Aus dem Institut für Normale und Pathologische Physiologie Geschäftsführender Direktor: Prof. Dr. Dr. Jürgen Daut Des Fachbereichs Medizin der Philipps-Universität Marburg Titel der Dissertation: BETEILIGUNG VON TANDEM-KALIUM- KANÄLEN IN DER DURCH UNGESÄTTIGTE FETTSÄUREN HERVORGERUFENEN KORONAREN VASODILATATION Inaugural-Dissertation zur Erlangung des Doktorgrades der gesamten Medizin dem Fachbereich Humanmedizin der Philipps-Universität Marburg vorgelegt von Marko Daniel Burmester geboren am 29.11.1979 in Hamburg erschienen in Marburg im Jahre 2007
Transcript
Aus dem Institut für Normale und Pathologische
Physiologie
Geschäftsführender Direktor: Prof. Dr. Dr. Jürgen Daut
Des Fachbereichs Medizin der Philipps-Universität Marburg
Titel der Dissertation:
BETEILIGUNG VON TANDEM-KALIUM-
KANÄLEN IN DER DURCH
UNGESÄTTIGTE FETTSÄUREN
HERVORGERUFENEN KORONAREN
VASODILATATION
Inaugural-Dissertation zur Erlangung des Doktorgrades der gesamten Medizin
dem Fachbereich Humanmedizin der Philipps-Universität Marburg
vorgelegt von
Marko Daniel Burmester
geboren am 29.11.1979 in Hamburg
erschienen in Marburg im Jahre 2007
Angenommen vom Fachbereich Medizin der Philipps-Universität Marburg am 25. Januar 2007.
Gedruckt mit Genehmigung des Fachbereichs.
Dekan: Prof. Dr. Bernhard Maisch
Referent: Prof. Dr. Dr. Jürgen Daut
Korreferent: Prof. Dr. Thomas Gudermann
ZUSAMMENFASSUNG
Der einfach ungesättigten Fettsäure Oleinsäure wird ebenso wie den mehrfach ungesättigten
Fettsäuren Eicosapentaensäure und Docosahexaensäure ein protektiver Einfluss auf die
menschliche Gesundheit zugesprochen. Ein vasodilatatorischer Einfluss dieser Substanzen im
Koronarsystem ist zwar seit längerem bekannt, doch sind die zugrundeliegenden Mechanismen
noch Gegenstand intensiver Forschung. Die Beteiligung von Kaliumkanälen in der Regulation der
koronaren Perfusion ist zwar ebenfalls bekannt, während die genaue Rolle von doppelporigen (2P)
sogenannten Tandem-K+-Kanälen – der neuesten Subgruppe von Kaliumkanälen – in diesem
Zusammenhang weitestgehend ungeklärt ist; bei TREK und TASK handelt es sich um bestimmte
Formen dieser 2P-K+-Kanäle. Wir haben am Langendorffschen Modell des isoliert perfundierten
Meerschweinchenherzens im Rahmen von 88 Experimenten die Wirkungen dieser Lipide
quantifizieren und eine Beteiligung von doppelporigen Kaliumkanälen nahelegen können.
Aufgrund ähnlichem Metabolismus wurden die Effekte mit Arachidonsäure und seinem Amid,
dem Endocannabinoidrezeptor-Agonisten Anandamid, verglichen; Anandamid blockiert
bekannterweise selektiv TASK1-Kanäle. Das Lokalanästhetikum Bupivacain inhibiert unter
anderem TREK- und TASK-Kanäle. Die als Reduktion des koronaren Perfusionsdruckes
gemessene mittlere maximal erreichbare Vasodilatation der einzelnen Lipide nach Erreichen eines
konstanten Perfusionsdruckes betrug 51,55 % für Oleinsäure; 40,37 % für Arachidonsäure; 47,95
% für Docosahexaensäure; 47,40 % für Eicosapentaensäure; 55,80 % für Anandamid; 72,11 % für
R-Bupivacain; 39,85 % für S-Bupivacain und 70,65 % für razemisches Bupivacain. Bei Zusatz vom
NO-Synthase-Inhibitor Nitroarginin [10 µM] zu allen Perfusionslösungen zeigte sich eine
Reduktion der maximalen Dilatation um 18,07 % für Oleinsäure; 13,10 % für Docosahexaensäure
und 7,55 % für Eicosapentaensäure, wobei nur der Effekt für Oleinsäure signifikant war. Der KATP-
Kanal-Blocker Glibenclamid zeigte in einer Konzentration von 2 µM keinen messbaren Effekt auf
die durch Olein-, Arachidon-, Docosahexaen- und Eicosapentaensäure sowie R-Bupivacain
hervorgerufene Vasodilatation; hierdurch konnte ein wesentlicher Einfluss der ansonsten häufig an
der Regulation des Koronartonus beteiligten ATP-abhängigen Kaliumkanäle in diesem
Zusammenhang praktisch ausgeschlossen werden. Auch der COX-Inhibitor Indometacin [10 µM]
hatte keinen Einfluss auf den vasodilatativen Effekt von Arachidon-, Docosahexaen-,
Eicosapentaensäure oder Anandamid. Razemisches Bupivacain und S-Bupivacain zeigten zunächst
eine rasche Reduktion des koronaren Perfusionsdruckes gefolgt von einem langsamen Anstieg,
während das R-Isomer nur eine Reduktion zeigte. In einer Konzentration von 25 µM zeigte S-
Bupivacain seinen maximalen vasokonstriktorischen Effekt mit einer mittleren Erhöhung des
Perfusionsdruckes um 22,10 %. Bei dieser Konzentration war also ein Maximum de 2P-K+-
blockierenden Wirkung anzunehmen. Die durch 25 µM S-Bupivacain erreichte durchschnittliche
Reduktion der Dilatation betrug 86,4 % für Eicosapentaensäure [10 µM], 73,3 % für
Eicosapentaensäure [100 µM], 82,1 % für Docosahexaensäure [10 µM] und 76,1 % für
Docosahexaensäure [100 µM]. Wir haben hiermit einen mit hoher Wahrscheinlichkeit durch
Kaliumkanäle (KV), 3. Kalzium-abhängige Kaliumkanäle (KCa) und 4. einwärts-gleichrichtende
Kaliumkanäle (Kir). Neben diesen Kanälen sind in letzter Zeit zunehmend die Zwei-Poren-
Kalium-Kanäle (K2P) erforscht worden, wobei sich diverse Subtypen unterscheiden liessen.
Neben der funktionellen Einteilung der Kaliumkanäle lassen sich diese auch nach ihrer
molekularen Struktur ordnen. Das gemeinsame molekulare Motiv aller Kaliumkanäle ist die
4
Poren-Domäne, welche in Säugetierzellen hochkonserviert vorliegt (Doyle et al. 1998).
Daneben besitzen die Untereinheiten der Kanäle eine unterschiedliche Anzahl an
Transmembrandomänen, wonach eine grobe Einteilung in zwei, vier oder sechs solcher
Domänen zählende Kaliumkanaluntereinheiten vorgenommen werden kann. So weisen
beispielsweise die Untereinheiten der einwärts-gleichrichtenden Kir-Kanäle zwei, diejenigen
der auswärts-gleichrichtenden KV-Kanäle dagegen sechs Transmembrandomänen auf; die
K2P-Kanal-Untereinheiten zeigen eine Molekülstruktur mit vier Transmembransegmenten
(Lesage et al. 1996, Guillemare et al. 1996, Fink et al. 1996). Die fertigen Kanäle entstehen
durch eine Zusammenlagerung der Poren-Domänen der jeweiligen Untereinheiten. Bei den
Untereinheiten mit zwei oder sechs Transmembrandomänen werden die fertigen Kanäle
durch Tetramere gebildet, während die K2P-Kanäle wahrscheinlich Dimere darstellen (Lesage
et al., Reyes et al., Fink et al. 1996).
K2P-Kanäle lassen sich weiterhin in fünf funktionelle Subtypen unterteilen:
1. Lipid-sensitive mechanisch beeinflussbare Kaliumkanäle: TREK-1, TREK-2 und TRAAK (Fink et al. 1996 & 2000, Bang et al. 2000)
2. pH-sensitive Auswärts-Gleichrichter: TASK-1 bis TASK-5 (Duprat et al. 1997, Kim et al. 2000, Rajan et al. 2000, Leonoudakis et al. 1998, Reyes et al. 1998, Kim et al. 1998, Kim et al. 2001, Decher et al. 2001)
3. Schwache Einwärts-Gleichrichter: TWIK-1 und TWIK-2 (Lesage et al. 1996, Patel et al. 2000, Pountney et al. 1999, Chavez et al. 1999, Salinas et al. 1999)
4. Alkalisch-aktivierte Hintergrundkaliumkanäle: TALK-1 und TALK-2 (Kang et al. 2004, Duprat et al. 2005)
5. Die Halothan-inhibierten Kaliumkanäle THIK-1 und THIK-2 (Rajan et al. 2001)
6. Die “Rückenmarks“-Kaliumkanäle TRESK-1 und TRESK-2, welche besonders hohe
Sensitivität gegenüber Inhalationsanästhetika wie Halothan zeigen (Keshavaprasad et
al. 2005, Liu et al. 2004)
Es hat sich gezeigt, dass mehrere dieser Subtypen eine elementare Rolle bei der Genese des
Ruhepotenzials unterschiedlichster Körperzellen einnehmen. Unter anderem wird die
Öffnungswahrscheinlichkeit dieser Kanäle durch Hypoxie, intra- und extrazellulären pH-
Wert, Temperatur, Druck, Neurotransmitter sowie bioaktive Lipide moduliert (Girard et al.
2004).
5
Im Rahmen der vorliegenden Abhandlung soll bevorzugt auf die lipid-sensitiven
Kaliumkanäle eingegangen werden. Diese Lipidsensitivität ist bislang teils auf direkte Effekte
auf Kaliumkanäle, teils jedoch auch auf indirekte – das heißt membranvermittelte –
mechanische Stimulierbarkeit zurückgeführt worden (s. a. Kap. 4). Die genannten Kanäle sind
aufgrund ihrer hohen Expression im Hirngewebe insbesondere für die Anästhesie von großer
Bedeutung. Neuere Untersuchungen von Bryan et al. (2006) haben jedoch ergeben, dass
TREK-1- und TRAAK-Kanäle auch im Endothel nachweisbar sind. Die mechanische
Stimulierbarkeit der Kanäle macht sie unter anderem für solche Substanzen empfindlich,
welche sich in der Lipid-Doppelschicht lösen und damit die Membranspannung modulieren
können. Hierzu gehört neben diversen Fettsäuren auch das Inhalationsanästhetikum
Halothan. Das Lokalanästhetikum Bupivacain zeigte
in verschiedenen Untersuchungen auch einen
direkten inhibitorischen Effekt auf TREK-1 (Punke
et al. 2003), TASK-2 (Kindler et al. 2003) und
TASK-3 (Meadows et al. 2001). Das mit Bupivacain
verwandte Ropivacain zeigt in unserem Modell
ähnliche Effekte wie Bupivacain, worauf jedoch im
Rahmen dieser Arbeit nicht weiter eingegangen werden soll (vgl. Burmester et al. 2005).
Während das Endocannabinoid Anandamid unabhängig von CB-Rezeptoren TASK1-Kanäle
direkt und selektiv blockiert, zeigt der klassische Kaliumkanalblocker Glibenclamid keinen
Effekt auf Tandemkanäle (Maingret et al. 2001, Skatchkov 2006). Es konnte weiterhin ein
CB1-Rezeptor-unabhängiger Calcium-Ionen freisetzender Effekt von Anandamid auf
menschliche Endothelzellen nachgewiesen werden (Mombouli et al. 1999). Somit erscheint
eine Beteiligung von Tandem-Kaliumkanälen im menschlichen Koronarkreislauf zwar
wahrscheinlich, ist jedoch bislang unzureichend erforscht worden. Angesichts der weiten
Verbreitung von Inhalationsanästhetika und der häufig postulierten kardioprotektiven
Wirkung diverser Fettsäuren besteht diesbezüglich ein deutlicher Abklärungsbedarf, welchem
mit dieser Arbeit nachgegangen werden soll.
1.4. Die Rolle der Fettsäuren im gesunden Herzen
Bereits in den Jahren 1950-1974 analysierte das Krankenhaus von Upernavik im Nordwesten
Grönlands die Inzidenz unterschiedlicher chronischer Erkrankungen bei den einheimischen
Bupivacain
6
Eskimos und verglich diese Daten mit einer entsprechenden mitteleuropäischen Bevölkerung.
Hierbei fiel vor allem auf, dass die Inzidenz von koronarer Herzerkrankung bei den Eskimos
signifikant niedriger war, was bereits zu diesem Zeitpunkt mit dem hohen Anteil an ω-3-
Fettsäuren in der Nahrung der Eskimos in Zusammenhang gebracht wurde (Kromann et al.
1980). Auch in weiterführenden Studien konnte diese Theorie insbesondere als Senkung des
Myokardinfarktrisikos bestätigt werden (Kromhout et al. 1985 & 1995, Norell et al. 1986,
Gramenzi et al. 1990, Siscovick et al. 1995, Daviglus et al. 1997).
Die als bedingt essentiell geltenden Fettsäuren Eicosapentaensäure (EPA) und
Docosahexaensäure (DHA) zählen zu den mehfach ungesättigten Fettsäuren (PUFAs) und
sind Abkömmlinge der α-Linolensäure (ALA). Durch mehrere enzymatische Reaktionen
kann ALA über EPA in DHA umgewandelt werden (Neddleman et al. 1979, Sprecher et al.
1999) (s. Abb. 1-1). Diese Fettsäuren werden als ω-3-Fettsäuren bezeichnet, da die erste
Doppelbindung vom höchstoxidierten Kohlenstoffatom (=Carboxygruppe) aus gezählt am
drittletzten Kohlenstoffatom anzutreffen ist. Häufig wird die Anzahl der C-Atome gefolgt
von einem Doppelpunkt und der Anzahl der Doppelbindungen angegeben: so zum Beispiel
“20:5“ für EPA oder “22:6“ für DHA. EPA und DHA werden ähnlich wie der klassische
Metabolit Arachidonsäure über die Zyklooxygenase (COX) abgebaut. Im Endothel wird so
aus EPA Prostazyklin I3 (PGI3) gebildet, aus Arachidonsäure entsteht dagegen PGI2; beide
Stoffe wirken vasodilatatorisch (Nishikawa et al. 1997, Lagarde et al. 1993, Hamazaki et al.
1988). Ein physiologischer Unterschied im Gegensatz zur Wirkung von AA zeigt sich
dagegen beim Umbau von EPA und DHA innerhalb der Thrombozyten: hier wird AA zum
stark vasokonstriktorisch wirksamen Thromboxan A2 (TXA2), während EPA und DHA in
das wesentlich schwächer vasokonstriktorische TXA3 umgewandelt werden (Das et al. 2005,
Papanicolaou et al. 1987).
ALA EPA DPA DHA 18:3 18:4 20:4 20:5 22:5 22:6
∆-6-Desaturase ∆-5-Desaturase 24:5 24:6
Peroxisom
Abb. 1-1, Sprecher-Pathway: Schematische Darstellung der metabolischen Umwandlung von
α-Linolensäure (ALA) in die höhergradig ungesättigten Fettsäuren. Der größte Teil dieses
Prozesses findet im endoplasmatischen Retikulum statt, wobei ein wechselhafter Anteil in den
7
Peroxisomen einen wichtigen regulatorischen Einfluss auf den PUFA-Gehalt der Lipidmembran
haben könnte (Sprecher et al. 1995).
Weitere bekannte Effekte der PUFAs beinhalten vor allem die Inhibiton der Bildung von
VLDL und LDL in der Leber, weswegen ein antiatherogener Effekt postuliert worden ist.
Ein positiver therapeutischer Effekt in der Behandlung der rheumatoiden Arthritis ist
ebenfalls in Verbindung mit einer vermehrten PUFA-Aufnahme assoziiert worden (Kojima et
al. 2005, Kraemer et al. 2005). Interessant ist auch der Effekt von Fischölen auf die
Insulinsekretion: nachdem nachgewiesen werden konnte, dass PUFAs die Glukoseverwertung
in Ratten verbessern können, wurden intensivierte Untersuchungen im Bereich der
Diabetesforschung durchgeführt. Hierbei wurde die Theorie aufgestellt, dass der vermehrte
Einbau von PUFAs in die Plasmamembran des Skelettmuskels dort die AA kompetitiv
verdrängt und somit weniger PGE2 gebildet wird (Arkhipova et al. 2005, Barnett et al. 1987,
Wu et al. 1988). Da PGE2 ein potenter Inhibitor der cAMP-Bildung und damit auch der
Insulinsekretion ist, wird diese durch PUFAs gesteigert.
αααα-Linolensäure (ALA)
Eicosapentaensäure (EPA)
Docosahexaensäure (DHA)
Arachidonsäure (AA)
Anandamid
8
Die essentielle Fettsäure Linolensäure findet sich physiologischerweise in besonders hoher
Konzentration in der Wand von Koronararterien. Weiterhin ist Linolensäure eine wichtige
Vorstufe von Arachidonsäure, welche ihrerseits als Substrat für die Synthese von
Prostaglandinen und Thromboxan A2 fungiert. Hiermit ist Linolensäure intensiv in die
Regulation des koronaren Gefäßtonus und der Hämostase involviert. Es konnte gezeigt
werden, dass diese Fettsäure eine endothel-unabhängige Relaxation von
Koronararterienringen induziert, die durch den Na+/K+-ATPase-Inhibitor Ouabain blockiert
wurde (Pomposiello et al. 1998). Somit ist ein Effekt von Linolensäure auf die Na+/K+-
ATPase von koronaren glatten Muskelzellen nahegelegt worden.
Membranständige Arachidonsäure kann physiologisch in den Endocannabinoid-Rezeptor-
Agonisten Anandamid umgebaut werden (Arreaza et al. 1997). Während die Wirkungen auf
das ZNS bereits seit längerem intensiv erforscht werden, sind die systemischen Auswirkungen
von Endocannabinoiden bislang nur sehr schlecht bekannt. In Studien konnten Grainger et
al. 2001 nachweisen, dass Anandamid in Koronararterien eine Vasodilatation bewirkt. Dieser
Effekt ließ sich nicht durch Cannabinoid-Rezeptor-Antagonisten blockieren, dafür aber durch
einen NO-Synthase-Inhibitor (L-NAME) und einen COX-Hemmer (Indometacin)
verringern. In dieser Studie wurde postuliert, dass die Konversion des Fettsäureamids
Anandamid in ein vasoaktives Prostanoid hierbei eine wichtige Rolle spiele, wodurch unter
anderem Kaliumkanäle geöffnet würden.
Ölsäure ist die häufigste in der Natur vorkommende Fettsäure und spielt damit auch eine
wesentliche Rolle in der menschlichen Ernährung. Oleinsäure ruft bekannterweise eine vom
Endothel abhängige Vasodilatation hervor (Ryan et al. 2000). Andererseits handelt es sich im
Gegensatz zu den bisher genannten um eine einfach ungesättigte Fettsäure, die ebenso wie
ALA, AA, EPA und DHA 2P-Kaliumkanäle vom TRAAK-Typ zu öffnen vermag. Obwohl
der Effekt von vielen Fettsäuren insbesondere auf TREK-1-, TREK-2- und TRAAK-Kanäle
nachgewiesen worden ist, ist anzunehmen dass auch andere 2P-Kaliumkanäle mechanisch
durch PUFAs aktiviert werden können (Patel et al. 2001).
Oleinsäure
9
1.5. Die Rolle der Fettsäuren im kranken Herzen
Pathologische Prozesse greifen über vielerlei direkte und indirekte Mechanismen in das
molekulare und elektrische Gleichgewicht einer Zelle ein. Das Säugetierherz ist durch seine
konstante Belastung und seine zentrale Rolle im Kreislauf einer enormen Vielfalt an solchen
Einflüssen ausgesetzt. Beim Menschen werden Schäden am Myokard in erster Linie durch
erhöhten arteriellen Blutdruck, Nikotinabusus oder ein metabolisches Ungleichgewicht zum
Beispiel im Rahmen eines Diabetes mellitus oder einer Hypercholesterinämie ursächlich
ausgelöst. Diese Störungen bewirken über unterschiedlichste Mechanismen (direkte Effekte
auf Ionenkanäle, Atherosklerose, toxische Metaboliten) in aller Regel eine Alteration des
koronaren Blutflusses, wodurch eine Myokardischämie und -nekrose provoziert werden
können. Im ischämischen Myokard kommt es zur Akkumulation zahlreicher Metabolite,
welche selbst nach erfolgreicher Reperfusion einen arrhythmogenen Einfluss auszuüben
vermögen. Es ist bereits seit längerem bekannt, dass während der Reperfusion sehr hohe
Konzentrationen an Fettsäuren im menschlichen Myokard anzutreffen sind (Hutter et al.
1992; Van der Vusse et al. 1997). Vor allem die einfach-ungesättigte Fettsäure Oleinsäure ist
hierbei mehrfach nachgewiesen worden (Svensson et al. 1990; Charnock et al. 1994;
Lopaschuk et al. 1994; al Makdessi et al. 1995). Weiterführende Experimente mit Oleat haben
gezeigt, dass diese Fettsäure den transienten Auswärtsstrom in menschlichen Myozyten
blockiert (Crumb et al. 1999), was mit den bereits in Kapitel 1.4 genannten Effekten von
Oleinsäure in Einklang gebracht werden kann.
Durch Medikamente können die endogenen Wege des PUFA-Katabolismus erheblich
modifiziert werden. So haben Serhan et al. 2002 nachweisen können, dass DHA unter dem
Einfluss des COX-Inhibitors Acetylsalicylsäure zu einer zuvor unbekannten Substanzgruppe
von 17R-Hydroxy-Docosanoiden konvertiert wird. Diesen später als Resolvine bezeichneten
Substanzen wird ein wichtiger antiinflammatorischer Effekt durch Unterbindung der
Genexpression von Zytokinen zugesprochen. Auch die anderen PUFAs können in Resolvine
umgewandelt werden und beteiligen sich hierdurch modulatorisch an unterschiedlichen
inflammatorischen Prozessen (Arita et al. 2005). Auf diesem Gebiet wird in den nächsten
Jahren hoffentlich sehr viel mehr Information zur Aufklärung der genaueren
Zusammenhänge zwischen PUFAs, Resolvinen, Herzinfarktrisiko und anderen teils
autoimmunologisch und teils infektiös bedingten pathogenetischen Prozessen erscheinen.
10
K a p i t e l 2
METHODIK
2.1 Herzpräparation und apparativer Aufbau
Die Meerschweinchen wurden von der Firma Charles River (Sulzfeld) geliefert. Es handelte
sich hierbei um weibliche Meerschweinchen des Stammes BFA bunt mit einem
Körpergewicht von 280-350 g welche mit dem Tierfutter Altromin 1320 und Wasser ernährt
worden waren. Die Tiere wurden nach ihrer Lieferung zwischenzeitlich in großen
Plastikwannen mit Pressspanflocken gehalten, wo sie stets Zugang zu Trinkwasser hatten.
Vor Versuchsbeginn wurde ein einzelnes Tier in einen kleineren Plastikbehälter von ca. 9
Liter Volumen umgelagert. Auf den Boden dieses Behälters wurde eine dünne Lage
saugfähigen Zellstoffs gelegt, auf die unmittelbar vor Versuchsbeginn das
Inhalationsanästhetikum appliziert werden konnte. Vor der Anästhesie wurde der Behälter
mindestens drei Minuten lang mit 100%igem Sauerstoff begast, um eine möglichst gute
Sauerstoffversorgung des Herzens zu gewährleisten. Als Inhalationsanästhetikum wurden 4
ml Sevofluran verwendet, welche mit einer Spritze auf den Zellstoff appliziert wurden.
Hiernach wurde der Behälter luftdicht verschlossen, bis der Muskeltonus der Tiere deutlich
erschlafft war und es zu einer spontanen Blasenentleerung kam.
Mit der Herzpräparation wurde unmittelbar nach eingetretener Sedation begonnen. Die Tiere
wurden decapitiert und das Abdomen wurde mit einer einzigen subxiphoidalen transversalen
Inzision eröffnet. Nach Durchtrennung des Diaphragmas wurde die vordere Thoraxhälfte
durch zwei parallel zum Sternum von kaudal nach kranial durchgeführte Schnitte mobilisiert
und das Perikard vorsichtig von der Thoraxwand getrennt. Auf diese Weise konnte eine
optimale Sicht auf das Herz und die Lungen innerhalb weniger Sekunden gewährleistet
werden. Daraufhin wurde der Aortenbogen vom umgebenden Fett- und Bindegewebe
freipräpariert und auf Höhe des Truncus brachiocephalicus mit einer kleinen anatomischen
Pinzette gefasst. Distal dieser Stelle wurde die Aorta durchtrennt und das Herz mit der
Pinzette nach ventro-kaudal gezogen, wobei die Pulmonalgefäße und Hohlvenen
nacheinander abgeschnitten wurden um so das Herz völlig abzupräparieren. Das Herz wurde
daraufhin sofort in ein kleines Gefäß mit der späteren Perfusionslösung komplett eingetaucht,
um Luftembolisationen zu vermeiden. Durch mehrere Kontrollexperimente konnte gezeigt
11
werden, dass die spätere Ansprechrate des Herzens auf vasomotorische Stimuli ebenso wie
die maximale Dauer der Experimente erheblich vor allem von zwei Faktoren abhängt;
nämlich erstens von der Vermeidung von Luftembolisationen und Minimierung der Zeit, in
der das Herz tatsächlich schlägt; und zweitens von der gesamten Präparationszeit, welche die
spezifische Hämostasezeit von 29 Sekunden nicht wesentlich überschreiten sollte (Hwang
und Wosilait, 1970). Bei der als erstes verwendeten physiologischen Salzlösung handelte es
sich um die gekühlte (8 °C) und oxygenierte spätere Perfusionslösung. Bei dieser ersten
Spülung wurde durch die Eigenaktivität des noch schlagenden Herzens bereits ein Großteil
des Blutes entfernt. Hiernach wurde eine T-förmige Kunststoffkanüle in den Aortenstumpf
vorgeschoben und mit zwei Fäden fest fixiert. Das zweite Ende dieser Kanüle wurde danach
am Rand der Präparationsschale befestigt, um eine sichere bimanuelle Präparation zu
ermöglichen. Das dritte Ende der Kanüle wurde an einen Schlauch angeschlossen, aus dem
die oxygenierte Perfusionslösung bei einer Temperatur von 22 °C und einem konstanten
Druck von 65 mmHg strömte. Unter diesen Bedingungen wurde es den Koronararterien
ermöglicht, die Flussrate entsprechend der metabolischen Bedürfnisse des Herzens durch ihre
Autoregulation anzupassen. Relativ hohe Pyruvatkonzentrationen wurden angewandt, um die
Regenerationszeit bis zum Erreichen eines konstanten Gefäßtonus zu verkürzen. Während
die Koronargefäße nun druckkonstant perfundiert wurden und das Myokard aufgrund der
hohen Kaliumkonzentration (15 mM) seine Kontraktionen einstellte, konnte die Präparation
sorgfältig zu Ende geführt werden. Hierbei wurde möglicherweise noch vorhandenes
Fremdgewebe wie Lunge und Haare entfernt und das Perikard abpräpariert, um eine spätere
Tamponade zu vermeiden.
Nach 15-minütiger druckkonstanter Perfusion wurde das Herz mitsamt der Kanüle aus der
Präparationsschale entfernt und an eine vierkanälige Peristaltikpumpe (“Perimax 16“, Spetec)
mit einem konstanten Fluss von 8,5 ml/min wie in Abb. 2-1 dargestellt angeschlossen. Um
optimale Versuchskonditionen zu generieren, wurden zwei abwechselnd pulsierende
Pumpenkanäle jeweils zu einem weitestgehend pulsationsfreien Kanal zusammengeschlossen.
Erst nach Sicherung dieser Perfusion wurde dann der Schlauch mit der druckkonstanten
Perfusion entfernt und durch ein Druckmessgerät (siehe unten) ersetzt. Die an die Pumpe
angeschlossenen Perfusionslösungen wurden innerhalb eines Wasserbades bei einer
konstanten Temperatur von 37,0 °C gehalten. Ein der Pumpe nachgeschalteter
Wärmeaustauscher wurde verwendet, um eventuelle während des Pumpendurchtritts
aufretende Wärmeverluste zu kompensieren. Hinter dem Wärmeaustauscher traten die
12
Lösungen durch eine kurze Glasröhre mit einem Luftfangsystem, um Luftembolisationen und
darauf folgende Perfusionsdefizite während des Experiments auszuschließen. Zwischen
verschiedenen Perfusaten konnte mittels eines 1 cm vor dem Herzen angebrachten Ventils
gewechselt werden. Das Herz selbst wurde in einen 10 ml fassenden doppelwandigen
Glasbehälter gehängt.
Abb. 2-1 Versuchsaufbau: An das System konnten stets zwei unterschiedlich begaste
Perfusionslösungen gleichzeitig angeschlossen werden. Die Flussregulation erfolgte über eine 4-
kanälige Peristaltikpumpe (1); Umschaltung zwischen den Lösungen mittels Drehventil (2); die
nicht in das Koronarsystem geleitete Lösung wurde verworfen (3); Übertragung der Daten zum
Computer durch einen Druckwandler (4); Meerschweinchenherz-Präparat (5); das im Wasserbad
(6) auf 37°C erwärmte Wasser wurde durch ein Schlauchsystem zum doppelwandigen
Organbehälter (7) und von dort zum Wärmetauscher (8) geleitet.
Der Raum zwischen den Glaswänden wurde ständig mit Wasser der Temperatur 37,0 °C
durchspült und so ein Abkühlen des Organs verhindert. Das aus den Koronargefäßen über
den rechten Vorhof und die offenen Hohlvenenstümpfe abfließende Perfusat füllte somit den
das Herz umgebenden Raum und floss letztendlich über die Ränder des Glasgefäßes ab.
Nun konnte der Druck innerhalb der Aorta über einen angeschlossenen A/D-Wandler
("Digidata 1200", Axon Instruments) und damit verbundenen Computer in Echtzeit gemessen
37 °C
Gas 2 Gas 1
1 2
3
4 5
6 7
8
13
werden. Die Daten wurden durch ein Computerprogramm von Axon Instruments ("Clampex"
7.0.0.86) aufgenommen und gespeichert. Dieser intraaortale Druck war dem koronaren
Gesamtwiderstand direkt proportional, da ein Abfließen des Perfusats bei geschlossener
Aortenklappe nur über die Herzkranzgefäße möglich war und die Flussgeschwindigkeit
während des Experimentes konstant gehalten wurde. Nun konnte also mit der Messung des
koronaren Gefäßwiderstandes in Abhängigkeit von den applizierten Substanzen begonnen
werden.
Wenn die Herzpräparation sorgfältig genug durchgeführt worden war, konnte man zu Beginn
eines jeden Experimentes eine rasche Zunahme des Perfusionsdruckes beobachten. Ein Steady
state wurde in der Regel in 4 bis 10 Minuten erreicht; bei länger dauernder Regenerationszeit
musste entweder von einem Leckstrom oder von einer ischämischen Schädigung des
Myokards ausgegangen und das Experiment abgebrochen werden. Ein Abbruch erfolgte
ebenso wenn der Perfusionsdruck im Steady state unter der Flussgeschwindigkeit von 8,5
ml/min nicht zwischen 70 und 120 mmHg lag. Nach dem Erreichen eines konstanten
Druckes im genannten Bereich wurde die Flussgeschwindigkeit so lange reduziert, bis ein
Wert zwischen 60 und 65 mmHg erreicht war. In diesem niedrigen Druckbereich konnte
einerseits noch von einer ausreichenden Versorgung des kontraktil inaktiven Myokards
ausgegangen werden, andererseit wurde aber auch die Neigung zur interstitiellen
Ödembildung minimiert.
Um das Reaktionsvermögen des Herzens zu überprüfen, wurde vor jeder Versuchsreihe ein
positiver Kotrollversuch mit dem KATP-Kanal-öffner Cromakalim (1,0 µM) durchgeführt und
ein Druckabfall von mindestens 50 % innerhalb von 5 Minuten zur Fortsetzung des
Versuches vorausgesetzt. Nach dieser Zeit oder nach Erreichen eines konstanten
Minimaldruckes wurde das Herz erneut mit Cromakalim-freier
Lösung perfundiert, bis wieder der Steady state –Druck vorlag.
Häufig kam es nach der ersten Cromakalim-Applikation zu einem
Ansteigen des Perfusionsdruckes über das ursprüngliche Niveau
hinaus. Dies kann jedoch durch den zusätzlichen regenerativen
Effekt der maximalen Vasodilatation erklärt werden und wird
durch die Beobachtung unterstützt, dass mehrmalige Cromakalim-
Administrationen den Steady state –Druck nicht noch weiter
steigern konnten. Aus diesem Grunde erfolgte nach einer einmaligen Cromakalim-induzierten
Vasodilatation eine nochmalige Korrektur der Perfusionsgeschwindigkeit, so dass der
Cromakalim
14
Perfusionsdruck wieder im Bereich 60 bis 65 mmHg lag. Für alle Experimente wurden somit
Flussgeschwindigkeiten von 3,5 bis 7,0 ml/min verwendet.
Eine Erhöhung der Cromakalim-Dosis hatte weder einen Effekt auf das Ausmaß der
Vasodilatation, noch wurde die Auswaschphase dadurch verlängert. Alternativ zur
Verwendung von Cromakalim beschreiben manche Autoren die Perfusion mit hypoxischen
Lösungen als gleichwertige oder gar überlegene Qualitätskontrolle. In den folgenden
Experimenten wurde der Effekt zwischen diesen beiden Methoden verglichen.
2.2 Perfusionlösungen
Die Stammlösung für jede Versuchsreihe wurde jeweils am Versuchstag aus durch reverse
Osmose ultrahochgereinigtem Wasser frisch hergestellt und enthielt folgende
Salzkonzentrationen:
NaCl 105 mM Sigma-Aldrich Chemie, Steinheim
NaHCO3 24 mM Sigma-Aldrich Chemie, Steinheim
KCl 15 mM Sigma-Aldrich Chemie, Steinheim
Glucose 10 mM Sigma-Aldrich Chemie, Steinheim
Na-Pyruvat 5 mM Sigma-Aldrich Chemie, Steinheim
NaH2PO4 1 mM Sigma-Aldrich Chemie, Steinheim
CaCl2 1 mM Sigma-Aldrich Chemie, Steinheim
MgCl2 800 µM Merck AG, Darmstadt
Die als oxygeniert bezeichneten Lösungen wurden mit Carbogen (95% O2, 5% CO2)
mindestens 10 Minuten vor Perfusionsbeginn begast; bei den als hypoxisch bezeichneten
Lösungen wurde analog eine Gasmischung aus 95% N2 und 5% CO2 verwendet. Der pH-
Wert wurde unter Begasung regelmäßig (jeweils vor Versuchsbeginn) gemessen und betrug
stets 7,41 ± 0,04.
Des weiteren wurden folgende Stoffe für einzelne Versuchsreihen verwendet: Anandamid,
Natrium-Eicosapentaenoat, Natrium-Oleat und Nω-Nitro-L-Arginin (Sigma-Aldrich Chemie,
Steinheim). Die jeweiligen Konzentrationen werden im Zusammenhang mit den
15
entsprechenden Versuchsreihen genannt. Die Stoffe wurden sorgfältig mittels langsamer
Verdünnung in Lösung gebracht, wobei auf löslichkeitsfördernde Stoffe wie DMSO
verzichtet werden konnte. Lediglich bei Oleat wurde bei den entsprechend bezeichneten
Versuchen die vom Hersteller an das Makromolekül Cyclodextrin gekoppelte Variante
gewählt, um bei längerer Perfusionsdauer eine vorzeitige Fettembolisation zu vermeiden. Bei
DHA, EPA und AA wurden die Lösungen zusätzlich 15 Minuten im Ultraschallbad
behandelt, um eine möglichst gute Löslichkeit zu erreichen. Die R- und S-Isomere von
Bupivacain wurden uns freundlicherweise von AstraZeneca (Södertälje, Schweden) zur
Verfügung gestellt.
2.3 Kalibrierung der Apparatur
Abb. 2-2 Kalibrierungsgerade: Abhängigkeit der mittels A/D-Konverter gemessenen
Spannung vom tatsächlich vorliegenden durch eine Wassersäule produzierten Druck.
0
1000
2000
3000
4000
5000
6000
7000
0 20 40 60 80 100 120
Druck [mmHg]
Spannung [mV]
16
Um die Umwandlung der gemessenen Druckwerte in elektrische Spannung und somit in
verwertbare Computerdaten zu kalibrieren, wurde eine 135,95 cm hohe Wassersäule (WS) mit
einem Glasröhrensystem hergestellt. Da 1 Meter WS in etwa 73,556 mm Quecksilbersäule
(mmHg) entspricht, konnte durch stufenweise Reduktion der Wassersäulenhöhe eine
Kalibrierung bis zu einem Äquivalentdruck von 100 mmHg erfolgen.
Bei der Kalibrierung zeigte sich eine ab einem Druck von circa 5 mmHg zuverlässig lineare
Proportionalität der Messwerte in Abhängigkeit vom tatsächlich vorliegenden Druck.
17
K a p i t e l 3
DURCHFÜHRUNG / RESULTATE
3.1 Vergleich hypoxischer mit Cromakalim-induzierter Vasodilatation
Zum Beginn einer jeden Versuchsreihe wurde durch Applikation von 1 µM Cromakalim eine
maximale Vasodilatation ausgelöst. Der typische Versuchsbeginn einer Messung mit der
Justierung des koronaren Perfusionsdrucks auf einen Wert von ca. 60 mmHg ist in Abb. 3-1
dargestellt. Da Cromakalim in diesem Konzentrationsbereich ein selektiver Öffner von ATP-
sensitiven Kaliumkanälen (KATP Kanälen) ist, war die Vasodilatation mit großer
Wahrscheinlichkeit auf die Öffnung der KATP-Kanäle, die daraus resultierende
Hyperpolarisation und den nachfolgenden Abfall der freien intrazellulären Ca2+
Konzentration in den glatten Muskelzellen der Koronargefäße zurückzuführen (Daut et al.
1990; von Beckerath et al. 1991). Zusätzlich wurde auch durch Perfusion des Herzens mit
Stickstoffbegaster Lösung eine hypoxische Vasodilatation durchgeführt, deren Amplitude sich
nicht signifikant von der der Cromakalim-induzierten Vasodilatation unterschied (Abb. 3.2).
Abb. 3-1, Induktion einer Cromakalim-induzierten (durch die Öffnung von KATP-
Kanälen vermittelten) Reduktion des KPD: Zunächst sind Druckschwankungen während
der späten Herzpräparation (Entfernung des Perikards) zu registrieren, danach erfolgt die
Einstellung eines konstanten Perfusionsdrucks von zunächst 61 mmHg. Unter Perfusion mit einer
2500 0
70 -
Zeit [s]
KPD [mmHg]
Cromakalim 1 µM
18
1 µM Cromakalim-Lösung kommt es zu einem Druckabfall bis zu einem Minimum von 20 mmHg.
Unter Reperfusion mit cromakalimfreier Lösung steigt der Druck erneut bis auf ein geringfügig
höheres Niveau als zuvor an (Regenerativer Effekt der maximalen Vasodilatation). Nach erneuter
Einstellung der Perfusionsrate konnte mit den eigentlichen Experimenten begonnen werden.
Um eine durch einen anderen Mechanismus vermittelte Vasodilatation hiermit zu vergleichen,
wurden zusätzlich fünf Experimente mit ATP-haltiger Lösung durchgeführt. Die ATP-
induzierte Gefäßerweitung wird durch P2X- und P2Y-Purinorezeptoren vermittelt
(Harrington et al. 2005). Alle drei Formen der Vasodilatation zeigten einen raschen, in etwa
exponentiellen Druckabfall, gefolgt von einem sigmoiden Druckanstieg während der
Reperfusionsphase mit oxygenierter Lösung (siehe Abb. 3-1). Um die Rolle von
Stickstoffmonoxid (NO) bei der hypoxischen Vasodilatation zu eruieren, wurde bei acht
Experimenten zur Blockade der endothelialen NO-Synthase Nω-Nitro-L-Arginin (NArg) in
einer Konzentration von 10 µM zu allen Lösungen hinzugefügt. Auf diese Weise konnte der
dilatative Effekt der verschiedenen Qualitätskontrollen verglichen werden, wobei sich das in
Abb. 3-2 dargestellte Ergebnis zeigte.
0
10
20
30
40
50
60
70
80
90
ATP [10 µM], n = 5
Cromakalim [1 µM],
n = 42 Hypoxie, n = 14
Hypoxie mit NArg
[10 µM], n = 8
Mittlerer KPD-Abfall [%]
Abb. 3-2, Vergleich vasodilatativer Stimuli: Nach Erreichen eines Steady state wurde der
Druck unter dem jeweiligen Stimulus mit dem Ausgangsdruck in Relation gesetzt (± eine
Standardabweichung).
Hiermit konnte gezeigt werden, dass NO die Vasodilatation nicht beeinflusst. Während ATP
den stärksten Effekt zeigte, konnte kein signifikanter Unterschied zwischen
cromakalimvermittelter und hypoxisch bedingter Vasodilatation nachgewiesen werden. Somit
19
wurde bei den folgenden Experimenten eine Qualitätskontrolle durch Cromakalim als
suffizientes Maß für die Reagibilität des Herzens gewertet.
3.2 Die Wirkung von Oleat auf den koronaren Widerstand
Um die Löslichkeit der Fettsäure Oleat zu verbessern, wurde sie zu sammen mit dem
Makromolekyl 2-Hydroxyethyl-β-Zyklodextrin appliziert. Zyklodextrine sind eine Gruppe
natürlich vorkommender Oligosaccharide welche aus mindestens sechs über 1,4-
Verbindungen verknüpften α-D-Glucopyranosyl-Einheiten bestehen; diese Einheiten werden
industriell aus Stärke gewonnen. Um einen selbständigen Einfluss von Zyklodextrin auf den
Koronartonus auszuschließen, wurde es Versuchsweise in verhältnismäßig hoher Dosierung
(8,7 mg/l) mit und ohne Cromakalim (2 µM) appliziert (Abb. 3-3).
Abb. 3-3, Wirkung von Cyclodextrin: Nach Cromakalim-vermittelter Vasodilatation und
darauffolgender Druckregeneration wurde Cyclodextrin hochdosiert mit und ohne Cromakalim
hinzugeführt. In vier aufeinanderfolgenden Experimenten zeigte sich kein signifikanter Einfluss
von Zyklodextrin auf den KPD oder die Reagibilität des Herzens.
Hiermit konnte eine Beeinflussung der Experimente durch Cyclodextrin weitestgehend
ausgeschlossen werden. In einer nachfolgenden Serie von Experimenten wurde orientierend
KPD [mmHg]
80
20
Zeit [s] 1000 2000
Cromakalim 2 µM Cromakalim 2 µM
2-Hydroxyethyl-β-Zyklodextrin 8,7 mg/l
0
20
untersucht, welchen Einfluss an Zyklodextrin gebundenes Oleat auf den koronaren
Widerstand hat und in welchem Konzentrationsbereich sich dieser Effekt beobachten lässt.
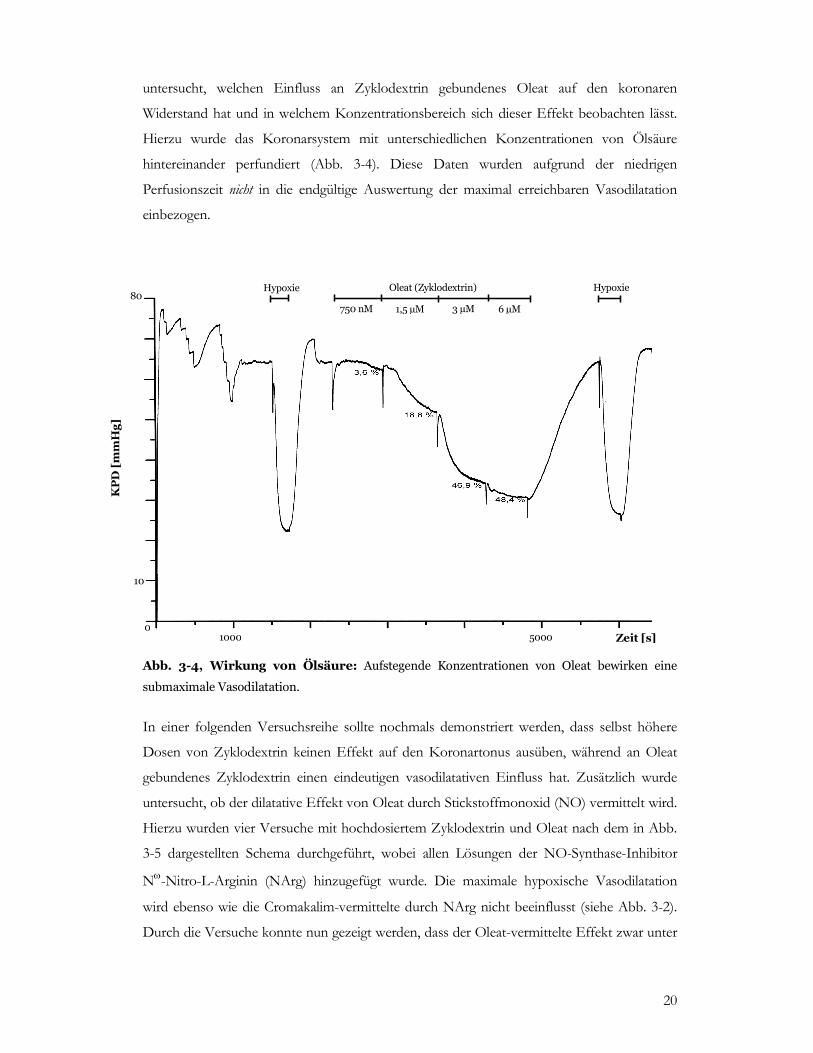
Hierzu wurde das Koronarsystem mit unterschiedlichen Konzentrationen von Ölsäure
hintereinander perfundiert (Abb. 3-4). Diese Daten wurden aufgrund der niedrigen
Perfusionszeit nicht in die endgültige Auswertung der maximal erreichbaren Vasodilatation
einbezogen.
Abb. 3-4, Wirkung von Ölsäure: Aufstegende Konzentrationen von Oleat bewirken eine
submaximale Vasodilatation.
In einer folgenden Versuchsreihe sollte nochmals demonstriert werden, dass selbst höhere
Dosen von Zyklodextrin keinen Effekt auf den Koronartonus ausüben, während an Oleat
gebundenes Zyklodextrin einen eindeutigen vasodilatativen Einfluss hat. Zusätzlich wurde
untersucht, ob der dilatative Effekt von Oleat durch Stickstoffmonoxid (NO) vermittelt wird.
Hierzu wurden vier Versuche mit hochdosiertem Zyklodextrin und Oleat nach dem in Abb.
3-5 dargestellten Schema durchgeführt, wobei allen Lösungen der NO-Synthase-Inhibitor
Nω-Nitro-L-Arginin (NArg) hinzugefügt wurde. Die maximale hypoxische Vasodilatation
wird ebenso wie die Cromakalim-vermittelte durch NArg nicht beeinflusst (siehe Abb. 3-2).
Durch die Versuche konnte nun gezeigt werden, dass der Oleat-vermittelte Effekt zwar unter
KPD [mmHg]
Zeit [s]
80
10
1000 5000
Hypoxie Hypoxie Oleat (Zyklodextrin)
750 nM 1,5 µM 3 µM 6 µM
0
21
NArg eine Abschwächung erfährt, jedoch auch bei hoher NArg-Dosierung nicht komplett
blockiert wird.
Um das Ausmaß der Oleat-bedingten Vasodilatation zu erfassen wurde eine weitere Reihe
von 21 Experimenten durchgeführt, bei denen Oleat in einer bestimmten Konzentration bis
zum Erreichen eines Steady state appliziert und danach mit Standardlösung bis zum Erreichen
des konstanten Ausgangsdruckes ausgewaschen wurde (nicht abgebildet, Prinzip wie in Abb.
3-8 und 3-9). Somit konnte die Vasodilatation mit (n=7) und ohne (n=14) NArg quantifiziert
und eine Konzentrations-Wirkungskurve erstellt werden (Abb. 3-6). Die durch OA
erreichbare maximale Vasodilatation betrug 51,55 ± 6,28 %, während dieser Wert unter
Zusatz von NArg [10 µM] im Durchschnitt 33,48 ± 6,94 % betrug.
Abb. 3-5, Effekt von Oleat unter NArg: Auch nach Blockade der NO-Synthase durch NArg
reagiert das Koronarsystem sowohl auf eine Hypoxie als auch auf eine Oleat-Applikation mit
Vasodilatation. Dennoch ist das Ausmaß dieser Dilatation geringer als ohne NArg. Zyklodextrin
alleine zeigte keinen Effekt.
2-Hydroxyethyl-β-Zyklodextrin 25,9 mg/l Hypoxie
Oleat 6 µM
Hypoxie
70
1000 4000
KPD [mmHg]
Zeit [s] 0
22
0
10
20
30
40
50
60
70
0 5000 10000 15000 20000
Konzentration [nM]
Vasodilatation [%]
OA + Zyklodextrin OA + Zyklodextrin + NArg
Die Oleat-bedingte Vasodilatation zeigte in allen Experimenten einen deutlich langsameren
Effekt als die durch maximale KATP-Kanal-Öffnung mit Cromakalim hervorgerufene.
Dennoch könnte die restliche (d.h. nicht NO-vermittelte) Vasodilatation durch KATP-Kanäle
vermittelt werden, da diese in einer Vielzahl von Effekten beteiligt sind. Um dies zu
überprüfen, wurden Versuche unter Zusatz von Glibenclamid, einem klassischen KATP-Kanal-
Blocker durchgeführt (Abb. 3-7).
Abb. 3-6, Quantifi-zierung der Oleat-vermittelten Vasodilatation mit (n=7) und ohne (n=14) NArg: Die Blockade der NO-Synthase bewirkt insbesondere im mittleren Konzentra-tionsbereich eine signifikante aber subtotale Verringe-rung der vasodilata-tiven Wirkung von Oleat. (± eine Stan-dardabweichung)
Glibenclamid
23
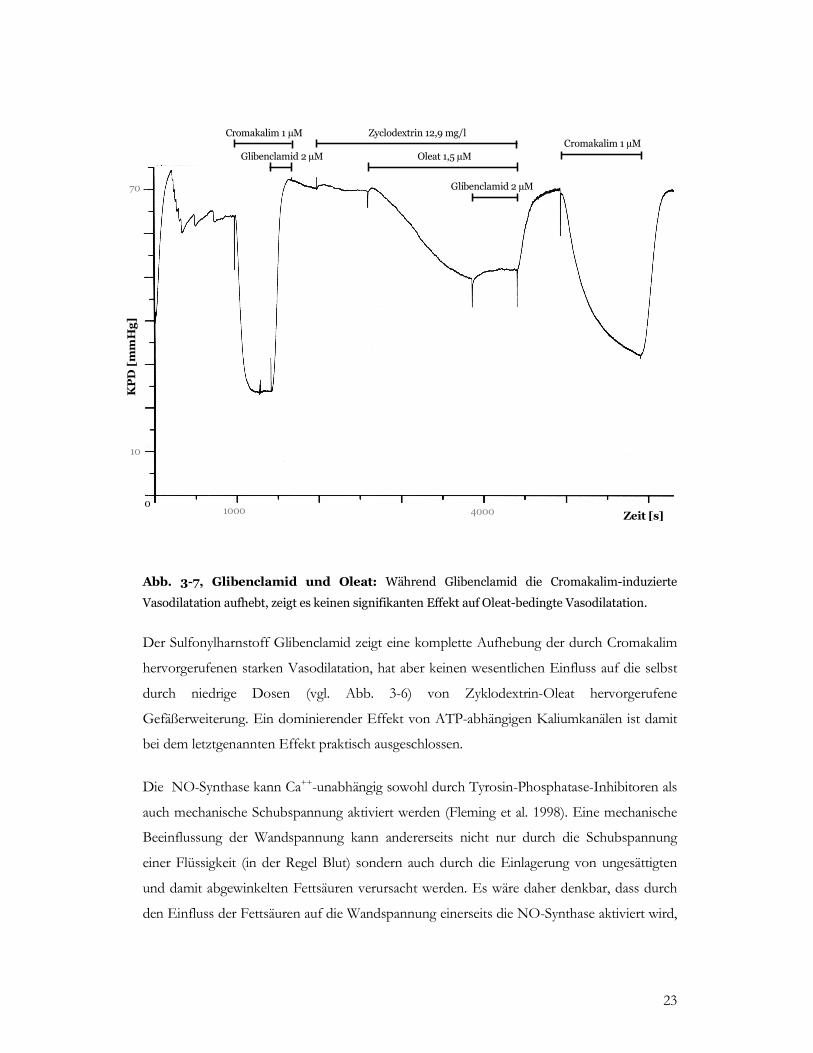
Abb. 3-7, Glibenclamid und Oleat: Während Glibenclamid die Cromakalim-induzierte
Vasodilatation aufhebt, zeigt es keinen signifikanten Effekt auf Oleat-bedingte Vasodilatation.
Der Sulfonylharnstoff Glibenclamid zeigt eine komplette Aufhebung der durch Cromakalim
hervorgerufenen starken Vasodilatation, hat aber keinen wesentlichen Einfluss auf die selbst
durch niedrige Dosen (vgl. Abb. 3-6) von Zyklodextrin-Oleat hervorgerufene
Gefäßerweiterung. Ein dominierender Effekt von ATP-abhängigen Kaliumkanälen ist damit
bei dem letztgenannten Effekt praktisch ausgeschlossen.
Die NO-Synthase kann Ca++-unabhängig sowohl durch Tyrosin-Phosphatase-Inhibitoren als
auch mechanische Schubspannung aktiviert werden (Fleming et al. 1998). Eine mechanische
Beeinflussung der Wandspannung kann andererseits nicht nur durch die Schubspannung
einer Flüssigkeit (in der Regel Blut) sondern auch durch die Einlagerung von ungesättigten
und damit abgewinkelten Fettsäuren verursacht werden. Es wäre daher denkbar, dass durch
den Einfluss der Fettsäuren auf die Wandspannung einerseits die NO-Synthase aktiviert wird,
Glibenclamid 2 µM
Zyclodextrin 12,9 mg/l
Oleat 1,5 µM
Glibenclamid 2 µM
Cromakalim 1 µM
Zeit [s]
KPD [mmHg]
10
70
1000 4000 0
Cromakalim 1 µM
24
andererseits aber auch andere Signaltransduktionswege eine wichtige Rolle spielen. Sollte
dieses Modell stimmen, müsste die Vasodilatation bei mehrfach ungesättigten Fettsäuren wie
DHA und EPA stärker ausgeprägt sein als bei der einfach ungesättigten Fettsäure OA.
3.3 Effekte von DHA und EPA auf koronaren Vasotonus
EPA und DHA wurden nicht an Zyklodextrin gebunden, sondern mittels Ultraschallbad in
Lösung gebracht. Um die Vergleichbarkeit mit Oleat zu gewährleisten, wurden sechs
Versuche mit nicht Zyklodextrin-gebundenem Oleat durchgeführt. Hierbei zeigte sich im
Konzentrationsbereich von 2 bis 20 µM kein signifikanter Druckunterschied zum
Zyklodextrin-gebundenen Oleat.
EPA und DHA wurden im Konzentrationsbereich von 125 nM bis 10 µM appliziert und der
jeweilige Perfusionsdruck nach 1-minütiger Druckkonstanz registriert. Für beide Substanzen
wurden fünf Versuchsreihen durchgeführt.
Abb. 3-8, EPA: Vasodilatation bei unterschiedlichen Konzentrationen.
EPA 10 µM EPA 5 µM
0
60 -
KPD [mmHg]
Zeit [s] 1000 3000
25
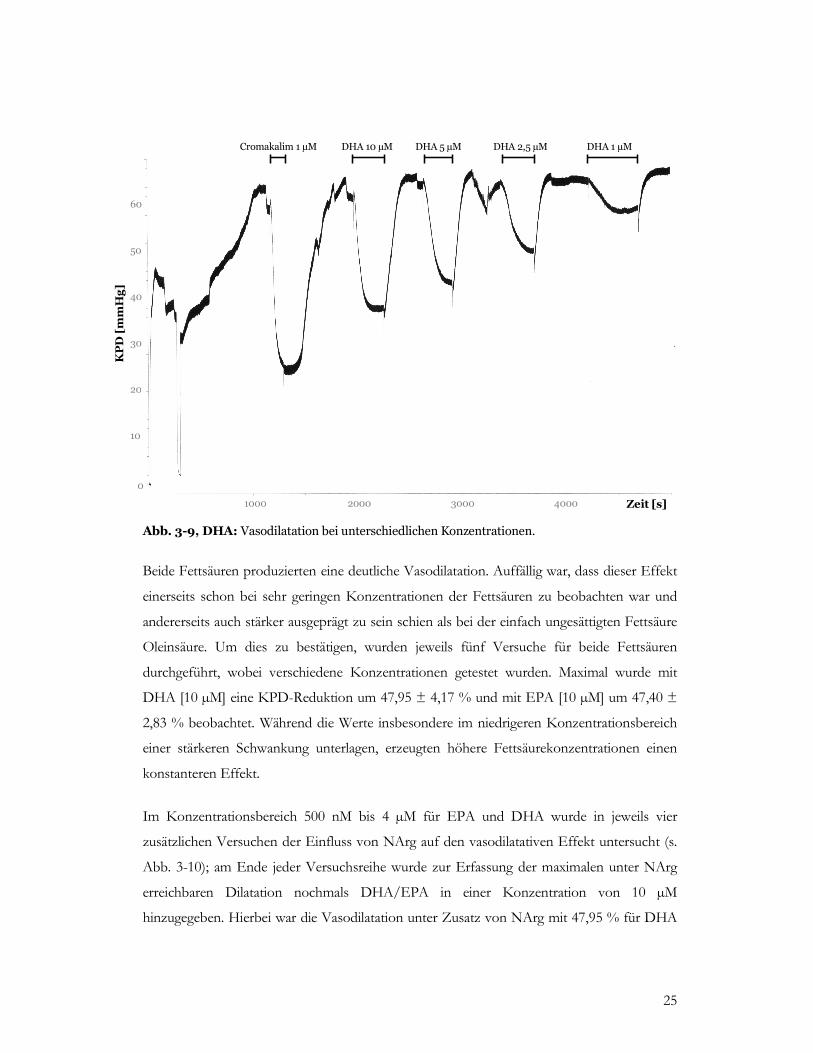
Abb. 3-9, DHA: Vasodilatation bei unterschiedlichen Konzentrationen.
Beide Fettsäuren produzierten eine deutliche Vasodilatation. Auffällig war, dass dieser Effekt
einerseits schon bei sehr geringen Konzentrationen der Fettsäuren zu beobachten war und
andererseits auch stärker ausgeprägt zu sein schien als bei der einfach ungesättigten Fettsäure
Oleinsäure. Um dies zu bestätigen, wurden jeweils fünf Versuche für beide Fettsäuren
durchgeführt, wobei verschiedene Konzentrationen getestet wurden. Maximal wurde mit
DHA [10 µM] eine KPD-Reduktion um 47,95 ± 4,17 % und mit EPA [10 µM] um 47,40 ±
2,83 % beobachtet. Während die Werte insbesondere im niedrigeren Konzentrationsbereich
einer stärkeren Schwankung unterlagen, erzeugten höhere Fettsäurekonzentrationen einen
konstanteren Effekt.
Im Konzentrationsbereich 500 nM bis 4 µM für EPA und DHA wurde in jeweils vier
zusätzlichen Versuchen der Einfluss von NArg auf den vasodilatativen Effekt untersucht (s.
Abb. 3-10); am Ende jeder Versuchsreihe wurde zur Erfassung der maximalen unter NArg
erreichbaren Dilatation nochmals DHA/EPA in einer Konzentration von 10 µM
hinzugegeben. Hierbei war die Vasodilatation unter Zusatz von NArg mit 47,95 % für DHA
Cromakalim 1 µM DHA 10 µM DHA 5 µM DHA 2,5 µM DHA 1 µM
0
60
50
40
30
20
10
Zeit [s] 1000 2000 3000 4000
KPD [mmHg]
26
und 47,40 % für EPA zwar geringer (Erhöhung des mittleren KPD um 13,10 ± 10,61 % für
DHA und 7,55 ± 6,08 % für EPA), doch war der Unterschied in beiden Fällen nicht
signifikant. Die durch die mehrfach ungesättigten Fettsäuren hervorgerufene Vasodilatation
ist also höchstens zu einem kleinen Teil durch NO vermittelt.
0
10
20
30
40
50
60
0 1000 2000 3000 4000 5000
EPA
EPA + NArg
0
10
20
30
40
50
0 1000 2000 3000 4000 5000
DHA
DHA + NArg
Abb. 3-10: Konzentrations-Wirkungskurve von EPA und DHA mit (n=4) und ohne (n=5) NArg
(100 % = Vasodilatation durch 1 µM Cromakalim).
Vasodilatation [%]
Konzentration [nM]
Konzentration [nM]
Vasodilatation [%]
27
Es wurde bereits erwähnt, dass die Bildung von vasoaktiven Prostanoiden aus PUFAs einen
wichtigen vasorelaxatorischen Mechanismus darstellen könnte. Da sowohl EPA und DHA
genauso wie AA durch Zyklooxygenasen umgebaut werden, müsste der Effekt durch COX-
Hemmer beeinflussbar sein. Um dies zu überprüfen wurde der COX-Inhibitor Indometacin
zu den Lösungen hinzugegeben (Abb. 3-11).
Abb. 3-11: Indometacin verringert nicht die DHA-vermittelte Vasodilatation.
Interessanterweise konnte keine Beeinflussung der vasodilatativen Wirkung von DHA
beobachtet werden. Dasselbe galt für eine Kontrolluntersuchung, bei der eine Vasodilatation
mit 9 µM AA hervorgerufen wurde und anschließend 10 µM Indometacin hinzugegeben
wurde. Bei dem akuten vasorelaxatorischen Effekt von AA und DHA scheint also auch die
COX nicht beteiligt zu sein. Auch dies erscheint angesichts der Annahme nicht überraschend,
dass die Fettsäulen zunächst mit membranständigen Strukturen in Wechselwirkung treten,
bevor sie im Zytosol weiterer Konversion unterliegen (vgl. Kap. 4). Letztendlich ließ sich
auch bei EPA und DHA (je 10 µM) ein KATP-Kanal-vermittelter Effekt ausschließen, indem
Glibenclamid (2 µM) hinzugegeben und keine Reduktion der Relaxation festgestellt wurde.
DHA 10 µM
DHA 10 µM +
Indometacin 10 µM DHA 5 µM
60 -
KPD [mmHg]
Zeit [s] 5000
Indometacin 10 µM
4000 6000 0 -
28
3.4 Anandamid und Arachidonsäure
Wie bereits erwähnt konnte von Grainger et al. 2001 ein relaxatorischer Effekt des
Endocannabinoids Anandamid auf durch U46619 (TXA2-Analogon) präkontrahierte
Koronararterienringe von Schafen nachgewiesen werden. Auch in früheren Versuchen war
eine Vasodilatation beschrieben worden. Die von Grainger et al. beobachtete Relaxation
zeigte jedoch eine starke Reduktion unter Indometacin-Zusatz: bei Anandamid-
Konzentrationen bis zu 5 µM wurde die Vasodilatation durch 10 µM Indometacin praktisch
komplett aufgehoben. Falls sich dies an unserem Modell des isoliert perfundierten
Meerschweinchenherzens bestätigen ließe, würde dies einen wesentlichen Unterschied zu den
anderen getesteten Lipiden darstellen, deren Effekt nicht unmittelbar von der
Prostanoidsynthese abhängig zu sein scheint. Vor allem der fehlende Effekt von Indometacin
auf AA-induzierte Gefäßerweiterung lässt an einer direkten COX-vermittelten
Anandamidreaktion zweifeln.
Anandamid (n=4) und AA (n=5) wurden analog zu den anderen Testsubstanzen in
unterschiedlichen Konzentrationen appliziert, bis ein Steady state erreicht wurde. Es zeigte sich
eine maximale Vasodilatation von 55,80 ± 2,57 % für Anandamid und 40,37 ± 12,51 % für
AA in einer Konzentration von je 10 µM, während Konzentrationen von 20 µM keine weitere
Verstärkung der KPD-Reduktion zeigten. Ausserdem wurden einzelne Lösungen mit Zusatz
von 10 µM Indometacin getestet und die mittlere Druckdifferenz ausgewertet (Abb.3-12).
29
Abb. 3-12: Anandamid 5 µM erzeugt unabhängig von Indometacin 10 µM eine ausgeprägte
Vasodilatation.
Auch in diesem Falle ließ sich bestätigen, dass Indometacin keinen Einfluss auf den von
Anandamid hervorgerufenen Effekt hatte. Glibenclamid [2 µM] wurde ebenfalls in jeweils
einem Experiment zu Anandamid und AA hinzugegeben und bewirkte keine Änderung des
KPD (Abb. 3-13).
Anandamid 5 µM
Anandamid 5 µM +
Indometacin 10 µM
0 -
60 -
1000 2000 3000 Zeit [s]
KPD [mmHg]
30
Abb. 3-13: Während 2 µM Glibenclamid den Cromakalim-Effekt blockiert, wird die durch 10 µM
AA hervorgerufene Dilatation nicht beeinflusst.
Es kann angenommen werden, dass mechanosensitive Kaliumkanäle der TREK/TRAAK
Familie hierbei beteiligt waren (s. Kap. 4). Da vor allem die TREK-Kanäle durch das
Lokalanästhetikum Bupivacain blockiert werden, sollte dies in den folgenden Versuchen
überprüft werden. Zuvor soll jedoch eine zusammenfassende Evaluation der bislang
gesammelten Messdaten der vasodilatatorischen Wirkung der getesteten Lipide durch
nachfolgende Tabelle geliefert werden (Abb. 3-14).
Zeit [s]
KPD [mmHg]
10
60
1000 2000 3000
Cromakalim 1 µM
Glibenclamid 2 µM Glibenclamid 2 µM
AA 10 µM
0
31
0
500
1000
1500
2000
2500
3000
3500
4000
OA (cyclo)
OA (cyclo) + NArg
EPA
EPA + NArg
DHA
DHA + NArg AA
Anandamid
c [nM]
Abb. 3-14: Mittlere Konzentrationen, die für 10 %ige Vasodilatation benötigt werden. (NArg =
Zusatz von 10 µM Nitroarginin; weitere Erläuterungen siehe Text)
In der Statistik sind die jeweils benötigten Konzentrationen der einzelnen Lipide angegeben,
um eine 10 %ige Vasodilatation zu erreichen. Es handelt sich hierbei um theoretisch
errechnete Konzentrationen, welche durch Interpolation der gemessenen Mittelwerte durch
logarithmische Funktionen errechnet wurden. Die Korrelationkoeffizienten (R²) dieser
logarithmischen Funktionen betrugen 0,93 für OA+Zyklodextrin; 0,72 für
OA+Zyklodextrin+NArg; 0,97 für EPA; 0,98 für EPA+NArg; 0,96 für DHA; 0,98 für
DHA+NArg (vgl. auch Abb. 3-10); 0,87 für Arachidonat und 0,93 für Anandamid. Die
niedrigeren Korrelationskoeffizienten haben nichts mit den Konfidenzintervallen zu tun,
welche beispielsweise bei DHA+Narg trotz besserem R² größer waren als bei OA+Narg (vgl.
Abb. 3-10 mit Abb. 3-6); vielmehr ist die Kurve für OA+NArg so flach, dass sie
bedauerlicherweise nicht gut mit einer logarithmischen Funktion abzubilden ist. Der
Vergleichbarkeit halber wurde dies dennoch einer polynomischen Darstellung bevorzugt. Zu
beachten ist weiterhin, dass in der Abbildung die kleineren Balken einen stärkeren Effekt
darstellen!
32
3.5 Bupivacain zeigt einen stereoselektiven Effekt auf den koronaren Vasotonus
Bupivacain ist ein weit verbreitetes Lokalanästhetikum vom Amid-Typ; im Gegensatz zu
Lidocain hält die anästhetische Wirkung jedoch länger an (ca. 6 statt 3 Stunden). Graf et al.
(1997, 2001 & 2002) gelang es, bei der Messung der AV-Überleitungszeit eine deutliche
stereoselektive Reduktion durch Bupivacain-Isomere im Bereich klinischer Konzentrationen
(5 bis 10 µM) nachzuweisen. Des weiteren wurden ein negativ inotroper Effekt und eine
Reduktion des Koronarflusses beobachtet. Iida et al. (2001) haben beschrieben, dass R-
Bupivacain und razemisches Bupivacain cerebrale piale Arteriolen von Hunden dilatierten, S-
Bupivacaine diese jedoch verengten. Eine mögliche stereoselektive Blockade von TREK- und
TASK-Kanälen könnte hierbei eine wichtige Rolle spielen.
In unseren Experimenten verwendeten wir einerseits Bupivacain-Razemat (n=6) und
andererseits die reinen R- und S-Isomere (jeweils n=5) von Bupivacain. Da das Razemat
einen deutlichen biphasischen Effekt mit einer Vasodilatation gefolgt von einer Konstriktion
zeigte, sollte dieser Effekt durch die verschiedenen Isomere weiter unterschieden werden
(Abb. 3-15 und 3-16).
Abb. 3-15: Bupivacain-Razemat (200 µM) generiert einen biphasischen Effekt.
Bupivacain 200 µM
10 -
60 -
KPD [mmHg]
Zeit [s] 1500 2000
33
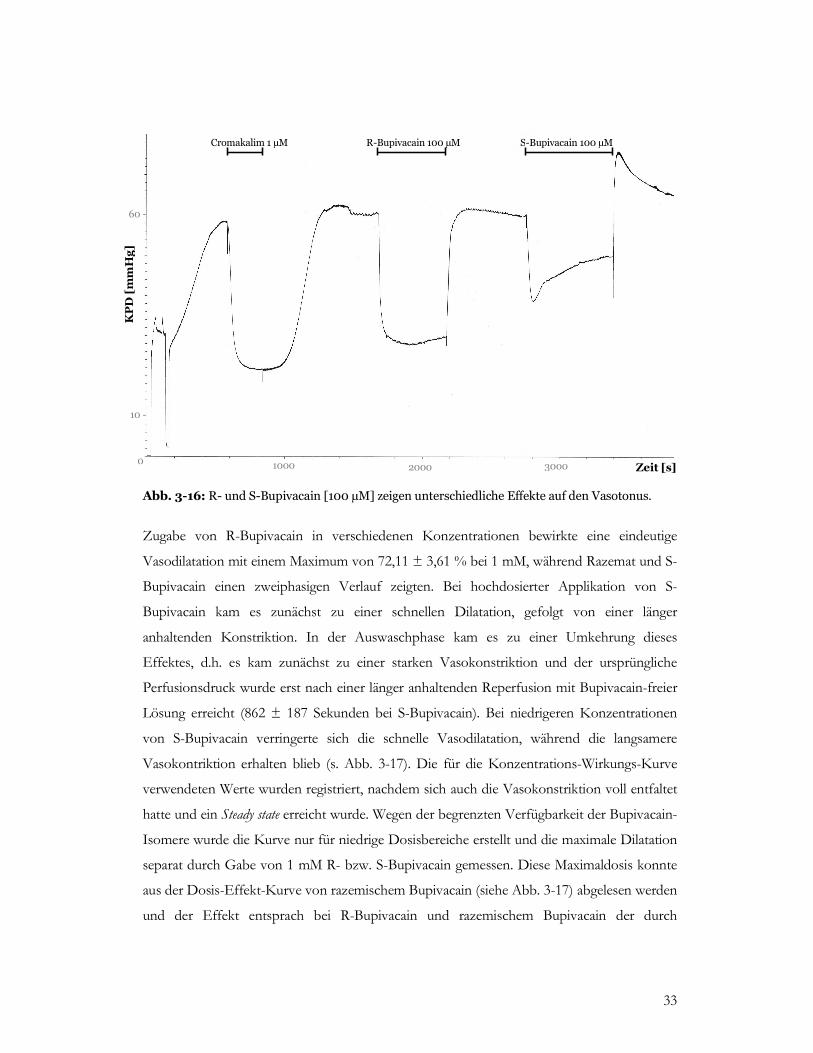
Abb. 3-16: R- und S-Bupivacain [100 µM] zeigen unterschiedliche Effekte auf den Vasotonus.
Zugabe von R-Bupivacain in verschiedenen Konzentrationen bewirkte eine eindeutige
Vasodilatation mit einem Maximum von 72,11 ± 3,61 % bei 1 mM, während Razemat und S-
Bupivacain einen zweiphasigen Verlauf zeigten. Bei hochdosierter Applikation von S-
Bupivacain kam es zunächst zu einer schnellen Dilatation, gefolgt von einer länger
anhaltenden Konstriktion. In der Auswaschphase kam es zu einer Umkehrung dieses
Effektes, d.h. es kam zunächst zu einer starken Vasokonstriktion und der ursprüngliche
Perfusionsdruck wurde erst nach einer länger anhaltenden Reperfusion mit Bupivacain-freier
Lösung erreicht (862 ± 187 Sekunden bei S-Bupivacain). Bei niedrigeren Konzentrationen
von S-Bupivacain verringerte sich die schnelle Vasodilatation, während die langsamere
Vasokontriktion erhalten blieb (s. Abb. 3-17). Die für die Konzentrations-Wirkungs-Kurve
verwendeten Werte wurden registriert, nachdem sich auch die Vasokonstriktion voll entfaltet
hatte und ein Steady state erreicht wurde. Wegen der begrenzten Verfügbarkeit der Bupivacain-
Isomere wurde die Kurve nur für niedrige Dosisbereiche erstellt und die maximale Dilatation
separat durch Gabe von 1 mM R- bzw. S-Bupivacain gemessen. Diese Maximaldosis konnte
aus der Dosis-Effekt-Kurve von razemischem Bupivacain (siehe Abb. 3-17) abgelesen werden
und der Effekt entsprach bei R-Bupivacain und razemischem Bupivacain der durch