, 217]
Zur Zuordnung des UV-Spektrums von CF3OCO ist ein Vergleich mit dem in der
Literatur beschriebenen Radikal CH3OCO[59, 218] naheliegend. Die energetisch tiefliegen-den
gefüllten Orbitale der CF3-Gruppe, die hauptsächlich an den Fluoratomen lokalisiert sind,
sollten keinen signifikanten Einfluß auf das �-System der chromophoren C=O-Ein-heit haben.
Die CH3-Gruppe verfügt über keine �-Orbitale, die mit dem chromophoren �-System in
Wechselwirkung treten können.
Für CH3OCO sind Berechnungen publiziert, die den Grundzustand und die angeregten
Zustände charakterisieren:[59, 218] Im Grundzustand mit Cs-Symmetrie befindet sich das
ungepaarte Elektron im wesentlichen am Carbonylkohlenstoff und besetzt ein �-Orbital, so
daß sich ein symmetrischer Zustand X2A' ergibt. Eine Anregung des Radikals durch Anheben
eines der nächst fester gebundenen Elektronen in einem a''-Orbital (�-Orbital der C=O
Bindung) in das halb besetzte �-Orbital führt zum 22A'' Zustand. Ebenfalls an der C=O
Bindung beteiligt ist ein zur Molekülspiegelebene symmetrisches �-Orbital. Wird also ein
Elektron aus diesem in das halb besetzte �-Orbital angeregt, ist der 32A'-Zustand realisiert.
Den Berechnungen folgend, ist aber auch eine Anregung des ungepaarten Elektrons selbst in
ein energetisch höheres, unbesetztes Orbital möglich. Die geringste Anregungsenergie
erfordert hierbei der Übergang in ein antibindendes a''-�*-Orbital, der zum 12A''-Zustand
führt.
Die Zuordnung der angeregten Zustände für CH3OCO wird auf CF3OCO übertragen:
Die verhältnismäßig intensitätsschwache Absorption, die ihr Maximum bei 327 nm aufweist,
wird dem 12A''�X2A' Übergang zugeordnet. Die Bande, die ihr Intensitätsmaximum bei 226
nm zeigt, ist wesentlich stärker, als die Berechnungen (für CH3OCO) für einen einzelnen
Übergang vorhersagen. Auch wenn man die Übergangs-momente und die damit verbundenen
Intensitäten der elektronischen Übergänge von CH3OCO und CF3OCO nicht vergleichen
kann, ist die Zuordnung dieser Absorption als Überlagerung zweier Übergänge 22A''�X2A'
und 32A'�X2A' naheliegend. Die Bande ist verhältnismäßig breit und zeigt im Gegensatz
zum 12A''�X2A' Übergang keine Gauß-kurvenkontur. In Tabelle 12.3 sind die UV-
spektroskopischen Daten für CF3OCO und CH3OCO dargestellt, zusammen mit berechneten
Daten für das Carbonylradikal HOCO.
Tabelle 12.3: Berechnete und gemessene elektronische Übergänge der Radikale CF3OCO,��CH3OCO und HOCO.
[a] Lage in nm, Intensität in 1020cm2 [b] Lage in nm; in Klammern: Intensität gemessen in1020cm2 bzw. berechnetes Übergangsmoment (CCSD(T)/cc-pVDZ, MRCI-CASSCF) in Debye, sieheReferenz [218] [c] Lage in nm; in Klammern: berechnetes Übergangsmoment (CCSD(T)/cc-pVDZ,MRCI-CASSCF) in Debye, siehe Referenz [219]
Tabelle 12.4: UV-Absorptionsquerschnitte von CF3OCO.
Auffällig sind hierbei die langen OO-Einfachbindungen (1.423 Å), der kleine Winkel (102.5°)
zwischen den einfach gebundenen Sauerstoffatomen in der Carbonat-Einheit (damit
verbunden sind die großen Winkel zwischen einfach und doppelt gebundenem Sauerstoff in
dieser Einheit), der fast perfekte Tetraederwinkel an dem an Kohlenstoff gebundenen
Sauerstoff der Trioxykette (109.4°) und der relativ spitze Winkel am zentralen Sauerstoff der
O3-Einheit (106.7°). Man findet eine geringe Abweichung von der idealen transoiden
Anordnung (=180°) in der Kette O-C-O-O (177.4°) und einen Diederwinkel bezüglich der
beiden OO-Bindungen in der O3-Einheit von je 90.2°, wie sie auch in anderen vergleichbaren
Verbindungen wie CF3OOCF3, FC(O)OOC(O)F oder CF3OONO2 angetroffen wird. Die
anderen Strukturparameter liegen ebenfalls in dem für C, F, O-Verbindungen typischen
Bereich.
Ein Vergleich mit dem Trioxid FC(O)OOOC(O)F zeigt, daß eine CF3O-Gruppe als
"Pseudofluor" angesehen werden kann. Die berechneten Strukturen weisen Gemeinsamkeiten
auf: Auch für FC(O)OOOC(O)F in Form seines stabilsten Rotamers, in dem die F-C-O-O-O-
C-F Kette mit Ausnahme der zentralen Diederwinkel transoid vorliegt, werden folgende
Werte für R (OO), � (OCO), � (COO) und � (OOO) berechnet: 1.427 Å, 103.8°, 109.6° und
106.6°. Die Diederwinkel F-C-O-O und C-O-O-O sind ebenfalls vergleichbar, sie liegen bei
176.9° bzw. 90.0°.
13.4 Die spektroskopische Charakterisierung von Bistrifluor-
methyltrioxydicarbonat CF3OC(O)OOOC(O)OCF3
13 CF3OC(O)OOOC(O)OCF2 141
Von reinem CF3OC(O)OOOC(O)OCF3 wurden IR Spektren sowohl in der Gas-phase
als auch isoliert in Edelgasmatrix aufgenommen. Darüber hinaus erfolgte die
Charakterisierung anhand eines bei -196°C aufgenommenen Ramanspektrums, eines Matrix-
UV-Spektrums und anhand von NMR-Daten.
Schwingungsspektren
Nach Aussage der quantenmechanischen Rechnungen besitzt
CF3OC(O)OOOC(O)OCF3 C2-Symmetrie und entsprechend lautet die irreduzible Darstellung
der Schwingungsfreiheitsgrade mit IR- und Ramanaktivitäten:
�vib = 23 A (IR, Ra p) + 22 B (IR, Ra dp) (13-14)
Demnach sind alle Schwingungen sowohl IR- als auch Raman-aktiv. Die beiden CF3OC(O)O-
Molekülteile sind symmetrieäquivalent: Jede Schwingungsbewegung innerhalb eines
Molekülteiles kann im anderen Molekülteil entweder in Phase oder mit gegenläufiger Phase
ablaufen, so daß beispielsweise die C=O Streckschwingungen parallel (in phase) oder
gegeneinander (off phase) ausgeführt werden. Demnach verteilen sich die 45
Schwingungsfreiheitsgrade auf 2·8 Valenzschwingungen, 2·9
Winkeldeformationsschwingungen, 2·1 out-of-plane Schwingungen und 2·4 Torsions-
Schwingungen, sowie die Deformation des zentralen OOO-Bindungswinkels auf. Die
Kopplung zwischen in phase und off phase Schwingungen sinkt, je weiter die jeweiligen
Bewegungen der bei den Schwingungen beteiligten Atome von der Molekülmitte entfernt
sind. Demnach ist nur bei den Schwingungen mit Beteiligung der OOO-Einheit mit einer
deutlichen (energetischen) Trennung von in phase (Rasse A) und off phase (Rasse B)
Schwingungen auszugehen. Mit Ausnahme der C=O Streckschwingungen zeigen die im
Molekül vorhandenen Bindungen ähnliche Stärken und die daran beteiligten Atome haben
vergleichbare Massen, was zu einer starken Vermischung der Schwingungs-koordinaten führt,
daher wird von einer Beschreibung der einzelnen Schwingungen abgesehen. In den
Abbildungen 13.2 bis 13.4 sind ein Tieftemperatur-Ramanspektrum des festen reinen
CF3OC(O)OOOC(O)OCF3, sowie IR-Spektren von gasförmigem und matrixisoliertem
Bistrifluormethyltrioxydicarbonat gezeigt.
13 CF3OC(O)OOOC(O)OCF2 142
Abbildung 13.2: Tieftemperatur-Ramanspektrum von CF3OC(O)OOOC(O)OCF3 im festen Zustand bei -196°C.
13 CF3OC(O)OOOC(O)OCF2 143
Abbildung 13.3: IR-Spektrum von CF3OC(O)OOOC(O)OCF3 isoliert in Neon-Matrix. Die mit p gekennzeichneten Banden stammen von wenigen mol-% CF3OC(O)OOC(O)OCF3.
Wie erwartet findet man selbst im Falle des Matrix-IR Spektrums die C=O
Streckschwingungen als nur eine nicht aufgelöste Bande mit einer Schulter an der
kurzwelligen Flanke. Die Berechnungen sagen voraus, daß die symmetrische
Carbonylstreckschwingung �1 mit geringer IR-Intensität nur etwa 4 cm-1 oberhalb der im IR-
Spektrum sehr starken Bande von �24 (antisymmetrische C=O Streckschwingung) zu finden
ist. Die Bande bei 1862 cm-1 stammt von einer Verunreinigung (CF3OC(O)OOC(O)OCF3).
Die Übereinstimmung von theoretisch berechnetem und experimentellem Spektrum
sowohl in Bandenposition als auch in Bandenintensität ist exzellent und schließt das
Vorliegen einer anderen Spezies als CF3OC(O)OOOC(O)OCF3 aus. Auch der Vergleich mit
dem korrespondierenden Peroxid zeigt, daß beide Verbindungen einen fast identischen
Aufbau besitzen müssen, da sich ihre IR-Spektren sehr ähneln. Zur Unterscheidung beider
Spezies läßt sich im IR-Spektrum vor allem die Bande bei 974 cm-1 heranziehen. Das
Peroxydicarbonat weist eine vergleichbare Bande bei 989 cm-1 auf, so daß in einer
Mischung der beiden Substanzen diese Banden deutlich voneinander getrennt detektiert
13 CF3OC(O)OOOC(O)OCF2 144
werden können. Im Ramanspektrum zeigt CF3OC(O)OOOC(O)OCF3 die stärkste Bande bei
923 cm-1, die zwei stärksten Banden des Peroxids CF3OC(O)OOC(O)OCF3 liegen bei 934
und 873 cm-1.[114]
Abbildung 13.4: IR-Spektrum von CF3OC(O)OOOC(O)OCF3 in der Gasphase bei 19°C.
Das IR-Spektrum von gasförmigem CF3OC(O)OOOC(O)OCF3 (Abb. 13.4) konnte
erhalten werden, indem das Trioxid schlagartig von -40°C auf +20°C erwärmt wird, in die
Meßzelle strömt und das Spektrum in wenigen Sekunden detektiert wird (8 scans). Da die
Halbwertszeit des CF3OC(O)OOOC(O)OCF3 unter diesen Bedingungen etwa 1 Minute
beträgt, ist nach einigen Sekunden noch kein nennenswerter Zerfall IR-spektroskopisch
feststellbar.
Eine Übersicht über die beobachteten und berechneten Fundamental-schwingungen
von CF3OC(O)OOOC(O)OCF3 im Vergleich zu CF3OC(O)OOC(O)OCF3 ist in Tabelle 13.2
gegeben.
Tabelle 13.2: Fundamentalschwingungen von CF3OC(O)OOOC(O)OCF3 und CF3OC(O)OOC(O)OCF3.
559.8 2.4 1.2 A �13 - - - - 546 4 547.0 545 0.10 563 s 555.9 0.54 2.7 B �36 556.6 541 5 541.9 541 0.07 544 s 552.7 1.8 3.2 A �14
563 555.6 442 11 450.2 448 0.89 449 w 449.9 19 1.2 B �37 460 460 460 460.9
427.4 0.21 0.5 B �38 426.1 433 m 427.2 0.16 0.8 A �15 431 434 428.5
381.4 0.03 1.5 A �16 383 386 383.7 386 s 379.2 1.1 0.3 B �39 379.8 339 m 329.5 0.90 0.6 B �40 327.1 286 m 274.3 0.29 1.3 A �17 343 340.4 252 s 243.4 0.50 2.9 A �18 242 231.8 180 vw 166.1 3.6 0.1 B �41 156.0
141.3 0.08 0.1 B �42 - - - - 121 s 116.6 0.04 0.6 A �19 161.5
100.5 0.34 0.7 A �20 108 111.4 88.8 0.47 0.5 B �43 100.7
72 w 70.9 0.12 0.4 A �21 74.7 57.7 0.07 0.4 B �44 56.7 29.8 0.23 0.1 B �45 33.0 27.8 0.20 0.2 A �22 39.1 21.4 0.00 0.3 A �23 26.5
[a]: Raman: Festkörper bei -196°C [b]: B3LYP/6-311G(d,p), diese Arbeit[c]: Ref. [114], Raman: flüssig [d]: relative integrierte Bandenintensitäten
13 CF3OC(O)OOOC(O)OCF2 147
UV Spektrum
Die Aufnahme eines UV-Spektrums von reinem CF3OC(O)OOOC(O)OCF3 in der
Gasphase gelingt nicht, da das Trioxid sich bei den in der Meßzelle herrschenden
Temperaturen von etwa 25°C während der Messung zu einem erheblichen Teil zersetzt und
das Folgeprodukt CF3OC(O)OOC(O)OCF3 ebenfalls ein UV-Spektrum zeigt. Die Ermittlung
von Absorptionsquerschnitten ist nicht möglich, da sich die Konzentration des Meßgases
verändert. Lediglich die Aussage, daß die Absorptionsquerschnitte des Trioxids größer sind,
als die des analogen Peroxids kann zweifelsfrei festgestellt werden. Es läßt sich ein UV-
Spektrum von CF3OC(O)OOOC(O)OCF3, isoliert in Neon-Matrix, aufnehmen. Auch in
diesem Fall ist die Ermittlung absoluter Absorptionsquerschnitte nicht exakt möglich, da die
Verdünnung der Probe mit Inertgas aufgrund der notwendigen Vorgehensweise nicht
bestimmbar ist und somit eine Kalibration der Spektren, etwa mit einem bekannten UV-
Absorber hinfällig wird. In Abbildung 13.5 ist ein UV-Spektrum (Extinktion gegen
Wellenlänge) dargestellt.
Abbildung 13.5: UV-Spektrum von CF3OC(O)OOOC(O)OCF3 isoliert in Ne-Matrix.
13 CF3OC(O)OOOC(O)OCF2 148
Man erkennt den langwelligen Teil einer breiten, unstrukturierten Absorptionsbande, deren
Maximum bei einer Wellenlänge unterhalb von 200 nm liegt und somit außerhalb des
Meßbereichs auftritt.
NMR-Spektren
Im 19F und 13C NMR-Spektrum beobachtet man Signale, die in den für CF3O-
bzw. C=O-Verbindungen typischen Bereichen liegen. Auch im Falle der NMR-Spektren
unterscheiden sich die Spezies CF3OC(O)OOOC(O)OCF3 und CF3OC(O)OOC(O)OCF3
kaum voneinander. Eine Übersicht gibt Tabelle 13.3.
Tabelle 13.3: NMR Daten fürCF3OC(O)OOOC(O)OCF3 und CF3OC(O)OOC(O)OCF3.
[a] diese Arbeit. 20 mg CF3OC(O)OOOC(O)OCF3 in 2 mL CFCl3 als interner Standard und 0.2 mLCD2Cl2 als interner Lock bei - 78°C. �-Werte beziehen sich auf: 19F-NMR �(CFCl3) bei - 78°C = 0;13C-NMR �(13CD2Cl2 ) bei -78°C = 54.0. [b] Ref. [114] CF3OC(O)OOC(O)OCF3 bei- 30°C mit externem Lock und Standard, Probe enthält einige mol-% CF3OC(O)OOOC(O)OCF3.
13.5 Eigenschaften von CF3OC(O)OOOC(O)OCF3
Bistrifluormethyltrioxydicarbonat zeigt eine geringe thermische Stabilität: Die
Zersetzung beginnt oberhalb von etwa - 35°C, bei + 25°C beträgt die Halbwertszeit der
Zersetzung etwa 1 Minute. Als bei Raumtemperatur stabile Endprodukte werden
CF3OC(O)OOC(O)OCF3 und ein nicht kondensierbares, IR-inaktives Gas (O2) beobachtet,
zudem finden sich geringe Mengen an COF2. Die Bruttogleichung des Zerfalls lautet:
CF3OC(O)OOOC(O)OCF3 � CF3OC(O)OOC(O)OCF3 + ½ O2 (13-15)
Die Bildung von Fluorphosgen kann mit Reaktionen an den Gefäßwänden erklärt werden. Die
thermische Instabilität des Trioxids kann bei Niederdruckpyrolyse-experimenten und
anschließender Matrixisolation der Reaktionsprodukte zur Erzeugung des Radikals
CF3OC(O)OO genutzt werden (siehe Kapitel 7), das neben CF3O und CO2 nachgewiesen
11/1998 - 05/1999 Diplomarbeit im Fach Anorganische Chemie
Thema:" CF3OO und FC(O)OO".
Referent: Prof. Dr. H. Willner
Korreferent: Prof. Dr. W. Uhrland
seit 07/1999 Promotion an der Gerhard-Mercator-Universität Duisburg im
Arbeitskreis Prof. Dr. H. Willner
Oberhausen, den 25.09.2002
Publikationsliste 199
Publikationen
Journal-Artikel
1 Vibrational spectra of cis and trans oxalyl fluoride and their site-selective IR inducedrotamerization in an Ar matrix.S. SANDER, H. WILLNER, L. KHRIACHTCHEV, M. PETTERSON,M. RÄSÄNEN, E. L. VARETTIJ. Mol. Spectrosc. 2000, 203(1), 145-150.
2 IR and UV spectra of the matrix isolated peroxy radicals CF3OO, trans FC(O)OO, andcis FC(O)OO.S. SANDER, H. PERNICE, H. WILLNERChemistry Eur. J. 2000, 6(19), 3645-3653.
3 Trifluormethylnitrat CF3ONO2.S. SANDER, H. WILLNER, H. OBERHAMMER, G. A. ARGÜELLOZ. Anorg. Allg. Chem. 2001, 627(4), 655-661.
4 High-resolution spectroscopy and preliminary analysis of the �1/�8 dyad of SF535Cl.
M. ROTGER, A. DECRETTE, V. BOUDON, M. LOETE, S. SANDER,H. WILLNERJ. Mol. Spec. 2001, 208(2), 169-179.
5 First high-resolution analysis of the six fundamental bands �1, �2, �3, �4, �5, and �6of C(O)35ClF in the 340 to 2000 cm-1 region.A. PERIN, J. M. FLAUD, H. BÜRGER, G. PAWELKE, S. SANDER,H. WILLNERJ. Mol. Spec. 2001, 209(1), 122-132.
6 Structure and conformation of �,�,�-trifluoroanisol, C6H5OCF3.D. FEDERSEL, A. HERMANN, D. CHRISTEN, S. SANDER, H. WILLNER,H. OBERHAMMERJ. Mol. Struct. 2001, 567, 127-136.
7 Structures and conformations of trifluormethyl fluoroformate CF3OC(O)F andperfluorodimethyl carbonate (CF3O)2CO.A. HERMANN, F. TRAUTNER, K. GHOLIVAND, S. v. AHSEN,E. L. VARETTI, C. O. DELLA VEDOVA, H. WILLNER, H. OBERHAMMERInorg. Chem. 2001, 40(16), 3979-3985.
8 The trifluoromethoxycarbonyl radical CF3OCO.S. v. AHSEN, J. HUFEN, H. WILLNER, J. S. FRANCISCOChemistry Eur. J. 2002, 8(5), 1189-1195.
9 The trifluoromethoxycarbonyl peroxy radical CF3OC(O)OO.S. v. AHSEN, H. WILLNER, J. S. FRANCISCOChemistry Eur. J., in Druck.
Publikationsliste 200
10 Experimental and calculated molecular structures and high symmetry ground state conformations of [Sb2F11]- (D4h) and 1,4-N2C4H4 � 2 SbF5 (D2h).I. H. T. SHAM, B. PATRICK, B. v. AHSEN, S. v. AHSEN, H. WILLNER,R. C. THOMPSON, F. AUBKEeingereicht.
11 Die Gasphasenstruktur und das Rotationsspektrum von FCO2.P. DREAN, Z. ZELINGER, M. BOGEY, H. PERNICE, S. v. AHSEN,H. WILLNER, J. BREIDUNG, W. THIELeingereicht.
12 The trifluoromethoxy trioxy radical CF3OOO.S. v. AHSEN, H. WILLNER, J. S. FRANCISCOin Vorbereitung.
13 Bis(trifluormethyl)peroxytricarbonate CF3OC(O)OOOC(O)OCF3.S. v. AHSEN, P. GARCIA, H. WILLNER, G. A. ARGÜELLOin Vorbereitung.
Poster und Vorträge
1 Fluorinated Peroxy Radicals. (Poster)S. SANDER, H. PERNICE, H. WILLNER16th Int. Symposium on Fluorine Chemistry, Durham, UK, 16.-21.07.2000.
2 CF3ONO2. (Vortrag)S. SANDER, H. WILLNER9. Deutscher Fluortag, Schmitten, 25.-27.09.2000.
3 High Resolution Rotational Spectra of the FCO2 Radical in its X2B2 State. (Poster)P. DREAN, A. WALTERS, M. BOGEY, Z. ZELINGER, S. SANDER,H. WILLNER, J. BREIDUNG, W. THIELInt. Symposium on Molecular Spectroscopy - 56th Meeting, Columbus (Ohio), USA,11.-15.06.2001.
4 C4v TDS Software and its Application to the �1/�8 Dyad of SF535Cl. (Poster)
M. ROTGER, A. DECRETTE, V. BOUDON, M. LOETE, H. BÜRGER,S. SANDER, H. WILLNER17th Int. Conference on High Resolution Molecular Spectroscopy, Prag, TschechischeRepublik, 1.-5.09.2001.